

Summary
Responses to developmental and environmental cues are dependent on precise spatiotemporal control of gene transcription. Enhancers, which comprise DNA elements bound by regulatory proteins, can activate target genes in response to these external signals. Recent studies have shown that enhancers are transcribed to produce enhancer RNAs (eRNAs). Do eRNAs play a functional role in activating gene expression or are they non-functional byproducts of nearby transcription machinery? The unstable nature of eRNAs and overreliance on knockdown approaches have made elucidating the possible functions of eRNAs challenging. Herein, we focus on studies that have cloned eRNAs to study their function as transcripts, revealing roles for eRNAs in enhancer-promoter looping, recruiting transcriptional machinery, and facilitating RNA polymerase pause release to regulate gene expression.
Keywords: Enhancer, enhancer RNA, eRNA, noncoding RNA, transcription, gene regulation
Regulation of Gene Expression by Enhancers
The ability of a cell to respond to specific developmental or environmental stimuli requires the tight regulation of gene expression, which is controlled by a variety of genomic regulatory elements. One class of these regulatory elements is enhancers (see Glossary), which are genomic DNA sequences that act as nucleation sites for the binding of sequence-specific transcription factors (TFs) and the formation of transcription regulatory complexes (Fig. 1). In 1981, a 72 base pair sequence from Simian virus 40 (SV40) was the first enhancer cloned and functionally examined; it was able to enhance the expression of a heterologous human β-globin gene in a fusion construct [1–3]. Subsequently, an enhancer was identified in the intron of the immunoglobulin (Ig) heavy-chain gene [4–6]. Analysis of this enhancer led to the observation that enhancer activity can be cell-type specific, with the IgG enhancer only activated in lymphoid cell lines [4, 5]. The development and application of a host of next-generation sequencing-based assays to the global study of enhancers (Box 1) have led to the suggestion that the expression of the ~20,000 protein-coding genes in the human genome is regulated by hundreds of thousands of enhancers [7–10]. Not surprisingly, mutations in enhancers have been implicated in the mechanisms of disease, such as the activation of oncogenes and development of drug resistance in cancer cells [11–17].
Figure 1. Architecture of enhancer-promoter interaction.
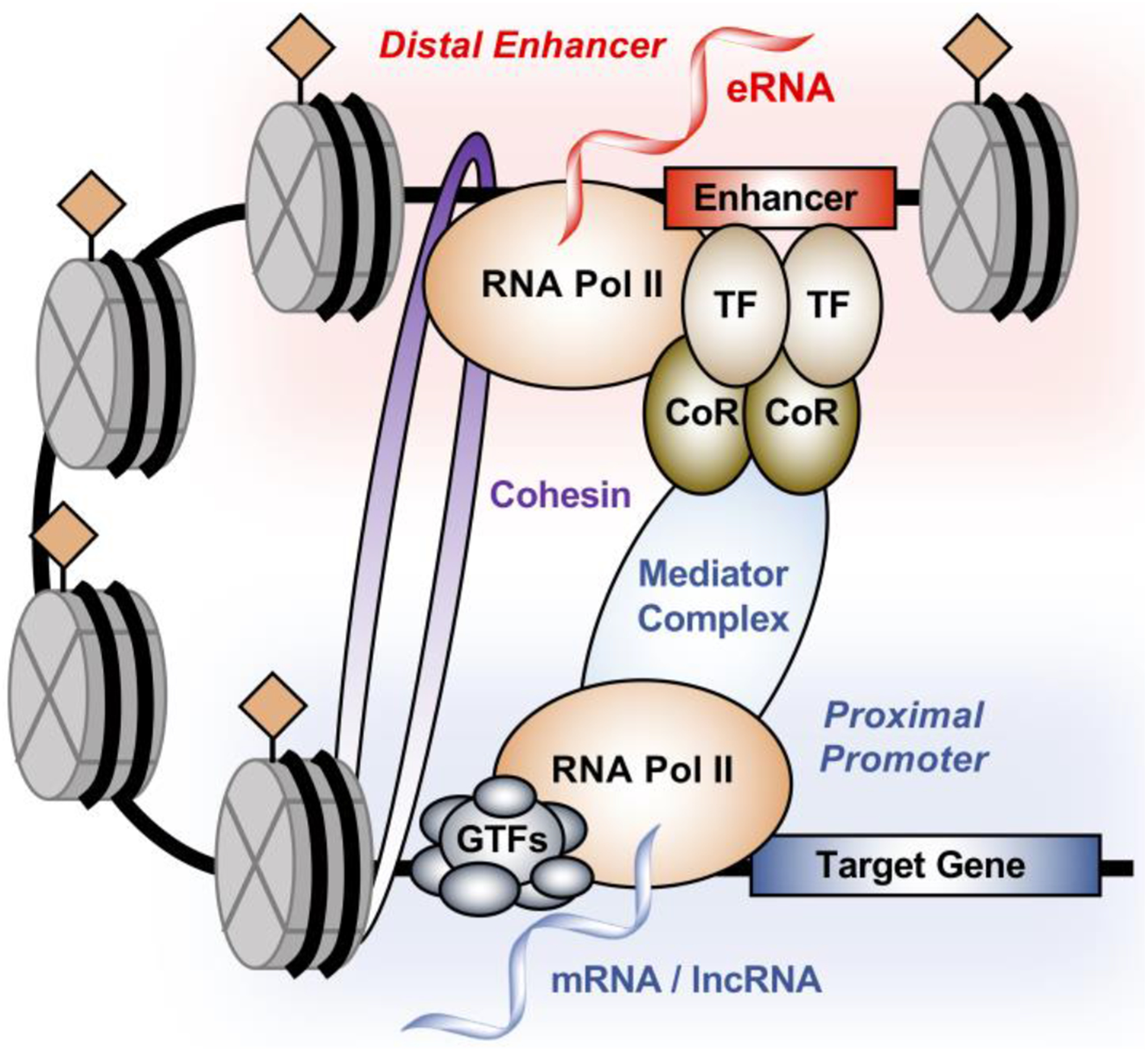
Enhancers are regulatory elements defined by genomic DNA sequences that act as nucleation sites for the binding of transcription factors (TFs) and coregulators (CoRs), leading to the formation of transcription regulatory complexes. Typically, these enhancers can also be characterized by an open chromatin environment, specific histone post-translational modifications, localization of RNA polymerase II (Pol II) and enhancer transcription to generate enhancer RNA (eRNA). The long-distance interaction between the enhancer and the promoter is mediated by proteins such as the Mediator and cohesion complexes, resulting in the stable formation of enhancer-promoter looping and facilitating the formation of regulatory complexes at the promoter of the target gene.
Box1. Common molecular features of active enhancers and their methods of detection.
Enhancers share a common set of molecular features, which can be explored using a variety of deep sequencing-based genomic assays.
Enhancers reside in an open or accessible chromatin environment and may be located within a gene (intragenic), in the intergenic regions between genes, or even on different chromosomes from where their target genes are located. The open regions of chromatin can be detected by genomic assays, such as DNase I hypersensitivity-sequencing (DNase-seq), formaldehyde-assisted isolation of regulatory elements-sequencing (FAIRE-seq), or transposase assisted measurements of chromatin accessibility-sequencing (ATAC-seq) [23, 24, 112–114].
Enhancers are enriched with a common set of histone modifications, as detected by chromatin immunoprecipitation-sequencing (ChIP-seq, see Glossary), including histone H3 lysine 4 monomethylation (H3K4me1) and histone H3 lysine 27 acetylation (H3K27ac) [25–27, 115, 116].
Enhancers are bound by coregulators and chromatin remodeling enzymes, as detected by ChIP-seq. These include p300/CBP and Mediator (coregulators) and Swi/Snf (chromatin remodeling enzymes) [28–30, 117–120].
Enhancers loop to target gene promoters, which in many cases requires architectural effects of cohesin and Mediator [22, 32–34]. Physical contact between enhancers and promoters, and long-range enhancer-promoter interactions forming chromatin loops, can be detected by assays such as ChIA-PET (chromatin interaction analysis with paired-end tag), chromosome conformation capture (3C), circular chromosome conformation capture (4C), chromosome conformation capture carbon copy (5C), and HiC [121–126].
Enhancers are actively transcribed, producing enhancer RNAs (‘eRNAs’) [44–48], which can be detected by assays such as GRO-seq, PRO-seq, and derivatives [12, 46, 58, 59, 68, 74]. This is reflected in the localization of RNA polymerase II (Pol II) to enhancers, as detected by ChIP-seq [26, 127]
Molecular Features of Enhancers
Although different TFs may exhibit cell type- or tissue-specific expression and regulate different repertoires of target genes, the enhancers that they form share several common molecular features [18–22] (Fig. 1, Box 1). For example, enhancers are typically (1) located in open regions of chromatin [23, 24], (2) enriched with a common set of histone modifications, including histone H3 lysine 4 monomethylation (H3K4me1, see Glossary) and histone H3 lysine 27 acetylation (H3K27ac, see Glossary) [25–27], and (3) bound by coactivators (e.g., p300/CBP, see Glossary) [28–30]. In addition, they loop to target gene promoters, which in many cases require the architectural effects of cohesin, a multisubunit protein complex that promotes enhancer-promoter interactions, and Mediator, a multisubunit coregulator complex that interacts with enhancers and promoters to drive chromatin looping and transcription initiation. [22, 31–34] (Fig. 1). Importantly, enhancers may be located far away (kilobases to megabases) from the target genes that they regulate via enhancer-promoter contacts driven by chromatin looping. Moreover, enhancers regulate target genes regardless of their sequence orientation [21, 22, 35].
