

Abstract
A series of new dihydrobenzooxophosphole-based Lewis Base organocatalysts were designed and synthesized. They are demonstrated effective in trichlorosilane-mediated stereoselective conjugate reductions of C=C bonds. DFT calculations reveal that the strong hydrogen bond between the amide linker and the chloride on silicon in the transition state contributes to the high reactivity of the catalyst 3a.
Keywords: P(O)NNP(O), Chiral Lewis Base, Organocatalyst, Enone Reduction, Hydrogen Bonding
Chiral Lewis base catalysts have emerged as powerful organocatalysts in the past decade for the construction of C‒C or C‒X bonds.1 Among them, the phosphine oxide-containing Lewis base catalysts (Figure 1) have been applied extensively in a number of enantioselective transformations including Mukaiyama aldol and double aldol reactions,2 trichlorosilane-mediated stereoselective reductions of C=N and C=C bonds,3 bromoaminocyclization,4 enantioselective allylation5 and epoxide ring-opening reactions.6
Figure 1:
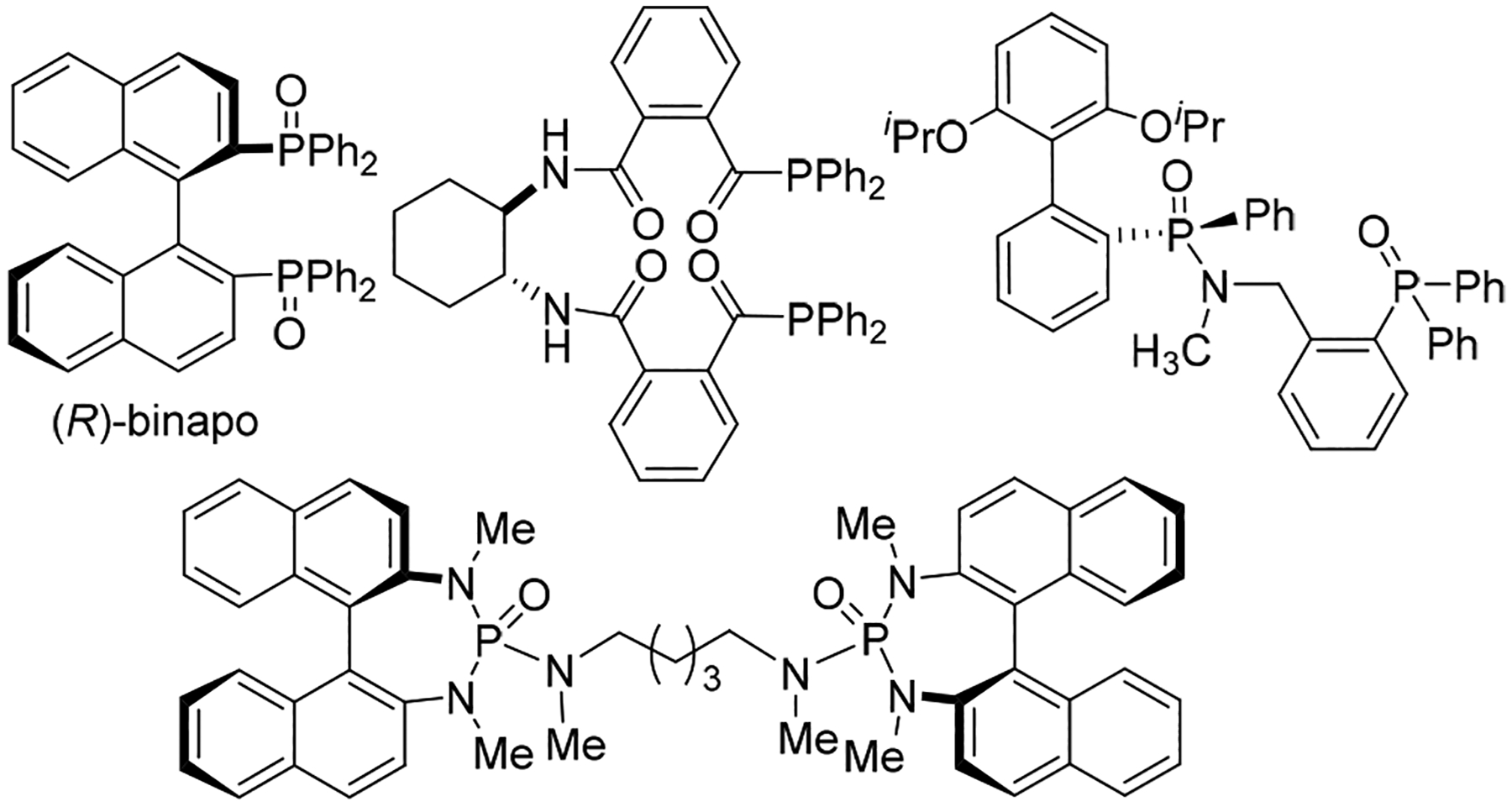
Selected P=O Containing Lewis Base Organocatalysts
The availability of diversified phosphine ligand libraries has enabled successful application of metal-catalyzed reactions on industrial scales. With increasing attention on developing organocatalyst-catalyzed asymmetric synthesis, the advancement of novel efficient catalyst systems remains an attractive endeavor. Recently, we reported a series of tunable P-stereogenic dihydrobenzooxophosphole-based bisphosphine ligands BIBOPs that are highly effective in a number of asymmetric transformations.7 We reasoned that the corresponding bis-phosphine oxide BIBOPOs might be applicable as Lewis base organocatalysts. The conjugate reduction of (E)-1,3-diphenylbutenone (1a) in the presence of trichlorosilane3 was selected as a model transformation to evaluate the effectiveness of such catalysts (Scheme 1). However, when BIBOPO was applied in the test reaction, only trace amount of product 2a was observed (Scheme 1a). We rationalized that an increased coordinating angle of the Si atom to bis P=O might be required to accommodate the substrate coordination (Scheme 1b). C2-symmetric tetradentate bisphosphine/diamine (PNNP) ligands with NH-functionality have been reported as highly efficient catalyst system for asymmetric hydrogenation/transfer hydrogenation of ketones8 and other enantioselective reactions.9 We therefore decided to apply the versatile features of a diamine linker and prepare new P(O)NNP(O)-based organocatalysts. We hypothesized that such linker would not only increase the bite angle, but also lead to potential hydrogen bonding of HSiCl3 with the hydrogen atoms on diamine.
