

Abstract
Degradation of mitochondria via a selective form of autophagy, named mitophagy, is a fundamental mechanism conserved from yeast to humans that regulates mitochondrial quality and quantity control. Mitophagy is promoted via specific mitochondrial outer membrane receptors, or ubiquitin molecules conjugated to proteins on the mitochondrial surface leading to the formation of autophagosomes surrounding mitochondria. Mitophagy‐mediated elimination of mitochondria plays an important role in many processes including early embryonic development, cell differentiation, inflammation, and apoptosis. Recent advances in analyzing mitophagy in vivo also reveal high rates of steady‐state mitochondrial turnover in diverse cell types, highlighting the intracellular housekeeping role of mitophagy. Defects in mitophagy are associated with various pathological conditions such as neurodegeneration, heart failure, cancer, and aging, further underscoring the biological relevance. Here, we review our current molecular understanding of mitophagy, and its physiological implications, and discuss how multiple mitophagy pathways coordinately modulate mitochondrial fitness and populations.
Keywords: autophagy, mitochondria, phosphorylation, quality and quantity control, ubiquitin
Subject Categories: Autophagy & Cell Death
This review describes the conserved pathways for mitochondrial degradation via selective autophagy across species, and how multiple mitophagy pathways cooperate to modulate mitochondrial fitness and number in normal or disease physiology.
Glossary
- ALLO‐1
Allophagy‐1
- ATG
Autophagy‐related protein
- BCL2L1/BCL‐XL
BCL2 like 1
- BCL2L13
B‐cell lymphoma 2‐like 13
- BNIP3
BCL2 and adenovirus E1B 19‐kDa‐interacting protein 3
- BNIP3L
Nip3‐like protein X (NIX)/BNIP3‐like protein
- CCCP
Carbonyl cyanide m‐chlorophenylhydrazone
- cGAS
Cyclic GMP‐AMP synthase
- CK2
Casein kinase 2
- CPS‐6
Mitochondrial endonuclease G
- DFCP1/ZFYVE1
DFCP1/zinc finger FYVE‐type containing 1
- FIP200/RB1CC1
FIP200/RB1‐inducible coiled‐coil protein 1
- Fis1
Fission, mitochondrial 1
- FKBP8/FKBP38
FK506‐binding protein 8
- FOXO1
Forkhead box O1
- FUNDC1
FUN14 domain‐containing protein 1
- GABARAP
GABA type A receptor‐associated protein
- GABARAPL1/2
GABA type A receptor‐associated protein‐like 1/2
- GFP
Green fluorescent protein
- HOPS
Homotypic fusion and vacuole protein sorting
- IGF‐1
Insulin‐like growth factor 1
- Keap1
Kelch‐like ECH‐associated protein 1
- LC3A/B/C
Microtubule‐associated protein 1 light chain 3 alpha/beta/gamma
- LIR
LC3‐interacting region
- MARCH5/MITOL
Membrane‐associated ring‐CH‐type finger 5
- MBP
Maltose‐binding protein
- Miro
Mitochondrial Rho
- mTORC1
Mechanistic target of rapamycin complex 1
- MUL1
mitochondrial E3 ubiquitin protein ligase 1
- NBR1
NBR1 autophagy cargo receptor
- NDP52/CALCOCO2
NDP52/calcium binding and coiled‐coil domain 2
- NLRP3
NLR family pyrin domain‐containing 3
- NOD
Nucleotide‐binding oligomerization domain
- NRF2
Nuclear factor, erythroid 2‐like 2
- OPTN
Optineurin
- p62/SQSTM1
p62/Sequestosome 1
- PARL
Presenilin‐associated rhomboid‐like protein
- PC
Phosphatidylcholine
- PE
Phosphatidylethanolamine
- PGAM5
PGAM family member 5, mitochondrial serine/threonine protein phosphatase
- PI
Phosphatidylinositol
- PI3K
Phosphatidylinositol 3‐kinase
- PI3P
Phosphatidylinositol‐3‐phosphate
- PINK1
PTEN induced kinase 1
- PLEKHM1
Pleckstrin homology and RUN domain‐containing M1
- RABGEF1
RAB guanine nucleotide exchange factor 1
- Rheb
Ras homolog, mTORC1 binding
- SNARE
Soluble N‐ethylmaleimide‐sensitive factor attachment protein receptor
- Src
SRC proto‐oncogene, non‐receptor tyrosine kinase
- STING
Stimulator of interferon genes
- TAX1BP1
Tax1 binding protein 1
- TBC1D15
TBC1 domain family member 15
- TBC1D17
TBC1 domain family member 17
- TBK1
TANK‐binding kinase 1
- TOMM/TOM
Translocase of the outer mitochondrial membrane
- TORC1
Target of rapamycin complex 1
- UBAN
Ubiquitin‐binding domain in ABIN proteins and NEMO
- ULK1
Unc‐51‐like autophagy activating kinase 1
- USP
Ubiquitin specific protease
- VDAC
Voltage‐dependent anion channel
- VPS
Vacuolar protein sorting
- WIPI
WD repeat domain, phosphoinositide interacting
Introduction
Mitochondria are double‐membrane‐bound subcellular compartments that function in fundamental processes such as ATP production, phospholipid biosynthesis/transport, calcium signaling, and iron homeostasis (Raffaello et al, 2016; Tamura & Endo, 2017; Spinelli & Haigis, 2018). These organelles act as platforms for various events including apoptosis, innate immune response, and cell differentiation (Mehta et al, 2017; Kalkavan & Green, 2018; Lisowski et al, 2018). Since mitochondria generate reactive oxygen species (ROS) from the electron transport chain, they are constantly challenged with oxidative stress that ultimately may lead to their structural and functional failure (Wong et al, 2017). Therefore, cells need sophisticated systems for maintaining mitochondrial fitness. Mitochondrial quality control relies on diverse pathways: ROS scavenging, DNA repair, and protein refolding/degradation (Scheibye‐Knudsen et al, 2015). In addition to these processes, mitochondrial fusion and fission play key roles in mitochondrial quality control (Eisner et al, 2018). While fusion promotes content mixing between healthy and partially dysfunctional mitochondria, fission separates damaged mitochondrial components from the mitochondrial pool.
The autophagic system targets impaired mitochondria and delivers them to lysosomes for degradation. This catabolic process, called mitophagy, contributes to maintaining mitochondrial quality control (Pickles et al, 2018) and mitochondrial quantity in multiple cell types. In tissues consuming a large amount of ATP such as brain, skeletal muscle, heart, liver, and kidney, mitochondria are highly developed in order to maintain the proper balance between energy demand and supply. When these cells are shifted from normoxia to hypoxia, mitophagy is induced to decrease mitochondrial quantity, thereby adapting cellular metabolism to anaerobic conditions (Wu & Chen, 2015). Thus, mitochondrial biogenesis and degradation are two opposing processes that determine mitochondrial quantity (Ploumi et al, 2017). In addition, mitochondria are almost completely eliminated during erythrocyte maturation (Ney, 2015). Furthermore, accumulating evidence reveals that maternal inheritance of mitochondrial DNA (mtDNA) depends on selective clearance of sperm‐derived paternal mitochondria during early embryogenesis (Sato & Sato, 2017).
Although autophagy is generally recognized as a bulk degradation process that non‐selectively transports cytoplasmic components such as nucleic acids, proteins, and organelles to lysosomes (Nakatogawa, 2020), it also acts as a selective system to mediate clearance of particular organelles (Gatica et al, 2018). Mitophagy is one of the organelle‐specific autophagy pathways that serves to maintain cell structure and function (Okamoto, 2014) (Fig 1). The term “mitophagy” was first coined in 2005 (Lemasters, 2005; Priault et al, 2005), and within a few years, major breakthroughs led to the discovery of key proteins that selectively mediate mitochondrial degradation in yeast (Okamoto et al, 2009; Kanki et al, 2009b) and mammalian cells (Schweers et al, 2007; Narendra et al, 2008; Sandoval et al, 2008). In this review, we will describe the molecular mechanisms underlying mitophagy in yeast, worms, Drosophila, and mammalian cells and cover its physiological and pathophysiological functions.
Figure 1. Overview of mitophagy.
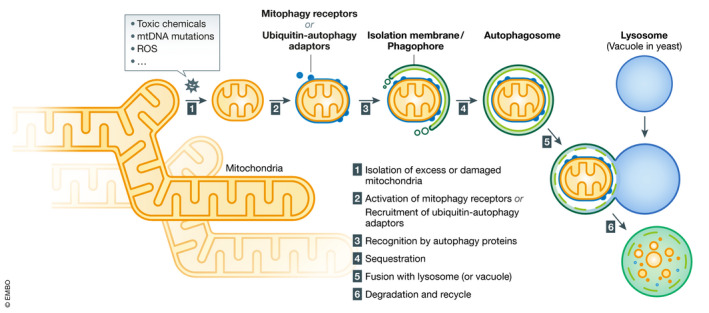
(1) Intra‐ and extracellular cues promote isolation of excess or damaged mitochondria via fragmentation of tubular networks. (2) Mitophagy receptors or ubiquitin–autophagy adaptors that confer selectivity for degradation are recruited and/or activated on the surface of mitochondria. (3) Core autophagy‐related proteins target to mitochondria and generate the isolation membrane/phagophore surrounding mitochondria. (4) Targeted mitochondria are enclosed and sequestrated by autophagosomes. (5) Autophagosomes are transported and fused with lytic compartments such as vacuoles in yeast or lysosomes in mammals. (6) Lysosomal or vacuolar acidic hydrolases flow into autophagosomes to degrade mitochondria, and the contents will be recycled.
Receptor‐mediated mitophagy in yeast
Regulation of mitophagy by Atg32
Mitophagy in the budding yeast Saccharomyces cerevisiae is mostly mediated by Atg32, a single‐pass transmembrane protein in the outer mitochondrial membrane (OMM) (Okamoto et al, 2009; Kanki et al, 2009) (Fig 2A). In this unicellular eukaryote, mitophagy is induced when cells are grown to stationary phase or upon nitrogen starvation (Tal et al, 2007; Kanki & Klionsky, 2008; Okamoto et al, 2009). Under such conditions, Atg32 expression is induced at the transcriptional level and accumulates on the OMM, forming a complex with Atg8 and Atg11 on the surface of mitochondria. Atg8 is localized to autophagosomes, and Atg11 acts as a scaffold for other Atg proteins to promote autophagosome formation. Loss of Atg32 almost completely abolishes mitophagy while its overexpression increases mitophagy activity, suggesting that this molecule is a rate‐limiting factor for regulating the number of mitochondria to be degraded. Atg32 is specifically important to degrade mitochondria and is dispensable for other types of autophagy‐related processes including bulk autophagy, the cytoplasm‐to‐vacuole targeting pathway, ER‐phagy, and pexophagy.
Figure 2. Receptor‐mediated mitophagy.
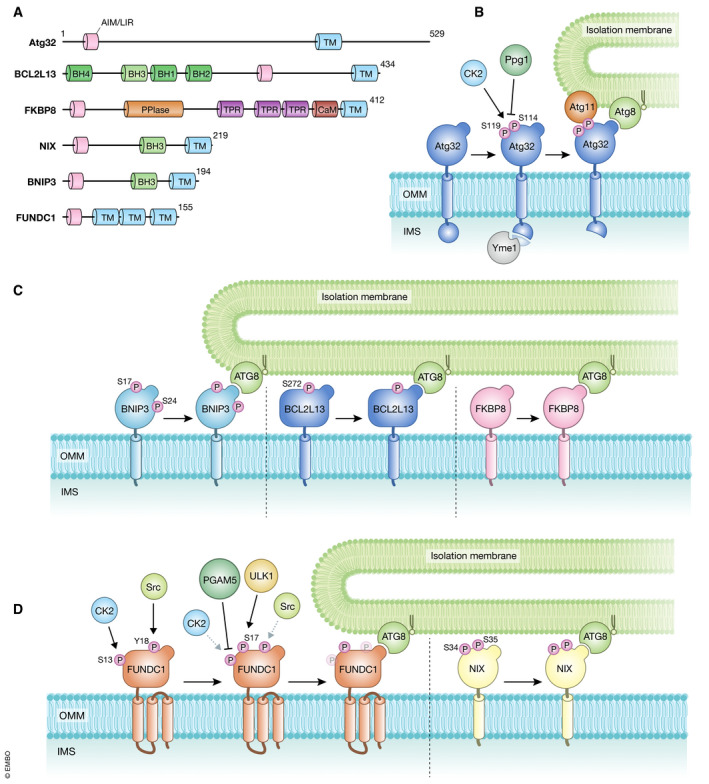
(A) Schematic representation of the domain structures of mitophagy receptors in yeast and mammals. AIM/LIR, Atg8‐family protein‐interacting motif/LC3‐interacting region (pink); TM, transmembrane domain (light blue); BH1‐4, Bcl‐2 homology 1‐4 domain (green and light green); PPlase, peptidyl‐prolyl cis‐trans isomerase domain (orange); TPR, tetratricopeptide repeat domain (purple); CaM, calmodulin‐binding domain (dark red). The protein size is indicated as the number of amino acids. (B‐D) Models for mitophagy receptor activation and protein recruitment on the mitochondrial surface. The yeast mitophagy receptor Atg32 (B), and the mammalian mitophagy receptors BNIP3, BCL2L13, FKBP8 (C), FUNDC1, and NIX (D) bind to ATG8 family proteins and then target the autophagy machinery to mitochondria. Phosphorylation and dephosphorylation serve as regulatory mechanisms to modulate the activity of mitophagy receptors. For details, see text.
Several lines of evidence reveal that phosphorylation is a key event for Atg32‐mediated mitophagy (Fig 2B). During respiration or upon a shift from respiration to starvation, Atg32 is phosphorylated in a manner dependent on its Atg11‐interacting motif containing Ser114 and Ser119 (Aoki et al, 2011; Kondo‐Okamoto et al, 2012). Importantly, this post‐translational modification is mediated by CK2, an evolutionarily conserved serine/threonine kinase that regulates a variety of cellular processes (Kanki et al, 2013). CK2 interacts with Atg32 in vivo and directly phosphorylates Atg32 in vitro (Kanki et al, 2013). Mutagenesis of Atg32 Ser114, Ser119, and other conserved residues in the Atg11‐interacting motif or impairment of CK2 function destabilizes Atg32‐Atg11 interactions and strongly suppresses mitophagy (Aoki et al, 2011; Kondo‐Okamoto et al, 2012; Kanki et al, 2013), suggesting that CK2‐dependent phosphorylation could act as a regulatory step to activate Atg32 for recruiting Atg11 to mitochondria.
A recent study has demonstrated that the protein phosphatase 2A (PP2A)‐like protein Ppg1 is critical for dephosphorylation of Atg32 and negatively regulates mitophagy (Furukawa et al, 2018) (Fig 2B). In cells lacking Ppg1, Atg32 is phosphorylated even at the respiratory log phase (stage prior to mitophagy induction), likely resulting in increased Atg32‐Atg11 interactions that accelerate mitochondrial degradation (Furukawa et al, 2018). Ppg1‐dependent mitophagy suppression also requires its binding partners Far proteins that have previously been suggested to form a complex critical for pheromone‐induced cell cycle arrest (Pracheil & Liu, 2013). These findings raise the possibility that the Ppg1‐Far complex dephosphorylates Atg32, competing against CK2‐mediated phosphorylation under mitophagy non‐inducing conditions.
Atg32 has been known to be proteolytically cleaved by Yme1, a catalytic subunit of metalloprotease in the inner mitochondrial membrane (IMM) that belongs to the ATPases associated with diverse cellular activities (AAA) protein family (Leonhard et al, 1996). Upon mitophagy, Atg32 is proteolytically processed at its C‐terminal portion in a Yme1‐dependent manner (Wang et al, 2013) (Fig 2B). Loss of Yme1 leads to a strong decrease in Atg32‐Atg11 interactions and mitophagy under nitrogen starvation (Wang et al, 2013). These findings support the idea that Yme1‐mediated proteolysis is required for efficient mitophagy. However, other studies suggest minor or no mitophagy deficiencies in cells lacking Yme1 (Welter et al, 2013; Gaspard & McMaster, 2015), raising the possibility that Yme1‐dependent processing may be relevant to Atg32‐mediated mitophagy in some specific strains and/or under some specific conditions.
Regulation of mitophagy via ER factors
In yeast, mitochondria and the ER are connected at contact sites via the ER–mitochondria encounter structure (ERMES) complex that facilitates phospholipid transfer between these two organelles (Kornmann et al, 2009). The ERMES complex is localized at discrete foci where the ER and mitochondria are closely positioned, and loss of ERMES leads to severe defects in starvation‐induced mitophagy (Bockler & Westermann, 2014). Under starvation conditions, the ERMES component Mmm1 forms foci that partially co‐localize with Atg8 dot‐like structures, suggesting that autophagosomes are associated with the ER–mitochondria contact sites (Bockler & Westermann, 2014). Ubiquitylation of the ERMES component Mdm12/34 by the E3 ligase Rsp5 has also been linked to mitophagy (Belgareh‐Touze et al, 2017).
Atg32‐mediated mitophagy is also regulated via Get1/2 complex and Opi3, two factors associated with the ER (Sakakibara et al, 2015; Onishi et al, 2018). The Get1/2 complex is important for insertion of tail‐anchored proteins into the ER membrane (Schuldiner et al, 2005; Schuldiner et al, 2008; Wang et al, 2014). Loss of Get1/2 causes defects in mitophagy under respiratory conditions, while other types of autophagy‐related pathways are slightly or hardly affected (Onishi et al, 2018). How Get1/2 acts in trans to promote mitochondrial clearance remains unclear. Surprisingly, loss of Opi3, a phospholipid methyltransferase localized in the ER, leads to suppression of Atg32 induction during respiration (Sakakibara et al, 2015). Opi3 acts in the phospholipid biosynthesis pathway for conversion of PE into PC. Depletion of Opi3 causes aberrant elevation of glutathione levels that reduces cellular oxidative stress and thus negatively affects induction of Atg32 and mitophagy (Deffieu et al, 2009; Okamoto et al, 2009; Sakakibara et al, 2015). These findings raise the possibility that respiring yeast cells coordinate phospholipid methylation and mitophagy through unknown mechanisms.
Receptor‐mediated mitophagy in mammals
In mammals, mitophagy is mechanistically more complex than in yeast and is induced by different cellular stress signals and developmental changes. Disruption of mitochondrial membrane potential is a potent trigger of mitophagy (Elmore et al, 2001). CCCP, a proton‐selective ionophore, and antimycin A (an inhibitor of the respiratory complex III) are commonly used to impair mitochondria and activate mitophagy. Because CCCP is highly toxic and induces non‐physiological levels of mitochondrial damage especially in neurons, antimycin A is often used to induce mitophagy in neuronal cells (Cai et al, 2012; Ashrafi et al, 2014). Both reagents trigger mitochondrial depolarization and promote accumulation of mitophagy receptors on the OMM. These receptors are integral membrane proteins that promote specific binding to mammalian Atg8 family members (LC3A/B/C, GABARAP, GABARAP‐L1/2) through a conserved LC3‐interacting regions (LIRs) and regulate the formation of isolation membranes enclosing mitochondria.
Two major types of receptors have been suggested to mediate elimination of mitochondria under physiological and pathological conditions in mammals (Fig 2A). One group includes BNIP3 and BNIP3L (also known as NIX) (Boyd et al, 1994; Matsushima et al, 1998; Chen et al, 1999; Vande Velde et al, 2000; Regula et al, 2002; Kubli et al, 2007; Schweers et al, 2007; Sandoval et al, 2008; Hanna et al, 2012), and the other group includes FUNDC1 (Liu et al, 2012). In addition, BCL2L13 is the mammalian functional counterpart of yeast receptor Atg32 (Murakawa et al, 2015) (Fig 2A). In the following part, we will discuss the molecular functions of mitophagy receptors in mammalian cells and the role of a family of receptors, namely FKBP proteins (Bhujabal et al, 2017).
BNIP3 and NIX
BNIP3 is required for efficient turnover of mitochondria under hypoxic conditions (Zhang et al, 2008). In response to hypoxia, BNIP3 is upregulated and anchored to the OMM via its C‐terminal transmembrane (TM) domain, exposing the N‐terminal domain to the cytosol (Hanna et al, 2012). BNIP3 is usually expressed as an inactive monomer in the cytosol, but following stress signals, it forms a stable homodimer via its C‐terminal TM domain and is integrated into the OMM (Chen et al, 1997; Ray et al, 2000; Kubli et al, 2008). BNIP3 mutations, which disrupt homodimerization but do not affect mitochondrial localization, cause a mitophagy defect, supporting the idea that homodimerization of BNIP3 is important for efficient degradation of mitochondria (Hanna et al, 2012). Similar to other mitophagy receptors, BNIP3 has a LIR motif at its N‐terminal region (Fig 2A) and mutations in this region block the interaction with LC3, leading to mitophagy defects. Phosphorylation of BNIP3 at Ser17 and Ser24 near the LIR motif is important for BNIP3‐LC3 interactions (Zhu et al, 2013) (Fig 2C).
