

Abstract
Objectives
The epidermal growth factor receptor variant type III (EGFRvIII) is the most common mutation of EGFR in glioblastoma multiforme (GBM) and is found in approximately 25% of all GBMs. Intriguingly, EGFRvIII is mostly found in GFAP+ astrocytic tumour cells in the brain, suggesting connection of EGFRvIII to astrogenesis. In this study, we explored whether EGFRvIII mutation facilitates astrogenesis in human development setting.
Materials and methods
Using CRISPR‐Cas9, we generated EGFRvIII mutations in H9‐hESCs. Wild type (wt) H9‐hESCs were used as an isogenic control. Next, we generated cerebral organoids using the wt and EGFRvIII‐hESCs and examined the astrogenic differentiation of the brain organoids.
Results
EGFRvIII‐organoids showed abundant astrocytes (GFAP+, S100β+), while no astrocytes were detected in wt hESC‐derived organoids at day 49. On the contrary, TUJ1+ neurons were more abundant in the wt‐organoids than the EGFRvIII‐organoids. This result suggested that constitutively active EGFRvIII promoted astrogenesis at the expense of neurogenesis. In addition, the EGFRvIII‐organoids were larger in size and retained more Ki67+ cells than wt‐organoids, indicating enhanced cell proliferation by the mutation. The EGFRvIII‐organoids displayed massive apoptotic cell death after treatment with temozolomide and hence, could be used for evaluation of anti‐GBM drugs.
Conclusions
EGFRvIII mutation‐induced astrogenesis and massive cell proliferation in a human brain development model. These results provide us new insights into the mechanisms relating EGFRvIII mutation‐mediated gliogenesis and gliomagenesis.
Keywords: astrogenesis, cancer modelling, cerebral organoid, EGFRvIII, glioblastoma, temozolomide
The epidermal growth factor receptor variant type III (EGFRvIII) mutation mostly found in high‐grade astrocytoma in the brain, suggesting connection of EGFRvIII to astrogenesis. Using human brain organoids, we demonstrated that EGFRvIII mutation facilitated astrogenesis at the expense of neurogenesis and cell proliferation. In summary, this study shows for the first time the close connection between EGFRvIII mutation and gliogenesis in human system.
![]()
1. INTRODUCTION
A growing body of evidence suggests that tumorigenesis is closely linked to developmental processes. Therefore, study of the function of tumour drivers (ie, gene mutations) in the setting of developmental biology may help us to better understand the process of tumorigenesis. 1 , 2 Brain tumours are generally classified into glioma and non‐glioma. There are three principle types of glioma: astrocytoma, oligodendroglioma and glioblastoma (GBM). 3 Among them, GBM is the most frequent and aggressive primary brain tumour, accounting for more than 60% of all brain tumours. 4 , 5 , 6 Unfortunately, despite intensive research efforts over the past few decades, the median survival rate has not changed significantly. 7 This may be at least partly attributed to the lack of an appropriate cancer model to study the generation of GBM.
In vitro 2‐dimentional (2‐D) culture models have frequently been used in studies of developmental biology and tumorigenesis due to their convenience. However, these 2‐D models do not accurately mimic the diversity of cell types and cell‐to‐cell and cell‐to‐extracellular environment interactions, often failing to recapitulate conditions seen in vivo.
To bridge this translational gap, patient‐derived xenograft (PDX) models have recently been developed. In PDX models, samples of tumour tissues from human cancer patients are grafted onto immunocompromised animals and used to study new treatments or basic biology. However, in addition to significant cost‐ and labour‐intensiveness, results from PDX models often fail to predict outcomes in human clinical trials. 8 , 9 , 10
Brain organoids have recently emerged as a promising 3‐D model that recapitulates many aspects of the human brain. 11 , 12 In this study, we used brain organoid models to investigate the role of a tumour driver mutation in GBM and to evaluate the therapeutic effects of temozolomide, an anti‐cancer drug. Using CRISPR/Cas9, 13 we introduced an epidermal growth factor receptor variant type III (EGFRvIII) mutation into hESCs, a mutation that is found in approximately 25% of all GBM patients. 14 , 15 We found that the EGFRvIII mutation, which generates a ligand‐independent and constitutively active form of EGFR, 16 promoted astrogenesis and also induced massive cell proliferation which was blocked by temozolomide, a drug used for the treatment of GBM.
This study adds new insights to the process of gliomagenesis and provides a useful platform to evaluate the efficacy of other anti‐GBM drugs.
2. MATERIALS AND METHODS
2.1. ESC culture
Human H9 (WA09) embryonic stem cells purchased from WiCell Research Institute, Inc hESCs were cultured on Matrigel (Thermo Fisher Scientific)‐coated culture dishes in the TeSR™‐E8™ medium (STEMCELL Technologies). Cells were passaged every 5 days by treating with 0.5 mmol/L EDTA (Thermo Fisher Scientific) for 3 minutes at 37°C and transfering at 1:20 ratio onto fresh matrigel‐coated plates containing TeSR™‐E8™ medium with 10 μmol/L Y‐27632 (Tocris). Next day, we changed the medium with fresh TeSR™‐E8™ medium without Y‐27632.
