

Abstract
Objective:
Metabolomic studies suggest plasma levels of bile acids (BAs) are elevated amongst subjects with non-alcoholic fatty liver disease (NAFLD) compared to healthy controls. However, it remains unclear whether or not specific BAs are associated with the clinically relevant transition from nonalcoholic fatty liver (i.e. simple steatosis) to non-alcoholic steatohepatitis (NASH), or enhanced progression of hepatic fibrosis, or genetic determinants of NAFLD/NASH.
Methods:
Among sequential subjects (n=102) undergoing diagnostic liver biopsy, we examined the associations of a broad panel of BAs with distinct histopathological features of NAFLD, the presence of NASH, and their associations with genetic variants linked to NAFLD and NASH.
Results:
Plasma BA alterations were observed through the entire spectrum of NAFLD, with several glycine conjugated forms of the BAs demonstrating significant associations with higher grades of inflammation and fibrosis. Plasma 7-Keto-DCA levels showed the strongest associations with advanced stages of hepatic fibrosis [odds ratio(95% confidence interval)], 4.2(1.2–16.4), NASH 24.5(4.1–473), and ballooning 18.7(4.8–91.9). Plasma 7-Keto-LCA levels were associated with NASH 9.4(1.5–185) and ballooning 5.9(1.4–28.8). Genetic variants at several NAFLD/NASH loci were nominally associated with increased levels of 7-Keto- and glycine-conjugated forms of BAs, and the NAFLD risk allele at the TRIB1 locus showed strong tendency toward increased plasma levels of GCA (p=0.02) and GUDCA (p=0.009).
Conclusions:
Circulating bile acid levels are associated with histopathological and genetic determinants of the transition from simple hepatic steatosis into NASH. Further studies exploring the potential involvement of bile acid metabolism in the development and/or progression of distinct histopathological features of NASH are warranted.
Keywords: Bile acids, Fibrosis, Metabolomics, NAFLD, NASH
1. INTRODUCTION
Non-alcoholic fatty liver disease (NAFLD) is the most prevalent form of chronic liver disease worldwide [1, 2]. NAFLD includes a broad spectrum of histopathologic abnormalities, ranging from simple steatosis (non-alcoholic fatty liver) to a more aggressive nonalcoholic steatohepatitis (NASH), which is characterized by steatosis, hepatocellular ballooning and inflammation, often with accompanying fibrosis, cirrhosis, and heightened risk for hepatocellular carcinoma [3]. Only a subset of subjects with simple steatosis will progress to NASH and cirrhosis through unclear mechanisms. Early detection of the transition from steatosis to NASH, and improved understanding of possible mechanisms involved are critical to the development of therapeutic interventions [4, 5].
Several mechanisms are believed to contribute to the progression of NAFLD [6, 7], from insulin resistance, to altered lipid metabolism, release of cytokines from adipocytes, and changes in gut-microbial composition [8, 9]. Genetic variants associated with NAFLD susceptibility have also recently been identified, and suggest numerous links to various aspects of lipid metabolism [10–14]. Despite intense interest in the mechanisms potentially contributing to development of NASH, examination of the association between NAFLD genetic susceptibility loci and systemic levels of BAs has not yet been reported. Increased circulating levels of bile acids (BAs) have been associated with the major risk factors for NAFLD, including insulin resistance and type 2 diabetes mellitus (T2DM) [15, 16]. Furthermore, initial metabolomic studies have shown global alterations in BA composition in individuals with either simple steatosis or NASH in comparison to healthy controls [9, 17, 18]. BAs have similarly been studied as a potential therapeutic target in NAFLD [19–21], given their signaling properties, including activation of the nuclear receptor, farnesoid X receptor (FXR), and the membrane Takeda G-protein coupled receptor 5 (TGR5) [22–26].
Despite intense interest in BAs and NASH, the potential association of structurally specific molecular species of BAs during the clinically relevant progression of simple steatosis into NASH remains largely unexplored. Moreover, the association of previously identified genetic variants associated with risk for NASH [14, 27–29] with BA signatures linked to histopathological contributors of NASH similarly remains unknown. BAs represent a large and diverse group of compounds whose analytical quantification can pose significant difficulties. We recently reported the development and use of a quantitatively robust stable isotope dilution liquid chromatography with on-line tandem mass spectrometry (LC/MS/MS) approach to enable simultaneous quantification of a large panel of primary and secondary BAs in biological matrices [30]. Herein we utilize this targeted metabolomic approach to interrogate the association of multiple specific molecular species of BAs with histopathological indices of hepatocellular fibrosis, advanced steatosis, ballooning and inflammation, in combination with exploration of their association with known genetic variants for NAFLD and NASH.
2. MATERIALS & METHODS
2.1. Patient characteristics and sample collection
Study protocols were approved by the Institutional Review Board of Cleveland Clinic. Written informed consent was obtained from all study participants. Our NAFLD cohort consisted of 102 consecutive patients from whom venous blood was drawn after an overnight fast on the day of elective diagnostic liver biopsy procedure at the Cleveland Clinic. Demographic, clinical, and laboratory data were obtained for each patient. Plasma was obtained by centrifugation of blood samples for 10 min at 1500×g at 4 °C and was immediately stored at −80 °C until further analyses. Patients were between 27 and 77 years of age and reported less than 20 grams/day of alcohol consumption for males and less than 10 grams/day for females. Patients with other liver diseases, including viral, drug-related, autoimmune, and metabolic/genetic liver diseases (e.g. hepatocellular carcinoma, viral hepatitis, hemochromatosis, Wilson’s disease and alpha-1 antitrypsin deficiency), were excluded from the study. Histologic scoring was done using the Nonalcoholic Steatohepatitis Clinical Research Network Histologic Scoring System [21]. According to this scoring system, the degree of steatosis, liver injury, and inflammatory activity are measured using a graded scale (steatosis, 0–3; lobular inflammation, 0–3; and ballooning, 0–2). The NAFLD activity score (NAS) is the unweighted sum of steatosis, lobular inflammation, and hepatocellular ballooning score. Definite NASH was defined by an NAS score ≥5, the presence of ballooning, and a pathological interpretation. Simple steatosis was defined by an NAS <3, and borderline NASH was defined by a NAS of 3–4 with ballooning. Patients were stratified according to the degree of fibrosis on liver biopsy as no fibrosis (F0) and those with fibrosis (F1–4). Clinical laboratory studies were performed on a Roche Cobas C311 analyzer, and complete blood cell count and differential were determined using a Bayer Advia hematology analyzer. A random sampling (n=50) of healthy control subjects (from a larger study designed to enroll healthy controls, and in which documented normal liver ultrasound was performed) were also analyzed for the BA panel LC/MS/MS analysis. To be considered for BA analysis as a control, subjects had to be healthy and without medical history of cardiovascular or metabolic disease (e.g. diabetes, chronic kidney disease), not on any chronic medications, and upon laboratory testing of plasma, confirmed to have normal liver function test panel (aspartate transaminase (AST), alanine transaminase (ALT), total bilirubin), basic metabolic panel, lipid profile and hemoglobin A1C.
2.2. Bile acid profiling
Stable isotope dilution LC/MS/MS was used for quantification of plasma BAs, as recently described [30]. Briefly, internal standard (IS) solution was prepared in methanol (IS composition: 0.1 μM each of D4-glycolithocholic acid, D4-glycoursodeoxycholic acid, D4-glycodeoxycholic acid, D4-glycocholic acid, D4-taurolithocholic acid, D4-tauroursodeoxycholic acid, D4-taurochenodeoxycholic acid, D4-taurodeoxycholic acid, and D4-taurocholic acid; 0.2 μM each of D4-lithocholic acid, D4-chenodeoxycholic acid, D4-deoxycholic acid, and D4-cholic acid; and 0.4 μM of D4-glycochenodeoxycholic acid). Plasma samples (20 μl) were mixed with ice-cold methanolic IS solution (80 μl), vortexed, and centrifuged at 4 °C, at which point the supernatant was transferred to HPLC vials for analysis. The LC/MS/MS system consisted of 4000 Q-Trap triple quadrupole tandem mass spectrometer (AB SCIEX, MA, USA), four binary pumps (LC-20 AD), autosampler operating at 10 °C (Nexera X2 SIL-30AC), controller (CBM-20A) (Shimadzu Scientific Instruments, Inc., MD, USA), and a dual column switching valve system Rheodyne (IDEX Health & Science, MA, USA). Analyses were performed using electrospray ionization in negative-ion mode with multiple reaction monitoring (MRM) of characteristic precursor and product ions for each BA. Chromatographic separations were performed on a reverse phase column (Kinetix C18, 2.6 μm, 150 mm × 4.6 mm ID; catalog # 00F-4462-E0; Phenomenex, Torrance, CA). Mobile phase A was 1 mM ammonium acetate and 0.1 % acetic acid in methnol:acetonitrile:water (1:1:3; v/v/v), and mobile phase B was 0.1 % acetic acid in methanol:acetonitrile:2-propanol (4.5:4.5:1; v/v/v). Injection volume was 10 μL.
Bile acids pools reported were defined as follows: Total 1° BA = the sum of taurocholic acid (TCA) + taurochenodeoxycholic acid (TCDCA) + glycocholic acid (GCA) + glycochenodeoxycholic acid (GCDCA) + glycohyocholic acid (GHCA) + chenodeoxycholic acid (CDCA) + cholic acid (CA) + hyocholic acid (HCA); Primary (1°) Conjugated BA = the sum of TCA + TCDCA + GCA + GCDCA + GHCA; 1° Free BA = CA + CDCA; Glycine 1° BA = the sum of GCA + GCDCA + GHCA; Taurine 1° BA = the sum of TCA + TCDCA; Total Secondary (2°) BA = the sum of deoxycholic acid (DCA) + glycodeoxycholic acid (GDCA) + taurodeoxycholic acid (TDCA) + lithocholic acid (LCA) + glycolithocholic acid (GLCA) + taurolithocholic acid (TLCA) + lithocholic acid-3-sulfate (LCA 3-Sulfate) + Isolithocholic acid (Iso-LCA) + ursodeoxycholic acid (UDCA) + glycoursodeoxycholic acid (GUDCA) + tauroursodeoxycholic acid (TUDCA) + hyodeoxycholic acid (HDCA) + glycohyodeoxycholic acid (GHDCA) + 7-Ketodeoxycholic acid (7-Keto-DCA) + 3-Ketodeoxycholic acid (3-Keto-DCA) + 7-Ketolithocholic acid (7-Keto-LCA) + Isodeoxycholic acid (Iso-DCA); Total BA = the sum of Total 1° BA + Total 2° BA; 2° Conjugated BA = the sum of GDCA + TDCA + GLCA + TLCA + GUDCA + TUDCA + GHDCA; 2° Free BA = the sum of DCA + LCA + LCA 3-Sulfate + Iso-LCA + UDCA + HDCA + 7-Keto-DCA + 3-Keto-DCA + 7-Keto-LCA + Iso-DCA; Glycine 2° BA = the sum of GDCA + GLCA; Taurine 2° BA = the sum of TDCA + TLCA; 2° Free BA = the sum of DCA + LCA, 2° Conjugated BA = the sum of GLCA +TLCA + GDCA + TDCA; Total CDCA = the sum of CDCA + GCDCA + TCDCA; Conjugated CDCA = the sum of GCDCA + TCDCA; Total CA = the sum of CA + TCA + GCA; Conjugated CA = the sum of TCA + GCA; Total DCA = the sum of DCA + GDCA + TDCA; Total LCA = the sum of LCA + GLCA + TLCA; Total ursodeoxycholic acid (UDCA) = the sum of UDCA + GUDCA + TUDCA; Total hyocholic acid (HCA) = the sum of HCA + GHCA.
