

Abstract
Autophagy, a cellular stress response to starvation and bacterial infection, is executed by double-membrane-bound organelles called autophagosomes. Autophagosomes transfer cytosolic material to acidified lysosomes for degradation following soluble N-ethylmaleimide-sensitive factor attachment receptor (SNARE)-dependent fusion processes. Many of the autophagy related disorders stem from defective end-step proteolysis inside lysosomes. Role of epithelial cystic fibrosis (CF) transmembrane conductance regulator (CFTR) chloride channel has been argued to be critical for efficient lysosomal clearance, however its context to autophagic clearance and the underlying mechanism is poorly defined. Here, we report that syntaxin17 (Stx17), an autophagic SNARE protein interacts with CFTR under nutritional stress and bacterial infection and incorporates it into mature autophagosomes to mediate an efficient lysosomal clearance. Lack of CFTR function and Stx17 and loss of CFTR-Stx17 interaction impair bacterial clearance. We discover a specialized role of Stx17-CFTR protein complex that is critical to prevent defective autophagy as has been the reported scenario in CF airway epithelial cells, infectious diseases and lysosomal clearance disorders.
Introduction
Macroautophagy, or simply autophagy, is a catabolic program for restoring cellular resources and energy balance as cells recognize damaged organelles, protein aggregates, and invading pathogens(1, 2). As new studies emerge, the role of autophagy has been expanded to include cell growth, cell development and aging, tumor suppression, and immunity among others(2). Cystic fibrosis transmembrane conductance regulator (CFTR) is a chloride channel present in the lung and intestinal epithelial cells and forms a key transporter to regulate fluid transit across the epithelium. Deficiency of CFTR dependent function due to genetic alterations in Cftr gene causes cystic fibrosis (CF). CF is a multi-organ disorder while the lung remains the most impacted organ due to recurrent episodes of infection that leads to inflammation. Role of autophagy in CF is now becoming increasingly recognized with some evidence of impaired autophagic clearance in CF macrophages causing low bactericidal activity, aggresome formation, and therapeutic benefit of autophagy modulators in the CF epithelial cells (3–5).
Autophagy can be stimulated by nutritional deprivation and pathogen insult (6). Autophagy begins with the formation of an isolation membrane (phagophore) at the endoplasmic reticulum (ER)-mitochondrial interface(7), with subsequent sequestering of a portion of cytoplasm and damaged organelles as autophagic intermediates continue to grow and form enclosed, double-membraned structures called autophagosomes. Autophagosomes fuse with late endosomes/lysosomes, the final degradative compartments (2, 6). Loss or perturbation of lysosomal acidification can cause lysosomal storage disorders that may be associative to autophagosomal clearance defect. Defective lysosomal clearance causing accumulation of harmful cargos can largely disturb cellular homeostasis inducting cells into apoptosis, depressed growth, persistent infection and inflammation (8, 9).
Autophagosome-lysosome fusion can be regulated by soluble N-ethylmaleimide-sensitive factor attachment receptors (SNARE) protein Syntaxin17 (Stx17) (10–12). Stx17’s strict localization to mature autophagosomes but not isolation membranes mandates fusion of only mature autophagosomes with lysosomes(10). In addition, Stx17 in concert with autophagy-related gene Atg14 orchestrates the formation of autophagosomes at the ER-mitochondrial contact and autophagosome-lysosome fusion (7, 12).
In this study, we define previously unknown mechanism of a direct participation of CFTR in autophagy being facilitated by its physical coupling with Stx17 and demonstrate that CFTR incorporation into autophagosomes accelerates clearance of cargo including bacteria. This specialized role of CFTR may become integral to maintenance of cellular homeostasis and bear relevance to CF, infectious diseases and lysosomal storage disorders.
Materials and Methods
Statistics
All experiments showing SEM were performed at least in triplicate. A minimum number of five cells was used for immunofluorescence microscopy-based analyses. The statistical significance was calculated using Student’s t-test or one-way ANOVA with adjustment for multiple comparisons. P<0.05 was considered significant.
Cell lines
For this study CFBE41o−, Calu 3, and T84 cells were used as previously described (13–15). Differentiated Primary human bronchial epithelial cells on the transwells were obtained from two sources: Cystic Fibrosis Center, CCHMC and Charles River laboratories, Wilmington, MA.
Chemicals, antibodies, and cell lines
Bafilomycin A1 was purchased from LC laboratories, spautin-1, dynasore and chloroquine were obtained from Sigma, Scrambled and Stx17, CLC stealth siRNAs were obtained from Thermo Scientific. For knocking down expression of Stx17 in CFBE41o− cells, lentiviral sh RNA particles were obtained from Dharmacon. Antibodies in this study included anti-CFTR (R1104) and anti-Stx17 (M212–3, MBL) for Western blots; anti-CFTR (R1104), anti-Stx17 (17815–1-AP, Proteintech) for immunofluorescence; anti-LC3 (7543, Sigma) for both Western blots and immunofluorescence, as well as biotin-FLAG-Ab (Sigma), anti-FLAG HRP (Sigma), anti-SNAP 29 (E-5) (Santa Cruz), anti-VAMP 8 (CST), and β-actin (Sigma). CFBE41o−, Calu 3, and T84 cells were previously described(13–15).
Differentiated Primary human bronchial epithelial cells on the transwells were obtained from two sources: Cystic Fibrosis Center, CCHMC and Charles River laboratories, Wilmington, MA.
Ready to transduce GIPZ lentiviral human Stx17 shRNA particles were obtained from GE Dharmacon. CFBE41o− cells were treated with the 1 X105 TU viral particles for 48 h before the experiment. Two Stx17 shRNAs were used in combination. A 60–80% knockdown efficiency was obtained using these shRNAs. For cDNA transfection, Lipofectamine 3000 (Thermo Scientific) was used according to manufacturer’s instructions.
Autophagy assays
Methods in this study for assessing autophagy were previously validated (16). Autophagy was induced under nutritional deprivation or starvation. For no starvation, cells were maintained in regular medium (DMEM/F-12 10% FBS for HEK 293, MEM 10% FBS for Calu 3 and CFBE41o−, and DMEM/F-12 50/50 10% FBS for T84 cells). For starvation, cells were washed with HBSS three times and incubated in HBSS (Gibco) (starvation medium) for 2 h at 37°C. To block autophagy flux, 100 nm BafA1 or CQ were added along with the starvation medium and incubated for 2 h at 37°C. Autophagy activity was assessed using following approaches: Quantitation of LC3+ puncta per cell (immunofluorescence) and LC3-II formation [Western blot using 1:10,000 dilution of the antibody against LC3 (Sigma)]. LC3 turnover was calculated by subtracting LC3-II (starvation) from LC3-II (starvation+bafilomycin A1). β-actin was used as a loading control in Western blots. Western blot quantitation was performed using ImageJ (NIH).
Cell lysate preparation
Whole-cell lysates were prepared in PBS 0.2% Triton X-100 containing a protease-inhibitor cocktail (1 μM aprotonin, 1 μM leupeptin, 1 mM phenylmethylsulfonyl fluoride). Lysates were mixed with 5X sample buffer, incubated for 10 min at 37°C, and subjected to SDS–PAGE and Western blot following standard protocols. CFTR protein cross-linking for the immunoprecipitation of protein complexes from total cell lysate has been described previously (17). GFP Vamp8 was immunoprecipitated from the cell lysate using GFP-nAb™ (ABP-NAB-GFPA100, Allele Biotechnology).
