

Abstract
Background:
Increased physical activity is a common feature of anorexia nervosa (AN). Although high activity levels are associated with greater risk of developing AN, particularly when combined with dieting, most individuals who diet and exercise maintain a healthy bodyweight. It is unclear why some individuals develop AN while most do not. A rodent model of resilience and vulnerability to AN would be valuable to research. Dopamine, which is believed to play a crucial role in AN, regulates both reward and activity and may modulate vulnerability.
Methods:
Adolescent and young adult female C57BL/6N mice were tested in the activity-based anorexia model (ABA), with an extended period of food restriction in adult mice. ABA was also tested in dopamine transporter knockdown mice and wild-type littermates. Mice that adapted to conditions and maintained a stable bodyweight were characterized as ‘resilient.’
Results:
In adults, vulnerable and resilient phenotypes emerged in both the ABA and food restricted mice without wheels. Vulnerable mice exhibited a pronounced increase in running throughout the light cycle, which dramatically peaked prior to requiring removal from the experiment. Resilient mice exhibited an adaptive decrease in total running, appropriate food anticipatory activity, and increased consumption, thereby achieving stable body weight. Hyperdopaminergia accelerated progression of the vulnerable phenotype.
Conclusions:
Our demonstration of distinct resilient and vulnerable phenotypes in mouse ABA significantly advances the utility of the model for identifying genes and neural substrates mediating AN risk and resilience. Modulation of dopamine may play a central role in the underlying circuit.
Keywords: activity-based anorexia, exercise, dopamine, hyperdopaminergic, starvation, food restriction, resilience, anorexia nervosa, vulnerability
INTRODUCTION
Anorexia nervosa (AN) is characterized by severe restriction of food intake and fear of gaining weight, leading to life-threatening weight loss. The disorder tends to be chronic, resistant to treatment and associated with high mortality (1–4). Neural mechanisms underlying the disorder remain poorly understood and there are no approved pharmacological treatments (5).
Excessive physical activity has been associated with AN since its earliest description (6), with 31-81% of AN patients exhibiting high activity levels, depending on how it is defined (7,8). Although characterized as compulsive (9–12) or compensatory (13) voluntary exercise, increased non-exercise activity, such as fidgeting, has also been observed (14). Exercise is associated with poorer outcomes, including greater risk of relapse, longer hospitalizations and increased chronicity (15–19), indicating a role for exercise in maintenance of the disorder. Additionally, higher levels of premorbid activity have been associated with greater risk of developing AN (20–22), even among athletes (23–25), supporting a role for physical activity in the development of AN. However, the majority of individuals who combine diet and exercise do not develop AN and the underlying factors mediating AN vulnerability or resilience are not understood.
Alterations in dopamine and associated changes in reward have been implicated in AN (26–31). Importantly, dopamine also modulates physical activity (32–34), as exemplified by increased psychomotor activity resulting from increased dopamine transmission with psychostimulants. The relationship between altered dopamine and the increased activity observed in AN has not been empirically characterized, though potentially important to understanding the disorder.
Activity-based anorexia (ABA), a widely used rodent model of AN, assesses the interaction between food restriction and physical activity. The model combines limited access to food with unlimited access to a running wheel, leading to hyperactivity, self-starvation, rapid weight loss and death unless removed from the experiment (35–37). We conducted a detailed analysis of running behavior of adolescent and adult female C57BL/6 mice in the ABA model and assessed the impact of genetically increasing dopamine. We discovered distinct vulnerable and resilient phenotypes, with the latter showing adaptation to ABA and weight stabilization. In contrast, vulnerable mice exhibit severely dysregulated running activity, inadequate consumption, and catastrophic weight loss. Vulnerability to ABA was increased in hyperdopaminergic mice, indicating that dopamine may play a central role in the development of AN.
METHODS AND MATERIALS
Animals
Female C57BL/6N mice (Taconic Biosciences, Germantown, NY) were purchased at postnatal day (PND) 21 and 56 for the adolescent and early adulthood behavioral studies, respectively. Male and female DAT-cre (DAT, dopamine transporter; cre, cre-recombinase) heterozygous mice (DAT knockdown, KD; DATcre/+; Cat# JAX: 020080; C57BL/6J genetic background) (38) and wild-type littermates (WT, DAT+/+) were bred in-house (New York State Psychiatric Institute; Hunter College). Only female KD and WT mice were used in behavioral studies. Prior to experiments, mice were group housed on a 12h light-dark cycle with ad libitum chow (Prolab Isopro 3000 5P75, WF Fisher & Son, Somerville, NY). Experiments were approved by the Institutional Animal Care and Use Committees of the New York State Psychiatric Institute, Hunter College, and Queen’s College.
ABA Procedure
Mice were distributed into four groups: activity-based anorexia (ABA; 2h/day food access; unlimited wheel access), wheel control (WH; unlimited food and wheel access), food restricted control (FR; 2h/day food access; unlimited access to a locked wheel), homecage control (HC; unlimited access to food and a locked wheel). Mice were weighed immediately preceding dark cycle onset and removed when they lost 25% of their baseline weight (supplemental methods). Mice that did not require removal after 10 days were characterized as “resilient.”
Fast-scan cyclic voltammetry (FSCV)
Evoked dopamine release was measured in DATcre/+ and WT littermates of both sexes using slice FSCV. For adolescent and young adult females (C57BL/6N), FSCV was done in intact, anesthetized animals (supplemental methods).
Statistical Analyses
Data were analyzed with a t test, ANOVA with Bonferroni post hoc correction, or Mantel-Cox log-rank for survival analyses. For analyses across days of restriction, where mice dropped out on different days, the linear mixed effect model was used (LME) with the LME4 package (39) in R (40) computing F values using lmerTest (41). We modeled ‘mouse’ as a random effect, including intercept and slope across experimental days. See supplemental materials for immunohistochemistry, mRNA, and protein quantification methods.
RESULTS
Adolescent C57 female mice are vulnerable to activity-based anorexia
We first tested adolescent female mice (PND 43 on ABA day 1). There were no baseline differences in bodyweight (F(3,60) = 0.23, p = 0.87). During the restriction phase, food restricted (FR; locked wheel) and ABA (food restricted; freely moving wheel) groups lost substantial weight compared to non-food restricted groups (Fig 1A, LME, day x group, F(3,66) = 13.89, p < 0.001). Although ABA and FR groups did not significantly differ in weight loss (LME, F(1,23), p = 0.39), the survival analysis revealed greater risk for the ABA group (Fig 1B, hazard ratio ABA/FR, 3.96, CI 1.65-9.50, p < 0.001). The LME analysis includes data points prior to removal and does not take into account time of removal reflected in the hazard ratio.
Figure 1.
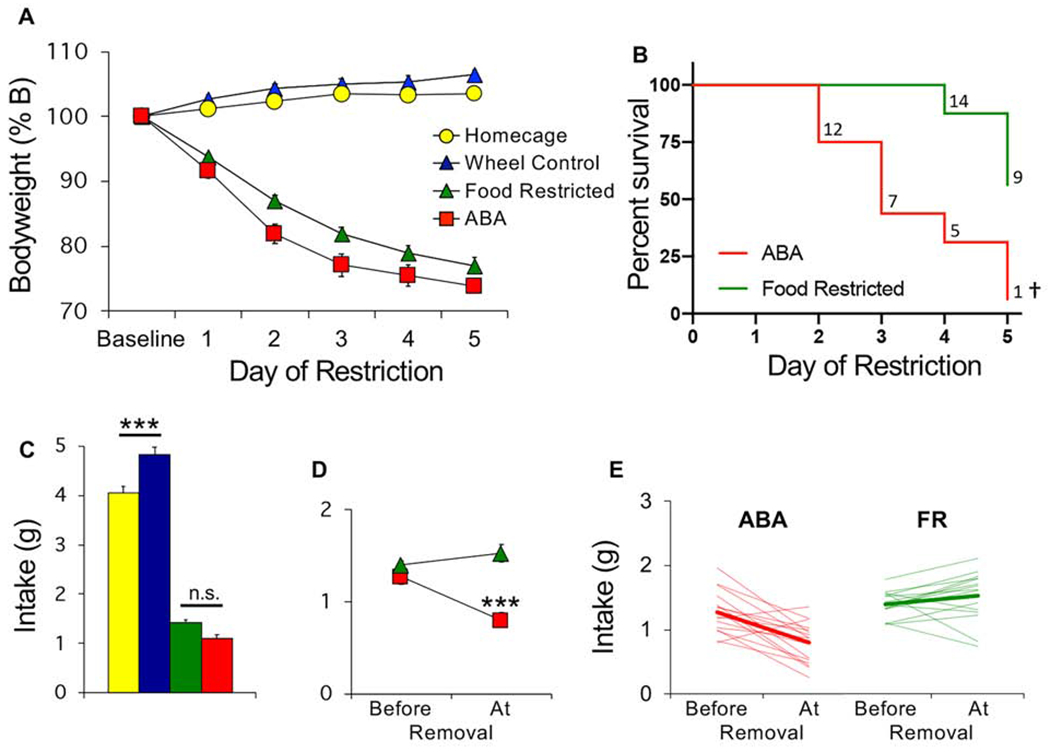
Adolescent female mice are vulnerable to activity-based anorexia. (A) Bodyweight across days of food restriction. (B) Survival curves for ABA and food restricted (FR) mice. Numbers indicate number of surviving mice that day. (C) Average food intake across 5 days of restriction. (D) Average and (E) individual (light traces) food intake of ABA and FR mice prior to removal versus the day of removal (bold lines in E show group average). n=16/group. ***p<0.001; †Survival curves, p<0.001. Error bars, ±SEM.
Food consumption differed between groups (Fig 1C, F(3,60) = 278.8, p < 0.001). Non-food restricted wheel controls (WH) increased their consumption compared to homecage controls (HC), presumably to compensate for running (Fig 1C, Bonferroni p < 0.001). In contrast, there was no difference in intake between the ABA and FR groups (Fig 1C, Bonferroni p = 0.29). However, ABA mice exhibited a drop in consumption on the day of removal, which was not observed in the FR mice (Fig 1D–E, time point x group, F(1,30) = 21.18 p < 0.001). That some FR mice required removal indicates that caloric restriction alone can induce life-threatening weight loss.
Abrupt increase in light cycle running precedes removal from the model in adolescent mice
There were no baseline differences between ABA and wheel controls (WH) in the amount (Fig 2A, LME, group, F(1,26) = .121, p = 0.73) or circadian distribution (Fig 2B1, LME, group x hour, F(1,26) = 0.28, p = 0.60) of wheel running. During food restriction, the ABA mice exhibited a sharp increase in light but not dark cycle running (Figs 3A, 3B1, bar graphs: group x cycle, F(1,26) = 19.50, p<0.001; light cycle, Bonferroni p<0.001, dark cycle, Bonferroni p = 0.99, also Fig 2). Though typically considered food anticipatory activity, this increase started near the onset of the light cycle (Fig 2B2, LME, group x hour, F(1,26) = 8.16, p < 0.001). This alteration in the distribution of circadian activity increased across days of food restriction (Fig 2A). Plotting the running of individual mice revealed that each mouse exhibited an abrupt increase in light cycle running (Fig 3B1). A comparison of the maximum increase in day-to-day running revealed that the peak change was higher in the ABA than the WH group during the light cycle (Fig 3C, group x cycle, F(1,26) = 130.4, p <0.001). This abrupt increase generally preceded removal from the model by one day in ABA mice (Fig 3D), suggesting a predictive relationship between peak change in light cycle running and removal. Light-cycle running was positively correlated with weight loss in ABA mice (Fig 3E1). Dark cycle running also correlated with weight loss in ABA mice (Fig 3E2), despite similar levels of dark cycle running between groups (Fig 3A, 3B2). We observed no correlation between running and change in bodyweight in wheel controls (Fig 3F1, 3F2).