2.3. Evaluation of genetic variants for association with BAs.
Twelve variants previously identified through genome-wide association studies (GWAS) for NAFLD, NASH, or for NAFLD-related traits, such as alcoholic fatty liver disease or blood levels of enzymes used to evaluate liver function [10], were selected for association testing with plasma BA levels. These analyses were carried out in the GeneBank Study (ClinicalTrials.gov Identifier: NCT00590200), a cohort of sequential stable subjects undergoing elective diagnostic coronary angiography at the Cleveland Clinic. Genotypes for rs12137855 (LYPLAL1), rs1260326 (GCKR), rs2241766 (ADIPOQ), rs1800591 (MTTP), rs4240624 (PPP1R3B), rs2954021 (TRIB1), rs11597086 (ERLIN1–CHUK–WF19L1), rs174546 (FADS1), rs7946 (PEMT), rs641738 (MBOAT7), rs58542926 (TM6SF2), and rs738409 (PNPLA3) were obtained for GeneBank subjects of European ancestry (n=1840) profiled on either the Affymetrix 6.0 GeneChip or Illumina Infinium Global Screening Array for whom plasma BA species were quantified by stable isotope dilution LC/MS/MS, as described above [30]. Linear regression analysis for each BA was carried out using natural-log or normal inverse transformed values, with adjustment for age and sex (STATA v15.0, StataCorp LP, Texas, USA). Associations were considered significant at a Bonferroni-corrected threshold of p<4.2×10−3 (0.05/12 SNPs).
2.4. Statistical Analysis
Primary and secondary BAs were detected and quantified in plasma samples of NAFLD patients. BA concentrations were calculated as median values (25th, 75th percentiles), and categorical variables were shown as counts and percentages. The statistical significance of the change between groups was determined by the Mann-Whitney test after normality testing. Significance of change across the groups was determined by the Kruskal-Wallis test with post hoc analysis using Dunn’s test (after the normality test), and by test for trend for increasing BAs or BA pools within each group; p < 0.05 deemed to be significant. Calculated p values were further adjusted for multiple testing, where indicated, by false discovery rate (FDR). Odds ratios (95% confidence intervals) unadjusted and adjusted by multivariable logistic regression model (adjusted for age, gender, body mass index (BMI), homeostatic model assessment for insulin resistance (HOMA-IR), and alanine aminotransferase (ALT)). Statistical analyses were performed with GraphPad Prism 8.0 (GraphPad Software, Inc., San Diego, CA), R (R-3.4.4 for Windows 64 bit) and XLSTAT version 2019 complete statistical add-in for Microsoft Excel®.
3. RESULTS
3.1. Clinical and histologic characteristics of the NAFLD patients, and summary BA results
A total of 102 participants with biopsy-proven NAFLD underwent fasting plasma BA assessment for the entire spectrum of NAFLD phenotypes. Participants (>85% Caucasian) had a mean (± S.D.) age of 52.3 (±10.6) years and a mean BMI of 32.8 ± 5.8 kg/m2. Clinical, demographic, biochemical, and histological data of this cohort are provided in Supplementary Tables 1 and 2. All participants had biopsy proven liver steatosis, 59% had hepatocellular ballooning, and histological evidence (any stage) of inflammation and fibrosis were observed in 86% and 73%, respectively. According to the NASH Clinical Research Network scoring system, 30% of participants had simple steatosis, 43% had borderline NASH and 27% had definite NASH. Patients with fibrosis had significantly decreased plasma total cholesterol and LDL cholesterol levels, higher AST/ALT ratio and were significantly older (P<0.05, each). When compared to healthy control subjects (n=50) (Methods), individuals in the NAFALD cohort had significantly higher levels of almost all tested circulating BAs, as demonstrated in Table 1 and Supplementary Tables 3 and 4. Collectively, these results demonstrated a generalized significant elevation in circulating levels of the majority of BA molecular species among subjects with NAFLD compared to healthy controls.
Table 1.
Plasma levels of circulating BAs that are significantly different in subject with versus without NAFLD
| Control (n=50) Median (μM) (25th,75th percentiles) |
NAFLD (n=102) Median (μM) (25th,75th percentiles) |
P value | P-FDR value | |
|---|---|---|---|---|
| Total BA | 1927.0(1038.4–3290.5) | 5426.4(3247.5–8169.1) | <0.001 | <0.001 |
| Total 1° BA | 871.5(523.9–1702.2) | 3074.6(1830.2–5517.9) | <0.001 | <0.001 |
| Total 2° BA | 1042.7(494.1–1609.4) | 1684.0(1092.1–2562.4) | <0.001 | <0.001 |
| Glycine 1° BA | 474.6(267.1–947.2) | 1994.6(1002.0–3683.8) | <0.001 | <0.001 |
| Glycine 2° BA | 245.9(127.2–450.4) | 742.5(391.3–1217.0) | <0.001 | <0.001 |
| Primary bile acids | ||||
| CDCA | 68.0 (22.5–366.3) | 227.8 (71.7–565.3) | 0.001 | 0.003 |
| GCDCA | 383.4 (193.9–762.2) | 1442.1 (713.4–2765.6) | <0.001 | <0.001 |
| TCDCA | 44.8 (20.6–86.6) | 211.5 (114.0–417.1) | <0.001 | <0.001 |
| GCA | 105.1 (52.0–207.9) | 522.0 (202.3–1009.3) | <0.001 | <0.001 |
| TCA | 12.8 (6.2–25.8) | 70.2 (28.6–155.1) | <0.001 | <0.001 |
| GHCA | 6.8 (2.5–13.2) | 12.9 (6.2–30.9) | <0.001 | <0.001 |
| Secondary bile acids | ||||
| GDCA | 136.7 (61.3–239.5) | 409.7 (158.7–933.7) | <0.001 | <0.001 |
| TDCA | 19.9 (8.5–40.0) | 63.4 (23.6–156.5) | <0.001 | <0.001 |
| LCA | 27.7 (19.1–34.2) | 18.2 (3.9–36.1) | 0.018 | 0.032 |
| LCA3-Sulfate | 8.3(3.6–14.0) | 5.0 (1.0–14.2) | 0.048 | 0.079 |
| GUDCA | 61.0 (33.3–119.9) | 197.1 (89.1–341.5) | <0.001 | <0.001 |
| HDCA | 2.5 (2.5–2.5) | 28.7 (8.0–102.6) | <0.001 | <0.001 |
| 7-Keto-DCA | 11.7 (9.5–20.7) | 3.5 (3.5–10.8) | <0.001 | <0.001 |
| 3-Keto-DCA | 1.5 (1.5–5.5) | 14.1 (5.5–31.1) | <0.001 | <0.001 |
| Iso-DCA | 2.5 (2.5–7.5) | 8.5 (2.5–8.5) | <0.001 | <0.001 |
Comparison of different bile acid (BA) pools and individual BAs significantly different in healthy individuals (control n=50) vs individuals with NAFLD (n=102). For complete list of all BAs in comparison between plasma levels from healthy controls vs NAFLD, see also Supplementary Tables 3 and 4.
Concentrations (μM) are presented as median (25th,75th percentiles). P values in bold denote significance (P <0.05), and were calculated by the Mann-Whitney test. P-FDR, false discovery rate adjusted P values.
For definitions of BA pools see Materials and Methods.
To aid in tracking the overall results, Table 2 summarizes the major findings of our studies, indicating the BAs whose circulating levels showed significant associations with more advanced stages of one or more histopathological features of NAFLD/NASH, as discussed in the detailed analyses that follow.
Table 2.
