

Abstract
Purpose:
CDK12 aberrations have been reported as a biomarker of response to immunotherapy for metastatic castration-resistant prostate cancer (mCRPC). Herein, we characterize CDK12-mutated mCRPC, presenting clinical, genomic, and tumor-infiltrating lymphocyte data.
Experimental Design:
Patients with mCRPC consented to the molecular analyses of diagnostic and metastatic CRPC biopsies. Genomic analyses involved targeted next generation (MiSeq™; Illumina) and exome sequencing (NovaSeq™; Illumina). Tumor-infiltrating lymphocytes (TIL) were assessed by validated immunocytochemistry coupled with deep learning-based artificial intelligence analyses including multiplex immunofluorescence assays for CD4, CD8, and FOXP3 evaluating TIL subsets. The control group comprised a randomly selected mCRPC cohort with sequencing and clinical data available.
Results:
Biopsies from 913 patients underwent targeted sequencing between Feb/15 and Oct/19. Forty-three patients (4.7%) had tumors with CDK12 alterations. CDK12 altered cancers had distinctive features, with some revealing high chromosomal break numbers in exome sequencing. Biallelic CDK12-aberrant mCRPC had shorter overall survival from diagnosis than controls (5.1 years [95% CI: 4.0, 7.9] vs 6.4 years [95% CI: 5.7, 7.8]; HR=1.65 [95% CI: 1.07, 2.53]; P=0.02). Median intratumoral CD3+ cell density was higher in CDK12 cancers, although this was not statistically significant (203.7 versus 86.7 cells/mm2, P=0.07). This infiltrate primarily comprised CD4+FOXP3− cells (50.5 versus 6.2 cells/mm2, P<0.0001), where high counts tended to be associated with worse survival from diagnosis (HR=1.64; 95% CI: [0.95, 2.84], P=0.077) in the overall population.
Conclusions:
CDK12-altered mCRPCs have worse prognosis with these tumors surprisingly being primarily enriched for CD4+FOXP3− cells that seem to associate with worse outcome and may be immunosuppressive.
Keywords: CDK12, prostate cancer, Tumor lymphocyte infiltration
INTRODUCTION
Cyclin-dependent kinase 12 (CDK12) is a transcription-associated CDK that forms a heterodimeric complex with Cyclin K (CycK) implicated in DNA repair (DDR), splicing, and differentiation [1–6]. CDK12 maps on chromosome 17q12 and generates two mRNA splice isoforms, differing only in the last exon with both having a central kinase region flanked by large N- and C-terminal extensions with arginine-serine-rich (RS) domains [3]. Recent genomic analyses of advanced prostate cancer (PCa) [7–11] and metastatic castration-resistant prostate cancer (mCRPC) [11–15] have identified recurrent, deleterious, CDK12 alterations in 2–4% of primary PCa [9,11] and in 4.7–11% of mCRPC [11, 14, 15]. Integrative genomic analysis of 360 mCRPC samples has demonstrated that CDK12-mutated PCa is genetically, transcriptionally, and phenotypically distinct from tumors with homologous recombination repair defects (HRD) and mismatch repair deficiency (MMRd) [16], being reported to have innumerable tandem duplications (TD) and genomic rearrangements, high neoantigen burdens, and increased tumor-infiltrating lymphocytes (TILs) [16]. This is postulated to confer a vulnerability to immunotherapy similar to cancers with MMRd [16,17], with this genomic profile probably also acquiring immune cell evasive strategies to allow tumor growth [18–20].
Understanding the clinical, molecular, and immune characteristics of these tumors is critically important to the development of successful clinical therapeutic strategies for this disease sub-class [20–22]. Herein, we characterize CDK12 mutated PCa, with both bi-allelic and mono-allelic aberrations, describing their genomic, clinical, histopathological, and immune infiltrate features.
MATERIALS AND METHODS
Patient population
This retrospective study included patients with metastatic PCa who consented for molecular characterization of their PCa biopsies at The Institute of Cancer Research (London, UK). Consent was obtained either within a Royal Marsden Hospital (RMH) specific protocol (Ethics Review Committee reference no. 04/Q0801/60) and/or the phase II trial TOPARP-B (ClinicalTrials.gov, NCT01682772). These studies were conducted in accordance with the Declaration oh Helsinki. Both studies allowed acquisition, whenever possible, of a metastatic CRPC biopsy and the diagnostic hormone-sensitive prostate cancer (HSPC) biopsy.
Demographic and clinical data for each patient were collected from the hospital electronic patient record or the clinical trial database. Clinical outcomes included overall survival (OS) from diagnosis, OS from castration resistance, and time to CRPC, respectively defined as the time between diagnosis of PCa and death or last follow up, between castration resistance and death or last follow up, and between diagnosis and castration resistance. Furthermore, time-on-treatment with abiraterone/enzalutamide (whichever came first) was investigated separately before and after treatment with docetaxel.
Tumor Tissue samples
Formalin-fixed-paraffin-embedded (FFPE) PCa tissue was obtained from prostate needle biopsies, transurethral resections of the prostate, prostatectomies, or PCa metastases within bone (bone marrow trephine), lymph node, soft tissue, or visceral metastases (needle biopsies). A subset of patients had matching, same-patient, primary tissue and CRPC biopsies available. All tissue blocks were freshly sectioned, tumor content was confirmed by examination of H&E stained samples sections by a trained pathologist (B.G.).
Next-Generation Sequencing (NGS)
Targeted NGS was performed on diagnostic and metastatic PCa samples. DNA was extracted from FFPE blocks positive for tumor content using the FFPE Tissue DNA kit (Qiagen) and quantified with the Quant-iT high-sensitivity PicoGreen double-stranded DNA Assay Kit (Invitrogen). The Illumina FFPE QC kit (WG-321–1001) was used for DNA quality control tests according to the manufacturer’s protocol. Libraries for next generation targeted sequencing were constructed from 40ng of DNA using a customized panel (Generead DNAseq Mix-n-Match Panel v2; Qiagen) covering 6025 amplicons across 113 genes. Libraries were run using the MiSeq Sequencer (Illumina). FASTQ files were generated using the Illumina MiSeq Reporter v2.5.1.3. Sequence alignment and mutation calling were performed using BWA tools and the GATK variant annotator by the Qiagen GeneRead Targeted Exon Enrichment Panel Data Analysis Web Portal. Biallelic alterations in CDK12 were prospectively defined as (a) a deleterious mutation with loss of heterozygosity (LOH) at the wild-type allele, (b) copy number loss (homozygous deletion), (c) ≥2 CDK12 deleterious genomic alterations in a given sample. Mutations with >5% allele frequency were reported.
Exome sequencing and Copy number break analysis
Libraries for whole exome sequencing (WES) were performed using Kapa Hyper Plus Library Prep Kits and the Agilent SureSelectXT V6 target enrichment kit. Paired-end sequencing was performed using the NovaSeq 6000 S2 flow cell (2×100 cycles, Illumina). FASTQ files were generated from the sequencer’s output using Illumina bcl2fastq2 software (v.2.17.1.14, Illumina) with the default chastity filter to select sequence reads for subsequent analyses. All reads were aligned to the human genome reference sequence (GRCh37-hg19) using the BWA-MEM algorithm (v. 0.7.12). Picard tools (v.2.8.2) were used to mark PCR duplicates and to calculate sequencing metrics for quality control check. The Genome Analysis Toolkit (GATK, v. 3.5–0) was applied to realign local indels, recalibrate base scores, and identify point mutations, small insertions and deletions. Somatic point mutations and indels were called using MuTect2 by comparing tumor DNA to germline DNA control. Copy-number estimation was obtained through a modified ASCAT2 package. CNA segmentation file was used to define the CNA breakpoint when there is a change of segment and/or CNA.
