

Abstract
Cancer cell metabolism is a targetable vulnerability; however, a precise understanding of metabolic heterogeneity is required. Inactivating mutations in BRCA1-associated protein 1 (BAP1) are associated with metastasis in uveal melanoma (UM), the deadliest adult eye cancer. BAP1 functions in UM remain unclear. UM patient sample analysis divided BAP1 mutant UM tumors into two subgroups based on oxidative phosphorylation (OXPHOS) gene expression suggesting metabolic heterogeneity. Consistent with patient data, transcriptomic analysis of BAP1 mutant UM cell lines also showed OXPHOShigh or OXPHOSlow subgroups. Integrated RNA sequencing, metabolomics and molecular analyses showed that OXPHOShigh BAP1 mutant UM cells utilize glycolytic and nucleotide biosynthesis pathways, whereas OXPHOSlow BAP1 mutant UM cells employ fatty acid oxidation. Furthermore, the two subgroups responded to different classes of metabolic suppressors. Our findings indicate that targeting cancer metabolism is a promising therapeutic option for BAP1 mutant UM; however, tailored approaches may be required to due metabolic heterogeneities.
Keywords: metabolic profiling, heterogeneity, BAP1 mutation, uveal melanoma
Introduction
Cancer cells exhibit altered metabolism, including increased glycolysis, lipid metabolism and nucleotide synthesis to meet increased bioenergetic and biosynthetic demands of high proliferation and survival under stressful conditions (1–3). Transformation induced by oncogene and/or tumor suppressor mutations often involves changes in nutrient signaling pathways and metabolic enzyme expression (3, 4). For example, Myc gain-of-function mutations alter mitochondrial metabolism and increase glucose and fatty acid (FA) utilization (5). Loss of expression of the TP53 leads to elevation of the glycolytic pathway, which promotes anabolism in cancer cells (6). It is critical to study the metabolic functions of cancer-related gene such as BAP1 to understand tumorigenesis and progression and broaden potential therapeutic options. Targeting cancer metabolism may provide promising therapeutic avenues, but these are likely to be challenged by metabolic heterogeneity. For example, variation in lipid metabolism due to MITF expression in cutaneous melanoma is associated with an invasive phenotype and differential response to drugs targeting lipid metabolism (7). Therefore, understanding the level of metabolic heterogeneity in cancer type will help optimize metabolic targeting approaches.
A cancer type in which metabolic inhibitors may provide treatment advances is uveal melanoma (UM), the most aggressive intraocular cancer in adults (8). Eye-localized primary tumors are efficiently treated with radiotherapy or enucleation but approximately 50% of patients develop metastasis within 10 years of initial treatment (9–11). The patients who develop metastasis have a one year survival rate of < 30% (12–14). Primary UM is divided into two prognosis subgroups: class 1 (low metastatic risk) and class 2 (high metastatic risk). BAP1 inactivating mutations are found in > 80% of class 2 UM tumors, suggesting that BAP1 mutations are strongly correlated with metastasis and poor prognosis in UM (15, 16).
BAP1 is a tumor suppressor that acts as a deubiquitinating enzyme (DUB) in key cellular pathways including cell cycle, DNA damage response, and differentiation (17). The BAP1 gene is located on chromosome 3p21.1. Deletion of chromosome 3 and BAP1 inactivating mutations cause loss of BAP1 expression and function in cancer (17). Germline BAP1 mutations are associated with an elevated risk of developing UM, mesothelioma and renal cell carcinoma, a disorder named “BAP1 cancer predisposition syndrome” (18, 19). The function of BAP1 as a tumor suppressor in UM is not well established and little is known about the role of BAP1 in cellular metabolism (20–22). Studying metabolism in UM could broaden our understanding of BAP1’s role in advanced UM, and potentially uncover novel therapeutic strategies.
Here, we show BAP1 mutant UM exhibits an upregulated gene set of OXPHOS, a major parameter of cancer cell metabolism, compared to BAP1 wild-type UM tumors. Within BAP1 mutant samples, there was metabolic heterogeneity. By integrating multiple approaches, two metabolic phenotypes in BAP1 mutant UM were characterized with either increased glycolysis and nucleotide biosynthesis (OXPHOShigh) or high dependency on FA oxidation (FAO, OXPHOSlow). The two metabolic subtypes of BAP1 mutant UM cells displayed differential sensitivity to metabolic inhibitors highlighting that different therapeutic approaches may be necessary for BAP1 mutant UM depending on the specific metabolic phenotype.
Results
OXPHOS gene expression profiling defines two different metabolic phenotypes in BAP1 mutant UM
OXPHOS is frequently elevated in tumor cells and associated with cancer metastasis and that metastatic cancer cells display metabolic heterogeneity (23, 24). We analyzed UM cases in The Cancer Genome Atlas (TCGA) (25) to investigate whether BAP1 alterations influence OXPHOS. Samples were separated into BAP1 mutant (n=40) and wild-type (n=40) groups. Gene set enrichment analysis (GSEA) was performed using the OXPHOS gene set, which was found to be positively enriched in the BAP1 mutant group compared to the wild-type group (Fig. 1a).
Fig 1. BAP1 mutant UM samples are divided into two distinct metabolic subpopulations based on OXPHOS gene set.
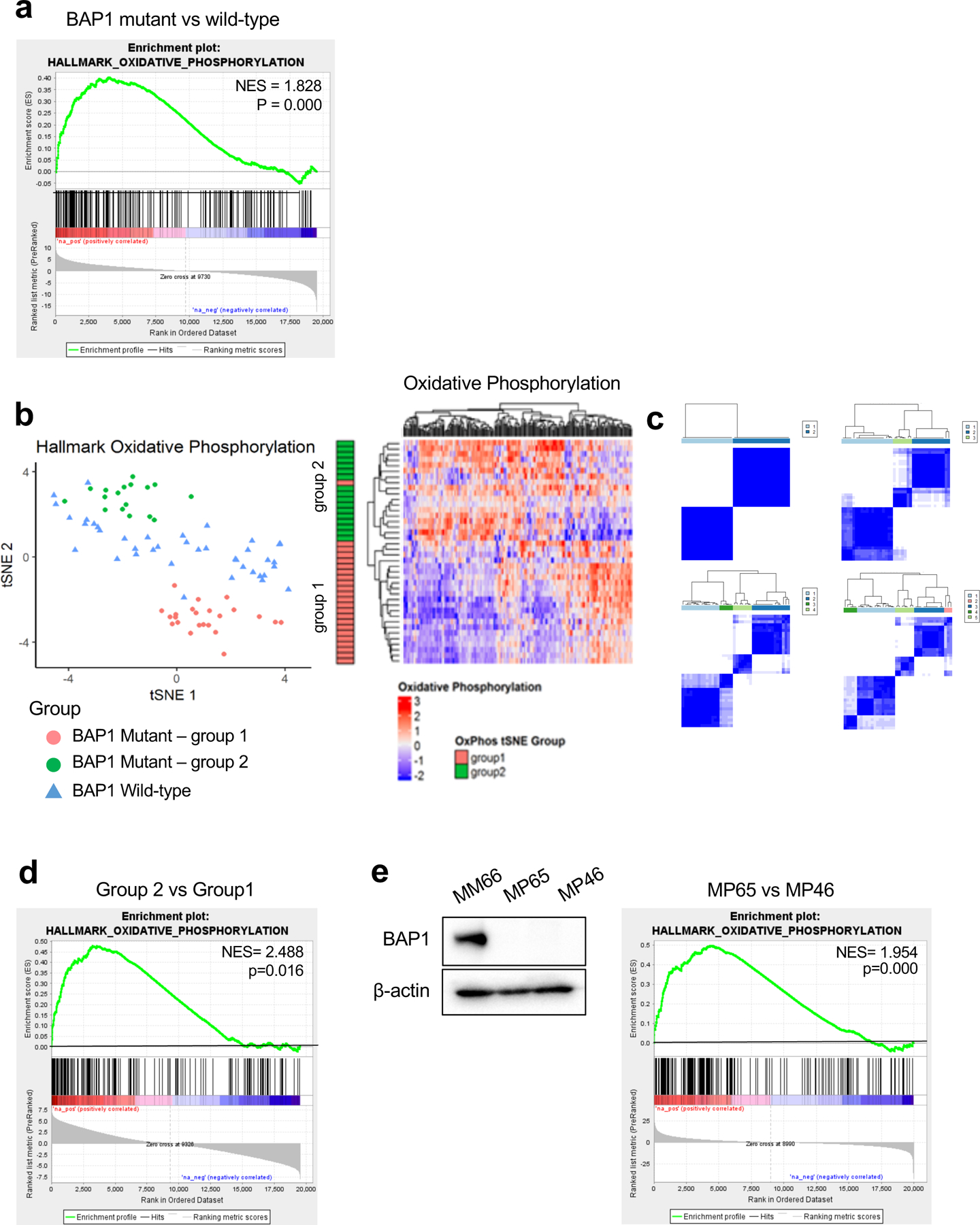
UVM RNA-seq V2 gene expression data from TCGA were retrieved from the latest Broad GDAC Firehose data run (stddata_2016_01_28). Based on BAP1 mutation and copy loss, samples were stratified into BAP1 mutant and wild-type groups. Differential expression analysis was performed between BAP1 mutant (n=40) and wild-type (n=40) and used for performing GSEA. a. GSEA enrichment plots of the OXPHOS hallmark gene set for comparison in BAP1 mutant vs wild-type group. b. tSNE plot of BAP1 mutant samples grouped into two subgroups (group 1 and group 2) and BAP1 wild-type samples based on OXPHOS genes. Heatmaps of OXPHOS gene expression from the RNA-seq data for BAP1 mutant samples with group indexing based on tSNE clustering Fig. 1b. c. Sample-group similarity matrices from consensus cluster plus k-means clustering analysis of BAP1 mutant UM samples, with the number of groups ranging from 2 to 5. d. GSEA enrichment plot of the top-ranked OXPHOS hallmark gene set for comparison in group 2 vs group 1. e. Protein expression of BAP1 in BAP1 wild-type (MM66) and two BAP1 mutant (MP65 and MP46) UM cell lines was analyzed by western blot. GSEA enrichment plot of the top-ranked OXPHOS hallmark gene set for comparison in MP65 vs MP46 cells.
