

Abstract
Purpose:
MET tyrosine kinase inhibitors (TKIs) can achieve modest clinical outcomes in MET exon 14-altered lung cancers, likely secondary to primary resistance. Mechanisms of primary resistance remain poorly characterized and comprehensive proteomic analyses have not previously been performed.
Experimental Design:
We performed hybrid capture-based DNA sequencing, targeted RNA sequencing, cell-free DNA sequencing, mass spectrometry (SRM-MS), and immunohistochemistry (IHC) on patient samples of MET exon 14-altered lung cancers treated with a MET TKI. Associations between overall response rate (ORR), progression free survival (PFS), and putative genomic alterations and MET protein expression were evaluated.
Results:
Seventy-five of 168 MET exon 14-altered lung cancers received a MET TKI. Previously undescribed (zygosity, clonality, whole genome duplication) and known (copy number focality, tumor mutational burden, mutation region/type) genomic factors were not associated with ORR/PFS (P > 0.05). In contrast, MET expression was associated with MET TKI benefit. Only cases with detectable MET expression by SRM-MS (N = 15) or IHC (N = 22) responded to MET TKI therapy, and cancers with H-score ≥ 200 had a higher PFS than cancers below this cutoff (10.4 vs 5.5 months, respectively; hazard ratio 3.87, P = 0.02).
Conclusions:
In MET exon 14-altered cancers treated with a MET TKI, a comprehensive analysis of previously unknown and known genomic factors did not identify a genomic mechanism of primary resistance. Instead, MET expression correlated with benefit, suggesting the potential role of interrogating the proteome in addition to the genome in confirmatory prospective trials.
INTRODUCTION
MET exon 14 alterations occur in 4% of non-small lung cancers (NSCLCs). Most MET exon 14 alterations involve splice acceptor or donor sites flanking exon 14 which lead to exclusion of exon 14 that contains the Y1003 ubiquitin-binding site. Other alterations include MET fusions and Y1003 substitutions that recapitulate the biology of MET exon 14 splice site mutations. This family of MET alterations is presumed to lead to impaired MET protein ubiquitination, degradation, and increased oncogenic signaling (1).
MET tyrosine kinase inhibitors (TKIs) such as the multikinase inhibitor crizotinib are clinically active in MET exon 14-altered NSCLCs. In these tumors, crizotinib achieved an overall response rate (ORR) of 32% and a median progression-free survival (PFS) of 7.3 months (2). The more selective and potent MET inhibitors, tepotinib and capmatinib, are also active (3,4) and were recently approved by Japanese regulatory authorities and the US FDA, respectively, for treatment of MET exon 14-altered NSCLCs (5,6). Unfortunately, response rates for many MET TKIs are modest relative to the activity of targeted therapy in other oncogene-driven lung cancers (e.g. those with sensitizing EGFR mutations) where ORRs are more consistently greater than 60%. For example, while capmatinib achieved an ORR of 68% in treatment-naïve patients, the ORR was only 41% in pre-treated patients (4). The ORR with tepotinib was 48%, and response was similar regardless of prior therapy (3). The response to savolitinib, another selective MET inhibitor, was 48%(7). The fact that most of the reported response rates fall below 50% suggests that primary resistance to MET inhibition may represent a major issue.
We and others have reported that genomic profiling has been unsuccessful at identifying biomarkers of resistance to MET inhibitors in MET exon 14-altered lung cancers (2–4,8). Variability in genomic factors, such as MET exon 14 splice region or mutation type, have not been associated with lack of clinical benefit. We hypothesized that primary therapeutic resistance is mediated by one or more previously unexplored pretreatment genomic or proteomic factors such as zygosity, clonality, mutational burden, or protein expression.
METHODS
Clinical Samples and Study Endpoints
All human tissues were obtained with Institutional Review Board approval and patients provided written informed consent. The study was conducted in accordance with the U.S. Common Rule. Patients were retrospectively included if they had advanced lung cancers with MET exon 14 alterations identified by DNA- or RNA-based next-generation sequencing diagnosed between 2008 and 2018. Clinicopathologic characteristics, including age, sex, histology, and smoking history were collected. Patients were followed until June 2019. Response to therapy was evaluated using RECIST v1.1.
DNA- and RNA-based Next-generation Sequencing
Tumor nucleic-acid testing was performed with targeted next-generation sequencing (NGS) of DNA using MSK-IMPACT or Foundation One (Foundation Medicine, Cambridge, MA) (9). Targeted NGS of tumor RNA was performed using an anchored multiplex polymerase chain reaction (MSK Solid Fusion Panel, NY, NY, USA) (10).
Clonality and Zygosity Analyses
Samples sequenced by MSK-IMPACT were analyzed for zygosity using FACETS (11). FACETS is an open- source integrated software tool designed to quantify several factors, including zygosity, tumor purity, ploidy, and clonal heterogeneity. Mutations called by the MSK-IMPACT pipeline were annotated for cancer cell fraction (CCF) using FACETS-suite (12); variants were classified as clonal if the upper-bound CCF was > 85%. MET zygosity was labelled as high amplification if ≥ 6 total copies. Focal amplifications were called if the MET copy number was ≥ 6 and ≥ 3 more than the calculated copies of chromosome 7q. For other genetic alterations, amplification was defined as > 2.0-fold change and deletion was defined as < −2.0-fold change.
Protein Expression Evaluation
Protein expression of pre-TKI tumor samples was performed using a targeted selected reaction monitoring-mass spectrometry panel (SRM-MS; NantOmics) as previously described (13). In brief, tissues from sectioned formalin-fixed paraffin-embedded (FFPE) blocks were placed onto DIRECTOR microdissection slides. These were deparaffinized and stained with hematoxylin, then microdissected and solubilized to tryptic peptides. These peptides were analyzed with TSQ Quantiva triple-quadrupole mass spectrometers (Thermo Scientific, San Jose, CA, USA). Standardization for quantification and quality control for MET were previously reported. Positive MET expression by SRM-MS was previously defined as > 150 amol/μg (13).