Immunohistochemistry (IHC)
Three-μm thick sections from FFPE tissue blocks were obtained using a microtome, floated in a water bath, and mounted in Superfrost slides. Slides were baked and dried overnight at 37°C. CD3 IHC was performed using a rabbit anti-CD3 antibody (#A0452; polyclonal; DAKO; Agilent Technologies: Santa Clara, California, United States) and performed using an automated staining platform (Bond-RX, Leica Microsystems). Antigen retrieval was achieved by using Bond Epitope Retrieval Solution 1 (#AR9961, Leica Biosystems) for 30 minutes prior to incubation with anti-CD3 antibody (1:150 dilution) for 15 minutes at room temperature. The reaction was visualized using the Bond Polymer refine kit (#DS9800, Leica Biosystems). Antibody specificity was confirmed in human appendix and prostate tissues (positive controls) and LNCaP cell pellets (negative control).
PTEN and ATM protein expression studies were conducted as previously described [23,24].
Nuclear staining (and cytoplasmic staining for PTEN) was semi-quantitatively assessed using an H-score (3 × % of strongly staining cells and 2 × the % of moderately staining cells, and the % of weakly staining cells, for a range of 0 to 300). Cases were considered as PTEN or ATM loss if they had an H-score≤10.
Slide digitization and computer assisted image analysis for IHC
CD3 IHC stained slides were scanned at high resolution (200x) using the ZEISS Axio Scan.Z1 digital slide scanner (Carl Zeiss AG). The digitized slides were then analyzed with the HALO™ image analysis suite (Indica Labs, New Mexico, USA). A supervised machine-learning algorithm was trained to recognize prostate cancer foci and surrounding stroma. Color deconvolution for DAB and hematoxylin stains was performed, cell recognition and nuclear segmentation was optimized for hematoxylin stain, and recognition of CD3 staining was optimized for the membranous and cytoplasmic compartments. The analysis algorithm was adjusted to provide optical density data for intensity of CD3 membranous and cytoplasmic staining in the automatically annotated tumor and stromal regions. A threshold was set to categorize each detected cell into CD3 positive or negative. The number of intratumoral and stromal CD3 positive cells was then divided by the total area of prostate tumor and stroma, respectively, providing intratumoral and stromal CD3 density values (CD3+ per mm2) for each sample. Each stained sample was also visually assessed and assigned an intratumoral and stromal chronic inflammation score, using a modified version of the histopathological classification system developed by Nickel et al [25].
Multiplex Immunofluorescence (IF) Assay
Intratumoral TILs were determined by IF for T cell subpopulations as previously described [26]. Briefly, multiplex IF staining was performed on 3μm FFPE tissue sections using an automated staining platform (BOND RX, Leica Microsystems). Antigen retrieval was achieved using BOND Epitope Retrieval Solution 2 (#AR9640, Leica Microsystems). Endogenous peroxidase was inactivated in 3% H2O2. Tissue sections were incubated with antibodies against CD4 (#ab133616, clone EPR6855, 1:100, Abcam) and CD8 (#M7103, clone C8/144B, 1:200, Dako, Agilent Technologies), and detected with AlexaFluor 555-conjugated IgG (H+L) goat anti-rabbit (#A21429, Invitrogen) and AlexaFluor 488-conjugated IgG (H+L) goat anti-mouse (#A-11029, Invitrogen) secondary antibodies, respectively. Endogenous biotin was blocked with Avidin/Biotin blocking kit according to the manufacturer’s protocol (#ab64212, Abcam). Next, tissue sections were incubated with antibodies against FOXP3 conjugated to biotin (#13-4777-82, clone 236A/E7, 1:100, eBioscience) and pan-cytokeratin (PanCK) conjugated to AlexaFluor 647 (#4528S, clone C11, 1:100, Cell Signaling Technology), followed by streptavidin peroxidase (HRP) (#K5001, Dako, Agilent Technologies) and TSA Coumarin detection system (#NEL703001KT, Akoya Biosciences). Nuclei were counterstained with DRAQ 7 (#DR71000, Biostatus) and tissue sections mounted with ProLong Gold antifade reagent (#P36930, Molecular Probes).
Slides were scanned using the Vectra Automated Multispectral Imaging System (Akoya Biosciences) and analysed using inForm v2.2.1 (Akoya Biosciences). Tissue and nuclear cell segmentation were performed using previously described methods (1). TIL phenotype determination was based on staining for CD8, CD4, and FOXP3 and were separated into bins as follows: CD8+, CD4+FOXP3+, and CD4+FOXP3− T cells. Quantification of the immune cell densities are presented as number of cells per mm2.
Statistical analysis
In this analysis, the prevalence of CDK12 aberrations observed in our cohort was reported. In a subset of patients with available clinical (n=36, 28 biallelic, 8 monoallelic) and TIL data (n=24, 17 biallelic, 7 monoallelic), tumors with CDK12 aberrations were compared to a control group that comprised a random selection of patients concurrently sequenced at our institution without such aberrations nor mismatch-repair alterations. Comparison of lymphocyte infiltration between CDK12 biallelic, monoallelic cases, and controls was performed using a t-test after density per mm2 was log10 transformed, whereas Wilcoxon rank sum test assessed differences between diagnostic and mCRPC samples in CDK12 cases. Wilcoxon rank sum test was also used for copy number breaks (CNBs) comparison between CDK12 biallelic, monoallelic cases, and controls. Kaplan-Meier estimator and Cox regression models were used for time-to-event analyses comparing controls and biallelic CDK12 cases only. In a sensitivity analysis, all time-to-event analyses were repeated with mono- and biallelic CDK12 cases being pooled into one group and compared to the control group. The significance level was set to 5% for all analyses and there was no multiplicity adjustment made as to the exploratory nature of this work. Analyses were carried out with R version 3.6.2.
RESULTS
CDK12 altered prostate cancers
Between March 2015 and October 2019, 913 PCa patients were analyzed by targeted next-generation-sequencing after passing quality control. All patients had a histological diagnosis of PCa and either diagnostic tissue and/or a mCRPC biopsy sample available for analysis; the patient characteristics and their treatment histories are described in Table 1 and Supplementary Table 1. We identified 43 patients with potentially pathogenic alterations of CDK12 (4.7%), of which 31 (3.4%) had biallelic genomic aberrations. One tumor had a concomitant MSH6 somatic mutation with associated MSH6 loss of expression on IHC and was not included in subsequent analyses being deemed to be an MMRd PCa. The remaining 11 patients were considered to have monoallelic CDK12 alterations (Supplementary Figure 1).
Table 1.