To ensure separation of patient tumor samples, we used the t-distributed stochastic neighbor embedding (tSNE) method to plot OXPHOS signature gene RNA-seq data. Based on OXPHOS gene expression, we observed that BAP1 mutant samples separated from wild-type UM samples (Fig. 1b). Interestingly, there was a clear division within BAP1 mutant samples into two subgroups (group1 and group 2) (Fig. 1b). We also observed two distinct subgroups within BAP1 mutant samples with consensus clustering analysis using the OXPHOS genes (Fig. 1c). We compared gene expression between the two BAP1 mutant subgroups. GSEA showed that group 2 has significantly upregulated OXPHOS genes expression compared to the group 1 (Fig. 1d and Supplementary Fig. 1a). For example, NDUFS6, NDUFS7 and NDUFS8 which encode enzymes involved in the mitochondrial electron transport chain were more highly expressed in group 2 compared to group 1. Compared to the BAP1 wild-type group, both group 1 and 2 had upregulated expression of OXPHOS genes, although only group 2 was significantly enriched (Supplementary Fig. 1b).
To further evaluate the metabolic properties in BAP1-proficient and -deficient UM, we performed studies utilizing two BAP1 mutant cell lines (MP65 and MP46) compared to a BAP1 wild-type cell line (MM66) (Fig. 1e). Bulk RNA-seq and GSEA in these cell lines showed that MP65 have a significant OXPHOS gene sets upregulation compared to MP46 and MM66, while MP46 display significant downregulation compared to MM66 (Fig. 1e and Supplementary Fig 1c). This indicates that MP65 and MP46 have different phenotypes that appear to resemble the two distinct subtypes observed in BAP1 mutant patient tumors using TCGA data. Analysis of additional BAP1 mutant UM cell lines showed that similar to MP65, MP38 have an OXPHOShigh gene signature and, similar to MP46, PDX4 have OXPHOSlow gene signature (Supplementary Fig 1d). Together, this indicates that BAP1 mutant UM is segregated from BAP1 wild-type UM based on OXPHOS gene expression profiles and BAP1 mutant UM is further divided into two distinct subtypes which are characterized as either high or low OXPHOS.
Characterization of the two distinct metabolic phenotypes in BAP1 mutant UM
Next, we compared metabolite profiles of MP65 (OXPHOShigh) and MP46 (OXPHOSlow) cells. MP65 and MP46 exhibited distinct metabolite trends that correlated with gene expression data (FC>2, adj. p-val. <0.05) (Fig. 2a). We further analyzed the metabolomics data to determine metabolic pathway differences between MP65 and MP46. When metabolite levels in MP65 were compared to MP46 and vice versa, we found 66% of metabolites upregulated in MP65 were related to nucleotide biosynthesis pathways and 64% of metabolites increased in MP46 were associated with FA metabolism (Fig. 2b and Supplementary Table 1). Additionally, we compared the metabolite changes in MP65 and MP46 to the BAP1 wild-type cell line, MM66. We found that metabolites upregulated in MP65 and MP46 were predominantly associated with nucleotide biosynthesis pathways (22 out of 34 identified metabolites or 65%) and FA metabolism (11 out of 32 identified metabolites or 34%), respectively (Supplementary Fig 2a, 2b, and Table 1). These observations align with findings from TCGA (Fig. 1d) and RNA-seq data of cell lines (Fig. 1e), which showed that two distinct gene expression profiles exist among BAP1 mutant tumors and cell lines. Metabolomics analysis indicates that OXPHOS gene expression patterns within BAP1 mutant UM are associated with distinguishable metabolic phenotypes: OXPHOShigh cells, which have increased nucleotide biosynthesis; and OXPHOSlow cells, which show elevated FA metabolism.
Fig 2. BAP1 status alters the metabolic pathways in UM cells.
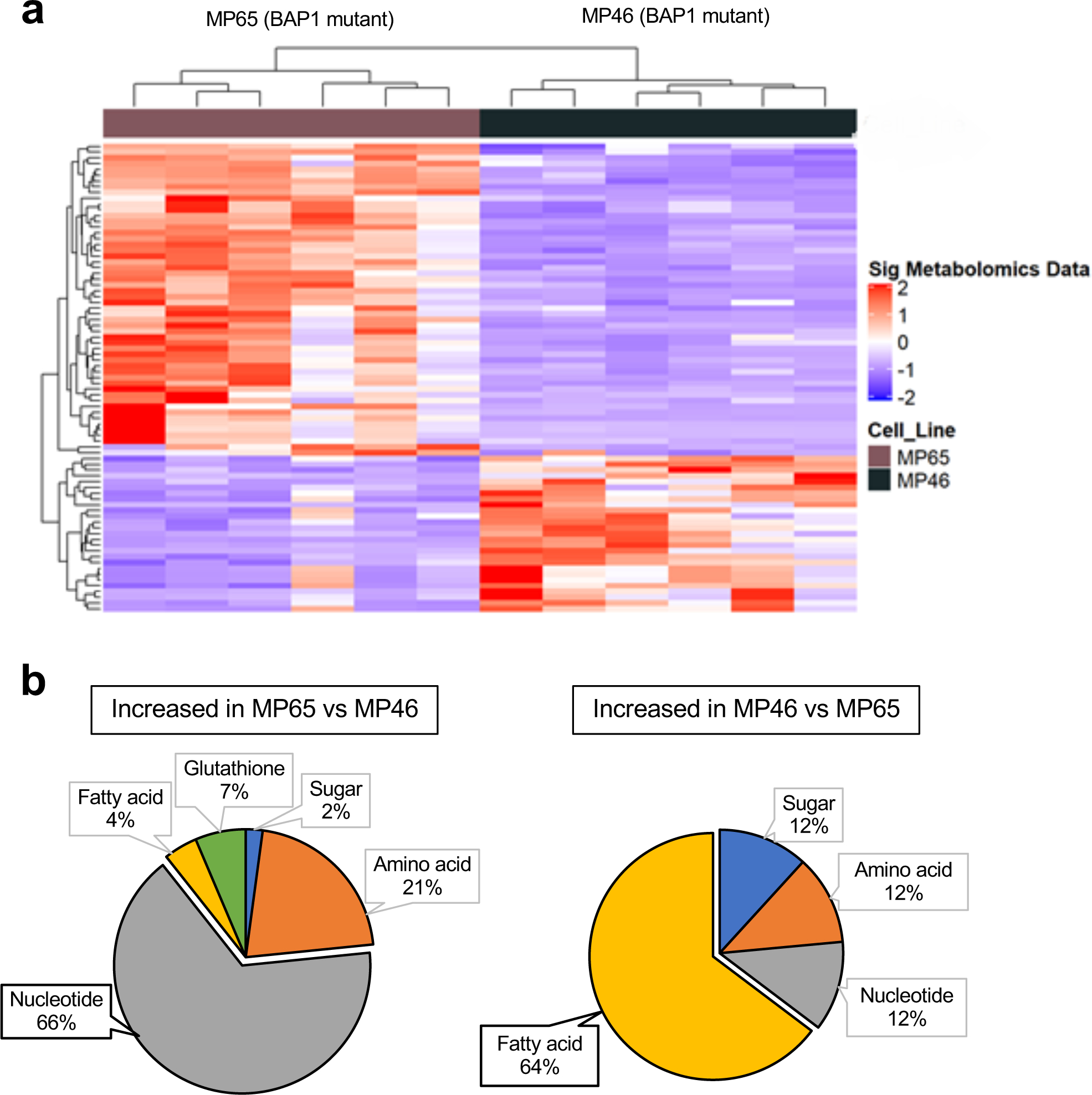
a. Heatmap showing z-scores for significantly different levels of metabolites (FC >2, adj. p-val. < 0.05) between two BAP1 mutant cell lines, MP65 and MP46. Unsupervised hierarchical clustering of samples and metabolites is shown. b. Percentage distributions of upregulated metabolites in MP65 or MP46 cells. Metabolites were categorized with specific metabolic pathways (FC >2, adj. p-val. < 0.05). Metabolite data were analyzed from whole cell extractions (n=6).
OXPHOShigh phenotype displays elevated glycolytic-nucleotide biosynthesis pathways
Cancer cells elevate glucose utilization to encourage substrate production for synthetic pathways. The pentose phosphate pathway (PPP) responsible for nucleotide biosynthesis is one of the main fates of glucose utilization (26). Since MP65 have increased metabolites associated with nucleotide biosynthesis, we examined key enzymes involved in glucose utilization and the PPP in OXPHOShigh BAP1 mutant cells. We observed that both MP65 and MP38 display up-regulation of glucose transporter 3 (GLUT3) and hexokinase 1 (HK1) compared to MM66 (Fig. 3a), while GLUT1 and GLUT 4 expression remained unchanged (Supplementary Fig 3a). Additionally, we observed that MP65 and MP38 express a markedly elevated level of glucose-6-phosphate dehydrogenase (G6PD), the rate-limiting enzyme of the PPP, and transketolase (TKT), the major enzyme involved in nucleotide synthesis (Fig. 3a).
Fig 3. OXPHOShigh BAP1 mutant phenotype is linked to increased glycolytic-nucleotide biosynthetic pathway.
