

Abstract
Tauopathies are a group of neurodegenerative diseases characterized by hyperphosphorylation of the microtubule-binding protein, tau, and typically feature axon impairment and synaptic dysfunction. Cyclin-dependent kinase5 (Cdk5) is a major tau kinase and its activity requires p35 or p25 regulatory subunits. P35 is subjected to rapid proteasomal degradation in its membrane-bound form and is cleaved by calpain under stress to a stable p25 form, leading to aberrant Cdk5 activation and tau hyperphosphorylation. The type Ib transmembrane protein RPS23RG1 has been implicated in Alzheimer’s disease (AD). However, physiological and pathological roles for RPS23RG1 in AD and other tauopathies are largely unclear. Herein, we observed retarded axon outgrowth, elevated p35 and p25 protein levels, and increased tau phosphorylation at major Cdk5 phosphorylation sites in Rps23rg1 knockout (KO) mice. Both downregulation of p35 and the Cdk5 inhibitor roscovitine attenuated tau hyperphosphorylation and axon outgrowth impairment in Rps23rg1 KO neurons. Interestingly, interactions between the RPS23RG1 carboxyl-terminus and p35 amino-terminus promoted p35 membrane distribution and proteasomal degradation. Moreover, P301L tau transgenic (Tg) mice showed increased tau hyperphosphorylation with reduced RPS23RG1 levels and impaired axon outgrowth. Overexpression of RPS23RG1 markedly attenuated tau hyperphosphorylation and axon outgrowth defects in P301L tau Tg neurons. Our results demonstrate the involvement of RPS23RG1 in tauopathy disorders, and implicate a role for RPS23RG1 in inhibiting tau hyperphosphorylation through homeostatic p35 degradation and suppression of Cdk5 activation. Reduced RPS23RG1 levels in tauopathy trigger aberrant Cdk5-p35 activation, consequent tau hyperphosphorylation, and axon outgrowth impairment, suggesting that RPS23RG1 may be a potential therapeutic target in tauopathy disorders.
Subject terms: Neural ageing, Neurological disorders
Introduction
Tau is a microtubule-binding protein enriched in axons, and functions to stabilize and regulate microtubule structure and neuronal connectivity [1, 2]. Tau comprises multiple phosphorylation sites, and its homeostatic phospho-regulation is essential for physiological tau function [3, 4]. In neurodegenerative tauopathy disorders such as Alzheimer’s disease (AD) and frontotemporal dementia, tau is hyperphosphorylated and accumulates in neuronal cells, thereby leading to impaired microtubule binding and axon dysfunction [5–8].
Cyclin-dependent kinase5 (Cdk5) is a proline-directed serine/threonine protein kinase that plays a significant role in axon growth, synapse development, and memory formation [9–12]. Although Cdk5 is ubiquitously expressed, its kinase activity is restricted in postmitotic neurons and requires interactions with regulatory p35 or p39 subunits for activation [13, 14]. Although both p35 and p39 are expressed in neurons, Cdk5 kinase activity is reduced by 90% in p35 knock out (KO) mouse brain, suggesting that p35 is the primary Cdk5 activator in the brain [15, 16]. The Cdk5-p35 complex phosphorylates cytoskeletal proteins such as tau, CRMP-2, and MAP1B to facilitate axon outgrowth [17–19]. P35 is myristoylated within an amino (N)-terminal glycine (G2) to facilitate its localization to membranes and promote homeostatic p35 proteasomal degradation [20, 21]. Moreover, Ca2+-dependent p35 cleavage by calpain generates a stable p25 fragment that constitutively binds and activates Cdk5 [22]. Under stress conditions such as β-amyloid (Aβ) exposure in AD, neuronal calpain activity is enhanced, thereby inducing p35 cleavage and consequent p25-mediated Cdk5 activation. Given that p25 lacks the N-terminal membrane-associated myristoyl moiety, cytosolic Cdk5-p25 distribution enables phosphorylation of an enhanced number of targets and features an extended half-life compared to Cdk5-p35. Aberrant activation of Cdk5 leads to tau hyperphosphorylation and induces neuronal dysfunction [23, 24].
Cdk5 is relatively stable, while p35 has a short half-life and is subjected to ubiquitin–dependent proteasomal degradation. In addition to myristoylation-mediated degradation mentioned above, p35 S-nitrosylation promotes its ubiquitination and degradation by PJA2 [25]. Furthermore, enhanced p35 turnover through the Sigma-1 receptor has been shown to regulate axon extension [18]. As p35 is a fundamental component in regulating tau phosphorylation during axon growth and neurodegeneration, a comprehensive understanding of p35 regulation may provide new insight into mechanisms underlying the pathogenesis of tauopathy disorders.
RPS23RG1 is a type Ib transmembrane protein abundantly expressed in the central nervous system, including neurons, astrocytes, and microglia. RPS23RG1 levels are found to be reduced in postmortem human AD brain and brain from AD mouse models. The RPS23RG1 transmembrane domain can interact with adenyl cyclases to enhance cAMP/PKA/GSK3β signaling, thereby inhibiting AD-associated amyloid-β (Aβ) generation and tau phosphorylation [26, 27]. The RPS23RG1 carboxyl (C)-terminus can interact with post-synaptic density (PSD) components PSD95 and PSD93 to inhibit their ubiquitination and proteasomal turnover [28]. Although these findings implicate a role for RPS23RG1 dysfunction in AD pathogenesis, how RPS23RG1 maintains physiological brain function and its role in related tauopathy disorders beyond AD has yet to be determined.
Herein, we identify a novel role for RPS23RG1 in tauopathy and provide a mechanism by which RPS23RG1 maintains proper tau phosphorylation and axon outgrowth through p35 interaction to promote homeostatic p35 degradation.
Materials and methods
Animals
Wild type (WT) C57BL/6 mice were obtained from the Xiamen University Laboratory Animal Center (Xiamen, China). Rps23rg1 knockout (KO) mice were generated and maintained as previously described [28]. P301L tau Tg mice were from The Jackson Lab and maintained by crossing a CaMKIIα-tTA line with the Tg(tetO-tauP301L) line to constitutively express human P301L tau primarily in forebrain neurons [29, 30]. Animal experiments were approved by the Animal Ethics Committee of Xiamen University and conducted following the Committee’s guidelines.
DNA constructs and siRNA
Plasmids expressing RPS23RG1 and its truncated forms were generated previously [28]. HA-tagged RPS23RG2 was cloned in the pCMV-HA plasmid (Clontech, Mountain View, CA, USA). Myc-tagged full-length p35 and its mutated and truncated forms, as well as tau and P301L tau were cloned into the pcDNA 3.1-Myc-His plasmid (Invitrogen, Carlsbad, CA, USA).
Scrambled control and p35-targeting siRNAs, as well as their FAM-labeled oligos, were synthesized by GenePharma (Shanghai, China). Their sequences were as follows: p35-siRNA#1 (sense: 5’-GCAAGAACGCCAAGGACAATT-3’, antisense: 5’-UUGUCCUUGGCGUUCUUGCTT-3’), p35-siRNA#2 (sense: 5’-GCAACAUCGCGCAUCUCAATT-3’, antisense: 5’-UUGAGAUGCGCGAUGUUGCTT-3’), p35-siRNA#3 (sense: 5’-CCCACACUAUUUCACACAATT-3’, antisense: 5’-UUGUGUGAAAUAGUGUGGGTT-3’), and scrambled control (sense: 5’-UUCUCCGAACGUGUCACGUTT-3’, antisense: 5’-ACGUGACACGUUCGGAGAATT-3’).