Summary of structurally specific BAs showing significant associations with NAFLD histology phenotypes
| Fibrosis | NASH | Inflammation | Ballooning |
|---|---|---|---|
| GCA | 7-Keto-DCA | GCA | 7-Keto-DCA |
| GCDCA | 7-Keto-LCA | TCA | Iso-DCA |
| 7-Keto-DCA | GCDCA | 7-Keto-LCA | |
| GUDCA | TCDCA | Iso-LCA |
A panel of structurally specific BAs were quantified in the plasma of serial consenting subjects undergoing elective diagnostic liver biopsy for suspected NAFLD / NASH as described under Methods. Shown are those BAs whose circulating levels showed significant associations with one or more histopathological features of NAFLD/NASH. NASH: Non-Alcoholic Steatohepatitis; GCA: glycocholic acid; GCDCA: glycochenodeoxycholic acid; 7-Keto-DCA: 7-Ketodeoxycholic acid; GUDCA: glycoursodeoxycholic acid; 7-Keto-LCA: 7-Ketolithocholic acid; TCA: taurocholic acid; TCDCA: taurochenodeoxycholic acid; Iso-DCA: isodeoxycholic acid; Iso-LCA: isolithocholic acid
3.2. Hepatic fibrosis is associated with larger circulating BA pool size, especially glycine conjugated primary Bas
To gain further insights into the relationship between BAs and NAFLD, we first evaluated BA patterns with severity of liver fibrosis amongst patients with NAFLD. Individuals with liver fibrosis (F1–F4) had higher levels of total plasma BA pool (p=0.03 vs no-fibrosis (F0); Supplementary Table 5). Given that we analyzed several dozen structurally specific individual BAs, for all analyses we also performed statistical adjustments for multiple testing (Methods), and observed a strong trend for higher total BA levels with higher indices of fibrosis (false discovery rate (FDR) p=0.08, Supplementary Table 5). In addition, the plasma concentration of total BAs showed a dose-dependent association with worsening stages of fibrosis (p=0.03 (KW)), which again showed strong trend but failed to meet significance after adjustment for multiple testing (FDR p=0.07 (KW), Fig. 1A and Supplementary Fig. 1). A significant proportion of the elevated plasma levels of total BAs noted with advancing stages of hepatic fibrosis was due to higher levels of primary BAs (p=0.003 and FDR adjusted p=0.02 for comparison of patients with versus without fibrosis; Supplementary Table 5). In contrast, secondary BAs showed no difference between the groups (p=0.48 fibrosis vs no-fibrosis; Supplementary Table 5). The increase in total primary BAs with worsening fibrosis was primarily due to glycine conjugated primary BAs (glyco-BAs) (p=0.005 (KW), Supplementary Fig. 1). In particular, glycochenodeoxycholic acid (GCDCA), and glycocholic acid (GCA) showed strong association with advancing fibrosis grade (p=0.003 (KW)/p=0.003 (for trend) and p=0.04 (KW)/p=0.001 (for trend), respectively, Fig. 1B). Also of interest, two specific secondary BAs, 7-Keto-deoxycholic acid (7-Keto-DCA; p=0.006 (KW) and p=0.0001 (for trend)) and glycoursodeoxycholic acid (GUDCA; p=0.02 (KW) and p=0.004 (for trend)), derived from cholic acid (CA) and chenodeoxycholic acid (CDCA), respectively, were significantly associated with advancing fibrosis (Fig. 1B). For each of the above indicated BAs, dose dependent associations with fibrosis level remained significant following adjustment for multiple testing (FDR adjusted p for trends of p=0.02 for GCDCA, p=0.01 for GCA, p=0.0001 for 7-Keto-DCA and p=0.02 for GUDCA). In summary, advancing histological grade of hepatic fibrosis was characterized by higher circulating levels of two glycine conjugated primary BAs (GCDCA and GCA) and two secondary BAs (7-Keto-DCA and GUDCA.
Fig. 1. Glycine conjugated primary BAs are predominantly associated with the progress of hepatic fibrosis.
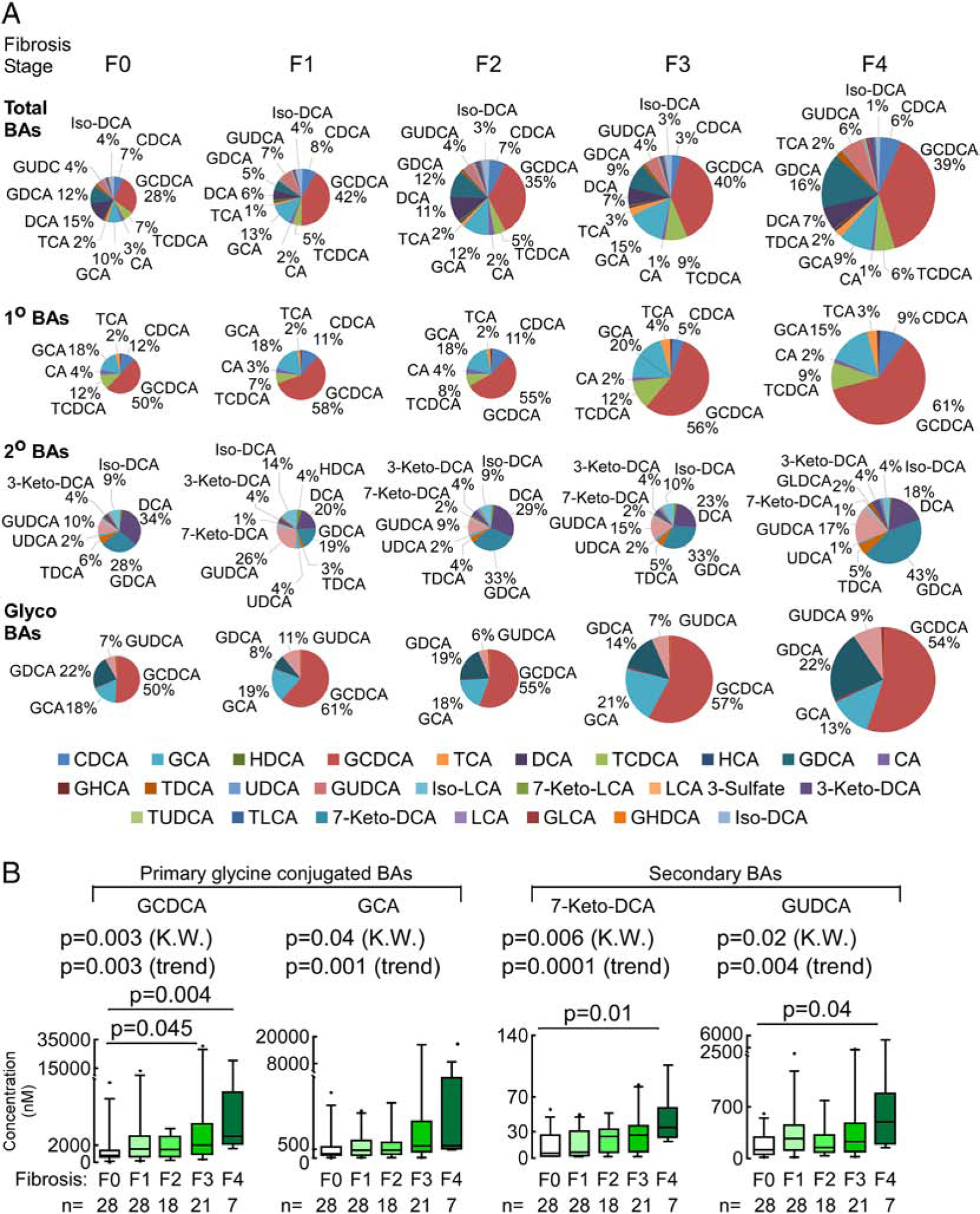
(A) Changes in total plasma BAs, primary (1°), secondary (2°) and glycine conjugated BAs through the progression of fibrosis. Pie chart plot representing the proportion of each bile acid in the plasma BA pool (expressed as a percentage of the total plasma BA pool) through the progression of fibrosis. Colors reflect the BAs patterns and pie sizes reflect changes in the sum of plasma BAs pool. Percentage values are for the most abundant or significant BAs in the circulating pool. (B) GCDCA, GCA, 7-Keto-DCA and GUDCA plasma levels increased significantly along with the increase in fibrosis severity. Box-whisker plots for individual plasma BAs showing significant differences between no fibrosis and fibrosis subjects, lower and upper lines of the box indicate the 25th and 75th percentiles, line in the middle indicates the median, upper and lower whiskers indicate 5th and 95th percentiles. P-values were calculated by the Kruskal-Wallis test (K.W.) with post hoc analysis using Dunn’s test and by test for trend for increasing BAs within each group.
3.3. Plasma level of 7-Keto secondary BAs are associated with NASH versus simple steatosis
We then evaluated BA distribution amongst NAFLD patients with simple steatosis, borderline, and definite NASH. Plasma BA pools in NASH vs no-NASH are summarized in Supplementary Table 5 (note – all “no-NASH” subjects still have NAFLD; i.e. do not include the healthy controls). Levels of total plasma BAs, primary BAs, or glycine conjugated primary BAs did not differ significantly in individuals with NASH vs no-NASH (p=0.93, p=0.80 and p=0.76, respectively, Supplementary Table 5). However, when compared to individuals with simple steatosis (NAS=1–2), patients with borderline NASH (NAS=3–4) had higher levels of total BAs, total primary BAs, and primary glycine conjugated BAs. There was no difference in levels of total BAs, total primary BAs, or primary glycine conjugated BAs between individuals with simple steatosis and those with definite NASH (NAS=5–8) (Fig. 2A and Supplementary Fig. 2) Among individual BA molecular species, two secondary BAs, 7-Keto-DCA (p<0.001) and 7-Keto-lithocholic acid (7-Keto-LCA; p=0.01), were higher in patients with definite NASH, with elevated 7-Keto-DCA levels remaining significant following multiple testing (FDR adjusted p=0.002, Supplementary Table 6). Both of these keto BAs are formed by one-step microbial transformation involving 7-dehydrogenation of the sterol ring of their corresponding precursor primary BAs, CA and CDCA, respectively. Plasma levels of both 7-Keto-DCA and 7-Keto-LCA were also associated with increasing NAS score (p<0.0001 and p=0.04 (KW), respectively, Fig. 2B), with 7-Keto-DCA remaining significant following multiple testing adjustments (FDR adjusted p<0.001 (KW)). In summary, subjects with NASH compared to early stage NAFLD (simple steatosis) are characterized by increased levels of two specific keto-BAs, 7-Keto-DCA and 7-Keto-LCA.
Fig. 2. Significant changes in plasma BA profile are associated with the severity of fibrosis in NAFLD.
