

Abstract
The human gene catalogue is essentially complete, but we lack an equivalently vetted inventory of bona fide human enhancers. Hundreds-of-thousands of candidate enhancers have been nominated by biochemical annotations; however, only a handful of these are validated and confidently linked to their target gene(s). Here we review emerging technologies for discovering, characterizing and validating human enhancers at scale. We furthermore propose a new framework for operationally defining enhancers that accommodates the heterogeneous and complementary results that are emerging from reporter assays, biochemical measurements, and CRISPR screens.
In this Review, Gasperini, Tome and Shendure discuss evolving definitions of transcriptional enhancers, as well as diverse, modern experimental tools to identify them. They describe how these diverse mindsets and methods provide differing but complementary insights into enhancers, each with notable strengths and caveats. They discuss how such views and approaches might be combined in a comprehensive catalogue of functional enhancers.
Introduction
The human genome is currently believed to harbour hundreds-of-thousands to millions of enhancers — stretches of DNA that bind transcription factors [G] (TFs) and enhance the expression of genes encoded in cis. Collectively, enhancers are thought to play a principal role in orchestrating the fantastically complex program of gene expression that underlies human development and homeostasis. Although most causal genetic variants for Mendelian disorders fall in protein-coding regions, the heritable component of common disease risk distributes largely to non-coding regions, and appears to be particularly enriched in enhancers that are specific to disease-relevant cell types. This observation has heightened interest in both annotating and understanding human enhancers. However, despite their clear importance to both basic and disease biology, there is a tremendous amount that we still do not understand about the repertoire of human enhancers, including where they reside, how they work, and what genes they mediate their effects through.
This is not from a lack of effort. Rather, our understanding of the core characteristics of enhancers, based largely on a few paradigmatic examples, is being challenged by studies that suggest a more heterogeneous landscape. New data types, e.g. based on massively parallel reporter assays (MPRAs) or genome editing, are further complicating the picture, particularly as biochemical annotations and functional data are not always in agreement. As a consequence, the field lacks a clear framework for identifying enhancers, with different subfields (e.g. biochemistry, genomics, etc.) using different definitions and criteria, even though we are all ostensibly studying the same underlying biological phenomenon. These challenges are critical to resolve, and also represent an excellent opportunity to gain further insight into the nature of enhancers, as well as the landscape and heterogeneity of gene regulatory mechanisms across the human genome.
Here we present a survey of emerging technologies for discovering, characterizing, and validating enhancers at scale. We begin with a history of the concept of an enhancer and its evolving operational definition. We then review contemporary and emerging technologies for characterizing enhancers at scale. Next, we propose a set of evidentiary standards for considering a candidate enhancer as strongly, moderately, or weakly supported. Finally, we look forward and highlight the key challenges in the field.
A brief history of the concept of an enhancer
The term ‘enhancer’ first appeared in the context of molecular biology in 1981 (Box 1). By this point in time, gene expression was already thought to be regulated by proteins1 that bound DNA2. But why do these proteins bind to specific locations, and how does their binding control gene expression? In eukaryotic systems, in addition to primary sequence itself, chromatin accessibility was suspected to have a role3,4, and distal, cell-type-specific regions of open chromatin [G] had already been identified far from genes’ promoters5. However, these distal sites had not yet been shown to affect gene expression.
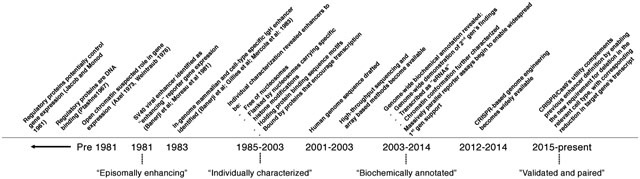
Box 1. A history of operational definitions of enhancers. [Contains a timeline figure].
Operational definitions of an enhancer, i.e. the practical criteria by which enhancers are distinguished from other sequences, have varied over time. This box summarizes the emergence of different operational definitions of enhancers (see the timeline figure).
‘Episomally enhancing’
The first reported enhancer sequence was described as a non-coding sequence that could enhance the expression of a cis encoded reporter gene (Figure 1b). The enhancer demonstrated activity from a number of locations on the same plasmid, both upstream and downstream of the promoter and in either orientation6,7.
‘Individually characterized’
New enhancers were discovered and experimentally characterized on a one-by-one basis. Shared features of such enhancers included that they were free of nucleosomes; flanked by nucleosomes with transcriptional-activity-associated histone modifications; contained sequence motifs for transcription factors (TFs), and were bound by these TFs; and likely to be accessing target promoters by looping in 3D space. Highly expressed genes located within the vicinity of the enhancer and exhibiting similar cell-type specificity were inferred to represent the target gene236,237.
‘Biochemically annotated’
Biochemical features associated with enhancers were measured genome-wide in selected cell types and tissues. These were used to annotate and define cell-type-specific enhancers on a genome-wide basis, generally without demonstration of enhancing activity. Enhancers were additionally found to be transcribed (enhancer RNAs (eRNAs)), and enriched for 3D proximity to putative target promoters. Massively parallel reporter assays (MPRAs) began to enable the scalable generation of supporting functional data similar to the original enhancer-defining work of Banerji et al.7 (1981).
‘Validated and target-linked’ (proposed)
With the emergence of CRISPR–Cas9 genome engineering, we propose that to reach the highest level of support as an enhancer, distally located elements should meet three criteria: first, deletion from its native genomic context results in altered expression of a potential target gene; second, evidence for a cis acting mechanism; and last, one line of orthogonal evidence that the underlying sequence is an enhancer (either in the form of a reporter assay or biochemical annotation) (Figure 4).
In 1981, these concepts culminated in the first demonstration of a non-coding DNA sequence that ‘enhanced’ the expression of a gene encoded in cis, in a manner that was distinct from transcriptional activation mediated by promoters6,7. Specifically, on an episomal reporter vector [G] , a non-coding region of the Simian Virus 40 (SV40) genome increased expression at a distance remote from the reporter gene’s promoter and independent of the enhancing region’s orientation. From this experiment came the original definition of an enhancer that is still widely quoted today: “the transcriptional enhancer element could act in either orientation at many positions… [even] downstream from the transcription initiation site”7. A few years later, using a similar in vitro method, the first endogenous, mammalian, cell-type-specific enhancer was identified within the IgH locus8-10. A few years after that, endogenous regulatory sequences were shown to have in vivo activity, enhancing the expression of the cancer-inducing large T antigen in a cell-type-specific manner11.
The genomic characteristics of typical enhancers were further fleshed out over the ensuing decade, and a few general principles emerged. First, enhancers are free of nucleosomes, as measured by hypersensitivity to DNase I12,13, but are flanked by nucleosomes with specific, transcription-associated histone modifications14-16. Second, enhancers contain clusters of TF binding motifs17, and binding of TFs to these motifs underlies their enhancing activity18,19. Third, enhancers are likely to loop in 3D space into proximity with their target promoters20,21.
Like many concepts in biology, these generalizations were based on a handful of examples that were studied in depth using tools available at the time. One paradigmatic example, then and now, is the mammalian β-globin locus control region (LCR), a non-coding region that controls the developmental timing of expression of a cluster of globin genes. First discovered as a distally located deleted region in patients with β-thalassemia who lacked mutations impacting the coding region of the β-globin gene22-24, the β-globin LCR was hypersensitive to DNase I12, contained motifs corresponding to relevant TFs (e.g. GATA1)25,26, was bound by these TFs18, and was proposed to loop in 3D space to regulate globin genes27. Of note, the pattern of evolutionary conservation of the β-globin LCR, e.g. in mouse28, rabbit29, goat30, and chicken31, critically supported its functional dissection.
Notwithstanding such exemplars, relatively few enhancers had been identified by the late 1990s, orders of magnitude fewer than the number of genes known at the time. Unlike genes, enhancers could not be identified by expressed sequence tag [G] (EST) sequencing, and moreover lacked a defined grammar that supported their assignment as actually functional (e.g. an open reading frame [G] ). Indeed, the dearth of discussion of distal regulatory elements in the initial report of the human genome illustrates the difficulty of this task at the time32,33.
One encouraging point was that nearly all of the enhancers that had been deeply characterized at the time were evolutionarily conserved34. Taking a ‘conservation first’ approach, Loots and colleagues35 identified non-coding regions regulating several interleukin genes by comparing 1 Mb of mouse–human orthologous sequence35. The global application of this strategy was one of the key motivations for the sequencing of the mouse genome34,36,37.
However, immediately upon comparing the human and mouse genomes, the field faced the opposite problem, as the number of conserved non-coding regions, each a potential regulatory element [G] , now vastly exceeded the number of genes38-41. How many of these conserved non-coding regions represented bona fide enhancers, as opposed to other kinds of functional elements? A further challenge was that the Human Genome Project had revealed the number of human genes to be about the same as that of the nematode, Caenorhabditis elegans. If the greater complexity of mammalian development was instead encoded by enhancers, a belief that took hold at the time and persists today, they too required cataloguing and characterization. In what cell types and at what developmental time points is each enhancer active? Which genes does each enhancer regulate?
To these and other ends, in the wake of the Human Genome Project, the field immediately shifted its attention to the genome-scale characterization of the epigenome, e.g. through the Encyclopedia of DNA Elements (ENCODE) Consortium and similar projects. To briefly summarize an immense amount of work, genome-wide chromatin accessibility was measured by DNase I hypersensitivity42-45, DNA methylation by bisulfite sequencing46,47,48, and genome-wide histone modifications49-51 and TF binding52-54 by chromatin immunoprecipitation. Each such biochemical assay was coupled to a genome-wide readout, initially microarrays and subsequently massively parallel DNA sequencing55. In a surprising finding, whole-transcriptome RNA sequencing revealed transcription of active enhancers (‘eRNAs’)56-59. Altogether, over the past 15 years, such biochemical methods have been applied to characterize the non-coding genome in hundreds of mammalian cell types and tissues58,60-63. This has resulted in the cataloguing of over one million candidate cis-regulatory elements with enhancer-like signatures; these collectively span ~16% of the human genome64. It is now widely recognized that a major component of the heritability of nearly all common diseases partitions to these regions, and in particular to regions that have enhancer-like signatures in disease-relevant cell types65.
What defines an enhancer?
In the current parlance of the field, the term ‘enhancer’ is often used interchangeably to refer to: first, DNA sequence elements that meet the original Banerji et al.7 (1981) definition, i.e. enhancing transcription in a reporter assay; second, DNA sequence elements that bear biochemical marks associated with enhancer activity; or third, endogenous, distally located DNA sequence elements that serve to enhance transcription of a cis-located gene, in vivo and in their native genomic context. But these definitions are not equivalent. There may be sequences that activate transcription in the context of a reporter assay but do not meaningfully do so in vivo. There may be sequences that bear enhancer-associated biochemical marks but do not actually function as enhancers in vivo. Finally, there may be in vivo enhancers that are non-canonically marked, or that have contextual dependencies that are not maintained in a reporter assay.
Which definition should we use? In our view, the first two are operational definitions, whereas the last is a biological definition. An operational definition is not what an enhancer is, but rather follows from the practical framework that we use to distinguish biological enhancers from other sequences. Much like blind men inspecting an elephant66, operational definitions are a means to characterize a phenomenon, but fall short of the phenomenon itself. Here, we use the term ‘enhancer’ to refer to the in vivo phenomenon, i.e. short regions of DNA that in their endogenous genomic, cellular and organismal context, bind proteins that increase the likelihood of transcription of one or more distally located genes through a cis-regulatory mechanism. We acknowledge that our viewpoint is not universally shared — subsets of the community may prefer to define enhancers as sequences exhibiting enhancing activity in an in vitro reporter assay, and indeed we ourselves have slipped into this definition in the past67. However, particularly as new assays proliferate, it is the elephant (i.e. the biological enhancer) that must remain the primary focus, rather than the angle at which we first bumped into it.
Indeed, the operational definitions used for enhancers have been anything but static (Box 1). The original operational definition, from Banerji et al. (1981), was relatively simple: sequences that increase expression of a reporter gene, when sequence and reporter gene are co-located on an episome7. However, this was quickly followed by efforts to characterize the biochemical features of such sequences in their native genomic and cellular context8-10. Our present understanding is that enhancers are bound by cell-type-specific TFs, are associated with regions of open chromatin, and are flanked by histones carrying H3K27ac and/or H3K4me1 modifications. They interact with their cognate promoters in 3D space, and can be latent, primed, or active68,69. Although the endogenous distributions of enhancer sizes and enhancer–gene distances remain an important topic of exploration, a typical enhancer is probably hundreds of base-pairs in length70,71, and acts over a few to tens of kilobases72. Although we have many clues, the mechanistic details of how enhancers activate expression of their target genes have yet to be fully worked out (Figure 1a).
Figure 1. Approaches for identifying, validating and characterizing enhancers.