Fluorescence resonance energy transfer (FRET) microscopy and data Analysis
For direct sensitized emission FRET, CFBE41o− cells were transiently transfected with Cyan Stx17 and YFP-CFTR using Lipofectamine. Single transfected cells were used to acquire CFP- or YFP- only images for bleedthrough calculations. The corrected FRET (FRETc) was normalized with donor CFP intensity (FRETc/CFP), yielding the normalized corrected FRET (N-FRETc), and the intensity of N-FRETc images was presented in pseudocolor and monochrome mode, stretched between the low and high normalization values, according to an intensity to color mapped lookup table. All calculations were performed using the Channel Math and FRET modules of SlideBook 4.2 software (Intelligent Imaging Innovations; Denver, CO) as described previously (18).
Opti-Prep-based fractionation of vesicles
Calu 3 cells were subjected to starvation for 4 h in the presence of HBSS containing protease inhibitors PepA (1 μg/ml)+ Leu (1 μg/ml). Cells were scraped in PBS and pelleted at 800g for 2 min. Cells were resuspended in 2 ml of homogenization buffer (250 mM sucrose, 1 mM EDTA, 10 mM HEPES, pH 7.2 plus protease inhibitors) and transferred to a prechilled Dounce tissue grinder. Cells were homogenized on ice with 20 Dounce strokes times 3 at 1-min intervals. Homogenate was centrifuged at 500g for 10 min at 4°C. Supernatant was collected and sampled as postnuclear supernatant (PNS). Supernatant was prepared for density gradient centrifugation. In an ultracentrifuge tube, series of Opti-Prep gradients prepared from 60% Opti-Prep gradients in PBS were overlayered in descending concentrations: 30% (bottom), 27%, 23%, 20%, and 17% (top) gradients. Supernatant was mixed with Opti-Prep to obtain a final concentration of 15% Opti-Prep and laid on top of the density gradients prepared earlier. Samples were ultracentrifuged at 145,000g for 2 h at 4°C. Several bands appeared in the gradient, and 200 μl of solution from the top layer was collected for a total of nine fractions (1–9). About 40 μl of the fraction volume was suspended in 5X sample buffer and subjected to Western blotting. The remaining volume of fractions 3–6 was pooled and used for immunoprecipitation with LC3-antibody-conjugated resin and probed for CFTR, Stx17, and LC3 proteins.
Immunofluorescence
After 48 h of transfection, cells were fixed using 3.7% formaldehyde for 10 min at 25°C. Cells were then washed three times with 1X PBS and permeabilized using PBS 0.3% Triton X-100 for 15 min. After a brief wash, cells were blocked with (2% BSA 0.05% Tween in PBS) for 1 h at 25°C. Cells were incubated with LC3 antibody (1:100) at 4°C overnight, washed with PBS 0.05% Tween buffer, and incubated with mouse secondary antibody (Alexa fluor 568, 1:500, Thermo Scientific) for 1 h at 25°C. Primary human bronchial epithelial, Calu-3, and T84 cells were stained with mouse anti-CFTR (R1104) and rabbit anti-Stx17 (Proteintech) primary antibodies. Stained cells were examined using a confocal microscope (Olympus FV1200).
Cloning, expression, and purification of recombinant proteins
The following constructs used in this study were obtained from Addgene, Inc.: pMRXIP GFP-Stx17 WT (Plasmid #45909), pEGFP VAMP8 (Plasmid #42311), FLAG SNAP 29 (Plasmid #45915), and pBABE-puro mCherry-EGFP-LC3B (Plasmid #22418). Full-length Stx17 (Stx171–302) was PCR amplified from pMRXIP GFP-Stx17 and inserted into EcoR I/Xho I restriction endonuclease sites in the pAmCyan vector (Clontech Laboratories) to generate the cyan Stx17 fusion protein. Stx17ΔC (Stx171–228) was generated from full-length Stx17 by inserting a premature stop codon. Full-length Stx17 and Stx17ΔC were inserted into the pET41 vector backbone (Novagen) by using ligation-independent cloning (TA Cloning®, Thermo Scientific). These constructs were transformed into BL21 competent cells to generate GST-His-S-tagged proteins. His-S-Vamp8 and His-S-SNAP 29 proteins were produced by generating respective pTriEx4 (Novagen) expression constructs followed by transformation into Origami competent cells. For GST-tagged and His-tagged proteins, lysis buffer composed of 50 mM Tris, 1 mM EDTA, and 10% sucrose pH 7.2 and resuspension buffer containing 25 mM HEPES-KOH, 400 mM KCl, and 5 mM β-mercaptoethanol (added fresh), respectively, were used followed by adding lysozyme (1 mg/ml) prepared in lysis or resuspension buffer for 30 min at 4°C. Next, 2.1% Triton X-100 was added to the lysate and mixed for another 30 min at 4°C. Lysate was transferred to clean centrifuge tubes and centrifuged at 10,000g for 30 min at 4°C. To the clear supernatant, 100 μl of glutathione beads (Thermo Scientific) for GST-tagged proteins and TALON® beads (Clontech) for His-tagged proteins were added and mixed for 2–3 h at 4°C. Glutathione beads and TALON® beads containing proteins were washed three to four times with PBS 0.2% Triton-X-100 and resuspension buffer, respectively, in the presence of protease inhibitors. GST-tagged proteins and His-tagged proteins were eluted with 20 mM glutathione and 200 mM imidazole, respectively, one to four times. Protein concentrations of the samples were quantified by using Bradford assay or by direct absorbance at 280 nm.
HEK 293 cells stably expressing FLAG-CFTR were plated on 10X100-mm dishes and grown to 100% confluency. The cells were then washed twice with PBS and lysed with PBS containing 0.2% Triton X-100 and protease inhibitor cocktail (phenylmethylsulfonyl fluoride 1 mM, Pep-A 1 Δg/ml, leupeptin 1 Δg/ml, and aprotinin 1 Δg/ml). The cell lysate was mixed on a shaker for 10 min at 4°C and centrifuged for 10 min at 10,000g. The supernatant containing total protein was immunoprecipitated using anti-FLAG beads (Sigma) overnight at 4°C. The beads with bound protein were washed, and the protein was eluted with 100 mM glycine (pH 2.2) and quickly neutralized with 150 mM Tris (pH 8.8). The eluted protein was subjected to SDS-PAGE and visualized using Coomassie blue stain.
GST pull-down assay
HEK 293 cells expressing FLAG-WT CFTR were lysed in PBS 0.2% Triton X-100 containing protease inhibitors. Purified GST-His-S-Stx17ΔC protein (0 to 2.76 μM) was added to the cell lysate and mixed for 1 h at 4°C. Glutathione beads (20 μl) were added to each sample and allowed to mix overnight at 4°C. Next day, samples were washed three times with PBS 0.2% Triton X-100, and proteins were eluted using sample buffer. Samples were subjected to Western blotting, and the blot was probed with anti-CFTR mouse antibody (R1104). In a similar manner, GST-His-S-Stx17 protein segments (1.38 μM), 1–60, 1–129, and 1–228 amino acids were used to immunoprecipiate CFTR from HEK 293 cells stably expressing FLAG-WT CFTR.
Purified GST-His-S-Stx17ΔC (0 to 0.69 μmoles), His-S-VAMP8 (400 pmoles), His-S-SNAP 29 (133 pmoles), and FLAG-CFTR (6.25 pmoles) proteins were mixed, and the protein complexes were immunoprecipitated using glutathione beads (20 μl) in an overnight binding at 4°C. The protein complexes were eluted using sample buffer, subjected to Western blotting, and probed with anti-FLAG, anti-VAMP8, and anti-SNAP 29 antibodies.
