

Abstract
Neurodegenerative disorders such as Alzheimer’s disease (AD), Lewy body diseases (LBD), and the amyotrophic lateral sclerosis and frontotemporal dementia (ALS-FTD) spectrum are defined by the accumulation of specific misfolded protein aggregates. However, the mechanisms by which each proteinopathy leads to neurodegeneration remain elusive. We hypothesized that there is a common “pan-neurodegenerative” gene expression signature driving pathophysiology across these clinically and pathologically diverse proteinopathies. To test this hypothesis, we performed a systematic review of human CNS transcriptomics datasets from AD, LBD, and ALS-FTD patients and age-matched controls in the Gene Expression Omnibus (GEO) and ArrayExpress databases, followed by consistent processing of each dataset, meta-analysis, pathway enrichment, and overlap analyses. After applying pre-specified eligibility criteria and stringent data pre-processing, a total of 2600 samples from 26 AD, 21 LBD, and 13 ALS-FTD datasets were included in the meta-analysis. The pan-neurodegenerative gene signature is characterized by an upregulation of innate immunity, cytoskeleton, and transcription and RNA processing genes, and a downregulation of the mitochondrial electron transport chain. Pathway enrichment analyses also revealed the upregulation of neuroinflammation (including Toll-like receptor, TNF, and NFκB signaling) and phagocytosis, and the downregulation of mitochondrial oxidative phosphorylation, lysosomal acidification, and ubiquitin-proteasome pathways. Our findings suggest that neuroinflammation and a failure in both neuronal energy metabolism and protein degradation systems are consistent features underlying neurodegenerative diseases, despite differences in the extent of neuronal loss and brain regions involved.
Keywords: Alzheimer’s disease, Amyotrophic lateral sclerosis, Frontotemporal dementia, Lewy body diseases, Meta-analysis, Mitochondrial energy metabolism, Neurodegeneration, Neuroinflammation, Proteostasis, Transcriptomics
1. Introduction
Neurodegenerative disorders such as Alzheimer’s disease (AD), Lewy body diseases (LBD), and the amyotrophic lateral sclerosis and frontotemporal dementia (ALS-FTD) spectrum are all defined by the accumulation of specific misfolded protein aggregates. AD is characterized by the presence of both amyloid plaques – which are extracellular deposits of the amyloid-β peptide (Aβ), a by-product cleaved from the amyloid-β precursor protein (AβPP) (Glenner and Wong, 1984) – and intraneuronal neurofibrillary tangles (NFTs) of hyperphosphorylated microtubule-associated protein tau (Brion et al., 1986; Grundke-Iqbal et al., 1986). Accumulation of α-synuclein (αSyn) in Lewy bodies and neurites is the hallmark feature of LBD, an umbrella term which includes Parkinson’s disease (PD), Parkinson’s disease dementia (PDD), and dementia with Lewy bodies (DLB) (Spillantini et al., 1997). Finally, an array of proteins form neuronal inclusions characteristic of ALS-FTD, including the RNA-binding proteins TAR DNA-binding protein 43 (TDP-43) (Neumann et al., 2006) and fused in sarcoma (FUS) (Kwiatkowski et al., 2009), as well as dipeptide repeats (DPRs) formed by RAN translation of the hexanucleotide repeat expansion in the C9orf72 gene (Mori et al., 2013).
One proposed common mechanism underlying all neurodegenerative proteinopathies is the intrinsically disordered nature of the aggregating proteins in native conditions (i.e., lack of a stable tertiary structure), which would render them prone to misfolding and subsequent aggregation in pathological conditions (Jarosz and Khurana, 2017), for example upon certain abnormal post-translational modifications (Toth-Petroczy et al., 2016; Wesseling et al., 2020). Another proposed mechanism is a failure in proteostasis, including the autophagy and ubiquitin-proteasome systems, which has been posited as a hallmark of aging (López-Otín et al., 2013; Walther et al., 2017). Although both protein misfolding and proteostatic stress mechanisms are supported by mounting evidence, our understanding of the extent to which these processes play analogous roles across multiple neurodegenerative diseases remains speculative. We reasoned that recent advances in -omics technologies, coupled with the public availability of a large number of neurodegenerative gene expression datasets, could enable the discovery of drivers of neurodegeneration across proteinopathies in large patient cohorts. Specifically, we hypothesized that there is a “pan-neurodegenerative” gene expression signature shared by different neurodegenerative diseases regardless of the underlying proteinopathy, together with disease-specific signatures which could be explained by the individual aggregating protein and/or neuronal type or CNS region involved. To test this hypothesis, we conducted a systematic review and meta-analysis of publicly available human brain transcriptomics datasets of AD, LBD, ALS-FTD and control individuals, followed by pathway enrichment and overlap analyses.
Previous meta-analyses of brain transcriptomic studies have focused on one neurodegenerative disease, such as AD (Ciryam et al., 2016; Patel et al., 2019; Wan et al., 2020) or PD (Kelly et al., 2019; Oerton and Bender, 2017; Wang et al., 2019; Zheng et al., 2010), in some instances comparing with other neurodegenerative diseases or mouse models (Das et al., 2020; Kelly et al., 2019; Patel et al., 2019; Wan et al., 2020), but did not pursue a common neurodegenerative signature. Moreover, prior studies investigating pan-neurodegenerative transcriptional changes have been constrained by comparatively small sample sizes (Durrenberger et al., 2015; Labadorf et al., 2018; Li et al., 2014). In this study, we analyzed 2600 samples from AD, LBD, and ALS-FTD patients and age-matched controls across 60 datasets to identify signatures of pan-neurodegeneration.
2. Materials and methods
Briefly, the data analysis pipeline consisted of (1) systematic review and dataset selection, (2) quality control, (3) differential expression analysis, (4) meta-analysis, and (5) gene set enrichment analysis (GSEA). Fig. 1 provides a graphical summary of the workflow applied in this study. Unless otherwise indicated, all analyses were performed in the R programming language and statistical computing environment (version 4.0.2).
