

Abstract
Skeletal muscle regeneration that follows acute injury is strongly influenced by interactions with immune cells that invade and proliferate in the damaged tissue. Discoveries over the past 20 years have identified many of the key mechanisms through which myeloid cells, especially macrophages, regulate muscle regeneration. In addition, lymphoid cells that include CD8+ T-cells and regulatory T-cells also significantly affect the course of muscle regeneration. During aging, the regenerative capacity of skeletal muscle declines, which can contribute to progressive loss of muscle mass and function. Those age-related reductions in muscle regeneration are accompanied by systemic, age-related changes in the immune system, that affect many of the myeloid and lymphoid cell populations that can influence muscle regeneration. In this review, we present recent discoveries that indicate that aging of the immune system contributes to the diminished regenerative capacity of aging muscle. Intrinsic, age-related changes in immune cells modify their expression of factors that affect the function of a population of muscle stem cells, called satellite cells, that are necessary for normal muscle regeneration. For example, age-related reductions in the expression of growth differentiation factor-3 (GDF3) or CXCL10 by macrophages negatively affect adult myogenesis, by disrupting regulatory interactions between macrophages and satellite cells. Those changes contribute to a reduction in the numbers and myogenic capacity of satellite cells in old muscle, which reduces their ability to restore damaged muscle. In addition, aging produces changes in the expression of molecules that regulate the inflammatory response to injured muscle, which also contributes to age-related defects in muscle regeneration. For example, age-related increases in the production of osteopontin by macrophages disrupts the normal inflammatory response to muscle injury, resulting in regenerative defects. These nascent findings represent the beginning of a newly-developing field of investigation into mechanisms through which aging of the immune system affects muscle regeneration.
1. Introduction.
With the possible exception of wisdom, biological functions progressively and inevitably decline beginning in late adulthood. In some cases, age-related changes can be rather abrupt (e.g., aging of hair follicles) or can be associated with a gradual, progressive loss of tissue that accompanies decline of function (e.g., aging of the brain; Skullerud, 1985). Regardless of the variable course of lost functions during aging, the progressive and detrimental changes in the expression of genes that regulate normal tissue homeostasis contribute significantly to aging. Those genetic changes result to some extent from an accumulation of genetic mutations that occur over the lifespan (Moskalev et al., 2013). However, the rate, frequency and magnitude of changes in gene expression are also strongly influenced by interactions of the aging tissue with its environment, including the external environment inhabited by the aging organism and the internal environment in which the aging tissue interacts with other cells (Jaenisch and Bird, 2003; Lopez-Otin et al., 2013).
Age-related changes in muscle are obvious, characterized by a loss of tissue mass and a decline in tissue function, such as reduction of maximum force production. Muscle strength tends to peak between the second and third decades and remains the same until about 45 to 50 years of age in men. Losses then begin to occur at the rate of approximately 12% to 15% per decade until the eighth decade (Hurley, 1995) (Figure 1). In addition, the ability of muscle to respond to the environment declines dramatically during aging. Exercising, elderly humans typically show less muscle hypertrophy than similarly-trained young subjects (Bickel et al., 2011). Likewise in mice, treadmill training in old mice produces 18% less increase in soleus mass than occurs in young mice (Soffe et al., 2016).
Figure 1.

Approximate, relative timelines for mice, rats and humans.
The loss of muscle mass and function during aging, called sarcopenia, reflects to some extent the gradual shift of muscle to a negative protein balance. However, muscle also experiences a progressive, age-related loss of its ability to regenerate following tissue damage (Guttmann and Carlson, 1976; Zacks and Sheff, 1982; Brooks and Faulkner, 1988; Carlson and Faulkner, 1989, 1996; Musaro et al., 2001), which may contribute age-related defects in muscle (Figure 2). Although young, injured muscle can recover to pre-injury muscle mass and function within weeks of an acute injury, the same injury in old muscle can lead to prolonged and perhaps permanent loss of muscle mass and function. This suggests that some of the progressive loss of muscle mass and function during aging could result from accumulated episodes of incomplete muscle regeneration that occur throughout adulthood and aging. Those age-related defects in muscle repair mechanisms can be intrinsic to the muscle itself or can result from age-related changes in regulatory systems that are external to muscle (recently reviewed by Munoz-Canoves et al., 2020).
Figure 2.

Time course of muscle regeneration after intramuscular BaCl2 injection. Hematoxylin-stained cross-sections of TA muscles from 3-month-old (A-C) and 24-month-old (D-F) mice, either non-injured (A, D), or 3-dpi (B, E) or 7-dpi (C, F). Representative fibers with central nuclei (white arrows) and areas of inflammation or edema between damaged fibers (*) are indicated. Note that 3-month-old muscle at 7-dpi shows numerous, relatively-large, regenerating fibers that contain multiple, central nuclei and few inflammatory cells between fibers (C). Muscle from 24-month-old mice at 7-dpi shows fewer and smaller central-nucleated fibers and more numerous inflammatory cells between fibers. Bars = 120 μm.
In this review, we present evidence that age-related changes in the immune system contribute to reductions in the regenerative capacity of muscle during aging. Research over the last few decades has shown the important role of the immune system in regulating muscle regeneration following acute injury and disease, primarily in young, adult animals (reviewed by Tidball, 2017). Other investigations have established that complex, age-related changes in the immune system can fundamentally modify the roles it plays, not only in responding to injury or disease, but also in maintaining normal function in other cells and tissues (reviewed by Zmora et al., 2017). The intersection of these fields has led to new insights into how aging of the immune system can influence muscle regeneration. Here, we first present an overview of key features of the process of muscle regeneration that follow injury and then provide an overview of the immune cells and molecules that influence muscle regeneration. We then review the age-related changes in specific myeloid and lymphoid cell populations that contribute to regenerative defects in aging muscle. Finally, we summarize findings of investigations that suggest the possibility that some of the beneficial effects of exercise on regeneration of aging muscle may be attributable in part to influences of exercise on the immune system.
2. Skeletal muscle regeneration following acute injury.
Muscle regeneration following damage relies on adult myogenesis, a developmental program that is similar, but not identical, to processes that drive myogenesis during embryonic development (Lepper et al., 2009). Several superb, recent reviews comprehensively present the current status of our knowledge of cellular, molecular and genetic controls of adult myogenesis (e.g., Dumont et al., 2015; Giordani et al., 2018). Here, we summarize some of the basic, fundamental features of adult myogenesis during muscle regeneration that pertain to the following discussions of the regeneration of aging muscle.
Adult myogenesis during muscle regeneration relies on activation of a population of muscle stem cells, called satellite cells, that normally reside in a quiescent state on the surface of muscle fibers, beneath the basal lamina that ensheathes each muscle fiber (Mauro, 1961; Zammit et al., 2002) (Figure 3). The quiescent state of satellite cells is maintained by the influence of the transcription factor Pax7, which represses the expression of transcripts required for the proliferation and differentiation of myogenic cells. Muscle damage that results from acute injury, intense exercise, or some diseases causes the release of factors from both muscle and non-muscle cells that leads to satellite cell activation. Following activation, satellite cells may escape the basal lamina sheath, enter the cell cycle, reduce the expression of Pax7 and then activate the expression of other muscle-specific transcription factors, such as MyoD, that participate in regulating changes in gene expression associated with activation (Allen and Boxhorn, 1989; Fuchtbauer and Westphal 1992; Grounds et al., 1992; Cornelison and Wold, 1997; Cornelison et al., 2000; Miller et al., 2000; Tatsumi et al., 2002; Charge and Rudnicki, 2004; Gattazzo et al., 2020). Thus, Pax7+/MyoD− myogenic cells are quiescent satellite cells; Pax7+/MyoD+ cells are activated satellite cells or recently activated myogenic cells that have escaped their satellite cell localization (Figure 4).
Figure 3.
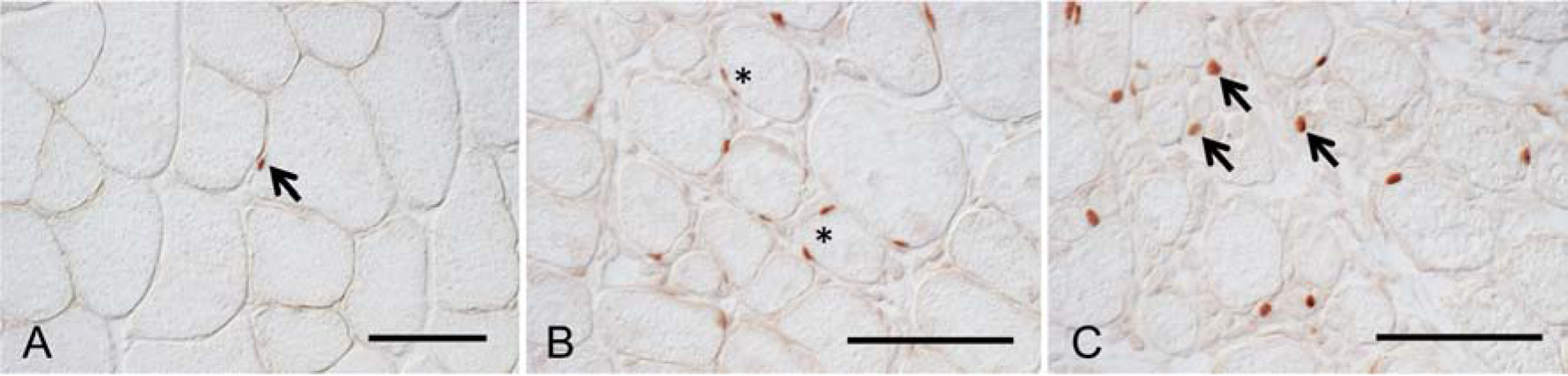
Pax7-expressing cells in non-injured (A) and BaCl2-injured TA muscles at 7-dpi (B, C) from 6-month-old mice. A. In non-injured muscle, Pax7+ cells satellite cells appear at relatively-low numbers in tissue sections, flattened on the surface of healthy muscle fibers. An anti-Pax7-labeled cell is stained red (arrow). B: At 7-dpi, areas of relatively little muscle damage show elevated numbers of satellite cells located on the surface of muscle fibers. Some fibers are associated with more than one satellite cell at the fiber surface (*), indicating satellite cell proliferation at the fiber surface. C: At 7-dpi, areas of more extensive muscle damage show elevated numbers of satellite cells, many of which have left the surface of muscle fibers, to occupy the extracellular space (arrows), where they may undergo further differentiation and fusion to form myotubes. Bars = 50 μm.
Figure 4.
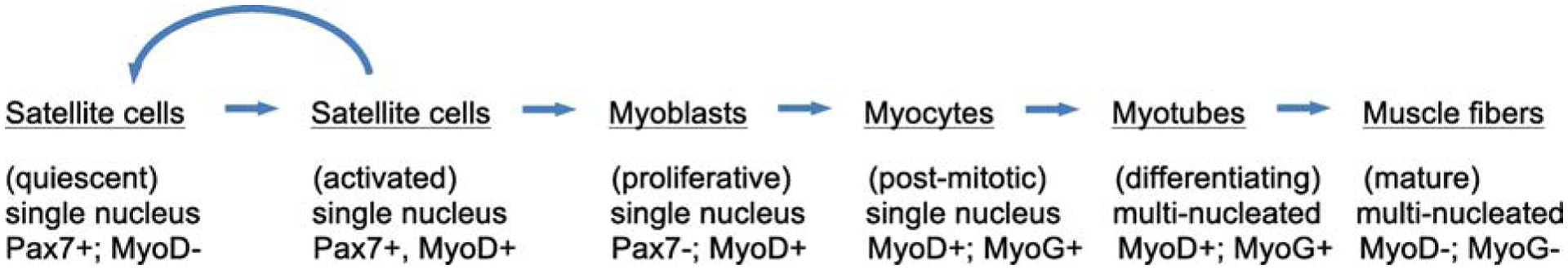
General overview of the stages of satellite cell activation and muscle cell differentiation and maturation following acute injury. Activated satellite cells can either return to quiescence or proceed along a course of differentiation to contribute to mature muscle fibers. A few of the key transcription factors that are expressed at different stages of adult myogenesis are indicated.
Activated satellite cells then proliferate and their daughter cells proceed to permanently withdraw from the cell cycle and continue differentiation, or they can return to quiescence and rejoin the satellite cell population (Halevy et al., 2004; Olguin and Olwin 2004; Zammit et al., 2004, 2006). The frequency distribution of these alternative developmental fates can profoundly affect the long-term health of muscle; if too few activated cells return to the satellite cell pool and if other potential sources of satellite cells such as mesoangioblasts are insufficient (Lolmede et al., 2009), the number of satellite cells will decline over time, leading to a reduction of the future regenerative capacity of muscle (Sambasivan et al., 2011).
Activated satellite cells that become differentiated myogenic cells exhibit a sequential change in the expression of muscle-specific transcription factors, called myogenic regulatory factors (MRFs), that regulate the course of muscle differentiation. Myogenin is an especially important MRF (Hasty et al., 1993) that is expressed by post-mitotic, myogenic cells entering terminal-differentiation and then fuse with other myogenic cells to generate multinucleated myotubes (Creuzet et al., 1998). The myotubes continue to increase in volume as myonuclear accretion progresses and differentiation proceeds, leading to tremendously elevated expression of contractile proteins and other muscle-specific proteins. Myonuclei lie along the central axis of the myotubes throughout myotube growth and differentiation, where they remain through late stages of development. As differentiation is completed, the central nuclei migrate to the periphery of fully-mature muscle fibers. Each fully-differentiated muscle fiber can range in size from hundreds to tens of thousands of micrometers in length and have diameters that exceed 50 micrometers and is occupied by hundreds of myonuclei. The location of nuclei at later stages of muscle growth and differentiation enable morphological identification of regenerating muscle fibers by their central nucleation (Schmalbruch, 1976) (Figure 2).
Each stage of myogenesis is strongly influenced by soluble factors, including cytokines, chemokines and growth factors that can be produced at high levels by immune cells. For example, interferon-gamma (IFNγ), interleukin-6 (IL6), and tumor necrosis factor (TNFα) are cytokines that are vigorously synthesized and secreted by immune cells, as well as some non-immune cells, and each can have powerful effects on myogenesis. IFNγ can serve as a potent regulator of both proliferation and differentiation of myogenic cells (Kalovidouris et al., 1993; Kelic et al., 1993). IL6 also demonstrates pleiotropic effects on myogenic cells, influencing both proliferation and differentiation, as well as muscle fiber growth (Haddad et al., 2005; Toth et al., 2011; Hoene et al., 2013; Muñoz-Cánoves et al., 2013). TNFα can maintain satellite cells in early proliferative stages of myogenesis, inhibit differentiation and affect muscle cell fusion (Guttridge et al., 1999; Langen et al., 2001, 2004; Li 2003; Wang et al., 2018). In addition, insulin-like growth factor-1 (IGF1) can be secreted at high levels by immune and non-immune cells, and has strong effects on muscle cell proliferation, differentiation and growth of mature muscle fibers (Jennische et al., 1987; Allen and Boxhorn, 1989; Palmer et al., 1997). Thus, every step of the course of myogenesis can be greatly influenced by inflammation.
3. Inflammation of regenerating muscle.
As with other acutely-injured tissues, skeletal muscle experiences a rapid invasion of leukocytes that are associated with the removal of damaged tissue and with modulating the repair, regeneration and regrowth of the injured tissue. Although recent reviews have addressed in depth the mechanisms through which the immune system can influence regeneration of adult muscle (Tidball, 2017), key aspects of those mechanisms that are important for understanding the influence of aging of the immune system on muscle regeneration are summarized here.
The presence of large numbers of inflammatory cells at sites of regeneration of injured muscle has been interpreted as indicating a role for the immune system in muscle regeneration for over 120 years (Volkmann, 1893). Although nearly all of the leukocytes that appear in acutely-injured muscle are myeloid cells, a much smaller population of lymphoid cells in the inflammatory infiltrate can also have significant, modulatory effects on regeneration. Most immune cells accumulate in the injured muscle as a consequence of invasion, not by proliferation of immune cells residing in the injured muscle; mice that were pulse labeled with tritiated thymidine at the time of acute muscle injury contained few labeled cells in the injured muscle at 2 days post-injury (dpi) (Bintliff and Walker,1960). Interestingly, the same investigators found that if the pulse labeling were performed 2.5 days before muscle injury, most inflammatory cells in the muscle at 2 dpi were radiolabeled. The observations supported the conclusion that proliferative, hematopoietic stem cells (HSCs) in the bone marrow at 2.5 days before muscle injury were the primary source of inflammatory cells located in the regenerative muscle at 2 dpi (Figure 5). Despite the prevalence of invasive leukocytes in regenerating muscle, immune cells that reside in healthy muscle also play an important role in the response of muscle to injury, by releasing chemokines that play a significant role in attracting the invading leukocytes (Brigitte et al., 2010),
Figure 5.

General overview of the stages of hematopoiesis that give rise to myeloid and lymphoid cells that can influence muscle regeneration. Hematopoietic stem cells that reside in the bone marrow enter either the myeloid or lymphoid cell lineage by differentiating to become either common myeloid progenitors (CMPs) or common lymphoid progenitors (CLPs). CLPs then further differentiate to enter either the B-cell or T-cell lineage. B-cells are not known to have any direct influences on adult myogenesis. T-cells then differentiate to become CD8+ or CD4+ T-cells, which have direct effects on muscle regeneration. CMPs differentiate to become granulocyte progenitors (GPs) or monocyte progenitors (MPs). MPs then further differentiate to monocytes and then become macrophages, which have the most prominent influence on muscle regeneration of all immune cell populations.
Neutrophils.
The initial population of leukocytes that accumulate in injured muscles is predominantly neutrophils, which are typically identified by their expression of Ly6G and CD11b, and by their characteristic, multi-lobed nucleus. Typically, neutrophil numbers are significantly elevated in muscle within 1 to 6 hours of injury and their numbers rapidly decline by 12- to 24-hours post-injury (Fielding et al., 1993; Tidball, 2005). Neutrophil functions in injured and regenerating muscle largely reflect their specialized roles in rapidly producing high levels of free radicals, phagocytosis of cellular debris caused by tissue damage and release of proinflammatory cytokines and chemokines that recruit other leukocyte populations that directly promote muscle regeneration (Table 1). Despite the apparent benefits of those functions, the net effect of neutrophil accumulation in acutely-injured muscle is typically to exacerbate damage. For example, experimental depletion of neutrophil numbers reduces muscle damage that is caused by muscle ischemia followed by reperfusion, or by forced, lengthening contractions, or by exhaustive exercise (Korthuis et al., 1988; Yokota et al., 1989; Carden et al., 1990; Kyriakides et al., 1999; Lockhart and Brooks, 2008; Kawanishi et al., 2016). In addition, genetic ablation of enzymes in neutrophils that generate free radicals (gp91phox or myeloperoxidase) can greatly reduce the extent of muscle fiber damage caused by modified muscle use (Nguyen and Tidball, 2003; Nguyen et al., 2005). Although many of the injury-exacerbating actions of neutrophils in damaged muscle are attributable to direct actions of free radicals on muscle fibers, they may also have indirect effects by increasing the cytotoxicity of other leukocyte populations (Nguyen et al., 2003). Surprisingly, the possibility that aging affects the function of neutrophils in injured muscle remains unexplored, despite the occurrence of age-related reductions in neutrophil functions, such as diminished phagocytosis and free radical production, that occur in neutrophils in vitro and in other, aging tissues (Fulop et al., 2004; Tortorella et al., 2007).