Antibodies and reagents
Antibodies used include anti-pS199 (44–734G), anti-pS396 (44–752G), anti-Tau5 (AHB0042), anti-pT205 (44–738G), and anti-AT8 (MN1020) from Thermo Fisher (Waltham, MA, USA); anti-pS404 (#20194), anti-p35 (#2680), anti-pS9-GSK-3β (#9336), and anti-β-actin (#8457) from Cell Signaling Technology (Danvers, MA, USA); anti-Myc (M20002L) and anti-HA (M20003L) from Abmart (Berkeley Heights, CA, USA); anti-Cdk5 (sc-173) from Santa Cruz Biotechnology (Dallas, TX, USA); anti-GSK-3β (51065-1-AP) from Proteintech (Chicago, IL, USA); anti-ubiquitin (ab7780) from Abcam (Cambridge, UK); and anti-Tuj1 (801201) from BioLegend (San Diego, CA, USA). A monoclonal antibody targeting mouse RPS23RG1 was generated using the RPS23RG1 “QQRNIGYFNHLK” epitope (Sino biological Inc., Beijing, China).
Roscovitine, TDZD-8, cycloheximide, and MG132 were from MedChemExpress (Monmouth Junction, NJ, USA). NH4Cl was from Sigma-Aldrich (Shanghai, China).
Cell culture and transfection
Human HEK293T cells and Hela cells were originally from ATCC (Manassas, VA, USA) and maintained in our laboratory. They were cultured in high-glucose DMEM (Thermo Fisher) with 10% fetal bovine serum (Thermo Fisher). HEK293/tau cells were maintained in the same media with additional 200 μg/mL G418 (Thermo Fisher). Plasmids were transfected into cells using Turbofect transfection reagent (Thermo Fisher), according to the manufacturer’s instructions.
Primary hippocampal neurons were prepared from postpartum day 0 (P0) mouse pups and cultured in neurobasal medium (Thermo Fisher) supplied with 2% B27 (Thermo Fisher) and 1 mM glutamine (Thermo Fisher). Primary neurons were transfected with plasmids using EntransterTM-H4000 transfection reagent (Engreen Biosystem, Beijing, China) at 1 or 3 day in vitro (DIV1 or DIV3), or transfected with siRNAs using Lipofectamine 2000 reagent (Thermo Fisher) at DIV0 or DIV2, following manufacturers’ protocols.
Western blot and co-immunoprecipitation (co-IP)
Proteins in mouse brain tissues or cells were assayed by western blot and/or co-IP as previously described [28]. Protein band intensity was quantified using the ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Immunofluorescence
Immunofluorescence of cells were carried out as previously described [28, 31]. Cell images were acquired by confocal microscopy (FV1000MPE-B, Olympus, Tokyo, Japan). Longest neurites in cultured primary neurons were defined as axon [32, 33]. Axon length was quantified using ImageJ.
Protein degradation assays
Primary neurons derived from Rps23rg1 KO and WT mice and HEK293T cells transfected with RPS23RG1 were treated with cycloheximide for indicated time periods, in the presence or absence of MG132 and NH4Cl.
Membrane and cytosol protein fractionation
Membrane and cytosol proteins were separated using a membrane/cytosol protein extraction kit from Beyotime Institute of Biotechnology (Shanghai, China), and detected by immunoblotting.
Statistics
All data were presented as mean ± SEM (standard error of mean). Statistical analyses were carried out using GraphPad Prism (GraphPad Software, San Diego, CA). Student’s t-test was used to evaluate statistical significance between groups.
Results
Rps23rg1 deletion results in increased tau phosphorylation and impaired axon extension
We previously observed a reduced number of synapses in primary neurons from Rps23rg1 KO mice compared to wild type (WT) control mice [28]. To further study a role for RPS23RG1 in neuronal development, we cultured primary neurons derived from postnatal day 0 (P0) Rps23rg1 KO and WT mice and characterized neurite outgrowth in vitro after 2 (DIV2) or 4 (DIV4) days in culture, where neurons were stained for Tuj-1 to show neuronal morphology. Rps23rg1 KO neurons at DIV2 (Fig. 1a) and DIV4 (Fig. 1b) exhibited significantly shorter axons (the longest neurite) compared to WT neurons. As tau hyperphosphorylation can impair axonal development [6, 18], we assessed the effects of Rps23rg1 deletion on tau phosphorylation. Total and phosphorylated tau levels in primary neurons (Fig. 1c) and hippocampal samples (Fig. 1d) from Rps23rg1 KO and WT mice were compared. Although no significant change in total tau (probed using the Tau5 antibody) was detected in Rps23rg1 KO samples, Rps23rg1 deficiency resulted in dramatically increased tau phosphorylation at major Cdk5 phosphorylation sites, S404 and S202/T205 (probed using the AT8 antibody) [34, 35] (Fig. 1c, d). Although Rps23rg1 deficiency also led to increased tau phosphorylation at two minor Cdk5 phosphorylation sites, S199 and S396, effects were less than observed with S404 and S202/T205 phospho-sites (Fig. 1c, d). These results collectively indicate that RPS23RG1 is important for tau phosphorylation regulation and axon outgrowth.
Fig. 1. Rps23rg1 KO mice feature increased tau phosphorylation and impaired axon elongation.

a, b Hippocampal neurons from WT and Rps23rg1 KO mice at postnatal day 0 (P0) were immunostained for Tuj-1 at 2 days in vitro (DIV2) (a) and DIV4 (b). Immunofluorescence images were obtained by confocal microscopy. Axon length was quantified using ImageJ for comparison. Data represent mean ± SEM (n = 30 neurons from 3 mice per group, with the mean of WT set to one arbitrary units, A.U.). Scale bars = 10 μm. c, d Protein lysates from P0 WT and Rps23rg1 KO mouse hippocampal neurons at DIV2 and DIV4 (c), and P0 and P7 hippocampal tissues (d) were subjected to immunoblotting for the proteins indicated. Protein levels were quantified by densitometry, normalized to β-actin levels, and compared to respective WT controls (set to one A.U., indicated by dashed lines). Data represent mean ± SEM (n = 3 per group). *p < 0.05, ***p < 0.001 (two-tailed Student’s t-test).
