

Abstract
In human airway smooth muscle (hASM), mitochondrial volume density is greater in asthmatic patients compared with normal controls. There is also an increase in mitochondrial fragmentation in hASM of moderate asthmatics associated with an increase in dynamin-related protein 1 (Drp1) and a decrease in mitofusin 2 (Mfn2) expression, mitochondrial fission, and fusion proteins, respectively. Proinflammatory cytokines such TNFα contribute to hASM hyperreactivity and cell proliferation associated with asthma. However, the involvement of proinflammatory cytokines in mitochondrial remodeling is not clearly established. In nonasthmatic hASM cells, mitochondria were labeled using MitoTracker Red and imaged in three dimensions using a confocal microscope. After 24-h TNFα exposure, mitochondria in hASM cells were more fragmented, evidenced by decreased form factor and aspect ratio and increased sphericity. Associated with increased mitochondrial fragmentation, Drp1 expression increased while Mfn2 expression was reduced. TNFα also increased mitochondrial biogenesis in hASM cells reflected by increased peroxisome proliferator-activated receptor-γ coactivator 1α expression and increased mitochondrial DNA copy number. Associated with mitochondrial biogenesis, TNFα exposure also increased mitochondrial volume density and porin expression, resulting in an increase in maximum O2 consumption rate. However, when normalized for mitochondrial volume density, O2 consumption rate per mitochondrion was reduced by TNFα exposure. Associated with mitochondrial fragmentation and biogenesis, TNFα also increased hASM cell proliferation, an effect mimicked by siRNA knockdown of Mfn2 expression and mitigated by Mfn2 overexpression. The results of this study support our hypothesis that in hASM cells exposed to TNFα mitochondria are more fragmented, with an increase in mitochondrial biogenesis and mitochondrial volume density resulting in reduced O2 consumption rate per mitochondrion.
Keywords: airway smooth muscle, fission, fusion, imaging, mitochondria
INTRODUCTION
Proinflammatory cytokines such as tumor necrosis factor-α (TNFα) have been shown to contribute to human airway smooth muscle (hASM) hypercontractility and cell proliferation associated with asthma (1–7). Mitochondria, the predominant source of ATP within a cell, are also involved in regulating intracellular Ca2+, cell proliferation, cell differentiation, and apoptosis (8–13). In this regard, mitochondria have tremendous potential to affect both hASM hyperreactivity and proliferation (14, 15). In many cell types, including hASM cells, a transient increase in cytosolic Ca2+ concentration ([Ca2+]cyt) leads to a transient increase in mitochondrial Ca2+ concentration ([Ca2+]mito) that stimulates ATP production through oxidative phosphorylation (14–20). This coupling between [Ca2+]cyt and [Ca2+]mito may reflect an “excitation-energy coupling” in ASM that matches ATP production via oxidative phosphorylation with the energy demand resulting from excitation-contraction coupling (14, 15, 21). We recently reported that TNFα increases force generation in porcine ASM, which is accompanied by an increase in ATP hydrolysis rate (3). Similarly, TNFα increases hASM cell proliferation (1, 6, 7), which is likely accompanied by an increase in ATP demand. Mitochondrial volume density is greater in hASM from asthmatic compared with nonasthmatic patients (22, 23). This increase in mitochondrial volume density in asthmatic hASM could reflect a response to an increase in energy demand within hASM cells with increased mitochondrial O2 consumption and ATP production (14, 15, 24, 25). The increase in mitochondrial volume density most likely results from increased mitochondrial biogenesis.
The mechanisms controlling mitochondrial biogenesis are tightly associated with those regulating mitochondrial network morphology (10, 26). The dynamic balance between mitochondrial fission and fusion regulates mitochondrial network morphology, which is usually described as tubular, reticular or filamentous. Dynamin-related protein 1 (Drp1) promotes mitochondrial fission or fragmentation by tethering to mitochondria at specific positions known as constriction sites (10, 27–29). In human cells, some studies have shown that fission 1 protein (hFis1) is involved in recruiting Drp1 (10, 30–33). The fusion of the outer membranes of adjacent mitochondria is dependent on the dimerization of two GTPase proteins, mitofusin 1 (Mfn1) and 2 (Mfn2), while the fusion of the inner membrane requires optic atrophy protein 1 (OPA1) (10, 27–29). Using two-dimensional (2-D) microscopic imaging, we recently reported that there is an increase in mitochondrial fission (i.e., fragmentation) in hASM of moderate asthmatics (34). This mitochondrial fragmentation in asthmatic hASM cells is associated with an increase in Drp1 and a decrease in Mfn2 expression (34). We also reported that TNFα exposure decreases Mfn2 expression in nonasthmatic hASM cells (35). Whether this TNFα-induced decrease in Mfn2 expression is associated with a concomitant increase in Drp1 expression and mitochondrial fragmentation was not explored. An increase in mitochondrial fission is generally associated with reduced mitochondrial O2 consumption (36–39), but in hASM, we found that knockdown of Mfn2 using small-interfering RNA (siRNA) was associated with an increase in total mitochondrial O2 consumption rate (40). However, in this study, we did not normalize O2 consumption rate for mitochondrial volume density. In the present study, we hypothesize that 24-h TNFα exposure increases Drp1 and reduces Mfn2 expression in hASM, resulting in increased mitochondrial fission, mitochondrial biogenesis and mitochondrial volume density and reduced O2 consumption rate per mitochondrion.
MATERIALS AND METHODS
Isolation of hASM Cells
Lung samples from third to sixth generation bronchi were obtained from anonymized female and male patients incidental to surgery at Mayo Clinic (approved and considered exempt by Mayo Institutional Review Board). The patients had no history of asthma or other chronic lung disease, and their surgeries related to resection of lung cancer, which included portions of otherwise normal lung tissue. As previously described (41, 42), hASM cells were dissociated from dissected airway tissue using papain and collagenase with ovomucoid/albumin separation as per manufacturer's instructions (Worthington Biochemical, Lakewood, NJ). hASM cells were maintained at 37°C (5% CO2-95% air) using phenol red-free DMEM/F-12 medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum in sterile culture flasks or eight-well glass-bottomed Lab-Tek chambers (Thermo Fisher Scientific, Waltham, MA). Freshly isolated hASM cells or from passages 1–3 of subcultures were used all experiments. Cell viability (tested by exclusion of Trypan Blue) and periodic assessment of hASM phenotype (smooth muscle actin, myosin, agonist receptors, and lack of fibroblast markers) were performed as previously described (1, 35, 43, 44).
Exposure of hASM Cells to TNFα
Before experiments, the medium was changed to DMEM/F-12 (no. 11039021, Thermo Fisher Scientific) lacking serum for 48 h. Two groups of hASM cells were then exposed to either TNFα (20 ng/mL) or left untreated for 24 h in DMEM/F-12 lacking serum at 37°C (5% CO2-95% air). Experiments were performed at 37°C, and pH was maintained at 7.4. Untreated and TNFα-treated groups were processed at the same time and under the same conditions.
Confocal Fluorescence Imaging of Mitochondria in hASM Cells
To visualize mitochondria in three-dimensional (3-D), hASM cells plated on eight-well glass-bottomed Labteks chambers were loaded with 300 nM MitoTracker Red CMXRos (no. M7512, Thermo Fisher Scientific; excitation wavelength: 568 nm; emission wavelength: 590 nm) for 5 min followed by extensive washing in dye-free solution. Mitochondria were visualized using a Nikon Eclipse A1 laser scanning confocal system (Nikon Instruments Inc., Melville, NY) at 12-bit resolution, 1,024 × 1,024 pixels, using a ×60/1.4 NA oil-immersion lens and experiments were performed at 37°C using a temperature-controlled stage. Optical sectioning was performed with a 0.5-µm step size (voxel dimensions: 0.207 × 0.207 × 0.5 µm) (41, 43, 45–47). Typically, multiple hASM cells were visualized within a single microscopic field, and optical sections included the full depth of each cell within the field. Because the large size of hASM cells leads to overlapping of some cell borders, we analyzed only those cells whose borders were not overlapping. Typically, this selection process resulted in our analyzing two to three cells per single microscopic field. A total of five complete individual hASM cells per sample was analyzed for each experimental group (untreated or TNFα-treated), generated from each bronchial sample (n = 25 cells per group), with each patient sample having equal numbers of control and TNFα-treated cells contributing to the data set.
Image Deconvolution, Processing, and 3-D Reconstruction
Each 2-D fluorescences image within the Z-stack was deconvolved using NIS-Elements software [Nikon Instruments Inc., Research Resource Identifier (RRID): SCR_014329] and a blind deconvolution algorithm (Point Scan Confocal, 3 iterations) as previously described (46). Deconvolution increases the spatial resolution nearly twofold, which was confirmed by assessment of point spread functions in a previous study (46). The deconvolved images were then processed for backgound correction, ridge filter detection, skeletonization, and thresholding using ImageJ software (https://imagej.nih.gov/ij/, Fiji, RRID: SCR_002285) as previously described (43, 46, 48–53). The morphological top hat transform and median filter were used to segment mitochondria (50–52), and images were then thresholded using the Default method in the ImageJ/Fiji software (48). The deconvolved/processed images were then reconstructed in 3-D using either ImageJ software for mitochondrial morphologic analysis or NIS-Elements software for volume measurements.
Mitochondrial Localization within hASM Cells
To examine if TNFα exposure affected the localization of mitochondria within hASM cells, thresholding was applied to the empty space corresponding to the nucleus and the centroid was computed using ImageJ software. Mitochondria were then assessed with respect to their distances (i.e., closest point) to the nuclear centroid. hASM cells used for mitochondrial localization (5 hASM cells for both untreated and TNFα-treated group per bronchial sample and five bronchial samples, n = 25 cells per group) were also used for mitochondrial morphology.
Mitochondrial Morphology
For 2-D and 3-D analysis of mitochondrial morphology, deconvolved Z-stacks images were processed using the Mitochondria Analyzer plugin for ImageJ/Fiji as previously described (48). Three morphological metrics were used to determine the balance between mitochondrial fusion and fission in hASM cells with or without exposure to TNFα as described previously (48, 50–52): 1) form factor defined as perimeter2/4π·area, which indicates mitochondrial length and degree of branching (50–52); 2) aspect ratio defined as the ratio of the major to minor axes, which indicates mitochondrial length (50–52); and 3) sphericity (36πV2/S3) (48) calculated from volume (V – determined from the number of voxels within defined cell boundaries) and surface area (S) measurements. To determine if mitochondrial orientation is anisotropic or isotropic, fluorescent images were optically sliced in the orthogonal XZ and YZ planes and analyzed using ImageJ software. Form factor, aspect ratio, and sphericity were measured for the same hASM cell (5 hASM cells for both untreated and TNFα-treated groups per bronchial sample and five bronchial samples; n = 25 cells per group).