Figure 2.
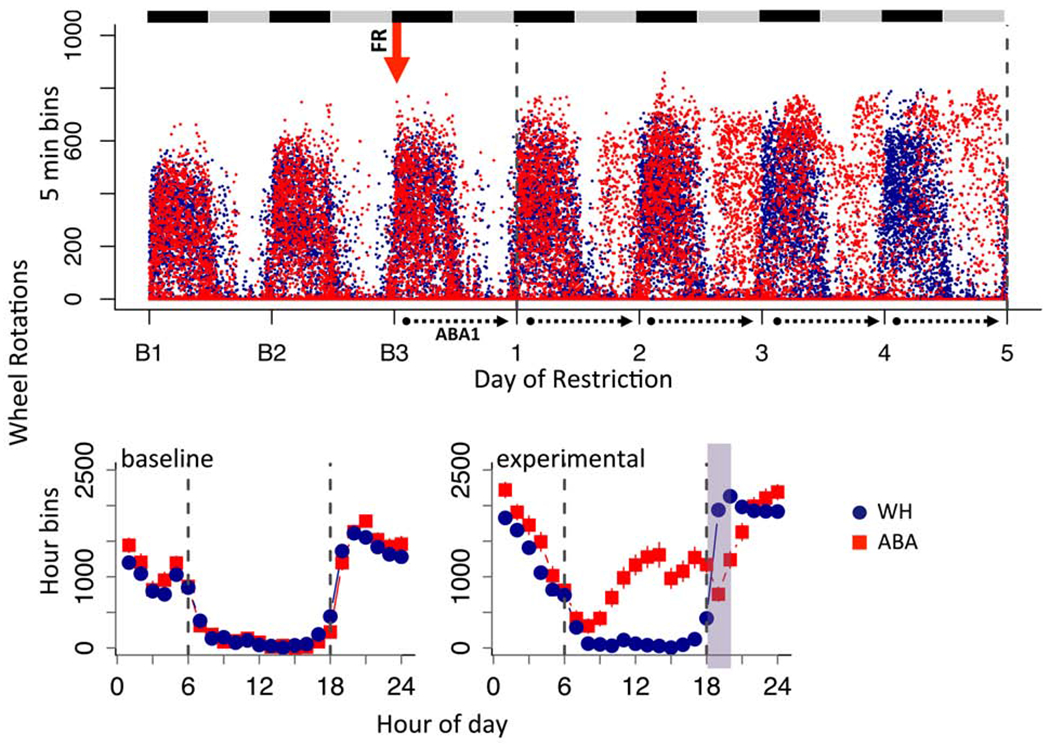
ABA increases light cycle running in adolescent mice. (A) Dot plot of wheel running across days of experiment for all mice in 5 min bins. Black and gray bars indicate dark and light cycles, respectively. Red arrow indicates start of food restriction. Dotted lines demarcate data collection days under ABA. (B) Group averages in hour bins across 24 hour period averaged for (B1) baseline days and (B2) ABA experimental days. Dotted line marks light cycle between 6 (lights on) and 18 (lights off) hrs and green shading indicates when food was available. Error bars, ±SEM.
Figure 3.
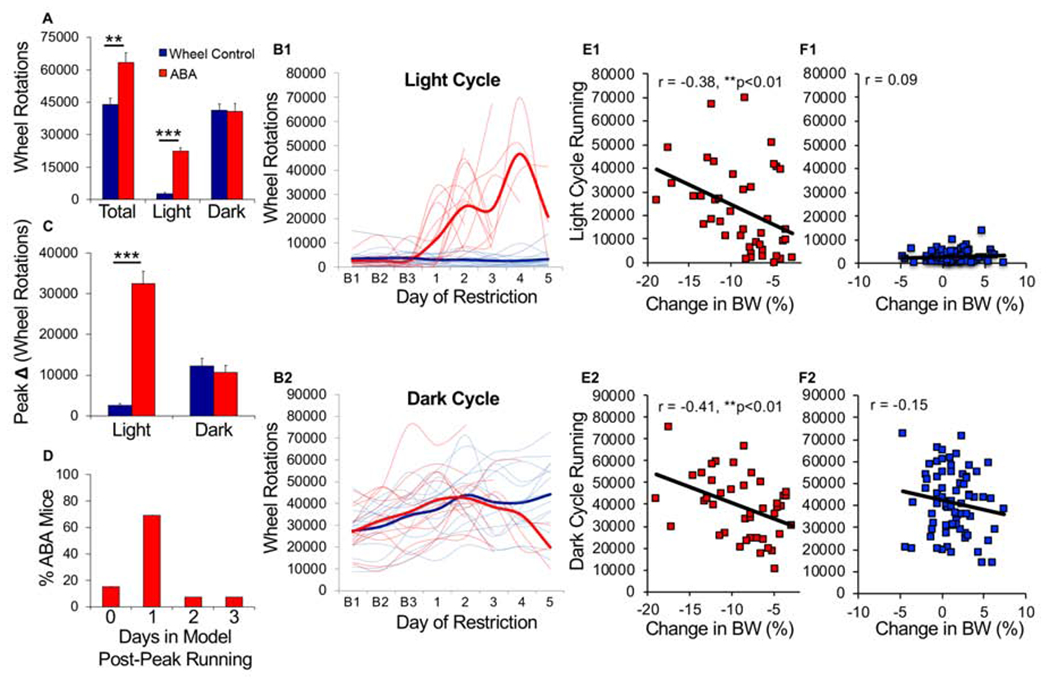
Abrupt increase in light cycle running precedes removal from model. (A) Average wheel activity across days of food restriction. (B) Wheel running of individual mice (light traces) and group mean (bold traces) across food restriction for (B1) light and (B2) dark cycle running. (C) Maximum increase in wheel running across two consecutive days (averaged by group) during the light cycle and dark cycle. (D) Histogram of number of days mice remained in model after maximum increase in light cycle running. (E-F) Correlation between light (top) or dark cycle running (bottom) and change in bodyweight the next day for ABA mice (E1-E2) and wheel control mice (F1-F2). Each symbol represents one animal on one experimental day. n=13 (ABA), n=15 (wheel controls); **p<0.01; ***p<0.001. Error bars, ± SEM.
Adult mice exhibit vulnerable and resilient phenotypes
We tested ABA in young adult mice (PND 68 on ABA day 1) using the same protocol, but extended food restriction to 10 days. Bodyweight was not different between groups at baseline (F(3,60) = 2.03, p = 0.12). In the first 5 days, both ABA and FR mice lost substantial weight with no group difference (Fig 4A, LME, group x day, F(1,30), p = 0.23). In days 6-10, weight stabilized for the remaining mice, who survived through the end of the experiment and were characterized as resilient (Fig 4A, 4C1–C2). Adult mice were less vulnerable to ABA than younger mice (Fig 4B, adult vs. adolescent, p<0.001), with ~50% of adults surviving (Fig 4D). Some FR mice (~30%) required removal (Fig 4B, 4D), indicating that a subset of both adult ABA and FR mice are vulnerable to catastrophic weight loss with prolonged caloric restriction, though ABA increases risk and accelerates progression (Fig 4B, hazard ratio ABA/FR, 2.22, CI 0.82-5.97, p=0.10). These results demonstrate that both ABA and FR mice exhibit vulnerable (ABA-V, FR-V) and resilient (ABA-R, FR-R) phenotypes. Interestingly, ABA-R mice gained more weight than FR-R mice (Fig 4A, LME, last 3 days, ABA vs FR, F(1,17) = 7.52 p = 0.01), suggesting that access to a running wheel promoted an adaptive response in resilient mice.
Figure 4.
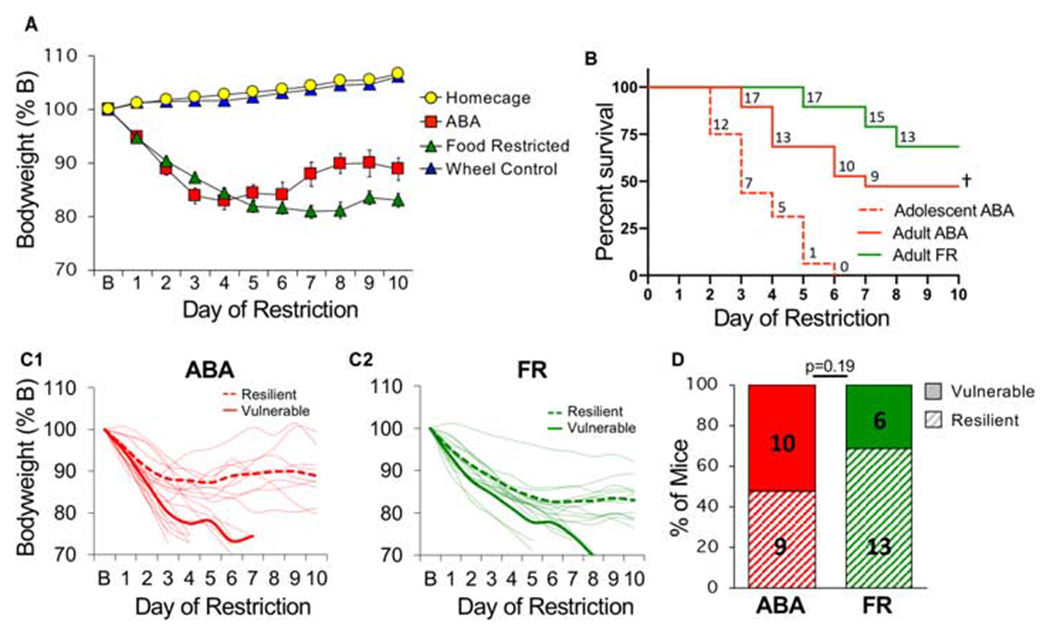
Young adult female mice exhibit vulnerable and resilient phenotypes. (A) Bodyweight across days of food restriction. (B) Survival curves for each group. Dotted red is replot of adolescent survival from Fig. 1 for reference. Numbers indicate number of surviving mice that day. (C) Body weight of individual mice (light traces) across days of food restriction in (C1) ABA and (C2) FR groups. Group averages for resilient (dashed line) and vulnerable (solid line) mice are shown in bold. (D) Percent of vulnerable (solid) and resilient (hatched) mice within ABA and FR groups (number of animals is indicated inside bars). n=19 (ABA), n=19 (FR), n=12 (homecage), n=14 (wheel controls). †Survival curves, difference between adolescent and adult ABA, p<0.0001. Error bars, ± SEM.