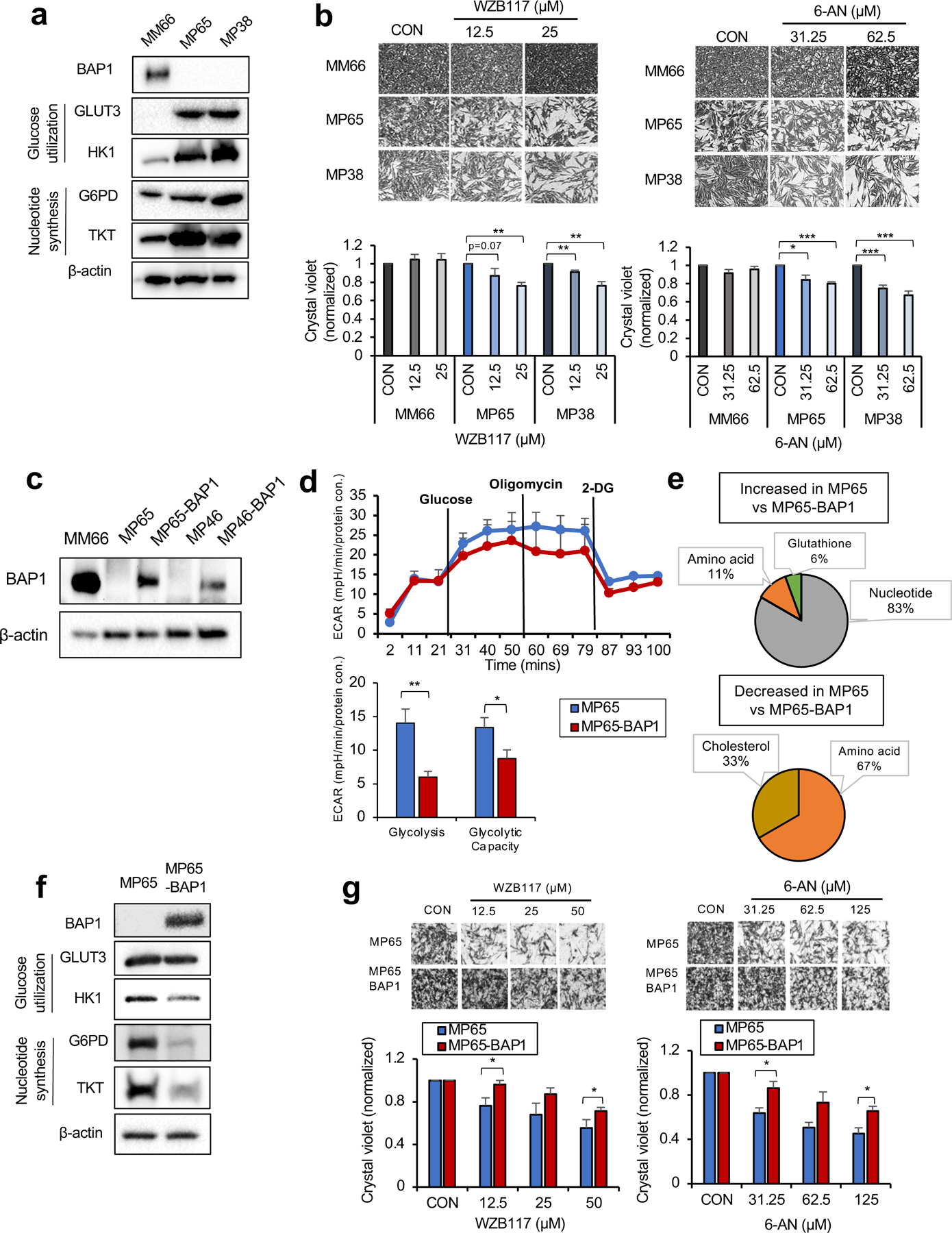
a. Protein expression of BAP1 and metabolic enzymes involved in glucose utilization and nucleotide synthesis in MM66, MP65 and MP38 cells. b. MM66, MP65 and MP38 cells were treated with WZB117 (12.5 or 25 μM) or 6-aminonicotinamide (6-AN, 31.25 or 62.5 μM) for 3 days. Cell viability changes were determined by crystal violet staining. Quantification of cell growth with WZB117 and 6-AN treatments in MM66, MP65 and MP38 cells presented as fold change in crystal violet staining of colonies. Representative crystal violet images of cell growth are shown. Scale bar: 100 μm. c. Re-expression of BAP1 in UM mutant cell clines, MP65 and MP46, was confirmed by western blot. MM66 serves as a positive control for BAP1 expression. d. The effect of BAP1 re-expression on the glycolytic capacity of MP65 cells was measured by ECAR using the Seahorse XF24 analyzer. Seahorse data were normalized to protein concentration and analyzed by Agilent Seahorse XF report generators. Data are shown as mean ± SEM (n=12). e. Metabolite percentage distributions within specific metabolic pathways that are increased or decreased in MP65 compared to MP65-BAP1 cells (FC >2, adj. p-val. < 0.05). f. Protein expression of metabolic enzymes involved in glucose utilization and nucleotide synthesis in MP65 and MP65-BAP1 cells. g. MP65 and MP65-BAP1 cells were treated with WZB117 (12.5, 25 or 50 μM) or 6-aminonicotinamide (6-AN, 31.25, 62.5 or 125 μM) for 5 days. Cell viability changes were determined by crystal violet staining. Quantification of cell growth with WZB117 and 6-AN treatments in MP65 and MP65-BAP1 cells presented as fold changes in crystal violet stain. Data are shown as mean ± SEM from biological replicate experiments (n=4). *p<0.05 and **p<0.01 unpaired t-test. ECAR; extracellular acidification rate, GLUT; glucose transporter, HK; hexokinase, G6PD; glucose-6-phosphate dehydrogenase and TKT; transketolase.
To test whether the increase in glycolysis and nucleotide biosynthesis enzymes represents a potential therapeutic vulnerability in OXPHOShigh BAP1 mutant cells, we treated MM66, MP65 and MP38 with WZB117 (a GLUT1, 3, 4 inhibitor) and 6-aminonicotinamide (6-AN, a G6PD inhibitor). WZB117 and 6-AN inhibited the MP65 and MP38 growth, while MM66 were not affected (Fig. 3b). In addition, we tested whether these metabolic features are unique to OXPHOShigh BAP1 mutant cells. As OXPHOSlow BAP1 mutant cells (e.g. MP46) exhibit FA metabolites over other metabolites, we hypothesized that OXPHOSlow BAP1 mutant cells would exhibit poor responses to WZB117 and 6-AN. Indeed, neither WZB117 nor 6-AN elicited a significant effect on the MP46 and PDX4 growth (Supplementary Fig 3b).
To investigate whether these metabolic features are dependent on BAP1 expression, we re-expressed wild-type BAP1 in MP65 and MP46 and BAP1 re-expression (MP65-BAP1 and MP46-BAP1) is confirmed by western blotting (Fig. 3c). Then, we compared glycolysis and OXPHOS by Seahorse analysis. When we re-expressed BAP1, glycolysis and glycolytic capacity of MP65 significantly decreased (Fig. 3d). MP65 had significantly lower basal respiration compared to MP65-BAP1, but higher spare respiratory capacity (SRC) (Supplementary Fig 3c). This indicates that the activity of the glycolytic metabolic pathways is predominant over OXPHOS in MP65 compared to MP65-BAP1. We also observed that the majority (88%) of metabolites that significantly changed in MP65 compared to MP65-BAP1 were involved in nucleotide biosynthesis metabolism (Fig. 3e and Supplementary Table 1), such as glucose-6-phosphate (G-6-P), a substrate of glycolysis and the PPP (Supplementary Fig 3d).
Since we have demonstrated MP65 have higher glycolysis, glycolytic capacity and increased nucleotide biosynthesis metabolites compared to MP65-BAP1, we examined key enzymes involved in glycolysis and the PPP. Re-expression of BAP1 in MP65 decreased GLUT3, HK1, G6PD and TKT levels (Fig. 3f).
We then tested whether BAP1 re-expression altered the vulnerability of MP65 to WZB117 and 6-AN. Both inhibitors diminished MP65 and MP65-BAP1 growth; however, MP65 were more sensitive to these compounds compared to MP65-BAP1 (Fig. 3g). Then, we hypothesized that MP46 would respond poorly to WZB117 and 6-AN according to our hypothesis of the dependency of MP46 on FA metabolism. As expected, neither WZB117 nor 6-AN had any significant effect on the MP46 and MP46-BAP1 growth (Supplementary Fig 3e).
Together, these observations show that OXPHOShigh BAP1 mutant cells represented by the MP65 and MP38 cell lines depend on glycolysis and nucleotide biosynthesis pathways, making them more vulnerable to inhibitors targeting those pathways and that this effect is partially lost when BAP1 is re-expressed. These observations present potential treatment options for OXPHOShigh BAP1 mutant UM.
OXPHOSlow phenotype has upregulated FA catabolic pathways
Increased FAO of cancer cells is associated with tumorigenesis by providing advantages in ATP production and reducing equivalents (i.e. NADPH) (27). Since MP46 have elevated FA metabolism, we probed key metabolic enzymes involved in FAO in OXPHOSlow BAP1 mutant cells. Both MP46 and PDX4 show increased expression of carnitine palmitoyltransferase1A (CPT1A), a rate-limiting enzyme of FAO (Fig. 4a). To investigate whether increased CPT1A expression represents a potential therapeutic vulnerability in OXPHOSlow BAP1 mutant cells, we targeted FAO with IACS (a selective inhibitor of mitochondrial ETC complex I, (28)) or etomoxir (ETO, a CPT1 inhibitor) and determined changes in cell growth. IACS and ETO significantly reduced MP46 and PDX4 growth, but have little to no effect on MM66 (Fig. 4b). Since OXPHOShigh BAP1 mutant cells have a metabolic dependency on glucose utilization and nucleotide biosynthesis over FA metabolism, we predicted that these cells would respond poorly to FAO inhibitors. As hypothesized, neither IACS nor ETO elicited a significant effect on the survival of MP65 and MP38 (Supplementary Fig 4a).
Fig 4. OXPHOSlow BAP1 mutant phenotype is associated with an elevated FA oxidation pathway.