Mitochondrial Volume/Density
The Z-stacks images were processed as described above, and after 3-D reconstruction and delimitating the boundaries of each hASM cell, voxels containing mitochondria were determined by thresholding MitoTracker Red fluorescence. The volume of the cell occupied by mitochondria was determined from the number of positive MitoTracker Red voxels and normalized to the total volume of the hASM cell. These measurements were performed using NIS-Elements software using the 3 D object measurement plug-in (Nikon Instruments, Inc.) (46, 54). A total of five individual hASM cells per patient were analyzed for each group (untreated or TNFα treated) for each bronchial sample with a total of 5 samples (n = 25 cells per group).
Automated Capillary Electrophoresis Western Analysis of Protein Expression
Human ASM cells plated on a six-well plate were lysed using Cell Lysis Buffer (no. 9803S, Cell Signaling Technology, Danvers, MA) supplemented with cOmplete Protease Inhibitor Cocktail (no. 11697498001, Sigma-Aldrich, St. Louis, MO) and PhosSTOP Phosphatase Inhibitor Cocktail (no. 4906845001, Roche Molecular Biochemicals, Mannheim, Germany). Protein (0.05–0.8 µg) was loaded into microplates provided by the manufacturer, and capillary electrophoresis Western analysis was carried out following the manufacturer’s instructions (ProteinSimple WES, San Jose, CA) (55) using anti-Drp1 (ab56788, RRID: AB_941306), anti-Mfn2 (ab50843, RRID: AB_2235186), anti-porin (VDAC1, ab15895, RRID: AB_2214787), anti-peroxisome proliferator-activated receptor-γ coactivator-1α (PGC1α, ab106814, RRID: AB_10865490) (Abcam, Cambridge, MA), and anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH, g9545, RRID: AB_796208) (Sigma-Aldrich) antibodies. The dilution for all antibodies was 1:25. Antibodies were validated in previous studies using siRNAs, overexpression vectors, and/or positive control lysates (34, 35, 40). The resulting data (area under the curve) were analyzed with the inbuilt Compass software (ProteinSimple). Drp1, Mfn2, porin, and PGC1α proteins expression were normalized relative to GAPDH expression as a control for protein loading. hASM cells from a total of five bronchial samples were used for protein expression analysis. Proteins from untreated and TNFα-treated groups (split from the same bronchial sample, n = 5 samples per group) were prepared and analyzed using automated capillary electrophoresis Western analysis in parallel and not in separate experiments.
Quantification of Mitochondrial DNA Copy Number
Total DNA from hASM cells was extracted and purified using QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). The relative number of copies of human mitochondrial DNA was examined using the human mitochondrial to nuclear DNA ratio kit (no. 7246, Takara Bio USA, Mountain View, CA) (56). Real-time PCR was carried out following the manufacturer’s instructions using two sets of genomic [solute carrier organic anion transporter family member 2B1 (SLCO2B1) and serpin family A member 1 (SERPINA1)] and mitochondrial [NADH dehydrogenase subunit 1 (ND1) and NADH:ubiquinone oxidoreductase core subunit 5 (ND5)] primers (Takara Bio USA) (56). hASM cells from a total of five bronchial samples were used for DNA analysis. DNA from untreated and TNFα-treated groups (split from the same bronchial sample; n = 5 samples per group) were prepared and analyzed in parallel and not in separate experiments.
Measurement of Mitochondrial Respiration
Approximately 50,000 hASM cells were seeded onto each well of 24-well XF-24 plates (no. 100777-004, SeaHorse Biosciences, Billerica, MA) in DMEM Complete medium supplemented with 10% fetal bovine serum. After the cells had adhered, the medium was changed to DMEM/F-12 lacking serum for 48 h. Two groups of hASM cells were then exposed to either TNFα (20 ng/mL) or left untreated for 24 h in DMEM/F-12 lacking serum. The XFe24 Extracellular Flux Analyzer (SeaHorse Biosciences) was used to measure oxygen Consumption Rate (OCR; an indicator of mitochondrial respiration), and measurements were performed in the presence of 10 mM glucose, 1 mM pyruvate, and 2 mM glutamine. Inhibitors (no. 103015-100, Seahorse XF Cell Mito Stress Test kit) used to test different aspects of mitochondrial respiration were as follows: 1 µM oligomycin (ATP uncoupler), 1.25 µM FCCP (accelerates electron transport chain), and 1 µM antimycin A (a Complex III inhibitor) with 1 µM rotenone (a Complex I inhibitor), allowing for determination of basal respiration, ATP production, maximum respiration, and spare respiratory capacity, respectively (57, 58). Values were normalized for total protein or mitochondrial volume density post hoc. hASM cells from a total of five bronchial samples were used for O2 consumption rate measurement. O2 consumption rate from untreated and TNFα-treated groups (from the same bronchial sample n = 5 samples per group) was measured simultaneously (same plate) and not in separate experiments.
Cell Transfection
Human ASM cells were plated on eight-well Labteks for real-time fluorescence imaging or six-well plate for protein expression analysis. Cells were grown to ∼80% confluence before being serum deprived for transfection with 200 pM small-interfering RNA (siRNA) against Mfn2 (Integrated DNA Technologies, Inc., Coralville, IA), using Lipofectamine transfection reagent (Thermo Fisher Scientific) as previously described (34, 40, 44). For overexpressing Mfn2, human ASM cells were transfected with a plasmid encoding YFP-tagged Mfn2 construct (plasmid no. 28010; RRID: Addgene_28010, Addgene, Cambridge, MA) (59) using Lipofectamine 2000 (no. 11668019, Thermo Fisher Scientific) (44). Transfection controls included vehicle-alone treatment, scrambled (nontargeting) RNA and empty plasmids.
CyQuant Cell Proliferation Assay
To quantify hASM cell proliferation, hASM cells (∼5,000 ASM) were grown in 96-well plates, serum deprived for 48 h, siRNA transfected, transfected with Mfn2 overexpression plasmid and/or exposed to TNFα (20 ng/mL) for 24 h, and exposed to CyQuant NF for 30 min as per manufacturer's instructions (no. C35006, Thermo Fisher Scientific). Proliferation of hASM cells based on DNA content was measured as described previously (1). Dye (CyQuant) binding to DNA (fluorescence) was measured using microplate reader (Molecular Devices, Sunnyvale, CA). Dye calibrations and normalization were performed as per manufacturer's instructions and as previously described (1). hASM cells from a total of five bronchial samples were used for cell proliferation assay. For each bronchial sample, hASM cells were split in several groups: untreated, TNFα treated, siRNA transfected, Mfn2 overexpression plasmid transfected, Mfn2 overexpression plasmid transfected and exposed to TNFα, vehicle-alone treatment, scrambled (nontargeting) RNA, and empty plasmid groups (from the same bronchial sample) and measurement were performed simultaneously (within the same 96-well plate).
Statistical Analysis
Human ASM cells were dissociated from bronchial samples from female and male patients and split into TNFα-treated and untreated groups. Twenty total bronchial samples were used with five samples used for each experimental protocol, and some samples used for several but not all experimental protocols as follows: imaging studies, protein expression analysis, DNA analysis, hASM cell proliferation, and O2 consumption. N values correspond to the numbers of samples and when appropriate n values represent the number of hASM cells and are provided in the figure legends. For imaging studies, measurements were performed for five cells per group (TNFα-treated and untreated) for each of hASM samples (N = 5 patients) (i.e., n = 25 hASM cells for both TNFα treated and untreated groups). In these imaging studies, TNFα-treated and untreated groups were compared using a paired t test. For biochemical measurements (protein expression analysis, mitochondrial DNA copy number), a paired t test was used to compare TNFα-treated and untreated groups (N = 5). For O2 consumption measurement and hASM cell proliferation, repeated measures were performed as per manufacturer's instructions and for hASM cell proliferation several groups were compared (TNFα-treated and untreated groups, siRNA transfected). Accordingly, one-way ANOVA (with repeated measures) was used. Bonferroni correction was applied for multiple comparisons. Statistical significance was established at P < 0.05. Data are presented as medians and interquartile range represented as a box-and-whisker plot.
RESULTS
Morphology of Dissociated hASM Cells
As previously observed (1, 35, 43, 44), dissociated hASM cells expressed contractile proteins (smooth muscle actin and myosin) and could be readily distinguished from other cells by their spindle shape (Figs. 1 and 2). Table 1 summarizes the morphology of hASM cells and their nuclei in TNFα treated and untreated groups. The length of dissociated hASM cells was ∼15- to 16-fold greater than the width or height (Z-axis) of the cells. The nucleus was centrally located and comprised ∼16% of total hASM cell volume. Following 24-h TNFα treatment, there was no difference in any of the morphometric parameters of hASM cells.
Figure 1.
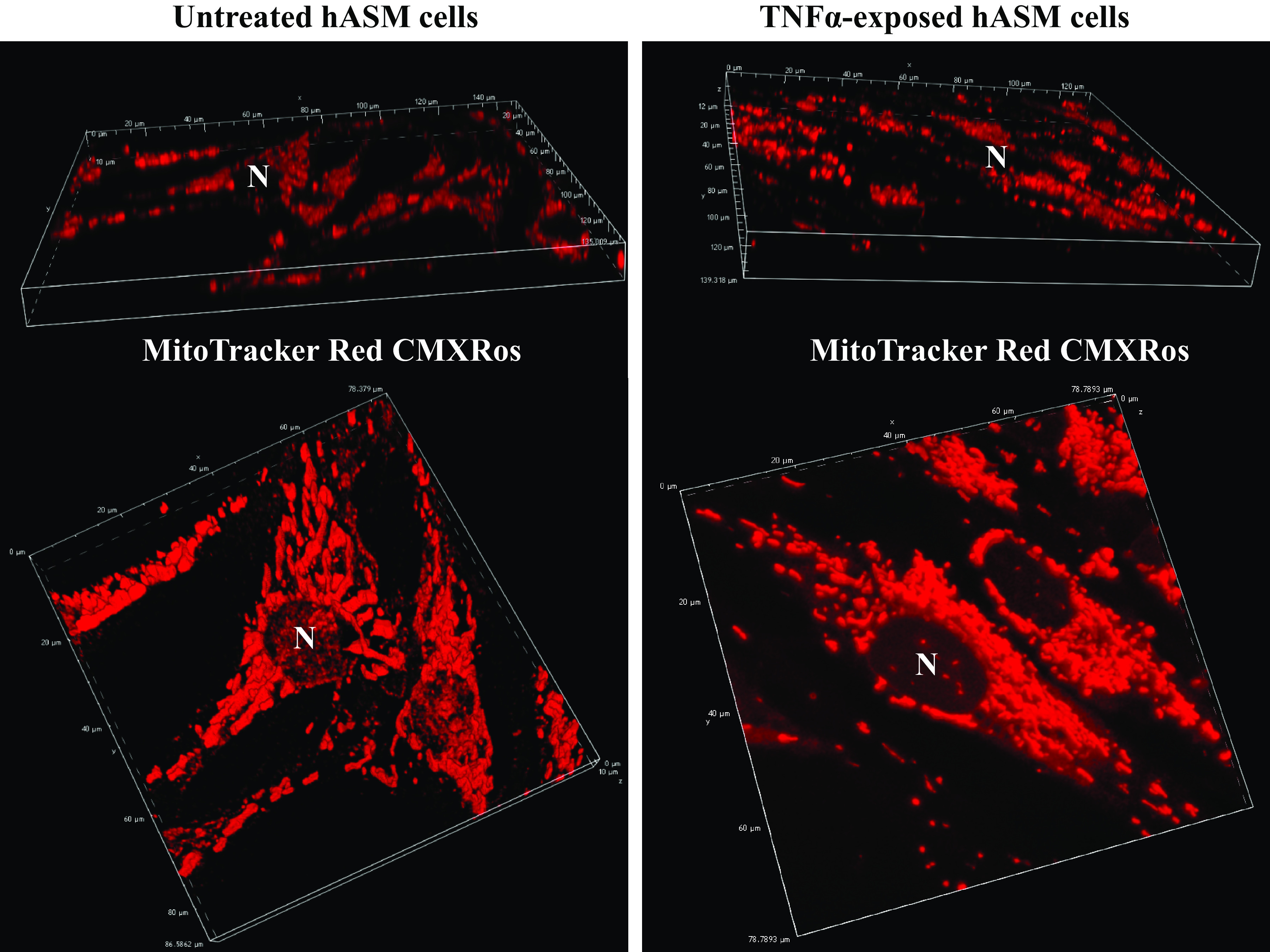
Representative 3-dimensional (3-D) reconstruction of Z-stacks fluorescent confocal images of human airway smooth muscle (hASM) cells untreated or treated with TNFα loaded with MitoTracker Red CMXRos to visualize mitochondria. Multiple hASM cells (top) were visualized within a single microscopic field and were used for mitochondrial distribution, mitochondrial morphology, and mitochondrial volume density measurements. Expanded view of hASM cells loaded with range represented MitoTracker Red CMXRos (bottom) showing that mitochondria are more fragmented in hASM cells treated with TNFα compared with untreated hASM cells. N, nucleus.