Resilient mice adapt food intake
Food restriction similarly reduced consumption in ABA and FR groups (Fig 5A, ABA vs. FR, Bonferroni p=0.99). Resilient mice consumed more food than vulnerable mice in both ABA and FR groups (Fig 5B–5C; Fig 5C1: LME, ABA phenotype x day, F(1,125) = 21.28, p<0.001; Fig 5C2: LME, FR phenotype x day, F(1,56) = 6.41, p < 0.05). Consistent with increased weight noted above, ABA-R mice exhibited a larger increase in consumption than FR-R mice (Fig 5C1–C2, LME, group x phenotype x day, F(1,235) = 3.94, p < 0.05). In contrast, vulnerable mice showed a decline in consumption that occurred earlier in ABA-V than FR-V mice (Fig 5C1–C2, days in model, t(13) = 2.86, p=0.01). This cannot be due to insufficient time to eat, because the resilient mice increased food intake in the same amount of time. Similar to adolescent mice, consumption decreased at time of removal for vulnerable mice of both groups (Fig 5D–E, time point x phenotype, F(1,33) = 54.12, p <0.001). In resilient mice, consumption correlated with changes in bodyweight, a correlation reduced in FR-V mice and absent in ABA-V mice (Fig 5F–G).
Figure 5.
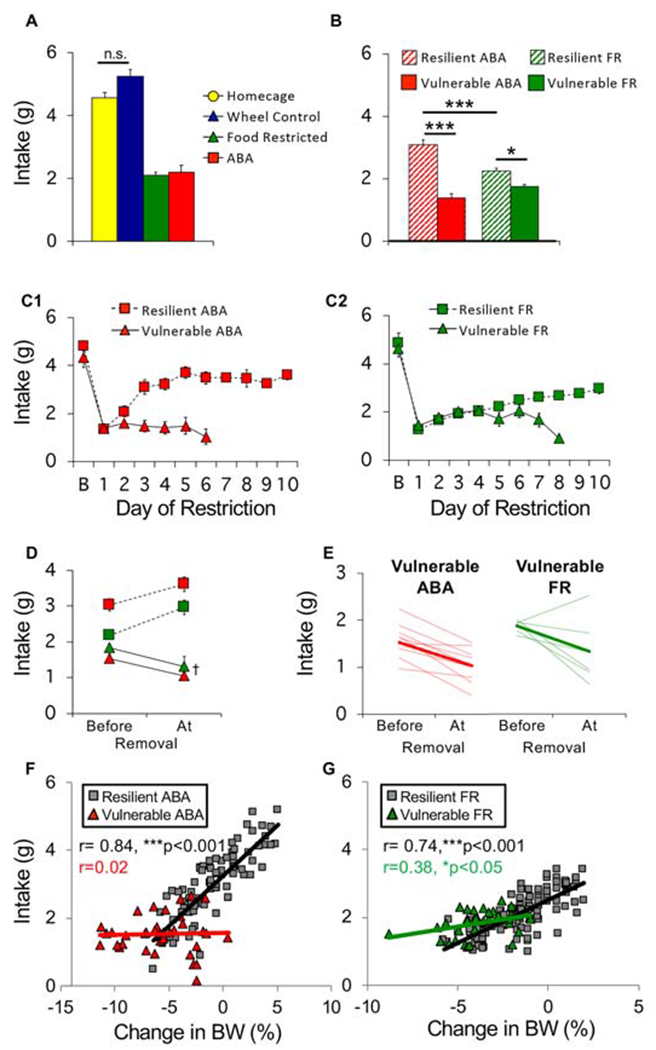
Resilient mice adapt food intake. (A) Average food intake across 10 days of restriction. (B) Average food intake of vulnerable (solid) and resilient (hatched) mice in ABA and food restricted (FR) groups. (C) Daily food intake of (C1) ABA and (C2) FR mice across days of restriction. (D) Average and (E) individual (light traces) food intake of ABA and FR mice prior to removal versus the day of removal (bold lines in E show group average). Correlation between consumption during restricted access to food and changes in body weight the next day for (F) ABA and (G) FR mice. Each symbol represents one animal on one experimental day. n=9 (resilient ABA), n=10 (vulnerable ABA), n=13 (resilient FR), n=6 (vulnerable FR), n=12 (homecage), n=14 (wheel controls). *p<0.05, ***p<0.001, †p<0.01. Error bars, ± SEM.
Vulnerable ABA mice exhibit maladaptive running behavior
There were no baseline differences between ABA-V, ABA-R and non-food restricted wheel controls (WH) in amount (Fig 6A, LME, group, F(2,30) = 0.398, p = 0.67) or circadian distribution of running (Fig 6B1, LME, group x hour, F(2,30) = 2.13, p = 0.13). During food restriction, ABA-R mice exhibited an adaptive reduction in total running, while ABA-V mice continued to run as much as wheel controls (Fig 7A, group, F(2,30)=5.21, p=0.01, ABA-R vs. WH Bonferroni p<0.05). As above, food restriction increased light cycle running, which was modest in ABA-R but dramatic in ABA-V mice (Fig 7B, 7D1, bar graphs: F(2,30) = 38.05, p<0.001, ABA-V vs. ABA-R, Bonferroni p<0.001, ABA-R vs. WH, Bonferroni p=0.05). In contrast, both ABA groups decreased dark cycle running (Fig 7C, 7D2, bar graphs: F(2,30) = 7.76 p < 0.01). These changes reflect a shift in running from dark to light cycle, an effect more pronounced in ABA-V and partially mitigated in ABA-R mice (Fig 7E1–E2, LME, light cycle: ABA phenotype x day, F(1,22) = 10.03, p < 0.01). As observed in adolescent mice, ABA-V mice exhibited an abrupt increase in light-cycle running (Fig 7D1, 7F, bar graphs: F(2,30) = 22.00, p<0.001; ABA-V vs. ABA-R, Bonferroni p<0.001) that preceded removal by 1-3 days (Fig 7G), an effect greatly reduced in ABA-R mice (7D1, 7F, ABA-R vs. WH, Bonferroni p=0.19). Light cycle running positively correlated with weight loss in the ABA-V but not ABA-R group (Fig 7H1). In contrast, dark cycle running positively correlated with weight loss in both groups (Fig 7H2). Like adolescent ABA mice, the ABA-V mice exhibited an altered distribution of circadian activity (Fig 6A), with running that began 2-3 hours into the light cycle (Fig 6B2). In contrast, the ABA-R mice exhibited a modest increase in activity prior to onset of the dark cycle, consistent with adaptive food anticipatory activity (Fig 6B2, ABA phenotype x hour, F(1,17) = 8.92, p < 0.01). These data highlight an association between dysregulated increases in light cycle running and vulnerability in the ABA model.
Figure 6.
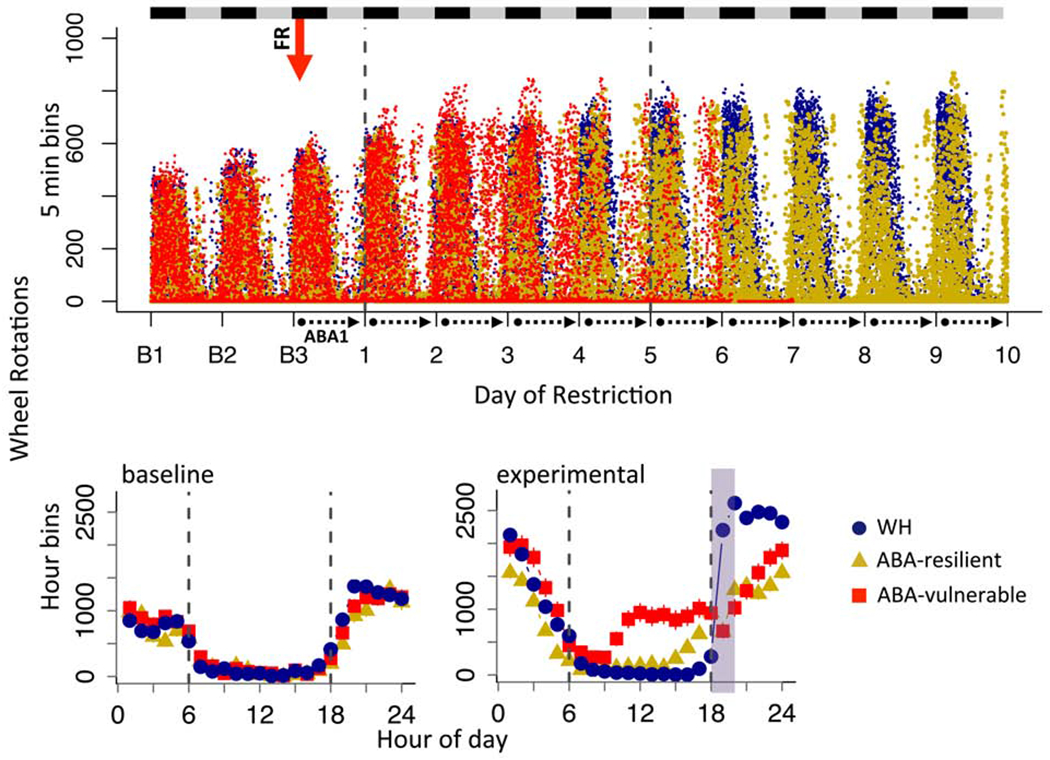
Vulnerable but not resilient ABA mice exhibit altered distribution of circadian running activity. (A) Dot plot of wheel running across days of experiment for all mice in 5 min bins. Black and gray bars indicate dark and light cycles, respectively. Red arrow indicates start of food restriction. Dotted lines demarcate data collection days under ABA. (B) Group averages in hour bins across 24 hour period averaged for (B1) baseline days and (B2) ABA experimental days. Dotted line marks light cycle between 6 (lights on) and 18 (lights off) hrs and green shading indicates when food was available. Error bars, ±SEM.
Figure 7.
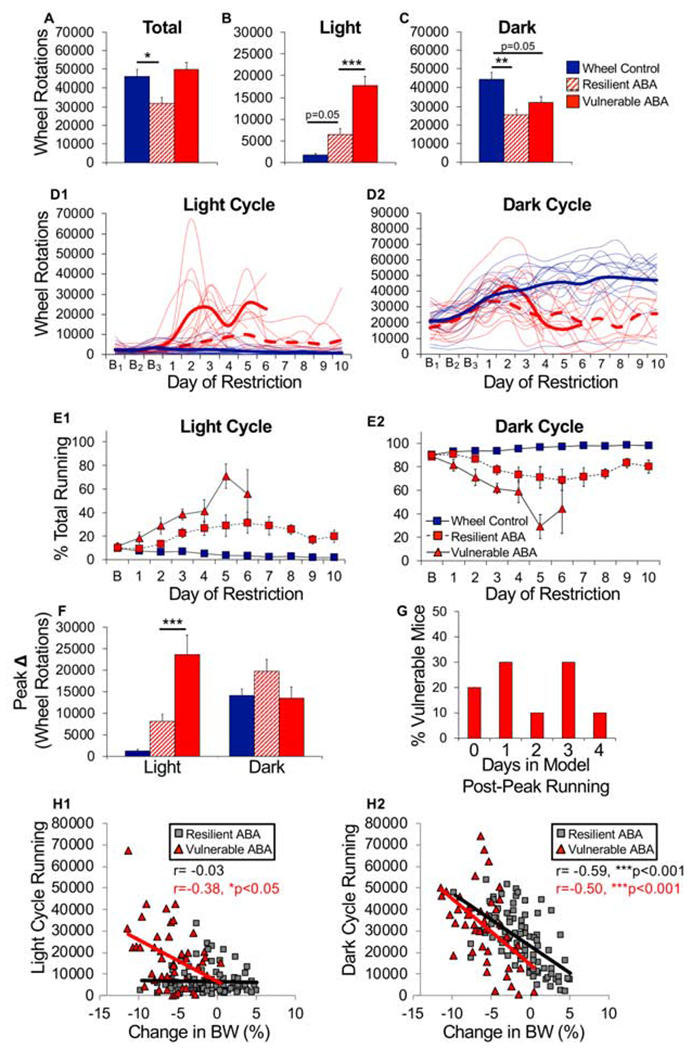
Vulnerable ABA mice exhibit maladaptive running behavior. (A) Total, (B) light cycle and (C) dark cycle wheel running averaged across days of food restriction. (D) Wheel running of individual running (light traces) and group mean (bold traces) across food restriction for (D1) light and (D2) dark cycle running (ABA-V, solid red; ABA-R, dashed red; WH, solid blue). (E) Percentage of total running during the (E1) light or (E2) dark cycle. (F) Maximum increase in wheel running across two consecutive days (averaged by group) during the light and dark cycle. (G) Histogram of number of days mice remained in model after maximum increase in light cycle running. Correlation between light (H1) or dark cycle running (H2) and change in bodyweight for ABA-V and ABA-R mice. Each symbol represents one animal on one experimental day. n=9 (resilient ABA), n=10 (vulnerable ABA), n=14 (wheel controls). *p<0.05; **p<0.01; ***p<0.001. Error bars, ±SEM.
