

Abstract
Animal mitochondrial genomic polymorphism occurs as low-level mitochondrial heteroplasmy and deeply divergent co-existing molecules. The latter is rare, known only in bivalvian mollusks. Here we show two deeply divergent co-existing mt-genomes in a vertebrate through genomic sequencing of the Tuatara (Sphenodon punctatus), the sole-representative of an ancient reptilian Order. The two molecules, revealed using a combination of short-read and long-read sequencing technologies, differ by 10.4% nucleotide divergence. A single long-read covers an entire mt-molecule for both strands. Phylogenetic analyses suggest a 7–8 million-year divergence between genomes. Contrary to earlier reports, all 37 genes typical of animal mitochondria, with drastic gene rearrangements, are confirmed for both mt-genomes. Also unique to vertebrates, concerted evolution drives three near-identical putative Control Region non-coding blocks. Evidence of positive selection at sites linked to metabolically important transmembrane regions of encoded proteins suggests these two mt-genomes may confer an adaptive advantage for an unusually cold-tolerant reptile.
Subject terms: Zoology, Genomics
Robert Macey et al. use deep sequencing to reveal two co-existing, deeply divergent, fully functional mitochondrial genomes in a single Tuatara from Lady Alice Island, New Zealand. This finding will stimulate further investigation, having profound implications for our understanding of mitochondrial evolution and function.
Introduction
Mitochondrial polymorphisms in animals exist as low-level mitochondrial heteroplasmy and as deeply divergent co-existing mitochondrial genomes. Low-level mitochondrial heteroplasmy typically arises via shallow historical mutation with the maternal passage leading to generational fixation1,2. Alternative mechanisms are mitochondrial degradation forming heteroplasmy2,3 that is generally not inherited and paternal leakage4. Instances of paternal leakage causing apparent heteroplasmy have been suggested to be mtDNA fragments recently integrated into the nuclear genome4. Deeply divergent co-existing mt-genomes are previously only known among bivalvian mollusks, where one mt-genome is typically maternally inherited and the other paternally inherited, a phenomenon known as doubly uniparental inheritance (DUI)5.
The vertebrate mt-genome is small in size (15–26 kb) with most taxa sharing a common gene order6. Among the more structurally complicated mt-genomes described is that of the Tuatara (Sphenodon punctatus, Fig. 1). A highly threatened reptile, the Tuatara remains on 32 small islands in New Zealand, and represents the sole-surviving taxon of a deep vertebrate lineage (Order: Rhynchocephalia), which diverged from squamate reptiles 220–250 million years ago (MYA)7. Previous studies reporting the first ‘complete’ mt-genome8 and subsequent 34 additional population-level Tuatara mt-genomes9,10 suggests the Tuatara mt-genome is missing three genes that encode ND5, tRNAHis, and tRNAThr. That finding is striking because absent tRNA genes have transcriptional implications for all mt-encoded proteins11.
Fig. 1. Mitochondrial gene organization in the Tuatara.

Arrangements are depicted as a linear bar for what is a circular molecule. A The standard vertebrate mt-gene order presumptive as ancestral among vertebrates. B The mt-gene order discovered in complete mt-genomes of the Tuatara. C The standard vertebrate mt-gene order as in “A” above. Abbreviations for genes are: 12 S and 16 S for small and large ribosomal RNAs; NDl–6 and 4 L for NADH dehydrogenase subunits; COI–III for cytochrome c oxidase subunits; Cytb for cytochrome b; 6 and 8 for ATPase subunits. All protein-coding genes are transcribed from the heavy strand with the exception of ND6 from the light strand. Transfer RNA genes are represented by their standard one-letter amino acid codes positioned on the strand encoded, with the top representing the heavy strand and the bottom representing the light strand. S1, S2, Ll, and L2 depict tRNASer(AGY), tRNASer(UCN), tRNALeu(UUR), and tRNALeu(CUN), respectively. The identical and duplicated tRNALeu(CUN) is labeled L2. The Control Region (OH), responsible for heavy-strand replication, standardly has one copy among vertebrates. Identified are three non-coding regions labeled NC1–3 and putative Control Regions. Black bars indicate protein-coding regions rearranged relative to the standard vertebrate mt-genic organization. Eight novel features of the Tuatara mt-genome are numbered with arrows. (1) The origin of light-strand replication (OL) is not present in the Tuatara19 but a stem-and-loop structure overlapping tRNA genes may supplement (Fig. 4), which is indicated by an arrow. (2) The bold italic K represents a duplicated tRNALys that appears to be a pseudogene8. (3) ND6 and tRNAGlu are moved relative to the standard vertebrate mt-genic organization. (4) tRNALeu(CUN) and ND5 are moved relative to the standard vertebrate mt-genic organization. (5) Cytb is moved relative to the standard vertebrate mt-genic organization. (6) tRNAHis is moved relative to the standard vertebrate mt-genic organization. (7) tRNAThr is moved relative to the standard vertebrate mt-genic organization. (8) tRNASer(AGY) is moved relative to the standard vertebrate mt-genic organization. The two identical or near identical copies of tRNALeu(CUN) flank the missing region of all previously reported Tuatara mt-genomes5–7 (horizontal green bar).
In order to investigate the true Tuatara mt-genome composition an array of sequencing techniques including Illumina whole-genome shotgun, Oxford Nanopore, PacBio, and PCR-based Sanger sequencing are used on individuals sampled across extremes of the Tuatara’s geographic distribution: Stephens Island (SI-1–4) and Lady Alice Island (LAI) samples, the latter sequenced for a whole-genome project12. In doing so, two deeply divergent mt-molecules are discovered in the Tuatara (LAI), each containing the three genes previously reported as missing, albeit bounded by a series of repeated Control Region copies. The Tuatara is exceptional in being a large-bodied reptile metabolizing in cold environments, in which mitochondrial ATP synthesis is conducted13,14. The likely time of divergence among these molecules and the selective pressures that may have led to maintain two deeply divergent, coexisting mitochondrial genomes is explored.
Results
The discovery of two mitochondrial genomes in the Tuatara
The discovery of two mt-genomes in the LAI individual is established via complementary Illumina (high coverage short-read) and Oxford Nanopore (long-read) data that identify two groups of DNA sequencing reads distinguishable by >5% sequence divergence. Illumina assembly of the LAI molecule 1 (M1) is 18,078 bases and molecule 2 (M2) 18,315 bases, each containing all 37 genes typical of animal mt-genomes. Molecular features implicate both genomes as mtDNA and not nuclear DNA. These include strong strand-bias against guanine (13.8% M1 and 14.6% M2; Supplementary Note 1), all protein-coding genes translate with no internal stop codons, tRNA genes encode tRNAs with stable secondary structures containing recognized anti-codons6, and no sequencing reads, whether short or long-read data, were flanked by nuclear DNA. The two mt-genomes are further confirmed via Oxford Nanopore sequencing of LAI total genomic DNA producing 9.46 Gb of sequence data in 7,229.48 K reads (Fig. 2, Table 1, and Supplementary Note 2). Filtering out mtDNA reads with assignments to M1 or M2, results in 114 mtDNA reads that map to 100% of the LAI M1 Illumina assembly reference. Thirty-two LAI M2 assigned reads map to 95.8% of the LAI M2 Illumina assembly reference. A single complete mt-genomic Oxford Nanopore read is obtained for LAI M1 from an mt-molecule naturally nicked, with both native DNA complement strands end-connected via laboratory manipulation, and sequenced in a single reaction (Supplementary Note 3 and Supplementary Fig. 1).
Fig. 2. Oxford Nanopore coverage of Lady Alice Island mt-genomes (M1 and M2).

Mapped Oxford Nanopore reads to the Illumina assembly of mt-genomes. Blue graphs represent mapping coverage. Gene annotations are shown below the coverage map (some gene names are truncated with the same gene order reported herein). A Molecule 1, with 100% of reference covered, averaging 2504.5 bp in length (std dev = 2409.2; min = 514; max = 16,978) with an average depth coverage of 16.9 (std dev = 3.4; min = 11; max = 25). The uppermost read covers the entire molecule. B Molecule 2, with 95.8% of reference covered, averaging 1990.2 bp in length (std dev = 1757.6; min = 551; max = 7026) with an average depth coverage of 3.7 (std dev = 3.4; min = 11; max = 25). Light gray arrows on the ends indicate reads that wrap around the molecules. See Supplementary Data 1 for a fasta file of LAI M1 Oxford Nanopore reads and Supplementary Data 2 for a fasta file of LAI M2 Oxford Nanopore reads, each a filtered product and mapped here.