Scheme 1:
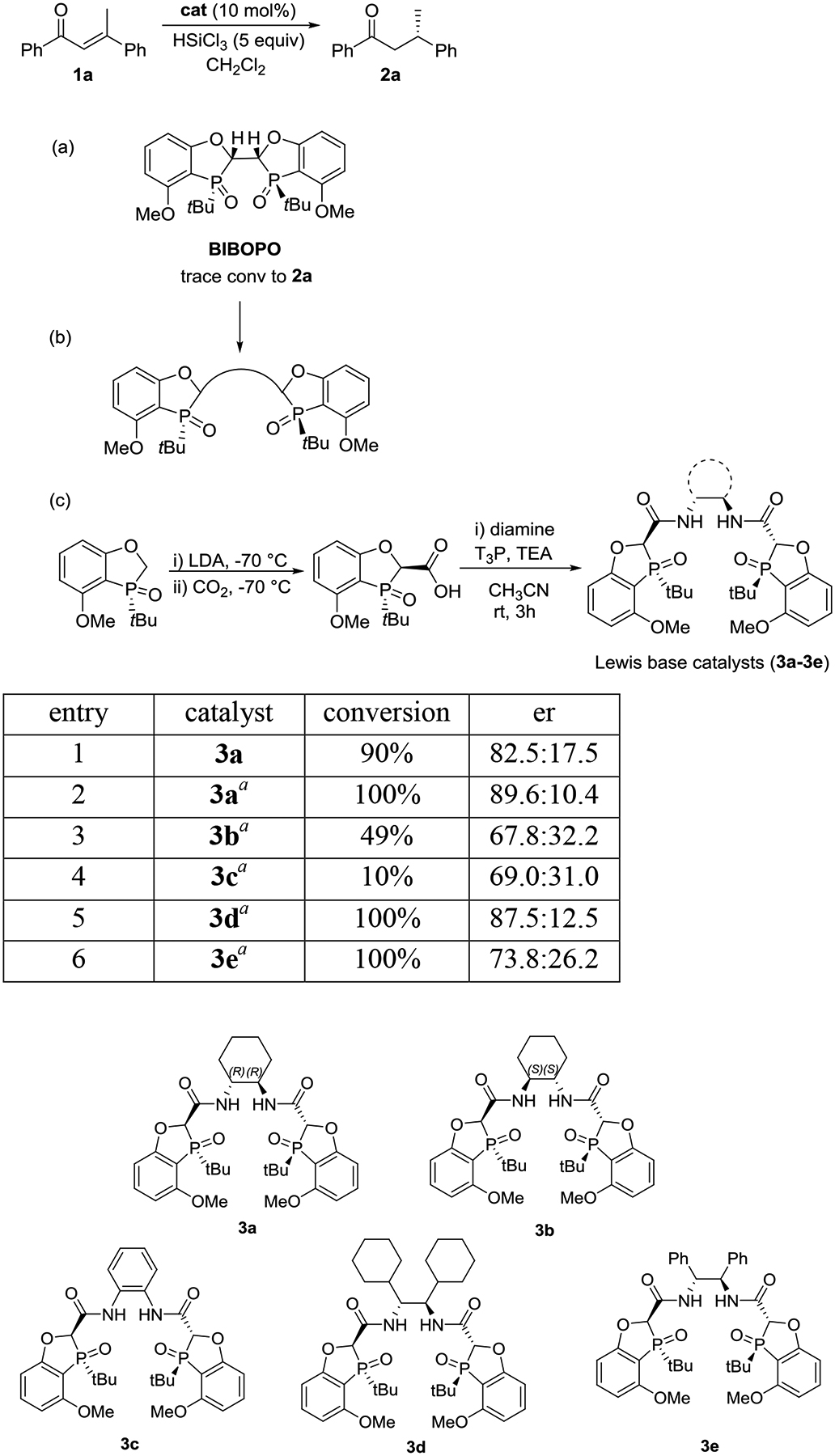
Conjugate reduction of 1a with P(O)NNP(O) Lewis Base catalysts. Reaction conditions: 1a (0.27 mmol), HSiCl3 (1.35 mmol), catalyst (0.027 mmol) in CH2Cl2 at 0 °C for 20 h. Conversion of 1a to 2a, and enantioselectivity were analyzed on SFC at 220 nm UV wavelength with a chiral stationary phase. (a) Application of BIBOPO catalyst; (b) rational design of new catalysts; (c) synthesis and evaluation of the new catalysts. aReaction was running in acetonitrile.