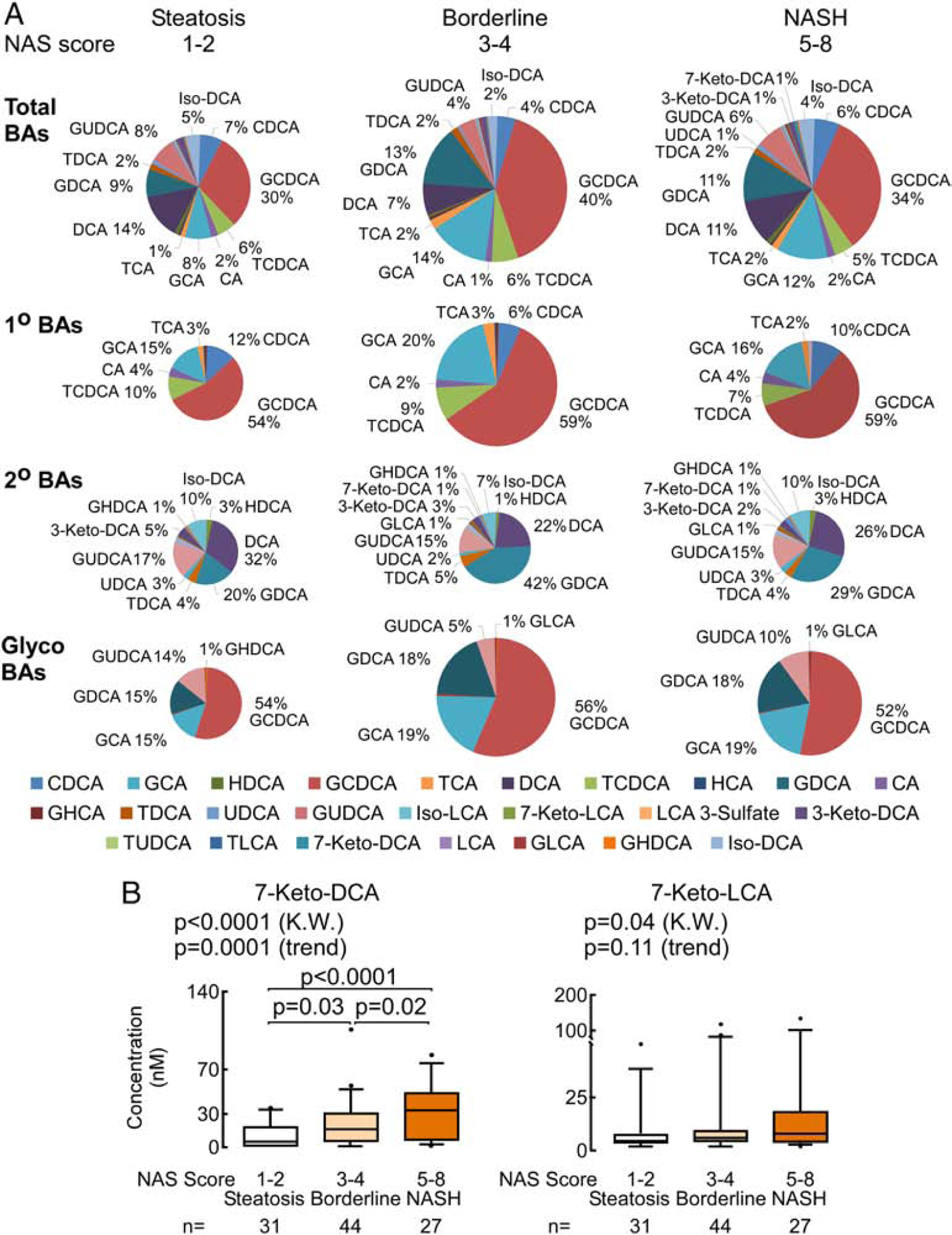
(A) Distinct changes in total plasma BAs through the progression from steatosis to NASH. Pie charts representing the proportion of each BA in the plasma BA pool (expressed as a percentage of the total plasma BA pool). The pie size is the measure of plasma BAs pool concentration. Percentage values are for the most abundant or significant BAs in the circulating pool. (B) Plasma levels of 7-Keto-DCA and 7-Keto-LCA are significantly increased in NASH compared to steatosis and both are both associated with increasing NAS. P values are calculated by the Kruskal-Wallis test (K.W.) with post hoc analysis using Dunn’s test and by test for trend for increasing BAs within each group.
3.4. Association of structurally-defined plasma BAs with inflammation, ballooning, hepatic steatosis, and metabolic phenotypes/diseases
We next examined the association of total plasma BA pools and individual BAs with specific histological components of NAFLD, namely, steatosis, lobular inflammation, and hepatocellular ballooning. Inflammation, similar to fibrosis, was associated with increased total BAs (p=0.004 and p(FDR)=0.02) (Supplementary Table 7). Further, plasma levels of primary BAs, but not secondary BAs, were significantly associated with advancing grades of inflammation (Fig. 3, Supplementary Table 7). Among the primary BAs, elevated total glycine and taurine conjugated (tauro) primary BAs were significantly associated with lobular inflammation (p(FDR)=0.02 and p(FDR)=0.02, respectively; Supplementary Table 7). Concentrations of total BAs, total primary BAs, and total primary glyco- and tauro- BAs were also higher with greater inflammation grade (all p(FDR)=0.049, KW) (Fig. 3A). Further examination of the individual BAs showed higher plasma levels of GCA (p=0.01), taurocholic acid (TCA; p=0.006), GCDCA (p=0.007), and taurochenodeoxycholic acid (TCDCA; p=0.02) were observed in patients with (versus without) histological evidence of inflammation, and with increasing inflammation grade (Fig. 3B, Supplementary Table 8).
Fig. 3. Significant changes in plasma total BA profile with the severity of inflammation in NAFLD.
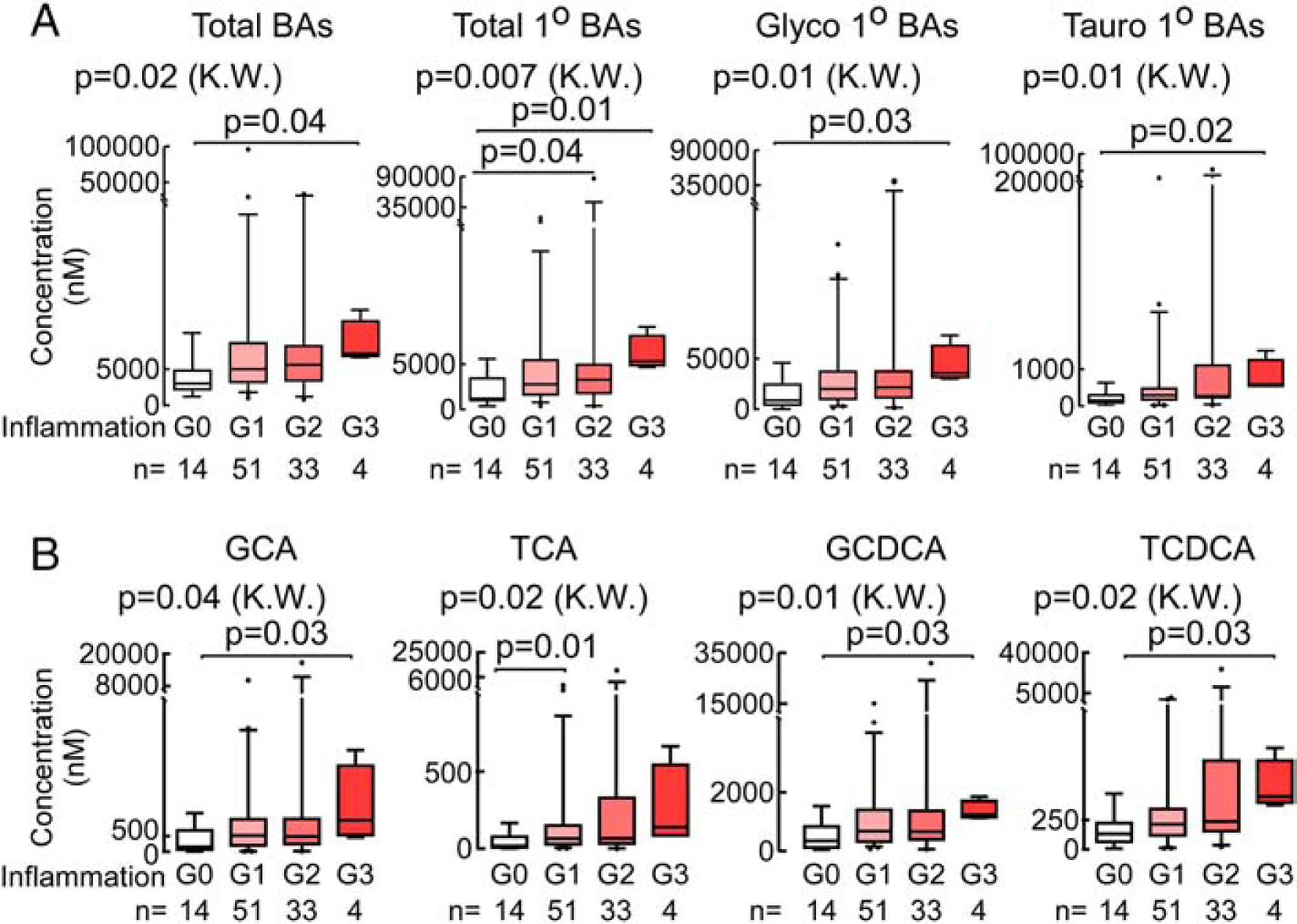
(A) Significantly increased plasma total BA, total primary BAs, glycine conjugated primary BAs (GCA+GCDCA) and taurine primary BAs (TCA+TCDCA) with higher grades of hepatic inflammation. (B) Significant increase in individual BAs (GCA, TCA, GCDCA and TCDCA) with severity of inflammation. Data presented as box-whiskers plots with the median. P values are calculated by the Kruskal-Wallis test (K.W.) with post hoc analysis using Dunn’s test.
Hepatocellular ballooning was not associated with statistically significant differences amongst BAs combinations (total, primary, secondary, glycine conjugated, taurine conjugated; Supplementary Table 7), but was associated with elevated plasma levels of 7-Keto-DCA (p<0.0001), 7-Keto-LCA (p=0.006), and their corresponding 6-OH isomers isodeoxycholic acid (Iso-DCA; p=0.03) and isolithocholic acid (Iso-LCA; p=0.03), respectively (Fig. 4A), with 7-Keto-DCA remaining significantly associated with ballooning even following adjustment for multiple testing (p(FDR)=0.0003). A stepwise increase in plasma concentration of 7-Keto-DCA and 7-Keto-LCA were each associated with advancing hepatocellular ballooning grade (Fig. 4B). We further stratified NAFLD patients into mild hepatic steatosis (G1) vs moderate-severe steatosis (G2,3) to assess changes in BA levels in relation to severity of hepatic steatosis. Patients with moderate-severe steatosis, when compared to mild steatosis alone, had no significant difference in any of the circulating BA pools (Supplementary Table 7), but had significantly higher plasma levels of secondary 7-Keto-LCA (p=0.03; Fig. 4C). The plasma level of 7-Keto-LCA was associated with greater histologic grade of hepatic steatosis (p=0.03 (KW); Fig. 4D), but this association failed to remain significant after multiple testing adjustments.
Fig. 4. Plasma BA association with hepatic ballooning and steatosis in NAFLD.