Table 1.
Data obtained from Oxford Nanopore runs both mined and retrieved mitochondrial DNA (see text for protocol information).
| Run | Modification to library preparation | Chemistry | Total # of reads (K) | Total yield (Gb) | mt-reads >500 bp | mtDNA yield (kb) | Mapped molecule 1 mt-reads | Mapped molecule 1 yield (kb) | Mapped molecule 2 mt-reads | Mapped molecule 2 yield (kb) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 10 kb shear | 1D, R9.4, SQK-108 | 165.43 | 0.99 | 7 | 25.31 | 7 | 25.31 | 0 | 0 |
| 2 | 10 kb shear, Trypsin | 1D, R9.4, SQK-108 | 353.58 | 2.06 | 16 | 94.42 | 9 | 43.87 | 5 | 24.54 |
| 3 | 2D | 2D, R9.4, SQK-208 | 18.79 | 0.26 | 10 | 27.83 | 9 | 45.02 | 0 | 0 |
| 4 | 5 kb shear | 1D, R9.4, SQK-108 | 34.64 | 0.31 | 0 | 0 | 0 | 0 | 0 | 0 |
| 5 | Trypsin | 1D, R9.4, SQK-208 | 265.17 | 0.98 | 8 | 46.46 | 6 | 14.16 | 1 | 4.836 |
| 6 | Trypsin | 1D, R9.4, SQK-108 | 18.31 | 0.11 | 1 | 3.43 | 1 | 3.39 | 0 | 0 |
| 7 | Trypsin | 1D, R9.4, LSK109 | 1224.00 | 1.30 | 36 | 74.99 | 31 | 64.60 | 4 | 8.22 |
| 8 | Trypsin | 1D, R9.4, LSK109 | 5149.57 | 3.45 | 80 | 112.71 | 51 | 74.69 | 22 | 26.09 |
| Totals | 7229.48 | 9.46 | 158 | 385.15 | 114 | 271.04 | 32 | 63.69 | ||
Run numbers are described in the text: library preparation methods refer to variations in protocols described in the methods section, chemistry is the type of sequencing run and the version of flow cell used. Run numbers 1–3 are SRA run numbers 1–3, run number 4 is SRA run number 5, run number 5 is SRA run number 6, run number 6 is SRA run number 10, run number 7 is SRA run number 14, and run number 8 is SRA run number 15.
A highly divergent second mt-genome via phylogenetics
Phylogenetic analysis of protein-coding sequences from LAI M1 and M2 with published9,10 mt-genome sequences (lacking ND5) places LAI M2 outside all extant populations (Fig. 3). The rooting position of M2 between northern and southern populations forms two groups of M1 separated by the Cook Strait with 1.0% sequence divergence. In contrast, all M1 sequences are on average 11.1% divergent to M2, further negating the possibility of M2 being a recent nuclear-integrated copy. Monophyly of LAI genomes (M1 and M2) is statistically rejected. The well-dated ND1–COI section in amphibians and reptiles is estimated to have a pair-wise sequence divergence of 1.3% per million years15–17. Application of this divergence rate to the Tuatara mt-genomes indicates a 7.8-million-year separation between M1 and M2 (10.1% sequence divergence), with a 1.2-million-year separation between northern and southern population M1 genomes (1.6% average sequence divergence), indicating a deeply divergent second mt-molecule (Supplementary Note 4).
Fig. 3. Phylogenetic relationships of Tuatara populations in relation to Lady Alice Island molecules (M1 and M2).
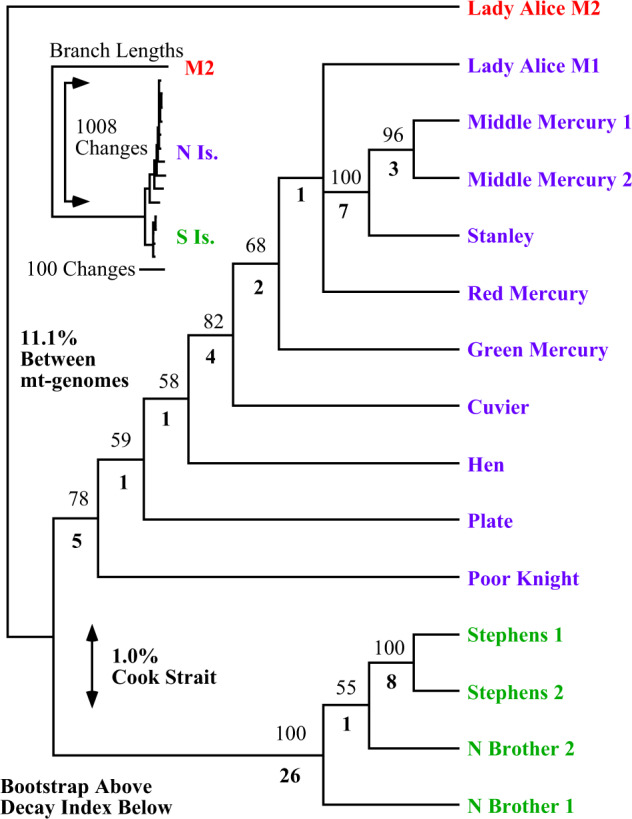
Analysis of 12 mt-protein-coding genes from published data9,10 not including ND5 in relation to both M1 and M2 sequences from LAI reported here. Bootstraps are depicted above with decay indices below. The tree has a length of 1302 steps from analysis of 9522 aligned sites of which 170 are parsimony informative; the entire tree collapses in 26 steps, hence the decay index below branches. Using LAI M2 as a root, it is implicated it is placed between northern and southern populations forming two groups separating in the Cook Strait. These northern and southern groups have only 1.0% sequence divergence between them, while sequence divergence to LAI M2 is 11.1%. The upper left corner shows relative branch lengths with the LAI M2 sample considerably different in nucleotide changes. Phylogram depiction of the shortest tree illustrates branch length DNA changes along the shortest path.
An extremely divergent genomic organization among vertebrates
The most complex and rearranged vertebrate mt-genome is discovered in the Tuatara. All 37 mt-genes (13 protein-coding, 2 rRNA, and 22 tRNA) are present with the addition of three putative Control Region non-coding blocks (NC1–3), and duplicate tRNALys and tRNALeu(CUN) copies (Fig. 1). A newly identified segment containing ND5, tRNAThr, tRNAHis, NC2, and tRNALeu(CUN) second copy is discovered that was not reported in all 35 previously published Tuatara mt-genomes8–10. In contrast to the standard vertebrate mt-gene order, the section between ND4 and tRNAPhe contains the highly rearranged section of ND6, tRNAGlu, NC1, tRNALeu(CUN) first copy, ND5, tRNAThr, tRNAHis, NC2, tRNALeu(CUN) second copy, Cytb, tRNAPro, tRNASer(AGY), and NC3. This large-scale regional genic rearrangement with NC duplications involves protein-coding genes and numerous tRNA gene movements that cannot be explained by a simple duplication and deletion of redundant sequence model6. The Tuatara mt-genome architecture is also confirmed in a PacBio sequenced individual (SI-3) and our mining of a transcriptome library18 (SI-4), Sanger sequencing of PCR-amplified products from SI-2 and SI-3 with forward and reverse primers extending inside ND5 further support that gene and other gene junctions are in the mt-genome (Supplementary Note 5).
