

Abstract
The renal-outer-medullary-potassium (ROMK) channel, mutated in Bartter’s syndrome, regulates ion exchange in kidney, but its extra-renal functions remain unknown. Additionally, ROMK was postulated to be the pore-forming subunit of the mitochondrial ATP-sensitive K+ channel (mitoKATP), a mediator of cardioprotection. Using global and cardiomyocyte-specific knockout mice (ROMK-GKO and ROMK-CKO respectively), we characterize the effects of ROMK knockout on mitochondrial ion handling, the response to pharmacological KATP channel modulators, and ischemia/reperfusion (I/R) injury. Mitochondria from ROMK-GKO hearts exhibited a lower threshold for Ca2+-triggered permeability transition pore (mPTP) opening but normal matrix volume changes during oxidative phosphorylation. Isolated perfused ROMK-GKO hearts exhibited impaired functional recovery and increased infarct size after I/R injury, particularly when I/R was preceded by an ischemic preconditioning (IPC) protocol. Because ROMK-GKO mice exhibited severe renal defects and cardiac remodeling, we further characterized ROMK-CKO hearts to avoid confounding systemic effects. Mitochondria from ROMK-CKO hearts had unchanged matrix volume responses during oxidative phosphorylation and still swelled upon addition of a mitoKATP opener, but exhibited a lower threshold for mPTP opening, similar to GKO mitochondrial. Nevertheless, I/R induced damage was not exacerbated in ROMK-CKO hearts, either ex vivo or in vivo. Lastly, we examined the response of ROMK-CKO hearts to ex vivo I/R injury with or without IPC and found that IPC still protected these hearts, suggesting that cardiomyocyte ROMK does not participate significantly in the cardioprotective pathway elicited by IPC. Collectively, our findings from these novel strains of mice suggest that cardiomyocyte ROMK is not a central mediator of mitoKATP function, although it can affect mPTP activation threshold.
Keywords: Kcnj1 or Kir1.1 or ROMK, mitochondrial ATP-sensitive potassium channel, ischemic preconditioning, mitochondrial permeability transition pore, renal potassium channel, Bartter’s syndrome
1. Introduction
ATP-regulated potassium channels (KATP) are important metabolic sensors that couple the energetic status of the cell to membrane excitability [1]. KATP channels in cell surface membranes have well-described roles in insulin secretion in pancreatic cells [2], in the regulation of vessel tone in vascular smooth muscle cells, and in the regulation of action potential duration (APD) in cardiac myocytes [3]. By analogy to the surface KATP, mitoKATP channels are present in the inner mitochondrial membrane (IMM) and exhibit overlapping, but not identical, pharmacological properties to the surface KATP channels [4, 5]. In cardiomyocytes, the mitoKATP channel opens when cellular ATP/ADP levels drop, e.g. during high workload or brief ischemic stress, and its role is to maintain mitochondrial matrix volume and preserve energy transfer [6, 7]. Extensive evidence suggests that mitoKATP is centrally implicated in the cardioprotection afforded by diazoxide and a number of other K+ channel openers (KCOs) [8, 9]. Early patch-clamp studies identified an inward rectifier type potassium channel in the mitochondrial inner membrane [10] and reconstitution studies suggested regulation by an associated sulfonylurea receptor protein [11, 12]; however, knockout studies of known plasma membrane KATP subunits yielded inconclusive results [9]. Toward this hypothesis, it has been recently demonstrated that reconstitution of a protein with a previously unknown function (CCDC51) together with a member of the ABC transporter family (ABCB8) form a channel with mitoKATP-like properties in reconstituted membranes [13]. Moreover, knockout of MITOK (CCDC51) resulted in impaired mitochondrial function, altered cristae structure, heightened sensitivity to infarction, and loss of diazoxide-induced cardioprotection, elevating this combination of proteins to the forefront as candidates for mitoKATP.
In a prior study, we used mass spectrometry to analyze the proteome of enriched IMM from bovine heart mitochondria and identified renal outer medullary K channel (ROMK, also known as Kcnj1 or Kir1.1) as a novel mitochondrial K+ channel [14]. Further investigation showed that ROMK localizes to mitochondria owing to a canonical mitochondrial targeting sequence (MTS). Matrix swelling downstream of mitoKATP opening by diazoxide was inhibited by the ROMK toxin tertiapin Q, which also reduced basal thallium (Tl+) transport into mitochondria (a proxy of K+ movement) in H9C2 cardiac myoblasts or neonatal rat cardiomyocytes (NRCM). Furthermore, knockdown of ROMK reduced the initial rate of Tl+ transport into mitochondria and exacerbated cell death in an assay of oxidant-induced cell death, while ROMK overexpression was cytoprotective [14]. Patch-clamp analysis in H9C2 mitoplasts overexpressing ROMK2 corroborated the participation of this isoform in the formation of the mitoKATP channel [15]. In addition to our study, other groups have identified ROMK in mitochondria of dermal fibroblasts [16] and in the cristae of liver and heart mitochondria [17]. ROMK protein was also detected in human embryonic stem cells undergoing maturation into cardiomyocytes [18]. Lastly, tertiapin Q (100–500 nM) was also found to reverse the protective effects of diazoxide in a cardioplegia cardiomyocyte stress model [19]. Thus, available evidence confirms that ROMK is present in cardiomyocytes, but its cardioprotective role, has not been directly tested in the mammalian heart using genetic tools.
ROMK was first identified through functional screening of kidney mRNAs [20]. Isoforms of ROMK are detectable in the thick ascending limb and the distal nephron where they participate in K+ reabsorption and secretion, respectively [21, 22]. Mutations in ROMK have been linked to Bartter’s syndrome type 2, a heritable renal disorder characterized by salt wasting, hypokalemia and low blood pressure [23]. ROMK knockout mice generated by classic gene targeting in embryonic stem (ES) cells (in the 129/SvJ × NIH Black swiss mixed genetic background) recapitulate several features of Bartter’s type 2 and also exhibit poor survival and growth deficits [24–26]. Nevertheless, the implications of ROMK deficiency on cardiac physiology and pathophysiology, and on sensitivity to cardiac ischemia/reperfusion (I/R) injury and ischemic preconditioning (IPC)-mediated cardioprotection have not been investigated.
Here, we utilize CRISPR/Cas9 genome editing to target ROMK in mice (in the pure C57Bl6/J background) and we characterize the phenotype of two distinct mouse mutants that are either globally deficient (GKO) or cardiomyocyte-deficient (CKO) for ROMK. We find that ROMK-GKO mice exhibit striking abnormalities in the renal system, whereas their hearts undergo mild remodeling characterized by lower mass and abnormal electrocardiogram (ECG) waveforms. ROMK-GKO hearts sustained greater cardiac injury after a combined IPC plus I/R protocol ex vivo. In contrast, in ROMK-CKO hearts, IPC-induced protection was intact and in vivo I/R injury did not differ from controls. Mitochondria from GKO hearts showed increased sensitivity to Ca2+ triggered mitochondrial permeability transition pore (mPTP) opening. Similar to the ROMK-GKO mitochondria, the Ca2+ threshold for mPTP activation was lower in mitochondria from ROMK-CKO hearts but the response to a mitoKATP opener was unchanged. Together, these results indicate that cardiomyocyte ROMK is dispensable for cardioprotection and mitoKATP responses, but does impact the sensitivity of the mPTP to Ca2+ activation. ROMK is, therefore, unlikely to be essential for the mitoKATP channel in mouse cardiomyocytes, leaving open the possibility that the recently proposed CCDC51 [13] or ATP synthase subunits [27] can serve as pore forming components of mitoKATP in this cell type.
2. Materials and Methods
See supplement for detailed materials and methods
3. Results
3.1. Generation of ROMK deficient mouse strains with CRISPR/Cas9 genome editing
The CRISPR/Cas9 strategy for generating the germline KCNJ1−/− knockout mouse (ROMK-GKO), the KCNJ1 floxed (KCNJ1fl/fl) mouse and the cardiomyocyte-specific knockout (ROMK-CKO; KCNJ1fl/fl × α-MHC-Cre mouse) are described in detail in the Supplemental Materials, along with full characterization of the mutations and mouse lines obtained (Suppl. Figures 1–12).
Consistent with previous reports [24, 25] the ROMK-GKO mice created herein exhibit early-onset features of kidney dysfunction, poor postnatal survival and severe kidney damage in the adult stage. A detailed description of the kidney, bladder, urine, blood and other phenotypes is included in the supplemental text accompanying this article and in Supplemental Figures 13–19.
3.2. Newborn ROMK-GKO mice display mild cardiac remodeling without overt cardiac dysfunction
To rule in or out preexisting cardiac insufficiency as a possible cause of early mortality, we examined the structure and function of the heart in WT and ROMK-GKO neonates using echocardiography. We found that the left-ventricle dimension was slightly smaller in GKO neonates (Suppl. Figure 14A–B) while contractility (measured by EF and FS) and heart rate remained unaltered (Suppl. Figure 14C–D). Moreover, no gross histological abnormalities were identified in the ROMK-GKO myocardium that would indicate dilated cardiomyopathy or excess fibrosis (Suppl. Figure 14E). Thus, despite growth differences the overall function is preserved and it is unlikely to have contributed to the observed early lethality.
