

Abstract
Advances in prenatal imaging, molecular diagnostic tools, and genetic screening have unlocked the possibility to treat congenital diseases in utero prior to the onset of clinical symptoms. While fetal surgery and in utero stem cell transplantation can be harnessed to treat specific structural birth defects and congenital hematological disorders, respectively, in utero gene therapy allows for phenotype correction of a wide range of genetic disorders within the womb. However, key challenges to realizing the broad potential of in utero gene therapy are biocompatibility and efficiency of intracellular delivery of transgenes. In this review, we outline the unique considerations to delivery of in utero gene therapy components and highlight advances in viral and non-viral delivery platforms that meet these challenges. We also discuss specialized delivery technologies for in utero gene editing and provide future directions to engineer novel delivery modalities for clinical translation of this promising therapeutic approach.
1. In Utero Therapy: Clinical Relevance and Therapeutic Avenues
Congenital disorders are a set of structural or functional anomalies that originate during prenatal development and result in significant morbidity, mortality, and health care resource utilization. These conditions affect ~2% of live births and are the leading cause of infant mortality in the United States, accounting for approximately 20% of all infant deaths.1,2,3 Globally, the World Health Organization estimates that 303,000 newborns perish prior to reaching 4 weeks of age as a result of congenital disorders or their associated complications.4 Structural and functional birth defects impact patients throughout their lives, leading to debilitating disabilities, disproportionally high healthcare costs, psychological trauma, and diminished quality of life.5,6
The majority of congenital disorders originate at conception and are due to genetic abnormalities. While cytogenic abnormalities and copy number variants (CNVs) are responsible for many congenital disorders, single-gene defects are also significant contributors. To date, more than 3500 monogenic congenital diseases have been characterized.7 Prenatal diagnosis of congenital diseases, including monogenic disorders, has expanded exponentially in recent years with the advent of high-resolution ultrasonography, ultrafast fetal MRI, and high sensitivity, high-throughput genetic testing.8,9 In particular, fetal whole exome sequencing (WES), coupled with image-guided ultrasound and digital PCR testing of cell-free DNA present in maternal blood, has emerged as a powerful diagnostic triad, unlocking the potential to diagnose and thus treat congenital disorders in utero prior to the onset of clinical symptoms.10
Scientific, clinical, and ethical considerations should be evaluated prior to prenatal treatment of genetic diseases.11 In brief, a strong correlation between genotype, phenotype, and clinical prognosis should exist before in utero therapy is considered. In addition, in utero therapy should be driven by the presence of a correctable anomaly, which, if untreated prenatally, would result in significant morbidity and/or mortality. Finally, there should be a favorable risk-to-benefit ratio for intervention, taking into account both maternal and fetal safety. If these clinical prerequisites are satisfied and diagnosis of congenital disease is confirmed, one of three in utero therapeutic avenues can be explored, as visualized in Figure 1.
Figure 1 |. Therapeutic avenues for fetal therapy of congenital disease.

Prenatal diagnosis of congenital disease has been facilitated by the development and sophistication of fetal ultrasound, exome sequencing of fetal DNA, and molecular testing of maternal-fetal blood. Fetal surgery offers the opportunity to correct structural anomalies prior to birth after they are identified via fetal ultrasound and MRI. Combining fetal imaging with genetic testing allows for early diagnosis of congenital disease. In utero stem cell transplantation (IUSCT) may be used to treat congenital disorders that result from a defect in a specific hematopoietic or mesenchymal cell type. For monogenic disorders, in utero gene therapy may be an effective tool to introduce physiologically deficient genes and even use editing to correct or remove disease-causing variants.
Open and minimally invasive fetal surgery for congenital anatomical malformations is a rapidly evolving field, born from the idea that irreversible end-organ damage resulting from a structural defect identified early in gestation can be alleviated by prenatal surgical correction.12 After nearly 40 years of translational small and large animal research studies and clinical practice, fetal surgery now exists as a viable and promising option for a select group of patients with anatomical malformations, including myelomeningocele (MMC), sacrococcygeal teratoma and congenital diaphragmatic hernia. The promise of fetal surgery for select patients is highlighted by the Management of Myelomeningocele Study (MOMS), a multi-institutional randomized control trial, which demonstrated improved outcomes following prenatal compared to postnatal repair in fetuses with a MMC in which specific maternal and fetal criteria were met.13 However, in utero surgical correction is highly invasive and poses risks to both mother and fetus, such as preterm delivery, chorioamnionitis, chorioamniotic membrane separation, placental abruption, uterine rupture, and the potential need for all subsequent pregnancies to be delivered via caesarian delivery. Moreover, fetal surgery is intrinsically limited to the correction of congenital structural abnormalities and does not address traditional genetic diseases.14
In utero stem cell transplantation (IUSCT) is a less invasive approach for the treatment of congenital disorders that often result from a defect in a specific hematopoietic or mesenchymal cell type. IUSCT takes advantage of normal developmental properties of the fetus, including its small size and immunologic immaturity, to facilitate allogenic stem cell engraftment and reconstitution of pathologically afflicted cell types. Transplantation of stem cells in utero circumvents typical immune barriers of postnatal bone marrow transplant via induction of donor-specific tolerance and avoids the toxic complications of myeloablative conditioning.15 In utero hematopoietic cell transplantation has been successful in the treatment of fetuses with X-linked severe combined immunodeficiency (SCID), but, up till now, has had minimal success in other hematologic disorders in which a selective donor cell engraftment advantage does not exist.16 More recently, the Boost Brittle Bones Before Birth (BOOSTB4) trial (NCT03706482) has sought to treat osteogenesis imperfecta with in utero mesenchymal stem cells, following promising preclinical and initial clinical cases, although data collection is still ongoing.17
In utero gene therapy offers the promise of a minimally invasive and broadly generalizable treatment for genetic disorders at their root causes. By transferring a functional exogenous copy of a gene to compensate for the dysfunction of a pathologic variant, in utero gene therapy aims to achieve sustained phenotype correction prior to the onset of disease pathogenesis. Building on this treatment modality, novel in utero gene editing technology presents an opportunity to therapeutically correct monogenic disorders.18 While the potential benefits of in utero gene therapy and gene editing are tremendous, efficient and long-term delivery of gene products to target locations in the body pose major challenges to clinical translation.19–21
In this review, we outline the unique advantages to and considerations for the delivery of gene therapy components in utero and highlight advancements in viral and non-viral delivery platforms that could be utilized to achieve fetal gene transfer. We also discuss specialized delivery technologies for in utero gene editing and provide future directions to engineer novel, non-viral delivery modalities to clinically translate this therapeutic approach.
2. Considerations for Delivery of In Utero Gene Therapy
2.1: Advantages of In Utero Gene Therapy
The delivery of genetic material to target cells in a developing fetus has several physiologic advantages, as depicted in Figure 2. The small size of the fetus (~100 g at 14-16 weeks) compared to a postnatal recipient (e.g. ~3.5 kg newborn, ~60 kg adult) maximizes delivery vector titer per weight of recipient, which facilitates efficient gene transduction.22 In addition, small recipient weight minimizes large-scale manufacturing constraints of delivery vectors.18
Figure 2 |. Advantages of the fetus for delivery of gene therapeutics.

The fetus possesses several unique characteristics that make it well-suited to receive gene therapy: (1) Small size provides a significant dose advantage; (2) Highly accessible population of progenitor cells allows for long-term proliferation of transduced cells; (3) Tolerogenic immune system limits a robust immune response to exogenous genetic material and delivery vehicle; (4) Fetal shunts maximize bioavailability of transgenes in the systemic circulation; and (5) Permeable blood-brain barrier (BBB) permits treatment of postnatally inaccessible central nervous system disorders.