2.2. CRISPR/Cas9‐mediated genome editing of hESC
CRISPR/Cas9‐mediated genome editing of hESCs was performed as follows: hESCs were dissociated into single cells using Accutase Dissociation Reagent (Thermo Fisher Scientific) and subjected to electroporation with 30 μg of Cas9 protein and 20 μg of sgRNA. Cells were then cultured on matrigel‐coated culture dishes and maintained in TeSR™‐E8™ medium. Y27632 (10 μmol/L) was added to the medium for the first 24 hours. After 5 days, the colonies were dissociated into single cells and seeded onto fresh matrigel‐coated dishes at a low density to obtain clones. Individual colonies grown for 5 days were transferred into each well of 48 well plates and expanded in TeSR™‐E8™ medium. Table S1 contains the list of sgRNA used in this study.
2.3. RNA extraction, reverse transcription and RT‐PCR
Total RNAs were extracted using the Nucleospin RNA kit (MACHEREY‐NAGEL), according to the manufacturer's protocol. RNA concentration was measured using the NanoDrop Spectrophotometer (Thermo Fisher Scientific) and cDNA was synthesized using a ReverTra Ace® qPCR RT kit (TOYOBO). The resulting cDNA was amplified by the polymerase chain reaction (PCR) using EGFR primers (Table S1).
2.4. Differentiation into the three germ layer
hESCs were detached with 2 mg/mL collagenase Type IV (Worthington Biochemical) for 30 minutes at 37°C. The detached cells were centrifuged at 300 g for 5 minutes and plated on Petri dishes containing DMEM/F12, 20% Knockout Serum Replacement, 1% NEAA and 55 μmol/L 2‐Mercaptoethanol (all from the Thermo Fisher Scientific) for embryoid body (EB) formation. After 7‐day culture, the EBs were attached to martigel‐coated dishes and subjected to spontaneous differentiation in DMEM/F12 medium containing 10% FBS, and 1% Penicillin/Streptomycin (all from the Thermo Fisher Scientific) for 1‐2 weeks.
2.5. Immunocytochemistry
Undifferentiated cells and differentiated cells into the three germ layer fixed in 4% paraformaldehyde (PFA) for 30 minutes at room temperature (RT), washed 2‐3 times with 1X phosphate‐buffered saline (PBS). Fixed cells permeabilized with 0.2% Triton‐X100/1X PBS for 10 minutes, were blocked with 5% normal goat serum for 1 hours at RT, followed by subsequent incubation with primary and secondary antibodies for 3 hours and 1 hour, respectively, at RT. The following primary antibodies were used: OCT4 (Santa Cruz; sc‐5279, 1:500), SSEA4 (Santa Cruz; sc‐21704, 1:500), TRA1‐81(Millipore; MAB4381, 1:500), SMA (Agilent; M0851, 1:500) and AFP (Agilent; A0008, 1:500). Secondary antibodies conjugated with Alexa‐488 and ‐594 (Thermo Fisher Scientific; 1:500), were used depending on the primary antibody used. DAPI (4',6‐Diamidino‐2‐Phenylindole, Dihydrochloride) was used for counter‐staining at a concentration of 1.0 μg/mL (Table S2).
2.6. Generation of brain organoids
A total of 10 000 cells were plated into each well of an ultra‐low‐attachment 96‐well plate (Thermo Fisher Scientific) containing E6 medium with 4 ng/mL bFGF (CHA Meditech) and 50 μmol/L Y27632. EBs were formed and maintained in the 96‐well plate for 6 days, followed by transferring into a Petri dish containing neural induction medium. Neural induction medium consisted of DMEM/F12, 1% N2 supplement, 1% NEAA, 1% GlutaMAX (all from Thermo Fisher Scientific) and heparin (1 μg/mL; Sigma‐Aldrich). On day 11, EBs were embedded in droplets of matrigel and then solidified at 37°C. Embedded EBs were subsequently incubated in neural differentiation medium containing 1:1 mixture of DMEM/F12 and Neurobasal, 1% N2 supplement, 2% B27 supplement Minus Vitamin A, 0.5% NEAA, 1% GlutaMAX, 25 μmol/L 2‐Mercaptoethanol (all from Thermo Fisher Scientific) and human Insulin (2.5 μg/mL; Sigma‐Aldrich). The tissue droplets in 6‐cm Petri dishes were cultured in stationary phase for 4 days, and then transferred to an orbital shaker (Daihan Scientific) that rotated continuously at 80 rpm. During the rotation culture, the neural differentiation medium was changed to a fresh neural differentiation medium that contained 2% B27 with ascorbic acid (Thermo Fisher Scientific).
2.7. Histology and imaging
Organoids were fixed in 4% paraformaldehyde (PFA) for 30 minutes at RT, washed 2‐3 times with 1× PBS and subject to dehydration overnight at 4°C with 30% sucrose solution. The dehydrated organoids were frozen in Optimal cutting temperature (OCT) compound (Sakura® Finetek), followed by sectioning (10‐15 μm thick) using Leica CM3050 S Research Cryostat (Leica). Sections on the slides were permeabilized with 0.2% Triton‐X100/1XPBS for 10 minutes, were blocked with 5% normal goat serum for 30 minutes at RT, followed by subsequent incubation with primary and secondary antibodies for 3 hours and 1 hour, respectively, at RT. The following primary antibodies were used: SOX2 (Millipore; MAB4343, 1:200), PAX6 (BioLegend; 901301, 1:200), NeuN (Millipore; MAB377, 1:200), MAP2 (Millipore; AB5622, 1:200), Nestin (Millipore; MAB5326, 1:200), TUJ1 (Convance; MMS‐435P, 1:500), GFAP (Thermo Fisher Scientific; MA5‐12023, 1:200; Abcam, ab68428, 1:200), S100β (Novus Biologicals; NBP1‐87102, 1:200), Ki67 (BD Biosciences; 550609, 1:500) and cleaved caspase‐3 (Cell Signaling; 9661, 1:500). Secondary antibodies conjugated with Alexa‐488 and ‐594 (Thermo Fisher Scientific; 1:500) and DAPI were stained. Fluorescent images were captured using a Zeiss LSM‐700 confocal microscope (Zeiss; Table S2).
