

Abstract
The immune microenvironment of tumors can play a critical role in promoting or inhibiting tumor progression depending on the context. We present evidence that tumor-associated macrophages/microglia (TAMs) can promote tumor progression in the sonic hedgehog subgroup of medulloblastoma (SHH-MB). By combining longitudinal manganese-enhanced magnetic resonance imaging (MEMRI) and immune profiling of a sporadic mouse model of SHH-MB, we found the density of TAMs is higher in the ~50% of tumors that progress to lethal disease. Furthermore, reducing regulatory T cells or eliminating B and T cells in Rag1 mutants does not alter SHH-MB tumor progression. As TAMs are a dominant immune component in tumors and are normally dependent on colony-stimulating factor 1 receptor (CSF1R), we treated mice with a CSF1R inhibitor, PLX5622. Significantly, PLX5622 reduces a subset of TAMs, prolongs mouse survival and reduces the volume of most tumors within four weeks of treatment. Moreover, concomitant with a reduction in TAMs the percentage of infiltrating cytotoxic T cells is increased, indicating a change in the tumor environment. Our studies in an immunocompetent preclinical mouse model demonstrate TAMs can have a functional role in promoting SHH-MB progression. Thus, CSF1R inhibition could have therapeutic potential for a subset of SHH-MB patients.
Introduction
Medulloblastoma (MB) is a highly aggressive, WHO-grade IV embryonal tumor most commonly seen in children under 16 years of age 18, 25. Current treatments, including surgery, radiation and chemotherapy yield five-year survival rates of ~70% 25. However, survivors suffer from treatment-related neurocognitive and neuroendocrine sequelae 38. Thus, less toxic therapeutic options are urgently needed. MB is divided into four major subgroups (WNT, Sonic Hedgehog [SHH], Group 3 and Group 4) 45. Genomic, epigenomic, transcriptomic and proteomic analysis of tumor samples combined with clinical data have further subdivide each subgroup into subtypes, emphasizing the heterogeneity of the disease and the importance of incorporating the subtypes into clinical risk stratification 1, 5, 31. Despite the large volume of data on the intrinsic cellular drivers of MB, the potential roles of the immune microenvironment in promoting or inhibiting human MB progression have not been extensively addressed.
Previous transcriptomic studies and immunohistochemical analysis of MB patient samples and mouse MB models revealed a higher level of immune cells in the SHH-subgroup compared to the other three MB subgroups, with tumor-associated macrophages/microglia (TAMs) being the major immune component 4, 22, 24, 33. TAMs, which can include both bone marrow-derived macrophages and brain-resident microglia, are considered a major contributor to immunosuppression through secretion of cytokines and inhibition of T cell functions and can thus promote cancer progression 20, 36, 50. Indeed, in studies of mouse models of glioblastoma (GBM), inhibition of colony-stimulating factor 1 receptor (CSF1R), a survival factor for normal macrophages/microglia, was found to reduce tumor progression, despite TAMs not being depleted 34, 35. A recent study showed that TAMs, and likely microglia-derived TAMs, promote Ptch+/−; Trp53−/− SHH-MB initiation at postnatal day (P) 35 by secreting insulin-like growth factor 1 (IGF1) 51. In contrast, another study found that knocking out Ccr2, which leads to a decrease of bone marrow-derived TAMs and not microglia 27, 47, is associated with reduced survival in a different model of SHH-MB (NeuroD2:SmoA1)24, and concluded an anti-tumor function of bone marrow-derived TAMs. Apart from TAMs, a T cell gene signature was found to be higher in SHH-MB than other subgroups 4. In mouse experiments, one study found that boosting CD8 T cells with a checkpoint inhibitor did not alter tumor progression in transplanted Ptch+/− tumors 33, whereas another study showed that inhibition of TGFβ signaling in T cells, which boosts T cell killing activity, prolonged mouse survival in the NeuroD2:SmoA1 model 9. Therefore, further research on the immune microenvironment of different SHH-MB models is critical to explore the potential use of immunotherapy on SHH-MB patients.
To investigate the role of immune cells in SHH-MB, we manipulated three different immune cell populations in an immunocompetent mouse model of sporadic SHH-MBs in which ~50% of mice die of disease. We discovered that tumors that progress to cancer have greater infiltration by TAMs. Furthermore, genetic elimination of mature B and T cells does not alter survival in our mouse model of SHH-MB. In contrast, CSF1R inhibition prolongs mouse survival, reduces tumor size by four weeks of treatment and diminishes the infiltration of a subset of TAMs and increases the percentage of in CD8 T cells in tumors. Our studies thus reveal a critical function of TAMs in promoting tumor progression in a mouse model of SHH-MB and suggest a translational potential for CSF1R inhibition in a subset of SHH-MB patients.
Results
Tumors that Progress to SHH-MB have Similar Transcriptional Profiles to Tumors that Regress
To study SHH-MB progression, we used an immunocompetent mouse model of sporadic SHH-MB in which the human oncogenic form of the SHH receptor Smoothened (SmoM2) is fused to YFP and expressed in rare granule cell progenitors (GCPs), the main cell of origin of SHH-MBs, starting at P2 by injection of a low dose of Tamoxifen (Tm) into Atoh1-CreER/+;R26SmoM2-YFP/+ mice (Atoh1-SmoM2) 43. Survival analysis of Atoh1-SmoM2 mice revealed that only a subset of mice died from advanced tumors starting at 8 weeks (52% or 68% depending on the low dose of Tm used) (Sup Fig. 1A). Previous longitudinal manganese-enhanced magnetic resonance imaging (MEMRI) analysis, however, revealed that all mice had one or two tumors in one or both hemispheres (lateral cerebellum) at 5 weeks, demonstrating that in approximately half the mice the tumors regressed 37, 43. By examining growth of the two separate tumors in individual Atoh1-SmoM2 mice from 7 weeks to 11 weeks we found their growth dynamics were not correlated (Sup Fig. 1B). Therefore, for all experiments we compared the tumor progression of individual tumors irrespective of the mouse.
As a means to test whether cell intrinsic differences could dictate tumor progression/regression we performed bulk RNA sequencing analysis of SmoM2-YFP+ tumor cells isolated by fluorescence activated cell sorting from progressing and regressing tumors at 11 weeks (Fig. 1A; Sup Fig. 1C). 161 genes were found to be differentially expressed between the two tumor types (p<0.05; fold change >1.5), with 131 genes up-regulated in the progressing tumors (Sup Table 1). Consistent with previous findings that IGF1R signaling promotes SHH-MB progression 8, 12, 39, 44, 49, 51, Gene Ontology (GO) analysis 2, 7, 26 of the genes up-regulated in the progressing tumors identified regulation of the IGF1R pathway among the top biological processes that are enriched and no enrichment for the 30 genes up-regulated in regressing tumors (Sup Fig. 1D; Sup Table 2). However, Gene Set Enrichment Analysis (GSEA)17, 42 on all differentially expressed genes showed no significant enrichment (FDR<0.25) within the hallmark gene sets for either progressing or regressing tumors. Unsupervised clustering using principal component analysis also revealed no distinct clusters of progressing and regressing tumors (Fig. 1B; Sup Fig. 1E). Thus, there appear to be few intrinsic differences between the tumor types.
Figure 1. Increased number of TAMs is associated with tumor progression in a sporadic SHH-MB mouse model.
