

Abstract
Uveal melanoma (UM) is a currently untreatable form of melanoma with a 50% mortality rate. Characterization of the essential signaling pathways driving this cancer is critical to develop target therapies. Activating mutations in the Gαq signaling pathway at the level of GNAQ, GNA11 or rarely CYSLTR2 or PLCβ4 are considered alterations driving proliferation in UM and several other neoplastic disorders. Here, we systematically examined the oncogenic signaling output of various mutations recurrently identified in human tumors. We demonstrate that CYSLTR2->GNAQ/11->PLCβ act in a linear signaling cascade that, via protein kinase C (PKC), activates in parallel the MAP-kinase and FAK/YAP pathways. Using genetic ablation and pharmacological inhibition, we show that the PKC/RasGRP3/MAPK signaling branch is the essential component that drives the proliferation of UM. Only inhibition of the MAPK branch but not the FAK branch synergizes with inhibition of the proximal cascade, providing a blueprint for combination therapy. All oncogenic signaling could be extinguished by the novel GNAQ/11 inhibitor YM-254890, in all UM cells with driver mutation in the Gαq subunit or the upstream receptor. Our findings highlight the GNAQ/11->PLCβ->PKC->MAPK pathway as the central signaling axis to be suppressed pharmacologically to treat for neoplastic disorders with Gαq pathway mutations.
Introduction
Uveal melanoma (UM) originates from melanocytes within the uvea of the eye, a structure comprised of the choroidal plexus, ciliary body, or iris of the eye and represent the most common intraocular malignancy in adults (1, 2). 50% of patients develop metastases, mainly to the liver (95% of patients) (1). The average survival for patients with metastatic UM is less than 6 months. Despite dramatic successes in other melanoma subtypes, immune checkpoint blockade and targeted therapies have been largely ineffective in UM (3–6), resulting in an urgent need to develop effective therapeutic regimens.
UMs do not have mutations in BRAF, NRAS and NF1 that are common in other melanoma types. Instead, more than 90% of uveal melanomas harbor constitutively active mutations in GNAQ and GNA11 (7–9), which encode the closely related α subunits Gq and G11. They are part of the Gαq family, which further comprises G14 and G15/16. Individual α subunits bind to β and γ subunits to form heterotrimeric G proteins, which transfer signaling from Gαq coupled GPCRs to downstream effectors. The mutations in UM mainly affect codons Q209 and less frequently codons R183 of either GNAQ or GNA11 and functionally compromise their GTPase catalytic activity. There is some variation between the mutation spectra of GNAQ and GNA11 (9, 10), and subtle differences in the tertiary structure and downstream signaling between GNAQQ209L and GNAQQ209P mutation are emerging (11). The 10% of UMs that do not have GNAQ or GNA11 mutations harbor recurrent mutations at codon Leu129 in CYSLTR2, a Gαq-coupled GPCR, or at Asp630 in PLCB4, encoding phospholipase C β4, the immediate downstream of Gαq (12, 13). Thus, constitutively activation of the Gαq pathway by somatic mutations can be considered disease-defining of UM. Mutations in the Gαq pathway are also found in additional neoplastic disorders, including blue nevus, and blue nevus-like melanoma, and mucosal melanoma (14), melanocytomas of the central nervous system (15), phakomatosis pigmentovascularis (16), and a range of vascular proliferations including congenital (17), and anastomosing hemangiomas (18), capillary malformations (19, 20), hepatic small vessel neoplasms (21), Sturge-Weber syndrome and port-wine stains(22, 23).
Similar to BRAF mutations in cutaneous melanomas, Gαq pathway mutations arise early during tumor evolution of melanocytic neoplasms and can already be found in benign lesions (7, 24). Additional mutations in genes including BAP1, SF3B1, or EIF1AX are required for full malignant transformation of UM (25–28).
Once activated by GTP-bound Gαq, PLCβ hydrolyses the membrane phospholipid phosphatidylinositol 4,5-bisphosphate (PIP2) into diacyl glycerol (DAG) and inositol 1,4,5-trisphosphate (IP3)(29). DAG and IP3, are important second messengers that mediate diverse cellular processes. DAG activates more than 30 proteins by binding to their C1 domains. These include conventional and novel PKC isoforms and RasGRPs (30). IP3 plays an important role in raising intracellular Ca2+ levels, which activates a plethora of signaling pathways including classic protein kinase C (PKC) isoforms. Together, PKC and RasGRPs activate the MAP-kinase pathway (31). In the setting of UM, MAPK signaling depends on two specific PKC isoforms, δ and ε, which in turn activate the RAS-exchange factor RasGRP3, which is highly abundant specifically in UM (32–34). Additional oncogenic effector pathways downstream implicated in UM include activation of the Hippo/YAP pathway via TRIO-RhoA-FAK, downstream of mutant Gαq independent of PLC β (35–37). The fact that somatic mutations in UM are highly concentrated on the CYSLTR2->Gαq->PLCβ4 pathway, however, highlights its particular importance in UM pathogenesis. Nevertheless, the knowledge of the signaling effects of the various individual mutations within this pathway is still incomplete. Specifically, it is not clear whether the different mutations in GNAQ/11 or mutations in CYSLTR2 and PLCB4 are functionally equivalent as some studies indicate that mutant Gαq may activate the MAP-kinase independent of PLCβ (38). A detailed understanding of the oncogenic signaling pathways and their branches is critical to meet the desperate need of rationally based therapies for UM and other neoplasms driven by aberrant Gαq signaling.
The goal of the current study was to characterize signaling pathways induced by mutations found in human tumors to determine paradigms for targeted therapy of neoplasms driven by mutations in the Gαq signaling pathway.
Results
Functional characterization of Gαq pathway mutations in UM
78 out of 80 (97.5%) of human UMs in The Cancer Genome Atlas (TCGA) have mutually exclusive mutations in either GNAQ (n=40, 56%), GNA11(n=36, 46%), CYSLTR2 (n=3, 4%) or PLCβ4 (n=2, 2.5%) (Figure 1A). The main hotspot in GNAQ and GNA11 is at codon 209, with Q209L and Q209P mutations accounting for 54.9% and 34.4% of all GNAQ mutations, respectively (sources TCGA and COSMIC), whereas for GNA11 92.1% are Q209L mutations (Figure 1B). A second minor hotspot affects the arginine at codon 183, with R183Q accounting for 4.3% of GNAQ mutations and R183C for 4.4% of GNA11 mutations. We systematically examined the functional characteristics of the recurring variants GNAQQ209P, GNAQQ209L, GNA11Q209L and GNAQR183Q, GNA11R183C, and the less frequent variants GNAQG48V, GNAQT175R, GNAQF228L, GNA11E191G, and GNA11E234K. The function of these latter mutations is undefined but they are classified as potentially pathogenic by PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/index.shtml). We also analyzed the rare and incompletely characterized oncogenes CYSLTR2L129Q and PLCβ4D630Y, found in the minority of UM without GNAQ/11 mutations.
Figure 1: Functional characterization of Gαq pathway mutations in UM.
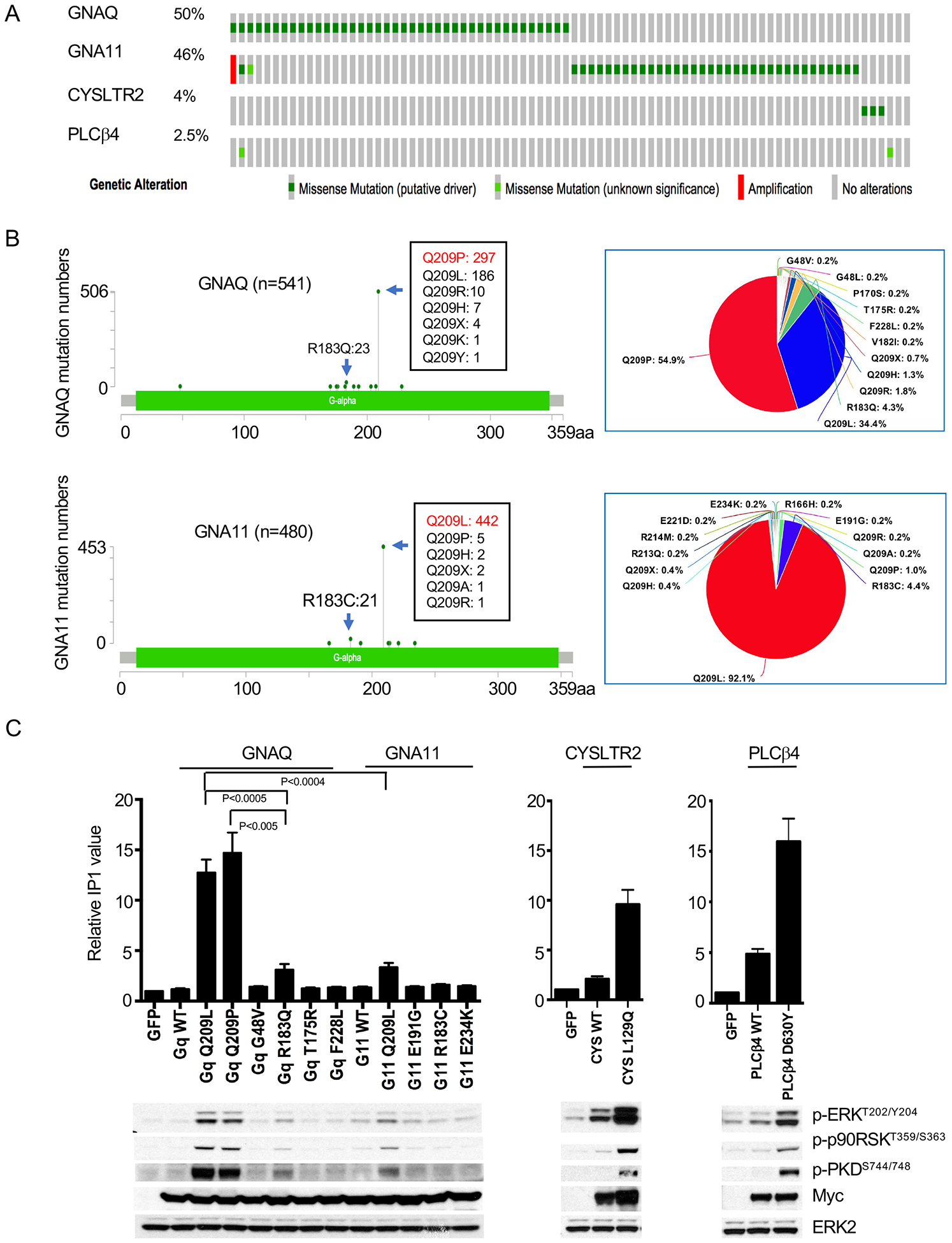
(A) Oncoprint of 80 UM samples from the TCGA UM project (https://www.cbioportal.org)(73, 74)
(B) Mutation spectra across the coding region of GNAQ and GNA11 from TCGA and COSMIC V91 (GNAQ: n=541; GNA11: n=480)
(C) IP1 accumulation to assess PLCβ activity induced by mutations in GNAQ, GNA11, and PLCB4. 293FT cells were transfected with 1μg of the respective cDNAs for 24 hours before measurement of IP1 (20,000 cells per sample) (top panel) and western blot (bottom panel). Error bars represent the SEM from at least three independent experiments. Representative western blot of three independent experiments is shown. Statistical significance was calculated by two-tailed unpaired Student t Test.
