

Summary
Following antigen-driven expansion in lymph node, transforming growth factor-β (TGFβ) is required for differentiation of skin-recruited CD8+ T cell effectors into epidermal resident memory T cells (Trm) and their epidermal persistence. We found that the source of TGFβ supporting Trm cells was autocrine. In addition, antigen-specific Trm cells that encountered cognate antigen in the skin, and bystander Trm cells that did not, both displayed long-term persistence in the epidermis under steady-state conditions. However, when the active-TGFβ was limited or when new T cell clones were recruited into the epidermis, antigen-specific Trm cells were more efficiently retained than bystander Trm cells. Genetically enforced TGFβR signaling allowed bystander Trm cells to persist in the epidermis as efficiently as antigen-specific Trm cells in both contexts. Thus, competition between T cells for active TGFβ represents an unappreciated selective pressure that promotes the accumulation and persistence of antigen-specific Trm cells in the epidermal niche.
eTOC blurb
Epidermal residence of CD8+ memory T cells requires TGFβ, but the source of this cytokine and the relevance of this requirement are unclear. Hirai et al. reveal that intraclonal competition for transactivation of autocrine TGFβ preferentially enriches for antigen-specific T cells at the skin barrier.
Graphical Abstract
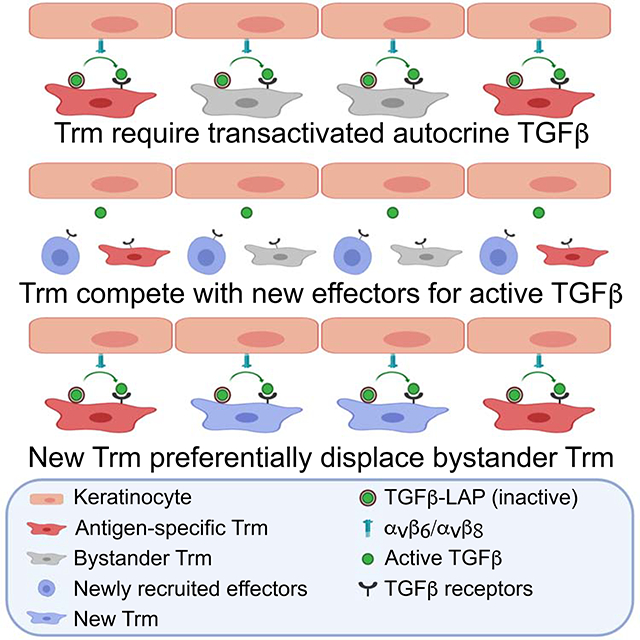
INTRODUCTION
Tissue resident CD8+ memory T cells (Trm) are a highly abundant, noncirculating subset of memory T cells that provide efficient peripheral immune surveillance (Masopust and Soerens, 2019). In the skin, cutaneous infection with vaccinia virus (VV) or herpes simplex virus is well described to result in the development of large numbers of Trm cells that preferentially reside in the epidermis (Gebhardt et al., 2011; Hirai et al., 2020; Jiang et al., 2012). In these models, viral-derived antigen is initially brought to skin-draining lymph nodes by dendritic cells or through lymph which drives the expansion of CD8+ T cell effectors (Allan et al., 2003; Bedoui et al., 2009; Liu et al., 2006; Reynoso et al., 2019). These effectors are then recruited into the sites of infected skin where they re-encounter antigen and some differentiate into long-lived Trm cells. In certain tissues such as the lung, re-encounter with cognate antigen in peripheral tissue is required for Trm cells differentiation (Lee et al., 2011; McMaster et al., 2018). In flank skin, however, a secondary encounter with cognate antigen is not required. For example, epicutaneous application of the sensitizing hapten 1-Fluoro-2,4-dinitrobenzene (DNFB) efficiently “pulls” non-antigen-specific, bystander effector cells expanded by viral infection or in vitro activation into the skin where they then differentiate into Trm cells (Mackay et al., 2012). Antigen-specific and bystander DNFB-pulled Trm cells appear phenotypically similar (Park et al., 2018) though potential functional differences have not been well explored.
The cytokine transforming growth factor β (TGFβ) is required for the development of Trm cells in many tissues such as skin, gut, and lung (Casey et al., 2012; Mackay et al., 2013; Sheridan et al., 2014; Wakim et al., 2015). TGFβ appears to be required at multiple steps in Trm cell development. Naïve steady-state CD8+ T cells require TGFβ in order to maintain their potential to differentiate into Trm cells (Mani et al., 2019). In the skin, CD8+ T cell effectors recruited by viral infection depend on TGFβ to differentiate into Trm cells (Mackay et al., 2013; Mackay et al., 2015). Exposure of CD8+ T cells to TGFβ in vitro or in vivo efficiently induces expression of the αE integrin which complexes with integrin β7 to form αEβ7 (CD103) and binds to E-cadherin expressed by epithelial cells (Casey et al., 2012; Mackay et al., 2013; Sheridan et al., 2014; Wakim et al., 2015). Although Trm cell expression of CD103 is not universal across all tissues, in skin at least, CD103 expression is induced by Trm cell precursors upon entry to epidermis and, along with CD69, is a good phenotypic marker for Trm cells (Casey et al., 2012; Mackay et al., 2013). Ablation of CD103 in CD8+ T cell prevents their long-term persistence in the epidermis (Mackay et al., 2013).
TGFβ is produced bound to the latency associate peptide that prevents bioactivity until the complex is activated (Worthington et al., 2011). In the epidermis, TGFβ activation is mediated exclusively by activation via the integrins αvβ6 and αvβ8 that are expressed by keratinocytes (KCs) (Aluwihare et al., 2009; Mohammed et al., 2016; Yang et al., 2007). Using a DNFB-pull Trm cell model in Itgb6−/−Itgb8ΔKC mice (Itgb6−/− x Itgb8fl/fl-Krt14-cre) lacking both integrins in the skin, we have previously shown that CD8+ T cell effectors are recruited into the epidermis at early time points, but Trm cells are not evident at late time points. In addition, blocking mAbs to αvβ6 could partially deplete fully differentiated Trm cells suggesting that continued access to integrin-activated TGFβ is required for persistence of epidermal Trm cells (Hirai et al., 2019; Mohammed et al., 2016). A formal demonstration that epidermal Trm cells require continued exposure to TGFβ, identification of the source of TGFβ supporting Trm cells and whether DNFB-pull bystander and antigen-specific Trm cells equally depend on TGFβ remain unexplored questions.
Herein, we show that fully differentiated epidermal Trm cells have an absolute dependence on TGFβ transactivated by αvβ6 and αvβ8 on KC for long-term persistence in the epidermis. In addition, the source of TGFβ supporting Trm cells was autocrine. We also found that both “antigen-specific” OT-I Trm cells generated by VV-OVA skin infection and bystander DNFB-pull Trm cells persisted long-term under steady-state conditions. However, when active-TGFβ was limited by administration of blocking αvβ6 and αvβ8 mAbs or by small molecule inhibition, antigen-specific Trm cells were able to persist in the epidermis while bystander Trm cells were depleted. Similarly, when new T cells were recruited into the skin by a subsequent challenge, antigen-specific Trm cells were better able to persist than bystander Trm cells. Genetically enforced TGFβR signaling rescued this phenotype and allowed bystander Trm cells to persist in the epidermis as efficiently as antigen-specific Trm cells in both contexts. Thus, we have found that Trm cells compete for integrin-activated TGFβ resulting in an unexpected selective pressure promoting the retention of antigen-specific Trm cells within the epidermal niche.
RESULTS
Epidermal Trm cells require TGFβ signaling for persistence
Despite strong evidence that active TGFβ is required for differentiation of CD8+ T cell effectors into Trm cells, it remains unclear whether continued exposure to TGFβ is required for their persistence in skin. To directly test a requirement for TGFβ in the memory phase, 105 CD8+ T cells from OT-I ROSA26-creERT2 x Tgfbr2fl/fl (Thy1.2/Thy1.2, CD45.2/CD45.2) and control OT-I ROSA26-creERT2 x Tgfbr2f/+ (Thy 1.1/Thy1.2, CD45.1/CD45.2) mice were mixed and adoptively transferred into naïve C57BL/6 CD45.1 recipient mice (Figure 1A). Recipients were then infected with recombinant vaccinia virus expressing OVA257–264 (VV-OVA) using the skin scarification method on the flank (Hirai et al., 2019). Equal expansion of OT-I was confirmed in blood (Figure 1B) and tamoxifen was administered i.p for 5 consecutive days starting on day 42 post-infection to ablate Tgfbr2. 23 days after tamoxifen treatment (day 70 post infection) the number of epidermal Tgfbr2−/− and sufficient OT-I was determined by immunofluorescent microscopic imaging of epidermal sheets. We enumerated epidermal Trm cells by direct immunofluorescent visualization because flow cytometry of enzymatically digested tissue greatly underestimates cell numbers in epidermis as well as other non-lymphoid tissues (Figure S1) (Collins et al., 2016; Steinert et al., 2015). Ablation of Tgfbr2 resulted in a significant decrease in the number of epidermal Trm cells compared with control (Figure 1C). From these data we conclude that Trm cells require TGFβ signaling in order to maintain epidermal persistence at a late timepoint post infection well after differentiation has occurred. Thus, skin Trm cells depend on TGFβ at all stages of their development: naïve (Mani et al., 2019), transition of effectors to memory (Mackay et al., 2013; Mackay et al., 2015), as well as memory maintenance.
Figure 1. Trm cells require TGFβ signaling and transactivated TGFβ for epidermal persistence.

(A) Experimental scheme for testing a TGFβ signaling requirement for Trm cell persistence. Naïve CD8+ T cells from OT-I ROSA26-creERT2 x Tgfbr2f/+ (Thy 1.1/Thy1.2, CD45.1/CD45.2) and OT-I ROSA26-creERT2 x Tgfbr2fl/fl (Thy1.2/Thy1.2, CD45.2/CD45.2) mice were adoptively transferred into CD45.1/CD45.1 recipient mice followed by skin VV-OVA infection. (B) Representative flow plot of peripheral blood at day 7 post infection. gated on live CD3+CD8α+ T cells is shown. (C) Quantification of OT-I cells in epidermal whole mount 23 days post tamoxifen are shown. (D) Thy1.1+ OT-I mice were transferred to WT, Itgb6−/−, or Itgb6−/−Itgb8ΔKC mice followed by VV-OVA skin infection. Representative immunofluorescence images of epidermal whole mount stained for Thy1.1+ OT-I cells are shown. (E) Summary data at the indicated time points from (D) is shown. (F) Representative flow plots and (G) summary data of epidermal cells gated on live CD45+Thy1.1+ OT-I cells are shown. Data are representative of two (B-C) or three separate experiments (D-G). Each symbol represents data from an individual animal (C, G) or pooled data with at least three mice per point (E) and lines indicate data from the same animal. Data are mean ± SEM. **p < 0.01, ***p < 0.001, and ****p < 0.0001. Scale bar represents 100 μm. TAM, tamoxifen; dpi, days post-infection; gMFI, geometric mean fluorescence intensity. See also Figure S1.