The proposed catalysts were synthesized by coupling of the corresponding carboxylic acid10 with diamines in the presence of propylphosphonic anhydride (T3P) in acetonitrile (Scheme 1c).11 In each case, only one diastereomer product was obtained under the coupling conditions without any racemization. We first synthesized the bis-amide catalyst 3a with a (R,R)-diaminocyclohexane linker. Gladly, (S)-1,3-diphenylbutan-1-one 2a12 was furnished in 90% conversion and an 82.5:17.5 enantiomeric ratio in CH2Cl2 (entry 1). Several environmentally friendly solvents were tested, and acetonitrile was identified to provide the highest selectivity in an 89.6:10.4 er (entry 2).13 Interestingly, the corresponding diastereomeric (S,S)-diaminocyclohexyl–containing catalyst 3b provided incomplete conversion and 67.8:32.2 er indicating that the conformation of the bis-amide linker is integral to both turnover and selectivity. When increasing the rigidity of the catalyst backbone linker with phenyl diamine, however, low reactivity and selectivity were observed for catalyst 3c. On the other hand, the (R,R)-1,2-dicyclohexyl ethylene diamine–containing catalyst 3d provided the product 2a with 100% conversion and an 87.5:12.5 er. When replacing the bis-cyclohexyl group with bis-phenyl group in the catalyst 3f; same reactivity of 100% conversion, but diminished enantioselecitivity of 74:26 er was observed.
We initiated a DFT study with Gaussian 1614 with B3LYP/6–31G(d)15 to understand the origin of the catalyst reactivity in these reductions. According to the proposed mechanism,16 the stereodetermining step involves a 1,4-addition of a silicon hydride to the enone via a six-membered transition state. In this transition state, the cationic silicon coordinates to the carbonyl oxygen, geometrically placing the silicon hydride near the β-carbon of the enone. Upon reduction, the enolate remains coordinated to the cationic silicon. Possible transition states differ in the position of the hydride on the catalyst, the position of the enone on the catalyst, the orientation of the hydride relative to the enone, and the geometry of the enone (since it can undergo isomerization under the reaction conditions). Detailed conformational analyses of these transition states were undertaken to identify the relevant lowest energy transition states for the different catalysts.
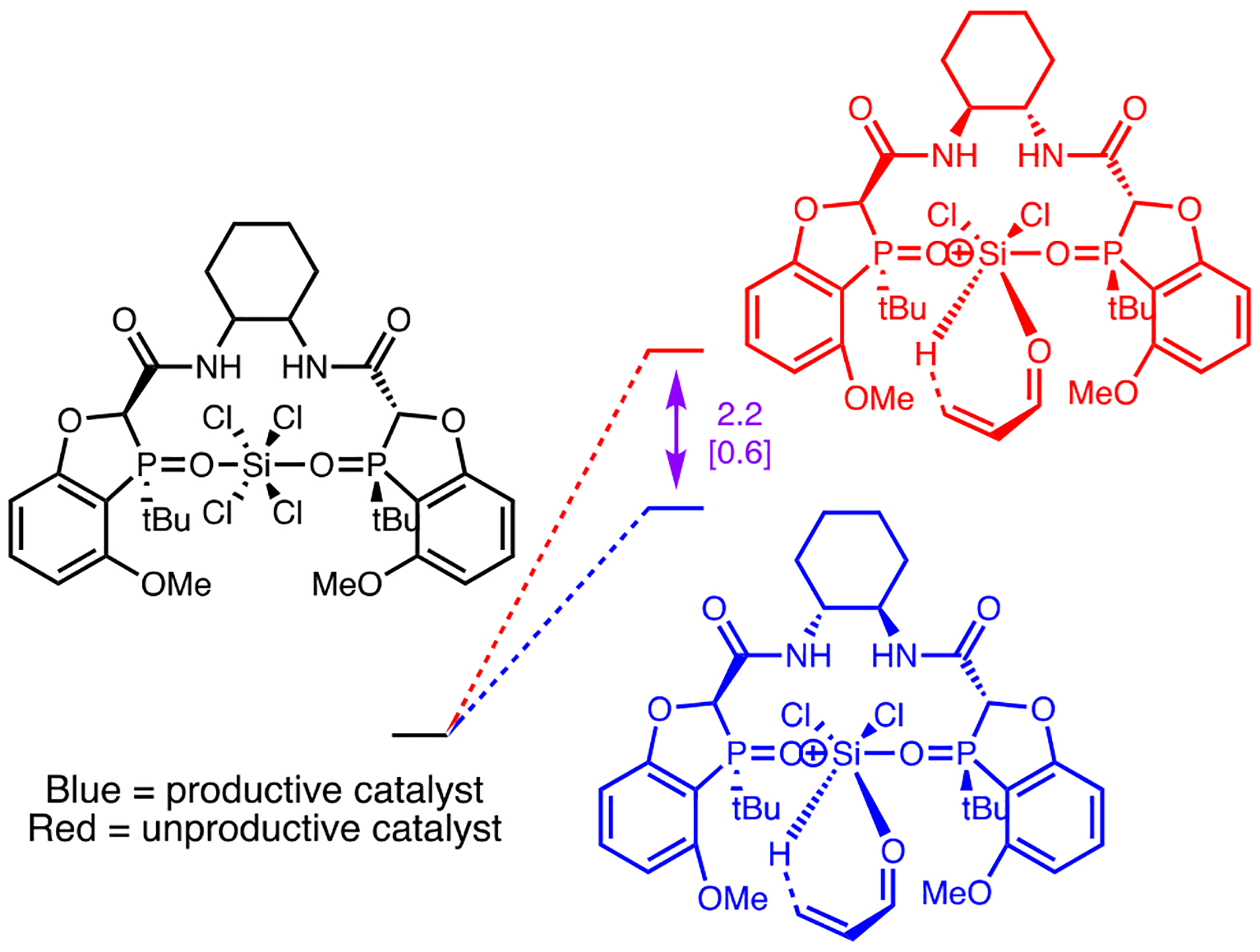
Amongst the catalysts screened, there was a notable difference in behavior of the diastereomeric compounds obtained from the two enantiomers of cyclohexane diamine (matched 3a, mismatched 3b). To access the reactivity of these diastereomeric catalysts, the activation energies for hydride transfer in respective systems were analyzed. Calculations showed that a higher activation energy is required for the reduction of the enone using the (S,S)-isomer of the catalyst 3b by 2.2 kcal/mol, which is consistent with the observed reactivity.