The immunologic immaturity of the fetus allows for introduction of antigens (e.g. vector materials, transgenes) without a limiting immune response and with the induction of antigen-specific immune tolerance.23 This advantage is demonstrated in a study by Chan et al., who utilized viral vectors to achieve curative levels of human factor IX in a fetal macaque model of Hemophilia B, while demonstrating a high degree of immune tolerance and no long-term adverse effects of vector or transgene expression after four years.24 For target diseases that require serial doses of therapeutic vector, tolerance to gene therapy components is also favorable, since it avoids diminishing returns due to a gradual immune blockade. In contrast, multiple animal and clinical studies have described the presence of serum anti-vector antibodies and immune cells in recipients treated with postnatal gene therapy.25 Of particular note, Charlesworth et al. identified a high prevalence of anti-SaCas9 and anti-SpCas9 antibodies and Cas9 specific T cells in adult serum, which may preclude postnatal gene editing with CRISPR-Cas9 systems.26
The fetus also has a highly accessible and abundant population of stem and/or progenitor cells, which are ideal targets for long-term therapeutic genetic correction given their enhanced potential for expansion with propagation of the genetic correction, migration, and distribution in the fetal microenvironment.27 These highly proliferative cell populations have been effectively targeted in several animal studies. For instance, Porada et al. demonstrated successful modification of hematopoietic cells and long-term transgene expression following direct injection of a retroviral vector in utero.28 Kim et al. delivered MsrB3, a key gene associated with auditory function, to inner ear otocysts in MsrB3−/− knockout mice with congenital hearing loss at embryonic day 12.5 and observed hearing recovery at postnatal day 28.29 Similarly, Ito et al. targeted neural stem cells via a vector expressing the PQBP1 gene to rescue a mouse model of microcephaly.30 In fetal sheep, Tran et al. displayed transgene expression 40 months post-injection after targeting hematopoietic stem cells in utero.31 In adults, the vast majority of stem cells undergo quiescence, reducing both the efficiency and longevity of postnatal gene transfer in target tissues.32
The unique anatomy that supports the developing fetus during pregnancy may assist in delivery of transgenes to specific tissues via various routes of administration. Direct access to fetal circulation has been safely established via ultrasound-guided puncture of the umbilical vein in the late second trimester or even direct intracardiac injections earlier in gestation.21 Due to fetal vascular shunting via the ductus venosus, umbilical vein injections not only target the fetal liver but also provide the potential for a more robust systemic delivery approach.33,34 Importantly, during development, the liver also functions as a major site of fetal hematopoiesis and thus prenatal hepatic targeting provides a potential route for in vivo targeting of hematopoietic stem cells.35 An alternative route of administration to a different set of tissues involves injection into the amniotic fluid. While intra-amniotic delivery takes advantage of fetal breathing and swallowing mechanisms to target the developing lungs and GI tract respectively, this method is limited by dilution of vector concentration in the large volume of amniotic fluid.21 In large animals and clinical applications, direct fetal intratracheal injections provides an option to bypass this potential limitation. The routes of administration for in utero gene therapy detailed above, along with other, less common modes of delivery, have distinct advantages and disadvantages in the fetus, as outlined in Table 1.
Table 1 |.
Routes of Administration for In Utero Gene Therapy
| Routes of Administration | Advantages | Disadvantages | Reference |
|---|---|---|---|
| Intra-venous | Vast biodistribution, established clinical procedure | Lack of specificity | 28, 30 |
| Intra-amniotic | High transduction to tissues bathed by amniotic fluid (e.g. skin, GI tract, lungs) | Limited set of transfectable tissues, high therapeutic dosage due to dilution | 30, 31 |
| Intra-tracheal | High lung transduction | Possibly high procedure risk | 39 |
| Intra-cardiac | High tissue specificity, targeted delivery | Possibly high procedure risk | 40 |
| Intra-muscular | Ease of administration, established clinical procedure | Variable absorption rates and limited gene transfer efficacy | 41 |
| Intra-peritoneal | Effective gene transfer | Limited organ selectivity | 31, 42 |
| Intra-organ | High organ specificity | Possibly high procedure risk | 43 |
Fetal permeability of the blood-brain barrier (BBB) permits potential treatment of central nervous system (CNS) disorders with gene therapy via systemic delivery, which is a difficult endeavor postnatally. Mattar et al. showed expression of green fluorescent protein (GFP) up to 14 weeks after birth in the majority of neurons transduced intravascularly with gene therapy; although, diminished response was observed in astrocytes and the peripheral nervous system.44 Systemic delivery across the BBB is particularly advantageous in the treatment of neurodegenerative disease. Massaro et al. rescued lethal neurodegeneration in a knockout mouse model of Gaucher disease via in utero systemic delivery of a vector with reconstituted neuronal glucocerebrosidase expression.45 Thus, fetal gene delivery could allow early enough correction to prevent irreversible pathological changes in the brain.
2.2: Considerations for Fetal Delivery of Gene Therapy
As shown in Figure 3, there are several unique considerations for the delivery of gene therapy in utero, which can be explored by following the therapeutic course of a gene transfer vector. The vector must first enter the fetal circulation in a manner that is non-disruptive to both the mother and the fetus. Given the importance of ensuring maternal safety during in utero gene therapy, fetal gene delivery platforms must also achieve a delicate balance of transduction between its two recipients, maximizing transfer to the fetus and minimizing transfer to the mother. Of particular note, the potential for transplacental trafficking of viral vectors has been disputed in the literature, highlighting the need for further studies to explore this phenomenon prior to clinical translation.24,46
Figure 3 |. Considerations for successful in utero delivery of gene therapeutics.

In utero gene therapies must be administered in a manner safe for both the mother and the fetus. Once inside the fetus, there is a potential for the delivery vehicle to disrupt the developmental processes of adjacent tissues, as well as to travel into the maternal blood stream via the placenta. At the cellular level, the delivery vehicle may also be sensitive to fetal pH or temperature variation, especially within the endosome. Once the encapsulated transgene is released and transported into the nucleus, there is potential for the transgene or delivery vehicle to modulate the function of nearby genes, for the transgene to be lost during rapid cellular division, or for the transgene to be silenced during fetal epigenetic reprogramming. Finally, clearance of gene transfer vectors may be hastened in the fetal environment.
Once in fetal circulation, gene therapy vectors must reach their designated target tissue(s). This is complicated by the close physical proximity of developing embryologic tissues and the sensitivity of progenitor cells to incremental changes in their environment. The current repertoire of delivery platforms for fetal gene transfer has a relatively broad biodistribution, evidenced by the results of multiple studies that aimed to transduce a specific population of cells but reported widespread, albeit lower levels, of transduction in other tissues.34,47 To avoid unintended disruption to other tissues, delivery of the gene therapy vector should be tightly regulated. Moreover, expression of a transgene at an inappropriate place or time within the fetal environment could have particularly deleterious consequences (e.g. carcinogenesis, germline modification), although this has been described in only a few studies.48–50 Nevertheless, these findings motivate the development of highly specific delivery platforms for fetal gene transfer, along with tissue or cell-type specific promoters to control the expression of transgenes.
The delivery vector must then be successfully taken up by the target fetal tissue, which is mediated by ligand-cell surface receptor interactions and subsequently endosomal processes. In the fetal environment, the distribution of cell membrane molecules, receptors, and tissue factors is in constant flux, given the dynamic nature of neonatal ontology. Moreover, the internal body temperature of the human fetus has been observed to be 0.2°C higher than that of the mother, which may contribute to the alteration of the membrane state.51 Endosomal internalization, transport, and uncoating are all pH dependent processes. Given the pH sensitivity of both the endosome and the delivery vectors themselves, the marginally lower pH of the fetus (7.25-7.35) in comparison to a human adult (7.35-7.45) may pose a barrier to maximal gene transfer.52
Following transport into the nucleus, the vector genome must persist within the fetal cell to achieve therapeutic efficacy for a gene therapy. Depending on the choice of vector, the gene product can exist as an episome, an active exogenous DNA molecule lost during cell division, or can integrate into the host chromosome and persist in daughter cells. Most conventional gene therapies utilize episomal vectors, since they can persist long-term if delivered to relatively quiescent tissues.53 However, given the highly proliferative nature of fetal cells, these vectors may be disadvantaged by rapid turnover of cells. While insertional vectors may last longer in target progenitor cells, they have the potential to activate or disrupt nearby genes via insertional mutagenesis.19 Although this is not well elucidated in the literature for prenatal delivery applications, one study has reported a high incidence of hepatocellular carcinoma in mice after in utero gene therapy.54 Thus, delivery vectors for in utero gene therapy must be optimized to maximize gene persistence and minimize risk of downstream complications. In contrast, episomal vectors are actually preferable vehicles for in utero gene editing, given the heightened risk of insertional vector integration into cut sites.
To achieve lifelong expression, the vector genome must sustain transcriptional expression in light of epigenetic modifications. Especially prominent in neonatal ontology, dynamic regulation of the epigenome underlies cellular plasticity and provides a response to developmental cues.55 Epigenetic programming has been observed in the transitions between different states of embryonic stem cells and during lineage differentiation of tissue-resident stem cells.56 Given this, there may be an enhanced likelihood of epigenetic modification to the vector genome in fetal tissues, along with indirect modulation of neighboring epigenetic signatures. While the interaction between the fetal epigenome and gene therapy has not been well characterized, studies of metabolic syndrome have implicated in utero epigenetic programming in disease pathogenesis.57
A final consideration for gene transfer delivery vehicles is their clearance from the body. A study of fetal morphine uptake in late gestation demonstrated that the fetus has an elevated intrinsic metabolic clearance for this substance.58 If this result holds true for gene therapy delivery vehicles, which is especially likely for non-viral vectors, the highly active clearance machinery in the fetal liver and bloodstream could decrease the potential for cellular uptake of a transgene and increase required therapeutic dosage. However, it should be acknowledged that no sophisticated pharmacokinetic studies have been conducted in utero with gene therapy delivery vehicles.
3. Viral Delivery Platforms for In Utero Gene Therapy
Therapeutic transgenes are unable to directly pass through the cell membrane because of their large size and negative charge.59 As such, transgenes are introduced via delivery vectors. The vast majority of gene therapies utilize viral delivery platforms, given their naturally high efficiency of gene transduction to eukaryotic cells, developed over the course of evolution.60–62 In fact, 70% of ongoing gene therapy clinical trials worldwide are using a viral vector.63 While the spectrum of viral vectors is very broad, there are four major classes of viral gene therapy methods: retroviruses, lentiviruses, adenoviruses, and adeno-associated viruses.