a ∣ Biochemical annotations of candidate enhancers. a schematic depiction of an enhancer and target gene marked with the biochemical annotations used to nominate candidate enhancers and other features of non-coding DNA. Although the enhancer has been depicted in 3D proximity to its target promoter, we note that the mechanistic importance of such enhancer–promoter proximity is far from settled. We refer the reader to the section “Emerging approaches for biochemical annotation: 3D conformation mapping” for a discussion of open questions of enhancer–promoter communication and the importance of chromatin looping. b ∣ Episomal reporter assay: a candidate enhancer and reporter gene located in cis on an episomal vector. The candidate enhancer may increase expression of the reporter gene by recruiting transcriptional machinery. The degree of enhancer-mediated activation is measured by the abundance of reporter transcripts or the quantity of the reporter-encoded protein. c ∣ Massively parallel reporter assays (MPRAs): many candidate enhancers can be interrogated simultaneously in a reporter assay if a barcode is encoded in the reporter transcript. The relative abundance of barcodes can be used to estimate the relative activities of the candidate enhancers to which they are linked. We show here just one of many formats of MPRAs that have been developed. 3C, chromosome conformation capture; 4C, chromosome conformation capture-on-chip; ATAC-seq, assay for transposase-accessible chromatin using sequencing; ChIP-seq, chromatin immunoprecipitation followed-by sequencing; DNase-seq, DNaseI hypersensitivity sequencing; MNase-seq, micrococcal nuclease digestion combined with sequencing; PRO-seq, precision run-on sequencing; POL, RNA polymerase; TF, transcription factor. Part a is adapted from REF69.
Particularly in recent years, operational definitions of enhancers based on biochemical annotations have been solidified by the ENCODE Consortium, as these are available throughout the genome and across many cell types. For example, enhancer-associated biochemical features have been used for regulatory variant effect prediction in the context of both rare73 and common74,75 disease. In human genomics, characterizations of the genome-wide sizes and distributions of enhancers rely heavily on biochemical datasets51,76. Many investigators are careful to qualify catalogues based on such annotations as ‘predicted’ or ‘candidate’ enhancers, but the qualification is often dropped, and such sequences simply referred to as enhancers.
However, much like in vitro reporter assays, a definition based purely on biochemical annotations has clear limitations. First, biochemical annotations are based on observations made in a sequence’s native genomic context but usually obtained on highly derived cell lines or tissues which represent mixtures of many cell types. Second, although these measurements may correlate with function, they fall short of demonstrating regulation of the expression of a cis encoded gene. In fact, it remains entirely possible that many biochemically identified enhancers may not be enhancing transcription of anything77. Third, biochemical annotations fail to specify which gene(s) a putative enhancer regulates, let alone the degree of activation conferred78,79. Fourth, many enhancer-associated biochemical features may have nothing to do with the enhancers’ mechanism of action. For example, the MLL3/4 complex has been shown to serve as an essential coactivator at some enhancers completely independent of its catalytic activity as a H3K4me1 writer80,81. Fifth, the coarseness of many biochemical features (e.g. broad peaks) fails to resolve which specific subsequence and nucleotides underlies any enhancing function. Finally, such annotations are often used in a ‘one size fits all’ manner, potentially dis-allowing bona fide enhancers that are non-canonically marked.
We do not mean to say that operational definitions of enhancers, whether of a reporter assay or based on biochemical features, have been anything less than tremendously useful. However, we should be continuously evolving towards a framework for discovering, characterizing and validating enhancers that is as close as possible to the biological phenomenon itself. To this point, new methods have recently emerged that overcome many of the key limitations of earlier technologies. These include single-cell (‘sc’) methods to identify cell-type-specific open chromatin in complex tissues82,83; higher resolution chromosome conformation capture [G] methods to more finely map enhancer–promoter contacts84; MPRAs to dissect or trap enhancer activity on a reporter vector at scale85; and high-throughput CRISPR screens to directly perturb enhancers in their native genomic context and link them to their target genes86.
The rapid maturation of these technologies should force us to reexamine how we operationally define enhancers. At the same time, given the heterogeneity of both biochemical and functional methods that can now be applied at scale, it is important to acknowledge that this is going to be a complicated task.
What features identify an enhancer?
Enhancers are only one class of non-coding DNA regulatory element, although they are widely presumed to be the most numerically prevalent (Box 2). For this Review, we focus on enhancers, and mammalian enhancers in particular, although many of the assays and concepts described are potentially applicable to other classes of non-coding DNA regulatory elements.
Box 2. Other types of regulatory elements.
Enhancers are only one class of non-coding DNA regulatory element. This is a brief list of other major classes, perhaps unified in that they generally correspond to open chromatin in cell types in which they are active. These are not covered in detail in this Review, but many of the assays described in this Review could also be applied to these other classes of regulatory elements. Furthermore, because all of these elements share many biochemical features and/or functional characteristics, the lines between them can be blurry59,238,239.
Promoter
An element that initiates transcription of a gene by RNA polymerase, by definition located at the 5ʹ end of the gene and encompassing its transcription start site (TSS). Composed of transcription factor (TF) binding sites that generally act independently of orientation, and core promoter elements (e.g. the TATA box) that tend to be oriented relative to the TSS240. Most promoters contain only a few discernable core promoter elements 241.
Silencer
An element similar to an enhancer but that acts to reduce expression of a target gene. It tends to bind repressive TFs242.
Insulator
A boundary element that restricts the ability of positive (enhancer) or negative (silencer) regulatory elements to modulate the expression of genes located on the other side of the boundary. Often bound by CTCF243.
Cis regulatory elements
Elements that regulate a target gene by a mechanism that depends on their residing on the same chromosome or episome. Regulatory elements located on the same chromosome and within 1 Mb of their target gene244 are often assumed to act through a cis regulatory mechanism.
Trans regulatory elements
Elements whose regulation of a target gene is mediated by a trans acting factor. Regulatory elements located over 1 Mb from their target gene on the same chromosome, or that are located on a different chromosome, are often assumed to act through a trans regulatory mechanism.
Enhancers are ‘punctate’ relative to broader chromatin domains (e.g. chromosomes, topologically associating domains [G] (TADs) and sub-compartments of TADs87), but their in vivo functionality is dependent on both the chromatin context in which they reside88 and on the trans milieu (e.g. cell-type-specific TFs)89. How do enhancers enhance expression of their target gene(s)? The classic model is that enhancers recruit cell- and condition-specific TFs and then loop in 3D space to interact with their target promoter90. The recruited TFs directly or indirectly (e.g. via a co-activator) facilitate chromatin remodelling and recruitment of the basal transcriptional machinery at the promoter (Figure 1a), thereby enhancing transcription91. However, it should be emphasized that this is not an inexorable chain of events. For example, stimulus-responsive enhancers may exhibit open chromatin and 3D interactions with their promoters before activation92,93. The production of a functional mRNA is a complex process, and which steps are rate limiting varies by gene and context94. Mammalian promoters are typically suboptimal in one or several ways95. Thus, from a mechanistic perspective, enhancers might tune transcription levels by affecting any number of steps. For example, some enhancers were recently shown to regulate the release of promoter-proximal paused RNA polymerase II96, and others to act through splicing-dependent mechanisms97. Further heterogeneity can be introduced by the same enhancers acting via different co-regulators at different times98.
Regardless of any such mechanistic heterogeneity, a common property is that the activity of individual enhancers is generally cell-type-specific or even condition-specific99, and this specificity is a function of the expression levels of the TFs that are able to bind to it89. But even this generality is complicated by the fact that the capacity of an expressed TF to interact with an enhancer may depend on the chromatin state of the region in which the enhancer resides100, which is in turn a function not only of a cell’s present state but also its developmental history. It may also depend on the nature of the TF, e.g. whether it is a pioneer factor [G] 101. Finally, there are multiple models for how enhancers interact with their target promoter(s), including tracking, linking/chaining, short- or long-range looping, transcription factory and hub/condensate models (reviewed in 102), more than one of which may be correct.
Well-established enhancers bear biochemical marks that are now routinely used to classify other sequences as enhancers. These include: sequence-level features (TF binding site motifs and conservation); 1D biochemical annotations (accessible chromatin; H3K27Ac and H3K4me1 modifications on flanking histones for active enhancers; H3K4me1 and HK27me3 for poised enhancers; closed chromatin that has been pre-marked by H3Kme1 for primed enhancers103); direct binding of TFs or secondary binding of cofactors (such as p300); and 3D biochemical annotations (nuclear spatial proximity to promoters as measured by 3C, 4C, 5C, or Hi-C) (reviewed in 69). Through projects such as ENCODE, these annotations underlie the classification of over one million candidate regulatory elements in the human genome as potential enhancers in one or more cell types60,64.
Yet, none of these features serve as perfect rules for identifying endogenous enhancers, as counterexamples can be found for each one. Not all distal conserved elements are detectably enhancers39 and far more of the gene-distal non-coding genome is annotated as a regulatory element than is conserved60,104. TF sequence motifs alone are poorly predictive, as only a small fraction of potential TF binding sites in the genome are typically bound in a cell type where the TF is expressed68. Although enriched, histone modification and cofactor binding is not completely predictive of enhancer activity54,58,72,77,105. Furthermore, to the extent that functional activity has been measured at scale, e.g. via MPRAs, its correlation with the annotations typically used to call enhancers is modest at best106-108. Many enhancers are spatially proximate to their target promoter in 3D109,110, but exceptions have been described111-114. Genes can be affected by a single enhancer or multiple enhancers acting in concert115; conversely, individual enhancers can regulate multiple genes72. Some enhancers reside in clusters of a handful116 to even hundreds (‘super enhancers’76,117), whereas many are solo. At least a few enhancers reside at great distances from their target gene (e.g. the ZRS enhancer located 1 Mb from the Shh gene118, and a MYC enhancer located 1.7 Mb downstream119), although most are much more proximal to their target promoters72. Enhancers regulating housekeeping genes may act via a distinct sets of TFs and cofactors than enhancers regulating developmentally specific genes79,120. Enhancers may have complex relationships with promoters, including feedback loops or competition with neighbouring genes121,122.
In light of this heterogeneity, a ‘one-size-fits-all’ set of annotations to catalogue enhancers seems problematic. Furthermore, as per their biological definition, enhancers are ultimately defined not by biochemical marks, but by their endogenous functional activity: increasing the likelihood of transcription of one or more distally located genes through a cis regulatory mechanism. It is also worth emphasizing that ruling out that a sequence is a biological enhancer may be far more difficult than proving it is. This is simply because it would be extremely impractical to test every possible developmental time-point, cell type and condition.
As touched on above, technologies for functionally characterizing non-coding regulatory elements at scale are rapidly evolving. This creates an opportunity to rethink our operational definition of enhancers. In the next several sections, we review current and emerging technologies for the scalable characterization of enhancers, and consider the evidence that each provides (Table 1).
Table 1.