Figure 2.
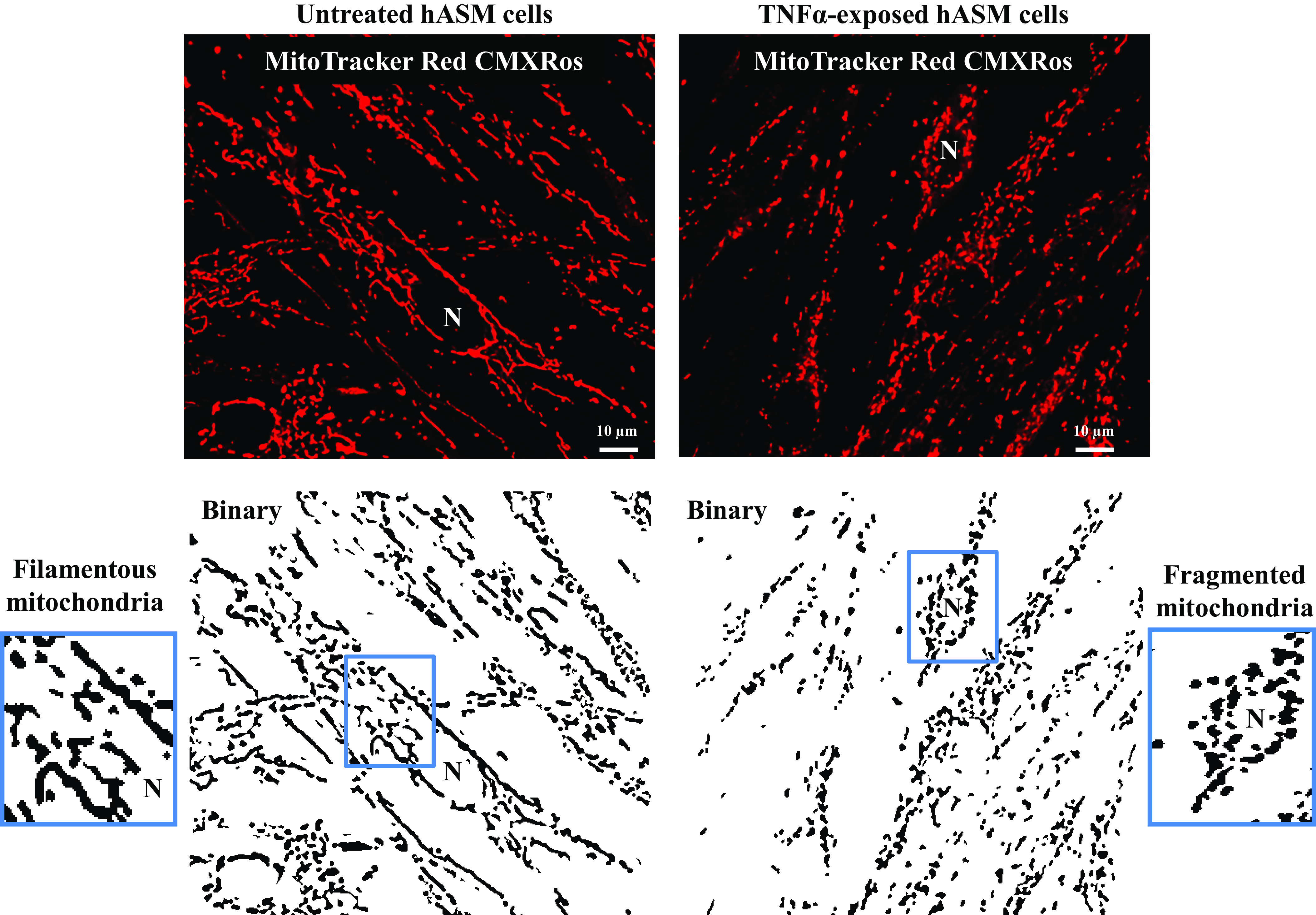
Representative 2-dimensional (2-D) fluorescent confocal images of human airway smooth muscle (hASM) cells untreated or treated with TNFα loaded with MitoTracker Red CMXRos to visualize mitochondria and their corresponding binary images showing mitochondrial morphology. In untreated hASM cells, mitochondria appear filamentous, while in TNFα-treated hASM cells, mitochondria appear fragmented or globular. N, nucleus.
Table 1.
Imaging metrics for untreated and TNFα-exposed hASM cells
| Untreated |
TNFα Treated |
|||
|---|---|---|---|---|
| Whole Cell | Nucleus | Whole Cell | Nucleus | |
| Major axis (length), µm | 243.75 ± 39.30 | 18.88 ± 3.09 | 288.55 ± 44.31 | 19.66 ± 2.77 |
| Minor axis (maximum), µm | 15.92 ± 3.69 | 11.11 ± 2.35 | 19.11 ± 2.36 | 13.33 ± 3.01 |
| Z axis (maximum), µm | 12.22 ± 1.98 | 11.01 ± 2.33 | 13.05 ± 1.59 | 11.01 ± 2.11 |
| Volume, µm3 | 5814.86 ± 332.47 | 942.69 ± 43.23 | 6140.26 ± 589.32 | 968.48 ± 51.36 |
Values are means ± SD; N = 5 patients (3 males and 2 females); n = 25 human airway smooth muscle (hASM) cells (freshly isolated or from passages 1–3 of subculture).
Mitochondrial Labeling and Location within hASM Cells
As previous reported (43), MitoTracker Red clearly labeled mitochondria within hASM cells (Figs. 1 and 2). Also as previously noted (43), there was a more distinct labeling of mitochondria near the nucleus (i.e., perinuclear) and mitochondria appeared dynamic with considerable movement. To analyze if mitochondria distribution within hASM cells was affected by TNFα exposure, mitochondria filaments were analyzed with respect to their distance from the nuclear centroid (Fig. 3, A and B). As can be seen, there was a predominance of perinuclear location of mitochondria within hASM cells as evidenced by the higher incidence of mitochondria at shorter distances from the centroid (Fig. 3, A and B). Approximately 60% of all mitochondrial filaments were located within 30 µm of the nuclear centroid in untreated hASM cells (Fig. 3, A and C). Following 24-h exposure to TNFα, there was a significant shift in the distribution of mitochondria away from the nucleus (Fig. 3B). In contrast to untreated hASM cells, only ∼48% of all mitochondria were located within 30 µm of the nuclear centroid in TNFα-treated hASM cells (Fig. 3, B and C). We also optically sliced mitochondrial images in the orthogonal XZ and YZ planes to determine if mitochondrial orientation in hASM cells is anisotropic or isotropic (Fig. 4). In hASM cells, the Z-axis is much shorter (∼10 µm) as compared with the X (∼100 µm) and Y (∼50 µm)-axes. In addition, as we have previously reported the spatial resolution in the Z axis is complicated by Z-axis distortion of ∼10%. With these caveats, we saw no evidence to suggest that mitochondrial orientation is anisotropic (Fig. 4). Indeed, the orientation appears to be isotropic along the long axis of hASM cells (Fig. 4).
Figure 3.

Effect of TNFα on mitochondrial distribution with respect to their distances to nuclear centroids. A and B: representative binary image showing mitochondrial morphology in untreated hASM cells (A) or TNFα-treated human airway smooth muscle (hASM) cells (B) (C: nuclear centroid computed using ImageJ). C: summary data showing the distribution of mitochondrial filaments as %total mitochondria in respect to their distances (in µm) to the nuclear centroid. Treated and untreated groups were collected at the same time and under the same conditions. Data are presented as medians and interquartile range represented as a box-and-whisker plot. Results were analyzed using a paired t test [N = 5 patients (3 males and 2 females), 5 cells per group (TNFα-treated and untreated) for each of hASM samples, n = 25].
Figure 4.