In addition to these molecular features, gene-specific and genomic assays have revealed that many enhancers are bound by RNA polymerase II (Pol II) and are actively transcribed, producing enhancer RNAs (‘eRNAs’, see Glossary) [36–49] (Fig. 1). Even before the development of next-generation sequencing technologies, enhancer transcription was observed at the locus control regions (LCR) of the β-globin gene [50–52], the human growth hormone gene [53], and the major histocompatibility complex class II locus [54]. The stable accumulation of eRNAs, however, may play a functional or structural role and may facilitate gene looping, as discussed in detail herein [32, 48, 55–62]. Currently, the field is actively debating the functions of eRNAs, but the act of enhancer transcription may also aid in the formation of an open chromatin environment and promote enhancer function [48, 63]. With respect to the latter, studies of eRNAs and long non-coding RNAs (lncRNAs; see Glossary) have suggested that the act of transcription itself, rather than the transcripts, may regulate neighboring genes in cis (see Glossary) [64, 65]. In fact, the lncRNA field is also exploring the act of transcription versus the stable accumulation of transcripts with respect to gene regulation by lncRNAs [66, 67]. Approaches that examine transcripts as molecular entities are needed for these types of analyses. In this review, we summarize the current state of the field and discuss the experimental approaches that are needed to further our understanding of the functions of eRNAs as molecular entities, including the use of cloned eRNAs in “addition” experiments.
Molecular Features of Enhancer RNAs
A variety of locus-specific and genomic assays have been used to characterize the chemical, molecular, and genomic properties of eRNAs, providing some clues to their functions. These include 5’ and 3’ RACE (rapid amplification of cDNA ends, see Glossary), which can be used to map the ends of eRNAs, and RT-qPCR (reverse transcription-quantitative PCR), which can be used to determine the expression levels of eRNAs. The most common representations of eRNAs, however, are read densities in genome browser tracks of genomic transcription data derived RNA sequencing (RNA-seq, see Glossary), global run-on and sequencing (GRO-seq, see Glossary), or related methods [44–49] (Fig. 2A, top). Although these browser track representations provide information about the genomic location, nucleotide sequence, length, and level of expression of eRNAs, they are “ghosts” (non-physical entities) that provide little information about the functional nature of eRNAs.
Figure 2. Properties of eRNAs.
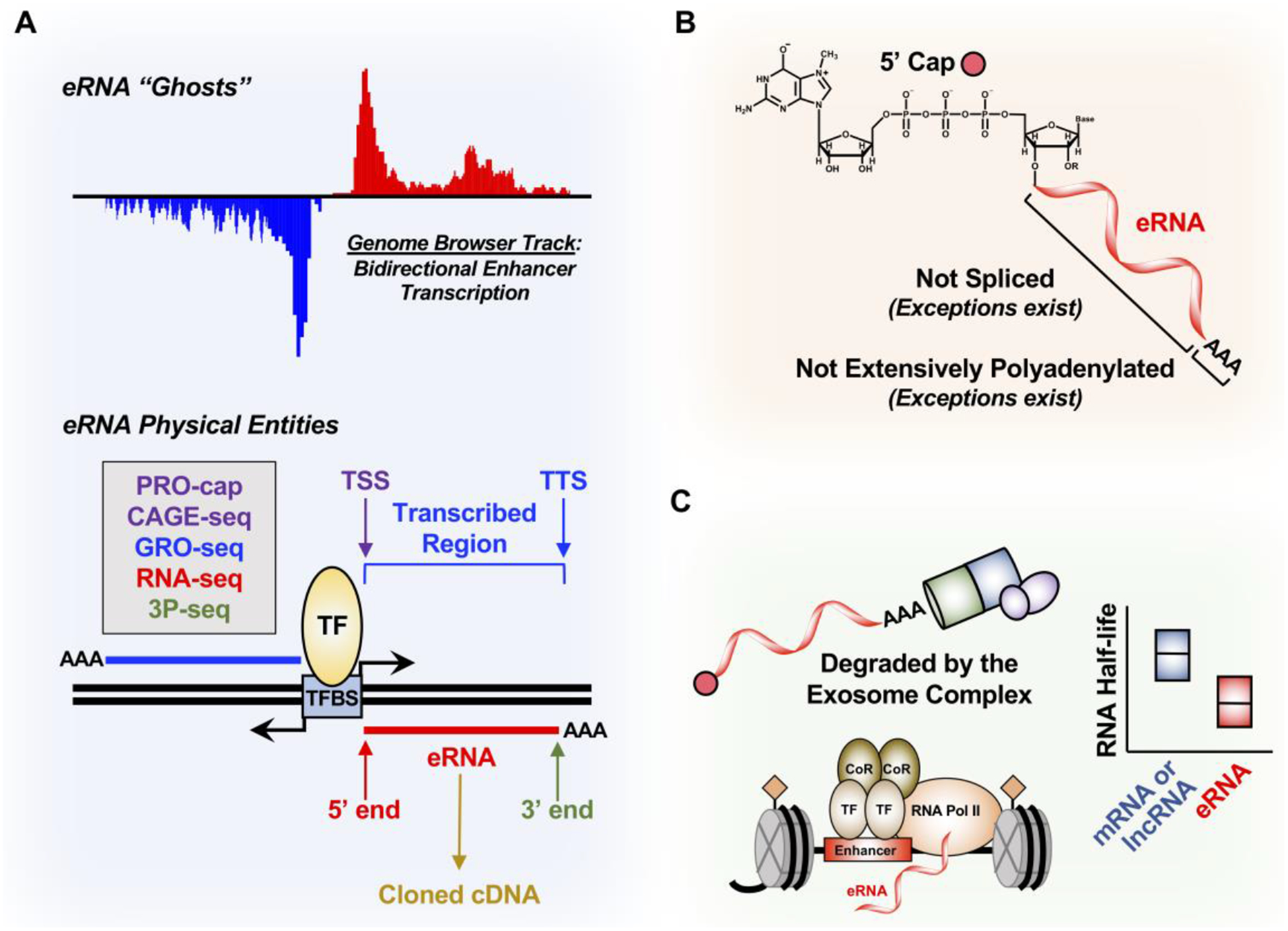
(A) eRNA ghosts have been mostly studied as genome browser tracks derived from bidirectional enhancer transcription. To start studying eRNAs as physical entities, techniques such as PRO-cap to define the 5’ end and the transcription start site (TSS), GRO-seq to define the transcribed enhancer region, RNA-seq to define the eRNA body, and poly(A)-position profiling by sequencing (3P-seq) to determine the 3’ end and the transcription termination site (TTS) are all needed to clone eRNAs into cDNAs.
(B) eRNAs are generally thought to have a 7’-methylguanosine cap at the 5’ end, not spliced, and not extensively polyadenylated, although there are exceptions.
(C) eRNAs are rapidly degraded by the nuclear 3’−5’ exoribonuclease exosome complex, which contributes to their shorter RNA half-life compared to mRNA and lncRNA. This may be correlated to the minimal polyadenylation of eRNAs.
Although some studies have used RNA-seq to detect steady-state eRNAs [37, 44], most have relied on methods that detect nascent eRNA transcription, such as GRO-seq and its successor precision run-on sequencing (PRO-seq, see Glossary), to annotate these unstable RNAs [12, 42, 46, 58, 59, 68–70] (Fig. 2A, bottom). Nascent transcription, however, does not reflect the mature RNA species after transcription termination. The lack of eRNA annotations beyond nascent transcription has impeded the cloning and functional analysis of these transcripts on a genomic scale, an approach that is undoubtedly needed to fully understand the function of eRNAs.
In general, eRNAs are (1) relatively short (100 nt to 1,000 nt) with variable 3’ ends, as mapped by GRO-seq or RNA-seq [44–46], (2) 5’ capped (see Glossary) with 7-methylguanosine, as determined by precision run-on of capped RNA and sequencing (PRO-cap) or 5’ GRO-seq [39, 58, 71–73], or by cap analysis gene expression (CAGE) [39, 72], (3) not extensively polyadenylated (see Glossary) [37, 45, 73, 74], and (4) not spliced [39, 73], although exceptions have been identified and characterized [32, 55, 75, 76] (Fig. 2A, bottom; Fig. 2B). eRNAs are relatively unstable compared to mRNAs and lncRNAs [45, 58, 60, 74, 77–79], and can be rapidly degraded by the exosome complex soon after synthesis [39, 73, 80, 81] (Fig. 2C). Estimates using transient transcriptome sequencing (TT-seq) suggest that eRNAs are approximately 100 times less stable than mRNAs and lncRNAs [77, 79] (Fig. 2C). This transience has made annotating and studying eRNAs more difficult.