We measured signaling output of PLCβ via the accumulation of inositol monophosphate (IP1), a stable metabolite of the second messenger inositol-1,4,5-trisphosphate (IP3), which phospholipase Cβ (PLCβ) generates upon activation by Gαq (39). We assessed PKC activity by monitoring phosphorylation of protein kinase D (PKD) at residue serine 744/748, a site phosphorylated specifically by PKC (40). We monitored MAP-kinase pathway activation downstream using pERK and pp90RSK levels.
As shown in Figure 1C, there was considerable variation in the accumulation of IP1 induced by different GNAQ/11 variants. GNAQQ209L and GNAQQ209P transfected cells had more than 12-fold increases in IP1, while the increase induced by GNAQR183Q and GNA11Q209L was lower. These differences were not attributable to variation in construct expression levels (myc tags, bottom panel of Figure 1C). By contrast, the Gαq variants of unknown significance had no effect, rendering it unlikely that they are bona fide driver mutations. Consistent with their role as alternative UM oncogenes, CYSLTR2L129Q and PLCβ4D630Y also induced accumulation of IP1 (Figure 1C). All IP1 inducing mutations - in GNAQ/11, CYSLTR2 or PLCβ4 - also increased pERK, pp90RSK and pPKD, supporting the notion that they all activate the PLCβ, PKC and MAPK signaling pathway (Figure 1C, bottom panel).
CYSLTR2, GNAQ/11 and PLCβ form a linear signaling cascade that drives MAPK signaling via PKC
In order to explore the functional relationship among CYSLTR2, GNAQ/11 and PLCβ, we utilized the Gαq inhibitor YM-254890 (41). As shown in Figure 2A, The YM-254890 compound inhibited IP1 accumulation induced by GNAQQ209L, GNAQQ209P, GNAQR183Q and GNA11Q209L in a dose dependent manner. It also inhibited IP1 accumulation in CYSLTR2L129Q transfected cells, confirming that CYSLTR2L129Q activates PLCβ via GNAQ/11. By contrast, it had no effect on PLCβ4D630Y, which confirms that YM-254890 acts directly on GNAQ/11, upstream of PLCβ. Notably, YM-254890 was less effective on GNAQQ209L compared to the other Gαq variants. As the Gαq family has additional members, GNA14 and GNA15/16 (42, 43), we tested the selectivity of YM-254890 acts on Gαq family members, using GNA14Q205L (44) and GNA15Q212L (45), constitutively active mutants corresponding to GNAQ/11Q209L. For comparison, we also included the GNASQ227L oncogene (46), a member of the Gs family as a control. As shown in Figure S1A, all three variants increased pERK, pp90RSK, but only GNA14Q205L and GNA15Q227L increased IP1 accumulation and pPKD. As expected, GNASQ227L increased cAMP levels. YM-254890 had no effect on GNA14Q205L and GNA15Q227L nor GNASQ227L (Figure 2A).
Figure 2. CYSLTR2, GNAQ/11, and PLCβ act in a linear signaling cascade that activates MAP-kinase via PKC.
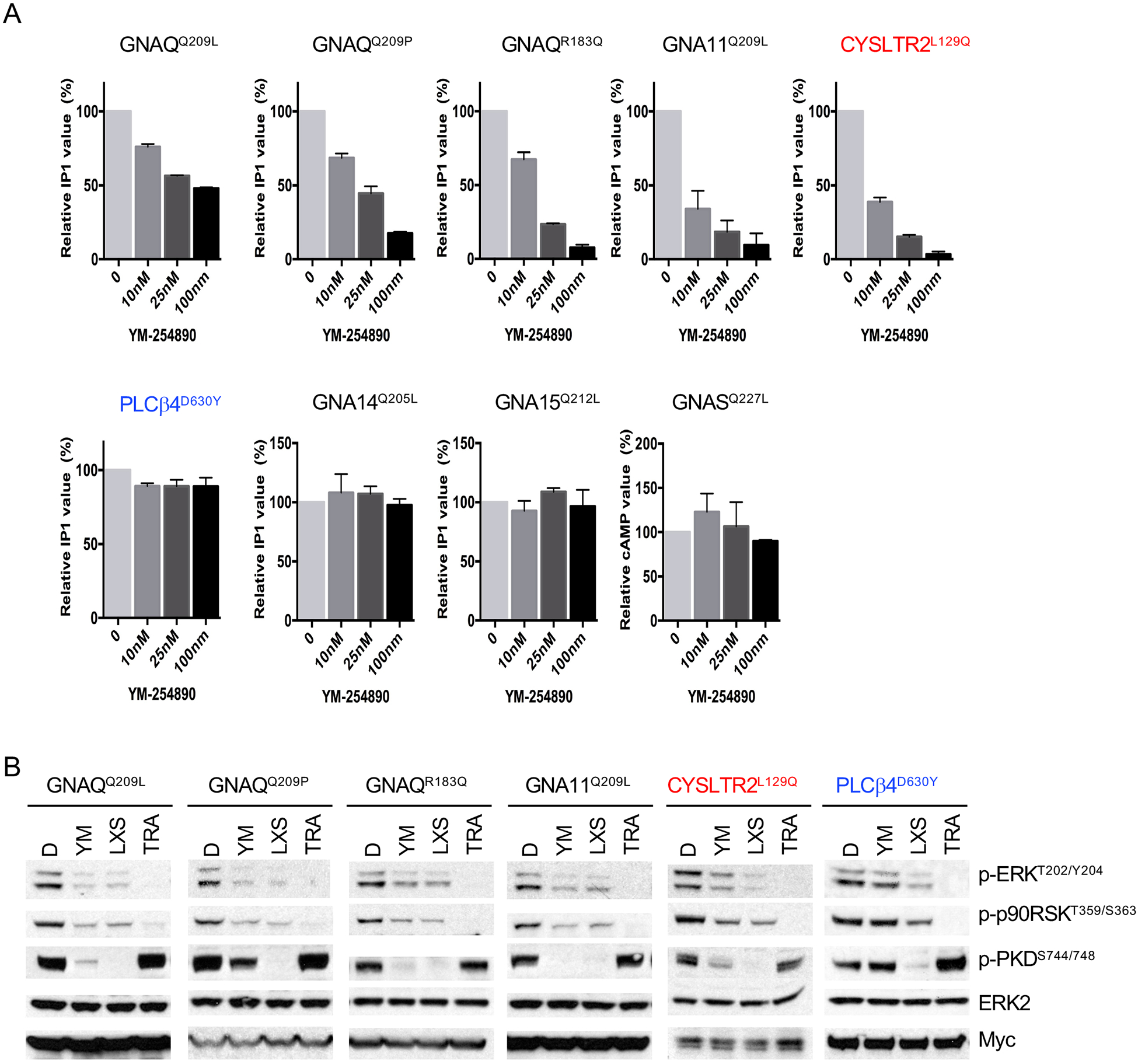
(A) The effect of YM-254890 on IP1 accumulation induced by different Gαq mutants. 293FT cells transfected with indicated cDNAs for 24 hours were treated with YM at indicated concentrations for 2h (20,000 cells per sample). Error bars represent the SEM.
(B) PKC and MAPK pathways are activated by mutations of GNAQ, GNA11, CYSLTR2, and PLCB4 and PKC activation is upstream of the MAP-kinase pathway. 293FT cells transfected with different Gαq variants were treated with DMSO (D), the Gαq inhibitor YM-254890 (YM) at 100nM, the PKC inhibitor LXS196 (LXS) at 1μM, or the MEK inhibitor Trametinib (TRA) at 100nM for 24 hours.
We probed the hierarchical order of the signaling components using inhibitors of GNAQ/11, PKC, and MEK. YM-254890 strongly inhibited PKC and MAPK in cells transfected with GNAQQ209L, GNAQQ209P, GNAQR183Q, GNA11Q209L, CYSLTR2L129Q as evidenced by a reduction of pERK, pp90RSK, and pPKD (Figure 2B) confirming that CYSLTR2 acts upstream of Gαq. By contrast, YM-254890 had no effect on cells transfected with PLCβ4D630Y, consistent with its position downstream of Gαq (Figure 2B and Figure S1B). By contrast, the PKC inhibitor LXS196 that is currently under clinical investigation (47) strongly inhibited pERK, pp90RSK and pPKD in all UM oncogenes. Trametinib suppressed pERK and pp90RSK levels across all settings but did not suppress PKC activity. In summary, these data show that CYSLTR2->Gαq->PLCβ represents a linear signaling module that activates PKC, which in turn activates MAPK signaling.
PLCβ in parallel activates both FAK signaling and MAPK signaling via PKC
Previous studies have proposed that oncogenic GNAQ/11 activates FAK signaling pathway and that this occurs independently of PLCβ in UM (37)(Figure 3A). In order to determine the branchpoint to FAK signaling, we introduced the PLCβ4D630Y into 293T cells. As shown in Figure 3B, mutant but not wild type PLCβ4 activated FAK as evidenced by increased phosphorylation at Y397, putting PLCβ4 upstream of FAK. This activation could be suppressed by the PKC inhibitor LXS196, in a dose-dependent manner (Figure 3C) and LXS196 also suppressed FAK phosphorylation in UM cells with GNAQ or GNA11 mutations, with no effect on BRAF mutant cells (Figure 3D). Similar results were obtained with AHT956, another PKC inhibitor (Figure S2). These data indicate that in the context of Gαq pathway mutations FAK activation occurs via PLCβ and involves PKC.
Figure 3: PLCβ activates FAK and MAPK signaling in parallel via PKC.