NIX shows homology to BNIP3 (53–56% amino acid sequence identity) (Matsushima et al, 1998; Chen et al, 1999) and promotes selective degradation of mitochondria during reticulocyte maturation (Schweers et al, 2007; Sandoval et al, 2008). During erythroid differentiation, cell nucleus, mitochondria, and other intracellular organelles are eliminated, so that erythrocytes can keep maximum space for hemoglobin that delivers oxygen (Koury et al, 2005; Yoshida et al, 2005; Fader & Colombo, 2006). With the high sequence similarity between these two proteins, expression of BNIP3 can restore mitochondrial clearance in reticulocytes lacking NIX (Zhang et al, 2012). NIX contains an LIR motif that promotes binding to LC3A, LC3B, GABARAP, GABARAP‐L1, and GABARAP‐L2 (Novak et al, 2010) (Fig 2A). In CCCP‐treated cells, NIX recruits GABARAP‐L1 to damaged mitochondria and promotes mitophagy in a manner dependent on its LIR motif (Novak et al, 2010). Phosphorylation of Ser34 and Ser35, two tandem serine residues near the LIR motif, stabilizes NIX‐LC3 interactions and promotes mitophagy (Rogov et al, 2017) (Fig 2D). Similar to BNIP3, dimerization of NIX, which is regulated by phosphorylation of its C‐terminal region, is important for efficient recruitment of the autophagic machinery to mitochondria (Marinkovic et al, 2020).
Accumulation of ROS (triggered by oxidative phosphorylation) promotes NIX‐mediated mitophagy via a recruitment of LC3 to mitochondria (Melser et al, 2013). Under conditions of oxidative phosphorylation, Rheb, a small GTPase of the Ras superfamily, translocates to mitochondria and forms a complex with NIX and LC3 to promote mitophagosome formation (Melser et al, 2013). Expression of Rheb in HeLa cells increases mitochondrial respiration, and loss of Rheb decreases the oxygen consumption capacity (Melser et al, 2013). Whether these phenotypes depend on Rheb‐induced mitophagy remains to be addressed. BNIP3 has also been shown to bind and inhibit Rheb, which is crucial for mTORC1 activation (Li et al, 2007). As mTORC1 negatively regulates bulk autophagy and mitophagy (Bartolome et al, 2017), BNIP3‐dependent mTORC1 inhibition might facilitate mitophagy induction or take part in a positive feedback loop to amplify the initiation signal of mitophagy.
Several studies have reported that BNIP3 and NIX act in PINK1/Parkin‐mediated mitophagy. NIX is ubiquitylated by Parkin, which in turn promotes targeting of the selective autophagy adaptor NBR1 that binds both ubiquitin and LC3/GABARAP to promote formation of autophagosomes surrounding mitochondria (Gao et al, 2015). In addition, BNIP3 interacts with PINK1 and facilitates accumulation of PINK1 on the OMM, resulting in Parkin translocation to mitochondria (Zhang et al, 2016a). NIX also contributes to CCCP‐induced mitochondrial depolarization, and accumulation of Parkin on damaged mitochondria (Ding et al, 2010b). Pathophysiological relevance of BNIP3 and NIX in Parkinson’s disease remains unknown.
FUNDC1
FUNDC1 is an integral OMM protein that functions as a receptor for hypoxia‐induced mitophagy. It contains a typical LIR motif near the N‐terminal region and three TM domains (Liu et al, 2012) (Fig 2A). Mutations in the LIR motif disrupt FUNDC1‐LC3 interactions and mitophagy induction (Liu et al, 2012). FUNDC1 protein levels are regulated in part by OMM‐anchored MARCH5/MITOL (Chen et al, 2017), an E3 ubiquitin ligase that is known to ubiquitylate several proteins acting in mitochondrial dynamics (Yonashiro et al, 2006; Sugiura et al, 2013; Park et al, 2014). FUNDC1 expression is decreased during hypoxia in a ubiquitin–proteasome‐dependent manner due to MARCH5‐mediated ubiquitylation of FUNDC1 at Lys119 (Chen et al, 2017). Knockdown of endogenous MARCH5 or overexpression of a MARCH5 catalytic mutant impairs ubiquitylation and degradation of FUNDC1, thereby enhancing hypoxia‐induced mitophagy (Chen et al, 2017). Similar to Atg32 in yeast cells, FUNDC1 is regulated via phosphorylation and dephosphorylation during mitophagy on residues Ser13 and Tyr18 that are located near the LIR motif. Under normoxia conditions, Ser13 is phosphorylated by CK2, while the Src tyrosine kinase mediates phosphorylation of Tyr18 to negatively regulate FUNDC1‐LC3 interactions (Liu et al, 2012; Chen et al, 2014) (Fig 2D). Upon hypoxia, Src becomes inactivated, causing decreased phosphorylation of Tyr18, stabilization of the interaction between FUNDC1 and LC3, and promotion of mitophagosome formation (Liu et al, 2012). The mitochondrial serine/threonine phosphatase PGAM5 dephosphorylates Ser13 and enhances FUNDC1‐LC3 interactions to promote mitophagy (Chen et al, 2014).
Hypoxia or mitochondrial depolarization induces ULK1 expression and its targeting to mitochondria, leading to FUNDC1 phosphorylation at Ser17 (near the LIR motif) and stabilization of its interaction with LC3 (Wu et al, 2014b). Expression of a FUNDC1 variant defective in ULK1 binding inhibits targeting of ULK1 to mitochondria and mitophagy, suggesting that FUNDC1 also acts as a receptor for ULK1 (Wu et al, 2014b). Under normoxic conditions, BCL2L1/Bcl‐xL, an antiapoptotic BH3 domain‐containing molecule, binds PGAM5 and inhibits PGAM5‐FUNDC1 interactions to prevent dephosphorylation of FUNDC1 Ser13 and mitophagy (Wu et al, 2014a).
BCL2L13
Atg32 homologs have so far not been identified in mammalian cells, but findings from yeast reveal that BCL2L13 can induce mitophagy in cells lacking Atg32, raising the possibility that BCL2L13 acts as a mammalian Atg32 functional counterpart (Murakawa et al, 2015). BCL2L13 is an OMM‐anchored single‐pass membrane protein containing two LIR motifs (Fig 2A). BCL2L13 also regulates mitochondrial morphology and its overexpression induces mitochondrial fragmentation, while its silencing causes mitochondrial elongation (Murakawa et al, 2015). BCL2L13‐dependent mitophagy in yeast cells lacking Atg32 is likely mediated via the conventional autophagy machinery as it requires Atg7, a core protein essential for Atg8 lipidation (Murakawa et al, 2015). In addition, mutations in the second LIR motif reduce BCL2L13‐dependent mitochondrial degradation in the absence of Atg32, supporting the notion that BCL2L13 promotes mitophagy via Atg8 in yeast (Murakawa et al, 2015). BCL2L13 phosphorylation also seems to contribute to regulation of BCL2L13‐LC3 interactions as the mutation at Ser272 near the second LIR motif reduces mitophagy (Murakawa et al, 2015) (Fig 2C). BCL2L13 also interacts with ULK1 to localize the autophagy initiation complex to mitochondria (Murakawa et al, 2019). However, under which physiological conditions BCL2L13 is induced and activated remains to be elucidated.
FKBP8
The immunosuppressant drug FK506 (also known as tacrolimus) binds to a conserved family of proteins called FKBP that functions in different cellular processes including transcription, protein folding/trafficking, signaling, and apoptosis (Bonner & Boulianne, 2017). Co‐overexpression of FKBP8 and LC3A promotes degradation of depolarized mitochondria in CCCP‐treated, Parkin‐depleted HeLa cells (Bhujabal et al, 2017). FKBP8 is an integral OMM protein containing a canonical LIR motif near the N‐terminus and a TM domain at the C‐terminus (Fig 2A). FKBP8 preferentially interacts with LC3A over other Atg8 family proteins in vivo, and this is critical for its mitophagy activity (Bhujabal et al, 2017) (Fig 2C). Moreover, FKBP8 can escape from degradation‐prone mitochondria and localizes to the ER via unknown mechanisms (Saita et al, 2013; Bhujabal et al, 2017). Given the complexity due to its versatile functions (Bonner & Boulianne, 2017), further studies are needed to clarify whether endogenous FKBP8 is directly involved in mitophagy.
Ubiquitin‐mediated mitophagy
PINK1 and Parkin
Parkinson’s disease (PD) is a major neurodegenerative disease characterized by cell death of dopaminergic neurons (Lotharius & Brundin, 2002). PD occurs sporadically in 1–2% of people above 65 years of age but can also arise earlier mostly due to genetic mutations. Common disease phenotypes observed in PD patients are motor symptoms (tremor, bradykinesia, rigidity, and postural instability) that result from dopaminergic neuronal loss in substantia nigra. Non‐motor symptoms such as autonomic dysfunction, neuropsychiatric problems, and sleep difficulties are also frequently observed. The relationship between sporadic PD and mitochondrial abnormality has been suggested since 1980s (Corti et al, 2011). The serine–threonine kinase PINK1 and the E3 ubiquitin ligase PARKIN were identified as causal genes for hereditary recessive PD with young onset (Kitada et al, 1998; Valente et al, 2004).
Parkin activation
In 2008, a key study revealed that loss of the mitochondrial membrane potential triggers recruitment of Parkin to mitochondria and that Parkin promotes degradation of damaged mitochondria through autophagy (Narendra et al, 2008). PINK1 has subsequently been reported to regulate Parkin E3 activity upon mitochondrial depolarization (Matsuda et al, 2010; Narendra et al, 2010). Since conversion of Parkin from inactive to active form requires PINK1, PINK1‐mediated phosphorylation should play an important role in Parkin activation. PINK1 directly phosphorylates and activates Parkin on Ser65 in its ubiquitin‐like (Ubl) domain, and this phosphorylation is important for Parkin function (Kondapalli et al, 2012; Shiba‐Fukushima et al, 2012; Iguchi et al, 2013). However, phosphomimetic mutation did not cause autoubiquitylation of GFP‐tagged phosphomimetic Parkin, suggesting that Parkin phosphorylation itself is insufficient for its activation. Three groups independently found another PINK1 target that is key for Parkin activation, and discovered that (i) PINK1 phosphorylates ubiquitin at Ser65; (ii) ubiquitin‐derived Ser65 phosphopeptide can be detected in cells accumulating PINK1 on depolarized mitochondria; and (iii) the phospho‐ubiquitin accelerates Parkin E3 ligase activity in vitro (Kane et al, 2014; Kazlauskaite et al, 2014; Koyano et al, 2014). Recent advances in structural information and molecular mechanisms underlying Parkin activation are described in detail in Box 1.
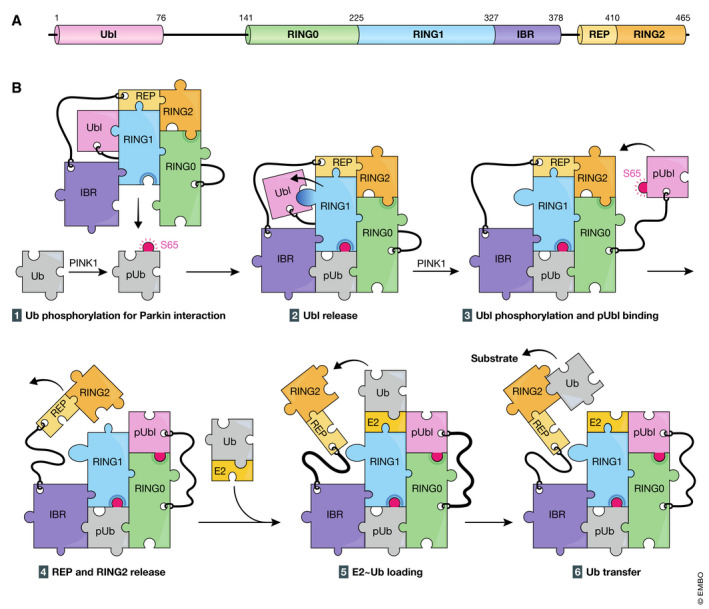
Box 1. Molecular mechanisms of Parkin activation on depolarized mitochondria.
Parkin consists of Ubl (ubiquitin‐like), RING0 (really interesting new gene 0), RING1, IBR (in‐between‐RING), REP (repressor element of Parkin), and RING2 domains (A). Structural analysis of Parkin alone (equivalent to a latent E3 form at steady‐state conditions) revealed that Parkin has an auto‐inhibited conformation mediated by multiple domain–domain interactions. Namely, the RING0 (also referred to as UPD; unique parkin domain) occludes the catalytic core residue Cys431 (ubiquitin acceptor site) in the RING2, and the REP binds the RING1 to block its E2‐binding interface (B‐1) (Riley et al, 2013; Trempe et al, 2013; Wauer & Komander, 2013; Kumar et al, 2015). Interestingly, when phosphorylated ubiquitin interacts with Parkin (B‐2), intramolecular structural remodeling takes place. Helix of the RING1 (H3) is straightened by phosphorylated ubiquitin, which induces conformational changes in RING1 and IBR, thereby releasing the Ubl (B‐2) (Kazlauskaite et al, 2015; Kumar et al, 2015; Sauve et al, 2015; Wauer et al, 2015a; Yamano et al, 2015). Consequently, the Ubl becomes more mobile and is phosphorylated more easily by PINK1 at Ser65 (B‐3). The phosphorylated Ubl localizes proximal to RING0/UPD as phosphorylated Ser65 of Ubl interacts with a positively charged pocket made by Lys161, Arg163, and Lys211 in RING0/UPD (B‐4) (Gladkova et al, 2018; Sauve et al, 2018). The RING2 is then flipped out and liberated from suppression by the RING0/UPD, and the catalytic center Cys431 (which is hitherto hidden in the molecule) becomes exposed (B‐4). Simultaneously, the E2 interaction surface in the RING1 (which is usually concealed by the REP) is also uncovered (B‐4). Ubiquitin‐carrying E2 binds the RING1 (B‐5), and the RING2 receives ubiquitin via a thioester linkage from E2, and finally, ubiquitin is transferred to a substrate (B‐6). By such cascading structural remodeling, Parkin is converted from a self‐inhibited dormant enzyme to an active E3.
Autophosphorylation of PINK1 is essential for ubiquitin recognition
PINK1 Ser228 and Ser402 residues are autophosphorylated upon decreased mitochondrial membrane potential, and this autophosphorylation is essential for Parkin recruitment onto damaged mitochondria (Okatsu et al, 2012). The significance of autophosphorylation at Ser402 is still unknown, and this phosphorylation site does not exist in insect PINK1. Autophosphorylation of Ser228 has been shown in both mammalian cells and insects (Woodroof et al, 2011). In the kinase domain, PINK1 has three unique insert regions called Insert 1, Insert 2, and Insert 3 (Fig 3A). Insert 1 varies in length from 35 to only 5 amino acids in human and in insect PINK1, respectively, and Insert 2 is not well‐conserved. By contrast, Insert 3 is highly conserved from insects to humans. The structures of the kinase and C‐terminal region (CTR) domains of insect PINKs—TcPINK (small beetle Tribolium castaneum) and PhPINK1 (Pediculus humanus corporis)—have been solved (Kumar et al, 2017; Schubert et al, 2017; Okatsu et al, 2018). The kinase domain consists of an N‐lobe containing five β‐sheets and a C‐lobe containing α‐helices that are connected by a hinge region. The ATP binding site and enzymatic catalytic center localize in groove between N‐lobe and C‐lobe. These features are basically common to other kinases. As a characteristic structure of PINK1, the CTR domain consists of four α‐helices that support the C‐lobe structure from backside. The structural analysis of the PhPINK1–ubiquitin complex revealed that Insert 3 is a key motif for PINK1 to recognize ubiquitin (Schubert et al, 2017). Phosphorylated PhPINK1 Ser202 (corresponding to human HsPINK1 Ser228) interacts with Insert 3 Arg282/Asn283 to proper position Insert 3 for ubiquitin recognition and subsequent phosphorylation (Fig 3B). As Ser202 locates on the upper side of N‐lobe and far from the enzymatic active center of PhPINK1, this seems not to involve intramolecular autophosphorylation but rather autophosphorylated in trans via intermolecular phosphorylation. Indeed, dimerization of HsPINK1 on depolarized mitochondria is thought to be important for autophosphorylation (Okatsu et al, 2013; Rasool et al, 2018).
Figure 3. PINK1 and ubiquitin‐mediated mitophagy.
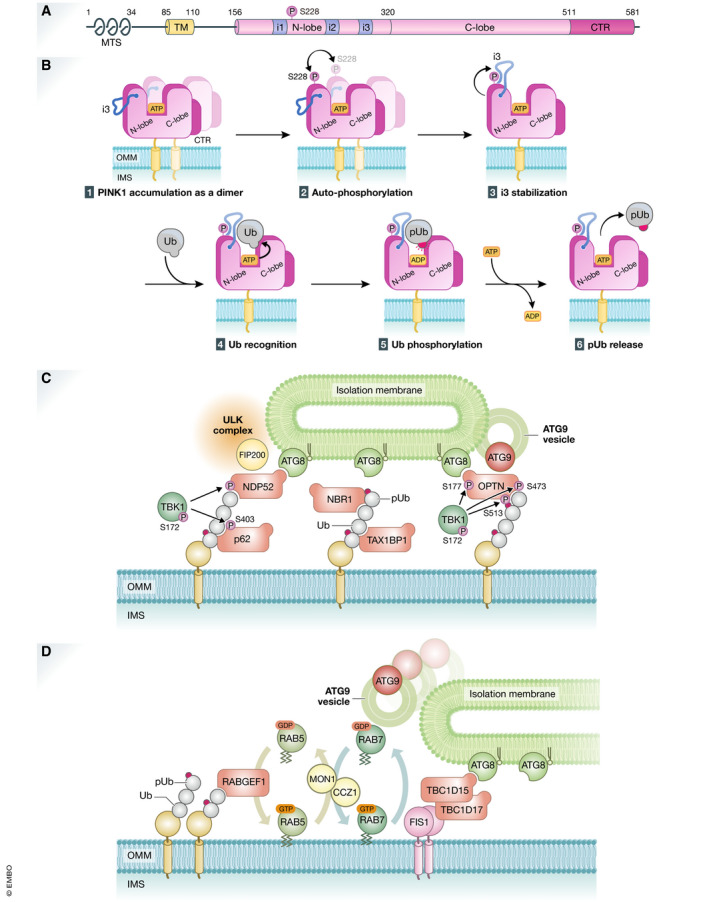
(A) Schematic depiction of the domain structures of PINK1. The protein and domain sizes are indicated as the number of amino acids. MTS, mitochondrial targeting signal; TM, transmembrane segment; N‐lobe and C‐lobe, N‐terminal and C‐terminal lobes found in a typical kinase, respectively. i1, i2, and i3, the insert regions unique to PINK1; CTR, C‐terminal region conserved among PINK1 homologs. (B) Molecular mechanisms underlying ubiquitin phosphorylation by PINK1 on depolarized mitochondria. (1) PINK1 forms a dimer on damaged mitochondria. (2) Ser228 is phosphorylated via intermolecular autophosphorylation in dimerized PINK1. (3) Ser228 phosphorylation stabilizes and underpins “insert 3 (i3)” at the correct position. (4) Ubiquitin (Ub) is recognized by PINK1 as a genuine substrate, (5) Ub Ser65 residue is phosphorylated via ATP hydrolysis, and (6) phosphorylated Ub (pUb) is released. (C) Recruitment of the core autophagy proteins and isolation membranes to mitochondria during PINK1/Parkin‐mediated mitophagy. Poly‐ubiquitin chains on damaged mitochondria are recognized directly by various autophagy adaptors. They are phosphorylated by TBK1 kinase, and the phosphorylation enhances the binding affinity to ubiquitin chains and ATG8 family proteins. NDP52 and OPTN specifically recruit ULK complex via FIP200 and ATG9 vesicles, respectively. (D) RABGEF1 recruited to mitochondria by poly‐ubiquitin chains triggers endosomal Rab cycles including RAB5 and the MON1/CCZ1 complex. MON1/CCZ1 directs RAB7 to mitochondria, and RAB7 facilitates the assembly of ATG9 vesicles to the autophagosome formation site. Mitochondrial Rab‐GAPs, TBC1D15 and TBC1D17, assist to complete RAB7 cycles and interact with ATG8 family proteins to recruit the isolation membrane.