Hyperdopaminergic mice show increased vulnerability to ABA
To test dopamine’s contribution to ABA vulnerability, we used heterozygote DAT-cre mice in which one allele of the dopamine transporter (DAT) was replaced with cre-recombinase (DATcre/+). DAT mRNA (Fig 8A, genotype, F(1,22) = 4.7, p<0.05) and protein (Fig 8B–C, t(27) = 2.06, p=0.049) were reduced without affecting expression of other key dopamine-related genes (Fig 8A, TH, p=0.81; VMAT2, p=0.50; D2, p=0.30), rendering the mice DAT knockdowns (KD). Evoked dopamine release measured by FSCV revealed reduced clearance and increased peak amplitude in KD mice across striatal regions (Fig 8D, genotype effects for single stimulation: tau, F(1,51) = 24.1, p<0.001; peak, F(1,51) = 23.1, p<0.001; burst stimulation: tau, F(1.51) = 27.6, p<0.001; peak, F(1,51) = 23.7, p<0.001; no genotype x region interactions), indicating mild hyperdopaminergia. We compared young adult (PND 62-78, ABA day 1) female KD and WT littermates in ABA, FR, and WH conditions separately .
Figure 8.
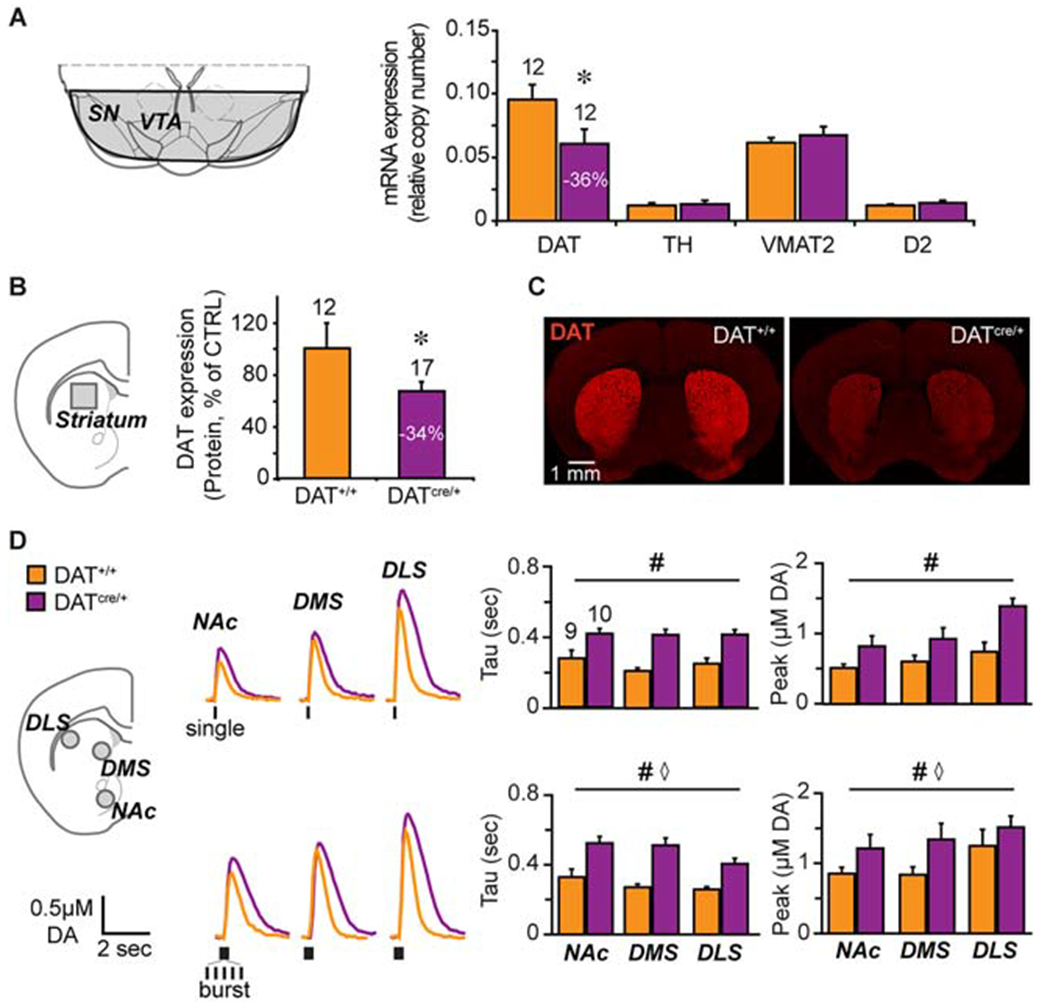
Characterizing hyperdopaminergic DATcre/+ knockdown mice. (A) Schematic - of a coronal brain slice showing the ventral midbrain dissection (shaded area) used for mRNA determinations (left). Relative mRNA expression (right) of dopamine transporter (DAT), tyrosine hydroxylase (TH), vesicular monoamine transporter 2 (VMAT2) and dopamine D2 receptor (D2R) in DATcre/+ and control DAT+/+ littermates. n= 5 (female DAT+/+), n=7 (male DAT+/+), n=5 (female DATcre/+), n=7 (male DATcre/+). (B) Schematic of a coronal brain slice showing the striatal dissection (left, grey square) used for protein determination (left). Relative DAT expression in DATcre/+ compared to DAT+/+ control mice (right). n= 6 (female DAT+/+), n= 6 (male DAT+/+), n=7 (female DATcre/+), n=10 (male DATcre/+). (C) Photomicrographs of coronal slices showing that DAT immunofluorescence in the striatum is diminished in the DATcre/+ mice. (D) Schematic of a coronal slice (left) showing the FSCV recordings sites: NAc core, dorsomedial- and dorsolateral- striatum (grey circles). Representative recordings (middle) of evoked DA release following single (top) or burst (20 Hz, bottom) stimulation (traces) and respective average DA clearance (Tau) and peak DA release (right). Number of animals is indicated above the bars. n=3 (female DAT+/+), n=6 (male DAT+/+), n=4 (female DATcre/+), n=6 (male DATcre/+). *p<0.05, # genotype effect p< 0.001, ⋄ region effect p<0.05. Error bars, ±SEM.
At baseline, genotypes were similar in weight (F(1,72) = 1.07, p = 0.31), food intake (F(1,61) = 0.87, p = 0.35), and light cycle running (F(1,45) = 0.05, p = 0.82). On the first day of baseline, dark cycle running was 36% higher in DATcre/+ than WT mice (t(45) = 2.95, p <0.01). This difference declined to non-significance by the third day of baseline (t(45) = 1.24, p = 0.22), indicating an increased response to novelty rather than sustained elevated running. This interpretation is supported by the lack of a difference between genotypes during 10 additional days in WH controls (Fig S1C–E). Combining data across test conditions revealed no main effect of genotype on weight (LME F(1,48) = 1.51, p = 0.23). Significant genotype interactions are described below.
Under ABA conditions, KD mice exhibited accelerated weight loss (Fig 9A–B, LME, genotype, F(1,28) = 6.64, p < 0.05) and poorer survival (Fig 9C, hazard ratio KD/WT, 2.23, CI 0.91-5.47, p<0.05). All KD mice exhibited the vulnerable phenotype, while some WT mice were resilient (Fig 9C). There were no differences between genotypes in consumption across the experiment (Fig 9D, LME, F(1,20) = 1.16, p = 0.21), but resilient WT mice exhibited a compensatory increase in consumption (Fig 9D, LME, WT across days 4-10, F(1,22) = 6.18, p < 0.05). Vulnerable mice of both genotypes decreased consumption on the day of removal (Fig 9E, time point, F(1,15) = 49.89, p<0.001; time point x genotype, F(1,15) = 3.43, p=0.08). Food intake was not correlated with body weight in KD mice, like the vulnerable mice described above (Fig 9F).
Figure 9.