Fig. 1. Methods Overview.
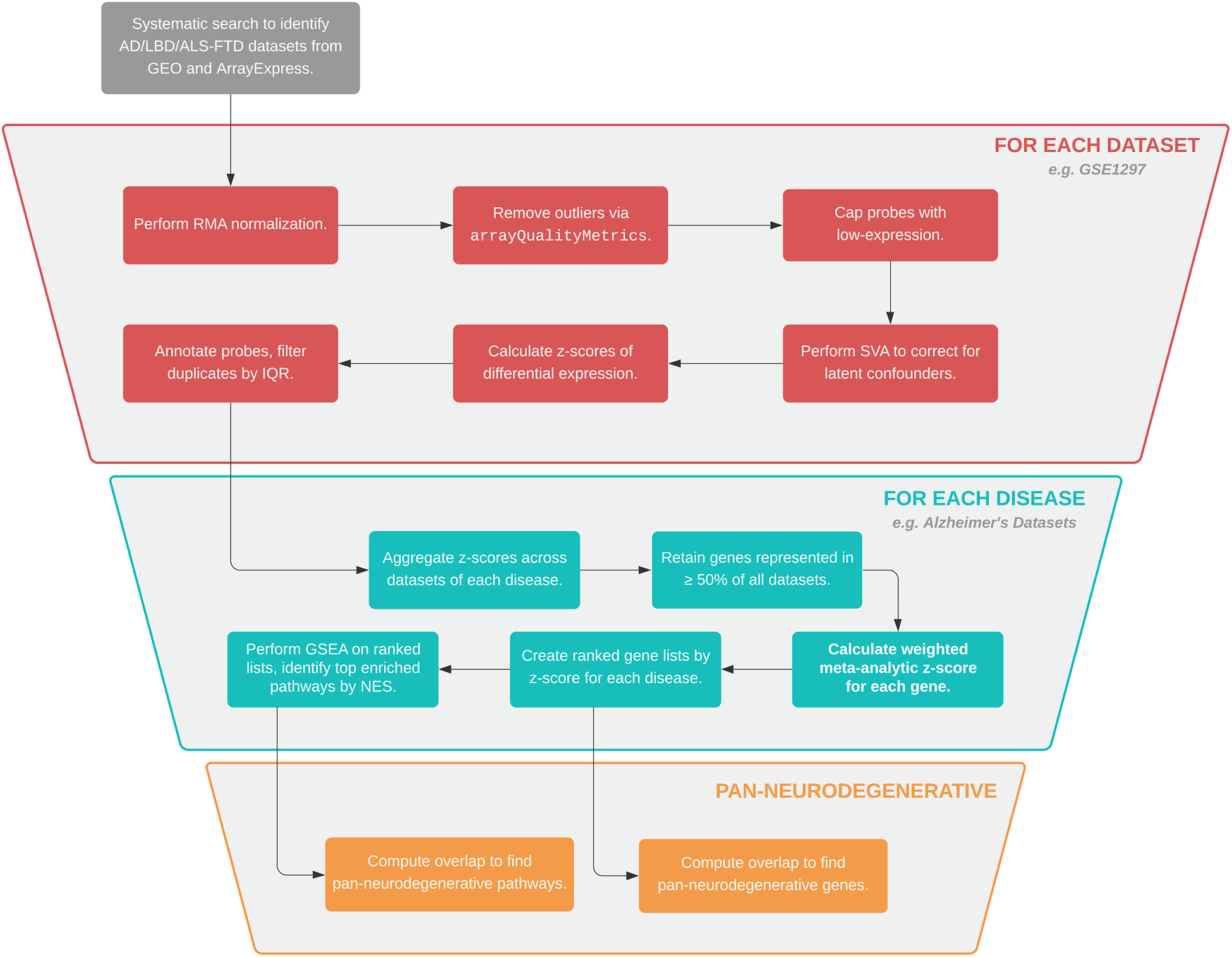
Summary of the data analysis pipeline applied in this study.
2.1. Systematic review
To identify datasets for inclusion in a comprehensive fashion, we followed the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines, as explained below (Liberati et al., 2009).
2.2. Eligibility criteria
Datasets were selected based on pre-specified inclusion and exclusion criteria. Inclusion criteria were: (1) original datasets and (2) human microarray expression profiling datasets from relevant CNS regions in AD, LBD, and ALS-FTD. Exclusion criteria were: (1) non-original datasets (e.g., duplicate studies, re-analyses of pre-existing datasets); (2) not human brain tissue; (3) not pertaining to disease in question (i.e., AD, LBD, or ALS-FTD); (4) patient-derived in vitro cell lines or disease models; (5) study design other than case/control; (6) brain region not significantly affected by neurodegeneration (e.g., cerebellum in AD); and (7) incompatible technologies.
2.3. Information sources
We interrogated two databases: The National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) and the European Bioinformatics Institute (EMBL-EBI) ArrayExpress.
2.4. Search strategies and dataset selection
We inquired the two databases on the same day (July 1, 2020) with no date filters applied. Full details of search terms used and screening rationale for each dataset are available in Tables S1 and S2 for GEO and ArrayExpress, respectively. Fig. 2 describes the systematic review according to PRISMA guidelines. Eligible datasets were those meeting all the inclusion criteria and none of the exclusion criteria. Duplicate records were removed in R, rendering n = 530 identified datasets. Within each dataset, only CNS regions relevant to neuropathology and involved in neurodegeneration were included, as listed in the exclusion criteria. To ensure the specificity of each disease expression signature, we excluded intermediate phenotypes whenever reported, i.e. only disease AD samples classified as Braak V/VI (corresponding to a neocortical NFT stage) were included. We also excluded familial AD and LBD datasets to increase data homogeneity, however, familial ALS-FTD datasets were considered eligible and included C9orf72, PGRN, SOD1, and CHMP2B mutations. Included datasets are described in Table S3.