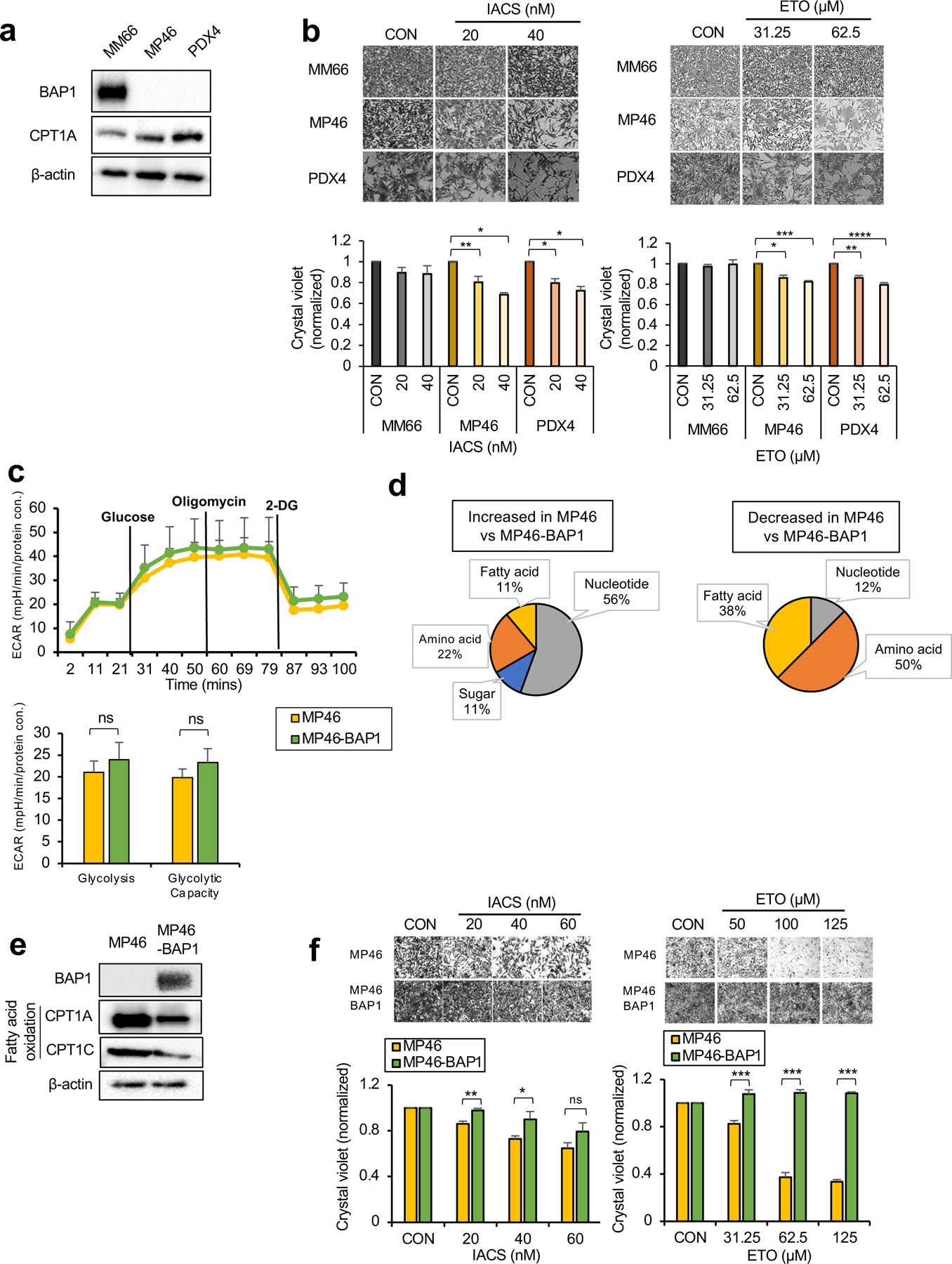
a. Protein expression of BAP1 and metabolic enzymes involved in FAO in MM66, MP46 and PDX4 cells. b. MM66, MP46 and PDX4 cells were treated with IACS (20 or 40 nM) or etomoxir (ETO, 31.25 or 62.5 μM) for 3 days. Cell viability changes were determined by crystal violet staining. Quantification of cell growth with IACS or ETO treatments in cells presented as fold change in crystal violet staining. Representative crystal violet images of cell growth are shown. Scale bar represents 100 μm. c. Effects of BAP1 re-expression on the glycolytic capacity of MP46 cells was measured by ECAR using the Seahorse XF24 Analyzer. Seahorse data were normalized to protein concentration and generated by Agilent Seahorse XF report generators. Data are shown as mean ± SEM (n=12). d. Metabolite percentage distribution within specific metabolic pathways that are upregulated or down-regulated in MP46 compared to MP46-BAP1 cells (FC >2, adj. p-val. < 0.05). e. Protein expression of metabolic enzymes involved in FA synthesis and oxidation in MP46 and MP46-BAP1 cells. f. MP46 and MP46-BAP1 cells were treated with IACS (20, 40 or 60 nM) or etomoxir (ETO, 31.25, 62.5 or 125 μM) for 5 days. Cell viability changes were determined by crystal violet staining. Quantification of cell growth with IACS and ETO treatments in cells presented as fold change in crystal violet staining. Data are shown as mean ± SEM from biological replicate experiments (n=4). *p<0.05, **p<0.01, ***p<0.001 and not significant (ns) unpaired t-test. ECAR; extracellular acidification rate, CPT1; carnitine palmitoyltransferase1, 2DG; 2-deoxyglucose and PHX; perhexiline.
Next, we determined the dependency of OXPHOSlow phenotype on BAP1. We did not observe significant differences in glycolysis, glycolytic capacity or OXPHOS parameters between MP46 and MP46-BAP1 (Fig. 4c), although SRC levels were lower in MP46 than MP46-BAP1 (Supplementary Fig 4b). In comparison to MP46-BAP1, the predominant (56%) metabolites increased in MP46 were related to nucleotide biosynthesis pathways, while most metabolite decreases were associated with amino acid and FA metabolism (Fig. 4d and Supplementary Table 1). We further investigated the metabolite levels related to FA metabolism and observed MP46 had significantly lower levels of FAs and palmitoylcarnitine, which is involved in FA transport into mitochondria for oxidation compared to MP46-BAP1, but higher acetyl-carnitine levels, a byproduct of FAO (Supplementary Fig 4c). We also found that BAP1 re-expression in MP46 reduced enzyme levels related to FAO, CPT1A and CPT1C (Fig. 4e). Together, these results imply that FAO is enhanced in MP46 and this effect is reversed following BAP1 re-expression.
We then compared FAO-oxygen consumption rate (OCR) levels between MP46 and MP46-BAP1 by Seahorse analysis. First, we measured FA utilization levels between MP46 and MP46-BAP1 by incubating cells with palmitate (PAL) as the only oxidizable energy substrate. Following PAL treatment, OCR levels increased significantly in MP46, but were unchanged in MP46-BAP1 (Supplementary Fig 4d), suggesting that MP46 utilized or metabolized FA more efficiently than MP46-BAP1. We also tested for FAO-dependent OCR between MP46 and MP46-BAP1 by treatment with the FAO inhibitor, perhexiline (PHX). OCR levels significantly decreased in MP46 after addition of PHX, suggesting that the high OCR levels in MP46 were maintained largely by FAO (Supplementary Fig 4e). Next, we treated MP46 and MP46-BAP1 with IACS or ETO. Both IACS and ETO significantly reduced MP46 growth but had little-to-no effect on cell growth when BAP1 was re-expressed (Fig. 4f). In addition, these inhibitors did not alter MP65 and MP65-BAP1 growth (Supplementary Fig 4f).
Overall, we observed that cell lines, which metabolically represent OXPHOSlow BAP1 mutant UM preferentially utilize FAO-dependent metabolic pathways. Moreover, these cells have specific vulnerabilities to inhibitors that suppress FAO that are partially reversed by BAP1 re-expression. These findings suggest that FAO inhibitors may be used to treat OXPHOSlow BAP1 mutant UM.
The two BAP1 mutant UM metabolic phenotypes respond differently to metabolic stress
The ability to cope with metabolic stress is important in promoting cancer cell survival and metastasis (29, 30). SRC is a measurement of ATP production through OXPHOS and can be used as an indicator of ATP synthesizing capacity under stressful conditions (i.e. nutrient and/or oxygen deprivation) (31). We observed that BAP1 re-expression significantly lowered the SRC level in MP65, but increased SRC in MP46 (Supplementary Fig 3c and 4b). This suggests that the two metabolic subtypes in BAP1 mutant UM cells may have different survival abilities under metabolic stress conditions.
To test this possibility, cells were grown under glucose deprivation (2.5mM glucose or 2DG treatment) and hypoxia (1% O2). We found that MP65 (OXPHOShigh) were more resistant to both metabolic stress environments than MP65-BAP1, whereas MP46 (OXPHOSlow) were more vulnerable to stressful conditions compared to MP46-BAP1 (Fig. 5a and 5b). Of note, BAP1 expression was increased in hypoxic conditions in MP46-BAP1 (Fig 5b), implying the association between hypoxic-induced HIF1α and exogenous BAP1 expression. This suggests that the distinct metabolic profiles (OXPHOShigh and OXPHOSlow) within BAP1 mutant UM is related to different patient survival outcomes. However, analysis did not show any differences in patient survival between the two BAP1 mutant phenotypes (Supplementary Fig 5a). Nevertheless, our studies overall show that OXPHOS is important in predicting vulnerabilities of BAP1 mutant UM to unique metabolic inhibitors.
Fig 5. The two metabolic phenotypes respond differently to metabolic stress.