Pros and cons of various strategies for identifying, validating and/or characterizing enhancers
| Type | Technologies | Single cell? | Pro | Con |
|---|---|---|---|---|
| Conservation | PhyloP 224; PhastCons 225 | Not applicable | Computable genome-wide; support for critical function | Not cell-type specific; not a measurement of enhancer activity; no target gene identified |
| Sequence motif | Databases: JASPAR 226; HOCOMOCO 227 | Not applicable | Computable genome-wide; informative as to potentially bound proteins | Limited cell-type specificity; not a measurement of enhancer activity; no target gene identified |
| Open chromatin | DNase-seq 43, MNase-seq 228, ATAC-seq 229 | Yes (e.g. sci-ATAC-seq 82) | High-throughput biochemical annotation; associated with enhancer activity; cell-type specific | Not a measurement of enhancer activity; no target gene identified; unknown specificity |
| Transcription | RNA-seq, PRO-seq 230, GRO-cap 59, CoPro 231 | Yes (e.g. scRNA-seq, although usually only mRNAs) | High-throughput biochemical ‘eRNA’ annotation; implies active RNA polymerase near enhancer | Transcription does not necessarily guarantee enhancer activity; no target gene identified |
| Histone marks | Enhancer-associated histone modifications on ChIP-seq | Emerging (e.g. scChIC-seq 232) | High-throughput biochemical annotation; can support poised, active, or silenced enhancers; cell-type specific | Not a measurement of enhancer activity; no target gene identified; unknown specificity |
| Protein Binding | Transcription Factor ChIP-seq, CUT&RUN 233 | Emerging (e.g. uliCut&Run 170) | High-throughput biochemical annotation; cell-type specific | Not a measurement of enhancer activity; no target gene identified; unknown specificity |
| eQTL | Many datasets available (e.g. GTEx Consortium 136) | Emerging (e.g. sc-eQTLGen Consortium 138) | In-genome; direct measurement from human tissues; can test all variants by all transcripts | Limited to common genetic variants; variants fall in linkage disequilibrium blocks |
| 3D proximity | Chromatin conformation ‘C’s (e.g. Hi-C 141, microscopy | Yes (e.g. microscopy, sci-Hi-C 163) | High-throughput biochemical annotation; cell-type specific; informs enhancer–gene links | Not a measurement of enhancer activity; unknown specificity |
| 3D proximity + live imaging | Microscopy 172 | Yes, microscopy is inherently single-cell | Live cells, dynamic imaging of 3D proximity and transcriptional bursting across time | Limited to a small number of loci at once |
| 3D proximity + biochemical annotation | ChIA-PET 147; HiChIP 148, DNase-HiC 150, PLAC-seq 149 | None yet | High-throughput biochemical annotation; cell-type specific; informs enhancer–gene links; more cost-effective than Hi-C | Not a measurement of enhancer activity; unknown specificity |
| Computational prediction | Example: ChromHMM 234; Segway 235 | Yes (e.g. Cicero 134) | Computable genome-wide; potentially cell-type specific, can nominate enhancer–gene links | Requires experimental functional validation |
| Reporter plasmid activity | Luciferase, MPRAs 67,173,184, lentiMPRAs 190 | None yet | High throughput; relatively straightforward to implement; provides functional support | Episomal; removed from genomic context; no target gene identified; unknown specificity |
| Single-gene CRISPR screens | ‘Indel’ scans 195, long-deletion scans 203,204, CRISPRi scans 105,208 | None yet | High throughput; in native genomic context; provides functional support; informs enhancer–gene links | Only tests candidate enhancers against one gene at a time; unknown sensitivity |
| Whole-transcriptome CRISPR screens | Mosaic-seq216; multiplexed scRNA-seq 72 | Yes | High throughput; in native genomic context; provides functional support; informs enhancer–gene links; many genes at a time | Currently only implemented using epigenetic perturbation; unknown sensitivity |
| In vivo model organism: transgenic reporter | Episomal or transgenic delivery 219,221 | None yet | In vivo test across many developmental contexts | Low throughput; does not test enhancer in native genomic context |
| In vivo model organism: sequence deletion | Direct genomic sequence deletion 115 | None yet | In vivo test across many developmental contexts; potential detection of organismal phenotypes | Low throughput; not all enhancers are conserved between mouse and humans |
ATAC-seq, assay for transposase-accessible chromatin using sequencing; ChIA-PET, chromatin interaction analysis with paired-end tag sequencing; scChIC-seq, single-cell chromatin immunocleavage sequencing; ChIP-seq; chromatin immunoprecipitation followed by sequencing; CoPro, coordinated precision run-on and sequencing; ChromHMM, a chromatin state annotator based on hidden Markov models; CRISPRi, CRISPR-based transcriptional interference; CUT&RUN, cleavage under targets and release using nuclease; DNase-seq, DNaseI hypersensitivity sequencing; eQTL, expression quantitative trait locus; eQTLGen, eQTL Genetics Consortium; eRNA, enhancer RNA; GTEx, Genotype–Tissue Expression Program; GRO-cap, cap-enriched global nuclear run-on sequencing; HOCOMOCO, Homo sapiens Comprehensive Model Collection; indel, insertion or deletion; lentiMPRAs, lentiviral MPRAs; MNase-seq, micrococcal nuclease digestion combined with sequencing; MPRAs, massively parallel reporter assays; PLAC-seq, proximity ligation-assisted ChIP-seq; PRO-seq, precision run-on sequencing; RNA-seq, RNA sequencing; sc, single-cell; sci, single-cell combinatorial indexing; uli, ultra-low input.
Methods for scalable enhancer characterization
Current technologies and their limitations
DNA sequence.
Primary sequence is modestly informative for distinguishing where enhancers lie. Evolutionary conservation can support the functional candidacy of a region39, but not all enhancers are conserved123-125,126. Surveying a genome or candidate regulatory elements for TF binding motifs can add further support127, but not all motifs are known or perfectly described128. Furthermore, the presence of a motif for an expressed TF does not mean that it is bound, and even if it is, not all binding is functional68. Consequent to these limitations, automatic sequence-based enhancer annotation is helpful and worthwhile129, but performs modestly for predicting enhancers and the contexts in which they are active. A further limitation is that primary sequence cannot identify the gene(s) an enhancer regulates, beyond predictions based purely on linear proximity.
Biochemical annotations.
Biochemical annotations that correlate with enhancer activity and are measurable on a genome-wide scale include assays for histone modifications or TF binding (e.g. ChIP-seq, CUT&RUN), open chromatin (e.g. DNase-seq, MNase-seq, ATAC-seq), DNA methylation (e.g. bisulfite sequencing), and the initiation and abundance of transcription (e.g. PRO-seq or RNA sequencing (RNA-seq)) (Figure 1a). Through the ENCODE Consortium and related efforts, such data have been collected in diverse cell types and tissues, to inform the cataloguing of cell-type-specific enhancers. Although unquestionably useful, it remains unknown what proportion of candidate enhancers identified solely by biochemical marks are technical false positives130-132 or a product of having enhancer-like biochemical features but no meaningful impact on the expression of cis encoded genes133. Furthermore, ‘1D’ biochemical annotations fail to inform us which gene(s) an enhancer regulates (biochemical annotations based on 3D chromosome conformation capture techniques are discussed further below). To some degree this can be overcome by correlative approaches (e.g. correlating open chromatin status between promoters and enhancers across large numbers of cell types) but such links remain inferential45,134,135.
eQTL mapping.
Expression quantitative trait locus (eQTL) studies in human populations can be used to validate and characterize distally located candidate regulatory elements. In brief, genome-wide genotypes in human cohorts (measured by microarrays and imputation, or by genome sequencing) are tested for correlation with the expression of genes located in cis (measured by bulk RNA-seq of an accessible tissue from those same individuals). Variants that are significantly associated with gene expression differences after appropriate corrections are called as eQTLs. The eQTL framework is very powerful, and for variants residing within distally located candidate enhancers, can provide in vivo validation of those enhancers while also linking them to their target genes136. On one hand, given the diversity of epigenomic contexts traversed during development, eQTL studies may represent our only hope for comprehensively observing the consequences of human enhancer disruption (as all engineered mutations will be in models such as cell lines, organoids, or mice). On the other hand, the framework has clear limitations, including its reliance on naturally occurring human genetic variation (most enhancers do not harbour common variants that substantially perturb their activity), linkage disequilibrium [G] (multiple variants in a haplotype block may equivalently explain an association), and restriction to cell types and tissues that can be practically obtained from large numbers of individuals for expression profiling (e.g. peripheral blood mononuclear cells)137,138.
Emerging approaches for biochemical annotation
3D conformation mapping.
A long-hypothesized model of enhancers involves their looping in 3D space to access target promoters139,140. In recent years, successively more powerful chromosome conformation capture (‘3C’) methods have yielded high-resolution 3D conformational maps of the human genome in a few cell types (Figure 1a). With 3C methods, genomic DNA fragments are ligated to other, physically proximate genomic DNA fragments within the nucleus141,142. The resulting datasets have led to the identification of large-scale compartments of genome organization at various scales, including A/B compartments141, TADs143-146, and possibly enhancer–promoter loops109. 3C methods have also been paired with biochemical assays to enrich for potentially functional interactions, e.g. methods including ChIA-PET147, HiChIP148, PLAC-seq149, DNase Hi-C150 and others.
Does physical proximity strongly predict enhancer–gene links? Is it necessary and/or sufficient? In an elegant recent study relying on live imaging, sustained proximity of an enhancer to its target was indeed required for activation151. Furthermore, a strong signal for distal chromatin interactions in bulk genomic assays such as Hi-C is associated with tissue-specific, presumably enhancer-dependent, expression152. On the other hand, proximity is sometimes maintained even when the gene or enhancer is inactive112,153. Other studies have found enhancer mobility, rather than proximity per se, to be a key determinant of activation154. Finally, the temporary disruption of 3D loops on a genome-wide scale through cohesin depletion was found to have minimal lasting effect on gene expression155,156. Overall, the precise mechanistic relevance of 3D proximity to enhancer-mediated gene regulation remains unclear.
Single-cell molecular profiling.
Conventional or ‘bulk’ biochemical assays of chromatin return the mean profile of their input cells, which due to Simpson’s paradox [G] is potentially representative of none of the cells therein157,158. Until recently, the field has dealt with cellular heterogeneity by either ignoring it or, where possible, resorting to physical dissection or cell sorting61,159. However, methods for profiling chromatin state in single cells are advancing quickly and have the potential to overcome this challenge. For example, single-cell ATAC-seq has enabled the in vivo profiling of accessible chromatin at the scale of a whole organism82,83.160,161. Single cell MNase-seq, ChIP-seq, Cut&Run and Hi-C methods have also been developed162,163-166,167,168,169,170. As touched on above, microscopy — the original single cell method — has revealed cases in which enhancer–promoter proximity either is or is not required for gene activation151,171. A major advantage of microscopy relative to genomic assays is the ability to study dynamic gene regulation in live cells172. Although currently limited to studying one or a few loci at a time, methods for multiplexing at the interface of microscopy and genomics are rapidly advancing.
Overall, single-cell methods have the potential to replace conventional bulk 1D and 3D biochemical assays. From datasets such as these, links between enhancers and promoters can be potentially nominated by their correlation across large numbers of cells, rather than large numbers of samples134. Single-cell methods may also enable the identification of candidate enhancers that appear to be active in extremely specific developmental contexts, or heterogeneously active within a single cell type. However, like the biochemical annotations on which they are based, any such candidate links will still lack functional validation.
Technologies for measuring enhancer activity
Massively parallel reporter assays.
An MPRA tests the functional activity of thousands of candidate regulatory sequences in a single experiment. The typical setup of MPRAs is very similar to the original demonstration of the properties of the SV40 enhancer, i.e. position-independent activity within an episomal vector7 (Figure 1b). Although first developed to dissect all possible single nucleotide variants of a promoter in 2009173, MPRAs have mostly been used to study enhancers (reviewed in 85). Enhancer-focused MPRAs involve cloning a library of candidate enhancers into a reporter vector, wherein they have the opportunity to enhance the expression of a reporter gene via a minimal promoter (Figure 1c). Each reporter gene transcript includes a barcode that is associated with a particular enhancer (or is the enhancer itself, in the case of STARR-seq174). The relative abundance of each RNA barcode, normalized to its DNA-based representation, is used to quantify the activity of its cognate candidate enhancer175.
A clear strength of MPRAs is their ability to simultaneously test large numbers of sequences for regulatory activity via a relatively straightforward, widely accessible toolkit (i.e. oligonucleotide synthesis, molecular biology, cell culture, and sequencing)175. MPRAs have been applied to assess biochemically annotated candidate enhancers77,176-178, candidate enhancers harbouring variants that potentially mediate eQTLs179-182, and even scans of the entire human genome108,183. A major advantage of MPRAs is that sequences to be tested can simply be synthesized, enabling straightforward saturation mutagenesis [G] of enhancers67,184,185 as well as programming synthetic enhancers to inform modelling of their properties186,187. In contrast with MPRAs relying on re-synthesis of candidate sequences, genome-wide ‘shotgun MPRAs’108,174,183,188 nicely avoid a priori assumptions about which sequences to test.
However, at least as they are usually implemented, MPRAs remain limited by several factors, including the length constraints and cost of DNA synthesis or the immense complexity of shotgun libraries, the confounding effect of the reporter’s minimal promoter, and the use of episomes whose chromatin may have different properties than that of the genome189. Specific types of MPRAs can address these concerns at least in part, e.g. by integrating MPRA reporters into the genome88,190,191. The fact that MPRAs test each sequence of interest entirely out of context is on one hand a strength, as it isolates that sequence in order to study its properties independently of that context. But it is also a weakness in that properties observed out-of-context may be irrelevant when that native context is restored. The fact that most MPRAs only test for enhancer activity using a single promoter, or at best a handful79, could contribute to a high false negative rate. To put it another way, most MPRAs assume that enhancers act in a promoter-generic fashion, when that in fact may not be the case. Conventional MPRAs also fail to capture how each sequence affects and is affected by its genomic neighbourhood, nor which promoter(s) an ‘active’ enhancer endogenously affects. Practitioners of MPRAs, including ourselves, typically fail to confirm that each ‘positive’ element fully meets the original Banerji definition (i.e. active in both orientations and from many positions).
CRISPR screens of non-coding sequence.
An exciting recent development in this space has been the emergence of pooled CRISPR [G] -based enhancer screens for in-genome perturbation (Figure 2). These studies springboard off CRISPR-based genome-wide screens of genes192-194, but instead with the aim of characterizing massive numbers of enhancers in their native genomic context. In brief, such screens entail delivery of a library of enhancer-targeting guide RNAs (gRNAs) to a pool of cells, followed by a phenotypic assay that informs which of those gRNAs impact expression of a target gene or genes. To date, all such screens use Cas9-induced perturbations, including active Cas9 for sequence disruption195, or nuclease dead-Cas9 (dCas9) tethered to an epigenetic repressor105 or activator domain92. As these genetic or epigenetic perturbations of enhancers are phenotyped by methods that directly or indirectly measure gene expression, they have the potential to functionally link enhancers to their target gene(s) at scale, potentially filling a longstanding gap in the field.