3.3. Cardiac Phenotype of adult ROMK-GKO mice
Next, we examined the baseline cardiac phenotype of the adult GKO mice. M-mode echocardiography on conscious mice revealed a small decrease in left ventricle (LV) inner dimension (LVID;d) in ROMK-GKO mice (Figure 1A–B). This mild structural defect was not associated with any functional abnormalities detectable by echocardiography; fractional shortening (FS), ejection fraction (EF) and heart rate (HR) were similar between WT and ROMK-GKO mice (Figure 1C–D). The ratio of cardiac mass normalized to tibia length was slightly, but significantly, decreased in ROMK-GKO mice (Figure 1E). Histological analysis did not reveal any significant alterations at the macro and microscopic levels, and Masson’s trichrome staining did not detect overt cardiac interstitial or perivascular fibrosis (Figure 1F–G). Transcript analysis showed that ROMK mRNA was absent from adult ROMK-GKO hearts (Figure 1H). Moreover, mRNA analysis for markers of fibrosis and myocardial remodeling was not significantly altered in ROMK-GKO hearts, apart from a moderate upregulation of alpha-smooth muscle actin (aSMA) (Figure 1I).
Figure 1. Baseline cardiac phenotype characterization in adult WT and ROMK-GKO mice.
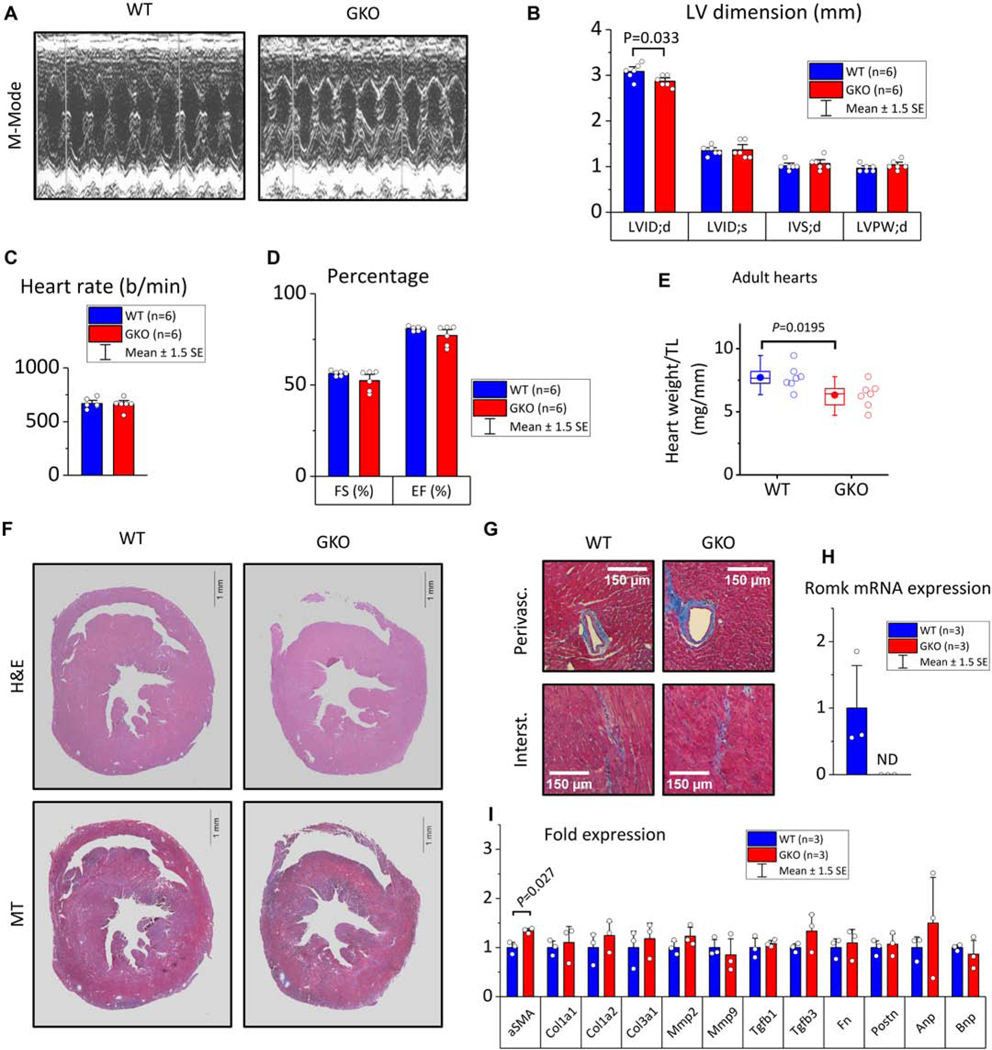
(A) Representative M-mode echocardiograms at the papillary muscle level, (B) Left-ventricle cavity dimensions and wall thickness in diastole (d) and systole (s). Starting from left: LVID;d left ventricle inner dimension in diastole, LVID;s left ventricle inner dimension in systole, IVS;d inter ventricular septum (thickness) in diastole, LVPW;d left ventricle posterior wall (thickness) in diastole. (C-D) Heart rate and the contractility indexes fractional shortening (FS) and ejection fraction (EF), (E) Morphometric assessment of cardiac mass in adult WT and GKO mice. HW; heart weight, TL; tibia length, n=7 mice per group. Boxes cover the 25–75% range of data, the mean is shown as a solid dot and the median as a line. Whiskers cover the 5–95% range, (F) Macroscopic appearance of WT and GKO hearts at the short axis. H&E, hematoxylin and eosin staining to visualize nuclei and cytoplasm, scale bar is 1 mm (G) Detailed microscopic appearance of WT and GKO myocardium stained with Masson’s trichrome to visualize perivascular (upper two panels) or interstitial (lower two panels) collagen deposition. Scale bars represent 150 μm. (H) Quantification of Romk expression in whole heart homogenate with real-time quantitative PCR in WT and GKO hearts. ND: not determined due to sub-detection amount, (I) qPCR analysis in WT and GKO hearts to assess expression across a panel of genes implicated in cardiac hypertrophy, remodeling and fibrosis (n=3 samples per genotype, two tailed t-tests used to assess statistical significance at P<0.05).
Additionally, we examined the function of ROMK-GKO hearts in situ by invasive hemodynamic pressure-volume (PV) analysis (Figure 2 and Supplemental Table 13). We found that the end-systolic (Pes) as well as the developed pressures were significantly decreased in ROMK-GKO mice (Figure 2A–C). The end-diastolic pressure, however, was unchanged (Figure 2D) as well as several other parameters of diastolic function (Supplemental Table 13). The observed decrease in Pes and increase in stroke volume in the ROMK-GKO hearts indicates that arterial elastance (Ea = Pes/SV) is decreased, perhaps reflecting decreased peripheral vascular resistance and lower blood pressure. Interestingly, examination of preload-independent indexes of contractility such as ESPVR (end-systolic pressure volume relationship, slope created by varying preload) and dp/dtmax/iP (peak rate of pressure increase normalized by instantaneous pressure) revealed a significant increase in contractility of the ROMK-GKO hearts (Figure 2E–F). In addition, electrocardiograms (ECGs) taken from lightly anesthetized WT and ROMK-GKO mice displayed abnormal waveform patterns (Figure 2G, M shapes resembling bundle branch block). In these ECGs, the overall heart rate appears to be unaffected, but the R wave amplitude tends to be lower in GKO hearts (Figure 2H–I).
Figure 2. Hemodynamics and electrophysiology in adult WT and ROMK-GKO mice (GKO).

(A) Representative pressure-volume (PV) loops obtained by in situ LV catherization and vena cava occlusion for progressively decreased preload. (B-D) Preload-dependent parameters of contractility in WT vs. GKO animals (E-F) Preload-independent parameters of contractility. Pes; pressure at end-systole, Ped; pressure at end-diastole, Pdev; developed pressure, Espvr; end-systolic pressure-volume relationship (preload independent), dp/dtmax/iP; dp/dt max normalized by instantaneous pressure (preload independent). n=3 for WT and GKO mice, Indicated P values are from two tailed t-tests (G) Representative electrocardiograms (ECG) obtained with a configuration-II surface lead placed on lightly anesthetized mice. The ECG in GKO animals appears strikingly different from WT presenting similarities to a right bundle branch block. (H-I) R-R interval and R wave amplitude in WT vs. GKO animals, n=9 WT and n=4 for GKO mice, P values are from two tailed t-tests. In the scatter box plots, boxes cover the 25–75% range of data, the mean is shown as a solid dot and the median as a line. Whiskers cover the 5–95% range.
3.4. Functional alterations in isolated mitochondria from ROMK-GKO mouse hearts
The kinetics of mitochondrial oxidative phosphorylation can be assessed by the addition of ADP to mitochondria respiring in state 4. During adenine nucleotide phosphorylation, a transient decrease in ΔΨm is observed, accompanied by an increase (matrix contraction) and decrease (swelling) in light scattering (Figure 3A–3B), which may reflect changes in transmembrane K+ flux. Therefore, we examined whether the absence of ROMK affects reversible matrix contraction in mitochondria phosphorylating limiting amounts of ADP. We found that the ADP effects on matrix contraction and re-swelling were similar between WT and ROMK-GKO mitochondria (Figure 3A–B), suggesting unaltered bioenergetic function or phosphorylation-coupled ion homeostasis. Mitochondrial calcium overload and aberrant ROS production are linked to mitochondrial dysfunction and I/R injury. Isolated heart mitochondria from ROMK-GKO mice showed a lower threshold for calcium-dependent activation of the mitochondrial permeability transition pore (mPTP) compared to WT mitochondria (Figure 3C–E). Additionally, the rate of ROS emission from either mitochondrial complex I or complex III was not significantly different between WT and ROMK-GKO mitochondria under either forward or reverse electron flow conditions (Figure 3F–H).