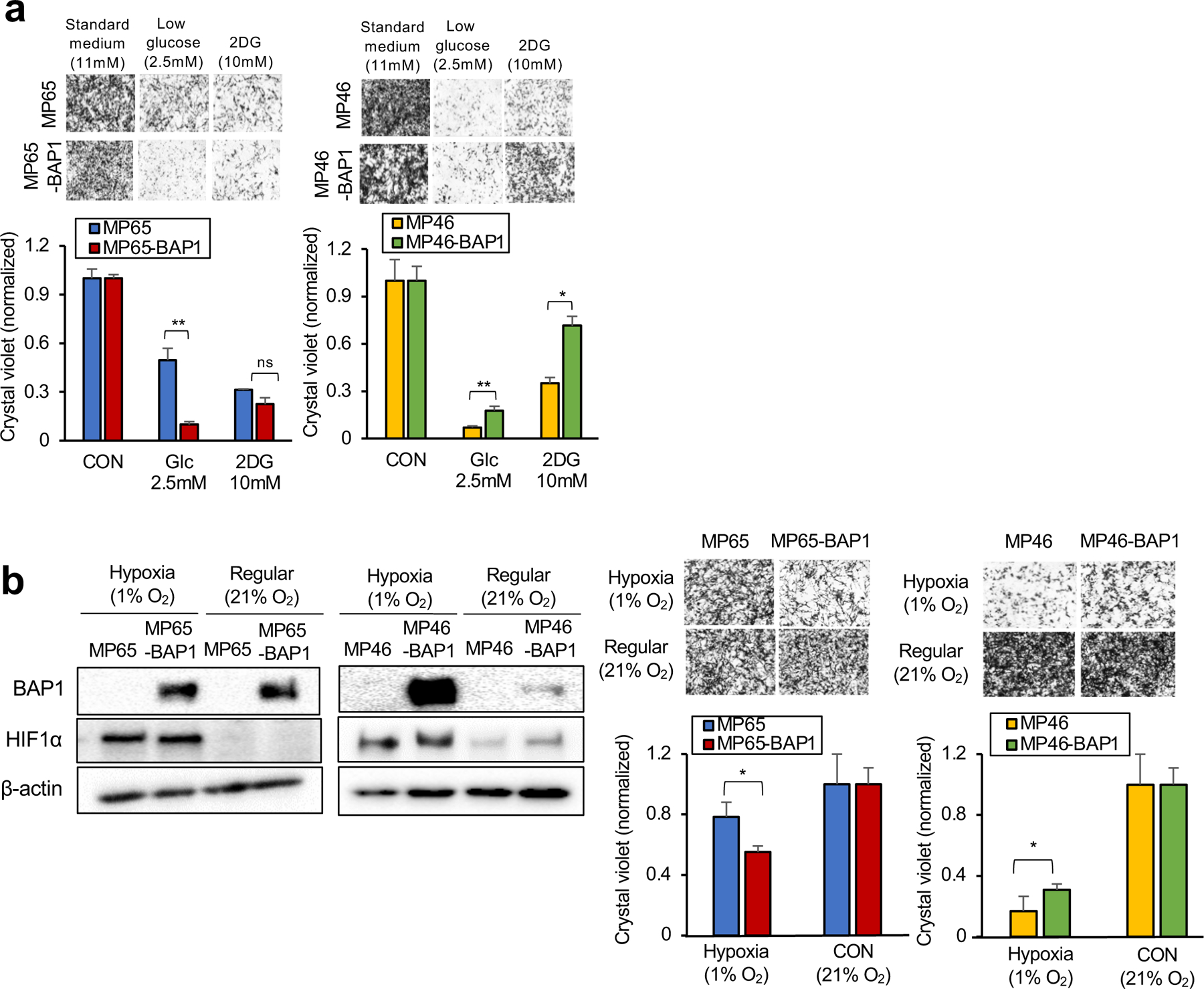
MP65, MP65-BAP1, MP46 and MP46-BAP1 cells were exposed to metabolic stress; glucose deprivation for 3 days or hypoxic conditions for 5 days. a. Cells were cultured in regular growth medium (11 mM glucose), medium with low glucose (2.5 mM) or medium containing 2-DG (10 mM) for 3 days. b. For 5 days, the cells were under either hypoxic (1% O2) or normoxic (21% O2) conditions. As a marker of hypoxia, expression of HIF1α was probed on day 3 by western blotting. Cell viability changes were measured by crystal violet staining. Representative crystal violet images of cell growth are shown. Scale bar: 100 μm. Data are shown as mean ± SEM from biological replicate experiments (n=4). The unpaired t-test was used for statistical significance. *p<0.0, **p<0.01 and not significant (ns).
BAP1 mutant UM patient subgroups differ in their nucleotide and FA metabolism
Our results thus far have shown that distinct metabolic phenotypes in BAP1 mutant UM, OXPHOShigh or OXPHOSlow, have increased glycolysis and nucleotide synthesis or increased FA metabolism, respectively. To visualize our findings regarding the metabolic heterogeneity in the context of pathways, we integrated RNA-seq and metabolite data of UM cell lines related to pyrimidine and purine metabolism pathways into the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database. We determined differences in gene expression and metabolite levels between MP65 and MP46 in pyrimidine and purine metabolism pathways, further supporting that the two BAP1 mutant cell lines have different profiles (Supplementary Fig 6a and 6b). We also explored gene expression profiles of nucleotide and FA metabolism in the OXPHOShigh and OXPHOSlow groups in UM TCGA patient dataset with indexing based on OXPHOS clustering analysis in Fig. 1b. Similar to findings in Fig. 1b, we observed that patient samples separated BAP1 mutant (group 1 and 2) and BAP1 wild-type samples (Fig. 6a). Moreover, there was a division of BAP1 mutant samples into two subpopulations with distinct gene expression profiles (Fig. 6a). Comparing the two subgroups within BAP1 mutant UM patient tumors and cell lines, GSEA plots showed that the OXPHOShigh subtype has upregulated gene expression of nucleotide and FA metabolism compared to OXPHOSlow subtype (Fig. 6b).
Fig 6. Nucleotide and FA metabolism gene expression separate BAP1 mutant samples into two distinct subgroups.
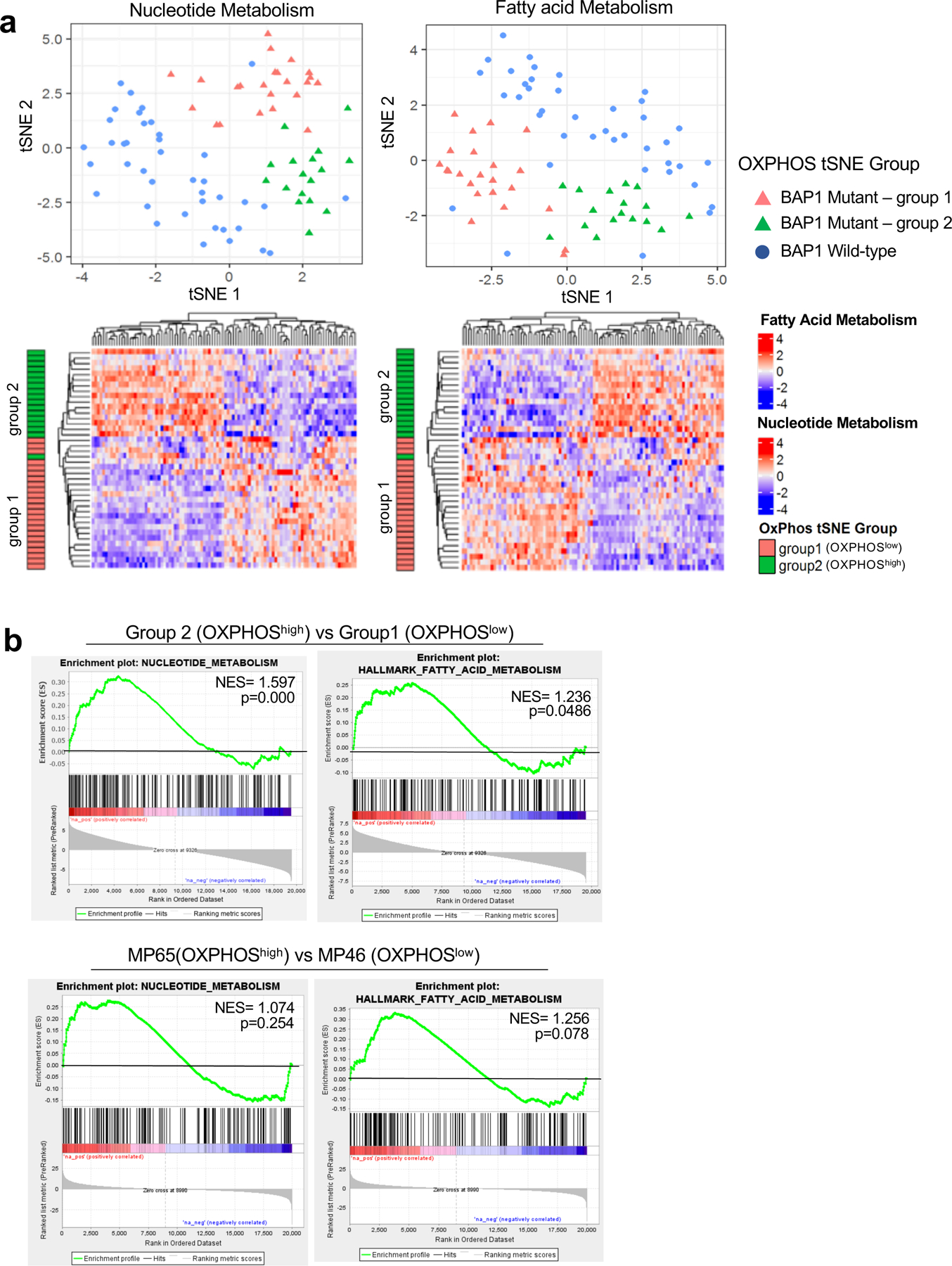
UM RNA-seq V2 gene expression data from the TCGA were retrieved from the latest Broad GDAC Firehose data run (stddata__2016_01_28). The samples were stratified into BAP1 mutant and wild-type groups based on BAP1 mutation and copy loss. Differential expression analysis was performed between BAP1 mutant (n=40) and wild-type (n=40). a. tSNE plot presenting BAP1 mutant samples with two subgroups (group 1 and group 2) and BAP1 wild-type samples based on nucleotide metabolism (left) and FA metabolism (right) genes. Below, heatmaps showing unsupervised hierarchical clustering of z-scores for each gene set in BAP1 mutant samples with group indexing based on OXPHOS clustering analysis in Fig. 1. b. GSEA plots of the nucleotide and FA metabolism gene set for comparison of group 2 vs group 1 in BAP1 mutant UM data from TCGA, and two BAP1 mutant cells, MP65 vs MP46.