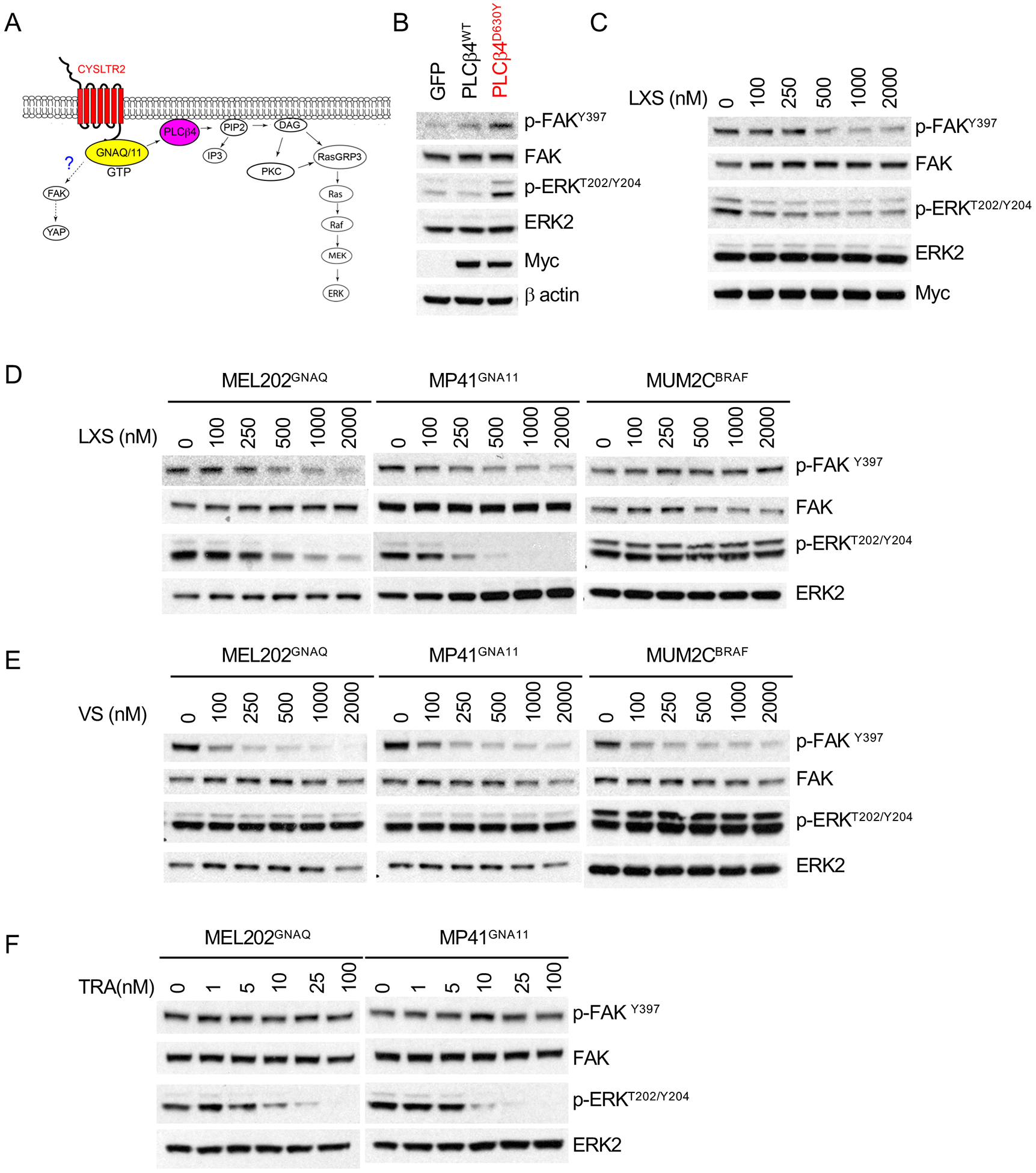
(A) Proposed model of FAK and YAP signaling in uveal melanoma(37)
(B) PLCβ4D630Y increased FAK phosphorylation. Western blot of 293T cells transfected with GFP, PLCβ4wt and PLCβ4D630Y for 24 hours.
(C) LXS196(LXS) inhibited FAK phosphorylation in 293T cells transfected with PLCβ4D630Y in a dose-dependent manner.
(D) LXS196(LXS) inhibited FAK phosphorylation in GNAQ/11 mutant UM cells but not in BRAF mutant cells. Cells were treated with indicated dose of LXS196 for 24 hours and subjected to western blot.
(E) VS-4718(VS) suppressed FAK phosphorylation in a dose-dependent manner but not ERK phosphorylation in GNAQ/11 mutant UM cells.
(F) Trametinib (TRA) inhibited ERK phosphorylation in dose-dependent manner but not FAK phosphorylation in GNAQ/11 mutant UM cells.
In the above experiments FAK activation occurred concomitantly with MAP-kinase pathway activation. We examined the relationship between these two pathways by using the FAK inhibitor VS-4718 (48) and the MEK inhibitor trametinib. VS-4718 suppressed FAK phosphorylation in a dose-dependent manner, but had no effect on p-ERK levels, even at higher concentrations in GNAQ (MEL202), GNA11 (MP41), BRAF mutant (MUM2C) cell lines (Figure 3E). By contrast, Trametinib had an inverse response pattern with dose-dependent inhibition of ERK phosphorylation and no effect on p-FAK levels (Figure 3F). These findings indicate that mutations in the Gαq pathway at the level of Gαq or PLC β activate PKC, after which the signal flux branches into the MAPK and FAK pathways.
PLCβ/PKC activity but not FAK/YAP is elevated in UM cell lines as a consequence of Gαq pathway mutations
The results above highlight that all oncogenic signaling in UM goes through PLCβ. Next, we investigated the intrinsic PLCβ activity in UM cell lines. Nearly all UM cell lines that are available to the research community harbor mutations in GNAQ or GNA11. One exception is the MEL290 line whose driver mutations are not known. We determined the intrinsic PLCβ activity in 12 UM cell lines, 11 with different Gαq mutations (GNAQ: Q209L, Q209P, R183Q; GNA11: Q209L) and MEL290. These cell lines also harbor a range of secondary driver mutations including those in SF3B1, EIF1AX or BAP1, representative of the genetic landscape of human UM (mutation details in supplemental table 1). The OMM1.3 and MM66 lines were originally derived from liver metastases and OMM1 from subcutaneous metastases whereas the remainder stem from primary UMs. Cutaneous melanoma (CM) cell lines have no mutations in the Gαq pathway and instead have MAP-kinase pathway mutations, mainly in BRAF or NRAS. We used five different cutaneous CM cell lines to represent the Gαq wild type state of the melanocytic lineage. As shown in Figure 4A, GNAQ/11-mutant UM cell lines had more than 20-fold higher levels of IP1 compared to CM cell lines. The ‘driver-less’ MEL290 UM cell line also had elevated IP1 amounts, albeit lower than UM cells with GNAQ/11 mutations. In UM cells, the total IP1 amount increased with the cell number (Figure S3A) and time (Figure S3B), but remained flat in CM cells, indicating that PLCβ activity is very low or absent in these cells. Taken together, these results indicate that UM cells invariably have high PLCβ activation generating high levels of the second messenger IP3, whereas CM cells do not. This pattern is independent of mutation of secondary driver mutations in BAP1, SF3B1, and EIF1AX.
Figure 4: PLCβ/PKC activity but not FAK/YAP is elevated in UM cell lines as a consequence of Gαq pathway mutations.
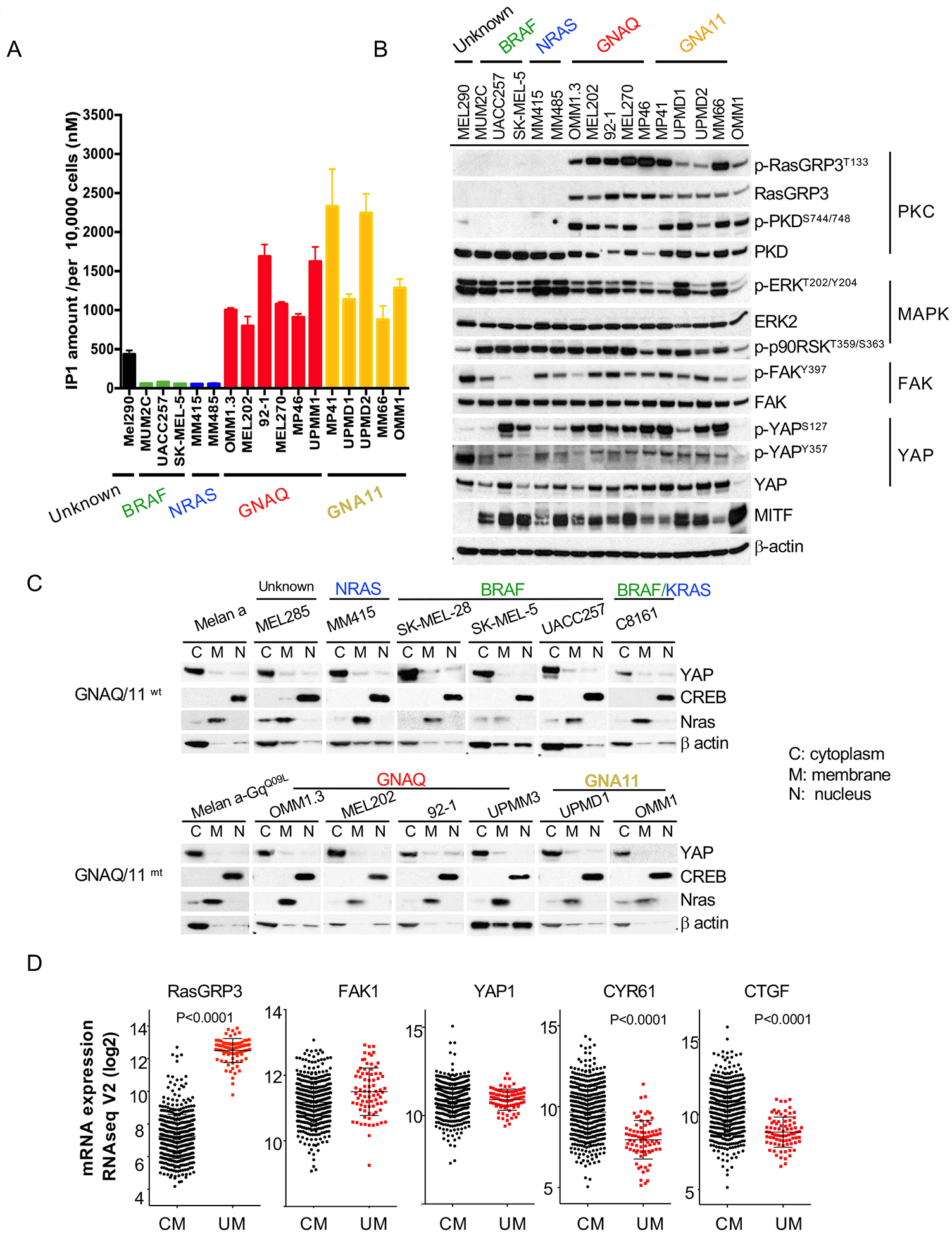
(A) Elevated levels of IP1 in cell lines with Gαq pathway mutations. IP1 accumulation was measured in UM and CM cell lines with different somatic mutations. 10,000 cells were subjected to each IP1 measurement.
(B) Western blot of melanoma cell lines with different driver mutations shows selective and consistent activation of the PKC pathway in UM cells, whereas FAK, YAP, and MAP-kinase pathways show similar activation in CM and UM.
(C) Western blots of subcellular fractions of cell lines with or without GNAQ/11 mutations probed with indicated antibodies show YAP1 mainly localized in cytoplasmic fractions for all cell lines regardless of genetic mutation status. CREB is used as a control for nuclear localization and NRAS for membrane localization.
(D) TCGA RNAseq data shows selectively increased expression of RasGRP3 in UM compared to cutaneous melanoma (CM), but similar expression levels of FAK1 and YAP1. The YAP targets CYR61 and CTGF show lower expression in UM tissues than in CM tissues. Error bars represent the SEM.