Bacterial clearance assay
Briefly, 1×105 primary human bronchial epithelial cells with normal CFTR and F508del CFTR mutation and polarized on costar transwells. To determine the effect of autophagy in bacterial clearance, cells were treated with chloroquine (20 μM, 2 h) before adding bacteria. Cells were infected with 100 μL of serum free MEM media with GFP labeled P. aeruginosa (PAO1, ATCC) at a 1∶25 MOI for 6 hours in the 37°C incubator. Extracellular bacteria were then killed with cell impermeable antibiotics (50 U/mL each of penicillin and streptomycin, 200 μg/mL gentamycin (Life Technologies) for 20 minutes to study only internalized bacteria. To test the effect of CFTR inhibitor on intracellular PAO1 load, non-CF primary human bronchial epithelial cells were incubated with CFTR inhibitor 172 (20 μM) over night. Media was replaced in the morning and new media with inhibitor was added right before adding PAO1 at 1:10 MOI for 6 hours at 37°C. Cells were imaged using a confocal microscope Olympus FV1200 and GFP intensity was measured to estimate the number of internalized bacteria.
For bacterial clearance assay in CFBE41o− cells, 1×106 primary human bronchial epithelial cells were plated in a 6-well dish. Cells were infected with 1 ml of serum free MEM media with GFP PAO1 at a 1∶25 MOI for 3 hours in the 37°C incubator. Extracellular bacteria were then killed with cell impermeable antibiotics (50 U/mL each of penicillin and streptomycin (Life Technologies), 200 μg/mL gentamycin (Life Technologies) for 20 minutes to study only internalized bacteria. Cell were lysed in PBS 0.2% Triton-X-100 and plated on the bacterial culture plates to estimate the bacterial load or colony forming units. Cells that were not infected with PAO1 were used for background subtraction.
Results
Stx17 and CFTR protein complexes transit as intracellular puncta upon autophagy induction
To explore the potential significance of CFTR and Stx17 in autophagy, we first studied the localization pattern of CFTR and Stx17 proteins in response to the induction of autophagy upon nutritional starvation. Starvation potently stimulated the formation of Stx17 puncta close to all occupied by CFTR (CFTR+Stx17+ puncta) in lung (Calu 3) and colon (T-84) epithelial cell lines and primary human bronchial epithelial cells (Fig. 1A-C). Please note that CFTR-Stx17 co-localization in the above described epithelial cells corresponds to endogenously expressed proteins detected using immunostaining. We attempted to recapitulate these observations in overexpression-based studies for quantitation. Consistent with the earlier findings, cyan Stx17 protein co-localized extensively with YFP CFTR under starvation while not co-localizing in the growing state (regular medium or no starvation) in cystic fibrosis bronchial epithelial cells (CFBE41o−) (Fig. 1D and 1E). Treatment of cells with spautin 1, a specific and potent autophagy inhibitor(19) resulted in a marked decline in the starvation induced formation of Stx17+ puncta and consequently CFTR+Stx17+ puncta (Fig. 1D and E). To mark the formation of CFTR+Stx17+ puncta as a true event and not an overexpression artifact, we confirmed that TMEM16a, a membrane protein with calcium-activated chloride-channel activity (20), did not coincide with Stx17+ puncta (Fig. 1D and E). Hence, formation of CFTR-Stx17 vacuolar complex is selective and autophagy induced.
Fig. 1. Stx17 and CFTR protein complexes coincide as intracellular puncta under nutritional starvation.

Confocal images demonstrate immunostaining with anti-CFTR and anti-Stx17 antibodies in A, Calu 3 and B, T84 cells kept in regular culture medium (Fed) or HBSS for 2 h (starved) and C, primary human bronchial epithelial cells starved in HBSS for 2 h. D, CFBE41o− cells co-expressing Cyan Stx17 and YFP CFTR or Cyan Stx17 and TMEM16a were kept in regular culture medium (Fed) or starvation medium (HBSS) for 2–6 h and analyzed using immunofluorescence microscopy. In a separate set of cells, autophagy inhibitor spautin-1 10 μM was added simultaneously with the starvation medium for 6 h. Note that the 6-h starvation period did not decrease the amount of CFTR-Stx17 co-localization, and these data have been averaged into the 2 h starvation period. E, Quantification of the red (YFP CFTR or TMEM16a) and green (Cyan Stx17) puncta per cell with SEM analyzed from immunofluorescent images of the n=5–10 cells per experiment from three to five independent experiments. P-value by ANOVA with Bonferroni’s multiple comparisons test.
We next considered the possibility of an interaction between CFTR and Stx17 based on their extensive intracellular co-localization. Indeed, there was a direct interaction between CFTR and Stx17 (both Stx17 full-length = Stx171–302 and Stx17ΔC = Stx171–228) (Fig. 2A and B; Supplemental Fig. 1A), also suggesting the dispensability of C-terminal transmembrane domains of Stx17 in constituting an interaction with CFTR. GST-His-S-Stx17ΔC was used to determine EC50 (0.6 μM), due to its high purity compared to GST-His-S-Stx17 full-length (Fig. 2A and B; Supplemental Fig. 1A). We determined that CFTR binds to a region between amino acid residues 129 and 228 of Stx17 which includes the SNARE domain (Supplemental Fig. 1A). We detected CFTR-Stx17 intracellular binding through determination of a positive FRET signal (~ 20%) between Cyan Stx17 and YFP CFTR under starvation in live CFBE41o− cells (Fig. 2C). There was no binding observed between CFTR with GFP-Stx17 TM, consisting only of the two transmembrane domains (TMDs) (including no SNARE domain) (Fig. 2D). Although, GFP-Stx17 TM formed starvation induced puncta but failed to coincide with CFTR (Fig. 2E and 2F). Hence, CFTR recruitment into Stx17 positive vesicles is dependent upon SNARE domain in Stx17. We screened interaction of FLAG-Stx17 against different regions of CFTR: N1–80, R595–860, NBD1-R350–850 and NBD2-C1150–1480. Previously, N-CFTR cytoplasmic tail has been found to interact with Stx1A that can be outcompeted by Stx1A binding with Munc18 to relieve the negative channel regulation of CFTR by Stx1A (21, 22). The R-domain is a unique feature of CFTR protein that forms the PKA/PKG dependent phosphorylation platform, key for channel opening (23). CFTR NBDs bind and hydrolyze ATP to control CFTR channel gating (24). The binding study of Stx17 with different domains of CFTR suggested that Stx17 predominantly binds N-CFTR but may form multiple contacts with different regions of CFTR (Fig. 2G and 2H). How this multi-contact binding of Stx17 with CFTR regulates various aspects of the channel function would be an interesting topic for future investigation. In conclusion, CFTR-Stx17 binding and their intensive co-localization upon autophagy induction suggests that there is a dedicated role of CFTR in autophagy which we further investigated.
Fig. 2. Stx17 interacts with CFTR and requires SNARE domain in Stx17.

A, GST-pull-down assay to determine that CFTR (whole-cell lysate expressing FLAG CFTR) interacts directly with GST-His-S-Stx17ΔC (Stx171–228 aa) tested over a range of concentrations of Stx17ΔC purified protein. B, Line graph corresponding to the detected intensity of CFTR protein band from the Western blot shown in a across increasing concentrations of Stx17ΔC purified protein to determine the EC50 of CFTR binding with Stx17ΔC. C, Pseudocolor and monochrome images from starved CFBE41o− cells co-expressing Cyan Stx17 and YFP CFTR represent NFRETc corresponding to CFTR-Stx 17 interaction. FRET efficiency (NFRETc)= 20% was calculated from n=3 independent experiments as represented in Fig. 2C. D, Western blot depict FLAG-tag immunoprecipitation from FLAG-tagged WT CFTR CFBE41o− cells with transient expression of GFP Stx17 or GFP Stx17 TM and probed using anti-GFP antibody. E, Confocal images of CFBE41o− cells co-expressing RFP CFTR and GFP Stx17 or RFP CFTR and GFP Stx17 TM and subjected to starvation for 2 h. F, Quantitation of RFP CFTR colocalization with GFP Stx17 and GFP Stx17 TM vesicles in CFBE41o− under starvation (n=3 independent experiments). Error bars represent SEM, P-value by t-test. G, Western blot depict FLAG-tag immunoprecipitation from FLAG-Stx17 expressing CFBE41o− cells that co-expressed various His-S-tagged CFTR regions as indicated and probed using anti-S-protein HRP antibody. H, Quantitation of interaction of FLAG-Stx17 with various His-S-tagged CFTR regions as indicated. I, Interaction between purified FLAG-CFTR and autophagic SNAREs Stx17, SNAP 29, and VAMP 8 by using an in vitro GST pull-down assay followed by Western blot (immunoblot) by using specific antibodies to CFTR, SNAP 29, and VAMP 8.