Transcriptional upregulation within the OXPHOShigh phenotype may explain the survival differences between OXPHOShigh and OXPHOSlow BAP1 mutant UM cells (Fig. 5a and 5b). Under metabolic stress, OXPHOShigh BAP1 mutant UM may increase utilization of nucleotide and FA metabolic pathways that allows them to be more resistant to metabolic stress than OXPHOSlow BAP1 mutant UM. Together, these observations suggest that OXPHOShigh phenotypes have transcriptional upregulation of both nucleotide and FA metabolism compared to OXPHOSlow phenotypes, although they rely more on nucleotide biosynthesis than FA metabolism.
Discussion
Here, we observed that BAP1 mutant UM exhibits metabolic heterogeneity based on OXPHOS gene expression profiles using both cell line and patient data. We further showed that the OXPHOShigh phenotype utilizes glucose and nucleotide biosynthesis pathways, whereas, the OXPHOSlow phenotype preferentially utilizes FA metabolism to support survival. Our studies demonstrate that metabolism can be a new therapeutic target for BAP1 mutant UM and that the two subtypes of tumor cells respond differently to selective inhibitors according to their metabolic profiles.
Understanding of UM-specific genomics has advanced by whole genome sequencing of primary and metastatic UM tumors (32). There are currently no FDA-approved treatment strategies for metastatic UM (33). BAP1 mutations are highly correlated with advanced UM and poor patient survival; moreover, loss of BAP1 shapes transcriptional features of metastatic UM (15, 34). BAP1 plays a key role in cellular pathways (17); however, BAP1 functions in metabolic regulation are less clear. Several studies demonstrated that BAP1 regulates reactive oxidative species (ROS), mitochondrial function, and lipid and glucose homeostasis in mouse liver (21, 35). In addition, BAP1 controls cell survival under glucose deprivation by participating in metabolic stress response and endoplasmic reticulum stress regulation (22). Cells that carry germline BAP1 mutations exhibit the Warburg effect, which renders these cells susceptible to malignancy (20). Although a few studies have investigated the metabolic functions of BAP1 (20–22, 35), little is known about the metabolic role of BAP1 in UM.
By analyzing RNA-seq data in TCGA UM cases, we observed that within BAP1 mutant samples, there are two distinct subpopulations in terms of their OXPHOS gene signature (OXPHOShigh and OXPHOSlow). We further profiled the metabolic phenotypes of two primary BAP1 mutant UM cell lines through RNA-seq and metabolomics analysis. Similar to our observations with patient data, we found that the BAP1 mutant cell lines have different OXPHOS gene expression and metabolic characteristics, thereby confirming that BAP1 mutations result in metabolic heterogeneity in UM. As a DUB, BAP1 interacts with transcriptional factors and chromatin-modifying enzymes including Hcfc1, Ogt, Asxl1/2 and FOXK1/2 (36–39), which are involved in nutrient homeostasis by regulating transcriptional pathways (40, 41). The detailed mechanisms that drive metabolic heterogeneity in BAP1 mutant UM remain unclear, although one possibility is the involvement of BAP1 as a DUB and its interactions with transcriptional factors. This line of investigation will be a focus in future studies.
Targeting cancer cell metabolism is a promising strategy to treat cancers and accumulating evidence have suggested that the metabolic heterogeneity identification is an essential factor in therapeutic outcomes. For example, selective inhibition of glucose or lipid metabolism based on distinct metabolic subpopulations elevated the efficacy of treatment agents in lung squamous cell carcinoma and pancreatic ductal adenocarcinoma (42, 43). Here, we similarly demonstrated that targeting BAP1 mutant UM requires precise therapeutic approaches based on metabolic features to increase efficacy. We identified two distinct metabolic phenotypes in BAP1 mutant UM cells, which have either increased glycolysis-nucleotide biosynthesis or FAO. Cancer cells remodel their metabolism to enhance bioenergetic and biosynthetic pathways based on OXPHOS signatures. In cancer cells, increased glucose utilization and the PPP is responsible for prompt substrate production for anabolic pathways such as nucleotide biosynthesis (26). On the other hand, upregulated FA metabolism increases the production of ATP via FAO compared to glucose oxidation (1, 27). Thus, glucose/PPP and FA metabolic pathways can be potential targets in cancer cells (44–46). Our data showed that inhibitors of nucleotide biosynthesis and FAO selectively suppressed BAP1 mutant cell growth according to their metabolic profiles with modest or no effect on cells that re-express BAP1 or wild-type UM cells. These findings led us to uncover novel potential therapeutic approaches for BAP1 mutant UM based on their unique metabolic profiles. Additionally, BAP1 mutations results in slower growth rate and doubling time compared with BAP1 wild-type UM cells (47, 48). Remodeling metabolism allows cancer cells to meet increased bioenergetic and biosynthetic needs for high growth rate; thus, it is possible that the different metabolic dependencies between BAP1 wild-type and BAP1 mutant UM cells are correlated to their different growth rates, requiring the further investigations in the future studies.
Altered metabolism is often correlated with clinical outcomes. For example, subtypes with elevated glycolysis and nucleotide metabolism are highly related to poor prognosis in melanoma, and adrenocortical carcinoma (49, 50). In addition, cancer cells manage metabolic stress within the tumor microenvironment to survive and metastasize (30). We found that OXPHOShigh BAP1 mutant cells survive better under metabolic stress than OXPHOSlow BAP1 mutant cells, which might come from transcriptional upregulation of both nucleotide and FA metabolic pathways. These observations suggest that metabolic inhibitors might yield different outcomes in normal and metabolically stressful conditions, requiring future studies to assess metabolic profiles in relevant local tumor microenvironments. In addition, we observed that hypoxic conditions increase exogenous BAP1 expression, raising the possibility of future studies to investigate HIF1α-BAP1 cross-talk, and its role in metabolic alterations. In contrast with our in vitro results, there was no significant difference in overall patient survival between OXPHOShigh and OXPHOSlow phenotypes within BAP1 mutant UM. These findings can likely be explained by the fact that the BAP1 mutation itself, through its role in metastasis, is a strong influencer of patient survival in UM (51). This highlights the importance of identifying BAP1-driven metabolic heterogeneity to improve the overall survival of BAP1 mutant UM.
In conclusion, this study is the first to demonstrate the role of BAP1 in metabolism in UM and to show metabolic heterogeneity in BAP1 mutant UM via bio-informatics, RNA-seq, metabolomics, and molecular analyses. We found two metabolically distinct subgroups in BAP1 mutant UM: OXPHOShigh, a glycolytic phenotype and OXPHOSlow, an FAO-dependent phenotype (Fig. 7). The OXPHOShigh phenotype have enhanced glucose utilization and nucleotide biosynthesis, making them more sensitive to glycolysis and nucleotide biosynthesis inhibitors. In contrast, FAO metabolism is upregulated in the OXPHOSlow phenotype, which was more vulnerable to OXPHOS and FAO inhibitors. Future studies will be required to evaluate the efficacy of these inhibitors in vivo and relevant in vitro stressful environments. Our findings indicate that targeting cancer cell metabolism can be a promising therapeutic option for BAP1 mutant UM, especially when tailored to the metabolic heterogeneity present in BAP1 mutant UM.
Fig 7. Two distinct metabolic phenotypes in BAP1 mutant UM.
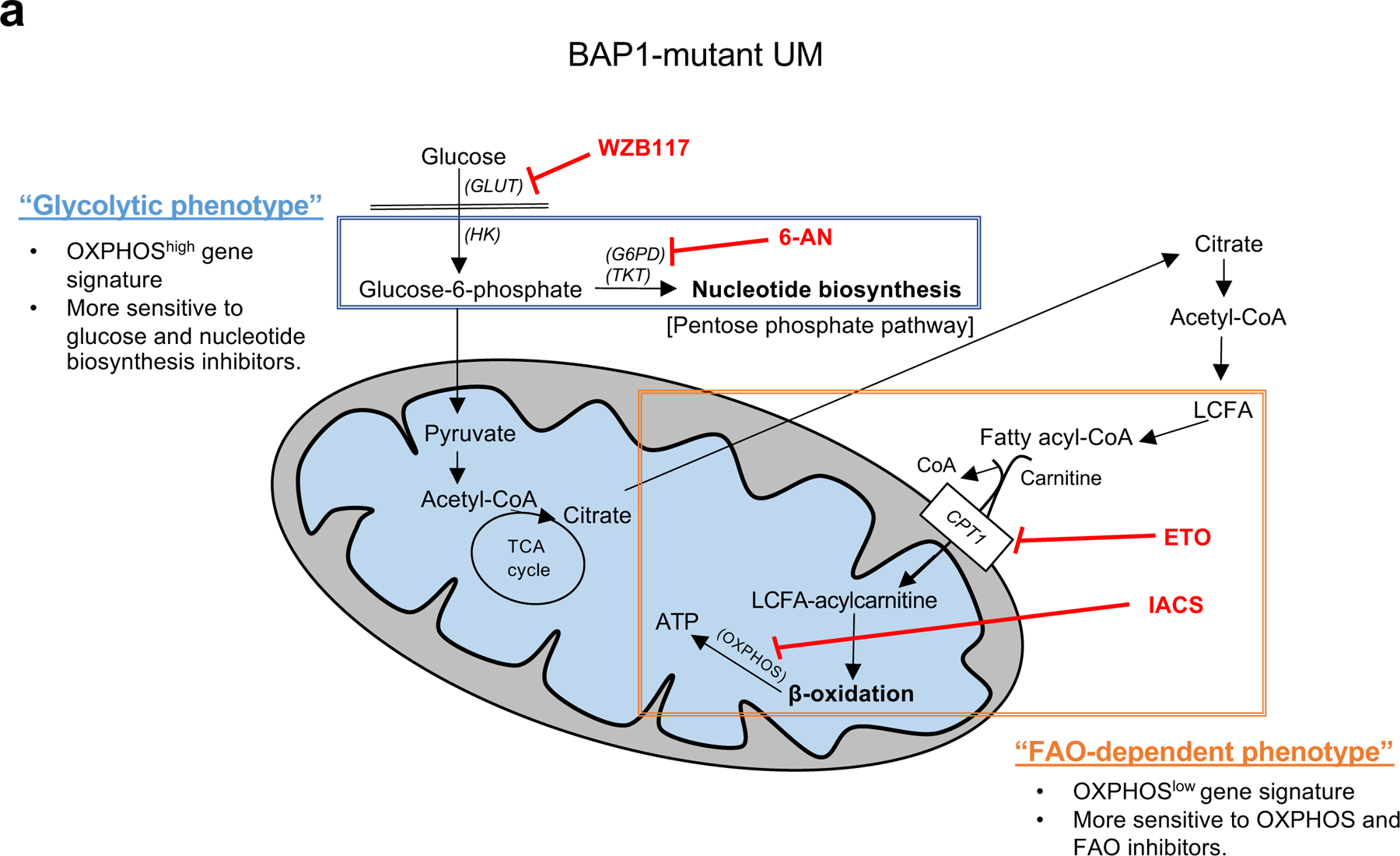
Summary figure showing two different metabolic phenotypes in BAP1 mutant UM. The glycolytic phenotype has a high OXPHOS gene signature and primarily relies on glucose utilization, which can upregulate oxidative PPP. This phenotype is specifically sensitive to glycolysis or nucleotide synthesis inhibitors. The second subtype has low OXPHOS gene expression and is more dependent on FAO. This subtype is vulnerable to either OXPHOS or FAO-specific inhibitors. FA; fatty acid, GLUT; glucose transporter, HK; hexokinase, G6PD; glucose-6-phosphate dehydrogenase, TKT; transketolase, LCFA; long chain fatty acid, CPT1; carnitine palmitoyltransferase1, 6-AN; 6-aminonicotinamide and ETO; etomoxir.