Table 1.
Aging modulates the expression in skeletal muscle of molecules that influence muscle inflammation and regeneration.
| Molecule | Species | Net change with age | Potential source in muscle | Effects on muscle regeneration | Effect on muscle inflammation | References |
|---|---|---|---|---|---|---|
| TNFα | human, mouse, rat | increase | macrophages, neutrophils, muscle cells, endothelial cells, fibroblasts | promotes muscle cell proliferation, inhibits myoblast differentiation, promotes protein catabolism, reduces protein synthesis rate, chemotactic recruitment of muscle cells to lesions | stimulates production of Th1 cytokines, promotes the M1 macrophage phenotype | Greiwe et al., 2001; van der Poel et al., 2011; Wang et al., 2015, 2018; Wehling-Henricks et al., 2018; Collins and Grounds, 2001; Sloboda et al., 2018; Warren et al., 2002; Li, 2003; Langen et al., 2001, 2004; Reid and Li, 2001, Lang et al., 2002; Torrente et al., 2003; Locati et al., 2003; Tidball and Villalta, 2010 |
| IFNγ | mouse | increase | macrophages, natural killer cells, T-cells | inhibits muscle cell proliferation, inhibits muscle cell differentiation | stimulates production of Th1 cytokines, promotes the M1 macrophage phenotype, represses the M2 macrophage phenotype | Wang et al., 2015; Wehling-Henricks et al., 2018; Cheng et al., 2008; Fu et al., 2015; Panduro et al., 2018; Kalovidouris et al., 1993; Kelić et al., 1993; Londhe and Davie et al., 2011; Villalta, et al., 2011a; Mills et al., 2000; Tidball, 2017; Tidball and Villalta, 2010 |
| IL6 | human, mouse, rat | unchanged | macrophages, muscle cells | promotes muscle cell proliferation, promotes muscle cell differentiation | promotes the M2 macrophage phenotype, macrophage chemoattractant | Przybyla et al., 2006; Wang et al., 2015; Hamada et al., 2015; Visser et al., 2002; Serrano et al., 2008; Kami and Senba, 1998; Zhang et al., 2013; Hoene et al., 2013, Toth et al., 2011; Haddad et al., 2005; Chen et al., 2018, Duluc et al., 2007 |
| IL1β | mouse, human | increase | macrophages | inhibits IGF-1-mediated differentiation of muscle cells | promotes Th1 cytokine production, macrophage chemoattractant | Przybyla et al., 2006; Wang et al., 2015; Arnold et al., 2007; Perdiguero et al., 2011, Wang et al., 2014; Broussard et al., 2004; Mortala et al., 2018; Rider et al., 2011 |
| IL10 | human, mouse | increase | macrophages, regulatory T-cells | IL-10-activated macrophages promote muscle cell proliferation, reduces M1 macrophage-mediated cytotoxicity, modulates muscle cell differentiation | deactivates the M1 macrophage phenotype, promotes the M2 macrophage phenotype | Przybyla et al., 2006; Wang et al., 2015; Deng et al., 2012; Villalta et al., 2009; Villalta et al., 2011b; Castiglioni et al., 2015; Villalta et al., 2014; Mosser and Zhang, 2008; Murray, 2016; Martinez-Pomares et al., 2003; Welc et al., 2020 |
| GDF3 | mouse | decrease (following injury) | macrophages | promotes muscle cell fusion, increases regenerating myofiber cross-sectional area | inhibits Th1 cytokine production | Patsalos et al., 2018; Varga et al., 2016; Wang et al., 2020 |
| OPN | mouse | increase (following injury) | macrophages, muscle cells, T-cells | promotes muscle cell fusion (ECM-bound OPN), promotes muscle cell differentiation (soluble OPN), inhibits muscle cell differentiation (soluble OPN), promotes muscle regeneration | promotes Th1 cytokine production, inhibits the M2 macrophage phenotype, macrophage chemoattractant, neutrophil chemoattractant | Paliwal et al., 2012; Denhardt et al., 2001; Pereira et al., 2006; Uaesoontrachoo n et al., 2008; Wasgewatte Wijesinghe et al., 2019; Patarca et al., 1989; Hirata et al., 2003; Marcondes et al., 2008; Shi et al., 2018; Zhu et al., 2004; Koh et al., 2007; Ashkar et al., 2000; Many et al., 2016 |
| Klotho | mouse | decrease | macrophages, muscle cells, fibro-adipogenic progenitor cells | promotes muscle cell proliferation, increases myofiber growth | promotes Th1 cytokine production, promotes Th2 cytokine production, increases M2-biased macrophage numbers | Kuro-o et al., 1997; Wehling-Henricks et al., 2016, 2018; Sahu et. al., 2018; Welc et al., 2020b; Sahu et al., 2018; Ahrens et al., 2018 |
| IGF1 | human, mouse, rat | decrease | macrophages, muscle cells | promotes muscle cell proliferation, promotes muscle cell differentiation, stimulates protein synthesis, promotes muscle fiber growth, increases aged muscle growth (exogenous administration) | promotes the M2 macrophage phenotype | Corpas et al., 1993; Kelijman, 1991; Lamberts et al., 1997; Lu et al., 2010; Tonkin et al., 2015; Jennische et al., 1987; Allen and Boxhorn, 1989; Coolican et al., 1997; Palmer et al., 1997; Barton-Davis et al., 1998 |
Macrophages.
Macrophages vastly exceed all other leukocyte populations in muscle following acute injury and, unlike neutrophils, they persist at elevated levels in muscle throughout the period of muscle repair and regeneration, which may extend for weeks (Figure 6). Macrophages also express CD11b but they can be distinguished from neutrophils and other leukocytes in muscle by their expression of CD68 and, in mice, by their expression of F4/80. Some macrophages can play roles in injured muscle that are similar to neutrophil functions; they release free radicals that can affect muscle damage and repair (Villalta et al., 2009; Vezzoli et al., 2010), they phagocytose debris at injury sites (McLennan, 1996; Summan et al., 2006; Zhao et al., 2016; Zhang et al., 2019), they produce chemokines and other factors that amplify the inflammatory response (Robertson et al., 1993; Bryer et al., 2008; Rigamonti et al., 2013; Wang et al., 2014; Nicholas et al., 2015) and they attract satellite cells and other stem cells to the site of muscle damage (Lolmede et al., 2009; Lemos et al., 2015). However, other macrophage populations release anti-inflammatory molecules and produce factors that influence the activation, proliferation and differentiation of myogenic cells and increase the growth of regenerating muscle fibers (e.g. IL10, IGF1, Klotho) (Tables 1 and 2). Macrophages demonstrating those functions increase in muscle between 2 and 4 dpi, and their numbers remain elevated over the remaining course of muscle repair and regeneration. During the resolution of inflammation in injured muscle, macrophages are eliminated, at least in part, by apoptosis (Tidball and St. Pierre, 1996).
Figure 6.
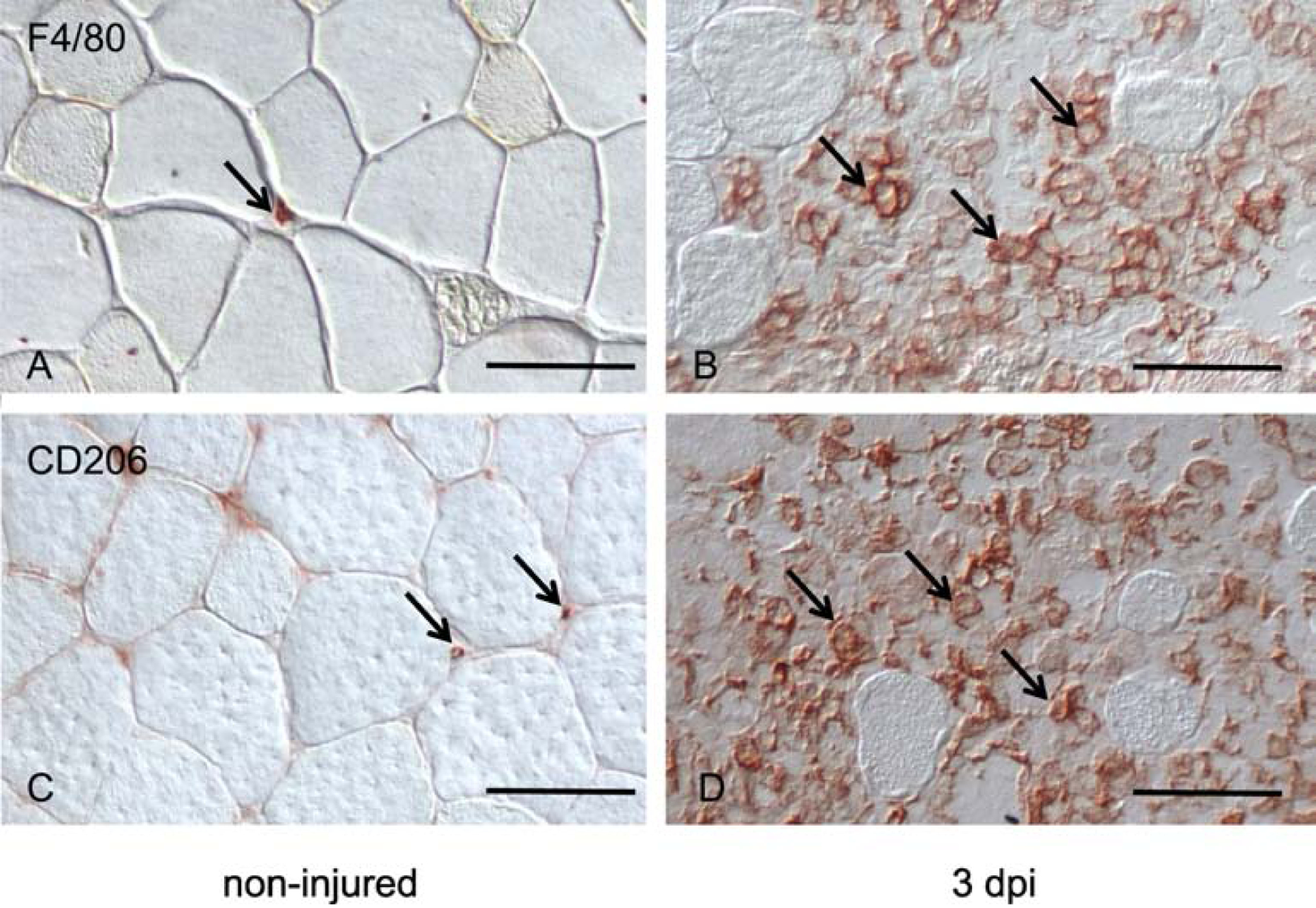
Non-injured and BaCl2-injured (3 dpi) TA muscles, immunolabeled for F/80+ macrophages (A, B) and CD206+ macrophages (C, D). Note that macrophages in non-injured muscle (A, C) are small, indicating lack of activation, versus macrophages in areas of extensive injury and inflammation where they are greatly enlarged, reflecting their activation (B, D). Representative macrophages in each sample are indicated by arrows. Bars = 50 μm.
Table 2.
Age-dependent defects in the regulation of inflammatory molecules that mediate regeneration are highlighted by perturbations in the levels of these molecules after muscle injuries.
| Injury models | |||||
|---|---|---|---|---|---|
| Molecule | Species | Injury protocol | Expression level changes in old muscle relative to young muscle | References | |
| Pre-injury | Post-injury | ||||
| TNFα | mouse | lengthening contractions | no change | increase | Sloboda et al., 2018 |
| rat | bupivacaine hydrochloride injection | no change | increase | van der Poel et al., 2001 | |
| IFNγ | mouse | CTX injection | no change | decrease | Zhang et al., 2020 |
| IL6 | rat | CTX injection | no change | increase | van der Poel et al., 2011 |
| IL1β | rat | bupivacaine hydrochloride injection | no change | increase | van der Poel et al., 2011 |
| IL10 | mouse | lengthening contraction | data not provided | decrease | Sloboda et al., 2018 |
| GDF3 | mouse | CTX injection | no change | decrease | Patsalos et al., 2018 |
| OPN | mouse | CTX injection | data not provided | increase | Paliwal et al., 2012 |
| Klotho | mouse | CTX injection | no change | decrease | Sahu et al., 2018 |
| IGF1 | mouse | unloading and reloading | no change | decrease | Reidy et al., 2019 |
| rat | Pneumatic tourniquet (ischemia-reperfusion) | no change | decrease | Hammers et al., 2008 | |
| Exercise models | |||||
| Molecule | Species | Exercise protocol | Expression level changes in old muscle relative to young muscle | References | |
| Pre-exercise | Post-exercise | ||||
| TNFα | human | 45-minutes of downhill running | data not provided | data not provided | Hamada et al., 2005 |
| 3-months of resistance training | increase | data not provided | Greiwe et al., 2001 | ||
| IL1β | human | single resistance training session | increase | no change | Przybyla et al., 2006 |
| 45-minutes of downhill running | data not provided | data not provided | Hamada et al., 2016 | ||
| IL10 | human | Single resistance training session | increase | no change | Przybyla et al., 2006 |
The plasticity of macrophage phenotypes is regulated by the presence of cytokines and growth factors that activate them to potentially innumerable different phenotypes that occur along a functional continuum between a quintessential, pro-inflammatory phenotype (M1-biased) and a quintessential, anti-inflammatory phenotype (M2-biased) (Mills et al., 2000; Locati et al., 2013; Weisser et al., 2013; Murray et al., 2014). Muscle macrophages in mice that are biased toward an M1-phenotype are typically identified by their expression of F4/80 and relatively high levels of CD11b or Ly6C. M2-biased macrophages in mouse muscle are typically identified by their expression of F4/80 and relatively low levels of CD11b or Ly6C or by their expression of CD163 or CD206. Although many cytokines can modulate macrophage phenotypes, several are particularly powerful at promoting the M1-biased, pro-inflammatory phenotype (IFNγ, TNFα) or the M2-biased, anti-inflammatory phenotype (IL4, IL10, IL13) (Mills et al., 2000; Locati et al., 2013; Weisser et al., 2013). Macrophages biased toward either end of the spectrum of phenotypes have positive influences on muscle regeneration, but the transition from the initial population of macrophages, dominated by cells biased toward the M1 phenotype, to the later population, dominated by an M2-biased phenotype, is particularly important in the progress of muscle regeneration. For example, depletion of F4/80+ macrophages at the time of M1/M2 phenotype transition from muscles injured by increased loading greatly slowed muscle repair, growth and regeneration, accompanied by disruptions in normal expression of myogenic transcription factors (Tidball and Wehling-Henricks, 2007). Muscle fiber growth following injury caused by injection of cardiotoxin (CTX) was also slowed in mice from which CD11b+ cells, which would predominantly be macrophages, were ablated at the time of peak muscle regeneration (Arnold et al., 2007).
Lymphoid cells.
In contrast to myeloid cells such as neutrophils and macrophages, lymphoid cells occur at relatively low numbers in muscle following acute injury (Orimo et al., 1991; McLennan, 1996; Castiglioni et al., 2015; Deyhle et al., 2018) and, likely as a result of their smaller numbers, their potential roles in muscle regeneration are less explored. Their invasion following acute muscle injury follows a time-course that is similar to changes in M2-biased macrophages in injured muscles, which suggests that they may also support regeneration. Following muscle damage by sterile injection of CTX, both CD4+ and CD8+ T-lymphocyte populations reached maximum numbers in muscle at ~3 dpi, after which their numbers slowly declined as muscle regeneration occurred (Zhang et al., 2014; Fu et al., 2015). Furthermore, ablation of all T-cell and B-cell populations significantly slowed muscle regeneration following CTX injury, although macrophage numbers were unaffected by the mutation (Fu et al., 2015). Surprisingly, CD8+ cytotoxic T-lymphocytes (CTLs) are responsible for a substantial proportion of the pro-regenerative role played by lymphoid cells. Although CTLs primarily serve as central components of the adaptive immune response, which is not typically activated in acute, sterile injuries, their numbers are elevated following acute injury. In addition, specifically deleting CTLs from mice by genetic ablation of Cd8a slowed muscle regeneration following sterile injection of CTX (Zhang et al., 2014), similar to the slowing achieved by Rag1 mutation (Fu et al., 2015). However, the extent to which CTLs or other lymphoid cells influence muscle regeneration through direct actions on myogenic cells in the injured muscle versus through effects mediated by myeloid cells is uncertain. On one hand, numbers of mature myeloid cells that expressed high levels of the Gr1 phenotypic marker were greatly reduced in injured muscles of Cd8a mutant mice, which corresponded to a reduction in the expression of chemokine ligand-2 (CCL2) (Zhang et al., 2014), the most potent and well-documented chemoattractant for myeloid cells to injured muscle (Warren et al., 2004; Shireman et al., 2006, 2007; Martinez et al., 2010; Lu et al., 2011). Because macrophages can promote proliferation of myogenic cells (Bencze et al., 2012), the findings supported the interpretation that CTLs promote muscle regeneration by attracting macrophages to the muscle via CCL2, and those macrophages then accelerate regeneration by more rapidly expanding the myogenic cell population. On the other hand, muscle regeneration was slowed without affecting numbers of F4/80+ macrophages in mice that were Rag1 mutants, preventing the production of mature T-cells or B-cells (Fu et al., 2015). In addition, pro-inflammatory cytokines (IL1α, TNFα and IFNγ) secreted by T-cells increased proliferation of myogenic cells in vitro (Fu et al., 2015). Those findings show that some of the pro-regenerative effects of T-cells on muscle regeneration can occur via direct actions on the myogenic cells, although those directly-acting, effector T-cells may be a population other than CTLs.
Although CD4+ T-cells are a diverse population, CD4+ regulatory T-cells (Treg cells) are now established as having a significant regulatory role in muscle regeneration (Burzyn et al., 2013; Villalta et al., 2014). Treg cells, which express the forkhead box protein3 (FOXP3), also accumulate in muscles subjected to acute sterile injury with kinetics similar to M2-biased macrophages, reaching peak numbers at about 4 dpi (Burzyn et al., 2013). In addition, their depletion during muscle regeneration slowed muscle growth and regeneration, prolonged muscle inflammation and disrupted the pattern of expression of myogenic transcription factors (Burzyn et al., 2013). These effects of Treg cell depletion were similar to the effects of depleting F4/80 macrophages from regenerating muscle at the time of macrophage phenotype transition (Tidball and Wehling-Henricks, 2007), suggesting that the effects of Treg cell depletion could be mediated in part by disrupting macrophage influences on muscle regeneration. That possibility was also supported by the finding that the normal transition between M1-biased and M2-biased macrophage phenotypes was disrupted by ablating Treg cells in regenerating muscle (Burzyn et al., 2013). A role for Treg cells in promoting the M2-biased phenotype is partly attributable to Treg cell-derived IL10 (Villalta et al., 2014; Castiglioni et al., 2015). However, muscle Treg cells are also a source of the growth factor amphiregulin which can act directly on myogenic cells in vitro to increase their differentiation, without affecting proliferation (Burzyn et al., 2013). Thus, Treg cells can also have regulatory influences on muscle regeneration by modulating the immune response and by directly acting on myogenic cells.