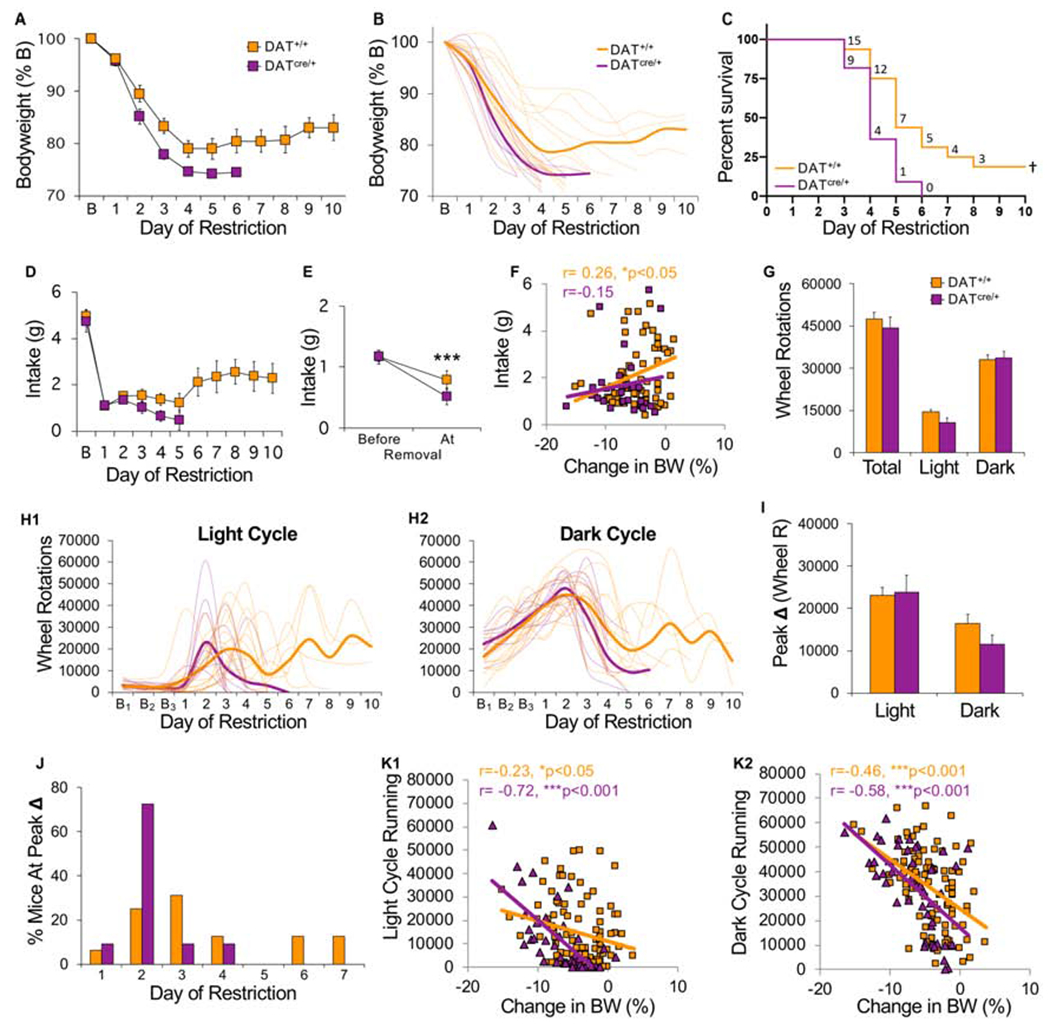
Hyperdopaminergic mice show increased vulnerability to ABA. All mice were tested in the ABA condition. (A) Bodyweight across days of food restriction. (B) Bodyweight of individual mice (light traces) across days of food restriction (bold lines indicate group average). (C) Survival curves for each group. Numbers indicate number of surviving mice that day. (D) Daily food intake across days of restriction. (E) Average food intake prior to removal versus the day of removal. (F) Correlation between consumption during restricted access to food and changes in body weight the next day. Each symbol represents one animal on one experimental day. (G) Average wheel activity across days of food restriction. (H) Wheel running of individual mice (light traces) and group mean (bold traces) across food restriction for (H1) light and (H2) dark cycle running. (I) Maximum increase in wheel running across two consecutive days (averaged by group) during the light and dark cycle. (J) Histogram of the percentage of mice that exhibited peak increase in light cycle running on each day of food restriction. (K) Correlation between light (left) or dark cycle running (right) and change in bodyweight the next day. Each symbol represents one animal on one experimental day. Food intake data: n=12 (DAT+/+), n=8 (DATcre/+); All other data: n=16 (DAT+/+), n=11 (DATcre/+). †Survival curves, p<0.05; ***p<0.001. Error bars, ±SEM.
During ABA, there were no differences between genotypes when light and dark cycle running were averaged across all days of food restriction (Fig 9G, genotype, F(1,25)=0.56, p=0.46). Vulnerable mice of both genotypes exhibited abrupt increases in light cycle running only (Fig 9H1–2), but these occurred earlier in KD mice. Most KD mice (82%) exhibited peak light cycle running by the second day of food restriction, while peak running was more distributed across days in WT mice (Fig 9J, Log-rank, p<0.05). That is, KD mice responded to food restriction with greater increases in light cycle running between days 1 and 2 (t(25) = 2.51, p < 0.05), and more total light cycle running on day 2 (t(25) = 2.02, p = 0.05). When it did occur, peak change in light cycle running was equally high in both groups (Fig 9I, genotype, F(1,25) =0.50, p=0.49), but preceded removal by 1-2 days in most KD mice (73%) and 1-4 days in WT mice (data not shown). Like ABA-V mice above, there was a positive correlation between light cycle running and weight loss that was greater in KD than WT mice (Fig 9K1), while the correlation between dark cycle running and weight loss was similar in both genotypes (Fig 9K2). These data demonstrate that increased dopamine promotes the vulnerable phenotype by accelerating increases in running that occur in response to caloric restriction.
In contrast to ABA, under FR conditions there were no differences between genotypes in weight loss (Fig 10A, LME, genotype, F(1,24) = 0.19, p = 0.66, genotype x day, F(1,23) = 1.20, p = 0.28) or survival (Fig 10B, hazard ratio KD/WT, 1.33, CI 0.41-4.34, p = 0.61). A subset of both KD and WT FR control mice exhibited resilient phenotypes (Fig 10C), with resilient mice of each genotype exhibiting the same pattern of weight stabilization (Fig 10D1–D2, LME phenotype x day F(1,23) = 66.43, p < 0.001; genotype x phenotype x day, F(1,23) = 0.05, p = 0.82) found in the FR-V group (Fig 4C2). Resilient mice consumed more food than vulnerable mice (LME phenotype x day F(1,23) = 10.42, p < 0.01), regardless of genotype (Fig 10E, LME, genotype x phenotype x day, F(1,23) = 1.19, p = 0.28). In both genotypes, consumption correlated with changes in bodyweight, a correlation reduced in vulnerable KD mice (Fig 10G1–G2).
Figure 10.
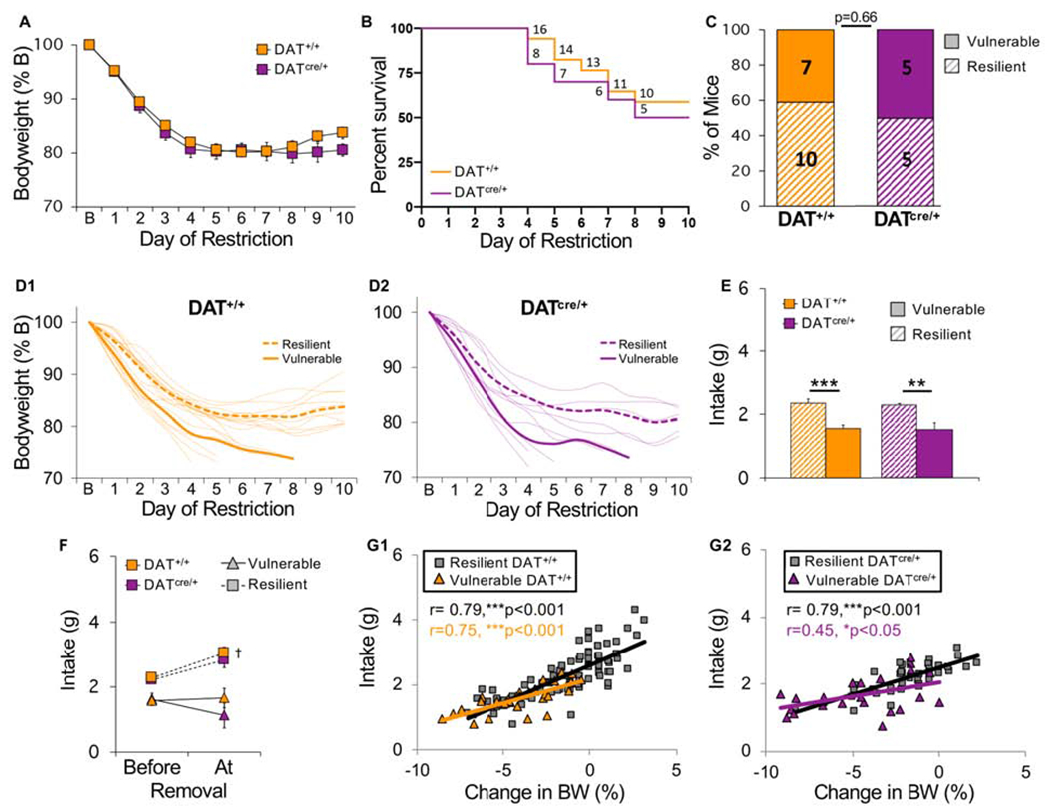
Hyperdopaminergic mice do not show increased vulnerability to food restriction in the absence of a running wheel. All mice were tested in the FR condition. (A) Bodyweight across days of food restriction. (B) Survival curves for each group. Numbers indicate number of surviving mice that day. (C) Percent of vulnerable (solid) and resilient (hatched) mice of each genotype (number of animals is indicated inside bars). (D) Bodyweight of individual (D1) control and (D2) hyperdopaminergic mice (light traces) across days of food restriction. Group averages for resilient (dashed line) and vulnerable (solid line) mice are shown in bold. (E) Average food intake of vulnerable (solid) and resilient (hatched) mice of each genotype. (F) Average food intake prior to removal versus the day of removal. (G) Correlation between consumption during restricted access to food and changes in bodyweight the next day for (G1) control and (G2) hyperdopaminergic mice. Each symbol represents one animal on one experimental day. n=10 (resilient DAT+/+), n=7 (vulnerable DAT+/+), n=5 (resilient DATcre/+), n=5 (vulnerable DATcre/+). ***p<0.001, **p<0.01, †p<0.05 . Error bars, ±SEM.
Under WH conditions, there were no differences between genotypes in bodyweight (Fig S1A), food intake (Fig S1B), or wheel running (Fig S1C–E) during 10 additional days of unlimited food access. This indicates that the observed genotype differences do not result from a generalized, non-specific elevation in activity, but are specific to ABA.
Basal dopamine function is similar in adolescent and young adult mice
To assess whether differences in basal dopamine function could account for differences in vulnerability between adolescent (PND 43) and young adult mice (PND 71), we compared striatal dopamine in female C57BL/6N mice at these ages. In adolescent mice, there was increased synaptic DAT in the nucleus accumbens (t(16) = 3.42, p<0.01) but not the dorsolateral striatum (Fig S2). There were no differences between age groups in synaptic D1 or D2 receptors or cytosolic TH in either region (Fig S2). Fast-scan cyclic voltammetry revealed no difference between adolescents and adults in evoked dopamine release or clearance in the nucleus accumbens (Fig S3). These data suggest age-related shifts in vulnerability do not emerge from developmental changes in basal dopamine function. However, this does not rule out differential dopamine response to environmental factors (e.g., 42).
DISCUSSION
It is unclear why combining diet and exercise leads to AN in some individuals, while most maintain a healthy bodyweight. One approach for studying this is to examine individual differences in ABA (43,44). Here, we demonstrate distinct vulnerable and resilient phenotypes, providing a robust animal model for investigating the physiological and neural adaptations underlying resilience and vulnerability to AN.
ABA resilience is associated with a progressive increase in food intake and decrease in overall wheel activity, leading to weight stabilization. Similar decreases have been reported in older C57BL/6 females (45,46), possibly reflecting an age-related increase in resilience. Moreover, we found that ABA resilient mice actually eat more than FR resilient mice and consequently gain more weight, despite energy expenditure from wheel running. Though counterintuitive, this parallels evidence from clinical studies demonstrating that appropriate exercise may be beneficial in clinical treatment (47–50)
In contrast, vulnerable mice fail to increase their consumption, reducing intake as caloric restriction continues. This failure cannot be attributed to insufficient time to eat, because resilient mice increase consumption in the same 2-hour period of food access. While low food intake correlated with daily weight loss in vulnerable FR mice, this was not found in vulnerable ABA mice. Instead, daily weight loss correlated with amount of wheel activity. Vulnerability did not arise from pre-existing differences in activity, as vulnerable and resilient mice ran similarly at baseline. During food restriction, running dramatically increased throughout the light cycle with vulnerable animals primarily running instead of sleeping. These findings are consistent with prior studies demonstrating that caloric restriction can induce activity that is partially diurnal (51–53) and may reflect exaggerated FAA or a shift in circadian activity to promote foraging (52). Disruption of circadian rhythms have been observed in AN (54). This, as well as hyperactivity could arise from activation of starvation-induced foraging mechanisms that promote physical activity (26,55–57). Individual differences in this response might arise from genetic variation in a human foraging gene (58).