Fig. 2. Dataset Selection Workflow.
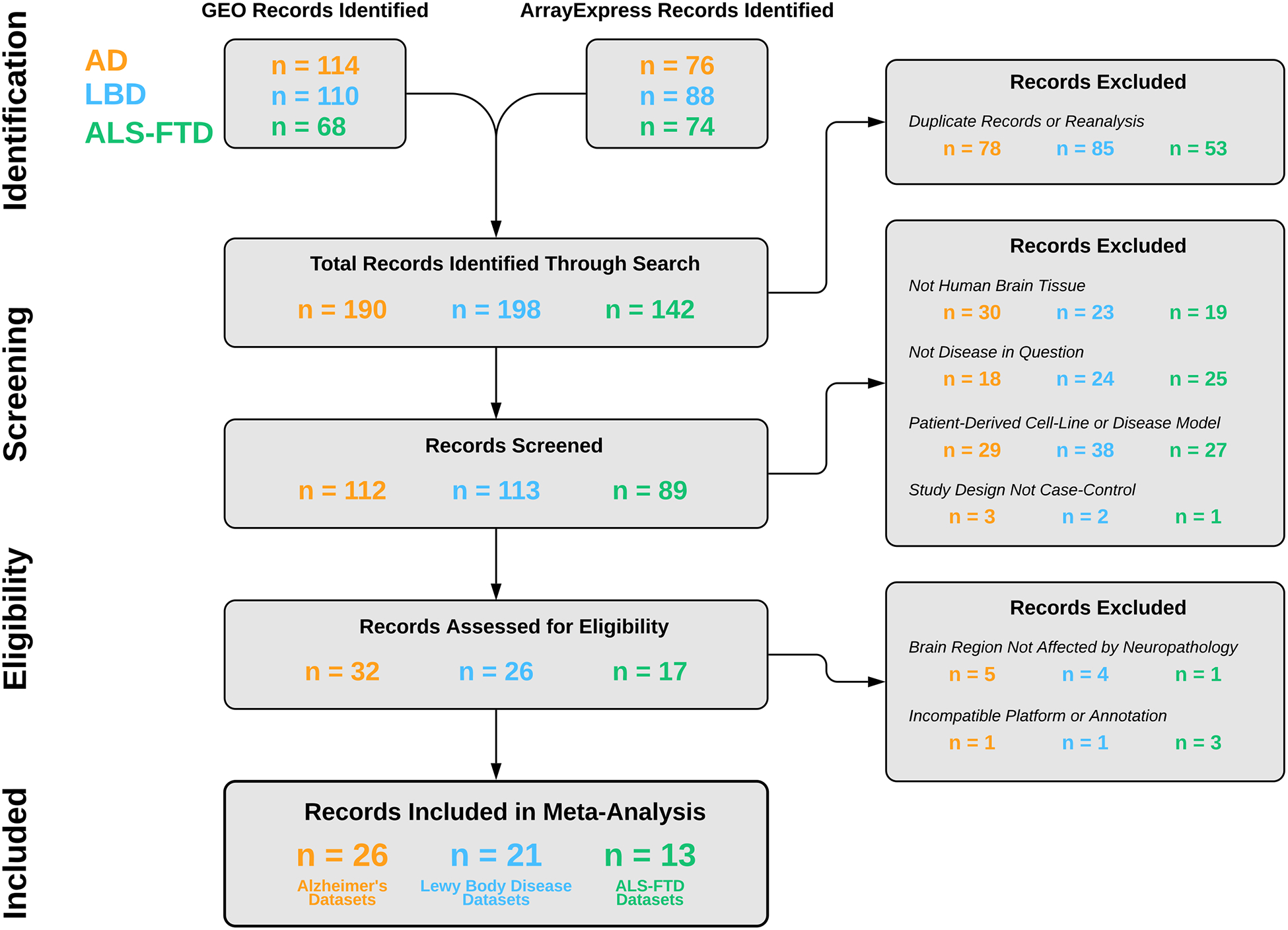
Description of the systematic review of human transcriptomics from AD, LBD, and ALS-FTD patients in the Gene Expression Omnibus (GEO) and ArrayExpress databases according to PRISMA guidelines. After applying pre-specified inclusion and exclusion criteria, our systematic review yielded 1677 control and 1563 disease samples from 26 AD, 21 LBD, and 13 ALS-FTD datasets.
2.5. Quality control
We categorized each included dataset by both disease and brain region and studied them individually, entailing 89 separate analyses. For each analysis, we normalized expression data as needed using the Robust Multichip Average approach implemented in the oligo package (Carvalho and Irizarry, 2010; Irizarry et al., 2003). We generated data quality reports with diagnostic plots via the arrayQualityMetrics package (Kauffmann et al., 2009; Kauffmann and Huber, 2010), which were used to discard outliers as described in our accompanying Data in Brief article. Probes were capped at the 20th percentile to filter for low expression. Finally, we used surrogate variable analysis to remove unknown latent variation in the data (Leek et al., 2012; Leek and Storey, 2007).
2.6. Differential expression analysis
For each dataset, differentially expressed genes (DEGs) were identified via the limma package in R (Ritchie et al., 2015). We used the mean difference between cases and controls and the empirical Bayes estimated variance from limma to calculate z-scores of differential expression (Phipson et al., 2016). Microarray probes were converted to ENTREZ IDs using Bioconductor annotation packages (Maglott et al., 2007). In the event that multiple probes mapped to the same gene, the single probe with the greatest interquartile range (IQR) was retained (Walsh et al., 2015; Wang et al., 2012).