3.1: Retroviruses
Retroviruses are a class of enveloped, single-stranded RNA viruses, which retrotranscribe their genome to DNA via reverse transcriptase.64 Simple retroviruses, such as the murine leukemia virus (MLV), were once thought to be ideal vectors for gene therapy, since they integrate into the genome and provide long-term stable expression of a large transgene in transduced cells and their progeny.53 Retroviruses also display minimal immunogenicity due to low viral reproduction and a predilection for immune evasion.65 Since the retrotranscribed genome of simple retroviruses is unable to pass the nuclear membrane, integration only occurs during mitosis, limiting viral transduction to dividing tissues.59 This characteristic of simple retroviruses is decidedly advantageous in the fetus due to the proliferative nature of constituent cell populations.
In the early 2000s, several in utero studies in animal models, including sheep and non-human primates, demonstrated successful transduction of fetal progenitor cells and amelioration of clinical phenotype with retroviral-based gene therapy.31,66 However, work was stymied following the results of the first gene therapy trial in children with X-linked severe combined immunodeficiency (X-SCID), which showed the oncogenicity of retroviral vectors that undergo insertional mutagenesis at gene regulatory sites.67 To reduce the genotoxicity of retroviral vectors, researchers have generated self-inactivating (SIN) vectors that lack enhancer or promoter regions of the long terminal repeat (LTR), decreasing the potential for insertional mutagenesis through transcriptional inactivation.68 Although SIN-MLV vectors have been shown to possess reduced transduction capability, their integration profile is shifted to disfavor cell growth genes, transcription start sites, and epigenetically-defined promoters.69 These vectors are currently being tested in clinical trials for SCID, and further studies are necessary to determine the suitability of simple retroviruses for fetal applications.70 As discussed in the next section, viral gene therapy within this family of vectors has moved towards the use and optimization of lentiviruses.
3.2: Lentiviruses
Lentiviruses are a subclass of retroviruses that are primarily distinguished by their ability to transduce both dividing and non-dividing cells, since they encode proteins required to permit nuclear localization of viral DNA.53 In addition, engineered complex lentiviruses, such as those constructed from a human immunodeficiency virus (HIV), are self-inactivating due a deletion of the LTR, which limits their genotoxicity.71 Taken together, these qualities make lentiviruses useful for gene therapy in adults, who have relatively quiescent tissues. Indeed, third-generation, self-inactivating lentiviral vectors have recently been used in multiple clinical trials to correct immunodeficiencies and hemoglobinopathies in vivo without adverse events.72 Lentiviruses have also been used to successfully facilitate ex-vivo gene therapy of autologous stem cells, although it is unlikely that such a procedure could be adapted in utero due to the inability to safely obtain adequate numbers of an autologous cell source.73
The safety and efficacy of in utero gene delivery using lentiviral vectors in multiple animal models has been well characterized.74–77 Recently, Shangaris et al. injected a humanized mouse model of β-thalassemia with a β globin-expressing lentiviral vector in utero and noted postnatal normalization of blood hemoglobin levels.36 While this result demonstrates the applicability of lentiviral vectors for fetal applications, the researchers found unintended integration sites, including the Peg12 gene, which is associated with tumorigenesis. In this study, animals were sacrificed 32 weeks after birth at which time no carcinoma was observed. Thus, while lentiviral vectors appear to hold promise for in utero gene therapy, further research into tissue-restricted promoters, enhanced efficiency, and overall safety is necessary prior to their usage in fetuses.72,78
3.3: Adenoviruses
Adenoviruses are large, nonenveloped viruses with icosahedral nucleocapsids and double-stranded DNA genomes.53 The vector genome of adenoviruses remains episomal unlike in integrating viruses.71 Without rapid turnover of cells, adenovirus gene constructs are able to endure long-term and sustain gene expression, making these viruses particularly useful in adult gene therapy. In fact, adenoviruses are the most frequently used vectors in adult clinical trials.79 However, rapid cell division in the fetus may make an adenovirus less desirable for gene therapy applications in which episomal persistence of the therapeutic transgene is required.21
The high immunogenicity of adenoviruses is another significant concern.37,47 While less toxic and less inflammatory third-generation, helper-dependent adenoviral vectors have been constructed, these delivery platforms still prompt an immediate innate immune response and a secondary antigen-dependent response.80 Thus, the clinical translation of adenoviruses for in utero applications may be limited, although it should be noted that the relatively immuno-naïve fetus may be able to better tolerate adenoviral antigens. Certainly, the large packing capacity and high gene transduction efficiency of adenoviruses make these delivery vectors strong candidates for proof-of-principle in utero studies in animal models.
3.4: Adeno-associated viruses (AAVs)
AAVs are single-stranded, episomal DNA viruses that can transduce both dividing and quiescent cells.81 AAVs are easily pseudotyped with 13 serotypes currently identified, each of which uses a different receptor repertoire on host cell surfaces for infection.53 Consequently, AAVs can be used to transduce specific sets of cells or tissue types. In comparison to adenoviruses, AAVs are non-pathogenic and are less immunogenic.82 As such, AAVs have been used in landmark adult gene therapy clinical trials for hemophilia A and B, and in utero therapy for these disorders has been shown to be promising in non-human primates.24,83,84 However, AAVs have a small packaging capacity of ~4 kb, which restricts the pool of transgenes that can be carried.85
Both the safety and long-term efficacy of AAVs have been well characterized in a number of in utero studies across target organs. For instance, an AAV-mediated in utero gene therapy was used to rescue a mouse model of acute neuronopathic Gaucher disease.86 In a macaque model, in utero transfer of AAVs expressing factor IX produced long-term gene expression without toxicity.24 The use of AAVs versus other viral vectors for in utero gene therapy has also been directly compared. In a mouse model, Joyeux et al. found that lung cells transduced with an AAV2/9 vector expressing GFP in utero had expression for up to 6 months, while lentiviral-mediated gene transfer resulted in no observed GFP expression.87 While these results are promising for clinical translation in utero, the potential for AAV integration in the fetal environment needs to be fully evaluated to ensure adequate safety.
4. Non-Viral Delivery Platforms for In Utero Gene Therapy
The underlying mutagenicity and immunogenicity of viruses are barriers to their use in postnatal gene therapy.63 Consequently, non-viral vectors have emerged as safer alternatives.88–90 Some of the potential benefits of postnatal non-viral gene therapy approaches are also highly applicable to in utero applications, making these delivery platforms useful for gene therapy and gene editing before birth. There are four large classes of non-viral gene delivery platforms: physical methods, inorganic nanoparticles, polymer-based nanoparticles, and lipid-based nanoparticles.
4.1: Physical Methods
A naked nucleic acid injection into local tissues or the systemic circulation without a carrier is the simplest fetal delivery platform. However, due to rapid degradation by endonucleases and clearance by the mononuclear phagocyte system, transfection efficacy is limited.91 Consequently, research has shifted toward a number of physical manipulations to improve the efficiency of gene delivery.92 For instance, electroporation introduces nucleic acids into cells via an electric field that causes transient destabilization of the cell membrane.93 New electroporation technologies, which could be adapted for in utero brain applications, deliver large transgenes rapidly with high protein expression and very little cellular toxicity.94 While this approach works well for solid tissues, delivery to the vast majority of soft organs is currently not possible and thus approach is not currently clinically relevant.95
Despite this limitation, in utero electroporation may be used in the delivery of transgenes to specific cell populations in the central nervous system. Tabata and Nakajima report gene expression of GFP in more than 80% of newborn mice after fetal electroporation.96 Similar work has shown transfection of other brain structures, including the hippocampus and the frontal cortex.97,98 The ability of this technique to specifically target progenitor neurons is a major advantage in comparison to other methods, which require cell-specific targeting factors. Extending the scope of this procedure, Takeda et al. utilized electroporation-mediated gene transfer in utero to partially rescue hearing and vestibular function in mice.99 However, large animal studies have not yet been performed, risk of electric shock to the fetus is ethically challenging, and low-level release of immune system-inducing cytokines has been reported.95
4.2: Inorganic Nanoparticles
Inorganic nanoparticles are well-suited for gene delivery due to their biocompatibility and unique physical and chemical properties.100,101 In particular, gold nanoparticles (AuNPs) are highly modular, have strong surface plasmon resonance, and are easily functionalized.102 Owing to their high payload, low toxicity, efficient uptake, and fast endosomal escape, AuNPs are increasingly being employed for adult gene therapy purposes in vitro and in vivo.103,104
For instance, Conde et al. developed DNA-complexed AuNPs that induced RNA interference against the C-MYC proto-oncogene in a mouse model.105 Moreover, dendrimer-entrapped gold nanoparticles (Au DENPs) complexed with HIC1 gene were shown to inhibit cancer cell migration and metastasis in vitro.106 Recent work has demonstrated the ability of a gold nanocarrier platform to deliver gene editing technology in vivo, as described in the subsequent section.107 However, to date, no gene therapy experimentation has been conducted in utero with these delivery vehicles.