Peripheral hepatocyte growth factor (HGF) Measurement
Peripheral blood was collected from select patients pre-, on-, and post-progression of a MET TKI and plasma was separated and frozen at −80°C until further evaluation. Healthy control human plasma from five subjects was obtained from the New York Blood Center, aliquoted into individual cryovials, and frozen at −80°C until further evaluation. Peripheral HGF levels from each patient with available sample were quantified with an HGF ELISA kit (SHG00B, R&D Systems, Minneapolis, MN, USA) (14). The data was normalized such that the mean level of five healthy control plasma samples were kept equivalent across experiments.
Statistics
Mann-Whitney test was used to compare ORR between groups. For ORR in 3 or more groups, Kruskal- Wallis test was used. P-value for PFS was determined using the log-rank test. P-values ≤ 0.05 were considered significant. Hazard ratios and confidence intervals were generated using the Mantel- Haenszel test. Statistical analyses were performed with Prism 7 (GraphPad Software).
RESULTS
Among 168 patients with MET exon 14-altered NSCLCs, 167 were identified by DNA-based NGS (Supplementary Fig. S1). The remaining case was identified by an RNA-based NGS assay (MSK-Fusion). Of cases identified by DNA with sufficient additional tissue, subsequent RNA testing confirmed exon skipping in 97% (N = 97/100). RNA testing helped identify deep intronic or exonic MET exon 14 alterations that were difficult to interpret from DNA sequencing alone (Fig. 1). For instance, MSK-Fusion confirmed two cases of MET exon 14 skipping that were deep into intron 13 (MET c.2888-32_2888-29delinsG, c.2888-46_2888- 22delCATGATAGCCGTCTTTAACAAGCTC) and one deep into exon 14 (c.2913_2962delCGATGCAAGAGTACACACTCCTCATTTGGATAGGCTTGTAAGTGCCCGAA). In contrast, MET exon 14 skipping was not confirmed in three samples, including two mutations deep within exon 14 (c.2993_3008delinsG and c.2967_2976delinsGGCAGTCCAA) and one mutation deep into intron 14 (c.3028+1221G > A). This underscores the complementary diagnostic utility of RNA analysis as a means of determining that select mutations detected by DNA analysis are unexpected to lead to splicing defects.
Figure 1. Landscape of MET exon 14-alterations in lung cancer and sequencing by RNA-based anchored multiple PCR.

For many patients, the likelihood of exon 14 being skipped in their cancer based on DNA-based sequencing was high (light-green). The rest of the cases were nominated for RNA-based confirmation via the MSK-Fusion panel. In patients, with sufficient tissue for MSK-Fusion testing, cases where exon 14 skipping was confirmed are shown in dark green, while those where exon 14 skipping was not observed in RNA are shown in red. Of note, one mutation not confirmed by RNA (c.3028+1221G > A) is deep into intron 14 and is not shown. Patients whose cancers were nominated for MSK-Fusion testing but did not have sufficient tissue for test completion are shown in gray. +, 2 patients; *, 3 patients.
Of these patients, 75 received a MET TKI. Demographics are summarized in Table 1. Most had lung adenocarcinoma (79%, N = 59/75) or sarcomatoid carcinoma (9%, N = 7/75). The majority of patients (88%, N = 66) received crizotinib. Most MET exon 14 alterations were detected by MSK-IMPACT (95%, N = 71/75); Foundation One detected the rest (5%, N = 4/75). The first major observation was that MET exon 14 alterations were highly heterogeneous (Fig. 2A, B) beyond previously described factors (e.g. splice site region/mutation type). Specifically, we examined novel genomic characteristics such as zygosity, clonality, and whole genome duplication, in addition to concurrent MET amplification, focality, and tumor mutational burden (Fig. 2B), the frequencies of which are in Table 2. Interestingly, none of these factors correlated with ORR (P > 0.05) or PFS (P > 0.05, Table 2, Supplementary Fig. S2, S3) with MET TKI therapy, although no responses were seen in subclonal tumors versus clonal ones (ORR 0% and 44%, respectively; P = 0.50). Tumor mutational burden ranged from 0.9-18 mutations per megabase (Fig. 2C) and did not affect MET TKI ORR (P = 0.44) or PFS (P = 0.80; Table 2, Supplementary Fig. S3B).
Table 1.
Clinicopathologic features of patients with advanced MET exon 14-altered lung cancers.
| Patients Treated with MET TKI (N = 75) | ||
|---|---|---|
| Age, median (range) | 73 years (44-91 years) | |
| Sex, % (N) | Female | 52% (39) |
| Cigarette smoking, % (N) | Never Former or Current |
44% (33) 56% (42) |
| Histology, % (N) | Adenocarcinoma Sarcomatoid Other |
79% (59) 9% (7) 12% (9) |
| Number of MET TKIs, % (N) | 1 2 or more |
75% (56) 25% (19) |
| First MET TKI received, % (N) | Crizotinib Cabozantinib Tepotinib |
88% (66) 1% (1) 11% (8) |
| DNA-based NGS, % (N) | MSK-IMPACT Foundation One |
95% (71) 5% (4) |
| MET Splice Site Region#, % (N) | Intron 13 Acceptor Intron 14 Donor Fusion Other Not Detected by DNA-based NGS |
33% (25) 60% (45) 3% (2) 3% (2) 1% (1) |
| MET Mutation Type#, % (N) | Base substitution Insertion/deletion Large deletion (>35 bp) Fusion Other Note Detected by DNA-based NGS |
48% (36) 35% (26) 8% (6) 3% (2) 3% (2) 4% (3) |
Percentages in select demographic groups do not add up to 100% due to rounding.
Figure 2. Pre-treatment genomic features of MET exon 14-altered lung cancers and MET inhibitor activity.