Figure 2. CRISPR-based approaches for perturbing enhancers.

The CRISPR system has been repurposed with four main perturbation methods that can disrupt enhancer activity a ∣ Single-cut small sequence insertion or deletion (indel). An active CRISPR nuclease such as Cas9 is directed to make a single-cut that by inaccurate repair will usually create a small indel <10 bp. This indel can sometimes disrupt an enhancer’s function if, for example, it overlaps a key transcription factor (TF) binding site. b ∣ Dual-cut long sequence deletions. To guarantee that a perturbation disrupts the enhancer’s functional sequence, the entire enhancer can be deleted by directing two flanking cuts to either side. In some cells, due to inaccurate repair, deletions may occur between the two cuts. However, this is inefficient and will only be one of several possible repair outcomes, which must be accounted for in experimental design. c ∣ CRISPRi-based epigenetic repression. The nuclease domain of the CRISPR enzyme is rendered inactive (‘dead’, such as dCas9) but is tethered to a repressive domain (e.g. KRAB) which is known to disrupt enhancer activity and expression. d ∣ CRISPRa-based epigenetic activation. A dead-CRISPR enzyme is tethered to an activating domain (e.g. a fusion of VP64, p65, and rtTA) that can potentially induce activation of a target gene when targeted to a primed enhancer. POL, RNA polymerase.
Nuclease-active genome editing screens.
The initial CRISPR screens of regulatory elements delivered an individual gRNA per cell195. The gRNA–Cas9 nuclease complex directed double-stranded breaks (DSBs) at target sites, which after repair by error-prone non-homologous end-joining (NHEJ)196,197, resulted in 1–10 bp deletions or 1 bp insertions (‘indels’) in as many as 90% of cells192,194 (Figure 2a). These were ‘single gene’ screens, in that the experiments were designed to detect expression perturbations of a specific gene. The first such screen targeted gRNAs to effectively tile small indels across a known cluster of enhancers of BCL11A195. The authors flow-sorted the edited cells on the basis of the BCL11A-dependent switch to fetal haemoglobin, sequenced guides enriched in cells that had or hadn’t switched, and on the basis of those enrichments, successfully identified a primate-specific GATA1 motif critical for that enhancer’s function. A transcription-activator-like effector nuclease (TALEN)-mediated indel scan of the same enhancer found the same motif, albeit via a much lower throughput experiment198. Additional single-locus CRISPR screens of regulatory elements quickly followed at larger scales199, including experiments perturbing thousands of candidate enhancers per experiment133,200-202.
Non-coding CRISPR screens present different challenges than coding CRISPR screens. In a coding screen, the indels resulting from NHEJ at a single DSB are likely to result in a frameshift and the gene’s complete loss of function. However, the rules of disrupting enhancer function are more nebulous. Although small indels are probably capable of disrupting TF binding sites within an enhancer, they might only do so if directly overlapping the binding site itself. In this vein, the ability of single guide scans to fully ‘tile’ a region is limited both by the distribution of protospacer adjacent motif [G] (PAM) sites and the non-random distribution of NHEJ-mediated mutations. Furthermore, disruption of a single TF binding site might be insufficient to detectably disrupt the function of the enhancer. To address all of these technical challenges at once, other CRISPR screens of regulatory elements have sought to program larger deletions to increase effect sizes and facilitate more complete tiling of regions of interest203-205 (Figure 2b). Such ‘long deletion’ scans deliver pairs of gRNAs per cell that target closely located sites, which can result in clean deletion of the intervening sequence. However, a challenge is that the further apart the pair of cuts induced by the gRNAs, the less often full deletion occurs, e.g. ~20% for a 365 bp deletion204.
In sum, although powerful, CRISPR screens of non-coding regulatory elements are currently limited by effect size, efficiency or both. Additional challenges include that the variability of NHEJ-mediated repair outcomes plagues these screens with unprogrammed editing outcomes204,206, and that in non-haploid cells, each allele of the targeted locus can be heterogeneously edited within each cell, complicating the interpretation of results.
Nuclease-inactive epigenome editing screens.
Relying on epigenetic perturbations, rather than genetic ones, bypasses many of these limitations, e.g. allowing all alleles in a given cell to be more consistently perturbed. The dCas9–KRAB repressor domain (CRISPR interference or ‘CRISPRi’) was the first construct shown to synthetically silence a target enhancer by inducing ~1-2 kb of repressive marks in the vicinity of the gRNA target207 (Figure 2c). CRISPRi has subsequently been used in multiple single-gene screens of regulatory elements105,208,209. Activating domains (dCas9–VPR or dCas9–p300) have also been used to scan for poised enhancers in an approach termed CRISPR activation or ‘CRISPRa’92,208(Figure 2d). Additional dCas9-tethered domains have been shown to disrupt enhancer activity (e.g. histone demethylase LSD1210, histone deacetylase 3211, and DNA methylators MQ212 or DNMT3A213-215), and could potentially be adapted to large-scale screens.
However, although nuclease-inactive epigenome scans of regulatory elements have some clear technical advantages, the synthetic nature of the perturbation leaves something to be desired. Although the epigenetic changes somewhat recapitulate how enhancers are physiologically turned on or off, the synthetic domains (e.g. KRAB or VPR) used in a CRISPRi or CRISPRa system are probably not perfectly recapitulating the subtleties of enhancer regulation. This may lead to false positives (e.g. through spreading of KRAB repressive effects, or unnatural activation by VPR) or false negatives (e.g. an active enhancer that is not susceptible to CRISPRi-mediated inactivation). By contrast, wholesale deletions of candidate enhancers are unambiguously disruptive of a bounded region.
Whole-transcriptome screens.
A shared limitation of single-gene screens, whether by CRISPR, CRISPRi, or CRISPRa, is that the phenotyping is restricted to one or a few genes per experiment, e.g. by engineering a reporter to the target gene133,201,203,208, by labelling mRNA products with fluorescence in situ hybridization (FISH)209, or by focusing on drug-responsive202,204, antibody-detectable92, or proliferation-related105,200 genes. Each such phenotyping assay requires a specific technical setup, which sharply limits scalability and ease of adoption (Figure 3a).
Figure 3. CRISPR-based screens of enhancer–gene links.

In all such screens, guide RNA (gRNA)-based perturbations are designed to candidate enhancers and delivered to mammalian cells as a pool. a ∣ In most screens, cells are separated by expression of a single or few genes, and perturbations are tested for enrichment in high or low expression bins. b ∣ In ‘whole-transcriptome’ screens, single-cell RNA sequencing (RNA-seq) is used to evaluate the expression of any gene against each perturbation. c ∣ The future of such screens would benefit from higher standards (and better methods) to validate screen results (e.g. by deletion of individual elements), investigating why all such screens have had a low ‘hit rate’ thus far, and comparison of results with massively parallel reporter assay (MPRA) readouts of activity.
Towards genome-wide functional maps of enhancer–gene interactions, several groups have developed ‘whole-transcriptome’ screens of regulatory elements, which circumvent the need for gene-specific assays to be developed (Figure 3b). In brief, a library of enhancer-targeting gRNAs and some form of Cas9 is still introduced to cells, but the phenotyping is performed by single-cell RNA-seq (scRNA-seq) of both mRNA and gRNAs. The subsets of cells with versus without each gRNA are then tested for expression differences. The first such screen delivered one CRISPRi perturbation per cell, targeting 71 candidate enhancers across 7 genomic loci216. As scRNA-seq is costly and individual enhancers most likely only regulate one or a few genes in cis, we developed a related approach wherein ~28 gRNAs were introduced per cell, enabling 5,779 candidate enhancers to be evaluated in a single experiment72. However, even with extensive multiplexing, such experiments are still expensive. For such screens to become routine, greater multiplexing and/or further reductions in the cost of scRNA-seq are needed. Furthermore, such multiplex screens may be limited to epigenetic perturbation, particularly if large numbers of DSBs are toxic to cells.
Future prospects for CRISPR-based screens of non-coding sequence.
Within just a few years, CRISPR and CRISPRi screens of non-coding elements have delivered clear progress in terms of validating enhancers in their native context while also linking them to their target gene(s). However, technical improvements are needed and many questions remain (Figure 3c). For example, validating each screen-based ‘hit’, such as by deleting it outside of a screen, remains challenging204,206 but should probably be the standard expectation for strong claims about enhancer functionality (see “Defining and cataloguing enhancers” for further discussion of this point). Also, as the number of unambiguous ‘positive control’ enhancer–gene links remains small, the false negative rates for these scans by and large remain unknown.
In the vein of the latter concern over false negatives, one of the larger surprises of these studies is that relatively high proportions of biochemically or MPRA-supported candidate enhancers tested do not detectably influence expression of a cis encoded gene, in both CRISPR or CRISPRi screens and even when assaying the whole transcriptome (e.g. ~90% in REF72). How should this be interpreted? Potential explanations include: epigenetic perturbations of enhancers have a high false negative rate for technical reasons; scRNA-seq fails to detect subtle changes in gene expression; shadow enhancers [G] are buffering regulatory effects217; most screens to date are in terminally differentiated, stable cell lines whose lack of dynamics masks regulatory effects; and finally, analogous to early estimates of the number of genes, there are many fewer bona fide enhancers than biochemical and MPRA-based annotations would have us believe.
On the other side of the balance sheet, putative enhancers identified by CRISPR screens may fail to show activity in MPRAs. Are such instances false positives of CRISPR screens, or false negatives of MPRAs? Of note, most conventional MPRAs utilize a single promoter for the reporter, that may not be sensitive to all enhancers79. Also, some established mechanisms of enhancer–gene interaction, such as high physical mobility154 or weak interaction networks218, may not translate well to an MPRA context. Finally, MPRAs would fail to recapitulate complex gene–enhancer networks121,122. Considerable further work is necessary to differentiate between these and other potential explanations.
Technologies for in vivo validation
All of the aforementioned methods (biochemical annotations, MPRAs, CRISPR screens, etc.) are performed in vitro on cell lines, and are therefore only capable of accessing a limited number of biological contexts. As discussed above, eQTL studies are powerful for assessing in vivo effects, but are limited in critical ways. Consequently, the mouse model will remain a crucial asset for the validation and characterization of human enhancers for the foreseeable future.
First, transgenic reporter assays continue to provide valuable information regarding the tissue specificity of candidate enhancers219. The advantages of in vivo transgenic reporter assays include that a much broader range of developmentally and physiologically relevant contexts is ‘accessed’ than will ever be possible in in vitro systems; and that the sequences tested experience the natural developmental history of these contexts, rather than being transfected or transduced into already differentiated cells. The disadvantages of in vivo reporter assays are similar to MPRAs, including that elements are tested outside of their native genomic context, and the elements are not linked to their endogenous target gene(s).
Second, CRISPR technology has recently made it much more straightforward to delete genomic sequences in the mouse, enabling new insights into aspects such as enhancer redundancy115 and the consequences of disrupting TADs220. Although observing phenotypic changes consequent to in vivo manipulation of an endogenous regulatory sequence is a powerful paradigm, a first disadvantage is that if the goal is to understand human enhancers, then such studies may be restricted to elements conserved across mammals. Furthermore, the organismal phenotypic defects caused by deleting regulatory elements can be subtle and challenging to detect221. Finally, similar to in vivo transgenic reporter assays, in vivo deletion of candidate enhancers will be challenging to scale beyond a handful of sequences. Despite these limitations, we envision that both murine in vivo CRISPR deletion and transgenic reporter assays of selected elements will be critical for benchmarking the validity of any emerging catalogue of functionally characterized human enhancers.
Defining and cataloguing enhancers
Ever since the Human Genome Project, a natural goal for the field of genomics has been to generate a catalogue of human enhancers. Indeed, this is one of the primary goals of the ENCODE Consortium, which has generated the vast majority of the aforementioned biochemical annotations. However, in order for such a catalogue to be both comprehensive and maximally useful, it should not simply comprise a list of sequences believed to be enhancers on the basis of biochemical annotations from cell lines and tissues, except perhaps in its very initial form. Rather, our goal should be to apply emerging, scalable biochemical and functional assays in order to generate a considerably more useful catalogue.
Spurred by efforts including ENCODE-4 [G] , the Human Cell Atlas [G] , and others, developments that we anticipate within the next few years include the following. First, single-cell profiling, of chromatin accessibility, histone marks, transcription factor binding, and 3D conformation, will yield genome-wide catalogues of enhancer-associated biochemical marks for nearly all human cell types, from tissues obtained in vivo and from nearly all developmental stages. Second, MPRAs will be applied to comprehensively test candidate regulatory elements in representative cell types, quantifying the transcriptional activation potential of each element in a uniform context. Third, CRISPR screens will be applied to these same candidates in these same cell types, validating a subset of elements in their native genomic context while also revealing the targets of enhancer regulation. Finally, the number of elements tested in mouse models, either by transgenic reporters or CRISPR-mediated deletion, will continue to grow as well, albeit at a much slower rate.