2.8. Temozolomide treatment
Brain organoids were grown for 50 days and then treated with 1 mmol/L Temozolomide for 10 days. DMSO was used as a negative control. Medium containing Temozolomide was changed daily during treatment period. The treated brain organoids were frozen and subjected to immunostaining with antibodies against cleaved caspase 3 (Cell Signaling Technology) and Ki67 (BD Biosciences).
2.9. Statistical analysis
Statistical analysis was performed using the graphpad prism Version 5 program. Unpaired two‐tailed Student's t test was used to compare two groups, while one‐way ANOVA with Turkey's test was used to compare the multiple groups. Only P‐values smaller than .05 were considered statistically significant.
3. RESULTS
3.1. Generation of brain organoids
We generated cerebral organoids from H9‐hESCs using a protocol established by the Knoblich group 11 with one modification: for the first 6 days of culture, we used E6 medium containing 4 ng/mL bFGF and 50 μmol/L Y27632 instead of DMEM/F12, 20% knockout serum replacement (KSR), 3% FBS, 1% Glutamax, 1% NEAA, 0.0007% 2‐Mercaptoethanol, 4 ng/mL bFGF and 50 μmol/L Y27632. This modification resulted in the generation of EB more efficiently.
Matrigel embedding was performed at day 11 of organoid culture, and shaking was initiated on day 15. As expected, the size of brain organoids increased as the incubation period went on. The detailed structures of the cerebral organoids were analysed at day 35‐40 (Figure 1A).
FIGURE 1.
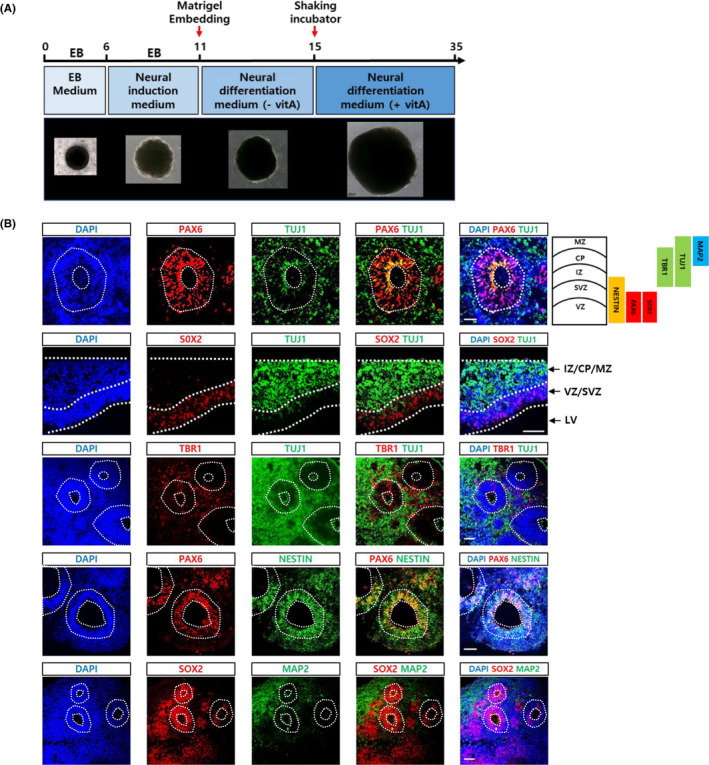
Generation of cerebral organoids. A, Schedule of brain organoid generation using an unguided method. 10 The shape and relative sizes of brain organoids at each step were shown at the bottom. B, Brain organoids at 6 weeks of culture were sectioned and analysed for the expression of various neural markers: Neural progenitor markers (PAX6, Nestin and SOX2), early neuronal markers (TUJ1), mature neuronal marker (MAP2) and postmitotic cortical neuronal marker (TBR1) were shown. Scale bar, 50 μm. Layers and representative markers of human cerebral cortex were shown on the top & right side. CP, cortical plate; IZ, intermediate zone; LV, lateral ventricles; MZ, marginal zone; SVZ, subventricular zone; VZ, ventricular zone
Several structures containing epithelial‐type cells surrounding the ventricle‐like space were frequently detected. In particular, cortical structures composed of ventricular and subventricular zones (VZ and SVZ) marked by SOX2 and PAX6 antibodies were often evident. NESTIN expression mostly overlapped with SOX2 and PAX6, but its expression extended further into the intermediate zone (IZ). TUJ1 (an early neuronal marker) marked the IZ, cortical plate (CP) and marginal zone (MZ). TBR1 (a deep‐layer cortical marker) was expressed in the IZ and CP. Finally, MAP2 (a mature marker) was mostly detected in the CP and MZ (Figure 1B).
These results suggested that our cerebral organoids recapitulated the typical features of early cerebral development.