Given that Stx17-SNAP 29 binary target and vesicle SNARE VAMP 8 complex is crucial for membrane-fusion activity between autophagosomes and endolysosomes(10, 12), using purified proteins, we performed a protein-complex assay and observed that CFTR can exist in the ternary complex of Stx17–SNAP 29–VAMP 8 through interaction with Stx17 or in the immunoprecipitates of GFP VAMP 8 (Fig. 2I; Supplemental Fig. 2A and B). Using CFTR-specific antibody based immunoprecipitation of CFTR from Calu-3 cells, we also detected endogenously expressed Stx17 and VAMP 8 in complex with CFTR (Supplemental Fig. 2C). Hence, CFTR is a component of late-autophagosomal SNARE complex.
Since CFTR is a plasma membrane protein with half-life > 24 h, and we observed dramatic reduction of surface levels of CFTR and its primary inclusion into Stx17 vesicles under an acute period of starvation, we asked whether endocytic machinery is involved in the formation of Stx17+CFTR+ vesicles (15). Dyansore is a cell-permeable potent inhibitor of dynamin, a protein essential for clathrin-dependent coated vesicle formation (25). Clathrin is a coat protein that comprises of three clathrin heavy chains (CLC) each associated with a light chain (26). Adaptor proteins such as AP-2 that bind to clathrin recognize membrane cargos and enable nucleation step of the coated pit following which the pit invaginates and acquires the omega shape to be nicked by the GTPase acitivity of dynamin. Pharmacological inhibition of endocytic scission using dyansore and knockdown of CLC significantly reduced the formation of Stx17+CFTR+ vesicles (Supplemental Fig. 3A and B). Treatment of cells with dynasore resulted in conspicuous blebbing of Stx17 vesicles near the plasma membrane suggestive of vacuolar stalling (Supplemental Fig. 3A and B). These data suggest that plasma membrane localized CFTR is the most likely candidate population for recruitment into autophagosomes. It is likely based on prior studies (27) that ATG16L may communicate with the endocytic components to facilitate recruitment of Stx17-CFTR complexes into autophagosomal membranes. We have not tested this possibility in the current study.
Stx17+CFTR+ vesicles localize into autophagosomes to advance clearance
Major insights into the molecular finesse of autophagy have come from studies in yeast (Saccharomyces cerevisiae) (28). Molecular screening in yeast has identified 32 different Atg proteins, most of them conserved across phylogeny, which work in a step-wise fashion to form autophagosomes. LC3 encoded by the mammalian homologue of Atg8 is the only Atg protein that does not detach from completed autophagosomes, making it a reliable autophagosome marker(1, 10). The presence of CFTR-Stx17 complexes in autophagosomes was confirmed through their colocalization with LC3+ puncta (Fig. 3A). Co-expression of Stx17 TM dramatically decreased CFTR colocalization into LC3+ puncta (Fig. 3B). This data also suggested that a significant proportion of the LC3 labeled autophagosomes (~60%) were occupied by CFTR (Fig. 3B). We occasionally captured Stx17 bridging CFTR vesicles and autophagosomes under starvation and mediating the fusion between CFTR vesicle and lysosomes marked by LAMP1 (Fig. 3C). Autophagosomal localization of CFTR dramatically increased with the concomitant expression of cyan Stx17 suggesting that Stx17 recruits CFTR-containing vesicles to autophagosomes (Fig. 3D). Knockdown of Stx17 expression consistently reduced the number of CFTR+LC3+ vesicles, including in the case of co-expression of Cyan Stx17 (Fig. 3D). Stx17+CFTR+ vesicles did not coincide with isolation membrane marker Atg16L (Supplemental Fig. 4A). Using optiprep-based vesicular fractionation, we identified CFTR and Stx17 in different-sized vesicles (fractions 1 through 9) and especially in the membrane from LC3-II-enriched vesicles (fractions 3–6) (Supplemental Fig. 4B). Fractions 3–6 were subsequently pooled to immunoprecipitate LC3-positive vesicles that were shown to contain CFTR and Stx17 (Supplemental Fig. 4B).
Fig. 3. Stx17-CFTR complex improves autophagic clearance.
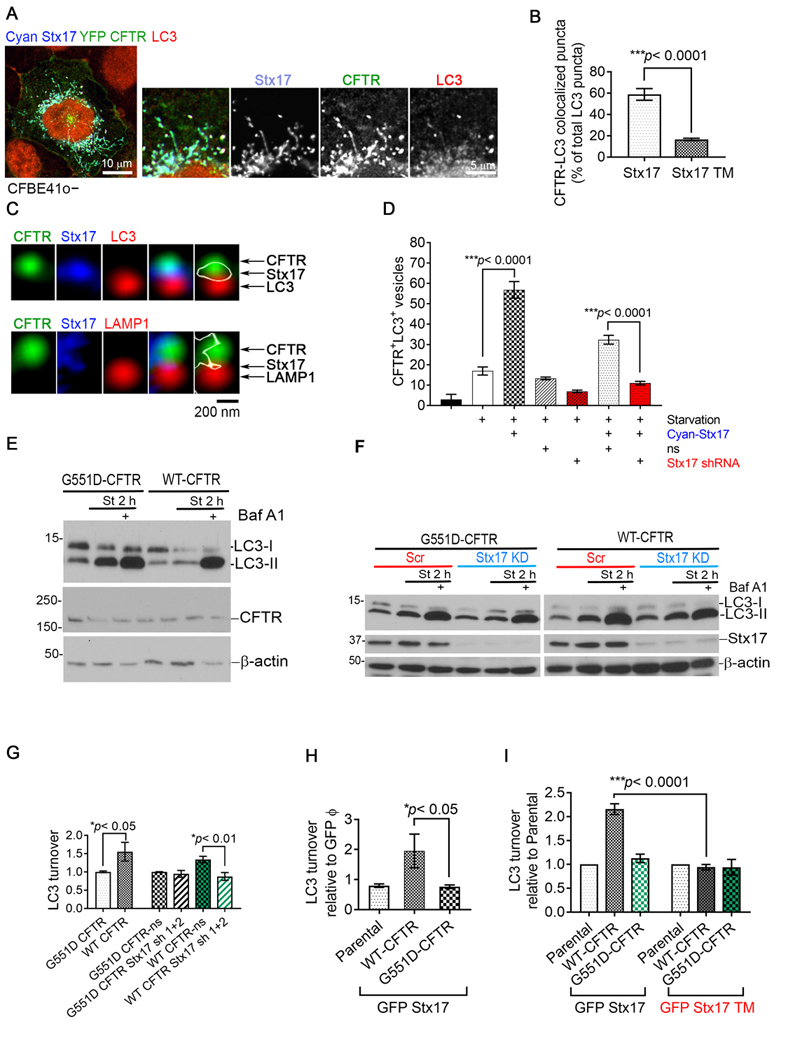
A, Confocal image of a region of starved CFBE41o− cells transfected with Cyan Stx17 and YFP CFTR and immunostained for LC3. B, Quantitation of RFP CFTR in LC3+ vesicles upon co-expression of GFP Stx17 or GFP Stx17 TM in starved CFBE41o− cells from n=3 independent experiments. Error bars represent SEM, P-value by t-test. C, Confocal images demonstrate vesicular structures corresponding to Cyan Stx17, YFP CFTR and LC3 at a region proximal to plasma membrane (top) and Cyan Stx17, YFP CFTR and LAMP1 (bottom) in starved CFBE41o− cell. White outline indicates the Stx17 protein. D, Quantitation of CFTR+LC3+ vesicles in CFBE41o− cells under various treatment conditions (± starvation ± Stx17 ± nonsilencing (ns) viral particles ± Stx17 sh RNA) from n=5 independent experiments. Error bars represent SEM, P-value by ANOVA with Bonferroni’s multiple comparisons test. E, Western blot to determine LC3 turnover in the presence YFP WT-CFTR, and functionally inactive YFP G551D-CFTR in CFBE41o−cells. YFP WT-CFTR and YFP G551D-CFTR could be detected in the CFTR overexpression samples by using anti-CFTR R1104 antibody. β-actin was used as a loading control. F, Western blot to determine LC3 turnover with (Stx17 shRNA 1 and 2) and without (ns) Stx17 knockdown in CFBE41o−cells with G551D CFTR or WT CFTR. β-actin was used as a loading control. G, Quantitation of LC3 turnover [(LC3-II under starvation+BafA1) - (LC3-II under starvation)] in CFBE41o−cells to determine the effect of loss of CFTR function with G551D CFTR and Stx17 knockdown on autophagic clearance from n=3 independent experiments. Error bars represent SEM, P-value by t-test. H, Quantitation of LC3 turnover in CFBE41o−cells in the presence WT-CFTR and G551D-CFTR co-expressed with GFP only and GFP Stx17 (n=3 independent experiments). Error bars represent SEM, P-value by t-test. I, Quantitation of LC3 turnover in CFBE41o−cells in the presence WT-CFTR and G551D-CFTR co-expressed with GFP Stx17 and GFP Stx17 TM (n=3 independent experiments). Error bars represent SEM, P-value by t-test.