Functional Analyses of eRNAs: From Ghosts to Physical Entities
Most of our understanding of eRNAs comes from the browser track representations noted above, plus associated chromatin features (Box 1), from which functions are inferred. More specific functional analyses of eRNAs have relied heavily on loss-of-function or “subtraction” approaches, such as small interfering RNA (siRNA, see Glossary)-mediated knockdown of eRNAs [32, 57–61, 63, 75, 76, 82–84], a strategy that presents problems from an experimental standpoint. Such approaches may be especially problematic with signal-regulated enhancers that are activated and produce eRNAs within minutes of a stimulus. In commonly used protocols, cells are treated with siRNAs targeting an eRNA for 24 to 48 hours before the stimulus, enhancer assembly, and expression of the eRNA. In these cases, endpoints (e.g., enrichment of active enhancer features, target gene expression) are typically examined within 20 to 60 minutes after administration of the stimulus [59–61, 85]. Thus, how such a “pre-knockdown” actually degrades an eRNA in the nucleus and produces rapid effects after the stimulus is unclear. Moreover, how further accelerating the turnover of highly unstable eRNAs by RNAi would impact their potential functions is unclear. Finally, siRNAs are thought to function primarily [86], but perhaps not exclusively [87, 88], in the cytoplasm, raising questions about whether RNAi could effectively perturb eRNAs. Antisense oligonucleotides (ASOs, see Glossary), which promote cleavage of nascent transcripts and induce premature transcription termination [89, 90], may suffer from similar caveats for induced enhancers, but may also have utility as rapidly-acting enhancer transcription terminators.
Another experimental approach to reduce eRNA expression is to mutate DNA elements within the enhancer to stop enhancer formation and consequently, enhancer transcription and eRNA synthesis. Indeed, a naturally occurring A-to-C mutation found in non-medullary thyroid carcinoma located in chromosome 4q32 impairs the binding of TFs POU2F and YY1, and causes decreased eRNA expression [91]. The precise mechanism by which enhancer function is disrupted by this mutation, however, is unclear. It can be attributed to disruption of TF binding and enhancer formation, suppression of enhancer transcription, or reduction of eRNA levels. This example illustrates why a gain-of-function, or “addition”, approaches are required to study the precise functions of eRNAs.
A limited subset of studies has used a gain-of-function or “addition” approach, which involves annotating the eRNA transcript and cloning an eRNA cDNA, which can then be ectopically expressed in the cell. This approach, in essence, turns the eRNA ghost into a physical entity whose properties and functions can be explored and understood. For the remainder of this review, we will focus on studies that have cloned eRNAs and examined them as physical entities in “addition” experiments to uncover the potential mechanisms by which eRNA transcripts regulate target gene expression (Table 1). In addition, we will review and discuss the methods and experimental approaches that have been used in these studies.
Table 1.
Studies discussed in this review that have cloned and functionally analyzed eRNAs using molecular, biochemical, and cell-based approaches.
| Studies | Potential eRNA Regulatory Mechanisms | Method for eRNA Annotation | Notes |
|---|---|---|---|
| A. eRNAs promote target gene transcription | |||
| Melo et al. (2013) | Required for efficient transcriptional enhancement of interacting target genes (detailed mechanisms not described). | Cloned 3 regions around the identified p53 enhancer. | Effects of eRNAs on target gene transcription were assessed using luciferase reporter assays. |
| Lam et al. (2013) | Required for efficient transcriptional enhancement of interacting target genes (detailed mechanisms not described). The Rev-Erb TF act to repress eRNA transcription. | Cloned a fragment based on 5’-GRO- seq GRO-seq data. | Effects of eRNAs on target gene transcription were assessed using luciferase reporter assays. |
| B. eRNAs drive enhancer-promoter looping | |||
| Li et al. (2013) | Increase the strength of enhancer- promoter looping initiated by ERa binding. Cohesin acts, in part, by stabilizing ERa/eRNA-induced enhancer-promoter looping. | Nascent RNA transcripts derived from GRO-seq data. | Effects of eRNAs on target gene transcription were assessed using luciferase reporter assays. Looping mechanism was tested using an eRNA knockdown approach. |
| Lai et al. (2013) | Interact with Mediator to regulate its chromatin localization and kinase activity towards histone H3 serine 10. | 5’ and 3’ RACE. | eRNAs, called ncRNA-a, were polyadenylated and spliced. Looping mechanism was tested using an eRNA knockdown approach. |
| Tsai et al. (2018) | Interact with cohesion to promote spatially appropriate cohesin loading in trans to regulate gene expression. | RNA-seq from ribosomal RNA- depleted fraction. | The eRNA tested (DKKeRNA) is polyadenylated and spliced. |
| Panigrahi et al. (2018) | No function was ascribed to the eRNA. | A portion of GREB1 enhancer transcribed region. | eRNA failed to stimulate transcription in a cell-free assay. |
| C. eRNAs promote the recruitment of transcription factors and coregulators, and regulate their activities | |||
| Sigova et al. (2015) | The YY1 TF binds to both enhancers and their cognate eRNAs across the genome. The eRNA stabilizes YY1 at the enhancers. | Fused 60 nt of the eRNA to a sgRNA and tethered the fusion construct to an a YY1 binding site using CRISPR/ Cas9. | RNA tested was derived from the promoter ofAridla. Reduced enhancer transcription diminished YY1 occupancy, whereas artificial tethering of the eRNA enhanced YY1 occupancy. |
| Bose et al. (2017) | Stimulate CBP/p300 acetyltransferase activity, resulting in eRNA-dependent changes in histone acetylation mediated by CBP, such as H3K27ac. | Nascent RNA transcripts derived from GRO-seq data. | Effect in cells was tested using a knockdown approach. |
| Rahnamoun et al. (2018) | eRNAs bind to BRD4 to increase BRD4 binding to acetylated histones, promoting transcriptional coregulatory activity. | Nascent RNA transcript derived from GRO-seq data. | BRD4 selectively associates with eRNAs produced from BRD4-bound enhancers. Bromodomains of BRD2, BRD3, BRDT, BRG1, and BRD7 interact directly with eRNAs. |
| D. eRNAs facilitate RNA Pol II pause-release to promote transcription elongation | |||
| Schaukowitch et al. (2014) | Bind to and promote the release of NELF-E, a negative regulator of RNA Pol II elongation, to facilitate RNA Pol II transition to elongation. | Circularized RACE to determine 5’ and 3’ ends. | eRNAs failed to promote enhancer-promoter looping. |
Molecular Functions of eRNA Transcripts in Gene Regulation
The results from the addition experiments with cloned eRNAs listed in Table 1 indicate clearly that some eRNAs can act to promote enhancer formation and target gene transcription, although the detailed molecular mechanisms have not always been elucidated (Table 1A). In cases where mechanisms have been defined, they have revealed three primary roles for eRNAs: (1) promoting enhancer-promoter looping (Table 1B, Fig. 3A); (2) promoting recruitment of transcription factors and coregulators, and regulating their activities, which may promote acetylation of H3K27 at the enhancers (Table 1C, Figs. 3B and 3C); and (3) facilitating RNA Pol II pause-release to promote transcription elongation (Table 1D, Fig. 3D). These mechanisms are not mutually exclusive. Methodologically, these mechanistic analyses begin with efforts to annotate the eRNAs (start, stop, transcribed region, etc.) (Fig. 2A) and characterize the epigenomic features of the enhancers from which they originate (Fig. 4).
Figure 3. Mechanism by which eRNA transcripts regulate target gene expression.
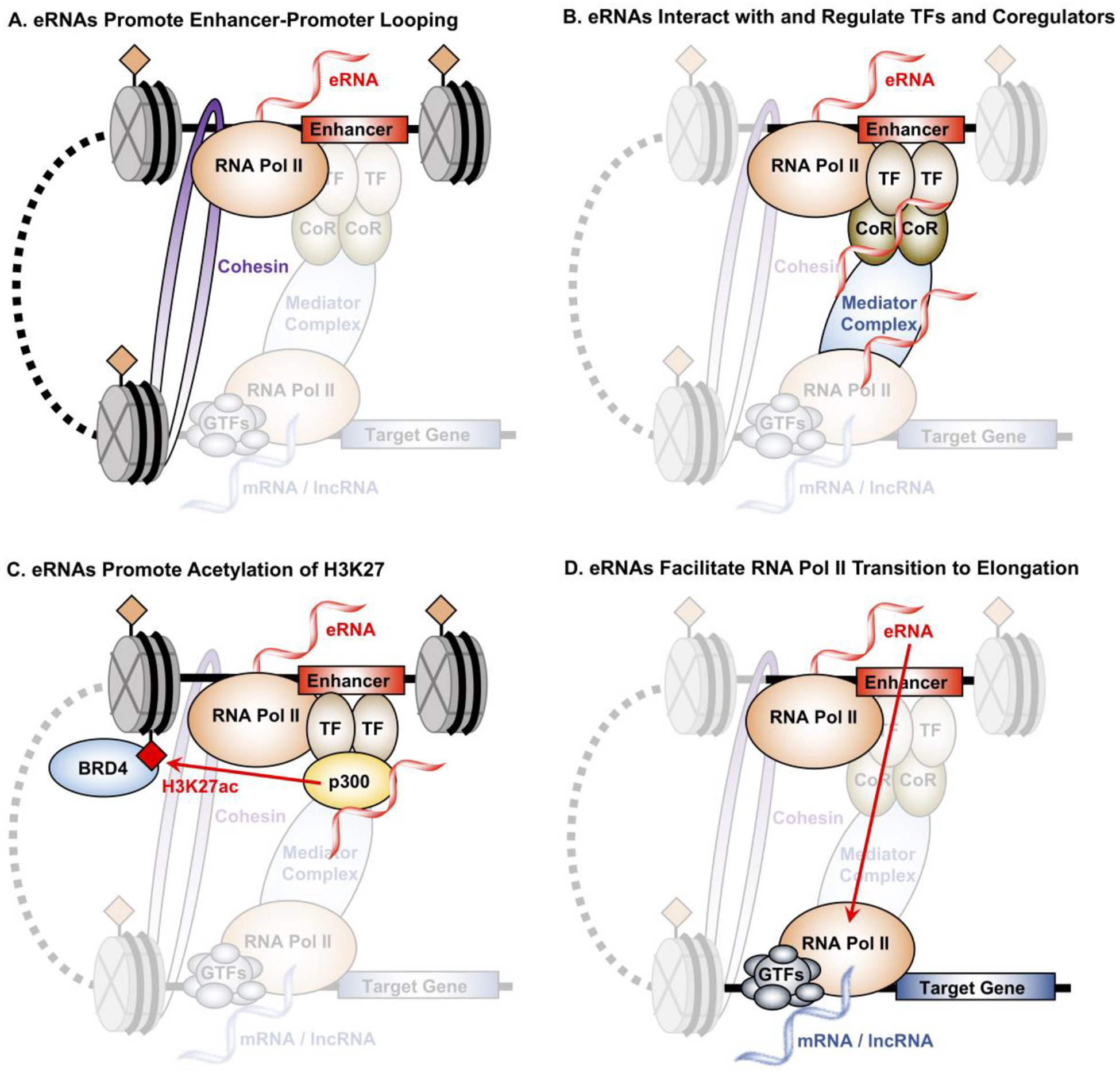
(A) eRNAs stabilize enhancer-promoter interaction by increasing DNA looping through interaction with the cohesion and Mediator complexes. Knockdown of eRNAs in several studies have shown that eRNAs are essential for the formation and stabilization of enhancer-promoter interaction.