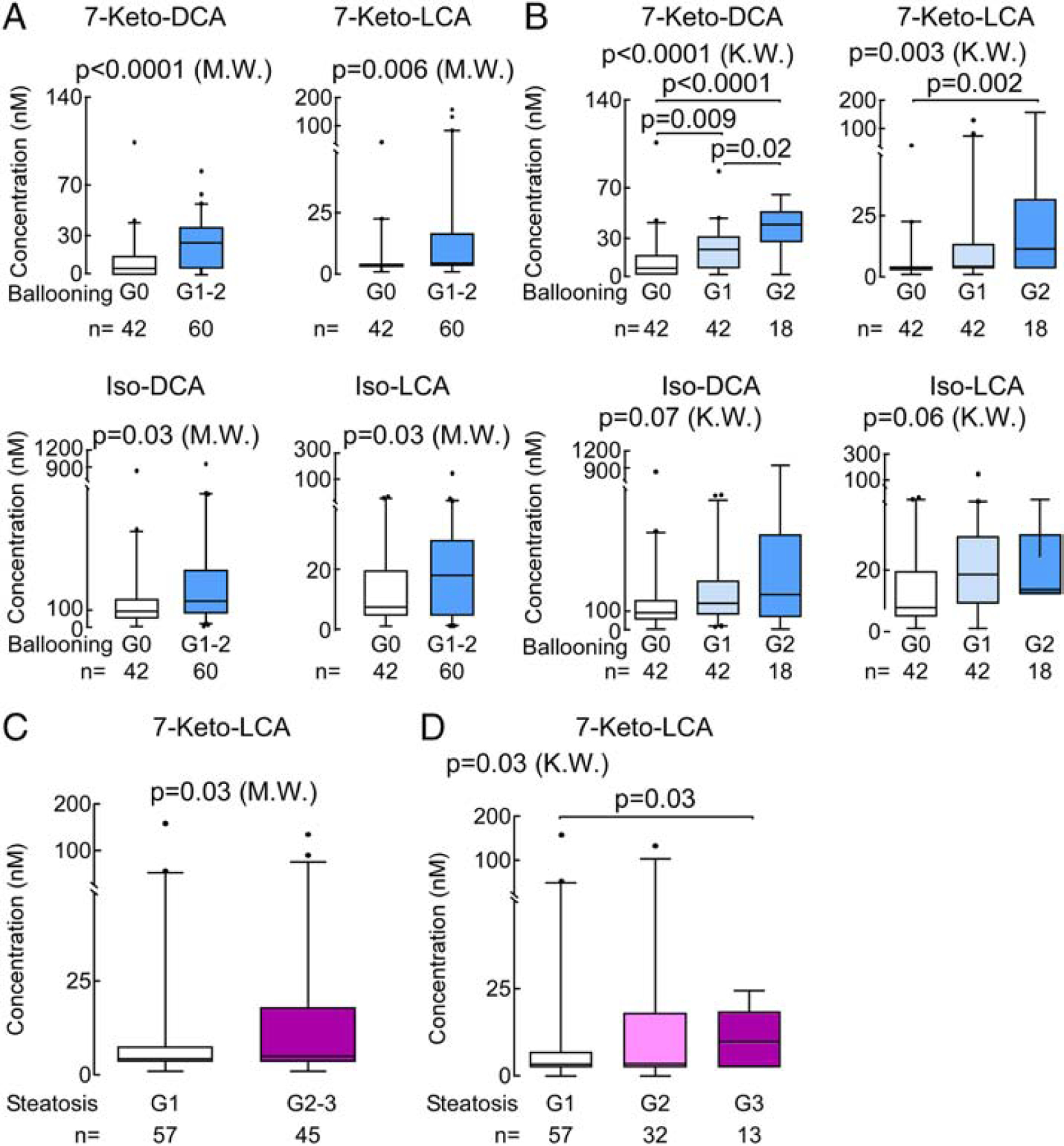
(A) Plasma levels of 7-Ketodeoxycholic acid (7-Keto-DCA), 7-Ketolithocholic acid (7-Keto-LCA), iso-deoxycholic acid (Iso-DCA) and isolithocholic acid (Iso-LCA) are significantly increased in ballooning. (B) Plasma 7-Keto-DCA, and 7-LCA are significantly associated with advanced ballooning grades. (C) Significantly increased plasma 7-Keto-LCA with advanced steatosis (G2,3) compared to simple steatosis (G1) in NAFLD and (D) its concentration was significantly associated with advanced grades. Data presented as box-whiskers plots with the median. P values are calculated by the Mann-Whitney test (M.W.) for two group comparions and Kruskal-Wallis test (K.W.) with post hoc analysis using Dunn’s test for muti group comparisons.
Thus far, our observations show an association between specific BA patterns and progression of NAFLD. Elevated levels of BAs were also reported in individuals with type 2 diabetes, as well as individuals with insulin resistance [30]. Insulin resistance is a common abnormality in NAFLD [31], and it is considered to be one of the multiple hits that contributes to progression from simple steatosis to NASH [7, 16, 32]. In the current NAFLD cohort, we observed significant correlations between fasting plasma insulin levels and total primary glycine and taurine BAs (Supplementary Table 9), as well as individual BAs: 7-Keto-DCA, TCA, TDCA, GDCA and GLCA (Fig. 5). Total conjugated BAs, 7-Keto-DCA, GDCA, and GLCA were also significantly associated with HOMA-IR (homeostatic model assessment of insulin resistance; Fig. 5; Supplementary Table 9). Components of metabolic syndrome that are significantly associated with NAFLD and risk factors were also significantly correlated with BA concentrations (Fig. 5 and Supplementary Table 9). In summary, amonst subjects with NAFLD, heightened hepatic inflammation was characterized with higher circulating levels of glycine and taurine conjugated primary BAs (GCA, TCA, GCDA and TCDCA), histopathological evidence of ballooning with elevated 7-Keto-DCA, 7-Keto-LCA and their corresponding isomers (Iso-DCA and Iso-LCA), while enhanced degrees of hepatic steatosis was characterized by higher plasma levels of 7-Keto-LCA.
Fig. 5. Relationship between individual plasma BAs levels and fibrosis risk.

Heat map showing Spearman correlation between plasma levels of individual BAs and age, body mass index (BMI), alanine aminotransferase (ALT), aspartate transaminase (AST), fasting glucose, insulin, hemoglobin A1c (HbA1c), homeostatic model assessment for insulin resistance (HOMA-IR), cholesterol (Chol), high-density lipoprotein cholesterol (HDL-C), low-density lipoprotein cholesterol (LDL-C) and triglycerides (TG). The correlation strength is shown by the color bar, with blue/red representing positive/negative association, respectively, and white no association. *P<0.05, **P<0.01. Rho values of Spearman’s rank correlation coefficient.
3.5. Overall summary of relationship between elevated plasma levels of individual molecular species of BAs and histopathological features of NASH
To better visualize the associations of fasting plasma levels of individual and combined plasma pools of BAs with severity of histopathological features of NAFLD, logistic regression models were constructed to determine the odds and 95% confidence intervals of elevated levels (4th vs 1st quartile) of each BA (Fig. 6) and circulating BA pools (Supplementary Fig. 4) versus the presence of fibrosis, definitive diagnosis of NASH (NAS≥5), or the presence of inflammation, ballooning or steatosis. In addition, models were adjusted for comorbidities and diagnostic laboratory analyses associated with NASH including age, gender, body mass index (BMI), ALT (alanine aminotransferase), and HOMA-IR. Fibrosis was associated with higher total BAs, total primary BAs, specifically glycine-primary BAs (Supplementary Fig. 4), and among the individual BAs, GDCA, GCA, 7-Keto-DCA and GUDCA. While the total level of primary conjugated, primary-glycine-BAs, GDCA and 7-Keto-DCA were significantly associated with severity of fibrosis, after adjustment, total BAs, GCA, and GUDCA were no longer significantly associated with severity of fibrosis (Fig. 6). Simple steatosis versus NASH was significantly associated with elevated levels of 7-Keto-derived secondary BAs, 7-Keto-DCA and 7-Keto-LCA both before and following comorbidity and laboratory studies adjustments (Fig. 6 and Supplementary Fig. 4). The patterns observed between BA molecular species and heightened levels of lobular inflammation were on the whole similar to those observed for fibrosis (Figs. 6, and S4). After adjusting for age, gender, BMI, ALT, and HOMA-IR, only total conjugated BAs, total primary BAs, and conjugated CA (GCA and TCA) stayed significantly associated with lobular inflammation (Fig. 6 and Supplementary Fig. 4). The patterns observed between higher levels of specific BAs and likelihood of NASH, and advanced ballooning, followed similar patterns (Figs 6 and Supplementary Fig. 4). Among the BAs associated with hepatocellular ballooning (7-Keto-DCA, 7-Keto-LCA, Iso-LCA and Iso-DCA), 7-Keto-BAs were significantly associated with the presence and severity of hepatocyte ballooning after adjustments, while neither the plasma BA pools nor individual BAs were significantly associated with steatosis after adjustments (Fig. 6 and Supplementary Fig. 4). Collectively, our results show all conjugated primary BAs (GCA, TCA, GCDCA and TCDCA) were associated with hepatic inflammation, while glycine conjugate primary BAs (GCA and GCDCA) also showed association with fibrosis. In addition, Iso-DCA and Iso-LCA were associated with ballooning and GUDCA with fibrosis. Most notably, plasma levels of 7-Keto-BAs were dose dependently associated with NASH and ballooning, with 7-Keto-DCA levels also showing association with increasing stages of hepatic fibrosis.
Fig. 6. Association of structurally defined plasma BAs histopathology components of NAFLD.