Parkin’s substrates and Ubiquitin chain amplification
PINK1‐mediated phosphorylation leads to Parkin activation and ubiquitination of substrates on damaged mitochondria that function as autophagy‐mediated degradation signals (Pickrell & Youle, 2015; Khaminets et al, 2016; Yamano et al, 2016). Upon mitophagy, several OMM proteins such as mitofusin, Miro, and VDAC have been identified as Parkin substrates (Gegg et al, 2010; Poole et al, 2010; Tanaka et al, 2010; Ziviani et al, 2010; Geisler et al, 2010a; Rakovic et al, 2011; Wang et al, 2011). Other OMM proteins that undergo Parkin‐mediated ubiquitylation have later been identified by mass spectrometry (Chan et al, 2011; Sarraf et al, 2013), suggesting that Parkin can ubiquitylate a large number of proteins on the surface of mitochondria. Although general E3 ligases have stringent substrate selectivity that prevents cross‐reaction among other E3s to ensure correct substrate ubiquitylation, Parkin seems to have rather low substrate selectivity. Instead, Parkin has evolved to have spatial selectivity for depolarized mitochondria rather than substrate selectivity. Artificial mitochondria‐targeted exogenous proteins such as GFP and MBP can be ubiquitylated by Parkin (Koyano et al, 2019). Such a unique specificity seems optimal for Parkin to achieve efficient and quick ubiquitylation of dysfunctional mitochondria. Even under steady‐state conditions, a small amount of ubiquitin is attached to proteins on the surface of mitochondria. When PINK1 phosphorylates such ubiquitin, the resultant phospho‐ubiquitin recruits Parkin from the cytosol and activates it on depolarized mitochondria to generate more ubiquitin chains. This Parkin‐catalyzed ubiquitylation then further drives PINK1‐catalyzed ubiquitin phosphorylation, leading to formation of a positive feedback loop for PINK1‐ and Parkin‐catalyzed ubiquitylation (Ordureau et al, 2014; Okatsu et al, 2015). Low substrate specificity of Parkin might facilitate this positive feedback cycle as only a small amount of PINK1 on the OMM is needed to recruit quite a few amount of Parkin to dysfunctional mitochondria (Matsuda, 2016; Matsuda & Yamano, 2020).
Autophagosome formation in PINK1/Parkin‐mediated mitophagy
In order to detach damaged mitochondria from a healthy network and to eliminate them, proper and selective encapsulation of damaged mitochondria by autophagosomes is required. In addition, autophagosomes containing damaged mitochondria must rapidly fuse with lysosomes to facilitate their degradation. To complete these processes, many molecules involved in autophagosome/autolysosome formation work cooperatively with PINK1 and Parkin. In starvation‐induced autophagy, formation of phagophore begins at a particular region of the ER (Hayashi‐Nishino et al, 2009), or at the contact sites between the ER and mitochondria (Hamasaki et al, 2013). Several autophagy‐related proteins are recruited to the autophagosome formation site in a hierarchical order (Itakura & Mizushima, 2010).
Autophagy adaptors in PINK1/Parkin‐mediated mitophagy
In selective autophagy‐related processes including mitophagy, a series of autophagy adaptors (p62/SQSTM1, NBR1, NDP52/CALCOCO2, TAX1BP1, and OPTN) play important roles in selective uptake of cargoes (Johansen & Lamark, 2011; Mizushima & Komatsu, 2011; Stolz et al, 2014; Zaffagnini & Martens, 2016). These autophagy adaptors contain both a ubiquitin‐binding domain that recognizes ubiquitin chains conjugated to the cargoes and an LC3‐interacting region that acts to recruit phagophore membranes coated with LC3. During mitophagy, all known autophagy adaptors are recruited to damaged mitochondria in a Parkin/PINK1‐dependent manner (Lazarou et al, 2015). Compared to the autophagic events under starvation, different cascading reactions occur during PINK1/Parkin‐mediated mitophagy. Upon mitochondrial membrane potential dissipation, the ULK1 complex and ATG9 vesicles are recruited near damaged mitochondria even in the absence of membrane‐bound LC3 (Itakura et al, 2012). Loss of autophagy adaptors impairs not only recruitment of the LC3‐labeled membrane to damaged mitochondria, but also recruitment of upstream autophagy‐related proteins such as ULK1 and WIPI1 during PINK1/Parkin‐mediated mitophagy. Among five autophagy adaptors, only NDP52 and OPTN can grow isolation membrane through an ATG8‐dependent positive feedback loop (Padman et al, 2019). In addition, NDP52 directly binds to FIP200/RB1CC1, a ULK1 complex subunit (Vargas et al, 2019), and OPTN can form a complex with ATG9 vesicles (Yamano et al, 2020). Therefore, NDP52 and OPTN bind to multiple core autophagy proteins. As compared to the hierarchy of autophagy under starvation conditions (mTORC1→ULK1→LC3), Parkin‐mediated mitophagy uses the following cascading reaction: ubiquitylation→NDP52→ULK1/LC3, and ubiquitylation→OPTN→ATG9/LC3.
TBK1 kinase in PINK1/Parkin‐mediated mitophagy
During PINK1/Parkin‐mediated mitophagy, TBK1 directly or indirectly mediates phosphorylation of all known autophagy receptors (Richter et al, 2016). TBK1 activity is required for efficient recruitment of OPTN and NDP52 to the ubiquitinated mitochondria (Heo et al, 2015) where TBK1 phosphorylates OPTN at Ser177 to increase LC3 binding affinity (Wild et al, 2011) and at Ser473 and Ser513 to further increase the binding of OPTN to ubiquitin chains (Heo et al, 2015) (Fig 3C). Thus, in addition to Parkin–PINK1–ubiquitin‐positive feedback loop, another feedback loop (ubiquitin–OPTN–TBK1) constitutes more landing sites for autophagy adaptors on damaged mitochondria. In addition, TBK1 during mitophagy blocks mitosis due to the sequestration of TBK1 from its physiological role at centrosomes (Sarraf et al, 2019).
Elongation of phagophore membranes during mitophagy
Unlike starvation‐induced autophagy by which cytoplasmic components are randomly encapsulated, mitophagy requires elongation of the phagophore membrane specifically surrounding damaged mitochondria. The LIR‐containing proteins TBC1D15 and TBC1D17 are important for expansion of the phagophore membrane during mitophagy (Yamano et al, 2014). TBC1D15 and TBC1D17 function as GTPase‐activating proteins (GAPs) for Rab‐type GTPases regulating membrane fusion processes in vesicular trafficking (Barr & Lambright, 2010). TBC1D15 and TBC1D17 target the OMM via their receptor Fis1 (Onoue et al, 2013). Abnormal LC3‐labeled tubular phagophore structures are formed upon loss of TBC1D15 or Fis1 during mitophagy, but not during starvation‐induced autophagy, in mammalian cultured cells (Yamano et al, 2014). In addition, loss of Fis1 in Caenorhabditis elegans causes a PINK1‐dependent accumulation of LC3 aggregates (Shen et al, 2014). Both Fis1 and TBC1D15 are required for efficient OXPHOS‐induced mitophagy and for elimination of paternal mitochondria in fertilized eggs (Rojansky et al, 2016). Fis1‐TBC1D15/17‐Rab may be additionally required for proper formation of autophagosomes during mitophagy. RABGEF1, an upstream factor of the endosomal Rab GTPase cascade, is recruited to damaged mitochondria via ubiquitin binding downstream of Parkin. RABGEF1 directs the Rab proteins RAB5 and RAB7 to damaged mitochondria. Furthermore, depletion of RAB7 or loss of TBK1‐mediated RAB7 phosphorylation inhibits ATG9 vesicle assembly and subsequent encapsulation of mitochondria by autophagic membranes (Yamano et al, 2014; Heo et al, 2018). These results suggest that the endosomal Rab cycle on damaged mitochondria acts as a crucial regulator of mitophagy via assembling ATG9 vesicles (Fig 3D). Furthermore, other Rab‐GAPs such as TBC1D5 target ATG9A vesicles around damaged mitochondria by regulating Rab7 activity during mitophagy (Jimenez‐Orgaz et al, 2018).
Autophagosome closure and autophagosome–lysosome fusion
The final step to eliminate damaged mitochondria requires fusion of autophagosomes with lysosomes. Although it has been thought that Atg8 family proteins and their conjugation systems are required for autophagosome formation, autophagosome‐like structures are formed in the absence of lipidated Atg8 family proteins (Tsuboyama et al, 2016). In mammals, Atg8 family consists of six different proteins divided into the LC3 (LC3A, LC3B, and LC3C) and GABARAP (GABARAP, GABARAP‐L1, and GABARAP‐L2) subfamilies. All six proteins are covalently linked to the PE via two ubiquitin‐like conjugation systems. PE‐conjugated Atg8 family proteins associate with both elongating isolation membranes and mature autophagosomes, and LC3B is widely used as an autophagic membrane marker (Kabeya et al, 2000; Kabeya et al, 2004). Atg8 family proteins are not essential for encapsulation of damaged mitochondria by autophagosomes, but required for autophagosome–lysosome fusion (Nguyen et al, 2016) or efficient degradation of the inner autophagosomal membrane in lysosomes (Tsuboyama et al, 2016). Although damaged mitochondria are properly sequestered by autophagosomal membranes in cells lacking all Atg8 family proteins, the size of autophagosomes is much smaller than that in wild‐type cells (Nakatogawa et al, 2007; Weidberg et al, 2010).
Unlike starvation‐induced autophagy, PINK1/Parkin‐mediated mitophagy may need PLEKHM1 rather than STX17, an autophagosome‐specific SNARE, for autophagosome–lysosome fusion (McEwan et al, 2015). PLEKHM1 contains multiple functional domains that directly bind Rab7, the HOPS complex, and Atg8 family proteins, and is required for selective and nonselective autophagy (McEwan et al, 2015). GABARAP subfamily proteins localize on mature autophagosome and associate with PLEKHM1 at the lysosome to facilitate autophagosome–lysosome fusion during PINK1/Parkin‐mediated mitophagy (Nguyen et al, 2016).
Deubiquitylating enzymes in PINK1/Parkin‐mediated mitophagy
Ubiquitylation is a reversible process as deubiquitylating enzymes can remove ubiquitin from ubiquitylated substrates. USP8, USP15, and USP30 regulate PINK1/Parkin‐mediated mitophagy positively and negatively (Bingol et al, 2014; Cornelissen et al, 2014; Durcan et al, 2014; Cunningham et al, 2015; Liang et al, 2015). USP15 and USP30 deubiquitylate mitochondrial substrates to counteract Parkin‐mediated ubiquitylation and subsequent mitophagy (Bingol et al, 2014; Cornelissen et al, 2014; Cunningham et al, 2015; Liang et al, 2015). In contrast, USP8 detaches ubiquitin from autoubiquitylated Parkin, acting as a positive regulator that promotes Parkin mitochondrial targeting and accelerates mitophagy (Durcan et al, 2014). Although USP8 can digest ubiquitin chains of any linkage in vitro (Faesen et al, 2011), it selectively removes K6‐linked ubiquitin chains from Parkin in mammalian cultured cells (Durcan et al, 2014). USP30 is thought to specifically digest K6‐linked ubiquitin chains through unique ubiquitin recognition mechanisms (Gersch et al, 2017; Sato et al, 2017). It remains unclear how K6‐linked ubiquitin chains of Parkin and OMM proteins are removed selectively by USP8 and USP30, respectively. Although USP15 has been suggested to trim K48‐ and K63‐linked ubiquitin chains on depolarized mitochondria (Cornelissen et al, 2014), the effect of USP15 on K6‐linked ubiquitin chains has not been examined. Moreover, PINK1‐mediated ubiquitin phosphorylation impedes the enzyme activities of USP8, USP15, and USP30 (Wauer et al, 2015b), adding a new layer of complexity to the deubiquitylation reactions. Although early studies suggested USP30 counteracts Parkin‐mediated ubiquitylation as described, recent two papers showed that ubiquitylation of the vast majority of Parkin targets is rather unaffected in USP30 knockout cells (Ordureau et al, 2020; Phu et al, 2020). Instead, elevated ubiquitylation is observed in components of the mitochondrial translocator and intramitochondrial substrates in USP30 knockout cells. It is possible that USP30 removes ubiquitin from import substrates and components of the mitochondrial translocator, and these processes are required for efficient translocation through the import channels. Future studies on the actions of PINK1, Parkin, and USP8/15/30 will shed light on the functions of deubiquitylating enzymes and the significance of K6‐linked ubiquitylation in PINK1/Parkin‐mediated mitophagy.
Parkin alternatives in mitophagy
Many papers reported that PINK1/Parkin‐catalyzed ubiquitylation induces mitophagy of damaged mitochondria. However, most of these data were obtained from experiments using cultured cells (e.g., Parkin‐expressing HeLa cells), and there is much less evidence for PINK1/Parkin‐mediated mitophagy in vivo. In the case of genetic studies using Drosophila, it is controversial whether PINK1/Parkin‐catalyzed ubiquitylation induces mitophagy or not. One study reported that age‐dependent rise in mitophagy activity is abrogated in PINK1‐ or Parkin‐deficient flies (Cornelissen et al, 2018), whereas another work showed that any substantial impact on basal mitophagy was not observed in pink1 or parkin‐null flies (Lee et al, 2018). Transgenic mice to monitor mitophagy have already been established, and loss of PINK1 did not influence basal mitophagy activities in such mice (McWilliams et al, 2018). This finding seemingly suggests that PINK1 and Parkin are not involved in mitophagy in vivo; however, the results can be interpreted in several ways. Unlike human, whose dysfunction of PINK1 or Parkin causes early‐onset Parkinsonism, disease‐relevant phenotypes have not been observed in pink1 or parkin knockout mice. It might not be surprising even if mitophagy activity is normal in pink1 KO mice without an obvious phenotype. To reconcile these conflicting findings, we have to consider functional redundancy of other mitochondrial E3 ligases. Indeed, ARIH1/HHARI (Villa et al, 2017), March5 (Chen et al, 2017), MAPL/MULAN/GIDE/MUL1 (Ambivero et al, 2014; Yun et al, 2014; Li et al, 2015; Igarashi et al, 2020), p62‐keap1‐Rbx1 axis (Yamada et al, 2018), and HUWE1 (Di Rita et al, 2018) have been reported to mediate Parkin‐independent mitophagy. These E3s could compensate for PINK1/Parkin‐mediated mitophagy and conceal the output when the PINK1/Parkin function is inhibited.
Mitophagy in worms and flies
PINK‐1/PDR‐1‐mediated mitophagy in somatic cells
In the nematode Caenorhabditis elegans, stress‐induced mitophagy is regulated by PINK‐1 and PDR‐1 (a worm Parkin homolog), supporting that the PINK1/Parkin‐dependent pathway has been conserved during evolution (Palikaras et al, 2015) (Fig 4A). The NIX and BNIP3 homolog DCT‐1 functions as an autophagy receptor for PINK‐1/PDR‐1‐mediated mitophagy (Palikaras et al, 2015). DCT‐1 is ubiquitylated on its Lys26 residue, and this modification is enhanced under mitophagy‐inducing conditions in a PINK‐1‐dependent manner (Palikaras et al, 2015). In addition to DCT‐1, the Bcl‐2 homolog CED‐9 interacts with DCT‐1 and may act in the same genetic pathway to control mitophagy (Palikaras et al, 2015).
Figure 4. Regulation of mitophagy in C. elegans .
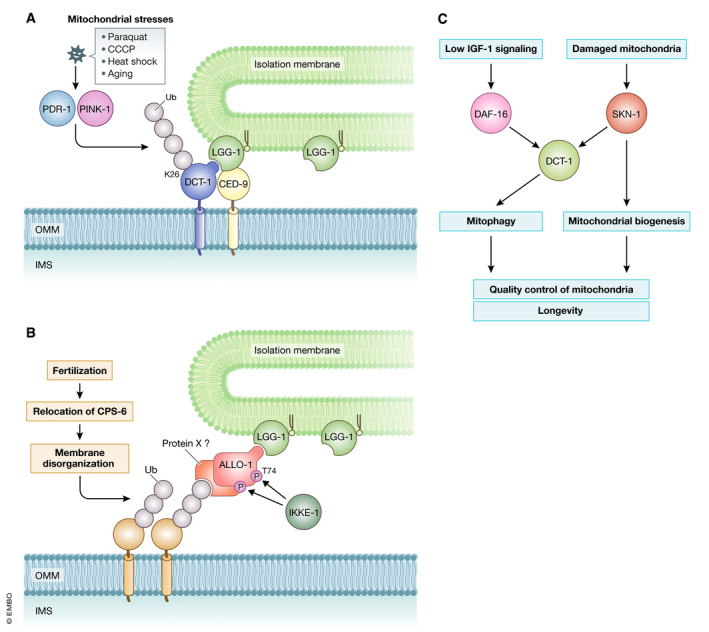
(A) PINK‐1/PDR‐1‐mediated mitophagy in somatic cells. DCT‐1 functions as an autophagy adaptor in association with CED‐9. DCT‐1 Lys26 (K26) is ubiquitinated in a PINK‐1/PDR‐1‐dependent manner. (B) Mechanism of allophagy in embryos. Fertilization triggers relocation of CPS‐6, the mitochondrial endonuclease, leading to membrane disorganization and ubiquitylation of paternal mitochondria. Ubiquitin (Ub) molecules on paternal mitochondria are recognized directly or indirectly by the autophagy adaptor ALLO‐1. IKKE‐1‐dependent phosphorylation of ALLO‐1 is also important for allophagy. (C) Transcriptional regulation of DCT‐1 contributes to mitochondrial homeostasis and longevity.
Clearance of paternal mitochondria
Besides mitophagy in somatic cells, paternal mitochondria provided by sperm are selectively degraded via autophagy in C. elegans fertilized embryos (Al Rawi et al, 2011; Sato & Sato, 2011). This type of mitophagy is a developmentally programmed process and does not require any artificial stimuli to be induced and is referred to as allogeneic (non‐self) organelle autophagy (allophagy) (Al Rawi et al, 2012; Sato & Sato, 2012) since the paternal organelle, so‐called membranous organelles (MOs), are also degraded in this process (Al Rawi et al, 2011; Sato & Sato, 2011). mtDNA is maternally inherited in many organisms including humans (Ankel‐Simons & Cummins, 1996; Sato & Sato, 2013; Sato & Sato, 2017). In worm mutants of core Atg genes, paternal mitochondria and their mtDNA persist in late‐stage embryos or even in F1 larvae, suggesting that allophagy is required to prevent transmission of paternal mtDNA to the progeny (Al Rawi et al, 2011; Sato & Sato, 2011). Autophagy‐dependent degradation of paternal mitochondria also occurs in Drosophila and mouse embryos (Politi et al, 2014; Rojansky et al, 2016).
Caenorhabditis elegans has two Atg8 family members, LGG‐1 and LGG‐2 that are both recruited to allophagosomes (autophagosomes containing paternal mitochondria and/or MOs). In lgg‐2 mutant embryos, LGG‐1‐positive allophagosomes are formed, but their turnover is delayed (Manil‐Segalen et al, 2014; Djeddi et al, 2015). LGG‐2 directly binds to VPS‐39, a subunit of the HOPS complex, and enhances fusion of autophagosomes with lysosomes (Manil‐Segalen et al, 2014). LGG‐2 is also required for microtubule‐dependent migration of autophagosomes toward the pericentrosomal region where lysosomes are concentrated (Djeddi et al, 2015). In addition to autophagy‐related genes, degradation of paternal mtDNA is delayed by knockdown of proteasome subunit genes, suggesting that the ubiquitin–proteasome system is involved in this process (Zhou et al, 2011).
Electron tomography has revealed that the inner membrane structure of paternal mitochondria starts to be disorganized quickly after fertilization (Zhou et al, 2016). The OMM rapture and reduced membrane potential were also observed (Zhou et al, 2016). These changes in paternal mitochondrial structure are initiated before autophagosome formation and lysosomal degradation. Such qualitative alteration of paternal mitochondria could be a trigger to promote their selective autophagic clearance. CPS‐6, a mitochondrial endonuclease G, is also linked to clearance of paternal mitochondria (Zhou et al, 2016). CPS‐6 was originally identified as an apoptotic factor that redistributes from mitochondria to the nucleus and mediates chromosome fragmentation during apoptosis (Parrish et al, 2001). When paternal cps‐6 is mutated, clearance of paternal mitochondria is delayed (Zhou et al, 2016). Since CPS‐6 in paternal mitochondria relocates from the mitochondrial intermembrane space to the matrix after fertilization, mtDNA digestion by matrix‐localized CPS‐6 might initiate degeneration of mitochondrial membranes (Zhou et al, 2016). The IMM protein prohibitin 2 also plays a role in the selective engulfment of paternal mitochondria by autophagosomes and functions as an autophagy receptor for damaged mitochondria in mammals and paternal mitochondria in C. elegans (Wei et al, 2017).