3.2. Generation of the EGFRvIII mutation
EGFRvIII is the most common mutation of EGFR and is found in ~25% of all cases of GBM. The mutation is characterized by the loss of exons 2‐7 of the EGFR gene, resulting in an in‐frame deletion of 267 amino acids in the extracellular domain. EGFRvIII has not been detected in normal brains but is found in glioma. To explore whether the EGFRvIII mutation alone could promote gliogenesis and cell proliferation, we first sought to generate the mutation by deleting exon 2 to 7 of EGFR in H9‐hESCs using CRISPR/Cas9 (Figure 2A). As a result of genome editing, we generated EGFRwt/vIII‐ and EGFRvIII/vIII‐hESC lines which had a monoallelic and biallelic EGFRvIII gene, respectively. These mutations were confirmed via RT‐PCR and DNA sequencing (Figure 2B,C).
FIGURE 2.
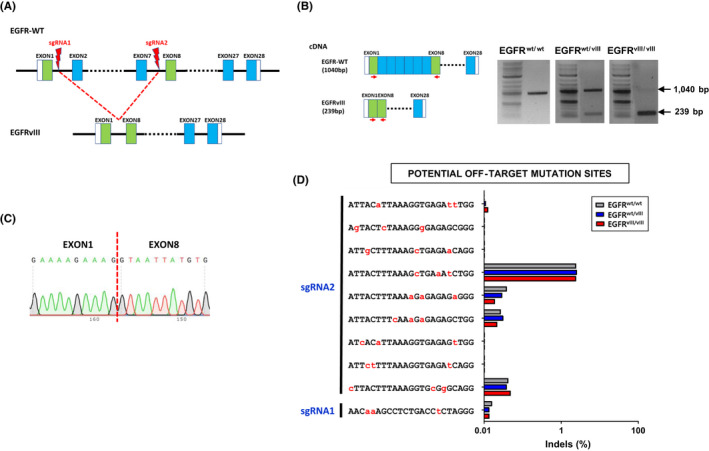
Generation of EGFRvIII‐hESC lines by CRISPR/Cas9 genome editing. A, Scheme of generation of EGFRvIII mutation by CRISRP/Cas9 system. Two sgRNA binding sites, one in intron 1 and the other in intron 7 of wild type EGFR locus, were indicated. B, Confirmation of monoallelic and biallelic EGFRvIII mutations was confirmed by RT‐PCR using primer pairs recognizing exon1 and exon 8: Primers were indicated as red arrows. Wild type EGFR cDNA produces 1040 bp PCR product, while EGFRvIII cDNA produces 239 bp DNA band. C, Sequencing analysis confirmed the deletion of exon 1 through exon 7 in EGFRvIII mutation. D, Targeted deep‐sequencing analysis revealed undetectable off‐target mutations in both EGFR‐edited clones, EGFRwt/vIII and EGFRvIII/vIII. Prediction of potential off‐target sites with up to three mismatches with the two sgRNA target sequences (sgRNA1‐targeting intron1 and sgRNA2‐targeting intron7 sequences) was performed with CRISPR RGEN Tools website (http://www.rgenome.net/about/): Our EGFR sgRNA target sequences had no potential off‐target sites with one or two mismatches, but had ten potential off‐target sites with three mismatches (one site in intron 1 and 9 sites in intron 7). The mismatched nucleotides were denoted in red and small capital letters in each sequence
To assess for potential off‐target editing effects, we compared the sgRNAs targeting intron 1 and 7 of the EGFR gene, sgRNA1 and 2, respectively, to the whole human genome. We found no off‐target sequences that matched these sgRNAs completely or with a 1‐nucleotide mismatch. However, a total of ten off‐target sequences had a 3‐base mismatch with one of the two sgRNAs, as shown in Figure 2D. Deep sequencing of the EGFRvIII (EGFRwt/vIII and EGFRvIII/vIII)‐hESC lines showed that no off‐target mutations were generated at any of these 10 3‐nt mismatching sequences after genome editing (Figure 2D).
We confirmed that the EGFRvIII‐hESC lines retained pluripotency through verifying expression of typical pluripotency markers (OCT4, SSEA4 and TRA1‐81; Figure 3A). Further, we found that the edited cell lines could still be differentiated into cells of all three lineages, mesoderm (smooth muscle actin, SMA), endoderm (alpha‐fetoprotein, AFP) and ectoderm (PAX6; Figure 3B). G‐banding analysis revealed no gross chromosomal aberrations in the EGFRvIII‐hESC clones (Figure 3C).
FIGURE 3.