4.3: Polymer-based Nanoparticles
Biodegradable polymers and their copolymers are co-blocked with poly(ethylene glycol) (PEG) to form polymer-based nanoparticles (PNPs) that encapsulate and transport nucleic acids.63 PNPs have great chemical diversity and are easily functionalized, priming them to be conjugated with basic chemistry to target specific cells.108 Nonetheless, common PNPs, like polyethyleneimine (PEI) and poly-L-lysine (PLL), are cytotoxic, limiting their application to the fetus.109
Recently, safer biodegradable polymers have been developed and tested. For instance, poly(b-amino esters) (PBAEs) are degradable poly-cations with demonstrated potential as delivery platforms for nucleic acids both in vitro and in vivo.110–113 Hyperbranched PBAEs have also been synthesized, which enable the nanoformulation of stable and concentrated mRNA polyplexes suitable for inhalation.114 These PNPs may be promising candidates for fetal delivery, although further animal studies are required to characterize their safety and efficacy. The potential use of PNPs for in utero gene editing is also being explored with poly(lactic-co-glycolic acid) (PLGA) nanoparticles, as detailed in the subsequent section.34
4.4: Lipid-based Nanoparticles
Lipid nanoparticles (LNPs) are self-assembled nanostructures with the ability to encapsulate and deliver nucleic acids.115 LNPs possess incredible modularity, are minimally immunogenic, and have high carrying capacity, which permits large nucleic acids and protein delivery.63 Moreover, like non-integrating viral vectors and other nonviral delivery systems, LNPs enable transgene expression without risk of insertional mutagenesis or germline transmission.116,117 In adult animal models, studies have exhibited safe LNP-assisted delivery of nucleic acids for cancer therapy.118–122 However, LNPs have low transfection efficacy owing to poor stability and rapid clearance.63
LNPs may have enhanced delivery efficacy in utero due to the distinct plasma lipoprotein profile of fetal blood, which is skewed towards high-density lipoprotein that contains a high proportion of apolipoprotein E (apoE).123 Given that apoE is the principal endogenous targeting ligand for LNPs, this delivery modality may overcome transfection limitations observed in adults.124 Strong biosafety also makes LNPs exciting candidates for fetal applications, although no studies have utilized these vehicles to deliver gene therapy in utero. In postnatal studies, it has been observed that LNPs tend to accumulate in the liver, which could be intrinsically beneficial for a wide range of congenital metabolic and hematopoietic disorders. For extrahepatic fetal applications, this accumulation in the liver could be a limitation, although a liposome designed to transiently occupy liver cells may allow for broader systemic delivery.125 The targeting specificity of LNPs may also be improved by the addition of chemical moieties, including cell adhesion molecules, antibodies, or tissue factors.126 LNPs are at the cutting-edge of biomaterial research, and their full potential for both gene delivery and gene editing applications has yet to be elucidated.
5. Delivery Platforms for In Utero Gene Editing: Progress and Prospect
The type II microbial clustered, regularly interspaced, palindromic repeats (CRISPR)-associated (Cas) system has been engineered into a powerful genome editing tool consisting of the Cas9 nuclease and a single guide RNA (sgRNA).127,128 Cas9 targets regions of the genome complementary to the sgRNA and generates double-stranded DNA breaks, allowing cellular DNA machinery to repair them via nonhomologous end-joining (NHEJ) or homology-directed repair (HDR). Since the sgRNA sequence can be engineered to target virtually any site in the genome with some sequence limitations, this system is distinguished by its incredible specificity and versatility.128 Importantly, CRISPR-Cas9 has made therapeutic editing of the genome a possibility and facilitated the development of new, more sophisticated technologies.129 For instance, base editing is a novel genome editing approach that uses components from CRISPR-Cas9 systems along with other enzymes to directly generate point mutations into cellular DNA without creating double-stranded DNA breaks, thereby reducing the potential for indels, translocations, and genomic rearrangements.130 The in vivo therapeutic potential of genome editing technology has been demonstrated for a broad spectrum of diseases in preclinical models, including Leber congenital amaurosis type 10 and Hutchinson-Gilford progeria syndrome.131,132
To maximize its potential, genome editing technology must be specifically and efficiently delivered within the body.133 Alongside conventional challenges for gene delivery, transport of genome editing technology is complicated by the large size and charge of its components.134 Furthermore, for fetal applications, targeted delivery of gene editing technology is paramount, as the consequences of off-target effects are potentially more consequential than in ex vivo or postnatal gene editing. However, if the system is safely delivered, genome editing technology holds tremendous potential to remove or correct pathogenic mutations early in development prior to the onset of irreversible pathology, especially given the highly proliferative nature of fetal cells and the ability to harness the relatively error-free HDR pathway. Several platforms have been developed to deliver genome editing technology in utero, as described below.
5.1: Current Delivery Platforms for In Utero Gene Editing
For proof-of-principle experiments, researchers have turned to adenoviral vectors for delivery of genome editing technology due to their larger carrying capacity and efficient transduction of multiple cell types. Specifically, Rossidis et al. utilized an adenoviral vector to deliver base editor 3, an SpCas9-based cytosine base editor, in utero to precisely introduce a nonsense mutation in the Hpd gene of a hereditary tyrosinemia type 1 mouse model and rescue its neonatal lethal phenotype. This study was one of the first to demonstrate the feasibility of CRISPR-mediated in utero gene editing.47 Similarly, Alapati et al. used an adenoviral delivery approach to excise the mutant SftpcI73T gene via CRISPR-mediated NHEJ in gestational day 16 fetuses in the mouse model of surfactant protein C deficiency, rescuing its neonatal lethal phenotype in a subset of mice.37 However, both studies acknowledge the immunogenicity of adenovirus vectors and their limitations for clinical translation, while pointing towards AAV and novel non-viral delivery platforms as potential replacements.
Multiple studies in neonatal and adult mouse models have demonstrated the feasibility of delivering Cas9 and gene editing technology for in vivo editing via AAVs, highlighting their potential as a prenatal delivery approach. Given the limited packaging capacity of AAVs, Nelson et al. used a dual-AAV approach to deliver Cas9 and two sgRNAs, designed to excise exon 23 from the DMD gene, in the neonatal Duchenne muscular dystrophy (DMD) mouse model.135 They showed successful genome editing and dystrophin protein restoration for 1 year after a single intravenous administration of the AAVs. Given that genome editing technology induces novel DNA breaks, the risk of AAV genotoxicity is heightened.136 Indeed, researchers in the aforementioned study detected several AAV genome integrations. Furthermore, the requisite multi-virus approach due to the low packaging capacity of AAV also has the potential for lower gene editing efficiency.
Given their large carrying capacity and low immunogenicity, non-viral platforms have tremendous potential for the delivery of genome editing technology in vivo and more specifically in utero. Shinmyo et al. used electroporation to deliver Cas9 in utero in order to knock out the Satb2 gene from the mouse brain.137 Their technique is efficient but is likely limited for human applications given the high voltage that must be applied. A clinically translatable recent innovation is CRISPR-Gold, a gold nanoparticle conjugated with DNA and complexed with donor DNA, Cas9 ribonucleoprotein, and an endosomal disruptive polymer.107 In a mouse model, CRISPR-Gold was shown to lower mGluR5 levels in the striatum and prevent exaggerated repetitive behaviors due to Fragile X syndrome.138
Nanoparticles are an alternative, generalizable delivery platform. Riccardi et al. exhibited in utero delivery of PLGA nanoparticles loaded with triplex-forming peptide nucleic acids and donor DNAs for complete postnatal amelioration of β-thalassemia in a humanized mouse model.34 This study noted a discernable phenotype improvement with gene editing frequency of ~6% in target cells. To produce higher gene editing frequencies, which may be required clinically, the investigators suggest the delivery of multiple treatments, which is made possible by the reported low toxicity of each dose. PBAEs have also recently been shown to efficiently co-deliver DNA plasmids encoding Cas9 and sgRNA in utero.139 It remains to be seen if this technology could be successful in utero. To date, there are no reported in utero gene editing studies that utilized LNPs. However, given their modularity, LNPs are potentially strong candidates to deliver therapeutic gene editing technology to the fetus.
5.2: Optimizing LNPs for In Utero Gene Editing
Studies in adult mice and non-human primates have reported successful LNP-mediated delivery of mRNA.121,140,141 A dual-delivery approach with LNP-mediated delivery of Cas9 mRNA and AAV-mediated delivery of sgRNA was applied to repair a Fah-splice mutation in a mouse model of hereditary tyrosinemia type 1.142 Recently, LNPs were shown to deliver both Cas9 mRNA and sgRNA to adult mice and rats to edit the Ttr gene and result in knock down of transthyretin serum protein to levels that would be therapeutically beneficial in amyloidosis.143 While these studies demonstrate that LNPs have the potential to deliver gene editing components, they highlight the need to improve on cell transfection and gene editing efficacy. In Figure 4, we outline a component-directed rationale for low LNP gene transfer and discuss optimization strategies to overcome these pitfalls.
Figure 4 |. Optimization strategies for enhanced LNP gene transfection.