Epidermal Trm cells require integrin-activated TGFβ for persistence
We have previously reported using both a VV-infection model and a “DNFB-pull” bystander model that epidermal Trm cells are absent in Itgb6−/−Itgb8ΔKC mice that lack KC expression of the TGFβ-activating integrins αvβ6 and αvβ8 (Hirai et al., 2019; Mohammed et al., 2016). To clarify if the defect of Trm cells in Itgb6−/−Itgb8ΔKC mice results from failure of differentiation and/or maintenance, we performed a kinetic analysis of Trm cells in WT, Itgb6−/−, or Itgb6−/−Itgb8ΔKC mice. We adoptively transferred 105 OT-I cells into naïve WT, Itgb6−/−, or Itgb6−/−Itgb8ΔKC mice followed by VV-OVA skin scarification infection. The number of epidermal OT-I cells was quantified by immunofluorescent microscopy on days 14, 28, and 56 days post VV-OVA infection. OT-I numbers were greatly reduced in Itgb6−/−Itgb8ΔKC starting on day 14 post infection (Figure 1D and 1E). Trm cell number in Itgb6−/− were intact on day 14 but declined on days 28 and 56 compared to WT mice. The early absence of Trm cells in Itgb6−/−Itgb8ΔKC likely represents a failure of differentiation of Trm cells due to the absence of active TGFβ while the delayed loss observed in Itgb6−/− likely represents a failure of persistence when active TGFβ is limited. This is supported by the presence of OT-I cells that lack expression of the Trm cell marker CD103 in Itgb6−/−Itgb8ΔKC mice (Figure 1F and 1G). In addition, even though Trm cell developed in Itgb6−/− mice, expression of CD103 was reduced compared to Trm cells in WT mice indicating that amounts of CD103 expression reflect the amounts of available active TGFβ.
To formally confirm that loss of Trm cells in Itgb6−/− mice resulted from reduced amounts of active TGFβ, we bred Itgb6−/− mice to mice with CD8-specific expression of creERT2 (E8I-creERT2) and mice expressing a constitutively active form of the TGFβ receptor I (TGFβRCA) behind a lox-stop-lox cassette (Bartholin et al., 2008; Hirai et al., 2019; Liu et al., 2020; Vincent et al., 2010). The resulting mice (Itgb6−/− x TGFβRCA) allow for tamoxifen (TAM)-induced expression of TGFβRCA in CD8+ T cells. Itgb6−/− x TGFβRCACD8 mice and control Itgb6+/−, Itgb6−/−, and Itgb6+/− x TGFβRCACD8 mice were infected with VV by skin scarification and treated with tamoxifen i.p. on days 7–11 post infection. On day 46 post infection, epidermal Trm cells were enumerated. As expected, Trm cell numbers and expression of CD103 were reduced in Itgb6−/− compared to Itgb6+/− mice (Figure 2A–D). Notably, in Itgb6−/− mice, tamoxifen-induced expression of TGFβRCA rescued Trm cell numbers and CD103 expression to amounts equivalent to those observed in control Itgb6+/− mice. Forced expression of TGFβRCA by CD8+ T cells in control Itgb6+/− mice increased expression of CD103 but did not increase Trm cell numbers. From these data we conclude that reduced expression of CD103 and reduced numbers of Trm cells in Itgb6−/− mice resulted from lower amounts of available active TGFβ.
Figure 2. TGFβR signaling rescues Trm cell numbers in absence of αvβ6.

(A-D) TGFβRCACD8 x Itgb6−/+, TGFβRCACD8 x Itgb6−/−, and control Itgb6−/+ and Itgb6−/− mice were infected with VV-OVA on flank skin and treated with tamoxifen i.p. on days 7–11 post infection. (A) Representative immunofluorescence images and (B) summary data of epidermal whole mount for stained for CD8α+ harvested on day 46 post infection are shown. (C) Representative flow plots gated on live CD45+TCRβ+CD8α+ CD8+ T cells in epidermis and (D) the percentage of CD8+ T cells expressing CD69 and CD103 as well as CD103 gMFI of CD69+CD103+ CD8+ T cells. (E) WT OT-I transferred mice were dual infected on ear and flank with VV-OVA. Representative immunofluorescence images and (F) summary data of epidermal whole mounts stained for Thy1.1+ OT-I cells are shown. (G) Representative flow plots gated on live CD45+Thy1.1+ OT-I cells in epidermis and (H) gMFI of CD103+ on OT-I cells is shown. (I) Representative flow plot of KCs from naive Itgb6−/− flank (gray), WT flank (green), and WT ear (red) gated on live CD45− cells and gMFI of αvβ6 are shown. (J) TGFβRCACD8 and control mice were dual infected on flank and ear with VV-OVA followed by tamoxifen i.p. on day 14-dpi. Representative immunofluorescence images of flank or ear epidermal whole mount for CD8α+ CD8+ T cells and (K) summary data are shown. Data are representative of two (E-I) or three (A-D) separate experiments. Each symbol represents data from an individual animal. **p < 0.01, ***p < 0.001, and ****p < 0.0001. Scale bar represents 100 μm. gMFI, geometric mean fluorescence intensity. See also Figure S2.
Some groups have reported relatively low numbers of Trm cells in the ear and many of the Trm cells were observed to express little to no CD103 (Collins et al., 2016; Gamradt et al., 2019; Hobbs and Nolz, 2019; Slutter et al., 2017). This is in contrast with our data showing uniformly high expression of CD103 in the flank. We hypothesized that the skin sites infected (i.e. flank vs. ear) could explain this discrepancy. To test this hypothesis, OT-I adoptive transfer WT mice were dual infected with VV-OVA on both the ear and flank skin. On day 34 post infection, the number of OT-I was significantly greater in flank than ear skin (Figure 2E and 2F). This could have potentially resulted from altered viral infection efficiency except that we noticed Trm cells in ear skin consistently expressed lower amounts of CD103 suggesting reduced availability of active TGFβ (Figure 2G and 2H). Comparison of αvβ6 expression on KCs by flow cytometry and RTqPCR revealed lower expression in ear compared with flank skin (Figure 2I and S2). We did not observe differential expression of Il7 or Il15 mRNA (Figure S2) (Adachi et al., 2015; Mackay et al., 2013; Richmond et al., 2018). Finally, TGFβRCACD8 were dual infected with VV on ear and flank followed by administration of tamoxifen i.p. starting on day 14. Analysis of epidermis 37 days after infection revealed that induction of constitutive TGFβR signaling significantly rescued Trm cell numbers in ear (Figure 2J and 2K). These data further support the concept that Trm cells are sensitive to amounts of available active TGFβ in the epidermis and that these amounts can vary between anatomical sites.
Epidermal Trm cells require autocrine TGFβ
Potential epidermal sources of TGFβ to support Trm cell persistence include keratinocytes (KC), Langerhans cells (LC), dendritic epidermal T cells (DETC), and Trm cells (Yang et al., 2019). Trm cells are unaffected by the absence of LC and DETC indicating these cell types are not obligate sources of TGFβ (Mohammed et al., 2016; Zaid et al., 2014). Since KC are the largest source of TGFβ in the epidermis (Yang et al., 2019), we hypothesized that KC-derived TGFβ is required to support Trm cells persistence. To test this, we used Krt14-creERT2 x Tgfb1fl/fl mice (TgfbΔKC) that allow for the inducible ablation of Tgfb1 from KCs. TgfbΔKC mice also have a ROSA26-YFP reporter, which accurately reports Tgfb1 ablation (Yang et al., 2019). TgfbΔKC and control mice were infected with VV followed by topical application of 4-OH-tamoxifen on day 42 post infection. The epidermis was analyzed on day 63 post infection (21 days after tamoxifen). We observed that approximately 80% of KC expressed YFP (green) indicative of loss of Tgfb1 (Figure 3A and 3B). Importantly, YFP− KC were clustered together resulting in large YFP+ areas lacking Tgfb1 expression. We observed that numbers of Trm cells (magenta) were unaffected by the absence of Tgfb1 (Figure 3C) and were distributed equally throughout YFP+ and YFP− areas (Figure 3A) indicating that KC-derived TGFβ is not required for Trm cell persistence.
Figure 3. Epidermal Trm cells require autocrine TGFβ.

(A) Control (Tgfb1fl/fl) and TgfbΔKC (Krt14-creERT2 x Tgfb1fl/fl) mice with a YFP reporter were skin infected with VV followed by topical treatment of 4-OH tamoxifen for two consecutive days starting at least on day 42 post infection. Representative immunofluorescence images of epidermal whole mounts harvested on day 21 after the last tamoxifen treatment stained for CD8α+ (magenta) and YFP (green). (B) Representative flow plot gated on live CD45− KCs is shown. (C) Summary data from (A) is shown. (D) Control (E8I-creERT2 x Tgfb1fl/+) and TgfbΔCD8 (E8I-creERT2 x Tgfb1fl/fl) mice with a human nerve growth factor receptor reporter (hNGFR) were infected with VV followed by i.p. tamoxifen starting at least day 42 post infection. Representative epidermal whole mounts harvested 21 days after the last tamoxifen treatment and stained for CD8α+ (magenta) and hNGFR (yellow) are shown. (E) Summary data from (D). Data are representative of two (D and E) or three (A-C) separate experiments. ****p < 0.0001. Scale bar represents 100 μm.
We next examined whether Trm cell persistence requires autocrine TGFβ. We bred E8I-creERT2 mice with a human nerve growth factor receptor reporter (hNGFR) and Tgfb1fl/fl mice. The resulting TgfbΔCD8 mice allow for tamoxifen-inducible ablation of Tgfb1 from CD8+ T cells that can be monitored by expression of hNGFR. TgfbΔCD8 and control hNGFRCD8 mice were infected with VV followed by i.p tamoxifen for 5 consecutive days starting on day 42 post infection. Epidermal sheets were evaluated by immunofluorescent microscopy for expression of CD8α (magenta) to identify all Trm cells and hNGFR (yellow) to identify those Trm cells that had successfully undergone cre-mediated excision. We observed that tamoxifen-mediated cre activation with hNGFR expression was observed in approximately 50% Trm cells in control hNGFRCD8 mice. The number of hNGFR+ CD8+ Trm cells in TgfbΔCD8 mice, however, was significantly reduced compared with control mice (Figure 3D and 3E). Notably, hNGFR− and hNGFR+ Trm cells were observed in close proximity to each other in control hNGFRCD8 mice but in TgfbΔCD8 mice, hNGFR− Trm cells were unaffected. Thus, we conclude that epidermal Trm cells are dependent on an autocrine source of TGFβ to maintain epidermal persistence.