Cdk5-p35 is important for RPS23RG1-mediated tau phosphorylation
To elucidate potential mechanisms underlying RPS23RG1-mediated tau phosphorylation, we assayed potential changes in total levels in tau kinases such as GSK-3β and Cdk5. We observed no change in protein levels of these components in Rps23rg1 KO neurons (Fig. 2a) or hippocampal samples (Fig. 2b) compared to WT. However, we observed a slight reduction in phosphorylated GSK-3β (S9) levels in Rps23rg1 KO samples, suggesting that GSK-3β activity may be enhanced with Rps23rg1 deletion (Fig. 2a, b); this is consistent with previous observations that overexpression of RPS23RG1 reduced GSK-3β activity [26, 27]. Interestingly, we found that protein levels of p35 and its cleavage product, p25 were markedly increased in Rps23rg1 KO samples (Fig. 2a, b), implicating potential involvement of the Cdk5-p35 complex in RPS23RG1-mediated tau phosphorylation. To confirm this, we overexpressed RPS23RG1 and p35 individually or together in a HEK293 cell line stably expressing the longest human tau isoform tau441 (HEK293/tau) [36]. As expected, p35 overexpression promoted tau phosphorylation at all sites studied here (S404, S202/T205, S199, and S396), with relatively higher effects on p35 major phosphorylation sites (S404 and S202/T205) compared to minor phosphorylation sites (S199 and S396) (Fig. 2c). RPS23RG1 overexpression attenuated tau phosphorylation at all sites studied in HEK293/tau cells; and its overexpression in p35-overexpressing cells also reversed tau hyperphosphorylation and reduced p35 protein levels (Fig. 2c). In cells overexpressing p35, treatment with the Cdk5-specific inhibitor roscovitine dose-dependently reversed tau phosphorylation mainly at S404 and S202/T205 without affecting p35 levels, whereas treatment with the GSK-3β inhibitor TDZD-8 dose-dependently reversed tau phosphorylation mainly at S199 and S396 (Fig. 2c). Analysis using immunofluorescence imaging also showed that RPS23RG1 overexpression reduced phospho-S404 tau fluorescence in HEK293/tau cells (Fig. 2d), and reversed the elevation of phospho-S404 tau signals induced by p35 overexpression (Fig. 2e). These results suggest that in addition to GSK-3β, the Cdk5-p35 pathway also plays an important role in RPS23RG1-mediated tau phosphorylation.
Fig. 2. RPS23RG1 attenuates tau phosphorylation through the Cdk5/p35 pathway.

a, b Protein lysates from postnatal day 0 (P0) WT and Rps23rg1 KO mouse hippocampal neurons at DIV2 and DIV4 (a), and P0 and P7 hippocampal tissues (b) were subjected to immunoblotting for the proteins indicated. Protein levels were quantified by densitometry, normalized to β-actin levels, and compared to respective WT controls (set to one arbitrary units, A.U., indicated by dashed lines). Data represent mean ± SEM (n = 3). c HEK293/tau cells were transfected with pcDNA3.1-Myc control or p35-Myc vectors for 6 h, and subsequently transfected with pCMV-HA control or HA-RPS23RG1 (RR1) vectors, or treated with increased doses of roscovitine (Ros, 5–25 μM) or TDZD-8 (5–25 μM) for another 18 h. Protein lysates were subjected to immunoblotting for the proteins indicated. Protein levels were quantified for comparison to respective controls (with the mean of control cells set to one A.U.). Data represent mean ± SEM (n = 3). d HEK293/tau cells were transfected with HA-RR1. After immunostaining of HA-RR1 (d1, red) and tau pS404 (d2, green), cells were imaged by confocal microscopy. Three areas in merged images (d3) were enlarged to show pS404 intensity in cells with (d3’’ and d3’’’, indicated by arrows) or without (d3’, indicated by arrowheads) HA-RR1 expression. Intensity of pS404 was quantified. Data represent mean ± SEM (n = 20 cells in each group, with the mean of the intensity in cells with no HA-RR1 expression set to one A.U.). e HEK293/tau cells were co-transfected with HA-RR1 and p35-Myc for 24 h. After immunostaining for HA-RR1 (e1, red), p35-Myc (e2, white), and tau pS404 (e3, green), cells were imaged by confocal microscopy. Two areas in merged images (e4) were enlarged to show pS404 intensity in cells expressing p35-Myc alone (e4’, indicated by arrowheads) and in cells co-expressing HA-RR1 and p35-Myc (e4’’, indicated by arrows). The intensity of pS404 was quantified for comparison. Data represent mean ± SEM (n = 26 cells in each group, with the mean of the intensity in cells with no HA-RR1 and p35-Myc expression set to one A.U.). Scale bars = 10 μm. *p < 0.05, **p < 0.01, ***p < 0.001 (two-tailed Student’s t-test).
RPS23RG1 mediates homeostatic proteasomal p35 turnover
In order to determine how RPS23RG1 regulates p35 levels, we performed quantitative real-time PCR (RT-PCR) analysis to characterize p35 mRNA levels with RPS23RG1 modulation. No significant differences in p35 mRNA levels were detected between Rps23rg1 KO and WT samples (Fig. S1), suggesting that RPS23RG1 does not affect p35 transcription. We then treated hippocampal neuron cultures derived from Rps23rg1 KO and WT mice with cycloheximide (CHX) to inhibit protein synthesis and quantified p35 protein degradation kinetics. We found that p35 degradation was significantly slower in Rps23rg1 KO neurons compared to WT controls (Fig. 3a). In contrast, in RPS23RG1-overexpressing cells, p35 showed faster degradation compared to control cells (Fig. 3b).
Fig. 3. RPS23RG1 promotes proteasomal p35 turnover.

a, b Cultured primary neurons from WT and Rps23rg1 KO mice (a), and HEK293T cells co-transfected with p35-Myc and pCMV-HA (Con) or HA-RPS23RG1 (RR1) (b) were treated with 30 μM cycloheximide (CHX) for the time indicated. P35 levels were determined by immunoblotting, quantified by densitometry, and normalized to β-actin levels (values at the 0 h time point were set to one arbitrary units, A.U.). Data represent mean ± SEM (n = 3). *p < 0.05, **p < 0.01 (two-tailed Student’s t-test). c, d Protein lysates from WT and Rps23rg1 KO mouse hippocampal tissues (c), and HEK293T cells co-transfected with p35-Myc and control (Con), HA-RR1, or HA-RR1△C plasmid for 24 h (d) were subjected to immunoprecipitation (IP) with an antibody against p35. Precipitates were determined by immunoblotting (IB) with an antibody against ubiquitin (Ubiq). Ten percent of IP lysates were used as input and subjected to immunoblotting for the proteins indicated.
Previous studies demonstrate that p35 is primarily degraded through the ubiquitin-proteasome pathway [37, 38]. We also confirmed that the degradation of endogenous p35 (Fig. S2a) and exogenous p35 upon RPS23RG1 co-expression (Fig. S2b) could be inhibited by the proteasomal inhibitor MG-132 but not the lysosomal inhibitor NH4Cl. Moreover, levels of high molecular weight, poly-ubiquitin conjugated p35 immunoprecipitates were reduced in Rps23rg1 KO mouse hippocampal lysates compared to WT controls (Fig. 3c). In contrast, increased high molecular weight, poly-ubiquitin conjugated p35 bands were observed with RPS23RG1 overexpression (Fig. 3d). These findings suggest that RPS23RG1 facilitates p35 turnover through the ubiquitin-proteasome pathway.