(B) The presence of eRNAs may also promote the localization and binding of transcription factors (TFs) and coregulators (CoRs) to enhancers through a mechanism coined “trapping”, such as the case for the TF YY1.
(C) eRNAs stimulate the acyltransferase activity of CBP, which increases the H3K27ac histone modification enriched at enhancers. eRNAs amplify this the interaction between H3K27ac and BRD4 by not only stimulating the deposition of H3K27ac at enhancers, but also the recruitment of BRD4, a coregulator that interacts with H3K27ac and promotes target gene transcription.
(D) eRNAs facilitate the transition of RNA Pol II at target gene promoter from pause to productive elongation through the binding and release of the NELF-E, a negative elongation factor that prevents RNA Pol II from elongation.
Figure 4. Methods for studying eRNAs.
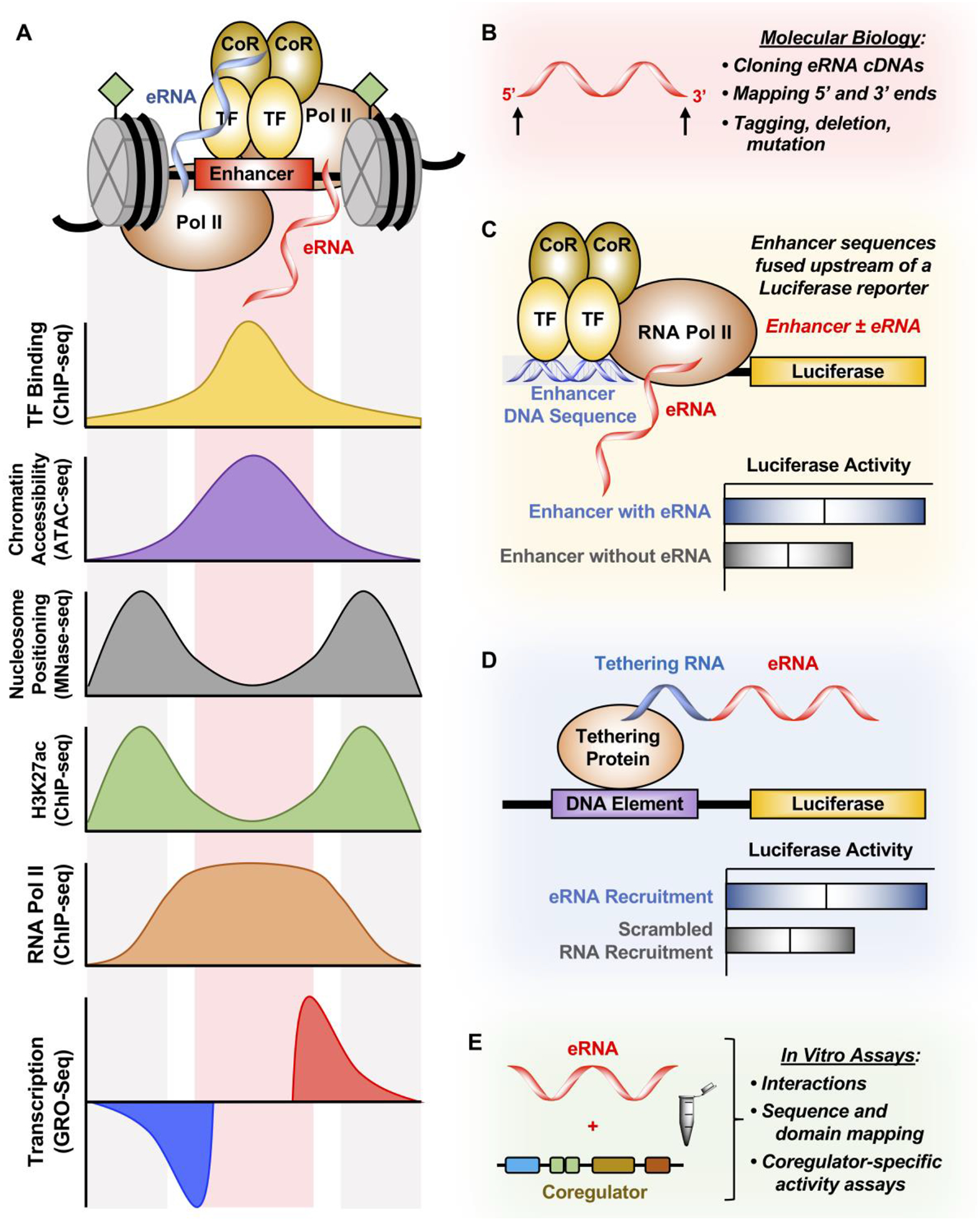
(A) Enhancers are characterized by TF binding (determined by ChIP-seq), increased chromatin accessibility (determined by ATAC-seq), depletion of nucleosomes at TF binding (determined by MNase-seq), enrichment of H3K27ac and RNA Pol II (determined by ChIP-seq), and finally enhancer transcription (determined by GRO-seq). These features are used to derive enhancer region and eRNA ghosts, resulting in a lack of cDNAs derived from eRNAs.
(B) 5’ and 3’ ends of eRNAs can be validated using RACE in order to clone eRNAs into cDNAs to study them by tagging, deleting, and mutating these transcripts.
(C) Enhancer DNA containing or excluding the eRNA sequence derived from genomic sequencing experiments can be cloned upstream of a luciferase reporter to determine the enhancing effect of including the eRNA sequence with the enhancer. However, this technique cannot distinguish between the act of enhancer transcription and the eRNA transcript itself in enhancing luciferase gene transcription.
(D) eRNA can also be tethered to the luciferase reporter construct by using the BoxB-λN or the CRISPR/dCas9 system. This method directly tests whether eRNAs have a function in regulating luciferase gene transcription.
(E) In vitro transcribed eRNAs can be tested for transcription factor binding or coregulator activation in in vitro assays. Additionally, mapping of both the eRNA and the interacting protein can be conducted to further understand how eRNA affects coregulator-specific activity.
eRNAs promote target gene transcription
One of the earliest studies to examine the role of eRNAs in regulating gene transcription focused on p53-bound enhancers in primary BJ/ET fibroblasts [57] (Table 1A). At these p53-bound enhancers, which are characterized by typical histone modifications observed at enhancers (i.e., elevated levels of H3K4me1 and H3K27ac, Fig. 4A), eRNAs are synthesized in a p53-dependent manner. The authors cloned seven different p53 enhancer regions and found that two of these enhancers (termed p53BER2 and p53BER4 in the study) could produce p53-induced eRNAs. To determine whether the eRNAs play a role in enhancing transcription, the authors cloned three regions around p53BER2 to generate an eRNA-MS2 hairpin chimeric RNA (Fig. 4D). They co-transfected these chimeric RNA constructs into cells expressing MS2-coat viral protein fused to the Gal4 DNA-binding domain (Gal4-MS2-CP), which was used to recruit the eRNA-MS2 chimeric RNA to the promoter of a luciferase reporter (see Glossary) construct. One of the three regions tested showed enhanced reporter activity, which was abolished upon the introduction of siRNAs targeting p53BER2. Although no mechanisms of action were determined in the study, it was one of the first reports demonstrating that eRNA transcripts promote target gene transcription.