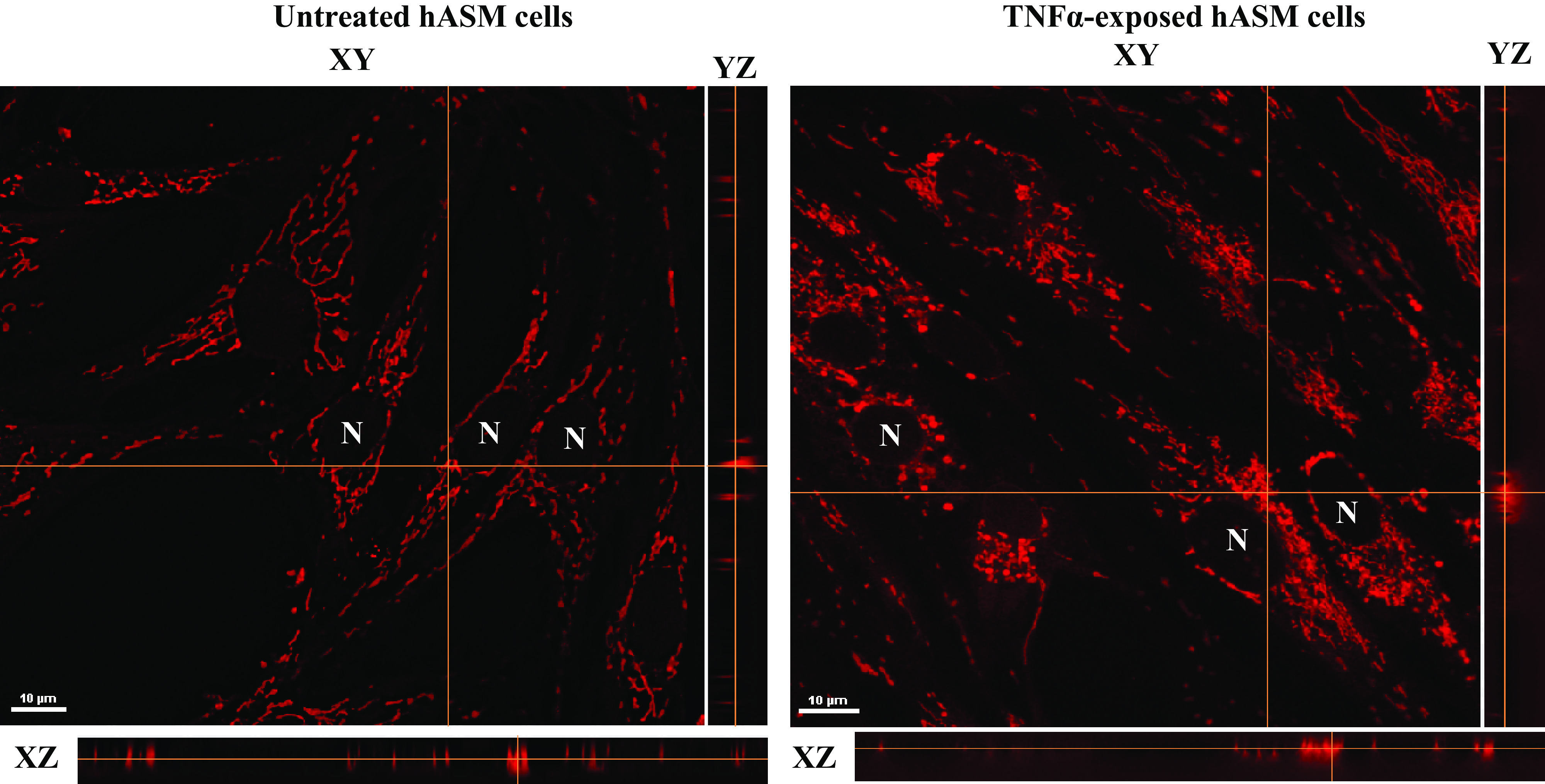
Representative 2-dimensional (2-D) fluorescent confocal images of human airway smooth muscle (hASM) cells untreated or treated with TNFα loaded with MitoTracker Red CMXRos to visualize mitochondria and their corresponding orthogonal XY and YZ slices showing that mitochondrial orientation is isotropic along the long axis of hASM cells. No differences were found between hASM cells untreated or treated with TNFα. N, nucleus.
Effect of TNFα on Mitochondrial Fission and Fusion
In untreated hASM cells, mitochondrial network usually appears filamentous and branched (Fig. 1 and 2) with few fragmented or globular mitochondria. By contrast, there were few filamentous mitochondria in hASM cells exposed to TNFα, and mitochondria appeared more fragmented or globular indicating mitochondrial fission (Fig. 1 and 2). Three mitochondrial morphological metrics, form factor, aspect ratio, and sphericity (48, 50–52, 60), were used to quantify the degree of mitochondrial fission and fusion (Fig. 5). In hASM cells exposed to TNFα, form factor and aspect ratio were reduced by ∼72% and ∼61%, respectively, compared with untreated hASM cells (Fig. 5, A and C; P < 0.05). In untreated hASM cells, the number of fragmented mitochondrial filaments (form factor <2 to 4) compared with longer filaments (form factor >4) was relatively evenly distributed (Fig. 5B). By contrast, more than 80% of mitochondria in hASM cells exposed to TNFα were fragmented as evidenced by a form factor of <2, with more than 90% having a form factor <4 (Fig. 5B). Comparable results were obtained from analysis of aspect ratio. In untreated hASM cells, ∼79% of mitochondria were relatively filamentous as evidenced by an aspect ratio of 4 or more (Fig. 5D). By contrast, ∼78% of mitochondria in hASM cells exposed to TNFα were fragmented as evidenced by an aspect ratio of <4 (Fig. 5D). Similarly, the sphericity of mitochondria in hASM cells exposed to TNFα was ∼50% greater than untreated hASM cells (Fig. 5, E and F; P < 0.05). The sphericity of ∼90% of all mitochondria in untreated hASM cells was <0.50 (Fig. 5F). By contrast, in hASM cells exposed to TNFα <50% of mitochondria had a sphericity of <0.50 (Fig. 5F).
Figure 5.

Effect of TNFα on mitochondrial morphology. Two- or three-dimensional (2-D or 3-D) fluorescent confocal images of human airway smooth muscle (hASM) cells untreated or treated with TNFα loaded with MitoTracker Red CMXRos to visualize mitochondria were analyzed to compute three morphological metrics: form factor, aspect ratio, and sphericity. A: summary data showing that form factor is decreased in hASM cells treated with TNFα compared with untreated hASM cells. B: distribution of mitochondrial filaments (%total) in respect to form factor showing that in hASM cells treated with TNFα, a majority of mitochondrial filaments had a form factor <2 while in untreated hASM cells mitochondrial filaments were relatively evenly distributed in respect to form factor. C: summary data showing that aspect ratio is decreased in hASM cells treated with TNFα compared with untreated hASM cells. D: distribution of mitochondrial filaments (%total) in respect to aspect ratio showing that in hASM cells treated with TNFα, a majority of mitochondrial filaments had an aspect ratio <4. By contrast, a majority of mitochondria in untreated hASM cells had an aspect ratio of 4 or more. E: summary data showing that sphericity is increased in hASM cells treated with TNFα compared with untreated hASM cells. F: distribution of mitochondrial filaments (% of total) in respect to sphericity showing that in hASM cells treated with TNFα, few mitochondrial filaments had a sphericity of 0.25 or less, while in untreated hASM cells, a majority of mitochondria had a sphericity of 0.25 or less or between 0.25 and 0.5. Treated and untreated groups were collected at the same time and under the same conditions. Data are presented as medians and interquartile range represented as a box-and-whisker plot. Results were analyzed using a paired t test. *Significant difference compared with untreated hASM cells [P < 0.05, N = 5 patients (3 males and 2 females), 5 cells per group (TNFα-treated and untreated) for each of hASM samples, n = 25].
Effect of TNFα on Drp1 and Mfn2 Expression
As we previously reported (34, 35), both Drp1 and Mfn2 were expressed in hASM cells (Fig. 6). Following 24-h exposure to TNFα, Drp1 expression in hASM cells increased by ∼42% compared with untreated hASM cells (Fig. 6B; P < 0.05). By contrast, 24-h TNFα exposure resulted in an ∼47% decrease in Mfn2 expression in hASM cells (Fig. 6C; P < 0.05).
Figure 6.

TNFα increases dynamin-related protein 1 (Drp1) and reduces mitofusin 2 (Mfn2) protein expression. Drp1 and Mfn2 protein expression was measured using automated capillary electrophoresis western analysis. A and B: a computer-generated image using Compass software showing Drp1, Mfn2, and GAPDH protein expression in human airway smooth muscle (hASM) cells treated with TNFα (T) or untreated (UT) (A) and representative densitometry tracings showing Drp1 and Mfn2 protein expression in hASM cells treated with TNFα or untreated (B). MW, molecular weight. C: summary data showing an increase in Drp1 relative to GAPDG expression in hASM cells treated with TNFα compared with untreated hASM cells. D: summary data showing a decrease in Mfn2 relative to GAPDG expression in hASM cells treated with TNFα compared with untreated hASM cells. Treated and untreated groups were collected at the same time and under the same conditions. Data are presented as medians and interquartile range represented as a box-and-whisker plot. Results were analyzed using a paired t test. *Significant difference compared with untreated hASM cells (P < 0.05, N = 5 patients, 3 males and 2 females).
TNFα Increases Mitochondrial Biogenesis
Mitochondrial biogenesis in hASM cells was assessed by measuring PGC1α protein expression and by determining the ratio of mitochondrial DNA to genomic DNA (Fig. 7). In hASM cells exposed to TNFα for 24 h, expression of PGC1α increased by ∼33% compared with untreated hASM cells (Fig. 7; P < 0.05). Further evidence that TNFα exposure induced mitochondrial biogenesis in hASM cells was provided by an ∼45% increase in the relative number of copies of human mitochondrial DNA (ND1/SLCO2B1 and ND5/SERPINA1) compared with untreated hASM cells (Fig. 8, A and B; P < 0.05).
Figure 7.

TNFα increases mitochondrial biogenesis. Peroxisome proliferator-activated receptor-γ coactivator 1α (PGC1α) protein expression and mitochondrial DNA copy number were examined as indicators of mitochondrial biogenesis in human airway smooth muscle (hASM) cells. A: computer generated image using Compass software showing PGC1α protein expression relative to GAPDH protein expression in hASM cells treated with TNFα (T) and untreated (UT) hASM cells. MW, molecular weight. B: representative densitometry tracings showing PGC1α protein expression in hASM cells treated with TNFα and untreated hASM cells. C: representative densitometry tracings showing GAPDG protein expression in hASM cells treated with TNFα and untreated hASM cells. D: summary data showing that TNFα significantly increases PGC1α protein expression in hASM cells compared with untreated hASM cells. Treated and untreated groups were collected at the same time and under the same conditions. Data are presented as medians and interquartile range represented as a box-and-whisker plot. Results were analyzed using a paired t test. *Significant difference compared with untreated hASM cells (P < 0.05, N = 5 patients, 3 males and 2 females).
Figure 8.

TNFα increases mitochondrial DNA copy number. Mitochondrial DNA copy number was examined using real-time PCR and 2 sets of genomic [solute carrier organic anion transporter family member 2B1 (SLCO2B1)] and [serpin family A member 1 (SERPINA1)] and mitochondrial ([solute carrier organic anion transporter family member 2B1 (SLCO2B1)] and [serpin family A member 1 (SERPINA1)] primers. A and B: summary data showing that TNFα significantly increases the ratio NADH dehydrogenase subunit 1 (ND1)/SLCO2 (A) and NADH:ubiquinone oxidoreductase core subunit 5 (ND5)/SERPINA1 (B) copy number in hASM cells compared with untreated hASM cells. Treated and untreated groups were collected at the same time and under the same conditions. Data are presented as medians and interquartile range represented as a box-and-whisker plot. Results were analyzed using a paired t test. *Significant difference compared with untreated hASM cells (P < 0.05, N = 5 patients, 3 males and 2 females).