2.7. Meta-analysis
Z-scores of differential expression were tabulated separately across AD, LBD, and ALS-FTD. For each disease, genes represented in at least 50% of the datasets were retained. Next, we calculated meta-analytic z-scores using Lipták’s weighted Z-test, viz. a weighted average of z-scores (Lipták, 1958; Zaykin, 2011). If multiple tissue samples were derived from the same patient (i.e., different CNS regions), we corrected for multiple comparisons using the Bonferroni method. The top 1000 upregulated and top 1000 downregulated genes in each ranked gene list comprised the disease-predominant gene signatures for each disease, which were intersected to identify possible pan-neurodegenerative genes (Table S4).
2.8. Gene set enrichment analysis
We performed Gene Set Enrichment Analysis (GSEA) on the ranked meta-analytic z-score lists of each disease (Subramanian et al., 2007, 2005). Enrichment was calculated against the Gene Ontology: Biological Processes (GO: BP) available from the Molecular Signatures Database (MSigDB) (Ashburner et al., 2000; Liberzon et al., 2011; The Gene Ontology Consortium, 2019).
For each disease, the top 200 enriched pathways by Normalized Enrichment Score (NES) were intersected to define the pan-neurodegenerative and disease-predominant pathways. To reduce redundancy of reported pathways and combine parent-child ontology terms describing the same biological phenomenon, the GO: Biological Processes were aggregated by hierarchical clustering based on the Jac-card similarity coefficient and were manually expert-annotated to assign representative labels. Genes with the top z-scores comprising these pathways were visualized in heatmaps as well as protein-protein interaction (PPI) functional networks constructed via the STRING biological database (version 11.0) (Szklarczyk et al., 2019).
2.9. Validation
To validate these results, we performed three separate sensitivity analyses. First, we repeated steps described in sections 2.5 to 2.8 for all datasets after randomly permuting the phenotype label prior to differential expression analysis, and hence removing control/disease classification information (Fig. S1). Second, we re-computed GSEA against the Reactome database also available via MSigDB (Jassal et al., 2020), and compared the resulting enriched pathways with those obtained from the GSEA against GO: BP.
Lastly, using a previously published dataset of immuno-panned human brain cell subpopulations (Zhang et al., 2016), we investigated whether neuronal loss was driving the downregulated pathways. Specifically, we identified a cassette of 280 neuron-predominant genes, each with an average expression of ≥5 times the sum of their expression in all other cell types. For each disease, we corrected the meta-analytic z-score of each gene by the median meta-analytic z-score of the neuron-predominant genes. We then re-performed GSEA against GO: BP with these corrected z-scores.
3. Results
3.1. Systematic review of AD, LBD, and ALS-FTD microarray datasets
After application of pre-specified inclusion and exclusion criteria following PRISMA guidelines, our systematic review in the GEO and ArrayExpress repositories yielded 1648 control and 1586 disease samples from 26 AD, 21 LBD, and 13 ALS-FTD datasets (Fig. 2, Table S3). During our stringent data pre-processing pipeline, 355 control and 279 disease samples failed to meet the data quality requirements and were discarded. Hence, a total of 2600 samples – 1293 control and 1307 disease – were included in the subsequent analyses.
3.2. Meta-analysis of AD, LBD, and ALS-FTD microarray datasets reveals a pan-neurodegenerative gene signature