RPS23RG1 interacts with p35 and mediates p35 distribution to membranes
P35 is distributed to membranes through N-terminal myristoylation at G2, and membrane localization has been previously shown to be prerequisite for p35 degradation [20, 21]. To determine whether RPS23RG1-dependent p35 turnover requires p35 localization at membranes, we compared p35 levels in the membrane and cytosolic fractions. We found that membrane-bound p35 and soluble p35 levels were significantly decreased and increased, respectively, in Rps23rg1 KO hippocampal neurons compared to WT controls (Fig. 4a). In contrast, RPS23RG1 overexpression significantly increased membrane-associated p35 levels and reduced soluble p35 levels (Fig. 4b).
Fig. 4. RPS23RG1 interacts with p35 to regulate p35 membrane anchoring.

a, b Protein lysates from cultured hippocampal WT and Rps23rg1 KO mouse neurons at DIV4 (a), and of HEK293T cells co-transfected with p35-Myc and control (Con), HA-RR1, or HA-RR1△C plasmid (b) were subjected to cytoplasm (Cyt) and membrane (Mem) fractionation. P35 levels were determined by immunoblotting and quantified by densitometry for comparison. Data represent mean ± SEM (n = 4 for neurons and 3 for HEK293T, with the mean of controls set to one arbitrary units, A.U.). ns p > 0.05, **p < 0.01, ***p < 0.001 (two-tailed Student’s t-test). c Hela cells were co-transfected with HA-RR1 and p35-Myc for 24 h. After immunostaining with antibodies against HA (for RR1, green) and Myc (for p35, red), cells were counterstained with DAPI and observed by confocal microscopy. Scale bars =10 μm. d WT mouse primary neurons were transfected with HA-RR1 at DIV3, and immunostained with antibodies against HA (for RR1, green) and p35 (red) and counterstained with DAPI at DIV4. Images were acquired by confocal microscopy. Scale bars = 10 μm. e Schematic depictions of human RR1 and p35 and their truncated constructs. Full-length RR1 comprises a single transmembrane (TM) domain. RR1△C lacks the carboxyl (C)-terminal intracellular region. P35 is cleaved into an amino (N)-terminal (1–98) N10 fragment and a C-terminal (99–307) p25 fragment by calpain. f–h HEK293T cells were co-transfected with p35-Myc and HA-RR1 (f), p35-Myc and HA-RR1△C (g), or p35-N10-Myc and HA-RR1 (h). Cell lysates were subjected to immunoprecipitation (IP) with antibodies against HA and Myc and control immunoglobulin G (IgG), and immunoblotted (IB) for the components indicated. i WT mouse brain lysates were subjected to IP with an antibody against mouse RR1 and immunoblotted with an antibody against both p35 and p25, or the antibody against RR1.
To elucidate how RPS23RG1 regulates p35 membrane localization, we characterized potential RPS23RG1 interactions with p35. Indeed, exogenously-expressed RPS23RG1 showed marked colocalization with exogenous p35 in Hela cells (Fig. 4c), and with endogenous p35 in hippocampal neurons (Fig. 4d). Moreover, we generated various RPS23RG1 and p35 truncated constructs (Fig. 4e) and assayed binding by co-immunoprecipitation. We found that full-length p35 interacted with full-length RPS23RG1 (Fig. 4f) but not RR1△C that lacks RPS23RG1 intracellular carboxyl-terminal region (Fig. 4g). Full-length RPS23RG1 interacted with a p35 N-terminal fragment (N10) (Fig. 4h). Moreover, we observed that endogenous RPS23RG1 interacted with full-length p35 but not p25 in mouse brain lysates (Fig. 4i), further indicating that the p35 N-terminus is essential for RPS23RG1 interaction. We also found that overexpression of RR1△C had no effect on promoting p35 poly-ubiquitination (Fig. 3d) and p35 membrane distribution (Fig. 4b). Together, these results demonstrate that the RPS23RG1 intracellular domain interacts with the p35 amino-terminus to facilitate p35 membrane anchoring. Interestingly, mutation of the p35 myristoylation site (G2A) did not affect its interaction with RPS23RG1 (Fig. S3), implying that the binding of RPS23RG1 to p35 does not require p35 myristoylation and that membrane-binding of p35 may be facilitated independently through its myristoylation and its interaction with RPS23RG1.
Mouse Rps23rg2 is a functional homolog to Rps23rg1. We found no change in Rps23rg2 expression in Rps23rg1 KO mice compared to controls, suggesting that there are no compensatory changes in Rps23rg2 levels with Rps23rg1 deletion (Fig. S4a). We also found that both mouse RPS23RG1 and RPS23RG2 interacted with p35 (Fig. S4b), and overexpression of both proteins enhanced p35 membrane distribution and reduced p35 levels (Fig. S4c). However, the effects of RPS23RG2 on modulating p35 levels and membrane distribution were weaker compared to mouse and human RPS23RG1 (Fig. S4c). One possible explanation may be that sequences in the longer carboxyl-terminal tail in RPS23RG2 reduces its binding to p35.
P35 downregulation and Cdk5 activity inhibition rescue tau hyperphosphorylation and impaired axon outgrowth in Rps23rg1 KO neurons
To verify that elevated p35 levels can induce tau hyperphosphorylation and axon outgrowth impairment in Rps23rg1 KO mice, we designed three p35-targeting siRNAs and assayed these siRNAs for p35 knockdown efficiency (Fig. 5a). Using two p35-targeting siRNA oligos to deplete p35 in Rps23rg1 KO neurons, we found that tau phosphorylation at major Cdk5 phospho-sites (S404 and S202/T205) was significantly reduced, and tau phosphorylation at minor Cdk5 phospho-sites (S199 and S396) was slightly reduced (Fig. 5b). Furthermore, treatment with roscovitine dose-dependently attenuated tau phosphorylation at S404 and S202/T205, and treatment with TDZD-8 dose-dependently attenuated tau phosphorylation at S199 and S396 (Fig. 5b). More importantly, we found that p35 downregulation significantly ameliorated impaired axon elongation in Rps23rg1 KO neurons (Fig. 5c). Treatment with roscovitine also enhanced axon outgrowth in Rps23rg1 KO neurons (Fig. 5d). In order to confirm that results using siRNAs were not due to off-target effects, we constructed a p35 expression construct comprising silent mutations (SM) refractory to targeting by siRNA #1 oligos. Overexpression of wild type and SM p35 plasmids comparably induced tau hyperphosphorylation in HEK293/tau cells. However, additional transfection of p35 siRNA #1 only reduced p35 levels and tau phosphorylation in cells expressing wild type p35 but not p35 SM mutant (Fig. S5). These results confirm that hyperphosphorylation and axon outgrowth impairment in Rps23rg1 KO mice is triggered by elevated p35 levels and enhanced Cdk5 activation.
Fig. 5. Downregulation of p35 and inhibition of Cdk5 activity attenuate tau phosphorylation and rescue impaired axon outgrowth in Rps23rg1 KO neurons.