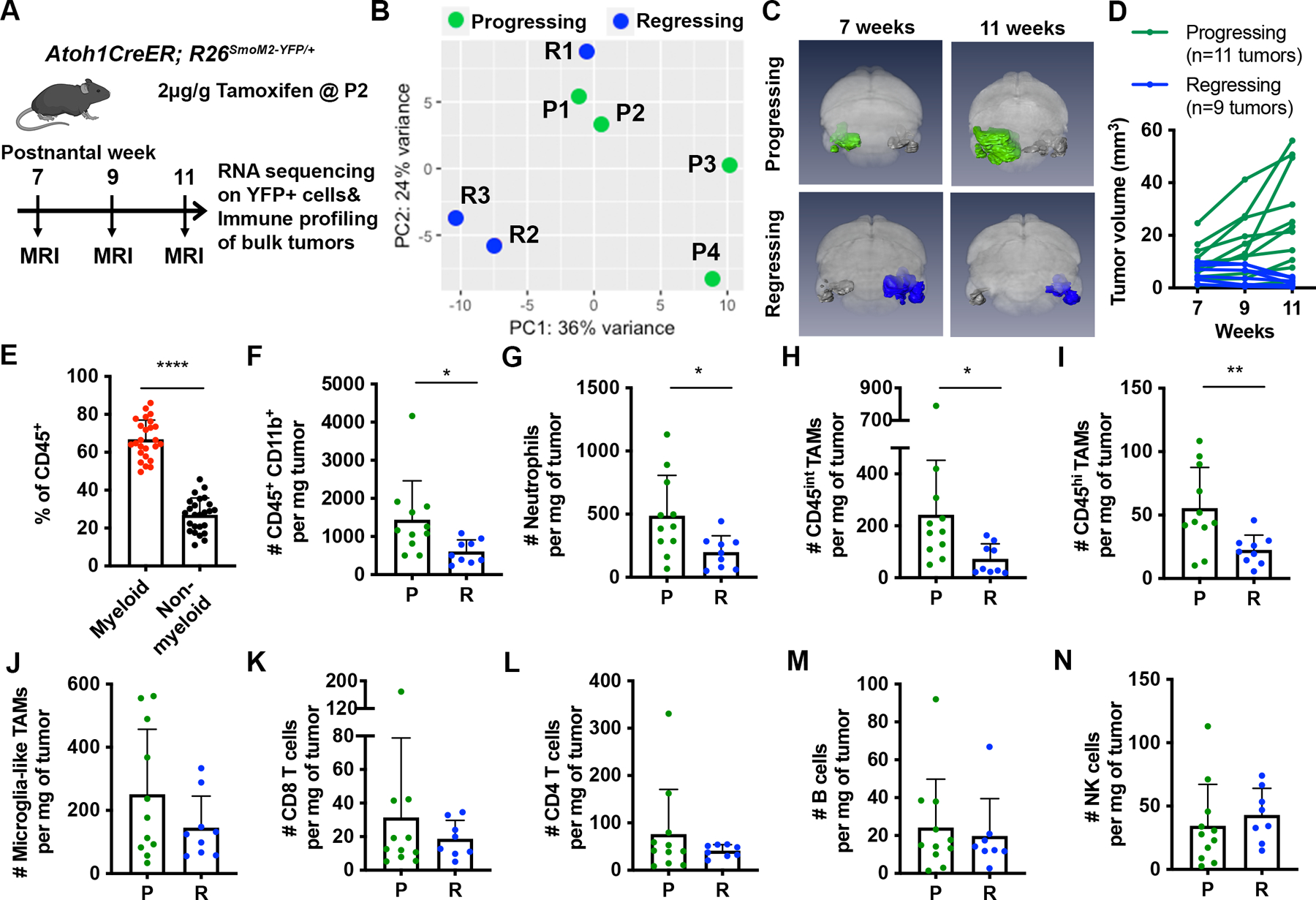
(A) Schematic representation of experimental design. (B) Principal component analysis (PCA) of progressing (P; n=4) and regressing tumors (R; n=3), which defined as having a >50% increase or >20% decrease in size between 7–11 weeks, respectively. (C) 3D MEMRI volume renderings of the brain of progressing (green) or regressing (blue) tumors in Atoh1-SmoM2 mice. (D) Tumor progression curves of progressing and regressing tumors. (E) Percentage of CD45+CD11B+ myeloid cells among all immune cells in 25 Atoh1-SmoM2 tumors (n=15 mice). (F) Number of CD45+CD11B+ myeloid cells per milligram (mg) of tumor in progressing and regressing tumors. (G–N) Number of indicated cell types per mg of tumor. Data are pooled from 3 FACS experiments. Mean ± SD. Significance was determined using unpaired t test, ****p<0.0001, **p<0.01, *p<0.05.
The Immune Microenvironment of Tumors that Progress to SHH-MB have more TAMs than Tumors that Regress
We next asked whether the tumor microenvironment (TME) could be different between progressing and regressing tumors. We first analyzed the RNA-seq data for differential expression of genes associated with influencing or responding to the microenvironment. Interestingly, while cytokine transcripts were present at low levels with little difference between the progressing and regressing tumor cells (Sup Fig. 1F), both tumor types expressed several cytokine receptors and interferon (IFN) receptors showing that tumor cells are capable of responding to cytokines secreted by immune cells (Sup Fig. 1G). Histological analysis of tumor sections showed that both tumor types have a classic MB histology (Sup Fig. 2A–C). Immunofluorescence staining revealed that regressing tumors have a higher amount of astrogliosis (GFAP+ cells) and have clusters of CD45+ cells, a pan-immune cell marker (Sup Fig. 2D–F). Furthermore, co-staining for CD45 and IBA1, a macrophage/microglia marker, showed a high density of scattered IBA1+ cells that have low expression of CD45 in the progressing tumors, whereas in regressing tumors IBA1+ cells were seen in clusters as well as scattered throughout the tumor (Sup Fig. 2G–I), suggesting a difference in the TME of progressing and regressing tumors.
To quantify the immune components of the two types of tumors, individual tumors were macro-dissected from Atoh1-SmoM2 mice at 11 weeks based on MEMRI and immune profiled by flow cytometry (Fig. 1A). As seen previously 37, 43, we found ~50% of tumors spontaneously regressed after 7 weeks based on longitudinal MEMRI of Atoh1-SmoM2 mice (n= 34 mice) (Fig. 1C–D). Consistent with human SHH-MB tumors, we observed that CD45+ CD11b+ myeloid cells constituted ~70% of all immune cells present in Atoh1-SmoM2 tumors (Fig. 1E). Furthermore, progressing tumors showed a higher number of CD45+ CD11b+ myeloid cells per milligram (mg) tumor compared to regressing tumors (Fig. 1F). Since the expression level of CD45 was previously used to distinguish CNS-infiltrating, bone marrow-derived myeloid (BMDM) cells (CD45high (hi)) from resident microglia (CD45intermediate (int)) in mouse models 10, 14, we analyzed the myeloid population in our tumors based on the expression levels of CD45, CD11b, and several additional myeloid cell markers (Sup Table 3). Interestingly we identified 3 clear cell cluster after gating on the CD45+ CD11b+ Ly6G− population: 1) a CD45hi CD11bhi mixed myeloid population that includes CD45hi TAMs, dendritic cells, and eosinophils, 2) a population of cells with CD45int CD11bhi similar to microglia in the normal brain (referred to as microglia-like TAMs), and 3) a CD45int CD11bint population not seen in the normal cerebellum (referred to as CD45int TAMs) (Sup Fig 3A–B and Sup Table 3). Since BMDMs have been found to down-regulate CD45 and acquire tissue resident (microglia) gene expression upon infiltration into the brain, and activated microglia in tumors up-regulate of CD45 3, 10, BMDM- and microglia-derived TAMs can be present in all three populations.
Flow cytometry analysis showed that the number of neutrophils, CD45int TAMs and CD45hi TAMs per mg of tumor was significantly higher in progressing tumors than in regressing tumors (Fig. 1G–I). Immunofluorescence staining of sections also indicated a higher density of neutrophils (Ly6G+) in progressing tumors (Sup Fig 4A–D). No significant changes were observed in the number of microglia-like TAMs, CD8+ T cells, CD4+ T cells, B cells or natural killer cells between the two tumor types (Fig. 1J–N). Taken together, our analysis reveals a positive correlation between tumor progression and the number of neutrophils, CD45hi and CD45int TAMs but not microglia-like TAMs.
SHH-MB Patient Samples with Different Driver Mutations have Similar Levels of T cell and TAM Infiltration
To expand upon previous studies showing that human SHH-MBs have a higher infiltration of TAMs compared to other MB subgroups in both patient samples and mouse models 4, 22, 24, 33, we asked whether the driver mutation in human SHH-MBs influences the infiltration of TAMs or T cells. We performed IHC staining for a T cell marker (CD3), two TAM markers (CD68 and CD163) and a proliferation marker (Ki67) on sections from 20 SHH-MB patient samples and analyzed the association between number and distribution of positive cells and driver mutations and/or clinical presentation (Fig. 2A–D). In SHH-MBs, there are four frequent mutually exclusive driver gene mutations (PTCH1, SUFU, SMO, and TP53), and each mutation is associated with a particular clinical presentation 15. Our results confirmed the presence of T cells and TAMs in all MB patient samples, but also showed no clear correlation between the immune cell types examined and driver mutation, patient age group or tumor histology (Fig. 2E–J, Sup Fig. 5A–H). Tumors belonging to the childhood age group (4–17 years old), however, had less Ki67 staining than those in the infant and adult age group indicating reduced proliferation in this tumor subtype (Sup Fig. 5A). Thus, our results indicate that T cells and TAMs infiltrate human SHH-MBs regardless of driver mutation or subtype.
Figure 2. The amount of immune cell infiltration in human SHH-MB samples does not correlate with driver mutation.