More recently, a novel LIR‐containing protein named ALLO‐1 has been identified as an autophagy adaptor for degradation of paternal mitochondria and MOs (Sato et al, 2018) (Fig 4B). In allo‐1 mutant embryos, allophagosomes are not formed, and paternal organelles and paternal mtDNA remain in the late embryos or larvae (Sato et al, 2018). ALLO‐1 is conserved only in nematode species; however, its function is very similar to that of known autophagy adaptors. Several lines of evidence suggest that ubiquitylation of targets is involved in ALLO‐1 localization (Al Rawi et al, 2011; Sato & Sato, 2011). It is also reported that simultaneous knockdown of ubc‐16 and ubc‐18 impairs allophagy (Molina et al, 2019). Since mutations in the pink‐1 or pdr‐1 gene do not significantly affect allophagy, it remains unknown how this ubiquitylation is regulated (Sato et al, 2018). In addition to ALLO‐1, the worm homolog of mammalian TBK1/IKKε kinases IKKE‐1 is essential for allophagy (Sato et al, 2018) (Fig 4B). IKKE‐1 phosphorylates ALLO‐1 on Thr74 although additional phosphorylation targets are likely to exist (Sato et al, 2018). This is reminiscent of TBK1 function in mitophagy and xenophagy, and phosphorylation of adaptor molecules could be a conserved mechanism regulating selective autophagy pathways (Fig 4B).
In Drosophila, paternal mitochondria form a very long shape parallel to the axoneme that is degraded by multiple‐step mechanisms (Politi et al, 2014). After fertilization, paternal mitochondria are dissociated from the axoneme and fragmented into small mitochondria, which are then engulfed by autophagosomes. Their degradation partly depends on p62, and accumulation of K63‐linked ubiquitin chains on paternal mitochondria has been observed. Although the precise mechanism remains unclear, this might also involve autophagy regulators during early dissociation or fragmentation steps (Politi et al, 2014).
In mouse embryos, ubiquitin and autophagy regulators such as LC3, GABARAP, and p62 are detected on paternal mitochondria (Sutovsky et al, 1999; Al Rawi et al, 2011). Knockdown of p62 or PINK1 in embryos impairs degradation of paternal mitochondria, supporting autophagy‐dependent degradation of ubiquitylated paternal mitochondria (Rojansky et al, 2016). Degradation of paternal mitochondria is also impaired by simultaneous knockdown of Parkin and MUL1, a mitochondria‐localized E3 ubiquitin ligase, suggesting that these E3 ligases may function redundantly (Rojansky et al, 2016). The fly and worm Parkin mutants exhibit slight or minor defects in degradation of paternal mitochondria (Politi et al, 2014; Sato et al, 2018), but possible redundancies with other E3 ligases function cannot be excluded. Similar to C. elegans, loss of inner membrane potential has been observed in paternal mitochondria in mouse embryos (Rojansky et al, 2016). Notably, Fis1 and TBC1D15 act in degradation of paternal mitochondria (Rojansky et al, 2016). These observations suggest a significant overlap between paternal mitochondria degradation and mitophagy of damaged mitochondria in somatic cells. However, in contrast to these studies, Luo et al argued that paternal mitochondria are not actively removed and persist in embryos at least until the morula stage (Luo et al, 2013). Further studies are needed to resolve when and how paternal mitochondria are removed.
Physiology and pathophysiology of mitophagy
A growing body of research has explored the pathophysiological functions of mitophagy mainly by using mammalian cells or mice lacking key mitophagy‐related factors. These studies also provide a framework for physiological functions of mitophagy and unveil previously unappreciated links to diverse biological processes (Fig 5).
Figure 5. Physiological functions of mitophagy against mitochondrial stresses.
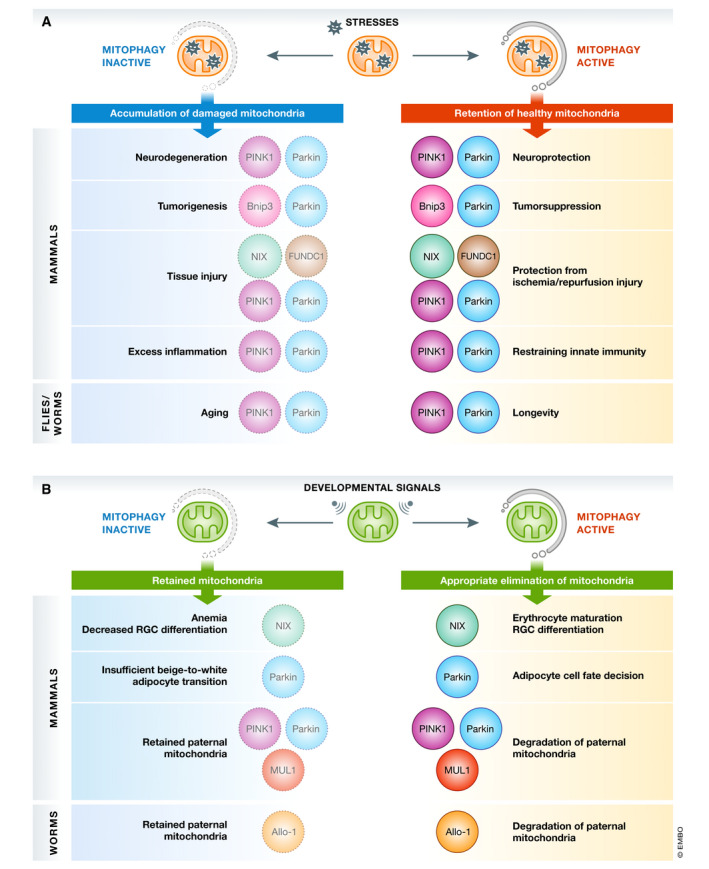
(A) Mitochondria are constantly challenged by a subset of mitochondrial stresses such as oxidative stress. Mitophagy contributes to mitochondrial quality control and prevention of pathologies including neurodegeneration, tumorigenesis, tissue injury, excess inflammation, and aging. (B) Physiological functions of mitophagy during development and differentiation. Mitochondrial elimination in response to developmental cues is crucial for maturation of cells and tissues.
Physiological functions of mitophagy in yeast
Since Atg32 is an essential protein for mitophagy in yeast, atg32‐null yeast cells have been used to explore the physiological significance of mitophagy. Under longevity‐extending conditions, loss of Atg32 causes accumulation of dysfunctional mitochondria and impaired mitochondrial network, leading to a shortened lifespan (Richard et al, 2013). Mitophagy also contributes to the maintenance of mtDNA (Kurihara et al, 2011; Karavaeva et al, 2017). In heteroplasmic zygotes containing wild‐type and mutant mtDNA molecules, mitophagy is activated and further accelerated by the treatment of mitochondrial uncouplers, suggesting that enhanced mitophagy in zygotes could prevent clonal expansion of mutant mtDNA (Karavaeva et al, 2017). Furthermore, during prolonged nitrogen starvation, cells lacking Atg32 exhibit mitochondrial ROS accumulation and mtDNA instability, indicating that mitophagy contributes to mitochondrial fitness under stress conditions (Kurihara et al, 2011).
Mitophagy in development, differentiation, and tissue protection
One example that illustrates the physiological function of mitophagy in mammals is NIX‐mediated mitochondrial elimination during erythrocyte maturation (Schweers et al, 2007; Sandoval et al, 2008). Intracellular organelles including mitochondria are removed when reticulocytes differentiate into mature erythrocytes. Electron microscopic analysis revealed that during early stage of erythrocyte differentiation, autophagic bodies accumulated in human peripheral blood cells, rat erythroblasts, and reticulocytes (Takano‐Ohmuro et al, 2000). Mice lacking the autophagy gene Atg7 in the hematopoietic system suffer from severe anemia, and Atg7‐deficient erythrocytes accumulate damaged or dysfunctional mitochondria with altered membrane potential (Mortensen et al, 2010). Consistent with these observations, loss of NIX in mice causes defects in mitochondrial clearance and anemia (Schweers et al, 2007; Sandoval et al, 2008). NIX is also involved in mouse retinal ganglion cell (RGC) differentiation (Esteban‐Martinez et al, 2017). During RGC differentiation, a shift from oxidative phosphorylation to glycolysis is needed in order to meet the metabolic demands of RGCs (Galvan‐Pena & O'Neill, 2014; Ng et al, 2015; Chandel et al, 2016). Retinas from NIX‐deficient mice show increased mitochondrial mass, reduced expression of glycolytic enzymes, and inefficient neuronal differentiation (Esteban‐Martinez et al, 2017).
Mitophagy has also been linked to the maturation of muscle tissue. During myogenesis and muscle regeneration, mitochondrial activity is drastically increased (Duguez et al, 2002; Sin et al, 2016), likely due to a shift in metabolism from glycolysis to oxidative phosphorylation which eventually increases mitochondrial oxidative stress. Suppression of the essential autophagy gene Atg5 leads to accumulation of abnormal mitochondria and inefficient differentiation into mature muscle tissue (Sin et al, 2016).
Parkin‐dependent degradation of mitochondria has also been linked to cell fate decision of adipocytes. Mice lacking Parkin retain mitochondrial abundance in beige adipocytes and show defects in beige‐to‐white adipocyte transition (Lu et al, 2018). While white adipocytes containing a small quantity of mitochondria serve as fat tissues to store energy, beige adipocytes contain a large quantity of mitochondria and act in thermogenesis by uncoupling mitochondrial proton gradient in response to various cues such as chronic cold exposure and exercise (Harms & Seale, 2013; Kajimura et al, 2015). After withdrawal of such stimuli, beige adipocytes acquire white adipocyte‐like characteristics in a manner dependent on autophagic mitochondrial turnover (Altshuler‐Keylin et al, 2016).
In addition to its roles in development and differentiation, mitophagy is also involved in tissue protection against several types of injuries. Studies in mice demonstrated autophagy‐ and mitophagy‐dependent protection against ischemia/reperfusion (I/R) injury in several tissues. While reperfusion to restore blood flow after ischemia is necessary to salvage the injured tissues, this can paradoxically lead to an excess ROS production from mitochondria (Pulsinelli & Duffy, 1983; Aronowski et al, 1997). Genetic suppression of core autophagy‐related genes aggravates neuronal injury and cellular death after I/R injury, mainly by increasing cytochrome c release from mitochondria (Zhang et al, 2013). NIX expression in Nix‐deficient neurons restored cell viability after I/R‐induced injury and knockout of NIX in mice exacerbated I/R brain injury as indicated by increased cerebral infarct volume (Yuan et al, 2017). FUNDC1 seems to function in cardioprotection through modulating the platelet activity (Zhang et al, 2016b). I/R‐induced platelet activation and release of platelet‐derived mediators aggravate tissue injury in the heart (Gawaz, 2004). Hypoxia induces FUNDC1‐mediated mitophagy in platelets, thereby promoting turnover of mitochondria and suppressing platelet activation (Zhang et al, 2016b). Parkin plays a protective role in heart against myocardial infarction, as Parkin‐deficient mice exhibit accumulation of dysfunctional mitochondria, a broader zone of the infarction, and reduced survival rates (Kubli et al, 2013). PINK1 KO mice also exhibit dysregulated mitochondrial functions and excess cardiomyocyte cell death (Billia et al, 2011).
In addition, mitophagy contributes to kidney homeostasis and protection against acute kidney injury (AKI). PINK1‐, Parkin‐, and double‐KO mice show increased mitochondrial damage, ROS generation, inflammatory response, and serum creatinine (an index of renal dysfunction), raising the possibility that PINK1/Parkin‐mediated mitophagy prevents cell death and maintains renal function against I/R‐induced AKI (Tang et al, 2018). The autophagy receptor optineurin has also been suggested to act in renal tissue protection (Chen et al, 2018). Deletion of optineurin drastically decreases mitophagosome formation during high glucose treatment and exacerbates RTEC (renal tubular epithelial cells) senescence (Chen et al, 2018), one of the factors contributing to renal injury in diabetic kidney.
Alcoholic liver disease (ALD) is caused by excess alcohol intake (Rehm et al, 2013). Several studies suggest that autophagy‐related processes, especially mitophagy, act in protection against alcohol‐induced liver injury (Ding et al, 2010a; Ding et al, 2011; Williams & Ding, 2015). Since ALD pathology is associated with ROS accumulation and mtDNA damage, it is conceivable that mitophagy is induced in the liver tissue of ALD patients. Consistent with this idea, Parkin KO mice display increased liver injury, oxidative stress, and steatosis after alcohol treatment, highlighting a protective role of mitophagy against tissue injury in the liver (Williams et al, 2015).
Nonalcoholic fatty liver disease (NAFLD) is the most common cause of liver disease and has also been linked with mitochondrial dynamics and Parkin‐independent mitophagy (Loomba & Sanyal, 2013; Masuoka & Chalasani, 2013; Yamada et al, 2018). In the NAFLD mice model, megamitochondria, which are extremely enlarged mitochondria, are observed (Wakabayashi, 2002; Neuman et al, 2014; Targher et al, 2018; Younossi et al, 2018). Deletion of Opa1 (an IMM protein required for mitochondrial fusion) in NAFLD model mice decreases mitochondrial size and ameliorates liver tissue damage, suggesting that restoring mitochondrial size can be a potent therapeutic treatment (Yamada et al, 2018). In hepatocytes, the autophagy adaptor p62 recruits Keap1, a component of the cullin–RING ubiquitin ligase complex containing the E3 enzyme Rbx1, to mitochondria and promotes ubiquitylation of OMM proteins and mitophagy in a manner independent of Parkin (Yamada et al, 2018).
Mitophagy and cancer
Since accumulation of dysfunctional mitochondria is involved in tumorigenesis, it is conceivable that mitophagy seems to be important as a tumor‐suppressive system (Gogvadze et al, 2008; Vara‐Perez et al, 2019). Expression of the mitophagy receptor BNIP3 declines in several types of cancer and is associated with cancer metastasis and chemoresistance (Erkan et al, 2005; Manka et al, 2005; Koop et al, 2009). Mice lacking Bnip3 display more rapid tumor growth than wild‐type mice, which can be caused by excess accumulation of dysfunctional mitochondria and elevated ROS production (Chourasia et al, 2015).
Expression of Parkin is also lost in many types of cancer (Bernardini et al, 2017). Overexpression of Parkin in breast and glioma cells retarded cellular proliferation (Tay et al, 2010). However, as Parkin is involved in proteasomal degradation of cyclins, which is fundamentally important for cell cycle control and tumor growth suppression (Staropoli et al, 2003; Veeriah et al, 2010; Gong et al, 2014), Parkin might act via multiple mechanisms to suppress tumor growth. Similar to Parkin, PINK1 overexpression is suggested to attenuate in vivo glioblastoma growth (Agnihotri et al, 2016).
Although mitophagy factors described above are shown to be dysregulated in cancer patients, it should be noted that whether they act as a tumor suppressor or promoter depends on cellular subtypes and cancer stages (e.g., BNIP3 is also suggested to support melanoma migration (Maes et al, 2014)). Further studies are needed to clarify the precise roles of mitophagy in tumorigenesis.
Mitophagy and neurodegeneration
Mitophagy may contribute to the prevention of neurodegeneration. As discussed above, PD is a major neurodegenerative disease characterized by loss of dopaminergic neurons in the substantia nigra (Lotharius & Brundin, 2002). Mutations in the Pink1 gene have been associated with PD pathogenesis (Valente et al, 2001; Valente et al, 2002; Geisler et al, 2010b). Dopaminergic neurons expressing Pink1 mutants show enlarged mitochondria and undergo cell death (Park et al, 2006). Overexpression of Parkin can eliminate mitochondria containing mtDNA mutations in heteroplasmic cybrid cells, ultimately leading to an increase in mitochondria containing wild‐type mtDNA (Suen et al, 2010). Loss of Parkin, which by itself does not cause obvious PD pathogenesis in mice (Palacino et al, 2004; Stichel et al, 2007), synergistically promotes dopaminergic neuron degeneration in mouse models that also contain mtDNA mutations. These mice accumulate dysfunctional mitochondria, supporting the idea that Parkin acts in mitochondrial quality control and neuroprotection (Pickrell et al, 2015; Song et al, 2017).
Mitophagy may also play a protective role against Alzheimer’s disease (AD), a progressive neuronal disorder characterized by a severe loss of memories and cognitive functions (Querfurth & LaFerla, 2010). Accumulation of insoluble β‐amyloid plaques and formation of neurofibrillary tangles (aggregates of hyperphosphorylated tau proteins) in brain are the major pathological hallmarks of AD (Small et al, 2006; Querfurth & LaFerla, 2010). PINK1 protein levels are decreased, and the number of mitochondria is increased in AD model mouse hippocampal neurons (Manczak et al, 2018). Overexpression of Parkin in AD model mice decreases β‐amyloid plaques and amyloid‐induced inflammation in hippocampus and cortex, contributing to amelioration of behavioral abnormalities (Hong et al, 2014). Pharmacological or genetic stimulation of mitophagy mitigates β‐amyloid plaque formation and tau hyperphosphorylation, and reverses memory impairment (Sorrentino et al, 2017; Fang et al, 2019).
Similarly, mitophagy may abate progression of amyotrophic lateral sclerosis (ALS), a disease characterized by degeneration of motor neurons, which leads to muscle weakness and paralysis (Evans & Holzbaur, 2018). Mutations in several autophagy‐related proteins including optineurin, which acts downstream of PINK1/Parkin‐mediated mitophagy, have been linked to ALS (Maruyama et al, 2010; Cirulli et al, 2015). Loss of optineurin decreases LC3 recruitment to damaged mitochondria and impairs subsequent autophagosome formation. One of the ALS‐associated mutations, E478G, is located in the UBAN domain for ubiquitin binding, disturbs targeting of optineurin to damaged mitochondria, and suppresses mitophagosome formation (Wong & Holzbaur, 2014). TBK1, which phosphorylates optineurin and enhances its binding to ubiquitin chains, is also mutated in ALS patients (Freischmidt et al, 2015; Oakes et al, 2017; Pozzi et al, 2017). A disease‐associated mutation in the TBK1 C‐terminal coiled‐coil domain disrupts its interaction with optineurin and possibly affects mitophagy and ALS pathogenesis (Freischmidt et al, 2015). It should be noted that optineurin and TBK1 are also crucial to eliminate cytosolic protein aggregates via autophagy and whether the phenotypes are due to impaired elimination of toxic protein aggregates, or mitophagy, or a combination of both is not currently clear.
Mitophagy and immune response
Autophagy and mitophagy have been linked to the immune response, suppressing overactivation of the NLRP3 inflammasome and subsequent immune response. Inflammasomes are multisubunit protein complexes consisting of NOD‐like receptor (NLR) that induce downstream immune signaling against microbial infection and intracellular damage (Schroder & Tschopp, 2010). As the NLRP3 inflammasome is activated by mitochondrial ROS and mtDNA (Nakahira et al, 2011), autophagy‐dependent clearance of mitochondria suppresses overactivation of NLRP3 inflammasome. Sestrin2, a conserved stress‐inducible metabolic protein, protects cells and tissues against excess activation of the NLRP3 inflammasomes (Kim et al, 2016). Macrophages isolated from Sestrin2‐deficient mice show hyperactivation of caspase‐1, leading to enhanced secretion of IL‐1β and IL‐18. Sestrin2 localizes to mitochondria upon lipopolysaccharide stimulation and promotes targeting of p62 to damaged mitochondria, thereby contributing to mitophagy during the immune response and preventing prolonged NLRP3 inflammasome activation (Kim et al, 2016).
PINK1 and Parkin KO mice have also been shown to be more sensitive to polymicrobial sepsis‐induced multiple organ failure and death (Kang et al, 2016). The enhanced sensitivity of these KO mice to lethal sepsis is alleviated by simultaneous depletion of Nlrp3 (Kang et al, 2016). PINK1/Parkin‐mediated mitophagy is also linked to the STING pathway, a major intracellular signaling pathway of the type I IFN in response to cytosolic DNA (Sliter et al, 2018). Disruption of mitochondria triggers inflammatory responses via the NLRP3 inflammasome and also via the cGAS‐STING pathway (Rongvaux et al, 2014). After exhaustive exercise, PINK1‐ and Parkin‐deficient mice show increased STING activation (Sliter et al, 2018). Importantly, inflammatory responses in these mutant mice are abolished by concurrent depletion of STING, supporting the idea that PINK1 and Parkin may prevent release of mtDNA from dysfunctional mitochondria, thereby inhibiting an excess inflammatory response via the STING pathway (Sliter et al, 2018). However, loss of Sting does not suppress mitochondrial dysfunctions in Drosophila Pink1/parkin mutants, raising the possibility that the Pink1/parkin‐mediated processes are not linked to the STING pathway in flies (Lee et al, 2020).