a Cultured hippocampal neurons from P0 Rps23rg1 KO mice were transfected with scrambled control (SC) or various p35 siRNAs (#1–3) at DIV0. Three days later, cell lysates were immunoblotted for p35. P35 protein levels were quantified by densitometry, normalized to β-actin levels, and compared to SC (set to one arbitrary units, A.U.). Data represent mean ± SEM (n = 4). b Hippocampal neurons from P0 Rps23rg1 KO mice were transfected with p35 siRNAs (#1 and #2) or SC siRNA for 72 h, or treated with increased doses of roscovitine (Ros, 5–25 μM) and TDZD-8 (5–25 μM) for 24 h. Cells lysates were immunoblotted for the proteins indicated. Protein levels were quantified and compared to SC siRNA (set to one A.U.). Data represent mean ± SEM (n = 3). c, d Hippocampal neurons from P0 Rps23rg1 KO mice were transfected with SC and p35 siRNAs (c), or treated with Ros (5 and 25 μM) (d) at DIV0. At DIV2 or DIV4, cells were immunostained for Tuj-1. Immunofluorescence images were acquired by confocal microscopy. Axon length was quantified using ImageJ for comparison. Data represent mean ± SEM (n = 30 neurons from 3 mice per group). Scale bars = 10 μm. ns p > 0.05, *p < 0.05, **p < 0.01, ***p < 0.001 (two-tailed Student’s t-test).
RPS23RG1 overexpression attenuates tau hyperphosphorylation and axon outgrowth defects in P301L tau Tg neurons
Cumulative evidence indicates that Cdk5 is aberrantly activated in brains from the murine P301L tau Tg tauopathy model, where Cdk5 inhibition attenuated tau pathology in these mice [39, 40]. Interestingly, we found that RPS23RG1 protein levels were reduced, whereas p35 and p25 protein levels were elevated in P301L tau Tg mouse hippocampal samples compared to littermate controls (Figs. 6a and S6a). In addition, P301L tau Tg mouse hippocampal neurons exhibited dramatically increased tau phosphorylation and impaired axon outgrowth compared to WT controls (Fig. 6b). Moreover, we found that overexpression of P301L tau but not WT tau dramatically reduced RPS23RG1 levels in HEK293T cells (Fig. S6b).
Fig. 6. RPS23RG1 overexpression mitigates tau hyperphosphorylation and promotes axon outgrowth in tau P301L Tg mouse neurons.

a Hippocampal neurons from P0 WT and P301L tau Tg mice at DIV4 were subjected to immunoblotting for the proteins indicated. Protein levels were quantified by densitometry, normalized to β-actin levels, and compared to WT (set to one arbitrary units, A.U.). Data represent mean ± SEM (n = 3). b Hippocampal neurons from P0 P301L tau Tg mice were immunostained for Tuj-1 (green) and pS404 (red) at DIV4. Immunofluorescence images were obtained by confocal microscopy. Axon length and pS404 intensity were quantified using ImageJ. Data represent mean ± SEM (n = 26 neurons from 3 mice per group, with the mean of WT set to one A.U.). Scale bars = 10 μm. c Hippocampal neurons from P0 P301L tau Tg mice were transfected with pCMV-HA control (Con) or HA-RPS23RG1 (RR1) at DIV3 for 24 h. Cell lysates were subjected to immunoblotting for the proteins indicated. Protein levels were quantified and compared to controls (set to one A.U., indicated by the dashed line). Data represent mean ± SEM (n = 3). d Hippocampal neurons from P0 P301L tau Tg mice were transfected with Con or HA-RR1 vectors. Total lysates were subjected to immunoprecipitation (IP) with an antibody against p35 and immunoblotting (IB) with an antibody against ubiquitin (Ubiq). Ten percent of IP lysates were used as input and immunoblotted for the proteins indicated. Alternatively, treated cells were subjected to membrane (Mem) fractionation and the proteins indicated in membrane fractions were detected by immunoblotting. e Hippocampal neurons from P0 P301L tau Tg mice were transfected with HA-RR1 at DIV3 for 24 h. Cells were co-immunostained for HA-RR1 (e2 and e2’, green) and pT205 (e3, red) or pS404 (e3’, red). Cells expressing HA-RR1 were identified by single-channel image merging (e1 and e1’). Representative cells with (e3-1 and e3’-1) and without (e3-2 and e3’-2) HA-RR1 expression were enlarged for pT205 (e3-1 and e3-2) and pS404 (e3’-1 and e3’-2) intensity quantitation using ImageJ. Data represent mean ± SEM (n = 15 cells for each group, with mean immunofluorescence intensity in cells without HA-RR1 expression set to one A.U.). Scale bars = 10 μm. f Hippocampal neurons from P0 P301L tau Tg mice were transfected with Con or HA-RR1 at DIV3 for 24 h. Cells were co-immunostained for HA-RR1 (red) and Tuj-1 (green). Immunofluorescence images were acquired by confocal microscopy. Axon length was quantified using ImageJ. Data represent mean ± SEM (n = 25 neurons from 3 mice in each group, with the mean of those of control cells set to one A.U.). Scale bars = 10 μm. *p < 0.05, **p < 0.01, ***p < 0.001 (two-tailed Student’s t-test).
To elucidate a potential role for RPS23RG1 in tau phosphoregulation in tauopathy, we overexpressed RPS23RG1 in HEK293/tau cells (Fig. S7) and P301L tau Tg mouse hippocampal neurons (Figs. 6c and S8). As expected, overexpression of RPS23RG1 significantly reduced tau phosphorylation at both major and minor Cdk5 phosphorylation sites, without affecting total tau levels. RPS23RG1 overexpression also increased high molecular weight, poly-ubiquitin conjugated p35 bands in P301L tau Tg mouse neurons (Fig. 6d). Consistent with these results, immunostaining assays showed that RPS23RG1 overexpression reduced tau phosphorylation at S404 and T205 phospho-sites in primary neurons from P301L tau Tg mice (Fig. 6e). Importantly, RPS23RG1 overexpression also increased axon length in P301L tau Tg neurons (Fig. 6f). Together, these findings demonstrate that RPS23RG1 overexpression can arrest tau hyperphosphorylation and promote axon outgrowth in P301L tau Tg mice.
Discussion
RPS23RG1 is a recently characterized membrane protein with physiological and pathological functions that have yet to be fully elucidated. Previous studies suggest that the RPS23RG1 transmembrane domain can interact with adenyl cyclases to activate the cAMP/PKA/GSK-3β pathway, thereby inhibiting Aβ generation and tau phosphorylation. The RPS23RG1 intracellular domain can also interact with PSD-93 and PSD-95 to maintain PSD architecture, implicating a role for RPS23RG1 in AD [26–28]. In this study, we identified an additional physiological function for RPS23RG1 in mediating homeostatic p35 proteasomal degradation, thereby restricting Cdk5-p35-mediated tau phosphorylation and facilitating axon outgrowth (Fig. 7). More importantly, we observed that RPS23RG1 levels were markedly decreased in P301L tau Tg mice; and RPS23RG1 overexpression ameliorated tau hyperphosphorylation and axon outgrowth deficits.
Fig. 7. Schematic diagram of RPS23RG1 (RR1) regulating tau phosphorylation by modulating p35/Cdk5 and GSK-3β.