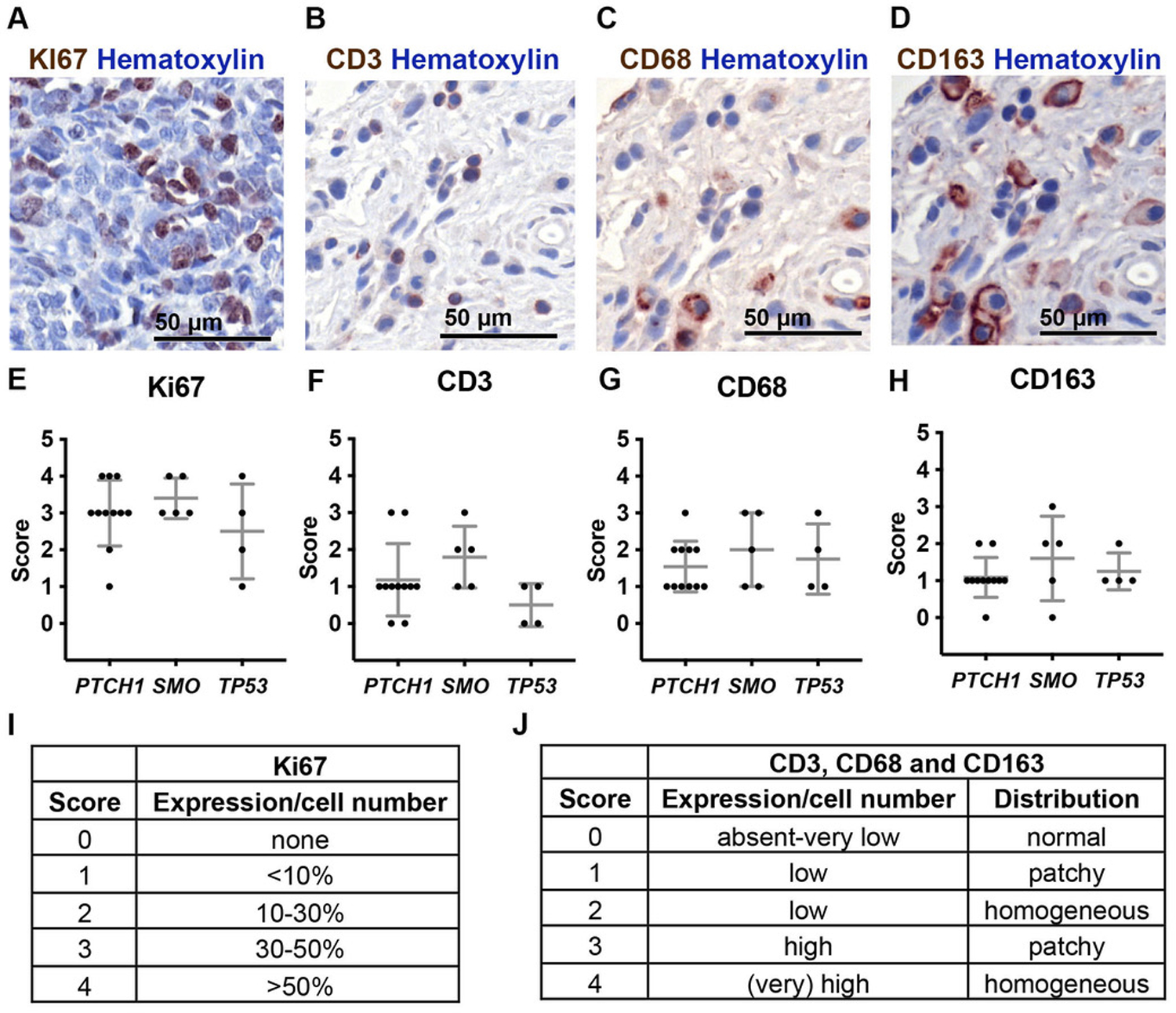
(A–D) Representative immunohistochemical staining of Ki67 (A), CD3 (B), CD68 (C) and CD163 (D) from 20 SHH-MB samples. (E–H) Correlation between the staining of Ki67 (E), CD3 (F), CD68 (G) and CD163 (H) and driver mutation. Quantification was done on the region that had the strongest positive signal and a score was given to each sample based on the expression and distribution of positive cells as indicated in (I–J). Mean ± SD. Significance was determined using one-way ANOVA with Tukey post hoc pairwise comparison.
Depletion of Lymphocytes Does Not Alter Tumor Progression in Atoh1-SmoM2 Mice
Given the contradictory findings of whether boosting T cell function reduces SHH-MB progression 9, 33, we reduced CD4+ CD25+ regulatory T cells (Tregs), which inhibit T cell function and thus can play an important role in immunosuppression in cancer 41, 46 in Atoh1-SmoM2 mice. We performed a survival analysis in which Atoh1-SmoM2 mice were treated with IgG or PC61 antibody, one dose every 3 weeks, starting at 3 weeks until mice became symptomatic from tumor burden or our experimental endpoint (150 days). Treg depletion, however, did not lead to a significant survival benefit or change in tumor volume at 11 weeks in Atoh1-SmoM2 mice (Fig. 3A–C). Immune profiling at 11 weeks confirmed an ~50% decrease in the percentage of Tregs among CD4+ T cells in the PC61-treated tumors compared to IgG control, although the percentage of CD8+ T cells among TCRβ+ cells and CD45+ CD11b+ myeloid cells among CD45+ cells remained similar between the two groups (Fig. 3D–F). We next asked whether depletion of mature T and B cells would promote progression of Atoh1-SmoM2 tumors by generating Atoh1-SmoM2 mice deficient for Rag1−/−28. Interestingly, survival analysis showed that Atoh1-SmoM2-Rag1−/− mice do not have a survival disadvantage when compared to mice heterozygous or wild type for a Rag1 null mutation (Fig. 3G; Sup Fig 6A). In summary, an ~50% reduction in Tregs or genetic loss of T and B cells does not alter mouse survival in Atoh1-SmoM2 mice.
Figure 3. Depletion of Tregs or T cells in Atoh1-SmoM2 mice does not alter mouse survival.
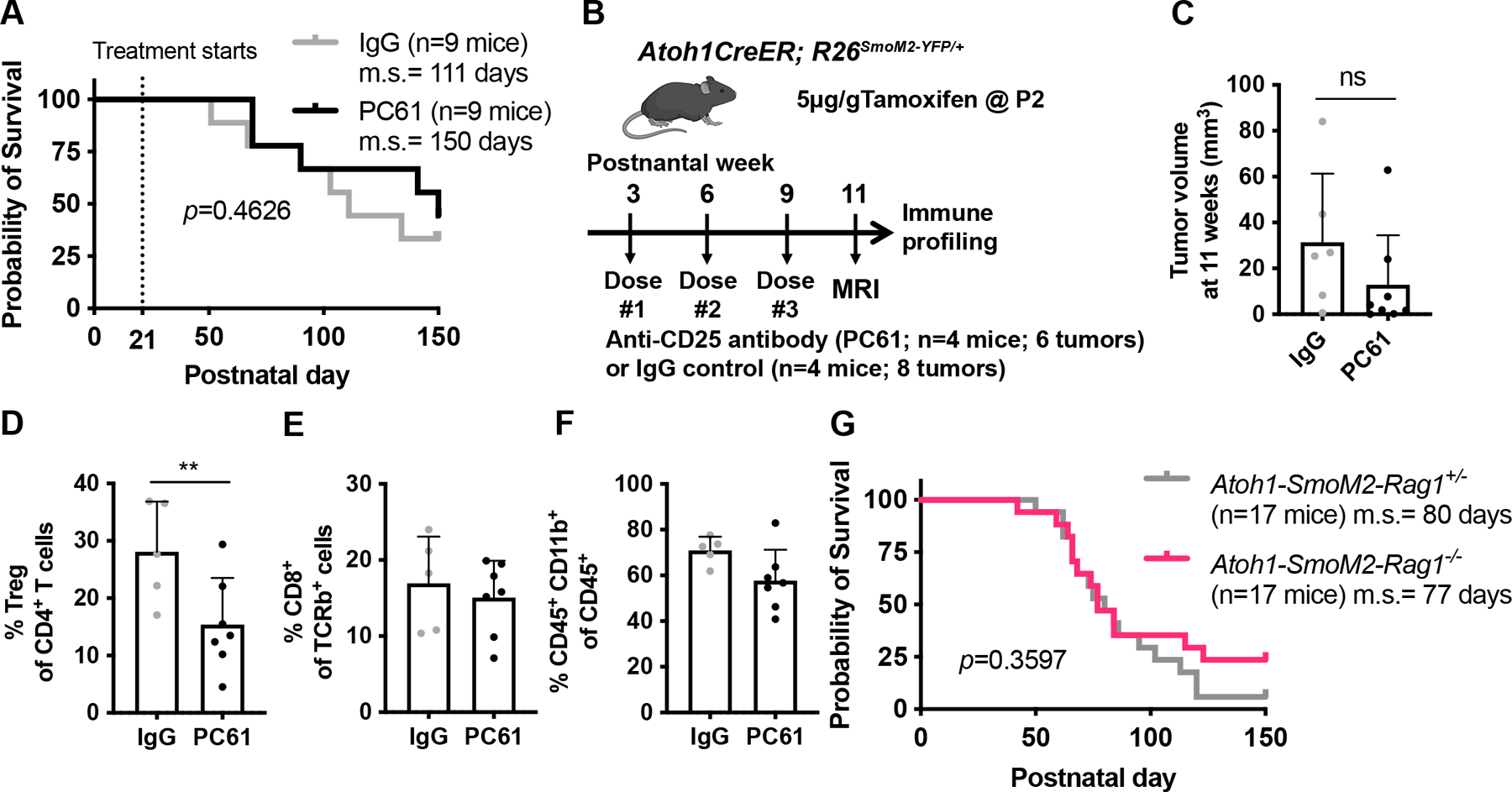
(A) Kaplan–Meier curves of Atoh1-SmoM2 mice treated with IgG control antibody or anti-CD25 antibody (PC61) antibody starting at 3 weeks. (B) Schematic representation of Treg depletion experimental design. (C) Tumor volume of mice after given 3 doses of PC61or IgG control antibody (8 weeks of treatment). (D–F) Flow cytometry analysis of control (n=3 mice, 5 tumors) and PC61-treated (n=4 mice, 7 tumors) showing percentage of Tregs among CD4+ T cells (C), percentage of CD8+ T cells among TCRb+ T cells (D) and percentage of CD45+ CD11b+ myeloid cells among CD45+ immune cells (E). (G) Kaplan–Meier curves of Atoh1-SmoM2-Rag1 heterozygous null (control) and Atoh1-SmoM2-Rag1 homozygous null (no mature B and T cells) littermates. m.s. = median survival. Significance was determined using Log-rank test for (A, G) and unpaired t test for (C–F). **p<0.01, ns = non-significant.