(A) Progression-free survival and best objective response with MET inhibition. Each column is an individual patient/biopsy (N = 75). (B) Splice site region, zygosity, whole genome duplication, copy number changes, focality, and clonality (clonal if > 80%). (C) Tumor mutational burden (TMB). (D) Sample origin, previous exposure to chemotherapy, and concurrent genomic alterations.
Table 2.
Objective Response and Progression-Free Survival by MET Genomic Aberrations.
| Factors | N (%) | Responses (ORR %) | P value | 95% CI | PFS (months) | P value | HR | 95% CI | |
|---|---|---|---|---|---|---|---|---|---|
| MET Zygosity | WT Copies | 23 (31%) | 7 (30%) | 0.88 | 15% to 51% | 7.9 | 0.84 | ||
| CN LOH | 24 (32%) | 9 (38%) | 21% to 57% | 7.2 | |||||
| Heterozygous loss | 5 (7%) | 1 (20%) | 2% to 64% | 9 | |||||
| Amplification | 9 (12%) | 3 (33%) | 12% to 65% | 6.5 | |||||
| NE | 14 (19%) | ||||||||
| Whole Genome Duplication (WGD) | Yes | 17 (23%) | 9 (53%) | 0.14 | 31% to 74% | 7.3 | 0.66 | 1.2 | 0.6 to 2.3 |
| No | 50 (67%) | 15 (30%) | 19% to 44% | 8.8 | |||||
| NE | 8 (11%) | ||||||||
| MET Clonality | Subclonal | 2 (3%) | 0 (0%) | 0.5 | 0% to 71% | 4.5 | 0.1 | 2.0 | 0.5 to 8.2 |
| Clonal | 43 (57%) | 19 (44%) | 30% to 59% | 8.9 | |||||
| NE | 30 (40%) | ||||||||
| MET Focality | Broad | 2 (3%) | 1 (50%) | 1.00 | 9% to 91% | 11.0 | 0.51 | 0.6 | 0.1 to 3.3 |
| Focal | 7 (9%) | 2 (29%) | 8% to 65% | 5.5 | |||||
| MET non-amplified§ | 65 (87%) | ||||||||
| NE | 1 (10%) | ||||||||
| Tumor Mutational Burden (TMB) | ≥4.5 mut/Mb | 33 (44%) | 14 (42%) | 0.44 | 27% to 59% | 7.4 | 0.80 | 0.9 | 0.5 to 1.7 |
| <4.5 mut/Mb | 33 (44%) | 10 (30%) | 17% to 47% | 7.2 | |||||
| NE | 9 (12%) | ||||||||
Percentages may not add up to 100% due to rounding.
ORR, objective response rate; PFS, progression-free survival; HR, hazard ratio; CI, confidence interval; WT, wildtype; CN LOH, copy neutral loss of heterozygosity; mut, mutations; Mb, megabase;
MET focality can only be evaluated in MET-amplified cases; NE, not evaluable (no evaluable biomarker and/or not RECIST-evaluable)
Pretreatment concurrent genomic alterations were then assessed. These commonly involved TP53 (41%), MDM2 (29%), CDK4 (21%), TERT (19%), and CDKN2A (17%, Fig. 2D). Most alterations did not impact ORR or PFS (Supplementary Table S1, Fig. S3C–G). Previous studies showed that acquired RAS activation was a mechanism of MET inhibitor resistance (15,16). However, RAS or NF1 alterations did not affect response (20% versus 39% in mutated versus wildtype, respectively; P = 0.64; Supplementary Table S1, Fig. S3H, S4A) or PFS (7.5 versus 7.3 months in mutated and wildtype, respectively; P = 0.36; Supplementary Table S1, Fig. S3H). In contrast, the ORR was numerically lower in tumors with PI3KCA or PTEN co-mutations (13% versus 41% in mutated versus wildtype, respectively; P = 0.24; Supplementary Table S1, Fig. S4B).
Conceptually, MET exon 14 alterations are thought to mediate oncogenesis by increasing MET expression. However, the degree of MET protein expression by targeted selected reaction monitoring mass spectrometry (SRM-MS) proteomics or immunohistochemistry (IHC; SP44 antibody, Ventana) was heterogeneous. Surprisingly, only 67% (N = 10/15) of cases had detectable MET expression by SRM-MS in pretreatment biopsies (Fig. 3A) despite the high sensitivity of this assay (13). MET IHC H-scores (% cells multiplied by 1/2/3+ MET staining intensity) were: 0 (N = 1), 1-149 (N = 3), 150-199 (N = 5), and ≥200 (N = 13) (Fig. 3B); MET expression by SRM-MS correlated with IHC H-scores (Spearman rho = 0.77; P = 0.008, Fig. 3C). High MET IHC expression (H-score ≥ 200) clustered with higher MET copy number (> 3 copies, Supplementary Fig. S5A).
Figure 3. MET expression in MET exon 14-altered lung cancers and acquired resistance.