On one hand, these developments are encouraging. They move us closer to a comprehensive catalogue of functionally supported human enhancers that is well-annotated in terms of the cell type(s) in which each element is active, the gene(s) that each element regulates, the degree of activation each element confers, etc. On the other hand, as compared with the current practice in which putative enhancers are operationally identified often solely based on biochemical marks, future enhancer catalogues are likely to be more nuanced. For example, it will probably more often than not be the case that specific elements are supported by some, but not all, forms of evidence. Which are we to interpret as ground truth?
As a starting point for dealing with this anticipated heterogeneity, we propose a relatively straightforward framework for how to describe the level of support for candidate enhancers. This is illustrated in Figure 4. At the very top are enhancers meeting a new ‘validated and target-linked’ operational definition, wherein a non-coding sequence has a demonstrated effect on a specific target gene’s expression in its endogenous context. To be more specific, validated and target-linked enhancers would meet the following evidentiary criteria:
Figure 4.

A tiered framework to describe the level of support for the enhancer candidacy of a non-coding sequence. We propose ‘validated and target-linked’ support as the degree of evidence that we should be aiming for in cataloguing non-coding sequences as bona fide human enhancers. If the evidence falls short of that, as it currently does for nearly all candidate enhancers, we propose strong, moderate, and weak tiers to describe candidate enhancers with less or conflicting evidence. The vast majority of candidate human enhancers are presently only weakly supported.
First, targeted deletion of the element in its native genomic context should result in altered expression of a distally located target gene. A deletion of the candidate enhancer in vivo or in a cell line should result in a measurable, reproducible change in the expression of one or more target genes. This would provide strong functional evidence that the sequence in question actually performs a regulatory function, while also linking it to at least one gene that it regulates. The deletion could be of only the element, or possibly in combination with other deletions or perturbations, in order to unmask any redundancy.
Second, there should be evidence for a cis acting mechanism: Perturbations of non-coding elements can have secondary effects, so there should be at least some rationale for concluding that an observed effect is mediated primarily by a cis regulatory mechanism. This could simply be linear proximity between the candidate enhancer and its target gene (e.g. <100 kilobases) or other experimental data (e.g. allelic imbalance or 3D proximity). Although they do not definitively demonstrate cis regulation, such lines of evidence at least support the possibility that observed effects are not secondary or trans.
Third, there should be at least one line of orthogonal evidence that the sequence is an enhancer. Because it is plausible that a deleted sequence could influence the mRNA abundance of a cis located gene through mechanisms other than serving as an enhancer, this criteria serves to add additional support. We propose that this evidence could come either in the form of the sequence episomally enhancing expression of a reporter gene on a plasmid (in accordance with the original 1981 definition7) or in the form of enhancer-associated biochemical marks (in accordance with the operational definition of the ENCODE Consortium and its successors). The flexibility, i.e. requiring one but not both of these lines of complementary support, allows for exceptions to the rule (e.g. bona fide enhancers that do not function in reporter assays, or bona fide enhancers that bear non-canonical biochemical marks). Of course, these assays are correlated, so in many cases there will be agreement across the board.
We emphasize that we propose these as inclusionary, rather than exclusionary, criteria for defining enhancers. As discussed above, it is very difficult to prove a sequence is not an enhancer. Additionally, we also note that our definition may not be easily adaptable to candidate enhancers that overlap promoters or protein-coding regions.
For candidate enhancers that fall short of ‘validated and target-linked’ status, we propose three additional tiers (Figure 4). ‘Strongly supported’ candidate enhancers should be supported by agreement of all three classes of experimental data, i.e. biochemical marks, episomal reporter activity, and CRISPRi/CRISPRa-based perturbation (but not necessarily deletion of the candidate enhancer, or else they would qualify as ‘validated and target-linked’; see “Nuclease-inactive epigenome editing screens” for related discussion). ‘Moderately supported’ enhancers would have two out of three of these, with the third either inconsistent, inconclusive or not performed. Finally, ‘weakly supported’ enhancers, a category that would presently apply to the vast majority of current human candidate enhancers, would be supported by only one of these three forms of evidence, with the other two either inconsistent, inconclusive or not performed.
We recognize that this scheme may be light in detail relative to the practical realities, e.g. standards will be needed for how to threshold the datasets underlying each form of support, specific biochemical marks will need to be defined as enhancer-associated, etc. However, particularly as the generation of such datasets accelerates, it seems critical that we have some framework in place for dealing with the inevitable heterogeneity in the confidence with which elements are named as enhancers, both in terms of the kinds of assays being used as well as in the results of those assays. In our view, the standard for declaring that an element is a biological enhancer should be better grounded in activity-based functional evidence, and the scheme in Figure 4 is consistent with that. Furthermore, particularly as the functional dissection of trait-associated genetic variants from genome-wide association studies (GWAS) is likely to be a major focus of the field for the coming decade, it seems key that future efforts should prioritize the linking of enhancers to their target genes. Such links will necessarily accompany all ‘validated and target-linked’ and ‘highly supported’ enhancers by the criteria above, as well as a subset of moderately and weakly supported enhancers.
Conclusions and future perspectives
Advances in scalable methods for the biochemical annotation and functional characterization of regulatory elements are paving the way to a comprehensive catalogue of human enhancers. In our view, such a catalogue can and should include knowledge of the cell-type-specificity of each element, at least some degree of functional support for its role as a bona fide enhancer, and knowledge of the element’s target gene(s) (Figure 5). Such a catalogue could prove to be a critical resource for furthering our understanding of the human genome and its role in disease.
Figure 5: The blind men and the elephant of human enhancer biology.

Much like blind men inspecting an elephant66, operational definitions of enhancers are merely a means to characterize the underlying biological phenomenon, but fall short of the phenomenon itself. As we work to develop a catalogue of bona fide biological enhancers, an updated operational definition that accommodates the heterogeneous and complementary results that are emerging from reporter assays, biochemical measurements, and CRISPR screens is likely to be necessary. In our view, the catalogue can and should aim for knowledge of the cell-type specificity of each element, strong and multifaceted support for each element's role as a bona fide enhancer, and knowledge of each element’s target gene(s).
A first challenge to this goal is that it is already clear that the results of different types of assays will frequently disagree. How are we to explain the fact that the vast majority of biochemically nominated candidate enhancers, when perturbed by CRISPRi, do not result in detectable changes in expression of genes located in cis72? As touched on above, there are numerous credible technical and biological explanations for this observation, and distinguishing between these seems key for the field to be able to move forward effectively. The broader point is that we remain largely in the dark regarding the sensitivity and specificity of most of these assays. Establishing a larger set of ‘true positives’ and ‘true negatives’ may be critical for adjudicating disagreements, which are trending towards being more prevalent than cases of agreement.
A further challenge is that although technologies are rapidly improving, it may simply not be realistic to test every candidate enhancer with every functional approach in every cell type of interest. However, as increasing numbers of elements are tested, our ability to quantitatively predict which sequences are bona fide enhancers, as well as the gene(s) each regulates, is likely to improve as well. For example, machine learning strategies to predict enhancer–gene links on the basis of 1D and 3D biochemical annotations are already advancing beyond the simple ‘nearest gene’ approach134,209,222. Particularly given the heterogeneous mechanisms by which enhancers might operate, establishing a community accepted set of strongly supported enhancer–gene links, ascertained by relatively unbiased methods, seems key in order to calibrate the performance of such predictive tools.
We note that many of the challenges highlighted here apply not only to candidate enhancers, but also to non-coding variants located within them. What standards of evidence should apply for a non-coding variant hypothesized to contribute to the association signal for a common disease? Heterogeneous classes of data will be available for many variants (e.g. biochemical annotations, molecular QTLs, computational predictions of variant effects, MPRAs, and CRISPR perturbation) but will not always agree. Further challenges include linkage disequilibrium, the possibility that more than one variant contributes to a given association, the need to match the cell type in which functional characterization and/or biochemical annotation is being carried out with the disease in question, and the fact that the mechanisms by which non-coding variants exert their effects on disease risk remain unclear223. Although a definitive map of cell-type-specific enhancers and enhancer–gene links is critical to accelerate efforts to move beyond GWAS associations to causal variants and genes, it will clearly not be enough.
Additionally, for the purposes of this Review and in line with how enhancers are broadly thought of in the field, we have focused on the modulation of gross transcript levels as an enhancer’s primary activity of relevance. However, we should remain open to the possibility that many enhancer or enhancer-like sequences have more nuanced or tightly orchestrated effects, such as effects on splicing, subtle effects on the spatiotemporal unfolding of gene expression programs during development, or other fine-grained effects. An evolving definition could also make room for surveys of enhancers’ impact on whole-cell or organismal phenotypes, although the effects on expression through which such effects were mediated would be important to know. Our overall point is that the operational definition of enhancers is likely to continue to evolve, alongside further advances in technology and biological understanding.
As we approach the 40th anniversary of their original definition6,7, fascinating questions remain about enhancer biology. How does an enhancer pick its target gene? Is 3D chromatin structure a determinant of gene regulation, or a residual feature? How do individual enhancers coordinate within a regulatory circuit, and how widespread is redundancy within these? What constitutes the differences between mechanisms underlying enhancer versus promoter activity? And last, what is (or what are) the true precise mechanism(s) of an enhancer’s activity at a target promoter? Although it will not be enough, we anticipate that confidently identifying thousands of bona fide enhancers, ideally through some relatively unbiased method, will facilitate efforts to answer these questions, while also advancing our understanding of how this class of elements orchestrates the remarkable program of mammalian development.
Acknowledgements
The authors thank Seungsoo Kim, Cole Trapnell, and Silvia Domcke, as well as other members of the Shendure Lab for helpful discussions. J.S. is an Investigator of the Howard Hughes Medical Institute.
Glossary terms
- Transcription factors
(TFs). A protein that binds DNA, typically a specific DNA sequence or motif, and contributes to the regulation of RNA transcription.
- Open chromatin
A nucleosome-loose packaging state of DNA that is permissive for transcription factor binding.
- Episomal reporter vector
Plasmid DNA that can be synthetically delivered, is autonomous from genomic DNA, and includes a reporter gene, typically downstream of a candidate regulatory element (e.g. an enhancer adjacent to a minimal promoter).
- Expressed sequence tag
(EST). In the early days of genomics, shotgun sequencing of cDNA was used as an efficient strategy for discovering genes, and subsequently to quantify their relative abundance.
- Open reading frame
The portion of a gene that is translatable by a ribosome. These are relatively straightforward to annotate by sequence alone due to the required start and stop codons.
- Regulatory element
A functional non-coding DNA sequence that regulates transcription. Classes of regulatory elements include enhancers, promoters, silencers, and insulators (further defined in Box 2).
- Chromosome conformation capture
Methods that map the 3D positioning, looping, and spatial organization of DNA within the nucleus, often relative to other segments of DNA.
- Topologically associating domains
(TADs). Broad regions of genomic DNA that are physically packaged together in the nucleus in 3D space. Typically at the scale of hundreds of kilobases to several megabases.
- Pioneer factor
A transcription factor (TF) that can directly interact with compact, closed chromatin. This class of TFs are thought to initiate (‘pioneer’) chromatin remodelling events.
- Linkage disequilibrium
The population genetics phenomenon by which genetic variants are non-randomly associated within a population. Variants are said to be in linkage disequilibrium if they are found to reside on a haplotype more frequently than one would expect by completely random assortment. Variants ‘in linkage disequilibrium’ are nearby on a genomic locus and hence co-inherited because they are rarely separated through meiotic recombination.
- Simpson’s paradox
A phenomenon in statistics in which different trends may exist in sub-groups of a dataset, but are undetectable when the groups are analyzed as a whole.
- Saturation mutagenesis
A molecular biology technique in which all possible sequence changes are generated from a parental sequence (e.g. all possible amino acids in an open reading frame, or all possible single nucleotide variants in an enhancer).
- CRISPR
The CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) system consists of the components of a bacterial immune system that have been adopted for synthetic genetic perturbation. The term is most often used in reference to the Type II Cas9 endonuclease version that can introduce a double-stranded break to genomic DNA as directed by a synthetic guide RNA.
- Protospacer adjacent motif
(PAM). In the original CRISPR bacterial immune system, fragments of previously encountered viral DNA are preserved in the bacterial genome. These ‘remembered’ sequences are processed into RNAs that guide the CRISPR nuclease to destroy newly invading viral DNA. But to prevent the nuclease from destroying the matching ‘remembered’ sequence in the bacteria’s own genome, a motif (the PAM) is required next to the target sequence in the viral genome. When genome editing is performed in eukaryotic cells, the presence of this sequence is still required by CRISPR nucleases.