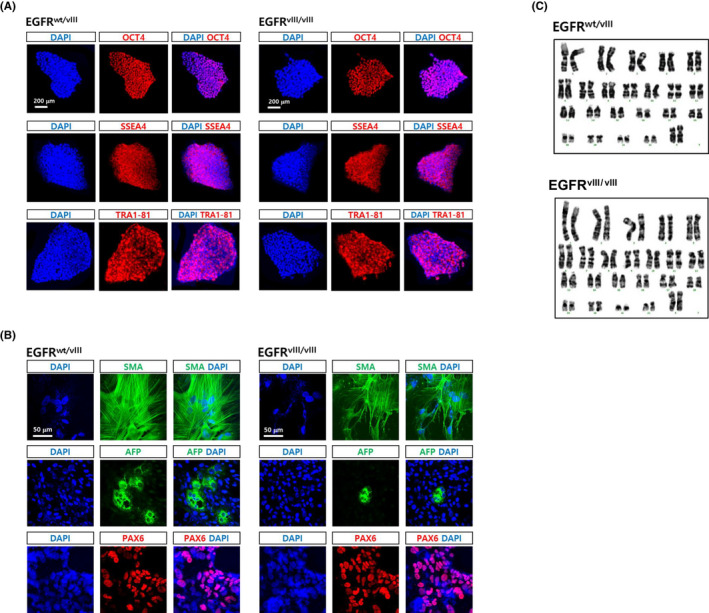
Characterization of EGFRvIII‐hESC clones. A, Both monoallelic and biallelic EGFRvIII clones were immunostained with antibodies recognizing typical hESC markers, Oct4, SSEA4, Oct4 and Tra1‐81. DAPI staining (blue) were also performed to expose the presence of cells. Scale bar, 200 μm. B, The EGFRvIII clones were spontaneously differentiated into derivatives of the three germ layers for 3 weeks via EB formation. Smooth muscle actin (SMA), alpha‐fetoprotein (AFP) and PAX6 were detected by immunostaining as markers for mesoderm, endoderm and ectoderm, respectively. Scale bar, 50 μm. C, G‐banding analysis of the EGFRvIII clones was performed to examine the presence of gross chromosomal abnormality
Taken together, these studies suggested that we successfully generated monoallelic (EGFRwt/vIII) and biallelic (EGFRvIII/vIII) EGFRvIII‐hESC lines that retained pluripotency, which could be used as a model of the role of the EGFRvIII mutation in gliogenesis.
3.3. EGFRvIII‐cerebral organoids
We generated cerebral organoids using monoallelic and biallelic EGFRvIII‐hESCs. Wild type (wt) H9‐hESCs (EGFRwt/wt‐hESCs) were used as an isogenic control. The size of both brain organoids that were generated from the EGFRvIII‐hESCs (EGFRwt/vIII and EGFRvIII/vIII‐hESCs) was much larger than those generated from wt‐hESCs (Figure 4A,B).
FIGURE 4.
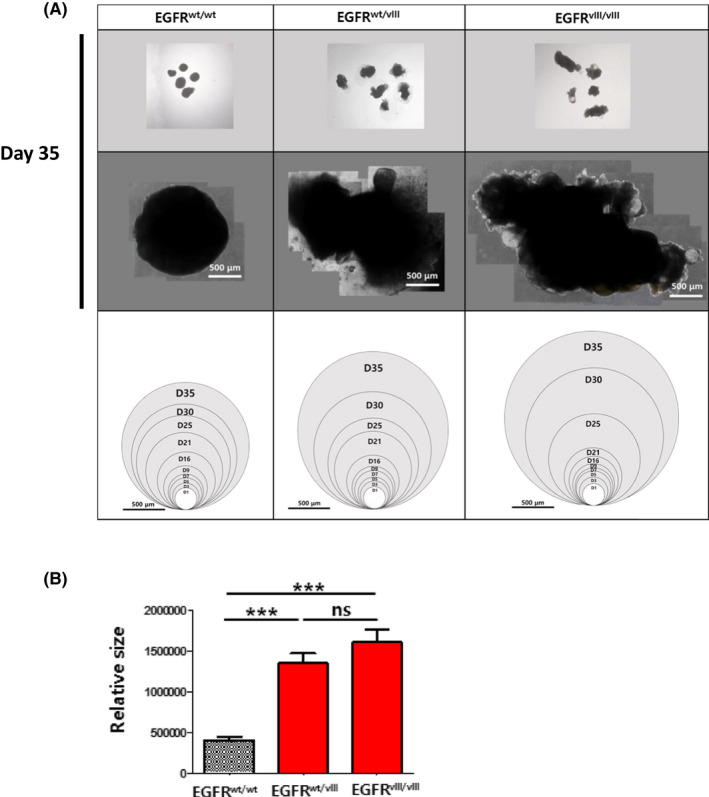
Size comparison of normal and EGFRvIII‐brain organoids. A, The bright‐field images of whole‐brain organoids were taken from day 1 through day 35. The representative images of brain organoids at day 35 were shown on top and middle panels. Total areas of brain organoids at individual days were measured by imagej software and were denoted as a circle corresponding to the area (bottom panels). Scale bar, 500 μm. B, The sizes of brain organoids of each group were shown in a graph. Results are presented as mean ± SEM. *P < .05; **P < .01; ***P < .001; ns, not significant
We used immunostaining to analyse the expression of representative neuronal (TUJ1) and glial (GFAP and S100β) markers. Beta‐tubulin III (TUJ1) was extensively detected in wt‐organoids, but significantly fewer TUJ1+ cells were detected in both EGFRwt/vIII and EGFRvIII/vIII‐organoids (Figure 5A,B, left‐most column). Two astrocyte markers, GFAP and S100β, were not detected in wt‐organoids. In contrast, the expression of GFAP and S100β was prominent in organoids derived from EGFRwt/vIII and EGFRvIII/vIII‐hESCs (Figure 5A,B, the second and third columns). GFAP immunostaining of the whole‐brain organoids showed extensive signal in EGFRwt/vIII and EGFRvIII/vIII‐organoids compared to sparse signal from wt‐organoids (Figures S1 and S2).
FIGURE 5.