Figure 3. Multiparametric analysis of mitochondrial function in isolated mitochondria from WT and GKO hearts.

(A-B) Representative results of mitochondria matrix contraction and recovery in respiring mitochondria from WT and GKO mitochondria (left panel and right panel respectively). Changes in matrix volume are monitored by light scattering (absorbance at 540 nm, black trace). Additionally recorded is mitochondrial membrane potential (determined from ratiometric measures of TMRM at ex./emm. 546/590 and 573/590, red trace) and NADH oxidation/reduction (ex./emm. 340/450 nm, blue trace). The incubation medium contains 0.5 mg mitochondria and 5 mM each of the substrates glutamate, malate and succinate. The medium also contains ~150 mM K+. At the indicated times (arrows), 200 μM ADP are added to the incubation medium and changes in mitochondrial light scattering are recorded along with the other two bioenergetic parameters. (C) Aliquots of mouse heart mitochondria (0.5 mg) were added to 2.0 ml assay buffer containing 100 nM Calcium Green 5N, 300 nM TMRM, and 5 mM each of glutamate and malate (G/M). The first calcium addition is 25 μM and each subsequent addition is 10 μM. Calcium green fluorescence units were converted to free [Ca2+] using a calibration curve constructed from separate experiments using identical incubation and acquisition conditions in the presence of depolarized mitochondria. Multiple known calcium additions were performed and free extramitochondrial [Ca2+] was calculated using MaxChelator to account for the presence of EGTA in the buffer. The curve was fitted to the equation [Ca2+]=kd(F-Fmin)/(Fmax-F), where F is fluorescence intensity of Calcium Green 5N (ex./emm. 505/535 nm). From this equation the apparent dissociation constant of calcium green for Ca was found to be kd = 27.03. Representative Calcium Green traces from 7 and 6 runs for WT and GKO mitochondria are shown. The end of the experiment is determined by the onset of mitochondrial permeability transition evident by rapid efflux of calcium into the incubation medium. (D-E) Per addition and total Ca taken up by energized mitochondria. Results are reported as nanomoles of taken up Calcium normalized to 1 mg mitochondrial protein. (F-G) Representative traces of Amplex Red fluorescence (ex./emm. 530/590 nm) to monitor mitochondrial H2O2 production in the presence of substrates (added to a final concentration 5 mM) and the inhibitors Rotenone and Antimycin A (final concentration 500 nM each). The reaction contained contains 100 μg mitochondria and rates of H2O2 production were determined from the linear part of each stage of the experiment (H) Summary of H2O2 production in WT and GKO mitochondria energized with substrates and inhibitors to manipulate ROS production from complex I or complex III.
3.5. Response of isolated-perfused ROMK-GKO hearts to ischemia-reperfusion injury
MitoKATP is cardioprotective in the context of ischemia-reperfusion injury and is thought to be a mediator of ischemic preconditioning, where the channel can be activated by short ischemia-reperfusion cycles or by KATP channel opener drugs. Using a constant flow configuration, we exposed hearts to I/R (30 minutes global ischemia and 1 hr reperfusion) or IPC (3 cycles, 5 minutes each of ischemia and 5 reperfusion) followed by I/R (Supplemental Fig. 20A–B). Hemodynamic parameters (systolic, diastolic and coronary perfusion pressures, heart rate and indices of contractility (dp/dtmax) and relaxation (dp/dtmin) were monitored throughout each experiment and infarct size was determined at the end of reperfusion (Supplemental Fig. 20C). LV function, monitored as rate-pressure product (RPP) was similar between WT and ROMK-GKO hearts throughout baseline perfusion (Figure 4A). ROMK-GKO hearts exhibited a slight, but not significant, impairment of post-ischemic recovery of RPP in the I/R protocol (Figure 4A). However, when the duration of ischemia was extended to include the 3 cycles of IPC, reperfused ROMK-GKO hearts exhibited significantly lower RPP recovery than WT (Figure 4B). Side-by-side comparison within each genotype revealed a trend for improvement in post-ischemic RPP recovery by IPC in WT hearts but a worsening in ROMK-GKO hearts (Figure 4C–D). Assessing recovery of function using dp/dtmax showed significantly lower recovery of function in the IPC-treated ROMK-GKO hearts compared to WT hearts (I/R or IPC-treated) (Figure 4E). In addition to recovery of function, we also examined infarct size by staining with TTC after the 60 minutes of reperfusion (Figure 4F). From this analysis, we found that reperfused ROMK-GKO hearts tend to have bigger infarct size and this is further exacerbated by IPC (Figure 4G).
Figure 4. Responses of WT and GKO to ex vivo IR with or without IPC.

(A) Across-protocol monitoring of rate pressure product (RPP) in the LV of hearts subjected to 35 min baseline perfusion and followed by 30 min. global ischemia (grey area) and 1 hr. of reperfusion (I/R protocol). (B) Across-protocol monitoring of RPP in perfused hearts subjected to 20 min baseline perfusion and followed by 3 cycles of 5min. ischemia and 5 min. of reperfusion (IPC protocol). The heart was then subjected to 30 min. global ischemia and 1 hr. of reperfusion. Dots represent within-group means and whiskers are standard errors of the mean. n indicates a different heart preparation randomly assigned to either the IR and IPC protocol. Comparisons of post-ischemic RPP recovery between WT and GKO groups were performed using 2way repeated measures ANOVA followed by Bonferroni’s multiple comparisons test (significance threshold at P< 0.05). (C-D) Re-plotting data from A and B to examine the effects of IPC on post-ischemic RPP recovery within each genotype. The x axis is shortened to show only the post-ischemic phase and the maximum for the y axis is changed to 17500 mmHg.beats.min-1. (E) Post-ischemic recovery of cardiac contractility assessed by dp/dtmax in the four experimental groups. * P<0.05 for WT IR vs GKO IPC and # P<0.05 for WT IPC vs GKO IPC by 2way repeated measures ANOVA and Bonferroni’s multiple comparisons test. (F) Representative photos of heart slices stained with TTC at the end of reperfusion. Infarct areas are shown delineated with yellow line. (G) Infarct size (%) across the four experimental groups. Boxes cover the 25–75% range of data, the mean is shown as a solid circle and the median as a line. Whiskers cover the 5–95% range. Data outside the range of 1.5-fold the interquartile range (1.5 IQR) were considered outliers and removed from the analysis. Means comparisons were performed with 2way ANOVA followed by Bonferroni’s post hoc tests and considered significant at the P<0.05 threshold.
To assess the effect of ROMK deficiency on I/R injury that exclusively focuses on the myocytes and permits measurement of ΔΨm responses, we employed a previously described coverslip ischemia/reperfusion injury model on neonatal mouse ventricular myocyte monolayers [28]. Mouse cardiomyocytes from 1–2-day old WT and ROMK-GKO mice underwent simulated ischemia by applying a glass coverslip over a region of cells for 1 hr, followed by removal of the coverslip and monitoring for another 1 hr to simulate reperfusion. The overall course of mitochondrial membrane potential loss and recovery (measured as TMRM dispersion) was not significantly altered in ROMK-GKO myocytes, either during ischemia or after reperfusion (Supplemental Fig. 21A and D). Similarly, intramitochondrial calcium overload (reported by mito-GCaMP6 fluorescence), and oxidation of the intramitochondrial glutathione pool (reported by the mito-Grx ratio), were not significantly impacted in ROMK-GKO myocytes undergoing this form of injury (Supplemental Fig. 21B–D).
3.6. Characterization of conditional cardiomyocyte-specific knockout: ROMK-CKO mice
Mice from both the F.23 and F.582 KCNJ1 flox lines were bred to α-Myosin Heavy Chain promoter-driven Cre mice (MHCcre, Jax laboratories stock No. 01103, background C57BL6J) to produce cardiomyocyte-specific ROMK knockout mice (ROMK-CKO). Examination of the recombination efficiency by quantitative PCR showed a nearly 90% recombination in the genome of myocytes isolated from mice homozygous for the conditional allele and positive for MHCcre (Suppl. Figure 11C). As with ROMK-GKO mice, to avoid any founder effects that might be related to CRISPR-related off-target events, we utilized mice from more than one founder that were collectively used as ROMK-CKO mice. Littermate KCNJ1fl/fl homozygous mice lacking the MHCcre transgene were used as controls and when available, we also used mice resulting from the same strains that were not floxed but were carriers of the MHCcre transgene (both genotypes collectively used as Ctrl, of which 10.3% were MHCcre-only controls). In contrast to ROMK-GKO mice, ROMK-CKO mice did not exhibit any survival or growth deficiencies, and no abnormalities were detected in kidney, bladder, spleen or brain (Supplemental Figure 22A–D). Moreover, the heart-rate and ECG appearance were normal in ROMK-CKO mice (Supplemental Figure 22E–H). Additionally, the examination of heart weight did not reveal statistically significant changes between the two groups (Supplemental Figure 22I).