As projected, both of the aforementioned catalysts form hydrogen bonds between the amide linker and the chlorine ligands of the silicon in the transition state. The productive catalyst 3a forms stronger hydrogen bonds than the unproductive catalyst 3b, as determined by bond lengths (Figure 3). These hydrogen bonds would be expected to enhance the Lewis acidity of the silicon center and thereby enhance reactivity. It thus appears that the mismatched catalyst, 3b, must engage in greater deformation (i.e. is more strained) to achieve such hydrogen bonds, which are also weaker causing less activation. Together, these factors account for the much lower reactivity of 3b (49% conv) relative to 3a (100% conv).
Figure 3.
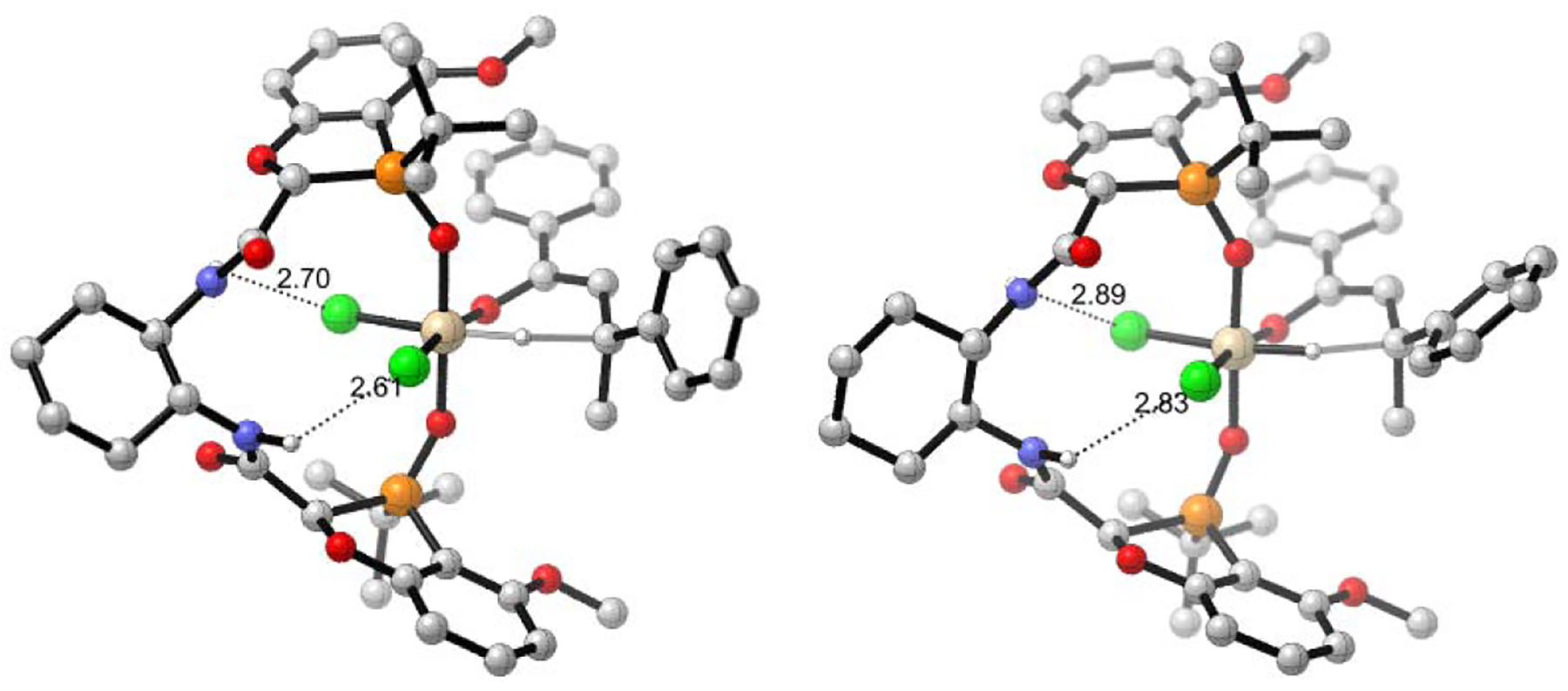
CYL view of the transition state structures with productive (3a, left) and unproductive (3b, right) diastereomeric catalysts. Hydrogen bonds between the amide linker and chlorine atoms are indicated with dashed lines, distances in angstroms.
The conjugate reduction conditions with catalyst 3a are compatible for (E)-1,3-diphenylbutenones with both electron-rich and electron-poor functional groups to give similar enantioselectivity (2a-2d) in acetonitrile (Scheme 2).