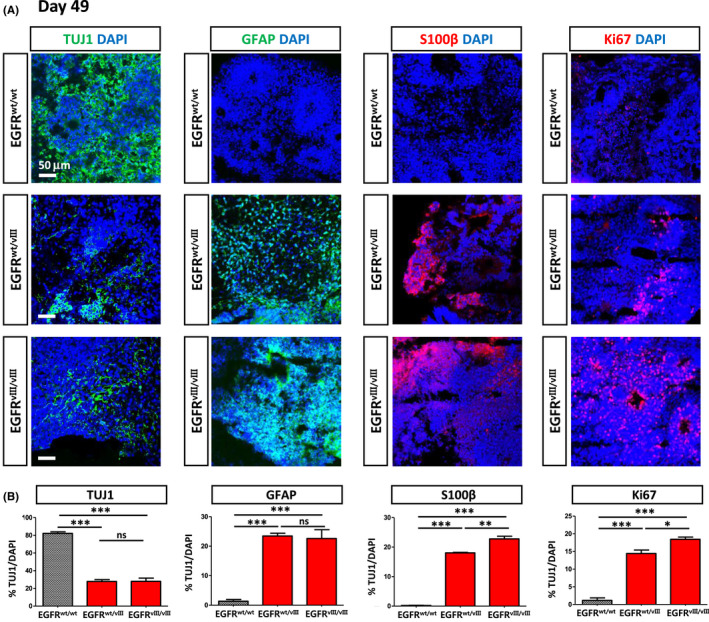
Immunofluorescence images of brain organoids. A, Brain organoids were generated using normal and EGFRvIII‐hESCs and were compared for the expression of representative markers for neurons (TUJ1), astrocytes (GFAP and S100β) and proliferating cells (Ki67) at day 49 by immunostaining. Scale bar, 50 μm. B, Expression of each marker among different organoids was quantified as % marker/DAPI. For this, the numbers of TUJ1+, GFAP+, S100β+ and Ki67+ cells in each brain organoid were quantified among total cells (DAPI+). Three independent experiments were performed
The number of proliferating cells (Ki67+ cells) was significantly higher in both EGFRwt/vIII and EGFRvIII/vIII‐organoids relative to wt‐organoids (Figure 5A,B, the right‐most column), consistent with the larger size of EGFRwt/vIII and EGFRvIII/vIII‐organoids compared with wt‐organoids.
Together, these results suggest that the EGFRvIII mutation promoted astrogenesis and massive cell proliferation in this organoid model.
3.4. Temozolomide treatment of the EGFRvIII‐brain organoids
Temozolomide (TMZ), an alkylating agent, is a chemotherapy drug that has been used to treat GBM and anaplastic astrocytoma. To test whether brain organoids are a good model to evaluate the efficacy of anti‐cancer drugs, we treated the EGFRvIII‐brain organoids with 1 mmol/L TMZ for 10 days (between days 50 and 60, Figure 6A).
FIGURE 6.
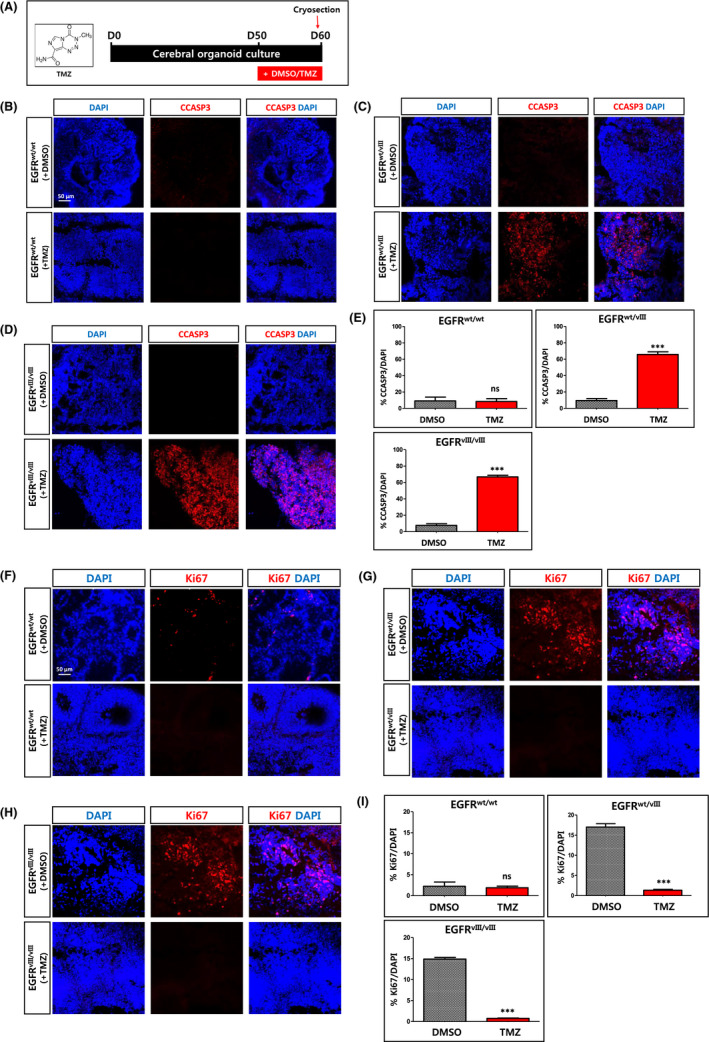
Effects of Temozolomide (TMZ) on EGFRvIII‐brain organoids. A, Scheme of treatment of brain organoids with Temozolomide (TMZ). As a negative control, DMSO was used. B‐D, After treatment with TMZ or DMSO for 10 days, the brain organoids were analysed by immunostaining using antibody recognizing cleaved caspase 3 (CCASP3). Brain organoids were generated using wt H9‐hESCs (B), monoallelic EGFRvIII (EGFRwt/vIII)‐hESCs (C) and biallelic EGFRvIII (EGFRvIII/vIII)‐hESCs (D). Scale bar, 50 μm. E, The percentages of CCASP3‐positive cells among total cells (DAPI+) were shown as graph. Results are presented as mean ± SEM. ***P < .001. N = 3 independent experiments. F‐H, After treatment with TMZ or DMSO for 10 days, the extent of proliferating cells in the brain organoids was analysed by immunostaining using antibody recognizing Ki67. Brain organoids were generated using wt H9‐hESCs (F), monoallelic EGFRvIII (EGFRwt/vIII)‐hESCs (G) and biallelic EGFRvIII (EGFRvIII/vIII)‐hESCs (H). Scale bar, 50 μm. I, The percentages of Ki67‐positive cells among total cells (DAPI+) were shown as a graph. Results are presented as mean ± SEM. **P < .01; ***P < .001; ns, not significant. N = 3 independent experiments
Immunostaining of the brain sections showed that cleaved caspase 3 (CCASP3), an apoptotic marker, was extensively detected in EGFRvIII (both EGFRwt/vIII and EGFRvIII/vIII)‐organoids, but not in wt‐organoids after TMZ treatment (Figure 6B‐E).
As seen in Figure 5 (at day 49), massive proliferation of Ki67+ cells was detected in EGFRvIII (both EGFRwt/vIII and EGFRvIII/vIII)‐organoids, but not in wt‐organoids at day 60 without TMZ treatment. On the other hand, a 10‐day treatment with TMZ robustly reduced the number of Ki67+ cells in EGFRwt/vIII and EGFRvIII/vIII‐organoids (Figure 6F‐I). This observation was in line with the significant increase in cell death in EGFRvIII‐organoids, but not in wt‐organoids, after TMZ treatment, as seen in Figure 6B‐E.