3.7. Functional assessment of isolated mitochondria from ROMK-CKO mouse hearts
Next, we assessed matrix contraction and recovery during oxidative phosphorylation by adding non-saturating aliquots of ADP to suspensions of Control and CKO mitochondria. Additions of ADP reproducibly induced the expected contraction and recovery in mitochondrial volume, but these did not differ between Control and ROMK-CKO mitochondria (Figure 5A–B).
Figure 5. Genetic and pharmacological analysis of mitochondrial matrix contraction and recovery in isolated Control and CKO mitochondria.

(A-B) Matrix contraction and recovery and concomitant monitoring of ΔΨm during mitochondrial respiration in Control and CKO mitochondria. Respiration is driven by sequential additions of ADP (200 μM). The reaction contained 1.0 mg mitochondria protein, 5 mM each of the substrates glutamate and malate and 300 nM TMRM. The K+- based incubation medium consists of 120 mM KCl, 10 mM NaCl, 2 mM MgCl2.6H2O, 2 mM KH2PO4, 20 mM MOPS, 0.7 mM CaCl2.2H2O and 1 mM EGTA, pH adjusted to 7.2 with 6 M KOH. Matrix contraction and recovery in response to ADP addition is closely similar between Control and CKO mitochondria. (C-D) Matrix contraction assay first with sequential additions of ADP followed by pharmacological activation and inhibition of mitoKATP with BMS-191095 and glibenclamide respectively. Treatment with the mitoKATP opener BMS (5 μM, green arrow) causes matrix swelling that is similar between Control and CKO mitochondria and reversible by ADP. Subsequent changes in matrix volume are abolished by the addition of the mitoKATP inhibitor glibenclamide (10 μM, orange arrow) in both groups. Representative traces from three independent observations are shown. (E-F) Matrix contraction assay starting with the addition of BMS-191095, followed by sequential additions of ADP followed by addition of the ROMK-specific inhibitor VU591 (5 μM, purple arrow and bar). The addition of VU591 abolishes matrix recovery in subsequent additions of ADP but it does so similarly in both Control and CKO mitochondria.
To directly determine if mitoKATP responses were impacted in ROMK-CKO mitochondria, we employed the potent mitoKATP channel opener BMS-191095 [29], and the classic mitoKATP inhibitor glibenclamide. As shown in Figure 5C, the BMS compound added to Control mitochondria during state 4 respiration induced mild matrix swelling (decreased light scattering) without depolarizing mitochondrial inner membrane potential. Two sequential additions of ADP elicited the expected matrix contraction and recovery responses, whereas a subsequent addition of glibenclamide induced matrix contraction back to pre-BMS levels (Figure 5C). Matrix volume responses during physiologic ADP-driven respiration (Figure 5D), or in response to the mitoKATP opener BMS191095, were not different in ROMK-CKO mitochondria compared to controls (Figure 5C–F). This indicates that ROMK is not the target of the mitoKATP opener in mouse cardiac mitochondria and that the ADP-mediated contraction and swelling cycle is also independent of ROMK. Additionally, the effects of the sulfonylurea KATP channel inhibitor glibenclamide were not altered by ROMK knockout (Figure 5D). Surprisingly, we found that the putatively specific ROMK-channel blocker VU591 (IC50 240 nM; 5 μM VU591) [30], also caused matrix contraction and mitochondrial depolarization (purple arrow), but this too was unchanged by ROMK knockout (Figure 5E–F), suggesting that this blocker has non-ROMK targets in mitochondria.
We further examined the function of isolated mitochondria from Control and ROMK-CKO mice in their predisposition to undergo mPTP opening. As in the ROMK-GKO mitochondria, isolated energized mitochondria from ROMK-CKO hearts exhibited a decreased threshold for calcium-triggered mPTP activation, evident during the fourth and fifth addition of Ca2+ and in total nmoles Ca2+ taken up by equal amounts of mitochondria (Figure 6A, C and D). Monitoring of the intramitochondrial Ca2+ showed that the earlier triggering of the mPTP was not due to an impaired capacity to buffer Ca2+ (Figure 6B). Finally, we also examined the oxygen consumption rates (OCR) of isolated mitochondria in the presence of glutamate/malate or succinate using respirometry in the absence or presence of ADP. This analysis showed that the respiratory capacity with different substrates and in the absence/presence of ADP was not significantly different between Control and ROMK-CKO mitochondria (Figure 6E–H).
Figure 6. Analysis of Ca2+–induced permeability transition pore (PTP) opening, intramitochondrial Ca2+ handling and respiratory capacity in Control and CKO mitochondria.

Isolated heart mitochondria (1.0 mg mitochondrial protein) were added to a stirred cuvette containing 100 nM Calcium Green 5N, 300 nM TMRM, and 5 mM each of glutamate and malate (G/M) and succinate in 2.0 ml assay buffer. The first calcium addition is 25 μM and each subsequent addition is 10 μM. Calcium green fluorescence units (ex./emm. 505/535 nm) were converted to free [Ca2+] using a calibration curve as described previously. For monitoring intramitochondrial calcium, mitochondria were preloaded with [20 μM] of the ratiometric calcium probe Fura-FF-AM for 25 min. Ratios (R) of Fura-FF fluorescence (ex./emm. 340/510 nm and 380/510 nm for calcium-bound and calcium-free probe respectively) were calculated and results applied to the equation [Ca2+]=kd(R-Rmin)/(Rmax-R) to convert fluorescence to free intramitochondrial [Ca2+]. The kd was calculated by fitting the equation to a calibration curve constructed with multiple pulses of 5mM CaCl2 added to a reaction identical to the one above and also in the presence of 2 μM of the ionophore A23187, 5 μg/ml Oligomycin and 5 μM FCCP. From this experiment we determined the Kd = 16.34 μM. (A) Representative Calcium Green traces converted to extramitochondrial [Ca2+] from Control and CKO mitochondria (blue and red traces respectively). The corresponding effects in intramitochondrial free calcium recorded simultaneously are shown below in panel D. The TMRM signal monitoring ΔΨm was omitted from these graphs for simplicity. The end of the experiment is determined when there is no more mitochondrial uptake and calcium is released into the medium. (B) Representative Fura-FF ratio traces converted to free (unbuffered) intramitochondrial [Ca2+] from Control and CKO mitochondria. The increases in free intramitochondrial calcium correspond to the additions of external calcium shown in A above. (C-D) Per addition and total Ca uptake in Control and CKO mitochondria. Results are reported as nanomoles calcium/mg mitochondrial protein. Each n represents a different mitochondrial preparation obtained from a pool of 3 hearts of the same group. P values indicated above the brackets represent assessment by two tailed t-tests. (E-F) Representative traces of oxygen consumption rate (OCR) normalized to mitochondrial protein concentration. Arrows indicate the sequential additions of substrates (Glutamate/Malate 5 mM each in E, Succinate/Rotenone 5 mM and 1 μM respectively in F), followed by ADP (1 mM), oligomycin (1 μg/ml) and the uncoupler dinitrophenol (DNP, 75 μM). Each datapoint represents the average of 16–24 wells within each group (± SEM) and three time points are collected for each stage of the experiment. Each run is conducted with mitochondria isolated from a combined pool of three hearts of the same genotype group. (G-H) Summarized results of OCR in Control and CKO mitochondria energized by Glutamate/Malate (G) or Succinate/Rotenone (H). Bars represent means ± SEM and each datapoint (open circle) indicates a separate mitochondrial prep obtained from a pool of three hearts of the same genotype (n ranges from 4–8 in the various conditions/groups). No significant differences are observed between the two groups within a given respiratory state.
3.8. Response of isolated-perfused ROMK-CKO hearts to ischemia-reperfusion injury
Control and ROMK-CKO hearts were subjected to ex vivo Langendorff-perfusion followed by I/R injury. Pre- and post-ischemic recovery of function expressed as RPP were not significantly different between Control and ROMK-CKO hearts (Figure 7A and C). Examination of CPP revealed largely similar post-ischemic coronary vascular dysfunction in the Control and CKO hearts (Figure 7B). Post-ischemic recovery of dp/dtmax and dp/dtmin were also not significantly different between Control and CKO perfused hearts (Figure 7D–E). Consistent with the functional outcomes, there were no significant differences in the infarct size between the two groups (Figure 7F and G). To further test this conclusion, we also subjected a cohort of Control and ROMK-CKO mice to in vivo I/R injury by LAD occlusion and reperfusion (45 minutes ischemia followed by 24 hrs of reperfusion) in a genotype-blinded study (performed in the laboratory of Dr. Lefer). The area at risk (AAR) normalized to left ventricle (LV) area was similar between Control and ROMK-CKO groups, confirming that equivalent ischemic stress was applied to both groups (Figure 7H and I). In these conditions, the infarct area (IA) normalized to AAR did not differ between Control and ROMK-CKO groups, indicating that in vivo I/R injury was not impacted by cardiomyocyte-specific ROMK knockout (Figure 7H and I). To examine whether compensatory increases in the expression of antioxidant genes were induced in ROMK-CKO hearts we assessed the expression of an array of relevant genes (Supplemental Figure 22J). There were few changes in gene expression in the CKO hearts, although a group of genes (Ptgs2 aka Cox-2, lipoxygenase and Ccl5) exhibited greater than two-fold upregulation in the CKO hearts.