Blockade of integrin-mediated TGFβ activation eliminates bystander Trm cells
As discussed above, antigen-specific Trm cells generated by VV skin infection and bystander Trm cells generated by a “DNFB-pull” are both absent from Itgb6−/−Itgb8ΔKC mice which lack integrin-mediated TGFβ activation. To test whether antigen-specific and bystander Trm cells are equally dependent on integrin-activated TGFβ, we compared the ability of VV-induced Trm cells and DNFB-pulled Trm cells to persist following administration of blocking anti-αvβ6 and αvβ8 mAbs. Thy1.1+ OT-I cells were adoptively transferred into naïve WT mice followed by skin VV-OVA infection on left flank skin. On day 5 post infection, the right flank was painted with DNFB to recruit expanded OT-I effectors into the site. Mice were injected weekly with anti-αvβ6 and αvβ8 mAbs starting on day 42–49 post infection for a total of 3 weeks followed by immunofluorescent microscopic evaluation of epidermal whole mounts (Figure 4A). As a positive control, we stained for MHC-II (green) to identify Langerhans cells and observed the expected reduction following administration of anti-αvβ6 and αvβ8 at VV-OVA, DNFB-pulled and non-treated (NT) sites (Figure 4B and 4C) (Mohammed et al., 2016). VV-OVA infection and DNFB-pulled treatment induced Trm cells (Thy1.1, cyan) to the sites in equal number and much more than NT sites. We observed a sizable reduction in the number of Trm cells in anti-αvβ6 and αvβ8 treated animals at the DNFB-pulled and NT sites but not at the VV-OVA sites. Notably, expression of CD103 was reduced in Trm cells at both in VV-OVA and DNFB-pulled indicating an equivalent reduction in available active TGFβ at both sites (Figure 4D). We obtained similar results using CWHM-12, a small molecule αvβ6 and αvβ8 inhibitor (Figure 4E and 4F) (Henderson et al., 2013). The loss of Trm cells from the VV-OVA site seen in Itgb6−/− mice but not following anti-αvβ6/αvβ8 or CHWM12 treatment likely resulted from only partial inhibition of integrin-mediated TGFβ activation by these agents. In the absence of TGFβ-blockade, Trm cells at both sites appeared functionally equivalent and were able to persist in the epidermis under steady state conditions for up to 460 days (Figure S3). To determine whether the lowered sensitivity to limiting amounts of TGFβ at the VV-OVA sites was related to viral-induced local inflammation, we repeated the experiment by dual infecting mice with VV-OVA and recombinant VV expressing an irrelevant antigen (nucleocapsid protein of vesicular stomatitis virus, VV-N) on opposite flanks. As expected, the number of OT-I cells at the VV-N infected sites was significantly lower than the VV-OVA sites due to antigen-mediated competition with endogenous VV-N-specific CD8+ T cells (Figure 4G and 4H) (Khan et al., 2016; Muschaweckh et al., 2016). Importantly, following administration of anti-αvβ6 and αvβ8, bystander OT-I cells at the VV-N sites were depleted but OT-I cells at the VV-OVA sites were unaffected. Thus, from these data we conclude that antigen-specific OT-I cells at the VV-OVA sites are better able to persist in the epidermis than bystander OT-I cells at either DNFB-pulled, NT, and VV-N sites under conditions where integrin-mediated TGFβ activation is limited.
Figure 4. Blockade of integrin-mediated TGFβ activation reduces bystander Trm cells.

(A) Experimental scheme. Thy1.1+ OT-I cells were adoptively transferred into naïve WT mice followed by skin VV-OVA infection on left flank. On day 5 post infection, the right flank was painted with 0.15% DNFB. The mice were injected weekly with anti-αvβ6 and αvβ8 mAbs starting on day 42–49 post infection for a total of 3 weeks. The epidermis from ‘not treated’ abdomen (NT), left flank (VV-OVA), and right flank (DNFB-pulled) were harvested 7 days after the last mAbs treatment. (B) Representative immunofluorescence images of epidermal whole mounts stained for MHCII+ (green, LC) and Thy1.1+ (cyan, OT-I) cells are shown. (C) Quantification of LC and OT-I numbers from control (open squares) and mAb treat mice (closed squares) are shown. (D) Frequency of CD69+CD103+ OT-I cells gated on live CD45+Thy1.1+ cells and CD103 gMFI of CD69+CD103+ OT-I cells are shown. (E) Mice were treated as in A, except CWHM-12 or control was delivered by implantable ALZET osmotic minipumps for 28 days instead of mAbs. Quantification of LC and OT-I cells from epidermal whole mounts are shown. (F) Frequency of CD69+CD103+ OT-I cells gated on live CD45+Thy1.1+ cells and CD103 gMFI of CD69+CD103+ OT-I cells are shown. (G) OT-I adoptive transfer mice were dual infected with VV-OVA and VV-N on opposite flanks and treated with anti-αvβ6 and αvβ8 mAbs for 3 weeks starting on day 49 post infection. Representative epidermal whole mounts from VV-OVA and VV-N sites harvested 7 days after the last mAb or control treatment are shown. (H) Quantification of the number of Thy1.1+ CD8+ OT-I and endogenous Thy 1.1− CD8+ T cells is shown. (I) Experimental scheme. As in (A) except that OVA257–264 peptide was applied to the DNFB-pulled sites 1 day after DNFB painting and treated with CWHM-12 instead of the mAbs. (J) Quantification of the number of MHC-II+ LC and Thy1.1+ OT-I cells in epidermal whole mounts is shown. Data are representative of two (E-J) or three (A-D) separate experiments. Each symbol represents data from an individual animal. *p < 0.05, **p < 0.01, and ****p < 0.0001. Scale bar represents 100 μm. gMFI, geometric mean fluorescence intensity. See also Figure S3.
We next sought to test the hypothesis that encounter with antigen in the skin renders Trm cells more resistant to limiting amounts of active TGFβ. WT OT-I adoptive transfer mice were infected with VV-OVA on the left flank followed by epicutaneous application of DNFB on day 5 post-infection (Figure 4I). One day later the DNFB-pulled sites were painted with OVA257–264 peptide. On Day 42, mice were treated with CWHM-12 or control for 4 weeks to inhibit integrin-mediated TGFβ activation. Addition of cognate antigen to the DNFB-pulled sites now made Trm cells at these sites behave similar to Trm cells at the VV-OVA sites as they persisted in the epidermis despite inhibition of TGFβ activation (Figure 4J). From these data we conclude that under conditions of limiting amounts of active TGFβ created by either blocking mAbs or small molecule inhibition, Trm cells that have encountered cognate antigen in the skin are better able than bystander Trm cells to persist in the epidermis.
TGFβ signaling provides a competitive advantage during Trm cell development
Since access to active TGFβ is required for Trm cells to establish in the epidermis and antigen-specific Trm cells are better able to persist during the memory phase than bystander Trm cells when amounts of active TGFβ are limited, we hypothesized that competition for active TGFβ may provide a selective pressure during the development of Trm cells. To test this hypothesis, we generated OT-I x TGFβRCACD8 mice. CD8+ T cells from these or control OT-I mice were adoptively transferred into WT recipients and dual infected with VV-OVA and VV-N on opposite flanks (Figure 5A). Mice were treated with tamoxifen i.p for 5 days starting on day 7 post-infection and the epidermis was analyzed on day 34 post-infection for the presence of OT-I (Thy1.1, cyan) and endogenous (CD8α, red) Trm cells (Figure 5B). As expected, large numbers of OT-I Trm cells were evident in control mice at the VV-OVA sites but these cells were out competed by endogenous VV-specific Trm cells at the VV-N sites (Figure 5B and 5C). OT-I TGFβRCACD8 formed Trm cells as efficiently as control OT-I at the VV-OVA sites where cognate OVA antigen was available. Importantly, at the VV-N sites where OVA antigen is absent, OT-I TGFβRCACD8 Trm cells were able to effectively compete with endogenous Trm cells resulting in equivalent numbers of endogenous and OT-I TGFβRCACD8 Trm cells. At the non-treated (NT) sites where the Trm cells are sparse and there is less inter-clonal competition, constitutive TGFβR signaling did not provide a competitive advantage. From these data we conclude that enforced TGFβR signaling is able to overcome the competitive advantage provided by antigen encounter in skin during Trm cell development. This suggests that competition for active TGFβ can participate in determining which clones occupy the epidermal niche.
Figure 5. TGFβ signaling provides a competitive advantage during Trm cell development.

(A) Experimental scheme. Thy1.1+ OT-I or OT-I TGFβRICA were transferred to WT mice followed by dual infection of VV-OVA and VV-N on opposite flanks. Mice were treated with tamoxifen i.p. for 5 consecutive days starting on day 7. Representative immunofluorescence images of epidermal whole mounts stained for Thy1.1 (cyan, OT-I cells) and CD8α (red) from left flank (VV-OVA), right flank (VV-N), and not treated abdomen (NT) sites harvested day 34 post infection are shown. (C) Summary data showing numbers of OT-I (Thy1.1+CD8α+) and endogenous CD8+ T cells (Thy1.1−CD8α+) from OT-I TGFβRICA (closed squares) and control OT-I (open squares) recipients are shown. Each symbol represents data from an individual animal (C). Data are representative of three separate experiments. *p < 0.05. Scale bar represents 100 μm.
Bystander Trm cells are preferentially displaced by newly recruited CD8+ T cells
If Trm cell clones compete for active TGFβ in order to persist in the epidermis, then one prediction is that bystander Trm cells will compete less efficiently than antigen-specific Trm cells when new CD8+ T cells are recruited into the skin during a subsequent challenge. To test this prediction, WT OT-I adoptive transfer mice were infected with VV-OVA on one flank followed by a DNFB-pulled on the opposite flank to establish antigen specific and bystander Trm cell sites, respectively (Figure 6A). After at least 40 days post-infection, mice were sensitized at a distant sites (abdomen) with the hapten oxazolone which is antigenically distinct from DNFB to generate oxazolone-specific effectors (Kish et al., 2009). Oxazolone effectors were then recruited into both the VV-OVA and DNFB-pulled sites by epicutaneous application of oxazolone. At least 30 days after oxazolone challenge, the epidermis at the VV-OVA and DNFB-pulled sites were analyzed. As noted earlier, without the oxazolone challenge equivalent numbers of OT-I Trm cells developed at the VV-OVA and DNFB-pulled sites (Figure 6B and 6C). Challenge with oxazolone, however, reduced the numbers of OT-I at both sites, but numbers of OT-I Trm cells at the DNFB-pulled sites were consistently lower than at the VV-OVA sites. This suggests that Trm cells at the VV-OVA sites were better able than Trm cells at the DNFB-pulled sites to compete with the newly recruited endogenous CD8+ T cells. To ensure that this is a result of competition between Trm cells and is not reflective of recruited circulating OT-I cells, we repeated the experiment but depleted circulating OT-I cells by injecting titrated anti-Thy1.1 just prior to oxazolone challenge (Figure S4). In the absence of circulating OT-I cells, we observed a similar phenotype where OT-I Trm cells at the VV-OVA sites are better able to compete with newly recruited T cells than OT-I Trm cells at the DNFB-pulled sites (Figure 6E). In addition, if the DNFB-pulled sites was treated with OVA257–264 during the initial seeding (day 6 p.i.), then Trm cells at this location showed a similar capacity as Trm cells at the VV-OVA sites to compete with newly recruited T cells (Figure 6E). Thus, Trm cells that have encountered Ag in the skin during initial Trm cell formation are better able to persist in the context of a subsequent challenge when new T cells are recruited into the skin.