A recent study reveals that hepatocyte‐specific FUNDC1 knockout promotes initiation of hepatocarcinogenesis (HCG), whereas FUNDC1 overexpression in hepatocytes suppresses it, suggesting that FUNDC1 acts in prevention of HCG (Li et al, 2019). Loss of FUNDC1 causes accumulation of damaged mitochondria and induces release of mtDNA into the cytosol, leading to aggravated activation of inflammasomes. Thus, dysregulated immune response in FUNDC1‐depleted hepatocytes seems to excessively promote hepatocellular proliferation.
Mitophagy and aging
Health issues associated with aging are of great concern, especially in aging societies. Autophagy has been suggested to be a convergent mechanism whose activity prevents aging and declines with age (Uddin et al, 2012; Hansen et al, 2018; Nakamura & Yoshimori, 2018). Similarly, the activity of mitophagy in specific brain region decreases during aging, as indicated by an in vivo study using transgenic mice expressing mt‐Keima (Sun et al, 2015). On the other hand, a study using Drosophila expressing mt‐Keima reveals that mitophagic activity increases in aged flight muscle, raising the possibility that age‐related changes in mitophagy vary in some species and/or tissues (Cornelissen et al, 2018). Nevertheless, accumulation of excess mtDNA mutations is one of the underlying factors in mammalian aging (Trifunovic et al, 2004; Kujoth et al, 2005), implying that mitophagy may function to eliminate mitochondria with mutated mtDNA and prevent aging.
The roles of PINK1 and Parkin in aging have extensively been studied in Drosophila (Greene et al, 2003; Pesah et al, 2004). Parkin‐null flies are viable but show significantly reduced longevity compared with wild‐type. Parkin‐null flies have degenerated muscle tissues and severe defects in locomotor functions, highlighting Parkin as a critical regulator of tissue homeostasis (Greene et al, 2003; Pesah et al, 2004). Ubiquitous overexpression of Parkin in flies leads to lifespan extension, likely via modulating intracellular proteostasis and mitochondrial dynamics (Rana et al, 2013). Notably, neuron‐specific overexpression of Parkin is sufficient to prolong lifespan (Rana et al, 2013), whereas PINK1 mutant flies display shortened lifespan and myopathology (Clark et al, 2006; Park et al, 2006; Yang et al, 2006) .
In C. elegans, disruption of mitophagy contributes to progressive accumulation of damaged mitochondria and decreased cellular functions during aging (Palikaras et al, 2015). The daf‐2 insulin/IGF‐1 receptor mutant is generally used as a model of extended lifespan. Strikingly, mitophagy in the long‐lived daf‐2 mutant is upregulated, and knockdown of mitophagy regulators shortens lifespans of the daf‐2 mutant, suggesting that mitophagy is critical for lifespan extension of the daf‐2 mutant (Palikaras et al, 2015). Simultaneous knockdown of DCT‐1 or PINK‐1 and SKN‐1, an ortholog of mammalian nuclear factor NRF2 involved in mitochondrial biogenesis, further shortens lifespan, suggesting that coordination of mitochondrial biogenesis and degradation is critical for longevity in worms (Fig 4C).
Conclusions and future perspectives
Mitophagy deficiency is emerging as a potential cause of various pathologies, and thus, interventions targeting mitophagy may possess therapeutic potential (Georgakopoulos et al, 2017). Pharmacological screens to identify chemical agents to modulate elimination of mitochondria are ongoing, and several synthetic and natural chemical compounds including Urolithin A have been shown to facilitate mitophagy (Ryu et al, 2016). Moreover, a recent study established AUTAC, an autophagy‐targeting chimera that contains S‐guanylation‐inspired degradation tag for autophagy and a warhead to provide target specificity (Ito et al, 2013; Takahashi et al, 2019). When AUTAC is targeted to mitochondria, selective clearance of mitochondria via autophagy is induced in a manner independent of PINK1/Parkin, and in turn, biogenesis of functional mitochondria is increased in cells from Down syndrome patients. Very recently, mito‐SRAI, a new mitophagy probe that can be applied to both live and fixed samples, has been developed as a tool for high‐throughput in vitro screen for mitophagy chemical inducers and in vivo histological analysis in mouse models of neurodegeneration (Katayama et al, 2020). Future attempts to identify small molecules that specifically bind and regulate mitophagic factors will aid therapeutic approaches to human disorders associated with mitochondrial dysfunction.
In yeast, Atg32‐mediated mitophagy seems to be the sole pathway that confers selectivity toward mitochondria versus other cellular constituents, acting as a quantity adaptation to the low‐energy demand in non‐dividing cells. In mammals, multiple mitophagy receptors/adaptors promote mitochondrial degradation in certain specific cell types and under particular conditions, but they may also function redundantly in reducing or completely eliminating mitochondria. Since mammalian cells contain a large number of mitochondria that are heterogeneous (e.g., membrane potential, respiratory activity, and oxidative damage), they may have needed to additionally evolve diverse ubiquitin‐mediated pathways that establish selectivity toward dysfunctional mitochondria versus healthy mitochondria, acting as a quality management system. These mitophagy‐dependent mitochondrial quantity and quality control mechanisms are not mutually exclusive, as the former can help improve mitochondrial fitness in cooperation with mitochondrial biogenesis that provides fresh mitochondria, and the latter can help decrease mitochondrial populations without wasting healthy mitochondria.
Over the last decade, numerous studies have contributed to establish the paradigm that mitophagy serves as a system to modulate mitochondrial fitness and populations in response to changes in intra‐ and extracellular environments. Studies using in vivo models have provided new insights into the physiological and pathological implications of mitophagy (see Box 2). Although loss of mitophagy is detrimental to mitochondrial homeostasis, it seems conceivable that aberrantly hyperactivated mitophagy could also be deleterious and may ultimately lead to cell death. Therefore, mitophagy must be tightly regulated by both accelerators and brakes. Several outstanding questions remain to be addressed: What are those pro‐ and anti‐mitophagic factors/mechanisms? How is basal mitochondrial turnover controlled? Are there additional mitophagy receptors that are ubiquitous or limited to specific tissues and cell types? How do cells coordinate mitochondrial biogenesis and degradation? Do other organelles promote and/or suppress mitophagy? Undoubtedly, many exciting discoveries and translational innovations are yet to come.
Box 2. Tools for monitoring mitophagy in vivo .
A. mt‐Keima
Mitochondrial degradation can be analyzed in vitro and in vivo by fluorescence microscopy using a mitochondrial matrix‐targeted Keima (mt‐Keima) whose excitation spectrum peaking depends on pH (Sun et al, 2017). Upon mitophagy, this red fluorescent protein is delivered to lysosomes and changes its excitation peak from 438 nm at neutral pH to 550 nm at acidic pH, which allows for dual‐excitation ratiometric imaging (Katayama et al, 2011). Since mt‐Keima is resistant against lysosomal proteases and stays fluorescent at acidic pH, it has been used to provide a readout of mitophagy in mammalian cells and tissues (Bingol et al, 2014; Kageyama et al, 2014; Mizumura et al, 2014; Hirota et al, 2015; Ikeda et al, 2015; Shirakabe et al, 2016a; Shirakabe et al, 2016b; Xu et al, 2018). Moreover, a transgenic mouse and fly expressing mt‐Keima has been established to evaluate mitophagy in vivo under a variety of experimental conditions, revealing that mitophagy activity varies from tissues to tissues (Sun et al, 2015; Cornelissen et al, 2018).
B. MitoTimer
Mitophagy and mitochondrial biogenesis involve a dynamic turnover of mitochondria. Hence, a time‐sensitive fluorescent protein is a good probe to chase changes in mitochondrial dynamics. Fluorescent timer, or DsRed1‐E5, is a redox‐sensitive variant, and its fluorescence shifts over time from green to red as it becomes mature (Terskikh et al, 2000). MitoTimer consisting of the timer fluorescent protein fused to the N‐terminal mitochondrial targeting sequence of COX8A has been established and expressed in different tissues (Ferree et al, 2013; Hernandez et al, 2013; Laker et al, 2014; Trudeau et al, 2014; Stotland & Gottlieb, 2016; Laker et al, 2017). Rates of mitochondrial turnover and MitoTimer fluorescence transmission depend on the balance between import of newly synthesized components and degradation of old materials. Transgenic mice expressing MitoTimer reveal that this fluorescent molecular clock is a good tool to monitor mitochondrial structure, function, oxidative stress, and mitophagy in vivo under physiological and pathophysiological conditions (Wilson et al, 2017).
C. Mito‐QC
Mito‐QC is a pH‐sensitive tandem mCherry‐GFP‐tagged fluorescent marker fused with the C‐terminal TM domain derived from the OMM‐anchored protein FIS1 (Allen et al, 2013; McWilliams & Ganley, 2019). Upon mitophagy, mitochondria containing mito‐QC (both mCherry‐ and GFP‐positive) are delivered to lysosomes where mCherry remains resistant against acidic pH and proteases, but GFP becomes quenched under acidic conditions. Thus, mCherry‐only (GFP‐negative) foci can be observed and quantified as indicators of mitochondrial degradation in lysosomes. Recent studies using transgenic mouse models expressing mito‐QC reveal high levels of mitophagy in the developing heart and adult kidney, and PINK1‐independent basal mitochondrial turnover in vivo (McWilliams et al, 2016; McWilliams et al, 2018).
Conflict of interest
The authors declare that they have no conflict of interest.
Acknowledgements
The authors apologize to the many colleagues whose work could not be cited owing to space limitations. We thank Soichiro Kakuta (Juntendo University, Tokyo, Japan) and Shotaro Saita (Osaka University, Suita, Japan) for helpful comments on this manuscript. This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI Grants JP18K06237 and JP18H05500 (to KY), JP18H02435 and JP19H05712 (to MS), JP18H02443 and JP19H05712 (to NM), and JP16H04784 and JP19H03222 (to KO); CREST, Japan Agency for Medical Research and Development (to NM) and Takeda Science Foundation (to MS and NM). MO was supported by Aid for JSPS Fellows (MEXT | JSPS) Grant JP19J10384.
The EMBO Journal (2021) 40: e104705.
See the Glossary for abbreviations used in this article.
Contributor Information
Miyuki Sato, Email: m-sato@gunma-u.ac.jp.
Noriyuki Matsuda, Email: matsuda-nr@igakuken.or.jp.
Koji Okamoto, Email: kokamoto@fbs.osaka-u.ac.jp.
References
- Agnihotri S, Golbourn B, Huang X, Remke M, Younger S, Cairns RA, Chalil A, Smith CA, Krumholtz SL, Mackenzie D et al (2016) PINK1 is a negative regulator of growth and the Warburg effect in glioblastoma. Cancer Res 76: 4708–4719 [DOI] [PubMed] [Google Scholar]
- Al Rawi S, Louvet‐Vallee S, Djeddi A, Sachse M, Culetto E, Hajjar C, Boyd L, Legouis R, Galy V (2011) Postfertilization autophagy of sperm organelles prevents paternal mitochondrial DNA transmission. Science 334: 1144–1147 [DOI] [PubMed] [Google Scholar]
- Al Rawi S, Louvet‐Vallee S, Djeddi A, Sachse M, Culetto E, Hajjar C, Boyd L, Legouis R, Galy V (2012) Allophagy: a macroautophagic process degrading spermatozoid‐inherited organelles. Autophagy 8: 421–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen GF, Toth R, James J, Ganley IG (2013) Loss of iron triggers PINK1/Parkin‐independent mitophagy. EMBO Rep 14: 1127–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altshuler‐Keylin S, Shinoda K, Hasegawa Y, Ikeda K, Hong H, Kang Q, Yang Y, Perera RM, Debnath J, Kajimura S (2016) Beige adipocyte maintenance is regulated by autophagy‐induced mitochondrial clearance. Cell Metab 24: 402–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambivero CT, Cilenti L, Main S, Zervos AS (2014) Mulan E3 ubiquitin ligase interacts with multiple E2 conjugating enzymes and participates in mitophagy by recruiting GABARAP. Cell Signal 26: 2921–2929 [DOI] [PubMed] [Google Scholar]
- Ankel‐Simons F, Cummins JM (1996) Misconceptions about mitochondria and mammalian fertilization‐ Implications for theories on human evolution. Proc Natl Acad Sci USA 93: 13859–13863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki Y, Kanki T, Hirota Y, Kurihara Y, Saigusa T, Uchiumi T, Kang D (2011) Phosphorylation of Serine 114 on Atg32 mediates mitophagy. Mol Biol Cell 22: 3206–3217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronowski J, Strong R, Grotta JC (1997) Reperfusion injury: demonstration of brain damage produced by reperfusion after transient focal ischemia in rats. J Cereb Blood Flow Metab 17: 1048–1056 [DOI] [PubMed] [Google Scholar]
- Ashrafi G, Schlehe JS, LaVoie MJ, Schwarz TL (2014) Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J Cell Biol 206: 655–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr F, Lambright DG (2010) Rab GEFs and GAPs. Curr Opin Cell Biol 22: 461–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartolome A, Garcia‐Aguilar A, Asahara SI, Kido Y, Guillen C, Pajvani UB, Benito M (2017) MTORC1 regulates both general autophagy and mitophagy induction after oxidative phosphorylation uncoupling. Mol Cell Biol 37: e00441–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belgareh‐Touze N, Cavellini L, Cohen MM (2017) Ubiquitination of ERMES components by the E3 ligase Rsp5 is involved in mitophagy. Autophagy 13: 114–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardini JP, Lazarou M, Dewson G (2017) Parkin and mitophagy in cancer. Oncogene 36: 1315–1327 [DOI] [PubMed] [Google Scholar]
- Bhujabal Z, Birgisdottir AB, Sjottem E, Brenne HB, Overvatn A, Habisov S, Kirkin V, Lamark T, Johansen T (2017) FKBP8 recruits LC3A to mediate Parkin‐independent mitophagy. EMBO Rep 18: 947–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billia F, Hauck L, Konecny F, Rao V, Shen J, Mak TW (2011) PTEN‐inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc Natl Acad Sci USA 108: 9572–9577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingol B, Tea JS, Phu L, Reichelt M, Bakalarski CE, Song Q, Foreman O, Kirkpatrick DS, Sheng M (2014) The mitochondrial deubiquitinase USP30 opposes parkin‐mediated mitophagy. Nature 510: 370–375 [DOI] [PubMed] [Google Scholar]
- Bockler S, Westermann B (2014) Mitochondrial ER contacts are crucial for mitophagy in yeast. Dev Cell 28: 450–458 [DOI] [PubMed] [Google Scholar]
- Bonner JM, Boulianne GL (2017) Diverse structures, functions and uses of FK506 binding proteins. Cell Signal 38: 97–105 [DOI] [PubMed] [Google Scholar]
- Boyd JM, Malstrom S, Subramanian T, Venkatesh LK, Schaeper U, Elangovan B, D'Sa‐Eipper C, Chinnadurai G (1994) Adenovirus E1B 19 kDa and Bcl‐2 proteins interact with a common set of cellular proteins. Cell 79: 341–351 [DOI] [PubMed] [Google Scholar]
- Cai Q, Zakaria HM, Simone A, Sheng ZH (2012) Spatial parkin translocation and degradation of damaged mitochondria via mitophagy in live cortical neurons. Curr Biol 22: 545–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, Hess S, Chan DC (2011) Broad activation of the ubiquitin‐proteasome system by Parkin is critical for mitophagy. Hum Mol Genet 20: 1726–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandel NS, Jasper H, Ho TT, Passegue E (2016) Metabolic regulation of stem cell function in tissue homeostasis and organismal ageing. Nat Cell Biol 18: 823–832 [DOI] [PubMed] [Google Scholar]
- Chen G, Cizeau J, Vande Velde C, Park JH, Bozek G, Bolton J, Shi L, Dubik D, Greenberg A (1999) Nix and Nip3 form a subfamily of pro‐apoptotic mitochondrial proteins. J Biol Chem 274: 7–10 [DOI] [PubMed] [Google Scholar]
- Chen G, Han Z, Feng D, Chen Y, Chen L, Wu H, Huang L, Zhou C, Cai X, Fu C et al (2014) A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor‐mediated mitophagy. Mol Cell 54: 362–377 [DOI] [PubMed] [Google Scholar]
- Chen G, Ray R, Dubik D, Shi L, Cizeau J, Bleackley RC, Saxena S, Gietz RD, Greenberg AH (1997) The E1B 19K:Bcl‐2–binding protein Nip3 is a dimeric mitochondrial protein that activates apoptosis. J Exp Med 186: 1975–1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Dai H, Yuan J, Chen J, Lin L, Zhang W, Wang L, Zhang J, Li K, He Y (2018) Optineurin‐mediated mitophagy protects renal tubular epithelial cells against accelerated senescence in diabetic nephropathy. Cell Death Dis 9: 105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Liu L, Cheng Q, Li Y, Wu H, Zhang W, Wang Y, Sehgal SA, Siraj S, Wang X et al (2017) Mitochondrial E3 ligase MARCH5 regulates FUNDC1 to fine‐tune hypoxic mitophagy. EMBO Rep 18: 495–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chourasia AH, Tracy K, Frankenberger C, Boland ML, Sharifi MN, Drake LE, Sachleben JR, Asara JM, Locasale JW, Karczmar GS et al (2015) Mitophagy defects arising from BNip3 loss promote mammary tumor progression to metastasis. EMBO Rep 16: 1145–1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS, Couthouis J, Lu YF, Wang Q, Krueger BJ et al (2015) Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 347: 1436–1441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M (2006) Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441: 1162–1166 [DOI] [PubMed] [Google Scholar]
- Cornelissen T, Haddad D, Wauters F, Van Humbeeck C, Mandemakers W, Koentjoro B, Sue C, Gevaert K, De Strooper B, Verstreken P et al (2014) The deubiquitinase USP15 antagonizes Parkin‐mediated mitochondrial ubiquitination and mitophagy. Hum Mol Genet 23: 5227–5242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelissen T, Vilain S, Vints K, Gounko N, Verstreken P, Vandenberghe W (2018) Deficiency of parkin and PINK1 impairs age‐dependent mitophagy in Drosophila. Elife 7: e35878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corti O, Lesage S, Brice A (2011) What genetics tells us about the causes and mechanisms of Parkinson's disease. Physiol Rev 91: 1161–1218 [DOI] [PubMed] [Google Scholar]
- Cunningham CN, Baughman JM, Phu L, Tea JS, Yu C, Coons M, Kirkpatrick DS, Bingol B, Corn JE (2015) USP30 and parkin homeostatically regulate atypical ubiquitin chains on mitochondria. Nat Cell Biol 17: 160–169 [DOI] [PubMed] [Google Scholar]
- Deffieu M, Bhatia‐Kissova I, Salin B, Galinier A, Manon S, Camougrand N (2009) Glutathione participates in the regulation of mitophagy in yeast. J Biol Chem 284: 14828–14837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Rita A , Peschiaroli A, D’Acunzo P, Strobbe D, Hu Z, Gruber J, Nygaard M, Lambrughi M, Melino G, Papaleo E et al (2018) HUWE1 E3 ligase promotes PINK1/PARKIN‐independent mitophagy by regulating AMBRA1 activation via IKKalpha. Nat Commun 9: 3755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding WX, Li M, Chen X, Ni HM, Lin CW, Gao W, Lu B, Stolz DB, Clemens DL, Yin XM (2010a) Autophagy reduces acute ethanol‐induced hepatotoxicity and steatosis in mice. Gastroenterology 139: 1740–1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding WX, Li M, Yin XM (2011) Selective taste of ethanol‐induced autophagy for mitochondria and lipid droplets. Autophagy 7: 248–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding WX, Ni HM, Li M, Liao Y, Chen X, Stolz DB, Dorn GW, Yin XM (2010b) Nix is critical to two distinct phases of mitophagy, reactive oxygen species‐mediated autophagy induction and Parkin‐ubiquitin‐p62‐mediated mitochondrial priming. J Biol Chem 285: 27879–27890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djeddi A, Al Rawi S, Deuve JL, Perrois C, Liu YY, Russeau M, Sachse M, Galy V (2015) Sperm‐inherited organelle clearance in C. elegans relies on LC3‐dependent autophagosome targeting to the pericentrosomal area. Development 142: 1705–1716 [DOI] [PubMed] [Google Scholar]