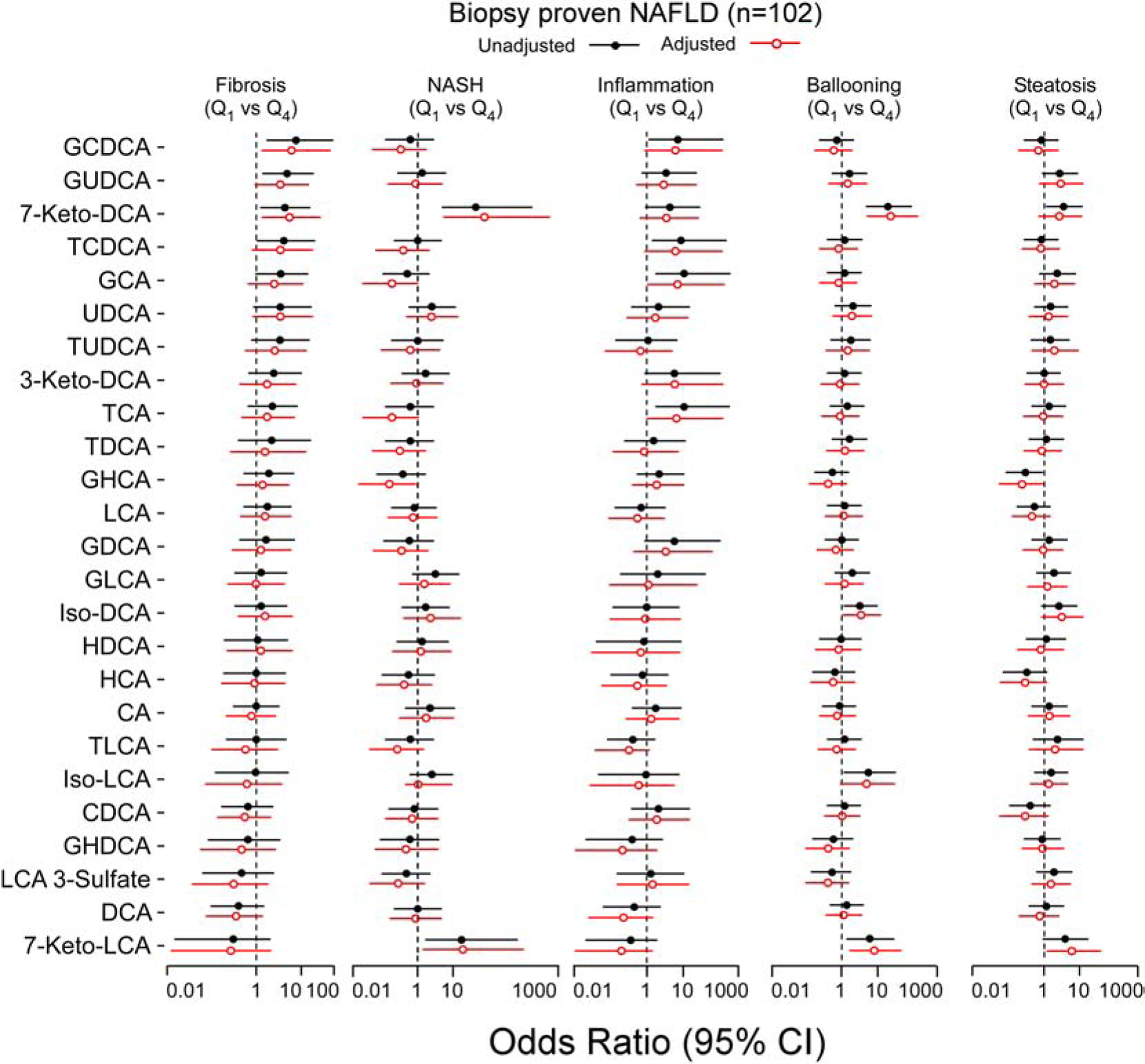
Forest plots of odds ratios (OR) of 4th quartile (Q4) versus 1st quartile (Q1) for fibrosis, NASH, inflammation, ballooning and steatosis phenotypes. Bars represent 95% confidence interval (CI). Black lines with closed/open circle represent unadjusted/adjusted OR, respectively. Adjustments are done by multivariable logistic regression model (adjusted for age, gender, body mass index (BMI), homeostatic model assessment for insulin resistance (HOMA-IR) and alanine aminotransferase (ALT).
3.6. Association of NAFLD and NASH genetic variants with fasting plasma BA levels.
We next examined whether previously reported genetic determinants of NAFLD, NASH, or related traits, such as liver enzymes levels or alcoholic fatty liver disease, were associated with plasma BA levels, as described under Methods. Since genotype data were not available in the 102 participants with biopsy-proven NAFLD and power to detect associations would be very limited due to the small sample size, we used the GeneBank cohort (n=1,840) for genetic analyses. These analyses also focused on the eight BAs shown to be associated with fibrosis, inflammation, ballooning, and/or NASH (Fig. 7), and tested 12 variants previously identified through genome wide association studies (Supplementary Table 10). Of the 12 regions tested, variants at five loci, including PPP1R3B, TRIB1, and LYPLAL1, yielded nominal associations (P<0.05) with 7-Keto-DCA, GCA, GUDCA, and Iso-DCA. Interestingly, both GCA and GUDCA were higher in carriers of the risk allele rs2954021 (A) at the TRIB1 locus (Table 3). Except for variants at the PPP1R3B and PNPLA3 loci, NAFLD/NASH risk alleles were associated with increased BA levels, consistent with the positive clinical association of these BAs with histological features and severity of NAFLD and NASH.
Fig. 7. The relationships between metabolic pathways of specific BAs and NAFLD histology phenotypes.
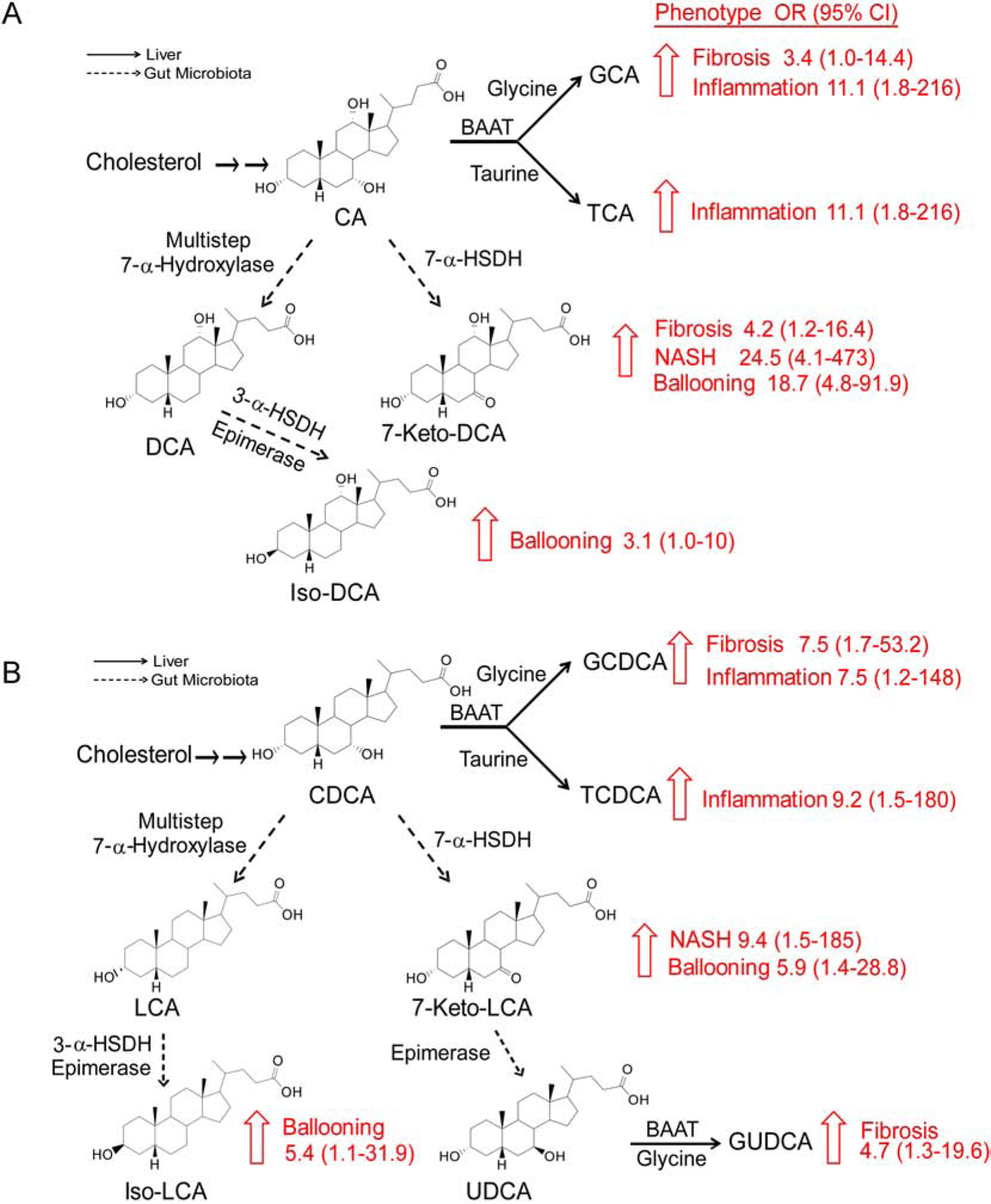
Upward arrows show significant changes in the progression of NAFLD phenotypes. (A) BAs originating from cholic acid (CA). (B) BAs originating from chenodeoxycholic acid (CDCA). Dashed arrows represent gut microbiota pathways involved in the synthesis of secondary BAs, and the solid arrows represent pathways in the host (liver). Gut microbiota enzymes: 7-α-HSDH, 7α-hydroxysteroid dehydrogenase; 3-α-HSDH, 3-α-hydroxysteroid dehydrogenase; liver enzyme BAAT, bile acid-CoA: amino acid N-acyltransferase.
Table 3.
Association of Genetic Determinants for NAFLD and NASH with Plasma BA Levels.
| Bile Acid | SNP | Locus | a EA/OA | b EAF | c Beta (SE) | d P-value |
|---|---|---|---|---|---|---|
| rs4240624 | PPP1R3B | A/G | 0.91 | −0.150 (0.048) | 0.002 | |
| 7-Keto-DCA | rs11597086 | ERLIN1-CHUK-CWF19L1 | A/C | 0.58 | 0.060 (0.028) | 0.03 |
| rs738409 | PNPLA3 | G/C | 0.22 | −0.084 (0.033) | 0.01 | |
| GCA | rs2954021 | TRIB1 | A/G | 0.52 | 0.087 (0.037) | 0.02 |
| GUDCA | rs12137855 | LYPLAL1 | C/T | 0.81 | 0.083 (0.041) | 0.04 |
| rs2954021 | TRIB1 | A/G | 0.52 | 0.105 (0.053) | 0.009 | |
| Iso-DCA | rs4240624 | PPP1R3B | A/G | 0.91 | 0.086 (0.033) | 0.049 |
Shown are the structurally specific individual BAs quantified whose plasma levels were found to be significantly associated with validated genetic variants of NAFLD or NASH.
EA/OA, effect allele/other allele; EA refers to allele previously reported to increase risk of NAFLD and/or NASH or was associated with increased liver enzyme levels.
EAF = effect allele frequency.
Betas refer to effect of EA on normal inverse transformed plasma values of the indicated BA.
P-values were obtained by linear regression analyses using normal inverse transformed values with adjustment for age and sex.