4. Mechanisms through which aging of the immune system can affect muscle regeneration.
a. Age-related defects in muscle regeneration are affected by factors extrinsic to muscle cells.
Satellite cell numbers in rodents and humans decline during aging (Allbrook et al., 1971; Snow, 1977; Gibson and Schultz, 1983; Renault et al., 2002) and, because adult myogenesis depends on the presence of satellite cells, their reduced numbers may contribute to the impaired regenerative capacity of muscle. This was demonstrated experimentally in young, adult mice in which Pax7-expressing satellite cells were nearly completely eliminated leading to greatly impaired histological signs of regeneration in TA muscles that were injured by CTX injection (Sambasivan et al., 2011). Similarly, genetic ablation of Pax7-expressing satellite cells in 17-month-old mice resulted in histological evidence of diminished regeneration in TA muscles injured by barium chloride (BaCl2) injection (Fry et al., 2015). Thus, normal muscle regeneration is influenced by the number of satellite cells, at least in cases of extreme reductions of their numbers. During aging of mice, satellite cell numbers decline by about 50% in mice by 20- to 24-months of age compared to young adults (Brack et al., 2005; Wang et al., 2019), which may be sufficient to impair muscle regenerative capacity with age. This reduction in cell numbers is attributable both to age-related changes that are intrinsic to myogenic cells and to age-related changes in their environment. For example, proliferation of myogenic cells isolated from soleus and extensor digitorum longus (EDL) muscles from mice ranging from 6-days to 30-months of age showed that older cells were less proliferative in vitro, reflecting an intrinsic, age-related change (Schultz and Lipton, 1982). On the other hand, exposure to serum from ~3-month-old mice increased the proliferative capacity of satellite cells on isolated muscle fibers obtained from 19- to 26-month-old mice in vitro (Conboy et al., 2005), showing that extrinsic factors also influence age-related changes in satellite cell proliferation. In addition, exposure of old satellite cells in injured muscle to circulating factors obtained from young mice reduced the age-related shift of old satellite cells from a myogenic phenotype to a more fibrogenic phenotype following their activation (Brack et al 2007). Collectively, these findings show that age-related changes in satellite cell number and function are attributable to both intrinsic and extrinsic factors that influence myogenesis.
Although age-related changes within satellite cells contribute to the loss of the regenerative capacity of aging muscles, numerous findings show that changes in extrinsic factors during aging may dominate the progressive decline in muscle’s ability to repair itself. Early, pioneering studies used age-matched (isochronic) and age-mismatched (heterochronic) muscle transplantations in which rat EDL muscles were transplanted between rats ~3-months of age and 24-months of age, to assess whether muscle regeneration following transplantation was most influenced by the age of the rat muscle that was transplanted or the age of the host rat that received the transplantation (Carlson and Faulkner 1989). Using increases in muscle mass, recovery of muscle force production and restoration of muscle histology as indices of regeneration, each assay showed that the most successful regeneration occurred if the host were young, regardless of the age of the transplanted muscle, and the worse regeneration occurred in old hosts, regardless of the age of the transplanted muscle. Subsequently, the circulatory system was shown to be the source of at least some of the host-derived, exogenous factors that affect muscle regeneration. After surgically-joining age-matched and age-mismatched mice between young (2- to 3-month-old) and old (19- to 24-month-old) mice so that they shared a common circulatory system, old mice that experienced acute injury by freeze-damage to the TA muscle demonstrated better myotube formation and less muscle fibrosis if they were conjoined with a young mouse than if they were conjoined with an old mouse (Conboy et al., 2005). Although these findings clearly demonstrate a role for an age-related change in circulating cells and factors in affecting the age-related change in muscle regeneration, we do not yet know the relative importance of those circulating factors versus age-related changes in other cells and tissues that influence muscle regeneration. For example, age-related changes in the neuromuscular system outside the muscle fiber can contribute substantially to the regenerative ability of acutely-injured muscle (Carlson and Faulkner, 1998), and we do not know whether the age-related changes in the motor nervous system are more important than age-related changes in circulating factors in influencing muscle regeneration. Furthermore, age-related changes in circulating cells and factors may influence muscle regeneration indirectly, by acting on other tissues involved in muscle repair, such as the nervous system and circulatory system. The relative importance of those direct versus indirect effects of circulating factors on age-related changes in muscle regeneration is unknown. Nevertheless, circulating factors definitively contribute to age-related changes in muscle regeneration to some extent, as shown by the discovery that the direct application of serum from young mice improved satellite cell proliferation, compared to serum from old mice (Conboy et al., 2005). Those findings provided the foundation for current investigations into the source and identity of the specific molecules that provide extrinsic regulatory controls on muscle regeneration that change with age, and introduced the possibility that the immune system was a significant component of the regulatory controls.
b. Do age-related changes in the numbers or functions of immune cells in injured muscle contribute to loss of muscle regenerative capacity?
i. Does aging cause defects in leukocyte recruitment to injured muscle?
Some of the earliest studies of the regeneration of aging muscle identified relationships between changes in muscle regeneration that coincided with disruptions of muscle inflammation, especially changes in the numbers of macrophages and other myeloid cells. Because most leukocytes in muscle following acute injuries are derived from cells recruited from the circulation, an age-related defect in recruitment would diminish the normal regulatory influences of the immune system on muscle regeneration. Several observations support that possibility. Injuries caused by injection of bupivicaine (which can cause analgesia in addition to muscle fiber damage) into rat tibialis anterior (TA) muscles at 3-months or 2-years of age showed that regeneration, assayed by the time of appearance of myotubes, was greatly delayed in old animals and accompanied by much slower growth of muscle fibers (Sadeh, 1988). The slowing of myogenesis was also paralleled by a slowing of the accumulation of inflammatory cells in the injured muscle and a prolongation of the inflammatory response (Sadeh, 1988). Together, the observations suggested that an age-related delay in the recruitment of inflammatory cells into old, injured muscle impaired regeneration. Similarly, more recent observations showed that EDL muscles that were injured by a series of forced, lengthening contractions experienced a larger increase of CD68+ macrophages in muscles at 5 dpi in young muscle (3- to 5-month-old mice) than in old (25- to 27-month-old) (Sloboda et al., 2018), consistent with an age-related defect in leukocyte recruitment or expansion after recruitment to the injured tissue. Importantly, age-matched mice in the same investigation that were not injured showed much higher numbers of monocytes in the circulation of old mice than in young (Sloboda et al., 2018), indicating that the diminished accumulation of CD68+ cells in the old mice was not a consequence of a reduction in the pool available for recruitment.
However, defects of inflammatory cell recruitment or migration into old, injured muscle are not apparent or are minimal in other acute injury models in mice. For example, transplantation of muscle grafts causes muscle damage, and transplantation of muscle from young mice (2 months) to young recipients, or transplantation from young mice to old recipients (21 months) or transplantation of old muscle to either young or old recipients produced similar courses of muscle inflammation, with only slight delays of inflammation in older recipients (Smythe et al., 2008). In addition, muscle injury that is caused by removing loading from hindlimb muscles (hindlimb suspension; HS) followed by a period of reloading the muscles by a return to weight-bearing (reloading) produces an inflammatory response that is qualitatively similar to inflammation caused by more extreme injuries such as toxin injection. However, muscles experiencing HS/reloading showed no impairment in the accumulation of Ly6C+ cells (neutrophils/monocytes/macrophages) during reloading of old gastrocnemius muscles (24- to 26-month-old mice) compared to young muscles (4- to 5-month-old), although muscle regeneration, assayed by fiber growth, was slower in old muscle (Reidy et al., 2019b). In addition, expression levels of Ccl2 were higher in old muscles during reloading (Reidy et al., 2019b), which is significant because CCL2 is the strongest driver of leukocyte chemoattraction into muscle following acute injury (Warren et al., 2004; Shireman et al., 2006, 2007; Martinez et al., 2010; Lu et al., 2011). Thus, aging of muscle and the immune system does not produce a universal, intrinsic defect in myeloid cell recruitment to injured muscles in mice. Similarly, quadriceps muscles in young humans (~32 years) that were injured by resistance exercise showed no difference in the increase of numbers of CD68+ macrophages in the muscles at 72 hours post-exercise, compared to old humans (~71 years) (Przybyla et al., 2006). Because CD68 is expressed by all circulating monocytes and by all macrophages in humans, and the vast majority of inflammatory cells in injured muscle are monocytes/macrophages, the observation indicates that aging does not cause a significant, universal defect in leukocyte recruitment to injured muscle, in humans as well as in mice.
Why does aging cause no defect in leukocyte recruitment following mild muscle damage (e.g., HS/reloading) but causes reduced leukocyte numbers following more severe muscle damage (e.g., myotoxin injection)? Although the cause of this difference in the effect of age on leukocyte recruitment in different injury models has not been identified, it could reflect age-related differences in the response of old muscle to injury. Old muscle experiences more extensive damage than young muscle as a consequence of the same perturbation (Zerba et al., 1990; Brooks and Faulkner 1996) and perhaps, in more severe injury models, the amplified damage to old muscle may impair the production or release of signals from the muscle that are necessary for leukocyte recruitment. Alternatively, the more extensive damage of old muscle may generate other obstacles for trafficking of leukocytes into injured tissue if, for example, there is more extensive damage to the vascular bed. However, no experimental findings are available to address this alternative possibility.
ii. Does aging cause intrinsic changes to macrophage phenotypes that affect muscle regeneration?
The tremendous preponderance of macrophages in injured muscle and the growing body of knowledge that demonstrates their central roles in regulating regeneration suggests that age-related defects in their function in muscle would significantly affect muscle regeneration. As outlined in Section 3 above, the shift in macrophages along the spectrum of phenotypes over the course of muscle regeneration is key to determining the success of regeneration. This point has been illustrated by investigations in which levels of IL10 in injured muscle have been disrupted. IL10 expression is elevated in regenerating muscle and it mediates the normal transition of macrophages from an M1-biased phenotype to an M2-biased, CD163+, CD206+ phenotype in muscle following HS/reloading (Deng et al., 2012) or BaCl2 injection (Welc et al., 2020a). If IL10 is not expressed, transition to an M2-biased phenotype does not occur and muscle regeneration is impaired. However, premature elevation of IL10 in early stages of regeneration also disrupted regeneration (Perdiguero et al., 2011), showing that disruption of the normal time course of macrophage transitions may also negatively affect regeneration.
Macrophage phenotype transitions are perturbed in aged muscle following injury. For example, injuries caused by repeated, forced, lengthening contractions (eccentric contractions) of EDL muscles showed a greater increase in CD163+, M2-biased macrophages in old (25- to 27-month-old) mice compared to young (3- to 5-month-old), although the increase in total CD68+ macrophages was greater in young mice at the same stage of regeneration (Sloboda et al., 2018). Similarly, human quadriceps muscles subjected to repeated, forced, eccentric contractions showed a reduction of CD11bhigh, M1-biased macrophages and an increase in CD206+, M2-biased macrophages in old muscles (~77 years) compared to young muscles (~22 years) during regeneration, although the number of total CD68+ macrophages was unchanged (Sorensen et al., 2019). Some of these changes in numbers of M2-biased macrophages in old versus young, injured muscle are also reflected in changes in gene expression in macrophages. FACS-isolated CD45+, CD11b+ cells from gastrocnemius muscles of mice experiencing HS followed by 4 days of reloading showed significantly higher expression of Retnia (Fizz1), a marker of the M2-biased phenotype, in cells sorted from old (24- to 26-month-old) mice compared to young (4- to 5-month-old), with no difference in the expression of pro-inflammatory genes (IL1β, TNFα, IFNγ) (Reidy et al., 2019b). However, expression levels of another M2-phenotypic marker (Mrc1 which encodes CD206) did not differ between cells sorted from old versus young muscles (Reidy et al., 2019b). Together, these findings indicate that there is an age-related shift of macrophages toward an M2-biased phenotype in regenerating muscle by increasing both the numbers of M2-biased macrophages in old regenerating muscle and by increasing the levels of expression of some M2 phenotypic markers in individual macrophages.
The greater prevalence of M2-biased macrophages in regenerating muscle of old mice and humans is surprising for a couple of reasons. First, aging of the immune system produces “inflammaging,” a condition in which there is a systemic shift of the immune system to a more pro-inflammatory environment characterized by higher levels of Th1 cytokines (TNFα, IL1β and IFNγ); that created the expectation that the inflammatory response in aged-muscle would also be shifted toward a pro-inflammatory, M1-biased macrophage population. In addition, the unexpected bias of intramuscular macrophages to an M2 phenotype is not only a consequence of injury; non-injured, old muscles also show an increased proportion of CD163+, CD206+ M2-biased macrophages (Wang et al., 2015; Sorensen et al., 2019; Reidy et al., 2019; Cui et al., 2019), despite an age-related shift of muscle toward the expression of pro-inflammatory cytokines. For example, non-injured muscle exhibits inflammaging, associated with elevated expression of IL1β and IFNγ (Dennis et al., 2004; Przybyla et al., 2006; Leger et al., 2008; Peake et al., 2010; Wang et al., 2015) and either elevated or unchanged levels of TNFα (Dennis et al., 2004; Hamada et al., 2005; Raue et al., 2007; Leger et al., 2008; Wang et al., 2015). Muscle aging is also associated with no change in the level of expression of IL6 (Hamada et al., 2005; Przybyla et al., 2006; Wang et al., 2015), a cytokine that promotes the M2 phenotype (Duluc et al., 2007) and a decline in IGF1 expression (Dennis et al., 2004), a growth factor that supports the M2-biased phenotype (Tonkin et al., 2015). Thus, the shift of macrophages toward an anti-inflammatory, M2-phenotype in non-injured muscle occurs contrary to the presence of a pro-inflammatory, inflammaging environment that mirrors systemic changes, at least in part. Injury can then further amplify that inclination (Sloboda et al., 2018).
We do not know why the aging, intramuscular environment favors an M2-biased macrophage phenotype when expression levels of Th1 cytokines in aging muscle reflect systemic inflammaging, but a possible explanation may lie in the large increases in expression of IL10 that occur during muscle aging (Leger et al., 2008; Wang et al., 2015). IL10 is a potent deactivator of the M1 macrophage phenotype in muscle and increases in its production are an established feature of aging macrophages (Chelvarajan et al., 2005). Its powerful role in regulating shifts in macrophages to an M2-biased phenotype in young, injured muscles (Deng et al., 2010; Welc et al., 2020a) suggests the untested possibility that the age-related shift in its expression in muscle contributes to the M2 bias of macrophages in aging muscle.
The greater prevalence of M2-biased macrophages in regenerating muscles of old mice and humans is also unexpected because M2-biased macrophages play important roles in promoting growth and regeneration of younger muscles following injury, but old muscles with a higher proportion of M2-biased macrophages (Wang et al., 2015; Sloboda et al., 2018) show impaired regeneration (Gutmann and Carlson, 1976; Zacks and Sheff 1982; Carlson and Faulkner, 1988, 1996; Musaro et al., 2001). The apparent discrepancy between the elevated numbers of M2-biased macrophages and impaired regeneration is also likely explained by the great, functional diversity of the M2 macrophage phenotype and the inadequacy of associating the expression of a small number of phenotypic markers (e.g., CD163, CD206, CD11blow, Ly6Clow) with a specific function (e.g., pro-regeneration or pro-reparative macrophages). Despite that limitation, other aspects of the age-related dysregulation of intramuscular macrophage phenotype are more clearly associated with another defect of muscle regeneration in old animals: increased muscle fibrosis (Brack et al., 2007; Hidestrand et al., 2008; Serrano et al., 2010; Fry et al., 2015; Wang et al., 2015; 2019; Lyu et al., 2019). In part, this increase in fibrosis is attributable to the age-related shift of satellite cells to a more fibrotic phenotype (Brack et al., 2007). However, elevated numbers of CD163+ M2-biased macrophages in aging muscle can also increase fibrosis (Wang et al., 2015, 2019).
Some of the age-related changes in macrophages that affect muscle regeneration are the consequence of intrinsic changes in the function of old macrophages that occur independent of the muscle environment. For example, human myoblasts that were treated with conditioned media obtained from macrophages that were derived from whole-blood monocytes from old humans showed less proliferation than myoblasts treated with conditioned media from macrophages from young humans (Sorensen et al., 2019). Furthermore, these intrinsic changes in macrophage function occurred during aging of hematopoietic stem cells (HSCs) that are progenitors of macrophages, while the HSCs still reside in the bone marrow. Myoblasts treated with conditioned media from macrophages that were differentiated from bone marrow cells (BMCs) from old mice (bone marrow derived macrophages; BMDMs) were less proliferative than myoblasts treated with conditioned media from young BMDMs (Wang et al., 2019). In addition, the myoblasts treated with media from old BMDMs showed lower levels of expression of MyoD and myogenin, indicating that aging of macrophage progenitors influences their capacity to directly affect both proliferation and differentiation of myogenic cells (Wang et al., 2019). Furthermore, this intrinsic, age-related shift of macrophage progenitor cells to a phenotype that is less able to support myogenesis is demonstrable in vivo. Transplantation of old BMCs (18-month-old) into young mice (2-month-old) that were then aged to 10-months yielded satellite cell numbers that were reduced to levels occurring in mice 24- to 28-month-old. Note that the BMCs in those mice at the time of tissue collection were 26-month-old while the host mice were 10-months. However, transplantation of young BMCs (2-month-old) into young mice (2-month-old) that were then aged to 10-months caused no change in numbers of satellite cells compared to the muscles of age-matched, non-transplanted controls (Wang et al., 2019). Thus, aging of the hematopoietic system leads to intrinsic changes in myeloid lineage cells that diminish their capacity to maintain satellite cell numbers in vivo. However, whether aging of HSCs also contributes to age-related defects in muscle regeneration has not been tested.
c. What macrophage-derived molecules influence age-related defects in muscle regeneration?