In another early study, Lam et al. explored the role of eRNAs in activating target gene expression by examining the ability of the nuclear receptors Rev-Erbα and -β to inhibit gene expression by the suppressing eRNA transcription in macrophages [58] (Table 1A). To determine whether eRNA transcripts contribute to enhancer function, the authors cloned a core enhancer region containing the key TF binding sites, as well as an expanded region consisting of the core enhancer region plus the upstream and downstream sequences for the sense and antisense eRNA transcripts (Fig. 4C). Inclusion of the transcribed sense eRNA increased promoter activity by two-fold compared to the core enhancer region alone. Flipping the sequence for the sense eRNA strand or using only the antisense eRNA strand resulted in a loss of transcriptional enhancement of the interacting target gene. These results suggest a sequence-dependence for eRNA effects on enhancer function. Depletion of the endogenous eRNAs using siRNAs or ASOs reduced target gene expression, but the mechanisms by which these eRNAs regulate gene transcription were not revealed.
eRNAs drive enhancer-promoter looping
Other studies have gone beyond demonstrating a role for eRNAs in enhancer function and target gene expression to elucidate specific molecular mechanisms underlying the functions of eRNAs (Table 1B). In this regard, a number of studies have observed a role of eRNAs in promoting enhancer-promoter looping (Fig. 3A). Li et al. fused the estrogen-induced FOXC1 eRNA to the BoxB viral RNA and recruited the FOXC1 eRNA-BoxB RNA chimera to a 5xUAS luciferase reporter construct using λN-Gal4 in estrogen-responsive MCF-7 breast cancer cells [59] (Fig. 4D). In this system, the FOXC1 sense eRNA, but not the antisense eRNA, stimulated reporter activity, again suggesting a sequence-dependent mechanism for eRNA function. They also observed that estrogen-induced eRNAs are essential for cohesin-dependent enhancer-promoter looping, since knockdown of eRNAs using siRNAs or locked nucleic acids decreased enhancer-promoter interactions [59]. This is consistent with their observation that the expression of estrogen-induced genes is suppressed upon knockdown of subunit of cohesin (SMC3) in MCF-7 cells. Together, these data indicate a role for eRNAs in regulating long-range enhancer-promoter interaction (Fig. 3A), although some of the conclusions from this study were made using knockdown approaches, rather than eRNA addition approaches.
A role for eRNAs in promoting enhancer-promoter interactions is further supported by studies from Lai et al. [32]. They examined eRNAs, which they called ‘noncoding RNA-activating’ (ncRNA-a), and their interaction with the Mediator complex, a multi-subunit transcriptional coactivator that is required for enhancer-promoter interactions (Fig. 3B). Like other eRNAs, the ncRNA-as were annotated as ncRNAs that do not overlap protein-coding genes, promoters and 3’-associated transcripts, or annotated lncRNAs [55]. But, unlike other eRNAs, ncRNA-as are spliced. Using an in vitro Mediator kinase assay (Fig. 4E), the authors used two ncRNA-as, annotated by 5’- and 3’-RACE (Fig. 4B), and showed they are capable of stimulating Mediator kinase activity, which is required for gene activation through the phosphorylation of histone H3 serine 10, a histone modification correlated with transcriptional activation. Depletion of Mediator subunits (MED1 or MED12) or the ncRNA-a in 293T cells abrogated chromosomal looping between ncRNA-a-producing enhancers and their target genes, consistent with a role for these eRNAs in regulating enhancer-promoter looping (Fig. 3A).
The major enhancer of the myogenic master regulator gene, Myod1 (encoding MyoD), generates a core eRNA that regulates transcription of Myod1 in cis and a distal regulatory region eRNA (termed DRReRNA) that regulates the transcription of Myog (encoding myogenin) in trans (see Glossary) on a separate chromosome [75]. Unlike conventional eRNAs, the ~2 kb DRReRNA is spliced, polyadenylated, and has a much longer half-life of 30 min than other eRNAs that have been characterized. Using CRISPR/Cas9 (see Glossary) technology to recruit the dCas9-KRAB repressor to inhibit transcription of the DRReRNA in C2C12 skeletal muscle myoblasts decreased the expression of the Myog without affecting the expression of Myod1 gene upon myoblast differentiation. The DRReRNA was recruited to the Myog locus, and proteomic analysis of DRReRNA-interacting proteins by mass spectrometry revealed components of RNA splicing and processing machinery, chromosome organization machinery, the cohesin complex, and the integrator complex (Fig. 3A and 3B) [75]. Depletion of the DRReRNA by siRNAs reduced the localization of SMC3 (a component of the cohesin complex) at the Myog locus upon differentiation. These results suggest that DRReRNA regulates myogenin in trans by recruiting cohesin complex to the locus. Since DRReRNA is polyadenylated, it has increased stability compared to non-polyadenylated eRNA, allowing for gene regulation in trans. DRReRNA is the only eRNA discussed herein that exerts its actions in trans; whether such a mechanism is generalizable to other eRNAs is unclear. In this regard, the level of eRNA polyadenylation may plays a role in specifying trans gene regulation or DRReRNA may be an outlier.
While the three aforementioned studies demonstrated a role for eRNAs in promoting enhancer-promoter looping, Panigrahi et al. failed to observe a role for eRNAs in chromosomal looping [92]. In their study, the authors developed a unique cell-free system to study enhancer-promoter interactions using estrogen-regulated chromosomal looping as a model. An eRNA derived from the GREB1 estrogen receptor enhancer failed to enhance target gene transcription in the cell-free assay. Surprisingly, incubating the GREB1 enhancer with the GREB1 promoter showed reciprocal transcription activation of each regulatory element. The authors concluded that mutual co-stimulation of enhancer and promoter transcription is not dependent on the eRNA transcript, but rather the act of transcription. One caution regarding these experiments, however, is that critical cellular biochemical and biophysical properties and contacts may be lost in cell-free assays. Nevertheless, further studies will be required to reconcile these differing observations on the role of eRNA transcripts in enhancer-promoter looping.
eRNAs promote the recruitment of transcription factors and coregulators, and regulate their activities
Another mechanism by which eRNAs regulate target gene transcription is through interaction with and regulation of TF and coregulators (Table 1C, Fig. 3B). For example, eRNAs interact with and regulate the TF Ying-Yang 1 (YY1) to trap it at regulatory elements [80]. Sigova et al. found that YY1 is bound at both active enhancers and promoters and interacts with RNAs produced from these regulatory elements in mouse embryonic stem cells as revealed by in vitro and cell-based assays [80]. Knockdown of the exosome complex that degrades eRNAs, leading to increased untethered eRNAs, titrated YY1 away from its binding sites in enhancers. To determine whether increasing the amount of tethered RNA influenced YY1 binding, the authors targeted a portion of the Arid1a RNA transcribed at the promoter to six different enhancers using a CRISPR/dCas9 approach. At all six enhancers tested, the presence of the targeted Arid1a promoter RNA fragment increased YY1 binding, indicating that increased RNA tethering at enhancers boosts YY1 occupancy. This study is one of the few to use a native enhancer environment in cells to examine the role of ncRNAs at enhancers.
Most enhancers contain nucleosomes enriched for H3K27ac, which is catalyzed by the histone acetyltransferases p300 and CBP (collective referred to as p300/CBP), coregulators that are commonly found at enhancers (Fig. 4A). Bose et al. asked whether CBP binds to eRNAs to regulate enhancer formation and function [84]. Using photoactivable ribonucleoside-enhanced crosslinking and immunoprecipitation (PAR-CLIP) to explore interactions between CBP and eRNAs in mouse embryonic fibroblasts, the authors identified ~8,000 CBP-RNA interactions, with 73% of these interactions localizing to RNAs produced from intergenic or intronic regions of the genome. Protein sequence analyses and biochemical assays narrowed the RNA-interacting domain of CBP to a highly basic region within the core histone acetyltransferase domain [84]. The interactions between CBP and RNA were not limited to eRNAs, however, since lncRNAs and exonic RNAs were also identified, suggesting sequence-independent interactions. Thus, the enrichment of CBP interactions with eRNAs is most likely due to the enrichment of CBP at enhancer regions rather than the sequence specificity of eRNAs. At the enzymatic level, RNA binding to CBP stimulated its acetyltransferase activity in vitro by displacing the activation loop from the active site. In contrast, depletion of eRNAs using ASOs reduced not only H3K27 acetylation, but also H3K18 acetylation in cells, suggesting a mechanism whereby eRNAs can positively regulate CBP and promote the enrichment of H3K27ac at enhancers (Table 1C, Fig. 3C) [84]. A subsequent study, however, failed to recapitulate the observation that eRNAs can stimulate p300 histone acetyltransferase activity [93], One possible explanation may be the extent of refolding or renaturation of the in vitro transcribed eRNAs used in the assays, although this is not clear from the reports. Additional studies will be required to reconcile this discrepancy.
H3K27ac is recognized by effector proteins often referred to as “readers.” Histone lysine acetylation is recognized by bromodomains, which are acetyl-lysine binding domains found in many chromatin-associated “reader” proteins [94]. Members of the bromodomain and extraterminal motif (BET) family of proteins, including BRD4, bind H3K27ac and act as acetylated lysine reader protein [95–97]. Using ultraviolet-RNA immunoprecipitation, Rahnamoun et al. found that eRNAs interact with BRD4 via its tandem bromodomains [82]. The authors used eRNAs from the MMP9 and CCL2 enhancers, which they annotated based on maximal GRO-seq peaks. They found that these eRNAs assist BRD4 in binding to acetylated histones, with the interactions among the eRNA, H3K27ac, and BRD4 reinforcing BRD4 occupancy at enhancers and activating target gene expression [82]. Interestingly, the interaction between eRNA and BRD4 appears to be sequence-independent, since BRD4 readily interacted with mRNAs. This observation is similar to the sequence-independent action of eRNAs in stimulating CBP acetyltransferase enzymatic activity described above [84]. The reinforcement of BRD4 occupancy at enhancers by eRNAs represents an opportunity for cells to utilize eRNA transcripts at enhancers and highlights a specific mechanism by which eRNAs can interact with and regulate coregulators at enhancers (Table 1C, Fig. 3C).
eRNAs facilitate RNA Pol II pause-release to promote transcription elongation
Transcription start site-proximal pausing of RNA Pol II at developmental and stimulus-responsive genes is a genome-wide regulatory mechanism by which cells can initiate rapid or synchronous responses to external stimuli at the transcriptional level [98, 99]. RNA Pol II pausing is induced by two factors, negative elongation factor (NELF) and DRB sensitivity-inducing factor (DSIF), which bind directly to RNA Pol II and the nascent mRNA transcript. Recent studies have shown that eRNAs facilitate RNA Pol II pause-release to promote transcription elongation (Table 1D, Fig. 3D).