Figure 7. Response of ROMK Control and CKO hearts subjected to IR injury.

(A) Monitoring of rate pressure product (RPP) in Control and CKO hearts undergoing 35 min baseline perfusion followed by 30 min. global ischemia (grey area) and 1 hr. of reperfusion (I/R protocol). (B) Monitoring of coronary perfusion pressure (CPP). Dots represent within-group means and whiskers are standard errors of the mean. (C) Re-plotting data from A to examine post-ischemic RPP recovery within each genotype. (D-E) Post-ischemic recovery of systolic and diastolic function assessed by dp/dtmax and dp/dtmin respectively. Means comparisons between Control and CKO hearts for the three indexes (RPP, dp/dtmax and dp/dtmin) were performed with 2way repeated measures ANOVA. (F) Representative photos of heart slices stained with TTC at the end of reperfusion. Infarct areas (white) are delineated. (G) Infarct size (%) in Control and CKO hearts. Boxes cover the 25–75% range of data, the mean is shown as a solid circle and the median as a line. Whiskers cover the 5–95% range (see also inset legend). The n values used in the analysis are indicated below each group. Means comparisons were performed with unpaired student’s t-test. (H) Representative photos of heart slices stained with Evans blue and TTC at the end of 24 hrs of in vivo reperfusion (ischemic injury induced by 45 min. of LAD occlusion and reopening in vivo). (I) Area percentages (%) for the two experimental groups. Bars show the mean and whiskers cover the SEM (1.5 coeff.). Unpaired t-tests assuming unequal variance were applied within each category to evaluate differences in means between Ctrl and CKO and P values are indicated. N=9 Ctrl and 8 CKO mice.
3.9. Response of isolated-perfused ROMK-CKO hearts to constant-pressure ischemia-reperfusion injury and ischemic preconditioning (IPC)
In the experiments discussed above using constant flow-perfused hearts (Figure 4), the protective effect of IPC in the WT hearts was marginal, therefore, we pursued an independent strategy to test the responses of ROMK-CKO hearts to I/R injury. Dr. Murphy’s laboratory carried out a genotype-blinded study of I/R (20 minutes ischemia and 90 minutes of reperfusion) and IPC (induced by 4 cycles of 5 minutes each of ischemia and reperfusion after 20 minutes of baseline) or IPC plus I/R in a constant pressure Langendorff configuration. Unblinding occurred only after the protocols were completed and infarct size determinations were made. A significant infarct-sparing effect of IPC was observed in the WT hearts (P=0.05 for I/R vs. IPC + I/R), and that was also the case in the CKO hearts (P=0.04 for I/R vs. IPC + I/R, Figure 8). The fact that infarct size was reduced by IPC in ROMK-CKO hearts suggests that cardiomyocyte ROMK is not required to observe this cardioprotective phenomenon.
Figure 8. Responses of ROMK Control and CKO hearts subjected to ex vivo IR injury with or without IPC.

Infarct size (%) across the four experimental groups of hearts perfused in the constant pressure mode. The I/R perfusion protocol is 20 min baseline perfusion followed by 20 min ischemia and 1.5 hr of reperfusion whereas the IPC + I/R protocol is 20 min baseline followed by 4 cycles of occlusion/reperfusion (5 min each) and then index ischemia and reperfusion as above. Means comparisons were performed with 2way ANOVA followed by Bonferroni’s post hoc tests to detect significant differences between groups as indicated on the graph. Box plots signify the 25–75% range of data and whiskers the 5–95% range of data. The horizonal line inside the box indicates the median and the dot the mean. The mean values are also indicated next to the dot. Data outside the 1.5 IQR were considered outliers and were removed from the analysis.
4. Discussion
This study is the first to evaluate the role of ROMK in heart function, cardiac I/R injury and IPC using novel CRISPR/Cas9-derived germline and cardiomyocyte-specific ROMK knockout mice. The main findings of the study are: 1) ROMK is essential for proper renal function throughout life, including the early perinatal period of development and growth, 2) global ROMK knockout significantly alters myocardial structure and function, reduces heart weight, alters in vivo contractility, and worsens I/R injury, 3) mitochondria from ROMK-GKO or ROMK-CKO hearts exhibit normal responses to ADP-driven matrix contraction and recovery, as well as matrix volume responses to KATP channel modulators, 4) mitochondria from both ROMK-GKO and ROMK-CKO hearts exhibit increased sensitivity to Ca2+ triggered mPTP opening, and 5) the responses to cardiac I/R and the protective effects of IPC are unchanged in ROMK-CKO mice compared to controls.
Elucidating the molecular identity of mitochondrial K+ channels has been the focus of intense research for over two decades, which has undoubtedly expanded our inventory of molecular targets forming mitochondrial K+ channels of variable conductance and modes of regulation (e.g. voltage, ATP, Na+ or Ca2+ [31–33]). Recently, novel additional proteins have been proposed to comprise the mitoKATP channel, including CCDC51, a coiled-coil domain containing mitochondrial protein that forms a non-specific monovalent cation pore when reconstituted in liposomes, which is inhibited by ATP and modulated by diazoxide, glibenclamide and 5-hydroxydecanoate only when co-assembled with a mitochondrial ABC protein (ABCB8) [13]. An alternative proposal has been put forward, implicating the mitochondrial ATP synthase as an energy-coupled K+ uniporter that can be modulated by KATP channel openers and inhibitors [27]. The collective observations presented here indicate that cardiomyocyte ROMK is not a major component involved in the formation of the cardioprotective mitoKATP channel in the mouse heart, which will inform future studies exploring the basic and translational implications of mitochondrial K+ transport. The findings also highlight the effects of global ROMK knockout on the heart, which may have clinical relevance to the pathophysiology of Bartter’s syndrome.
4.1. Systemic impact of global ROMK ablation
The striking phenotype of the ROMK-GKO mice underscores the fundamental importance of maintaining salt and water balance in the nephron during development. For example, early lethality due to disruption of the epithelial Na+ channel subunits, βENaC and γENaC, occurs within 24–48 hours after birth due to electrolyte imbalance and hyperkalemia [34, 35]. The ROMK-GKO mice begin to die within the first five days of life, and a large proportion, but not all, perish by the second week of life. Thus, in contrast to the phenotype observed due to ENac deficiency, the lethality observed with ROMK deficiency occurs later in postnatal life, it is not fully penetrant, and it is associated with the inability to grow rather than acute organ failure. Moreover, the reduction in body size in ROMK-GKO mice is apparently not due to incapacity to receive nourishment, because there is milk in the stomachs of ROMK-GKO mice a few hours after birth and later. A similar phenotype of partial early lethality and growth retardation was also observed in mice deficient for Kcnma1 (the pore-forming subunit of the voltage and Ca2+-activated “BK”-type K+ channel) [36]. In fact, the expression of BK and ROMK channels in the kidney is developmentally coordinated, with ROMK channels expressed first and BK channels following [37, 38]. It is interesting to note that in the early postnatal kidney, low K+ secretion is maintained to preserve K+ levels essential for somatic growth [39]. Given the evidence of renal dysfunction and polyuria observed within the first 5 days of life in ROMK-GKO mice, it is possible that excessive K+ loss early might contribute to an initial growth defect at postnatal days 3–5 that becomes more severe and compromises competition for nourishment and survival.
In addition to kidney, ROMK has been detected in the lower urinary tract, including the ureters and bladder (umbrella cells and muscular layers) [40]. Consistently, a recent study detected ROMK in bladder umbrella cells and the detrusor muscle, and ROMK−/− mice exhibited increased bladder volume and hypertrophy of the urothelium [41]. We observed significant enlargement of the bladder and hydronephrosis in the ROMK-GKO mice that could at least in part be explained by impaired contraction and relaxation in the ureters and bladder [40], in a manner similar to that proposed for BK channels that regulate nerve-evoked bladder contractions [42]. Alternatively, the bladder enlargement could be a direct consequence of excess water filtration through the kidneys. By methylene blue diffusion, we observed no physical obstruction to bladder emptying. Expression of ROMK in the smooth muscle cells of the lower urinary tract raises the possibility that ROMK is also expressed in other groups of smooth muscle cells, including those of blood vessels, which could explain the low, but ubiquitous, expression of ROMK transcripts in tissues outside the renal system [40, 43, 44]. For example, immunohistochemical analysis detected ROMK in rat pulmonary veins proximal to the left atrium [45]. In this regard, it would be interesting to consider whether ROMK has roles in vascular tone regulation. A possible role of ROMK in peripheral vascular tone regulation could explain at least in part the hypotension observed in ROMK−/− mice.
ROMK expression has been reported in glial-like cells of taste buds [46] and also the brain, specifically in the cortex and the hippocampus [47]. Interestingly, our study revealed that ROMK-GKO mice have disproportionally smaller brains (normalized to tibial length) than WT controls, though it remains to be determined whether this effect is due to absence of brain-specific ROMK expression or due to global systemic defects. ROMK is also reported to be functionally expressed during cytokine-induced differentiation of primitive hematopoietic stem cells [48]. In this regard, we found that ROMK-GKO mice have increased platelet number and reduced erythrocyte volume, as well as increased leukocyte counts, which might reflect either a specific defect at a developmental step during commitment to the white blood lineage or an inflammatory response to the systemic abnormalities occurring in ROMK-GKO mice. The splenomegaly we detected in the ROMK-GKO mice might be consistent with the latter. Of note, ROMK transcripts, more specifically the variants ROMK1 and ROMK6, have been detected in spleen of healthy animals [43].