4. DISCUSSION
Despite the growing interest in metabolomics as a discovery tool in studies focused on NAFLD and NASH, studies employed thus far, for the most part, have not utilized quantitative stable isotope dilution LC-MS/MS approaches to examine the association between plasma levels of distinct molecular species of BAs and the clinically relevant transition between simple steatosis, and various histopathological indices related to NASH. In addition, the limited studies present thus far have typically used semi-quantitative approaches, or predominantly focused on comparisons of healthy controls versus NAFLD or NASH [9, 17, 18, 33, 34]. Finally, the association of BAs with previously identified genetic variants associated with risk for NASH remains unknown. In the present study, we used a robust and validated stable isotope dilution LC/MS/MS method to investigate the association between plasma levels of BAs and various histological components of NAFLD/NASH (fibrosis, inflammation, ballooning and steatosis) in a large, well-characterized cohort of patients with biopsy-proven NAFLD. Our major findings are summarized in Fig. 7. Plasma levels of the secondary BAs 7-Keto-DCA and 7-Keto-LCA were dose dependently related to ballooning and NASH, while the levels of conjugated primary BAs were related to inflammation and fibrosis. Others have reported increased TCA and GCA in NAFLD patients with fibrosis stages F0–F1 vs F≥2 [34], and elevated total BAs across the F0–F4 [35]. Our observations are consistent with these reports, yet substantially extend these data to both identify specific molecular species whose levels are associated with distinct histopathological features of NASH (Figure 7). Moreover, our studies show that the total plasma BA pool is related to the fibrosis stages. By comparison, a large Chinese study did not observe an association of total BAs, as measured by an enzymatic assay, with ultrasound-based diagnoses of NAFLD [36]. However, differences in the ethnicities of patients (European vs. Asian), our LC/MS/MS method to quantify BAs, and most importantly, our reliance on hepatic biopsy proven NAFLD and NASH likely account for the observed differences. Importantly, our use of liver biopsy proven NAFLD and NASH classification enabled discovery of BA molecular species whose circulating levels are dose dependently related to histological features of NAFLD/NASH. We also observed that of the glycine conjugated primary BAs (GCDCA and GCA), GCDCA remained significantly associated with fibrosis after adjusting for the predefined multiple risk factors for NAFLD. Interestingly, the increase in individual tauro- primary BAs were associated with severity of lobular inflammation but not with the stage of fibrosis. The cause for the association between elevated plasma levels of the observed BAs amongst subjects at risk for progression of NAFLD, especially with heightened stage of inflammation and severe fibrosis stages, is unknown.
In contrast to a previous report of an inverse correlation between total free secondary BAs and fibrosis score in NAFLD patients [35], by examining individual molecular species of BAs in detailed fashion, we observed patterns of specific BA metabolic pathways associated with distinct aspects of NAFLD of interestaore advanced fibrotic disease, suggesting possible therapeutic targets for future investigation. For example, an increase in 7-Keto-DCA with more severe fibrosis was observed. 7-Keto-DCA, synthetized by microbial 7α-hydroxysteroid dehydrogenase (7α-HSDH) from CA, was associated with worsening severity of NAFLD. 7-Keto-LCA, BAs generated by the same enzyme as the primary BA CDCA, was also associated with progression of NASH, hepatocellular ballooning, and steatosis. These observations suggest that a common biochemical signature of NAFLD progression is increased circulating levels of 7-Keto-BA. It is possible that increased circulating 7-Keto-BA levels amongst those with more advanced NAFLD may be due to heightened microbial production of this BA, either due to an increase in substrate (CA) availability to the microbes as subjects with NAFLD have been reported to have increased CA and CDCA in feces, and/or a shift in microbial community structure (dysbiosis), which also has been reported in subjects with NAFLD or NASH [37]. In addition to oxidation on position 7 to generate 7-Keto-BA, BAs can lose the same hydroxyl group by gut microbiota via a multi-step reaction to generate DCA and LCA from CA and CDCA, respectively. Both DCA and LCA can further undergo isomerization of the hydroxyl group at position 6 and form Iso-DCA and Iso-LCA, respectively. Consistent with this interpretation, both Iso-BAs in our cohort were related to higher hepatocyte ballooning grade (Fig. 7). Moreover, elevated plasma levels of GUDCA reflect the intestinal conversion of GCDCA excess to their respective hydrophilic UDCA conjugate [33] in more severe forms of NAFLD to moderate BA mediated hepatotoxicity.
It is notable that recently, obeticholic acid (6α-ethyl-chenodeoxycholic acid), a semi-synthetic primary bile acid analogue, is being evaluated by the FDA for NASH [38, 39]. Obeticholic acid is a highly selective agonist of liver nuclear BA receptor; farnesoid X receptor (FXR). It has been found that activation of FXR by BAs will decrease hepatic lipid synthesis and plasma triglycerides, and will also promote insulin sensitivity by decreasing gluconeogenesis [40, 41]. In addition, FXR regulates other pathways with direct anti-inflammatory and anti-fibrotic effects that make obeticholic acid effective in reversing NASH and reducing fibrosis [19]. The numerous BA profile changes observed in the present studies further raise the question of whether associations observed are a consequence of the disease, versus a contributor to the disease – and the reported clinical beneficial effects of obeticholic acid therapy on NASH progression argues for a possible causal contribution.
Since clinical association studies cannot directly address cause vs effect, we sought to determine if genetic variants previously reported to increase susceptibility to NAFLD and NASH were associated with any of the candidate molecular species of BAs whose circulating levels are associated with histopathological features of NASH. Evaluation of genetic susceptibility factors for NAFLD/NASH revealed multiple directionally consistent associations with increased plasma levels of several of the eight clinically relevant BAs (Table 3). In particular, 7-Keto-DCA, which showed multiple significant associations including hepatic fibrosis, NASH and ballooning (Fig. 7), was nominally associated with variants in the patatin-like phospholipase domain-containing 3 (PNPLA3)I148M gene, which has been linked to inter-individual differences in liver steatosis and susceptibility to progressive NASH [14, 27–29]. Functionally, PNPLA3 is involved in multiple lipid metabolic processes, including lipid droplet remodeling, VLDL secretion, triglyceride and retinyl-palmitate esterase activity in hepatic stellate cells, and also has been linked to hepatic fibrosis [42]. Of note, prior studies have also demonstrated that perturbation of PNPLA3 expression in hepatoma cells alters levels of another secondary BA, taurolithocholate [43]. Taken together with our present findings, these observations support the notion that PNPLA3 may play a potentially broader role in BA metabolism.
In further genetic analyses, additional validated genetic variants for NAFLD/NASH were found to be associated with differences in levels of other specific BA molecular species. For example, elevated 7-Keto-DCA levels were also associated with genetic variants at the ERLIN1-CHUK-CWF19L1 locus, which has previously been identified as influencing both simple steatosis and hepatic steatosis with inflammation (ALT elevation) [28]. Another interesting observation is the associations observed between plasma GCA and GUDCA levels and both hepatic fibrosis and inflammation (Fig. 7). Increased plasma levels of both GCA and GUDCA are similarly associated with the NAFLD/NASH risk allele of rs2954021 (A) at the TRIB1 locus (Table 3). Experimental evidence suggests that TRIB1 may play a role in reducing hepatic triglyceride synthesis and secretion in humans, possibly through a direct interaction with hepatocyte nuclear factor 4-alpha (HNF4A) [44]. Among its many metabolic functions, HNF4A is essential for BA metabolism, since mice deficient for this nuclear receptor have markedly elevated levels of glycine conjugated BAs [45]. Taken together, the present and prior published observations suggest that one potential mechanism for association of genetic variation at TRIB1 specifically with plasma BAs conjugated with glycine may be mediated through interactions with HNF4A. However, we note that nearly all of the association signals observed, while intriguing, do not exceed a Bonferroni-corrected threshold for testing 12 variants (p=0.05/12=4.2×10−3) or an even more conservative approach that further takes into account the eight BA species that were evaluated. Therefore, replication of these genetic findings will be necessary before definitive conclusions can be drawn with respect to BA metabolism being one mechanism through which these variants are associated with NAFLD and NASH-related traits.
In conclusion, our results demonstrate numerous intriguing associations between changes in plasma BA profile and the severity of NAFLD. While genetic data partially support potential contributions of BAs to NAFLD progression, they will require independent replication in additional cohorts, as well as further mechanistic investigation. The detailed exploration of specific BA molecular species and NAFLD/NASH relevant phenopytes in the present studies lays the foundation for hypotheses that can be tested in future studies. Microbial transplantation studies will be necessary before a direct causal contribution of gut microbiota to a specific BA and NAFLD/NASH relevant phenotypes can be made. In particular, our studies point to 7-Keto-BAs as molecular species worthy of further investigation. Manipulating the exposure to 7-Keto-DCA or other BAs in different NAFLD models could clarify whether 7-Keto-DCA participates in the pathogenesis of the disease. Our data also lay the foundation for use of several circulating BAs as relatively minimally invasive (compared to liver biopsy) biomarkers of simple steatosis progression risk into NASH. Such approaches will require independent validation, as this was not our initial intent when designing the present studies. Instead, we aimed to gain potential insights into candidate BAs linked to adverse liver remodeling during the transformation of simple steatosis to NASH. Finally, our studies suggest that examination of a comprehensive BA profile may provide useful information in the future when evaluating potential NASH targeting therapeutics, since discovery of non-invasive, circulating biomarkers to monitor both candidacy for initiation of therapy and therapeutic efficacy of agents are needed.
Supplementary Material
HIGHLIGHTS.
Plasma bile acid (BA) alterations are observed through the entire spectrum of NAFLD
Glycine conjugated primary BAs are associated with liver fibrosis
7-Keto-BAs are a common biochemical signature of NAFLD progression
Iso-deoxycholic acid and Iso-lithocholic acid are associated with ballooning
Financial support:
This work is supported by grants from the NIH and Office of Dietary Supplements (P01 HL147823, R01 HL103866, R01 HL126827, R01 DK120679, P50 AA024333, R01 HL133169, R01 HL148110, U01 DK061732, R01 GM119174, DP1 DK113598) and the Foundation Leducq (17CVD01). Mass spectrometry studies were performed on instrumentation supported in part through a Shimadzu Center of Excellence award. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
ABBREVIATIONS:
- 1°
primary
- 2°
secondary
- 7-αHSDH
7α-hydroxysteroid dehydrogenase
- 3-Keto-DCA
3-ketodeoxycholic acid
- 7-Keto-DCA
7-ketodeoxycholic acid
- 7-Keto-LCA
7-ketolithocholic acid
- ALT
alanine aminotransferase
- BA
bile acid
- CA
cholic acid
- CDCA
chenodeoxycholic acid
- DCA
deoxycholic acid
- FXR
farnesoid X receptor
- GCA
glycocholic acid
- GCDCA
glycochenodeoxycholic acid
- GDCA
glycodeoxycholic acid
- GHCA
glycohyocholic acid
- GHDCA
glycohyodeoxycholic acid
- GLCA
glycolithocholic acid
- glyco-BA
glycine-conjugated bile acid
- GUDCA
glycoursodeoxycholic acid
- HOMO-IR
homeostatic model assessment for insulin resistance
- HCA
hyocholic acid
- HDCA
hyodeoxycholic acid
- Iso-DCA
isodeoxycholic acid
- Iso-LCA
isolithocholic acid
- LCA
lithocholic acid
- LCA 3-sulfate
lithocholic acid 3-sulfate
- TCA
taurocholic acid
- TCDCA
taurochenodeoxycholic acid
- TDCA
taurodeoxycholic acid
- TGR5
Takeda G-protein coupled receptor 5
- TLCA
taurolithocholic acid
- TUDCA
tauroursodeoxycholic acid
- UDCA
ursodeoxycholic acid
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest:
Drs. Wang and Hazen report being named as co-inventor on pending and issued patents held by the Cleveland Clinic relating to cardiovascular diagnostics and therapeutics. Dr. Hazen also reports being a paid consultant for Procter & Gamble, having received research funds from Procter & Gamble, and Roche Diagnostics, and being eligible to receive royalty payments for inventions or discoveries related to cardiovascular diagnostics or therapeutics from Cleveland HeartLab and Procter & Gamble. The other authors have reported that they have no relationships relevant to the contents of this paper to disclose.