Schaukowitch et al. explored whether eRNAs facilitate the release of paused RNA Pol II upon induction of immediate early genes in neurons [60]. Knockdown of the eRNAs from neuronal gene enhancers [e.g. Activity-regulated cytoskeletal protein (Arc) and Growth arrest and DNA-damage-inducible beta (Gadd45b) enhancers] decreased induction of Arc and Gadd45b mRNA expression, respectively, in neurons subjected to potassium chloride-mediated membrane depolarization. In both cases, the decrease in mRNA transcripts upon eRNA knockdown led to decreased protein expression, showing a direct downstream effect of the eRNAs. Contrary to other reports highlighted above, knockdown of the Arc or Gadd45b eRNAs did not affect enhancer-promoter interactions or the recruitment of the Mediator and cohesion complexes to the enhancers and promoters. Rather, knockdown of the eRNA caused the retention of NELF-E at the gene promoters, inhibiting the release of the NELF complex from paused RNA Pol II [60]. The authors annotated the Arc or Gadd45b eRNAs using circularized RACE (Fig. 4B), cloned them, and observed that they interact directly with NELF-E in a sequence-specific manner. The NELF-eRNA interactions regulated target gene expression by stimulating the release of paused RNA Pol II release into productive elongation (Fig. 3D) [60]. Like other general mechanisms proposed for eRNAs, it will be interesting to determine (1) how many eRNAs are capable of initiating a mechanism such as this, (2) whether the interactions with the NELF complex to facilitate pause-release is a general property of eRNAs and, if so, (3) the molecular and structural mechanisms that underlie this regulation. Interestingly, androgen receptor-regulated eRNAs found in prostate cancer cells bind to cyclin T1 and activate P-TEFb to release paused RNA Pol II, supporting a role for eRNAs in regulating RNA Pol II pause-release [100].
Concluding Remarks
As highlighted in this review, recent studies have revealed new information on the potential mechanisms by which eRNAs contribute to enhancer function and the regulation of target gene transcription. The studies described in detail in this review are distinguished from others because they have cloned and tested the functions of eRNAs in “addition” experiments. The disparate ways in which the eRNAs have been annotated, the various ways in which they have been studied, and the limited number that have been examined thus far makes generalizing about potential mechanisms of regulation difficult (see Outstanding Questions). Furthermore, the lack of independent confirmation from different groups for the mechanisms described herein have limited the conclusions that can be drawn and the confidence that they are correct.
Outstanding Questions.
“Spirits in the Material World: Enhancer RNAs in Transcriptional Regulation”
Tim Y. Hou and W. Lee Kraus
Why do some enhancers produce stable eRNAs, while others are transcribed, but do not produce stable transcripts?
What fraction of eRNAs in a cell are functional and what fraction is transcriptional noise?
What are additional mechanisms by which eRNAs might regulate target gene expression?
Do structural features of eRNAs underlie the specific mechanisms by which eRNAs activate target gene expression?
How can the sequence-specific features and the sequence-independent features of eRNAs be distinguished?
What fraction of eRNAs act in cis and what fraction acts in trans?
How can polyadenylated versus non-polyadenylated eRNAs, or spliced versus unspliced eRNAs, be better defined?
Do eRNAs play a role in diseases, or are they merely markers of enhancers that are activated in a given disease state?
Is there therapeutic potential in targeting eRNAs?
Do single nucleotide polymorphisms in eRNAs drive gains- or losses-of-function that may lead to the development of disease?
These issues are compounded by the diversity of the biological systems in which the eRNAs have been studied. Consistent and comprehensive genome-wide annotations of eRNAs across many cell types will be required in the future, not only to dissect the mechanisms, but also to guide the field in exploring important facets of eRNAs. For example, these large-scale eRNA annotations could lead to studies on structural determinants of functional eRNAs and their evolutionary conservation (Fig. 4B). Importantly, many of the studies mentioned herein examined the mechanisms of eRNA function using in vitro assays or reporter assays. Only a limited set of studies have examined the mechanisms of eRNAs function in their endogenous contexts, typically using the CRISPR/dCas9-based assays to guide eRNAs to their cognate enhancers [76, 80]. Additional studies exploring eRNAs in their endogenous contexts will help reconcile the discrepancies observed in current studies.
Another important area to investigate will be how eRNAs participate in liquid-liquid phase separation, a concept of emerging importance to the field of enhancer biology. The high concentration of TF and coactivators, especially at super-enhancers (clusters of enhancers in proximity to one another), lead to the compartmentalization of the transcriptional machinery to allow activation of target genes [101–106]. Typically, weak multivalent interactions of intrinsically disordered domains between various TF [102, 105], coregulators [103, 104], and even RNA Pol II [106] lead to liquid-liquid phase separation to form non-membrane-bound compartments. The unique biophysical properties of phase separated domains may allow for a high concentration of transcriptional machinery to coalesce at enhancers and promoters to activate target genes. Intriguingly, both RNA transcription [107] and RNA itself modulate phase separation [108–111]. Thus, it is tempting to speculate that eRNAs may drive regulatory phase separation at enhancers to activate target gene expression. Perhaps this contributes to the sequence-specific and sequence-independent mechanisms of eRNAs highlighted in this review.
Beyond the biochemical and molecular functions of eRNAs, an important question that remains unanswered is whether eRNAs are dysregulated or mutated in diseases. Mutations found in enhancers can lead to activation of oncogenes in cancer [11–17]. One intriguing possibility is that mutations in enhancers cause differences in eRNA expression capable of driving disease development. An equally interesting prospect is that single-nucleotide polymorphisms, deletions, or insertions in eRNA transcripts may cause gains- or losses-of-function that lead to disease. In this regards, deletion of a CAAA tract in a polyadenylated non-coding RNA transcribed from a super-enhancer active during myoblast differentiation results in the loss-of-function and inability to regulate target gene expression [76]. Although more studies are required to answer these questions, it is clear that turning these eRNAs from mere ghosts visualized in browser tracks into physical entities will facilitate the study of their mechanisms and functions, as well as their roles in diseases.
Highlights.
“Spirits in the Material World: Enhancer RNAs in Transcriptional Regulation”
Tim Y. Hou and W. Lee Kraus
Enhancers are regulatory regions in the genome that bind sequence-specific DNA-binding transcription factors. They are transcribed to produce enhancer RNAs (eRNAs).
Current studies have relied on depletion of eRNAs (“subtraction” experiments), rather than expressing them as physical entities from cloned cDNAs (“addition” experiments). This has left open questions about whether these non-coding transcripts have functions in regulating gene expression, or whether they are just transcriptional noise.
A limited number of studies that have expressed eRNAs from cloned cDNAs suggest that they function by facilitating enhancer-promoter looping, stimulating the recruitment and activity of regulatory proteins, establishing a permissive chromatin environment for gene activation, and aiding in the release of paused RNA polymerase II.
Future studies using genome-targeting approaches to study eRNAs in endogenous chromatin will further define the functions of eRNAs in the regulation of target gene expression.
Acknowledgments
The authors would like to thank Andrew M. Kelleher, David M. Owen, and other members of the Kraus lab for insightful feedback during the preparation of this manuscript. The authors would also like to acknowledge the funding to W.L.K. that supports the enhancer-related studies in the Kraus lab: a grant from the NIH/NIDDK (DK058110), grants from the Cancer Prevention and Research Institute of Texas (CPRIT; RP160318 and RP190235), and funds from the Cecil H. and Ida Green Center for Reproductive Biology Sciences Endowment.
Glossary
- Antisense oligonucleotide (ASO)
Single-stranded deoxyribonucleotides, which are complementary to a target mRNA, used to downregulate their target.
- Chromatin immunoprecipitation and sequencing (ChIP-seq)
A method coupling chromatin immunoprecipitation with next-generation sequencing to identify binding sites of a protein of interest across the genome.
- Cis
Action at the site of origin. With respect to eRNAs, this refers to an eRNA that is produced from and acts at the same enhancer, or at a proximal target gene of that enhancer.
- CRISPR/(d)Cas9
An RNA-guided endonuclease system derived from bacteria. The Cas9 nuclease can be directed to a locus in the genome for editing by a single guide RNA (sgRNA). Variants of Cas9 can introduce a myriad of modifications to the DNA, while a catalytically dead Cas9 (dCas9) can be used to tether RNA to regions of interest.
- Enhancer
DNA sequences in the genome that act as a nucleation site for the binding of sequence-specific TFs and the formation of transcription regulatory complexes.
- Enhancer RNA (eRNA)
Short non-coding RNAs that are transcribed from enhancer loci. The recruitment of RNA Pol II to enhancers results in enhancer transcription and the production of eRNAs.
- Five-prime cap (5’-cap)
The addition of a 7-methylguanylate cap to the 5’-end of RNA.
- Global run-on sequencing (GRO-seq) and precision run-on sequencing (PRO-seq)
Global nuclear run-on assays used to determine the position, amount, and orientation of transcriptionally active RNA polymerases in the genome. PRO-seq allows the mapping of RNA polymerases at single nucleotide resolution.