Patients Characteristics.
| Overall | control | monoallelic | biallelic | p | |
|---|---|---|---|---|---|
| n | 180 | 144 | 8 | 28 | |
| Age at diagnosis (median [IQR]) | 61.0 [56.0, 64.2] | 61.0 [55.8, 64.0] | 59.0 [55.0, 63.5] | 62.5 [60.0, 65.0] | 0.4011 |
| T group (%) | 0.2812 | ||||
| T1 | 2 (1.1) | 1 (0.7) | 1 (12.5) | 0 (0.0) | |
| T2 | 20 (11.1) | 18 (12.5) | 0 (0.0) | 2 (7.1) | |
| T3 | 76 (42.2) | 57 (39.6) | 4 (50.0) | 15 (53.6) | |
| T4 | 34 (18.9) | 26 (18.1) | 2 (25.0) | 6 (21.4) | |
| TX | 48 (26.7) | 42 (29.2) | 1 (12.5) | 5 (17.9) | |
| T3/4 = Yes (%) | 110 (83.3) | 83 (81.4) | 6 (85.7) | 21 (91.3) | 0.6232 |
| N (%) | 0.1972 | ||||
| N0 | 42 (36.8) | 28 (33.3) | 5 (83.3) | 9 (37.5) | |
| N1 | 65 (57.0) | 51 (60.7) | 1 (16.7) | 13 (54.2) | |
| N2 | 7 (6.1) | 5 (6.0) | 0 (0.0) | 2 (8.3) | |
| N1/2 = Yes (%) | 72 (63.2) | 56 (66.7) | 1 (16.7) | 15 (62.5) | 0.0592 |
| M = M1 (%) | 96 (56.5) | 76 (56.3) | 2 (28.6) | 18 (64.3) | 0.2312 |
| Total Gleason score (G) (median [IQR]) | 9.0 [7.0, 9.0] | 8.0 [7.0, 9.0] | 9.0 [7.8, 9.0] | 9.0 [9.0, 9.0] | <0.0011 |
| G≤7vs G>8 = Yes (%) | 117 (72.2) | 84 (66.1) | 6 (75.0) | 27 (100.0) | <0.0012 |
| PSA at diagnosis (median [IQR]) | 63.0 [15.4, 176.1] | 53.5 [10.9, 162.0] | 106.0 [34.5, 137.0] | 72.0 [25.5, 229.2] | 0.4491 |
| Prostatectomy Y/N = Yes (%) | 22 (12.2) | 18 (12.5) | 2 (25.0) | 2 (7.1) | 0.3182 |
| Radiotherapy Y/N = Yes (%) | 67 (37.2) | 57 (39.6) | 2 (25.0) | 8 (28.6) | 0.4502 |
IQR= interquartile range; N= presence of nodal disease; M= presence of metastatic disease; PSA= prostate specific antigen.
Kruskal-Wallis test.
Fisher Exact test.
For the 31 biallelic cases identified, a total of 34 diagnostic and metastatic PCa samples were sequenced by NGS; three patients had matched primary and metastatic samples available for analyses, which were concordant with the presence of the same CDK12 genomic alterations. The biallelic group comprised 11 missense alterations, 13 nonsense alterations, 32 frameshift mutations or indels, and two splice site mutations. We discovered a CDK12 deep deletion on copy number analysis in one case (Figure 1A; Supplementary Table 2). The majority of these aberrations were truncating mutations, predominantly located in the region between the amino-terminus and the kinase domain or within the kinase domain, whereas the missense mutations were predominantly clustered in the CDK12 kinase domain. Two patients had three CDK12 aberrations, including V463G, one of the missense mutations not located in the kinase domain (lollipop plot shown in Figure 1A).
Figure 1.
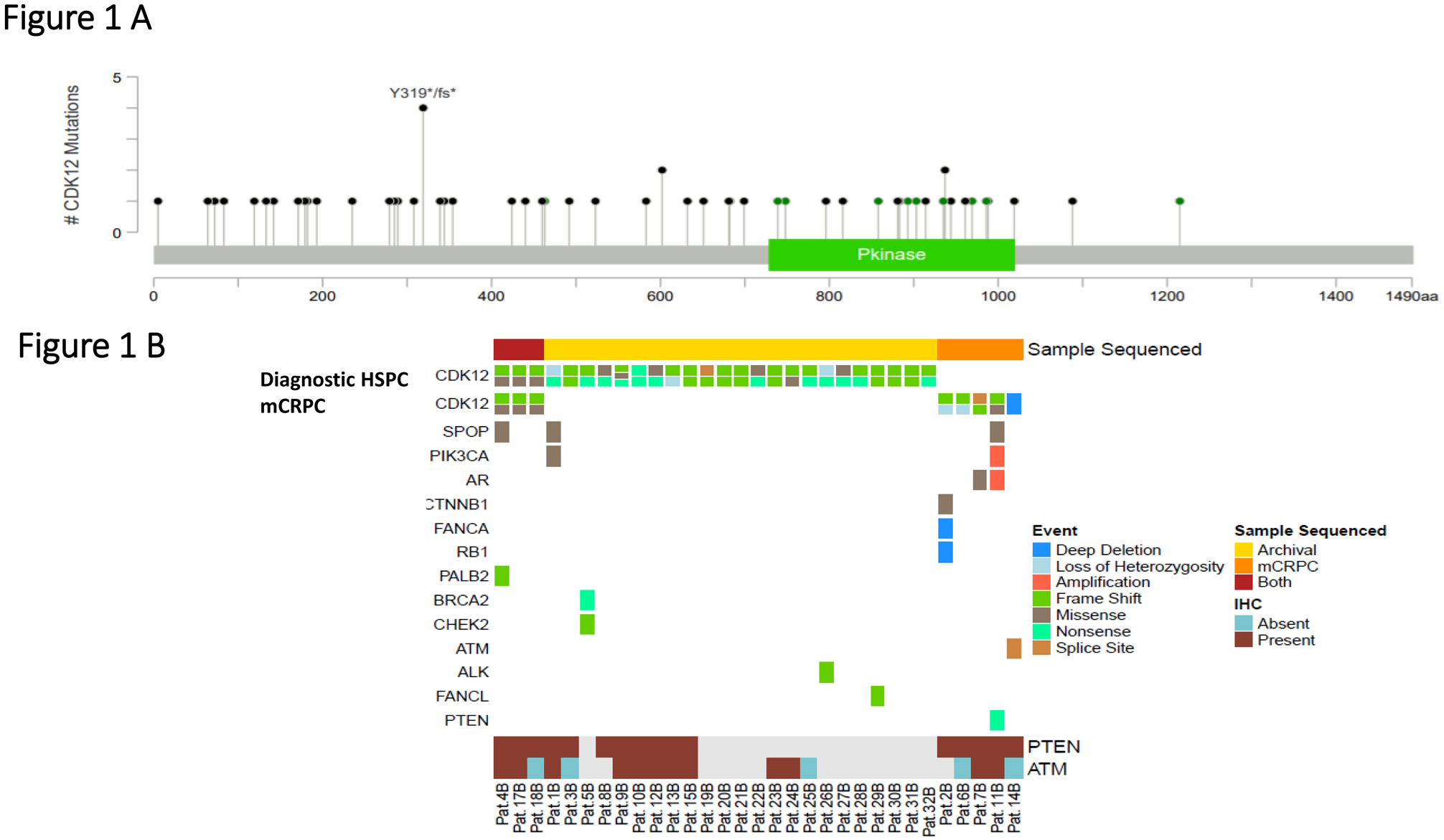
A. Lollipop plot depicting mutations in CDK12 in this prostate cancer cohort with biallelic alterations (n=31); missense mutations are depicted in green, frameshift and non-sense mutations in black.
B. OncoPrint figure representing CDK12 mutations for the bi-allelic tumors in hormone sensitive prostate cancer (HSPC) and metastatic castration-resistant (mCRPC) samples (top figures).
Middle panel shows concomitant alterations in other DNA repair genes and other relevant pathways in prostate cancer, detected on targeted and exome sequencing in diagnostic and/or mCRPC samples. Bottom panel depicts the available immunohistochemistry (IHC) data for PTEN and ATM on the samples sequenced. PTEN and ATM data were respectively available for 16 and 17 samples of the 31 CDK12 biallelic cases.