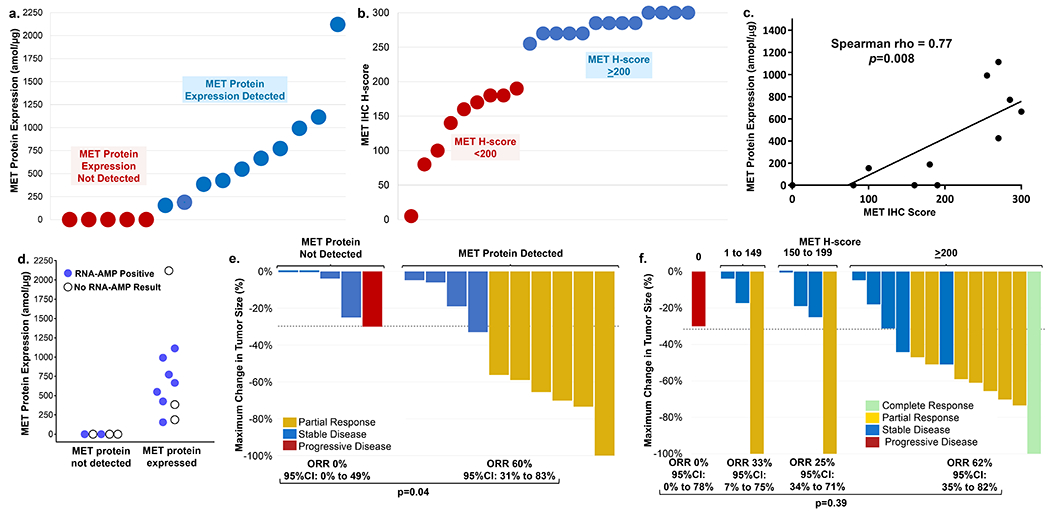
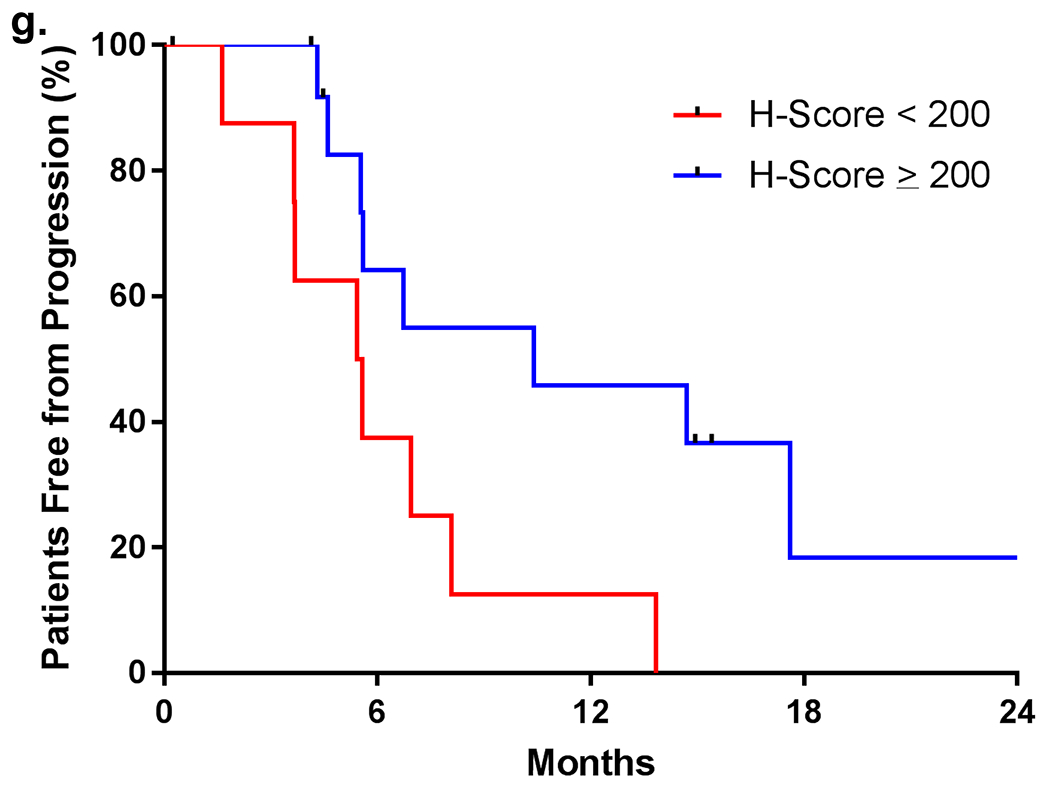
(a) MET protein expression by mass spectrometry (SRM-MS). (b) MET expression by immunohistochemistry (IHC). (c) Correlation of MET protein expression between SRM-MS and IHC. (d) MET exon 14 skipping detected by RNA-based anchored multiplex PCR (blue – skipping present; open circle– insufficient tissue). (e) Best response to MET inhibition by IHC expression. (f) Best response to MET inhibition by protein expression (SRM-MS). (g) Progression-free survival in cancers stratified by H-score.
Factors responsible for undetectable MET expression were explored. DNA-based NGS, MSK-Fusion, and SRM-MS were performed to determine if DNA-level exon 14 alteration resulted in RNA-level loss of exon 14 and decreased MET protein expression. MET expression was undetectable in two tumors with DNA- and RNA-confirmed exon 14 skipping (Fig. 3D).
Response to MET inhibition was not observed in cancers with undetectable MET expression by SRM-MS. The ORR was 60% (N = 6 of 10) versus 0% (N = 0 of 5) in cases with detectable versus undetectable MET (P = 0.04; Fig. 3E). ORRs were higher (62%, N = 8 of 13) in cancers with an H-score ≥ 200 than with an H-score of 150-199 (25%, N = 1 of 4) or 1-149 (33%, N = 1 of 3; P = 0.39; Fig. 3F). No response was seen in the one case without MET expression by IHC. Depth of response was not associated with the degree of MET protein expression by SRM-MS or IHC. The median PFS was longer in tumors with an H-score ≥ 200 than an H-score < 200 (10.4 versus 5.5 months, respectively; hazard ratio 3.87, P = 0.02; Fig. 3G). The median PFS with crizotinib alone in tumors with an H-score ≥ 200 compared to an H-score < 200 was 6.7 versus 5.4 months, respectively; hazard ratio 2.65, P = 0.10), recognizing that the latter group included tumors that expressed MET, albeit at lower levels. No significant differences in clinicopathologic characteristics, including treatment with crizotinib or tepotinib, were seen in H-score ≥ 200 compared to H- score < 200 groups (Supplementary Table S2).