Scheme 2:
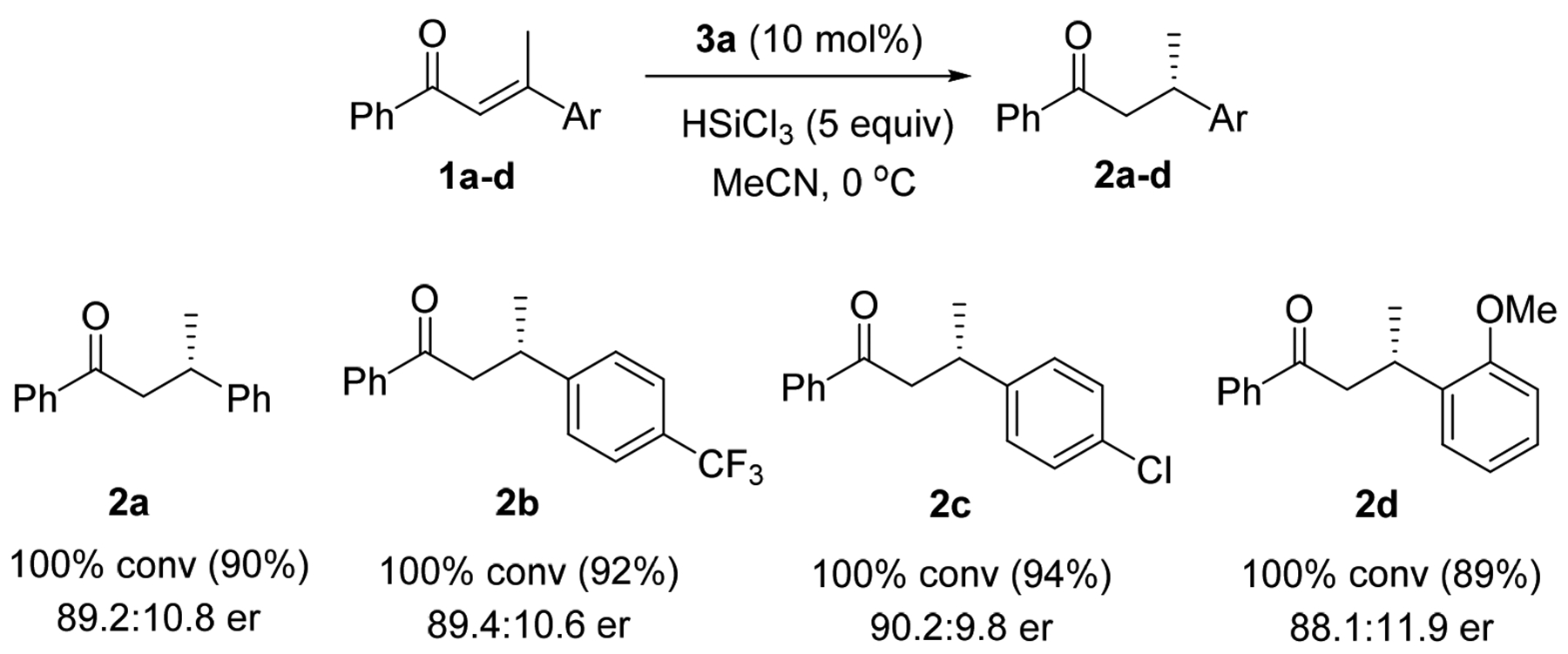
Conjugate 1,4-Enone Reduction Using Organocatalyst 3aa
a Reaction conditions: enone 1 (0.27 mmol), HSiCl3 (1.35 mmol), catalyst 3a (0.027 mmol) in acetonitrile at 0 °C for 20 h; the yields in parenthesis are isolated yields after silica gel purification.
In conclusion, we have developed new Lewis base organocatalysts derived from P‒stereogenic dihydrobenzooxophosphole core structure by coupling with (R,R)-dicyclohexyl ethylene diamine, which provides a conformational match for the enantioselective reduction of enone derivatives mediated with HSiCl3. The asymmetric transformation is affected by the P‒stereogenic center and the selectivity is tunable by the substituents on the catalyst. This class of catalysts has potential as tunable Lewis base catalysts in terms of reactivity and selectivity. Further work is to identify catalysts that are more efficient; such tuning will be reported in due course.
Supplementary Material
Figure 2.
Relative activation energies of the reduction of the simplified enone by both diastereomeric catalysts 3a (blue) and 3b (red). Computed using B3LYP/6–31G(d). Free energy values are reported in kcal/mol, and enthalpies are in brackets. Acrolein was used as a model for the enone.
Acknowledgment
M. C. K. thanks the NIH (GM087605) and Boehringer Ingelheim Pharmaceuticals for financial support. Computational support was provided by XSEDE (TG-CHE120052). The authors also thank Mr. Scott Pennino for HRMS analysis.
References:
- 1.a) Denmark SE; Stavenger RA Acc. Chem. Res 2000, 33, 432. [DOI] [PubMed] [Google Scholar]; b) Guizzetti S; Benaglia M Eur. J. Org. Chem 2010, 5529. [Google Scholar]
- 2.a) Shimoda Y; Kubo T; Sugiura M; Kotani S; Nakajima M Angew. Chem. Int. Ed 2013, 52, 3461. [DOI] [PubMed] [Google Scholar]; b) Aoki S; Kotani S; Sugiura M; Nakajima M Chem. Commun 2012, 48, 5524. [DOI] [PubMed] [Google Scholar]
- 3.(a) Han ZS; Zhang L; Xu Y; Sieber JD; Marsini MA; Li Z; Reeves JT; Kandrick KR; Patel ND; Desrosiers J-N; Qu B; Chen A; Rudzinski DM; Samankumara LP; Ma S; Grinberg N; Roschangar F; Yee NK; Wang G; Song JJ; Senanayake CH Angew. Chem. Int. Ed 2015, 54, 5474. [DOI] [PubMed] [Google Scholar]; (b) Sugiura M; Sato N; Kotani S; Nakajima M Chem. Commun 2008, 4309. [DOI] [PubMed] [Google Scholar]
- 4.Li Z; Shi Y Org. Lett 2015, 17, 5752. [DOI] [PubMed] [Google Scholar]
- 5.Ogasawara M; Kotani S; Nakajima H; Furusho H; Miyasaka M; Shimoda Y; Wu W-Y; Sugiura M; Takahashi T; Nakajima M Angew. Chem. Int. Ed 2013, 52, 13798. [DOI] [PubMed] [Google Scholar]
- 6.Kotani S; Hashimoto S; Nakajima M Tetrahedron 2007, 63, 3122. [Google Scholar]
- 7.a) Chong E; Qu B; Zhang Y; Cannone ZP; Leung JC; Tcyrulnikov S; Nguyen KD; Haddad N; Biswas S; Hou X; Kaczanowska K; Chwalba M; Tracz A; Czarnocki S; Song JJ; Kozlowski MC; Senanayake CH Chem. Sci 2019, 10, 4339. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tang W; Qu B; Capacci AG; Rodriguez S; Wei X; Haddad N; Narayanan B; Ma S; Grinberg N; Yee NK; Krishnamurthy D; Senanayake CH Org. Lett 2010, 12, 176. [DOI] [PubMed] [Google Scholar]