Depletion of TAMs Prolongs Mouse Survival
As a means to test whether TAMs promote tumor progression in SHH-MBs, we treated Atoh1-SmoM2 mice with the CSF1R inhibitor PLX5622 (PLX) 13, 32 . Recent studies in several mouse tumor models, including GBM, showed that targeting TAMs with CSF1R inhibitors can reduce tumor growth through either depleting or reprograming TAMs 34, 40, 52. We therefore first determined whether CSF1R inhibition reduces the number of TAMs in Atoh1-SmoM2 tumors. We preselected Atoh1-SmoM2 mice at 7 weeks for those with growing tumors with a >7 mm3 volume using MEMRI, and then fed the mice for 2 or 6 weeks PLX or control chow. Interestingly, unlike the findings in a GBM model 34, we observed an ~60% reduction in the density of IBA1+ TAMs on sections of SHH-MBs from mice fed PLX chow for 2 or 6 weeks compared to control (CTRL) chow (Sup Fig. 7A–B). There was also a 50–60% reduction in the density of microglia in the forebrain of PLX-treated mice at both stages, confirming the drug crosses the blood brain barrier (Sup Fig. 7C).
We next tested whether PLX treatment offers a survival benefit to mice with SHH-MBs. Atoh1-SmoM2 mice were randomly placed on CTRL chow or PLX chow at 7 weeks and treated for 100 days or until the mice died. Significantly, 65% (17/26) of CTRL mice died with a median survival of 49 days post-treatment, whereas only 33% (8/24) of PLX mice died by the experiment endpoint (Fig. 4A). CSF1R blockade should not have a direct effect on Atoh1-SmoM2 tumor cells since CSF1R protein or RNA expression was not be detected (Sup Fig. 8A–C). Indeed, when Atoh1-SmoM2 granule cell precursors were grown in serial doses of PLX, growth was not reduced although the highest dose was toxic (Sup Fig.8D–F). Thus, depletion of TAMs by CSF1R inhibition significantly increases overall survival of mice in our SHH-MB model.
Figure 4. Depletion of TAMs prolongs mouse survival and blocks tumor progression in most Atoh1-SmoM2 mice.
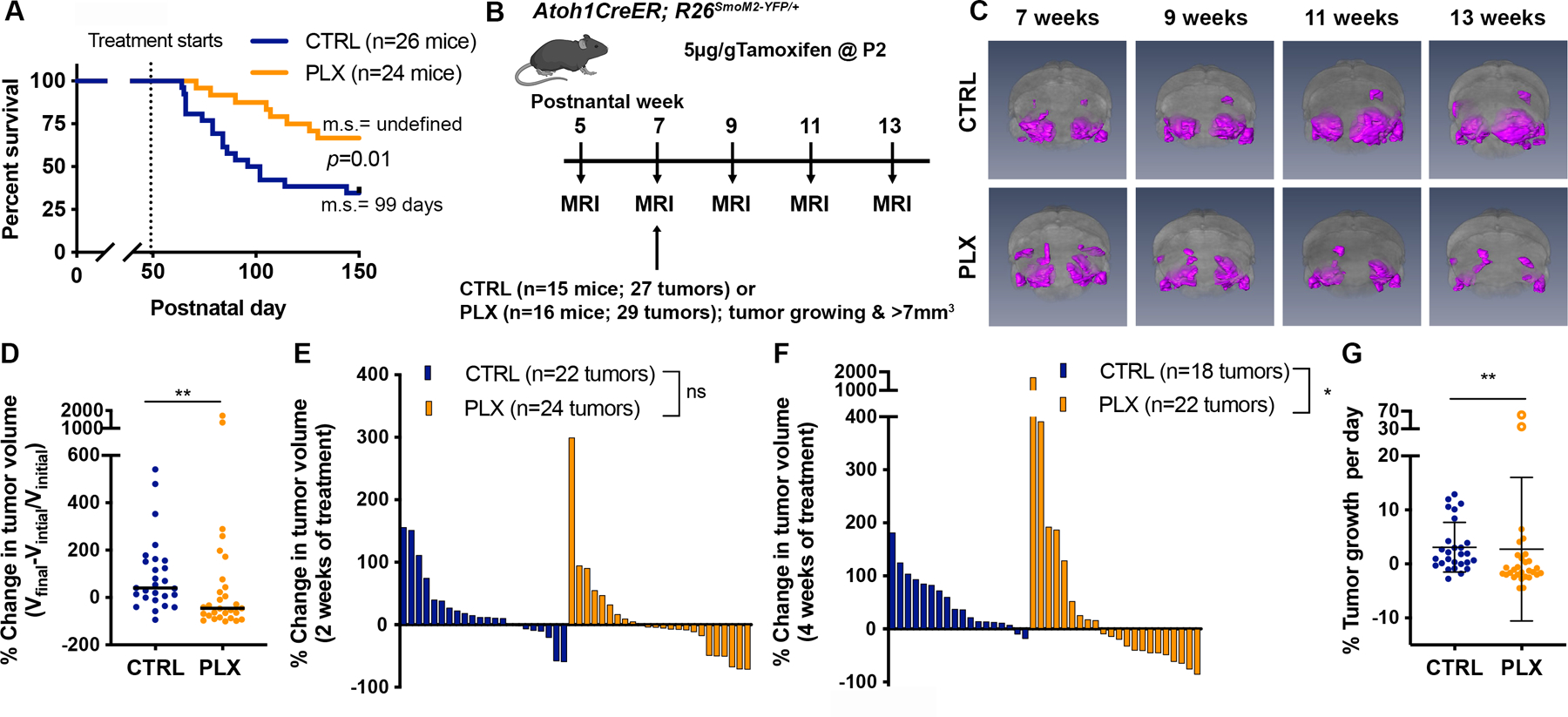
(A) Kaplan–Meier curves of control (CTRL)- or PLX5622 (PLX)-treated Atoh1-SmoM2 mice. m.s. = median survival. (B) Schematic representation of experimental design. (C) 3D MEMRI volume renderings of the brain (grey) and tumors (magenta) in CTRL and PLX-treated mice at the indicated time points. (D) The overall change in tumor volume between the pre-treatment time point (Vinitial; 7 weeks) and the experimental endpoint (Vfinal; mice became symptomatic or at 13 weeks). Black line indicates median. (E–F) Waterfall plots showing the change in tumor volume from the pre-treatment time to after 2 weeks (E) and 4 weeks (F) of CTRL or PLX treatment. (G) The growth rate calculated as the percentage tumor volume change per day. The open circles indicate outlier PLX tumors that grew faster than the controls. Mean ± SD. Significance was determined using Log-rank test for (A) and Mann-Whitney test for (D–G). **p<0.01, *p<0.05, ns = non-significant.
Depletion of TAMs Attenuates Tumor Progression
To understand how CSF1R inhibition affects tumor progression we excluded mice with small or regressing tumors using MEMRI, and identified Atoh1-SmoM2 mice with tumors that were likely to progress based on the criteria of growth in at least one tumor between 5 and 7 weeks and a size >7mm3 37. Tumor progression was tracked every two weeks using MEMRI until mice showed tumor symptoms or reached the experimental endpoint of 13 weeks (Fig. 4B–C). Strikingly, we found PLX treatment significantly attenuated tumor growth in most mice. Whereas there was a 40% increase in the median overall tumor volume change for CTRL mice, there was instead a 45% decrease for PLX mice (Fig. 4D). MEMRI showed that 2 weeks of PLX treatment was not sufficient to block tumor progression, but that 4 weeks of PLX treatment produced a significant reduction in tumor volume (Fig. 4E–F). Overall, we observed that 20/27 (74%) of tumors in the CTRL group had a positive growth rate whereas only 10/29 (34%) tumors did in the PLX group. Curiously, two of the PLX-treated tumors in different mice (2/16 mice) did not respond to PLX by reducing growth, but instead grew faster than tumors in the CTRL mice (Fig. 4G; referred to as outliers). Interestingly, each mouse had a second tumor in the contralateral cerebellar hemisphere that did regress, further showing that independent tumors in mice have different responses to drug treatment. Our results thus demonstrate that CSF1R inhibition reduces growth of most but not all Atoh1-SmoM2 MBs. Furthermore, for rare tumors, PLX treatment might promote tumor growth and thus have an adverse effect (see Supplemental Discussion).