Figure 6. Bystander Trm cells are preferentially displaced by newly recruited CD8+ T cells.

(A) Experimental scheme. Thy1.1+ OT-I cells were adoptively transferred into naïve WT mice followed by skin VV-OVA infection on left flank. On day 5 post infection, the right flank was painted with 0.15% DNFB. More than 35 days after DNFB treatment, mice were sensitized at abdomen with oxazolone and 5 days later both flanks were challenged by oxazolone. (B) Representative immunofluorescence images of epidermal whole mounts harvested at least 30 days post oxazolone challenge stained for Thy1.1+ (cyan) and CD8α+ (red) are shown. (C) Summary data showing numbers of OT-I (Thy1.1+, CD8α+) and total CD8+ T cells (Thy1.1−/+CD8α+). (D) The frequency of CD69+CD103+ OT-I cells gated on live CD45+Thy1.1+ and CD103 gMFI of CD69+CD103+ OT-I cells from VV-OVA sites (open squares) and DNFB-pulled sites (closed circles) are shown. (E) WT mice were treated as in (A), but all mice were treated i.p. with titrated anti-Thy1.1 depleting antibody before oxazolone challenge and some groups were treated with epicutaneous OVA257–264 peptide on DNFB-treated flank day 6 post infection. Quantification of OT-I from epidermal whole mounts is shown. Each symbol represents data from an individual animal. Data are representative of two (E) or three (B-D) separate experiments. **p < 0.01. Scale bar represents 100 μm. Oxa, oxazolone; gMFI, geometric mean fluorescence intensity. See also Figure S4.
We repeated this experiment to analyze the epidermis at an earlier timepoint—day 10 post-oxazolone challenge—in order to better visualize clonal competition when Trm cell differentiation is still occurring. In addition, to determine whether loss of OT-I cells following oxazolone-challenge required recruitment of CD8+ T cells, we depleted all circulating CD8+ T cells by titrated anti-CD8β antibody before oxazolone challenge (Figure 7A and Figure S4). As expected, in control mice treated with anti-Thy1.1 to deplete circulating OT-I cells, we observed a large influx of endogenous CD8+ T cells at both VV-OVA and DNFB-pulled sites. Numbers of OT-I Trm cells at the DNFB-pulled were decreased but not at the VV-OVA sites (Figure 7B and 7C). In mice treated with anti-CD8β antibody, recruited endogenous T cells were not observed and, importantly, numbers of OT-I Trm cells were not reduced indicating that competition from newly recruited CD8+ T cells drives the loss of OT-I cells. Furthermore, we observed significant decrease of CD103 expression by OT-I Trm cells from both oxazolone VV-OVA and DNFB-pulled sites that did not occur in mice treated with anti-CD8β antibody (Figure 7D). These data suggest that when newly recruited CD8+ T cells enter the epidermis, the amount of available active TGFβ is reduced and that competition for active TGFβ is a key factor determining persistence of Trm cells.
Figure 7. Competition for active TGFβ allows for preferential retention of antigen-specific Trm cells.

(A) Experimental scheme. Cohorts of mice were treated as in Figure 6A except titrated anti-Thy1.1 or anti-CD8β depleting antibodies were administered prior to oxazolone challenge. (B) Representative immunofluorescence images of epidermal whole mounts harvested at 10 days post oxazolone challenge stained for Thy1.1+ (cyan) and CD8α+ (red) are shown. (C) Summary data showing numbers of OT-I (Thy1.1+CD8α+) and endogenous CD8+ T cells (Thy1.1−CD8α+) from mice challenged with oxazolone (closed circles) or vehicle (open circles). (D) The frequency of CD69+CD103+ OT-I cells gated on live CD45+Thy1.1+ and gMFI CD103+ from VV-OVA are shown. (E) Experimental scheme. As in (A) except that WT mice were adoptively transferred with control OT-I (E8I-creERT2) or OT-I TGFβRCA (E8I-creERT2 x TGFβRCA). Prior to oxazolone sensitization, mice were treated i.p. tamoxifen for 5 consecutive days. (F) Representative immunofluorescence images of epidermal whole mounts harvested at least 34 days post oxazolone challenge stained for Thy1.1+ (cyan) and CD8α+ (red) are shown. (G) Summary data showing numbers of OT-I (Thy1.1+CD8α+) and total CD8+ T cells (Thy1.1+/−CD8α+) from mice transferred with control OT-I (open squares) or OT-I TGFβRCA (closed squares) is shown. Data are representative of two (B-D) or three (F and G) separate experiments. *p < 0.05, **p < 0.01, and ***p < 0.001. Scale bar represents 100 μm. Oxa, oxazolone; gMFI, geometric mean fluorescence intensity. See also Figure S4, S5, and Table S1.
To determine whether competition for active TGFβ determines persistence of established Trm cells when new CD8+ T cells are recruited, we repeated the above experiment but compared OT-I with OT-I TGFβRCACD8 Trm cells (Figure 7E). CD8+ T cells from OT-I or OT-I TGFβRCACD8 mice were adoptively transferred into WT recipients. Trm cells were established using VV-OVA followed by a DNFB-pull on opposite flanks. Mice were then treated on day 42 post-infection with tamoxifen i.p. followed by oxazolone sensitization on the abdomen and challenge on VV-OVA and DNFB-pulled sites. The epidermis was analyzed at least 30 days following oxazolone challenge. As before, we observed that oxazolone challenge reduced numbers of OT-I Trm cells but numbers of OT-I TGFβRCACD8 Trm cells were largely intact following oxazolone challenge (Figure 7F and 7G). Thus, forced TGFβR signaling allows Trm cells to better compete with newly recruited CD8+ T cells. From these data we conclude that competition for active TGFβ is a key factor determining epidermal residency of Trm cells.
To explore molecular pathways that could facilitate the increased persistence of antigen-specific over bystander Trm cells in the epidermal niche, we performed RNA-seq analysis on paired epidermal OT-I Trm cells that were FACS sorted from the VV-OVA or DNFB treated sites of individual WT animals at 57–70 days after infection. The paired analysis revealed 100 differentially expressed genes (DEGs) (RPKM>4, p-value<0.05, log2FC>0.58) (Table S1). These DEGs did not include any in the TGFβ signaling pathway (Figure S5). In addition, gene ontology (GO) enrichment analysis of the DEGs did not identify alteration in TGFβ-associated pathways. Notably, expression of histone variants associated with chromosome silencing and chromosome organization was increased in bystander Trm cells. Thus, antigen-specific and bystander Trm cells manifest highly similar transcriptionally states but may have epigenetic differences that could determine their differing capacity to persist when active TGFβ becomes limited.
DISCUSSION
In summary, we demonstrated that long-term epidermal persistence of Trm cells under steady-state conditions requires intrinsic TGFβR signaling, autocrine TGFβ, and TGFβ transactivation by integrin αvβ6 and αvβ8 expressed on KC. We also showed that when integrin-mediated TGFβ activation is impaired by mAbs or small molecule blockade, antigen-specific Trm cells persist in the epidermis while bystander Trm cells are partially depleted. Similarly, when new CD8+ effectors are recruited into the epidermis, expression of CD103 on Trm cells is decreased indicative of a reduced availability of active TGFβ and bystander Trm cells are preferentially replaced by newly recruited CD8+ T cells. In both contexts, forced TGFβR signaling was sufficient to rescue bystander Trm cells. Based on these data we propose a model in which access to active TGFβ in the epidermis is a key factor in determining successful colonization of the epidermal niche by Trm cells and intraclonal competition for active TGFβ represents an unappreciated mechanism to preferentially enrich antigen-specific clones at the skin barrier.
The importance of TGFβ and TGFβ-induced CD103 for development of skin Trm cells has been well demonstrated using adoptive transfer of the gene deficient CD8+ T cells (Mackay et al., 2013). By conditionally ablating Tgfbr2 after epidermal Trm cell differentiation and by demonstrating that forced TGFβR signaling retains Trm cells in αvβ6-deficient mice, we now formally demonstrate that ongoing TGFβ signaling is required for maintenance of Trm cells in the epidermis. During the memory phase, Trm cells depend on transactivation of autocrine TGFβ. We favor an autocrine over a paracrine mechanism since physically adjacent Trm cells that had not excised Tgfb1 were not affected by the loss of Tgfb1 by their neighbors. Naïve CD8+ T cells also require TGFβ activation by αv-expressing integrins expressed on DC in lymph nodes to maintain Trm cell differentiation potential (Mani et al., 2019). This parallel and the dependence of Trm cells on TGFβ at all stages of development suggests that transactivation of autocrine TGFβ may also be required by naïve and effector cells. LC and regulatory T (Treg) cells also depend on autocrine TGFβ (Bobr et al., 2012). Treg cells express glycoprotein-A repetitions predominant protein (GARP) that binds surface autocrine TGFβ (Tran et al., 2009). CD8+ Trm cells, however, express low amounts of GARP but relatively high amounts of a GARP homologue, Lrrc33 (Mackay et al., 2013; Pan et al., 2017; Qin et al., 2018) (Figure S5). We speculate that LRRC33 may participate in binding autocrine TGFβ on Trm cells thereby facilitating transactivation.