We next performed differential expression analysis of diseased versus control individuals in each of the 60 included datasets, followed by a meta-analysis of differentially expressed genes (DEGs) across datasets of each disease. This rendered meta-analytic gene expression signatures for AD, LBD, and ALS-FTD. Overlap analysis of the top genes in each disease signature revealed an intersection of 88 upregulated and 45 downregulated genes (Fig. 3, Table S4). To examine cell-type enrichment of these genes, we used a public dataset of immuno-panned human brain cell subpopulations as reference (Zhang et al., 2016). In Fig. 3, the color of each gene corresponds to the cell-type with the highest expression for that gene. As expected, upregulated pan-neurodegenerative genes were predominantly glial, while downregulated genes were predominantly neuronal.
Fig. 3. Pan-Neurodegenerative Genes.
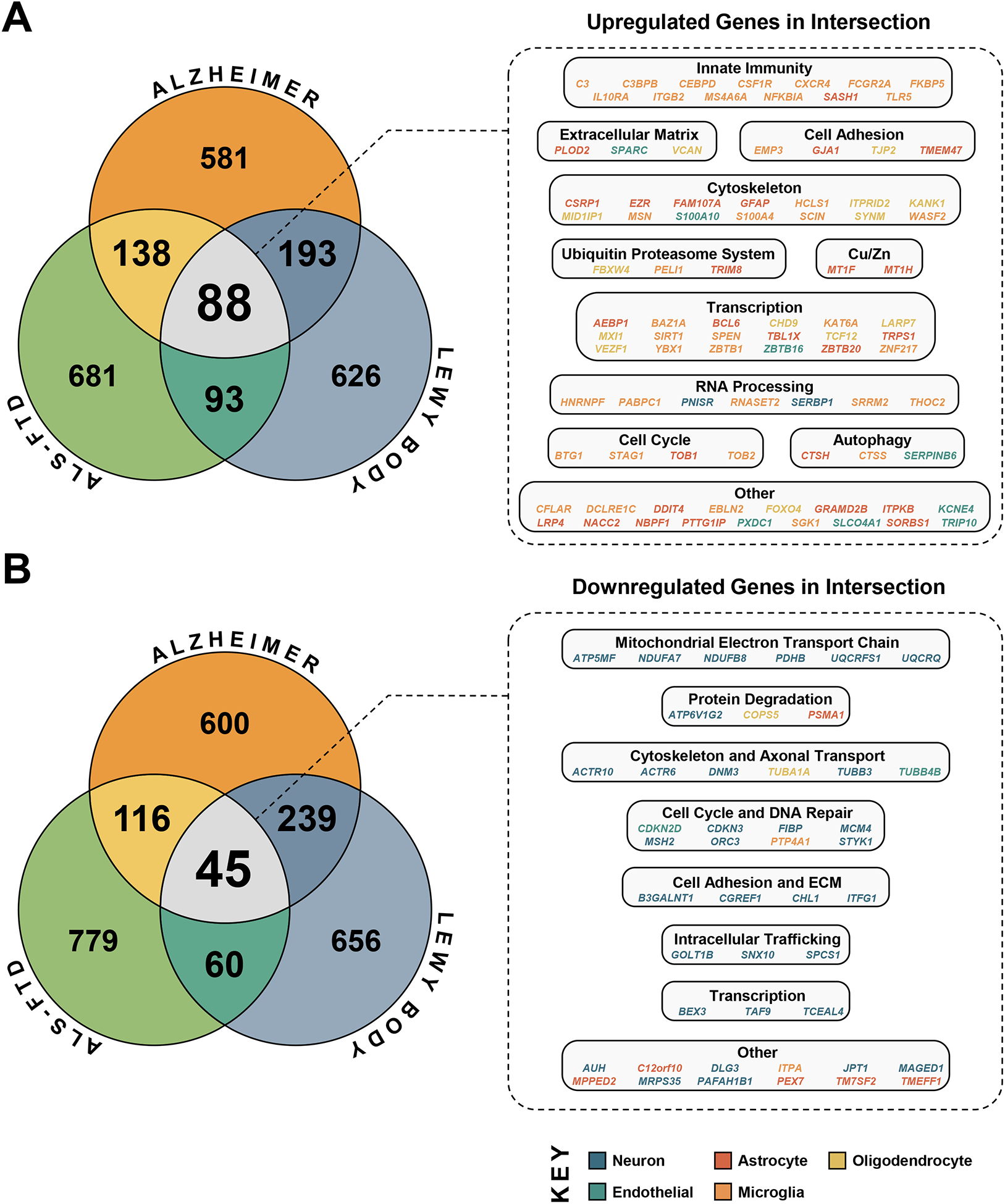
The top 1000 upregulated and top 1000 downregulated genes by meta-analytic z-score for AD, LBD, and ALS-FTD were intersected to define the pan-neurodegenerative gene signature, which was composed of 88 upregulated and 45 downregulated genes. The color for each gene corresponds to the cell-type with the highest expression for that gene by average FPKM (Zhang et al., 2016).
A close inspection of these 88 upregulated genes revealed gene cassettes related to innate immunity (C3, CEBPB, CEBPD, CSF1R, CXCR4, FCGR2A, FKBP5, IL10RA, ITGB2, MS4A6A, NFKBIA, SASH1, TLR5); extracellular matrix and cell adhesion/tight junction (EMP3, GJA1, PLOD2, SPARC, TJP2, TMEM47, VCAN); cytoskeleton, including actin (CSRP1, EZR, FAM107A, HCLS1, KANK1, ITPRID2, MSN, S100A4, S100A10, SCIN, WASF2), intermediate filament (GFAP, SYNM) and microtubule (MID1IP1); ubiquitin-proteasome system (FBXW4, PELI1, TRIM8) and autophagy (CTSH, CTSS, SERPINB6); copper/zinc homeostasis (MT1F and MT1H); cell cycle/apoptosis (BTG1, CFLAR, STAG1, TOB1, TOB2); RNA processing (HNRNPF, PABPC1, PNISR, RNASET2, SERBP1, SRRM2, THOC2); and regulation of transcription (AEBP1, BAZ1A, BCL6, CHD9, KAT6A, LARP7, MXI1, SIRT1, SPEN, TBL1X, TCF12, TRPS1, VEZF1, YBX1, ZBTB1, ZBTB16, ZBTB20, ZNF217).
Similarly, the 45 pan-neurodegenerative downregulated genes included mitochondrial electron transport chain and energy metabolism (ATP5MF, NDUFA7, NDUFB8, PDHB, UQCRFS1, UQCRQ); protein degradation genes, including ubiquitin-proteasome system (COPS5, PSMA1) and lysosome acidification (ATP6V1G2); cytoskeleton/axon transport (ACTR10, ACTR6, TUBA1A, DNM3, TUBB3, TUBB4B); cell cycle and DNA replication and repair (CDKN2D, CDKN3, FIBP, MCM4, MSH2, ORC3, PTP4A1, STYK1); intracellular trafficking (GOLT1B, SNX10, SPCS1); cell adhesion/extracellular matrix (B3GALNT1, CGREF1, CHL1, ITFG1); and regulation of transcription (BEX3, TCEAL4, TM7SF2).
3.3. Gene set enrichment analysis identifies pan-neurodegenerative functional pathways
Because transcriptional changes are more likely to be consistent across datasets at a pathway rather than at an individual gene level, and since genes are co-expressed in a pathway, we investigated the overlap between these three disease signatures at the functional pathway level. We performed Gene Set Enrichment Analysis (GSEA) (Subramanian et al., 2007, 2005) on the ranked meta-analytic gene sets across AD, LBD, and ALS-FTD. Top enriched pathways were then intersected to obtain the pan-neurodegenerative pathway signature. These pathways were grouped and expert-annotated based on the similarity of their constituent genes (see Methods) to obtain specific up- and downregulated functions relevant to neurodegeneration (Table S5). Genes with the top z-scores in these pathways were visualized in heatmaps (Fig. 4) and protein-protein functional interaction networks (Fig. S2).
Fig. 4. Pan-Neurodegenerative Pathways.