Materials and Methods
Analysis of TCGA data:
TCGA uveal melanoma RNA-seq V2 expression data were retrieved from the latest Broad GDAC Firehose data run (stddata__2016_01_28). BAP1 mutation and copy number data were collected using the RTCGA package (version 1.10.0). Samples with a copy number segment mean below −0.5 were classified as BAP1 copy loss. Samples were stratified into BAP1 mutant and wild-type groups based on BAP1 mutation and copy loss data. Differential expression analysis was performed between BAP1 mutant (n=40) and wild-type (n=40) groups using the DESeq2 package (version 1.20.0) (52). The Oxidative Phosphorylation gene set from the mSigDB Hallmark gene set collection (53) was used to cluster samples. The tSNE method was implemented using the Rtsne package (v0.13 https://github.com/jkrijthe/Rtsne). Scatter plots were generated using the ggplot2 package (v3.0.0 https://ggplot2.tidyverse.org/). Clustering of samples into groups was performed using the ConsensusClusterPlus package (v1.48.0) (54). Data analyses were performed in R (v3.5.1 http://www.R-project.org/).
Cell culture and modification of cell lines:
MM66, MP46, MP65, and MP38 cell lines were obtained from Dr. Sergio Roman-Roman (Paris, France) and confirmed to harbor GNAQ, GNA11, or BAP1 mutation by Sanger DNA sequencing (47). PDX4 was from Thomas Jefferson University and confirmed GNA11 or BAP1 mutation by Sanger sequencing. The detailed information of cell lines, growth media and detail protocol for re-expressing BAP1 are in Supplementary Materials. All cell lines were routinely tested for mycoplasma and authenticated by STR analysis. The most recent STR analysis was conducted in April 2018.
Inhibitors:
WZB117 (Tocris, Minneapolis, MN), 6-aminonicotinamide (Cayman Chemical Company, Ann Arbor, MI), Etomoxir (Sigma-Aldrich, St. Louis, MO), IACS-010759 (ChemieTek, Indianapolis, IN) were dissolved in DMSO.
Cell viability assay:
Preparation of cell plate is described in Supplementary Materials. Cell viability was determined by crystal violet staining as described previously (55).
Western blotting:
Sample preparation is described in Supplementary Materials. Primary antibodies were: BAP1 (#13271), HK 1 (#2024), GLUT1 (#12939), GLUT4 (#2213), and CPT1A (#12252) from Cell Signaling Technology (Danvers, MA); CPT1C (ab123794) Abcam (Cambridge, UK); GLUT3 (sc-30107), G6PD (sc-373886), and TKT (sc-390179) from Santa Cruz Biotechnology (Santa Cruz, CA) and β-actin from Sigma-Aldrich (St. Louis, MO).
Oxygen consumption, extracellular acidification flux experiment:
XF24 Analyzer (Agilent, Santa Clara, CA) was used to measure the OCR and ECAR. The detailed protocol and product information are described in Supplementary Materials. Data were normalized to protein concentration and analyzed by Agilent Seahorse XF report generators (mitochondrial and glycolysis stress test).
RNA-seq analysis:
RNA sample preparation is described in Supplementary Materials. RNA-seq data were aligned to the human reference genome (GRCh 38) using Star aligner (56). RSEM (57) was used to quantify gene and transcript level expression. Gene differential expression analysis was performed using DESeq2 (52). Data analyses were performed in R (v3.5.1 http://www.R-project.org/). Gene Set Enrichment Analysis (GSEA) (58, 59) was used to determine enriched pathways in the MSigDB Hallmark gene set collection (53). Pathview (60) was used to visualize RNA Seq and metabolite data on KEGG pathway maps (61).
Metabolites analysis:
Detailed protocol is described in Supplementary Materials. Comparison between cell types were based on evaluation of six biological replicates per cell line and changes were considered significant if the adjusted p-values was <0.05 and the differences was least 2-fold. Metabolites are organized into specific metabolic pathways (i.e. sugar, amino acid, nucleotide, FA, glutathione and cholesterol) using the chemical taxonomy from the Human Metabolome Database (62).
Statistical analysis:
Cell viability assay and flux experiments were repeated 3 times for quantitative in vitro data. The mean and standard error of the mean (SEM) of data are determined for trials done at least in triplicate. Statistical significance was calculated using the unpaired t-test. Metabolites analyses were conducted once using six biological replicates. The Student’s T-test was used to calculate differences between groups, and p-values were adjusted to account for multiple testing using Benjamini-Hochberg false-discovery rate method.
Data and code availability:
Code availability: Computational analyses were done using publicly-available software and R packages. Data availability: Bulk RNA-Sequencing data have been deposited to the Gene Expression Omnibus (GEO) database with accession code GSE149920.
Supplementary Material
Acknowledgments
We thank Dr. Sergio Roman-Roman (Uveal Melanoma Translational Group, Department of Translational Research, Institute Curie, PSL Research University, Paris, France) for cell lines. We thank Dr. Michele Carbone (University of Hawaii Cancer Center, Honolulu, HI, USA) for BAP1 cDNA. This work was supported by a Melanoma Research Alliance team science award (#559058) to A.E. Aplin and J.W. Harbour. Further support was from National Institutes of Health (NIH)/National Cancer Institute (NCI), R01 CA196278 to A.E. Aplin, P50CA174523 to D.W. Speicher and fellowships from the National Cancer Center and American Association for Cancer Research (AACR)/Ocular Melanoma Foundation (OMF) awarded to A Han and V. Chua. This work was also supported by grant from NCI P01 CA114046. The Wistar Proteomics and Metabolomics Facility was supported by P30CA010815 and S10OD023586.
Footnotes
Declaration of Interests:
A.E. Aplin reports receiving a commercial research grant from Pfizer Inc. (2013–2017) and has ownership interest in patent number 9880150. No potential conflicts of interest are disclosed by the other authors. J.W. Harbour is the inventor of intellectual property related to prognostic testing for uveal melanoma. He is a paid consultant for Castle Biosciences, licensee of this intellectual property, and he receives royalties from its commercialization.