Macrophages are rich sources of cytokines and growth factors that can influence muscle regeneration and the expression levels of many of those molecules change in muscle during aging (Table 1). However, we frequently do not know whether the age-related changes in expression of molecules that influence myogenesis occur in leukocytes or in non-immune cells within the aging muscle. For example, there is a systemic reduction in IGF1 expression during aging (Kelijman, 1991) and induction of IGF1 expression in muscle by injury is greatly reduced by aging (Hammers et al., 2008; Reidy et al., 2019a,b). IGF1 can influence both the proliferation and differentiation of muscle cells (Coolican et al., 1997), it can increase growth of embryonic and aging muscle (Powell-Braxton et al., 1993; Barton-Davis et al., 1998) and its expression by macrophages improves repair of injured, non-aged muscle (Lu et al., 2011; Tonkin et al., 2015). However, whether an age-related reduction in IGF1 expression in macrophages contributes to defects in regeneration of aged muscle is unknown, but offers a promising hypothesis for future experimentation. Nevertheless, some insights acquired over the last several years into the identity of specific, macrophage-derived molecules that influence muscle aging and regeneration provide more definitive evidence concerning how aging of the immune system affects muscle regeneration (Figure 7).
Figure 7.
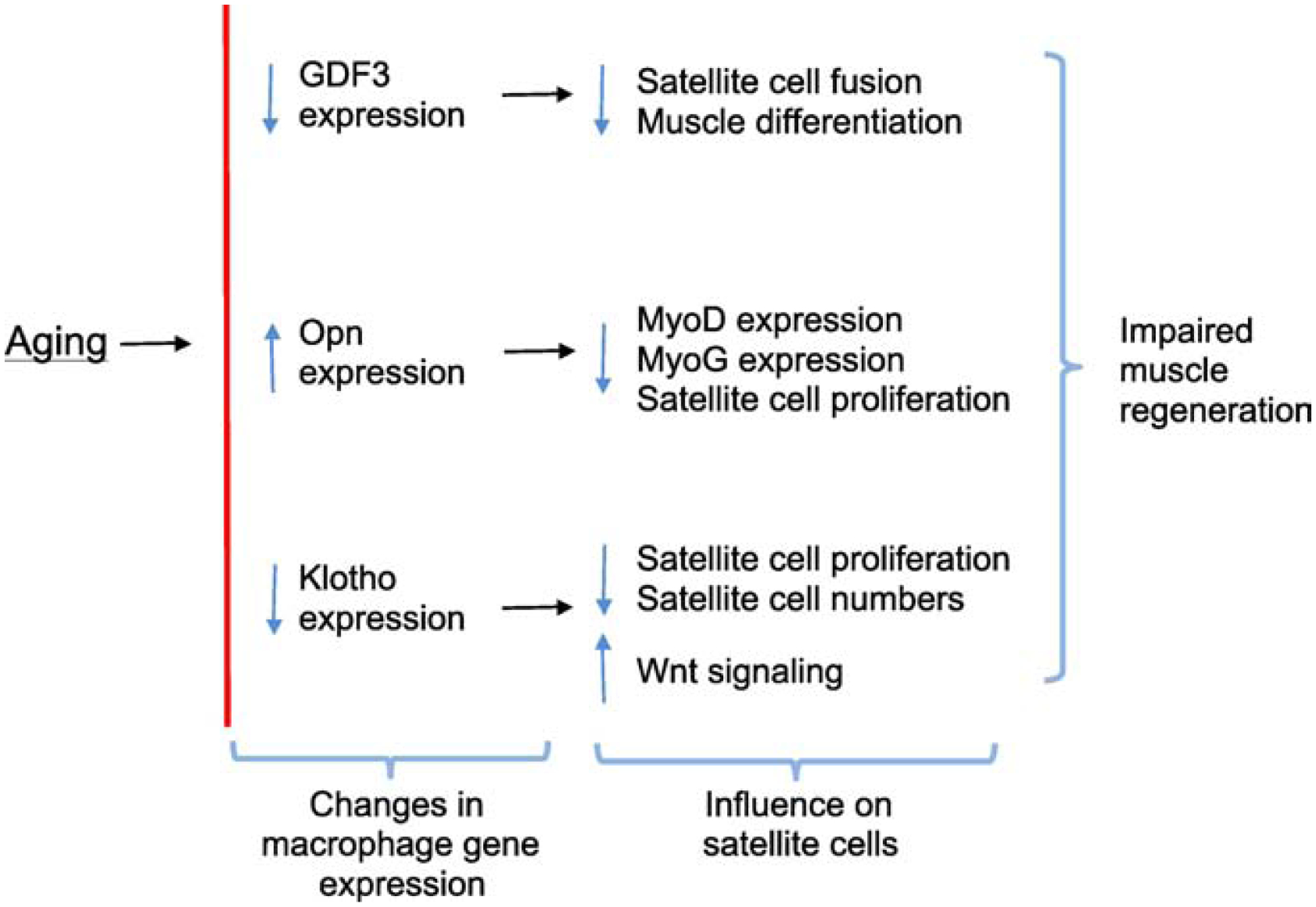
Summary diagram of changes in gene expression in intramuscular macrophages that occur during aging and the consequence of those changes on satellite cell function and numbers.
i. Growth differentiation factor 3 (GDF3).
GDF3 is a TGFβ family member (Levine and Brivanlou 2006) that is now established as a significant, macrophage-derived cytokine that modulates muscle regeneration (Varga et al., 2016; Patsalos et al., 2018). GDF3 is secreted by macrophages and is primarily expressed in muscles by CD45+ cells in the hematopoietic lineage, suggesting that macrophages are the major source of GDF3 in whole muscle tissue (Varga et al., 2016; Patsalos et al., 2018). Treatment of myogenic cells in vitro with GDF3 slightly reduced their proliferation and increased their rate of cell fusion (Varga et al., 2016), indicating a role for the protein in promoting muscle differentiation. Several observations provide strong support that GDF3 influences muscle regeneration in vivo and show that age-related reductions in GDF3 expression by macrophages contribute to defects in regeneration of old muscle. First, Gdf3−/− mice in which muscles were acutely injured by CTX injection experienced slower muscle fiber growth during regeneration than the growth that occurred in non-mutant mice (Varga et al., 2016). In addition, wild-type mice that received bone marrow transplantations (BMT) from Gdf3−/− donors showed similarly slowed growth (Varga et al., 2016), indicating that the GDF3 deletion specifically in myeloid cells was sufficient to delay regeneration. In contrast to a large induction of GDF3 expression by injury in young mouse muscles (2-month-old), old mice (23-month-old) showed little induction of GDF3, which was accompanied by reductions in growth of regenerative fibers and slowed repair of damaged fibers (Patsalos et al., 2018). However, the reduction of GDF3 in old injured muscle was not a consequence of reductions in inflammation; the old mice showed greater numbers of Ly6Chigh cells in the muscle, suggesting that old macrophages produced lower levels of GDF3, and that loss contributed to age-related defects in regeneration.
ii. Osteopontin.
Osteopontin (Opn; encoded by Spp1) is a pro-inflammatory cytokine expressed by macrophages, T-cells and natural killer (NK) cells that can be induced by IL1 and TNFα (reviewed by Denhardt and Noda, 1998; O’Regan et al., 2000; Denhardt et al., 2001). Much of the pro-inflammatory role of Opn in muscle is attributable to its strong chemoattractant function for neutrophils, monocytes and macrophages to sites of injury and inflammation (Zhu et al., 2004; Koh et al., 2007; Marcondes et al., 2008; Many et al., 2016; Shi et al., 2018) and its induction of Th1 cytokines and inhibition of Th2 cytokine production via its interactions with CD44 and integrins (Ashkar et al., 2000). In particular, Opn binding to CD44 inhibits the induction of IL10 (Ashkar et al,. 2000). However, despite the induction of pro-inflammatory cytokines and inhibition of IL10 expression by Opn, Spp1 mutant mice showed a lower proportion of CD206+ M2-biased macrophages in mouse muscles injured by transplantation (Wasgewatte Wijesinghe et al., 2019), suggesting that Opn increases inflammation through its chemotactic role but also affects macrophage phenotype in injured muscle through an unknown mechanism. Although many leukocyte populations (Denhardt et al., 2001) and muscle cells (Pereira et al., 2006) can express Opn, its expression is elevated in CTX-injured muscle primarily in F4/80+ macrophages, although not all F4/80+ macrophages, with peak expression levels occurring at 2 dpi (Hirata et al., 2003).
Although Opn’s most well-defined roles are as an immunomodulatory molecule, it also acts directly on myogenic cells in vitro and influences muscle regeneration in vivo. Opn treatment of myogenic cells in vitro inhibits their migration and differentiation (Uaesoontrachoon et al., 2008) and neutralization of Opn in vivo perturbs muscle regeneration following CTX injection (Paliwal et al., 2012). Interestingly, these influences of macrophage-derived Opn on muscle regeneration are affected by aging of the immune system. Following acute muscle injury, CD11b+ cells that were isolated from injured TA and gastrocnemius muscles of old mice (22- to 24-month-old) showed much higher levels of Opn expression than in CD11b+ cells from young (2- to 3-month-old) muscles (Paliwal et al., 2012), accompanying the delay in regeneration in old muscles. Furthermore, injection of recombinant Opn (rOpn) into young, injured muscles reduced histological signs of regeneration and injection of neutralizing antibodies to Opn into old, injured muscles improved histological signs of regeneration and increased expression levels of MyoD and myogenin (Paliwal et al., 2012). In vitro observations also support the conclusion that these in vivo effects are attributable to age-related changes in the interaction between muscle and macrophages. Although young satellite cells cultured with young macrophages in the presence of young serum were more proliferative in vitro than old satellite cells cultured with old macrophages and old serum, those higher levels of proliferation in the young cultures were reduced by adding rOpn. Conversely, adding anti-Opn to cultures of old cells increased proliferation of old satellite cells and, similarly, replacing old macrophages with young macrophages in old cultures restored proliferation (Paliwal et al., 2012).
iii. Tumor necrosis factor-α.
TNFα is a pro-inflammatory cytokine that is a quintessential representative of a pro-inflammatory mediator that is expressed at higher levels during aging. In addition, its systemic elevation has been associated with the loss of muscle mass and function during aging and disease. For example, serum TNFα concentration increases in men during aging (Léger et al., 2008) which correlates with their loss of muscle mass and strength (Greiwe et al., 2001; Visser et al., 2002). Furthermore, the systemic elevation of TNFα coincides with an increase in TNFα signaling in muscle (Phillips and Leeuwenburgh, 2005) and treatment of patients with Crohn’s disease with neutralizing antibodies to TNFα reduced sarcopenia associated with inflammation that characterizes that disease (Subramaniam et al., 2015). In part, the association between increases in TNFα and sarcopenia is attributable to a TNFα-induced shift of muscle to a negative protein balance via an NFκB-mediated pathway (Langen et al., 2001). However, elevation of TNFα reduces muscle regenerative capacity, as well as increasing muscle wasting (Coletti et al., 2005; Song et al., 2015). These negative effects of TNFα on muscle regeneration may reflect direct actions on satellite cells. Application of TNFα to myoblasts in vitro increases their proliferation at early stages of myogenesis while repressing their differentiation (Langen et al., 2001; Chen et al., 2005; Palacios et al., 2010) which is attributable in part to transcriptional activation of NF- κB and decreased stability of MyoD (Guttridge et al., 2000). Thus, increases in TNFα levels during inflammaging have the potential to disrupt adult myogenesis, which could then contribute to age-related defects in muscle regeneration.
The systemic elevation of TNFα during aging is mirrored by increases in its expression in aging muscle and those increases are further amplified following muscle injury. For example, muscles of old mice (25- to 27-month-old) showed higher levels of TNFα induction following injury by a series of lengthening contractions than occurred in young injured muscles (3- to 5-month-old) (Sloboda et al., 2018). However, whether those injury-induced elevations of TNFα expression occurred in macrophages is unknown; using a less damaging injury protocol (HS/reloading), CD45+, CD11b+ cells that were sorted from injured muscle showed no difference in the levels of TNFα expression in young (4- to 5-month-old) compared to old (24- to 26-month-old) macrophages (Reidy et al., 2019b).
Because non-immune cells can be significant sources of TNFα, determining whether macrophages are important sources of TNFα that affect age-related changes in muscle wasting and regeneration has been challenging to address. In particular, satellite cells and muscle fibers also express and secrete TNFα (de Rossi et al., 2000) and in situ hybridization studies showed TNFα mRNA located in muscle fibers and in mononucleated cells between muscle fibers, and showed that the apparent quantity of TNFα mRNA in those cells increased in humans during aging (Griewe et al., 2001). Despite the occurrence of TNFα expression by muscle cells and intramuscular leukocytes, more recent studies show that macrophage-derived TNFα plays a significant role in influencing muscle aging and affects the number of satellite cells present in aging muscle. First, the importance of systemic TNFα in age-related changes in muscle was demonstrated by the systemic ablation of TNFα which prevented sarcopenia that occurred by 20-months of age in mice (Wang et al., 2018). However, transplantation of wild-type BMCs into 12-months-old Tnf mutants yielded intramuscular CD68+ macrophages that expressed TNFα located in an otherwise TNFα-free intramuscular environment and that transplantation led to levels of sarcopenia that resembled 20-month-old wild-type mice (Wang et al 2018). This indicates that macrophage-derived TNFα contributes significantly to muscle wasting in old age. The transplantation of TNFα expressing BMCs into Tnf mutants also reduced the numbers of myonuclei and increased the numbers of satellite cells, which suggested that TNFα generated by intramuscular macrophages inhibits satellite cell differentiation and fusion. Although these observations support the conclusion that macrophage-derived TNFα can influence age-related changes in muscle and affect satellite cell numbers, they do not show that those influences also affect defects in the regeneration of aged muscle, which remains to be definitively tested.
iv. Klotho.
Klotho is a transmembrane protein that can be cleaved and released or expressed as a truncated form that is secreted to function as a hormone (Kuro-o et al., 1997; Matsumura et al, 1998; Li et al 2004; Kurosu et al., 2005). Klotho expression declines in many tissues during aging (Kuro-o et al., 1997) and in mouse muscles its expression progressively declines between 1- and 24 - months of age during normal development, maturation and aging (Wehling-Henricks et al., 2016, 2018). Delivery of Klotho to injured or diseased muscles that is achieved by expression of a Klotho transgene improves muscle growth and regeneration (Wehling-Henricks et al., 2016, 2018; Welc et al., 2020b). In part, the pro-regenerative effects of Klotho on injured muscle are attributable to its ability to act directly on myoblasts to increase their proliferation through a TNFα-mediated autocrine loop (Wehling-Henricks et al., 2016, 2018) and to protect them against mitochondrial DNA damage (Sahu et al., 2018) and to regulate Wnt signaling (Ahrens et al., 2018). In vivo, several observations show that the primary effect of Klotho on healthy, injured or diseased muscle is to function as a positive regulator of satellite cell numbers, which would have implications for the regenerative capacity of muscle. For example, genetic restoration of Klotho expression to dystrophic muscle in which Klotho is pathologically reduced, prevents the pathological reduction in satellite cell numbers that occurs during muscle aging (Wehling-Henricks et al., 2016). In addition, transplantation of BMCs expressing a Klotho transgene into dystrophic mice increased satellite cell numbers and improved muscle regeneration, confirming that myeloid-cell mediated delivery of Klotho to muscle can improve regeneration (Wehling-Henricks et al., 2018). Similarly, mice that are hypomorphic mutants for Klotho showed reduced numbers of satellite cells in non-injured muscles and displayed impaired muscle regeneration (Ahrens et al., 2018; Sahu et al., 2018) and reduced numbers of MyoD+ cells at the site of muscle damage by CTX-injection (Sahu et al., 2018).
Following skeletal muscle damage either by BaCl2 or CTX injection or during muscular dystrophy, Klotho expression is elevated and the Klotho that is detectible by immunohistochemistry is present in CD206+ M2-biased macrophages (Wehling-Henricks et al., 2018; Welc et al., 2020b) and in MyoD+ satellite cells (Sahu et al., 2018). In addition, the accumulation of Klotho-expressing M2-biased macrophages and MyoD+ satellite cells occurs at sites of muscle regeneration (Wehling-Henricks et al., 2018; Sahu et al., 2018) which indicates that Klotho expressed by both cell types may promote muscle regeneration. However, old, injured muscle did not experience an increase in Klotho (Sahu et al., 2018) and the age-related reduction of Klotho expression occurred in both M2-biased macrophages and MyoD+ satellite cells in regenerating, aged muscle (Wehling-Henricks et al., 2018; Sahu et al., 2018). Those findings show that aging of the immune system and intrinsic, age-related changes in satellite cells produce reductions in Klotho expression and suggest that their Klotho deficiency is a significant factor in the impaired regenerative capacity of aging muscle. However, current evidence is inadequate to address whether the age-related loss of Klotho in either macrophages or in muscle cells alone produces a sufficient Klotho deficiency to cause impaired regeneration.
d. Aging perturbs interactions between lymphoid cells and other immunoregulatory cells, contributing to defects in regeneration of aging muscle.
Although changes in macrophage interactions with muscle cells have been examined most extensively in studies of inflammaging and muscle regeneration, important regulatory roles for lymphocytes in muscle regeneration are also affected by aging. As noted in Section 3, both Treg cells and CTLs can influence regeneration of young muscle and depletion of either population slows regeneration (Burzyn et al., 2013; Zhang et al., 2014; Villalta et al., 2014; Panduro et al., 2018) which is partially attributable to reduced (Zhang et al., 2014) or increased (Burzyn et al., 2013) muscle inflammation, to disruption of the normal regulation of macrophage phenotype (Burzyn et al., 2013; Panduro et al., 2018) and to lost production of T-cell derived growth factors that promote myogenesis (amphiregulin; Burzyn et al., 2013). Thus, age-related defects in the recruitment or function of either Treg cells or CTLs could contribute to diminished regeneration of injured, old muscle. Indeed, the proportion of CD4+ T-cells that is comprised of Treg cells is lower in CTX-injured muscles of old mice (24-months-old) than in young (2-months) and increasing Treg cell recruitment into old, injured muscle by injection of IL33 improved regeneration (Kuswanto et al., 2016), supporting the conclusion that sufficient numbers of Treg cells in injured muscle are necessary for normal regeneration. Although IL33 is produced by many cell types, including macrophages, dendritic cells and endothelial cells, most detectable IL33 in old muscle is present in fibro/adipogenic progenitor cells (FAPs) and IL33+ cell numbers decline in aging muscle (Kuswanto et al., 2016). This indicates that some of the age-related defects involving perturbations of Treg cell influences on muscle regeneration are not intrinsic defects in Treg cells, but are primarily attributable to unknown factors that diminish numbers of IL33-expressing cells in injured, aging muscle.
Aging also produces defects in signaling between CTLs, Treg cells and macrophages that lead to impaired muscle regeneration, particularly involving perturbations of IFNγ-mediated pathways. CTLs in injured muscles are a primary source of IFNγ (Panduro et al., 2018; Zhang et al., 2020), a cytokine that has strong but variable influences on muscle regeneration. For example, null mutation of IFNγ in dystrophic muscles that experience chronic-injury can reduce muscle damage (Villalta et al., 2011) but ablation of IFNγ expression impairs regeneration of young, acutely-injured muscle (Panduro et al., 2018; Zhang et al., 2020). Much of the muscle regenerative defect in young, injured, IFNγ-null mice is rescued by the adoptive transfer of wild-type CD8+ T cells into the IFNγ mutant mice experiencing muscle injury (Zhang et al., 2020), emphasizing the importance of CTL-derived IFNγ in muscle regeneration. In part, the regenerative defects of reduced IFNγ signaling in injured muscle may result from disruptions of the normal inflammatory response to muscle damage because IFNγ ablation skews the inflammatory response in damaged muscle toward a Th2 response with elevated numbers of CD206+, M2-biased macrophages (Villalta et al., 2011). However, IFNγ also acts directly on myogenic cells to influence their proliferation and differentiation (Kalovidouris et al., 1993; Kelic et al., 1993), which could also exert important influences on muscle regeneration if IFNγ production were impaired.