The enhanced ability of antigen-specific Trm cells at the VV-OVA sites compared to bystander Trm cells at the DNFB-pulled sites to persist in the epidermis when new CD8+ T cells are recruited by oxazolone indicates that encounter to cognate antigen in the epidermis renders Trm cells more fit and better able to compete with new immigrants. The data indicating that it is competition for TGFβ comes from the observations that expression of CD103 is reduced on Trm cells shortly after oxazolone challenge only when CD8+ T cells are recruited into the skin but not when this is prevented by administration of anti-CD8β. In addition, forced expression of TGFβR signaling allowed bystander Trm cells to compete with newly recruited CD8+ T cells as efficiently as antigen-specific Trm cells. Moreover, when forced TGFβR signaling occurred during Trm cell differentiation, bystander OT-I recruited into a site without antigen (VV-N) were better able to compete with endogenous T cells. Thus, competition for active TGFβ appears to occur both during memory and during Trm cell differentiation. This suggests that competition for TGFβ is a mechanism to preferentially maintain antigen-specific over bystander Trm cells in the epidermis during sequential challenges. It may also participate in the enrichment of antigen-specific Trm cells during an initial challenge to naïve skin.
To explore why antigen-specific Trm cells have a competitive advantage over bystander Trm cells when active TGFβ is limited, we compared their transcriptional states. Somewhat unexpectedly, we found both types of Trm cells to manifest highly similar transcriptional states, including genes associated with the TGFβ signaling pathway. This is consistent with the observation that both bystander and antigen-specific Trm cells show a similar reduction of CD103 expression when new T cells are recruited into the skin, indicative of reduced access to active TGFβ due to competition. A key difference between antigen-specific and bystander Trm cells is that the former encounter cognate antigen in the epidermis that activates TCR signaling. The importance of a secondary TCR signaling input in distinguishing the cellular fate dynamics of antigen-specific and bystander Trm cells after their arrival in the epidermis is reinforced by the observations that 1) antigen encounter in the epidermis results in a durable alteration in Trm cells and 2) exposure of bystander Trm cells to cognate antigen in the epidermis enables them to persist despite inhibition of TGFβ activation. The RNA-seq analysis of antigen-specific and bystander Trm cells revealed histone gene variants associated with chromosome silencing and chromosome organization that were more highly expressed in bystander Trm cells. Collectively, these findings lead us to propose that antigen-specific and bystander Trm cells may not differ in their TGFβ sensing and TGFβR signaling capacities but rather that the presence or absence of TCR stimulation during their transition into the tissue-resident memory state may drive distinct epigenetic changes that affect their capacity to persist when active TGFβ becomes limiting.
An implication of our model is that the epidermis represents an environmental niche capable of maintaining a finite number of Trm cells possibly driven by limited physical space or survival factor(s). Circulating memory T cells can be sequentially expanded through multiple rounds of heterologous prime-boost immunizations (Vezys et al., 2009). In contrast, we do not observe expanded numbers of Trm cells following a subsequent challenge with oxazolone suggesting that the epidermis can support a finite number of T cells. This is consistent with previous reports showing that Trm cells replace DETC to maintain an invariable number of total epidermal T cells (Zaid et al., 2014) and that adoptive transfer of increasing numbers of in vitro activated T cells pulled by DNFB to skin does not increase Trm cells numbers beyond a set limit (Park et al., 2018). It is unlikely that the epidermal niche size is determined by active TGFβ since forced TGFβR signaling in T cells augmented intraclonal competition but did not increase overall numbers of epidermal Trm cells. Rather, we speculate that the epidermal niche is determined either by limitations of physical space or by limited amounts of T cell survival and/or homeostatic proliferation factors such as IL-7 or IL-15 (Adachi et al., 2015; Richmond et al., 2018). It remains unclear whether a limited niche exists only in the epidermis or whether it extends to other compact monolayers such as the intestinal epithelium, or solid organs and stromal compartments as well. We note that expression of CD103 by Trm cells is not universal and is relatively enriched in barrier epithelia like epidermis and intestinal epithelium (Steinert et al., 2015). The high frequency of CD103− Trm cells in tissues such as liver suggests that T cell competition for active TGFβ may be limited to certain barrier epithelia.
In conclusion, T cell competition for transactivated autocrine TGFβ participates in shaping the epidermal niche. Though we have focused exclusively on epidermal Trm cells, we think it is likely that a similar mechanism occurs in other active TGFβ-rich tissues where Trm cells express high amounts of CD103. In addition, we have focused on T cells recruited to the skin by transient infection or sensitizing hapten. Whether or not this mechanism also occurs in the context of chronic antigen such as in the tumor micro-environment or commensal-specific T cells and an exploration into the possible epigenetic alterations that distinguish bystander from antigen-specific Trm cells remain to be explored.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Daniel H. Kaplan (dankaplan@pitt.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The RNA-seq datasets reported in this paper can be found at GEO: GSE156668.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Itgb6−/− and Itgb8loxP mice were kindly provided by Dean Sheppard (University of California, San Francisco). E8I-creERT2 and ROSA26.LSL.hNGFR reporter mice were developed by Dario A.A. Vignali (University of Pittsburgh) (Hirai et al., 2019). TgfbloxP and TGFβRICA mice have been previously described (Bartholin et al., 2008; Liu et al., 2020; Marie et al., 2006). C57BL/6 (WT), B6.SJL-Ptprca Pepcb/BoyJ (CD45.1), Tg(KRT14-cre)1Amc/J (Krt14-cre), C57BL/6-Tg(TcraTcrb)1100Mjb/J (OT-I), B6.129S7-Rag1tm1Mom/J (Rag−/−), CBy.PL(B6)-Thy1a/ScrJ (Thy1.1) mice were purchased from Jackson Laboratories. We crossed Krt14-cre mice with Itgb8loxP and Itgb6−/− mice to obtain Itgb6−/−Itgb8ΔKC mice (Mohammed et al., 2016). E8I-creERT2 mice were crossed with TGFβRI-CA mice to obtain TGFβRICACD8 mice. TGFβRI-CACD8 mice were further crossed with Itgb6−/− mice to generate Itgb6−/− x TGFβRI-CACD8 mice. E8I-creERT2 mice were crossed with ROSA26.LSL.hNGFR reporter mice and TgfbloxP mice to obtain TgfbΔCD8 mice. Krt14-creERT2 mice were bred with TgfbloxP mice and ROSA26.LSL.YFP (Jackson Laboratories) reporter mice resulting TgfbΔKC mice. We generated Thy1.1+Rag−/−OT-I mice by crossing OT-I mice with Rag−/− and Thy1.1 mice. TGFβRICACD8 mice and Thy1.1+Rag−/−OT-I mice were bred to generate OT-I TGFβRI-CA mice. OT-I cells carrying ROSA26-creERT2 and floxed TGFβ receptor II gene (OT-I creERT2 TGFbRIIfl/fl) were kindly provided by Thorsten Mempel (Harvard University). We used age- and sex-matched (males and female) mice that were between 6 and 12 weeks of age in all experiments. All mice were maintained under specific-pathogen-free conditions and all animal experiments were approved by University of Pittsburgh Institutional Animal Care and Use Committee.
METHOD DETAILS
Trm cell models and blocking TGFβ activation treatments
Mice were infected by skin scarification (skin infection) with 3 × 106 plaque-forming units recombinant vaccinia virus expressing the SIINFEKL peptide of ovalbumin (VV-OVA). For skin scarification, 45 μL of VV was applied to shaved left flank (4–5 cm2) and the skin were gently scratched 100 times with 27 G needle under anesthesia. In some experiment, 3 × 106 plaque-forming units recombinant vaccinia virus expressing the nucleocapsid protein of vesicular stomatitis virus (VV-N) were applied to shaved right flank at the same day of VV-OVA infection. In some experiments VV-OVA infected mice were further treated with 0.15% 1-Fluoro-2,4-dinitrobenzene (DNFB, D1529: Sigma-Aldrich) in 4:1 acetone : olive oil (Sigma-Aldrich) on the flank opposite the site of infection (40 μL) 5 days post infection. In some experiments, 4:1 acetone and olive oil was added to the stock solution of OVA257–264 (Anaspec, Fremont, CA) in DMSO (D2650, Sigma-Aldrich) (10 mg/mL) for a final concentration of OVA257–264 at 0.2 mg/mL and 50 μL was painted to the DNFB-treated skin 1 day after DNFB-treatment. For blocking of TGFβ activation by antibodies, anti-αvβ6 (6.3g9) and anti-αvβ8 (ADWA-11) (kindly provided by Dean Sheppard) or Vehicle (PBS) were administered i.p. at a dose of 200 μg/mouse weekly for up to 3 weeks. For blocking TGFβ activation with a small molecule integrin inhibitor, compound CWHM-12 (kindly provided by Indalo therapeutics, Cambridge, MA) was solubilized in 100% DMSO. Further dilution to 50% DMSO was made in sterile PBS and dosed to 100 mg per kg body weight per day for 28 days. CWHM-12 or vehicle (control) was delivered by implantable ALZET osmotic minipumps (model 2004, Durect, Cupertino, CA). For sensitization of 4-Ethoxymethylene-2-phenyl-2-oxazolin-5-one (Oxazolone, 862207: Sigma-Aldrich), 40 μL of 20 mg/mL Oxazolone (in 4:1 acetone : olive oil) was administered twice with 5 days interval to shaved abdomen of Trm cell mice. For challenge, mice were received 40 μl Oxazolone (10 mg/mL 4:1 acetone : olive oil)/each side of shaved flank. In some experiment (Figure 2D–2J), mice infected VV-OVA on left flank were also infected with 1 × 106 VV-OVA (15 μL) on left ear on the same day as flank infection.
Adoptive transfers
OT-I cells were purified from spleen and lymph nodes by MojoSort Mouse CD8 T Cell Isolation Kit (Biolegend) according to the manufacturer’s instructions and 1 × 105 OT-I cells were intravenously transferred is all experiments. All mice were allowed to rest for 1 day before further experimentation.
Tamoxifen treatment
Tamoxifen (T5648; Sigma-Aldrich) was dissolved in 1/10th volume of 200 proof ethanol following incubation at 37 °C for 15–30 min with 300 rpm shaking. Corn oil (Sigma-Aldrich) was added for a final concentration of Tamoxifen at 10 mg/ml and was administered to mice for 5 consecutive days by intraperitoneal injection at 0.05 mg per g body weight. 5 mg of 4-Hydroxytamoxifen (4-OH tamoxifen) (H7904–5MG; Sigma-Aldrich) was dissolved in 1.25 mL ≥99.5% acetone following incubation at 37 °C for 5–10 min with occasional vortexing. Corn oil (Sigma-Aldrich) was added for a final concentration of 2 mg/mL and was administered to mice for 2 consecutive days by topical application (40 μL/shaved left flank).