Grey boxes indicate sample with no available PTEN or ATM results by IHC,
Beyond the MMRd cancer with a concomitant CDK12 deleterious alteration, we identified multiple PCas with biallelic CDK12 deleterious alterations contemporaneously harboring other DNA repair defects (Figure 1B). The PCa with CDK12 deep deletion also had an ATM alteration and ATM loss by IHC. Other samples presented with pathogenic PALB2 alteration (I265Tfs*12 in diagnostic and CRPC biopsies), BRCA2 (S744*) and CHEK2 (L464fs*16) mutations, FANCA deep deletion, and FANCL frame-shift mutation (T372Ifs*2). Other alterations in CDK12 biallelic tumors included PTEN mutations, the androgen receptor (AR) L702H point mutation, PIK3CA activating mutation (E545K) and SPOP alterations (Supplementary Table 2). We had IHC data for PTEN and ATM available respectively for 16 and 17 of the 31 CDK12 biallelic cases. Interestingly, none of the cancers with biallelic CDK12 alterations had PTEN loss by IHC; conversely, 4 of the biallelic CDK12 tumors (4/17, 23.5%) had ATM loss by IHC.
Clinical outcomes of PCa with CDK12 alterations
Clinical data were available for 36 of the 42 patients with CDK12 aberrations (28 biallelic, 8 monoallelic) and were compared to the data of 144 controls. Patient characteristics and treatment histories are described in Table 1 and Supplementary Table 1. The two groups were balanced for age, staging, and presence of metastatic disease as well as for PSA levels and initial treatment received. However, CDK12 tumors had higher Gleason scores at diagnosis (P<0.001). Biallelic CDK12 cases experienced worse survival than the control group with a median OS from diagnosis of 6.4 years (95% CI: [5.7, 7.8]) for the control group and 5.1 years (95% CI: [4.0, 7.9]) for the biallelic CDK12 group (HR=1.65; 95% CI: [1.07, 2.53]; P=0.02). Similar results were obtained when monoallelic and biallelic cases were combined into one group and compared to controls (HR=1.50; 95% CI: [1.02, 2.20]; P=0.04) (Figures 2A and Figure 2B) and when OS was estimated from the date of CRPC (Figure 2C and 2D). However, in the multivariable analysis (MVA), CDK12 mutational status was no longer independently prognostic (HR=0.94, CI: [0.6, 1.6]; P=0.81) (Supplementary Table 3). Median time to from diagnosis CRPC was not different between biallelic and control cohorts being 1.9 years (95% CI: [1.4, 2.6]) in the control group and 2.2 years (95% CI: [1.5, 2.7]) in the biallelic CDK12 group (HR=1.25; 95% CI: [0.82, 1.91]; P=0.3) (Supplementary Figure 2A). Almost all patients had received docetaxel chemotherapy, and abiraterone or enzalutamide; 61% of patients in the control group received cabazitaxel, but only 36% in the combined mono/biallelic CDK12 group (Supplementary Table 1). There was no statistically significant difference in time on abiraterone/enzalutamide between the groups with median duration of treatment in the pre-docetaxel setting being 7.6 months (95% CI: [6.0, 9.3]) in the control group and 8.2 months (95% CI: [4.7, NA]) in the biallelic-CDK12 group (HR=1.07; 95% CI: [0.58, 2.11]; P=0.75) with similar findings in the post-docetaxel setting (Supplementary Figure 2B and 2C) and when monoallelic and biallelic cases were combined into one group (data not shown).
Figure 2.
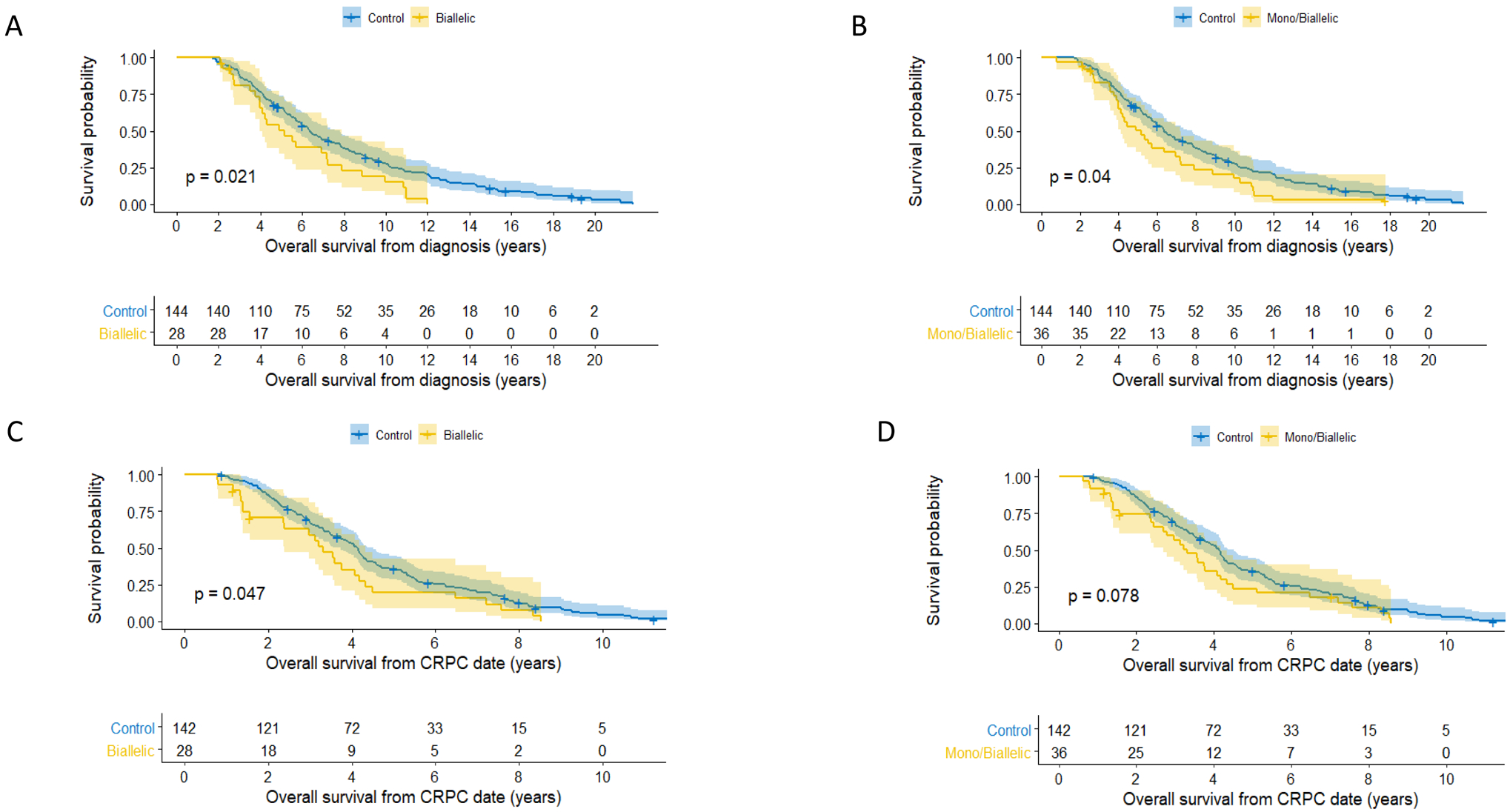
Survival curves: A. Kaplan-Meier curves for overall survival from date of diagnosis. CDK12 biallelic mutation cases in yellow, controls in blue.
B. Kaplan-Meier curves for overall survival from date of diagnosis. CDK12 biallelic and monoallelic mutated cases in yellow, controls in blue.
C. Kaplan-Meier curves for overall survival from date of castration resistant prostate cancer (CRPC) diagnosis. CDK12 biallelic mutated cases in yellow, controls in blue.
D. Kaplan-Meier curves for overall survival from date of castration resistant prostate cancer (CRPC) diagnosis. CDK12 biallelic and monoallelic mutated cases in yellow, controls in blue.