- Shadow enhancers
Redundant enhancers, often located far away from its target gene. Enhancer redundancy is thought to enable robust buffered expression of the target gene, and to provide a versatile platform for evolution of new regulatory functions.
- ENCODE-4
The fourth generation of projects funded by the Encyclopedia of DNA Elements (ENCODE) Consortium began in 2017 and includes a new component focused on the implementation of high-throughput functional assays
- Human Cell Atlas
An international scientific community to coordinate the generation of human single cell datasets with the goal of generating a reference map of every cell type in the human body.
Footnotes
Competing interests
The authors declare no competing interests.
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Jacob F & Monod J Genetic regulatory mechanisms in the synthesis of proteins. J. Mol. Biol 3, 318–356 (1961). [DOI] [PubMed] [Google Scholar]
- 2.Ptashne M Specific Binding of the λ Phage Repressor to λ DNA. Nature 214, 232–234 (1967). [DOI] [PubMed] [Google Scholar]
- 3.Axel R, Cedar H & Felsenfeld G Synthesis of globin ribonucleic acid from duck-reticulocyte chromatin in vitro. Proc. Natl. Acad. Sci. U. S. A 70, 2029–2032 (1973). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weintraub H & Groudine M Chromosomal subunits in active genes have an altered conformation. Science 193, 848–856 (1976). [DOI] [PubMed] [Google Scholar]
- 5.Stalder J et al. Tissue-specific DNA cleavages in the globin chromatin domain introduced by DNAase I. Cell 20, 451–460 (1980). [DOI] [PubMed] [Google Scholar]
- 6.Moreau P et al. The SV40 72 base repair repeat has a striking effect on gene expression both in SV40 and other chimeric recombinants. Nucleic Acids Res. 9, 6047–6068 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Banerji J, Rusconi S & Schaffner W Expression of a beta-globin gene is enhanced by remote SV40 DNA sequences. Cell 27, 299–308 (1981).The first episomal observation of in vitro enhancer activity, this work coined the term 'enhancer'.
- 8.Mercola M, Wang X, Olsen J & Calame K Transcriptional enhancer elements in the mouse immunoglobulin heavy chain locus. Science 221, 663–665 (1983). [DOI] [PubMed] [Google Scholar]
- 9.Banerji J, Olson L & Schaffner W A lymphocyte-specific cellular enhancer is located downstream of the joining region in immunoglobulin heavy chain genes. Cell 33, 729–740 (1983). [DOI] [PubMed] [Google Scholar]
- 10.Gillies SD, Morrison SL, Oi VT & Tonegawa S A tissue-specific transcription enhancer element is located in the major intron of a rearranged immunoglobulin heavy chain gene. Cell 33, 717–728 (1983). [DOI] [PubMed] [Google Scholar]
- 11.Hanahan D Heritable formation of pancreatic beta-cell tumours in transgenic mice expressing recombinant insulin/simian virus 40 oncogenes. Nature 315, 115–122 (1985). [DOI] [PubMed] [Google Scholar]
- 12.Tuan D, Solomon W, Li Q & London IM The ‘beta-like-globin’ gene domain in human erythroid cells. Proceedings of the National Academy of Sciences 82, 6384–6388 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gross DS & Garrard WT Nuclease hypersensitive sites in chromatin. Annu. Rev. Biochem 57, 159–197 (1988). [DOI] [PubMed] [Google Scholar]
- 14.Hebbes TR, Thorne AW & Crane-Robinson C A direct link between core histone acetylation and transcriptionally active chromatin. EMBO J. 7, 1395–1402 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee DY, Hayes JJ, Pruss D & Wolffe AP A positive role for histone acetylation in transcription factor access to nucleosomal DNA. Cell 72, 73–84 (1993). [DOI] [PubMed] [Google Scholar]
- 16.Hebbes TR, Clayton AL, Thorne AW & Crane-Robinson C Core histone hyperacetylation co-maps with generalized DNase I sensitivity in the chicken beta-globin chromosomal domain. The EMBO Journal 13, 1823–1830 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Serfling E, Jasin M & Schaffner W Enhancers and eukaryotic gene transcription. Trends Genet. 1, 224–230 (1985). [Google Scholar]
- 18.Ikuta T & Kan YW In vivo protein-DNA interactions at the beta-globin gene locus. Proc. Natl. Acad. Sci. U. S. A 88, 10188–10192 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Forsberg M & Westin G Enhancer activation by a single type of transcription factor shows cell type dependence. EMBO J. 10, 2543–2551 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ptashne M Gene regulation by proteins acting nearby and at a distance. Nature 322, 697–701 (1986). [DOI] [PubMed] [Google Scholar]
- 21.Müeller-Storm HP, Sogo JM & Schaffner W An enhancer stimulates transcription in trans when attached to the promoter via a protein bridge. Cell 58, 767–777 (1989). [DOI] [PubMed] [Google Scholar]
- 22.Van der Ploeg LHT et al. γ-β-Thalassaemia studies showing that deletion of the γ- and δ-genes influences β-globin gene expression in man. Nature 283, 637–642 (1980). [DOI] [PubMed] [Google Scholar]
- 23.Kioussis D, Vanin E, deLange T, Flavell RA & Grosveld FG β-Globin gene inactivation by DNA translocation in γβ-thalassaemi. Nature 306, 662–666 (1983). [DOI] [PubMed] [Google Scholar]
- 24.Driscoll MC, Dobkin CS & Alter BP Gamma delta beta-thalassemia due to a de novo mutation deleting the 5’beta-globin gene activation-region hypersensitive sites. Proceedings of the National Academy of Sciences 86, 7470–7474 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Philipsen S, Talbot D, Fraser P & Grosveld F The beta-globin dominant control region: hypersensitive site 2. EMBO J. 9, 2159–2167 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Talbot D, Philipsen S, Fraser P & Grosveld F Detailed analysis of the site 3 region of the human beta-globin dominant control region. EMBO J. 9, 2169–2177 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grosveld F et al. The regulation of human globin gene switching. Philos. Trans. R. Soc. Lond. B Biol. Sci 339, 183–191 (1993). [DOI] [PubMed] [Google Scholar]
- 28.Moon AM & Ley TJ Conservation of the primary structure, organization, and function of the human and mouse beta-globin locus-activating regions. Proc. Natl. Acad. Sci. U. S. A. 87, 7693–7697 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Margot JB, Demers GW & Hardison RC Complete nucleotide sequence of the rabbit beta-like globin gene cluster. Analysis of intergenic sequences and comparison with the human beta-like globin gene cluster. J. Mol. Biol 205, 15–40 (1989). [DOI] [PubMed] [Google Scholar]
- 30.Li Q, Zhou B, Powers P, Enver T & Stamatoyannopoulos G Primary structure of the goat β-globin locus control region. Genomics 9, 488–499 (1991). [DOI] [PubMed] [Google Scholar]
- 31.Reitman M & Felsenfeld G Developmental regulation of topoisomerase II sites and DNase I-hypersensitive sites in the chicken beta-globin locus. Mol. Cell. Biol 10, 2774–2786 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dunham I et al. The DNA sequence of human chromosome 22. Nature 402, 489–495 (1999). [DOI] [PubMed] [Google Scholar]
- 33.International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001). [DOI] [PubMed] [Google Scholar]
- 34.Hardison RC, Oeltjen J & Miller W Long Human–Mouse Sequence Alignments Reveal Novel Regulatory Elements: A Reason to Sequence the Mouse Genome. Genome Res. 7, 959–966 (1997). [DOI] [PubMed] [Google Scholar]
- 35.Loots GG et al. Identification of a coordinate regulator of interleukins 4, 13, and 5 by cross-species sequence comparisons. Science 288, 136–140 (2000).This study identified non-coding regions regulating several interleukin genes by comparing 1 Mb of mouse–human orthologous sequence. The global application of this strategy was one of the key motivations for the sequencing of the mouse genome.
- 36.Hardison RC Conserved noncoding sequences are reliable guides to regulatory elements. Trends Genet. 16, 369–372 (2000). [DOI] [PubMed] [Google Scholar]
- 37.Pennacchio LA & Rubin EM Genomic strategies to identify mammalian regulatory sequences. Nat. Rev. Genet 2, 100–109 (2001). [DOI] [PubMed] [Google Scholar]
- 38.Nobrega MA, Ovcharenko I, Afzal V & Rubin EM Scanning human gene deserts for long-range enhancers. Science 302, 413 (2003). [DOI] [PubMed] [Google Scholar]
- 39.Pennacchio LA et al. In vivo enhancer analysis of human conserved non-coding sequences. Nature 444, 499–502 (2006). [DOI] [PubMed] [Google Scholar]
- 40.Mouse Genome Sequencing Consortium et al. Initial sequencing and comparative analysis of the mouse genome. Nature 420, 520–562 (2002). [DOI] [PubMed] [Google Scholar]
- 41.Dermitzakis ET et al. Numerous potentially functional but non-genic conserved sequences on human chromosome 21. Nature 420, 578–582 (2002). [DOI] [PubMed] [Google Scholar]
- 42.Sabo PJ et al. Genome-scale mapping of DNase I sensitivity in vivo using tiling DNA microarrays. Nat. Methods 3, 511–518 (2006). [DOI] [PubMed] [Google Scholar]
- 43.Boyle AP et al. High-resolution mapping and characterization of open chromatin across the genome. Cell 132, 311–322 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hesselberth JR et al. Global mapping of protein-DNA interactions in vivo by digital genomic footprinting. Nat. Methods 6, 283–289 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thurman RE et al. The accessible chromatin landscape of the human genome. Nature 489, 75–82 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lister R et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462, 315–322 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cokus SJ et al. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 452, 215–219 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Laurent L et al. Dynamic changes in the human methylome during differentiation. Genome Res. 20, 320–331 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barski A et al. High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Mikkelsen TS et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448, 553–560 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Heintzman ND et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 39, 311–318 (2007).One of the earliest uses of genome-wide biochemical annotation datasets to annotate candidate enhancers.
- 52.Robertson G et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat. Methods 4, 651–657 (2007). [DOI] [PubMed] [Google Scholar]
- 53.Johnson DS, Mortazavi A, Myers RM & Wold B Genome-wide mapping of in vivo protein-DNA interactions. Science 316, 1497–1502 (2007). [DOI] [PubMed] [Google Scholar]
- 54.Visel A et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 457, 854–858 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shendure J et al. DNA sequencing at 40: past, present and future. Nature 550, 345–353 (2017). [DOI] [PubMed] [Google Scholar]
- 56.De Santa F et al. A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS Biol. 8, e1000384 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim T-K et al. Widespread transcription at neuronal activity-regulated enhancers. Nature 465, 182–187 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Andersson R et al. An atlas of active enhancers across human cell types and tissues. Nature 507, 455–461 (2014).This paper reported integrated analysis of cap analysis of gene expression (CAGE) datasets across hundreds of cell types and tissues to generate biochemical annotations of thousands of cell-type-specific enhancers by their signature of bidirectional transcription.
- 59.Core LJ et al. Analysis of nascent RNA identifies a unified architecture of initiation regions at mammalian promoters and enhancers. Nat. Genet. 46, 1311–1320 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).Summary of the wealth of data generated by the ENCODE consortium. Hundreds of genome-wide datasets are used across hundreds of cell types/tissues to ascribe a function to the majority of the genome via biochemical annotation.
- 61.Roadmap Epigenomics Consortium et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stunnenberg HG, International Human Epigenome Consortium & Hirst M The International Human Epigenome Consortium: A Blueprint for Scientific Collaboration and Discovery. Cell 167, 1897 (2016). [DOI] [PubMed] [Google Scholar]
- 63.Arner E et al. Transcribed enhancers lead waves of coordinated transcription in transitioning mammalian cells. Science 347, 1010–1014 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.SCREEN: Search Candidate Regulatory Elements by ENCODE. Available at: http://screen.encodeproject.org/index/about. (Accessed: 19th September 2019)
- 65.Maurano MT et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Contributors to Wikimedia projects. Blind men and an elephant - Wikipedia. Wikimedia Foundation, Inc; (2006). Available at: https://en.wikipedia.org/wiki/Blind_men_and_an_elephant. (Accessed: 24th September 2019) [Google Scholar]
- 67.Patwardhan RP et al. Massively parallel functional dissection of mammalian enhancers in vivo. Nat. Biotechnol 30, 265–270 (2012).Along with Melnikov et al 2012, the first application of an MPRA to enhancer sequences.