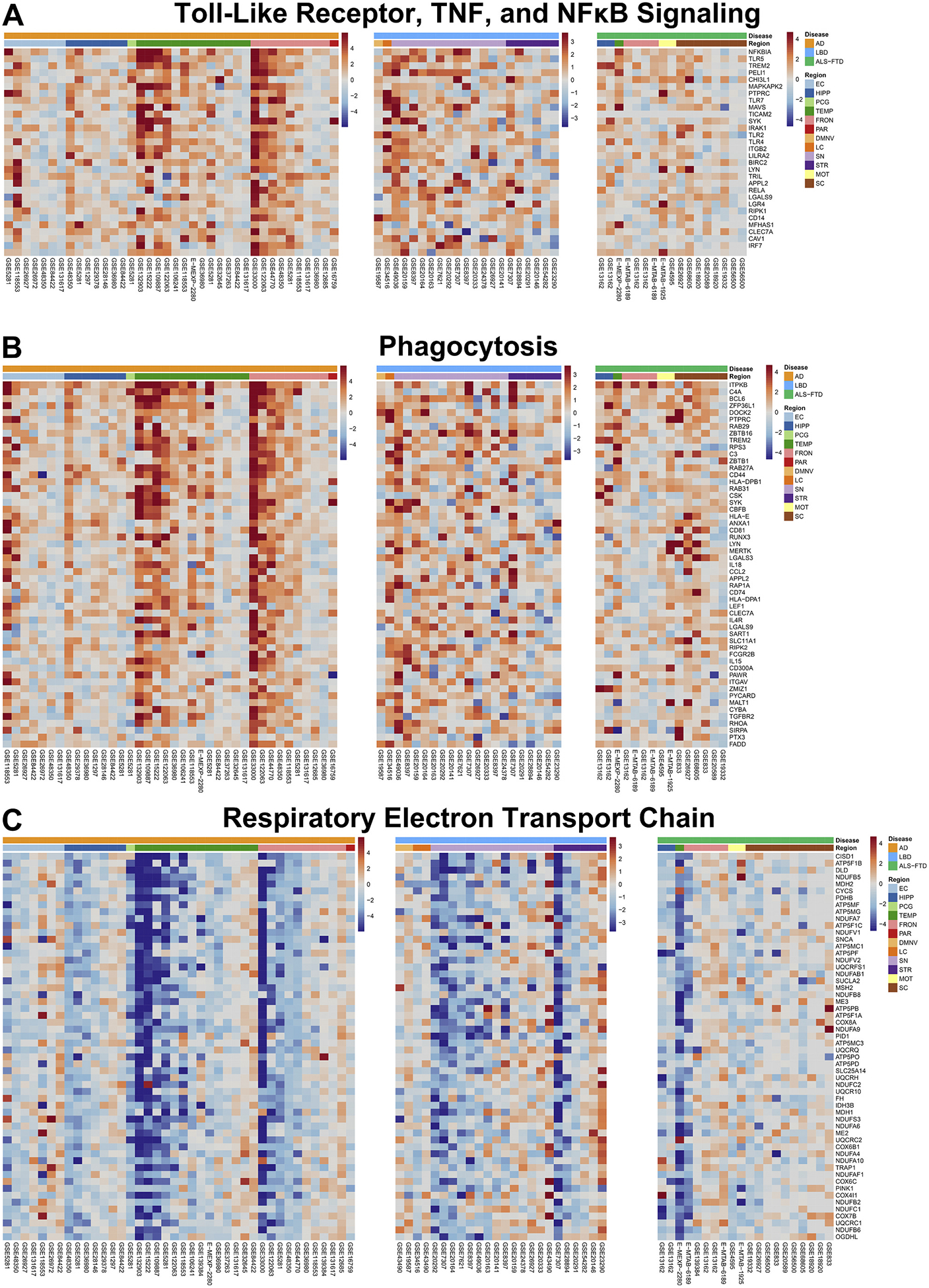
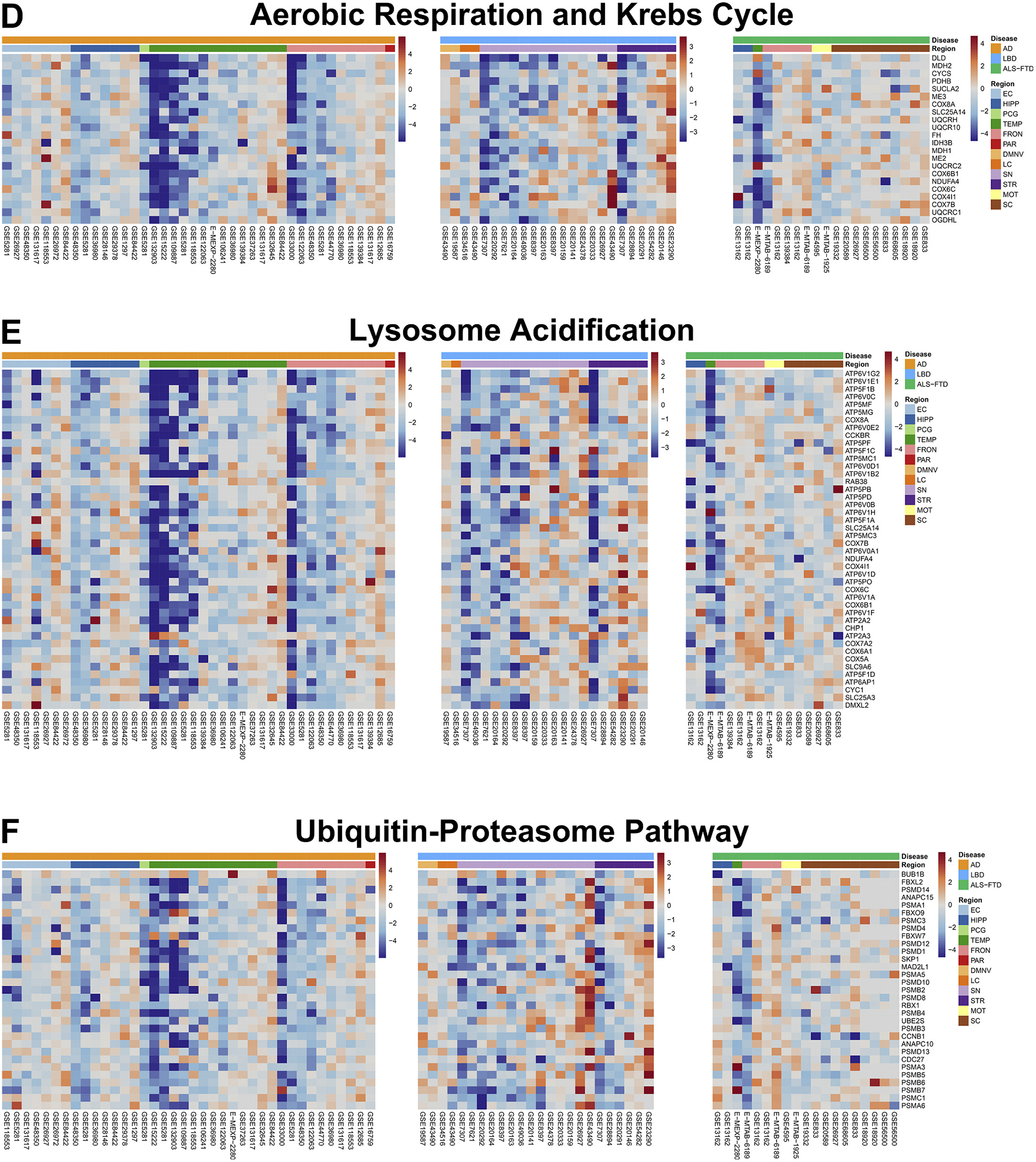
Genes with the top z-scores of the pan-neurodegenerative pathways were visualized in heatmaps, where columns represent individual datasets included in the meta-analysis, and rows represent moderated z-scores of differential expression for a specific gene across these datasets. Annotation bars represent z-scores, disease label, and CNS region (EC = entorhinal cortex, HIPP = hippocampus, PCG = posterior cingulate gyrus, TEMP = temporal cortex, FRONT = frontal cortex, PAR = parietal cortex, DMNV = dorsal motor nucleus of the vagus, LC = locus coeruleus, SN = substantia nigra, STR = striatum, MOT = motor cortex, SC = spinal cord).
Upregulated pathways in the pan-neurodegenerative signature were “response to zinc,” “toll-like receptor, TNF, and NFκB signaling,” “phagocytosis,” and “vasculature development.” Thus, phagocytosis and inflammatory responses emerge as the main functional gains in all three neurodegenerative diseases. The majority of the constituent genes of these pathways are enriched in glial cells: 58.5% of the genes have highest expression in either microglia (39.2%) or astrocytes (19.3%), followed by endothelial cells (14.4%), neurons (12.4%), and oligodendrocytes (5.1%); the remaining 9.6% were not enriched in any particular cell-type (Zhang et al., 2016).
In contrast, downregulated pathways included “respiratory electron transport chain,” “NAD metabolism,” “ATP biosynthesis,” “aerobic respiration and Krebs cycle,” “mitochondrial translation,” “aerobic electron transport chain,” “mitochondrion organization and axonal transport,” “calcium ion regulated catecholamine exocytosis,” “lysosome acidification,” and “ubiquitin-proteasome pathway.” Hence, the main functional losses are related to energy metabolism and proteostasis (both autophagy and ubiquitin-proteasome system). Many of these genes are neuronal: 38.9% of these genes have highest expression in neurons, followed by astrocytes (23.2%), microglia (9.9%), endothelial cells (8.2%), and oligodendrocytes (7.3%); the remaining 12.5% were not enriched in any particular cell-type (Zhang et al., 2016).