Reduction of TAMs is Associated with an Increase in CD8+ Cytotoxic T cells
Immune profiling by flow cytometry was performed to determine whether cellular changes in the immune populations other than IBA1+ TAMs were induced after 4 weeks of treatment, a time point that we observed a significant decrease in tumor volume in PLX-treated mice. We selected Atoh1-SmoM2 mice with tumors that were likely to progress with MEMRI and started PLX treatment at 7 weeks. We performed MEMRI again to evaluate tumor progression right before immune profiling at 11 weeks (Fig. 5A–B). Of note, one of the PLX-treated mice for which the immune cell composition was profiled had an outlier tumor that grew faster than CTRL-treated tumors (indicated by an open circle in Fig. 5B). Flow cytometry analysis of blood samples from 11-week-old CTRL/PLX-treated mice showed a significant reduction of Ly6Chi monocytes and an apparent although not significant reduction of Ly6Clow monocytes and macrophages (Sup Fig. 9A–C). No significant changes were observed in neutrophils, dendritic cells and lymphocyte populations in blood samples (Sup Fig. 9D–J). Analysis of CTRL and PLX tumors showed a significant reduction in the percentage of CD45int TAMs and microglia-like TAMs among all immune cells and CD45int TAMs among the CD45+ CD11b+ Ly6G− population in the PLX-treated tumors, indicating functional CSF1R inhibition in the PLX-treated tumors (Fig. 5C–F; Sup Fig 10A–B). Interestingly, the outlier tumor had a greater percentage of CD45int TAMs, even higher than in the controls (Fig. 5E). Furthermore, we found a positive correlation between tumor growth and the percentage of CD45int TAMs and microglia-like TAMs in the PLX-treated tumors but not in the CTRL-treated tumors that all have a high percentage of TAMs (Sup Fig 11A–B). No significant changes or correlations were observed in CD45hi TAMs and other myeloid cell types (Fig. 5G; Sup Fig. 10C–F, 11C.). In contrast, 4 weeks of PLX treatment led to a significant increase of CD8+ cytotoxic T cells and a decrease of B cells among lymphocytes in tumors, although there was no correlation between tumor growth and the percentage of these two immune cells types (Fig. 5H–I, K; Sup Fig 11D–E). No significant changes were observed in the percentage of CD4+ T cells, PD-1 expression on T cells and other lymphocyte populations in tumors (Fig. 5J; Sup Fig. 10G–J). Thus, CSF1R inhibition changes the immune microenvironment in a specific way that reduces a subset of TAMs and B cells but increases cytotoxic T cells. Furthermore, the correlation seen between the percentage of TAMs in a tumor treated with PLX and tumor growth might explain the heterogeneity in PLX treatment response, further supporting that the TAMs are a key immune cell influencing tumor growth.
Figure 5. Atoh1-SmoM2 tumors treated for 4 weeks with PLX show a decrease of monocytes/macrophages and an increase of CD8 cytotoxic T cells.
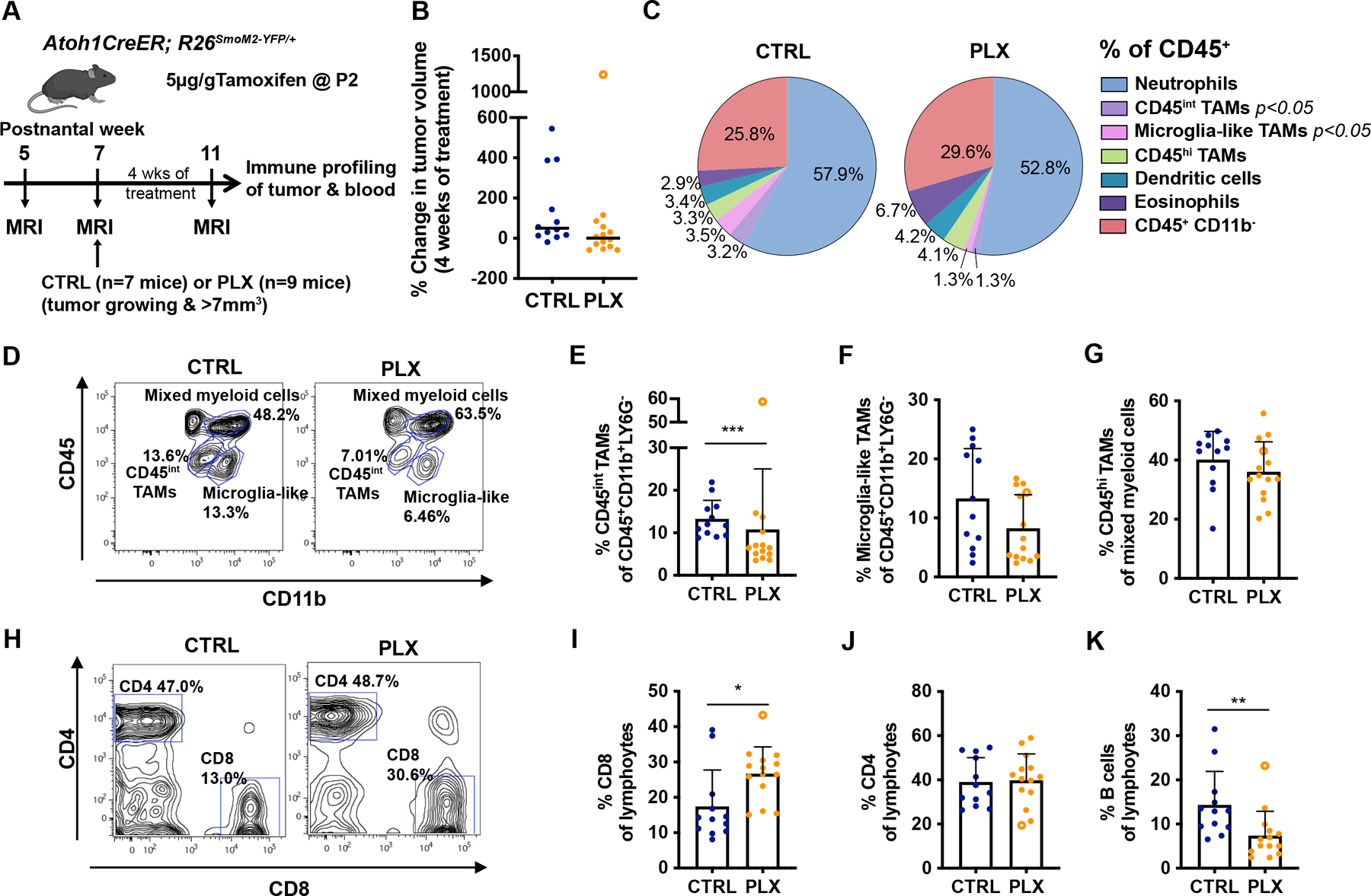
(A) Experimental design. (B) Change in tumor volume between pre-treatment and 4 weeks of PLX treatment. The open circle in (B, E–G, I–K) indicates an outlier PLX tumor. Black line indicates median. (C) Flow cytometry quantification of myeloid cell composition in CTRL (n=12) and PLX (n=14) treated tumors. Significance was found in the CD45int TAM and microglia-like TAMs populations (See Sup Fig. 10). (D–G) Representative flow cytometry panels from the CD45+CD11B+Ly6G− population (D) showing the proportion of CD45int TAMs (E) and microglia-like TAMs (F). (G) Percentage of CD45hi TAMs of mixed myeloid population. (H–J) Representative flow cytometry panel from the lymphocytes (CD45+CD19+TCRβ+) showing the proportion of CD8+ T cells (I), CD4 T+ cells (J). (K) Percentage of B cells (CD45+CD19+B220+) of the lymphocyte population. Data are pooled from 3 FACS experiments. Mean ± SD. Significance was determined using Mann-Whitney test for (B), and performed excluding the outlier tumor by using unpaired t test for (C), (E–G) and (I–K), ***p<0.001, **p<0.01, *p<0.05.
Given that TAMs have been reported to inhibit cytotoxic T cell proliferation 11, we asked whether the reduction of TAMs in PLX-treated tumors led to an increase in T cell activity. Flow cytometry was performed on CTRL tumors and PLX tumors, selected by MEMRI, to analyze T cell activation after 4 weeks of drug treatment (Fig. 6A). The number of CD8+ and CD4+ T cells per mg of PLX tumor was slightly but not significantly increased compared to the CTRL and we did not observe a significant difference in the number of CD45+ cells between CTRL and PLX tumors, possibly due to the concomitant decrease of TAMs in PLX tumors. (Fig. 6B–D). We observed an apparent increase of Granzyme B+ CD107+ CD8+ T cells per gm tumor and among CD8+ T cells indicating a higher level of cytotoxic activity of CD8+ T cells in PLX-treated tumors (Fig. 6E–F). Importantly, the number of IFNγ+ CD8+ T cells per mg of tumor and the percentage IFNγ+ cells among CD4+ T cells both were significantly increased, indicating a pro-inflammatory TME in PLX-treated tumors (Fig. 6G–L). Furthermore, immune profiling of PLX-treated tumors revealed a significant increase in the percentage of CD45int TAMs expressing MHC class II molecules, a gene induced by IFNγ in macrophages 6 (Fig. 7A–B). The percentage of MHC class II expressing microglia-like TAMs and CD45hi TAMs in tumors were similar between CTRL and PLX-treated mice (Fig. 7C–F). Interestingly, the outlier tumor had the lowest percentage of MHC class II+ CD45int TAMs. Our analysis thus shows that the decrease of TAMs in PLX treated tumors is associated with an increase of intratumoral CD8+ T cells with cytotoxic capacity, and a higher level of IFNγ+ production in a majority of mice. Taken together, the increase in cytotoxic T cell trafficking into the brain could contribute to an anti-tumor immune microenvironment in SHH-MBs in addition to a decrease in pro-tumor growth TAMs.