Unusual replication origins
The replication origin for the light strand (OL), missing between tRNAAsn and tRNACys6,19, has an OL-like structure overlapping 14 bases with tRNAAsn (normally none) and two bases with tRNACys (normally two to four). An associated structure of the adjacent tRNACys encoding a tRNA lacking a D-arm that instead contains a D-arm replacement loop19,20 may provide an alternative OL initiation site6 (Fig. 4). Concerted evolution of two Control Region sequences in which evolution of duplicated segments of DNA undergo rapid replacement during replication to either create identical or near identical copies through time21 is observed in several independent reptiles lineages22–24. The Tuatara three non-coding blocks (NC1–3) show features consistent with Control Region copies that are nearly identical suggestive of concerted evolution (Supplementary Note 6). Phylogenetic analysis of duplicated non-coding blocks of the two LAI mt-genomes and the SI-4 mt-genome produces two equally parsimonious trees showing duplicated regions within an mt-genome clustering (Fig. 5). Monophyly of NC1, NC2 or NC3 sequences is each statistically rejected, a result consistent with concerted evolution in which identical or near identical sequences in repeated regions are propagated21.
Fig. 4. Secondary structures in the region of the light strand replication origin (OL) in the Tuatara (Sphenodon punctatus).

Top right is a stem-and-loop structure that may serve as OL but is highly unusual (presented as heavy-strand template DNA sequence). This OL-like structure overlaps 14 bases with tRNAAsn (normally no overlap) and two bases with tRNACys (normally two to four), (see arrows and lower right for OL comparison). Putative light-strand encoded tRNAs are presented to the left and are the same sequence as heavy-strand DNA except for the use of U instead of T. The Tuatara has an unusual tRNACys that lacks a D-stem and instead contains a D-arm replacement loop (arrow)19,20. Stems are as follows: AA = amino-acid acceptor stem, D = dihydrouridine stem, AC = anticodon stem, and T = TphiC stem. Consensus heavy-strand sequence of putative OL from squamate lizards is presented in the lower right. This sequence is based on representatives from the Iguanidae (nine genera), Gekkonidae (Teratoscincus), Dibamidae (Dibamus), Lacertidae (Eremias), Teiidae (Cnemidophorus), Cordylidae (Platysaurus), Anguidae (Elgaria), Xenosauridae (Xenosuurus), and Varanidae (Varanus);6,54 Dibamus novaeguineae, U71327, was previously reported as Lialis jicari6. Bases in capitals are conserved pairings and downstream sequences. Bases in lower case are often paired. Variable positions are labeled with their standard one-letter codes: R = G or A; Y = C or T; B = G, C, or T; and V = G, C, or A. The 3’-GCC-5’ heavy-strand template sequence identified as the point of light-strand elongation in mouse55 is indicated in green font (arrow). The heavy-strand sequence 3’-GBCCB-5’ in tRNACys related to the downstream 3’-GGCCG-5’ sequence found to be required for in vitro replication in humans56 is underlined. Note the OL-like stem-and-loop structure in the Tuatara (upper right) has a G-C rich heavy-strand sequence of 3’-CGG-5’ (bases in bold and arrow), which is the complement of the 3’-GCC-5’ heavy-strand template sequence identified as the point of light-strand elongation in mouse55. No obvious sequence related to the downstream 3’-GGCCG-5’ sequence found to be required for in vitro replication in humans56 is identifiable in the Tuatara.
Fig. 5. Phylogenetic tree depicting relationships among non-coding blocks (NC1–3) using Lady Alice Island M1 and M2 with Stephans Island sample 4.
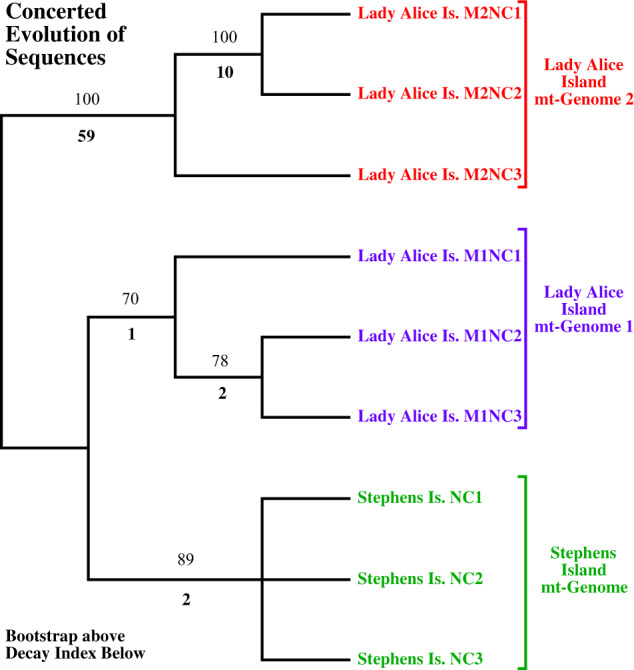
Strict consensus of two equally parsimonious trees with lengths of 107 steps from analysis of 737 aligned sites of which 80 are parsimony informative. Duplicated regions within an mt-genome cluster, as opposed to all NC1 sequences grouping, all NC2 sequences grouping, and all NC3 sequences grouping; a result consistent with concerted evolution of sequences in which identical or near identical sequences in repeated regions are propagated. Bootstraps are presented above and decay indices are depicted below.
Inheritance of two mt-genomes
The LAI individual exhibiting two mt-genomes (M1, M2) is a male, which received genome-wide sequencing efforts12, while all other samples were female and not as extensively sequenced. Mined Illumina mtDNA reads totaling 209,650 from LAI, identify 30,005 as M2, and 176,980 as M1, resulting in 206,985 reads assignable to either M1 or M2. Thus, M2 is represented by 14.5% (1/7th concentration) of assignable reads, relative to the dominant M1 copy. Among bivalves, males inherit and carry mtDNA from both parents, while females only carry mtDNA from the mother5. In males, somatic tissues are variable for the concentration of the male-type mt-genome within individuals and between species25, but generally in low concentration in somatic tissues5,26. A male inherited Tuatara copy is possible. The Tuatara M2 genome is detected only in the single male sampled and DNA was extracted from blood. No other male samples were available, and the M2 genome was not detected in any females sampled, although not subjected to genome-level sequencing.
Structural content of amino acids in two molecules
The Tuatara is a large-bodied unusually cold-tolerant reptile with a low standard metabolic rate (13 °C)13,14. Two co-occurring mt-genomes may therefore be advantageous for metabolic flexibility in cool environments. Adaptations in transmembrane proteins have been shown to have a high bearing on environmental fitness in extreme environments27. Selection analysis of codon positions with PAML (Supplementary Table 1) identifies a consistent pattern of purifying selection in all mitochondrial-encoded proteins, with an average ratio of non-synonymous to synonymous changes of ω = 0.096 ± 0.016 (Supplementary Table 2). However, PAML and the fixed effects approach in FEL detect a series of amino acids under putative positive selection in LAI M2 and show that the majority of them lie in transmembrane regions. A concentration of 43 out of 55 detected amino acid sites under putative positive selection (78%) reside in transmembrane regions of encoded ND1, ND2, ND4, COII, COIII, and Cytb (FDR ≤ 0.05; Fig. 6; Supplementary Table 3), contrasting typical patterns of mammalian evolution with strong enrichment of adaptive variation in loop regions28. As amino acid changes in transmembrane regions have a stronger impact on protein function than in loop regions29, the selective enrichment of transmembrane amino acid changes suggests divergence of molecules in relation to the species’ life history. Further work is needed to understand the biological implications of two deeply divergent Tuatara mt-genomes.
Fig. 6. Positive selection of amino acids in protein structure.

Protein structure of LAI M2 proteins depicting putative positive selection (orange) in encoded A ND1, B ND2, C ND4, D COII, E COIII, and F Cytb. Structures are estimated with the ConSurf method51 based on homologs of proteins in the UNIREF-90 database, and visualized using PyMOL. Amino acid changes under putative positive selection in LAI M2 as detected by PAML and FEL are colored in orange, with the majority of them residing in transmembrane regions.
Discussion
The discovery of two deeply-divergent mt-genomes in the Tuatara has profound implications for our understanding of animal mitochondrial genome organization, inheritance, and evolution. The Tuatara is the sole surviving member of an ancient vertebrate lineage (Rhynchocephalia) providing a long period of isolated evolution (220–250 million years)7. Whether duplication of Tuatara mt-genomes arose via uni- or biparental mechanisms, and how they have been maintained for a 7–8 million-year period through a potential adaptive advantage is a dynamic new area for research.