Our studies with hyperdopaminergic mice indicate that increased dopamine promotes vulnerability to ABA by accelerating increases in activity that occur in response to caloric restriction. Vulnerability was not increased in KD mice tested under FR conditions, suggesting a critical interaction between dopamine, physical activity, and vulnerability to caloric restriction. As the KD is global, such effects may arise from increased striatal dopamine or dopamine actions elsewhere. This could include the hypothalamus or prefrontal cortex (PFC), though dopamine reuptake in the PFC is mediated primarily by the norepinephrine transporter rather than DAT (59–61).
ABA studies have primarily used rats with fewer studies testing mice (45,46,62–64). In contrast to the stable bodyweight typically found in FR control rats (35,65,66 but see 67,68), we found that FR control mice can exhibit life-threatening weight loss (supplemental discussion). This suggests that caloric restriction is the primary driver of vulnerability, with wheel access accelerating and augmenting expression of the vulnerable phenotype. Increased homecage activity may have promoted weight loss in the FR mice, which would further suggest a central role for activity in the emergence of the vulnerable phenotype.
Consistent with rat studies (69) and AN in humans (70), we find reduced vulnerability to ABA with age. Smaller animals generally have higher metabolic rates, which in combination with less body mass may increase vulnerability in younger mice. The rapidity of decline in adolescent mice may preclude emergence of a resilient phenotype, while greater initial bodyweight in young adult mice may slow the decline sufficiently for a resilient phenotype to emerge. Extending daily food restriction allowed detection of the vulnerable phenotype in FR mice. Single housing likely exacerbates vulnerability in smaller animals, as the energetic cost of maintaining body temperature is increased without group huddling, compounding increased susceptibility associated with low body mass and high metabolic rate.
Although ABA vulnerability is increased by dopamine, the greater vulnerability we observe in adolescent mice is not due to differences in basal levels of striatal dopamine (Fig S3). Vulnerability during adolescence may instead result from age-related differences in how caloric restriction and exercise induce dopamine adaptations (see below). Alternatively, maturation of dopamine-prefrontal cortical innervation, which continues through young adulthood, may play a role by affecting inhibitory control. The smaller size of the adolescent mouse may be an additional factor.
Dopamine and AN vulnerability
Dopamine has been implicated in AN, but its contribution remains poorly understood. Recovered AN patients exhibit reduced dopamine metabolites, suggesting decreased turnover (71), but increased dopamine D2 receptor binding, reflecting increased D2R expression and/or decreased dopamine transmission (72). Patients often remain symptomatic after recovery (73), making it difficult to determine whether differences in dopamine observed in clinical studies represent pre-existing risk factors or potentially reversible abnormalities induced by starvation (26). Our finding that hyperdopaminergic mice are more vulnerable to ABA suggests enhanced dopamine increases AN risk. However, this does not necessarily mean that vulnerability is mediated by basal or trait differences in dopamine function. In AN, several factors arising from caloric restriction can upregulate dopamine, including increased glucocorticoids (74–78), enhanced insulin sensitivity (79–82), increased ghrelin (83–85) and decreased leptin (86–91). Indeed, studies have linked decreased leptin with hyperactivity (88,91–94 but see 95), with some implicating dopamine (88,91,94). Such adaptations to caloric restriction presumably arise to promote physical activity required to forage for food to obtain needed calories (26), making them potential therapeutic targets for reducing hyperactivity in AN. Dopamine also contributes to thermoregulation (96–98), which is altered in AN (6,99) and ABA (95,100). Hypothermia arising from weight loss may also induce changes in dopamine. Vulnerability to AN might vary due to individual differences in any of these responses to caloric restriction, differences that could result from variation in dopamine genes or genes that modulate dopamine (e.g., leptin, insulin, ghrelin, glucocorticoid, orexin, etc.). Of course, factors independent of dopamine may also modulate vulnerability (e.g. 101).
Dopamine function may change over the course of AN, as observed in addiction where dopamine contributes differently in different stages of the disorder (102–105). Such dynamic changes may account for why a recent study found that pharmacogenetic activation of dopamine protects rats from ABA (65). Artificial activation of dopamine cells could have blocked normally occurring changes in dopamine and progressive adaptations to caloric restriction that underlie ABA, dynamic changes facilitated in our hyperdopaminergic mice. The pharmacogenetic activation of some non-dopaminergic cells may have also contributed to the discrepancy between our findings. The nature of progressive changes in dopamine across AN/ABA is unknown but merits further study.
A robust model of vulnerability and resilience, as reported here, could aid in the discovery of genes that mediate AN risk and/or contribute to the progression of the disorder. Such work may lead to the identification of biomarkers for early diagnosis and the discovery of novel therapeutic targets.
Supplementary Material
KEY RESOURCES TABLE
| Resource Type | Specific Reagent or Resource | Source or Reference | Identifiers | Additional Information |
|---|---|---|---|---|
| Add additional rows as needed for each resource type | Include species and sex when applicable. | Include name of manufacturer, company, repository, individual, or research lab. Include PMID or DOI for references; use “this paper” if new. | Include catalog numbers, stock numbers, database IDs or accession numbers, and/or RRIDs. RRIDs are highly encouraged; search for RRIDs at https://scicrunch.org/resources. | Include any additional information or notes if necessary. |
| Antibody | rabbit polyclonal anti-DAT | Abcam | ab111468 | |
| Antibody | goat polyclongal anti-rabbit Alexa Fluor 594 | Fisher | A32740 | |
| Bacterial or Viral Strain | N/A | |||
| Biological Sample | N/A | |||
| Cell Line | N/A | |||
| Chemical Compound or Drug | QIAzol lysis reagent | Qiagen | 79306 | |
| Commercial Assay Or Kit | WES Separation Module | ProteinSimple | SM-W004 | |
| Commercial Assay Or Kit | Anti Rabbit Detection Module | ProteinSimple | DM-001 | |
| Commercial Assay Or Kit | Total Protein Detection Module | ProteinSimple | DM-TPO1 | |
| Commercial Assay Or Kit | Bicine/CHAPS extraction kit | ProteinSimple | CBC 403 | |
| Commercial Assay Or Kit | Rneasy RNA extraction kit | Qiagen | 74034 | |
| Commercial Assay Or Kit | RNeasy Lipid Tissue Mini Kit | Qiagen | 74804 | |
| Deposited Data; Public Database | N/A | |||
| Genetic Reagent | N/A | |||
| Organism/Strain | mouse, females (wild-type) | Taconic Biosciences | C57BL/6N | |
| Organism/Strain | mouse, females (DAT KD) | Jackson Laboratories | 20080 | |
| Peptide, Recombinant Protein | N/A | |||
| Recombinant DNA | N/A | |||
| Sequence-Based Reagent | N/A | |||
| Software; Algorithm | N/A | |||
| Transfected Construct | N/A | |||
| Other | mouse chow | WF Fisher and Son | Prolab Isopro 3000 5P75 |
ACKNOWLEDGEMENTS
This work was supported by NIDA, DA046058 (JB), NIMH, R21MH114182 (NB), T32MH19970 (AK), US National Institute on Minority Health and Health Disparities of the NIH, G12MD007599 (NB), PSC-CUNY Awards jointly funded by The Professional Staff Congress and The City University of New York (JB & NB) and a Conte Grant, P50 MH086404 (SR).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
FINANCIAL DISCLOSURES
All authors report no biomedical financial interests or potential conflicts of interest.
REFERENCES
- 1.Steinhausen H-C (2002): The Outcome of Anorexia Nervosa in the 20th Century. Am J Psychiatry 159: 1284–1293. [DOI] [PubMed] [Google Scholar]
- 2.Khalsa SS, Portnoff LC, McCurdy-McKinnon D, Feusner JD (2017): What happens after treatment? A systematic review of relapse, remission, and recovery in anorexia nervosa. J Eat Disord 5: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hay P, Mitchison D, Collado AEL, González-Chica DA, Stocks N, Touyz S (2017): Burden and health-related quality of life of eating disorders, including Avoidant/Restrictive Food Intake Disorder (ARFID), in the Australian population. J Eat Disord 5: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fichter MM, Quadflieg N, Crosby RD, Koch S (2017): Long-term outcome of anorexia nervosa: Results from a large clinical longitudinal study: FICHTER et al. Int J Eat Disord 50: 1018–1030. [DOI] [PubMed] [Google Scholar]
- 5.Frank GKW, Shott ME (2016): The Role of Psychotropic Medications in the Management of Anorexia Nervosa: Rationale, Evidence and Future Prospects. CNS Drugs 30: 419–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gull W (1888): Clinical notes: medical, surgical, obstetrical, and therapeutical. Anorexia Nervosa. Lancet 516–517. [Google Scholar]
- 7.Davis C, Katzman DK, Kaptein S, Kirsh C, Brewer H, Kalmbach K, et al. (1997): The prevalence of high-level exercise in the eating disorders: Etiological implications. Compr Psychiatry 38: 321–326. [DOI] [PubMed] [Google Scholar]
- 8.Rizk M, Lalanne C, Berthoz S, Kern L, EVHAN Group, Godart N (2015): Problematic Exercise in Anorexia Nervosa: Testing Potential Risk Factors against Different Definitions ((A. Stengel, editor)). PLOS ONE 10: e0143352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holland LA, Brown TA, Keel PK (2014): Defining features of unhealthy exercise associated with disordered eating and eating disorder diagnoses. Psychol Sport Exerc 15: 116–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cook BJ, Hausenblas HA (2008): The Role of Exercise Dependence for the Relationship between Exercise Behavior and Eating Pathology: Mediator or Moderator? J Health Psychol 13: 495–502. [DOI] [PubMed] [Google Scholar]