To extend this analysis, we next assessed PKC, MAPK and FAK signaling downstream of PLCβ in these cell lines. As PKC phosphorylates RasGRP3 at residue T133 in UM and induces its expression (32), we included it as marker for PKC activity. As shown in Figure 4B, all 10 melanoma cell lines with GNAQ/GNA11 mutations expressed RasGRP3, p-RasGRP3T133, and p-PKDS744/748, whereas CM cell lines did not. MEL290 expressed only trace levels of p-PKDS744/748. These data indicate that across the board of UM cells with diverse secondary mutations but not in melanoma cell lines with other mutations, PKC is invariably activated. By contrast, MAPK activation was observed in all melanoma cell lines, irrespective of mutation status. Interestingly, FAK signaling also showed no difference across melanoma cell lines and had no specific associations with mutations in the Gαq pathway.
The transcription coactivator Yes-associated protein (YAP), a major downstream effector of the Hippo pathway (49), is activated by FAK in UM cells (37). When the Hippo signaling pathway is inactivated, YAP translocates into the nucleus and interacts with TEAD family transcription factors and others to stimulate gene transcription (50). YAP’s transcriptional activity is regulated by phosphorylation. Phosphorylation at S127 is inhibitory as it sequesters it in the cytoplasm (50, 51), whereas phosphorylation at Y357 is activating as it increases its stability and nuclear localization (52–54). As shown in Figure 4B, the expression levels of p-YAPS127, p-YAPY357 and total YAP1 were similar in melanoma cell lines regardless of their genetic status. We further evaluated the subcellular localization of YAP, which is important for its transcriptional activity. In the immortalized mouse melanocyte line melan-a YAP remained cytoplasmic in the setting of GNAQQ209L mutation (Figure 4C). Similar results were observed in a panel of melanoma cells and no increased nuclear localization was seen in cells with Gαq pathway mutations. In the 80 UM cases in TCGA the expression levels of the YAP-target genes CTGF and CYR61 is decreased compared to CM cases in TCGA, whereas RasGRP3 expression is significantly higher in UM tissues than in CM samples as previously described (Figure 4D). In aggregate, these data affirm the selective activation of PLCβ and PKC in melanomas with Gαq pathway mutations, and do not specifically implicate FAK/YAP signaling in this context.
PKC/MAPK signaling but not FAK/YAP signaling is essential for proliferation of UM cells with Gαq pathway mutations
Our above data indicate that in UM cells PLCβ->PKC activates the FAK/YAP and MAPK signaling as two parallel branches. We next evaluated, the individual contribution of these two branches for UM proliferation and cell survival by comparing their inhibition to inhibition at the level of PKC. PKC δ and ε mediate MAPK activation in UM(32), and their simultaneous knock-down reduced cell survival by 50–75% at 6 days in UM cells with GNAQ or GNA11 mutations (Figure 5A, top panel). Comparable effects were obtained after ERK1/2 knock-down. In contrast, FAK knock-down reduced cell viability by no more than 25% and YAP1 knock-down had no effect. We confirmed that expression levels of the targeted genes remained suppressed during the course of these experiments (Figure 5A, bottom panel). Similar results were observed with a doxycycline-inducible CRISPR/Cas9 system(55). gRNAs targeting human YAP1, RasGRP3 or GNAQ were introduced into OMM1.3 cells, stably expressing inducible Cas9. The expression of targeted protein was significantly decreased after doxycycline treatment in these cell lines (Figure 5B). Knock-out of YAP1 has no effect on long term cell proliferation, whereas genetic depletion of RasGRP3 and GNAQ significantly inhibited cell proliferation.
Figure 5. PKC/MAPK and not FAK/YAP activity is essential for proliferation of UM cells with Gαq pathway mutations.
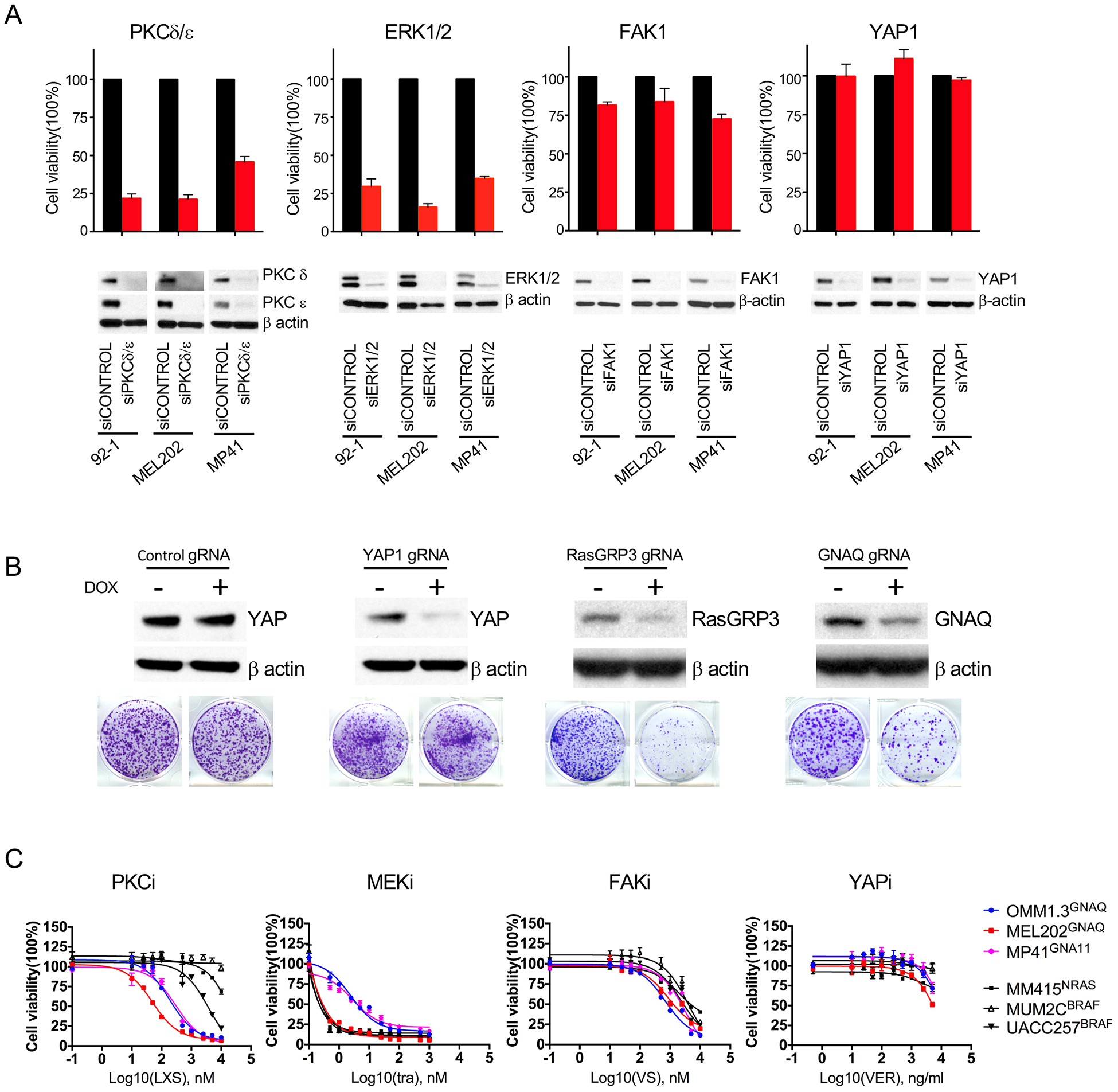
(A) siRNA mediated knock-down of PKC δ and ε or ERK1/2 but not FAK1 or YAP1 affects UM cell viability. UM cells (92–1, MEL202, MP41) were transfected with indicated siRNAs for 6 days and subjected to cell counting and western blot.
(B) CRISPR-Cas9 mediated knock-out of GNAQ, and RasGRP3 but not YAP1 inhibited cell proliferation in GNAQ- mutant OMM1.3 cells. Cells stably expressing lenti-iCas9-Neo were transduced with lenti-guide carrying indicated gRNA. After puromycin selection, cells were treated with or without 1μg/ml doxycycline (Dox) for 72h for western blot (top panels), or cultured for 10 days and stained with crystal violet (bottom panel).
(C) UM cells express selective sensitivity to the PKC inhibitor LXS196 (LXS) but not to the FAK inhibitor VS-4718(VS) and the YAP inhibitor verteportin (VER). Cell viability analysis after treated with indicated inhibitors with different dosages.
Error bars represent the SEM.
Similar results were obtained with pharmacological inhibitors of the respective pathways. Three GNAQ/11 mutant UM (OMM1.3, MEL202 and MP41) and three BRAF/NRAS mutant CM (MM415, MUM2C and UACC257) cell lines were treated with PKC, FAK, MEK, and YAP inhibitors, respectively. UM cell lines only expressed selective sensitivity to LXS196 (Figure 5C), with IC50 ranging from 49nM to 303nM. By contrast, there was no difference in the drug response curves between UM and CM cells for VS-4718, Verteporfin, and CM cell lines were more sensitive to MEK inhibitor Trametinib.
Together, these findings show that the PKC/RasGRP3/MAPK signaling pathway drives cell proliferation in UM cells and fail to confirm a specific role of FAK or YAP signaling in these cells.
Combined inhibition of PKC & MEK but not FAK & MEK or FAK & PKC synergistically reduces cell viability in UM cells
Because PKC in parallel activates both FAK and MAPK signaling, we also evaluated whether combined inhibition of FAK and MAPK would increase the therapeutic effect of PKC inhibition. We exposed one GNAQ mutant (OMM1.3) and one GNA11 mutant cell line (MP41) to pair-wise combinations of three inhibitors (VS-471 for FAK, Trametinib for MEK, and LXS196 for PKC) across different concentration ranges and determined the effect on cell viability after four days. The 64 different combinations for each drug combination are depicted in a dose matrix for each cell line (Figure 6 and Figure S4). Synergy was calculated using the Combenefit platform software (56). Bliss model (Figure 6 and Figure S4) and Loewe model (Figure S5) analyses revealed that the effects of LXS196 and Trametinib were strongly synergistic in both cell lines. By contrast, limited synergy was observed between VS-4718 when combined with either Trametinib or LXS196.
Figure 6. Combined inhibition of PKC & MEK but not FAK & MEK or FAK & PKC synergistically reduces cell viability in UM cells.