DUI is the only well-studied mode of co-existing, deeply divergent mt-molecules found in single individuals, and thus far only confirmed in bivalvian mollusks5. In mollusks, the maternal copy is present in somatic tissues, whereas the paternal copy is found primarily in germ-line cells5,26. Our discovery of two deeply divergent (10.4% sequence divergence) mt-genomes in the Tuatara is intriguing, but the origin, inheritance mechanism, and maintenance of the two molecules remains unknown. Thus, for now, our results do not conclusively support, nor refute a DUI hypothesis.
The two divergent mt-genomes were identified in a single male (the second mt-molecule at 1/7th the concentration of the first) and their discovery was only achievable in combination with genome-wide sequencing12 of DNA from blood. Additional samples were screened for a second mt-genome via long-read PacBio sequencing and molecule-specific PCR primers, yet were unsuccessful. These additional individuals were female, high molecular weight DNA extracted from liver was used, and these samples were not subjected to the same exhaustive genome-scale sequencing as our male sample. Additionally, the published transcriptome library18 was consolidated from multiple individual embryos sequenced at modest depth, and no signal of a second mt-genome was herein detected. The lack of detection in female samples could be attributable to simple unsuccessful attempts of PCR-primer specificity/preferential annealing and/or non-exhaustive genomic sequencing efforts.
Future studies seeking to confirm the widespread presence of two mt-molecules in the Tuatara should examine multiple individuals, of both sexes, across the geographic range of the Tuatara. Ideally, studies would be conducted in pedigrees to explore the inheritance of these molecules, and across multiple tissues to explore any tissue-specific patterns. Such a study is not trivial for a species that is protected under New Zealand and international law, and that is a taonga, or special treasure for those Maori iwi that are the kaitiaki, guardians, of the Tuatara. Our genome sequencing work was undertaken in partnership with Ngatiwai iwi, but any future studies in other locations would require further partnerships with Ngatiwai and another iwi across Aotearoa New Zealand to gain permissions to work on, sample, and sequence these taonga.
Tuatara mt-genomes show extreme rearrangement relative to other vertebrate mt-genomes. Tuatara mt-gene rearrangements include tRNA genes often found in mt-genomic rearrangements, but in the Tuatara with drastic distant locations relative to the standard vertebrate gene order6. Unlike most rearrangements among vertebrates, protein-coding and tRNA genes are switched and shuffled among duplicated origins of replication for the heavy strand (putative Control Regions, OH). A third of the mt-genome is significantly rearranged, two protein-coding genes are switched (ND5 and ND6), tRNA genes are drastically reshuffled, and putative origins of replication are duplicated and inserted in this region (Fig. 1). Putative Control Regions are observed in triplicate going through concerted evolution, and an unusual stem-and-loop structure is discovered with overlapping sequences of tRNAAsn and tRNACys in the location of light-strand replication (OL). These observations in totality reinforce the idea of replication errors driving mt-genomic rearrangement6.
Tandem complementary efforts of high-throughput short-read sequencing with newly advanced long-read sequencing identify two deeply divergent mt-genomes escaping science for decades. The genomic structure is elucidated via these techniques with a single Oxford Nanopore read covering both DNA strands, signaling a new era of organellar genomics with whole molecule sequencing.
Methods
Sampling
The LAI sample is a male and NCBI Biosample SAMN08793959 with the four Stephens Island samples all being female: (SI-1) SAMN10598677, (SI-2) SAMN10598679, (SI-3) SAMN10598680, and (SI-4) SAMN00855319 with published transcriptome data in SRA05164718. Blood of LAI was collected with ethical permissions from Victoria University of Wellington and under permits supplied by the Department of Conservation, New Zealand.
Sanger sequencing of Stephens Island samples (SI-1–3)
Shotgun Sanger sequencing of SI-1 PCR fragments from COIII to 12 S rRNA follows methods previously described30, with amplifications conducted using L9940 5’-GCAGCATGATACTGACACTTYGT-3’ and H10676. A MegaBACE 1000 (Amersham) DNA sequencer ran 768-reads that were assembled using Phrap. Sanger sequencing of SI-2 and SI-3 used two forward REX26_ND5F1 5’-GTGCACTAACACAAAACGATATC-3’ and REX27_ND5F1 5’-GCGCACTGACACAAAATGATATT-3’, and two reverse primers REX26_ND5R1 5’-GGATTCCTCCTATTTTTCGAATG-3’ and REX27_ND5R1 5’- GGATTCCTCCTATTTTTCAGATA-3’ designed in ND5 from SI-1 sequences. Amplifications applied forward ND5 primers with H10676 and reverse ND5 primers with L9940. End-sequencing was done on all PCR fragments with internal reactions using “ND4” 31 in ND4 and “IguaCytBR2”32 in Cytb. Reactions were run on an ABI3730 Sequencer (2011 Life Technologies) with 900 chemistry (Supplementary Method 1).
Illumina data collection of the LAI sample
Total genomic DNA was extracted using proteinase K digestion and Phenol-Chloroform extraction from blood. Sequencing was undertaken using the Illumina HiSeq 2000 and 2500 as well as MiSeq sequencing platforms (Illumina, San Diego). Sequencing libraries consisted of paired-end (PE) libraries with estimated insert sizes of 180 bp, 350 bp, and 550 bp and three mate-paired (MP) libraries with estimated insert sizes of 2500 bp, 5000 bp, and 8000 bp. The PE libraries were prepared using the Illumina TruSeq PCR-Free DNA library kit, while the mate-pair libraries were prepared using the Illumina TruSeq DNA library kit as per the manufacturer’s instructions. These libraries were normalized and pooled across 32 lanes on an Illumina HiSeq 2000 or 2500 using 2 × 100 bp PE sequencing at New Zealand Genomics Ltd., Dunedin. We further supplemented these data with additional Illumina TruSeq and Kappa DNA libraries, with insert sizes of 400 bp and 480 bp, respectively. These libraries were normalized and pooled across five Illumina MiSeq 2 × 250 bp runs via New Zealand Genomics Ltd., Dunedin (Supplementary Method 2).
Illumina assembly of LAI
Illumina reads were mined from total genomic shotgun data for mtDNA reads using the first previously published mt-genome8 (AF53439) and initial contigs from two PCR fragments obtained from Sanger sequencing of SI-1 with ND5, tRNAHis, and tRNAThr herein reported. Illumina HiSeq reads were extracted from eleven PE 100 bp data-sets with an insert size of 180 bp (totaling ~1.9 billion read-pairs) and two additional data-sets with an insert size of 2500 bp (totaling ~47 million read-pairs), and one data-set of 5000 bp (~128 million read-pairs), respectively, using Bowtie 233. Extracted reads were cleaned and only properly paired read-pairs were kept for further analysis. In total, 156,012 reads were obtained that could be used for the assembly of mtDNA. First, the insert size for all mapped reads was calculated and only reads where the determined insert size ranged between 100–200 bp (for the 180 bp library) and 2000–3000 bp (for the 2500 bp library) were used to perform the assembly. The initial assembly was created using MaSuRCA assembler34. Next, all contigs mapping to the two PCR fragments obtained from Sanger sequencing of SI-1 reported here were used to fine-tune the Illumina assembly using Minimus35. This draft assembly of Illumina HiSeq reads had an average coverage of 739.2 reads (std dev = 200.7; min = 14, max = 1625). In order to evaluate the Minimus assembly, 71mers were counted with one PE 180 bp data-set using Jellyfish36 that allowed the identification of a repeat region missed by the initial assembly. After including the repeat into the mtDNA sequence, we obtained a final sequence containing 18,078 bp. The final assembly had an average coverage of 723.2 reads (std dev = 178.3; min = 12, max = 1502). Further examination of Illumina data suggested there may be a second copy of the mt-genome. Initially, all reads mentioned above were de novo assembled in Geneious v10.2.4 (Biomatters, Auckland) with the highest sensitivity producing 937 contigs. Recovered contigs were backmapped to the initial Illumina LAI M1 assembly. Three of the 937 contigs nearly covered the entire molecule (M1) with ~98% identity. Twenty-seven additional contigs that mapped were within 90% identity to the initial LAI M1 mt-genome. These contigs were manually assembled to construct a nearly complete second mt-genome (M2). Therefore, all available Illumina reads were re-mined for mtDNA from Illumina HiSeq PE 100 bp reads from (a) eleven data-sets with an insert size of 180 bp (totaling ~1.9 billion read-pairs), (b) eight data-sets with an insert size of 350 bp (totaling ~446 million read-pairs), (c) eight data-sets with an insert size of 550 bp (totaling ~316 million read-pairs), (d) one data-set with an insert size of 2500 bp (~47 million read-pairs), (e) three data-sets with an insert size of 5000 bp (totaling ~307 million read-pairs), (f) four data-sets with an insert size of 8000 bp (totaling ~490 million read-pairs); and Illumina MiSeq PE 250 bp reads from (g) four data-sets with an insert size of 400 bp (totaling ~50 million read-pairs), and (h) one data-set with an insert size of 480 bp (~17 million read-pairs). This produced 104,825 paired-read sets with a total of 209,650 reads matching mtDNA. The 209,650 reads recovered were de novo assembled in Geneious with the highest sensitivity level, using all paired-read information that produced 856 contigs. Consensus sequences from these contigs were then backmapped to the 18,078 bp draft LAI M1 mt-genome obtained from the above assembly and to the draft M2 in Geneious. Contigs were screened for a minimum of 5% sequence divergence to either the 18,078 bp draft LAI M1 or the draft M2. This produced 19 contigs assigned to M2 for further evaluation. The 19 contigs containing raw reads were separately assembled producing 5 contigs that were manually assembled following manual trimming. These final contigs were backmapped to the draft M2, forming a contig containing 30,005 reads with an average coverage of 169.2 reads (std dev = 102.7; min = 3, max = 508). Ambiguities in M2 were resolved and a final second mt-genome was identified and annotated. Of the 856 contigs discovered above not assigned to M2, nine contigs were deemed unusable. The remaining 828 contigs were backmapped to LAI M1 producing a contig with 176,980 reads having an average coverage of 1007.5 reads (std dev = 213.8; min = 25, max = 1791).