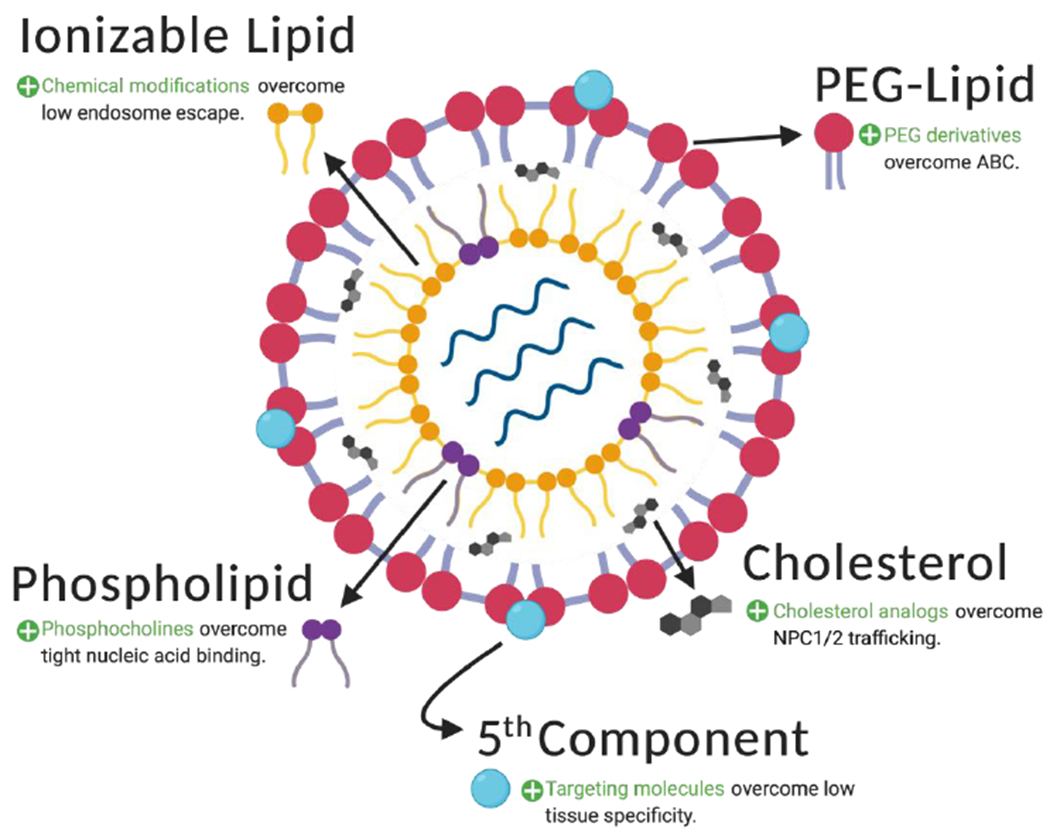
LNPs are synthesized from four components: ionizable lipids, phospholipids, cholesterol, and PEG-lipids. These combine to form liposome-like nanocarriers that encapsulate nucleic acids. While each of these components contributes to the overall functionality of the LNP, they have distinct drawbacks that may limit gene transfection of genome editing systems in utero. To improve the efficiency of gene transfer, an array of modified components has been proposed, as described in the text and summarized above. Additionally, a targeting molecule can be added to LNPs in order to improve the targeting specificity of gene delivery.
Conventionally, LNPs are formulated with four components: ionizable lipids, phospholipids, cholesterol, and polyethylene glycol (PEG)-lipid conjugates. Ionizable lipids are responsible for LNP cellular uptake via endocytosis.144 However, the limited capacity of LNPs to undergo endosomal escape diminishes their transfection efficacy, since only a small fraction (<2%) of their nucleic acid cargo is able to reach the cytoplasm.145 In recent work, our group screened a library of 24 ionizable lipids for delivery potential, optimized the top performer for transfection of primary T cells, and encapsulated chimeric antigen receptor (CAR) mRNA to generate functional CAR-T cells.146 A similar systematic methodology could be used to facilitate Cas9 mRNA endosomal escape in order to enhance delivery to a user-defined subset of fetal progenitor cells. At a cellular level, the interaction between ionizable lipids and the endosome is beginning to be understood. Maugeri et al. have posited that a neutrally charged 1:1 molar ratio between ionizable lipid and mRNA could facilitate an alternative extracellular vesicle pathway to improve endosomal escape and enhance cellular transfection.147
Phospholipids provide aid in endosomal escape and provide structure to the LNP bilayer.148 There are a variety of phospholipids used for LNP synthesis, each with its own encapsulation efficiency. While higher encapsulation efficiency may be beneficial during LNP production, it is a hindrance to release of mRNA into the cytosol and inhibits the onset of translation. These competing effects are captured in work by Kauffman et al., who reported that LNP formulations that utilized DSPC (36% encapsulation efficiency) instead of DOPE (51% encapsulation efficiency) have significantly higher transfection efficiency in vivo.149 Inclusion of phosphocholine-containing phospholipids and minimization of the scaffold length difference between phospholipids and ionizable lipids appear to improve LNP delivery potency. Such an optimization strategy may facilitate transfer of gene editing components in utero.150
Cholesterol enhances LNP stability and promotes membrane fusion. Recently, Patel et al. showed that incorporating cholesterol analogs, such as β-sitosterol, into LNPs can improve their nucleic acid delivery by facilitating alternative endosomal escape pathways.151 Structural analysis of these enhanced LNPs (eLNPs) revealed that they had a polyhedral surface morphology, instead of spherical, which may facilitate fusion with cellular membranes. Moreover, eLNPs were hypothesized to modulate the activity of cholesterol trafficking machinery, reducing eLNP efflux into the extracellular space, improving intracellular availability, and increasing delivery. Optimization of LNPs with naturally occurring materials, especially those with potential health benefits like β-sitosterol, is favorable for fetal applications.
PEG-lipid conjugates coat LNPs to reduce LNP aggregation, improve stability, and increase circulation time via reduced particle uptake by the mononuclear phagocyte system.152 However, a limitation of PEGylated delivery systems is the induction of accelerated blood clearance (ABC), a phenomenon of immune activation and rapid clearance upon repeated administration that results in massive accumulation of LNPs in the liver.153 Minimizing ABC would be beneficial since, despite improvements to transfection efficiency, LNP-mediated treatments to the fetus may have to be repeated for long-term therapeutic effect. Moreover, ABC is presumably counterproductive to LNP targeting to extrahepatic fetal tissues. PEG-lipid derivatives with varying pharmacokinetic profiles have been synthesized to meet this challenge. For example, increasing PEG molecular weight alleviated production of anti-PEG IGM and reduced the ABC phenomenon but lowered circulation times.154 As seen in this study, PEG modulation may result in a direct conflict between desirable and undesirable properties in LNPs.
Recent studies have motivated the addition of a fifth component to the standard LNP formulation to optimize targeting specificity and increase transfection efficiency to a selected tissue. Supplementation of a well-known quaternary amino lipid, DOTAP, during LNP synthesis altered the in vivo RNA delivery profile and mediated tissue-specific gene delivery as a function of the percentage of added DOTAP.155 The investigators hypothesized that this novel selective organ targeting (SORT) strategy may be due to a change in LNP pKa and surrounding protein corona. While further work is required to fully characterize this technology, these findings clearly demonstrate that the optimal solution to deliver in utero gene editing components may require more sophisticated LNP engineering than the current four-factor methodology.
6. Conclusion
In utero therapies have the potential to cure monogenic disorders prior to birth. While fetal surgery and IUSCT have exciting clinical applications, they are limited in scope. In contrast, in utero gene therapy offers a minimally invasive solution to prevent pathogenesis of congenital genetic disease. The fetus is uniquely equipped to receive gene therapy, although there are several physiological barriers that challenge therapeutic delivery. Viral vectors have been developed to overcome these barriers, possessing strong gene transfer profiles but also potential genotoxicity. Given their success in clinical gene therapy trials for monogenic diseases, it is likely that viral vectors will be among the first delivery modalities to be used in utero. However, non-viral vectors are also promising and potentially safer candidates for fetal delivery, given several intrinsic advantages in comparison to viral vectors. With further optimization of their safety and transfection efficacy, non-viral vectors may allow in utero gene therapy to become a clinical reality.
Acknowledgements:
M.J.M. acknowledges support from a Burroughs Wellcome Fund Career Award at the Scientific Interface (CASI), a US National Institutes of Health (NIH) Director’s New Innovator Award (DP2 TR002776), a grant from the American Cancer Society (129784- IRG-16-188-38-IRG), the National Institutes of Health (NCI R01 CA241661, NCI R37 CA244911, and NIDDK R01 DK123049), an Abramson Cancer Center (ACC)-School of Engineering and Applied Sciences (SEAS) Discovery Grant (P30 CA016520), and a 2018 AACR-Bayer Innovation and Discovery Grant, Grant Number 18-80- 44-MITC (to M.J.M.).
W.H.P. acknowledges support from the NIH Director’s New Innovator Award (DP2HL152427), the NIH (NIDDK R01DK123049 and NHLBI R01HL151352), and generous family gifts to the Children’s Hospital of Philadelphia.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Interests:
The authors declare no competing interests.