4.2. Cardiac impact of global and conditional ROMK ablation
No bradycardia or ECG abnormalities were noted in the ROMK-GKO neonates, hence, it is unlikely that the early lethality was due to arrhythmic sudden cardiac death. There were, however, significant abnormalities in the surface ECG obtained from adult ROMK-GKO mice. It is likely that these abnormalities are caused by systemic electrolyte imbalances. For example, T wave amplitude changes are often clinically associated with hypo- or hyperkalemia [49, 50]. Consistently, we did not find ECG abnormalities in the cardiomyocyte-specific ROMK-CKO mice that are free from renal dysfunction. Similarly, moderate cardiac structural remodeling was observed in the ROMK-GKO but not ROMK-CKO mice, implying that a systemic defect underlies the observation in GKO hearts. Distributed effects of ROMK deficiency on the cardiovascular system have been previously reported. Heterozygous carriers of ROMK inactivating mutations, or carriers of ROMK single nucleotide polymorphisms (SNPs) are protected from hypertension [51, 52], as are ROMK+/− rats [53]. ROMK−/− mice exhibit reduced blood pressure at baseline [24], consistent with our observations of a decreased afterload and altered arterial elastance in ROMK-GKO mice using in situ hemodynamic pressure-volume measurement. While LV systolic and developed pressures were decreased, preload-independent indexes indicated that intrinsic cardiac contractility was increased (increased ESPVR slope and dp/dtmax/iP). Regardless of whether the underlying mechanisms of increased cardiac contractility in ROMK-GKO mice involves direct effects on cardiomyocytes or are secondary systemic effects of the cardio-renal axis, our findings raise the possibility that short-term ROMK inhibition could increase cardiac inotropy in addition to lowering blood pressure, a combinatorial effect that might have beneficial effects in the context of heart failure and hypertension. In this regard, development and testing of pharmacologic inhibitors of ROMK is already underway for the treatment of hypertension [30, 54–56].
4.3. ROMK and cardioprotection
To test the hypothesis that ROMK underlies mitoKATP activity and serves as a key player in protecting the heart from I/R injury, we exposed isolated perfused ROMK-GKO or ROMK-CKO hearts to I/R injury. An array of LV functional indexes and infarct size assessment indicated that the global absence of ROMK had a detrimental effect in the ex vivo global I/R injury model, which was most striking after a combined IPC plus I/R protocol. Nevertheless, cardiomyocyte-specific ROMK knockout had no effect on either ex vivo or in vivo cardiac I/R injury. The most straightforward interpretation is that additional negative effects are introduced by global ROMK knockout, such as cardiac remodeling secondary to kidney dysfunction, or the possibility that ROMK contributes to protection through non-myocytes in the isolated perfused heart. For example, ROMK may exert cardioprotective effects in the reperfused heart through actions in the coronary circulation [57, 58]. Another alternative to be considered is that chronic ROMK knockout could induce an adaptive protective phenotype through hormesis. We explored one aspect of this possibility by examining antioxidant gene expression using a PCR array, which showed 3 genes changing by more than two-fold, namely, Ptgs2 (cyclooxygenase-2, Cox2), lipoxygenase and Ccl5. The upregulation of Ptgs2 in ROMK-deficient hearts that we observed may be in agreement with the finding that PGE2 (prostaglandin E2) and thromboxane (TXA2), both downstream products of Ptgs2, were found to be upregulated in ROMK−/− mice [59].
While we could not demonstrate a significant effect of ROMK knockout on infarct size or functional recovery after I/R, we were able to detect alterations in important functions in cardiac mitochondria from both the ROMK-GKO and CKO mice. Firstly, we found increased sensitivity for Ca2+-induced mitochondrial permeability transition pore (mPTP) opening in ROMK-deficient mitochondria. While significant, this increased mPTP sensitivity was apparently not sufficient to tip the balance toward a worsened ischemic outcome in either the ROMK-GKO or ROMK-CKO perfused hearts. The underlying causes of increased mPTP sensitivity in the absence of ROMK remain to be determined, although our observations in monitoring intramitochondrial Ca2+ levels rule out a potential defect in Ca2+ buffering [60]. The increased mPTP sensitivity is compatible with previously reported effects of mitoKATP, as it has been shown that mitoKATP opening counteracts mitochondrial Ca2+ overload and mPTP opening [61], yet this shift in mPTP threshold by ROMK knockout is not evidence for a link between ROMK and mitoKATP.
4.4. ROMK and mitoKATP
It has been previously reported that mitoKATP conductance is restricted to very low levels in mitochondria to prevent excess uncoupling [62]. MitoKATP channels are therefore assumed to be rare [62]. From patch clamp analyses on fused giant mitoplasts [10], we estimate the mitoKATP to be at a density of 0.2 mitoKATP channels/μm2 or 1 × 1010 channels/mg liver mitochondrial protein. This is about 125 times lower than estimates of mitochondrial Ca 2+ uniporter (MCU) channels but close to the range of uncoupling protein 2 (UCP2) in the same tissue [63, 64]. It is therefore reasonable to assume that the mitoKATP components are present in low quantities in tissues. We found that ROMK mRNA is expressed in low copy numbers in the heart which is compatible with the requirement of a low abundance molecular component of mitoKATP.
Several assays have been developed to quantify mitoKATP activity based on effects on mitochondrial light scattering and flavoprotein oxidation, or by direct monitoring of K+ fluxes in reconstituted proteoliposomes, planar lipid bilayers and patch clamping mitoplasts [5, 10, 11, 65, 66]. A proposed physiological role for the mitoKATP channel is to reverse matrix contraction caused by oxidative phosphorylation [67]. Tight regulation of matrix volume and preservation of the architecture of the cristae and intermembrane space (IMS) are essential for avoiding respiratory inhibition and to promote efficient energy transfer during substrate oxidation [68, 69]. We examined matrix contraction and recovery in WT and ROMK-GKO mitochondria during oxidative phosphorylation but found that the responses did not differ significantly between the two groups. Similarly, in ROMK-CKO mitochondrial, OxPhos-induced matrix volume regulation was not significantly different from that of Control mitochondria. Moreover, matrix swelling was similar between Control and ROMK-CKO mitochondria in the presence of the potent mitoKATP opener BMS-191095, indicating that ROMK is not the pharmacological target of BMS-191095. Surprisingly, the ROMK specific inhibitor VU591 reversed BMS-mediated matrix swelling in ROMK-CKO mitochondria in a manner similar to glibenclamide. This suggests that even though VU591 is, reportedly, a selective inhibitor of ROMK [30], it could be inhibiting other mitochondrial K+ channels with a similar pore structure [70], or that these inhibitors have off target effects that coincidentally offset the protection conferred by K+ channel openers.
Taken together, the evidence indicates that ROMK is not the pore-forming component of mitoKATP in the mouse heart, despite our earlier findings that its knockdown in H9c2 myoblasts suppressed mitochondrial K+ uptake and responses to KATP modulators [14]. It is possible that the profile of proteins responding to chemical K+ channel openers in mitochondria is cell-type specific and comprised of more than one player. It will be interesting to employ the mitoKATP assays and pharmacological agents described herein to gain-or-loss of function models of the recently described alternative candidates for mitoKATP in future studies.
5. Conclusions
ROMK plays an essential role in kidney development and regulates electrolyte and water homeostasis. Our findings show that ROMK knockout affects multiple extra-renal organs including the bladder, heart, spleen and brain. Cardiac effects include abnormalities in cardiac electrophysiology, contractility and I/R injury. Experiments designed to address the direct actions of ROMK specifically in cardiomyocytes and mitochondria implicate ROMK in Ca2+ induced mPTP opening but do not support a role for ROMK in mitoKATP activity in the mouse heart, or in the mechanism of ischemic preconditioning, negating our initial hypothesis.
Supplementary Material
7. Acknowledgements
This work was supported by an AHA Midwest Affiliate Postdoctoral fellowship (15POST24700006) to KNP and an AHA Scientist Development Grant (12SDG12060056) to DBF. KNP also acknowledges support from NIH grant K12 HL141952. DA is supported by NIH F31 grant HL134198. Work in the lab of EM is supported by ZIA-HL002066. Work in the lab of NP is supported by R01HL136918. Work in the lab of DBF is supported by an AHA Transformational Project Award (18TPA34170575) and R01HL134821. Work in the lab of BOR is supported by grants R01HL137259 and R01HL134821. We thank Chip Hawkins and the Johns Hopkins Transgenic core for embryo microinjections. We thank Agnes Sidor for preparing adenoviruses, Djahida Bedja for performing and analyzing echocardiograms, Guangshuo Zhu for performing and analyzing pressure-volume loops and Ophelia Rogers for performing complete blood counts.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests
The authors declare no competing interests
Statement for the use of animals
All procedures involving animals were approved by the local Institutional Animal Care and Use Committees (IACUC) at Johns Hopkins School of Medicine, the LSUHSC New Orleans and the National Heart Lung and Blood Institute, Bethesda, Maryland.