DNA preparation for Oxford Nanopore sequencing of LAI
Oxford Nanopore work relied on the sample being subsequently shipped between the University of Otago, New Zealand and San Diego Zoo, California, USA using CITES institutional transfers, together with supporting permits to export and import from the New Zealand Department of Conservation and US Fish and Wildlife Service, respectively. A partial sample was shipped to Dovetail (Santa Cruz, CA) for work on the Tuatara genome. Dovetail extracted DNA from ~100 µl of snap-frozen blood using the Qiagen Blood & Cell Culture DNA Midi Kit (Hilden, Germany), yielding 242 ng/µl provided in TE buffer (runs 1–6). A partial blood sample was shipped to SeqMatic LLC (Fremont, CA) for work on the Tuatara genome. SeqMatic (Fremont, CA) extracted DNA from ~50 µl of snap-frozen blood using an enzymatic DNA extraction with (run 7) and without phenol-chloroform (run 8), yielding 131 ng/µl and 79 ng/µl in TE buffer. Following extractions, DNA was stored and transferred to the Peralta Genomics Laboratory (Alameda, CA) at 4°C and never frozen (Supplementary Method 3).
Oxford Nanopore sequencing of LAI
Eight Oxford Nanopore runs using R9.4 chemistry were conducted applying Minion protocol version GDE_9002_v108_revT_18Oct2016 using a nick-repair enzyme (NEBNext FFPE Repair Mix). To increase library yield, NEBNext Ultra II End Repair/dA-Tailing Module was used to eliminate an additional Solid Phase Reversible Immobilization (SPRI)37 cleanup step with Agencourt AMPure XP beads. To increase library yield, 2 µl of concentrated NEB T4 DNA ligase was added during adaptor ligation. The general protocol consists of four steps. Step 1, DNA Nick-Repair, Blunt-Ending, and End Repair: Add in a clean low bind PCR tube, (a) ~ 2 µg genomic Tuatara DNA as described above of 8.27 µl, (b) NEBNext FFPE Repair Mix of 3 µl, (c) NEBNext Ultra II End Repair/dA-Tailing Mix of 3 µl, (d) NEBNext Ultra II End Repair/dA-Tailing Buffer of 7 µl, (e) 100X NAD + of 0.6 µl, and (f) 10 mM Tris HCl pH 8.5 buffer of 38.13 µl for a total volume of 60 µl. This is mixed gently via inversion and incubated at 20 °C for 60 min and 65 °C for 30 min in a thermocycler. Step 2, SPRI Cleanup: add to the above (a) Agencourt AMPure XP beads of 60 µl and let stand for 2 min followed by discarding supernatant while on the magnet, (b) wash with 70% EtOH of 140 µl X 2, and (c) elute in Nuclease-free water of 31 µl. Step 3, Adaptor Ligation: add to (a) eluted end-prepped DNA above of 30 µl, (b) 1D Adapter Mix of 20 µl, and (c) NEB Instant Sticky End Ligase of 50 µl, for a total volume of 100 µl. This is mixed gently via inversion and incubated at 20 °C for 10 min in a thermocycler. Step 4, Library Purification, with additional SPRI Cleanup: add to the above (a) Agencourt AMPure XP beads of 40 µl and let stand for 2 min followed by discarding supernatant while on the magnet, (b) wash with Adaptor Bead Binding Buffer of 140 µl X 2, and (c) elute in Oxford Nanopore Elution Buffer of 25 µl for DNA sequencing. Deviations to the above protocol are described. Deviations I, DNA Integrity and Sizing: DNA was kept in high integrity for runs 3, 5, 6, 7, and 8 but DNA shearing was conducted in two ways for runs 1, 2, and 4. Run number 4 sheared DNA to a target of 5 kb from ~ 2 µg of Tuatara DNA using a Covaris LE220R with (a) Peak Incident Power (W) 100, (b) Duty Factor 20%, (c) Cycles per Burst 1000, and (d) Treatment Time (s) 600. Runs 1 and 2 sheared DNA to a target of 10 kb from ~ 2 µg of Tuatara DNA using a Covaris g-tube. It is noteworthy that while subjecting DNA to shearing a direct following step implementing nick-repair was applied as described above. Final products from shearing procedures required volume adjustments between input DNA (from shearing in buffer) and 10 mM Tris HCl pH 8.5 buffer in the starting protocol of step 1 above to keep relative concentrations equivalent. Deviations II, Enzymatic Cleanup with Additional SPRI Cleanup: For runs 2, 5, 6, 7, and 8, replacements steps from steps above apply an activated immobilized trypsin resin (ThermoFisher Scientific). Step 2.1, to activate trypsin resin, the resin was gently mixed adding 20 µl of resin to 100 µl of 10 mM Tris HCl pH 8.5 buffer in a 1.5 ml tube. This was inverted for mixing followed by a 6000 rpm spin, with the supernatant discarded; this was repeated two additional times, with 20 µl of 10 mM Tris HCl pH 8.5 buffer added to the end product. Step 2.2, the (a) end-repaired DNA from step 1 of 60 µl, and (b) activated trypsin resin of 20 µl from step 2.1 were (c) incubated at 37 °C for 30 min. Only 60 µl of the total volume of 80 µl was retained leaving 20 µl of pelleted resin behind. Step 3, add to (a) eluted end-prepped DNA above of 60 µl, (b) 1D Adapter Mix of 20 µl, and (c) NEB Instant Sticky End Ligase of 80 µl, for a total volume of 160 µl. This is mixed gently via inversion and incubated at 20 °C for 10 min in a thermocycler. Step 4, add to the above (a) Agencourt AMPure XP beads of 64 µl and let stand for 2 min followed by discarding supernatant while on the magnet, (b) wash with Adaptor Bead Binding Buffer of 140 µl X 2, and (c) elute in Oxford Nanopore Elution Buffer of 25 µl for DNA sequencing. Deviations III, 2D Ligation and Library Preparation: For run 3, 2D chemistry requires replacement steps. Step 3, add to (a) eluted end-prepped DNA above of 30 µl, (b) 2D Adapter Mix of 10 µl, (c) dH2O of 5 µl, (d) HP Adapter of 2 µl with (d) NEB Instant Sticky End Ligase of 50 µl and (e) incubated for 10 min at room temp; followed with (f) HP tether of 1 µl and (g) 2 µl of concentrated NEB T4 DNA ligase added with (h) additional incubation at room temp for 10 min. The 2 µl of concentrated NEB T4 DNA ligase was added during adaptor ligation with the HP Tether to increase library yield. The total yield is 100 µl for this step. Step 4.1, MyOne C1 Streptavidin bead preparation. Add in a 1.5 ml Eppendorf DNA LoBind tube (a) MyOne C1 Streptavidin beads (Invitrogen) of 50 µl and (b) pellet beads on the magnet for 2 min; (c) discard supernatant and (d) add Oxford Nanopore Bead Binding Buffer of 140 µl; (e) vortex until homogeneous and (f) pellet on a magnet for 2 min; (g) discard supernatant and (h) repeat Oxford Nanopore Bead Binding Buffer wash step of 140 µl with pelleting on a magnet for 2 min X 2; (i) add Bead Binding Buffer of 100 µl and (j) label tube as “Washed Beads” for binding step. Step 4.2, for binding add (a) Washed Beads from previous step 4.1 of 100 µl to the tube containing the Ligated DNA in step 3 of 100 µl and (b) incubate at room temperature for 5 min; for elution add (c) Oxford Nanopore elution buffer to DNA-bound beads of 25 µl, (d) incubate tube on the hot block at 37 °C for 10 min, (e) pellet beads on the magnet for 2 min, and (f) transfer supernatant containing library into a clean 1.5 ml Eppendorf DNA LoBind tube for DNA sequencing. Libraries were run on SQK-108, SQK-208, and SQK-LSK-109 flow-cells.