Exome Sequencing data
Overall, out of the 913 patients with PCa evaluated by targeted NGS, 211 (diagnostic=93; CRPC=118) had samples suitable for exome sequencing including 26 samples harboring CDK12 genomic aberrations, 23 of which were biallelic and three monoallelic. Although 73% of the CDK12-aberrant cases (19/26) had CNB values higher than the median, which was 513 [Q1:405.5, Q3: 617.5], only 10 of the 26 (38.5%) CDK12-aberrant cases were above the CNB upper quartile (Figure 3A). In general, however, CDK12 tumors had significantly higher CNBs than the controls (P=0.01) (Figure 3B).
Figure 3.
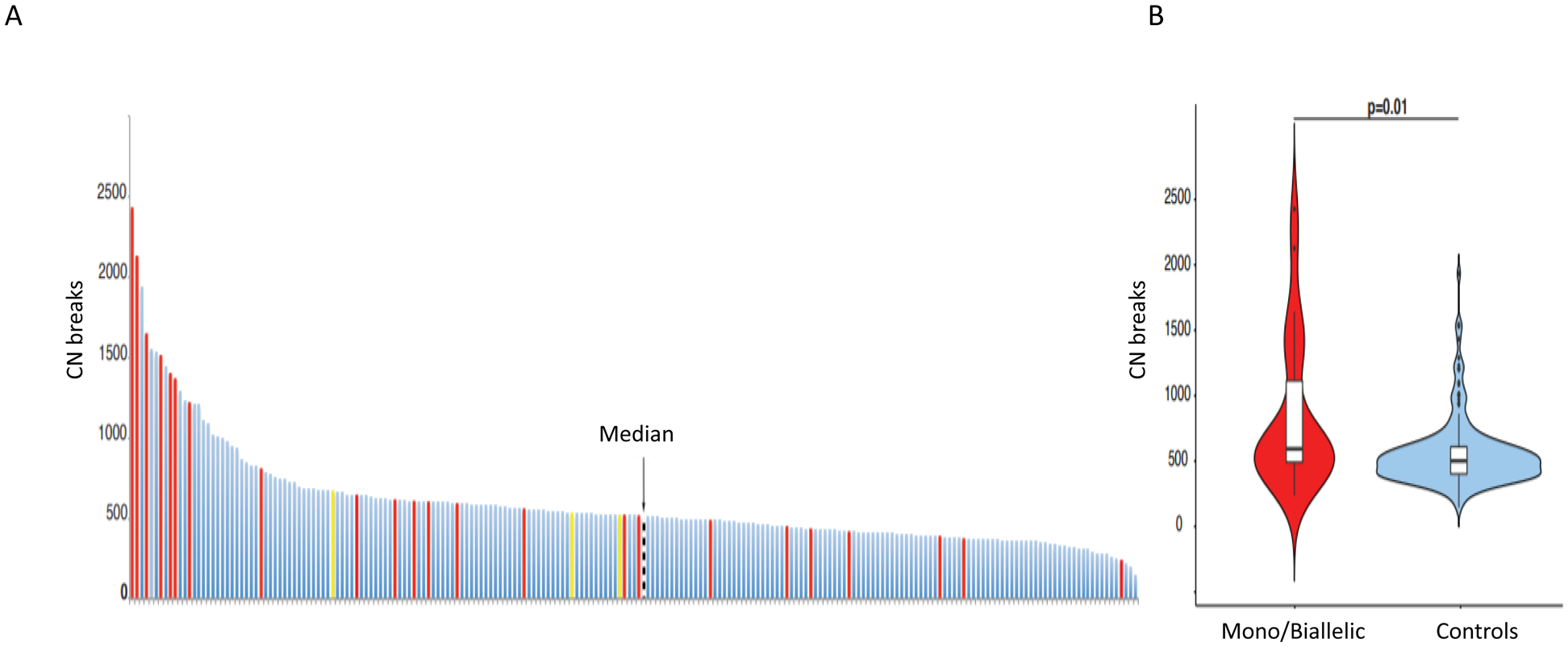
Genomic analyses: A. Copy number breaks (CNBs) determined from exome sequencing data in n=211 patients. CDK12 biallelic mutation cases (n=23) in red, monoallelic cases (n=3) in yellow, and cancers with no CDK12 alterations (controls) in blue. The black line indicates the median number of CNBs (median =513) in the whole cohort.
B. Violin plot depicting CNB numbers in CDK12 mutated in red and controls in blue.
Tumor-infiltrating-lymphocyte (TIL) analyses
For the analysis of intratumor CD3+ TIL density (D-TIL), there were 122 samples for controls as well as 17 samples with biallelic CDK12 alterations, and 7 monoallelic CDK12 alteration cases (tot n=146) (Figures 4A and 4B). Median intratumoral total CD3+ cell density was higher in CDK12 biallelic loss samples than in controls, although this was not statistically significant (203.7 versus 86.7 CD3+/mm2, P=0.073) (Figure 4B). Two of the CDK12 biallelic loss PCa cases had more than 1200 CD3+ cells per mm2, which is above the 90% centile (Figure 4A); however, multiple samples with biallelic CDK12 aberrations had CD3+ counts that were below the median. D-TIL was not statistically different between diagnostic and mCRPC samples for all CDK12 cases (Supplementary Figure 3). Interestingly, there was no association found in our analyses between copy number breaks and TIL density, neither in diagnostic nor in CRPC samples. The samples with the highest CD3+ density also had the highest CD4+and CD8+ infiltrates. The two samples with the highest CD3+ density had modest CNB scores of 613 and 600 (just above the median of 513).
Figure 4.
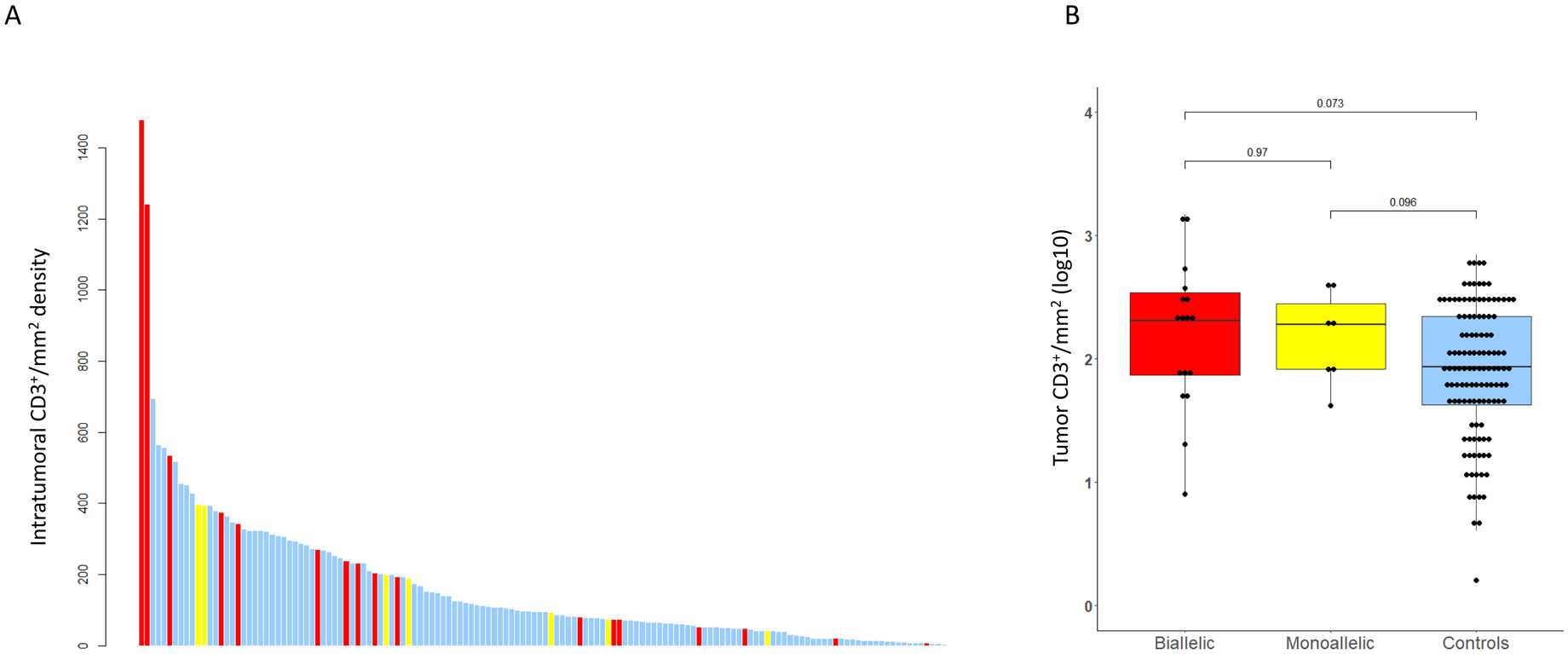
Tumor infiltrating lymphocytes (TILs) analyses: A. Intratumoral CD3+ TILs per mm2. Bar Chart depicting intratumoral CD3+/mm2 density (y-axis) in CDK12 biallelic cases in red, monoallelic cases in yellow, and controls in blue.