Disruption of IFNγ expression and signaling in old, injured muscle plays a significant role in impaired regeneration. Although IFNγ expression in old, uninjured muscles is either greater (Dennis et al., 2004; Przybyla et al., 2006; Leger et al., 2008; Peake et al., 2010; Wang et al., 2015) or does not differ from young muscles (Zhang et al., 2020), IFNγ expression is significantly lower in old muscle following injury than in young, injured muscle (Zhang et al., 2020). Whether this reduction results from reduction in the numbers or function of CTLs or other IFNγ-expressing cells in the injured muscle has not been established. In addition, aging produces a specific attrition of macrophages in injured muscle that are responsive to IFNγ (Zhang et al., 2020). These IFN-responsive macrophages (IFNRMs) are characterized by elevated expression of IFNγ response genes (e.g., Irf7, Ifit3, Isg15, Rsad2, Ifitm3) as well as transcripts traditionally associated with both the M2-biased phenotype (Mrc1 encoding CD206) and the M1-biased phenotype (Ly6c2) (Zhang et al., 2020). Several observations indicate that the reduction of IFNRMs in old, injured muscle can directly affect muscle regeneration. First, upon activation, IFNRMs secrete cytokine C-X-C motif chemokine 10 (CXCL10) that increases the proliferation and differentiation of satellite cells in vitro via binding its receptor, CXCR3 (Zhang et al., 2020). In addition, injection of recombinant CXCL10 into old, injured muscle increased satellite cell proliferation, increased muscle growth and reduced fibrosis, showing recovery of many regenerative defects of old muscle (Zhang et al., 2020).
Some of the down-stream effects of CTL-derived IFNγ production in injured muscle may be exerted by Treg cells, perhaps by their actions on IFNRMs or an IFNRM-related population. Although depletion of Treg cells from young mice that experienced acute muscle injury by CTX injection increased total numbers of CD45+ cells in the muscle, the loss of Treg cells reduced the proportion of macrophages in a subpopulation characterized by low levels of major histocompatibility complex class II (MHCII) expression (MHCIIlow macrophages) and by expression of transcripts associated with both an M2-biased phenotype (e.g., Mrc1, Arg2, GDF3) and associated with an M1-biased phenotype (e.g., LPS responsive genes) (Panduro et al., 2018). Particularly significant, the MHCIIhigh macrophage subpopulation that was elevated in the injured muscles of Treg cell depleted mice showed elevated expression of IFNγ response genes (e.g, Stat1, Soc1, Jak2, Irf1) and elevated expression of transcripts associated with the M1-biased macrophage phenotype (e.g., Nos2, IL1a) (Panduro et al., 2018). The correlations that Treg cell deficiency led to an elevated presence of a macrophage subpopulation that expressed IFNγ response genes and that injured muscles in the Treg cell depleted mice showed poorer regeneration, supported the interpretation that loss of Treg cell regulation of IFNγ-mediated signaling in macrophages caused the regeneration defect. The interpretation was also supported by the finding that injection of IFNγ into mice experiencing muscle injury worsened muscle repair (Panduro et al., 2018). Whether the MHCIIhigh macrophages that express IFNγ response genes are the same macrophage population as the IFNRMs has not been tested.
Although the investigations noted above support the general conclusion that disruption of a Treg cell / CTL / macrophage IFNγ-signaling network in aging muscle underlies some of the regenerative defects in aging muscle, they also illustrate a chronic limitation of our understanding of the role of IFNγ in muscle growth, injury and repair: on one hand, loss of IFNRMs is associated with impaired regeneration (Zhang et al 2020) and on the other hand, increases in IFNγ-responsive macrophages are also associated with impaired regeneration (Panduro et al, 2018). The apparently conflicting results may reflect the dichotomous, dose-dependent effects of IFNγ on myogenesis. For example, IFNγ can act directly on muscle cells in vitro and inhibit their proliferation (Kalovidouris et al., 1993; Villalta et al., 2011). However, treating myoblasts in vitro with neutralizing Abs to IFNγ reduced myoblast proliferation (Cheng et al., 2008), suggesting that IFNγ promotes myoblast proliferation. These differing outcomes may reflect the dose dependency of IFNγ influences on muscle proliferation in which low concentrations increase proliferation (Kelic et al 1993), but high doses of IFNγ inhibit proliferation (Fisher et al., 1983).
Nevertheless, the significance of the Treg cell / CTL / macrophage IFNγ-signaling network in regeneration of aged muscle is that aging produces a selective loss in IFNRMs, which yields a reduction of CXCL10, a protein that improves regeneration and reduces fibrosis of injured muscle (Zhang et al., 2020). Thus, inflammaging has the potential to disrupt this pro-regenerative network in old skeletal muscle not only through the age-related loss of IFNRMs (Zhang et al., 2020), but also through the loss of CD133-expressing cells that are necessary for the normal recruitment of Treg cells to injured muscle (Kuswanto et al., 2016), in which they can regulate this newly identified, pro-regenerative signaling pathway.
5. Are beneficial effects of exercise on regeneration of aging muscle attributable in part to the influence of exercise on the immune system?
Because age-related changes in immune cells influence the regenerative capacity of aging muscle, any life-style changes that act on the immune system could also affect the ability of old muscle to regenerate. This possibility has been explored little, but nascent findings support speculation that some of the effects of exercise on the regeneration of aging muscle may partially reflect the influence of exercise on the immune system. Definitively, exercise improves the regenerative capacity of old muscles and much of the restorative activity of exercise reflects changes in the number and activation of satellite cells. Satellite cell numbers increase after exercise in aged, healthy muscles of rodents (Shefer et al., 2010; Shefer et al., 2013; Fujimaki et al., 2014; Joanisse et al., 2016; Cisterna et al., 2016; Englund et al., 2020) and those exercise-induced increases are associated with improved regeneration. For example, this was demonstrated using an 8-week progressive treadmill exercise protocol that increased Pax7+ cells in muscles of aged mice (Joanisse et al., 2016). Following the exercise protocol, TA muscles were injured by CTX injections and cross-sectional areas of muscle fibers in exercised, aging muscle were found significantly larger than non-exercised aging muscle and similar to that of young, non-exercised mice at 28 dpi (Joanisse et al., 2016). Similarly, aged TA muscles injured by BaCl2 injection after 4-weeks of voluntary wheel running showed increased numbers of muscle fibers expressing embryonic myosin heavy chain, an index of regeneration, compared to age-matched muscle without running (Brett et al., 2020).
But, are any of these beneficial effects of exercise on old muscle mediated by the immune system? Exercise has substantial effects on the levels of expression in aging muscle of cytokines that can influence muscle regeneration and satellite cell proliferation and differentiation, including IL6 and TNFα (Lavin et al., 2020). In addition, the age-related increases in TNFα mRNA and protein in human muscle were decreased by 3-months of resistance exercise (Greiwe et al., 2001). However, muscle cells themselves are major sources of both IL6 and TNFα whether the exercise-effects acted significantly on leukocytes within the muscle is unknown. Nevertheless, more specific effects of exercise on leukocyte-derived cytokines that influence regeneration have been demonstrated. Aged human subjects that were lifelong aerobic exercisers (over 50 years) showed higher levels of IL-10 mRNA in their muscles compared to aged, non-exercisers (Lavin et al., 2020). Given the powerful influence of IL10 in regulating the phenotype of intramuscular macrophages that influence muscle regeneration, the finding suggests a link between exercise, immunity and muscle regeneration. Although exercise can influence numbers of both M1-biased and M2-biased macrophages in old muscle (Reidy et al., 2018; Jensen et al., 2020), and both acute and chronic exercise induce shifts of macrophages toward an M2-biased phenotype in young, healthy muscle (Gordon et al., 2012; Minari et al., 2015) and adipose tissue (Kawanishi et al., 2010), whether shifts in macrophage phenotype in aged muscle is similarly affected by exercise is unknown. Likewise, whether different types of exercise, for example resistance versus endurance, can exert different influences on the regulatory role of the immune system on the regeneration of aging muscle remains wholly unexplored.
6. Surprises and beginnings in a newly-developing area of muscle biology.
We are at the very early stages of understanding how aging of the immune system can affect muscle regeneration and much of what we have learned thus far would have been unpredictable just a few years ago. Prior to the first investigations into muscle aging, regeneration and inflammation, the systemic changes in the immune system during inflammaging that produce a shift toward elevated production of Th1 cytokines (IL1β, IFNγ, TNFα) and a prevalence of pro-inflammatory leukocytes would have also been expected to occur in muscles of old animals. We have now learned that immune-related changes in old muscle only partly mirror those systemic changes. Although uninjured muscle shows elevated expression of some pro-inflammatory cytokines (IL1β) during muscle aging, the expression of some anti-inflammatory cytokines that are powerful modulators of muscle inflammation (IL10) are even more greatly increased. In addition, the primary population of immune cells in the non-injured, old muscle consists of macrophages that express phenotypic markers typically associated with an M2-biased, “pro-regenerative” phenotype. Furthermore, old, injured muscles in which regeneration is impaired display a greater amplification of M2-biased macrophage populations than occurs in young muscle. These observations are broadly significant to our understanding of the influence of aging of the immune system on muscle regeneration because they tell us that changes that occur systemically are not predictive of those that we will discover in aging muscle. They also show us the limitations of associating the expression of a small number of transcripts with a phenotypic descriptor that indicates function, such as “pro-inflammatory,” “pro-regenerative” or “reparative” macrophage populations.
We have also learned that age-related perturbations in the action or expression of some immune-cell-derived signaling molecules that were predicted to affect age-related changes in muscle regeneration (e.g., IFNγ and TNFα) do actually contribute to some of the defects in repair and function of old muscle. However, other molecules that were completely unexpected to influence adult myogenesis were discovered to undergo changes in expression or function in aging immune cells and thereby influence muscle regeneration (e.g., GDF3, osteopontin, CXCL10, Klotho). Those new discoveries not only add to our knowledge of mechanisms through which aging of the immune system affects muscle regeneration, they also highlight the possibility that those mechanisms that were initially discovered in muscle may also be involved in regenerative defects in other aging or diseased tissues that have not yet been explored.
As studies of the influences of immune system aging on muscle regeneration advance, many basic questions will need to be addressed. For example, aging affects the earliest stages of lymphopoiesis and myelopoiesis which can have implications on the function of the immune system in old animals (Dorshkind et al., 2020). Do those age-related changes in the development of the immune system affect muscle regeneration? Does T-cell senescence or T-cell exhaustion during aging influence muscle regeneration? In addition, epigenetic modifications to genes that occur over the lifetime of an organism can produce persistent alterations in immune system function (van Beek et al., 2019). Could those epigenetic effects bear on defects in muscle regeneration in old age? Furthermore, age-related changes in the function of immune cells that have not yet been explored in the context of regenerative defects in old muscle, such as myeloid-derived suppressor cells, natural killer cells and dendritic cells, may also play significant but unknown roles in regenerative defects in aging muscle. Adding further to the immense list of unanswered questions, is the recently identified influence of the aging, gut microbiota that can affect muscle strength (Fielding et al., 2019). Could aging of the gut-muscle axis also affect muscle regeneration? Much work remains in the emerging field of muscle immunobiology.
Acknowledgements
The authors received support from the National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Institutes of Health Awards R01AR066036 and 1RO1AR075768 and National Institute on Aging, National Institutes of Health Award R01AG04114 (all to J.G.T.) and from National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Institutes of Health Award F31AR071783 (to IF) and from the Muscular Dystrophy Association, USA, Development Award #603201 (to SSW) and from the Fund for the Promotion of Joint International Research, Fostering Joint International Research, JSPS KAKENHI Grant No. JP17KK0073 (to EO).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahrens HE, Huettemeister J, Schmidt M, Kaether C, von Maltzahn J, 2018. Klotho expression is a prerequisite for proper muscle stem cell function and regeneration of skeletal muscle. Skeletal Muscle 8, 20 10.1186/s13395-018-0166-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allbrook DB, Han MF, Hellmuth AE, 1971. Population of muscle satellite cells in relation to age and mitotic activity. Pathology 3, 223–243. 10.3109/00313027109073739. [DOI] [PubMed] [Google Scholar]
- Allen RE, Boxhorn LK, 1989. Regulation of skeletal muscle satellite cell proliferation and differentiation by transforming growth factor-beta, insulin-like growth factor I, and fibroblast growth factor. J. Cell. Physiol 138, 311–315. 10.1002/jcp.1041380213. [DOI] [PubMed] [Google Scholar]
- Arnold L, Henry A, Poron F, Baba-Amer Y, van Rooijen N, Plonquet A, Gherardi RK, Chazaud B, 2007. Inflammatory monocytes recruited after skeletal muscle injury switch into 38nti-inflammatory macrophages to support myogenesis. J. Exp. Med 204, 1057–1069. 10.1084/jem.20070075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashkar S, Weber GF, Panoutsakopoulou V, Sanchirico ME, Jansson M, Zawaideh S, Rittling SR, Denhardt DT, Glimcher MJ, Cantor H, 2000. Eta-1 (osteopontin): an early component of type-1 (cell-mediated) immunity. Science 287, 860–864. 10.1126/science.287.5454.860. [DOI] [PubMed] [Google Scholar]
- Barton-Davis ER, Shoturma DI, Musaro A, Rosenthal N, Sweeney HL, 1998. Viral mediated expression of insulin-like growth factor I blocks the aging-related loss of skeletal muscle function. Proc. Natl. Acad. Sci. U S A 95, 15603–15607. 10.1073/pnas.95.26.15603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bencze M, Negroni E, Vallese D, Yacoub-Youssef H, Chaouch S, Wolff A, Aamiri A, Di Santo JP, Chazaud B, Butler-Browne G, et al. , 2012. Proinflammatory macrophages enhance the regenerative capacity of human myoblasts by modifying their kinetics of proliferation and differentiation. Mol. Ther 20, 2168–2179. 10.1038/mt.2012.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bickel CS, Cross JM, Bamman MM, 2011. Exercise dosing to retain resistance training adaptations in young and older adults. Med. Sci. Sports Exerc 43, 1177–1187. 10.1249/MSS.0b013e318207c15d.PMID:21131862. [DOI] [PubMed] [Google Scholar]
- Bintliff S, Walker BE, 1960. Radioautographic study of skeletal muscle regeneration. Amer. J. Anat 106, 233–245. 10.1002/aja.1001060304. [DOI] [Google Scholar]
- Brack AS, Bildsoe H, Hughes SM, 2005. Evidence that satellite cell decrement contributes to preferential decline in nuclear number from large fibres during murine age-related muscle atrophy. J. Cell Sci 118, 4813–4821. 10.1242/jcs.02602. [DOI] [PubMed] [Google Scholar]
- Brack AS, Conboy MJ, Roy S, Lee M, Kuo CJ, Keller C, Rando TA, 2007. Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science 317, 807–810. 10.1126/science.1144090. [DOI] [PubMed] [Google Scholar]
- Brett JO, Arjona M, Ikeda M, Quarta M, de Morrée A, Egner IM, Perandini LA, Ishak HD, Goshayeshi A, Benjamin DI, et al. , 2020. Exercise rejuvenates quiescent skeletal muscle stem cells in old mice through restoration of Cyclin D1. Nat. Metab 2, 307–317. 10.1038/s42255-020-0190-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brigitte M, Schilte C, Plonquet A, Baba-Amer Y, Henri A, Charlier C, Tajbakhsh S, Albert M, Gherardi RK, Chrétien F, 2010. Muscle resident macrophages control the immune cell reaction in a mouse model of notexin-induced myoinjury. Arthritis. Rheum 62, 268–279. 10.1002/art.27183. [DOI] [PubMed] [Google Scholar]