Oxford Nanopore data and evaluation of LAI
Oxford Nanopore 2D reads were extracted using Nanopolish38. By default, Nanopolish extracts reads for export either as a 2D consensus read or a 1D template read; hence there is only a single read per double-stranded (2D) or single-stranded (1D) read. Guppy (Oxford Nanopore) was only used to process 1D reads. All Oxford Nanopore reads obtained from genomic data were compiled into a single database and the Tuatara mt-genomic data were blasted against this database using the default parameters for Blastn and Megablast. This was done with both LAI M1 and M2 Illumina data assemblies reported here in separate searches, as the Rest et al8. mt-genome was discovered to lack a segment of the mt-genome. Following this 1.5 kb, 3 kb, and 6 kb random seeds were applied from the LAI M1 and M2 Illumina data assemblies reported here in separate searches to further maximize mitochondrial Oxford Nanopore read recovery. Reads less than 500 bp in length were no longer considered. To evaluate if recovered mt-genomic Oxford Nanopore reads were M1 or M2, matches were made to both the LAI Illumina assembly of M1 or the LAI Illumina assembly of M2, separately. This was done in three separate iterations per molecule using NCBI (a) Blastn, (b) Megablast, and (c) Discontiguous Megablast. A cut-off of 5% difference between molecules was adopted because the two Illumina draft molecules are ~10% sequence divergence and Oxford Nanopore reads have a bias of including numerous gaps representing unread bases. Oxford Nanopore sequences assigned to M1 or M2 were separately backmapped to their respective Illumina draft mt-genome molecules using Geneious with default parameters to evaluate depth coverage.
PacBio sequencing and assembly of Stephen Island sample 3 (SI-3)
Genomic DNA was extracted using the Qiagen Genomic Tip DNA extraction kit from ~20 mg of liver tissue. A PacBio SMRTbell library was prepared using the SMRTbell Express Template Preparation Kit (Pacific Biosciences). A 12 kb and above size selection with the Sage Science BluePippin system was implemented. Prepared libraries were run on the PacBio Sequel platform using version 2.1 chemistry. Two Single-Molecule Real-Time (SMRT) Cells were sequenced, where each library was sequenced on one SMRT cell with 360 min movie lengths. PacBio raw reads were assembled in CANU version 1.8 (Supplementary Method 4).
Transcriptome library mining
The published Tuatara transcriptome library (SI-4)18 was downloaded. Initial work discovered contaminated Rattus leucopus DNA sequences. The mt-genome of R. leucopus39 was used to purge Rattus mt-DNA from the Tuatara transcriptome library by using the ‘Map to Reference’ function in Geneious and saving a list of unused reads. The Rattus-free mt-DNA reads were mapped to LAI M1 producing a full-coverage of the mt-genome.
Mt-genome alignment for sequence divergence
Complete mt-genome alignments for pair-wise sequence divergence were conducted in Geneious using MUSCLE version 3.8.425, with adjustments to comply with secondary structures of encoding tRNA genes40. The end of the third non-coding block was not alignable and excluded (last 404 bp of NC3 in LAI M2, positions 17912–18315; last 230 bp of NC3 in LAI M1, positions 17849–18078; last 230 bp of NC3 in SI-3, positions 17849–18078; last 230 bp of NC3 in SI-4, positions 17842–18071).
Phylogenetic analysis of duplicate mt-genomes and duplicate non-coding blocks
Phylogenetic sampling used LAI M1 and M2 with samples from eight northern islands (Plate, Cuvier, Green Mercury, Middle Mercury, Stanley, Red Mercury, Hen, and Poor Knight) and two southern islands (Stephens and North Brother); GenBank numbers KP996609–19, 40–419,10 (Supplementary Method 5). The LAI M2 is 7 encoded amino acid positions longer in Cytb than sequences deemed molecule 1. Gaps were placed in all molecule 1 Cytb sequences: (a) after encoded amino acid position 3 (nucleotide position 9) 5 encoded amino acid positions were entered as gaps (15 nucleotide positions), and (b) after encoded amino acid position 291 (nucleotide position 873), and 2 encoded amino acid positions were entered as gaps (6 nucleotide positions). ND5 was not included because it was not reported in all previous studies8–10. Overlapping regions of bicistronic-encoded genes were excluded: Atp8 and Atp6 (last 96 nucleotides of Atp8), and ND4L and ND4 (last 7 nucleotides of ND4L). To evaluate duplicated non-coding block sequences (NC1–3) from both LAI mt-genomes, they were compared to sequences from SI-4 of the published transcriptome library (Supplementary Method 6). The SI-3 sample was not included because of ambiguities in sequence. Sequences were aligned in Geneious using MUSCLE version 3.8.425. Only areas in common between NC1–3 sequences were evaluated. Phylogenetic estimates were conducted using branch-and-bound searches in PAUP* version 4.041 with bootstraps generated via 1000 replicates. Decay indices were generated with successive searches retaining suboptimal trees, and computing tree length difference to the overall shortest tree(s). Two-tailed Wilcoxon signed-ranks tests42,43 incorporating a correction for tied ranks were used to examine the statistical significance of alternative phylogenetic hypotheses with the shortest estimate(s). Alternative phylogenetic estimates were discovered via searches applying constraint trees that saved suboptimal trees compatible with an alternative phylogenetic hypothesis, using MacClade version 4.0344 for constraint tree construction and search implementation in PAUP* version 4.041.
Protein evaluation
Aligned protein-coding regions of LAI M1 & M2, SI-3, SI-4, and GenBank KP996609–19, 40–419,10 were subjected to PAML-CodeML45 (Supplementary Table 1) using ETE346 and to FEL47 using HyPhy48 to detect selective pressures on individual sites of the LAI M2 lineage. For ND5, the only available LAI M1 & M2, SI-3, and SI-4 sequences were used. Within PAML, we used Bayes Empirical Bayes (BEB)49 with a threshold of P > 95% as evidence for sites under putative positive selection. Within FEL, the resulting p values were adjusted for multiple testing using FDR (false discovery rate50) across sites. For protein structure visualization, ConSurf Server51,52 uses HMMER to search for homologs in the UNIREF-90 database (E-value cut-off of 0.0001), and 150 sampled protein sequences (min/max identity of 35%/95%) are aligned using MAFFT-L-INS-i. HHPred and MODELER then apply the best evolutionary substitution model (JTT) to predict the 3D structure of proteins52. The structures are visualized using PyMOL53 (see Supplementary Method 7 and Supplementary Tables 1–3).