- H3K4me1 and H3K27ac
Post-translational modifications of histone H3 lysine 4 with monomethylation and histone H3 lysine 27 with acetylation, which are enriched at enhancers and serve as binding sites for the recruitment of coactivators to enhancers.
- Long non-coding RNAs (lncRNAs)
Transcribed RNAs that are not translated into proteins; they are spliced and polyadenylated.
- Luciferase reporter
A DNA-based activity reporter to monitor transcriptional output and gene expression. The amount of light emitted from luciferase is proportional to the amount of transcribed and translated luciferase present in a sample.
- p300/CBP
Two closely related transcriptional coactivators (p300 and CBP, CREB-binding protein), which are commonly referred to collectively as p300/CBP. They bind at enhancers and activate gene expression by their histone acetyltransferase activity and ability to act as scaffolds. The recruitment of p300/CBP to enhancers leads to the acetylation of H3K27.
- Polyadenylation
The addition of a poly(A) tail to the 3’-end of mRNA and lncRNA to increase transcript stability.
- Rapid amplification of cDNA ends (RACE)
A technique used to identify 5’ and 3’ ends of RNA. In 5’ RACE, the RNA serves as a template for first round reverse transcription. In 3’ RACE, the natural polyA tail is used as a template.
- RNA-seq
RNA sequencing allows for identification and quantification of the whole transcriptome. Protocols are available to enrich and identify mRNAs or non-coding RNAs.
- Small interfering RNAs (siRNAs)
Short (<25 nt) RNAs that hybridize to target regions to elicit RNA interference response. Usually, the timescale for siRNAs can take hours.
- Trans
Action at a site distal to the site of origin. With respect to eRNAs, this refers to an eRNA that is produced at one enhancer and acts at another enhancer or a gene located distally on the same chromosome or another chromosome.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Benoist C and Chambon P (1981) In vivo sequence requirements of the SV40 early promotor region. Nature 290 (5804), 304–10. [DOI] [PubMed] [Google Scholar]
- 2.Banerji J et al. (1981) Expression of a beta-globin gene is enhanced by remote SV40 DNA sequences. Cell 27 (2 Pt 1), 299–308. [DOI] [PubMed] [Google Scholar]
- 3.Moreau P et al. (1981) The SV40 72 base repair repeat has a striking effect on gene expression both in SV40 and other chimeric recombinants. Nucleic Acids Res 9 (22), 6047–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Banerji J et al. (1983) A lymphocyte-specific cellular enhancer is located downstream of the joining region in immunoglobulin heavy chain genes. Cell 33 (3), 729–40. [DOI] [PubMed] [Google Scholar]
- 5.Gillies SD et al. (1983) A tissue-specific transcription enhancer element is located in the major intron of a rearranged immunoglobulin heavy chain gene. Cell 33 (3), 717–28. [DOI] [PubMed] [Google Scholar]
- 6.Neuberger MS (1983) Expression and regulation of immunoglobulin heavy chain gene transfected into lymphoid cells. EMBO J 2 (8), 1373–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Consortium EP (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489 (7414), 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thurman RE et al. (2012) The accessible chromatin landscape of the human genome. Nature 489 (7414), 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buenrostro JD et al. (2015) Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523 (7561), 486–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cusanovich DA et al. (2018) A Single-Cell Atlas of In Vivo Mammalian Chromatin Accessibility. Cell 174 (5), 1309–1324 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xi Y et al. (2018) Histone modification profiling in breast cancer cell lines highlights commonalities and differences among subtypes. BMC Genomics 19 (1), 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Franco HL et al. (2018) Enhancer transcription reveals subtype-specific gene expression programs controlling breast cancer pathogenesis. Genome Res 28 (2), 159–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen H et al. (2018) A Pan-Cancer Analysis of Enhancer Expression in Nearly 9000 Patient Samples. Cell 173 (2), 386–399 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Northcott PA et al. (2014) Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature 511 (7510), 428–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Z et al. (2018) HEDD: Human Enhancer Disease Database. Nucleic Acids Res 46 (D1), D113–D120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maurano MT et al. (2012) Systematic localization of common disease-associated variation in regulatory DNA. Science 337 (6099), 1190–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Loven J et al. (2013) Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153 (2), 320–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Calo E and Wysocka J (2013) Modification of enhancer chromatin: what, how, and why? Mol Cell 49 (5), 825–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shlyueva D et al. (2014) Transcriptional enhancers: from properties to genome-wide predictions. Nat Rev Genet 15 (4), 272–86. [DOI] [PubMed] [Google Scholar]
- 20.Heinz S et al. (2015) The selection and function of cell type-specific enhancers. Nat Rev Mol Cell Biol 16 (3), 144–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim TK and Shiekhattar R (2015) Architectural and Functional Commonalities between Enhancers and Promoters. Cell 162 (5), 948–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bulger M and Groudine M (2011) Functional and mechanistic diversity of distal transcription enhancers. Cell 144 (3), 327–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crawford GE et al. (2006) Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS). Genome Res 16 (1), 123–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sheffield NC et al. (2013) Patterns of regulatory activity across diverse human cell types predict tissue identity, transcription factor binding, and long-range interactions. Genome Res 23 (5), 777–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heintzman ND et al. (2009) Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459 (7243), 108–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heintzman ND et al. (2007) Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 39 (3), 311–8. [DOI] [PubMed] [Google Scholar]
- 27.Rajagopal N et al. (2014) Distinct and predictive histone lysine acetylation patterns at promoters, enhancers, and gene bodies. G3 (Bethesda) 4 (11), 2051–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Visel A et al. (2009) ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 457 (7231), 854–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Z et al. (2009) Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 138 (5), 1019–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Allen BL and Taatjes DJ (2015) The Mediator complex: a central integrator of transcription. Nat Rev Mol Cell Biol 16 (3), 155–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bulger M and Groudine M (2010) Enhancers: the abundance and function of regulatory sequences beyond promoters. Dev Biol 339 (2), 250–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lai F et al. (2013) Activating RNAs associate with Mediator to enhance chromatin architecture and transcription. Nature 494 (7438), 497–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kagey MH et al. (2010) Mediator and cohesin connect gene expression and chromatin architecture. Nature 467 (7314), 430–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gorkin DU et al. (2014) The 3D genome in transcriptional regulation and pluripotency. Cell Stem Cell 14 (6), 762–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ong CT and Corces VG (2011) Enhancer function: new insights into the regulation of tissue-specific gene expression. Nat Rev Genet 12 (4), 283–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rahnamoun H et al. (2017) Mutant p53 shapes the enhancer landscape of cancer cells in response to chronic immune signaling. Nat Commun 8 (1), 754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koch F et al. (2011) Transcription initiation platforms and GTF recruitment at tissue-specific enhancers and promoters. Nat Struct Mol Biol 18 (8), 956–63. [DOI] [PubMed] [Google Scholar]
- 38.Meers MP et al. (2018) Transcription start site profiling uncovers divergent transcription and enhancer-associated RNAs in Drosophila melanogaster. BMC Genomics 19 (1), 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Andersson R et al. (2014) An atlas of active enhancers across human cell types and tissues. Nature 507 (7493), 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Core LJ et al. (2014) Analysis of nascent RNA identifies a unified architecture of initiation regions at mammalian promoters and enhancers. Nat Genet 46 (12), 1311–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arner E et al. (2015) Transcribed enhancers lead waves of coordinated transcription in transitioning mammalian cells. Science 347 (6225), 1010–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Franco HL et al. (2015) TNFalpha signaling exposes latent estrogen receptor binding sites to alter the breast cancer cell transcriptome. Mol Cell 58 (1), 21–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Flynn RA et al. (2011) Antisense RNA polymerase II divergent transcripts are P-TEFb dependent and substrates for the RNA exosome. Proc Natl Acad Sci U S A 108 (26), 10460–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim TK et al. (2010) Widespread transcription at neuronal activity-regulated enhancers. Nature 465 (7295), 182–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.De Santa F et al. (2010) A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS Biol 8 (5), e1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hah N et al. (2011) A rapid, extensive, and transient transcriptional response to estrogen signaling in breast cancer cells. Cell 145 (4), 622–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Melgar MF et al. (2011) Discovery of active enhancers through bidirectional expression of short transcripts. Genome Biol 12 (11), R113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Natoli G and Andrau JC (2012) Noncoding transcription at enhancers: general principles and functional models. Annu Rev Genet. [DOI] [PubMed] [Google Scholar]
- 49.Henriques T et al. (2018) Widespread transcriptional pausing and elongation control at enhancers. Genes Dev 32 (1), 26–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Collis P et al. (1990) Definition of the minimal requirements within the human beta-globin gene and the dominant control region for high level expression. EMBO J 9 (1), 233–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tuan D et al. (1992) Transcription of the hypersensitive site HS2 enhancer in erythroid cells. Proc Natl Acad Sci U S A 89 (23), 11219–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ashe HL et al. (1997) Intergenic transcription and transinduction of the human beta-globin locus. Genes Dev 11 (19), 2494–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ho Y et al. (2006) Locus control region transcription plays an active role in long-range gene activation. Mol Cell 23 (3), 365–75. [DOI] [PubMed] [Google Scholar]
- 54.Masternak K et al. (2003) Chromatin remodeling and extragenic transcription at the MHC class II locus control region. Nat Immunol 4 (2), 132–7. [DOI] [PubMed] [Google Scholar]
- 55.Orom UA et al. (2010) Long noncoding RNAs with enhancer-like function in human cells. Cell 143 (1), 46–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Orom UA and Shiekhattar R (2011) Noncoding RNAs and enhancers: complications of a long-distance relationship. Trends Genet 27 (10), 433–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Melo CA et al. (2013) eRNAs are required for p53-dependent enhancer activity and gene transcription. Mol Cell 49 (3), 524–35. [DOI] [PubMed] [Google Scholar]