As intra-autophagosomal components are degraded by lysosomal hydrolases upon constitution of autolysosomes, LC3-II is concomitantly degraded(16). Lysosomal turnover of LC3-II therefore reflects starvation-induced autophagic activity and efficiency of lysosomal turnover. Using this assay we determined that loss of CFTR function with gating mutation in G551D CFTR caused a decline in LC3 turnover (Fig. 3E and 3G). Please note that G551D CFTR produces a properly folded protein which traffics normally to the plasma membrane unlike the most common CFTR mutant F508del CFTR which goes minimally past ER due to an inherent folding defect. Due to a complex biology of F508del CFTR processing and previously reported involvement of F508del CFTR protein in retarding autophagy and tendency to form aggresomes (3), G551D CFTR was preferred as a loss of CFTR function candidate to study CFTR dependent autophagy clearance. Downregulation of Stx17 expression lead to a decrease in CFTR-mediated LC3-II turnover (Fig. 3F and 3G). Overexpression of Stx17 enhanced LC3 turnover in WT CFTR but not in G551D CFTR expressing cells (Fig. 3H). This effect was not seen when Stx17 TM was coexpressed with WT CFTR (Fig. 3I). Hence, Stx17 mediated recruitment of CFTR to autophagosomes accelerates autolysosomal clearance and that CFTR-Stx17 complex formation is essential for this effect.
CFTR improves bacterial clearance dependent upon Stx17 mediated recruitment into autophagosomes
The process of clearing intracellular pathogens is a selective form of autophagy, termed as xenophagy (29). The vacuoles for entrapping pathogens in xenophagy also require core autophagy machinery. Bacteria containing autophagosomal compartments can also be marked by LC3. Pseudomonas aeruginosa is the major bacteria found in CF associated lung infection (30). Prolonged presence of P. aeruginosa in CF lung causes bio-film formation and tissue damage that presents a serious threat towards a complete lung function collapse. Indeed, a perpetual cycle of infection and inflammation is the primary cause of CF associated mortality. Recent studies suggest that the bacterium acquires a long-term ability to colonize the host immune cells and that bronchial epithelial cells may perpetually carry reserves of intracellular bacteria during chronic infection (31, 32). Impaired autophagy is now potentially seen as one of the major cause of defect in innate immune responses in CF (32, 33). Therefore, we considered the possibility that CFTR-Stx17 autophagosomal complex may play an essential role in bacterial clearance. To test this hypothesis, we challenged primary bronchial epithelial cells with laboratory strain of GFP labeled P. Aeruginosa PAO1 to be studied in the context of CFTR-Stx17 complex. We observed that CFTR-Stx17-LC3 completely co-localized with the internalized bacteria in primary bronchial epithelial cells (Fig. 4A). Also, inhibition of autophagosomal clearance using chloroquine increased bacterial load in normal epithelial cells (Fig. 4B and 4C). There was a dramatic defect of bacterial clearance in F508del CFTR primary bronchial cells which did not exacerbate upon treatment of cells with chloroquine (Fig. 4B and 4C). Treatment of non-CF primary human bronchial epithelial cells with CFTR inhibitor 172 lead to a ~ 9-fold increase in the intracellular PAO1 load compared to non-treated cells suggesting that defective CFTR function hinders bacterial clearance (Supplementary figure 5A and 5B). Significance of CFTR-Stx17 functional coupling in bacterial resolution was recapitulated in CFBE41o− cells expressing WT CFTR which was impaired in parental (no CFTR) and CFTR mutant cells and upon downregulation of Stx17 expression in WT CFTR expressing cells (Fig. 4D and 4E). Overexpression of GFP-Stx17 improved bacterial clearance only in CFBE41o− cells expressing WT CFTR but not mutant CFTR (F508del and G551D CFTR) (Fig. 4F). Overexpression of GFP-Stx17 TM was unable to improve bacterial clearance in WT CFTR cells which is most likely caused by an absence of interaction of WT CFTR with GFP Stx17 TM (Fig. 4F). Hence, CFTR-Stx17 complex plays an important role in intracellular bacterial clearance.
Fig. 4. Stx17-CFTR complex improves clearance of infectious bacteria inside the autophagosomes.

Confocal image of primary human bronchial epithelial cells infected with GFP labeled P. Aeruginosa PAO1 (1:10 MOI) and immunostained for A, CFTR and Stx17, and CFTR and LC3. B, Confocal image of normal and CF (homozygous for F508del CFTR mutation) primary human bronchial epithelial cells on the transwells infected with GFP labeled P. Aeruginosa PAO1 (1:25 MOI) with and without a pretreatment with CQ (20 μM, 2 h). C, Quantitation of GFP PAO1 intensity in normal and CF bronchial epithelial cells under the experimental conditions described in Fig. 4C (n=3 transwells per condition). Error bars represent SEM, P-value by t-test. D, Confocal images depicting green signal corresponding to PAO1 in CFBE41o− cells with no CFTR, F508del CFTR, G551D CFTR ± Stx17 sh, WT CFTR ± Stx17 sh infected with the bacteria at 1:25 MOI. E, Quantitation of GFP PAO1 load in CFBE41o− cells under the experimental conditions described in Fig. 4D (n=3 independent experiments). P-value by ANOVA with Bonferroni’s multiple comparisons test. F, Quantitation of GFP PAO1 load in CFBE41o− cells with WT CFTR ± GFP Stx17 or GFP Stx17 TM, F508del CFTR ± GFP Stx17 or GFP Stx17 TM, and G551D CFTR ± GFP Stx17 or GFP Stx17 TM, infected with the bacteria at 1:25 MOI. (n=3 independent experiments). P-value by ANOVA with Bonferroni’s multiple comparisons test.