6. References
- 1.Nichols CG. KATP channels as molecular sensors of cellular metabolism. Nature. 2006;440(7083):470–6. [DOI] [PubMed] [Google Scholar]
- 2.Miki T, Nagashima K, Tashiro F, Kotake K, Yoshitomi H, Tamamoto A, et al. Defective insulin secretion and enhanced insulin action in KATP channel-deficient mice. Proceedings of the National Academy of Sciences. 1998;95(18):10402–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Flagg TP, Enkvetchakul D, Koster JC, Nichols CG. Muscle KATP channels: recent insights to energy sensing and myoprotection. Physiological reviews. 2010;90(3):799–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garlid KD, Paucek P, Yarov-Yarovoy V, Sun X, Schindler PA. The mitochondrial K channel as a receptor for potassium channel openers. Journal of Biological Chemistry. 1996;271(15):8796–9. [DOI] [PubMed] [Google Scholar]
- 5.Liu Y, Sato T, O’Rourke B, Marban E. Mitochondrial ATP-dependent potassium channels: novel effectors of cardioprotection? Circulation. 1998;97(24):2463–9. [DOI] [PubMed] [Google Scholar]
- 6.Costa AD, Garlid KD. MitoK ATP activity in healthy and ischemic hearts. Journal of bioenergetics and biomembranes. 2009;41(2):123–6. [DOI] [PubMed] [Google Scholar]
- 7.O’Rourke B. Myocardial KATP channels in preconditioning. Circulation research. 2000;87(10):845–55. [DOI] [PubMed] [Google Scholar]
- 8.Garlid KD, Paucek P, Yarov-Yarovoy V, Murray HN, Darbenzio RB, D’Alonzo AJ, et al. Cardioprotective effect of diazoxide and its interaction with mitochondrial ATP-sensitive K+ channels: possible mechanism of cardioprotection. Circulation research. 1997;81(6):1072–82. [DOI] [PubMed] [Google Scholar]
- 9.O’Rourke B. Evidence for mitochondrial K+ channels and their role in cardioprotection. Circulation research. 2004;94(4):420–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Inoue I, Nagase H, Kishi K, Higuti T. ATP-sensitive K+ channel in the mitochondrial inner membrane. 1991. [DOI] [PubMed] [Google Scholar]
- 11.Mironova GD, Negoda AE, Marinov BS, Paucek P, Costa AD, Grigoriev SM, et al. Functional distinctions between the mitochondrial ATP-dependent K+ channel (mitoKATP) and its inward rectifier subunit (mitoKIR). Journal of Biological Chemistry. 2004. [DOI] [PubMed] [Google Scholar]
- 12.Bajgar R, Seetharaman S, Kowaltowski AJ, Garlid KD, Paucek P. Identification and properties of a novel intracellular (mitochondrial) ATP-sensitive potassium channel in brain. Journal of Biological Chemistry. 2001;276(36):33369–74. [DOI] [PubMed] [Google Scholar]
- 13.Paggio A, Checchetto V, Campo A, Menabò R, Di Marco G, Di Lisa F, et al. Identification of an ATP-sensitive potassium channel in mitochondria. Nature. 2019:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Foster DB, Ho AS, Rucker J, Garlid AO, Chen L, Sidor A, et al. Mitochondrial ROMK channel is a molecular component of mitoK(ATP). Circulation research. 2012;111(4):446–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laskowski M, Augustynek B, Bednarczyk P, Żochowska M, Kalisz J, O’Rourke B, et al. Single-Channel Properties of the ROMK-Pore-Forming Subunit of the Mitochondrial ATP-Sensitive Potassium Channel. International Journal of Molecular Sciences. 2019;20(21):5323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bednarczyk P, Kicinska A, Laskowski M, Kulawiak B, Kampa R, Walewska A, et al. Evidence for a mitochondrial ATP-regulated potassium channel in human dermal fibroblasts. Biochimica et Biophysica Acta (BBA)-Bioenergetics. 2018;1859(5):309–18. [DOI] [PubMed] [Google Scholar]
- 17.Talanov EY, Pavlik LL, Mikheeva IB, Murzaeva SV, Ivanov AN, Mironova GD. Ultrastructural localization of the ROMK potassium channel in rat liver and heart. Biochemistry (Moscow) Supplement Series A: Membrane and Cell Biology. 2016;10(3):195–8. [Google Scholar]
- 18.Keung W, Ren L, Li S, Wong AO- T, Chopra A, Kong C- W, et al. Non-cell autonomous cues for enhanced functionality of human embryonic stem cell-derived cardiomyocytes via maturation of sarcolemmal and mitochondrial KATP channels. Scientific reports. 2016;6:34154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Henn MC, Janjua MB, Kanter EM, Makepeace CM, Schuessler RB, Nichols CG, et al. Adenosine Triphosphate‐Sensitive Potassium Channel Kir Subunits Implicated in Cardioprotection by Diazoxide. Journal of the American Heart Association. 2015;4(8):e002016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ho K, Nichols CG, Lederer WJ, Lytton J, Vassilev PM, Kanazirska MV, et al. Cloning and expression of an inwardly rectifying ATP-regulated potassium channel. Nature. 1993;362(6415):31–8. [DOI] [PubMed] [Google Scholar]
- 21.Boim MA, Ho K, Shuck ME, Bienkowski MJ, Block JH, Slightom JL, et al. ROMK inwardly rectifying ATP-sensitive K+ channel. II. Cloning and distribution of alternative forms. The American journal of physiology. 1995;268(6 Pt 2):F1132–40. [DOI] [PubMed] [Google Scholar]
- 22.Welling PA, Ho K. A comprehensive guide to the ROMK potassium channel: form and function in health and disease. American Journal of Physiology-Renal Physiology. 2009;297(4):F849–F63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Simon DB, Karet FE, Rodriguez-Soriano J, Hamdan JH, DiPietro A, Trachtman H, et al. Genetic heterogeneity of Barter’s syndrome revealed by mutations in the K+ channel, ROMK. Nature genetics. 1996;14(2):152. [DOI] [PubMed] [Google Scholar]
- 24.Lorenz JN, Baird NR, Judd LM, Noonan WT, Andringa A, Doetschman T, et al. Impaired renal NaCl absorption in mice lacking the ROMK potassium channel, a model for type II Bartter’s syndrome. The Journal of biological chemistry. 2002;277(40):37871–80. [DOI] [PubMed] [Google Scholar]
- 25.Lu M, Wang T, Yan Q, Yang X, Dong K, Knepper MA, et al. Absence of small conductance K+ channel (SK) activity in apical membranes of thick ascending limb and cortical collecting duct in ROMK (Bartter’s) knockout mice. The Journal of biological chemistry. 2002;277(40):37881–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang T. Renal outer medullary potassium channel knockout models reveal thick ascending limb function and dysfunction. Clinical and experimental nephrology. 2012;16(1):49–54. [DOI] [PubMed] [Google Scholar]
- 27.Juhaszova M, Kobrinsky E, Zorov DB, Nuss HB, Yaniv Y, Fishbein KW, et al. ATP synthase K+-and H+-flux drive ATP synthesis and enable mitochondrial K+-uniporter function. bioRxiv. 2019:355776. [Google Scholar]
- 28.Zhou L, Solhjoo S, Millare B, Plank G, Abraham MR, Cortassa S, et al. Effects of regional mitochondrial depolarization on electrical propagation: implications for arrhythmogenesis. Circulation: Arrhythmia and Electrophysiology. 2014;7(1):143–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grover GJ, D’Alonzo AJ, Garlid KD, Bajgar R, Lodge NJ, Sleph PG, et al. Pharmacologic characterization of BMS-191095, a mitochondrial KATP opener with no peripheral vasodilator or cardiac action potential shortening activity. Journal of Pharmacology and Experimental Therapeutics. 2001;297(3):1184–92. [PubMed] [Google Scholar]
- 30.Bhave G, Chauder BA, Liu W, Dawson ES, Kadakia R, Nguyen TT, et al. Development of a selective small-molecule inhibitor of Kir1. 1, the renal outer medullary potassium channel. Molecular pharmacology. 2010:mol. 110.066928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Szabo I, Zoratti M. Mitochondrial channels: ion fluxes and more. Physiological reviews. 2014;94(2):519–608. [DOI] [PubMed] [Google Scholar]
- 32.Laskowski M, Augustynek B, Kulawiak B, Koprowski P, Bednarczyk P, Jarmuszkiewicz W, et al. What do we not know about mitochondrial potassium channels? Biochimica et Biophysica Acta (BBA)-Bioenergetics. 2016;1857(8):1247–57. [DOI] [PubMed] [Google Scholar]
- 33.Smith CO, Nehrke K, Brookes PS. The Slo (w) path to identifying the mitochondrial channels responsible for ischemic protection. Biochemical Journal. 2017;474(12):2067–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McDonald FJ, Yang B, Hrstka RF, Drummond HA, Tarr DE, McCray PB, et al. Disruption of the β subunit of the epithelial Na+ channel in mice: hyperkalemia and neonatal death associated with a pseudohypoaldosteronism phenotype. Proceedings of the National Academy of Sciences. 1999;96(4):1727–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barker PM, Nguyen MS, Gatzy JT, Grubb B, Norman H, Hummler E, et al. Role of gammaENaC subunit in lung liquid clearance and electrolyte balance in newborn mice. Insights into perinatal adaptation and pseudohypoaldosteronism. The Journal of clinical investigation. 1998;102(8):1634–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Halm ST, Bottomley MA, Almutairi MM, Di Fulvio M, Halm DR. Survival and growth of C57BL/6J mice lacking the BK channel, Kcnma1: lower adult body weight occurs together with higher body fat. Physiological reports. 2017;5(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zolotnitskaya A, Satlin LM. Developmental expression of ROMK in rat kidney. American Journal of Physiology-Renal Physiology. 1999;276(6):F825–F36. [DOI] [PubMed] [Google Scholar]