Statistics and reproducibility
Phylogenetic statistical analyses are described in Methods. Mt-genome coverage statistics (e.g., average, min/max, depth) were calculated in Geneious. Protein evaluation statistical analyses used Python (http://www.python.org), applying multiple tests library for multiple testing correction. ETE346 was used to perform PAML-CodeML45 selection analyses, and HyPhy48 to perform FEL47 selection analyses. Additional details of all experiments are presented in Supplementary Information.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Description of Additional Supplementary Files
Acknowledgements
We thank Clive Stone (Ngatiwai) cultural liaison for the Tuatara genome project for his support and cultural leadership. We thank Ngatiwai, tangata whenua of the rohe from which the Tuatara for genome sequencing was sampled, for supporting the permits and permissions needed to collect, export and sequence Tuatara blood samples. We also thank Lindsay Anderson and Dr. Nicola Nelson (VUW) who collected the blood sample used for the Tuatara genome. Tuatara genome sequencing was supported by grants from the Allan Wilson Center for Molecular Ecology and Evolution, and the University of Otago to NJG. We thank Oliver Ryder (San Diego Zoo) for facilitating sample import to the USA and Richard E. Green, Margot Hartley and Michelle Vierra of Dovetail Genomics (Santa Cruz, CA) for providing samples and details on DNA extraction. Steven L. Salzberg and Art Delcher provided valuable assistance in the assembly of Illumina data during early stages. Danny Lee at Seqmatic (Fremont, CA) provided laboratory assistance. Alex Copeland provided computational assistance. Laboratory advice was provided by Chris Bolds (Sage Science, Boston) and Damon Tigh (BioRad, CA). Marco Passamonti provided valuable information on DUI inheritance in mollusks. We are grateful to Johnathan B. Losos (Living Earth Collaborative, St. Louis) and the St. Louis Zoo for providing Stephens Island samples. We thank staff of the Laboratories of Analytical Biology, National Museum of Natural History (NMNH), Smithsonian Institution for the use of their computer resources, particularly Matt Kweskin. We thank Robert Costello, Smithsonian’s Q?rius, and Sydney Bergman for supporting ESB in this project. Kim Rutherford provided critical data transfer and associated files.
Author contributions
Study design and supervision: J.R.M., D.G.M., and N.J.G. Writing: J.R.M., D.G.M., L.U., S.Pa., V.L.G., and N.J.G. Long-read sequencing: C.G.B., J.R.M., E.S.B., V.L.G., and D.G.M. Transcriptome analysis: E.S.B., V.L.G., and D.G.M. Protein analysis: L.U., J.R.M., D.G.M., and N.J.G. Data handling, deposition, and analysis: S.Pa., D.G.M., C.G.B., D.P.D., E.S.B., V.L.G., P.M.H., S.Pr., A.N.E., J.R.M., and N.J.G.
Data availability
New DNA sequences generated in this study are deposited as GenBank MN864228– MN864230 and Sequence Read Archive (SRA) PRJNA445603. Published transcriptome sequences18 used in this study are from SRA SRA051647 and the complete mt-genome from that transcriptome is deposited as a Third Party Annotation in the DDBJ/ENA/GenBank databases as TPA: BK012001.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information is available for this paper at 10.1038/s42003-020-01639-0.
References
- 1.Breton S, Stewart DT. Atypical mitochondrial inheritance patterns in eukaryotes. Genome. 2015;58:423–431. doi: 10.1139/gen-2015-0090. [DOI] [PubMed] [Google Scholar]
- 2.Hedberg A, et al. Cancer-specific SNPs originate from low-level heteroplasmic variants in human mitochondrial genomes of a matched cell line pair. Mitochondrial DNA. 2019;30:82–91. doi: 10.1080/24701394.2018.1461852. [DOI] [PubMed] [Google Scholar]
- 3.Bratic A, Larsson N-G. The role of mitochondria in aging. J. Clin. Invest. 2013;123:951–957. doi: 10.1172/JCI64125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wei W, et al. Nuclear-mitochondrial DNA segments resemble paternally inherited mitochondrial DNA in humans. Nat. Commun. 2020;11:1740. doi: 10.1038/s41467-020-15336-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zouros E. Biparental inheritance through uniparental Transmission: the doubly uniparental inheritance (DUI) of mitochondrial DNA. Evol. Biol. 2013;40:1–31. [Google Scholar]
- 6.Macey JR, Larson A, Ananjeva NB, Fang Z, Papenfuss TJ. Two novel gene orders and the role of light-strand replication in rearrangement of the vertebrate mitochondrial genome. Mol. Biol. Evol. 1997;14:91–104. doi: 10.1093/oxfordjournals.molbev.a025706. [DOI] [PubMed] [Google Scholar]
- 7.Daugherty CH, Patterson GB, Hitchmough RA. Taxonomic and conservation review of the New Zealand herpetofauna. N.Z. J. Zool. 1994;21:317–323. [Google Scholar]
- 8.Rest JS, et al. Molecular systematics of primary reptilian lineages and the Tuatara mitochondrial genome. Mol. Phylogenet. Evol. 2003;29:289–297. doi: 10.1016/s1055-7903(03)00108-8. [DOI] [PubMed] [Google Scholar]
- 9.Mohandesan E, Subramanian S, Millar CD, Lambert DM. Complete mitochondrial genomes of Tuatara endemic to different islands of New Zealand. Mitochondrial DNA. 2015;26:25–26. doi: 10.3109/19401736.2013.840613. [DOI] [PubMed] [Google Scholar]
- 10.Subramanian S, Mohandesan E, Millar CD, Lambert DM. Distance-dependent patterns of molecular divergences in Tuatara mitogenomes. Sci. Rep. 2015;5:8703. doi: 10.1038/srep08703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pett W, Lavrov DV. Cytonuclear interactions in the evolution of animal mitochondrial tRNA metabolism. Genome Biol. Evol. 2015;7:2089–2101. doi: 10.1093/gbe/evv124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gemmell, et al. The tuatara genome reveals ancient features of amniote evolution. Nature. 2020;584:403–409. doi: 10.1038/s41586-020-2561-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thompson MB, Daugherty CH. Metabolism of Tuatara, Sphenodon punctatus. Comp. Biochem. Physiol. 1998;119A:519–522. [Google Scholar]
- 14.Jarvie J, Jowett T, Thompson MB, Seddon PJ, Cree A. Effects of warm temperatures on metabolic rate and evaporative water loss in Tuatara, a cool-climate Rhynchocephalian survivor. Physiol. Biochem. Zool. 2018;91:950–966. doi: 10.1086/698495. [DOI] [PubMed] [Google Scholar]
- 15.Macey JR, et al. Phylogenetic relationships among Agamid lizards of the Laudakia caucasia species group: testing hypotheses of biogeographic fragmentation and an area cladogram for the Iranian Plateau. Mol. Phylogenet. Evol. 1998;10:118–131. doi: 10.1006/mpev.1997.0478. [DOI] [PubMed] [Google Scholar]
- 16.Macey JR, et al. A molecular phylogenetic hypothesis for the Asian agamid lizard genus Phrynocephalus reveals discrete biogeographic clades implicated by plate tectonics. Zootaxa. 2018;4467:1–81. doi: 10.11646/zootaxa.4467.1.1. [DOI] [PubMed] [Google Scholar]
- 17.Weisrock DW, Macey JR, Ugurtas IH, Larson A, Papenfuss TJ. Molecular phylogenetics and historical biogeography among salamandrids of the "true" salamander clade: rapid branching of numerous highly divergent lineages in Mertensiella luschani associated with the rise of Anatolia. Mol. Phylogenet. Evol. 2001;18:434–448. doi: 10.1006/mpev.2000.0905. [DOI] [PubMed] [Google Scholar]
- 18.Miller HC, Biggs PJ, Voelckel C, Nelson NJ. De novo sequence assembly and characterisation of a partial transcriptome for an evolutionarily distinct reptile, the tuatara (Sphenodon punctatus) BMC. Genom. 2012;13:439. doi: 10.1186/1471-2164-13-439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seutin G, Lang BE, Mindell DI, Morais R. Evolution of the WANCY region in amniote mitochondrial DNA. Mol. Biol. Evol. 1994;11:329–340. doi: 10.1093/oxfordjournals.molbev.a040116. [DOI] [PubMed] [Google Scholar]