- Brooks SV, Faulkner JA, 1988. Contractile properties of skeletal muscles from young, adult and aged mice. J. Physiol 404, 71–82. 10.1113/jphysiol.1988.sp017279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks SV, Faulkner JA, 1996. The magnitude of the initial injury induced by stretches of maximally activated muscle fibres of mice and rats increases in old age. J. Physiol 497, 573–580. 10.1113/jphysiol.1996.sp021790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broussard SR, McCusker RH, Novakofski JE, Strle K, Shen WH, Johnson RW, Dantzer R, Kelley KW, 2004. IL-1β impairs insulin-like growth factor I-induced differentiation and downstream activation signals of the insulin-like growth factor I receptor in myoblasts. J. Immunol 172, 7713–7720. 10.4049/jimmunol.172.12.7713. [DOI] [PubMed] [Google Scholar]
- Bryer SC, Fantuzzi G, Van Rooijen N, Koh TJ, 2008. Urokinase-type plasminogen activator plays essential roles in macrophage chemotaxis and skeletal muscle regeneration. J. Immunol 180, 1179–1188. 10.4049/jimmunol.180.2.1179. [DOI] [PubMed] [Google Scholar]
- Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, Sefik E, Tan TG, Wagers AJ, Benoist C, et al. , 2013. A special population of regulatory T cells potentiates muscle repair. Cell 155, 1282–1295. 10.1016/j.cell.2013.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carden DL, Smith JK, Korthuis RJ, 1990. Neutrophil-mediated microvascular dysfunction in postischemic canine skeletal muscle. Role of granulocyte adherence. Circ. Res 66, 1436–1444. 10.1161/01.res.66.5.1436. [DOI] [PubMed] [Google Scholar]
- Carlson BM, Faulkner JA, 1989. Muscle transplantation between young and old rats: age of host determines recovery. Am. J. Physiol 19256, C1262–C1266. 10.1152/ajpcell.1989.256.6.C1262. [DOI] [PubMed] [Google Scholar]
- Carlson BM, Faulkner JA, 1996. The regeneration of noninnervated muscle grafts and marcaine-treated muscles in young and old rats. J. Gerontol. A. Biol. Sci. Med. Sci 51, B43–B49. 10.1093/gerona/51a.1.b43. [DOI] [PubMed] [Google Scholar]
- Carlson BM, Faulkner JA, 1998. Muscle regeneration in young and old rats: effects of motor nerve transection with and without marcaine treatment. J. Gerontol. A. Biol. Sci. Med. Sci 53, B52–B57. 10.1093/gerona/53a.1.b52. [DOI] [PubMed] [Google Scholar]
- Castiglioni A, Corna G, Rigamonti E, Basso V, Vezzoli M, Monno A, Almada AE, Mondino A, Wagers AJ, Manfredi AA, et al. , 2015. FOXP3+ T Cells recruited to sites of sterile skeletal muscle injury regulate the fate of satellite cells and guide effective tissue regeneration. PLoS One 10, e0128094 10.1371/journal.pone.0128094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chargé SB, Rudnicki MA, 2004. Cellular and molecular regulation of muscle regeneration. Physiol. Rev 84, 209–238. 10.1152/physrev.00019.2003. [DOI] [PubMed] [Google Scholar]
- Chelvarajan RL, Collins SM, Van Willigen JM, Bondada S, 2005. The unresponsiveness of aged mice to polysaccharide antigens is a result of a defect in macrophage function. J. Leukoc. Biol 77, 503–512. 10.1189/jlb.0804449. [DOI] [PubMed] [Google Scholar]
- Chen L, Wang S, Wang Y, Zhang W, Ma K, Hu C, Zhu H, Liang S, Liu M, Xu N, 2018. IL-6 influences the polarization of macrophages and the formation and growth of colorectal tumor. Oncotarget 9, 17443–17454. 10.18632/oncotarget.24734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SE, Gerken E, Zhang Y, Zhan M, Mohan RK, Li AS, Reid MB, Li YP, 2005. Role of TNF-α signaling in regeneration of cardiotoxin-injured muscle. Am. J. Physiol. Cell Physiol 289, C1179–C1187. 10.1152/ajpcell.00062.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng M, Nguyen MH, Fantuzzi G, Koh TJ, 2008. Endogenous interferon-gamma is required for efficient skeletal muscle regeneration. Am. J. Physiol. Cell Physiol 294, C1183–C1191. 10.1152/ajpcell.00568.2007. [DOI] [PubMed] [Google Scholar]
- Cisterna B, Giagnacovo M, Costanzo M, Fattoretti P, Zancanaro C, Pellicciari C, Malatesta M, 2016. Adapted physical exercise enhances activation and differentiation potential of satellite cells in the skeletal muscle of old mice. J. Anat 228, 771–783. doi: 10.1111/joa.12429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coletti D, Moresi V, Adamo S, Molinaro M, Sassoon D, 2005. Tumor necrosis factor-alpha gene transfer induces cachexia and inhibits muscle regeneration. Genesis 43, 120–128. 10.1002/gene.20160. [DOI] [PubMed] [Google Scholar]
- Collins RA, Grounds MD, 2001. The role of tumor necrosis factor-alpha (TNF-α) in skeletal muscle regeneration: studies in TNF-α(−/−) and TNF-α(−/−)/LT-α(−/−) mice. J. Histochem. Cytochem 49, 989–1001. 10.1177/002215540104900807. [DOI] [PubMed] [Google Scholar]
- Conboy IM, Conboy MJ, Wagers AJ, Girma ER, Weissman IL, Rando TA, 2005. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature 433, 760–764. 10.1038/nature03260. [DOI] [PubMed] [Google Scholar]
- Coolican SA, Samuel DS, Ewton DZ, McWade FJ, Florini JR, 1997. The mitogenic and myogenic actions of insulin-like growth factors utilize distinct signaling pathways. J. Biol. Chem 272, 6653–6662. 10.1074/jbc.272.10.6653. [DOI] [PubMed] [Google Scholar]
- Cornelison DD Olwin BB, Rudnicki MA, Wold BJ, 2000. MyoD(−/−) satellite cells in single-fiber culture are differentiation defective and MRF4 deficient. Dev. Biol 224, 122–137. 10.1006/dbio.2000.9682. [DOI] [PubMed] [Google Scholar]
- Cornelison DDW, Wold BJ, 1997. Single-cell analysis of regulatory gene expression in quiescent and activated mouse skeletal muscle satellite cells. Dev. Biol 191, 270–283. 10.1006/dbio.1997.8721. [DOI] [PubMed] [Google Scholar]
- Corpas E, Harman SM, Blackman MR, 1993. Human growth hormone and human aging. Endocr. Rev 14, 20–39. 10.1210/edrv-14-1-20. [DOI] [PubMed] [Google Scholar]
- Creuzet S, Lescaudron L, Li Z, Fontaine-Pérus J, 1998. MyoD, myogenin, and desmin-nls-lacZ transgene emphasize the distinct patterns of satellite cell activation in growth and regeneration. J. Exp. Cell Res 243, 241–53. 10.1006/excr.1998.4100. [DOI] [PubMed] [Google Scholar]
- Cui CY, Driscoll RK, Piao Y, Chia CW, Gorospe M, Ferrucci L, 2019. Skewed macrophage polarization in aging skeletal muscle. Aging Cell 18, e13032 10.1111/acel.13032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng B, Wehling-Henricks M, Villalta SA, Wang Y, Tidball JG, 2012. IL-10 triggers changes in macrophage phenotype that promote muscle growth and regeneration. J. Immunol 189, 3669–3680. 10.4049/jimmunol.1103180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denhardt DT, Noda M, 1998. Osteopontin expression and function: role in bone remodeling. J. Cell. Biochem S30–S31, 92–102. . [DOI] [PubMed] [Google Scholar]
- Denhardt DT, Noda M, O’Regan AW, Pavlin D, Berman JS, 2001. Osteopontin as a means to cope with environmental insults: regulation of inflammation, tissue remodeling, and cell survival. J. Clin. Invest 107, 1055–1061. 10.1172/JCI12980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis RA, Trappe TA, Simpson P, Carroll C, Huang BE, Nagarajan R, Bearden E, Gurley C, Duff GW, Evans WJ, et al. , 2004. Interleukin-1 polymorphisms are associated with the inflammatory response in human muscle to acute resistance exercise. J. Physiol 560, 617–626. 10.1113/jphysiol.2004.067876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rossi M, Bernasconi P, Baggi F, de Waal Malefyt R, Mantegazza R, 2000. Cytokines and chemokines are both expressed by human myoblasts: possible relevance for the immune pathogenesis of muscle inflammation. Int. Immunol 12, 1329–1335. 10.1093/intimm/12.9.1329. [DOI] [PubMed] [Google Scholar]
- Deyhle MR, Hyldahl RD, 2018. The role of T lymphocytes in skeletal muscle repair from traumatic and contraction-induced injury. Front. Physiol 9, 768 10.3389/fphys.2018.00768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorshkind K, Höfer T, Montecino-Rodriguez E, Pioli PD, Rodewald HR, 2020. Do haematopoietic stem cells age? Nature Rev. Immunol 20, 196–202. doi: 10.1038/s41577-019-0236-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duluc D, Delneste Y, Tan F, Moles MP, Grimaud L, Lenoir J, Preisser L, Anegon I, Catala L, Ifrah N, et al. , 2007. Tumor-associated leukemia inhibitory factor and IL-6 skew monocyte differentiation into tumor-associated macrophage-like cells. Blood 110, 4319–4330. 10.1182/blood-2007-02-072587. [DOI] [PubMed] [Google Scholar]
- Dumont NA, Bentzinger CF, Sincennes MC, Rudnicki MA, 2015. Satellite cells and skeletal muscle regeneration. Compr. Physiol 5, 1027–1059. 10.1002/cphy.c140068. [DOI] [PubMed] [Google Scholar]
- Englund DA, Murach KA, Dungan CM, Figueiredo VC, Vechetti IJ Jr., Dupont-Versteegden EE, McCarthy JJ, Peterson CA, 2020. Depletion of resident muscle stem cells negatively impacts running volume, physical function, and muscle fiber hypertrophy in response to lifelong physical activity. Am. J. Physiol. Cell. Physiol 318, C1178–C1188. 10.1152/ajpcell.00090.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fielding RA, Manfredi TJ, Ding W, Fiatarone MA, Evans WJ, Cannon JG,1993. Acute phase response in exercise. III. Neutrophil and IL-1 beta accumulation in skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol 265, R166–R172. 10.1152/ajpregu.1993.265.1.R166. [DOI] [PubMed] [Google Scholar]
- Fielding RA, Reeves AR, Jasuja R, Liu C, Barrett BB, Lustgarten MS, 2019. 2019 Muscle strength is increased in mice that are colonized with microbiota from high-functioning older adults. Exp. Gerontol 127:110722. doi: 10.1016/j.exger.2019.110722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher PB, Miranda AF, Babiss LE, Pestka S, Weinstein IB, 1983. Opposing effects of interferon produced in bacteria and of tumor promoters on myogenesis in human myoblast cultures. Proc. Natl. Acad. Sci. USA 80, 2961–2965. 10.1073/pnas.80.10.2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry CS, Lee JD, Mula J, Kirby TJ, Jackson JR, Liu F, Yang L, Mendias CL, Dupont-Versteegden EE, McCarthy JJ, et al. , 2015. Inducible depletion of satellite cells in adult, sedentary mice impairs muscle regenerative capacity without affecting sarcopenia. Nat. Med 21, 76–80. 10.1038/nm.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X, Xiao J, Wei Y, Li S, Liu Y, Yin J, Sun K, Sun H, Wang H, Zhang Z, et al. , 2015. Combination of inflammation-related cytokines promotes long-term muscle stem cell expansion. Cell Res. 25, 655–673. 10.1038/cr.2015.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchtbauer EM, Westphal H, 1992. MyoD and myogenin are co-expressed in regenerating skeletal muscle of the mouse. Dev. Dyn 193, 34–39. 10.1002/aja.1001930106. [DOI] [PubMed] [Google Scholar]
- Fujimaki S, Hidaka R, Asashima M, Takemasa T, Kuwabara T, 2014. Wnt proteinmediated satellite cell conversion in adult and aged mice following voluntary wheel running. J. Biol. Chem 289, 7399–7412. 10.1074/jbc.M113.539247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulop T, Larbi A, Douziech N, Fortin C, Guerard KP, Lesur O, Khalil A, Dupuis G, 2004. Signal transduction and functional changes in neutrophils with aging. Aging Cell 3, 217–226. 10.1111/j.1474-9728.2004.00110.x [DOI] [PubMed] [Google Scholar]
- Gattazzo F, Laurent B, Relaix F, Rouard H, Didier N, 2020. Distinct Phases of Postnatal Skeletal Muscle Growth Govern the Progressive Establishment of Muscle Stem Cell Quiescence. Stem Cell Rep. 15, 597–611. 10.1016/j.stemcr.2020.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson MC, Schultz E, 1983. Age-related differences in absolute numbers of skeletal muscle satellite cells. Muscle Nerve 6, 574–580. 10.1002/mus.880060807. [DOI] [PubMed] [Google Scholar]
- Giordani L, Parisi A, Le Grand F, 2018. Satellite cell self-renewal. Curr. Top. Dev. Biol 126, 177–203. 10.1016/bs.ctdb.2017.08.001. [DOI] [PubMed] [Google Scholar]
- Gordon PM, Liu D, Sartor MA, IglayReger HB, Pistilli EE, Gutmann L, Nader GA, Hoffman EP, 2012. Resistance exercise training influences skeletal muscle immune activation: a microarray analysis. J. Appl. Physiol 112, 443–453. 10.1152/japplphysiol.00860.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greiwe JS, Cheng B, Rubin DC, Yarasheski KE, Semenkovich CF, 2001. Resistance exercise decreases skeletal muscle tumor necrosis factor alpha in frail elderly humans. FASEB J. 15, 475–482. 10.1096/fj.00-0274com. [DOI] [PubMed] [Google Scholar]
- Grounds MD, Garrett KL, Lai MC, Wright WE, Beilharz MW, 1992. Identification of skeletal muscle precursor cells in vivo by use of MyoD1 and myogenin probes. Cell Tissue Res. 267, 99–104. 10.1007/BF00318695. [DOI] [PubMed] [Google Scholar]
- Gutmann E, Carlson BM, 1976. Regeneration and transplantation of muscles in old rats and between young and old rats. Life Sci. 18, 109–114. 10.1016/00243205(76)90280-0. [DOI] [PubMed] [Google Scholar]
- Guttridge DD, Albanese C, Reuther JY, Pestell RG, Baldwin AS Jr., 1999. NF-kappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol. Cell. Biol 19, 5785–5799. 10.1128/mcb.19.8.5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttridge DC, Mayo MW, Madrid LV, Wang CY, Baldwin AS Jr., 2000. NF-kappaB-induced loss of MyoD messenger RNA: possible role in muscle decay and cachexia. Science 289, 2363–2366. doi: 10.1126/science.289.5488.2363. [DOI] [PubMed] [Google Scholar]
- Haddad F, Zaldivar F, Cooper DM, Adams GR, 2005. IL-6-induced skeletal muscle atrophy. J. Appl. Physiol 98, 911–917. 10.1152/japplphysiol.01026.2004. [DOI] [PubMed] [Google Scholar]
- Halevy O, Piestun Y, Allouh MZ, Rosser BWC, Rinkevich Y, Reshef R, Rozenboim I, Wleklinski-Lee M, Yablonka-Reuveni Z, 2004. Pattern of Pax7 expression during myogenesis in the posthatch chicken establishes a model for satellite cell differentiation and renewal. Dev. Dyn 231, 489–502. 10.1002/dvdy.20151. [DOI] [PubMed] [Google Scholar]
- Hamada K, Vannier E, Sacheck JM, Witsell AL, Roubenoff R, 2005. Senescence of human skeletal muscle impairs the local inflammatory cytokine response to acute eccentric exercise. FASEB J. 19, 264–266. 10.1096/fj.03-1286fje. [DOI] [PubMed] [Google Scholar]
- Hammers DW, Merritt EK, Matheny RW Jr., Adamo ML, Walters TJ, Estep JS, Farrar RP, 2008. Functional deficits and insulin-like growth factor-I gene expression following tourniquet-induced injury of skeletal muscle in young and old rats. J. Appl. Physiol 105, 1274–1281. 10.1152/japplphysiol.90418.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasty P, Bradley A, Morris JH, Edmondson DG, Venuti JM, Olson EN, Klein WH, 1993. Muscle deficiency and neonatal death in mice with a targeted mutation in the myogenin gene. Nature 364, 501–506. 10.1038/364501a0. [DOI] [PubMed] [Google Scholar]
- Hidestrand M, Richards-Malcolm S, Gurley CM, Nolen G, Grimes B, Waterstrat A, Zant GV, Peterson CA, 2008. Sca-1-expressing nonmyogenic cells contribute to fibrosis in aged skeletal muscle. J. Gerontol. A Biol. Sci. Med. Sci 63, 566–579. 10.1093/gerona/63.6.566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata A, Masuda S, Tamura T, Kai K, Ojima K, Fukase A, Motoyoshi K, Kamakura K, Miyagoe-Suzuki Y, Takeda S, 2003. Expression profiling of cytokines and related genes in regenerating skeletal muscle after cardiotoxin injection: a role for osteopontin. Am. J. Pathol 163, 203–215. 10.1016/S0002-9440(10)63644-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoene M, Runge H, Haring HU, Schleicher ED, Weigert C, 2013. Interleukin-6 promotes myogenic differentiation of mouse skeletal muscle cells: role of the STAT3 pathway. Am. J. Physiol. Cell Physiol 304, C128–C136. 10.1152/ajpcell.00025.2012. [DOI] [PubMed] [Google Scholar]
- Hurley BF, 1995. Age, gender, and muscular strength. J. Gerontol. A Biol. Sci. Med. Sci 50 Spec No:41–44. 10.1093/gerona/50a.special_issue.41. [DOI] [PubMed] [Google Scholar]
- Jaenisch R, Bird A, 2003. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat. Genet 33 Suppl, 245–254. 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- Jennische E, Skottner A, Hansson HA, 1987. Satellite cells express the trophic factor IGF-I in regenerating skeletal muscle. Acta Physiol. Scand 129, 9–15. 10.1111/j.1748-1716.1987.tb08034.x. [DOI] [PubMed] [Google Scholar]
- Jensen SM, Bechshøft CJL, Heisterberg MF, Schjerling P, Andersen JL, Kjaer M, Mackey AL, 2020. Macrophage subpopulations and the acute inflammatory response of elderly human akeletal muscle to physiological resistance exercise. Front. Physiol 11:811. doi: 10.3389/fphys.2020.00811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joanisse S, Nederveen JP, Baker JM, Snijders T, Iacono C, Parise G, 2016. Exercise conditioning in old mice improves skeletal muscle regeneration. FASEB J. 30, 3256–3268. 10.1096/fj.201600143RR. [DOI] [PubMed] [Google Scholar]
- Kalovidouris AE, Plotkin Z, Graesser D, 1993. Interferon-gamma inhibits proliferation, differentiation, and creatine kinase activity of cultured human muscle cells. II. A possible role in myositis. J. Rheumatol 20, 1718–1723. [PubMed] [Google Scholar]
- Kawanishi N, Mizokami T, Niihara H, Yada K, Suzuki K, 2016. Neutrophil Depletion Attenuates Muscle Injury after Exhaustive Exercise. Med. Sci. Sports. Exerc 48, 1917–1924. 10.1249/MSS.0000000000000980. [DOI] [PubMed] [Google Scholar]
- Kawanishi N, Yano H, Yokogawa Y, Suzuki K, 2010. Exercise training inhibits inflammation in adipose tissue via both suppression of macrophage infiltration and acceleration of phenotypic switching from M1 to M2 macrophages in high-fat-diet-induced obese mice. Exerc. Immunol. Rev 16, 105–118. [PubMed] [Google Scholar]
- Kelić S, Olsson T, Kristensson K, 1993. Interferon-g promotes proliferation of rat skeletal muscle cells in vitro and alters their AChR distribution. J. Neurol. Sci 114, 62–67. 10.1016/0022-510x(93)90050-9. [DOI] [PubMed] [Google Scholar]