Discussion
End-step clearance by lysosomes is integral to degradation, replenishment and necessary modifications of cellular substrates to maintain cellular homeostasis within endosomal–autophagic–lysosomal system. Defects in the end-step clearance associate molecularly with lysosomal storage disorders and defective autophagy (34). In this study, we described a specialized role of CFTR in proceeding more efficient end-step clearance in the autophagy cycle through physical coupling with autophagosomal SNARE Stx17 (Fig. 5). This binding also establishes the fidelity of CFTR towards regulating autophagy. Association of CFTR with Stx17, a late -stage autophagosomal SNARE, would ensure that CFTR becomes recruited into the autophagy process only close to the stage of lysosomal clearance.
Fig. 5. Model of the study.

Diagram to depict the essence of this study: Stx17 associates with CFTR at the late-step of autophagy cycle in response to nutritional starvation and bacterial infection. In this whole process, CFTR becomes integrated into the autophagosomal membrane, and accelerates substrate clearance (cytoplasmic cargos and bacteria).
Di and colleagues reaffirmed much earlier reports of CF-derived cells being slightly alkaline by demonstrating that CFTR-null alveolar macrophages exhibit poorer phagosomal acidification, which impedes clearance, than do normal cells (4, 35). On a different note, Luciani and colleagues suggested the role of reactive oxygen species affecting early-step Atg proteins(3). These studies confirm that autophagy is impacted in CF most likely by different mechanisms and helped shape the idea that CF disease phenotype can potentially be rescued through restoration of an impaired autophagy. While the role of CFTR as a vesicular pH modulator is debatable (4, 36, 37), it has been argued to be related to the type of the cargo and sub-cellular compartments carrying those cargos. Regulation of autolysosomal pH by CFTR could be a mechanism of improved autophagosomal clearance that is not the scope of the current investigation. Several of the autophagy related proteins express at lower levels in CF relative to the healthy epithelial cells (our unpublished data). In the wake of the earlier studies, our findings, and due to multitudinous and complexity of defects identified in F508del CFTR, we anticipate that a defunct CFTR inside autolysosomes is one among plausibly several factors leading to a defective autophagy in CF.
CFTR not being a ubiquitously present protein and largely expressed in epithelial cells with limited amounts identified in macrophages, clearly raises the question of its relevance to a ubiquitous pathway of autophagy. Several human disorders have been traced to autophagy defects in the epithelial cells (29, 38, 39). In the lung, epithelium forms a crucial component of the innate defense system to protect and elicit downstream responses against air-borne challenges including microbes (40, 41). Since autophagy plays an important role in the elimination of internalized bacteria as a specialized function and can mediate their bulk removal unlike other degradative processes, CFTR-Stx17 autophagy axis would be important for tuning up autophagy and protecting airway epithelial cells from serious infectious threats and avert their perpetuation. According to our data, nearly 60% of the total autophagosomes are occupied by CFTR-Stx17 complexes. Also, 80–100% of the internalized bacterial localized into CFTR-Stx17 vesicles. This represents a significant contribution of this complex towards autophagosomal clearance. As mentioned earlier that the common flora like P.aeruginosa found in CF can continue to propagate inside CF epithelial cells. One of the fundamental challenges in controlling CF airway disease is the vicious cycle of infection and inflammation that in most CF cases manifest as the cause of mortality. Based on our data, it is clear that CF epithelia cannot clear bacteria with at least one possibility of the lack of CFTR function inside autophagosomes.
Macrophages compose the first line of host defense and therefore, play crucial role in clearing bacterial pathogens commonly encountered in CF (42). Macrophage dysfunction in CF has been demonstrated to be integral to the failure of CF lungs in clearing bacterial infections (42). With some evidence of CFTR expression in the macrophages, there is sufficient data to support that lack of CFTR expression and/or CFTR function in the macrophages could also be primary to the bacterial clearance defect in the CF lungs (4, 43). We demonstrated that CFTR dependent autophagy is defective in CF epithelial cells, and there is prevailing evidence that CF patients and CF mice exhibit limited autophagy activity although no prior study determined the clear mechanism of how CFTR is involved in autophagy. Autophagy has been shown to be essential in clearing P. Aeruginosa infection in lung macrophages (44). Based on our data, it is clear that the binding ability of CFTR with an autophagy protein-Stx17 ensures the specific action of CFTR in autophagy to overall improve the efficiency of autophagic clearance. This will be our expected mechanism of autophagy in macrophages. In all, our data adds a new piece of information on how CFTR is an important regulator of host defense response via autophagy.
Autophagy is a highly conserved process and Atg genes have been found to be highly conserved among lancelet, lamprey and zebrafish (45). Phagocytosis and autophagy could be triggered in lamprey leukocytes when challenged with live E. coli, to degrade pathogenic bacteria, suggesting the role of autophagy in the host defense in these ancient species. An interesting study by Cui at al. revealed the evolution of CFTR as an effective chloride channel from more of an anion transporter (46). The authors used sea lamprey (Lp) CFTR as a template to compare with human CFTR and cited the bicarbonate transport to be a more important component relative to the chloride transport of CFTR function in sea lamprey for pH regulation in the intestine. Future studies will suggest whether the bicarbonate or chloride transport is essential for CFTR assisted clearance in the autophagosomes. It would be interesting if the pH mediated regulation by CFTR dependent bicarbonate secretion is involved in autophagic clearance. In this aforementioned study, the authors reported an extended N terminus in Lp-CFTR. Based on our data, N-terminus in CFTR has the highest affinity to bind to Stx17. Future studies will suggest how an extended N-terminus as in sea lamprey would affect the formation of CFTR-Stx17 complexes and contribute to autophagy.
An important question arises: At what step of PAO1 clearance might the CFTR/STX17 complex facilitate? We have evidence that Stx17 binds and recruits membrane localized CFTR into the autophagosomes as in the presence of inhibitor of endocytosis (dynasore) and upon knockdown of clathrin protein expression, CFTR fails to become recruited inside Stx17 positive vesicles and stays at the plasma membrane (Supplementary figure 3). Multiple studies have suggested that CFTR and the endocytic machinery is important for Pseudomonas uptake and ingestion inside the cells (47, 48). We speculate with caution based on these earlier studies that CFTR could be important for the initial step of ingesting the bacteria to be later directed into autophagosomes for clearance inside the lysosomes by CFTR-Stx17 complex. Based on our data, presence of CFTR would enhance the efficiency of bacterial clearance inside autophagolysosomes.
The current study looks at PAO1 clearance that happens via the autophagosomal/lysosomal (phagosomal) pathway (please see Fig 5). Phagocytic cells including macrophages commonly sort to clear P. aeruginosa inside the acidified vesicles. Although, there is growing evidence that P. aeruginosa is capable of escaping vacuolar space, come in contact with the cytoplasm and survive intracellularly (49). In fact, there are studies showing that intracellular GFP-labelled PAO1 (which was used in this study) increasing encounters intracellular environment (i.e., cytosol) in diverse mammalian cell types including macrophages (50). If the bacterial clearance is inefficient (e.g., in CF), one could predict that there would be significantly high incidence of vacuolar escape and increased intracellular load of the bacteria. Additionally, there is a mechanism of direct bacterial invasion and the ability of the bacteria to thrive intracellularly being consistently reported in the epithelial cells (49). It is argued that via this mechanism of intracellular penetration, the bacteria survive better and the epithelial cells could essentially become bacterial reserves leading to chronic infection (51). According to the authors of the first report that discovered Stx17 as the major SNARE protein for autophagosomes, it is very likely that cytosol is the major source for STX17 (52). Future studies will be needed to investigate whether Stx17 could also recognize cytosolic PAO1 and mediate its incorporation into mature autophagosomes containing CFTR.