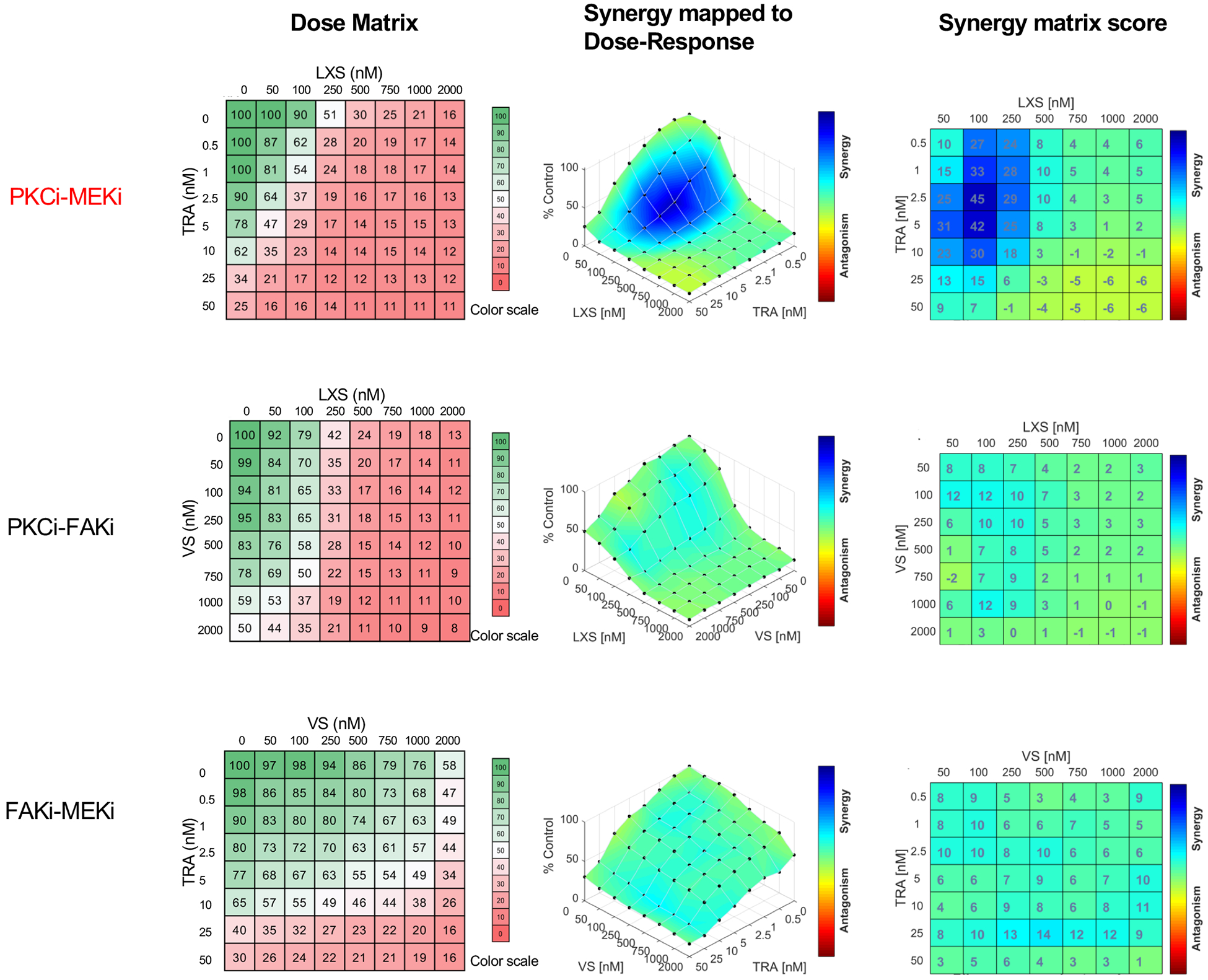
Left panel: Drug dose matrix data of MP41 cell line (left panel). The numbers in the individual cells indicate the percentage of viability of cells treated for 96 h with the corresponding compound combination relative to vehicle control-treated cells. The data were visualized over matrix using a color scale. Synergy was calculated using Bliss model with Combenefit software and indicates strong synergy for the LXS196 and Trametinib combination (middle and right panel).
YM-254890 selectively suppresses essential oncogenic signaling and growth in GNAQ/11 mutant UM cells
The prior studies nominate the GPCR-> GNAQ/11-> PLC β ->PKC module as the central conduit for oncogenic signaling in UM. We investigated whether the GNAQ/11 specific inhibitor YM-254890, can suppress growth in a broad range of UM cells with different secondary driver mutations and primary and metastatic origin. We found that YM-254890 selectively inhibited IP1 production in a dose-dependent manner in all 10 UM cell lines with GNAQ/11 mutations, irrespective of their pattern of additional mutations (Figure 7A, Figure S6A, supplemental table 1) and had no effect on CM cell lines and on the MEL290 cell line. Similarly, YM-254890 dose-dependently extinguished RasGRP3/MAPK signaling (Figure 7B and Figure S6B) in all UM cell lines irrespective of mutations in EIF1AX, SF3B1 or BAP1, but had no effect on CM cell lines. FAK signaling was also suppressed by YM-254890 (Figure 7B and Figure S6B). At concentration above 10nM, YM-254890 induced apoptosis in GNAQ/11 mutant cells, as evidenced by PARP cleavage (Figure 7B and Figure S6B).
Figure 7: YM-254890 selectively suppresses essential oncogenic signaling in GNAQ/11 mutant UM cells.
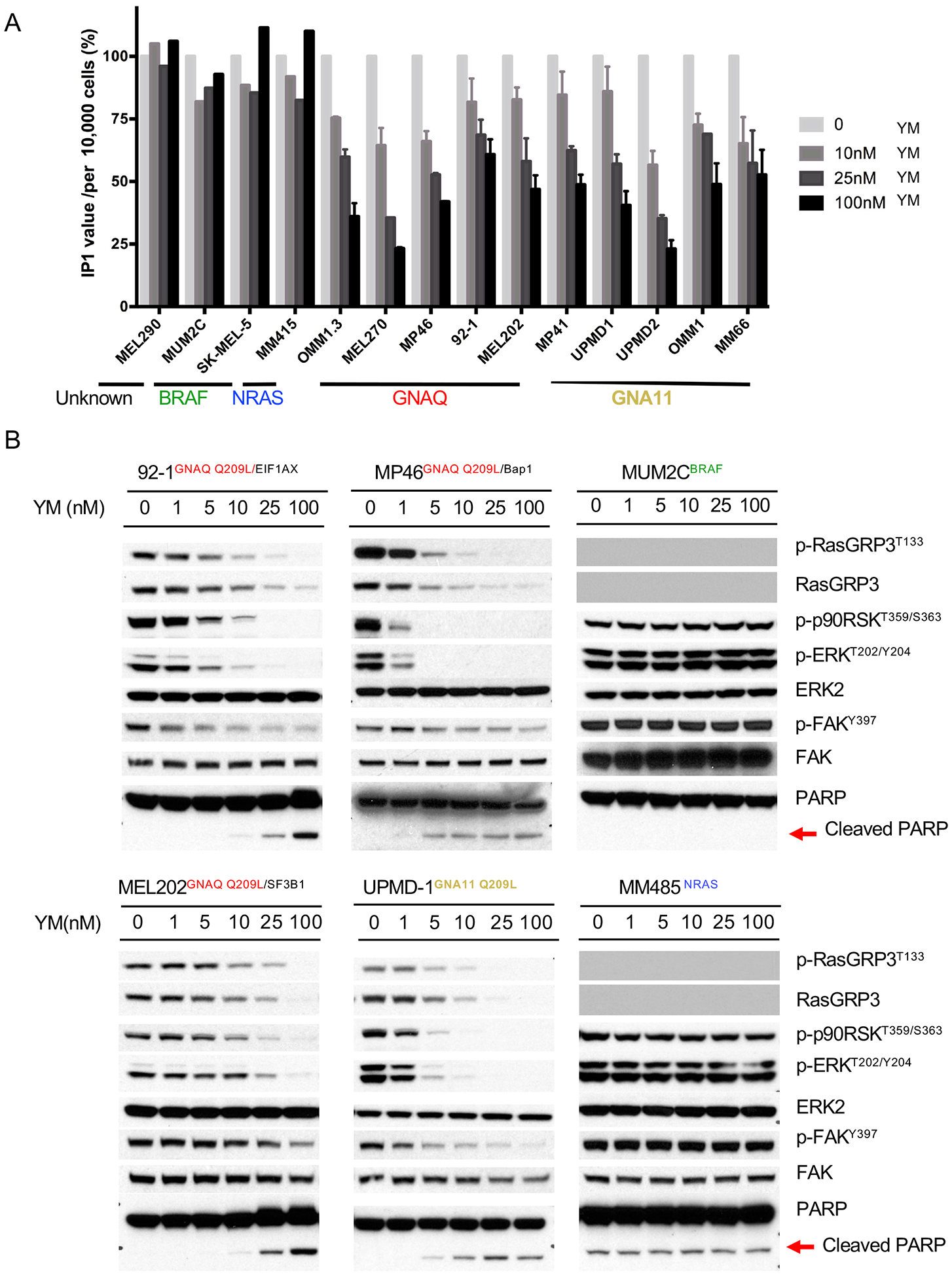
(A) YM-254890 reduces IP1 production in melanoma cell lines with Gαq pathway mutations but not with other mutations. 104 Cells were treated with indicated concentration of YM for 2 hours and then subjected to IP1 measurement. The level of IP1 in cells without YM treatment was normalized to 100%. The absolute IP1 changes in cells see supplemental Figure S6A.
(B) YM-254890 blocked RasGRP3/MAPK and FAK signaling in a concentration-dependent manner in cell lines with Gαq pathway but not BRAF or NRAS mutations. Western blots of GNAQ (92–1, MP46, MEL202) or GNA11 (UPMD-1) mutant cell lines and cell lines with BRAF mutation (MUM2C) or NRAS mutation (MM485) treated with increasing concentrations of YM for 24 hours. As noted, RasGRP3 expression was undetectable in MUM2C and MM485 cells.
Error bars reflect the SEM as above.
Growth assays showed a similar pattern in that all UM cell lines were highly sensitive to YM-254890 (IC50 0.5nM - 84nM), whereas the compound had no effect on CM cell lines (Figure 8A). Interestingly, GNA11 mutant cells were more sensitive (IC50: 0.5–2.7nM) than GNAQ mutant cells (IC50: 8.2 to 84.7nM) (p-value <0.05) (Figure 8 A,B). YM-254890 also markedly inhibited colony formation of UM cells with GNAQ (92–1, MEL270) or GNA11 mutations (MP41, UPMD1), with no effect on CM cells (Figure 8C).
Figure 8: YM-254890 selectively inhibits proliferation of melanoma cell lines with Gαq pathway mutations.