7. References
- 1.Center for Health Statistics, N. NCHS Data Brief, Number 279, March 2017. 1–8 (2005). [Google Scholar]
- 2.Feldkamp ML, Carey JC, Byrne JLB, Krikov S & Botto LD Etiology and clinical presentation of birth defects: Population based study. BMJ 357, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xu J, Murphy SL, Kochanek KD, Bastian B & Arias E Death: Final Report for 2016. Natl. Vital Stat. Rep 67, 1–76 (2018). [PubMed] [Google Scholar]
- 4.Liu L et al. MCEE-WHO methods and data sources for child causes of death 2000-2015. World Heal. Organ 1, 20 (2016). [Google Scholar]
- 5.Apfeld JC et al. The disproportionate cost of operation and congenital anomalies in infancy. Surg. (United States) 165, 1234–1242 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Kolaitis GA, Meentken MG & Utens EMWJ Mental health problems in parents of children with congenital heart disease. Front. Pediatr 5, 1–7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Amberger JS, Bocchini CA, Scott AF & Hamosh A OMIM.org: Leveraging knowledge across phenotype-gene relationships. Nucleic Acids Res. 47, D1038–D1043 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lord J et al. Prenatal exome sequencing analysis in fetal structural anomalies detected by ultrasonography (PAGE): a cohort study. Lancet 393, 747–757 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bianchi DW et al. DNA sequencing versus standard prenatal aneuploidy screening. N. Engl. J. Med 370, 799–808 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Yang Y et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N. Engl. J. Med 369, 1502–1511 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Almeida-Porada G et al. In Utero Gene Therapy Consensus Statement from the IFeTIS. Mol. Ther 27, 705–707 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adzick NS Prospects for fetal surgery. Early Hum. Dev 89, 881–886 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adzick NS et al. A randomized trial of prenatal versus postnatal repair of myelomeningocele. N. Engl. J. Med 364, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Farmer D Fetal surgery. BMJ 326, 461–462 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sagar R, Götherström C, David AL & Westgren M Fetal stem cell transplantation and gene therapy. Best Pract. Res. Clin. Obstet. Gynaecol 58, 142–153 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Flake AW et al. Treatment of X-Linked Severe Combined Immunodeficiency By In Utero Tranplantation of Paternal Bone Marrow. N. Engl. J. Med 335, 1806–1810 (1996). [DOI] [PubMed] [Google Scholar]
- 17.Götherström C et al. Pre- and Postnatal Transplantation of Fetal Mesenchymal Stem Cells in Osteogenesis Imperfecta: A Two-Center Experience. Stem Cells Transl. Med 3, 255–264 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peranteau WH & Flake AW The Future of In Utero Gene Therapy. Mol. Diagnosis Ther 24, 135–142 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Almeida-Porada G, Atala A & Porada CD In utero stem cell transplantation and gene therapy: rationale, history, and recent advances toward clinical application. Molecular Therapy - Methods and Clinical Development 5, 16020 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McClain LE & Flake AW In utero stem cell transplantation and gene therapy: Recent progress and the potential for clinical application. Best Pract. Res. Clin. Obstet. Gynaecol 31, 88–98 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Waddington SN et al. In utero gene therapy: Current challenges and perspectives. Molecular Therapy 11, 661–676 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Witt R, MacKenzie TC & Peranteau WH Fetal stem cell and gene therapy. Semin. Fetal Neonatal Med 22, 410–414 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Colletti E, Lindstedt S, Park PJ, Almeida-Porada G & Porada CD Early Fetal Gene Delivery Utilizes both Central and Peripheral Mechanisms of Tolerance Induction. Exp. Hematol 36, 816–822 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chan JKY et al. Therapeutic expression of human clotting factors IX and × following adeno-associated viral vector-mediated intrauterine gene transfer in early-gestation fetal macaques. FASEB J. 33, 3954–3967 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mingozzi F & High KA Immune responses to AAV vectors: Overcoming barriers to successful gene therapy. Blood 122, 23–36 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Charlesworth CT et al. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat. Med 25, 249–254 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hartman HA, Rossidis AC & Peranteau WH In Utero Gene Therapy and Genome Editing. Curr. Stem Cell Reports 4, 52–60 (2018). [Google Scholar]
- 28.Porada CD et al. In Utero Gene Therapy: Transfer and Long-Term Expression of the Bacterial neor Gene in Sheep after Direct Injection of Retroviral Vectors into Preimmune Fetuses. Hum. Gene Ther 9, 1571–1585 (1998). [DOI] [PubMed] [Google Scholar]
- 29.Kim MA et al. Methionine Sulfoxide Reductase B3-Targeted in Utero Gene Therapy Rescues Hearing Function in a Mouse Model of Congenital Sensorineural Hearing Loss. Antioxidants Redox Signal. 24, 590–602 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ito H et al. In utero gene therapy rescues microcephaly caused by Pqbp1-hypofunction in neural stem progenitor cells. Mol. Psychiatry 20, 459–471 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tran ND et al. Induction of stable prenatal tolerance to β-galactosidase by in utero gene transfer into preimmune sheep fetuses. Blood 97, 3417–3423 (2001). [DOI] [PubMed] [Google Scholar]
- 32.Engle KM; Mei T-S; Wasa M; Yu J-Q. Molecular regulation of stem cell quiescence. Acc. Chem. Res 45, 788–802 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gonin P & Gaillard C Gene transfer vector biodistribution: Pivotal safety studies in clinical gene therapy development. Gene Ther. 11, S98–S108 (2004). [DOI] [PubMed] [Google Scholar]
- 34.Ricciardi AS et al. In utero nanoparticle delivery for site-specific genome editing. Nat. Commun 9, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Popescu DM et al. Decoding human fetal liver haematopoiesis. Nature 574, 365–371 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shangaris P et al. In Utero Gene Therapy (IUGT) Using GLOBE Lentiviral Vector Phenotypically Corrects the Heterozygous Humanised Mouse Model and Its Progress Can Be Monitored Using MRI Techniques. Sci. Rep 9, 1–17 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alapati D et al. In utero gene editing for monogenic lung disease. Sci. Transl. Med 11, 1–14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Endo M et al. The developmental stage determines the distribution and duration of gene expression after early intra-amniotic gene transfer using lentiviral vectors. Gene Ther. 17, 61–71 (2010). [DOI] [PubMed] [Google Scholar]
- 39.Carlon M et al. Efficient gene transfer into the mouse lung by fetal intratracheal injection of rAAV2/6.2. Mol. Ther 18, 2130–2138 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sampaolesi M et al. Cell therapy of α-sarcoglycan null dystrophic mice through intraarterial delivery of mesoangioblasts. Science 301, 487–492 (2003). [DOI] [PubMed] [Google Scholar]
- 41.MacKenzie TC et al. Transduction of satellite cells after prenatal intramuscular administration of lentiviral vectors. J. Gene Med 7, 50–58 (2005). [DOI] [PubMed] [Google Scholar]
- 42.David AL et al. Recombinant adeno-associated virus-mediated in Utero gene transfer gives therapeutic transgene expression in the sheep. Hum. Gene Ther 22, 419–426 (2011). [DOI] [PubMed] [Google Scholar]
- 43.Mehta V, Nader KA, Waddington S & David AL Organ targeted prenatal gene therapy—how far are we? Prenat. Diagn 31, 720–734 (2011). [DOI] [PubMed] [Google Scholar]
- 44.Mattar CN et al. Systemic delivery of scAAV9 in fetal macaques facilitates neuronal transduction of the central and peripheral nervous systems. Gene Ther 20, 69–83 (2013). [DOI] [PubMed] [Google Scholar]
- 45.Massaro G et al. Systemic AAV9 gene therapy using the synapsin I promoter rescues a mouse model of neuronopathic Gaucher disease but with limited cross-correction potential to astrocytes. Hum. Mol. Genet 29, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mattar CNZ et al. In Utero Transfer of Adeno-Associated Viral Vectors Produces Long-Term Factor IX Levels in a Cynomolgus Macaque Model. Mol. Ther 25, 1843–1853 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rossidis AC et al. In utero CRISPR-mediated therapeutic editing of metabolic genes. Nat. Med 24, 1513–1518 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gonzaga S et al. Cystic adenomatoid malformations are induced by localized FGF10 overexpression in fetal rat lung. Am. J. Respir. Cell Mol. Biol 39, 346–355 (2008). [DOI] [PubMed] [Google Scholar]
- 49.Lee CCI, Jimenez DF, Kohn DB & Tarantal AF Fetal gene transfer using lentiviral vectors and the potential for germ cell transduction in rhesus monkeys (Macaca mulatta). Hum. Gene Ther 16, 417–425 (2005). [DOI] [PubMed] [Google Scholar]
- 50.Porada CD et al. Male germ-line cells are at risk following direct-injection retroviral-mediated gene transfer in utero. Mol. Ther 12, 754–762 (2005). [DOI] [PubMed] [Google Scholar]
- 51.Randall NJ, Bond K, Macaulay J & Steer PJ Measuring fetal and maternal temperature differentials: a probe for clinical use during labour. J. Biomed. Eng 13, 481–485 (1991). [DOI] [PubMed] [Google Scholar]