Considering the important physiological roles of autophagy, many components of the autophagy pathway are being pursued for therapeutic modulation in autoimmune disorders including inflammatory bowel disorders, slow onset inflammation associated metabolic diseases and several infectious diseases (53). This approach is currently under investigation in CF as well. Recently, Cysteamine, a transglutaminase-2 (TGM2) inhibitor is being explored to restore autophagy defect in CF patients (5, 54, 55). Cysteamine in combination with epigallocatechin gallate improved sweat chloride concentration plausibly through its action on autophagy proteins beclin 1 and p62 (5, 55). As new studies emerge, the idea that improving autophagy would help restore several CF lung associated defects is being strongly supported.
Supplementary Material
Acknowledgements
The authors thank late Dr. David L Armbruster for review and editing of the manuscript. We are thankful to Sunitha Yarlagadda, CCHMC for designing the recombinant cDNA constructs. This research was funded by the National Institutes of Health (APN; HL147351, DK080834 and DK093045) and CF foundation grants to APN (NAREN19RO) and KA (ARORA16F0).
References
- 1.Glick D, Barth S, and Macleod KF (2010) Autophagy: cellular and molecular mechanisms. J Pathol 221, 3–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mizushima N. (2007) Autophagy: process and function. Genes Dev 21, 2861–2873 [DOI] [PubMed] [Google Scholar]
- 3.Luciani A, Villella VR, Esposito S, Brunetti-Pierri N, Medina D, Settembre C, Gavina M, Pulze L, Giardino I, Pettoello-Mantovani M, D’Apolito M, Guido S, Masliah E, Spencer B, Quaratino S, Raia V, Ballabio A, and Maiuri L. (2010) Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nat Cell Biol 12, 863–875 [DOI] [PubMed] [Google Scholar]
- 4.Di A, Brown ME, Deriy LV, Li C, Szeto FL, Chen Y, Huang P, Tong J, Naren AP, Bindokas V, Palfrey HC, and Nelson DJ (2006) CFTR regulates phagosome acidification in macrophages and alters bactericidal activity. Nat Cell Biol 8, 933–944 [DOI] [PubMed] [Google Scholar]
- 5.Tosco A, De Gregorio F, Esposito S, De Stefano D, Sana I, Ferrari E, Sepe A, Salvadori L, Buonpensiero P, Di Pasqua A, Grassia R, Leone CA, Guido S, De Rosa G, Lusa S, Bona G, Stoll G, Maiuri MC, Mehta A, Kroemer G, Maiuri L, and Raia V. (2016) A novel treatment of cystic fibrosis acting on-target: cysteamine plus epigallocatechin gallate for the autophagy-dependent rescue of class II-mutated CFTR. Cell Death Differ 23, 1380–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Russell RC, Yuan HX, and Guan KL (2014) Autophagy regulation by nutrient signaling. Cell Res 24, 42–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y, Amano A, and Yoshimori T. (2013) Autophagosomes form at ER-mitochondria contact sites. Nature 495, 389–393 [DOI] [PubMed] [Google Scholar]
- 8.Wolfe DM, Lee JH, Kumar A, Lee S, Orenstein SJ, and Nixon RA (2013) Autophagy failure in Alzheimer’s disease and the role of defective lysosomal acidification. Eur J Neurosci 37, 1949–1961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Campoy E, and Colombo MI (2009) Autophagy in intracellular bacterial infection. Biochim Biophys Acta 1793, 1465–1477 [DOI] [PubMed] [Google Scholar]
- 10.Itakura E, Kishi-Itakura C, and Mizushima N. (2012) The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 151, 1256–1269 [DOI] [PubMed] [Google Scholar]
- 11.Takats S, Nagy P, Varga A, Pircs K, Karpati M, Varga K, Kovacs AL, Hegedus K, and Juhasz G. (2013) Autophagosomal Syntaxin17-dependent lysosomal degradation maintains neuronal function in Drosophila. J Cell Biol 201, 531–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diao J, Liu R, Rong Y, Zhao M, Zhang J, Lai Y, Zhou Q, Wilz LM, Li J, Vivona S, Pfuetzner RA, Brunger AT, and Zhong Q. (2015) ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature 520, 563–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arora K, Sinha C, Zhang W, Moon CS, Ren A, Yarlagadda S, Dostmann WR, Adebiyi A, Haberman Y, Denson LA, Wang X, and Naren AP (2015) Altered cGMP dynamics at the plasma membrane contribute to diarrhea in ulcerative colitis. Am J Pathol 185, 2790–2804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li C, Krishnamurthy PC, Penmatsa H, Marrs KL, Wang XQ, Zaccolo M, Jalink K, Li M, Nelson DJ, Schuetz JD, and Naren AP (2007) Spatiotemporal coupling of cAMP transporter to CFTR chloride channel function in the gut epithelia. Cell 131, 940–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arora K, Moon C, Zhang W, Yarlagadda S, Penmatsa H, Ren A, Sinha C, and Naren AP (2014) Stabilizing rescued surface-localized deltaf508 CFTR by potentiation of its interaction with Na(+)/H(+) exchanger regulatory factor 1. Biochemistry 53, 4169–4179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mizushima N, Yoshimori T, and Levine B. (2010) Methods in mammalian autophagy research. Cell 140, 313–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li C, Dandridge KS, Di A, Marrs KL, Harris EL, Roy K, Jackson JS, Makarova NV, Fujiwara Y, Farrar PL, Nelson DJ, Tigyi GJ, and Naren AP (2005) Lysophosphatidic acid inhibits cholera toxin-induced secretory diarrhea through CFTR-dependent protein interactions. J Exp Med 202, 975–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Penmatsa H, Zhang W, Yarlagadda S, Li C, Conoley VG, Yue J, Bahouth SW, Buddington RK, Zhang G, Nelson DJ, Sonecha MD, Manganiello V, Wine JJ, and Naren AP (2010) Compartmentalized cyclic adenosine 3’,5’-monophosphate at the plasma membrane clusters PDE3A and cystic fibrosis transmembrane conductance regulator into microdomains. Mol Biol Cell 21, 1097–1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu J, Xia H, Kim M, Xu L, Li Y, Zhang L, Cai Y, Norberg HV, Zhang T, Furuya T, Jin M, Zhu Z, Wang H, Yu J, Li Y, Hao Y, Choi A, Ke H, Ma D, and Yuan J. (2011) Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell 147, 223–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang F, Rock JR, Harfe BD, Cheng T, Huang X, Jan YN, and Jan LY (2009) Studies on expression and function of the TMEM16A calcium-activated chloride channel. Proc Natl Acad Sci U S A 106, 21413–21418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Naren AP, Nelson DJ, Xie W, Jovov B, Pevsner J, Bennett MK, Benos DJ, Quick MW, and Kirk KL (1997) Regulation of CFTR chloride channels by syntaxin and Munc18 isoforms. Nature 390, 302–305 [DOI] [PubMed] [Google Scholar]
- 22.Naren AP, Quick MW, Collawn JF, Nelson DJ, and Kirk KL (1998) Syntaxin 1A inhibits CFTR chloride channels by means of domain-specific protein-protein interactions. Proc Natl Acad Sci U S A 95, 10972–10977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma J, Zhao J, Drumm ML, Xie J, and Davis PB (1997) Function of the R domain in the cystic fibrosis transmembrane conductance regulator chloride channel. J Biol Chem 272, 28133–28141 [DOI] [PubMed] [Google Scholar]
- 24.Gadsby DC, and Nairn AC (1999) Control of CFTR channel gating by phosphorylation and nucleotide hydrolysis. Physiol Rev 79, S77–S107 [DOI] [PubMed] [Google Scholar]