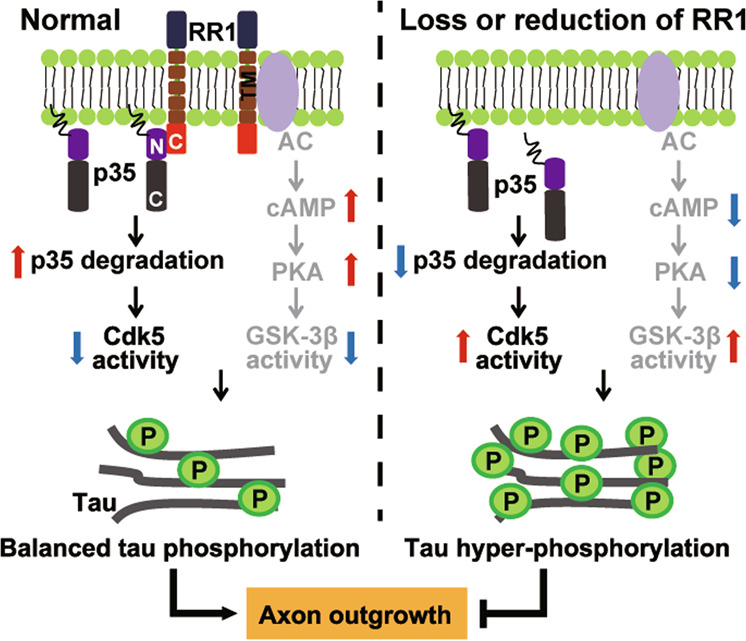
Under physiological conditions, the RR1 C-terminus interacts with the p35 N-terminus to facilitate p35 membrane-binding and degradation, thus suppressing Cdk5 activity. In addition, the trans-membrane domain of RR1 interacts with adenylate cyclases (AC) to promote cAMP production, leading to upregulated protein kinase A (PKA) activity to reduce GSK-3β activity. Thus, RR1 controls normal tau phosphorylation and facilitates physiological axon outgrowth. Rps23rg1 deletion in mice or reduced RR1 in tauopathies triggers membrane-bound p35 release into the cytosol, impairs p35 degradation, and downregulates AC/cAMP/PKA signaling. Consequent pathogenic activation of Cdk5 and GSK-3β results in tau hyperphosphorylation and impaired axon outgrowth.
Hyperphosphorylated tau is neurotoxic, and aggregates to form intracellular neurofibrillary tangles that pathologically manifest in AD and related tauopathy disorders [41, 42]. Therefore, inhibiting tau hyperphosphorylation is a therapeutically relevant strategy in limiting neurodegenerative effects in these disorders. Tau hyperphosphorylation is primarily induced by aberrant activation of Cdk5 and GSK-3β [35, 43, 44]. Herein, we provide both in vivo and in vitro evidence that RPS23RG1 can also regulate tau phosphorylation by modulating Cdk5-p35 activity. Cdk5 and its relatively unstable regulatory p35 subunit have been shown to play an important role in cytoskeletal organization and neurite outgrowth [45, 46]. P35-deficient mice feature abnormal neuronal layer development and as well as defects in cognition [47, 48]. Enhanced conversion of p35 to p25 with neurotoxic insult results in aberrant Cdk5 activation, leading to tau pathological hyperphosphorylation and cell death in AD and other tauopathic disorders [49–51]. These findings indicate that the homeostatic regulation of p35 protein levels is vital in tau phospho-regulation and neurite extension. To this point, molecular mechanisms underlying p35 regulation remain largely unknown. Our current study reveals a new route for p35 homeostasis maintenance by RPS23RG1.
As an additional kinase characterized in pathological tau phosphorylation, GSK-3β has been previously shown to act downstream of RPS23RG1, where RPS23RG1 overexpression reduces tau phosphorylation by attenuating GSK-3β activity [26, 27]. Indeed, we observed a slight increase in GSK-3β activity through reduced phosphorylation at GSK-3β S9 in Rps23rg1 KO mice. Although all tau sites characterized in this study are potential GSK-3β phospho-sites, Rps23rg1 deletion potentiated phosphorylation to a greater extent at major Cdk5 phospho-sites (S404 and S202/T205; >3 fold increase) compared to minor Cdk5 phospho-sites (S199 and S396; <2 fold increase) (Fig. 1). These results suggest that loss of RPS23RG1 may substantially promote Cdk5 activity and that this may have stronger effects than that seen with enhancing GSK-3β activity. Furthermore, overexpression of full-length RPS23RG1 dramatically reduced tau phosphorylation at phospho-sites described here. While overexpression of an RPS23RG1 C-terminal deletion construct (RR1ΔC) refractory to p35 binding had little effect on reducing tau phosphorylation at S404 and S202/T205 phospho-sites, this construct still significantly reduced tau phosphorylation at S199 and S396 which are also GSK-3β phospho-sites (Fig. S8). These findings demonstrate that RPS23RG1 can regulate tau phosphorylation by modulating both GSK-3β and Cdk5-p35 activity. However, loss of RPS23RG1 may have greater effects on Cdk5-p35 over GSK-3β. Given that cross-talk between GSK-3β and Cdk5-p35 pathways have been revealed previously [52, 53], regulatory effects of RSP23RG1 on these two kinases may be more complicated than what we describe here, and their interplay under various pathophysiological conditions warrants further scrutiny.
We previously found that RPS23RG1 expression was decreased in the postmortem AD brain and that Aβ treatment markedly reduced RPS23RG1 levels [28]. Herein, we found that RPS23RG1 levels were significantly decreased in P301L tau Tg mice compared to WT mice and in cells overexpressing P301L but not WT tau. Moreover, we noticed that RPS23RG1 levels were further decreased in P301L tau Tg mice during aging (Fig. S6c). Therefore, RPS23RG1 may be reduced in response to Aβ and pathologic tau proteotoxicity. As RPS23RG1 signaling modulates Aβ generation, tau phosphorylation, and synaptic function, pathological changes in Aβ, tau, and RPS23RG1 may form a vicious cycle which drives pathogenesis in AD and other tauopathies.
In summary, we demonstrate that RPS23RG1-p35 interactions promote proteasomal p35 turnover which limits physiological tau phosphorylation and enhances axon outgrowth in the brain. Since RPS23RG1 levels are decreased in P301L tau Tg mouse brain and upon P301L tau expression, and RPS23RG1 overexpression can attenuate tau hyperphosphorylation and axon outgrowth deficits in P301L tau Tg mouse neurons, RPS23RG1 and its downstream pathways may be potential therapeutic targets for tauopathy.
Supplementary information
Acknowledgements
We thank Dr. Jianzhi Wang for providing HEK293/tau cells. This work was supported by grants from National Key Research and Development Program of China (2016YFC1305903 and 2018YFC2000400 to Y-wZ), National Natural Science Foundation of China (81771377, U1705285, 91332112, and 81225008 to Y-wZ), Fundamental Research Funds for the Central Universities (20720180049 to Y-wZ), the Fujian Provincial Health Commission-Education Department Joint Tackling Plan (WKJ2016-2-18 to F-rL), and Postdoctoral Science Foundation of China (2020M671948 to DZ).
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Edited by G. Melino
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version of this article (10.1038/s41418-020-00620-y) contains supplementary material, which is available to authorized users.