- 52.Carbonne B, Pons K & Maisonneuve E Foetal scalp blood sampling during labour for pH and lactate measurements. Best Pract. Res. Clin. Obstet. Gynaecol 30, 62–67 (2016). [DOI] [PubMed] [Google Scholar]
- 53.Goswami R et al. Gene therapy leaves a vicious cycle. Front. Oncol 9, 1–25 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Themis M et al. Oncogenesis following delivery of a nonprimate lentiviral gene therapy vector to fetal and neonatal mice. Mol. Ther 12, 763–771 (2005). [DOI] [PubMed] [Google Scholar]
- 55.Geraghty AA, Lindsay KL, Alberdi G, McAuliffe FM & Gibney ER Nutrition during Pregnancy Impacts Offspring’s Epigenetic Status—Evidence from Human and Animal Studies. Nutr. Metab. Insights 8s1, NMI.S29527 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Atlasi Y & Stunnenberg HG The interplay of epigenetic marks during stem cell differentiation and development. Nat. Rev. Genet 18, 643–658 (2017). [DOI] [PubMed] [Google Scholar]
- 57.Zhu Z, Cao F & Li X Epigenetic Programming and Fetal Metabolic Programming. Front. Endocrinol. (Lausanne). 10, 1–15 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Garland M et al. Fetal morphine metabolism and clearance are constant during late gestation. Drug Metab. Dispos 34, 636–646 (2006). [DOI] [PubMed] [Google Scholar]
- 59.Giacca M & Zacchigna S Virus-mediated gene delivery for human gene therapy. J. Control. Release 161, 377–388 (2012). [DOI] [PubMed] [Google Scholar]
- 60.Thomas CE, Ehrhardt A & Kay MA Progress and problems with the use of viral vectors for gene therapy. Nat. Rev. Genet 4, 346–358 (2003). [DOI] [PubMed] [Google Scholar]
- 61.Robbins PD & Ghivizzani SC Viral vectors for gene therapy. Pharmacol. Ther 80, 35–47 (1998). [PubMed] [Google Scholar]
- 62.Waehler R, Russell SJ & Curiel DT Engineering targeted viral vectors for gene therapy. Nat. Rev. Genet 8, 573–587 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yin H et al. Non-viral vectors for gene-based therapy. Nat. Rev. Genet 15, 541–555 (2014). [DOI] [PubMed] [Google Scholar]
- 64.Vargas JE et al. Retroviral vectors and transposons for stable gene therapy: Advances, current challenges and perspectives. J. Transl. Med 14, 1–15 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dzutsev A et al. Development of T cell. Annu. Rev. Immunol 36, 199–230 (2017). [Google Scholar]
- 66.Tarantal AF et al. Rhesus monkey model for fetal gene transfer: Studies with retroviral-based vector systems. Mol. Ther 3, 128–138 (2001). [DOI] [PubMed] [Google Scholar]
- 67.Kohn DB, Sadelain M & Glorioso JC Occurrence of leukaemia following gene therapy of X-linked SCID. Nat. Rev. Cancer 3, 477–488 (2003). [DOI] [PubMed] [Google Scholar]
- 68.Kraunus J et al. Self-inactivating retroviral vectors with improved RNA processing. Gene Ther. 11, 1568–1578 (2004). [DOI] [PubMed] [Google Scholar]
- 69.Cavazza A et al. Self-inactivating MLV vectors have a reduced genotoxic profile in human epidermal keratinocytes. Gene Ther. 20, 949–957 (2013). [DOI] [PubMed] [Google Scholar]
- 70.Hacein-Bey-Abina S et al. A modified γ-retrovirus vector for X-linked severe combined immunodeficiency. N. Engl. J. Med 371, 1407–1417 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kotterman MA, Chalberg TW & Schaffer DV Viral Vectors for Gene Therapy: Translational and Clinical Outlook. Annu. Rev. Biomed. Eng 17, 63–89 (2015). [DOI] [PubMed] [Google Scholar]
- 72.Milone MC & O’Doherty U Clinical use of lentiviral vectors. Leukemia 32, 1529–1541 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ellison SM et al. Pre-clinical Safety and Efficacy of Lentiviral Vector-Mediated Ex Vivo Stem Cell Gene Therapy for the Treatment of Mucopolysaccharidosis IIIA. Mol. Ther. - Methods Clin. Dev 13, 399–413 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tarantal AF et al. Lentiviral vector gene transfer into fetal rhesus monkeys (Macaca mulatta): Lung-targeting approaches. Mol. Ther 4, 614–621 (2001). [DOI] [PubMed] [Google Scholar]
- 75.Waddington SN et al. Long-term transgene expression by administration of a lentivirus-based vector to the fetal circulation of immuno-competent mice. Gene Ther. 10, 1234–1240 (2003). [DOI] [PubMed] [Google Scholar]
- 76.Toelen J et al. Fetal gene transfer with lentiviral vectors: long-term in vivo follow-up evaluation in a rat model. Am. J. Obstet. Gynecol 196, 352.e1–352.e6 (2007). [DOI] [PubMed] [Google Scholar]
- 77.Skarsgard ED, Huang L, Reebye SC, Yeung AY & Jia WW Lentiviral vector-mediated, in vivo gene transfer to the tracheobronchial tree in fetal rabbits. J. Pediatr. Surg 40, 1817–1821 (2005). [DOI] [PubMed] [Google Scholar]
- 78.Schambach A, Zychlinski D, Ehrnstroem B & Baum C Biosafety features of lentiviral vectors. Hum. Gene Ther 24, 132–142 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee CS et al. Adenovirus-mediated gene delivery: Potential applications for gene and cell-based therapies in the new era of personalized medicine. Genes Dis. 4, 43–63 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Alba R, Bosch A & Chillon M Gutless adenovirus: Last-generation adenovirus for gene therapy. Gene Ther. 12, S18–S27 (2005). [DOI] [PubMed] [Google Scholar]
- 81.Naso MF, Tomkowicz B, Perry WL & Strohl WR Adeno-Associated Virus (AAV) as a Vector for Gene Therapy. BioDrugs 31, 317–334 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li C & Samulski RJ Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet 21, 255–272 (2020). [DOI] [PubMed] [Google Scholar]
- 83.George LA et al. Hemophilia B gene therapy with a high-specific-activity factor IX variant. N. Engl. J. Med 377, 2215–2227 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pasi KJ et al. Multiyear follow-up of aav5-hfviii-sq gene therapy for hemophilia a. N. Engl. J. Med 382, 29–40 (2020). [DOI] [PubMed] [Google Scholar]
- 85.Colella P, Ronzitti G & Mingozzi F Emerging Issues in AAV-Mediated In Vivo Gene Therapy. Mol. Ther. - Methods Clin. Dev 8, 87–104 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Massaro G et al. Fetal gene therapy for neurodegenerative disease of infants. Nat. Med 24, 1317–1323 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Joyeux L et al. In utero lung gene transfer using adeno-associated viral and lentiviral vectors in mice. Hum. Gene Ther. Methods 25, 197–205 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Irvin-Choy NS, Nelson KM, Gleghorn JP & Day ES Design of nanomaterials for applications in maternal/fetal medicine. J. Mater. Chem. B 8, 6548–6561 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Haley RM, Gottardi R, Langer R & Mitchell MJ Cyclodextrins in drug delivery: applications in gene and combination therapy. Drug Deliv. Transl. Res 10, 661–677 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fenton OS, Olafson KN, Pillai PS, Mitchell MJ & Langer R Advances in Biomaterials for Drug Delivery. Adv. Mater 30, 1–29 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mehier-Humbert S & Guy RH Physical methods for gene transfer: Improving the kinetics of gene delivery into cells. Adv. Drug Deliv. Rev 57, 733–753 (2005). [DOI] [PubMed] [Google Scholar]
- 92.Stewart MP et al. In vitro and ex vivo strategies for intracellular delivery. Nature 538, 183–192 (2016). [DOI] [PubMed] [Google Scholar]
- 93.Dal Maschio M et al. High-performance and site-directed in utero electroporation by a triple-electrode probe. Nat. Commun 3, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cao Y et al. Nontoxic nanopore electroporation for effective intracellular delivery of biological macromolecules. Proc. Natl. Acad. Sci. U. S. A 116, 7899–7904 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Young JL & Dean DA Electroporation-Mediated Gene Delivery. Adv. Genet 89, 49–88 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tabata H & Nakajima K Efficient in utero gene transfer system to the developing mouse brain using electroporation: Visualization of neuronal migration in the developing cortex. Neuroscience 103, 865–872 (2001). [DOI] [PubMed] [Google Scholar]
- 97.Navarro-Quiroga I, Chittajallu R, Gallo V & Haydar TF Long-term, selective gene expression in developing and adult hippocampal pyramidal neurons using focal in utero electroporation. J. Neurosci 27, 5007–5011 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Niwa M et al. Knockdown of DISC1 by In Utero Gene Transfer Disturbs Postnatal Dopaminergic Maturation in the Frontal Cortex and Leads to Adult Behavioral Deficits. Neuron 65, 480–489 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Takeda H et al. Prenatal electroporation-mediated gene transfer restores Slc26a4 knock-out mouse hearing and vestibular function. Sci. Rep 9, 1–12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Giljohann DA et al. Gold nanoparticles for biology and medicine. Angew. Chemie - Int. Ed 49, 3280–3294 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Veiseh O, Gunn JW & Zhang M Design and fabrication of magnetic nanoparticles for targeted drug delivery and imaging. Adv. Drug Deliv. Rev 62, 284–304 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mendes R, Fernandes AR & Baptista PV Gold nanoparticle approach to the selective delivery of gene silencing in cancer-The case for combined delivery? Genes (Basel). 8, 94 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ray M, Lee YW, Scaletti F, Yu R & Rotello VM Intracellular delivery of proteins by nanocarriers. Nanomedicine 12, 941–952 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhang Y, Røise JJ, Lee K, Li J & Murthy N Recent developments in intracellular protein delivery. Curr. Opin. Biotechnol 52, 25–31 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Conde J et al. Design of multifunctional gold nanoparticles for in vitro and in vivo gene silencing. ACS Nano 6, 8316–8324 (2012). [DOI] [PubMed] [Google Scholar]