- Kelijman M, 1991. Age-related alterations of the growth hormone/insulin-like-growth-factor I axis. J. Am. Geriatr. Soc 39, 295–307. 10.1111/j.1532-5415.1991.tb01654.x. [DOI] [PubMed] [Google Scholar]
- Koh A, da Silva AP, Bansal AK, Bansal M, Sun C, Lee H, Glogauer M, Sodek J, Zohar R, 2007. Role of osteopontin in neutrophil function. Immunology 122, 466–475. 10.1111/j.1365-2567.2007.02682.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korthuis RJ, Grisham MB, Granger DN, 1988. Leukocyte depletion attenuates vascular injury in postischemic skeletal muscle. Am. J. Physiol. Heart Circ. Physiol 254, H823–H827. 10.1152/ajpheart.1988.254.5.H823. [DOI] [PubMed] [Google Scholar]
- Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, et al. , 1997. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature, 390, 45–51. 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- Kurosu H, Yamamoto M, Clark JD, Pastor JV, Nandi A, Gurnani P, McGuinness OP, Chikuda H, Yamaguchi M, Kawaguchi H, et al. , 2005. Suppression of aging in mice by the hormone Klotho. Science 309, 1829–1833. 10.1126/science.1112766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuswanto W, Burzyn D, Panduro M, Wang KK, Jang YC, Wagers AJ, Benoist C, Mathis D, 2016. Poor repair of skeletal muscle in aging mice reflects a defect in local, interleukin-33-dependent accumulation of regulatory T cells. Immunity 44, 355–367. 10.1016/j.immuni.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyriakides C, Austen W, Wang Y, Favuzza J , Kobzik L, Moore FD, Hechtman HB, 1999. Skeletal muscle reperfusion injury is mediated by neutrophils and the complement membrane attack complex. Am. J. Physiol. Cell Physiol 277, C1263–C1268. 10.1152/ajpcell.1999.277.6.C1263. [DOI] [PubMed] [Google Scholar]
- Lamberts SWJ, Beld A.W. van den, Lely A.J. Van Der, 1997. The endocrinology of aging. Science 278, 419–424. 10.1126/science.278.5337.419. [DOI] [PubMed] [Google Scholar]
- Lang CH, Frost RA, Nairn AC, MacLean DA, Vary TC, 2002. TNF-alpha impairs heart and skeletal muscle protein synthesis by altering translation initiation. Am. J. Physiol. Endocrinol. Metab 282, E336–347. 10.1152/ajpendo.00366.2001. [DOI] [PubMed] [Google Scholar]
- Langen RC, Schols AM, Kelders MC, Wouters EF, Janssen-Heininger YM, 2001. Inflammatory cytokines inhibit myogenic differentiation through activation of nuclear factor-kapaB. FASEB J. 15, 1169–1180. 10.1096/fj.00-0463. [DOI] [PubMed] [Google Scholar]
- Langen RC, Van der Velden JL, Schols AM, Kelders MC, Wouters EF, Janssen-Heininger YM, 2004. Tumor necrosis factor-alpha inhibits myogenic differentiation through MyoD protein destabilization. FASEB J. 18, 227–237. 10.1096/fj.03-0251com. [DOI] [PubMed] [Google Scholar]
- Lavin KM, Perkins RK, Jemiolo B, Raue U, Trappe SW, Trappe TA, 2020. Effects of aging and lifelong aerobic exercise on basal and exercise-induced inflammation. J. Appl. Physiol 128, 87–99. https://doi.org/.1152/japplphysiol.00495.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Léger B, Derave W, De Bock K, Hespel P, Russell AP., 2008. Human sarcopenia reveals an increase in SOCS-3 and myostatin and a reduced efficiency of Akt phosphorylation. Rejuvenation Res. 11, 163–175B. 10.1089/rej.2007.0588. [DOI] [PubMed] [Google Scholar]
- Lemos DR, Babaeijandaghi F, Low M, Chang CK, Lee ST, Fiore D, Zhang RH, Natarajan A, Nedospasov SA, Rossi FM, 2015. Nilotinib reduces muscle fibrosis in chronic muscle injury by promoting TNF-mediated apoptosis of fibro/adipogenic progenitors. Nat Med. 21, 786–794. doi: 10.1038/nm.3869. [DOI] [PubMed] [Google Scholar]
- Lepper C, Conway SJ, Fan CM, 2009. Adult satellite cells and embryonic muscle progenitors have distinct genetic requirements. Nature 460, 627–631. 10.1038/nature08209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AJ, Brivanlou AH, 2006. GDF3 at the crossroads of TGF-beta signaling. Cell Cycle 5, 1069–1073. 10.4161/cc.5.10.2771. [DOI] [PubMed] [Google Scholar]
- Li S-A, Watanabe M, Yamada H, Nagai A, Kinuta M, Takei K, 2004. Immunohistochemical localization of Klotho protein in brain, kidney, and reproductive organs of mice. Cell Struct. Funct 29, 91–99. 10.1247/csf.29.91. [DOI] [PubMed] [Google Scholar]
- Li YP, 2003. TNF-alpha is a mitogen in skeletal muscle. Am. J. Physiol. Cell Physiol 285, C370–C376. 10.1152/ajpcell.00453.2002. [DOI] [PubMed] [Google Scholar]
- Locati M, Mantovani A, Sica A, 2013. Macrophage activation and polarization as an adaptive component of innate immunity. Adv. Immunol 120, 163–184. 10.1016/B978-0-12-417028-5.00006-5. [DOI] [PubMed] [Google Scholar]
- Lockhart NC, Brooks SV, 2008. Neutrophil accumulation following passive stretches contributes to adaptations that reduce contraction-induced skeletal muscle injury in mice. J. Appl. Physiol 104, 1109–1115. 10.1152/japplphysiol.00850.2007. [DOI] [PubMed] [Google Scholar]
- Lolmede K, Campana L, Vezzoli M, Bosurgi L, Tonlorenzi R, Clementi E, Bianchi ME, Cossu G, Manfredi AA, Brunelli S, Rovere-Querini P, 2009. Inflammatory and alternatively activated human macrophages attract vessel-associated stem cells, relying on separate HMGB1- and MMP-9-dependent pathways. J. Leukoc. Biol 85, 779–787. 10.1189/jlb.0908579. [DOI] [PubMed] [Google Scholar]
- Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G, 2013. The hallmarks of ageing. Cell 153, 1194–1217. 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Huang D, Saederup N, Charo IF, Ransohoff RM, Zhou L, 2011. Macrophages recruited via CCR2 produce insulin-like growth factor-1 to repair acute skeletal muscle injury. FASEB J. 25, 358–369. 10.1096/fj.10-171579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyu AK, Zhu SY, Chen JL, Zhao YX, Pu D, Luo C, Lyu Q, Fan Z, Sun Y, Wu J, et al. , 2019. Inhibition of TLR9 attenuates skeletal muscle fibrosis in aged sarcopenic mice via the p53/SIRT1 pathway. Exp. Gerontol 122, 25–33. 10.1016/j.exger.2019.04.008. [DOI] [PubMed] [Google Scholar]
- Many GM, Yokosaki Y, Uaesoontrachoon K, Nghiem PP, Bello L, Dadgar S, Yin Y, Damsker JM, Cohen HB, Kornegay JN, et al. , 2016. OPN-a induces muscle inflammation by increasing recruitment and activation of pro-inflammatory macrophages. Exp. Physiol 101, 1285–1300. 10.1113/EP085768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcondes MC, Poling M, Watry DD, Hall D, Fox HS, 2008. In vivo osteopontin-induced macrophage accumulation is dependent on CD44 expression. Cell. Immunol 254, 56–62. 10.1016/j.cellimm.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez CO, McHale MJ, Wells JT, Ochoa O, Michalek JE, McManus LM, Shireman PK, 2010. Regulation of skeletal muscle regeneration by CCR2-activating chemokines is directly related to macrophage recruitment. Am. J. Physiol. Regul. Integr. Comp. Physiol 299, R832–R842. 10.1152/ajpregu.00797.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura Y, Aizawa H, Shiraki-Iida T, Nagai R, Kuro-o M, Nabeshima Y, 1998. Identification of the human Klotho gene and its two transcripts encoding membrane and secreted Klotho protein. Biochem. Biophys. Res. Comm 242, 626–630. 10.1006/bbrc.1997.8019. [DOI] [PubMed] [Google Scholar]
- Mauro A, 1961. Satellite cell of skeletal muscle fibers. J. Biophys. Biochem. Cytol 9, 493–495. 10.1083/jcb.9.2.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLennan IS, 1996. Degenerating and regenerating skeletal muscles contain several subpopulations of macrophages with distinct spatial and temporal distributions. J. Anat 188, 17–28. [PMC free article] [PubMed] [Google Scholar]
- Miller KJ, Thaloor D, Matteson S, Pavlath GK, 2000. Hepatocyte growth factor affects satellite cell activation and differentiation in regenerating skeletal muscle. Am. J. Physiol. Cell Physiol 278, C174–C181. 10.1152/ajpcell.2000.278.1.C174. [DOI] [PubMed] [Google Scholar]
- Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM, 2000. M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol 164, 6166–6173. 10.4049/jimmunol.164.12.6166. [DOI] [PubMed] [Google Scholar]
- Minari AL, Oyama LM, Dos Santos RV, 2015. Downhill exercise-induced changes in gene expression related with macrophage polarization and myogenic cells in the triceps long head of rats. Inflammation 38, 209–217. 10.1007/s10753-014-0024-x. [DOI] [PubMed] [Google Scholar]
- Moratal C, Raffort J, Arrighi N, Rekima S, Schaub S, Dechesne CA, Chinetti G, Dani C, 2018. IL-1β- and IL-4-polarized macrophages have opposite effects on adipogenesis of intramuscular fibro-adipogenic progenitors in humans. Sci. Rep 8, 17005 10.1038/s41598-018-35429-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskalev AA, Shaposhnikov MV, Plyusnina EN, Zhavoronkov A, Bu-dovsky A, Yanai H, Fraifeld VE, 2013. The role of DNA damage and repair in aging through the prism of Koch-like criteria. Ageing Res. Rev 12, 661–684. 10.1016/j.arr.2012.02.001. [DOI] [PubMed] [Google Scholar]
- Mosser DM, Zhang X, 2008. Interleukin-10: new perspectives on an old cytokine. Immunol. Rev 226, 205–218. 10.1111/j.1600-065X.2008.00706.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz-Cánoves P, Neves J, Sousa-Victor P, 2020. Understanding muscle regenerative decline with aging: new approaches to bring back youthfulness to aged stem cells. FEBS J. 287, 406–416. 10.1111/febs.15182. [DOI] [PubMed] [Google Scholar]
- Muñoz-Cánoves P, Scheele C, Pedersen BK, Serrano AL, 2013. Interleukin-6 myokine signaling in skeletal muscle: a double-edged sword? FEBS J. 280, 4131–4148. 10.1111/febs.12338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, et al. , 2014. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41, 14–20. 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musarò A, McCullagh K, Paul A, Houghton L, Dobrowolny G, Molinaro M, Barton ER, Sweeney HL, Rosenthal N, 2001. Localized Igf-1 transgene expression sustains hypertrophy and regeneration in senescent skeletal muscle. Nat. Genet 27, 195–200. 10.1038/84839. [DOI] [PubMed] [Google Scholar]
- Nguyen HX, Lusis AJ, Tidball JG, 2005. Null mutation of myeloperoxidase in mice prevents mechanical activation of neutrophil lysis of muscle cell membranes in vitro and in vivo. J. Physiol 565, 403–413. 10.1113/jphysiol.2005.085506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HX, Tidball JG, 2003a. Null mutation of gp91phox reduces muscle membrane lysis during muscle inflammation in mice. J. Physiol 553, 833–841. 10.1113/jphysiol.2003.051912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HX, Tidball JG, 2003b. Interactions between neutrophils and macrophages promote macrophage killing of rat muscle cells in vitro. J. Physiol 547, 125–132. 10.1113/jphysiol.2002.031450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholas J, Voss JG, Tsuji J, Fulkerson ND, Soulakova J, Schneider BS, 2015. Time course of chemokine expression and leukocyte infiltration after acute skeletal muscle injury in mice. Innate Immun. 21, 266–274. 10.1177/1753425914527326. [DOI] [PubMed] [Google Scholar]
- Olguin HC, Olwin BB, 2004. Pax-7 up-regulation inhibits myogenesis and cell cycle progression in satellite cells: a potential mechanism for self-renewal. Dev. Biol 275, 375–388. 10.1016/j.ydbio.2004.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Regan AW, Nau GJ, Chupp GL, Berman JS, 2000. Osteopontin (Eta-1) in cell-mediated immunity: teaching an old dog new tricks. Immunol. Today 21, 475–478. 10.1016/s0167-5699(00)01715-1. [DOI] [PubMed] [Google Scholar]
- Orimo S, Hiyamuta E, Arahata K, Sugita H, 1991. Analysis of inflammatory cells and complement C3 in bupivacaine-induced myonecrosis. Muscle Nerve 14, 515–520. 10.1002/mus.880140605. [DOI] [PubMed] [Google Scholar]
- Palacios D, Mozzetta C, Consalvi S, Caretti G, Saccone V, Proserpio V, Marquez VE, Valente S, Mai A, Forcales SV, et al. 2010. TNF/p38α/polycomb signaling to Pax7 locus in satellite cells links inflammation to the epigenetic control of muscle regeneration. Cell Stem Cell 7, 455–469. 10.1016/j.stem.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paliwal P, Pishesha N, Wijaya D, Conboy IM, 2012. Age dependent increase in the levels of osteopontin inhibits skeletal muscle regeneration. Aging 4, 553–566. 10.18632/aging.100477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer RM, Thompson MG, Knott RM, Campbell GP, Thom A, Morrison KS, 1997. Insulin and insulin-like growth factor-I responsiveness and signalling mechanisms in C2C12 satellite cells: effect of differentiation and fusion. Biochim. Biophys. Acta 1355, 167–176. 10.1016/s0167-4889(96)00127-9. [DOI] [PubMed] [Google Scholar]
- Panduro M, Benoist C, Mathis D, 2018. T(reg) cells limit IFN-γ production to control macrophage accrual and phenotype during skeletal muscle regeneration. Proc. Natl. Acad. Sci. USA 115, E2585–E2593. 10.1073/pnas.1800618115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patarca R, Freeman GJ, Singh RP, Wei FY, Durfee T, Blattner F, Regnier DC, Kozak CA, Mock BA, Morse HC, 1989. Structural and functional studies of the early T lymphocyte activation 1 (Eta-1) gene. Definition of a novel T cell-dependent response associated with genetic resistance to bacterial infection. J. Exp. Med 170, 145–161. 10.1084/jem.170.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patsalos A, Simandi Z, Hays TT, Peloquin M, Hajian M, Restrepo I, Coen PM, Russell AJ, Nagy L, 2018. In vivo GDF3 administration abrogates aging related muscle regeneration delay following acute sterile injury. Aging Cell 17, e12815 10.1111/acel.12815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peake J, Della Gatta P, Cameron-Smith D, 2010. Aging and its effects on inflammation in skeletal muscle at rest and following exercise-induced muscle injury. Am. J. Physiol. Regul. Integr. Comp. Physiol 298, R1485–R1495. 10.1152/ajpregu.00467.2009. [DOI] [PubMed] [Google Scholar]
- Perdiguero E, Sousa-Victor P, Ruiz-Bonilla V, Jardí M, Caelles C, Serrano AL, Muñoz-Cánoves P, 2011. p38/MKP-1-regulated AKT coordinates macrophage transitions and resolution of inflammation during tissue repair. J. Cell Biol 195, 307–322. 10.1083/jcb.201104053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira RO, Carvalho SN, Stumbo AC, Rodrigues CAB, Porto LC, Moura AS, Carvalho L, 2006. Osteopontin expression in coculture of differentiating rat fetal skeletal fibroblasts and myoblasts. In Vitro Cell. Dev. Biol. Anim 42, 4–7. 10.1007/s11626-006-0003-0. [DOI] [PubMed] [Google Scholar]
- Phillips T, Leeuwenburgh C, 2005. Muscle fiber specific apoptosis and TNF-alpha signaling in sarcopenia are attenuated by life-long calorie restriction. FASEB J. 19, 668–670. 10.1096/fj.04-2870fje. [DOI] [PubMed] [Google Scholar]
- Powell-Braxton L, Hollingshead P, Warburton C, Dowd M, Pitts-Meek S, Dalton D, Gillett N, Stewart TA, 1993. IGF-I is required for normal embryonic growth in mice. Genes Dev. 7, 2609–2617. 10.1101/gad.7.12b.2609. [DOI] [PubMed] [Google Scholar]
- Przybyla B, Gurley C, Harvey JF, Bearden E, Kortebein P, Evans WJ, Sullivan DH, Peterson CA, Dennis RA, 2006. Aging alters macrophage properties in human skeletal muscle both at rest and in response to acute resistance exercise. Exp. Gerontol 41, 320–327. 10.1016/j.exger.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Raue U, Slivka D, Jemiolo B, Hollon C, Trappe S, 2007. Proteolytic gene expression differs at rest and after resistance exercise between young and old women. J. Gerontol. A Biol. Sci. Med. Sci 62, 1407–1412. 10.1093/gerona/62.12.1407. [DOI] [PubMed] [Google Scholar]
- Reid MB, Li YP, 2001. Tumor necrosis factor-α and muscle wasting: a cellular perspective. Respir. Res 2, 269–272. 10.1186/rr67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reidy PT, Lindsay CC, McKenzie AI, Fry CS, Supiano MA, Marcus RL, LaStayo PC, Drummond MJ, 2018. Aging-related effects of bed rest followed by eccentric exercise rehabilitation on skeletal muscle macrophages and insulin sensitivity. Exp. Gerontol 107:37–49. doi: 10.1016/j.exger.2017.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reidy PT, Dupont-Versteegden EE, Drummond MJ, 2019a. Macrophage regulation of muscle regrowth from disuse in aging. Exerc. Sport Sci. Rev 47, 246–250. 10.1249/JES.0000000000000201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reidy PT, McKenzie AI, Mahmassani ZS, Petrocelli JJ, Nelson DB, Lindsay CC, Gardner JE, Morrow VR, Keefe AC, Huffaker TB, et al. , 2019b. Aging impairs mouse skeletal muscle macrophage polarization and muscle-specific abundance during recovery from disuse. Am. J. Physiol. Endocrinol. Metab 317, E85–E98. 10.1152/ajpendo.00422.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renault V, Thornell LE, Eriksson PO, Butler-Browne G, Mouly V, 2002. Regenerative potential of human skeletal muscle during aging. Aging Cell 1, 132–139. 10.1046/j.1474-9728.2002.00017.x. [DOI] [PubMed] [Google Scholar]
- Rider P, Carmi Y, Guttman O, Braiman A, Cohen I, Voronov E, White MR, Dinarello CA, Apte RN, 2011. IL-1α and IL-1β recruit different myeloid cells and promote different stages of sterile inflammation. J. Immunol 187, 4835–4843. 10.4049/jimmunol.1102048. [DOI] [PubMed] [Google Scholar]
- Rigamonti E, Touvier T, Clementi E, Manfredi AA, Brunelli S, Rovere-Querini P, 2013. Requirement of inducible nitric oxide synthase for skeletal muscle regeneration after acute damage. J. Immunol 190, 1767–1777. 10.4049/jimmunol.1202903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson TA, Maley MA, Grounds MD, Papadimitriou JM, 1993. The role of macrophages in skeletal muscle regeneration with particular reference to chemotaxis. Exp. Cell Res 207, 321–31. 10.1006/excr.1993.1199. [DOI] [PubMed] [Google Scholar]