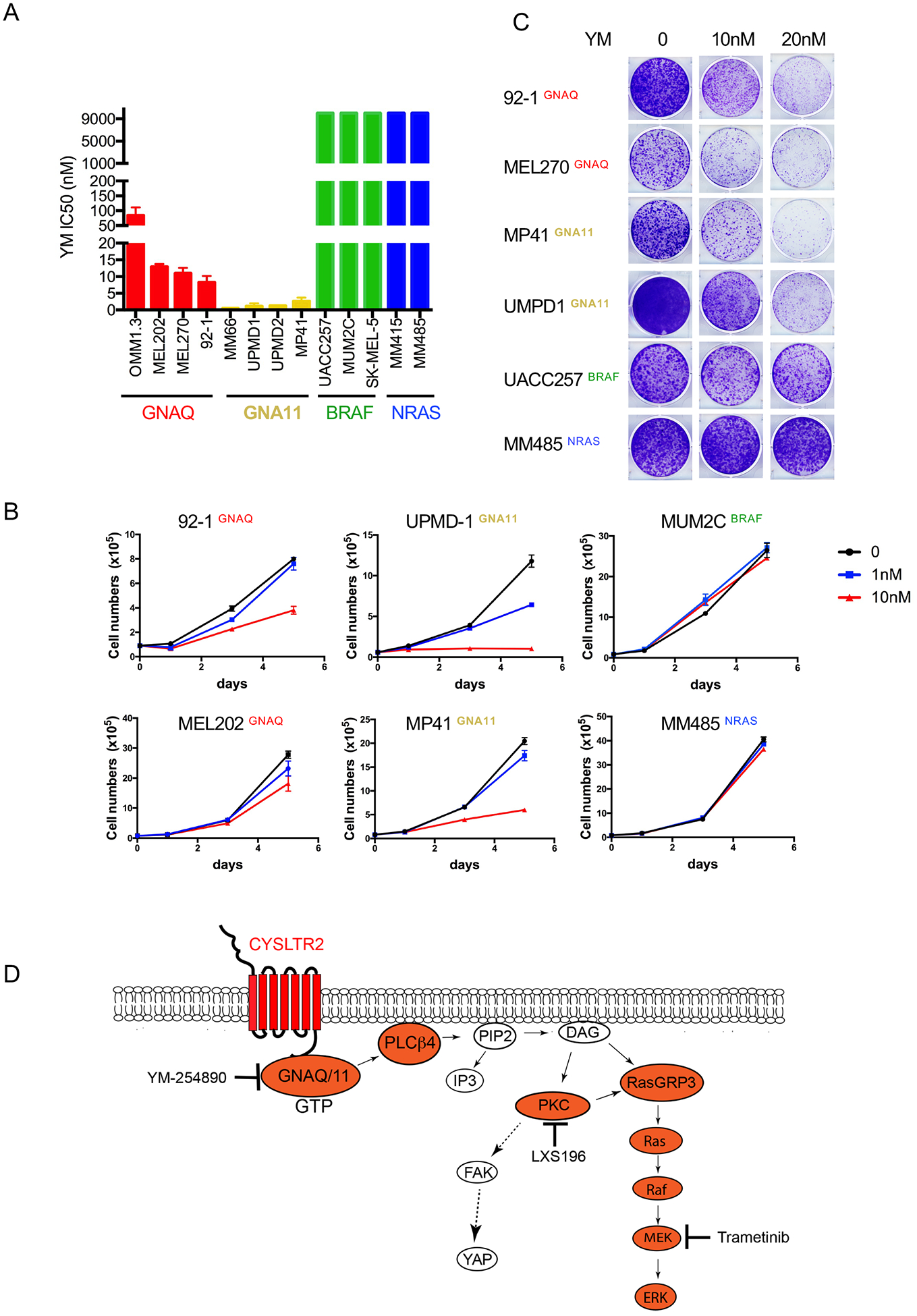
(A) IC50 of cell lines treated for 4 days with YM-254890, grouped by mutation status shows selective inhibition of melanoma cell lines with Gαq mutations.
(B) Growth kinetics of cell lines with or without Gαq mutations, treated with YM-254890 at 1 nM or 10 nM, compared to vehicle (DMSO) over 5 days. Cells were counted at day 0, 1, 3 and 5.
(C) Cells treated with different dosages of YM for 10–14 days and stained with crystal violet.
(D) Model of Gαq signaling in uveal melanoma. CYSLTR2->Gαq->PLCβ is a linear signaling cascade that activates PKC and then branches into the MAP-kinase and FAK/YAP pathway. While the PKC->RasGRP3->MAPK signal axis is essential for UM proliferation, the FAK/YAP pathway is not. The Gαq->PLCβ->PKC-> MAP-kinase pathway is the core signaling module for targeted therapy in UM.
In sum, YM-254890 effectively suppressed signaling downstream of Gαq and cell proliferation across a broad range of UM cells with different genetic backgrounds and irrespective of primary or metastatic provenance.
Discussion
Targeted therapy has become a mainstay of therapy in melanomas with BRAF mutations. This is despite the fact of considerable genetic diversity of secondary genetic alterations due to the high mutation burden in these tumors. UM from the perspective of the pattern of somatic mutations is a considerably less complex tumor type, with a very low mutation burden and few chromosomal aberrations(9, 24). Nevertheless, it has to date resisted attempts to developing effective targeted therapies.
The pattern of somatic mutations strongly points towards the Gαq signaling pathway as a therapeutic target. Over 90% of uveal melanomas have mutations at the level of the the Gαq family members GNAQ and GNA11 and the remainder mostly have mutations in CYSLTR2, a GPCR known to be Gαq-coupled, or PLCB4, encoding the Gαq effector PLCβ4. This distribution of mutations on its own implicates the CYSLTR2->Gαq->PLCβ4 module as critical for oncogenic signaling in UM. Our detailed in vitro studies confirm this notion. We show that mutant CYSLTR2 indeed activates GNAQ/11 and activates PLCβ. This and the finding of activating mutations in PLCB4 in the few UMs without GPCR or Gαq mutations points to PLC β activation as the central signaling node in UM.
We confirm this by showing that, across the board, UM cell lines show activation of PLCβ, whereas cutaneous melanomas do not. We note that the MEL290 line included in our panel is attributed to a uveal melanoma arising within a nevus of Ota but does not show any of the common genetic attributes of UM, such as mutations in the Gαq pathway or affecting BAP1, SF3B1, or EIF1AX. It also does not express melanocyte lineage markers such as HMB45 or Melan-A/MART1 (57), or RasGRP3 or MITF that is detected in all Gαq mutant UM cells. While it also showed increased PLC β activity in our study compared to cutaneous melanoma cell lines, its provenance remains to be confirmed genetically.
While activation of PLCβ was uniform across UM cell lines in our experiments, we also noted considerable variation in the level of activity as a function of the specific genetic alteration in the pathway, when individual driver mutations were compared in an isogenic setting. This was unexpected as GNAQ and GNA11 share 90% protein sequence similarity. GNAQQ209L and GNAQQ209P increased IP1 levels 12-fold compared to a merely 3-fold increase by GNA11Q209L. R183 mutants of GNA11 also resulted in lower IP1 accumulation than corresponding mutations in GNAQ. The finding that R183 mutants had an overall weaker signaling output than Q209 mutants was expected, as the mutations at R183 are known to only partially impede GTPase activity (58, 59). The difference between PLC β induction of mutant GNAQ and GNA11 might be attributable to variation in their effector spectrum resulting from subtle changes in their molecular conformations. The PLC β family consists of four homologues (PLCβ1–4)(29) and Gαq family members stimulate homologues with different potency (60–62). In our experiments, the difference in IP1 accumulation was only apparent in the isogenic setting using transfected 293T cells, carefully controlling for transgene expression levels. This contrasted with markedly reduced variation in IP1 levels among UM cell lines despite similar variations in GNAQ and GNA11 mutations and therefore indicates additional complexity in how cancer cells with driver mutations that vary in their ability to activate PLC β regulate optimal second messenger production. Future studies will reveal whether this is due to differences in the spectrum PLC β homologues, their expression, and/or enzymatic activity.
The variation among the biologic effects of the various mutations in the CYSLTR2->Gαq->PLC β pathway was also reflected in their response to YM-254890. The compound was less effective on GNAQQ209L compared to the other Gαq variants including GNAQQ209P. Differential effects on GNAQQ209L and GNAQR183Q have already been reported (41). The variation is possibly due to the unique conformation of GNAQQ209L resulting in lower affinity for YM-254890 compared to GNAQQ209P (11).
Several studies have implicated additional pathways to be specifically relevant in UM, specifically as YAP and FAK signaling. Some have proposed that activation of these pathways occurs independent of PLC β(36, 37). Our results do not support a PLCβ-independent branch to the FAK pathway. While we confirm its activation in UM, we show that it is activated downstream of PLC β and can be suppressed by PKC inhibition. Most importantly, we find no difference in the level of activation compared to cutaneous melanoma and that FAK inhibition alone or in combination with MEK or PKC inhibition does not cause selective or synergistic suppression of UM cells viability.
The same holds true for the YAP pathway, which has been previously implicated in UM oncogenesis downstream of Gαq and positioned downstream of FAK(35, 36). In our hands, genetic depletion or pharmacological inhibition of YAP signaling did not affect UM cell viability or proliferation, and UM tumors in TCGA do not show expression signatures of increased YAP activity. Our results are in line with a recent study that also found no effect of YAP depletion on UM cell lines and a lack of correlation between YAP activation levels and outcomes in UM patients (63). These findings along with the observation that genetic analyses of human UMs have failed to identify mutations in the FAK or YAP pathways (9, 24), make these pathway unlikely targets for therapeutic intervention in this disease.
Our results refocus the attention on the CYSLTR2->Gαq->PLCβ is a linear signaling cascade as the core signaling module driving oncogenic signaling in UM. The cascade continues via PKC to RasGRP3 to the MAP-kinase pathway. Targeted cancer therapy has been most successful in situations, in which a proliferation-driving gain of function mutation can be targeted directly. This is exemplified by therapeutic successes with kinase inhibitors directed against the BRAF, EGFR, KIT, BCR-ABL fusion and ALK oncogenes. However, it has been difficult to target oncogenes, whose activation is due to loss of function of their intrinsic ability to switch themselves off. Ras family members most prominently exemplify this conundrum. The mechanism of mutational activation of GNAQ/11 is essentially similar to that of RAS oncogenes but the prospect of their direct pharmacological inhibition has moved within reach with the recent identification of direct inhibitors (41, 64). In this study, we provide evidence that YM-254890 is effective in blocking signaling and proliferation across the board in a broad panel of UM cells carrying different activating mutation in Gαq pathway, independent of secondary mutations and tumor cell origin (primary or metastasis). Our results are consistent with other studies with another Gαq inhibitor FR900359 (38, 65, 66).
Although YM-254890 and FR900359 have similar structures, differing only by one amino acid and acyl group, we demonstrate that YM-254890 is GNAQ/11 specific. In contrast to FR900359 it had no inhibitory activity against GNA14 (64). Several recent studies show the two compounds have different properties such as differential isomer ratios, dissociation rates from Gαq, inhibitory effects on other Gαq family proteins (67–69), thus potentially resulting in divergent biological effects. Therefore, YM-254890 as a GNAQ/11 specific inhibitor would be expected to have less toxicity due to specific G protein inhibition than FR900359.
In summary our findings identify the GPCR-> Gαq->PLCβ->PKC cascade as the core signaling pathway in UM. We demonstrate that this pathway can be targeted successfully at the levels of Gαq or PKC and the therapeutic effect augmented by combination with MAP-kinase pathway inhibitors.
Materials and methods
Plasmid and Reagents
Wild type GNAQ and GNA11, GNAQQ209L, GNAQQ209P, GNA11Q209L, wild type CYSLTR2 full-length cDNAs were generated from mRNAs isolated from human UM cell lines (92–1, OMM1.3, UPMD1) by using reverse transcription polymerase chain reaction and cloned into pLenti-MYC-DDK vector from Origene (Rockville, MD). The human wild type PLCB4 full-length cDNA was obtained from GeneCopoeia (Rockville, MD) and was cloned into pLenti-MYC-DDK vector. Other GNAQ and GNA11 variants (GANQG48V, GNAQR183Q, GNAQT175R, GNAQF228L, GNA11E191G, GNA11R183C, GNA11E234K), CYSLTR2L129Q and PLCβ4D630Y constructs were generated by site-directed mutagenesis using the Quikchange II kit from Agilent Technologies (Santa Clara, CA). All constructs were confirmed by Sanger sequencing. Human wild type GNA14, GNA15, GNAS, GNA14Q205L, GNA15Q212L, and GNASQ227L cDNA constructs were from cDNA Resource Center (Bloomsburg, Pennsylvania). The luciferase construct was obtained from the William A Weiss laboratory at UCSF. Lenti-iCas9-neo plasmid was purchased from Addgene (#85400)(55). LentiGuide-Puro was obtained from Addgene (#52963)(70).