- 106.Xiong Z et al. Zwitterion-functionalized dendrimer-entrapped gold nanoparticles for serum-enhanced gene delivery to inhibit cancer cell metastasis. Acta Biomater. 99, 320–329 (2019). [DOI] [PubMed] [Google Scholar]
- 107.Lee K et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat. Biomed. Eng 1, 889–901 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rai R, Alwani S & Badea I Polymeric nanoparticles in gene therapy: New avenues of design and optimization for delivery applications. Polymers (Basel). 11, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kaczmarek JC et al. Polymer–Lipid Nanoparticles for Systemic Delivery of mRNA to the Lungs. Angew. Chemie - Int. Ed 55, 13808–13812 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mukalel AJ, Riley RS, Zhang R & Mitchell MJ Nanoparticles for nucleic acid delivery: Applications in cancer immunotherapy. Cancer Lett. 458, 102–112 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tzeng SY & Green JJ Polymeric nucleic acid delivery for immunoengineering. Curr. Opin. Biomed. Eng 7, 42–50 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rui Y, Wilson DR & Green JJ Non-Viral Delivery To Enable Genome Editing. Trends Biotechnol. 37, 281–293 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mastorakos P et al. Highly compacted biodegradable DNA nanoparticles capable of overcoming the mucus barrier for inhaled lung gene therapy. Proc. Natl. Acad. Sci. U. S. A 112, 8720–8725 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Patel AK et al. Inhaled Nanoformulated mRNA Polyplexes for Protein Production in Lung Epithelium. Adv. Mater 31, 1–7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhao Y & Huang L Lipid nanoparticles for gene delivery. Adv. Genet 88, 13–36 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hajj KA & Whitehead KA Tools for translation: Non-viral materials for therapeutic mRNA delivery. Nat. Rev. Mater 2, 1–17 (2017). [Google Scholar]
- 117.Guimaraes PPG et al. lonizable lipid nanoparticles encapsulating barcoded mRNA for accelerated in vivo delivery screening. J. Control. Release 316, 404–417 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhang R, Billingsley MM & Mitchell MJ Biomaterials for vaccine-based cancer immunotherapy. J. Control. Release 292, 256–276 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mitchell MJ, Jain RK & Langer R Engineering and physical sciences in oncology: Challenges and opportunities. Nat. Rev. Cancer 17, 659–675 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Riley RS, June CH, Langer R & Mitchell MJ Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov 18, 175–196 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Oberli MA et al. Lipid Nanoparticle Assisted mRNA Delivery for Potent Cancer Immunotherapy. Nano Lett. 17, 1326–1335 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Miao L et al. Delivery of mRNA vaccines with heterocyclic lipids increases anti-tumor efficacy by STING-mediated immune cell activation. Nat. Biotechnol 37, 1174–1185 (2019). [DOI] [PubMed] [Google Scholar]
- 123.Augsten M et al. Fetal HDL/apoE: A novel regulator of gene expression in human placental endothelial Cells. Physiol. Genomics 43, 1255–1262 (2011). [DOI] [PubMed] [Google Scholar]
- 124.Akinc A et al. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol. Ther 18, 1357–1364 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Saunders NRM et al. A Nanoprimer To Improve the Systemic Delivery of siRNA and mRNA. Nano Lett. 6, 4264–4269 (2020). [DOI] [PubMed] [Google Scholar]
- 126.Marcos-Contreras OA et al. Selective targeting of nanomedicine to inflamed cerebral vasculature to enhance the blood–brain barrier. Proc. Natl. Acad. Sci. U. S. A 117, 3405–3414 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Doudna JA & Charpentier E The new frontier of genome engineering with CRISPR-Cas9. Science 346 (2014). [DOI] [PubMed] [Google Scholar]
- 128.Hsu PD, Lander ES & Zhang F Development and applications of CRISPR-Cas9 for genome engineering. Cell vol. 157 1262–1278 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yin H et al. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat. Biotechnol 32, 551–553 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Rees HA & Liu DR Base editing: precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet 19, 770–788 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Jo DH et al. CRISPR-Cas9–mediated therapeutic editing of Rpe65 ameliorates the disease phenotypes in a mouse model of Leber congenital amaurosis. Sci. Adv 5, 1–10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Beyret E et al. Single-dose CRISPR–Cas9 therapy extends lifespan of mice with Hutchinson–Gilford progeria syndrome. Nat. Med 25, 419–422 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Yin H, Kauffman KJ & Anderson DG Delivery technologies for genome editing. Nat. Rev. Drug Discov 16, 387–399 (2017). [DOI] [PubMed] [Google Scholar]
- 134.Lino CA, Harper JC, Carney JP & Timlin JA Delivering crispr: A review of the challenges and approaches. Drug Deliv. 25, 1234–1257 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Nelson CE et al. In vivo editing improves muscle function in mouse of DMD. Science 351, 403–407 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hanlon KS et al. High levels of AAV vector integration into CRISPR-induced DNA breaks. Nat. Commun 10, 1–11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Shinmyo Y et al. CRISPR/Cas9-mediated gene knockout in the mouse brain using in utero electroporation. Sci. Rep 6, 1–13 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lee B et al. Nanoparticle delivery of CRISPR into the brain rescues a mouse model of fragile X syndrome from exaggerated repetitive behaviours. Nat. Biomed. Eng 2, 497–507 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Rui Y et al. Poly(beta-amino ester) nanoparticles enable non-viral delivery of CRISPR/Cas9 plasmids for gene knockout and gene deletion. Mol. Ther. - Nucleic Acids 20, 661–672 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hajj KA et al. Branched-Tail Lipid Nanoparticles Potently Deliver mRNA In Vivo due to Enhanced Ionization at Endosomal pH. Small 15, 1–7 (2019). [DOI] [PubMed] [Google Scholar]
- 141.Zhu X et al. Systemic mRNA Therapy for the Treatment of Fabry Disease: Preclinical Studies in Wild-Type Mice, Fabry Mouse Model, and Wild-Type Non-human Primates. Am. J. Hum. Genet 104, 625–637 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yin H et al. Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat. Biotechnol 34, 328–333 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Finn JD et al. A Single Administration of CRISPR/Cas9 Lipid Nanoparticles Achieves Robust and Persistent In Vivo Genome Editing. Cell Rep. 22, 2227–2235 (2018). [DOI] [PubMed] [Google Scholar]
- 144.Ramezanpour M et al. Ionizable amino lipid interactions with POPC: Implications for lipid nanoparticle function. Nanoscale 11, 14141–14146 (2019). [DOI] [PubMed] [Google Scholar]
- 145.Gilleron J et al. Image-based analysis of lipid nanoparticle-mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat. Biotechnol 31, 638–646 (2013). [DOI] [PubMed] [Google Scholar]
- 146.Billingsley MM et al. Ionizable Lipid Nanoparticle-Mediated mRNA Delivery for Human CAR T Cell Engineering. Nano Lett. 20, 1578–1589 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Maugeri M et al. Linkage between endosomal escape of LNP-mRNA and loading into EVs for transport to other cells. Nat. Commun 10, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Allen TM & Cullis PR Liposomal drug delivery systems: From concept to clinical applications. Adv. Drug Deliv. Rev 65, 36–48 (2013). [DOI] [PubMed] [Google Scholar]
- 149.Kauffman KJ et al. Optimization of Lipid Nanoparticle Formulations for mRNA Delivery in Vivo with Fractional Factorial and Definitive Screening Designs. Nano Lett. 15, 7300–7306 (2015). [DOI] [PubMed] [Google Scholar]
- 150.Sato Y et al. Hydrophobic scaffolds of pH-sensitive cationic lipids contribute to miscibility with phospholipids and improve the efficiency of delivering short interfering RNA by small-sized lipid nanoparticles. Acta Biomater. 102, 341–350 (2020). [DOI] [PubMed] [Google Scholar]
- 151.Patel S et al. Naturally-occurring cholesterol analogues in lipid nanoparticles induce polymorphic shape and enhance intracellular delivery of mRNA. Nat. Commun 11, 1–13 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Jokerst JV, Lobovkina T, Zare RN & Gambhir SS Nanoparticle PEGylation for imaging and therapy. Nanomedicine 6, 715–728 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ishida T & Kiwada H Accelerated blood clearance (ABC) phenomenon upon repeated injection of PEGylated liposomes. Int. J. Pharm 354, 56–62 (2008). [DOI] [PubMed] [Google Scholar]
- 154.Liu M et al. Accelerated Blood Clearance of Nanoemulsions Modified with PEG-Cholesterol and PEG-Phospholipid Derivatives in Rats: The Effect of PEG-Lipid Linkages and PEG Molecular Weights. Mol. Pharmaceutics 17, 1059–1070 (2020). [DOI] [PubMed] [Google Scholar]
- 155.Cheng Q et al. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR–Cas gene editing. Nat. Nanotechnol 15, 313–320 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]