References
- 1.Dehmelt L, Halpain S. The MAP2/Tau family of microtubule-associated proteins. Genome Biol. 2005;6:204. doi: 10.1186/gb-2004-6-1-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scholz T, Mandelkow E. Transport and diffusion of Tau protein in neurons. Cell Mol Life Sci. 2014;71:3139–50. doi: 10.1007/s00018-014-1610-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martin L, Latypova X, Wilson CM, Magnaudeix A, Perrin ML, Terro F. Tau protein phosphatases in ’Alzheimer’s disease: the leading role of PP2A. Ageing Res Rev. 2013;12:39–49. doi: 10.1016/j.arr.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 4.Billingsley ML, Kincaid RL. Regulated phosphorylation and dephosphorylation of tau protein: effects on microtubule interaction, intracellular trafficking and neurodegeneration. Biochem J. 1997;323:577–91. doi: 10.1042/bj3230577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alonso AC, Zaidi T, Grundke-Iqbal I, Iqbal K. Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc Natl Acad Sci USA. 1994;91:5562–6. doi: 10.1073/pnas.91.12.5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hatch RJ, Wei Y, Xia D, Gotz J. Hyperphosphorylated tau causes reduced hippocampal CA1 excitability by relocating the axon initial segment. Acta Neuropathol. 2017;133:717–30. doi: 10.1007/s00401-017-1674-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DeTure MA, Dickson DW. The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegener. 2019;14:32. doi: 10.1186/s13024-019-0333-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guo T, Zhang D, Zeng Y, Huang TY, Xu H, Zhao Y. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol Neurodegener. 2020;15:40. doi: 10.1186/s13024-020-00391-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Piccini A, Perlini LE, Cancedda L, Benfenati F, Giovedi S. Phosphorylation by PKA and Cdk5 mediates the early effects of synapsin III in neuronal morphological maturation. J Neurosci. 2015;35:13148–59. doi: 10.1523/JNEUROSCI.1379-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang H, Lin X, Liang Z, Zhao T, Du S, Loy MMT, et al. Cdk5-dependent phosphorylation of liprinalpha1 mediates neuronal activity-dependent synapse development. Proc Natl Acad Sci USA. 2017;114:E6992–7001. doi: 10.1073/pnas.1708240114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Odajima J, Wills ZP, Ndassa YM, Terunuma M, Kretschmannova K, Deeb TZ, et al. Cyclin E constrains Cdk5 activity to regulate synaptic plasticity and memory formation. Dev Cell. 2011;21:655–68. doi: 10.1016/j.devcel.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lai KO, Wong AS, Cheung MC, Xu P, Liang Z, Lok KC, et al. TrkB phosphorylation by Cdk5 is required for activity-dependent structural plasticity and spatial memory. Nat Neurosci. 2012;15:1506–15. doi: 10.1038/nn.3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dhavan R, Tsai LH. A decade of CDK5. Nat Rev Mol Cell Biol. 2001;2:749–59. doi: 10.1038/35096019. [DOI] [PubMed] [Google Scholar]
- 14.Cheung ZH, Ip NY. The roles of cyclin-dependent kinase 5 in dendrite and synapse development. Biotechnol J. 2007;2:949–57. doi: 10.1002/biot.200700056. [DOI] [PubMed] [Google Scholar]
- 15.Ohshima T, Ogawa M, Veeranna, Hirasawa M, Longenecker G, Ishiguro K, et al. Synergistic contributions of cyclin-dependant kinase 5/p35 and Reelin/Dab1 to the positioning of cortical neurons in the developing mouse brain. Proc Natl Acad Sci USA. 2001;98:2764–9. doi: 10.1073/pnas.051628498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hisanaga S, Endo R. Regulation and role of cyclin-dependent kinase activity in neuronal survival and death. J Neurochem. 2010;115:1309–21. doi: 10.1111/j.1471-4159.2010.07050.x. [DOI] [PubMed] [Google Scholar]
- 17.Cole AR, Soutar MP, Rembutsu M, van Aalten L, Hastie CJ, McLauchlan H, et al. Relative resistance of Cdk5-phosphorylated CRMP2 to dephosphorylation. J Biol Chem. 2008;283:18227–37. doi: 10.1074/jbc.M801645200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsai SY, Pokrass MJ, Klauer NR, Nohara H, Su TP. Sigma-1 receptor regulates Tau phosphorylation and axon extension by shaping p35 turnover via myristic acid. Proc Natl Acad Sci USA. 2015;112:6742–7. doi: 10.1073/pnas.1422001112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Crews L, Ruf R, Patrick C, Dumaop W, Trejo-Morales M, Achim CL, et al. Phosphorylation of collapsin response mediator protein-2 disrupts neuronal maturation in a model of adult neurogenesis: Implications for neurodegenerative disorders. Mol Neurodegener. 2011;6:67. doi: 10.1186/1750-1326-6-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Minegishi S, Asada A, Miyauchi S, Fuchigami T, Saito T, Hisanaga S. Membrane association facilitates degradation and cleavage of the cyclin-dependent kinase 5 activators p35 and p39. Biochemistry. 2010;49:5482–93. doi: 10.1021/bi100631f. [DOI] [PubMed] [Google Scholar]
- 21.Asada A, Yamamoto N, Gohda M, Saito T, Hayashi N, Hisanaga S. Myristoylation of p39 and p35 is a determinant of cytoplasmic or nuclear localization of active cyclin-dependent kinase 5 complexes. J Neurochem. 2008;106:1325–36. doi: 10.1111/j.1471-4159.2008.05500.x. [DOI] [PubMed] [Google Scholar]
- 22.Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999;402:615–22. doi: 10.1038/45159. [DOI] [PubMed] [Google Scholar]
- 23.Vagnozzi AN, Giannopoulos PF, Pratico D. Brain 5-lipoxygenase over-expression worsens memory, synaptic integrity, and tau pathology in the P301S mice. Aging Cell. 2018;17:e12695. doi: 10.1111/acel.12695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Currais A, Prior M, Dargusch R, Armando A, Ehren J, Schubert D, et al. Modulation of p25 and inflammatory pathways by fisetin maintains cognitive function in Alzheimer’s disease transgenic mice. Aging Cell. 2014;13:379–90. doi: 10.1111/acel.12185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang P, Fu WY, Fu AK, Ip NY. S-nitrosylation-dependent proteasomal degradation restrains Cdk5 activity to regulate hippocampal synaptic strength. Nat Commun. 2015;6:8665. doi: 10.1038/ncomms9665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang X, Chen Y, Li WB, Cohen SN, Liao FF, Li L, et al. The Rps23rg gene family originated through retroposition of the ribosomal protein s23 mRNA and encodes proteins that decrease Alzheimer’s beta-amyloid level and tau phosphorylation. Hum Mol Genet. 2010;19:3835–43. doi: 10.1093/hmg/ddq302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang YW, Liu S, Zhang X, Li WB, Chen Y, Huang X, et al. A functional mouse retroposed gene Rps23r1 reduces Alzheimer’s beta-amyloid levels and tau phosphorylation. Neuron. 2009;64:328–40. doi: 10.1016/j.neuron.2009.