Cases with concomitant MET amplification had increased MET expression. Almost all cases with greater than neutral MET copy numbers by FACETS (copy number > 2) had H-scores of ≥ 250 (Supplementary Fig. S5A). Since MET expression increased with MET copy number gain in these cancers, we evaluated whether utilizing both MET copy number and H-score could further select for therapeutic benefit. MET exon 14-altered cases that either had low MET expression (H-score < 200) with neutral MET copy numbers (copy number = 2) or lost a mutated or wildtype MET allele (copy number < 2) were less likely to respond to MET TKI therapy (0%, N = 0/6; Supplementary Fig. S5B). Those that had gained MET alleles (copy number > 2) or had an H-score ≥ 200 were more likely to respond (64%, 9 of 14; P = 0.0141). The median PFS was also significantly longer in cases with MET copy number > 2 or MET IHC ≥ 200 (13.8 months, N = 14) compared to cases with MET copy number ≤ 2 and IHC < 200 (4.6 months; HR 10.5, 95% CI 2.3 to 47.8; P = 0.003; Supplementary Fig. S5C).
We then evaluated whether low or high levels of MET expression by IHC were associated with the presence of co-alterations. Tumors that expressed MET at low levels were more likely to have a concomitant mutation in the RAS or PIK3CA/PTEN pathways (44%; N = 4/9; Supplementary Table S3) than tumors that expressed MET at higher levels (0%; N = 0/13, P = 0.017). These results suggest that low MET-expressing tumors may be reliant on more than one oncogenic pathway and can bypass MET inhibition.
Although HGF is the ligand for MET (1), peripheral HGF was not associated with benefit from a MET inhibitor. HGF levels were obtained from 28 patients with MET exon14-altered lung cancers pretreatment (N = 9), on therapy (N = 15), and post-progression (N = 15; Supplementary Fig. S5D). These plasma levels were then pooled at each timepoint of therapy for statistical analysis. Compared to normal healthy controls (N = 5), HGF was elevated in patients treated with MET exon 14-altered lung cancers regardless of treatment with a MET TKI (P = 0.009). HGF levels did not differ across treatment phase (pre-TKI, on-TKI, post- progression; P = 0.91). While plasma HGF levels were higher in patients with MET exon 14-altered NSCLCs than healthy controls (P = 0.018), the degree of HGF elevation did not correlate with MET inhibitor response (P = 1.0; Supplementary Table S1).
Having explored primary resistance to MET inhibitor therapy, factors mediating acquired resistance were analyzed. Paired pre- and post-MET TKI biopsies were obtained. On-target acquired resistance (METD1228N or HGF amplification) was observed in 20% (3/15) of cases (Fig. 4A). Off-target resistance was found in 33% (5/15) of cases (Fig. 4A). No resistance mechanism was identified in the remaining cases (47%, 7/15). Plasma cell-free DNA collected post-resistance (Fig. 4B) identified no on-target resistance; however, a hotspot KRAS mutation was found (9%, 1/11).
Figure 4.
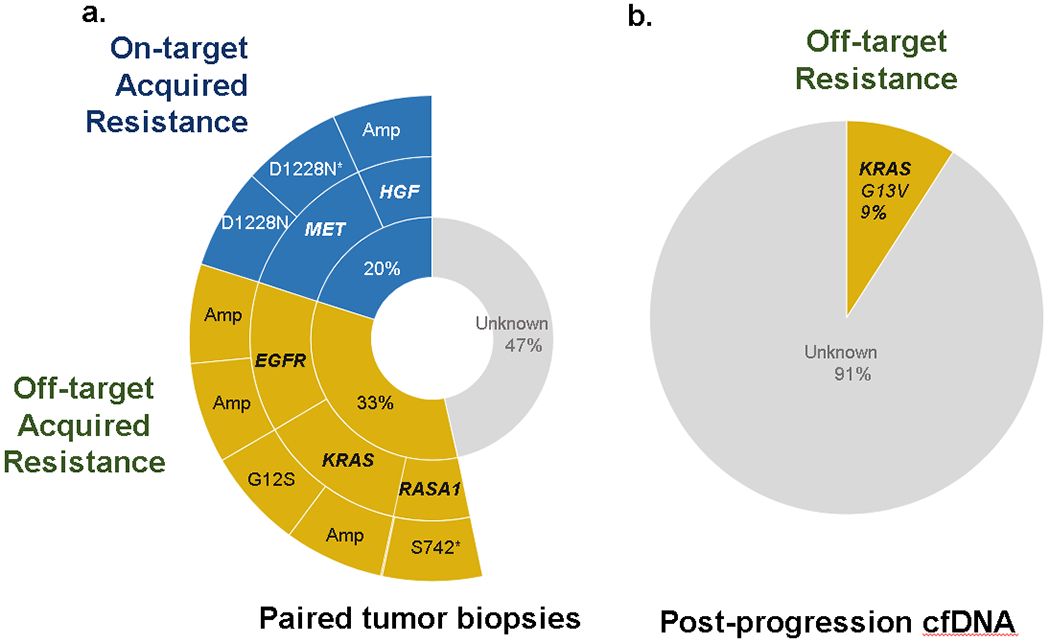
(a) On-/off-target mechanisms of acquired resistance in paired tumor biopsies. (b) Resistance detected in post-progression circulating tumor DNA.
Clonal heterogeneity was also seen in one acquired resistance sample (Supplementary Fig. S6). One clone (clone #1) had two MET exon 14 splice site mutations (c.3028G > C and c.2887+1G > A) prior to therapy and acquired a MET D1228N on-target mutation at progression. A second clone (clone #2) detected at progression was found to have a unique MET c.2888-22_2888-10delinsT exon 14 splice site mutation. The genomic profiles of each clone were distinct on NGS. These clones also had different histologies: clone #1 displayed predominantly a solid pattern whereas clone #2 displayed an acinar/lepidic pattern.