Figure 6. Depletion of TAMs in Atoh1-SmoM2 mice leads to an increase of pro-inflammatory T cells.
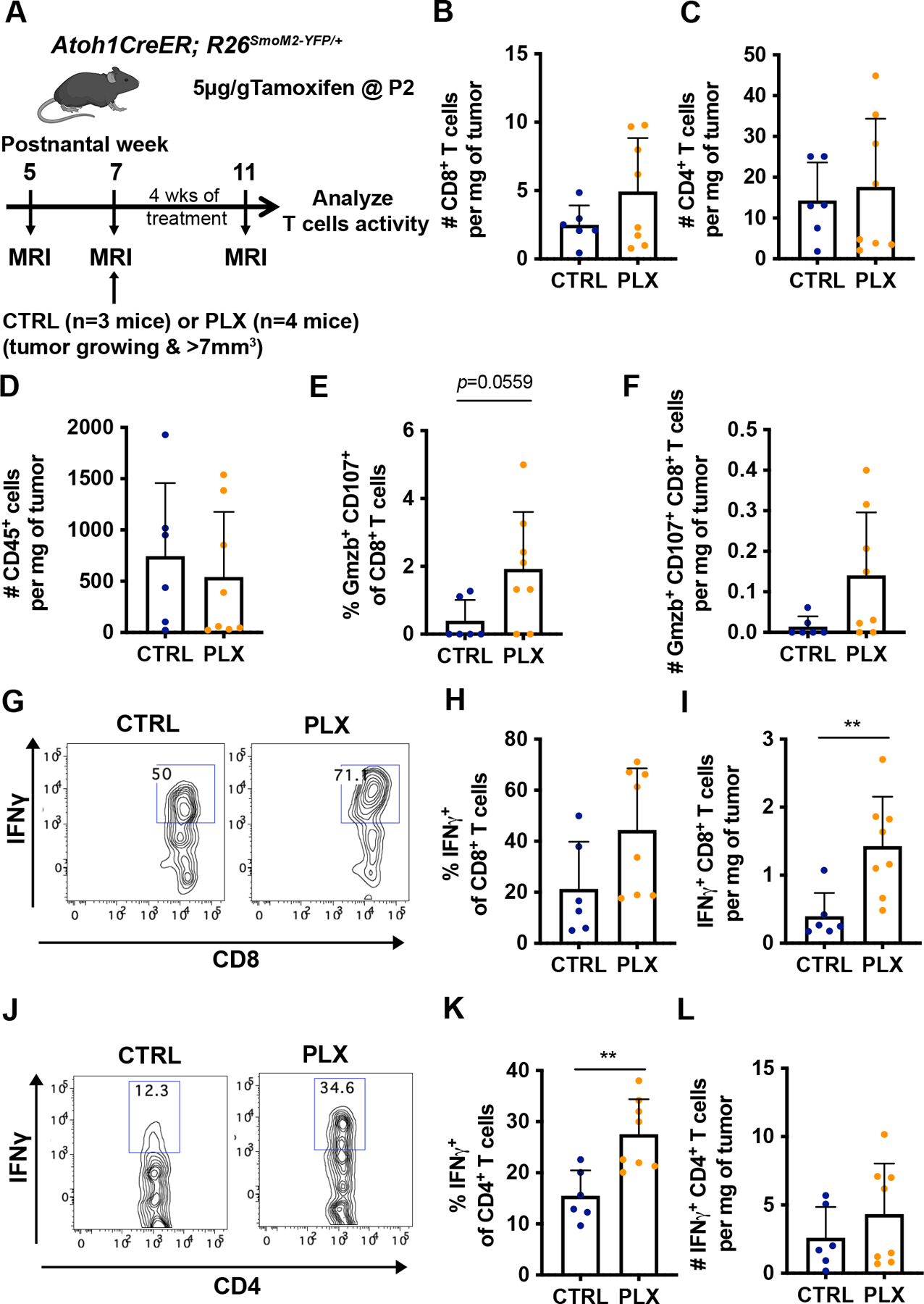
(A) Schematic representation of experimental design. (B–D) Number of CD8+ T cells (B), CD4+ T cells (C), and CD45+ immune cells (D) per milligram (mg) of tumor in control (CTRL)- and PLX-treated tumors. (E) Percentage of Granzyme B+ (Gzmb) CD107+CD8+ T cells of total CD8+ T cells. (F) Number of Gzmb+ CD107+CD8+ T cells per mg of tumor. (G–I) Representative flow cytometry panels from the CD45hi TCRβ+ CD8+ population (G) showing the proportion (H) and the number (I) of IFNγ+CD8+ T cells. (J–L) Representative flow cytometry panels from the CD45hiTCRβ+CD4+ population (J) showing the proportion (K) and the number (L) of IFNγ+CD4+ T cells. Mean ± SD. Significance was determined using unpaired t test for (B–F), (H–I) and (K–L). **p<0.01.
Figure 7. Depletion of TAMs in Atoh1-SmoM2 mice leads to a pro-inflammatory tumor microenvironment.
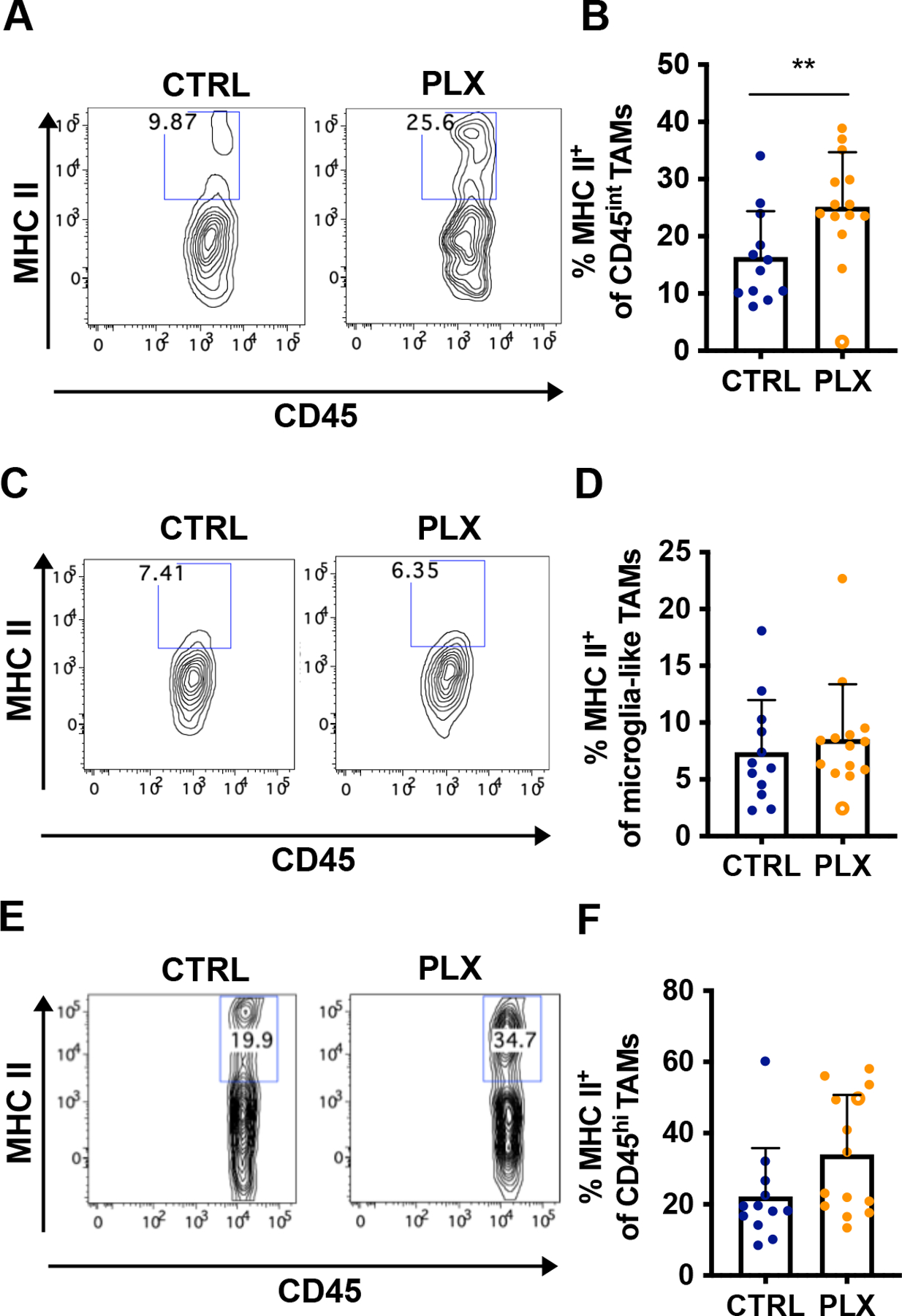
(A–F) Analysis of MHC II expression in 12 CTRL tumors and 14 PLX tumors (see Fig. 5). The open circle indicates an outlier PLX tumor. Representative flow cytometry panels from the CD45int TAMs population (A) showing the proportion of MHC II+ CD45int TAMs (B). Representative flow cytometry panels from the microglia-like TAMs population (C) showing the proportion of MHC II+ microglia-like TAMs (D). Representative flow cytometry panels from the CD45hi TAMs population (E) showing the proportion of MHC II+ CD45hi TAMs (F). Mean ± SD. Significance was performed excluding the outlier tumor by using unpaired t test. **p<0.01.