TNFα Increases Mitochondrial Volume Density
Mitochondrial volume density in hASM cells was determined from independent determinations of mitochondrial volume (number of MitoTracker Red-positive voxels; Fig. 1) and hASM cell volume (from 3-D reconstructed hASM cells; Table 1). The volume of hASM cells was unaffected by TNFα exposure (Table 1). However, after TNFα exposure and mitochondrial biogenesis, mitochondrial volume density in hASM cells exposed to TNFα (7.6 ± 0.8%) was increased by 3.3-fold (Fig. 9A; P < 0.05). The increase in mitochondrial volume density in hASM cells induced by TNFα exposure was also evidenced by a 2.5-fold increase in porin (VDAC) protein expression (Fig. 9, A and B; P < 0.05).
Figure 9.
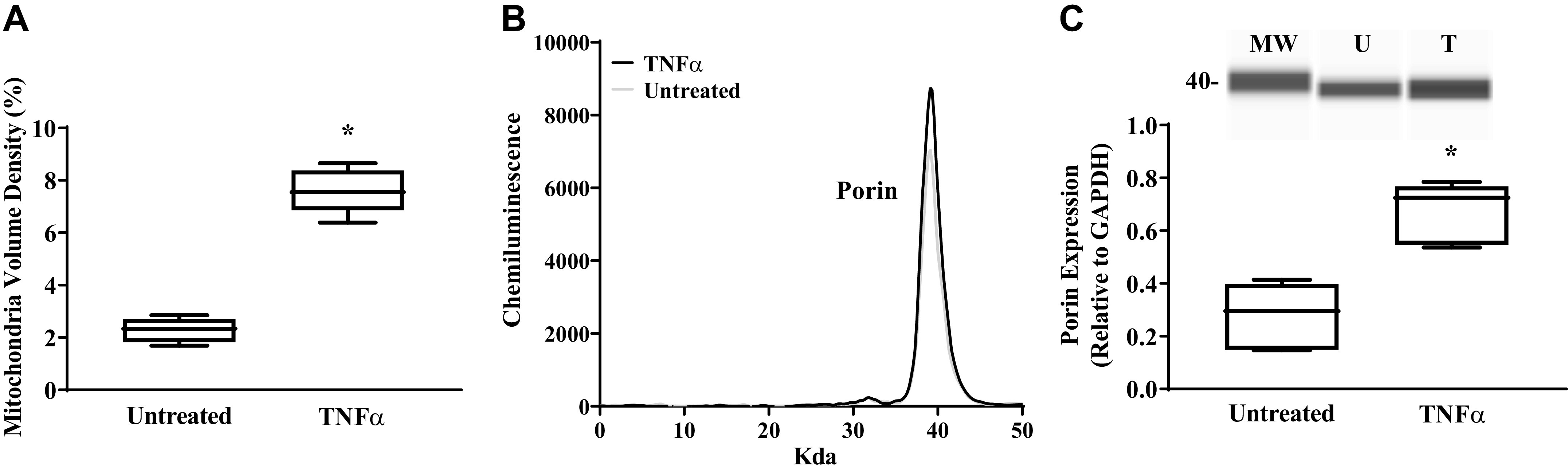
TNFα increases mitochondrial volume density and porin protein expression. A: Mitochondrial volume density was measured using Z-stacks fluorescent images of human airway smooth muscle (hASM) cells and 3-dimensional reconstruction using NIS-Elements software. Mitochondrial volume density was significantly higher in hASM cells treated with TNFα than in untreated hASM cells. B: porin protein expression was measured using automated capillary electrophoresis Western analysis. Representative densitometry tracings showing porin protein expression in hASM cells treated with TNFα and untreated hASM cells. C: a computer-generated image using Compass software (top) and summary data (bottom) showing an increase in porin expression in hASM cells treated (T) with TNFα compared with untreated (UT) hASM cells. MW, molecular weight. Treated and untreated groups were collected at the same time and under the same conditions. Data are presented as medians and interquartile range represented as a box-and-whisker plot. Results were analyzed using a paired t test for mitochondrial volume density (N = 5 patients (3 males and 2 females), 5 cells per group (TNFα-treated and untreated) for each of hASM samples, n = 25) and for porin protein expression (n = 5 patients, 3 males and 2 females). MW, molecular weight; U, untreated; T, treated. *Significant difference (P < 0.05) compared with untreated hASM cells.
Effect of TNFα on Maximum Mitochondrial Respiration
The increase in mitochondrial volume density in hASM cells induced by TNFα exposure was reflected by an ∼2.6-fold increase in maximum mitochondrial O2 consumption measured using the XFe24 Extracellular Flux Analyzer (Fig. 10A; P < 0.05). In contrast, when mitochondrial O2 consumption was normalized for mitochondrial volume density, maximum O2 consumption rate was ∼22% lower in hASM cells exposed to TNFα compared with untreated hASM cells (Fig. 10B; P < 0.05). This reflected a TNFα-induced decrease in maximum O2 consumption rate per mitochondrion.
Figure 10.

Effect of TNFα on maximum O2 consumption rate. O2 consumption rate in human airway smooth muscle (hASM) cells was measured using the XFe24 Extracellular Flux Analyzer and inhibitors of mitochondrial respiration [1 µM oligomycin (ATP uncoupler), 1.25 µM FCCP (accelerates electron transport chain), and 1 µM antimycin A (a Complex III inhibitor) with 1 µM rotenone (a Complex I inhibitor)], allowing for determination of maximum respiration. A: maximum O2 consumption rate in hASM cells treated with TNFα or left untreated were normalized for total protein post hoc. Maximum O2 consumption rate was significantly higher in hASM cells treated with TNFα than in untreated hASM cells. B: maximum O2 consumption rate in hASM cells treated with TNFα or left untreated were normalized for mitochondrial volume density post hoc. Maximum O2 consumption rate per mitochondrion was significantly higher in hASM cells treated with TNFα than in untreated hASM cells Treated and untreated groups were collected at the same time and under the same conditions. Data are presented as medians and interquartile range represented as a box-and-whisker plot. Results were analyzed using one-way ANOVA (with repeated measures). *Significant difference (P < 0.05) compared with untreated hASM cells (N = 5 patients, 3 males and 2 females).
TNFα Induced ASM Cell Proliferation: Role of Reduced Mfn2 Expression
The effect of 24-h TNFα exposure on the proliferation of hASM cells was examined by assessing the binding of the fluorescent dye CyQuant to DNA (Fig. 11). Twenty-four-hour TNFα exposure to TNFα caused an ∼80% increase in hASM cell proliferation compared with untreated hASM cells or hASM cells transfected with nontargeting siRNA (Fig. 11; P < 0.05). This effect of TNFα exposure was mimicked to some extent by siRNA knockdown of Mfn2 and mitigated by overexpressing Mfn2 (Fig. 11; P < 0.05).
Figure 11.

Effect of siRNA knockdown or overexpression of mitofusin 2 (Mfn2) on human airway smooth muscle (hASM) cell proliferation. After siRNA knockdown of Mfn2 expression, or treatment with TNFα with or without transfection with Mfn2 overexpression plasmid, hASM cell proliferation was examined using CyQuant cell proliferation assay and expressed as %untreated hASM cell proliferation. hASM cell proliferation was significantly increased with TNFα treatment and siRNA knockdown of Mfn2 expression has a similar effect on hASM cell proliferation. By contrast, overexpression of Mfn2 expression abolished the effect of TNFα on hASM cell proliferation. Nonsense (NS) RNA has no effect on hASM cell proliferation and was used as control for transfection. Treated and untreated groups were collected at the same time and under the same conditions. Data are presented as medians and interquartile range represented as a box-and-whisker plot. Results were analyzed using one-way ANOVA (with repeated measures). *Significant difference (P < 0.05) compared with untreated hASM cells (N = 5 patients, 2 males and 3 females).
DISCUSSION
The results of this present study indicate that the proinflammatory cytokine TNFα induces mitochondrial fragmentation in hASM, which is mediated through a decrease in Mfn2 and an increase in Drp1 protein expression. TNFα-induced mitochondrial fragmentation is accompanied by mitochondrial biogenesis in hASM cells, evidenced by an increase in PGC1α protein expression and mitochondrial DNA copy number. As a result, mitochondrial volume density increases in TNFα-exposed hASM cells. In hASM cells exposed to TNFα, overall maximum O2 consumption rate increases; however, when normalized for the increase in mitochondrial volume density, maximum O2 consumption rate per mitochondrion is reduced following 24-h TNFα exposure. In addition to mitochondrial fragmentation and biogenesis, TNFα also induces hASM cell proliferation, which is mitigated by overexpressing Mfn2, whereas siRNA knockdown of Mfn2 mimics the effect of TNFα on hASM cell proliferation.
TNFα Has No Effect on hASM Cell Morphology and Dimensions
In the present study, we confirmed that dissociated hASM cells have a typical fusiform or spindle-like shape and that 24-h TNFα exposure had no effect on hASM cell morphology. The length, width, and height of dissociated hASM cells were comparable between TNFα-treated and untreated groups. Thus, changes in cell shape and or volume, which have been reported to be associated with changes in mitochondrial morphology and function (for review, see Ref. 61) were not observed in the present study.
TNFα Induces Mitochondrial Fragmentation
In the present study, we used 3-D confocal imaging to evaluate mitochondrial morphology. Most previous studies, including our own (1, 40), used 2-D microscopic imaging techniques, which may under- or overestimate the extent of mitochondrial fragmentation depending of the plane of focus within the cell and the orientation of the mitochondrial network as recently reported in a study comparing 2-D versus 3-D methods (48). The effect of TNFα on mitochondrial fragmentation was not examined in these studies. Using 3-D confocal imaging, we observed that mitochondrial morphology is highly heterogeneous within hASM cells. The mitochondrial network within a hASM cell comprises both long and short mitochondrial filaments as well as fragmented/globular mitochondria, which contribute to considerable variance in the metrics of mitochondrial morphology (form factor, aspect ratio and sphericity). In hASM cells exposed to TNFα, we found a pronounced shift of the distribution of mitochondrial morphology toward lower form factors and aspect ratios and higher sphericity indicating that mitochondria are more globular and fragmented. Using 2-D confocal imaging, we previously reported that in hASM cells exposed to cigarette smoke mitochondria are more fragmented (40). We also found that mitochondria in hASM cells from asthmatic patients are more fragmented compared with those from nonasthmatic patients (34).