- 25.Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, and Kirchhausen T. (2006) Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell 10, 839–850 [DOI] [PubMed] [Google Scholar]
- 26.Royle SJ (2013) Protein adaptation: mitotic functions for membrane trafficking proteins. Nat Rev Mol Cell Biol 14, 592–599 [DOI] [PubMed] [Google Scholar]
- 27.Ravikumar B, Moreau K, Jahreiss L, Puri C, and Rubinsztein DC (2010) Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol 12, 747–757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakatogawa H, Suzuki K, Kamada Y, and Ohsumi Y. (2009) Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol 10, 458–467 [DOI] [PubMed] [Google Scholar]
- 29.Levine B, Mizushima N, and Virgin HW (2011) Autophagy in immunity and inflammation. Nature 469, 323–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davies JC (2002) Pseudomonas aeruginosa in cystic fibrosis: pathogenesis and persistence. Paediatr Respir Rev 3, 128–134 [DOI] [PubMed] [Google Scholar]
- 31.Schroeder TH, Reiniger N, Meluleni G, Grout M, Coleman FT, and Pier GB (2001) Transgenic cystic fibrosis mice exhibit reduced early clearance of Pseudomonas aeruginosa from the respiratory tract. J Immunol 166, 7410–7418 [DOI] [PubMed] [Google Scholar]
- 32.Junkins RD, Shen A, Rosen K, McCormick C, and Lin TJ (2013) Autophagy enhances bacterial clearance during P. aeruginosa lung infection. PLoS One 8, e72263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Junkins RD, McCormick C, and Lin TJ (2014) The emerging potential of autophagy-based therapies in the treatment of cystic fibrosis lung infections. Autophagy 10, 538–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lieberman AP, Puertollano R, Raben N, Slaugenhaupt S, Walkley SU, and Ballabio A. (2012) Autophagy in lysosomal storage disorders. Autophagy 8, 719–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barasch J, Kiss B, Prince A, Saiman L, Gruenert D, and al-Awqati Q. (1991) Defective acidification of intracellular organelles in cystic fibrosis. Nature 352, 70–73 [DOI] [PubMed] [Google Scholar]
- 36.Barriere H, Bagdany M, Bossard F, Okiyoneda T, Wojewodka G, Gruenert D, Radzioch D, and Lukacs GL (2009) Revisiting the role of cystic fibrosis transmembrane conductance regulator and counterion permeability in the pH regulation of endocytic organelles. Mol Biol Cell 20, 3125–3141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haggie PM, and Verkman AS (2009) Unimpaired lysosomal acidification in respiratory epithelial cells in cystic fibrosis. J Biol Chem 284, 7681–7686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Festa BP, Chen Z, Berquez M, Debaix H, Tokonami N, Prange JA, Hoek GV, Alessio C, Raimondi A, Nevo N, Giles RH, Devuyst O, and Luciani A. (2018) Impaired autophagy bridges lysosomal storage disease and epithelial dysfunction in the kidney. Nat Commun 9, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, Kishi C, Kc W, Carrero JA, Hunt S, Stone CD, Brunt EM, Xavier RJ, Sleckman BP, Li E, Mizushima N, Stappenbeck TS, and Virgin H. W. t. (2008) A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 456, 259–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hoiby N, Ciofu O, and Bjarnsholt T. (2010) Pseudomonas aeruginosa biofilms in cystic fibrosis. Future Microbiol 5, 1663–1674 [DOI] [PubMed] [Google Scholar]
- 41.Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhaes JG, Yuan L, Soares F, Chea E, Le Bourhis L, Boneca IG, Allaoui A, Jones NL, Nunez G, Girardin SE, and Philpott DJ (2010) Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol 11, 55–62 [DOI] [PubMed] [Google Scholar]
- 42.Bonfield TL (2015) Macrophage Dysfunction in Cystic Fibrosis: A Therapeutic Target to Enhance Self-Immunity. Am J Respir Crit Care Med 192, 1406–1407 [DOI] [PubMed] [Google Scholar]
- 43.Barnaby R, Koeppen K, Nymon A, Hampton TH, Berwin B, Ashare A, and Stanton BA (2018) Lumacaftor (VX-809) restores the ability of CF macrophages to phagocytose and kill Pseudomonas aeruginosa. Am J Physiol Lung Cell Mol Physiol 314, L432–L438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yuan K, Huang C, Fox J, Laturnus D, Carlson E, Zhang B, Yin Q, Gao H, and Wu M. (2012) Autophagy plays an essential role in the clearance of Pseudomonas aeruginosa by alveolar macrophages. J Cell Sci 125, 507–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Luo L, Lu J, Wang Q, Chen S, Xu A, and Yuan S. (2018) Autophagy participates in innate immune defense in lamprey. Fish Shellfish Immunol 83, 416–424 [DOI] [PubMed] [Google Scholar]
- 46.Cui G, Hong J, Chung-Davidson YW, Infield D, Xu X, Li J, Simhaev L, Khazanov N, Stauffer B, Imhoff B, Cottrill K, Blalock JE, Li W, Senderowitz H, Sorscher E, McCarty NA, and Gaggar A. (2019) An Ancient CFTR Ortholog Informs Molecular Evolution in ABC Transporters. Dev Cell 51, 421–430 e423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pier GB, Grout M, and Zaidi TS (1997) Cystic fibrosis transmembrane conductance regulator is an epithelial cell receptor for clearance of Pseudomonas aeruginosa from the lung. Proc Natl Acad Sci U S A 94, 12088–12093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bajmoczi M, Gadjeva M, Alper SL, Pier GB, and Golan DE (2009) Cystic fibrosis transmembrane conductance regulator and caveolin-1 regulate epithelial cell internalization of Pseudomonas aeruginosa. Am J Physiol Cell Physiol 297, C263–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lovewell RR, Patankar YR, and Berwin B. (2014) Mechanisms of phagocytosis and host clearance of Pseudomonas aeruginosa. Am J Physiol Lung Cell Mol Physiol 306, L591–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garai P, Berry L, Moussouni M, Bleves S, and Blanc-Potard AB (2019) Killing from the inside: Intracellular role of T3SS in the fate of Pseudomonas aeruginosa within macrophages revealed by mgtC and oprF mutants. PLoS Pathog 15, e1007812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Garcia-Medina R, Dunne WM, Singh PK, and Brody SL (2005) Pseudomonas aeruginosa acquires biofilm-like properties within airway epithelial cells. Infect Immun 73, 8298–8305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Itakura E, and Mizushima N. (2013) Syntaxin 17: the autophagosomal SNARE. Autophagy 9, 917–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rubinsztein DC, Codogno P, and Levine B. (2012) Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 11, 709–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shrestha CL, Assani KD, Rinehardt H, Albastroiu F, Zhang S, Shell R, Amer AO, Schlesinger LS, and Kopp BT (2017) Cysteamine-mediated clearance of antibiotic-resistant pathogens in human cystic fibrosis macrophages. PLoS One 12, e0186169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.De Stefano D, Villella VR, Esposito S, Tosco A, Sepe A, De Gregorio F, Salvadori L, Grassia R, Leone CA, De Rosa G, Maiuri MC, Pettoello-Mantovani M, Guido S, Bossi A, Zolin A, Venerando A, Pinna LA, Mehta A, Bona G, Kroemer G, Maiuri L, and Raia V. (2014) Restoration of CFTR function in patients with cystic fibrosis carrying the F508del-CFTR mutation. Autophagy 10, 2053–2074 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.