Several sensitivity analyses validated our pan-neurodegenerative signature. First, to ensure that our pan-neurodegenerative pathway signature was not merely the result of chance, we randomly permuted the disease and control labels corresponding to the individual samples in each dataset. After re-running all analyses, we obtained zero pathways at the intersection between the three diseases, supporting the robustness of our pan-neurodegenerative pathway signature (Fig. S1). Second, to demonstrate the reproducibility of our results with an independent pathway database, we conducted pathway enrichment analysis against the Reactome database and obtained very similar results to those from GO: BP (Table S6). Third, to account for any influence of neuron loss in our bulk RNA meta-analysis, we corrected the z-scores of the meta-analytic gene signatures using a set of genes expressed predominantly in neurons as described in the Methods section. These analyses yielded nearly identical results to those obtained without this correction, thus suggesting that our pan-neurodegenerative signature cannot be merely explained by a shift in cell-type proportions.
3.4. Meta-analysis of AD, LBD, and ALS-FTD microarray datasets also reveals disease-predominant transcriptomic signatures
Besides identifying a shared expression signature, this meta-analysis offered the opportunity to evaluate the transcriptomic changes characteristic of each of these three neurodegenerative diseases. GSEA followed by similarity-based clustering and expert annotation of resulting grouped pathways revealed distinct functional gains and losses in each disease (Table S5). Average normalized enrichment scores corresponding to key disease-predominant pathways are shown in Fig. 5.
Fig. 5. Disease-Predominant Pathways.