Using the nuclear centroid as a reference point, we found that the proportion of mitochondria near the nucleus decreases in hASM cells exposed to TNFα. Previously, we reported that in the perinuclear region in hASM cells, mitochondrial movement is more pronounced compared with the more distal compartment (43). Mitochondrial movement is impaired following 24-h TNFα exposure (43). We also found that mitochondrial Ca2+ influx is more pronounced in perinuclear mitochondria as compared with those in the more distal compartment (45). The proximity of mitochondria to the endo/sarcoplasmic reticulum (ER) decreases in hASM exposed to TNFα, which may relate to the TNFα-induced decrease in Mfn2 protein and reduced tethering of mitochondria to the ER (43). In this regard, the results of this present study are consistent with an effect of TNFα treatment on mitochondria distribution within hASM cells.
Mitochondria are highly dynamic organelles and undergo cycles of fission/fragmentation and fusion, which allow mitochondria to adapt to changes such as energy demand and maintain mitochondrial DNA stability (31, 38, 62–64). Dynamic mitochondrial remodeling is mediated by the balance between Drp1 and Mfn1/2. Drp1 promotes mitochondrial fission (10, 27–29); thus the TNFα-induced increase in Drp1 expression in hASM cells could by itself explain the increase in mitochondrial fragmentation after TNFα exposure. Phosphorylation of Drp1 at serine 616 promotes Drp1-mediated mitochondrial fission and could potentially provide more information in the involvement of Drp1 in TNFα-induced mitochondria fragmentation than total Drp1 expression measurement (65). However, we have been unable at the present time to validate a commercially available antibody for phospho-Drp1 in human airway smooth muscle. The fusion of mitochondria is dependent on dimerization of the mitofusin proteins Mfn1 and Mfn2 (10, 27–29); thus, the TNFα-induced decrease in Mfn2 expression in hASM cells could also explain the increase in mitochondrial fragmentation after TNFα exposure. In hASM cells from asthmatic patients, we reported that mitochondria are more fragmented as compared with hASM from nonasthmatic patients (34). The greater extent of mitochondrial fragmentation in asthmatic hASM cells is associated with an increase in Drp1 and a decrease in Mfn2 expression (34).
It has been suggested that in asthmatic hASM cells or hASM cells exposed to cigarette smoke, increased production of reactive oxygen species (ROS) is responsible for mitochondrial fragmentation (34, 40). Recently, we showed that TNFα induces an increase in ROS production in hASM within 1 h, which is maintained for 24 h (35). Furthermore, the TNFα-induced decrease in Mfn2 expression in hASM cells is mitigated by treatment with the ROS scavenger Tempol (35). The exact source of ROS production in TNFα-exposed hASM cells is not clearly established nor is it clear if the increase in ROS production mediates the reduction in Mfn2 expression. In a previous study, we found that TNFα induces selective activation of the pIRE1α/XBP1s ER stress pathway, which we suggested was triggered by the accumulation of unfolded proteins due to the effect of ROS formation. In support, we found that Tempol mitigated the effect of TNFα on ER stress in hASM cells. Whether TNFα-induced mitochondrial remodeling is 1) a homeostatic or stress response, or 2) dynamic and continuing after 24 h is not known. We found that activation of the pIRE1α/XBP1s ER stress pathway in hASM cells peaked at ∼12 h after TNFα exposure and remained elevated after 24 h before decreasing at 48 h (35). We believe that part of the homeostatic response triggered by the pIRE1α/XBP1s ER stress pathway is mitochondrial biogenesis and a reduction in O2 consumption per mitochondrion, which would reduce reactive oxygen species (ROS) production per mitochondrion. It is likely that other cytokines than TNFα induce ER stress and affect Drp1 and/or Mfn2 expression and mitochondrial fragmentation.
Mitochondria are a major source of ROS, a by-product of mitochondrial respiration and ATP production. Interestingly, TNFα increases ASM force generation and ATP hydrolysis (5), which would promote an increase in mitochondrial O2 consumption and ATP production. Previous studies also showed that TNFα exposure increases ASM cell proliferation (1, 66), which would also stimulate mitochondrial ATP synthesis (2, 21, 63, 64, 67). An increase in ATP production and ROS formation could be achieved by either an increase in mitochondrial biogenesis and volume density and/or an increase O2 consumption in individual mitochondrion but at the expense of increased ROS formation. Mitochondrial fragmentation is in this respect an essential step toward increasing mitochondrial volume density, thereby increasing the number of mitochondria producing ATP and lessening the burden of O2 consumption and ROS formation per mitochondrion. Thus mitochondrial fragmentation may reflect an important adaptive response to protect mitochondria from an increase in ROS production and oxidative (ER) stress.
TNFα Increases Mitochondrial Biogenesis and Volume Density
The results of our study provided converging evidence that 24-h TNFα exposure induces mitochondrial biogenesis in hASM cells. First, we observed a TNFα-induced increase in PGC1α expression in hASM cells. The role of PGC1α in regulating mitochondrial biogenesis is well established (68, 69). Interestingly, previous studies have reported that PGC1α expression is higher in asthmatic hASM with an associated increase in mitochondrial mass (22, 23). In fact, these studies correlated the severity of asthma with mitochondrial mass in hASM biopsies. The authors of these studies suggested a potential role of inflammation in underlying the increase in mitochondrial mass (22, 23). Mitochondrial DNA copy number, usually 2–10 per mitochondrion, is also extensively used as an indicator of mitochondrial biogenesis (56, 70–72). The results of the present study show that 24-h TNFα exposure increases the ratio of mitochondrial DNA to genomic DNA copy number in hASM cells, a second line of evidence indicating an increase mitochondrial biogenesis. As a third indicator of increased mitochondrial biogenesis, we found that mitochondrial volume density (mitochondrial mass) also increases in hASM cells exposed to TNFα. The increase in mitochondrial volume density was also indicated by an increase in porin/VDAC the expression in TNFα exposed hASM cells.
TNFα Decreases Maximum O2 Consumption per Mitochondrion
In a previous study, we reported that 24-h TNFα exposure increases porcine ASM force generation and ATP hydrolysis (5), which should also stimulate mitochondrial O2 consumption and ATP production. In the present study, total maximum O2 consumption of hASM cells increased after TNFα exposure. However, when maximum O2 consumption was normalized to mitochondrial volume density, O2 consumption per mitochondrion was reduced. Our results indicate that an increase in force and ATP hydrolysis induced by exposing hASM to TNFα is met by an increase in overall mitochondrial O2 consumption but at the expense of increased ROS formation. Subsequently, mitochondrial biogenesis increases mitochondrial volume density and thereby reducing O2 consumption and ROS formation per individual mitochondrion.
Mitochondrial O2 consumption and ATP synthesis is stimulated by mitochondrial Ca2+ influx (73–78), which is dependent on mitochondrial proximity to the ER mediated by Mfn2-dependent tethering of the mitochondria to the ER (79–83). Consistent with the TNFα-induced reduction in Mfn2, we previously found that mitochondrial Ca2+ influx is reduced in hASM cells exposed to TNFα (45). Furthermore, mitochondrial proximity to the ER is reduced in hASM cells exposed to TNFα (43).
TNFα Increases hASM Cell Proliferation
Mitochondrial biogenesis is not only associated with an increase in total cellular O2 consumption, ATP production and ROS formation, it is also associated with cell proliferation. In the present study, we found that TNFα exposure increases hASM cell proliferation. In the most fundamental sense, mitochondrial fission and fusion are required for mitochondrial biogenesis and to ensure that mitochondrial number is maintained subsequent to cell division (84). As a first step in mitochondrial biogenesis, fragmentation may reflect or induce an increased proliferative state of the cell (10, 84). The results of the present study showing a TNFα-induced increase in hASM cell proliferation are consistent with previous studies (1, 66). The potential involvement of the TNFα-induced decrease in Mfn2 is supported by the observation that siRNA knockdown of Mfn2 expression mimics the effect of TNFα on hASM cell proliferation, whereas overexpressing Mfn2 mitigates the effect of TNFα on hASM cell proliferation. Previous evidence in cell types other than hASM suggests that Mfn2 expression and the dynamic balance of mitochondrial fragmentation and fusion are involved in pro-proliferative pathways and cell division (10, 84). These investigators suggested that Mfn2 inhibits pro-proliferative kinases such as extracellular signal-regulated kinase (ERK1/2) (10). Importantly, several studies have shown that ERK1/2 activation mediates ASM cell proliferation (85–88). In a previous study, we found that cigarette smoke exposure induces mitochondrial fission in hASM and a decrease in Mfn2 expression (34). We also found that inhibition of ERK1/2 attenuated the effect of cigarette smoke exposure on mitochondrial fission (34). While the impact of cigarette smoke exposure on hASM cell proliferation was not examined in the present study, the results are consistent with a role of reduced Mfn2 in hASM cell proliferation.
In summary, the results of the present study indicate that in hASM cells, 24-h TNFα exposure increases overall mitochondrial O2 consumption to meet the ATP demand of increased force generation but at the expense of increased ROS production. It is likely that this heightened energetic demand following TNFα exposure leads to a coordinated adaptive response in hASM cells involving mitochondrial fragmentation, increased mitochondrial biogenesis and volume density, and impaired tethering of mitochondria to the ER. As a result, the maximum O2 consumption and ROS production per mitochondrion is reduced, while the demand for ATP synthesis is still met.
GRANTS
This work was supported by the National Heart, Lung, and Blood Institute Grants R01HL126451 and R01HL150890 (to G.C.S.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
P.D. and G.C.S. conceived and designed research; P.D. and N.M.M. performed experiments; P.D., N.M.M., and G.C.S. analyzed data; P.D., N.M.M., and G.C.S. interpreted results of experiments; P.D. and G.C.S. prepared figures; P.D. and G.C.S. drafted manuscript; G.C.S. edited and revised manuscript; P.D. and G.C.S. approved final version of manuscript..