YM-254890 was purchased from Adipogen (San Diego, CA). TPA and Verteporfin were from Sigma (St Louis, MO). Trametinib and VS-4718 were obtained from Selleckchem (Houston, TX). LXS196 was got from Chemie TEK (Indianapolis, IN). AHT956 was synthesized at Novartis Pharma AG (East Hanover, NJ).
Cell culture and cell line generation
The sources of UM cell lines, CM cell lines and mouse Melan -a cells expressing GNAQQ209L have been previously described (32, 71). MP46, MP41 and MM66 UM cell lines were kindly provided by Dr. Roman-Roman from Institut Curie, France (72). All melanoma cell lines were maintained in RPMI 1640 with 10% FBS. 293FT cells were obtained from Invitrogen (Grand Island, NY) and were cultured in DMEM with 10% PBS, 5% NEAA and 5% pyruvate.
Transient transfection, lentiviral transduction and siRNA mediated knock-down
For transient transfection, 293FT cells were transfected using lipofectamine 2000 Invitrogen (Grand Island, NY) following the manufacturer’s instruction. Cells were processed 24–48 hours after transfection. Lentiviral transductions were performed as previously described (32). Cells were transfected with 30nM siRNAs with RNAiMAX (Invitrogen, Grand Island, NY) following manufacture’s instruction for indicated times. The human gene ON-TARGET plus SMARTpool siRNAs were used: Non targeting siRNAs pool (D-001810-10-05), GNAQ (L-008562-00-0005), YAP1 (L-012200-00-0005), PKC δ (L-003524-00-0005), PKC ε (L-004653-00-0005), FAK1 (L-003164-00-0005), ERK1 (L-003592-00-0005), ERK2 (L-003555-00-0005) were all from Dharmacon, Inc. (Chicago, IL).
CRSPR/Cas9 knockout and doxycycline induction
The sequences of single guide RNA (gRNA) for each gene were designed using benchling.com. Single gRNA was cloned into LentiGuide-Puro vector. OMM1.3 cells were transduced with lentivirus expressing Lenti-iCas9-neo and then selected with neomycin. To further enhance the gene silencing efficiency, selected cells were then enriched for cells with tighter DOX-controlled expression of Cas9. Briefly, iCas9 cells were treated with doxycycline(DOX) followed by FACS sorting for EGFP positive cells. OMM1.3 expressing iCas9 then were transduced with lentivirus expressing single gRNA of indicated genes and then selected with puromycin. To induce gRNA expression, doxycycline was added to culture medium at a final concentration of 1 μg/mL for 72 hours.
Synergy analysis of drug combinations
Cells were plated in triplicate into 96 well tissue culture plates at 2000 cells per well. On the next day, mixtures of inhibitors were added to the cells according to the planned dose matrices. Cell viability was analyzed 96 h later by using CyQUANT NF cell proliferation assay (Life technologies Corporation, Eugene, Oregon) according to the manufacturer’s instruction. Plates were read in a SpectraMax M2 plate reader(Molecular Devices, Sunnyvale, CA). Synergy analysis was performed using the Combenefit Software(56).
Western blot, IP1, cAMP, cell proliferation, cell fractionation assays details see supplemental text.
STATISTICAL ANALYSIS
Statistical significance was assessed using a standard 2-tailed unpaired t-test, Mann-Whitney Test using prism 6.0 software (GraphPad software). p<0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
This work was supported by R35 grant CA220481, R01 grant CA142873 from the National Cancer Institute and the Gerson and Barbara Baker Distinguished Professorship (all to BCB). We thank Han Yue for providing help for IP1 assay. We thank the Preclinical Therapeutics, Laboratory for Cell Analysis of University of California, San Francisco for technical support and analyses.
Footnotes
Conflict interest: The authors declare no potential conflicts of interest
Reference
- 1.Singh AD, Bergman L, and Seregard S. Uveal melanoma: epidemiologic aspects. Ophthalmol Clin North Am. 2005;18(1):75–84, viii. [DOI] [PubMed] [Google Scholar]
- 2.Arnesen K The neural crest origin of uveal melanomas. Int Ophthalmol. 1985;7(3–4):143–7. [DOI] [PubMed] [Google Scholar]
- 3.Carvajal RD, Sosman JA, Quevedo JF, Milhem MM, Joshua AM, Kudchadkar RR, et al. Effect of selumetinib vs chemotherapy on progression-free survival in uveal melanoma: a randomized clinical trial. JAMA. 2014;311(23):2397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carvajal RD, Piperno-Neumann S, Kapiteijn E, Chapman PB, Frank S, Joshua AM, et al. Selumetinib in Combination With Dacarbazine in Patients With Metastatic Uveal Melanoma: A Phase III, Multicenter, Randomized Trial (SUMIT). J Clin Oncol. 2018;36(12):1232–9. [DOI] [PubMed] [Google Scholar]
- 5.Piperno-Neumann S, Larkin J, Carvajal RD, Luke JJ, Schwartz GK, Hodi FS, et al. Genomic Profiling of Metastatic Uveal Melanoma and Clinical Results of a Phase I Study of the Protein Kinase C Inhibitor AEB071. Mol Cancer Ther. 2020;19(4):1031–9. [DOI] [PubMed] [Google Scholar]
- 6.Schank TE, and Hassel JC. Immunotherapies for the Treatment of Uveal Melanoma-History and Future. Cancers (Basel). 2019;11(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Raamsdonk CD, Bezrookove V, Green G, Bauer J, Gaugler L, O’Brien JM, et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009;457(7229):599–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van Raamsdonk CD, Griewank KG, Crosby MB, Garrido MC, Vemula S, Wiesner T, et al. Mutations in GNA11 in uveal melanoma. N Engl J Med. 2010;363(23):2191–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robertson AG, Shih J, Yau C, Gibb EA, Oba J, Mungall KL, et al. Integrative Analysis Identifies Four Molecular and Clinical Subsets in Uveal Melanoma. Cancer Cell. 2017;32(2):204–20 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schneider B, Riedel K, Zhivov A, Huehns M, Zettl H, Guthoff RF, et al. Frequent and Yet Unreported GNAQ and GNA11 Mutations are Found in Uveal Melanomas. Pathol Oncol Res. 2017. [DOI] [PubMed] [Google Scholar]
- 11.Maziarz M, Leyme A, Marivin A, Luebbers A, Patel PP, Chen Z, et al. Atypical activation of the G protein Galphaq by the oncogenic mutation Q209P. J Biol Chem. 2018;293(51):19586–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moore AR, Ceraudo E, Sher JJ, Guan Y, Shoushtari AN, Chang MT, et al. Recurrent activating mutations of G-protein-coupled receptor CYSLTR2 in uveal melanoma. Nat Genet. 2016;48(6):675–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johansson P, Aoude LG, Wadt K, Glasson WJ, Warrier SK, Hewitt AW, et al. Deep sequencing of uveal melanoma identifies a recurrent mutation in PLCB4. Oncotarget. 2016;7(4):4624–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sheng X, Kong Y, Li Y, Zhang Q, Si L, Cui C, et al. GNAQ and GNA11 mutations occur in 9.5% of mucosal melanoma and are associated with poor prognosis. Eur J Cancer. 2016;65:156–63. [DOI] [PubMed] [Google Scholar]
- 15.Murali R, Wiesner T, Rosenblum MK, and Bastian BC. GNAQ and GNA11 mutations in melanocytomas of the central nervous system. Acta Neuropathol. 2012;123(3):457–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thomas AC, Zeng Z, Riviere JB, O’Shaughnessy R, Al-Olabi L, St-Onge J, et al. Mosaic Activating Mutations in GNA11 and GNAQ Are Associated with Phakomatosis Pigmentovascularis and Extensive Dermal Melanocytosis. J Invest Dermatol. 2016;136(4):770–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ayturk UM, Couto JA, Hann S, Mulliken JB, Williams KL, Huang AY, et al. Somatic Activating Mutations in GNAQ and GNA11 Are Associated with Congenital Hemangioma. Am J Hum Genet. 2016;98(4):789–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bean GR, Joseph NM, Gill RM, Folpe AL, Horvai AE, and Umetsu SE. Recurrent GNAQ mutations in anastomosing hemangiomas. Mod Pathol. 2017;30(5):722–7. [DOI] [PubMed] [Google Scholar]
- 19.Couto JA, Ayturk UM, Konczyk DJ, Goss JA, Huang AY, Hann S, et al. A somatic GNA11 mutation is associated with extremity capillary malformation and overgrowth. Angiogenesis. 2017;20(3):303–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Couto JA, Huang L, Vivero MP, Kamitaki N, Maclellan RA, Mulliken JB, et al. Endothelial Cells from Capillary Malformations Are Enriched for Somatic GNAQ Mutations. Plast Reconstr Surg. 2016;137(1):77e–82e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Joseph NM, Brunt EM, Marginean C, Nalbantoglu I, Snover DC, Thung SN, et al. Frequent GNAQ and GNA14 Mutations in Hepatic Small Vessel Neoplasm. Am J Surg Pathol. 2018;42(9):1201–7. [DOI] [PubMed] [Google Scholar]
- 22.Nakashima M, Miyajima M, Sugano H, Iimura Y, Kato M, Tsurusaki Y, et al. The somatic GNAQ mutation c.548G>A (p.R183Q) is consistently found in Sturge-Weber syndrome. J Hum Genet. 2014;59(12):691–3. [DOI] [PubMed] [Google Scholar]
- 23.Shirley MD, Tang H, Gallione CJ, Baugher JD, Frelin LP, Cohen B, et al. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med. 2013;368(21):1971–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shain AH, Bagger MM, Yu R, Chang D, Liu S, Vemula S, et al. The genetic evolution of metastatic uveal melanoma. Nat Genet. 2019;51(7):1123–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Furney SJ, Pedersen M, Gentien D, Dumont AG, Rapinat A, Desjardins L, et al. SF3B1 mutations are associated with alternative splicing in uveal melanoma. Cancer Discov. 2013;3(10):1122–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harbour JW, Roberson ED, Anbunathan H, Onken MD, Worley LA, and Bowcock AM. Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat Genet. 2013;45(2):133–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martin M, Masshofer L, Temming P, Rahmann S, Metz C, Bornfeld N, et al. Exome sequencing identifies recurrent somatic mutations in EIF1AX and SF3B1 in uveal melanoma with disomy 3. Nat Genet. 2013;45(8):933–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harbour JW, Onken MD, Roberson ED, Duan S, Cao L, Worley LA, et al. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science. 2010;330(6009):1410–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rhee SG, and Bae YS. Regulation of phosphoinositide-specific phospholipase C isozymes. J Biol Chem. 1997;272(24):15045–8. [DOI] [PubMed] [Google Scholar]