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao D, Meng J, Zhao Y, Huo Y, Liu Y, Zheng N, et al. RPS23RG1 is required for synaptic integrity and rescues Alzheimer’s disease-associated cognitive deficits. Biol Psychiatry. 2019;86:171–84. doi: 10.1016/j.biopsych.2018.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramsden M, Kotilinek L, Forster C, Paulson J, McGowan E, SantaCruz K, et al. Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L) J Neurosci. 2005;25:10637–47. doi: 10.1523/JNEUROSCI.3279-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–81. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zheng Q, Zheng X, Zhang L, Luo H, Qian L, Fu X, et al. The neuron-specific protein TMEM59L mediates oxidative stress-induced cell death. Mol Neurobiol. 2017;54:4189–200. doi: 10.1007/s12035-016-9997-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dotti CG, Sullivan CA, Banker GA. The establishment of polarity by hippocampal neurons in culture. J Neurosci. 1988;8:1454–68. doi: 10.1523/JNEUROSCI.08-04-01454.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takano T, Tomomura M, Yoshioka N, Tsutsumi K, Terasawa Y, Saito T, et al. LMTK1/AATYK1 is a novel regulator of axonal outgrowth that acts via Rab11 in a Cdk5-dependent manner. J Neurosci. 2012;32:6587–99. doi: 10.1523/JNEUROSCI.5317-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wen Y, Planel E, Herman M, Figueroa HY, Wang L, Liu L, et al. Interplay between cyclin-dependent kinase 5 and glycogen synthase kinase 3 beta mediated by neuregulin signaling leads to differential effects on tau phosphorylation and amyloid precursor protein processing. J Neurosci. 2008;28:2624–32. doi: 10.1523/JNEUROSCI.5245-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kimura T, Ishiguro K, Hisanaga S. Physiological and pathological phosphorylation of tau by Cdk5. Front Mol Neurosci. 2014;7:65. doi: 10.3389/fnmol.2014.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen LM, Xiong YS, Kong FL, Qu M, Wang Q, Chen XQ, et al. Neuroglobin attenuates Alzheimer-like tau hyperphosphorylation by activating Akt signaling. J Neurochem. 2012;120:157–64. doi: 10.1111/j.1471-4159.2011.07275.x. [DOI] [PubMed] [Google Scholar]
- 37.Takasugi T, Minegishi S, Asada A, Saito T, Kawahara H, Hisanaga S. Two degradation pathways of the p35 Cdk5 (Cyclin-dependent Kinase) activation subunit, dependent and independent of ubiquitination. J Biol Chem. 2016;291:4649–57. doi: 10.1074/jbc.M115.692871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Patrick GN, Zhou P, Kwon YT, Howley PM, Tsai LH. p35, the neuronal-specific activator of cyclin-dependent kinase 5 (Cdk5) is degraded by the ubiquitin-proteasome pathway. J Biol Chem. 1998;273:24057–64. doi: 10.1074/jbc.273.37.24057. [DOI] [PubMed] [Google Scholar]
- 39.Seo J, Kritskiy O, Watson LA, Barker SJ, Dey D, Raja WK, et al. Inhibition of p25/Cdk5 attenuates tauopathy in mouse and iPSC models of frontotemporal dementia. J Neurosci. 2017;37:9917–24. doi: 10.1523/JNEUROSCI.0621-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rao MV, McBrayer MK, Campbell J, Kumar A, Hashim A, Sershen H, et al. Specific calpain inhibition by calpastatin prevents tauopathy and neurodegeneration and restores normal lifespan in tau P301L mice. J Neurosci. 2014;34:9222–34. doi: 10.1523/JNEUROSCI.1132-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takashima A. Hyperphosphorylated tau is a cause of neuronal dysfunction in tauopathy. J Alzheimers Dis. 2008;14:371–5. doi: 10.3233/JAD-2008-14403. [DOI] [PubMed] [Google Scholar]
- 42.Hu W, Zhang X, Tung YC, Xie S, Liu F, Iqbal K. Hyperphosphorylation determines both the spread and the morphology of tau pathology. Alzheimers Dement. 2016;12:1066–77. doi: 10.1016/j.jalz.2016.01.014. [DOI] [PubMed] [Google Scholar]
- 43.Noble W, Olm V, Takata K, Casey E, Mary O, Meyerson J, et al. Cdk5 is a key factor in tau aggregation and tangle formation in vivo. Neuron. 2003;38:555–65. doi: 10.1016/S0896-6273(03)00259-9. [DOI] [PubMed] [Google Scholar]
- 44.Hernandez F, Lucas JJ, Avila J. GSK3 and tau: two convergence points in Alzheimer’s disease. J Alzheimers Dis. 2013;33:S141–4. doi: 10.3233/JAD-2012-129025. [DOI] [PubMed] [Google Scholar]
- 45.Furusawa K, Asada A, Urrutia P, Gonzalez-Billault C, Fukuda M, Hisanaga SI. Cdk5 regulation of the GRAB-mediated Rab8-Rab11 cascade in axon outgrowth. J Neurosci. 2017;37:790–806. doi: 10.1523/JNEUROSCI.2197-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Song JH, Wang CX, Song DK, Wang P, Shuaib A, Hao C. Interferon gamma induces neurite outgrowth by up-regulation of p35 neuron-specific cyclin-dependent kinase 5 activator via activation of ERK1/2 pathway. J Biol Chem. 2005;280:12896–901. doi: 10.1074/jbc.M412139200. [DOI] [PubMed] [Google Scholar]
- 47.Engmann O, Hortobagyi T, Pidsley R, Troakes C, Bernstein HG, Kreutz MR, et al. Schizophrenia is associated with dysregulation of a Cdk5 activator that regulates synaptic protein expression and cognition. Brain. 2011;134:2408–21. doi: 10.1093/brain/awr155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ohshima T, Ogura H, Tomizawa K, Hayashi K, Suzuki H, Saito T, et al. Impairment of hippocampal long-term depression and defective spatial learning and memory in p35 mice. J Neurochem. 2005;94:917–25. doi: 10.1111/j.1471-4159.2005.03233.x. [DOI] [PubMed] [Google Scholar]
- 49.Hsiao YH, Chen PS, Chen SH, Gean PW. The involvement of Cdk5 activator p35 in social isolation-triggered onset of early Alzheimer’s disease-related cognitive deficit in the transgenic mice. Neuropsychopharmacology. 2011;36:1848–58. doi: 10.1038/npp.2011.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Seo J, Giusti-Rodriguez P, Zhou Y, Rudenko A, Cho S, Ota KT, et al. Activity-dependent p25 generation regulates synaptic plasticity and Abeta-induced cognitive impairment. Cell. 2014;157:486–98. doi: 10.1016/j.cell.2014.01.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang L, Liu W, Szumlinski KK, Lew J. p10, the N-terminal domain of p35, protects against CDK5/p25-induced neurotoxicity. Proc Natl Acad Sci USA. 2012;109:20041–6. doi: 10.1073/pnas.1212914109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hallows JL, Chen K, DePinho RA, Vincent I. Decreased cyclin-dependent kinase 5 (cdk5) activity is accompanied by redistribution of cdk5 and cytoskeletal proteins and increased cytoskeletal protein phosphorylation in p35 null mice. J Neurosci. 2003;23:10633–44. doi: 10.1523/JNEUROSCI.23-33-10633.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zheng YL, Li BS, Kanungo J, Kesavapany S, Amin N, Grant P, et al. Cdk5 modulation of mitogen-activated protein kinase signaling regulates neuronal survival. Mol Biol Cell. 2007;18:404–13. doi: 10.1091/mbc.e06-09-0851. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.