- 8.Sui-Seng C; Freutel F; Lough AJ; Morris RH Angew. Chem., Int. Ed 2008, 47, 940. [DOI] [PubMed] [Google Scholar]; b) Gao JX; Ikariya T; Noyori R Organometallics 1996, 15, 1087. [Google Scholar]
- 9.Li Y-Y; Yu S-L; Shen W-Y; Gao J-X Acc. Chem. Res 2015, 48, 2587. [DOI] [PubMed] [Google Scholar]; (b) Trost BM Tetrahedron 2015, 71, 5708. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Trost BM; Van Vranken DL Angew. Chem. Int. Ed 1992, 21, 228. [Google Scholar]
- 10.Qu B; Samankumara LP; Ma S; Fandrick KR; Desrosiers J-N; Rodriguez S; Li Z; Haddad N; Han ZS; McKellop K; Pennino S; Grinberg N; Connella NC; Song JJ; Senanayake CH Angew. Chem. Int. Ed 2014, 53, 14428. [DOI] [PubMed] [Google Scholar]
- 11.General procedure for catalysts preparation: to a solution of carboxylic acid phosphine oxide (5.0 g, 17.6 mmol, 2.2 equiv), diamine (8.0 mmol, 1 equiv) and triethylamine (32.0 mmol, 4 equiv) in acetonitrile at room temperature was added propylphosphonic anhydride (T3P) solution in DMF (16.0 mmol, 2 equiv) in portions over 3 h. The reaction was stopped after complete consumption of the carboxylic acid. The mixture was then treated with 50% aqueous NaOH (10 mL) and stirred at 35 °C for 3 h. The suspension was diluted with water and the resulted clear solution was extracted three times with EtOAc; and the combined organic layer was washed with brine, dried with MgSO4 and concentrated. The crude mixture was purified by chromatography on silica (100% EtOAc to 10% MeOH/EtOAc) to obtain white solid after dryness.(2R,2’R,3S,3’S)-N,N’-((1R,2R)-cyclohexane-1,2-diyl)bis(3-(tert-butyl)-4-methoxy-2H-benzo[d][1,3]oxaphosphole-2-carboxamide 3-oxide) (3a): 3.67 g, 71% yield; 1H NMR (500 MHz, CDCl3) δ 7.46 (t, J = 8.23 Hz, 2H), 6.80 (br d, J = 6.70 Hz, 2H), 6.71 (dd, J = 8.3, 2.8 Hz, 2H), 6.54 (dd, J = 8.2, 4.4 Hz, 2H), 4.93 (s, 2H), 3.88 (s, 6H), 3.80 (br s, 2H), 2.11 (br d, J = 7.3 Hz, 2H), 1.65 (br s, 2H), 1.37 (d, J = 17.0 Hz, 18H), 1.25–1.23 (m, 4H); 13C NMR (125 MHz, CDCl3) 165.2 (d, J = 2.7 Hz), 164.5 (d, J = 14.2 Hz), 161.1 (d, J = 2.4 Hz), 136.9 (d, J = 0.87 Hz), 106.6 (d, J = 5.3 Hz), 104.1 (d, J = 5.7 Hz), 102.70 (d, J = 90.8 Hz), 75.4 (d, J = 48.4 Hz), 55.6, 53.7, 34.07 (d, J = 74.5 Hz), 32.4, 25.1 (d, J = 0.96 Hz), 24.5; 31P NMR (202 MHz, CDCl3) δ 62.83; HRMS (ESI) m/z 647.26495 (M + H+), calcd for C32H45O8N2P2 647.26457(2R,2’R,3S,3’S)-N,N’-((1S,2S)-cyclohexane-1,2-diyl)bis(3-(tert-butyl)-4-methoxy-2H-benzo[d][1,3]oxaphosphole-2-carboxamide 3-oxide) (3b): 3.36 g, 65% yield; 1H NMR (400 MHz, CDCl3) δ 7.43 (t, J = 8.2 Hz, 2H), 6.86 (br d, J = 6.0 Hz, 2H), 6.62 (dd, J = 8.2, 3.0 Hz, 2H), 6.50 (dd, J = 8.1, 4.4 Hz, 2H), 5.22 (s, 2H), 3.80 (s, 