Taken together, these results show that the great increases in cell proliferation due to the EGFRvIII mutation were reversed by treatment with TMZ.
4. DISCUSSION
The outcome of any developmental process is the production of mature cells of various lineages that contribute to the physiological function of tissues. In malignancy, the processes that generate the fully matured cells are subverted, resulting in the aberrant production of great numbers of lineage‐associated cells that eventually become malignant. Emerging from these observations is a molecular and functional interrelationship of the genetic drivers (ie, mutations) at the interface of development and malignancy.
One of the most common genetic aberrations in GBM is an EGFR mutation called EGFRvIII, caused by in‐frame deletion of exons 2‐7 (amino acids 6‐273) of the EGFR gene. Due to the large deletion of exons 2‐7, a significant portion of the extracellular domain of EGFR is lost, resulting in a constitutively active tyrosine kinase. Intriguingly, the EGFRvIII mutation is prevalent in gliomas but not in other types of brain tumours, suggesting a specific role of EGFRvIII in gliomagenesis.
In this study, using a brain organoid model, we explored whether the EGFRvIII mutation‐induced gliogenesis and contributed to gliomagenesis. To this end, we used CRISPR/Cas9‐genome editing to generate monoallelic (EGFRwt/vIII) and biallelic (EGFRvIII/vIII) conditions by removing exons 2‐7 of the EGFR gene from one or both alleles of hESCs, respectively, and generated brain organoids using the resulting cell lines. Wild type hESCs were used as an isogenic control.
We observed that EGFRvIII (both EGFRwt/vIII and EGFRvIII/vIII)‐organoids grew larger than wt‐organoids after 25 days of culture. In addition to the increased size, the shape of the EGFRvIII‐organoids became irregular, but wt‐organoids retained a round shape. Consistently, we detected a substantial number of proliferating (Ki67+) cells in EGFRvIII‐organoids, but not in wt‐organoids at day 49 of culture. This result suggested that the EGFRvIII mutation promoted abnormal proliferation of the cells in the EGFRvIII‐organoids, which may eventually lead to malignancy.
Strikingly, cells expressing astrocyte markers (GFAP+ or S100β+ cells), were abundant throughout the EGFRwt/vIII and EGFRvIII/vIII‐organoids, but no such cells were detected in wt‐organoids at day 49. In contrast, TUJ1+ neurons were much more abundant in the brain organoids derived from wt (EGFRwt/wt)‐hESCs relative to the EGFRvIII‐hESCs. This result suggested that the constitutive activation due to EGFRvIII promoted astrogenesis at the expense of neurogenesis. In line with our result, previous reports have demonstrated that stimulation of EGFR signalling induces preferential differentiation toward glial lineages both in vitro and in vivo experiments in rats. 17 , 18 , 19
Our human brain organoid model showed that EGFRvIII mutation tended to steer neural differentiation toward gliogenesis (ie, astrogenesis) and induced massive proliferation, which may contribute to the formation of glioma. To our knowledge, this is the first study directly showing a connection between the EGFRvIII mutation and gliogenesis/gliomagenesis in a human system, let alone in brain organoids.