Immunofluorescence of epidermis
Epidermal sheets were prepared as previously described (Mohammed et al., 2016). Briefly, epidermal side of shaved defatted flank skin or splitted ear skin was affixed to slides with double-sided adhesive (3M, St. Paul, MN). Slides were incubated in 10 mM EDTA in PBS for 90 min at 37°C, followed by physical removal of the dermis. The epidermal sheets were fixed in 4% PFA at RT for 15 min. The eidermal sheets were blocked with PBS containing 0.1% tween-20, 2% BSA and 2% rat serum or rabbit serum for 1 hour at RT before staining 1hour with antibodies at RT in PBS containing 0.1% tween-20 and 0.5% BSA. The slides were mounted with ProLong™ Gold Antifade Mountant (Thermo). Images were captured on an IX83 fluorescent microscope (Olympus Tokyo, Japan) using a x10 objective; image analysis was performed using cellSens Dimension software (Olympus). For enumeration of cells, three images from distant sites within an epidermal sheet from a mouse were counted (total 3 mm2) manually or automatically in ImageJ64 after image processing by Adobe Photoshop (version 6) and the average number per mm2 epidermis was calculated as representative of the epidermal sheet. Anti-CD45.2 (104), CD8α (53–6.7), Thy1.1 (OX-7), MHCII (M5/114.15.2), hNGFR (ME20.4), YFP (FM264G) were purchased from Biolegend.
Flow cytometry
Preparation of single cells suspension from tissues were performed as previously described (Mohammed et al., 2016). Briefly, for preparing single cells suspension from the epidermis, shaved skin was harvested and fat tissues was removed mechanically by forceps. Float their dermal side down in the Petri dish containing pre-warmed 37°C 0.3% trypsin (Sigma-Aldrich) solution in 150 mM NaCl, 0.5 mM KCl and 0.5 mM glucose. Incubate in CO2 incubator at 37° for 50–60 minutes. The epidermis was physically separated from the dermis and disrupted by mincing and vigorous pipetting in RPMI1640 (Gibco, Grand Island, NY) with 0.02 M HEPES (Sigma-Aldrich), and 10% FBS (Hyclone). The resulting cells were filtered through a 40 μm cell strainer (BD Biosciences). Blood were collected with heparin (Sigma-Aldrich) and treated with red blood cell lysing buffer (Sigma-Aldrich). Single-cell suspensions were blocked with 2.4G2 culture supernatant (American Type Culture Collection) and were stained with antibodies to extracellular markers and Fixable Viability Dye (eFluor 780) (eBioscience) in PBS with 2% FBS (Hyclone) and 0.05% Azide for 15 min at 4°C. For intracellular cytokines and Granzyme B staining, cells were fixed and permeabilized with Cytofix/Cytoperm kit (BD Biosciences) according to the manufacturer’s instructions. Anti- CD8α (53–6.7), CD44 (IM7), TCRβ (H57–597), CD3 (17A2), Thy1.2 (30-H12), Thy1.1 (OX-7), granzyme B (GB11), CD69 (H1.2F3), CD103 (2E7), CD45.1 (A20), CD45.2 (104), IL-2 (JES6–5H4), and TNF-α (MP6-XT22) were purchased from Biolegend. Anti-CD8α (53–6.7), CD44 (IM7), CD69 (H1.2F3) and Thy1.1 (OX-7) were purchased from BD Biosciences. Anti-IFN-γ (XMG1.2) was purchased from TONBO bioscience (San Diego, CA). A BD LSRFORTESSA (BD Biosciences) and Flowjo software (TreeStar, Ashland, OR) were used for analysis. Enumeration of cells by flow cytometry was made by adding AccuCheck Counting Beads (Thermo Fisher) to samples.
RTqPCR
Epidermis from ear or flank skin was separated from dermis as described for the flow cytometry. The epidermis was minced finely with scissors and processed using the Qiagen RNeasy mini Kit (Thermo Fisher Scientific) following the manufacturer’s instructions. RNA to cDNA conversion was performed using a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Carlsbad, CA). cDNA was analyzed for Intb6, Il7, and Il15 using TaqMan Gene Expression assays. Expression amounts of each gene were normalized to Gapdh.
In vivo recall response
OVA257–264 was purchased from Anaspec and the stock solution was prepared in DMSO (10 mg/mL) (D2650, Sigma-Aldrich). 4:1 acetone and olive oil was added for a final concentration of OVA257–264 at 1 mg/mL and 50 μL was painted to each shaved skin sites of Trm cell mice. 10 mg/ml stock solution of Brefeldin A (B6542, Sigma-Aldrich) was prepared in DMSO. Further dilution to 0.5 mg/mL was made in PBS, and 500 μL was injected i.p. 6 hours after topical OVA257–264 challenge. The skin was harvested 12 hours after the challenge and single suspension of epidermal cells were prepared as described above in the presence of Brefeldin A (2.5 μg/mL) for flow cytometry.
Circulating CD8+ T cells depletion
For depletion of circulating memory OT-I cells, Oxa-sensitized Trm cell mice were injected i.p. with 0.3–1 μg anti-Thy1.1 (HIS51; ThermoFisher) in 200 μL PBS for 2 consecutive days before Oxa challenge. For depletion of circulating total CD8+ T cells, mice were injected i.p. with 10 μg anti-CD8β (H35–17.2; ThermoFisher) in 200 μL PBS for 2 consecutive days before Oxa challenge. Depletion of OT-I or total CD8+ T cells (≤ 0.5% of total CD8+ T cells and ≤ 1% of TCRβ+ T cells, respectively) were confirmed by staining with anti-Thy1.1 (OX-7) or anti-CD8α (53–6.72), respectively using blood 3 days after Oxa challenge.
RNA-seq
Single cell suspension from VV-OVA infected or DNFB-treated epidermis 57–70 days after the infection from the same animal were prepared as described above. Samples from 2 animals were pooled to form a single sample. Live CD45+ Thy1.2− Thy1.1+ OT-I Trm cells were double FACS sorted to ensure high purity and directly lysed using the Clontech SMART-Seq v.4 kit for complimentary DNA synthesis. 200–1000 OT-I cells were collected in 12.5 uL of the lysis buffer. mRNA libraries were generated using Illumina Truseq Stranded mRNA Library Prep kit, followed by 75bp single indexed sequencing on an Illumina NextSeq500 to obtain 40–50 million reads per sample. RNA-seq analysis was performed as described previously (Chaudhri et al., 2020). Reads were mapped on mm9 genome assembly using Tophat2 (Kim et al., 2013; Langmead and Salzberg, 2012). Transcript abundances were calculated using cufflinks (Trapnell et al., 2010) as Reads Per Kilobase of transcript, per Million mapped reads (RPKM). RPKM values were imported in R and transformed as log2(RPKM+1). A paired t-test was then performed on this log transformed RPKM values. Genes with 50 percent fold increase or decrease (log2FC>0.58) with p-values<0.05 were considered differentially expressed and subjected to conditional gene ontology enrichment analysis using “GOstats” package (Falcon and Gentleman, 2007).
QUANTIFICATION AND STATISTICAL ANALYSIS
Groups were compared with Prism software (GraphPad) using the two-tailed unpaired Student’s t test for comparison of two groups or Dunnett’s test for comparisons of more than two groups with the control. Data are presented as each data point and mean or mean ± standard error of the mean (s.e.m.). p < 0.05 was considered significant.
Supplementary Material
Supplemental Table 1. Related to Figure 7. Paired RNAseq analysis of sorted OT-I Trm cells isolated from VV-OVA infected and DNFB-pull sites.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Brilliant Violet 605 anti-mouse CD8a (53–6.7) | BioLegend | Cat# 100744, RRID:AB_2562609 |
| Alexa Fluor 488 anti-mouse CD8a (53–6.7) | BioLegend | Cat# 100723, RRID:AB_ 389304 |
| Alexa Fluor 594 anti-mouse CD8a (53–6.7) | BioLegend | Cat# 100758, RRID:AB_2563237 |
| Alexa Fluor 647 anti-mouse CD8a (53–6.7) | BioLegend | Cat# 100724, RRID:AB_2722580 |
| BUV737 anti-mouse CD8a (53–6.7) | BD Biosciences | Cat# 564297, RRID:AB_2563103 |
| anti-mouse CD8b (H35–17.2) | Thermo Fisher Scientific | Cat# 14–0083-82, RRID:AB_657757 |
| Alexa Fluor 700 anti-mouse CD3 (17A2) | BioLegend | Cat# 100216, RRID:AB_493697 |
| PerCP/Cyanine5.5 anti-mouse CD3 (17A2) | BioLegend | Cat# 100218, RRID:AB_1595492 |
| Brilliant Violet 510 anti-mouse/human CD44 (IM7) | BD Biosciences | Cat# 563114, RRID:AB_2738011 |
| PerCP/Cyanine5.5 anti-mouse/human CD44 (IM7) | BioLegend | Cat# 103032, RRID:AB_2076204 |
| PE/Dazzle 594 anti-mouse TCR β chain (H57–597) | BioLegend | Cat# 109240, RRID:AB_2565655 |
| Alexa Fluor 700 anti-mouse CD90.2 (30-H12) | BioLengend | Cat# 105320, RRID:AB_493725 |
| Alexa Fluor 647 anti-mouse CD90.1 (OX-7) | BioLengend | Cat# 202508, RRID:AB_492884 |
| BUV 395 anti-mouse CD90.1 (OX-7) | BD Biosciences | Cat# 740261, RRID:AB_2721773 |
| anti-mouse CD90.1 (HIS51) | Thermo Fisher Scientific | Cat# 14–0900-81, RRID:AB_467373 |
| BUV737 anti-mouse CD69 (H1.2F3) | BD Biosciences | Cat# 564684, RRID:AB_2738891 |
| PE/Cy7 anti-mouse CD69 (H1.2F3) | BioLegend | Cat# 104512, RRID:AB_493564 |
| Brilliant Violet 510 anti-mouse CD103 (2E7) | BioLegend | Cat# 121423, RRID:AB_2562713 |
| Alexa Fluor 647 anti-mouse CD103 (2E7) | BioLegend | Cat# 121410, RRID:AB_535952 |
| PerCP/Cyanine5.5 anti-mouse CD45.2 (104) | BioLegend | Cat# 109828, RRID:AB_893350 |
| Alexa Fluor 488 anti-mouse CD45.2 (104) | BioLegend | Cat# 109816, RRID:AB_492868 |
| APC anti-mouse CD45.2 (104) | BioLegend | Cat# 109814, RRID:AB_389211 |
| Brilliant Violet 605 anti-mouse CD45.2 (104) | BioLegend | Cat# 109841, RRID:AB_2563485 |
| PerCP/Cyanine5.5 anti-mouse CD45.1 (A20) | BioLegend | Cat# 110728, RRID:AB_893346 |
| Brilliant Violet 605 anti-mouse IFN gamma (XMG1.2) | BioLegend | Cat# 505840, RRID:AB_2734493 |