- Duguez S, Feasson L, Denis C, Freyssenet D (2002) Mitochondrial biogenesis during skeletal muscle regeneration. Am J Physiol Endocrinol Metab 282: E802–809 [DOI] [PubMed] [Google Scholar]
- Durcan TM, Tang MY, Perusse JR, Dashti EA, Aguileta MA, McLelland GL, Gros P, Shaler TA, Faubert D, Coulombe B et al (2014) USP8 regulates mitophagy by removing K6‐linked ubiquitin conjugates from parkin. EMBO J 33: 2473–2491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisner V, Picard M, Hajnoczky G (2018) Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat Cell Biol 20: 755–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmore SP, Qian T, Grissom SF, Lemasters JJ (2001) The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J 15: 2286–2287 [DOI] [PubMed] [Google Scholar]
- Erkan M, Kleeff J, Esposito I, Giese T, Ketterer K, Buchler MW, Giese NA, Friess H (2005) Loss of BNIP3 expression is a late event in pancreatic cancer contributing to chemoresistance and worsened prognosis. Oncogene 24: 4421–4432 [DOI] [PubMed] [Google Scholar]
- Esteban‐Martinez L, Sierra‐Filardi E, McGreal RS, Salazar‐Roa M, Marino G, Seco E, Durand S, Enot D, Grana O, Malumbres M et al (2017) Programmed mitophagy is essential for the glycolytic switch during cell differentiation. EMBO J 36: 1688–1706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans CS, Holzbaur ELF (2018) Autophagy and mitophagy in ALS. Neurobiol Dis 122: 35–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fader CM, Colombo MI (2006) Multivesicular bodies and autophagy in erythrocyte maturation. Autophagy 2: 122–125 [DOI] [PubMed] [Google Scholar]
- Faesen AC, Luna‐Vargas MP, Geurink PP, Clerici M, Merkx R, van Dijk WJ, Hameed DS, El Oualid F, Ovaa H, Sixma TK (2011) The differential modulation of USP activity by internal regulatory domains, interactors and eight ubiquitin chain types. Chem Biol 18: 1550–1561 [DOI] [PubMed] [Google Scholar]
- Fang EF, Hou Y, Palikaras K, Adriaanse BA, Kerr JS, Yang B, Lautrup S, Hasan‐Olive MM, Caponio D, Dan X et al (2019) Mitophagy inhibits amyloid‐beta and tau pathology and reverses cognitive deficits in models of Alzheimer's disease. Nat Neurosci 22: 401–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferree AW, Trudeau K, Zik E, Benador IY, Twig G, Gottlieb RA, Shirihai OS (2013) MitoTimer probe reveals the impact of autophagy, fusion, and motility on subcellular distribution of young and old mitochondrial protein and on relative mitochondrial protein age. Autophagy 9: 1887–1896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freischmidt A, Wieland T, Richter B, Ruf W, Schaeffer V, Muller K, Marroquin N, Nordin F, Hubers A, Weydt P et al (2015) Haploinsufficiency of TBK1 causes familial ALS and fronto‐temporal dementia. Nat Neurosci 18: 631–636 [DOI] [PubMed] [Google Scholar]
- Furukawa K, Fukuda T, Yamashita SI, Saigusa T, Kurihara Y, Yoshida Y, Kirisako H, Nakatogawa H, Kanki T (2018) The PP2A‐like protein phosphatase Ppg1 and the Far complex cooperatively counteract CK2‐mediated phosphorylation of Atg32 to inhibit mitophagy. Cell Rep 23: 3579–3590 [DOI] [PubMed] [Google Scholar]
- Galvan‐Pena S, O'Neill LA (2014) Metabolic reprogramming in macrophage polarization. Front Immunol 5: 420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F, Chen D, Si J, Hu Q, Qin Z, Fang M, Wang G (2015) The mitochondrial protein BNIP3L is the substrate of PARK2 and mediates mitophagy in PINK1/PARK2 pathway. Hum Mol Genet 24: 2528–2538 [DOI] [PubMed] [Google Scholar]
- Gaspard GJ, McMaster CR (2015) The mitochondrial quality control protein Yme1 is necessary to prevent defective mitophagy in a yeast model of Barth syndrome. J Biol Chem 290: 9284–9298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatica D, Lahiri V, Klionsky DJ (2018) Cargo recognition and degradation by selective autophagy. Nat Cell Biol 20: 233–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gawaz M (2004) Role of platelets in coronary thrombosis and reperfusion of ischemic myocardium. Cardiovasc Res 61: 498–511 [DOI] [PubMed] [Google Scholar]
- Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH, Taanman JW (2010) Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin‐dependent manner upon induction of mitophagy. Hum Mol Genet 19: 4861–4870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W (2010a) PINK1/Parkin‐mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 12: 119–131 [DOI] [PubMed] [Google Scholar]
- Geisler S, Holmstrom KM, Treis A, Skujat D, Weber SS, Fiesel FC, Kahle PJ, Springer W (2010b) The PINK1/Parkin‐mediated mitophagy is compromised by PD‐associated mutations. Autophagy 6: 871–878 [DOI] [PubMed] [Google Scholar]
- Georgakopoulos ND, Wells G, Campanella M (2017) The pharmacological regulation of cellular mitophagy. Nat Chem Biol 13: 136–146 [DOI] [PubMed] [Google Scholar]
- Gersch M, Gladkova C, Schubert AF, Michel MA, Maslen S, Komander D (2017) Mechanism and regulation of the Lys6‐selective deubiquitinase USP30. Nat Struct Mol Biol 24: 920–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladkova C, Maslen S, Skehel J, Komander D (2018) Mechanism of parkin activation by PINK1. Nature 559: 410–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogvadze V, Orrenius S, Zhivotovsky B (2008) Mitochondria in cancer cells: what is so special about them? Trends Cell Biol 18: 165–173 [DOI] [PubMed] [Google Scholar]
- Gong Y, Zack TI, Morris LG, Lin K, Hukkelhoven E, Raheja R, Tan IL, Turcan S, Veeriah S, Meng S et al (2014) Pan‐cancer genetic analysis identifies PARK2 as a master regulator of G1/S cyclins. Nat Genet 46: 588–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ (2003) Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci USA 100: 4078–4083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y et al (2013) Autophagosomes form at ER‐mitochondria contact sites. Nature 495: 389–393 [DOI] [PubMed] [Google Scholar]
- Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson AB (2012) Microtubule‐associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem 287: 19094–19104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen M, Rubinsztein DC, Walker DW (2018) Autophagy as a promoter of longevity: insights from model organisms. Nat Rev Mol Cell Biol 19: 579–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms M, Seale P (2013) Brown and beige fat: development, function and therapeutic potential. Nat Med 19: 1252–1263 [DOI] [PubMed] [Google Scholar]
- Hayashi‐Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A (2009) A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol 11: 1433–1437 [DOI] [PubMed] [Google Scholar]
- Heo JM, Ordureau A, Paulo JA, Rinehart J, Harper JW (2015) The PINK1‐PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol Cell 60: 7–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo JM, Ordureau A, Swarup S, Paulo JA, Shen K, Sabatini DM, Harper JW (2018) RAB7A phosphorylation by TBK1 promotes mitophagy via the PINK‐PARKIN pathway. Sci Adv 4: eaav0443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez G, Thornton C, Stotland A, Lui D, Sin J, Ramil J, Magee N, Andres A, Quarato G, Carreira RS et al (2013) MitoTimer: a novel tool for monitoring mitochondrial turnover. Autophagy 9: 1852–1861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota Y, Yamashita S, Kurihara Y, Jin X, Aihara M, Saigusa T, Kang D, Kanki T (2015) Mitophagy is primarily due to alternative autophagy and requires the MAPK1 and MAPK14 signaling pathways. Autophagy 11: 332–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong X, Liu J, Zhu G, Zhuang Y, Suo H, Wang P, Huang D, Xu J, Huang Y, Yu M et al (2014) Parkin overexpression ameliorates hippocampal long‐term potentiation and beta‐amyloid load in an Alzheimer's disease mouse model. Hum Mol Genet 23: 1056–1072 [DOI] [PubMed] [Google Scholar]
- Igarashi R, Yamashita SI, Yamashita T, Inoue K, Fukuda T, Fukuchi T, Kanki T (2020) Gemcitabine induces Parkin‐independent mitophagy through mitochondrial‐resident E3 ligase MUL1‐mediated stabilization of PINK1. Sci Rep 10: 1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iguchi M, Kujuro Y, Okatsu K, Koyano F, Kosako H, Kimura M, Suzuki N, Uchiyama S, Tanaka K, Matsuda N (2013) Parkin‐catalyzed ubiquitin‐ester transfer is triggered by PINK1‐dependent phosphorylation. J Biol Chem 288: 22019–22032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J, Nomura M, Mihara K, Egashira K, Ohishi M et al (2015) Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res 116: 264–278 [DOI] [PubMed] [Google Scholar]
- Itakura E, Kishi‐Itakura C, Koyama‐Honda I, Mizushima N (2012) Structures containing Atg9A and the ULK1 complex independently target depolarized mitochondria at initial stages of Parkin‐mediated mitophagy. J Cell Sci 125: 1488–1499 [DOI] [PubMed] [Google Scholar]
- Itakura E, Mizushima N (2010) Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 6: 764–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito C, Saito Y, Nozawa T, Fujii S, Sawa T, Inoue H, Matsunaga T, Khan S, Akashi S, Hashimoto R et al (2013) Endogenous nitrated nucleotide is a key mediator of autophagy and innate defense against bacteria. Mol Cell 52: 794–804 [DOI] [PubMed] [Google Scholar]
- Jimenez‐Orgaz A, Kvainickas A, Nagele H, Denner J, Eimer S, Dengjel J, Steinberg F (2018) Control of RAB7 activity and localization through the retromer‐TBC1D5 complex enables RAB7‐dependent mitophagy. EMBO J 37: 235–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen T, Lamark T (2011) Selective autophagy mediated by autophagic adapter proteins. Autophagy 7: 279–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 19: 5720–5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Yamamoto A, Oshitani‐Okamoto S, Ohsumi Y, Yoshimori T (2004) LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form‐II formation. J Cell Sci 117: 2805–2812 [DOI] [PubMed] [Google Scholar]
- Kageyama Y, Hoshijima M, Seo K, Bedja D, Sysa‐Shah P, Andrabi SA, Chen W, Hoke A, Dawson VL, Dawson TM et al (2014) Parkin‐independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. EMBO J 33: 2798–2813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajimura S, Spiegelman BM, Seale P (2015) Brown and beige fat: physiological roles beyond heat generation. Cell Metab 22: 546–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalkavan H, Green DR (2018) MOMP, cell suicide as a BCL‐2 family business. Cell Death Differ 25: 46–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ (2014) PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol 205: 143–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang R, Zeng L, Xie Y, Yan Z, Zhou B, Cao L, Klionsky DJ, Tracey KJ, Li J, Wang H et al (2016) A novel PINK1‐ and PARK2‐dependent protective neuroimmune pathway in lethal sepsis. Autophagy 12: 2374–2385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanki T, Klionsky DJ (2008) Mitophagy in yeast occurs through a selective mechanism. J Biol Chem 283: 32386–32393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanki T, Kurihara Y, Jin X, Goda T, Ono Y, Aihara M, Hirota Y, Saigusa T, Aoki Y, Uchiumi T et al (2013) Casein kinase 2 is essential for mitophagy. EMBO Rep 14: 788–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanki T, Wang K, Cao Y, Baba M, Klionsky DJ (2009) Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev Cell 17: 98–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karavaeva IE, Golyshev SA, Smirnova EA, Sokolov SS, Severin FF, Knorre DA (2017) Mitochondrial depolarization in yeast zygotes inhibits clonal expansion of selfish mtDNA. J Cell Sci 130: 1274–1284 [DOI] [PubMed] [Google Scholar]
- Katayama H, Hama H, Nagasawa K, Kurokawa H, Sugiyama M, Ando R, Funata M, Yoshida N, Homma M, Nishimura T et al (2020) Visualizing and modulating mitophagy for therapeutic studies of neurodegeneration. Cell Cell 181: 1176–1187.e16 [DOI] [PubMed] [Google Scholar]
- Katayama H, Kogure T, Mizushima N, Yoshimori T, Miyawaki A (2011) A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem Biol 18: 1042–1052 [DOI] [PubMed] [Google Scholar]
- Kazlauskaite A, Martinez‐Torres R, Wilkie S, Kumar A, Peltier J, Gonzalez A, Johnson C, Zhang J, Hope AG, Peggie M et al (2015) Binding to serine 65‐phosphorylated ubiquitin primes Parkin for optimal PINK1‐dependent phosphorylation and activation. EMBO Rep 16: 939–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazlauskaite A, Kondapalli C, Gourlay R, Campbell DG, Ritorto MS, Hofmann K, Alessi DR, Knebel A, Trost M, Muqit MM (2014) Parkin is activated by PINK1‐dependent phosphorylation of ubiquitin at Ser65. Biochem J 460: 127–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaminets A, Behl C, Dikic I (2016) Ubiquitin‐dependent and independent signals in selective autophagy. Trends Cell Biol 26: 6–16 [DOI] [PubMed] [Google Scholar]
- Kim MJ, Bae SH, Ryu JC, Kwon Y, Oh JH, Kwon J, Moon JS, Kim K, Miyawaki A, Lee MG et al (2016) SESN2/sestrin2 suppresses sepsis by inducing mitophagy and inhibiting NLRP3 activation in macrophages. Autophagy 12: 1272–1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392: 605–608 [DOI] [PubMed] [Google Scholar]
- Kondapalli C, Kazlauskaite A, Zhang N, Woodroof HI, Campbell DG, Gourlay R, Burchell L, Walden H, Macartney TJ, Deak M et al (2012) PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol 2: 120080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo‐Okamoto N, Noda NN, Suzuki SW, Nakatogawa H, Takahashi I, Matsunami M, Hashimoto A, Inagaki F, Ohsumi Y, Okamoto K (2012) Autophagy‐related protein 32 acts as an autophagic degron and directly initiates mitophagy. J Biol Chem 287: 10631–10638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koop EA, van Laar T, van Wichen DF, de Weger RA, Wall E, van Diest PJ (2009) Expression of BNIP3 in invasive breast cancer: correlations with the hypoxic response and clinicopathological features. BMC Cancer 9: 175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornmann B, Currie E, Collins SR, Schuldiner M, Nunnari J, Weissman JS, Walter P (2009) An ER‐mitochondria tethering complex revealed by a synthetic biology screen. Science 325: 477–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koury MJ, Koury ST, Kopsombut P, Bondurant MC (2005) In vitro maturation of nascent reticulocytes to erythrocytes. Blood 105: 2168–2174 [DOI] [PubMed] [Google Scholar]
- Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T et al (2014) Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510: 162–166 [DOI] [PubMed] [Google Scholar]
- Koyano F, Yamano K, Kosako H, Tanaka K, Matsuda N (2019) Parkin recruitment to impaired mitochondria for nonselective ubiquitylation is facilitated by MITOL. J Biol Chem 294: 10300–10314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubli DA, Quinsay MN, Huang C, Lee Y, Gustafsson AB (2008) Bnip3 functions as a mitochondrial sensor of oxidative stress during myocardial ischemia and reperfusion. Am J Physiol Heart Circ Physiol 295: H2025–2031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubli DA, Ycaza JE, Gustafsson AB (2007) Bnip3 mediates mitochondrial dysfunction and cell death through Bax and Bak. Biochem J 405: 407–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubli DA, Zhang X, Lee Y, Hanna RA, Quinsay MN, Nguyen CK, Jimenez R, Petrosyan S, Murphy AN, Gustafsson AB (2013) Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J Biol Chem 288: 915–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA et al (2005) Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 309: 481–484 [DOI] [PubMed] [Google Scholar]
- Kumar A, Aguirre J, Condos T, Martinez‐Torres R, Chaugule V, Toth R, Sundaramoorthy R, Mercier P, Knebel A, Spratt D et al (2015) Disruption of the autoinhibited state primes the E3 ligase parkin for activation and catalysis. EMBO J 34: 2506–2521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Tamjar J, Waddell AD, Woodroof HI, Raimi OG, Shaw AM, Peggie M, Muqit MM, van Aalten DM (2017) Structure of PINK1 and mechanisms of Parkinson's disease‐associated mutations. Elife 6: e29985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurihara Y, Kanki T, Aoki Y, Hirota Y, Saigusa T, Uchiumi T, Kang D (2011) Mitophagy plays an essential role in reducing mitochondrial production of reactive oxygen species and mutation of mitochondrial DNA by maintaining mitochondrial quantity and quality in yeast. J Biol Chem 287: 3265–3272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laker RC, Drake JC, Wilson RJ, Lira VA, Lewellen BM, Ryall KA, Fisher CC, Zhang M, Saucerman JJ, Goodyear LJ et al (2017) Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise‐induced mitophagy. Nat Commun 8: 548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laker RC, Xu P, Ryall KA, Sujkowski A, Kenwood BM, Chain KH, Zhang M, Royal MA, Hoehn KL, Driscoll M et al (2014) A novel MitoTimer reporter gene for mitochondrial content, structure, stress, and damage in vivo . J Biol Chem 289: 12005–12015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ (2015) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524: 309–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JJ, Andreazza S, Whitworth AJ (2020) The STING pathway does not contribute to behavioural or mitochondrial phenotypes in Drosophila Pink1/parkin or mtDNA mutator models. Sci Rep 10: 2693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JJ, Sanchez‐Martinez A, Zarate AM, Beninca C, Mayor U, Clague MJ, Whitworth AJ (2018) Basal mitophagy is widespread in Drosophila but minimally affected by loss of Pink1 or parkin. J Cell Biol 217: 1613–1622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemasters JJ (2005) Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res 8: 3–5 [DOI] [PubMed] [Google Scholar]
- Leonhard K, Herrmann J, Stuart R, Mannhaupt G, Neupert W, Langer T (1996) AAA proteases with catalytic sites on opposite membrane surfaces comprise a proteolytic system for the ATP‐dependent degradation of inner membrane proteins in mitochondria. EMBO J 15: 4218–4229 [PMC free article] [PubMed] [Google Scholar]
- Li J, Qi W, Chen G, Feng D, Liu J, Ma B, Zhou C, Mu C, Zhang W, Chen Q et al (2015) Mitochondrial outer‐membrane E3 ligase MUL1 ubiquitinates ULK1 and regulates selenite‐induced mitophagy. Autophagy 11: 1216–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Li Y, Siraj S, Jin H, Fan Y, Yang X, Huang X, Wang X, Wang J, Liu L et al (2019) FUN14 domain‐containing 1‐mediated mitophagy suppresses hepatocarcinogenesis by inhibition of inflammasome activation in mice. Hepatology 69: 604–621 [DOI] [PubMed] [Google Scholar]
- Li Y, Wang Y, Kim E, Beemiller P, Wang CY, Swanson J, You M, Guan KL (2007) Bnip3 mediates the hypoxia‐induced inhibition on mammalian target of rapamycin by interacting with Rheb. J Biol Chem 282: 35803–35813 [DOI] [PubMed] [Google Scholar]
- Liang JR, Martinez A, Lane JD, Mayor U, Clague MJ, Urbe S (2015) USP30 deubiquitylates mitochondrial Parkin substrates and restricts apoptotic cell death. EMBO Rep 16: 618–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisowski P, Kannan P, Mlody B, Prigione A (2018) Mitochondria and the dynamic control of stem cell homeostasis. EMBO Rep 19: e45432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W et al (2012) Mitochondrial outer‐membrane protein FUNDC1 mediates hypoxia‐induced mitophagy in mammalian cells. Nat Cell Biol 14: 177–185 [DOI] [PubMed] [Google Scholar]
- Loomba R, Sanyal AJ (2013) The global NAFLD epidemic. Nat Rev Gastroenterol Hepatol 10: 686–690 [DOI] [PubMed] [Google Scholar]
- Lotharius J, Brundin P (2002) Pathogenesis of Parkinson's disease: dopamine, vesicles and alpha‐synuclein. Nat Rev Neurosci 3: 932–942 [DOI] [PubMed] [Google Scholar]