DISCUSSION
To date, trials of MET inhibitors in MET exon 14-altered NSCLCs have only performed genomic profiling to determine mechanisms of sensitivity or resistance, and these factors (MET exon 14 mutation region and type) did not predict benefit (2,8). We evaluated additional genomic factors (zygosity, clonality, whole genome duplication and tumor mutational burden) and did not find a correlation with response or survival. High MET expression level by IHC or SRM-MS, in contrast, was associated with benefit from MET inhibition.
In identifying patients for this study, we noted the significant heterogeneity of MET exon 14 alterations. Some MET alterations were deep into the intron 13 or exon 14 and thus pathogenicity was difficult to identify from DNA sequencing alone. Of these six cases, RNA-based testing with MSK-Fusion confirmed MET exon skipping in only half of the cases. Additionally, RNA-based testing uncovered MET exon 14 skipping in one case that was not found on DNA sequencing. This patient benefitted from crizotinib for 6.7 months. Previous studies have demonstrated the utility of using RNA sequencing in confirming MET exon 14 skipping or identifying these MET alterations in NSCLCs that were thought to be driver negative (10,17). These results reflect that DNA-based approaches are limited in their ability to capture the breadth of MET exon 14 alterations that complementary RNA-based approaches can detect (17,18).
We then evaluated whether pre-treatment genomic co-alterations could identify primary resistance to MET inhibition. While previous research showed that acquired RAS activation is a mechanism of MET inhibitor resistance, in this cohort, concomitant RAS/NF1 alterations did not impact response or survival (15,16). Common co-alterations, including TP53, MDM2, CDK4, TERT, and CDKN2A also did not influence these outcomes.
Beyond genomic heterogeneity, MET exon 14-altered NSCLCs were variable at the level of the proteome. Although MET exon 14 skipping leads to MET overexpression in pre-clinical models (19,20), one study demonstrated that MET protein expression was heterogeneous in early stage MET exon 14-altered lung cancers (21). Here, we show that MET protein is also heterogeneously expressed in metastatic MET exon 14-altered lung cancers. RNA transcripts were still measurable in cases with absent protein expression, implying the role of other regulatory factors (i.e. post-translational modification) in mediating low MET protein levels.
Interestingly, pretreatment MET protein expression by IHC or SRM-MS identified primary resistance to MET inhibition. Lung cancers with undetectable MET expression did not respond to MET TKI therapy. Pending further exploration, these findings are likely generalizable to more selective TKIs such as capmatinib or tepotinib that, like crizotinib, bind the MET kinase domain in a type I fashion; the lack of MET expression on the cell surface is likely to represent a shared liability for these drugs (19). In addition, high MET expression by IHC (or detectable MET protein levels by SRM-MS) correlated with an improved response and longer survival. The addition of MET copy number to IHC appeared to further isolate poor responders. In tumors with H-scores < 200, responses were still seen. However, no responses were seen in those with both low MET expression (H-score < 200) and either a neutral MET copy number or loss of a mutated or wildtype MET allele. H-score appeared to increase with MET copy number in this context.
MET protein expression is likely a surrogate marker of MET dependency in MET exon 14-altered lung cancers. In addition to correlating with response, tumors with low MET protein expression were more likely to have concomitant alterations in the RAS or PI3KCA/PTEN pathways than tumors with high MET protein expression. These results are in line with recent pre-clinical work showing primary resistance to MET inhibition in MET exon 14-altered cell lines that also had KRAS or PI3KCA/PTEN co-alterations (15,16,22). Thus, these low MET expressing tumors may rely on other oncogenic pathways, bypassing the effect of MET inhibition alone. In tumors that are less reliant on MET, other regulatory factors (e.g. post- translational modification) may contribute to the apparent absence of MET expression despite the presence of DNA- and RNA-level MET exon 14 skipping.
In the acquired resistance setting, recent studies have demonstrated that on-target mechanisms of acquired resistance to MET inhibition are uncommon in MET exon 14-altered NSCLCs (16,23). In our cohort, 20% of paired tumor tissue cases acquired de novo METD1228N or HGF amplification at resistance. Off-target mechanisms of resistance are more common and appear to be predominantly mediated by the RAS pathway. Here, 33% of paired biopsy cases developed new EGFR, KRAS, or RASA1 alteration at acquired resistance.
Our data suggests that even a thorough analysis of both known and previously undescribed factors fails to strongly nominate a genomic factor that mediates MET TKI benefit beyond the singular identification of a MET exon 14 alteration. In contrast, this series exposes a potential need to perform prospective proteomic profiling as a supplement to genomic testing to identify patients with MET-expressing cancers that may be more poised to benefit from MET TKI therapy, recognizing that this was a single-center experience that did not feature a large proportion of patients diagnosed with widely used assays in the community. In clinical trials, pre-treatment tumor biopsies should be tested for MET expression by both IHC, a practical assay that can be run in clinical laboratories, and a proteomic assay such as SRM-MS, with potentially increased reproducibility. In terms of limitations, it is important to recognize that this was a single-center experience that did not feature a large proportion of patients diagnosed with widely used assays in the community. Notably, the small sample size of this study is insufficient to support routine MET protein analysis as a standard of care test to exclude patients with MET exon 14-altered and non-MET-expressing cancers from MET TKI therapy, although it’s reasonable to closely monitor patients whose cancers fit this phenotype for primary progression.
In summary, pre-treatment genomic heterogeneity, including zygosity, clonality, or tumor mutational burden did not correlate with MET inhibitor benefit in MET exon 14-altered NSCLCs. Only undetectable MET protein expression resulted in decreased benefit from MET inhibition, a finding that should be validated in ongoing and future trials.
Supplementary Material
Translational Relevance.