REFERENCES
- [1].Fazel Y, Koenig AB, Sayiner M, Goodman ZD, Younossi ZM. Epidemiology and natural history of non-alcoholic fatty liver disease. Metabolism. 2016;65:1017–25. [DOI] [PubMed] [Google Scholar]
- [2].Sayiner M, Koenig A, Henry L, Younossi ZM. Epidemiology of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis in the United States and the Rest of the World. Clin Liver Dis. 2016;20:205–14. [DOI] [PubMed] [Google Scholar]
- [3].Rinella ME. Nonalcoholic fatty liver disease: a systematic review. JAMA. 2015;313:2263–73. [DOI] [PubMed] [Google Scholar]
- [4].Younossi Z, Tacke F, Arrese M, Chander Sharma B, Mostafa I, Bugianesi E. Global Perspectives on Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Hepatology. 2019;69:2672–82. [DOI] [PubMed] [Google Scholar]
- [5].Younossi ZM, Stepanova M, Rafiq N, Makhlouf H, Younoszai Z, Agrawal R. Pathologic criteria for nonalcoholic steatohepatitis: interprotocol agreement and ability to predict liver-related mortality. Hepatology. 2011;53:1874–82. [DOI] [PubMed] [Google Scholar]
- [6].Neuman MG, French SW, French BA, Seitz HK, Cohen LB, Mueller S, et al. Alcoholic and non-alcoholic steatohepatitis. Exp Mol Pathol. 2014;97:492–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Buzzetti E, Pinzani M, Tsochatzis EA. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism. 2016;65:1038–48. [DOI] [PubMed] [Google Scholar]
- [8].Boursier J, Mueller O, Barret M, Machado M, Fizanne L, Araujo-Perez F, et al. The severity of NAFLD is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology. 2016;63:764–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Dasarathy S, Yang Y, McCullough AJ, Marczewski S, Bennet C, Kalhan SC. Elevated hepatic fatty acid oxidation, high plasma fibroblast growth factor 21, and fasting bile acids in nonalcoholic steatohepatitis. Eur J Gastroenterol Hepatol. 2011;23:382–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Brouwers M, Simons N, Stehouwer CDA, Koek GH, Schaper NC, Isaacs A. Relationship Between Nonalcoholic Fatty Liver Disease Susceptibility Genes and Coronary Artery Disease. Hepatol Commun. 2019;3:587–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Dongiovanni P, Valenti L. Genetics of nonalcoholic fatty liver disease. Metabolism. 2016;65:1026–37. [DOI] [PubMed] [Google Scholar]
- [12].Kahali B, Halligan B, Speliotes EK. Insights from Genome-Wide Association Analyses of Nonalcoholic Fatty Liver Disease. Semin Liver Dis. 2015;35:375–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Macaluso FS, Maida M, Petta S. Genetic background in nonalcoholic fatty liver disease: A comprehensive review. World J Gastroenterol. 2015;21:11088–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Anstee QM, Day CP. The genetics of nonalcoholic fatty liver disease: Spotlight on PNPLA3 and TM6SF2. Semin Liver Dis. 2015;35:270–90. [DOI] [PubMed] [Google Scholar]
- [15].Ma H, Patti ME. Bile acids, obesity, and the metabolic syndrome. Best Pract Res Clin Gastroenterol. 2014;28:573–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Manco M Insulin Resistance and NAFLD: A Dangerous Liaison beyond the Genetics. Children (Basel). 2017;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kalhan SC, Guo L, Edmison J, Dasarathy S, McCullough AJ, Hanson RW, et al. Plasma metabolomic profile in nonalcoholic fatty liver disease. Metabolism. 2011;60:404–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bechmann LP, Kocabayoglu P, Sowa J-P, Sydor S, Best J, Schlattjan M. Free fatty acids repress small heterodimer partner (SHP) activation and adiponectin counteracts bile acid-induced liver injury in superobese patients with nonalcoholic steatohepatitis. Hepatology. 2013;57:1394–406. [DOI] [PubMed] [Google Scholar]
- [19].Neuschwander-Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. 2015;385:956–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Younossi ZM, Ratziu V, Loomba R, Rinella M, Anstee QM, Goodman Z, et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet. 2019;394:2184–96. [DOI] [PubMed] [Google Scholar]
- [21].Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–21. [DOI] [PubMed] [Google Scholar]
- [22].Kuipers F, Bloks VW, Groen AK. Beyond intestinal soap--bile acids in metabolic control. Nat Rev Endocrinol. 2014;10:488–98. [DOI] [PubMed] [Google Scholar]
- [23].Mencarelli A, Renga B, Distrutti E, Fiorucci S. Antiatherosclerotic effect of farnesoid X receptor. Am J Physiol Heart Circ Physiol. 2009;296:H272–81. [DOI] [PubMed] [Google Scholar]
- [24].Pols TW, Nomura M, Harach T, Lo Sasso G, Oosterveer MH, Thomas C, et al. TGR5 activation inhibits atherosclerosis by reducing macrophage inflammation and lipid loading. Cell Metab. 2011;14:747–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Sayin SI, Wahlstrom A, Felin J, Jantti S, Marschall HU, Bamberg K, et al. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013;17:225–35. [DOI] [PubMed] [Google Scholar]
- [26].Thomas C, Gioiello A, Noriega L, Strehle A, Oury J, Rizzo G, et al. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009;10:167–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40:1461–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Dongiovanni P, Romeo S, Valenti L. Genetic factors in the pathogenesis of nonalcoholic fatty liver and steatohepatitis. Biomed Res Int. 2015;2015:460190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kozlitina J, Smagris E, Stender S, Nordestgaard BG, Zhou HH, Tybjaerg-Hansen A, et al. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2014;46:352–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Choucair I, Nemet I, Li L, Cole MA, Skye SM, Kirsop JD. Quantification of bile acids: A mass spectrometry platform for studying gut microbe connection to metabolic diseases. J Lipid Res. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Khan RS, Bril F, Cusi K, Newsome PN. Modulation of insulin resistance in nonalcoholic fatty liver disease. Hepatology. 2019;70:711–24. [DOI] [PubMed] [Google Scholar]
- [32].Finck BN. Targeting metabolism, insulin resistance, and diabetes to treat nonalcoholic steatohepatitis. Diabetes. 2018;67:2485–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ferslew BC, Xie G, Johnston CK, Su M, Stewart PW, Jia W, et al. Altered bile acid metabolome in patients with nonalcoholic steatohepatitis. Dig Dis Sci. 2015;60:3318–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Puri P, Daita K, Joyce A, Mirshahi F, Santhekadur PK, Cazanave S. The presence and severity of nonalcoholic steatohepatitis is associated with specific changes in circulating bile acids. Hepatology. 2018;67:534–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Caussy C, Hsu C, Singh S, Bassirian S, Kolar J, Faulkner C, et al. Serum bile acid patterns are associated with the presence of NAFLD in twins, and dose-dependent changes with increase in fibrosis stage in patients with biopsy-proven NAFLD. Aliment Pharmacol Ther. 2019;49:183–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zhang Z, Dai W, Weng S, Luo M, Fu J, Zadroga JA, et al. The association of serum total bile acid with non-alcoholic fatty liver disease in Chinese adults: a cross sectional study. Lipids Health Dis. 2020; 19:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Mouzaki M, Wang AY, Bandsma R, Comelli EM, Arendt BM, Zhang L. Bile acids and dysbiosis in non-alcoholic fatty liver disease. PLOS ONE. 2016;11:e0151829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Hindson J Obeticholic acid for the treatment of NASH. Nat Rev Gastroenterol Hepatol. 2020;17:66. [DOI] [PubMed] [Google Scholar]
- [39].Shah RA, Kowdley KV. Obeticholic acid for the treatment of nonalcoholic steatohepatitis. Expert Rev Gastroenterol Hepatol. 2020. [DOI] [PubMed] [Google Scholar]
- [40].Porez G, Prawitt J, Gross B, Staels B. Bile acid receptors as targets for the treatment of dyslipidemia and cardiovascular disease. J Lipid Res. 2012;53:1723–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Cipriani S, Mencarelli A, Palladino G, Fiorucci S. FXR activation reverses insulin resistance and lipid abnormalities and protects against liver steatosis in Zucker (fa/fa) obese rats. J Lipid Res. 2010;51:771–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Valenti L, Al-Serri A, Daly AK, Galmozzi E, Rametta R, Dongiovanni P, et al. Homozygosity for the patatin-like phospholipase-3/adiponutrin I148M polymorphism influences liver fibrosis in patients with nonalcoholic fatty liver disease. Hepatology. 2010;51:1209–17. [DOI] [PubMed] [Google Scholar]
- [43].Kasper M, Karsten U, Stosiek P, Moll R. Distribution of intermediate-filament proteins in the human enamel organ: unusually complex pattern of coexpression of cytokeratin polypeptides and vimentin. Differentiation. 1989;40:207–14. [DOI] [PubMed] [Google Scholar]
- [44].Soubeyrand S, Martinuk A, McPherson R. TRIB1 is a positive regulator of hepatocyte nuclear factor 4-alpha. Sci Rep. 2017;7:5574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Inoue Y, Yu AM, Inoue J, Gonzalez FJ. Hepatocyte nuclear factor 4alpha is a central regulator of bile acid conjugation. J Biol Chem. 2004;279:2480–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.