- Lu X, Altshuler‐Keylin S, Wang Q, Chen Y, Henrique Sponton C, Ikeda K, Maretich P, Yoneshiro T, Kajimura S (2018) Mitophagy controls beige adipocyte maintenance through a Parkin‐dependent and UCP1‐independent mechanism. Sci Signal 11: eaap8526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo SM, Ge ZJ, Wang ZW, Jiang ZZ, Wang ZB, Ouyang YC, Hou Y, Schatten H, Sun QY (2013) Unique insights into maternal mitochondrial inheritance in mice. Proc Natl Acad Sci USA 110: 13038–13043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes H, Van Eygen S, Krysko DV, Vandenabeele P, Nys K, Rillaerts K, Garg AD, Verfaillie T, Agostinis P (2014) BNIP3 supports melanoma cell migration and vasculogenic mimicry by orchestrating the actin cytoskeleton. Cell Death Dis 5: e1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manczak M, Kandimalla R, Yin X, Reddy PH (2018) Hippocampal mutant APP and amyloid beta‐induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of Alzheimer's disease. Hum Mol Genet 27: 1332–1342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manil‐Segalen M, Lefebvre C, Jenzer C, Trichet M, Boulogne C, Satiat‐Jeunemaitre B, Legouis R (2014) The C. elegans LC3 acts downstream of GABARAP to degrade autophagosomes by interacting with the HOPS subunit VPS39. Dev Cell 28: 43–55 [DOI] [PubMed] [Google Scholar]
- Manka D, Spicer Z, Millhorn DE (2005) Bcl‐2/adenovirus E1B 19 kDa interacting protein‐3 knockdown enables growth of breast cancer metastases in the lung, liver, and bone. Cancer Res 65: 11689–11693 [DOI] [PubMed] [Google Scholar]
- Marinkovic M, Sprung M, Novak I (2020) Dimerization of mitophagy receptor BNIP3L/NIX is essential for recruitment of autophagic machinery. Autophagy 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama H, Morino H, Ito H, Izumi Y, Kato H, Watanabe Y, Kinoshita Y, Kamada M, Nodera H, Suzuki H et al (2010) Mutations of optineurin in amyotrophic lateral sclerosis. Nature 465: 223–226 [DOI] [PubMed] [Google Scholar]
- Masuoka HC, Chalasani N (2013) Nonalcoholic fatty liver disease: an emerging threat to obese and diabetic individuals. Ann N Y Acad Sci 1281: 106–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda N (2016) Phospho‐ubiquitin: upending the PINK‐Parkin‐ubiquitin cascade. J Biochem 159: 379–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F et al (2010) PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol 189: 211–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda N, Yamano K (2020) Two sides of a coin: Physiological significance and molecular mechanisms for damage‐induced mitochondrial localization of PINK1 and Parkin. Neurosci Res 19: 30171 [DOI] [PubMed] [Google Scholar]
- Matsushima M, Fujiwara T, Takahashi E, Minaguchi T, Eguchi Y, Tsujimoto Y, Suzumori K, Nakamura Y (1998) Isolation, mapping, and functional analysis of a novel human cDNA (BNIP3L) encoding a protein homologous to human NIP3. Genes Chromosomes Cancer 21: 230–235 [PubMed] [Google Scholar]
- McEwan DG, Popovic D, Gubas A, Terawaki S, Suzuki H, Stadel D, Coxon FP, Miranda de Stegmann D, Bhogaraju S, Maddi K et al (2015) PLEKHM1 regulates autophagosome‐lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol Cell 57: 39–54 [DOI] [PubMed] [Google Scholar]
- McWilliams TG, Ganley IG (2019) Investigating mitophagy and mitochondrial morphology in vivo using mito‐QC: a comprehensive guide. Methods Mol Biol 1880: 621–642 [DOI] [PubMed] [Google Scholar]
- McWilliams TG, Prescott AR, Allen GF, Tamjar J, Munson MJ, Thomson C, Muqit MM, Ganley IG (2016) mito‐QC illuminates mitophagy and mitochondrial architecture in vivo . J Cell Biol 214: 333–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWilliams TG, Prescott AR, Montava‐Garriga L, Ball G, Singh F, Barini E, Muqit MMK, Brooks SP, Ganley IG (2018) Basal mitophagy occurs independently of PINK1 in mouse tissues of high metabolic demand. Cell Metab 27: 439–449.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta MM, Weinberg SE, Chandel NS (2017) Mitochondrial control of immunity: beyond ATP. Nat Rev Immunol 17: 608–620 [DOI] [PubMed] [Google Scholar]
- Melser S, Chatelain EH, Lavie J, Mahfouf W, Jose C, Obre E, Goorden S, Priault M, Elgersma Y, Rezvani HR et al (2013) Rheb regulates mitophagy induced by mitochondrial energetic status. Cell Metab 17: 719–730 [DOI] [PubMed] [Google Scholar]
- Mizumura K, Cloonan SM, Nakahira K, Bhashyam AR, Cervo M, Kitada T, Glass K, Owen CA, Mahmood A, Washko GR et al (2014) Mitophagy‐dependent necroptosis contributes to the pathogenesis of COPD. J Clin Invest 124: 3987–4003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Komatsu M (2011) Autophagy: renovation of cells and tissues. Cell 147: 728–741 [DOI] [PubMed] [Google Scholar]
- Molina P, Lim Y, Boyd L (2019) Ubiquitination is required for the initial removal of paternal organelles in C. elegans . Dev Biol 453: 168–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen M, Ferguson DJ, Edelmann M, Kessler B, Morten KJ, Komatsu M, Simon AK (2010) Loss of autophagy in erythroid cells leads to defective removal of mitochondria and severe anemia in vivo . Proc Natl Acad Sci USA 107: 832–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakawa T, Okamoto K, Omiya S, Taneike M, Yamaguchi O, Otsu K (2019) A mammalian mitophagy receptor, Bcl2‐L‐13, recruits the ULK1 complex to induce mitophagy. Cell Rep 26: 338–345.e336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakawa T, Yamaguchi O, Hashimoto A, Hikoso S, Takeda T, Oka T, Yasui H, Ueda H, Akazawa Y, Nakayama H et al (2015) Bcl‐2‐like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat Commun 6: 7527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP et al (2011) Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12: 222–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura S, Yoshimori T (2018) Autophagy and longevity. Mol Cells 41: 65–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatogawa H (2020) Mechanisms governing autophagosome biogenesis. Nat Rev Mol Cell Biol 21: 439–458 [DOI] [PubMed] [Google Scholar]
- Nakatogawa H, Ichimura Y, Ohsumi Y (2007) Atg8, a ubiquitin‐like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 130: 165–178 [DOI] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183: 795–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ (2010) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 8: e1000298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuman MG, French SW, French BA, Seitz HK, Cohen LB, Mueller S, Osna NA, Kharbanda KK, Seth D, Bautista A et al (2014) Alcoholic and non‐alcoholic steatohepatitis. Exp Mol Pathol 97: 492–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ney PA (2015) Mitochondrial autophagy: Origins, significance, and role of BNIP3 and NIX. Biochim Biophys Acta 1853: 2775–2783 [DOI] [PubMed] [Google Scholar]
- Ng SK, Wood JP, Chidlow G, Han G, Kittipassorn T, Peet DJ, Casson RJ (2015) Cancer‐like metabolism of the mammalian retina. Clin Exp Ophthalmol 43: 367–376 [DOI] [PubMed] [Google Scholar]
- Nguyen TN, Padman BS, Usher J, Oorschot V, Ramm G, Lazarou M (2016) Atg8 family LC3/GABARAP proteins are crucial for autophagosome‐lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J Cell Biol 215: 857–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, Rogov V, Lohr F, Popovic D, Occhipinti A et al (2010) Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep 11: 45–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakes JA, Davies MC, Collins MO (2017) TBK1: a new player in ALS linking autophagy and neuroinflammation. Mol Brain 10: 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K (2014) Organellophagy: eliminating cellular building blocks via selective autophagy. J Cell Biol 205: 435–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Kondo‐Okamoto N, Ohsumi Y (2009) Mitochondria‐anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev Cell 17: 87–97 [DOI] [PubMed] [Google Scholar]
- Okatsu K, Koyano F, Kimura M, Kosako H, Saeki Y, Tanaka K, Matsuda N (2015) Phosphorylated ubiquitin chain is the genuine Parkin receptor. J Cell Biol 209: 111–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okatsu K, Oka T, Iguchi M, Imamura K, Kosako H, Tani N, Kimura M, Go E, Koyano F, Funayama M et al (2012) PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat Commun 3: 1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okatsu K, Sato Y, Yamano K, Matsuda N, Negishi L, Takahashi A, Yamagata A, Goto‐Ito S, Mishima M, Ito Y et al (2018) Structural insights into ubiquitin phosphorylation by PINK1. Sci Rep 8: 10382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okatsu K, Uno M, Koyano F, Go E, Kimura M, Oka T, Tanaka K, Matsuda N (2013) A dimeric PINK1‐containing complex on depolarized mitochondria stimulates Parkin recruitment. J Biol Chem 288: 36372–36384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onishi M, Nagumo S, Iwashita S, Okamoto K (2018) The ER membrane insertase Get1/2 is required for efficient mitophagy in yeast. Biochem Biophys Res Commun 503: 14–20 [DOI] [PubMed] [Google Scholar]
- Onoue K, Jofuku A, Ban‐Ishihara R, Ishihara T, Maeda M, Koshiba T, Itoh T, Fukuda M, Otera H, Oka T et al (2013) Fis1 acts as a mitochondrial recruitment factor for TBC1D15 that is involved in regulation of mitochondrial morphology. J Cell Sci 126: 176–185 [DOI] [PubMed] [Google Scholar]
- Ordureau A, Paulo JA, Zhang J, An H, Swatek KN, Cannon JR, Wan Q, Komander D, Harper JW (2020) Global landscape and dynamics of Parkin and USP30‐dependent ubiquitylomes in iNeurons during mitophagic signaling. Mol Cell 77: 1124–1142.e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordureau A, Sarraf SA, Duda DM, Heo JM, Jedrychowski MP, Sviderskiy VO, Olszewski JL, Koerber JT, Xie T, Beausoleil SA et al (2014) Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol Cell 56: 360–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padman BS, Nguyen TN, Uoselis L, Skulsuppaisarn M, Nguyen LK, Lazarou M (2019) LC3/GABARAPs drive ubiquitin‐independent recruitment of Optineurin and NDP52 to amplify mitophagy. Nat Commun 10: 408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacino JJ, Sagi D, Goldberg MS, Krauss S, Motz C, Wacker M, Klose J, Shen J (2004) Mitochondrial dysfunction and oxidative damage in parkin‐deficient mice. J Biol Chem 279: 18614–18622 [DOI] [PubMed] [Google Scholar]
- Palikaras K, Lionaki E, Tavernarakis N (2015) Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans . Nature 521: 525–528 [DOI] [PubMed] [Google Scholar]
- Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM et al (2006) Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441: 1157–1161 [DOI] [PubMed] [Google Scholar]
- Park YY, Nguyen OT, Kang H, Cho H (2014) MARCH5‐mediated quality control on acetylated Mfn1 facilitates mitochondrial homeostasis and cell survival. Cell Death Dis 5: e1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish J, Li L, Klotz K, Ledwich D, Wang X, Xue D (2001) Mitochondrial endonuclease G is important for apoptosis in C. elegans . Nature 412: 90–94 [DOI] [PubMed] [Google Scholar]
- Pesah Y, Pham T, Burgess H, Middlebrooks B, Verstreken P, Zhou Y, Harding M, Bellen H, Mardon G (2004) Drosophila parkin mutants have decreased mass and cell size and increased sensitivity to oxygen radical stress. Development 131: 2183–2194 [DOI] [PubMed] [Google Scholar]
- Phu L, Rose CM, Tea JS, Wall CE, Verschueren E, Cheung TK, Kirkpatrick DS, Bingol B (2020) Dynamic regulation of mitochondrial import by the ubiquitin system. Mol Cell 77: 1107–1123.e10 [DOI] [PubMed] [Google Scholar]
- Pickles S, Vigie P, Youle RJ (2018) Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol 28: R170–R185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell AM, Huang CH, Kennedy SR, Ordureau A, Sideris DP, Hoekstra JG, Harper JW, Youle RJ (2015) Endogenous Parkin preserves dopaminergic substantia nigral neurons following mitochondrial DNA mutagenic stress. Neuron 87: 371–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell AM, Youle RJ (2015) The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson's disease. Neuron 85: 257–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploumi C, Daskalaki I, Tavernarakis N (2017) Mitochondrial biogenesis and clearance: a balancing act. FEBS J 284: 183–195 [DOI] [PubMed] [Google Scholar]
- Politi Y, Gal L, Kalifa Y, Ravid L, Elazar Z, Arama E (2014) Paternal mitochondrial destruction after fertilization is mediated by a common endocytic and autophagic pathway in Drosophila . Dev Cell 29: 305–320 [DOI] [PubMed] [Google Scholar]
- Poole AC, Thomas RE, Yu S, Vincow ES, Pallanck L (2010) The mitochondrial fusion‐promoting factor mitofusin is a substrate of the PINK1/parkin pathway. PLoS One 5: e10054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozzi L, Valenza F, Mosca L, Dal Mas A, Domi T, Romano A, Tarlarini C, Falzone YM, Tremolizzo L, Soraru G et al (2017) TBK1 mutations in Italian patients with amyotrophic lateral sclerosis: genetic and functional characterisation. J Neurol Neurosurg Psychiatry 88: 869–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pracheil T, Liu Z (2013) Tiered assembly of the yeast Far3‐7‐8‐9‐10‐11 complex at the endoplasmic reticulum. J Biol Chem 288: 16986–16997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priault M, Salin B, Schaeffer J, Vallette FM, di Rago JP, Martinou JC (2005) Impairing the bioenergetic status and the biogenesis of mitochondria triggers mitophagy in yeast. Cell Death Differ 12: 1613–1621 [DOI] [PubMed] [Google Scholar]
- Pulsinelli WA, Duffy TE (1983) Regional energy balance in rat brain after transient forebrain ischemia. J Neurochem 40: 1500–1503 [DOI] [PubMed] [Google Scholar]
- Querfurth HW, LaFerla FM (2010) Alzheimer's disease. N Engl J Med 362: 329–344 [DOI] [PubMed] [Google Scholar]
- Raffaello A, Mammucari C, Gherardi G, Rizzuto R (2016) Calcium at the center of cell signaling: interplay between endoplasmic reticulum, mitochondria, and lysosomes. Trends Biochem Sci 41: 1035–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakovic A, Grunewald A, Kottwitz J, Bruggemann N, Pramstaller PP, Lohmann K, Klein C (2011) Mutations in PINK1 and Parkin impair ubiquitination of Mitofusins in human fibroblasts. PLoS One 6: e16746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rana A, Rera M, Walker DW (2013) Parkin overexpression during aging reduces proteotoxicity, alters mitochondrial dynamics, and extends lifespan. Proc Natl Acad Sci USA 110: 8638–8643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasool S, Soya N, Truong L, Croteau N, Lukacs GL, Trempe JF (2018) PINK1 autophosphorylation is required for ubiquitin recognition. EMBO Rep 19: e44981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray R, Chen G, Velde CV, Cizeau J, Park JH, Reed JC, Gietz RD, Greenberg AH (2000) BNIP3 heterodimerizes with Bcl‐2:Bcl‐XL and induces cell death independent of a Bcl‐2 homology 3 (BH3) domain at both mitochondrial and nonmitochondrial sites. J Biol Chem 275: 1439–1448 [DOI] [PubMed] [Google Scholar]
- Regula KM, Ens K, Kirshenbaum LA (2002) Inducible expression of BNIP3 provokes mitochondrial defects and hypoxia‐mediated cell death of ventricular myocytes. Circ Res 91: 226–231 [DOI] [PubMed] [Google Scholar]
- Rehm J, Samokhvalov AV, Shield KD (2013) Global burden of alcoholic liver diseases. J Hepatol 59: 160–168 [DOI] [PubMed] [Google Scholar]
- Richard VR, Leonov A, Beach A, Burstein MT, Koupaki O, Gomez‐Perez A, Levy S, Pluska L, Mattie S, Rafesh R et al (2013) Macromitophagy is a longevity assurance process that in chronologically aging yeast limited in calorie supply sustains functional mitochondria and maintains cellular lipid homeostasis. Aging 5: 234–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter B, Sliter DA, Herhaus L, Stolz A, Wang C, Beli P, Zaffagnini G, Wild P, Martens S, Wagner SA et al (2016) Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci USA 113: 4039–4044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley B, Lougheed J, Callaway K, Velasquez M, Brecht E, Nguyen L, Shaler T, Walker D, Yang Y, Regnstrom K et al (2013) Structure and function of Parkin E3 ubiquitin ligase reveals aspects of RING and HECT ligases. Nat Commun 4: 1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogov VV, Suzuki H, Marinkovic M, Lang V, Kato R, Kawasaki M, Buljubasic M, Sprung M, Rogova N, Wakatsuki S et al (2017) Phosphorylation of the mitochondrial autophagy receptor Nix enhances its interaction with LC3 proteins. Sci Rep 7: 1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojansky R, Cha MY, Chan DC (2016) Elimination of paternal mitochondria in mouse embryos occurs through autophagic degradation dependent on PARKIN and MUL1. Elife 5: e17896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rongvaux A, Jackson R, Harman CC, Li T, West AP, de Zoete MR, Wu Y, Yordy B, Lakhani SA, Kuan CY et al (2014) Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 159: 1563–1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu D, Mouchiroud L, Andreux PA, Katsyuba E, Moullan N, Nicolet‐Dit‐Felix AA, Williams EG, Jha P, Lo Sasso G, Huzard D et al (2016) Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat Med 22: 879–888 [DOI] [PubMed] [Google Scholar]
- Saita S, Shirane M, Nakayama KI (2013) Selective escape of proteins from the mitochondria during mitophagy. Nat Commun 4: 1410 [DOI] [PubMed] [Google Scholar]
- Sakakibara K, Eiyama A, Suzuki SW, Sakoh‐Nakatogawa M, Okumura N, Tani M, Hashimoto A, Nagumo S, Kondo‐Okamoto N, Kondo‐Kakuta C et al (2015) Phospholipid methylation controls Atg32‐mediated mitophagy and Atg8 recycling. EMBO J 34: 2703–2719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, Wang J (2008) Essential role for Nix in autophagic maturation of erythroid cells. Nature 454: 232–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarraf SA, Raman M, Guarani‐Pereira V, Sowa ME, Huttlin EL, Gygi SP, Harper JW (2013) Landscape of the PARKIN‐dependent ubiquitylome in response to mitochondrial depolarization. Nature 496: 372–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarraf SA, Sideris DP, Giagtzoglou N, Ni L, Kankel MW, Sen A, Bochicchio LE, Huang CH, Nussenzweig SC, Worley SH et al (2019) PINK1/Parkin influences cell cycle by sequestering TBK1 at damaged mitochondria, inhibiting mitosis. Cell Rep 29: 225–235.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K, Sato M (2017) Multiple ways to prevent transmission of paternal mitochondrial DNA for maternal inheritance in animals. J Biochem 162: 247–253 [DOI] [PubMed] [Google Scholar]
- Sato M, Sato K (2011) Degradation of paternal mitochondria by fertilization‐triggered autophagy in C. elegans embryos. Science 334: 1141–1144 [DOI] [PubMed] [Google Scholar]
- Sato M, Sato K (2012) Maternal inheritance of mitochondrial DNA: degradation of paternal mitochondria by allogeneic organelle autophagy, allophagy. Autophagy 8: 424–425 [DOI] [PubMed] [Google Scholar]
- Sato M, Sato K (2013) Maternal inheritance of mitochondrial DNA by diverse mechanisms to eliminate paternal mitochondrial DNA. Biochim Biophys Acta 1833: 1979–1984 [DOI] [PubMed] [Google Scholar]
- Sato M, Sato K, Tomura K, Kosako H, Sato K (2018) The autophagy receptor ALLO‐1 and the IKKE‐1 kinase control clearance of paternal mitochondria in Caenorhabditis elegans . Nat Cell Biol 20: 81–91 [DOI] [PubMed] [Google Scholar]
- Sato Y, Okatsu K, Saeki Y, Yamano K, Matsuda N, Kaiho A, Yamagata A, Goto‐Ito S, Ishikawa M, Hashimoto Y et al (2017) Structural basis for specific cleavage of Lys6‐linked polyubiquitin chains by USP30. Nat Struct Mol Biol 24: 911–919 [DOI] [PubMed] [Google Scholar]
- Sauve V, Lilov A, Seirafi M, Vranas M, Rasool S, Kozlov G, Sprules T, Wang J, Trempe J, Gehring K (2015) A Ubl/ubiquitin switch in the activation of Parkin. EMBO J 34: 2492–2505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauve V, Sung G, Soya N, Kozlov G, Blaimschein N, Miotto L, Trempe J, Lukacs G, Gehring K (2018) Mechanism of parkin activation by phosphorylation. Nat Struct Mol Biol 25: 623–630 [DOI] [PubMed] [Google Scholar]
- Scheibye‐Knudsen M, Fang EF, Croteau DL, Wilson DM 3rd, Bohr VA (2015) Protecting the mitochondrial powerhouse. Trends Cell Biol 25: 158–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder K, Tschopp J (2010) The inflammasomes. Cell 140: 821–832 [DOI] [PubMed] [Google Scholar]