MET inhibitors are active in MET exon 14-altered lung cancers. Crizotinib is listed in the National Cancer Center Guidelines and selective MET inhibitors are approved for this indication. For many of these agents, however, response and progression-free survival are modest compared to targeted therapy for other oncogene-driven lung cancers. We performed genomic and proteomic testing on pre-treatment samples from patients treated with a MET tyrosine kinase inhibitor. Genomic factors outside of an activating MET exon 14-alteration by next generation sequencing did not predict benefit with a MET tyrosine kinase inhibitor. In contrast, we show that MET expression by immunohistochemistry or mass spectrometry may predict benefit. These findings highlight that proteomic factors may modify response to targeted therapy in an oncogene-driven cancer.
Acknowledgements
We thank Dr. Marc Ladanyi for his comments and critical reading of the manuscript and Dr. Clare Wilhelm for his editorial support. We thank Dr. Emiliano Cocco and Dr. Maurizio Scaltriti for their scholarship and input in the manuscript.
Funding: This work was supported by the National Cancer Institute at the National Institutes of Health [grant numbers P30-CA-008748, P01-CA-129243, T32-CA-009207 to A.C.]
Disclosures:
JC, AC, LD, JG, OW, CM, AM, CF, KS, DA, RS, NR, AZ: declare no competing interests. RG: reports honoraria/speaker fees from Taiwan Lung Cancer Society. MO reports honoraria/speaker fees from PharmaMar, Novartis, Targeted Oncology, Bristol-Meyers Squibb, and Merck Sharp and Dohme. ARB reports stock holdings in Johnson and Johnson. YT is an employee of the FDA. FC is an employee of Astrazeneca. TH has a consulting or advisory role with NantOmics, LLC, and mProbe, Inc; and is an inventor of multiple issued and filed patents which are assigned to NantOmics, LLC. RB declares research funding from ArcherDx. BTL declares consulting fees from Genentech Roche, Thermo Fisher Scientific, Hengrui Therapeutics and Guardant Health. CMR declares honoraria/speaker/consulting fees from AbbVie, Amgen, Ascentage, Bicycle, Celgene, Daiichi Sankyo, Genentech Roche, Ipsen, Loxo, and PharmaMar; and has served on the advisory board for Harpoon and Elucida. MGK declares honoraria/speaker fees from AstraZeneca, Pfizer, Regeneron, WebMD, OncLive, Physicians Education Resources, Prime Oncology, Intellisphere, Creative Educational, Concepts, Peerview, i3 Health, Paradigm Medical Communications, AXIS, Carvive Systems, Astra Zeneca, and Research to Practive; fees for travel and lodging from AstraZeneca, Pfizer, Regeneron, and Genentech; research support from the National Cancer Institute of the National Institutes of Health, the Lung Cancer Research Foundation, Genentech Roche, and PUMA Biotechnology. MA declares honoraria/speaker fees from Biocartis and Invivoscribe relating to activities outside of the submitted work. PP declares honoraria/speaker fees from Takeda, AbbVie, Lilly, Bristol Meyers Squibb, Boehringer-Ingelheim, and Celgene; research funding from Celgene. AD has received honoraria/speaker fees from Medscape, OncLive, PeerVoice, Physicians Education Resources, Targeted Oncology, Research to Practice, TP Therapeutics, AstraZeneca, grants, Pfizer, Blueprint Medicines, Ignyta/Genentech/Roche, Takeda/Ariad/Millenium, Helsinn, Beigene, BergenBio, Hengrui Therapeutics, Exelixis, Loxo/Bayer/Lilly, Tyra Biosciences, Verastem, MORE Health, Wolters Kluwer, Loxo, Lilly, Abbvie; served/received fees for advisory board membership from TP Therapeutics, AstraZeneca, Pfizer, Blueprint Medicines, Ignyta/Genentech/Roche, Takeda/Ariad/Millenium, Helsinn, Beigene, BergenBio, Hengrui Therapeutics, Exelixis, Loxo/Bayer/Lilly, Tyra Biosciences, Verastem, MORE Health, Loxo, Abbvie; received research funding from the National Cancer Institute, Pfizer, Exelixis, Teva, Taiho, GlaxoSmithKline, PharmaMar, Foundation Medicine, Ignyta/Roche; non-financial support from Merck, Puma, Ignyta/Roche.
Footnotes
A. Zehir and A. Drilon co-supervised this article.
REFERENCES
- 1.Drilon A, Cappuzzo F, Ou SI, Camidge DR. Targeting MET in Lung Cancer: Will Expectations Finally Be MET? J Thorac Oncol 2017;12(1):15–26 doi 10.1016/j.jtho.2016.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Drilon A, Clark JW, Weiss J, Ou SI, Camidge DR, Solomon BJ, et al. Antitumor activity of crizotinib in lung cancers harboring a MET exon 14 alteration. Nat Med 2020;26(1):47–51 doi 10.1038/s41591-019-0716-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paik PK, Felip E, Veillon R, Sakai H, Cortot AB, Garassino MC, et al. Tepotinib in Non-Small-Cell Lung Cancer with MET Exon 14 Skipping Mutations. N Engl J Med 2020. doi 10.1056/NEJMoa2004407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wolf J, Seto T, Han JY, Reguart N, Garon EB, Groen HJM, et al. Capmatinib in MET Exon 14-Mutated or MET-Amplified Non-Small-Cell Lung Cancer. N Engl J Med 2020;383(10):944–57 doi 10.1056/NEJMoa2002787. [DOI] [PubMed] [Google Scholar]
- 5.KGaA M 2020. TEPMETKO (Tepotinib) Approved in Japan for Advanced NSCLC with METex14 Skipping Alterations. <https://www.emdgroup.com/en/news/tepotinib-25-03-2020.html>.
- 6.US Food and Drug Administration. 2020. FDA Approves First Targeted Therapy to Treat Aggressive Form of Lung Cancer. US Food and Drug Administration, <https://www.fda.gov/news-events/press-announcements/fda-approves-first-targeted-therapy-treat-aggressive-form-lung-cancer>.