B. Box and Whisker plot of log10-transformed intratumoral CD3+ cell density per mm2 depicting CDK12 biallelic in red, monoallelic cases in yellow, and control cases in blue.
We next quantified D-TILs, based on T-cell subtypes (Figures 5A and 5B; Supplementary Figure 4 A–C); tissue sites included prostate tissue (n=9), lymph node biopsies (n=31), bone (n=10), liver (n=1) and soft tissue metastases (n=1) (Supplementary Table 4). In the overall population, total CD3+ cell numbers were correlated with CD4+FOXP3− (R=0.51; P<0.001) and CD8+ cell numbers (R=0.45; P<0.001) (Figure 5A). Moreover, our analyses revealed that CDK12 cancers were significantly enriched for CD4+FOXP3− cells (median CD4+FOXP3−/mm2 50.5 in CDK12 biallelic cases versus 6.2 in controls, P<0.001), with a higher CD4/CD8 ratio (r) compared to controls (CD4/CD8 r=1.22 for CDK12 biallelic cases versus 0.4 for controls) without, significantly higher CD8+ or CD4+FOXP3+ (Figure 5B). Higher CD4+FOXP3− infiltration appeared to be associated with shorter OS (HR=1.64; 95% CI: [0.95, 2.84], P=0.077), with similar trends for CD4+FOXP3+ and CD8+ cells (respectively, HR=1.14, 95% CI: [0.73, 1.76], P=0.6 HR=1.42, 95% CI: [0.94, 2.16], P=0.0967).
Figure 5.
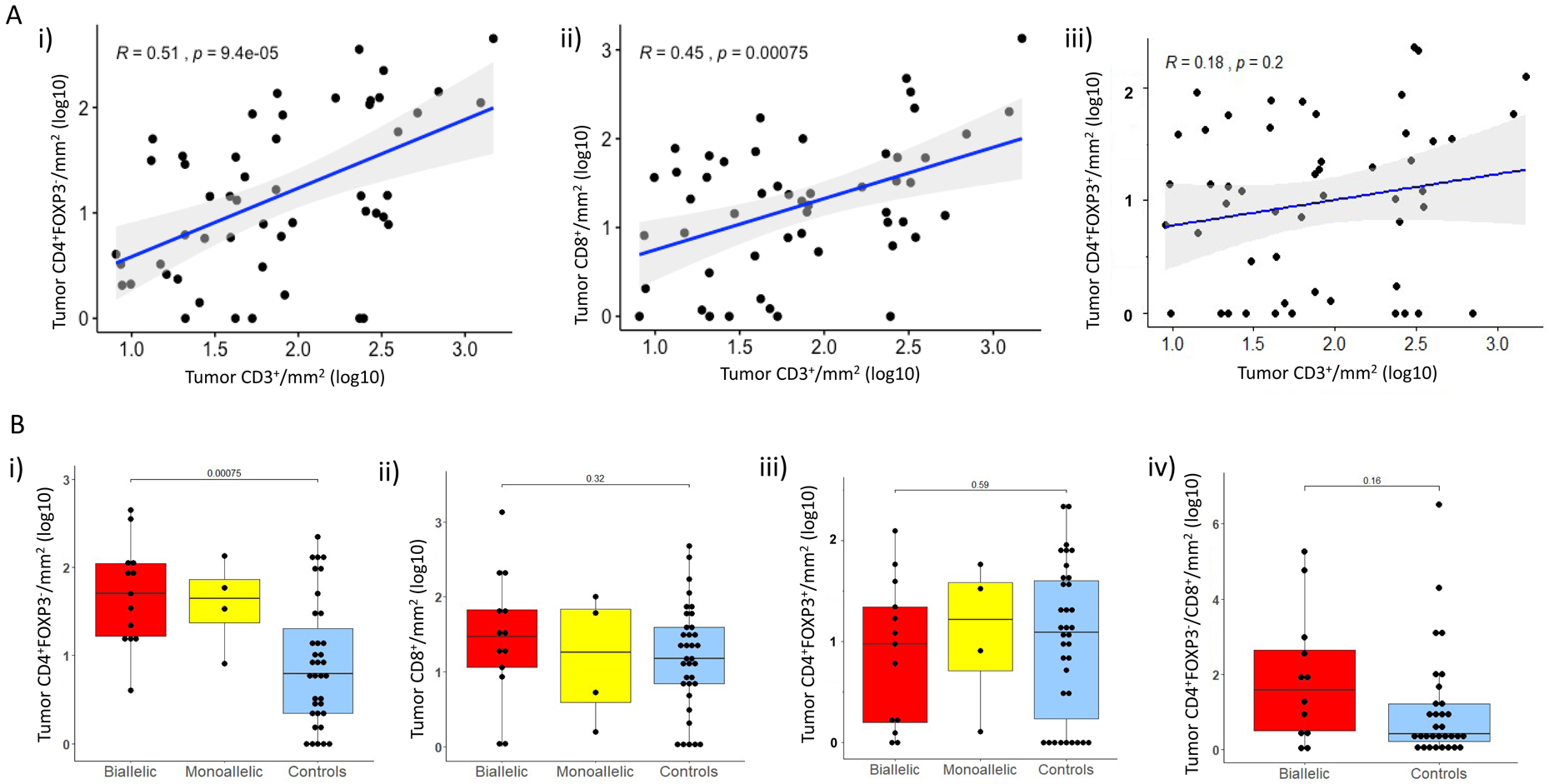
Tumor infiltrating lymphocyte (TIL) subset analyses: A. i) Scatter plots depicting associations between log-transformed CD3+ cells/mm2 TILs and CD4+FOXP3− cells/mm2, ii) CD3+ cells/mm2 and CD8+cells/mm2, as well as iii) CD3+ cells/mm2 and CD4+FOXP3+cells/mm2.
B. Box and whisker plots representing intratumoral infiltrate density as log10-transformed CD4+FOXP3− cells/mm2 (i), CD8+cells/mm2 (ii), CD4+ FOXP3+cells/mm2 (iii) in CDK12 biallelic (red), monoallelic (yellow), and control cases (blue); iv) Tumor CD4+FOXP3−/CD8+ ratio between biallelic cases (red) and controls (blue).