- 58.Lam MT et al. (2013) Rev-Erbs repress macrophage gene expression by inhibiting enhancer-directed transcription. Nature 498 (7455), 511–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li W et al. (2013) Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature 498 (7455), 516–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schaukowitch K et al. (2014) Enhancer RNA facilitates NELF release from immediate early genes. Mol Cell 56 (1), 29–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hsieh CL et al. (2014) Enhancer RNAs participate in androgen receptor-driven looping that selectively enhances gene activation. Proc Natl Acad Sci U S A 111 (20), 7319–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ilott NE et al. (2014) Long non-coding RNAs and enhancer RNAs regulate the lipopolysaccharide-induced inflammatory response in human monocytes. Nat Commun 5, 3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mousavi K et al. (2013) eRNAs promote transcription by establishing chromatin accessibility at defined genomic loci. Mol Cell 51 (5), 606–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Engreitz JM et al. (2016) Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature 539 (7629), 452–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cinghu S et al. (2017) Intragenic enhancers attenuate host gene expression. Mol Cell 68 (1), 104–117 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kopp F and Mendell JT (2018) Functional classification and experimental dissection of long noncoding RNAs. Cell 172 (3), 393–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bassett AR et al. (2014) Considerations when investigating lncRNA function in vivo. Elife 3, e03058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang D et al. (2011) Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature 474 (7351), 390–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Murakami S et al. (2017) Dynamic assembly and activation of estrogen receptor alpha enhancers through coregulator switching. Genes Dev 31 (15), 1535–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Core LJ et al. (2008) Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 322 (5909), 1845–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Djebali S et al. (2012) Landscape of transcription in human cells. Nature 489 (7414), 101–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kouno T et al. (2019) C1 CAGE detects transcription start sites and enhancer activity at single-cell resolution. Nat Commun 10 (1), 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Andersson R et al. (2014) Nuclear stability and transcriptional directionality separate functionally distinct RNA species. Nat Commun 5, 5336. [DOI] [PubMed] [Google Scholar]
- 74.Hah N et al. (2013) Enhancer transcripts mark active estrogen receptor binding sites. Genome Res 23 (8), 1210–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tsai PF et al. (2018) A Muscle-Specific Enhancer RNA Mediates Cohesin Recruitment and Regulates Transcription In trans. Mol Cell 71 (1), 129–141 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhao Y et al. (2019) MyoD induced enhancer RNA interacts with hnRNPL to activate target gene transcription during myogenic differentiation. Nat Commun 10 (1), 5787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schwalb B et al. (2016) TT-seq maps the human transient transcriptome. Science 352 (6290), 1225–8. [DOI] [PubMed] [Google Scholar]
- 78.Rabani M et al. (2014) High-resolution sequencing and modeling identifies distinct dynamic RNA regulatory strategies. Cell 159 (7), 1698–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Michel M et al. (2017) TT-seq captures enhancer landscapes immediately after T-cell stimulation. Mol Syst Biol 13 (3), 920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sigova AA et al. (2015) Transcription factor trapping by RNA in gene regulatory elements. Science 350 (6263), 978–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pefanis E et al. (2015) RNA exosome-regulated long non-coding RNA transcription controls super-enhancer activity. Cell 161 (4), 774–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rahnamoun H et al. (2018) RNAs interact with BRD4 to promote enhanced chromatin engagement and transcription activation. Nat Struct Mol Biol 25 (8), 687–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kim YJ et al. (2018) Global transcriptional activity dynamics reveal functional enhancer RNAs. Genome Res 28 (12), 1799–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bose DA et al. (2017) RNA Binding to CBP Stimulates Histone Acetylation and Transcription. Cell 168 (1–2), 135–149 e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nair SJ et al. (2019) Phase separation of ligand-activated enhancers licenses cooperative chromosomal enhancer assembly. Nat Struct Mol Biol 26 (3), 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wilson RC and Doudna JA (2013) Molecular mechanisms of RNA interference. Annu Rev Biophys 42, 217–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Castel SE and Martienssen RA (2013) RNA interference in the nucleus: roles for small RNAs in transcription, epigenetics and beyond. Nat Rev Genet 14 (2), 100–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gagnon KT et al. (2014) RNAi factors are present and active in human cell nuclei. Cell Rep 6 (1), 211–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lee JS and Mendell JT (2020) Antisense-mediated transcript knockdown triggers premature transcription termination. Mol Cell 77 (5), 1044–1054 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lai F et al. (2020) Directed RNase H cleavage of nascent transcripts causes transcription termination. Mol Cell 77 (5), 1032–1043 e4. [DOI] [PubMed] [Google Scholar]
- 91.He H et al. (2013) Ultra-rare mutation in long-range enhancer predisposes to thyroid carcinoma with high penetrance. PLoS One 8 (5), e61920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Panigrahi AK et al. (2018) SRC-3 Coactivator Governs Dynamic Estrogen-Induced Chromatin Looping Interactions during Transcription. Mol Cell 70 (4), 679–694 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ortega E et al. (2018) Transcription factor dimerization activates the p300 acetyltransferase. Nature 562 (7728), 538–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fujisawa T and Filippakopoulos P (2017) Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat Rev Mol Cell Biol 18 (4), 246–262. [DOI] [PubMed] [Google Scholar]
- 95.Lee JE et al. (2017) Brd4 binds to active enhancers to control cell identity gene induction in adipogenesis and myogenesis. Nat Commun 8 (1), 2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.De Raedt T et al. (2014) PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature 514 (7521), 247–51. [DOI] [PubMed] [Google Scholar]
- 97.Kanno T et al. (2014) BRD4 assists elongation of both coding and enhancer RNAs by interacting with acetylated histones. Nat Struct Mol Biol 21 (12), 1047–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jonkers I and Lis JT (2015) Getting up to speed with transcription elongation by RNA polymerase II. Nat Rev Mol Cell Biol 16 (3), 167–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liu X et al. (2015) Ready, pause, go: regulation of RNA polymerase II pausing and release by cellular signaling pathways. Trends Biochem Sci 40 (9), 516–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhao Y et al. (2016) Activation of P-TEFb by Androgen Receptor-Regulated Enhancer RNAs in Castration-Resistant Prostate Cancer. Cell Rep 15 (3), 599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hnisz D et al. (2017) A Phase Separation Model for Transcriptional Control. Cell 169 (1), 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Boija A et al. (2018) Transcription Factors Activate Genes through the Phase-Separation Capacity of Their Activation Domains. Cell 175 (7), 1842–1855 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sabari BR et al. (2018) Coactivator condensation at super-enhancers links phase separation and gene control. Science 361 (6400). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cho WK et al. (2018) Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science 361 (6400), 412–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chong S et al. (2018) Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science 361 (6400). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lu H et al. (2018) Phase-separation mechanism for C-terminal hyperphosphorylation of RNA polymerase II. Nature 558 (7709), 318–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Berry J et al. (2015) RNA transcription modulates phase transition-driven nuclear body assembly. Proc Natl Acad Sci U S A 112 (38), E5237–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Langdon EM et al. (2018) mRNA structure determines specificity of a polyQ-driven phase separation. Science 360 (6391), 922–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Jain A and Vale RD (2017) RNA phase transitions in repeat expansion disorders. Nature 546 (7657), 243–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Maharana S et al. (2018) RNA buffers the phase separation behavior of prion-like RNA binding proteins. Science 360 (6391), 918–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ries RJ et al. (2019) m(6)A enhances the phase separation potential of mRNA. Nature 571 (7765), 424–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Boyle AP et al. (2008) High-resolution mapping and characterization of open chromatin across the genome. Cell 132 (2), 311–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Buenrostro JD et al. (2013) Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10 (12), 1213–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Giresi PG et al. (2007) FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res 17 (6), 877–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Creyghton MP et al. (2010) Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A 107 (50), 21931–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Rada-Iglesias A et al. (2011) A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470 (7333), 279–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Whyte WA et al. (2013) Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153 (2), 307–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Blow MJ et al. (2010) ChIP-Seq identification of weakly conserved heart enhancers. Nat Genet 42 (9), 806–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Heinz S et al. (2010) Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38 (4), 576–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Heinz S et al. (2013) Effect of natural genetic variation on enhancer selection and function. Nature 503 (7477), 487–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Cullen KE et al. (1993) Interaction between transcription regulatory regions of prolactin chromatin. Science 261 (5118), 203–6. [DOI] [PubMed] [Google Scholar]
- 122.Dekker J et al. (2002) Capturing chromosome conformation. Science 295 (5558), 1306–11. [DOI] [PubMed] [Google Scholar]
- 123.Zhao Z et al. (2006) Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions. Nat Genet 38 (11), 1341–7. [DOI] [PubMed] [Google Scholar]
- 124.Simonis M et al. (2006) Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nat Genet 38 (11), 1348–54. [DOI] [PubMed] [Google Scholar]
- 125.Dostie J et al. (2006) Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Res 16 (10), 1299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lieberman-Aiden E et al. (2009) Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326 (5950), 289–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kim TH et al. (2005) A high-resolution map of active promoters in the human genome. Nature 436 (7052), 876–80. [DOI] [PMC free article] [PubMed] [Google Scholar]