- 11.Dittmer N, Jacobi C, Voderholzer U (2018): Compulsive exercise in eating disorders: proposal for a definition and a clinical assessment. J Eat Disord 6: 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schlegl S, Dittmer N, Hoffmann S, Voderholzer U (2018): Self-reported quantity, compulsiveness and motives of exercise in patients with eating disorders and healthy controls: differences and similarities. J Eat Disord 6: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Colleen Stiles-Shields E, Labuschagne Z, Goldschmidt AB, Doyle AC, Grange DL (2012): The use of multiple methods of compensatory behaviors as an indicator of eating disorder severity in treatment-seeking youth. Int J Eat Disord 45: 704–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Belak L, Gianini L, Klein DA, Sazonov E, Keegan K, Neustadt E, et al. (2017): Measurement of fidgeting in patients with anorexia nervosa using a novel shoe-based monitor. Eat Behav 24: 45–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Solenberger SE (2001): Exercise and eating disorders A 3-year inpatient hospital record analysis. Eat Behav 18. [DOI] [PubMed] [Google Scholar]
- 16.Carter JC, Blackmore E, Sutandar-Pinnock K, Woodside DB (2004): Relapse in anorexia nervosa: a survival analysis. Psychol Med 34: 671–679. [DOI] [PubMed] [Google Scholar]
- 17.Strober M, Freeman R, Morrell W (n.d.): The long-term course of severe anorexia nervosa in adolescents: Survival analysis of recovery, relapse, and outcome predictors over 10–15 years in a prospective study. 22. [DOI] [PubMed] [Google Scholar]
- 18.Casper RC, Jabine LN (1996): An eight-year follow-up: Outcome from adolescent compared to adult onset anorexia nervosa. J Youth Adolesc 25: 499–517. [Google Scholar]
- 19.Kostrzewa E, van Elburg AA, Sanders N, Sternheim L, Adan RAH, Kas MJH (2013): Longitudinal Changes in the Physical Activity of Adolescents with Anorexia Nervosa and Their Influence on Body Composition and Leptin Serum Levels after Recovery ((Z. Andrews, editor)). PLoS ONE 8: e78251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kostrzewa E, Eijkemans MJC, Kas MJ (2013): The expression of excessive exercise co-segregates with the risk of developing an eating disorder in women. Psychiatry Res 210: 1123–1128. [DOI] [PubMed] [Google Scholar]
- 21.Davis C, Kennedy SH, Ravelski E, Dionne M (1994): The role of physical activity in the development and maintenance of eating disorders. Psychol Med 24: 957–967. [DOI] [PubMed] [Google Scholar]
- 22.Davis C, Blackmore E, Katzman DK, Fox J (2005): Female adolescents with anorexia nervosa and their parents: a case-control study of exercise attitudes and behaviours. Psychol Med 35: 377–386. [DOI] [PubMed] [Google Scholar]
- 23.Holm-Denoma JM, Scaringi V, Gordon KH, Van Orden KA, Joiner TE (2009): Eating disorder symptoms among undergraduate varsity athletes, club athletes, independent exercisers, and nonexercisers. Int J Eat Disord 42: 47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bratland-Sanda S, Sundgot-Borgen J (2013): Eating disorders in athletes: Overview of prevalence, risk factors and recommendations for prevention and treatment. Eur J Sport Sci 13: 499–508. [DOI] [PubMed] [Google Scholar]
- 25.Smolak L, Murnen SK, Ruble AE (2000): Female athletes and eating problems: A meta-analysis. Int J Eat Disord 27: 371–380. [DOI] [PubMed] [Google Scholar]
- 26.Södersten P, Bergh C, Leon M, Zandian M (2016): Dopamine and anorexia nervosa. Neurosci Biobehav Rev 60: 26–30. [DOI] [PubMed] [Google Scholar]
- 27.O’Hara CB, Campbell IC, Schmidt U (2015): A reward-centred model of anorexia nervosa: A focussed narrative review of the neurological and psychophysiological literature. Neurosci Biobehav Rev 52: 131–152. [DOI] [PubMed] [Google Scholar]
- 28.Frank GKW, DeGuzman MC, Shott ME (2019): Motivation to eat and not to eat – The psycho-biological conflict in anorexia nervosa. Physiol Behav 206: 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berner LA, Brown TA, Lavender JM, Lopez E, Wierenga CE, Kaye WH (2018): Neuroendocrinology of reward in anorexia nervosa and bulimia nervosa: Beyond leptin and ghrelin. Mol Cell Endocrinol S0303720718303137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cowdrey FA, Park RJ, Harmer CJ, McCabe C (2011): Increased Neural Processing of Rewarding and Aversive Food Stimuli in Recovered Anorexia Nervosa. Biol Psychiatry 70: 736–743. [DOI] [PubMed] [Google Scholar]
- 31.Scaife JC, Godier LR, Reinecke A, Harmer CJ, Park RJ (2016): Differential activation of the frontal pole to high vs low calorie foods: The neural basis of food preference in Anorexia Nervosa? Psychiatry Res Neuroimaging 258: 44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kravitz AV, O’Neal TJ, Friend DM (2016): Do Dopaminergic Impairments Underlie Physical Inactivity in People with Obesity? Front Hum Neurosci 10. 10.3389/fnhum.2016.00514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beeler JA, Frazier CRM, Zhuang X (2012): Putting desire on a budget: dopamine and energy expenditure, reconciling reward and resources. Front Integr Neurosci 6. 10.3389/fnint.2012.00049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang S, Tan Y, Zhang J-E, Luo M (2013): Pharmacogenetic activation of midbrain dopaminergic neurons induces hyperactivity. Neurosci Bull 29: 517–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Routtenberg A, Kuznesof AW (1967): Self-starvation of rats living in activity wheels on a restricted feeding schedule. J Comp Physiol Psychol 64: 414–421. [DOI] [PubMed] [Google Scholar]
- 36.Epling WF, Pierce WD, Stefan L (1983): A theory of activity-based anorexia. Int J Eat Disord 3: 27–46. [Google Scholar]
- 37.Gutierrez E (2013): A rat in the labyrinth of anorexia nervosa: Contributions of the activity-based anorexia rodent model to the understanding of anorexia nervosa. Int J Eat Disord 46: 289–301. [DOI] [PubMed] [Google Scholar]
- 38.Zhuang X, Masson J, Gingrich JA, Rayport S, Hen R (2005): Targeted gene expression in dopamine and serotonin neurons of the mouse brain. J Neurosci Methods 143: 27–32. [DOI] [PubMed] [Google Scholar]
- 39.Bates D, Maechler M, Bolkler B, Walker S (2015): Fitting linear mixed-effects models using lme4. J Stat Softw 67: 1–48. [Google Scholar]
- 40.R Core Team (2019): R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing. Retrieved from https://www.R-project.org/ [Google Scholar]
- 41.Kuznetsova A, Brockhoff P, Christensen R (n.d.): lmerTest package: tests in linear mixed effecs models. J Stat Softw 82: 1–26. [Google Scholar]
- 42.Montagud-Romero S, Nunez C, Blanco-Gandia MC, Martinez-Laorden E, Aguilar MA, Navarro-Zaragoza J, et al. (2017): Repeated social defeat and the rewarding effects of cocaine in adult and adolescent mice: dopamine transcription factors, proBDNF signaling pathways, and the TrkB receptor in the mesolimbic system. Psychopharmacology (Berl) 234: 2063–2075. [DOI] [PubMed] [Google Scholar]
- 43.Chen Y-W, Wable GS, Chowdhury TG, Aoki C (2016): Enlargement of Axo-Somatic Contacts Formed by GAD-Immunoreactive Axon Terminals onto Layer V Pyramidal Neurons in the Medial Prefrontal Cortex of Adolescent Female Mice Is Associated with Suppression of Food Restriction-Evoked Hyperactivity and Resilience to Activity-Based Anorexia. Cereb Cortex 26: 2574–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barbarich-Marsteller NC, Underwood MD, Foltin RW, Myers MM, Walsh BT, Barrett JS, Marsteller DA (2013): Identifying novel phenotypes of vulnerability and resistance to activity-based anorexia in adolescent female rats: Vulnerability to Activity-Based Anorexia. Int J Eat Disord 46: 737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gelegen C, Collier DA, Campbell IC, Oppelaar H, van den Heuvel J, Adan RAH, Kas MJH (2007): Difference in susceptibility to activity-based anorexia in two inbred strains of mice. Eur Neuropsychopharmacol 17: 199–205. [DOI] [PubMed] [Google Scholar]
- 46.Gelegen C, van den Heuvel J, Collier DA, Campbell IC, Oppelaar H, Hessel E, Kas MJH (2008): Dopaminergic and brain-derived neurotrophic factor signalling in inbred mice exposed to a restricted feeding schedule. Genes Brain Behav 7: 552–559. [DOI] [PubMed] [Google Scholar]
- 47.Dittmer N, Voderholzer U, von der MQhlen M, Marwitz M, Fumi M, Monch C, et al. (2018): Specialized group intervention for compulsive exercise in inpatients with eating disorders: feasibility and preliminary outcomes. J Eat Disord 6: 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cook BJ, Wonderlich SA, Mitchell JE, Thompson R, Sherman R, Mccallum K (2016): Exercise in Eating Disorders Treatment: Systematic Review and Proposal of Guidelines. Med Sci Sports Exerc 48: 1408–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rizk M, Kern L, Lalanne C, Hanachi M, Melchior J-C, Pichard C, et al. (2018): High-intensity exercise is associated with a better nutritional status in anorexia nervosa. Eur Eat Disord Rev. 10.1002/erv.2661 [DOI] [PubMed] [Google Scholar]
- 50.Schlegel S (2015): The Freiburg sport therapy program for eating disordered outpatients: a pilot study. Eat Weight Disord 9. [DOI] [PubMed] [Google Scholar]
- 51.Challet E (2010): Interactions between light, mealtime and calorie restriction to control daily timing in mammals. J Comp Physiol B 180: 631–644. [DOI] [PubMed] [Google Scholar]
- 52.van der Vinne V, Riede SJ, Gorter JA, Eijer WG, Sellix MT, Menaker M, et al. (2014): Cold and hunger induce diurnality in a nocturnal mammal. Proc Natl Acad Sci 111: 15256–15260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Acosta-Rodriguez VA, de Groot MHM, Rijo-Ferreira F, Green CB, Takahashi JS (2017): Mice under Caloric Restriction Self-Impose a Temporal Restriction of Food Intake as Revealed by an Automated Feeder System. Cell Metab 26: 267–277. e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Menculini G, Brufani F, Bello VD, Moretti P, Tortorella A (2019): Cirdcadian rhythms disruptions and eating disorders: clinical impact and possible psychopathological correlates. Psychiatr Danub 31: 497–502. [PubMed] [Google Scholar]
- 55.Scheurink AJW, Boersma GJ, Nergardh R, Sodersten P (2010): Neurobiology of hyperactivity and reward: Agreeable restlessness in Anorexia Nervosa. Physiol Behav 100: 490–495. [DOI] [PubMed] [Google Scholar]
- 56.Sodersten P, Brodin U, Zandian M, Bergh C (2019): Eating Behavior and the Evolutionary Perspective on Anorexia Nervosa. Front Neurosci 13: 596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guisinger S (2003): Adapted to flee famine: Adding an evolutionary perspective on anorexia nervosa. Psychol Rev 110: 745–761. [DOI] [PubMed] [Google Scholar]
- 58.Struk AA, Mugon J, Huston A, Scholer AA, Stadler G, Higgins ET, et al. (2019): Self-regulation and the foraging gene ( PRKG1 ) in humans. Proc Natl Acad Sci 116: 4434–4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sesack SR, Hawrylak VA, Guido MA, Levey AI (1998): Cellular and subcellular localization of the dopamine transporter in rat cortex. Adv Pharmacol San Diego Calif 42: 171–174. [DOI] [PubMed] [Google Scholar]
- 60.Moron JA, Brockington A, Wise RA, Rocha BA, Hope BT (2002): Dopamine Uptake through the Norepinephrine Transporter in Brain Regions with Low Levels of the Dopamine Transporter: Evidence from Knock-Out Mouse Lines. J Neurosci 22: 389–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mundorf ML, Joseph JD, Austin CM, Caron MG, Wightman RM (2008): Catecholamine release and uptake in the mouse prefrontal cortex: Catecholamines in mouse prefrontal cortex. J Neurochem 79: 130–142. [DOI] [PubMed] [Google Scholar]