- 7.Lu S, Fang J, Li X, Cao L, Zhou J, Guo Q, et al. Phase II study of savolitinib in patients (pts) with pulmonary sarcomatoid carcinoma (PSC) and other types of non-small cell lung cancer (NSCLC) harboring MET exon 14 skipping mutations (METex14+). Journal of Clinical Oncology 2020;38(15_suppl):9519- doi 10.1200/JCO.2020.38.15_suppl.9519. [DOI] [Google Scholar]
- 8.Lu S, Fang J, Cao L, Li X, Guo Q, Zhou J, et al. Abstract CT031: Preliminary efficacy and safety results of savolitinib treating patients with pulmonary sarcomatoid carcinoma (PSC) and other types of non-small cell lung cancer (NSCLC) harboring MET exon 14 skipping mutations. Cancer Research 2019;79(13 Supplement):CT031-CT doi 10.1016/j.jmoldx.2014.12.006. [DOI] [Google Scholar]
- 9.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. The Journal of molecular diagnostics : JMD 2015;17(3):251–64 doi 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Benayed R, Offin M, Mullaney K, Sukhadia P, Rios K, Desmeules P, et al. High Yield of RNA Sequencing for Targetable Kinase Fusions in Lung Adenocarcinomas with No Mitogenic Driver Alteration Detected by DNA Sequencing and Low Tumor Mutation Burden. Clin Cancer Res 2019;25(15):4712–22 doi 10.1158/1078-0432.CCR-19-0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shen R, Seshan VE. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res 2016;44(16):e131 doi 10.1093/nar/gkw520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bielski CM, Donoghue MTA, Gadiya M, Hanrahan AJ, Won HH, Chang MT, et al. Widespread Selection for Oncogenic Mutant Allele Imbalance in Cancer. Cancer Cell 2018;34(5):852–62 e4 doi 10.1016/j.ccell.2018.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Catenacci DV, Liao WL, Thyparambil S, Henderson L, Xu P, Zhao L, et al. Absolute quantitation of Met using mass spectrometry for clinical application: assay precision, stability, and correlation with MET gene amplification in FFPE tumor tissue. PLoS One 2014;9(7):e100586 doi 10.1371/journal.pone.0100586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Llovet JM, Pena CE, Lathia CD, Shan M, Meinhardt G, Bruix J, et al. Plasma biomarkers as predictors of outcome in patients with advanced hepatocellular carcinoma. Clin Cancer Res 2012;18(8):2290–300 doi 10.1158/1078-0432.CCR-11-2175. [DOI] [PubMed] [Google Scholar]
- 15.Suzawa K, Offin M, Lu D, Kurzatkowski C, Vojnic M, Smith RS, et al. Activation of KRAS Mediates Resistance to Targeted Therapy in MET Exon 14-mutant Non-small Cell Lung Cancer. Clin Cancer Res 2019;25(4):1248–60 doi 10.1158/1078-0432.CCR-18-1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rotow JK, Gui P, Wu W, Raymond VM, Lanman RB, Kaye FJ, et al. Co-occurring Alterations in the RAS-MAPK Pathway Limit Response to MET Inhibitor Treatment in MET Exon 14 Skipping Mutation-Positive Lung Cancer. Clin Cancer Res 2020;26(2):439–49 doi 10.1158/1078-0432.CCR-19-1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davies KD, Lomboy A, Lawrence CA, Yourshaw M, Bocsi GT, Camidge DR, et al. DNA-Based versus RNA-Based Detection of MET Exon 14 Skipping Events in Lung Cancer. J Thorac Oncol 2019;14(4):737–41 doi 10.1016/j.jtho.2018.12.020. [DOI] [PubMed] [Google Scholar]
- 18.Poirot B, Doucet L, Benhenda S, Champ J, Meignin V, Lehmann-Che J. MET Exon 14 Alterations and New Resistance Mutations to Tyrosine Kinase Inhibitors: Risk of Inadequate Detection with Current Amplicon-Based NGS Panels. J Thorac Oncol 2017;12(10):1582–7 doi 10.1016/j.jtho.2017.07.026. [DOI] [PubMed] [Google Scholar]
- 19.Kong-Beltran M, Seshagiri S, Zha J, Zhu W, Bhawe K, Mendoza N, et al. Somatic mutations lead to an oncogenic deletion of met in lung cancer. Cancer Res 2006;66(1):283–9 doi 10.1158/0008-5472.CAN-05-2749. [DOI] [PubMed] [Google Scholar]
- 20.Abella JV, Peschard P, Naujokas MA, Lin T, Saucier C, Urbe S, et al. Met/Hepatocyte growth factor receptor ubiquitination suppresses transformation and is required for Hrs phosphorylation. Mol Cell Biol 2005;25(21):9632–45 doi 10.1128/MCB.25.21.9632-9645.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Awad MM, Oxnard GR, Jackman DM, Savukoski DO, Hall D, Shivdasani P, et al. MET Exon 14 Mutations in Non-Small-Cell Lung Cancer Are Associated With Advanced Age and Stage-Dependent MET Genomic Amplification and c-Met Overexpression. J Clin Oncol 2016;34(7):721–30 doi 10.1200/JCO.2015.63.4600. [DOI] [PubMed] [Google Scholar]
- 22.Jamme P, Fernandes M, Copin MC, Descarpentries C, Escande F, Morabito A, et al. Alterations in the PI3K Pathway Drive Resistance to MET Inhibitors in NSCLC Harboring MET Exon 14 Skipping Mutations. J Thorac Oncol 2020;15(5):741–51 doi 10.1016/j.jtho.2020.01.027. [DOI] [PubMed] [Google Scholar]
- 23.Recondo G, Bahcall M, Spurr LF, Che J, Ricciuti B, Leonardi GC, et al. Molecular mechanisms of acquired resistance to MET tyrosine kinase inhibitors in patients with MET exon 14 mutant NSCLC. Clin Cancer Res 2020. doi 10.1158/1078-0432.CCR-19-3608. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.