- 38.Woda CB, Miyawaki N, Ramalakshmi S, Ramkumar M, Rojas R, Zavilowitz B, et al. Ontogeny of flow-stimulated potassium secretion in rabbit cortical collecting duct: functional and molecular aspects. American Journal of Physiology-Renal Physiology. 2003;285(4):F629–F39. [DOI] [PubMed] [Google Scholar]
- 39.Satlin LM. Developmental regulation of expression of renal potassium secretory channels. Current opinion in nephrology and hypertension. 2004;13(4):445–50. [DOI] [PubMed] [Google Scholar]
- 40.Spector DA, Yang Q, Klopouh L, Deng J, Weinman EJ, Steplock DA, et al. The ROMK potassium channel is present in mammalian urinary tract epithelia and muscle. American Journal of Physiology-Renal Physiology. 2008;295(6):F1658–F65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim JM, Xu S, Guo X, Hu H, Dong K, Wang T. Urinary Bladder Hypertrophy is Susceptible in Male but Protected in Female ROMK Bartter’s Mouse. American journal of physiology Regulatory, integrative and comparative physiology. 2017:ajpregu 00315 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meredith AL, Thorneloe KS, Werner ME, Nelson MT, Aldrich RW. Overactive bladder and incontinence in the absence of the BK large conductance Ca2+-activated K+ channel. Journal of Biological Chemistry. 2004;279(35):36746–52. [DOI] [PubMed] [Google Scholar]
- 43.Kondo C, Isomoto S, Matsumoto S, Yamada M, Horio Y, Yamashita S, et al. Cloning and functional expression of a novel isoform of ROMK inwardly rectifying ATP-dependent K+ channel, ROMK6 (Kir1.1f). FEBS letters. 1996;399(1–2):122–6. [DOI] [PubMed] [Google Scholar]
- 44.Shuck ME, Bock JH, Benjamin CW, Tsai T- D, Lee KS, Slightom JL, et al. Cloning and characterization of multiple forms of the human kidney ROM-K potassium channel. Journal of Biological Chemistry. 1994;269(39):24261–70. [PubMed] [Google Scholar]
- 45.Michelakis ED, Weir EK, Wu X, Nsair A, Waite R, Hashimoto K, et al. Potassium channels regulate tone in rat pulmonary veins. American Journal of Physiology-Lung Cellular and Molecular Physiology. 2001;280(6):L1138–L47. [DOI] [PubMed] [Google Scholar]
- 46.Dvoryanchikov G, Sinclair MS, Perea-Martinez I, Wang T, Chaudhari N. Inward rectifier channel, ROMK, is localized to the apical tips of glial-like cells in mouse taste buds. The Journal of comparative neurology. 2009;517(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kenna S, Ro J, Ho K, Hebert S, Ashcroft SJ, Ashcroft FM. Differential expression of the inwardly-rectifying K-channel ROMK1 in rat brain. Molecular brain research. 1994;24(1–4):353–6. [DOI] [PubMed] [Google Scholar]
- 48.Shirihai O, Attali B, Dagan D, Merchav S. Expression of two inward rectifier potassium channels is essential for differentiation of primitive human hematopoietic progenitor cells. Journal of cellular physiology. 1998;177(2):197–205. [DOI] [PubMed] [Google Scholar]
- 49.Mattu A, Brady WJ, Robinson DA. Electrocardiographic manifestations of hyperkalemia. The American journal of emergency medicine. 2000;18(6):721–9. [DOI] [PubMed] [Google Scholar]
- 50.Diercks DB, Shumaik GM, Harrigan RA, Brady WJ, Chan TC. Electrocardiographic manifestations: electrolyte abnormalities. The Journal of emergency medicine. 2004;27(2):153–60. [DOI] [PubMed] [Google Scholar]
- 51.Ji W, Foo JN, O’Roak BJ, Zhao H, Larson MG, Simon DB, et al. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nature genetics. 2008;40(5):592–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tobin MD, Tomaszewski M, Braund PS, Hajat C, Raleigh SM, Palmer TM, et al. Common variants in genes underlying monogenic hypertension and hypotension and blood pressure in the general population. Hypertension. 2008;51(6):1658–64. [DOI] [PubMed] [Google Scholar]
- 53.Zhou X, Zhang Z, Shin MK, Horwitz SB, Levorse JM, Zhu L, et al. Heterozygous disruption of renal outer medullary potassium channel in rats is associated with reduced blood pressure. Hypertension. 2013;62(2):288–94. [DOI] [PubMed] [Google Scholar]
- 54.Lewis LM, Bhave G, Chauder BA, Banerjee S, Lornsen KA, Redha R, et al. High-throughput screening reveals a small-molecule inhibitor of the renal outer medullary potassium channel and Kir7. 1. Molecular pharmacology. 2009;76(5):1094–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tang H, Zhu Y, Teumelsan N, Walsh SP, Shahripour A, Priest BT, et al. Discovery of MK-7145, an Oral Small Molecule ROMK Inhibitor for the Treatment of Hypertension and Heart Failure. ACS Med Chem Lett. 2016;7(7):697–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hampton C, Zhou X, Priest BT, Pai L- Y, Felix JP, Thomas-Fowlkes B, et al. The renal outer medullary potassium channel inhibitor, MK-7145, lowers blood pressure, and manifests features of Bartter’s syndrome Type II phenotype. Journal of Pharmacology and Experimental Therapeutics. 2016;359(1):194–206. [DOI] [PubMed] [Google Scholar]
- 57.Heusch G. The coronary circulation as a target of cardioprotection. Circulation research. 2016;118(10):1643–58. [DOI] [PubMed] [Google Scholar]
- 58.Niccoli G, Montone RA, Ibanez B, Thiele H, Crea F, Heusch G, et al. Optimized treatment of ST-elevation myocardial infarction: the unmet need to target coronary microvascular obstruction as primary treatment goal to further improve prognosis. Circulation research. 2019;125(2):245–58. [DOI] [PubMed] [Google Scholar]
- 59.Yan Q, Yang X, Cantone A, Giebisch G, Hebert S, Wang T. Female ROMK null mice manifest more severe Bartter II phenotype on renal function and higher PGE2 production. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology. 2008;295(3):R997–R1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wei AC, Liu T, Winslow RL, O’Rourke B. Dynamics of matrix-free Ca2+ in cardiac mitochondria: two components of Ca2+ uptake and role of phosphate buffering. The Journal of general physiology. 2012;139(6):465–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Holmuhamedov EL, Wang L, Terzic A. ATP‐sensitive K+ channel openers prevent Ca2+ overload in rat cardiac mitochondria. The Journal of physiology. 1999;519(2):347–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Paucek P, Mironova G, Mahdi F, Beavis A, Woldegiorgis G, Garlid K. Reconstitution and partial purification of the glibenclamide-sensitive, ATP-dependent K+ channel from rat liver and beef heart mitochondria. Journal of Biological Chemistry. 1992;267(36):26062–9. [PubMed] [Google Scholar]
- 63.Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. 2004;427(6972):360. [DOI] [PubMed] [Google Scholar]
- 64.Brookes PS, Parker N, Buckingham JA, Vidal-Puig A, Halestrap AP, Gunter TE, et al. UCPs—unlikely calcium porters. Nature cell biology. 2008;10(11):1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Beavis A, Brannan R, Garlid K. Swelling and contraction of the mitochondrial matrix. I. A structural interpretation of the relationship between light scattering and matrix volume. Journal of Biological Chemistry. 1985;260(25):13424–33. [PubMed] [Google Scholar]
- 66.Bednarczyk P, Dołowy K, Szewczyk A. Matrix Mg2+ regulates mitochondrial ATP-dependent potassium channel from heart. FEBS letters. 2005;579(7):1625–32. [DOI] [PubMed] [Google Scholar]
- 67.Kowaltowski AJ, Seetharaman S, Paucek P, Garlid KD. Bioenergetic consequences of opening the ATP-sensitive K+ channel of heart mitochondria. Am J Physiol-Heart C. 2001;280(2):H649–H57. [DOI] [PubMed] [Google Scholar]
- 68.Halestrap AP. The regulation of the oxidation of fatty acids and other substrates in rat heart mitochondria by changes in the matrix volume induced by osmotic strength, valinomycin and Ca2+. Biochemical Journal. 1987;244(1):159–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Halestrap AP. The regulation of the matrix volume of mammalian mitochondria in vivo and in vitro and its role in the control of mitochondrial metabolism. Biochimica et Biophysica Acta (BBA)-Bioenergetics. 1989;973(3):355–82. [DOI] [PubMed] [Google Scholar]
- 70.Swale DR, Sheehan JH, Banerjee S, Husni AS, Nguyen TT, Meiler J, et al. Computational and functional analyses of a small-molecule binding site in ROMK. Biophysical journal. 2015;108(5):1094–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.