- Sadeh M, 1988. Effects of aging on skeletal muscle regeneration. J. Neurol. Sci 87, 67–74. 10.1016/0022-510x(88)90055-x. [DOI] [PubMed] [Google Scholar]
- Sahu A, Mamiya H, Shinde SN, Cheikhi A, Winter LL, Vo NV, Stolz D, Roginskaya V, Tang WY, St Croix C, et al. , 2018. Age-related declines in α-Klotho drive progenitor cell mitochondrial dysfunction and impaired muscle regeneration. Nature Commun. 9, 4859 10.1038/s41467-018-07253-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambasivan R, Yao R, Kissenpfennig A, Van Wittenberghe L, Paldi A, Gayraud-Morel B, Guenou H, Malissen B, Tajbakhsh S, Galy A, 2011. Pax7-expressing satellite cells are indispensable for adult skeletal muscle regeneration. Development 138, 3647–3656. 10.1242/dev.067587. [DOI] [PubMed] [Google Scholar]
- Schmalbruch H, 1976. The morphology of regeneration of skeletal muscles in the rat. Tissue Cell. 8, 673–692. 10.1016/0040-8166(76)90039-2. [DOI] [PubMed] [Google Scholar]
- Schultz E, Lipton BH, 1982. Skeletal muscle satellite cells: changes in proliferation potential as a function of age. Mech. Ageing Dev 20, 377–383. 10.1016/00476374(82)90105-1. [DOI] [PubMed] [Google Scholar]
- Serrano AL, Baeza-Raja B, Perdiguero E, Jardí M, Muñoz-Cánoves P, 2008. Interleukin-6 is an essential regulator of satellite cell-mediated skeletal muscle hypertrophy. Cell Metab. 7, 33–44. 10.1016/j.cmet.2007.11.011. [DOI] [PubMed] [Google Scholar]
- Serrano AL, Muñoz-Cánoves P, 2010. Regulation and dysregulation of fibrosis in skeletal muscle. Exp. Cell Res 316, 3050–3058. 10.1016/j.yexcr.2010.05.035. [DOI] [PubMed] [Google Scholar]
- Shefer G, Rauner G, Stuelsatz P, Benayahu D, Yablonka-Reuveni Z, 2013. Moderate-intensity treadmill running promotes expansion of the satellite cell pool in young and old mice. FEBS J. 280, 4063–4073. 10.1111/febs.12228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shefer G, Rauner G, Yablonka-Reuveni Z, Benayahu D, 2010. Reduced satellite cell numbers and myogenic capacity in aging can be alleviated by endurance exercise. PLoS One 5, e13307 10.1371/journal.pone.0013307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Shi L, Wang X, He J, 2018. Regulatory roles of osteopontin in production of monocyte-origin MCP-1. Cell Transplant 27, 1185–1194. 10.1177/0963689718756070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shireman PK, Contreras-Shannon V, Ochoa O, Karia BP, Michalek JE, McManus LM, 2007. MCP-1 deficiency causes altered inflammation with impaired skeletal muscle regeneration. J. Leukoc. Biol 81, 775–785. 10.1189/jlb.0506356. [DOI] [PubMed] [Google Scholar]
- Shireman PK, Contreras-Shannon V, Reyes-Reyna SM, Robinson SC, McManus LM, 2006. MCP-1 parallels inflammatory and regenerative responses in ischemic muscle. J. Surg. Res 134, 145–157. 10.1016/j.jss.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Skullerud K, 1985. Variations in the size of the human brain. Influence of age, sex, body length, body mass index, alcoholism, Alzheimer changes, and cerebral atherosclerosis. Acta Neurol. Scand Suppl. 102, 1–94. [PubMed] [Google Scholar]
- Sloboda DD, Brown LA, Brooks SV, 2018. Myeloid cell responses to contraction-induced injury differ in muscles of young and old mice. J. Gerontol. A Biol. Sci. Med. Sci 73,1581–1590. 10.1093/gerona/gly086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smythe GM, Shavlakadze T, Roberts P, Davies MJ, McGeachie JK, Grounds MD, 2008. Age influences the early events of skeletal muscle regeneration: studies of whole muscle grafts transplanted between young (8 weeks) and old (13–21 months) mice. Exp. Gerontol 43, 550–562. 10.1016/j.exger.2008.02.005. [DOI] [PubMed] [Google Scholar]
- Snow MH, 1977. The effects of aging on satellite cells in skeletal muscles of mice and rats. Cell Tissue Res. 185, 399–408. 10.1007/bf00220299. [DOI] [PubMed] [Google Scholar]
- Soffe Z, Radley-Crabb HG, McMahon C, Grounds MD, Shavlakadze T, 2016. Effects of loaded voluntary wheel exercise on performance and muscle hypertrophy in young and old male C57Bl/6J mice. Scand. J. Med. Sci. Sports 26, 172–188. 10.1111/sms.12416. [DOI] [PubMed] [Google Scholar]
- Song J, Saeman MR, De Libero J, Wolf SE, 2015. Skeletal muscle loss is associated with TNF mediated insufficient skeletal myogenic activation after burn. Shock 44, 479–486. 10.1097/shk.0000000000000444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen JR, Kaluhiokalani JP, Hafen PS, Deyhle MR, Parcell AC, Hyldahl RD, 2019. An altered response in macrophage phenotype following damage in aged human skeletal muscle: implications for skeletal muscle repair. FASEB J. 33, 10353–10368. 10.1096/fj.201900519r. [DOI] [PubMed] [Google Scholar]
- Subramaniam K, Fallon K, Ruut T, Lane D, McKay R, Shadbolt B, Ang S, Cook M, Platten J, Pavli P, Taupin D 2015. Infliximab reverses inflammatory muscle wasting (sarcopenia) in Crohn’s disease. Aliment. Pharmacol. Ther 41(5):419–28. doi: 10.1111/apt.13058 [DOI] [PubMed] [Google Scholar]
- Summan M, Warren GL, Mercer RR, Chapman R, Hulderman T, VanRooijen N, Simeonova PP, 2006. Macrophages and skeletal muscle regeneration: a clodronatecontaining liposome depletion study. Am. J. Physiol. Regul. Integr. Comp. Physiol 290, R1488–R1495. 10.1152/ajpregu.00465.2005. [DOI] [PubMed] [Google Scholar]
- Tatsumi R, Hattori A, Ikeuchi Y, Anderson JE, Allen RE, 2002. Release of hepatocyte growth factor from mechanically stretched skeletal muscle satellite cells and role of pH and nitric oxide. Mol. Biol. Cell 13, 2909–2918. 10.1091/mbc.e02-01-0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidball JG 2005. Inflammatory processes in muscle injury and repair. Am. J. Physiol. Regul. Integr. Comp. Physiol 288, R345–R353. 10.1152/ajpregu.00454.2004. [DOI] [PubMed] [Google Scholar]
- Tidball JG, 2017. Regulation of muscle growth and regeneration by the immune system. Nat. Rev. Immunol 17, 165–178. 10.1038/nri.2016.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidball JG, St. Pierre BA, 1996. Apoptosis of macrophages during the resolution of muscle inflammation. J. Leukoc. Biol 59(3):380–388. doi: 10.1002/jlb.59.3.380 [DOI] [PubMed] [Google Scholar]
- Tidball JG, Villalta SA, 2010. Regulatory interactions between muscle and the immune system during muscle regeneration. Am. J. Physiol. Regul. Integr. Comp. Physiol 298, R1173–R1187. 10.1152/ajpregu.00735.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidball JG, Wehling-Henricks M, 2007. Macrophages promote muscle membrane repair and muscle fibre growth and regeneration during modified muscle loading in mice in vivo. J. Physiol 578, 327–336. 10.1113/jphysiol.2006.118265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonkin J, Temmerman L, Sampson RD, Gallego-Colon E, Barberi L, Bilbao D, Schneider MD, Musarò A, Rosenthal N, 2015. Monocyte/Macrophage-derived IGF-1 Orchestrates murine skeletal muscle regeneration and modulates autocrine polarization. Mol. Ther 23,1189–1200. 10.1038/mt.2015.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrente Y, Fahime EE, Caron NJ, Del Bo R, Belicchi M, Pisati F, Tremblay JP, Bresolin N, 2003. Tumor necrosis factor-α (TNF-α) stimulates chemotactic response in mouse myogenic cells. Cell Transplant 12, 91–100. 10.3727/000000003783985115. [DOI] [PubMed] [Google Scholar]
- Tortorella C, Simone O, Piazzolla G, Stella I, Antonaci S, 2007. Age-related impairment of GM-CSF-induced signalling in neutrophils: role of SHP-1 and SOCS proteins. Ageing Res. Rev 6, 81–93. 10.1016/j.arr.2006.10.001. [DOI] [PubMed] [Google Scholar]
- Toth KG, McKay BR, De Lisio M, Little JP, Tarnopolsky MA, Parise G, 2011. IL-6 induced STAT3 signalling is associated with the proliferation of human muscle satellite cells following acute muscle damage. PLoS One 6, e17392 10.1371/journal.pone.0017392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uaesoontrachoon K, Yoo HJ, Tudor EM, Pike RN, Mackie EJ, Pagel CN, 2008. Osteopontin and skeletal muscle myoblasts: association with muscle regeneration and regulation of myoblast function in vitro. Int. J. Biochem. Cell Biol 40, 2303–2314. 10.1016/j.biocel.2008.03.020. [DOI] [PubMed] [Google Scholar]
- van Beek AA, Van den Bossche J, Mastroberardino PG, de Winther MPJ, Leenen PJM, 2019. Metabolic alterations in aging macrophages: ingredients for inflammaging? Trends Immunol. 40(2):113–127. doi: 10.1016/j.it.2018.12.007. [DOI] [PubMed] [Google Scholar]
- Van der Poel C, Gosselin LE, Schertzer JD, Ryall JG, Swiderski K, Wondemaghen M, Lynch GS, 2011. Ageing prolongs inflammatory marker expression in regenerating rat skeletal muscles after injury. J. Inflamm 8, 41 10.1186/1476-9255-8-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga T, Mounier R, Patsalos A, Gogolák P, Peloquin M, Horvath A, Pap A, Daniel B, Nagy G, Pintye E, et al. , 2016. Macrophage PPARγ, a Lipid Activated Transcription Factor Controls the Growth Factor GDF3 and Skeletal Muscle Regeneration. Immunity 45, 1038–1051. 10.1016/j.immuni.2016.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzoli M, Castellani P, Campana L, Corna G, Bosurgi L, Manfredi AA, Bianchi ME, Rubartelli A, Rovere-Querini P, 2010. Redox remodeling: a candidate regulator of HMGB1 function in injured skeletal muscle. Ann. N.Y. Acad. Sci 1209, 83–90. 10.1111/j.1749-6632.2010.05748.x. [DOI] [PubMed] [Google Scholar]
- Villalta SA, Deng B, Rinaldi C, Wehling-Henricks M, Tidball JG, 2011. IFN-γ promotes muscle damage in the mdx mouse model of Duchenne muscular dystrophy by suppressing M2 macrophage activation and inhibiting muscle cell proliferation. J. Immunol 187, 5419–5428. 10.4049/jimmunol.1101267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalta SA, Nguyen HX, Deng B, Gotoh T, Tidball JG, 2009. Shifts in macrophage phenotypes and macrophage competition for arginine metabolism affect the severity of muscle pathology in muscular dystrophy. Hum. Mol. Genet 18, 482–496. 10.1093/hmg/ddn376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalta SA, Rosenthal W, Martinez L, Kaur A, Sparwasser T, Tidball JG, Margeta M, Spencer MJ, Bluestone JA, 2014. Regulatory T cells suppress muscle inflammation and injury in muscular dystrophy. Sci. Transl. Med 6, 258ra142 10.1126/scitranslmed.3009925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visser M, Pahor M, Taaffe DR, Goodpaster BH, Simonsick EM, Newman AB, Nevitt M, Harris TB, 2002. Relationship of interleukin-6 and tumor necrosis factor-alpha with muscle mass and muscle strength in elderly men and women: the Health ABC Study. J. Gerontol. A Biol. Sci. Med. Sci 57, M326–M332. 10.1093/gerona/57.5.m326. [DOI] [PubMed] [Google Scholar]
- Volkmann R, 1893. Uber die regeneration des quergestreiften muskelgewebes beim menschen und saugetier. Beitr. Pathol. Anat 12, 233–332. [Google Scholar]
- Wang H, Melton DW, Porter L, Sarwar ZU, McManus LM, Shireman PK, 2014. Altered macrophage phenotype transition impairs skeletal muscle regeneration. Am. J. Pathol 184, 1167–1184. 10.1016/j.ajpath.2013.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Li Y, Wang X, Wang P, Essandoh K, Cui S, Huang W, Mu X, Liu Z, Wang Y, et al. , 2020. GDF3 protects mice against sepsis-induced cardiac dysfunction and mortality by suppression of macrophage pro-Inflammatory phenotype. Cells 9, 120 10.3390/cells9010120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Wehling-Henricks M, Samengo G, Tidball JG, 2015. Increases of M2a macrophages and fibrosis in aging muscle are influenced by bone marrow aging and negatively regulated by muscle-derived nitric oxide. Aging Cell 14, 678–688. 10.1111/acel.12350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Wehling-Henricks M, Welc SS, Fisher AL, Zuo Q, Tidball JG, 2019. Aging of the immune system causes reductions in muscle stem cell populations, promotes their shift to a fibrogenic phenotype, and modulates sarcopenia. FASEB J. 33, 1415–1427. 10.1096/fj.201800973r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Welc SS, Wehling-Henricks M, Tidball JG, 2018. Myeloid cell-derived tumor necrosis factor-alpha promotes sarcopenia and regulates muscle cell fusion with aging muscle fibers. Aging Cell 17, e12828 10.1111/acel.12828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren GL, O’Farrell L, Summan M, Hulderman T, Mishra D, Luster MI, Kuziel WA, Simeonova PP, 2004. Role of CC chemokines in skeletal muscle functional restoration after injury. Am. J. Physiol. Cell Physiol 286, C1031–C1036. 10.1152/ajpcell.00467.2003. [DOI] [PubMed] [Google Scholar]
- Wasgewatte Wijesinghe DK, Mackie EJ, Pagel CN, 2019. Normal inflammation and regeneration of muscle following injury require osteopontin from both muscle and non-muscle cells. Skelet. Muscle 9, 6 10.1186/s13395-019-0190-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehling-Henricks M, Li Z, Lindsay C, Wang Y, Welc SS, Ramos JN, Khanlou N, Kuro-O M, Tidball JG, 2016. Klotho gene silencing promotes pathology in the mdx mouse model of Duchenne muscular dystrophy. Hum. Mol. Genet 25, 2465–2482. 10.1093/hmg/ddw111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehling-Henricks M, Welc SS, Samengo G, Rinaldi C, Lindsay C, Wang Y, Lee J, Kuro-O M, Tidball JG, 2018. Macrophages escape Klotho gene silencing in the mdx mouse model of Duchenne muscular dystrophy and promote muscle growth and increase satellite cell numbers through a Klotho-mediated pathway. Hum. Mol. Genet 27, 14–29. 10.1093/hmg/ddx380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisser SB, McLarren KW, Kuroda E, Sly LM, 2013. Generation and characterization of murine alternatively activated macrophages. Methods Mol. Biol 946, 225–239. 10.1007/978-1-62703-128-8_14. [DOI] [PubMed] [Google Scholar]
- Welc SS, Wehling-Henricks M, Antoun J, Ha TT, Tous I, Tidball JG, 2020a. Differential effects of myeloid cell PPARδ and IL-10 in regulating macrophage recruitment, phenotype, and regeneration following acute muscle Injury. J. Immunol 205, 1664–1677. 10.4049/jimmunol.2000247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welc SS, Wehling-Henricks M, Kuro-O M, Thomas KA, Tidball JG, 2020b. Modulation of Klotho expression in injured muscle perturbs Wnt signalling and influences the rate of muscle growth. Exp. Physiol 105, 132–147. 10.1113/ep088142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokota J, Minei JP, Fantini GA, Shires GT, 1989. Role of leukocytes in reperfusion injury of skeletal muscle after partial ischemia. Am. J. Physiol 257, H1068–H1075. 10.1152/ajpheart.1989.257.4.h1068. [DOI] [PubMed] [Google Scholar]
- Zacks SI, Sheff MF, 1982. Age-related impeded regeneration of mouse minced anterior tibial muscle. Muscle Nerve 5, 152–161. 10.1002/mus.880050213. [DOI] [PubMed] [Google Scholar]
- Zammit PS, Golding JP, Nagata Y, Hudon V, Partridge TA, Beauchamp JR, 2004. Muscle satellite cells adopt divergent fates: A mechanism for self-renewal? J. Cell Biol 166, 347–357. 10.1083/jcb.200312007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zammit PS, Heslop L, Hudon V, Rosenblatt JD, Tajbakhsh S, Buckingham ME, Beauchamp JR, Partridge TA, 2002. Kinetics of myoblast proliferation show that resident satellite cells are competent to fully regenerate skeletal muscle fibers. Exp. Cell Res 281, 39–49. 10.1006/excr.2002.5653. [DOI] [PubMed] [Google Scholar]
- Zammit PS, Relaix F, Nagata Y, Ruiz AP, Collins CA, Partridge TA, Beauchamp JR, 2006. Pax7 and myogenic progression in skeletal muscle satellite cells. J. Cell Sci 119, 1824–32. 10.1242/jcs.02908. [DOI] [PubMed] [Google Scholar]
- Zerba E, Komorowski TE, Faulkner JA, 1990. Free radical injury to skeletal muscles of young, adult, and old mice. Am. J. Physiol 258, C429–C435. 10.1152/ajpcell.1990.258.3.c429. [DOI] [PubMed] [Google Scholar]
- Zhang C, Cheng N, Qiao B, Zhang F, Wu J, Liu C, Li Y, Du J, 2020. Age-related decline of interferon-gamma responses in macrophage impairs satellite cell proliferation and regeneration J. Cachexia Sarcopenia Muscle Advanced online publication; 10.1002/jcsm.12584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Li Y, Wu Y, Wang L, Wang X, Du J, 2013. Interleukin-6/signal transducer and activator of transcription 3 (STAT3) pathway is essential for macrophage infiltration and myoblast proliferation during muscle regeneration. J. Biol. Chem 288, 1489–1499. 10.1074/jbc.M112.419788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Qu C, Li T, Cui W, Wang X, Du J, 2019. Phagocytosis mediated by scavenger receptor class BI promotes macrophage transition during skeletal muscle regeneration. J. Biol. Chem 294, 15672–15685. 10.1074/jbc.ra119.008795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Xiao Z, Qu C, Cui W, Wang X, Du J, 2014. CD8 T cells are involved in skeletal muscle regeneration through facilitating MCP-1 secretion and Gr1(high) macrophage infiltration. J. Immunol 193, 5149–5160. 10.4049/jimmunol.1303486. [DOI] [PubMed] [Google Scholar]
- Zhao W, Lu H, Wang X, Ransohoff RM, Zhou L, 2016. CX3CR1 deficiency delays acute skeletal muscle injury repair by impairing macrophage functions. FASEB J. 30, 380–393. 10.1096/fj.14-270090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu B, Suzuki K, Goldberg HA, Rittling SR, Denhardt DT, McCulloch CA, Sodek J 2004. Osteopontin modulates CD44-dependent chemotaxis of peritoneal macrophages through G-protein-coupled receptors: evidence of a role for an intracellular form of osteopontin. J. Cell. Physiol 198, 155–167. 10.1002/jcp.10394. [DOI] [PubMed] [Google Scholar]
- Zmora N, Bashiardes S, Levy M, Elinav E, 2017. The role of the immune system in metabolic health and disease. Cell Metab. 25, 506–521. 10.1016/j.cmet.2017.02.006. [DOI] [PubMed] [Google Scholar]