More than 90% of GBMs are primary brain tumours that develop rapidly de novo in adults. Recent studies have demonstrated that GBMs arise from neural stem cells (NSCs) that acquire driver mutations in the subventricular zone (SVZ), both in rodents and humans. 20 , 21 Therefore, it is plausible that the EGFRvIII mutation occurring in NSCs in the SVZ induces astrogenesis at the expense of neurogenesis and leads to active proliferation, eventually causing GBM. Likewise, cancer stem cells harbouring the EGFRvIII mutation may generate GBM.
Because of their intense proliferation, we examined whether EGFRvIII‐organoids could be used as a model to test the efficacy of GBM drugs. We found that treatment with TMZ significantly induced apoptosis in EGFRvIII‐organoids, but not in wt‐organoids. Therefore, the EGFRvIII‐brain organoids may provide a useful system to evaluate the efficacy of potential anti‐glioma drugs, especially those that target the EGFRvIII receptor.
This work suggests a clear role for EGFRvIII in gliomagenesis, laying a foundation for further investigation of its role in various aspects of glioma such as initiation and invasiveness, as well as in the development of novel anti‐glioma therapeutics.
CONFLICT OF INTEREST
All authors of this paper declare no potential conflicts of interest.
AUTHOR CONTRIBUTIONS
H.K., J.L., J.Y., D.H. designed the project. H.K. performed development of methodology. H.K., D.H. performed acquisition of data, analysis and interpretation and wrote the paper. H.K., S.L., J.L., J.Y., D.H. performed review, and/or revision of the manuscript. S.L. supported technical, or material support. D.H. performed study supervision.
Supporting information
Fig S1‐S2
Table S1
Table S2
ACKNOWLEDGEMENTS
This work was funded by grants from the Ministry of Health and Welfare, Korea (HI16C1559, HI18C0096).
Kim H‐M, Lee S‐H, Lim J, Yoo J, Hwang D‐Y. The epidermal growth factor receptor variant type III mutation frequently found in gliomas induces astrogenesis in human cerebral organoids. Cell Prolif.2021;54:e12965 10.1111/cpr.12965
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Laug D, Glasgow SM, Deneen B. A glial blueprint for gliomagenesis. Nat Rev Neurosci. 2018;19(7):393‐403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wainwright EN, Scaffidi P. Epigenetics and Cancer Stem Cells: Unleashing, Hijacking, and Restricting Cellular Plasticity. Trends Cancer. 2017;3(5):372‐386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016;131(6):803‐820. [DOI] [PubMed] [Google Scholar]
- 4. Brandes A, Soesan M, Fiorentino MV. Medical treatment of high grade malignant gliomas in adults: an overview. Anticancer Res. 1991;11(2):719‐727. [PubMed] [Google Scholar]
- 5. Brandes AA, Tosoni A, Franceschi E, Reni M, Gatta G, Vecht C. Glioblastoma in adults. Crit Rev Oncol Hematol. 2008;67(2):139‐152. [DOI] [PubMed] [Google Scholar]
- 6. Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60(5):277‐300. [DOI] [PubMed] [Google Scholar]
- 7. Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987‐996. [DOI] [PubMed] [Google Scholar]
- 8. Shanks N, Greek R, Greek J. Are animal models predictive for humans? Philos Ethics Humanit Med. 2009;4:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tentler JJ, Tan AC, Weekes CD, et al. Patient‐derived tumour xenografts as models for oncology drug development. Nat Rev Clin Oncol. 2012;9(6):338‐350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. van der Worp HB, Howells DW, Sena ES, et al. Can animal models of disease reliably inform human studies? PLoS Medicine. 2010;7(3):e1000245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lancaster MA, Knoblich JA. Generation of cerebral organoids from human pluripotent stem cells. Nat Protoc. 2014;9(10):2329‐2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lancaster MA, Renner M, Martin CA, et al. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501(7467):373‐379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR‐Cas9 system. Nat Protoc. 2013;8(11):2281‐2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brennan CW, Verhaak RG, McKenna A, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155(2):462‐477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wong AJ, Ruppert JM, Bigner SH, et al. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc Natl Acad Sci U S A. 1992;89(7):2965‐2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nishikawa R, Ji XD, Harmon RC, et al. A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc Natl Acad Sci U S A. 1994;91(16):7727‐7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Burrows RC, Wancio D, Levitt P, Lillien L. Response diversity and the timing of progenitor cell maturation are regulated by developmental changes in EGFR expression in the cortex. Neuron. 1997;19(2):251‐267. [DOI] [PubMed] [Google Scholar]
- 18. Jori FP, Galderisi U, Piegari E, et al. EGF‐responsive rat neural stem cells: molecular follow‐up of neuron and astrocyte differentiation in vitro. J Cell Physiol. 2003;195(2):220‐233. [DOI] [PubMed] [Google Scholar]
- 19. Kuhn HG, Winkler J, Kempermann G, Thal LJ, Gage FH. Epidermal growth factor and fibroblast growth factor‐2 have different effects on neural progenitors in the adult rat brain. J Neurosci. 1997;17(15):5820‐5829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Alcantara Llaguno S, Chen J, Kwon CH, et al. Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer Cell. 2009;15(1):45‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lee JH, Lee JE, Kahng JY, et al. Human glioblastoma arises from subventricular zone cells with low‐level driver mutations. Nature. 2018;560(7717):243‐247. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S2
Table S1
Table S2
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.