- 20.Macey JR, Larson A, Ananjeva NB, Papenfuss TJ. Replication slippage may cause parallel evolution in the secondary structures of mitochondrial transfer RNAs. Mol. Biol. Evol. 1997;14:30–39. doi: 10.1093/oxfordjournals.molbev.a025699. [DOI] [PubMed] [Google Scholar]
- 21.Arnheim N, et al. Molecular evidence for genetic exchanges among ribosomal genes on nonhomologous chromosomes in man and apes. Proc. Natl Acad. Sci. USA. 1980;77:7323–7327. doi: 10.1073/pnas.77.12.7323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumazawa Y, Ota H, Nishida M, Ozawa T. Gene rearrangements in snake mitochondrial genomes: highly concerted evolution of control-region-like sequences duplicated and inserted into a tRNA gene cluster. Mol. Biol. Evol. 1996;13:1242–1254. doi: 10.1093/oxfordjournals.molbev.a025690. [DOI] [PubMed] [Google Scholar]
- 23.Castoe TA, Jiang ZJ, Gu W, Wang ZO, Pollock DD. Adaptive evolution and functional redesign of core metabolic proteins in snakes. PLoS One. 2008;3(5):e2201. doi: 10.1371/journal.pone.0002201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ujvari B, Dowton M, Madsen T. Mitochondrial DNA recombination in a free-ranging Australian lizard. Biol. Lett. 2007;3:189–192. doi: 10.1098/rsbl.2006.0587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Passamonti M. An unusual case of gender-associated mitochondrial DNA heteroplasmy: the mytilid Musculista senhousia (Mollusca Bivalvia) BMC Evol. Biol. 2007;7(Suppl. 2):S7. doi: 10.1186/1471-2148-7-S2-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghiselli F, Milani L, Passamonti M. Strict sex-specific mtDNA segregation in the germ line of the DUI species Venerupis philippinarum (Bivalvia: Veneridae) Mol. Biol. Evol. 2011;28:949–961. doi: 10.1093/molbev/msq271. [DOI] [PubMed] [Google Scholar]
- 27.Morin PA, et al. Demography or selection on linked cultural traits or genes? Investigating the driver of low mtDNA diversity in the sperm whale using complementary mitochondrial and nuclear genome analyses. Mol. Ecol. 2018;27:2604–2619. doi: 10.1111/mec.14698. [DOI] [PubMed] [Google Scholar]
- 28.da Fonseca RR, Johnson WE, O’Brien SJ, Ramos MJ, Antunes A. The adaptive evolution of the mammalian mitochondrial genome. BMC Genomics. 2008;9:119. doi: 10.1186/1471-2164-9-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saier MH. Computer-aided analyses of transport protein sequences: gleaning evidence concerning function, structure, biogenesis, and evolution. Microbiol. Rev. 1994;58:71–93. doi: 10.1128/mr.58.1.71-93.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Macey JR, Papenfuss TJ, Kuehl JV, Fourcade HM, Boore JL. Phylogenetic relationships among amphisbaenian reptiles based on complete mitochondrial genomic sequences. Mol. Phylogenet. Evol. 2004;33:22–31. doi: 10.1016/j.ympev.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 31.Arevalo E, Davis SK, Sites JW. Mitochondrial DNA sequence divergence and phylogenetic relationships among eight chromosome races of the Sceloporus grammicus complex (Phrynosomatidae) in central Mexico. Syst. Biol. 1994;43:387–418. [Google Scholar]
- 32.Corl A, Davis AR, Kuchta SR, Comendant T, Sinervo B. Alternative mating strategies and the evolution of sexual size dimorphism in the side-blotched lizard, Uta stansburiana: a population-level comparative analysis. Evolution. 2010;64:79–96. doi: 10.1111/j.1558-5646.2009.00791.x. [DOI] [PubMed] [Google Scholar]
- 33.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zimin AV, et al. The MaSuRCA genome assembler. Bioinformatics. 2013;29:2669–2677. doi: 10.1093/bioinformatics/btt476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sommer DD, Delcher AL, Salzberg SL, Pop M. Minimus: a fast, lightweight genome assembler. BMC Bioinformatics. 2007;8:64. doi: 10.1186/1471-2105-8-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marçais G, Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011;27:764–770. doi: 10.1093/bioinformatics/btr011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hawkins TL, O’Connor-Morin T, Roy A, Santillan C. DNA purification and isolation using a solid-phase. Nucleic Acids Res. 1994;22:4543–4544. doi: 10.1093/nar/22.21.4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Loman NJ, Quick J, Simpson JT. A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat. Methods. 2015;12:733–735. doi: 10.1038/nmeth.3444. [DOI] [PubMed] [Google Scholar]
- 39.Robins JH, et al. Evolutionary relationships and divergence times among the native rats of Australia. BMC Evol. Biol. 2010;10:375. doi: 10.1186/1471-2148-10-375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Macey JR, Verma A. Homology in phylogenetic analysis: alignment of transfer RNA genes and the phylogenetic position of snakes. Mol. Phylogenet. Evol. 1997;7:272–279. doi: 10.1006/mpev.1997.0379. [DOI] [PubMed] [Google Scholar]
- 41.Swofford DL. PAUP* Phylogenetic Analysis Using Parsimony (* and Other Methods), Beta Version 4.0. Sunderland, MA: Sinauer; 2002. [Google Scholar]
- 42.Templeton AR. Phylogenetic inference from restriction endonuclease cleavage site maps with particular reference to the evolution of humans and the apes. Evolution. 1983;37:221–244. doi: 10.1111/j.1558-5646.1983.tb05533.x. [DOI] [PubMed] [Google Scholar]
- 43.Felsenstein J. Confidence limits on phylogenies with a molecular clock. Syst. Zool. 1985;34:152–161. [Google Scholar]
- 44.Maddison WP, Maddison DR. Evolution, Version 4.03. Sunderland, MA: Sinauer; 2001. MacClade, Analysis of Phylogeny and Character. [DOI] [PubMed] [Google Scholar]
- 45.Yang ZH. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 46.Huerta-Cepas J, Serra F, Bork P. ETE 3: reconstruction, analysis, and visualization of phylogenomic data. Mol. Biol. Evol. 2016;33:1635–1638. doi: 10.1093/molbev/msw046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kosakovsky Pond SL, Frost SDW. Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005;22:1208–1222. doi: 10.1093/molbev/msi105. [DOI] [PubMed] [Google Scholar]
- 48.Pond SL, Frost SD, Muse SV. HyPhy: hypothesis testing using phylogenies. Bioinformatics. 2005;21:676–679. doi: 10.1093/bioinformatics/bti079. [DOI] [PubMed] [Google Scholar]
- 49.Yang Z, Wong WS, Nielsen R. Bayes empirical bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 2005;22:1107–1118. doi: 10.1093/molbev/msi097. [DOI] [PubMed] [Google Scholar]
- 50.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. 1995;57:289–300. [Google Scholar]
- 51.Landau M, et al. ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 2005;33:W299–W302. doi: 10.1093/nar/gki370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ashkenazy H, et al. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016;44:W344–W350. doi: 10.1093/nar/gkw408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC.
- 54.Macey JR, Larson A, Ananjeva NB, Papenfuss TJ. Evolutionary shifts in three major structural features of the mitochondrial genome among Iguanian lizards. J. Mol. Evol. 1997;44:660–674. doi: 10.1007/pl00006190. [DOI] [PubMed] [Google Scholar]
- 55.Brennicke A, Clayton DA. Nucleotide assignment of alkali-sensitive sites in mouse mitochondrial DNA. J. Biol. Chem. 1981;256:10613–10617. [PubMed] [Google Scholar]
- 56.Hixson JE, Wong TW, Clayton DA. Both the conserved stem-loop and divergent 5'-flanking sequences are required for initiation at the human mitochondrial origin of light-strand DNA replication. J. Biol. Chem. 1986;261:2384–2390. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
New DNA sequences generated in this study are deposited as GenBank MN864228– MN864230 and Sequence Read Archive (SRA) PRJNA445603. Published transcriptome sequences18 used in this study are from SRA SRA051647 and the complete mt-genome from that transcriptome is deposited as a Third Party Annotation in the DDBJ/ENA/GenBank databases as TPA: BK012001.