Discussion
Macrophages are the most abundant immune cell population found in SHH-MB, but have been found to have both pro- and anti-tumor functions 4, 22, 24, 51. While two separate studies found no correlation between the number of TAMs and survival4, 22, one study with a small number of patients found a higher level of CD86+ TAMs was associated with a worse overall survival in SHH-MB patients16. The conflicting results might be attributed to the high survival rate (~75%) for patients in the SHH subgroup and/or intertumoral heterogeneity within SHH-MBs 5, 30. Indeed, a recent study showed that high expression of the macrophage gene AIF1 correlates with better survival specifically in the SHH-alpha subtype that includes TP53 mutations 24. Thus, SHH-MBs with different mutation profiles might have distinct immune microenvironments.
In our study, we characterized the tumor immune environment of a mouse SHH-MB model that expresses an activating mutation in SMO, a type of mutation commonly found in adult SHH-MBs. We found that a higher number of myeloid cells is associated with tumors that progress than regress, raising the possibility of a pro-tumor growth function of TAMs. Given the increase in IGFR signaling in progressing tumor cells identified by RNA-seq and the previous finding that microglia-derived TAMs appear to promote tumor initiation via secretion of IGF1 in the Ptch+/−; Tp53−/− SHH-MB model 51, the lower number of TAMs in Atoh1-SmoM2 regressing tumors might result in altered IGF signaling that influences tumor progression.
A recent study found that deletion of Ccr2 reduces a subset of TAMs and enhances tumor progression, which on the surface indicates an opposite role of TAMs in NeuroD2:SmoA1 and Atoh1-SmoM2 tumors 24. Likely of significance, the tumors in the two SHH-MB models have distinct immune profiles, indicating that tumor cells can shape the TME and determine the response to TAM depletion. The predominant immune cell types in NeuroD2:SmoA1 tumors are CD45hi Ly6C+ TAMs (30% of all immune cells in the NeuroD2:SmoA1 model and 62% in the transplantation model) and CD45int Ly6Clow TAMs (51% and 29%), with relatively low percentages of neutrophils (9% and 4.6%) and lymphocytes (10% and 4.4%). In contrast, Atoh1-SmoM2 tumors have relatively less CD45hi myeloid cells (9.7% including CD45hi TAMs, dendritic cells, and eosinophils) and CD45int myeloid cells (6.7% including CD45int TAMs and microglia-like TAMs) and more neutrophils (57.9%) and lymphocytes (25.8%). CSF1R inhibition in the Atoh1-SmoM2 model led to a significant decrease of CD45int but not CD45hi myeloid cells and also an increase of pro-inflammatory T cells in tumors. It is possible that the increase in the ratio of T cells to CD45int myeloid cells resulted in the anti-tumor effect of CSF1R inhibition in Atoh1-SmoM2 mice. Since immune profiling was not performed when CSF1R was inhibited in NeuroD2:SmoA1 transplanted tumors using two inhibitors (GW2580, BLZ945), it is not known whether CD45hi and/or CD45int myeloid cells were depleted or T cells increased in this model. Immune profiling of NeuroD2:SmoA1 mice lacking Ccr2 or after orthotopic transplantation of SmoA1-Ccr2+/+ tumor cells into Ccr2 null mice did however show a decrease in CD45hi Ly6C+ TAMs but an increase of CD45int Ly6Clow TAMs. Since Ccr2 knockout mice display reduced recruitment of monocytes into inflamed tissue and Ccr2 is not required for the development of microglia 27, 47, it was assumed that the decrease of monocyte-derived TAMs led to a worse survival in the Ccr2 null mice. An alternative interpretation is that the CD45int Ly6Clow TAMs had a pro-tumor function, which is consistent with our study. However, the developmental origin of the CD45hi and CD45int myeloid cells in the various models remain to be elucidated and whether lineage has functional consequences.
The number of tumor-infiltrating lymphocytes is highly variable between the 4 subgroups of MBs, with SHH-MBs having a relatively higher amount of T cells 4, 23, 29, 48. We found no survival advantage of removing mature T and B cells in Atoh1-SmoM2 mice by deleting Rag1. Furthermore, reducing Tregs by ~50% (PC61 antibody) did not prolonged survival of Atoh1-SmoM2 mice, although cytotoxic T cells were also not increased. Increasing T cell infiltration using immune checkpoint blockade also was not found to reduce the tumor progression in a syngeneic Ptch1+/− SHH-MB model 33. In contrast, decreasing Tregs and boosting T cell killing activity by inhibiting TGFβ signaling in T cells was shown to prolong mouse survival in NeuroD2: SmoA1 mice 9. The different effect on survival might be explained by either the ways that T cells were manipulated or as discussed above by the different TMEs in each model. Overall, the studies support the conclusion that TAMs play a major role in shaping the immune microenvironment of SHH-MBs, raising the possibility that targeting pro-tumor TAMs could be efficacious to a subset of SHH-MB patients.
Materials and Methods
Animals
All animal experiments were performed with the approval of the Institutional Animal Care and Use Committees at MSKCC and NYU. Atoh1-CreER (Swiss Webster)19 and R26LSL-SmoM2-YFP /LSL-SmoM2-YFP (C57BL/6)21 mouse lines were bred to generate SHH-MBs (Atoh1-SmoM2 model), and to Rag1tm1Mom (C57BL/6)28 to generate tumors homozygous or heterozygous for Rag1. Both sexes were used for all studies. Animals were housed on a 12-hour (hr) light/dark cycle and were given access to food and water ad libitum. The number of tumors and/or mice analyzed are provided in the Results section and/or in the Figure legends. Mice from each litter in the survival studies were randomized into control or experimental groups to generate littermate controls. No blinging was performed for all animal studies.
CSF1R inhibitor treatment
PLX5622 powder, PLX5622 chow (1200 mg drug per kg chow) and control chow were provided by Plexxikon under a Materials Transfer Agreement. For all experiments, mice were given control or PLX5622 chow at 7 weeks of age.
Statistical analyses
All statistical analyses were performed using Prism software (GraphPad) and significance was determined as p< 0.05. Summary of all the statistical analysis and p values are in the Supplementary Materials and Methods. No statistical methods were used to predetermine the sample size, but our sample sizes are similar to those generally employed in the field.
Supplementary Material
Acknowledgments
We thank the Joyner lab members for helpful discussions and comments on the manuscript. We are grateful to Andrey Rymar and Parmveer Singh at Plexxikon for providing the PLX5622 used in these studies. We appreciate the technical assistance from Daniel Stephen, and Yu-Jung Chen. We also thank the Center for comparative medicine and pathology and the Integrated Genomics Operation (IGO) core facilities of MSKCC for outstanding technical support. This work was supported by the following grants: R01CA192176 (ALJ), MSKCC Brain Tumor Center fellowship (to AW), and a National Cancer Institute Cancer Center Support Grant [P30 CA008748-48].
Footnotes
Conflict of Interest: The authors declare they have no conflicts of interest.