- 68.Spitz F & Furlong EEM Transcription factors: from enhancer binding to developmental control. Nat. Rev. Genet 13, 613–626 (2012). [DOI] [PubMed] [Google Scholar]
- 69.Shlyueva D, Stampfel G & Stark A Transcriptional enhancers: from properties to genome-wide predictions. Nat. Rev. Genet 15, 272–286 (2014). [DOI] [PubMed] [Google Scholar]
- 70.Blackwood EM & Kadonaga JT Going the distance: a current view of enhancer action. Science 281, 60–63 (1998). [DOI] [PubMed] [Google Scholar]
- 71.Li L & Wunderlich Z An Enhancer’s Length and Composition Are Shaped by Its Regulatory Task. Front. Genet 8, 63 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gasperini M et al. A Genome-wide Framework for Mapping Gene Regulation via Cellular Genetic Screens. Cell (2019). doi: 10.1016/j.cell.2018.11.029 [DOI] [PubMed] [Google Scholar]
- 73.Smedley D et al. A Whole-Genome Analysis Framework for Effective Identification of Pathogenic Regulatory Variants in Mendelian Disease. Am. J. Hum. Genet 99, 595–606 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Corradin O & Scacheri PC Enhancer variants: evaluating functions in common disease. Genome Med. 6, 85 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gjoneska E et al. Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer’s disease. Nature 518, 365–369 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Whyte WA et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang X et al. High-resolution genome-wide functional dissection of transcriptional regulatory regions and nucleotides in human. Nature Communications 9, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ghavi-Helm Y et al. Highly rearranged chromosomes reveal uncoupling between genome topology and gene expression. Nat. Genet 51, 1272–1282 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zabidi MA et al. Enhancer-core-promoter specificity separates developmental and housekeeping gene regulation. Nature 518, 556–559 (2015).STARR-seq utilizing different classes of promoters in D. melanogaster showed that enhancers often do not work in a promoter-generic fashion.
- 80.Dorighi KM et al. Mll3 and Mll4 Facilitate Enhancer RNA Synthesis and Transcription from Promoters Independently of H3K4 Monomethylation. Mol. Cell 66, 568–576.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rickels R et al. Histone H3K4 monomethylation catalyzed by Trr and mammalian COMPASS-like proteins at enhancers is dispensable for development and viability. Nat. Genet 49, 1647–1653 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cusanovich DA et al. Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 348, 910–914 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Buenrostro JD et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Eagen KP Principles of Chromosome Architecture Revealed by Hi-C. Trends Biochem. Sci 43, 469–478 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Inoue F & Ahituv N Decoding enhancers using massively parallel reporter assays. Genomics 106, 159–164 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Klein JC, Chen W, Gasperini M & Shendure J Identifying Novel Enhancer Elements with CRISPR-Based Screens. ACS Chem. Biol 13, 326–332 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hnisz D, Day DS & Young RA Insulated Neighborhoods: Structural and Functional Units of Mammalian Gene Control. Cell 167, 1188–1200 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Maricque BB, Chaudhari HG & Cohen BA A massively parallel reporter assay dissects the influence of chromatin structure on cis-regulatory activity. Nat. Biotechnol (2018). doi: 10.1038/nbt.4285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Heinz S, Romanoski CE, Benner C & Glass CK The selection and function of cell type-specific enhancers. Nat. Rev. Mol. Cell Biol. 16, 144–154 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dixon JR et al. Chromatin architecture reorganization during stem cell differentiation. Nature 518, 331–336 (2015).Compelling use of Hi-C contact maps to study the dynamics of enhancer–promoter interactions through cellular differentiation.
- 91.Thanos D & Maniatis T Virus induction of human IFNβ gene expression requires the assembly of an enhanceosome. Cell 83, 1091–1100 (1995). [DOI] [PubMed] [Google Scholar]
- 92.Simeonov DR et al. Discovery of stimulation-responsive immune enhancers with CRISPR activation. Nature 549, 111–115 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ray J et al. Chromatin conformation remains stable upon extensive transcriptional changes driven by heat shock. bioRxiv 527838 (2019). doi: 10.1101/527838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fuda NJ, Ardehali MB & Lis JT Defining mechanisms that regulate RNA polymerase II transcription in vivo. Nature 461, 186–192 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Juven-Gershon T, Cheng S & Kadonaga JT Rational design of a super core promoter that enhances gene expression. Nat. Methods 3, 917–922 (2006). [DOI] [PubMed] [Google Scholar]
- 96.Chen FX et al. PAF1 regulation of promoter-proximal pause release via enhancer activation. Science 357, 1294–1298 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Engreitz JM et al. Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature 539, 452–455 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Murakami S, Nagari A & Kraus WL Dynamic assembly and activation of estrogen receptor α enhancers through coregulator switching. Genes Dev. 31, 1535–1548 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gosselin D et al. An environment-dependent transcriptional network specifies human microglia identity. Science 356, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Vihervaara A et al. Transcriptional response to stress is pre-wired by promoter and enhancer architecture. Nat. Commun. 8, 255 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Iwafuchi-Doi M et al. The Pioneer Transcription Factor FoxA Maintains an Accessible Nucleosome Configuration at Enhancers for Tissue-Specific Gene Activation. Mol. Cell 62, 79–91 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Furlong EEM & Levine M Developmental enhancers and chromosome topology. Science 361, 1341–1345 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Calo E & Wysocka J Modification of enhancer chromatin: what, how, and why? Mol. Cell 49, 825–837 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lindblad-Toh K et al. A high-resolution map of human evolutionary constraint using 29 mammals. Nature 478, 476–482 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fulco CP et al. Systematic mapping of functional enhancer-promoter connections with CRISPR interference. Science 354, 769–773 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Henriques T et al. Widespread transcriptional pausing and elongation control at enhancers. Genes Dev. 32, 26–41 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kwasnieski JC, Fiore C, Chaudhari HG & Cohen BA High-throughput functional testing of ENCODE segmentation predictions. Genome Res. 24, 1595–1602 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Muerdter F et al. Resolving systematic errors in widely used enhancer activity assays in human cells. Nat. Methods 15, 141–149 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rao SSP et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Weintraub AS et al. YY1 Is a Structural Regulator of Enhancer-Promoter Loops. Cell 171, 1573–1588.e28 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Williamson I et al. Anterior-posterior differences in HoxD chromatin topology in limb development. Development 139, 3157–3167 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ghavi-Helm Y et al. Enhancer loops appear stable during development and are associated with paused polymerase. Nature 512, 96–100 (2014). [DOI] [PubMed] [Google Scholar]
- 113.Benabdallah NS et al. PARP mediated chromatin unfolding is coupled to long-range enhancer activation. bioRxiv 155325 (2017). doi: 10.1101/155325 [DOI] [Google Scholar]
- 114.Alexander JM et al. Live-cell imaging reveals enhancer-dependent Sox2 transcription in the absence of enhancer proximity. Elife 8, e41769 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Osterwalder M et al. Enhancer redundancy provides phenotypic robustness in mammalian development. Nature 554, 239–243 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Levings PP & Bungert J The human beta-globin locus control region. Eur. J. Biochem 269, 1589–1599 (2002). [DOI] [PubMed] [Google Scholar]
- 117.Hnisz D et al. Super-enhancers in the control of cell identity and disease. Cell 155, 934–947 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lettice LA et al. A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum. Mol. Genet 12, 1725–1735 (2003). [DOI] [PubMed] [Google Scholar]
- 119.Bahr C et al. Author Correction: A Myc enhancer cluster regulates normal and leukaemic haematopoietic stem cell hierarchies. Nature 558, E4 (2018). [DOI] [PubMed] [Google Scholar]
- 120.Haberle V et al. Transcriptional cofactors display specificity for distinct types of core promoters. Nature (2019). doi: 10.1038/s41586-019-1210-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Cho SW et al. Promoter of lncRNA Gene PVT1 Is a Tumor-Suppressor DNA Boundary Element. Cell 173, 1398–1412.e22 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Cinghu S et al. Intragenic Enhancers Attenuate Host Gene Expression. Mol. Cell 68, 104–117.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Blow MJ et al. ChIP-Seq identification of weakly conserved heart enhancers. Nat. Genet. 42, 806–810 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Schmidt D et al. Five-Vertebrate ChIP-seq Reveals the Evolutionary Dynamics of Transcription Factor Binding. Science 328, 1036–1040 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.King MC & Wilson AC Evolution at two levels in humans and chimpanzees. Science 188, 107–116 (1975). [DOI] [PubMed] [Google Scholar]
- 126.Danko CG et al. Dynamic evolution of regulatory element ensembles in primate CD4+ T cells. Nat Ecol Evol 2, 537–548 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kulakovskiy IV et al. HOCOMOCO: a comprehensive collection of human transcription factor binding sites models. Nucleic Acids Res. 41, D195–202 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lambert SA et al. The Human Transcription Factors. Cell 172, 650–665 (2018). [DOI] [PubMed] [Google Scholar]
- 129.Van Loo P & Marynen P Computational methods for the detection of cis-regulatory modules. Brief. Bioinform 10, 509–524 (2009). [DOI] [PubMed] [Google Scholar]
- 130.Teytelman L, Thurtle DM, Rine J & van Oudenaarden A Highly expressed loci are vulnerable to misleading ChIP localization of multiple unrelated proteins. Proc. Natl. Acad. Sci. U. S. A 110, 18602–18607 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Worsley Hunt R & Wasserman WW Non-targeted transcription factors motifs are a systemic component of ChIP-seq datasets. Genome Biol. 15, 412 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Jain D, Baldi S, Zabel A, Straub T & Becker PB Active promoters give rise to false positive ‘Phantom Peaks’ in ChIP-seq experiments. Nucleic Acids Research 43, 6959–6968 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Diao Y et al. A new class of temporarily phenotypic enhancers identified by CRISPR/Cas9-mediated genetic screening. Genome Res. 26, 397–405 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Pliner HA et al. Cicero Predicts cis-Regulatory DNA Interactions from Single-Cell Chromatin Accessibility Data. Mol. Cell 71, 858–871.e8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ernst J et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473, 43–49 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Consortium, G. & GTEx Consortium. Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Strober BJ et al. Dynamic genetic regulation of gene expression during cellular differentiation. Science 364, 1287–1290 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.van der Wijst MGP et al. Single-cell RNA sequencing identifies celltype-specific cis-eQTLs and co-expression QTLs. Nat. Genet 50, 493–497 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ptashne M How eukaryotic transcriptional activators work. Nature 335, 683–689 (1988). [DOI] [PubMed] [Google Scholar]
- 140.Schleif R DNA looping. Annu. Rev. Biochem 61, 199–223 (1992). [DOI] [PubMed] [Google Scholar]
- 141.Lieberman-Aiden E et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Dekker J, Rippe K, Dekker M & Kleckner N Capturing chromosome conformation. Science 295, 1306–1311 (2002). [DOI] [PubMed] [Google Scholar]
- 143.Nora EP et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485, 381–385 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.de Laat W & Duboule D Topology of mammalian developmental enhancers and their regulatory landscapes. Nature 502, 499–506 (2013). [DOI] [PubMed] [Google Scholar]
- 145.Dixon JR et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Sexton T et al. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell 148, 458–472 (2012). [DOI] [PubMed] [Google Scholar]
- 147.Fullwood MJ & Ruan Y ChIP‐based methods for the identification of long‐range chromatin interactions. J. Cell. Biochem (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Mumbach MR et al. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nat. Methods 13, 919–922 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Fang R et al. Mapping of long-range chromatin interactions by proximity ligation-assisted ChIP-seq. Cell Res. 26, 1345–1348 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ma W et al. Fine-scale chromatin interaction maps reveal the cis-regulatory landscape of human lincRNA genes. Nat. Methods 12, 71–78 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Chen H et al. Dynamic interplay between enhancer-promoter topology and gene activity. Nat. Genet 50, 1296–1303 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Schmitt AD et al. A Compendium of Chromatin Contact Maps Reveals Spatially Active Regions in the Human Genome. Cell Rep. 17, 2042–2059 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Williamson I, Lettice LA, Hill RE & Bickmore WA Shh and ZRS enhancer colocalisation is specific to the zone of polarising activity. Development 143, 2994–3001 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Gu B et al. Transcription-coupled changes in nuclear mobility of mammalian cis-regulatory elements. Science 359, 1050–1055 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Rao SSP et al. Cohesin Loss Eliminates All Loop Domains. Cell 171, 305–320.e24 (2017).Rapid cohesin depletion in cultured cells results in a widespread loss of the genome’s 3D organization, with minimal changes in gene expression. This caused the field to question the importance of genome organization (i.e. stable enhancer–promoter loops as they had been conceptualized).