| PE anti-mouse IL-2 (JES6–5H4) | BioLegend | Cat# 503808, RRID:AB_315302 |
| Brilliant Violet 421 anti-mouse TNF-α (MP6-XT22) | BioLegend | Cat# 506328, RRID:AB_2562902 |
| Alexa Fluor 488 anti-mouse I-A/I-E (M5/114.15.2) | BioLegend | Cat# 107616, RRID:AB_493523 |
| Alexa Fluor 647 anti-human CD271 (NGFR) (ME20.4) | BioLegend | Cat# 345114, RRID:AB_2572059 |
| PE anti-human CD271 (NGFR) (ME20.4) | BioLegend | Cat# 345106, RRID:AB_2152647 |
| Alexa Fluor 488 anti-GFP (FM264G) | BioLegend | Cat# 338008, RRID:AB_2563288 |
| PE/Dazzle 594 anti-human GranzymeB (GB11) | BioLegend | Cat# 562462, RRID:AB_2737618 |
| Phycoerythrin Polyclonal Antibody | Thermo Fisher Scientific | Cat# PA5–35006, RRID:AB_ 2552333 |
| Anti-avb6 (6.3G9) | Dr. D. Sheppard, University of California | N/A |
| Anti-avb8 (ADWA-11) | Dr. D. Sheppard, University of California | N/A |
| Bacterial and Virus Strains | ||
| Vaccinia virus- Western Reserve strain expressing OVA257–264 (VV-OVA) | Dr. J. Yewdell, National Institute of Allergy and Infectious Diseases | N/A |
| Vaccinia virus- Western Reserve strain expressing nucleocapsid protein of vesicular stomatitis virus (VV-N) | Dr. J. Yewdell, National Institute of Allergy and Infectious Diseases | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| OVA257–264 (SIINFEKL) | ANASPEC | Cat# AS-62572–5 |
| CWHM-12 | Indalo therapeutics | N/A |
| Brefeldin A | Sigma-Aldrich | Cat# B6542 |
| 4-Ethoxymethylene-2-phenyl-2-oxazolin-5-one | Sigma-Aldrich | Cat# 862207 |
| Deoxyribonuclease I | Sigma-Aldrich | Cat# D5025 |
| Dimethyl sulfoxide | Sigma-Aldrich | Cat# D2650 |
| Fetal Bovine Serum, Heat Inactivated | HyClone | Cat# 89133–096 |
| 2-Mercaptoethanol | Sigma-Aldrich | Cat# M6250 |
| Penicillin-Streptomycin | Gibco | Cat# 15140122 |
| MEM Nonessential Amino Acids | Corning | Cat# 25025CI |
| Sodium Azide, 5% (w/v) Aqueous Solution | Ricca Chemical | Cat# 71448–16 |
| 0.5M EDTA, pH 8.0 | Invitrogen | Cat# 15–575–020 |
| 32% paraformaldehyde | Thermo Fisher Scientific | Cat# 50–980–495 |
| Protein Transport Inhibitor Cocktail | eBioscience | Cat# 00–4980–03 |
| Fixable Viability Dye eFluor™ 780 | eBioscience | Cat# 65–0865–14 |
| RPMI1640 | Corning | Cat# 10040CV |
| Red Blood Cell Lysing Buffer Hybri-Max | Sigma Aldrich | Cat# R7757 |
| Heparin sodium salt from porcine intestinal mucosa | Sigma Aldrich | Cat# H3393 |
| HEPES solution | Sigma Aldrich | Cat# H0887 |
| Corn oil | Sigma Aldrich | Cat# C8267 |
| Tamoxifen | Sigma Aldrich | Cat# T5648 |
| 4-OH Tamoxifen | Sigma Aldrich | Cat# H7904–5MG |
| Critical Commercial Assays | ||
| MojoSort™ Mouse CD8 T Cell Isolation Kit | BioLegend | Cat# 480035 |
| TaqMan™ Gene Expression Master Mix | Applied Biosystems | Cat# 4369016 |
| RNeasy Mini Kit | QIAGEN | Cat# 74104 |
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat# 4368814 |
| Alexa Fluor™ 555 Antibody Labeling Kit | Thermo Fisher Scientific | Cat# A20187 |
| TaqMan Gene Expression Assay Gapdh | Thermo Fisher Scientific | Cat# Mm99999915_g1 |
| TaqMan Gene Expression Assay Itgb6 | Thermo Fisher Scientific | Cat# Mm01269869_m1 |
| TaqMan Gene Expression Assay Il7 | Thermo Fisher Scientific | Cat# Mm01295803_m1 |
| TaqMan Gene Expression Assay Il15 | Thermo Fisher Scientific | Cat# Mm00434210_m1 |
| Experimental Models: Organisms/Strains | ||
| Mouse: WT: C57BL/6J | The Jackson Laboratory | Cat# JAX:000664, RRID:IMSR_JAX:000664 |
| Mouse: K14-Cre: Tg(KRT14-cre)1Amc/J | The Jackson Laboratory | Cat# JAX:004782, RRID:IMSR_JAX:004782 |
| Mouse: K14-CreERT : Tg(KRT14-cre/ERT)20Efu/J | The Jackson Laboratory | Cat# JAX: 005107, RRID:IMSR_JAX: 005107 |
| Mouse: ROSA26.LSL.YFP: B6.129X1-Gt(ROSA)26Sortm1(EYFP)Cos/J | The Jackson Laboratory | Cat# JAX: 006148, RRID:IMSR_JAX: 006148 |
| Mouse: OT-I: C57BL/6-Tg(TcraTcrb)1100Mjb/J | The Jackson Laboratory | Cat# JAX:003831, RRID:IMSR_JAX:003831 |
| Mouse: Rag−/−: B6.129S7-Rag1tm1Mom/J | The Jackson Laboratory | Cat# JAX:002216, RRID:IMSR_JAX:002216 |
| Mouse: Thy1.1: CBy.PL(B6)-Thy1a/ScrJ | The Jackson Laboratory | Cat# JAX:005443, RRID:IMSR_JAX:005443 |
| Mouse: CD45.1: B6.SJL-Ptprca Pepcb/BoyJ | The Jackson Laboratory | Cat# JAX: 002014, RRID:IMSR_JAX:002014 |
| Mouse: CD45.1: B6.SJL-Ptprca Pepcb/BoyJ | The Jackson Laboratory | Cat# JAX: 002014, RRID:IMSR_JAX:002014 |
| Mouse: CD45.1: B6.SJL-Ptprca Pepcb/BoyJ | The Jackson Laboratory | Cat# JAX: 002014, RRID:IMSR_JAX:002014 |
| Mouse: OT-I CreERT2 TGFbRIIfl/fl | Dr. Thorsten Mempel Harvard University | N/A |
| Mouse: Itgb6−/− | Dr. D. Sheppard, University of California | N/A |
| Mouse: Itgb8loxP | Dr. D. Sheppard, University of California | N/A |
| Mouse: TGFβRI-CA | Dr. L. Bartholin, Centre de Recherche en Cancérologie de LYON | N/A |
| Mouse: E8ICreERT2 | Dr. D.A. Vignali, University of Pittsburgh | N/A |
| Mouse: ROSA26.LSL.hNGFR | Dr. D.A. Vignali, University of Pittsburgh | N/A |
| Oligonucleotides | ||
| Software and Algorithms | ||
| Prism 8 | Graphpad Inc | RRID:SCR_002798 |
| Flowjo v10 | Treestar Inc | RRID:SCR_008520 |
| ImageJ 64 | RRID: SCR_003070 | |
| Paired RNAseq Data of Trm from VV-ova and DNFB sites | NCBI GEO | GSE156668 |
Key Points.
Long-term persistence of epidermal Trm cells requires transactivated autocrine TGFβ
Epidermal bystander Trm cells are depleted when active TGFβ is limited
Competition for TGFβ allows newly recruited effectors to replace bystander Trm cells
Intraclonal competition for active TGFβ shapes Trm occupancy in the epidermal niche
Acknowledgments
We thank Kate M. Vignali and Andrea L. Workman for constructing E8I-creERT2 and ROSA26.LSL.hNGFR reporter mice. We thank the members of the Kaplan and Vignali laboratory and members throughout the departments of Dermatology and Immunology for helpful discussions. We also thank the Division of Laboratory Animal Resources of the University of Pittsburgh for excellent animal care. The graphical abstract was created with BioRender.com. This work benefitted from SPECIAL BD LSRFORTESSATM funded by NIH 1S10OD011925-01.TH was supported by JSPS Overseas Research Fellowships; JDC by NIH T32 AI089443, AR060744S1; JDC by NIH T32AI089443; DHK by NIH R01AR060744; DAAV by NIH P01AI108545
Footnotes
Competing interests
D.G. is a consultant and equity holder of Indalo Therapeutics which provided compound CWHM-12. All other authors state no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adachi T, Kobayashi T, Sugihara E, Yamada T, Ikuta K, Pittaluga S, Saya H, Amagai M, and Nagao K (2015). Hair follicle-derived IL-7 and IL-15 mediate skin-resident memory T cell homeostasis and lymphoma. Nat Med 21, 1272–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allan RS, Smith CM, Belz GT, van Lint AL, Wakim LM, Heath WR, and Carbone FR (2003). Epidermal viral immunity induced by CD8alpha+ dendritic cells but not by Langerhans cells. Science 301, 1925–1928. [DOI] [PubMed] [Google Scholar]
- Aluwihare P, Mu Z, Zhao Z, Yu D, Weinreb PH, Horan GS, Violette SM, and Munger JS (2009). Mice that lack activity of alphavbeta6- and alphavbeta8-integrins reproduce the abnormalities of Tgfb1- and Tgfb3-null mice. J Cell Sci 122, 227–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartholin L, Cyprian FS, Vincent D, Garcia CN, Martel S, Horvat B, Berthet C, Goddard-Leon S, Treilleux I, Rimokh R, and Marie JC (2008). Generation of mice with conditionally activated transforming growth factor beta signaling through the TbetaRI/ALK5 receptor. Genesis 46, 724–731. [DOI] [PubMed] [Google Scholar]
- Bedoui S, Whitney PG, Waithman J, Eidsmo L, Wakim L, Caminschi I, Allan RS, Wojtasiak M, Shortman K, Carbone FR, et al. (2009). Cross-presentation of viral and self antigens by skin-derived CD103+ dendritic cells. Nat Immunol 10, 488–495. [DOI] [PubMed] [Google Scholar]
- Bobr A, Igyarto BZ, Haley KM, Li MO, Flavell RA, and Kaplan DH (2012). Autocrine/paracrine TGF-beta1 inhibits Langerhans cell migration. Proc Natl Acad Sci U S A 109, 10492–10497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey KA, Fraser KA, Schenkel JM, Moran A, Abt MC, Beura LK, Lucas PJ, Artis D, Wherry EJ, Hogquist K, et al. (2012). Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J Immunol 188, 4866–4875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhri VK, Dienger-Stambaugh K, Wu Z, Shrestha M, and Singh H (2020). Charting the cis-regulome of activated B cells by coupling structural and functional genomics. Nat Immunol 21, 210–220. [DOI] [PubMed] [Google Scholar]
- Collins N, Jiang X, Zaid A, Macleod BL, Li J, Park CO, Haque A, Bedoui S, Heath WR, Mueller SN, et al. (2016). Skin CD4(+) memory T cells exhibit combined cluster-mediated retention and equilibration with the circulation. Nat Commun 7, 11514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcon S, and Gentleman R (2007). Using GOstats to test gene lists for GO term association. Bioinformatics 23, 257–258. [DOI] [PubMed] [Google Scholar]
- Gamradt P, Laoubi L, Nosbaum A, Mutez V, Lenief V, Grande S, Redoules D, Schmitt AM, Nicolas JF, and Vocanson M (2019). Inhibitory checkpoint receptors control CD8(+) resident memory T cells to prevent skin allergy. J Allergy Clin Immunol 143, 2147–2157 e2149. [DOI] [PubMed] [Google Scholar]
- Gebhardt T, Whitney PG, Zaid A, Mackay LK, Brooks AG, Heath WR, Carbone FR, and Mueller SN (2011). Different patterns of peripheral migration by memory CD4+ and CD8+ T cells. Nature 477, 216–219. [DOI] [PubMed] [Google Scholar]