References
- 1.Pacella I, Procaccini C, Focaccetti C, Miacci S, Timperi E, Faicchia D, et al. Fatty acid metabolism complements glycolysis in the selective regulatory T cell expansion during tumor growth. PNAS. 2018;115(28):E6546–E55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Phan LM, Yeung S-CJ, Lee M-H. Cancer metabolic reprogramming: importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer biology & medicine. 2014;11(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. 2016;2(5):e1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jones NP, Schulze A. Targeting cancer metabolism–aiming at a tumour’s sweet-spot. Drug Discov Today. 2012;17(5–6):232–41. [DOI] [PubMed] [Google Scholar]
- 5.Stine ZE, Walton ZE, Altman BJ, Hsieh AL, Dang CV. MYC, metabolism, and cancer. Cancer Discov. 2015;5(10):1024–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kruiswijk F, Labuschagne CF, Vousden KH. p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat Rev Mol Cell Biol. 2015;16(7):393. [DOI] [PubMed] [Google Scholar]
- 7.Vivas-García Y, Falletta P, Liebing J, Louphrasitthiphol P, Feng Y, Chauhan J, et al. Lineage-Restricted Regulation of SCD and Fatty Acid Saturation by MITF Controls Melanoma Phenotypic Plasticity. Mol Cell. 2020;77(1):120–37. e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singh N, Bergman L, Seregard S, Singh AD. Uveal melanoma: Epidemiologic aspects Clinical Ophthalmic Oncology: Springer; 2014. p. 75–87. [Google Scholar]
- 9.Shields JA, Shields CL, Donoso LA. Management of posterior uveal melanoma. Surv Ophthalmol. 1991;36(3):161–95. [DOI] [PubMed] [Google Scholar]
- 10.Singh AD, Turell ME, Topham AK. Uveal melanoma: trends in incidence, treatment, and survival. Ophthalmology. 2011;118(9):1881–5. [DOI] [PubMed] [Google Scholar]
- 11.Jager MJ, Shields CL, Cebulla CM, Abdel-Rahman MH, Grossniklaus HE, Stern M-H, et al. Uveal melanoma. Nature Reviews Disease Primers. 2020;6(1):1–25. [DOI] [PubMed] [Google Scholar]
- 12.Luke JJ, Triozzi PL, McKenna KC, Van Meir EG, Gershenwald JE, Bastian BC, et al. Biology of advanced uveal melanoma and next steps for clinical therapeutics. Pigment cell & melanoma research. 2015;28(2):135–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Postow MA, Kuk D, Bogatch K, Carvajal RD. Assessment of overall survival from time of metastastasis in mucosal, uveal, and cutaneous melanoma. American Society of Clinical Oncology; 2014. [Google Scholar]
- 14.Seedor RS, Eschelman DJ, Gonsalves CF, Adamo RD, Orloff MM, Amjad A, et al. Liver-directed treatment for patients with uveal melanoma hepatic metastasis: A retrospective analysis of overall survival. American Society of Clinical Oncology; 2018. [Google Scholar]
- 15.Harbour JW, Onken MD, Roberson ED, Duan S, Cao L, Worley LA, et al. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science. 2010;330(6009):1410–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kalirai H, Dodson A, Faqir S, Damato B, Coupland S. Lack of BAP1 protein expression in uveal melanoma is associated with increased metastatic risk and has utility in routine prognostic testing. Br J Cancer. 2014;111(7):1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang A, Papneja A, Hyrcza M, Al-Habeeb A, Ghazarian D. BAP1: gene of the month. J Clin Pathol. 2016:jclinpath-2016–203866. [DOI] [PubMed] [Google Scholar]
- 18.Klebe S, Driml J, Nasu M, Pastorino S, Zangiabadi A, Henderson D, et al. BAP1 hereditary cancer predisposition syndrome: a case report and review of literature. Biomark Res. 2015;3(1):14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rai K, Pilarski R, Cebulla C, Abdel-Rahman M. Comprehensive review of BAP1 tumor predisposition syndrome with report of two new cases. Clin Genet. 2016;89(3):285–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bononi A, Yang H, Giorgi C, Patergnani S, Pellegrini L, Su M, et al. Germline BAP1 mutations induce a Warburg effect. Cell Death Differ. 2017;24(10):1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baughman JM, Rose CM, Kolumam G, Webster JD, Wilkerson EM, Merrill AE, et al. NeuCode proteomics reveals Bap1 regulation of metabolism. Cell Rep. 2016;16(2):583–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dai F, Lee H, Zhang Y, Zhuang L, Yao H, Xi Y, et al. BAP1 inhibits the ER stress gene regulatory network and modulates metabolic stress response. PNAS. 2017;114(12):3192–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Porporato PE, Payen VL, Pérez-Escuredo J, De Saedeleer CJ, Danhier P, Copetti T, et al. A mitochondrial switch promotes tumor metastasis. Cell Rep. 2014;8(3):754–66. [DOI] [PubMed] [Google Scholar]
- 24.Yu L, Lu M, Jia D, Ma J, Ben-Jacob E, Levine H, et al. Modeling the genetic regulation of cancer metabolism: interplay between glycolysis and oxidative phosphorylation. Cancer Res. 2017;77(7):1564–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robertson AG, Shih J, Yau C, Gibb EA, Oba J, Mungall KL, et al. Integrative analysis identifies four molecular and clinical subsets in uveal melanoma. Cancer Cell. 2017;32(2):204–20. e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hsu PP, Sabatini DM. Cancer cell metabolism: Warburg and beyond. Cell. 2008;134(5):703–7. [DOI] [PubMed] [Google Scholar]
- 27.Carracedo A, Cantley LC, Pandolfi PP. Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer. 2013;13(4):227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Molina JR, Sun Y, Protopopova M, Gera S, Bandi M, Bristow C, et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med. 2018;24(7):1036–46. [DOI] [PubMed] [Google Scholar]
- 29.Jones RG, Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev. 2009;23(5):537–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lehuédé C, Dupuy F, Rabinovitch R, Jones RG, Siegel PM. Metabolic plasticity as a determinant of tumor growth and metastasis. Cancer Res. 2016;76(18):5201–8. [DOI] [PubMed] [Google Scholar]
- 31.Desler C, Hansen TL, Frederiksen JB, Marcker ML, Singh KK, Juel Rasmussen L. Is there a link between mitochondrial reserve respiratory capacity and aging? Journal of aging research. 2012;2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johansson PA, Brooks K, Newell F, Palmer JM, Wilmott JS, Pritchard AL, et al. Whole genome landscapes of uveal melanoma show an ultraviolet radiation signature in iris tumours. Nat Commun. 2020;11(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chua V, Aplin AE. Novel therapeutic strategies and targets in advanced uveal melanoma. Curr Opin Oncol. 2018;30(2):134–41. [DOI] [PubMed] [Google Scholar]
- 34.Karlsson J, Nilsson LM, Mitra S, Alsén S, Shelke GV, Sah VR, et al. Molecular profiling of driver events in metastatic uveal melanoma. Nat Commun. 2020;11(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hebert L, Bellanger D, Guillas C, Campagne A, Dingli F, Loew D, et al. Modulating BAP1 expression affects ROS homeostasis, cell motility and mitochondrial function. Oncotarget. 2017;8(42):72513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dey A, Seshasayee D, Noubade R, French DM, Liu J, Chaurushiya MS, et al. Loss of the tumor suppressor BAP1 causes myeloid transformation. Science. 2012;337(6101):1541–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Misaghi S, Ottosen S, Izrael-Tomasevic A, Arnott D, Lamkanfi M, Lee J, et al. Association of C-terminal ubiquitin hydrolase BRCA1-associated protein 1 with cell cycle regulator host cell factor 1. Mol Cell Biol. 2009;29(8):2181–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scheuermann JC, de Ayala Alonso AG, Oktaba K, Ly-Hartig N, McGinty RK, Fraterman S, et al. Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature. 2010;465(7295):243–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu H, Mashtalir N, Daou S, Hammond-Martel I, Ross J, Sui G, et al. The ubiquitin carboxyl hydrolase BAP1 forms a ternary complex with YY1 and HCF-1 and is a critical regulator of gene expression. Mol Cell Biol. 2010;30(21):5071–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma J, Hart GW. Protein O-GlcNAcylation in diabetes and diabetic complications. Expert review of proteomics. 2013;10(4):365–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Izawa T, Rohatgi N, Fukunaga T, Wang Q-T, Silva MJ, Gardner MJ, et al. ASXL2 regulates glucose, lipid, and skeletal homeostasis. Cell Rep. 2015;11(10):1625–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goodwin J, Neugent ML, Lee SY, Choe JH, Choi H, Jenkins DM, et al. The distinct metabolic phenotype of lung squamous cell carcinoma defines selective vulnerability to glycolytic inhibition. Nat Commun. 2017;8:15503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Daemen A, Peterson D, Sahu N, McCord R, Du X, Liu B, et al. Metabolite profiling stratifies pancreatic ductal adenocarcinomas into subtypes with distinct sensitivities to metabolic inhibitors. PNAS. 2015;112(32):E4410–E7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. PNAS. 2007;104(49):19345–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vander Heiden MG. Targeting cancer metabolism: a therapeutic window opens. Nature reviews Drug discovery. 2011;10(9):671. [DOI] [PubMed] [Google Scholar]
- 46.Camarda R, Zhou AY, Kohnz RA, Balakrishnan S, Mahieu C, Anderton B, et al. Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat Med. 2016;22(4):427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Amirouchene-Angelozzi N, Nemati F, Gentien D, Nicolas A, Dumont A, Carita G, et al. Establishment of novel cell lines recapitulating the genetic landscape of uveal melanoma and preclinical validation of mTOR as a therapeutic target. Mol Oncol. 2014;8(8):1508–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Matatall KA, Agapova OA, Onken MD, Worley LA, Bowcock AM, Harbour JW. BAP1 deficiency causes loss of melanocytic cell identity in uveal melanoma. BMC Cancer. 2013;13(1):371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Peng X, Chen Z, Farshidfar F, Xu X, Lorenzi PL, Wang Y, et al. Molecular characterization and clinical relevance of metabolic expression subtypes in human cancers. Cell Rep. 2018;23(1):255–69. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gaude E, Frezza C. Tissue-specific and convergent metabolic transformation of cancer correlates with metastatic potential and patient survival. Nat Commun. 2016;7(1):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Van Essen TH, van Pelt SI, Versluis M, Bronkhorst IH, Van Duinen SG, Marinkovic M, et al. Prognostic parameters in uveal melanoma and their association with BAP1 expression. Br J Ophthalmol. 2014;98(12):1738–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The molecular signatures database hallmark gene set collection. Cell systems. 2015;1(6):417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wilkerson MD, Hayes DN. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics. 2010;26(12):1572–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cheng H, Chua V, Liao C, Purwin TJ, Terai M, Kageyama K, et al. Co-targeting HGF/cMET Signaling with MEK Inhibitors in Metastatic Uveal Melanoma. Mol Cancer Ther. 2017;16(3):516–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12(1):323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. PNAS. 2005;102(43):15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mootha VK, Lindgren CM, Eriksson K-F, Subramanian A, Sihag S, Lehar J, et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34(3):267. [DOI] [PubMed] [Google Scholar]
- 60.Luo W, Brouwer C. Pathview: an R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics. 2013;29(14):1830–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wishart DS, Feunang YD, Marcu A, Guo AC, Liang K, Vázquez-Fresno R, et al. HMDB 4.0: the human metabolome database for 2018. Nucleic Acids Res. 2018;46(D1):D608–D17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Code availability: Computational analyses were done using publicly-available software and R packages. Data availability: Bulk RNA-Sequencing data have been deposited to the Gene Expression Omnibus (GEO) database with accession code GSE149920.