DISCUSSION
Deleterious CDK12 mutations have been reported in multiple tumor types, but are not common although they may have especial clinical importance in view of their immunogenomic properties. A subset of mCRPC has deleterious CDK12 aberrations [16,27,28], resulting in characteristic genomic profiles with innumerable focal tandem duplications, gene-fusions, and neoantigens [16,27]. These PCa CDK12 alterations, like MMRd, have been postulated to represent a predictive biomarker of response to immunotherapy [17], making their study of huge interest [29,30]. Our analysis is arguably one of the largest integrated efforts interrogating their genomic, pathologic, and clinical characteristics and also investigating their TIL landscape.
We confirm that approximately 5% of mCRPC have CDK12 deleterious aberrations [11,16], although fewer (3.4%) are definitely biallelic with the majority being frameshift or nonsense mutations and a small minority single nucleotide aberrations of the kinase domain with a high likelihood of impact on protein function. Interestingly, these alterations are invariably present at diagnosis in matched same patient HSPC and mCRPC biopsies suggesting that they are clonal events. Exome analyses have identified a specific genomic pattern of CDK12-aberrant cancers characterized by a high number of copy number breaks, and this highly fragmented genome has been shown to be correlated with a tandem duplicator phenotype [16]. In our analysis, although high CNBs are characteristic of these tumors, not all PCa with biallelic CDK12 alterations had this genomic signature indicating that this may not be useful as the sole test to identify this subset. However, in keeping with the PROfound olaparib Phase III genomic analyses data we did observe concomitant mutations in other DNA repair genes in CDK altered tumors including PALB2, BRCA2, FANCA, ATM, MSH6 and in AR, PTEN and PI3K/AKT pathway genes, with 23.5% of these cancers presenting concomitant ATM loss but no PTEN loss by IHC [31]. We also show that PCa with CDK12 biallelic alterations associate with higher Gleason scores and shorter survival from diagnosis, although we did not observe any differences in outcome on abiraterone/enzalutamide which has been previously reported – maybe due to the retrospective nature of our investigation [29,30].
We also show for the first time that PCa with biallelic CDK12 aberrations are predominantly enriched for CD4+FOXP3− cells with these tumors surprisingly not having significantly higher CD4+FOXP3+ or CD8+ TILs compared to controls. Interestingly, multiple studies have reported on immunosuppressive CD4+FOXP3− TIL subsets [32,33], with some of these cells being reported to express high PD-1 (4PD1hi) and being able to inhibit cytotoxic T cell function in a PD-1/PD-L1 dependent fashion as well as limit immunotherapy antitumor activity [32]. The data herein suggest that CD4+FOXP3− cells, which CDK12-altered PCa seem to be enriched for, might have an immunosuppressive role, associating with worse survival and might be relevant to immunotherapy trials as a biomarker of interest [30, 34–35].
We acknowledge that significant limitations of our data include the fact that this was a single institution retrospective study, and that, while we screened almost a thousand patients, our CDK 12 analyses are based on a relatively small cohort, even though this comprises one of the largest studies of mCRPC subjects. Furthermore, we acknowledge that more effort is now needed to elucidate why not all PCas with biallelic CDK12 alterations have the tandem duplication signature, and studies need to evaluate whether monoallelic loss can result in any biological relevance perhaps due to haploinsufficiency and a gene dose effect. Moreover, further studies to characterize the CD4+FOXP3− TIL subset are now crucial to drive therapeutic advances for this subset.
In conclusion, our data indicate that biallelic CDK12 aberrations in PCa are associated with higher Gleason grade and poorer prognosis, and are infiltrated by CD4+FOXP3− T-lymphocytes that appear to be associated with worse outcome and are likely to be immunosuppressive, with this being relevant to immunotherapy approaches.
Supplementary Material
Statement of translational relevance:
Prostate cancers harboring CDK12 alterations have a worse prognosis and are enriched primarily in CD4+FOXP3-tumor-infiltrating lymphocytes that associate with a worse prognosis.
ACKNOWLEDGEMENTS
PR and JM are supported by a Prostate Cancer Foundation (PCF) Young Investigator Award. Work in the JdB laboratory is supported by Prostate Cancer UK and the Movember Foundation through the London Movember Centre of Excellence (CEO13_2-002), the US Department of Defense, the Prostate Cancer Foundation (20131017 and 20131017-1), Stand Up To Cancer (SU2C-AACR-DT0712), Cancer Research UK (CRM108X-A25144), and the UK Department of Health through an Experimental Cancer Medicine Centre grant (ECMC-CRM064X). AS has been supported by the Medical Research Council, the Academy of Medical Sciences, Prostate Cancer UK and the Prostate Cancer Foundation. TOPARP-B is an investigator-initiated study supported by Cancer Research UK (CRUK/11/029, C12540 A12829, C12540/A13230, and C12540/A20447) and conducted with support from the Investigator-Sponsored Study Collaboration between AstraZeneca and the National Institute for Health Research Cancer Research Network.
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
JSdB has served on advisory boards and received fees from many companies including Astra Zeneca, Astellas, Bayer, Bioxcel Therapeutics, Boehringer Ingelheim, Cellcentric, Daiichi, Eisai, Genentech/Roche, Genmab, GSK, Janssen, Merck Serono, Merck Sharp & Dohme, Menarini/Silicon Biosystems, Orion, Pfizer, Qiagen, Sanofi Aventis, Sierra Oncology, Taiho, Vertex Pharmaceuticals. He is an employee of The ICR, which have received funding or other support for his research work from AZ, Astellas, Bayer, Cellcentric, Daiichi, Genentech, Genmab, GSK, Janssen, Merck Serono, MSD, Menarini/Silicon Biosystems, Orion, Sanofi Aventis, Sierra Oncology, Taiho, Pfizer, Vertex, and which has a commercial interest in abiraterone, PARP inhibition in DNA repair defective cancers and PI3K/AKT pathway inhibitors (no personal income). He was named as an inventor, with no financial interest, for patent 8,822,438. He has been the CI/PI of many industry sponsored clinical trials. JSdB is a National Institute for Health Research (NIHR) Senior Investigator. The views expressed in this article are those of the author(s) and not necessarily those of the NHS, the NIHR, or the Department of Health. JM has served on advisory boards and received fees from companies including Astra Zeneca, Clovis Oncology, Janssen, Amgen, Roche and Merck-MSD. He received grant as PI from Pfizer, and served as Speakers Bureau for Astellas. MDF has received grants fees from “Fundación Cris contra el Cáncer” outside the submitted work. She has received travel fees from Astellas, AstraZeneca, Pfizer, Pierre Fabre, Roche, Bristol Meiers Squibb, Novartis, MSD, Janssen, and Bayer Pharmamar outside the submitted work. She has also received personal fees from Janssen, Pierre Fabre and Roche outside the submitted work. WY has received a meeting travel grant from Jilin Huarui Gene Technology Ltd.
Footnotes
No relevant conflicts of interest were disclosed by other authors.