- 62.Avraham Y, Hao S, Mendelson S, Berry EM (2001): Tyrosine improves appetite, cognition, and exercise tolerance in activity anorexia. Med Sci Sports Exerc 33: 2104–2110. [DOI] [PubMed] [Google Scholar]
- 63.Klenotich SJ, Seiglie MP, McMurray MS, Roitman JD, Le Grange D, Dugad P, Dulawa SC (2012): Olanzapine, but Not Fluoxetine, Treatment Increases Survival in Activity-Based Anorexia in Mice. Neuropsychopharmacology 37: 1620–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chowdhury TG, Wable GS, Sabaliauskas NA, Aoki C (2013): Adolescent female C57BL/6 mice with vulnerability to activity-based anorexia exhibit weak inhibitory input onto hippocampal CA1 pyramidal cells. Neuroscience 241: 250–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Foldi CJ, Milton LK, Oldfield BJ (2017): The Role of Mesolimbic Reward Neurocircuitry in Prevention and Rescue of the Activity-Based Anorexia (ABA) Phenotype in Rats. Neuropsychopharmacology. 10.1038/npp.2017.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Scharner S, Prinz P, Goebel-Stengel M, Kobelt P, Hofmann T, Rose M, Stengel A (2016): Activity-Based Anorexia Reduces Body Weight without Inducing a Separate Food Intake Microstructure or Activity Phenotype in Female Rats—Mediation via an Activation of Distinct Brain Nuclei. Front Neurosci 10. 10.3389/fnins.2016.00475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Aoki C, Sabaliauskas N, Chowdhury T, Min J-Y, Colacino AR, Laurino K, Barbarich-Marsteller NC (2012): Adolescent female rats exhibiting activity-based anorexia express elevated levels of GABAA receptor α4 and δ subunits at the plasma membrane of hippocampal CA1 spines. Synapse 66: 391–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Aoki C, Chowdhury TG, Wable GS, Chen Y-W (2017): Synaptic changes in the hippocampus of adolescent female rodents associated with resilience to anxiety and suppression of food restriction-evoked hyperactivity in an animal model for anorexia nervosa. Brain Res 1654: 102–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Woods SC, Routtenberg A (1971): “Self-starvation ” in activity wheels: developmental and chlorpromazine interactions. J Comp Physiol Psychol 76: 84–93. [DOI] [PubMed] [Google Scholar]
- 70.Bulik CM (2002): Eating disorders in adolescents and young adults. Child Adolesc Psychiatr Clin N Am 11: 201–218. [DOI] [PubMed] [Google Scholar]
- 71.Kaye W (1999): Altered Dopamine Activity after Recovery from Restricting-Type Anorexia Nervosa. Neuropsychopharmacology 21: 503–506. [DOI] [PubMed] [Google Scholar]
- 72.Frank GK, Bailer UF, Henry Se, Drevets W, Meltzer CC, Price JC, et al. (2005): Increased Dopamine D2/D3 Receptor Binding After Recovery from Anorexia Nervosa Measured by Positron Emission Tomography and [11C]Raclopride. Biol Psychiatry 58: 908–912. [DOI] [PubMed] [Google Scholar]
- 73.Wagner A, Barbarich-Marsteller NC, Frank GK, Bailer UF, Wonderlich SA, Crosby RD, et al. (2006): Personality traits after recovery from eating disorders: Do subtypes differ? Int J Eat Disord 39: 276–284. [DOI] [PubMed] [Google Scholar]
- 74.Holly EN, DeBold JF, Miczek KA (2015): Increased mesocorticolimbic dopamine during acute and repeated social defeat stress: modulation by corticotropin releasing factor receptors in the ventral tegmental area. Psychopharmacology (Berl) 232: 4469–4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wanat MJ, Hopf FW, Stuber GD, Phillips PEM, Bonci A (2008): Corticotropin-releasing factor increases mouse ventral tegmental area dopamine neuron firing through a protein kinase C-dependent enhancement of I h: CRF increases VTA dopamine neuron firing. J Physiol 586: 2157–2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Graf EN, Wheeler RA, Baker DA, Ebben AL, Hill JE, McReynolds JR, et al. (2013): Corticosterone Acts in the Nucleus Accumbens to Enhance Dopamine Signaling and Potentiate Reinstatement of Cocaine Seeking. J Neurosci 33: 11800–11810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Saal D, Dong Y, Bonci A, Malenka RC (2003): Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron 37: 577–582. [DOI] [PubMed] [Google Scholar]
- 78.Wei N-L, Quan Z-F, Zhao T, Yu X-D, Xie Q, Zeng J, et al. (2019): Chronic stress increases susceptibility to food addiction by increasing the levels of DR2 and MOR in the nucleus accumbens. Neuropsychiatr Dis Treat Volume 15: 1211–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Caravaggio F, Borlido C, Hahn M, Feng Z, Fervaha G, Gerretsen P, et al. (2015): Reduced Insulin Sensitivity Is Related to Less Endogenous Dopamine at D2/3 Receptors in the Ventral Striatum of Healthy Nonobese Humans. Int J Neuropsychopharmacol 18: pyv014–pyv014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cai W, Xue C, Sakaguchi M, Konishi M, Shirazian A, Ferris HA, et al. (2018): Insulin regulates astrocyte gliotransmission and modulates behavior. J Clin Invest 128: 2914–2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Konner AC, Hess S, Tovar S, Mesaros A, Sanchez-Lasheras C, Evers N, et al. (2011): Role for Insulin Signaling in Catecholaminergic Neurons in Control of Energy Homeostasis. Cell Metab 13: 720–728. [DOI] [PubMed] [Google Scholar]
- 82.Stouffer MA, Woods CA, Patel JC, Lee CR, Witkovsky P, Bao L, et al. (2015): Insulin enhances striatal dopamine release by activating cholinergic interneurons and thereby signals reward. Nat Commun 6: 8543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cone JJ, Roitman JD, Roitman MF (2015): Ghrelin regulates phasic dopamine and nucleus accumbens signaling evoked by food-predictive stimuli. J Neurochem 133: 844–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jerlhag E (2008): Systemic administration of ghrelin induces conditioned place preference and stimulates accumbal dopamine. Addict Biol 13: 358–363. [DOI] [PubMed] [Google Scholar]
- 85.Jerlhag E, Egecioglu E, Dickson SL, Douhan A, Svensson L, Engel JA (2007): Ghrelin administration into tegmental areas stimulates locomotor activity and increases extracellular concentration of dopamine in the nucleus accumbens. Addict Biol 12: 6–16. [DOI] [PubMed] [Google Scholar]
- 86.KrQgel U, Schraft T, Kittner H, Kiess W, Illes P (2003): Basal and feeding-evoked dopamine release in the rat nucleus accumbens is depressed by leptin. Eur J Pharmacol 482: 185–187. [DOI] [PubMed] [Google Scholar]
- 87.Hommel JD, Trinko R, Sears RM, Georgescu D, Liu ZW, Gau XB, et al. (2006): Leptin receptor signaling in midbrain dopamine neurons regulates feeding. Neuron 51: 678–680. [DOI] [PubMed] [Google Scholar]
- 88.Verhagen LAW, Luijendijk MCM, Adan RAH (2011): Leptin reduces hyperactivity in an animal model for anorexia nervosa via the ventral tegmental area. Eur Neuropsychopharmacol 21: 274–281. [DOI] [PubMed] [Google Scholar]
- 89.Davis JF, Choi DL, Benoit SC (2010): Insulin, leptin and reward. Trends Endocrinol Metab 21: 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fulton S, Pissios P, Manchon RP, Stiles L, Frank L, Pothos EN, et al. (2006): Leptin Regulation of the Mesoaccumbens Dopamine Pathway. Neuron 51: 811–822. [DOI] [PubMed] [Google Scholar]
- 91.Fernandes MFA, Matthys D, Hryhorczuk C, Sharma S, Mogra S, Alquier T, Fulton S (2015): Leptin Suppresses the Rewarding Effects of Running via STAT3 Signaling in Dopamine Neurons. Cell Metab 22: 741–749. [DOI] [PubMed] [Google Scholar]
- 92.Exner C, Hebebrand J, Remschmidt H, Wewetzer C, Ziegler A, Herpertz S, et al. (2000): Leptin suppresses semi-starvation induced hyperactivity in rats: implications for anorexia nervosa. Mol Psychiatry 5: 476–481. [DOI] [PubMed] [Google Scholar]
- 93.Hillebrand JJG, Koeners MP, de Rijke CE, Kas MJH, Adan RAH (2005): Leptin Treatment in Activity-Based Anorexia. Biol Psychiatry 58: 165–171. [DOI] [PubMed] [Google Scholar]
- 94.Evans MC, Kumar NS, Inglis MA, Anderson GM (2018): Leptin and insulin do not exert redundant control of metabolic or emotive function via dopamine neurons. Horm Behav 106: 93–104. [DOI] [PubMed] [Google Scholar]
- 95.Fraga A, Carreira MC, Gonzalez-Izquierdo A, Dieguez C, Lopez M, Gutierrez E (2020): Temperature but not leptin prevents semi-starvation induced hyperactivity in rats: implications for anorexia nervosa treatment. Sci Rep 10: 5300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Folgueira C, Beiroa D, Porteiro B, Duquenne M, Puighermanal E, Fondevila MF, et al. (2019): Hypothalamic dopamine signalling regulates brown fat thermogenesis. Nat Metab 1: 811–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Balthazar CH, Leite LHR, Ribeiro RMM, Soares DD, Coimbra CC (2010): Effects of blockade of central dopamine D1 and D2 receptors on thermoregulation, metabolic rate and running performance. Pharmacol Rep 62: 54–61. [DOI] [PubMed] [Google Scholar]
- 98.Hasegawa H, Meeusen R, Sarre S, Diltoer M, Piacentini MF, Michotte Y (2005): Acute dopamine/norepinephrine reuptake inhibition increases brain and core temperature in rats. J Appl Physiol 99: 1397–1401. [DOI] [PubMed] [Google Scholar]
- 99.Nishita JK, Knopes KD, Ellinwood EH Jr, Rockwell WJK (1986): Hypothermia and abnormalities in thermoregulation in patients with anorexia nervosa. Int J Eat Disord 5: 713–725. [Google Scholar]
- 100.Gutierrez E, Vazquez R, Boakes RA (2002): Activity-based anorexia: Ambient temperature has been a neglected factor. Psychon Bull Rev 9: 239–249. [DOI] [PubMed] [Google Scholar]
- 101.Aoki C, Chen Y-W, Chowdhury TG, Piper W (2018): α4βδ-GABAA receptors in dorsal hippocampal CA1 of adolescent female rats traffic to the plasma membrane of dendritic spines following voluntary exercise and contribute to protection of animals from activity-based anorexia through localization at excitator. J Neurosci Res 96: 1450–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Caprioloi D, Calu D, Shaham Y (2014): Loss of phasic dopamine: a new addiction marker? Nat Neurosci 17: 644–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Carelli RM, West EA (2014): When a good taste turns bad: Neural mechanisms underlying the emergence of negative affect and associated natural reward devaluation by cocaine. Neuropharmacology 76: 360–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rose EJ, Ross TJ, Salmeron BJ, Lee M, Shakleya DM, Huestis M, Stein EA (2012): Chronic Exposure to Nicotine Is Associated with Reduced Reward-Related Activity in the Striatum but not the Midbrain. Biol Psychiatry 71: 206–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Willuhn I, Burgeno LM, Groblewski PA, Phillips PEM (2014): Excessive cocaine use results from decreased phasic dopamine signaling in the striatum. Nat Neurosci 17: 704–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.