- Henderson NC, Arnold TD, Katamura Y, Giacomini MM, Rodriguez JD, McCarty JH, Pellicoro A, Raschperger E, Betsholtz C, Ruminski PG, et al. (2013). Targeting of alphav integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat Med 19, 1617–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai T, Whitley SK, and Kaplan DH (2020). Migration and Function of Memory CD8(+) T Cells in Skin. J Invest Dermatol 140, 748–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai T, Zenke Y, Yang Y, Bartholin L, Beura LK, Masopust D, and Kaplan DH (2019). Keratinocyte-Mediated Activation of the Cytokine TGF-beta Maintains Skin Recirculating Memory CD8(+) T Cells. Immunity 50, 1249–1261 e1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs SJ, and Nolz JC (2019). Targeted Expansion of Tissue-Resident CD8(+) T Cells to Boost Cellular Immunity in the Skin. Cell Rep 29, 2990–2997 e2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Clark RA, Liu L, Wagers AJ, Fuhlbrigge RC, and Kupper TS (2012). Skin infection generates non-migratory memory CD8+ T(RM) cells providing global skin immunity. Nature 483, 227–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan TN, Mooster JL, Kilgore AM, Osborn JF, and Nolz JC (2016). Local antigen in nonlymphoid tissue promotes resident memory CD8+ T cell formation during viral infection. J Exp Med 213, 951–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kish DD, Li X, and Fairchild RL (2009). CD8 T cells producing IL-17 and IFN-gamma initiate the innate immune response required for responses to antigen skin challenge. J Immunol 182, 5949–5959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YT, Suarez-Ramirez JE, Wu T, Redman JM, Bouchard K, Hadley GA, and Cauley LS (2011). Environmental and antigen receptor-derived signals support sustained surveillance of the lungs by pathogen-specific cytotoxic T lymphocytes. J Virol 85, 4085–4094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Somasundaram A, Manne S, Gocher AM, Szymczak-Workman AL, Vignali KM, Scott EN, Normolle DP, John Wherry E, Lipson EJ, et al. (2020). Neuropilin-1 is a T cell memory checkpoint limiting long-term antitumor immunity. Nat Immunol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Fuhlbrigge RC, Karibian K, Tian T, and Kupper TS (2006). Dynamic programming of CD8+ T cell trafficking after live viral immunization. Immunity 25, 511–520. [DOI] [PubMed] [Google Scholar]
- Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon ML, Vega-Ramos J, Lauzurica P, Mueller SN, Stefanovic T, et al. (2013). The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat Immunol 14, 1294–1301. [DOI] [PubMed] [Google Scholar]
- Mackay LK, Stock AT, Ma JZ, Jones CM, Kent SJ, Mueller SN, Heath WR, Carbone FR, and Gebhardt T (2012). Long-lived epithelial immunity by tissue-resident memory T (TRM) cells in the absence of persisting local antigen presentation. Proc Natl Acad Sci U S A 109, 7037–7042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay LK, Wynne-Jones E, Freestone D, Pellicci DG, Mielke LA, Newman DM, Braun A, Masson F, Kallies A, Belz GT, and Carbone FR (2015). T-box Transcription Factors Combine with the Cytokines TGF-beta and IL-15 to Control Tissue-Resident Memory T Cell Fate. Immunity 43, 1101–1111. [DOI] [PubMed] [Google Scholar]
- Mani V, Bromley SK, Aijo T, Mora-Buch R, Carrizosa E, Warner RD, Hamze M, Sen DR, Chasse AY, Lorant A, et al. (2019). Migratory DCs activate TGF-beta to precondition naive CD8(+) T cells for tissue-resident memory fate. Science 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marie JC, Liggitt D, and Rudensky AY (2006). Cellular mechanisms of fatal early-onset autoimmunity in mice with the T cell-specific targeting of transforming growth factor-beta receptor. Immunity 25, 441–454. [DOI] [PubMed] [Google Scholar]
- Masopust D, and Soerens AG (2019). Tissue-Resident T Cells and Other Resident Leukocytes. Annu Rev Immunol 37, 521–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMaster SR, Wein AN, Dunbar PR, Hayward SL, Cartwright EK, Denning TL, and Kohlmeier JE (2018). Pulmonary antigen encounter regulates the establishment of tissue-resident CD8 memory T cells in the lung airways and parenchyma. Mucosal Immunol 11, 1071–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammed J, Beura LK, Bobr A, Astry B, Chicoine B, Kashem SW, Welty NE, Igyarto BZ, Wijeyesinghe S, Thompson EA, et al. (2016). Stromal cells control the epithelial residence of DCs and memory T cells by regulated activation of TGF-beta. Nat Immunol 17, 414–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muschaweckh A, Buchholz VR, Fellenzer A, Hessel C, Konig PA, Tao S, Tao R, Heikenwalder M, Busch DH, Korn T, et al. (2016). Antigen-dependent competition shapes the local repertoire of tissue-resident memory CD8+ T cells. J Exp Med 213, 3075–3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, Tian T, Park CO, Lofftus SY, Mei S, Liu X, Luo C, O’Malley JT, Gehad A, Teague JE, et al. (2017). Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature 543, 252–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SL, Zaid A, Hor JL, Christo SN, Prier JE, Davies B, Alexandre YO, Gregory JL, Russell TA, Gebhardt T, et al. (2018). Local proliferation maintains a stable pool of tissue-resident memory T cells after antiviral recall responses. Nat Immunol 19, 183–191. [DOI] [PubMed] [Google Scholar]
- Qin Y, Garrison BS, Ma W, Wang R, Jiang A, Li J, Mistry M, Bronson RT, Santoro D, Franco C, et al. (2018). A Milieu Molecule for TGF-beta Required for Microglia Function in the Nervous System. Cell 174, 156–171 e116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynoso GV, Weisberg AS, Shannon JP, McManus DT, Shores L, Americo JL, Stan RV, Yewdell JW, and Hickman HD (2019). Lymph node conduits transport virions for rapid T cell activation. Nat Immunol 20, 602–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond JM, Strassner JP, Zapata L Jr., Garg M, Riding RL, Refat MA, Fan X, Azzolino V, Tovar-Garza A, Tsurushita N, et al. (2018). Antibody blockade of IL-15 signaling has the potential to durably reverse vitiligo. Sci Transl Med 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan BS, Pham QM, Lee YT, Cauley LS, Puddington L, and Lefrancois L (2014). Oral infection drives a distinct population of intestinal resident memory CD8(+) T cells with enhanced protective function. Immunity 40, 747–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slutter B, Van Braeckel-Budimir N, Abboud G, Varga SM, Salek-Ardakani S, and Harty JT (2017). Dynamics of influenza-induced lung-resident memory T cells underlie waning heterosubtypic immunity. Sci Immunol 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinert EM, Schenkel JM, Fraser KA, Beura LK, Manlove LS, Igyarto BZ, Southern PJ, and Masopust D (2015). Quantifying Memory CD8 T Cells Reveals Regionalization of Immunosurveillance. Cell 161, 737–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran DQ, Andersson J, Wang R, Ramsey H, Unutmaz D, and Shevach EM (2009). GARP (LRRC32) is essential for the surface expression of latent TGF-beta on platelets and activated FOXP3+ regulatory T cells. Proc Natl Acad Sci U S A 106, 13445–13450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, and Pachter L (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezys V, Yates A, Casey KA, Lanier G, Ahmed R, Antia R, and Masopust D (2009). Memory CD8 T-cell compartment grows in size with immunological experience. Nature 457, 196–199. [DOI] [PubMed] [Google Scholar]
- Vincent DF, Kaniewski B, Powers SE, Havenar-Daughton C, Marie JC, Wotton D, and Bartholin L (2010). A rapid strategy to detect the recombined allele in LSL-TbetaRICA transgenic mice. Genesis 48, 559–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakim LM, Smith J, Caminschi I, Lahoud MH, and Villadangos JA (2015). Antibody-targeted vaccination to lung dendritic cells generates tissue-resident memory CD8 T cells that are highly protective against influenza virus infection. Mucosal Immunol 8, 1060–1071. [DOI] [PubMed] [Google Scholar]
- Worthington JJ, Klementowicz JE, and Travis MA (2011). TGFbeta: a sleeping giant awoken by integrins. Trends Biochem Sci 36, 47–54. [DOI] [PubMed] [Google Scholar]
- Yang Y, Zenke Y, Hirai T, and Kaplan DH (2019). Keratinocyte-derived TGFbeta is not required to maintain skin immune homeostasis. J Dermatol Sci 94, 290–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Mu Z, Dabovic B, Jurukovski V, Yu D, Sung J, Xiong X, and Munger JS (2007). Absence of integrin-mediated TGFbeta1 activation in vivo recapitulates the phenotype of TGFbeta1-null mice. J Cell Biol 176, 787–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaid A, Mackay LK, Rahimpour A, Braun A, Veldhoen M, Carbone FR, Manton JH, Heath WR, and Mueller SN (2014). Persistence of skin-resident memory T cells within an epidermal niche. Proc Natl Acad Sci U S A 111, 5307–5312. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1. Related to Figure 7. Paired RNAseq analysis of sorted OT-I Trm cells isolated from VV-OVA infected and DNFB-pull sites.
Data Availability Statement
The RNA-seq datasets reported in this paper can be found at GEO: GSE156668.