- 156.Nora EP et al. Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell 169, 930–944.e22 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Trapnell C Defining cell types and states with single-cell genomics. Genome Res. 25, 1491–1498 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Novick A & Weiner M ENZYME INDUCTION AS AN ALL-OR-NONE PHENOMENON. Proc. Natl. Acad. Sci. U. S. A 43, 553–566 (1957). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Bonn S et al. Tissue-specific analysis of chromatin state identifies temporal signatures of enhancer activity during embryonic development. Nat. Genet 44, 148–156 (2012). [DOI] [PubMed] [Google Scholar]
- 160.Cusanovich DA et al. A Single-Cell Atlas of In Vivo Mammalian Chromatin Accessibility. Cell 174, 1309–1324.e18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Cusanovich DA et al. The cis-regulatory dynamics of embryonic development at single-cell resolution. Nature 555, 538–542 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lai B et al. Publisher Correction: Principles of nucleosome organization revealed by single-cell micrococcal nuclease sequencing. Nature 564, E17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ramani V et al. Massively multiplex single-cell Hi-C. Nat. Methods 14, 263–266 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Flyamer IM et al. Single-nucleus Hi-C reveals unique chromatin reorganization at oocyte-to-zygote transition. Nature 544, 110–114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Nagano T et al. Cell-cycle dynamics of chromosomal organization at single-cell resolution. Nature 547, 61–67 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Nagano T et al. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 502, 59–64 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Tan L, Xing D, Chang C-H, Li H & Xie XS Three-dimensional genome structures of single diploid human cells. Science 361, 924–928 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Lee DS, Luo C, Zhou J, Chandran S & Rivkin A Single-cell multi-omic profiling of chromatin conformation and DNA methylome. bioRxiv (2018). [Google Scholar]
- 169.Rotem A et al. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat. Biotechnol 33, 1165–1172 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Hainer SJ, Boskovic A, Rando OJ & Fazzio TG Profiling of pluripotency factors in individual stem cells and early embryos. doi: 10.1101/286351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Benabdallah NS et al. Decreased Enhancer-Promoter Proximity Accompanying Enhancer Activation. Mol. Cell (2019). doi: 10.1016/j.molcel.2019.07.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Fukaya T, Lim B & Levine M Enhancer Control of Transcriptional Bursting. Cell 166, 358–368 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Patwardhan RP et al. High-resolution analysis of DNA regulatory elements by synthetic saturation mutagenesis. Nat. Biotechnol 27, 1173–1175 (2009).The first demonstration of an MPRA coupled to sequencing-based readout. It was used to evaluate all possible single nucleotide variants of bacteriophage and mammalian core promoters.
- 174.Arnold CD et al. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science 339, 1074–1077 (2013).The first paper describing STARR-seq as well as the first whole-genome shotgun MPRA. Quantitative, genome-wide maps of enhancer potential generated in two different D. melanogaster cell lines provided insight into general characteristics of enhancers and enabled analysis of cell-type specificity.
- 175.Gasperini M, Starita L & Shendure J The power of multiplexed functional analysis of genetic variants. Nat. Protoc 11, 1782–1787 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Vockley CM et al. Direct GR Binding Sites Potentiate Clusters of TF Binding across the Human Genome. Cell 166, 1269–1281.e19 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Vanhille L et al. High-throughput and quantitative assessment of enhancer activity in mammals by CapStarr-seq. Nature Communications 6, (2015). [DOI] [PubMed] [Google Scholar]
- 178.Klein JC, Keith A, Agarwal V, Durham T & Shendure J Functional characterization of enhancer evolution in the primate lineage. Genome Biol. 19, 99 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Ulirsch JC et al. Systematic Functional Dissection of Common Genetic Variation Affecting Red Blood Cell Traits. Cell 165, 1530–1545 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Tewhey R et al. Direct Identification of Hundreds of Expression-Modulating Variants using a Multiplexed Reporter Assay. Cell 172, 1132–1134 (2018). [DOI] [PubMed] [Google Scholar]
- 181.Vockley CM et al. Massively parallel quantification of the regulatory effects of noncoding genetic variation in a human cohort. Genome Res. 25, 1206–1214 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Klein JC et al. Functional testing of thousands of osteoarthritis-associated variants for regulatory activity. Nat. Commun. 10, 2434 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Liu Y et al. Functional assessment of human enhancer activities using whole-genome STARR-sequencing. Genome Biol. 18, 219 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Melnikov A et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat. Biotechnol 30, 271 (2012).Along with Patwardhan et al 2012, the first application of an MPRA to variants of enhancer sequences. Coined the term massively parallel reporter assay (‘MPRA’).
- 185.Kircher M et al. Saturation mutagenesis of twenty disease-associated regulatory elements at single base-pair resolution. Nat. Commun 10, 3583 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Grossman SR et al. Systematic dissection of genomic features determining transcription factor binding and enhancer function. Proc. Natl. Acad. Sci. U. S. A 114, E1291–E1300 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Smith RP et al. Massively parallel decoding of mammalian regulatory sequences supports a flexible organizational model. Nat. Genet 45, 1021–1028 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.van Arensbergen J et al. Genome-wide mapping of autonomous promoter activity in human cells. Nat. Biotechnol 35, 145–153 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Gilbert N & Allan J Supercoiling in DNA and chromatin. Curr. Opin. Genet. Dev 25, 15–21 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Inoue F et al. A systematic comparison reveals substantial differences in chromosomal versus episomal encoding of enhancer activity. Genome Res. 27, 38–52 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Akhtar W et al. Chromatin position effects assayed by thousands of reporters integrated in parallel. Cell 154, 914–927 (2013). [DOI] [PubMed] [Google Scholar]
- 192.Shalem O et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Zhou Y et al. High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature 509, 487–491 (2014). [DOI] [PubMed] [Google Scholar]
- 194.Wang T, Wei JJ, Sabatini DM & Lander ES Genetic screens in human cells using the CRISPR-Cas9 system. Science 343, 80–84 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Canver MC et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 527, 192–197 (2015).The first CRISPR-based screen to identify functional non-coding sequence within an enhancer.
- 196.van Overbeek M et al. DNA Repair Profiling Reveals Nonrandom Outcomes at Cas9-Mediated Breaks. Mol. Cell 63, 633–646 (2016). [DOI] [PubMed] [Google Scholar]
- 197.Chen W, McKenna A, Schreiber J, Yin Y & Agarwal V Massively parallel profiling and predictive modeling of the outcomes of CRISPR/Cas9-mediated double-strand break repair. bioRxiv (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Vierstra J et al. Functional footprinting of regulatory DNA. Nat. Methods 12, 927–930 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Wright JB & Sanjana NE CRISPR Screens to Discover Functional Noncoding Elements. Trends Genet. 32, 526–529 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Korkmaz G et al. Functional genetic screens for enhancer elements in the human genome using CRISPR-Cas9. Nat. Biotechnol 34, 192–198 (2016). [DOI] [PubMed] [Google Scholar]
- 201.Rajagopal N et al. High-throughput mapping of regulatory DNA. Nat. Biotechnol 34, 167–174 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Sanjana NE et al. High-resolution interrogation of functional elements in the noncoding genome. Science 353, 1545–1549 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Diao Y et al. A tiling-deletion-based genetic screen for cis-regulatory element identification in mammalian cells. Nat. Methods 14, 629–635 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Gasperini M et al. CRISPR/Cas9-Mediated Scanning for Regulatory Elements Required for HPRT1 Expression via Thousands of Large, Programmed Genomic Deletions. Am. J. Hum. Genet 101, 192–205 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Aparicio-Prat E et al. DECKO: Single-oligo, dual-CRISPR deletion of genomic elements including long non-coding RNAs. BMC Genomics 16, 846 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Kosicki M, Tomberg K & Bradley A Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol (2018). doi: 10.1038/nbt.4192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Thakore PI et al. Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat. Methods 12, 1143–1149 (2015).The first demonstration that nuclease-inactivated Cas9 tethered to the KRAB repressor and targeted to an enhancer can mediate repression of a target gene.
- 208.Klann TS et al. CRISPR–Cas9 epigenome editing enables high-throughput screening for functional regulatory elements in the human genome. Nat. Biotechnol 35, 561 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Fulco CP, Nasser J, Jones TR, Munson G & Bergman DT Activity-by-Contact model of enhancer specificity from thousands of CRISPR perturbations. bioRxiv (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Kearns NA et al. Functional annotation of native enhancers with a Cas9-histone demethylase fusion. Nat. Methods 12, 401–403 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Kwon DY, Zhao Y-T, Lamonica JM & Zhou Z Locus-specific histone deacetylation using a synthetic CRISPR-Cas9-based HDAC. Nat. Commun 8, 15315 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Lei Y et al. Targeted DNA methylation in vivo using an engineered dCas9-MQ1 fusion protein. Nature Communications 8, 16026 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Vojta A et al. Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res. 44, 5615–5628 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Liu XS et al. Editing DNA Methylation in the Mammalian Genome. Cell 167, 233–247.e17 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Huang Y-H et al. DNA epigenome editing using CRISPR-Cas SunTag-directed DNMT3A. Genome Biology 18, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Xie S, Duan J, Li B, Zhou P & Hon GC Multiplexed Engineering and Analysis of Combinatorial Enhancer Activity in Single Cells. Mol. Cell 66, 285–299.e5 (2017).The first use of single-cell RNA-seq to phenotype pools of candidate enhancer perturbations in a ‘whole-transcriptome’ screen.
- 217.Hong J-W, Hendrix DA & Levine MS Shadow enhancers as a source of evolutionary novelty. Science 321, 1314 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Sabari BR et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science 361, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Visel A, Minovitsky S, Dubchak I & Pennacchio LA VISTA Enhancer Browser—a database of tissue-specific human enhancers. Nucleic Acids Res. 35, D88–D92 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Lupiáñez DG et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 161, 1012–1025 (2015).Demonstration that the rearrangement across TAD boundaries can cause a pathogenic phenotype in humans. The study also uses CRISPR–Cas9 to effectively reconstruct human patient genome rearrangements in mouse models.
- 221.Dickel DE et al. Ultraconserved Enhancers Are Required for Normal Development. Cell 172, 491–499.e15 (2018).This paper demonstrated that the knockout of ultraconserved enhancers in the mouse model had subtle but consequential effects on organismal development that were not readily detected by gross phenotyping.
- 222.Zeng W, Wu M & Jiang R Prediction of enhancer-promoter interactions via natural language processing. BMC Genomics 19, 84 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Boyle EA, Li YI & Pritchard JK An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell 169, 1177–1186 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Pollard KS, Hubisz MJ, Rosenbloom KR & Siepel A Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 20, 110–121 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Siepel A et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 15, 1034–1050 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Khan A et al. JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 46, D1284 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Kulakovskiy IV et al. HOCOMOCO: towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-Seq analysis. Nucleic Acids Res. 46, D252–D259 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Schones DE et al. Dynamic regulation of nucleosome positioning in the human genome. Cell 132, 887–898 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Buenrostro JD, Giresi PG, Zaba LC, Chang HY & Greenleaf WJ Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Mahat DB et al. Base-pair-resolution genome-wide mapping of active RNA polymerases using precision nuclear run-on (PRO-seq). Nat. Protoc 11, 1455–1476 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Tome JM, Tippens ND & Lis JT Single-molecule nascent RNA sequencing identifies regulatory domain architecture at promoters and enhancers. Nat. Genet. 50, 1533–1541 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Ku WL et al. Single-cell chromatin immunocleavage sequencing (scChIC-seq) to profile histone modification. Nat. Methods 16, 323–325 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Skene PJ & Henikoff S An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife 6, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Ernst J & Kellis M ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods 9, 215–216 (2012).A computational framework for utilizing ChIP-seq for histone modifications to classify regions of the genome by their likely biological function (e.g. strong-enhancer or weak-promoter).
- 235.Hoffman MM et al. Unsupervised pattern discovery in human chromatin structure through genomic segmentation. Nat. Methods 9, 473–476 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Gill LL, Karjalainen K & Zaninetta D A transcriptional enhancer of the mouse T cell receptor δ gene locus. Eur. J. Immunol 21, 807–810 (1991). [DOI] [PubMed] [Google Scholar]
- 237.Greaves DR, Wilson FD, Lang G & Kioussis D Human CD2 3’-flanking sequences confer high-level, T cell-specific, position-independent gene expression in transgenic mice. Cell 56, 979–986 (1989). [DOI] [PubMed] [Google Scholar]
- 238.Raab JR & Kamakaka RT Insulators and promoters: closer than we think. Nat. Rev. Genet 11, 439–446 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Mikhaylichenko O et al. The degree of enhancer or promoter activity is reflected by the levels and directionality of eRNA transcription. Genes Dev. 32, 42–57 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Weingarten-Gabbay S et al. Systematic interrogation of human promoters. Genome Res. 29, 171–183 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Ngoc LV, Wang Y-L, Kassavetis GA & Kadonaga JT The punctilious RNA polymerase II core promoter. Genes & Development 31, 1289–1301 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Jayavelu ND, Jajodia A, Mishra A & David Hawkins R An atlas of silencer elements for the human and mouse genomes. doi: 10.1101/252304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.West AG, Gaszner M & Felsenfeld G Insulators: many functions, many mechanisms. Genes Dev. 16, 271–288 (2002). [DOI] [PubMed] [Google Scholar]
- 244.Stranger BE et al. Patterns of cis regulatory variation in diverse human populations. PLoS Genet. 8, e1002639 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]