REFERENCES
- 1.Aravamudan B, Thompson M, Pabelick C, Prakash YS. Brain-derived neurotrophic factor induces proliferation of human airway smooth muscle cells. J Cell Mol Med 16: 812–823, 2012. doi: 10.1111/j.1582-4934.2011.01356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Delmotte P, Sieck GC. Endoplasmic reticulum stress and mitochondrial function in airway smooth muscle. Front Cell Dev Biol 7: 374, 2020. doi: 10.3389/fcell.2019.00374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dogan M, Han YS, Delmotte P, Sieck GC. TNFα enhances force generation in airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 312: L994–L1002, 2017. doi: 10.1152/ajplung.00550.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khan MA Inflammation signals airway smooth muscle cell proliferation in asthma pathogenesis. Multidiscip Respir Med 8: 11, 2013. doi: 10.1186/2049-6958-8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sieck GC, Dogan M, Young-Soo H, Osorio Valencia S, Delmotte P. Mechanisms underlying TNFα-induced enhancement of force generation in airway smooth muscle. Physiol Rep 7: e14220, 2019. doi: 10.14814/phy2.14220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stamatiou R, Paraskeva E, Gourgoulianis K, Molyvdas PA, Hatziefthimiou A. Cytokines and growth factors promote airway smooth muscle cell proliferation. ISRN Inflamm 2012: 1–13, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tliba O, Tliba S, Da Huang C, Hoffman RK, DeLong P, Panettieri RA Jr, Amrani Y. Tumor necrosis factor alpha modulates airway smooth muscle function via the autocrine action of interferon beta. J Biol Chem 278: 50615–50623, 2003. doi: 10.1074/jbc.M303680200. [DOI] [PubMed] [Google Scholar]
- 8.Bhandary B, Marahatta A, Kim HR, Chae HJ. Mitochondria in relation to cancer metastasis. J Bioenerg Biomembr 44: 623–627, 2012. doi: 10.1007/s10863-012-9464-x. [DOI] [PubMed] [Google Scholar]
- 9.Estaquier J, Vallette F, Vayssiere JL, Mignotte B. The mitochondrial pathways of apoptosis. Adv Exp Med Biol 942: 157–183, 2012. doi: 10.1007/978-94-007-2869-1_7. [DOI] [PubMed] [Google Scholar]
- 10.Liesa M, Palacin M, Zorzano A. Mitochondrial dynamics in mammalian health and disease. Physiol Rev 89: 799–845, 2009. doi: 10.1152/physrev.00030.2008. [DOI] [PubMed] [Google Scholar]
- 11.Sola S, Morgado AL, Rodrigues CM. Death receptors and mitochondria: two prime triggers of neural apoptosis and differentiation. Biochim Biophys Acta 1830: 2160–2166, 2013. doi: 10.1016/j.bbagen.2012.09.021. [DOI] [PubMed] [Google Scholar]
- 12.Tait SW, Green DR. Mitochondria and cell signalling. J Cell Sci 125: 807–815, 2012. doi: 10.1242/jcs.099234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wagatsuma A, Sakuma K. Mitochondria as a potential regulator of myogenesis. ScientificWorldJournal 2013: 1–9, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Delmotte P, Jia L, Sieck GC. The role of mitochondria in calcium regulation in airway smooth muscle. In: Calcium Signaling in Airway Smooth Muscle Cells, edited by Wang YX. New York: Springer International Publishing, 2014, p. 211–234. doi: 10.1007/978-3-319-01312-1_11. [DOI] [Google Scholar]
- 15.Delmotte P, Sieck GC. Interaction between endoplasmic/sarcoplasmic reticulum stress (ER/SR stress), mitochondrial signaling and Ca(2+) regulation in airway smooth muscle (ASM). Can J Physiol Pharmacol 93: 97–110, 2015. doi: 10.1139/cjpp-2014-0361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol 287: C817–C833, 2004. doi: 10.1152/ajpcell.00139.2004. [DOI] [PubMed] [Google Scholar]
- 17.Macaskill AF, Rinholm JE, Twelvetrees AE, Arancibia-Carcamo IL, Muir J, Fransson A, Aspenstrom P, Attwell D, Kittler JT. Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron 61: 541–555, 2009. doi: 10.1016/j.neuron.2009.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saotome M, Safiulina D, Szabadkai G, Das S, Fransson A, Aspenstrom P, Rizzuto R, Hajnoczky G. Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc Natl Acad Sci U S A 105: 20728–20733, 2008. doi: 10.1073/pnas.0808953105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schwarz TL Mitochondrial trafficking in neurons. Cold Spring Harb Perspect Biol 5, a011304, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang CC, Lin WN, Lee CW, Lin CC, Luo SF, Wang JS, Yang CM. Involvement of p42/p44 MAPK, p38 MAPK, JNK, and NF-κB in IL-1β-induced VCAM-1 expression in human tracheal smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 288: L227–L237, 2005. doi: 10.1152/ajplung.00224.2004. [DOI] [PubMed] [Google Scholar]
- 21.Schaible N, Delmotte P, Sieck GC. Mitochondrial excitation-energy coupling in airway smooth muscle. In: Mitochondrial Function in Lung Health and Disease, edited by Natarajan V, NL Parinandi. Totowa, NJ: Humana Press, Inc, 2014, p. 93–116. doi: 10.1007//978-1-4939-0829-5_5. [DOI] [Google Scholar]
- 22.Girodet PO, Ozier A, Bara I, Tunon de Lara JM, Marthan R, Berger P. Airway remodeling in asthma: new mechanisms and potential for pharmacological intervention. Pharmacol Ther 130: 325–337, 2011. doi: 10.1016/j.pharmthera.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 23.Trian T, Benard G, Begueret H, Rossignol R, Girodet PO, Ghosh D, Ousova O, Vernejoux JM, Marthan R, Tunon-de-Lara JM, Berger P. Bronchial smooth muscle remodeling involves calcium-dependent enhanced mitochondrial biogenesis in asthma. J Exp Med 204: 3173–3181, 2007. doi: 10.1084/jem.20070956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Denton RM, McCormack JG. Ca2+ as a second messenger within mitochondria of the heart and other tissues. Annu Rev Physiol 52: 451–466, 1990. doi: 10.1146/annurev.ph.52.030190.002315. [DOI] [PubMed] [Google Scholar]
- 25.Liu T, O’Rourke B. Regulation of mitochondrial Ca2+ and its effects on energetics and redox balance in normal and failing heart. J Bioenerg Biomembr 41: 127–132, 2009. doi: 10.1007/s10863-009-9216-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science 337: 1062–1065, 2012. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Palmer CS, Osellame LD, Stojanovski D, Ryan MT. The regulation of mitochondrial morphology: intricate mechanisms and dynamic machinery. Cell Signal 23: 1534–1545, 2011. doi: 10.1016/j.cellsig.2011.05.021. [DOI] [PubMed] [Google Scholar]
- 28.Ranieri M, Brajkovic S, Riboldi G, Ronchi D, Rizzo F, Bresolin N, Corti S, Comi GP. Mitochondrial fusion proteins and human diseases. Neurol Res Int 2013: 293893, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Song Z, Ghochani M, McCaffery JM, Frey TG, Chan DC. Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol Biol Cell 20: 3525–3532, 2009. doi: 10.1091/mbc.e09-03-0252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.James DI, Parone PA, Mattenberger Y, Martinou JC. hFis1, a novel component of the mammalian mitochondrial fission machinery. J Biol Chem 278: 36373–36379, 2003. [Erratum in J Biol Chem 27934: 36166, 2004. doi: 10.1074/jbc.M303758200. [DOI] [PubMed] [Google Scholar]
- 31.Lee YJ, Jeong SY, Karbowski M, Smith CL, Youle RJ. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell 15: 5001–5011, 2004. doi: 10.1091/mbc.e04-04-0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sheridan C, Martin SJ. Mitochondrial fission/fusion dynamics and apoptosis. Mitochondrion 10: 640–648, 2010. doi: 10.1016/j.mito.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 33.Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell 12: 2245–2256, 2001. doi: 10.1091/mbc.12.8.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aravamudan B, Kiel A, Freeman M, Delmotte P, Thompson M, Vassallo R, Sieck GC, Pabelick CM, Prakash YS. Cigarette smoke-induced mitochondrial fragmentation and dysfunction in human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 306: L840–L854, 2014. doi: 10.1152/ajplung.00155.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yap J, Chen X, Delmotte P, Sieck GC. TNFα selectively activates the IRE1alpha/XBP1 endoplasmic reticulum stress pathway in human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 318: L483–L493, 2020. doi: 10.1152/ajplung.00212.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol 13: 589–598, 2011. doi: 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jheng HF, Tsai PJ, Guo SM, Kuo LH, Chang CS, Su IJ, Chang CR, Tsai YS. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol Cell Biol 32: 309–319, 2012. doi: 10.1128/MCB.05603-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prakash YS, Pabelick CM, Sieck GC. Mitochondrial dysfunction in airway disease. Chest 152: 618–626, 2017. doi: 10.1016/j.chest.2017.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schrepfer E, Scorrano L. Mitofusins, from mitochondria to metabolism. Mol Cell 61: 683–694, 2016. doi: 10.1016/j.molcel.2016.02.022. [DOI] [PubMed] [Google Scholar]
- 40.Aravamudan B, Thompson M, Sieck GC, Vassallo R, Pabelick CM, Prakash YS. Functional Effects of cigarette smoke-induced changes in airway smooth muscle mitochondrial morphology. J Cell Physiol 232: 1053–1068, 2017. doi: 10.1002/jcp.25508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prakash YS, Iyanoye A, Ay B, Sieck GC, Pabelick CM. Store-operated Ca2+ influx in airway smooth muscle: Interactions between volatile anesthetic and cyclic nucleotide effects. Anesthesiology 105: 976–983, 2006. doi: 10.1097/00000542-200611000-00019. [DOI] [PubMed] [Google Scholar]
- 42.Prakash YS, Kannan MS, Sieck GC. Regulation of intracellular calcium oscillations in porcine tracheal smooth muscle cells. Am J Physiol Cell Physiol 272: C966–C975, 1997. doi: 10.1152/ajpcell.1997.272.3.C966. [DOI] [PubMed] [Google Scholar]
- 43.Delmotte P, Zavaletta VA, Thompson MA, Prakash YS, Sieck GC. TNFα decreases mitochondrial movement in human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 313: L166–L176, 2017. doi: 10.1152/ajplung.00538.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jia L, Delmotte P, Aravamudan B, Pabelick CM, Prakash YS, Sieck GC. Effects of the inflammatory cytokines TNF-alpha and IL-13 on stromal interaction molecule-1 aggregation in human airway smooth muscle intracellular Ca(2+) regulation. Am J Respir Cell Mol Biol 49: 601–608, 2013. doi: 10.1165/rcmb.2013-0040OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Delmotte P, Yang B, Thompson MA, Pabelick CM, Prakash YS, Sieck GC. Inflammation alters regional mitochondrial Ca2+ in human airway smooth muscle cells. Am J Physiol Cell Physiol 303: C244–C256, 2012. doi: 10.1152/ajpcell.00414.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rana S, Mantilla CB, Sieck GC. Glutamatergic input varies with phrenic motor neuron size. J Neurophysiol 122: 1518–1529, 2019. doi: 10.1152/jn.00430.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sathish V, Leblebici F, Kip SN, Thompson MA, Pabelick CM, Prakash YS, Sieck GC. Regulation of sarcoplasmic reticulum Ca2+ reuptake in porcine airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 294: L787–L796, 2008. doi: 10.1152/ajplung.00461.2007. [DOI] [PubMed] [Google Scholar]