References
- 1.Archer TC, Ehrenberger T, Mundt F, Gold MP, Krug K, Mah CK et al. Proteomics, Post-translational Modifications, and Integrative Analyses Reveal Molecular Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 2018; 34: 396–410 e398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature genetics 2000; 25: 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB et al. New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci U S A 2016; 113: E1738–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bockmayr M, Mohme M, Klauschen F, Winkler B, Budczies J, Rutkowski S et al. Subgroup-specific immune and stromal microenvironment in medulloblastoma. Oncoimmunology 2018; 7: e1462430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cavalli FMG, Remke M, Rampasek L, Peacock J, Shih DJH, Luu B et al. Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 2017; 31: 737–754 e736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Celada A, Klemsz MJ, Maki RA. Interferon-gamma activates multiple pathways to regulate the expression of the genes for major histocompatibility class II I-A beta, tumor necrosis factor and complement component C3 in mouse macrophages. European journal of immunology 1989; 19: 1103–1109. [DOI] [PubMed] [Google Scholar]
- 7.Consortium TGO. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic acids research 2019; 47: D330–d338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Corcoran RB, Bachar Raveh T, Barakat MT, Lee EY, Scott MP. Insulin-like growth factor 2 is required for progression to advanced medulloblastoma in patched1 heterozygous mice. Cancer research 2008; 68: 8788–8795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gate D, Danielpour M, Rodriguez J Jr., Kim GB, Levy R, Bannykh S et al. T-cell TGF-beta signaling abrogation restricts medulloblastoma progression. Proc Natl Acad Sci U S A 2014; 111: E3458–3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Greter M, Lelios I, Croxford AL. Microglia Versus Myeloid Cell Nomenclature during Brain Inflammation. Frontiers in immunology 2015; 6: 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gyori D, Lim EL, Grant FM, Spensberger D, Roychoudhuri R, Shuttleworth SJ et al. Compensation between CSF1R+ macrophages and Foxp3+ Treg cells drives resistance to tumor immunotherapy. JCI Insight 2018; 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hahn H, Wojnowski L, Specht K, Kappler R, Calzada-Wack J, Potter D et al. Patched target Igf2 is indispensable for the formation of medulloblastoma and rhabdomyosarcoma. The Journal of biological chemistry 2000; 275: 28341–28344. [DOI] [PubMed] [Google Scholar]
- 13.Huang Y, Xu Z, Xiong S, Sun F, Qin G, Hu G et al. Repopulated microglia are solely derived from the proliferation of residual microglia after acute depletion. Nature neuroscience 2018; 21: 530–540. [DOI] [PubMed] [Google Scholar]
- 14.Kana V, Desland FA, Casanova-Acebes M, Ayata P, Badimon A, Nabel E et al. CSF-1 controls cerebellar microglia and is required for motor function and social interaction. J Exp Med 2019; 216: 2265–2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kool M, Jones DT, Jager N, Northcott PA, Pugh TJ, Hovestadt V et al. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell 2014; 25: 393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee C, Lee J, Choi SA, Kim SK, Wang KC, Park SH et al. M1 macrophage recruitment correlates with worse outcome in SHH Medulloblastomas. BMC Cancer 2018; 18: 535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell systems 2015; 1: 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 2016; 131: 803–820. [DOI] [PubMed] [Google Scholar]
- 19.Machold R, Fishell G. Math1 is expressed in temporally discrete pools of cerebellar rhombic-lip neural progenitors. Neuron 2005; 48: 17–24. [DOI] [PubMed] [Google Scholar]
- 20.Mantovani A, Schioppa T, Porta C, Allavena P, Sica A. Role of tumor-associated macrophages in tumor progression and invasion. Cancer metastasis reviews 2006; 25: 315–322. [DOI] [PubMed] [Google Scholar]
- 21.Mao J, Ligon KL, Rakhlin EY, Thayer SP, Bronson RT, Rowitch D et al. A novel somatic mouse model to survey tumorigenic potential applied to the Hedgehog pathway. Cancer research 2006; 66: 10171–10178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Margol AS, Robison NJ, Gnanachandran J, Hung LT, Kennedy RJ, Vali M et al. Tumor-associated macrophages in SHH subgroup of medulloblastomas. Clin Cancer Res 2015; 21: 1457–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martin AM, Nirschl CJ, Polanczyk MJ, Bell WR, Nirschl TR, Harris-Bookman S et al. PD-L1 expression in medulloblastoma: an evaluation by subgroup. Oncotarget 2018; 9: 19177–19191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maximov V, Chen Z, Wei Y, Robinson MH, Herting CJ, Shanmugam NS et al. Tumour-associated macrophages exhibit anti-tumoural properties in Sonic Hedgehog medulloblastoma. Nat Commun 2019; 10: 2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McNeill KA. Epidemiology of Brain Tumors. Neurol Clin 2016; 34: 981–998. [DOI] [PubMed] [Google Scholar]
- 26.Mi H, Muruganujan A, Ebert D, Huang X, Thomas PD. PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic acids research 2019; 47: D419–d426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mizutani M, Pino PA, Saederup N, Charo IF, Ransohoff RM, Cardona AE. The fractalkine receptor but not CCR2 is present on microglia from embryonic development throughout adulthood. Journal of immunology (Baltimore, Md : 1950) 2012; 188: 29–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-deficient mice have no mature B and T lymphocytes. Cell 1992; 68: 869–877. [DOI] [PubMed] [Google Scholar]
- 29.Murata D, Mineharu Y, Arakawa Y, Liu B, Tanji M, Yamaguchi M et al. High programmed cell death 1 ligand-1 expression: association with CD8+ T-cell infiltration and poor prognosis in human medulloblastoma. J Neurosurg 2018; 128: 710–716. [DOI] [PubMed] [Google Scholar]
- 30.Northcott PA, Jones DT, Kool M, Robinson GW, Gilbertson RJ, Cho YJ et al. Medulloblastomics: the end of the beginning. Nature reviews Cancer 2012; 12: 818–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Northcott PA, Buchhalter I, Morrissy AS, Hovestadt V, Weischenfeldt J, Ehrenberger T et al. The whole-genome landscape of medulloblastoma subtypes. Nature 2017; 547: 311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peranzoni E, Lemoine J, Vimeux L, Feuillet V, Barrin S, Kantari-Mimoun C et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc Natl Acad Sci U S A 2018; 115: E4041–e4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pham CD, Flores C, Yang C, Pinheiro EM, Yearley JH, Sayour EJ et al. Differential Immune Microenvironments and Response to Immune Checkpoint Blockade among Molecular Subtypes of Murine Medulloblastoma. Clin Cancer Res 2016; 22: 582–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med 2013; 19: 1264–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Quail DF, Bowman RL, Akkari L, Quick ML, Schuhmacher AJ, Huse JT et al. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science (New York, NY) 2016; 352: aad3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Quail DF, Joyce JA. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017; 31: 326–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rallapalli H, Tan IL, Volkova E, Wojcinski A, Darwin BC, Lerch JP et al. MEMRI-based imaging pipeline for guiding preclinical studies in mouse models of sporadic medulloblastoma. Magn Reson Med 2020; 83: 214–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramaswamy V, Taylor MD. Medulloblastoma: From Myth to Molecular. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2017; 35: 2355–2363. [DOI] [PubMed] [Google Scholar]
- 39.Rao G, Pedone CA, Del Valle L, Reiss K, Holland EC, Fults DW. Sonic hedgehog and insulin-like growth factor signaling synergize to induce medulloblastoma formation from nestin-expressing neural progenitors in mice. Oncogene 2004; 23: 6156–6162. [DOI] [PubMed] [Google Scholar]
- 40.Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 2014; 25: 846–859. [DOI] [PubMed] [Google Scholar]
- 41.Schmidt A, Oberle N, Krammer PH. Molecular mechanisms of treg-mediated T cell suppression. Frontiers in immunology 2012; 3: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005; 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tan IL, Wojcinski A, Rallapalli H, Lao Z, Sanghrajka RM, Stephen D et al. Lateral cerebellum is preferentially sensitive to high sonic hedgehog signaling and medulloblastoma formation. Proc Natl Acad Sci U S A 2018; 115: 3392–3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tanori M, Santone M, Mancuso M, Pasquali E, Leonardi S, Di Majo V et al. Developmental and oncogenic effects of insulin-like growth factor-I in Ptc1+/− mouse cerebellum. Molecular cancer 2010; 9: 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC et al. Molecular subgroups of medulloblastoma: the current consensus. Acta neuropathologica 2012; 123: 465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Togashi Y, Shitara K, Nishikawa H. Regulatory T cells in cancer immunosuppression - implications for anticancer therapy. Nature reviews Clinical oncology 2019; 16: 356–371. [DOI] [PubMed] [Google Scholar]
- 47.Tsou CL, Peters W, Si Y, Slaymaker S, Aslanian AM, Weisberg SP et al. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. The Journal of clinical investigation 2007; 117: 902–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vermeulen JF, Van Hecke W, Adriaansen EJM, Jansen MK, Bouma RG, Villacorta Hidalgo J et al. Prognostic relevance of tumor-infiltrating lymphocytes and immune checkpoints in pediatric medulloblastoma. Oncoimmunology 2018; 7: e1398877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang JY, Del Valle L, Gordon J, Rubini M, Romano G, Croul S et al. Activation of the IGF-IR system contributes to malignant growth of human and mouse medulloblastomas. Oncogene 2001; 20: 3857–3868. [DOI] [PubMed] [Google Scholar]
- 50.Yang M, McKay D, Pollard JW, Lewis CE. Diverse Functions of Macrophages in Different Tumor Microenvironments. Cancer research 2018; 78: 5492–5503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yao M, Ventura PB, Jiang Y, Rodriguez FJ, Wang L, Perry JSA et al. Astrocytic trans-Differentiation Completes a Multicellular Paracrine Feedback Loop Required for Medulloblastoma Tumor Growth. Cell 2020. [DOI] [PMC free article] [PubMed]
- 52.Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer research 2014; 74: 5057–5069. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.