- 30.Teixeira C, Stang SL, Zheng Y, Beswick NS, and Stone JC. Integration of DAG signaling systems mediated by PKC-dependent phosphorylation of RasGRP3. Blood. 2003;102(4):1414–20. [DOI] [PubMed] [Google Scholar]
- 31.Goldsmith ZG, and Dhanasekaran DN. G protein regulation of MAPK networks. Oncogene. 2007;26(22):3122–42. [DOI] [PubMed] [Google Scholar]
- 32.Chen X, Wu Q, Depeille P, Chen P, Thornton S, Kalirai H, et al. RasGRP3 Mediates MAPK Pathway Activation in GNAQ Mutant Uveal Melanoma. Cancer Cell. 2017;31(5):685–96 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen X, Wu Q, Tan L, Porter D, Jager MJ, Emery C, et al. Combined PKC and MEK inhibition in uveal melanoma with GNAQ and GNA11 mutations. Oncogene. 2014;33(39):4724–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moore AR, Ran L, Guan Y, Sher JJ, Hitchman TD, Zhang JQ, et al. GNA11 Q209L Mouse Model Reveals RasGRP3 as an Essential Signaling Node in Uveal Melanoma. Cell Rep. 2018;22(9):2455–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu FX, Luo J, Mo JS, Liu G, Kim YC, Meng Z, et al. Mutant Gq/11 promote uveal melanoma tumorigenesis by activating YAP. Cancer Cell. 2014;25(6):822–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feng X, Degese MS, Iglesias-Bartolome R, Vaque JP, Molinolo AA, Rodrigues M, et al. Hippo-independent activation of YAP by the GNAQ uveal melanoma oncogene through a trio-regulated rho GTPase signaling circuitry. Cancer Cell. 2014;25(6):831–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feng X, Arang N, Rigiracciolo DC, Lee JS, Yeerna H, Wang Z, et al. A Platform of Synthetic Lethal Gene Interaction Networks Reveals that the GNAQ Uveal Melanoma Oncogene Controls the Hippo Pathway through FAK. Cancer Cell. 2019;35(3):457–72 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Annala S, Feng X, Shridhar N, Eryilmaz F, Patt J, Yang J, et al. Direct targeting of Galphaq and Galpha11 oncoproteins in cancer cells. Sci Signal. 2019;12(573). [DOI] [PubMed] [Google Scholar]
- 39.Trinquet E, Fink M, Bazin H, Grillet F, Maurin F, Bourrier E, et al. D-myo-inositol 1-phosphate as a surrogate of D-myo-inositol 1,4,5-tris phosphate to monitor G protein-coupled receptor activation. Anal Biochem. 2006;358(1):126–35. [DOI] [PubMed] [Google Scholar]
- 40.Rozengurt E, Rey O, and Waldron RT. Protein kinase D signaling. J Biol Chem. 2005;280(14):13205–8. [DOI] [PubMed] [Google Scholar]
- 41.Takasaki J, Saito T, Taniguchi M, Kawasaki T, Moritani Y, Hayashi K, et al. A novel Galphaq/11-selective inhibitor. J Biol Chem. 2004;279(46):47438–45. [DOI] [PubMed] [Google Scholar]
- 42.Kamato D, Thach L, Bernard R, Chan V, Zheng W, Kaur H, et al. Structure, Function, Pharmacology, and Therapeutic Potential of the G Protein, Galpha/q,11. Front Cardiovasc Med. 2015;2:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wilkie TM, Scherle PA, Strathmann MP, Slepak VZ, and Simon MI. Characterization of G-protein alpha subunits in the Gq class: expression in murine tissues and in stromal and hematopoietic cell lines. Proc Natl Acad Sci U S A. 1991;88(22):10049–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lim YH, Bacchiocchi A, Qiu J, Straub R, Bruckner A, Bercovitch L, et al. GNA14 Somatic Mutation Causes Congenital and Sporadic Vascular Tumors by MAPK Activation. Am J Hum Genet. 2016;99(2):443–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Heasley LE, Storey B, Fanger GR, Butterfield L, Zamarripa J, Blumberg D, et al. GTPase-deficient G alpha 16 and G alpha q induce PC12 cell differentiation and persistent activation of cJun NH2-terminal kinases. Mol Cell Biol. 1996;16(2):648–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Masters SB, Miller RT, Chi MH, Chang FH, Beiderman B, Lopez NG, et al. Mutations in the GTP-binding site of GS alpha alter stimulation of adenylyl cyclase. J Biol Chem. 1989;264(26):15467–74. [PubMed] [Google Scholar]
- 47.Ellen Kapiteijn MC, Valentina Boni, Delphine Loirat, Frank Speetjens, John Park, Emiliano Calvo, Richard Carvajal, Marta Nyakas, Juan Gonzalez-Maffe, Xu Zhu, Ramu Thiruvamoor, Padmaja Yerramilli-Rao, Sophie Piperno-Neumann. A Phase I trial of LXS196, a novel PKC inhibitor for metastatic uveal melanoma Cancer Res 2019;79(13 Suppl):Abstract nr CT068 2019. [Google Scholar]
- 48.Tanjoni I, Walsh C, Uryu S, Tomar A, Nam JO, Mielgo A, et al. PND-1186 FAK inhibitor selectively promotes tumor cell apoptosis in three-dimensional environments. Cancer Biol Ther. 2010;9(10):764–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, et al. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell. 2007;130(6):1120–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21(21):2747–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hao Y, Chun A, Cheung K, Rashidi B, and Yang X. Tumor suppressor LATS1 is a negative regulator of oncogene YAP. J Biol Chem. 2008;283(9):5496–509. [DOI] [PubMed] [Google Scholar]
- 52.Levy D, Adamovich Y, Reuven N, and Shaul Y. Yap1 phosphorylation by c-Abl is a critical step in selective activation of proapoptotic genes in response to DNA damage. Mol Cell. 2008;29(3):350–61. [DOI] [PubMed] [Google Scholar]
- 53.Li B, He J, Lv H, Liu Y, Lv X, Zhang C, et al. c-Abl regulates YAPY357 phosphorylation to activate endothelial atherogenic responses to disturbed flow. J Clin Invest. 2019;129(3):1167–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sugihara T, Werneburg NW, Hernandez MC, Yang L, Kabashima A, Hirsova P, et al. YAP Tyrosine Phosphorylation and Nuclear Localization in Cholangiocarcinoma Cells Are Regulated by LCK and Independent of LATS Activity. Mol Cancer Res. 2018;16(10):1556–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cao J, Wu L, Zhang SM, Lu M, Cheung WK, Cai W, et al. An easy and efficient inducible CRISPR/Cas9 platform with improved specificity for multiple gene targeting. Nucleic Acids Res. 2016;44(19):e149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Di Veroli GY, Fornari C, Wang D, Mollard S, Bramhall JL, Richards FM, et al. Combenefit: an interactive platform for the analysis and visualization of drug combinations. Bioinformatics. 2016;32(18):2866–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.van Dinten LC, Pul N, van Nieuwpoort AF, Out CJ, Jager MJ, and van den Elsen PJ. Uveal and cutaneous melanoma: shared expression characteristics of melanoma-associated antigens. Invest Ophthalmol Vis Sci. 2005;46(1):24–30. [DOI] [PubMed] [Google Scholar]
- 58.Martins L, Giovani PA, Reboucas PD, Brasil DM, Haiter Neto F, Coletta RD, et al. Computational analysis for GNAQ mutations: New insights on the molecular etiology of Sturge-Weber syndrome. J Mol Graph Model. 2017;76:429–40. [DOI] [PubMed] [Google Scholar]
- 59.Kostenis E, Pfeil EM, and Annala S. Heterotrimeric Gq proteins as therapeutic targets? J Biol Chem. 2020;295(16):5206–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jhon DY, Lee HH, Park D, Lee CW, Lee KH, Yoo OJ, et al. Cloning, sequencing, purification, and Gq-dependent activation of phospholipase C-beta 3. J Biol Chem. 1993;268(9):6654–61. [PubMed] [Google Scholar]
- 61.Lee CW, Lee KH, Lee SB, Park D, and Rhee SG. Regulation of phospholipase C-beta 4 by ribonucleotides and the alpha subunit of Gq. J Biol Chem. 1994;269(41):25335–8. [PubMed] [Google Scholar]
- 62.Lee CH, Park D, Wu D, Rhee SG, and Simon MI. Members of the Gq alpha subunit gene family activate phospholipase C beta isozymes. J Biol Chem. 1992;267(23):16044–7. [PubMed] [Google Scholar]
- 63.Kim YJ, Lee SC, Kim SE, Kim SH, Kim SK, and Lee CS. YAP Activity is Not Associated with Survival of Uveal Melanoma Patients and Cell Lines. Sci Rep. 2020;10(1):6209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schrage R, Schmitz AL, Gaffal E, Annala S, Kehraus S, Wenzel D, et al. The experimental power of FR900359 to study Gq-regulated biological processes. Nat Commun. 2015;6:10156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lapadula D, Farias E, Randolph CE, Purwin TJ, McGrath D, Charpentier TH, et al. Effects of Oncogenic Galphaq and Galpha11 Inhibition by FR900359 in Uveal Melanoma. Mol Cancer Res. 2019;17(4):963–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Onken MD, Makepeace CM, Kaltenbronn KM, Kanai SM, Todd TD, Wang S, et al. Targeting nucleotide exchange to inhibit constitutively active G protein alpha subunits in cancer cells. Sci Signal. 2018;11(546). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tietze D, Kaufmann D, Tietze AA, Voll A, Reher R, Konig G, et al. Structural and Dynamical Basis of G Protein Inhibition by YM-254890 and FR900359: An Inhibitor in Action. J Chem Inf Model. 2019;59(10):4361–73. [DOI] [PubMed] [Google Scholar]
- 68.Malfacini D, Patt J, Annala S, Harpsoe K, Eryilmaz F, Reher R, et al. Rational design of a heterotrimeric G protein alpha subunit with artificial inhibitor sensitivity. J Biol Chem. 2019;294(15):5747–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kuschak M, Namasivayam V, Rafehi M, Voss JH, Garg J, Schlegel JG, et al. Cell-permeable high-affinity tracers for Gq proteins provide structural insights, reveal distinct binding kinetics and identify small molecule inhibitors. Br J Pharmacol. 2020;177(8):1898–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sanjana NE, Shalem O, and Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods. 2014;11(8):783–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Griewank KG, Yu X, Khalili J, Sozen MM, Stempke-Hale K, Bernatchez C, et al. Genetic and molecular characterization of uveal melanoma cell lines. Pigment Cell Melanoma Res. 2012;25(2):182–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Amirouchene-Angelozzi N, Nemati F, Gentien D, Nicolas A, Dumont A, Carita G, et al. Establishment of novel cell lines recapitulating the genetic landscape of uveal melanoma and preclinical validation of mTOR as a therapeutic target. Mol Oncol. 2014;8(8):1508–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.