- Schubert AF, Gladkova C, Pardon E, Wagstaff JL, Freund SMV, Steyaert J, Maslen SL, Komander D (2017) Structure of PINK1 in complex with its substrate ubiquitin. Nature 552: 51–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuldiner M, Collins SR, Thompson NJ, Denic V, Bhamidipati A, Punna T, Ihmels J, Andrews B, Boone C, Greenblatt JF et al (2005) Exploration of the function and organization of the yeast early secretory pathway through an epistatic miniarray profile. Cell 123: 507–519 [DOI] [PubMed] [Google Scholar]
- Schuldiner M, Metz J, Schmid V, Denic V, Rakwalska M, Schmitt HD, Schwappach B, Weissman JS (2008) The GET complex mediates insertion of tail‐anchored proteins into the ER membrane. Cell 134: 634–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, Kundu M, Opferman JT, Cleveland JL, Miller JL et al (2007) NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci USA 104: 19500–19505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Q, Yamano K, Head BP, Kawajiri S, Cheung JT, Wang C, Cho JH, Hattori N, Youle RJ, van der Bliek AM (2014) Mutations in Fis1 disrupt orderly disposal of defective mitochondria. Mol Biol Cell 25: 145–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba‐Fukushima K, Imai Y, Yoshida S, Ishihama Y, Kanao T, Sato S, Hattori N (2012) PINK1‐mediated phosphorylation of the Parkin ubiquitin‐like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci Rep 2: 1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirakabe A, Fritzky L, Saito T, Zhai P, Miyamoto S, Gustafsson AB, Kitsis RN, Sadoshima J (2016a) Evaluating mitochondrial autophagy in the mouse heart. J Mol Cell Cardiol 92: 134–139 [DOI] [PubMed] [Google Scholar]
- Shirakabe A, Zhai P, Ikeda Y, Saito T, Maejima Y, Hsu CP, Nomura M, Egashira K, Levine B, Sadoshima J (2016b) Drp1‐dependent mitochondrial autophagy plays a protective role against pressure overload‐induced mitochondrial dysfunction and heart failure. Circulation 133: 1249–1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sin J, Andres AM, Taylor DJ, Weston T, Hiraumi Y, Stotland A, Kim BJ, Huang C, Doran KS, Gottlieb RA (2016) Mitophagy is required for mitochondrial biogenesis and myogenic differentiation of C2C12 myoblasts. Autophagy 12: 369–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sliter DA, Martinez J, Hao L, Chen X, Sun N, Fischer TD, Burman JL, Li Y, Zhang Z, Narendra DP et al (2018) Parkin and PINK1 mitigate STING‐induced inflammation. Nature 561: 258–262 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Small GW, Kepe V, Ercoli LM, Siddarth P, Bookheimer SY, Miller KJ, Lavretsky H, Burggren AC, Cole GM, Vinters HV et al (2006) PET of brain amyloid and tau in mild cognitive impairment. N Engl J Med 355: 2652–2663 [DOI] [PubMed] [Google Scholar]
- Song L, McMackin M, Nguyen A, Cortopassi G (2017) Parkin deficiency accelerates consequences of mitochondrial DNA deletions and Parkinsonism. Neurobiol Dis 100: 30–38 [DOI] [PubMed] [Google Scholar]
- Sorrentino V, Romani M, Mouchiroud L, Beck JS, Zhang H, D'Amico D, Moullan N, Potenza F, Schmid AW, Rietsch S et al (2017) Enhancing mitochondrial proteostasis reduces amyloid‐beta proteotoxicity. Nature 552: 187–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinelli JB, Haigis MC (2018) The multifaceted contributions of mitochondria to cellular metabolism. Nat Cell Biol 20: 745–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staropoli JF, McDermott C, Martinat C, Schulman B, Demireva E, Abeliovich A (2003) Parkin is a component of an SCF‐like ubiquitin ligase complex and protects postmitotic neurons from kainate excitotoxicity. Neuron 37: 735–749 [DOI] [PubMed] [Google Scholar]
- Stichel CC, Zhu XR, Bader V, Linnartz B, Schmidt S, Lubbert H (2007) Mono‐ and double‐mutant mouse models of Parkinson's disease display severe mitochondrial damage. Hum Mol Genet 16: 2377–2393 [DOI] [PubMed] [Google Scholar]
- Stolz A, Ernst A, Dikic I (2014) Cargo recognition and trafficking in selective autophagy. Nat Cell Biol 16: 495–501 [DOI] [PubMed] [Google Scholar]
- Stotland A, Gottlieb RA (2016) alpha‐MHC MitoTimer mouse: in vivo mitochondrial turnover model reveals remarkable mitochondrial heterogeneity in the heart. J Mol Cell Cardiol 90: 53–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suen DF, Narendra DP, Tanaka A, Manfredi G, Youle RJ (2010) Parkin overexpression selects against a deleterious mtDNA mutation in heteroplasmic cybrid cells. Proc Natl Acad Sci USA 107: 11835–11840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura A, Nagashima S, Tokuyama T, Amo T, Matsuki Y, Ishido S, Kudo Y, McBride HM, Fukuda T, Matsushita N et al (2013) MITOL regulates endoplasmic reticulum‐mitochondria contacts via Mitofusin2. Mol Cell 51: 20–34 [DOI] [PubMed] [Google Scholar]
- Sun N, Malide D, Liu J, Rovira II, Combs CA, Finkel T (2017) A fluorescence‐based imaging method to measure in vitro and in vivo mitophagy using mt‐Keima. Nat Protoc 12: 1576–1587 [DOI] [PubMed] [Google Scholar]
- Sun N, Yun J, Liu J, Malide D, Liu C, Rovira II, Holmstrom KM, Fergusson MM, Yoo YH, Combs CA et al (2015) Measuring in vivo mitophagy. Mol Cell 60: 685–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutovsky P, Moreno RD, Ramalho‐Santos J, Dominko T, Simerly C, Schatten G (1999) Ubiquitin tag for sperm mitochondria. Nature 402: 371–372 [DOI] [PubMed] [Google Scholar]
- Takahashi D, Moriyama J, Nakamura T, Miki E, Takahashi E, Sato A, Akaike T, Itto‐Nakama K, Arimoto H (2019) AUTACs: cargo‐specific degraders using selective autophagy. Mol Cell 76: 797–810.e10 [DOI] [PubMed] [Google Scholar]
- Takano‐Ohmuro H, Mukaida M, Kominami E, Morioka K (2000) Autophagy in embryonic erythroid cells: its role in maturation. Eur J Cell Biol 79: 759–764 [DOI] [PubMed] [Google Scholar]
- Tal R, Winter G, Ecker N, Klionsky DJ, Abeliovich H (2007) Aup1p, a yeast mitochondrial protein phosphatase homolog, is required for efficient stationary phase mitophagy and cell survival. J Biol Chem 282: 5617–5624 [DOI] [PubMed] [Google Scholar]
- Tamura Y, Endo T (2017) Role of intra‐ and inter‐mitochondrial membrane contact sites in yeast phospholipid biogenesis. Adv Exp Med Biol 997: 121–133 [DOI] [PubMed] [Google Scholar]
- Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ (2010) Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol 191: 1367–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang C, Han H, Yan M, Zhu S, Liu J, Liu Z, He L, Tan J, Liu Y, Liu H et al (2018) PINK1‐PRKN/PARK2 pathway of mitophagy is activated to protect against renal ischemia‐reperfusion injury. Autophagy 14: 880–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Targher G, Lonardo A, Byrne CD (2018) Nonalcoholic fatty liver disease and chronic vascular complications of diabetes mellitus. Nat Rev Endocrinol 14: 99–114 [DOI] [PubMed] [Google Scholar]
- Tay SP, Yeo CW, Chai C, Chua PJ, Tan HM, Ang AX, Yip DL, Sung JX, Tan PH, Bay BH et al (2010) Parkin enhances the expression of cyclin‐dependent kinase 6 and negatively regulates the proliferation of breast cancer cells. J Biol Chem 285: 29231–29238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terskikh A, Fradkov A, Ermakova G, Zaraisky A, Tan P, Kajava AV, Zhao X, Lukyanov S, Matz M, Kim S et al (2000) "Fluorescent timer": protein that changes color with time. Science 290: 1585–1588 [DOI] [PubMed] [Google Scholar]
- Trempe J, Sauve V, Grenier K, Seirafi M, Tang M, Menade M, Al‐Abdul‐Wahid S, Krett J, Wong K, Kozlov G et al (2013) Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science 340: 1451–1455 [DOI] [PubMed] [Google Scholar]
- Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly YM, Gidlof S, Oldfors A, Wibom R et al (2004) Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429: 417–423 [DOI] [PubMed] [Google Scholar]
- Trudeau KM, Gottlieb RA, Shirihai OS (2014) Measurement of mitochondrial turnover and life cycle using MitoTimer. Methods Enzymol 547: 21–38 [DOI] [PubMed] [Google Scholar]
- Tsuboyama K, Koyama‐Honda I, Sakamaki Y, Koike M, Morishita H, Mizushima N (2016) The ATG conjugation systems are important for degradation of the inner autophagosomal membrane. Science 354: 1036–1041 [DOI] [PubMed] [Google Scholar]
- Uddin MN, Nishio N, Ito S, Suzuki H, Isobe K (2012) Autophagic activity in thymus and liver during aging. Age 34: 75–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente EM, Abou‐Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG et al (2004) Hereditary early‐onset Parkinson's disease caused by mutations in PINK1. Science 304: 1158–1160 [DOI] [PubMed] [Google Scholar]
- Valente EM, Bentivoglio AR, Dixon PH, Ferraris A, Ialongo T, Frontali M, Albanese A, Wood NW (2001) Localization of a novel locus for autosomal recessive early‐onset parkinsonism, PARK6, on human chromosome 1p35‐p36. Am J Hum Genet 68: 895–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente EM, Brancati F, Caputo V, Graham EA, Davis MB, Ferraris A, Breteler MM, Gasser T, Bonifati V, Bentivoglio AR et al (2002) PARK6 is a common cause of familial parkinsonism. Neurol Sci 23(Suppl 2): S117–118 [DOI] [PubMed] [Google Scholar]
- Vande Velde C, Cizeau J, Dubik D, Alimonti J, Brown T, Israels S, Hakem R, Greenberg AH (2000) BNIP3 and genetic control of necrosis‐like cell death through the mitochondrial permeability transition pore. Mol Cell Biol 20: 5454–5468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vara‐Perez M, Felipe‐Abrio B, Agostinis P (2019) Mitophagy in cancer: a tale of adaptation. Cells 8: 493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas JNS, Wang C, Bunker E, Hao L, Maric D, Schiavo G, Randow F, Youle RJ (2019) Spatiotemporal control of ULK1 activation by NDP52 and TBK1 during selective autophagy. Mol Cell 74: 347–362.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeriah S, Taylor BS, Meng S, Fang F, Yilmaz E, Vivanco I, Janakiraman M, Schultz N, Hanrahan AJ, Pao W et al (2010) Somatic mutations of the Parkinson's disease‐associated gene PARK2 in glioblastoma and other human malignancies. Nat Genet 42: 77–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa E, Proics E, Rubio‐Patino C, Obba S, Zunino B, Bossowski JP, Rozier RM, Chiche J, Mondragon L, Riley JS et al (2017) Parkin‐independent mitophagy controls chemotherapeutic response in cancer cells. Cell Rep 20: 2846–2859 [DOI] [PubMed] [Google Scholar]
- Wakabayashi T (2002) Megamitochondria formation ‐ physiology and pathology. J Cell Mol Med 6: 497–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Chan C, Weir NR, Denic V (2014) The Get1/2 transmembrane complex is an endoplasmic‐reticulum membrane protein insertase. Nature 512: 441–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Jin M, Liu X, Klionsky DJ (2013) Proteolytic processing of Atg32 by the mitochondrial i‐AAA protease Yme1 regulates mitophagy. Autophagy 9: 1828–1836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, LaVoie MJ, Schwarz TL (2011) PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147: 893–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wauer T, Komander D (2013) Structure of the human Parkin ligase domain in an autoinhibited state. EMBO J 32: 2099–2112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wauer T, Simicek M, Schubert A, Komander D (2015) Mechanism of phospho‐ubiquitin‐induced PARKIN activation. Nature 524: 370–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wauer T, Swatek KN, Wagstaff JL, Gladkova C, Pruneda JN, Michel MA, Gersch M, Johnson CM, Freund SM, Komander D (2015) Ubiquitin Ser65 phosphorylation affects ubiquitin structure, chain assembly and hydrolysis. EMBO J 34: 307–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Chiang WC, Sumpter R Jr, Mishra P, Levine B (2017) Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell 168: 224–238.e10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidberg H, Shvets E, Shpilka T, Shimron F, Shinder V, Elazar Z (2010) LC3 and GATE‐16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J 29: 1792–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welter E, Montino M, Reinhold R, Schlotterhose P, Krick R, Dudek J, Rehling P, Thumm M (2013) Uth1 is a mitochondrial inner membrane protein dispensable for post‐log‐phase and rapamycin‐induced mitophagy. FEBS J 280: 4970–4982 [DOI] [PubMed] [Google Scholar]
- Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, Richter B, Korac J, Waidmann O, Choudhary C et al (2011) Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 333: 228–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JA, Ding WX (2015) A mechanistic review of mitophagy and its role in protection against alcoholic liver disease. Biomolecules 5: 2619–2642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JA, Ni HM, Ding Y, Ding WX (2015) Parkin regulates mitophagy and mitochondrial function to protect against alcohol‐induced liver injury and steatosis in mice. Am J Physiol Gastrointest Liver Physiol 309: G324–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RJ, Drake JC, Cui D, Zhang M, Perry HM, Kashatus JA, Kusminski CM, Scherer PE, Kashatus DF, Okusa MD et al (2017) Conditional MitoTimer reporter mice for assessment of mitochondrial structure, oxidative stress, and mitophagy. Mitochondrion 44: 20–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong HS, Dighe PA, Mezera V, Monternier PA, Brand MD (2017) Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J Biol Chem 292: 16804–16809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YC, Holzbaur EL (2014) Optineurin is an autophagy receptor for damaged mitochondria in parkin‐mediated mitophagy that is disrupted by an ALS‐linked mutation. Proc Natl Acad Sci USA 111: E4439–4448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodroof HI, Pogson JH, Begley M, Cantley LC, Deak M, Campbell DG, van Aalten DM, Whitworth AJ, Alessi DR, Muqit MM (2011) Discovery of catalytically active orthologues of the Parkinson's disease kinase PINK1: analysis of substrate specificity and impact of mutations. Open biology 1: 110012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Chen Q (2015) Hypoxia activation of mitophagy and its role in disease pathogenesis. Antioxid Redox Signal 22: 1032–1046 [DOI] [PubMed] [Google Scholar]
- Wu H, Xue D, Chen G, Han Z, Huang L, Zhu C, Wang X, Jin H, Wang J, Zhu Y et al (2014a) The BCL2L1 and PGAM5 axis defines hypoxia‐induced receptor‐mediated mitophagy. Autophagy 10: 1712–1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Tian W, Hu Z, Chen G, Huang L, Li W, Zhang X, Xue P, Zhou C, Liu L et al (2014b) ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep 15: 566–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Li T, Chen J, Li C, Zhao H, Yao C, Dong H, Wen K, Wang K, Zhao J et al (2018) Autosomal dominant retinitis pigmentosa‐associated gene PRPF8 is essential for hypoxia‐induced mitophagy through regulating ULK1 mRNA splicing. Autophagy 14: 1818–1830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Murata D, Adachi Y, Itoh K, Kameoka S, Igarashi A, Kato T, Araki Y, Huganir RL, Dawson TM et al (2018) Mitochondrial stasis reveals p62‐mediated ubiquitination in Parkin‐independent mitophagy and mitigates nonalcoholic fatty liver disease. Cell Metab 28: 588–604.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamano K, Fogel AI, Wang C, van der Bliek AM, Youle RJ (2014) Mitochondrial Rab GAPs govern autophagosome biogenesis during mitophagy. Elife 3: e01612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamano K, Kikuchi R, Kojima W, Hayashida R, Koyano F, Kawawaki J, Shoda T, Demizu Y, Naito M, Tanaka K et al (2020) Critical role of mitochondrial ubiquitination and the OPTN‐ATG9A axis in mitophagy. J Cell Biol 219: e201912144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamano K, Matsuda N, Tanaka K (2016) The ubiquitin signal and autophagy: an orchestrated dance leading to mitochondrial degradation. EMBO Rep 17: 300–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamano K, Queliconi B, Koyano F, Saeki Y, Hirokawa T, Tanaka K, Matsuda N (2015) Site‐specific interaction mapping of phosphorylated ubiquitin to uncover Parkin activation. J Biol Chem 290: 25199–25211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Gehrke S, Imai Y, Huang Z, Ouyang Y, Wang JW, Yang L, Beal MF, Vogel H, Lu B (2006) Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc Natl Acad Sci USA 103: 10793–10798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonashiro R, Ishido S, Kyo S, Fukuda T, Goto E, Matsuki Y, Ohmura‐Hoshino M, Sada K, Hotta H, Yamamura H et al (2006) A novel mitochondrial ubiquitin ligase plays a critical role in mitochondrial dynamics. EMBO J 25: 3618–3626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Kawane K, Koike M, Mori Y, Uchiyama Y, Nagata S (2005) Phosphatidylserine‐dependent engulfment by macrophages of nuclei from erythroid precursor cells. Nature 437: 754–758 [DOI] [PubMed] [Google Scholar]
- Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, George J, Bugianesi E (2018) Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol 15: 11–20 [DOI] [PubMed] [Google Scholar]
- Yuan Y, Zheng Y, Zhang X, Chen Y, Wu X, Wu J, Shen Z, Jiang L, Wang L, Yang W et al (2017) BNIP3L/NIX‐mediated mitophagy protects against ischemic brain injury independent of PARK2. Autophagy 13: 1754–1766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun J, Puri R, Yang H, Lizzio MA, Wu C, Sheng ZH, Guo M (2014) MUL1 acts in parallel to the PINK1/parkin pathway in regulating mitofusin and compensates for loss of PINK1/parkin. Elife 3: e01958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaffagnini G, Martens S (2016) Mechanisms of selective autophagy. J Mol Biol 428: 1714–1724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Bosch‐Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, Gonzalez FJ, Semenza GL (2008) Mitochondrial autophagy is an HIF‐1‐dependent adaptive metabolic response to hypoxia. J Biol Chem 283: 10892–10903 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhang J, Loyd MR, Randall MS, Waddell MB, Kriwacki RW, Ney PA (2012) A short linear motif in BNIP3L (NIX) mediates mitochondrial clearance in reticulocytes. Autophagy 8: 1325–1332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Xue L, Li L, Tang C, Wan Z, Wang R, Tan J, Tan Y, Han H, Tian R et al (2016a) BNIP3 protein suppresses PINK1 kinase proteolytic cleavage to promote mitophagy. J Biol Chem 291: 21616–21629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Ren H, Xu C, Zhu C, Wu H, Liu D, Wang J, Liu L, Li W, Ma Q et al (2016b) Hypoxic mitophagy regulates mitochondrial quality and platelet activation and determines severity of I/R heart injury. Elife 5: e21407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Yan H, Yuan Y, Gao J, Shen Z, Cheng Y, Shen Y, Wang RR, Wang X, Hu WW et al (2013) Cerebral ischemia‐reperfusion‐induced autophagy protects against neuronal injury by mitochondrial clearance. Autophagy 9: 1321–1333 [DOI] [PubMed] [Google Scholar]
- Zhou Q, Li H, Li H, Nakagawa A, Lin JL, Lee ES, Harry BL, Skeen‐Gaar RR, Suehiro Y, William D et al (2016) Mitochondrial endonuclease G mediates breakdown of paternal mitochondria upon fertilization. Science 353: 394–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Li H, Xue D (2011) Elimination of paternal mitochondria through the lysosomal degradation pathway in C. elegans . Cell Res 21: 1662–1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Massen S, Terenzio M, Lang V, Chen‐Lindner S, Eils R, Novak I, Dikic I, Hamacher‐Brady A, Brady NR (2013) Modulation of serines 17 and 24 in the LC3‐interacting region of Bnip3 determines pro‐survival mitophagy versus apoptosis. J Biol Chem 288: 1099–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziviani E, Tao RN, Whitworth AJ (2010) Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc Natl Acad Sci USA 107: 5018–5023 [DOI] [PMC free article] [PubMed] [Google Scholar]