6H), 3.73 (br s, 2H), 2.12 (br d, J = 6.6 Hz, 2H), 1.69 (br s, 2H), 1.31–1.26 (overlapping d, J = 17.0 Hz, and m, 22 H); 13C NMR (100 MHz, CDCl3) 165.7 (d, J = 2.0 Hz), 165.3 (d, J = 15.3 Hz), 161.3 (d, J = 2.1 Hz), 136.8, 106.4 (d, J = 5.4 Hz), 103.8 (d, J = 5.8 Hz), 101.6 (d, J = 91.6 Hz), 74.6 (d, J = 48.3 Hz), 55.6, 53.5, 34.3 (d, J = 74.0 Hz), 32.2, 24.6, 24.5; 31P NMR (162 MHz, CDCl3) δ 62.36; HRMS (ESI) m/z 647.26433 (M + H+), calcd for C32H45O8N2P2 647.26457.(2R,2’R,3S,3’S)-N,N’-(1,2-phenylene)bis(3-(tert-butyl)-4-methoxy-2H-benzo[d][1,3]oxaphosphole-2-carboxamide 3-oxide) (3c): 3.07 g, 60% yield; 1H NMR (400 MHz, CDCl3) δ 8.65 (s, 2H), 7.54 (m, 2H), 7.44 (t, J = 8.2 Hz, 2H), 7.16 (m, 2H), 6.68 (dd, J = 8.3, 2.8 Hz, 2H), 6.54 (dd, J = 8.2, 4.4 Hz, 2H), 5.33 (s, 2H), 3.86 (s, 6H), 1.34 (d, J = 17.1 Hz, 18 H); 13C NMR (100 MHz, CDCl3) 165.2 (d, J = 14.9 Hz), 164.5 (d, J = 2.6 Hz), 161.3 (d, J = 2.2 Hz), 137.0, 129.9, 126.7, 126.2, 106.5 (d, J = 5.4 Hz), 104.1 (d, J = 5.8 Hz), 102.0 (d, J = 91.6 Hz), 75.5 (d, J = 47.5 Hz), 55.7, 34.4 (d, J = 73.8 Hz), 24.7; 31P NMR (202 MHz, CDCl3) δ 63.9; HRMS (ESI) m/z 641.21767 (M + H+), calcd for C32H39O8N2P2 641.21762.(2R,2’R,3S,3’S)-N,N’-((1R,2R)-1,2-dicyclohexylethane-1,2-diyl)bis(3-(tert-butyl)-4-methoxy-2H-benzo[d][1,3]oxaphosphole-2-carboxamide 3-oxide) (3d): 3.03 g, 50% yield; 1H NMR (400 MHz, CDCl3) δ 7.45 (t, J = 8.2 Hz, 2H), 6.72–6.68 (overlapping s and dd, J = 8.2, 3.0 Hz, 4H), 6.54 (dd, J = 8.1, 4.3 Hz, 2H), 4.92 (d, J = 0.48 Hz, 2H), 4.06–4.00 (m, 6H), 3.88 (s, 6H), 1.74–1.67 (m, 9H), 1.54–1.34 (overlapping d, J = 17.0 Hz, and m, 24 H), 1.22–1.02 (m, 10 H), 0.9–0.81 (m, 2H); 13C NMR (100 MHz, CDCl3) 165.1 (d, J = 2.3 Hz), 164.4 (d, J = 13.9 Hz), 161.1 (d, J = 2.1 Hz), 136.7, 106.5 (d, J = 5.2 Hz), 104.1 (d, J = 5.7 Hz), 103.1 (d, J = 90.5 Hz), 75.6 (d, J = 48.9 Hz), 55.6, 54.7, 38.9, 34.1 (d, J = 74.4 Hz), 30.5, 27.3, 26.2, 26.16, 26.11, 25.2; 31P NMR (162 MHz, CDCl3) δ 61.64; HRMS (ESI) m/z 757.37417 (M + H+), calcd for C40H59O8N2P2 757.37412.(2R,2’R,3S,3’S)-N,N’-((1R,2R)-1,2-diphenylethane-1,2-diyl)bis(3-(tert-butyl)-4-methoxy-2H-benzo[d][1,3]oxaphosphole-2-carboxamide 3-oxide) (3e): 2.98 g, 50% yield; 1H NMR (500 MHz, CDCl3) δ 7.52 (br d, J = 2.8 Hz, 2H), 7.43 (t, J = 8.2 Hz, 2H), 7.11–7.06 (m, 10 H), 6.70 (dd, J = 8.3, 2.5 Hz, 2H), 6.50 (dd, J = 8.1, 4.3 Hz, 2H), 5.40–5.38 (m, 2H), 5.00 (s, 2H), 3.85 (s, 6H), 1.35 (d, J = 16.9 Hz, 18 H); 13C NMR (100 MHz, CDCl3) 165.5 (d, J = 2.4 Hz), 164.5 (d, J = 14.1 Hz), 161.1 (d, J = 2.3 Hz), 137.7, 136.8, 128.5, 127.74, 127.69, 106.5 (d, J = 5.3 Hz), 104.0 (d, J = 5.8 Hz), 102.7 (d, J = 91.1 Hz), 75.3 (d, J = 48.8 Hz), 59.1, 55.6, 34.1 (d, J = 74.5 Hz), 25.1 (d, J = 0.8 Hz); 31P NMR (202 MHz, CDCl3) δ 62.13; HRMS (ESI) m/z 745.28012 (M + H+), calcd for C40H47O8N2P2 745.28022.
- 12.(S)-isomer was produced by comparing to the literature data in ref. 3b.