- 48.Chaudhry A, Shi R, Luciani DS. A pipeline for multidimensional confocal analysis of mitochondrial morphology, function, and dynamics in pancreatic β-cells. Am J Physiol Endocrinol Metab 318: E87–E101, 2020. doi: 10.1152/ajpendo.00457.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Donoho DL, Johnstone IM. Ideal spatial adaptation via wavelet shrinkage. Biometrika 81: 425–455, 1994. doi: 10.1093/biomet/81.3.425. [DOI] [Google Scholar]
- 50.Koopman WJ, Distelmaier F, Esseling JJ, Smeitink JA, Willems PH. Computer-assisted live cell analysis of mitochondrial membrane potential, morphology and calcium handling. Methods 46: 304–311, 2008. doi: 10.1016/j.ymeth.2008.09.018. [DOI] [PubMed] [Google Scholar]
- 51.Koopman WJ, Visch HJ, Smeitink JA, Willems PH. Simultaneous quantitative measurement and automated analysis of mitochondrial morphology, mass, potential, and motility in living human skin fibroblasts. Cytometry A 69: 1–12, 2006. [DOI] [PubMed] [Google Scholar]
- 52.Koopman WJ, Visch HJ, Verkaart S, van den Heuvel LW, Smeitink JA, Willems PH. Mitochondrial network complexity and pathological decrease in complex I activity are tightly correlated in isolated human complex I deficiency. Am J Physiol Cell Physiol 289: C881–C890, 2005. doi: 10.1152/ajpcell.00104.2005. [DOI] [PubMed] [Google Scholar]
- 53.Sternberg SR Biomedical image processing. Computer 16: 22–34, 1983. doi: 10.1109/MC.1983.1654163, . [DOI] [Google Scholar]
- 54.Sieck GC, Mantilla CB, Prakash YS. Volume measurements in confocal microscopy. Methods Enzymol 307: 296–315, 1999. doi: 10.1016/S0076-6879(99)07019-6. [DOI] [PubMed] [Google Scholar]
- 55.Harris VM Protein detection by Simple Western analysis. Methods Mol Biol 1312: 465–468, 2015. doi: 10.1007/978-1-4939-2694-7_47. [DOI] [PubMed] [Google Scholar]
- 56.Yu Y, Liu H, Ikeda Y, Amiot BP, Rinaldo P, Duncan SA, Nyberg SL. Hepatocyte-like cells differentiated from human induced pluripotent stem cells: relevance to cellular therapies. Stem Cell Res 9: 196–207, 2012. doi: 10.1016/j.scr.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dranka BP, Benavides GA, Diers AR, Giordano S, Zelickson BR, Reily C, Zou L, Chatham JC, Hill BG, Zhang J, Landar A, Darley-Usmar VM. Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free Radic Biol Med 51: 1621–1635, 2011. doi: 10.1016/j.freeradbiomed.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhou W, Choi M, Margineantu D, Margaretha L, Hesson J, Cavanaugh C, Blau CA, Horwitz MS, Hockenbery D, Ware C, Ruohola-Baker H. HIF1alpha induced switch from bivalent to exclusively glycolytic metabolism during ESC-to-EpiSC/hESC transition. EMBO J 31: 2103–2116, 2012. doi: 10.1038/emboj.2012.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Santel A, Fuller MT. Control of mitochondrial morphology by a human mitofusin. J Cell Sci 114: 867–874, 2001. [DOI] [PubMed] [Google Scholar]
- 60.Valente AJ, Maddalena LA, Robb EL, Moradi F, Stuart JA. A simple ImageJ macro tool for analyzing mitochondrial network morphology in mammalian cell culture. Acta Histochem 119: 315–326, 2017. doi: 10.1016/j.acthis.2017.03.001. [DOI] [PubMed] [Google Scholar]
- 61.Anesti V, Scorrano L. The relationship between mitochondrial shape and function and the cytoskeleton. Biochim Biophys Acta 1757: 692–699, 2006. doi: 10.1016/j.bbabio.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 62.Knott AB, Perkins G, Schwarzenbacher R, Bossy-Wetzel E. Mitochondrial fragmentation in neurodegeneration. Nat Rev Neurosci 9: 505–518, 2008. doi: 10.1038/nrn2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pan S, Conaway S, Jr, Deshpande DA. Mitochondrial regulation of airway smooth muscle functions in health and pulmonary diseases. Arch Biochem Biophys 663: 109–119, 2019. doi: 10.1016/j.abb.2019.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pan S, Shah SD, Panettieri RA Jr, Deshpande DA. Bnip3 regulates airway smooth muscle cell focal adhesion and proliferation. Am J Physiol Lung Cell Mol Physiol 317: L758–L767, 2019. doi: 10.1152/ajplung.00224.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Breitzig MT, Alleyn MD, Lockey RF, Kolliputi N. A mitochondrial delicacy: dynamin-related protein 1 and mitochondrial dynamics. Am J Physiol Cell Physiol 315: C80–C90, 2018. doi: 10.1152/ajpcell.00042.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McKay S, Hirst SJ, Haas MB, de Jongste JC, Hoogsteden HC, Saxena PR, Sharma HS. Tumor necrosis factor-alpha enhances mRNA expression and secretion of interleukin-6 in cultured human airway smooth muscle cells. Am J Respir Cell Mol Biol 23: 103–111, 2000. doi: 10.1165/ajrcmb.23.1.3765. [DOI] [PubMed] [Google Scholar]
- 67.Salazar-Roa M, Malumbres M. Fueling the cell division cycle. Trends Cell Biol 27: 69–81, 2017. doi: 10.1016/j.tcb.2016.08.009. [DOI] [PubMed] [Google Scholar]
- 68.Handschin C, Spiegelman BM. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr Rev 27: 728–735, 2006. doi: 10.1210/er.2006-0037. [DOI] [PubMed] [Google Scholar]
- 69.Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98: 115–124, 1999. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- 70.Lee HC, Wei YH. Mitochondrial biogenesis and mitochondrial DNA maintenance of mammalian cells under oxidative stress. Int J Biochem Cell Biol 37: 822–834, 2005. doi: 10.1016/j.biocel.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 71.Ploumi C, Daskalaki I, Tavernarakis N. Mitochondrial biogenesis and clearance: a balancing act. FEBS J 284: 183–195, 2017. doi: 10.1111/febs.13820. [DOI] [PubMed] [Google Scholar]
- 72.Reddy PH Is the mitochondrial outermembrane protein VDAC1 therapeutic target for Alzheimer's disease? Biochim Biophys Acta 1832: 67–75, 2013. doi: 10.1016/j.bbadis.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Franzini-Armstrong C ER-mitochondria communication. How privileged? Physiology (Bethesda) 22: 261–268, 2007. doi: 10.1152/physiol.00017.2007. [DOI] [PubMed] [Google Scholar]
- 74.Hajnoczky G, Saotome M, Csordas G, Weaver D, Yi M. Calcium signalling and mitochondrial motility. Novartis Found Symp 287: 105–117, 2007. doi: 10.1002/9780470725207.ch8. [DOI] [PubMed] [Google Scholar]
- 75.Maack C, O'Rourke B. Excitation-contraction coupling and mitochondrial energetics. Basic Res Cardiol 102: 369–392, 2007. doi: 10.1007/s00395-007-0666-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Parekh AB Mitochondrial regulation of intracellular Ca2+ signaling: more than just simple Ca2+ buffers. News Physiol Sci 18: 252–256, 2003. doi: 10.1152/nips.01458.2003. [DOI] [PubMed] [Google Scholar]
- 77.Rizzuto R, Bernardi P, Pozzan T. Mitochondria as all-round players of the calcium game. J Physiol 529: 37–47, 2000. doi: 10.1111/j.1469-7793.2000.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Romagnoli A, Aguiari P, De Stefani D, Leo S, Marchi S, Rimessi A, Zecchini E, Pinton P, Rizzuto R. Endoplasmic reticulum/mitochondria calcium cross-talk. Novartis Found Symp 287: 122–131, 2007. doi: 10.1002/9780470725207.ch9. [DOI] [PubMed] [Google Scholar]
- 79.Filadi R, Theurey P, Pizzo P. The endoplasmic reticulum-mitochondria coupling in health and disease: Molecules, functions and significance. Cell Calcium 62: 1–15, 2017. doi: 10.1016/j.ceca.2017.01.003. [DOI] [PubMed] [Google Scholar]
- 80.Hajnoczky G, Csordas G, Yi M. Old players in a new role: mitochondria-associated membranes, VDAC, and ryanodine receptors as contributors to calcium signal propagation from endoplasmic reticulum to the mitochondria. Cell Calcium 32: 363–377, 2002. doi: 10.1016/S0143416002001872. [DOI] [PubMed] [Google Scholar]
- 81.Patergnani S, Suski JM, Agnoletto C, Bononi A, Bonora M, De Marchi E, Giorgi C, Marchi S, Missiroli S, Poletti F, Rimessi A, Duszynski J, Wieckowski MR, Pinton P. Calcium signaling around Mitochondria Associated Membranes (MAMs). Cell Commun Signal 9: 19, 2011. doi: 10.1186/1478-811X-9-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Raturi A, Simmen T. Where the endoplasmic reticulum and the mitochondrion tie the knot: the mitochondria-associated membrane (MAM). Biochim Biophys Acta 1833: 213–224, 2013. doi: 10.1016/j.bbamcr.2012.04.013. [DOI] [PubMed] [Google Scholar]
- 83.van Vliet AR, Verfaillie T, Agostinis P. New functions of mitochondria associated membranes in cellular signaling. Biochim Biophys Acta 1843: 2253–2262, 2014. doi: 10.1016/j.bbamcr.2014.03.009. [DOI] [PubMed] [Google Scholar]
- 84.Antico Arciuch VG, Elguero ME, Poderoso JJ, Carreras MC. Mitochondrial regulation of cell cycle and proliferation. Antioxid Redox Signal 16: 1150–1180, 2012. doi: 10.1089/ars.2011.4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dragon S, Hirst SJ, Lee TH, Gounni AS. IL-17A mediates a selective gene expression profile in asthmatic human airway smooth muscle cells. Am J Respir Cell Mol Biol 50: 1053–1063, 2014. doi: 10.1165/rcmb.2012-0267OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lee JH, Johnson PR, Roth M, Hunt NH, Black JL. ERK activation and mitogenesis in human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 280: L1019–L1029, 2001. doi: 10.1152/ajplung.2001.280.5.L1019. [DOI] [PubMed] [Google Scholar]
- 87.Movassagh H, Shan L, Halayko AJ, Roth M, Tamm M, Chakir J, Gounni AS. Neuronal chemorepellent Semaphorin 3E inhibits human airway smooth muscle cell proliferation and migration. J Allergy Clin Immunol 133: 560–567, 2014. doi: 10.1016/j.jaci.2013.06.011. [DOI] [PubMed] [Google Scholar]
- 88.Yu ZH, Wang YX, Song Y, Lu HZ, Hou LN, Cui YY, Chen HZ. Up-regulation of KCa3.1 promotes human airway smooth muscle cell phenotypic modulation. Pharmacol Res 77: 30–38, 2013. doi: 10.1016/j.phrs.2013.09.002. [DOI] [PubMed] [Google Scholar]