REFERENCES
- 1.Blazek D, Kohoutek J, Bartholomeeusen K, Johansen E, Hulinkova P, Luo Z, et al. The Cyclin K/Cdk12 complex maintains genomic stability via regulation of expression of DNA damage response genes. Genes Dev 2011;25(20):2158–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cheng SW, Kuzyk MA, Moradian A, Ichu TA, Chang VC, Tien JF, et al. Interaction of cyclin-dependent kinase 12/CrkRS with cyclin K1 is required for the phosphorylation of the C-terminal domain of RNA polymerase II. Mol Cell Biol 2012;32(22):4691–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chilà R, Guffanti F, Damia G. Role and therapeutic potential of CDK12 in human cancers. Cancer Treat Rev 2016;50:83–88. [DOI] [PubMed] [Google Scholar]
- 4.Juan HC, Lin Y, Chen HR, Fann MJ. Cdk12 is essential for embryonic development and the maintenance of genomic stability. Cell Death Differ 2016;23(6):1038–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tien JF, Mazloomian A, Cheng SG, Hughes CS, Chow CCT, Canapi LT, et al. CDK12 regulates alternative last exon mRNA splicing and promotes breast cancer cell invasion. Nucleic Acids Res 2017;45(11):6698–6716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ekumi KM, Paculova H, Lenasi T, Pospichalova V, Bösken CA, Rybarikova J, et al. Ovarian carcinoma CDK12 mutations misregulate expression of DNA repair genes via deficient formation and function of the Cdk12/CycK complex. Nucleic Acids Res 2015;43(5):2575–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet 2012;44(6):685–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fraser M, Sabelnykova VY, Yamaguchi TN, Heisler LE, Livingstone J, Huang V, et al. Genomic hallmarks of localized, non-indolent prostate cancer. Nature 2017;541(7637):359–364. [DOI] [PubMed] [Google Scholar]
- 9.Cancer Genome Atlas Research Network. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015;163(4):1011–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Linch M, Goh G, Hiley C, Shanmugabavan Y, McGranahan N, Rowan A, et al. Intratumoral evolutionary landscape of high-risk prostate cancer: the PROGENY study of genomic and immune parameters. Ann Oncol 2017;28(10):2472–2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abida W, Armenia J, Gopalan A, Brennan R, Walsh M, Barron D, et al. Prospective Genomic Profiling of Prostate Cancer Across Disease States Reveals Germline and Somatic Alterations That May Affect Clinical Decision Making. JCO Precis Oncol 2017;2017: 10.1200/PO.17.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med 2016;22(3):298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012;487(7406):239–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015;161(5):1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Armenia J, Wankowicz SAM, Liu D, Gao J, Kundra R, Reznik E, et al. The long tail of oncogenic drivers in prostate cancer. Nat Genet 2018;50(5):645–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu YM, Cieślik M, Lonigro RJ, Vats P, Reimers MA, Cao X, et al. Inactivation of CDK12 Delineates a Distinct Immunogenic Class of Advanced Prostate Cancer. Cell 2018;173(7):1770–1782.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rodrigues DN, Rescigno P, Liu D, Yuan W, Carreira S, Lambros MB, et al. Immunogenomic analyses associate immunological alterations with mismatch repair defects in prostate cancer. J Clin Invest 2018; 128(10):4441–4453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science 2011;331(6024):1565–70. [DOI] [PubMed] [Google Scholar]
- 19.Fridman WH, Pagès F, Sautès-Fridman C, Galon J. The immune contexture in human tumors: impact on clinical outcome. Nat Rev Cancer 2012;12(4):298–306. [DOI] [PubMed] [Google Scholar]
- 20.Hendry S, Salgado R, Gevaert T, Russell PA, John T, Thapa B, et al. Assessing Tumor-infiltrating Lymphocytes in Solid Tumors: A Practical Review for Pathologists and Proposal for a Standardized Method From the International Immunooncology Biomarkers Working Group: Part 1:. Assessing the Host Immune Response, TILs in Invasive Breast Carcinoma and Ductal Carcinoma In Situ, Metastatic Tumor Deposits and Areas for Further Research. Adv Anat Pathol 2017;24(5):235–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hendry S, Salgado R, Gevaert T, Russell PA, John T, Thapa B, et al. Assessing Tumor-Infiltrating Lymphocytes in Solid Tumors: A Practical Review for Pathologists and Proposal for a Standardized Method from the International Immuno-Oncology Biomarkers Working Group: Part 2:. TILs in Melanoma, GAstrointestinal Tract Carcinomas, Non-Small Cell Lung Carcinoma and Mesothelioma, Endometrial and Ovarian Carcinomas, Squamous Cell Carcinoma of the Head and Neck, Genitourinary Carcinomas, and Primary Brain Tumors. Adv Anat Pathol 2017;24(6):311–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barnes TA, Amir E. HYPE or HOPE: the prognostic value of infiltrating immune cells in cancer. Br J Cancer 2017;117(4):451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sundar R, Miranda S, Rodrigues DN, Chénard-Poirier M, Dolling D, Clarke M, et al. Ataxia Telangiectasia Mutated Protein Loss and Benefit From Oxaliplatin-based Chemotherapy in Colorectal Cancer. Clin Colorectal Cancer 2018;17(4):280–284. [DOI] [PubMed] [Google Scholar]
- 24.Rescigno P, Lorente D, Dolling D, Ferraldeschi R, Rodrigues DN, Riisnaes R, et al. Docetaxel Treatment in PTEN- and ERG-aberrant Metastatic Prostate Cancers. Eur Urol Oncol 2018;1(1):71–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nickel JC, True LD, Krieger JN, Berger RE, Boag AH, Young ID. Consensus development of a histopathological classification system for chronic prostatic inflammation. BJU Int 2001;87(9):797–805. [DOI] [PubMed] [Google Scholar]
- 26.Minchom A, Yuan W, Crespo M, Gurel B, Figueiredo I, Wotherspoon A, et al. Molecular and immunological features of a prolonged exceptional responder with malignant pleural mesothelioma treated initially and rechallenged with pembrolizumab. J Immunother Cancer 2020;8(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sokol ES, Pavlick D, Frampton GM, et al. : Pan-cancer analysis of CDK12 loss-of-function alterations and their association with the focal tandem-duplicator phenotype. Oncologist 2019;24:1526–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marshall CH, Imada EL, Tang Z, Ross JS, Miller VA, Ali SM, et al. CDK12 inactivation across solid tumors: An actionable genetic subtype. Oncoscience 2019;6:312–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reimers MA, Yip SM, Zhang L, Cieslik M, Dhawan M, Montgomery B, et al. Clinical outcomes in cyclin-dependent kinase 12 mutant advanced prostate cancer. Eur Urol 2020;77:333–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Antonarakis ES, Isaacsson Velho P, Fu W, Wang H, Agarwal N, Santos VS, et al. CDK12-Altered Prostate Cancer: Clinical Features and Therapeutic Outcomes to Standard Systemic Therapies, Poly (ADP-Ribose) Polymerase Inhibitors, and PD-1 Inhibitors. JCO Precision Oncology 2020;4:370–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Bono JS, Fizazi K, Saad F, Shore N, Sandhu SK, Mehra N, et al. Central prospective detection of homologous recombination repair gene mutations (HRRm) in tumor tissue from .4000 men with metastatic castration-resistant prostate cancer (mCRPC) screened for the PROfound study. Ann Oncol 2019;30:v325–v355. [Google Scholar]
- 32.Gagliani N, Magnani CF, Huber S, Gianolini ME, Pala M, Licona-Limon P, et al. Coexpression of CD49b and LAG-3 identifies human and mouse T regulatory type 1 cells. Nat Med 2013;19:739–46. [DOI] [PubMed] [Google Scholar]
- 33.Liu Y, Carlsson R, Comabella M, Wang J, Kosicki M, Carrion B, et al. FoxA1 directs the lineage and immunosuppressive properties of a novel regulatory T cell population in EAE and MS. Nat Med 2014;20:272–82. [DOI] [PubMed] [Google Scholar]
- 34.Zappasodi R, Budhu S, Hellmann MD, Postow MA, Senbabaoglu Y, Manne S, et al. Non-conventional Inhibitory CD4+Foxp3−PD-1hi T Cells as a Biomarker of Immune Checkpoint Blockade Activity. Cancer Cell 2018;34(4):691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schweizer MT, Ha G, Gulati R, Brown LC, McKay RR, Dorff T, et al. CDK12-Mutated Prostate Cancer: Clinical Outcomes With Standard Therapies and Immune Checkpoint Blockade. JCO Precis Oncol 2020;4:382–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.