- 13.General procedure for enone reduction: to a stirring solution of 1a (60 mg, 0.27 mmol) and catalyst 3a (0.027 mmol, 10 mol%) in acetonitrile (2 mL) at 0 °C was added HSiCl3 (1.35 mmol, 5 equiv) drop-wise, and stirred at 0 °C for 20 h. The reaction was then quenched with a solution of 5N aqueous NaOH (2 mL), and warmed to room temperature. The mixture was diluted with EtOAc and water. The phases were separated and the aqueous phase was further extracted once with EtOAc. The combined organic layers were washed with water followed by brine, dried with Na2SO4 and concentrated. The product was purified on silica with a mixture of hexanes/EtOAc (10:1) to obtain colorless oil after dryness. (S)-1,3-diphenylbutan-1-one (2a): 90% yield, 89.2:10.8 er; SFC ES Industries CCA column 4.6 × 100 mm, 3 μm: 35 °C, A: CO2, B: isopropanol; gradient: 1% B to 3% B in 3 min, to 50% B in 5 min; 3 mL/min, λ = 220 nm, tmajor = 2.50 min, tminor = 2.80 min. NMR data match those reported in the literature.17 1H NMR (500 MHz, CDCl3) δ 7.96 (d, J = 7.9 Hz, 2H), 7.57 (t, J = 7.3 Hz, 1H), 7.47 (t, J = 7.6 Hz, 2H), 7.35–7.21 (m, 4H), 7.35–7.28 (m, 2H), 7.23 (t, J = 6.8 Hz, 1H), 3.54 (sextet, J = 6.9 Hz, 1H), 3.33 (dd, J = 16.5 and 5.7 Hz, 1H), 3.22 (dd, J = 16.5 and 8.3 Hz, 1H), 1.37 (d, J = 6.9 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 199.1, 146.6, 137.2, 133.0, 128.6, 128.5, 128.1, 126.9, 126.3, 47.0, 35.6, 21.9.(S)-1-phenyl-3-(4-(trifluoromethyl)phenyl)butan-1-one (2b): 92% yield, 89.4:10.6 er; SFC ES Industries CCA column 4.6 × 100 mm, 3 μm: 35 °C, A: CO2, B: isopropanol; gradient: 1% B to 3% B in 3 min, to 50% B in 5 min; 3 mL/min, λ = 220 nm, tmajor = 1.69 min, tminor = 1.91 min. NMR data match those reported in the literature.18 1H NMR (400 MHz, CDCl3) δ 7.97–7.92 (m, 2H), 7.60–7.56 (m, 3H), 7.50–7.45 (m, 2H), 7.41 (d, J = 8.3 Hz, 2H), 3.69 (sextet, J = 6.9 Hz, 1H), 3.35 (dd, J = 16.8 and 6.4 Hz), 3.25 (dd, J = 16.9 and 7.5 Hz, 1H), 1.39 (d, J = 7.0 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 198.4, 150.6, 137.0, 133.2, 128.6, 128.0, 127.3, 125.5 (q, J = 3.8 Hz) 46.5, 35.3, 21.9.(S)-3-(4-chlorophenyl)-1-phenylbutan-1-one (2c): 94% yield, 90.2:9.8 er; SFC ES Industries CCA column 4.6 × 100 mm, 3 μm: 35 °C, A: CO2, B: methanol; gradient: 1% B to 3% B in 3 min, to 50% B in 5 min; 3 mL/min, λ = 220 nm, tmajor = 2.93 min, tminor = 3.51 min. NMR data match those reported in the literature.18 1H NMR (400 MHz, CDCl3) δ 7.95–7.92 (m, 2H), 7.60–7.56 (m, 1H), 7.50–7.45 (m, 2H), 7.30–7.27 (m, 2H), 7.25–7.21 (m, 2H), 3.52 (sextet, J = 6.9 Hz, 1H), 3.29 (dd, J = 16.7 and 6.3 Hz), 3.2 (dd, J = 16.7 and 7.7 Hz, 1H), 1.35 (d, J = 7.0 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 198.7, 145.0, 137.1, 133.1, 131.9, 128.6, 128.3, 128.0, 46.8, 35.0, 22.0.(S)-3-(2-methoxyphenyl)-1-phenylbutan-1-one (2d): 89% yield, 88.1:11.9 er; SFC Lux Cel 1 column 4.6 × 100 mm, 3 μm: 35 °C, A: CO2, B: methanol; gradient: 1% B to 3% B in 3 min, to 50% B in 5 min; 3 mL/min, λ = 220 nm, tmajor = 3.94 min, tminor = 4.27 min. NMR data match those reported in the literature.19 1H NMR (500 MHz, CDCl3) δ 7.90 (d, J = 7.8 Hz, 2H), 7.45 (t, J = 7.3 Hz, 1H), 7.36 (t, J = 7.6 Hz, 2H), 7.15 (d, J = 7.3 Hz, 1H), 7.11 (t, J = 7.7 Hz, 1H), 6.85 (t, J = 7.4 Hz, 1H), 6.77 (d, J = 8.1 Hz, 1H) 3.78–3.73 (m, 1H), 3.73 (s, 3H), 3.27 (dd, J = 15.8, 4.8 Hz, 1H), 2.97 (dd, J = 15.7, 9.2 Hz, 1H), 1.23 (d, J = 7.0 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 199.7, 156.9, 137.3, 134.5, 132.8, 128.5, 128.2, 127.2, 126.9, 120.7, 110.6, 55.3, 46.0, 29.6, 19.7.
- 14.Gaussian 16, Revision B.01, Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X, Caricato M, Marenich AV, Bloino J, Janesko BG, Gomperts R, Mennucci B, Hratchian HP, Ortiz JV, Izmaylov AF, Sonnenberg JL, Williams-Young D, Ding F, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski VG, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery JA Jr., Peralta JE, Ogliaro F, Bearpark MJ, Heyd JJ, Brothers EN, Kudin KN, Staroverov VN, Keith TA, Kobayashi R, Normand J, Raghavachari K, Rendell AP, Burant JC, Iyengar SS, Tomasi J, Cossi M, Millam JM, Klene M, Adamo C, Cammi R, Ochterski JW, Martin RL, Morokuma K, Farkas O, Foresman JB, and Fox DJ, Gaussian, Inc., Wallingford CT, 2016. [Google Scholar]
- 15.(a) Becke AD J. Chem. Phys 1993, 98, 5648. [Google Scholar]; (b) Lee C; Yang W; Parr RG Phys. Rev. B 1988, 37, 785. [DOI] [PubMed] [Google Scholar]
- 16.Sugiura M; Ashikari Y; Takahashi Y; Yamaguchi K; Kotani S; Nakajima MJ Org. Chem 2019, 84, 11458. [DOI] [PubMed] [Google Scholar]
- 17.Kanazawa Y; Tshchiya Y; Kobayashi K; Shiomi T; Itoh J-I; Kikuchi M; Yamamoto Y; Nishiyama H Chem. Eur. J 2006, 12, 63. [DOI] [PubMed] [Google Scholar]
- 18.Miaskiewicz S; Reed JH; Donets PA; Oliveira CC; Cramer N Angew. Chem. Int. Ed 2018, 57, 4039. [DOI] [PubMed] [Google Scholar]
- 19.Yamamoto Y; Kurihara K; Takahashi Y; Miyaura N Molecules 2013, 18, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.