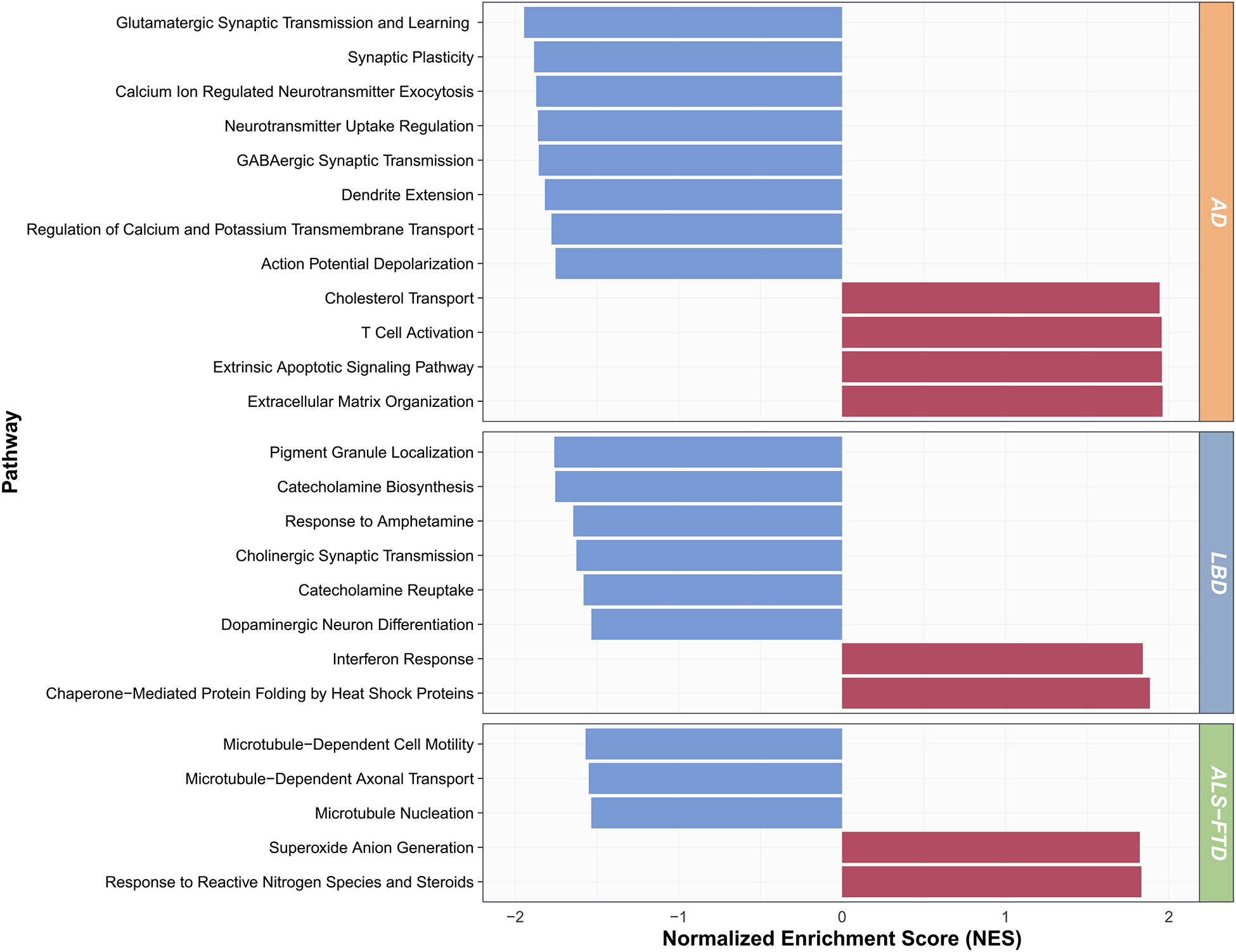
Gene set enrichment analysis (GSEA) normalized enrichment scores (NES) for representative disease-predominant pathways, averaged across the grouped GO: Biological Processes, are shown.
The AD-predominant signature was characterized by an upregulation of “cholesterol transport,” “extracellular matrix organization,” “extrinsic apoptotic signaling pathway,” and “T cell activation” among other functions, and a downregulation of “GABAergic transmission,” “glutamatergic synaptic transmission and learning,” “calcium ion regulated neurotransmitter exocytosis,” “neurotransmitter uptake regulation,” “action potential depolarization,” “regulation of calcium and potassium transmembrane transport,” “dendrite extension,” and “synaptic plasticity.”
The LBD-predominant signature consisted of an upregulation of “chaperone-mediated protein folding by heat shock proteins” and “interferon response,” and a downregulation of “catecholamine biosynthesis,” “response to amphetamine,” “catecholamine reuptake,” “pigment granule localization,” “dopaminergic neuron differentiation,” and “cholinergic synaptic transmission,” based on the most salient grouped pathways.
Finally, the ALS-FTD-predominant signature consisted of an upregulation of “response to reactive nitrogen species and steroids” and “superoxide anion generation,” and a downregulation of “microtubule-dependent axonal transport,” “microtubule-dependent cell motility,” and “microtubule nucleation,” among other pathways. Importantly, these pathways were largely conserved after correcting for neuron loss (see Methods), supporting the disease specificity of neurodegenerative processes in particular neuron types (i.e., glutamatergic in AD, cholinergic and catecholaminergic in LBD, and motor neurons – in which axonal transport is critical – in ALS-FTD).
4. Discussion
The use of bioinformatics tools on a large number of AD, LBD, and ALS-FTD transcriptomic datasets comprising a total of 1293 control and 1307 disease samples from diverse, pathologically relevant CNS regions (e.g., hippocampus, entorhinal cortex, and association cortex in AD; substantia nigra and striatum in LBD; and spinal cord and motor cortex in ALS-FTD) enabled the identification of a pan-neurodegenerative expression signature common to all three neurodegenerative diseases. This pan-neurodegenerative gene signature consisted of an upregulation of genes involved in innate immunity, cytoskeleton, RNA processing, and transcriptional regulation mainly in microglia, astrocytes and, to a lesser extent, oligodendrocytes, and a downregulation of genes involved in mitochondrial electron transport chain and energy metabolism, cytoskeleton/axon transport, and cell cycle/DNA repair, mainly in neurons. The overrepresentation of genes encoding for innate immune, actin-interacting, and other cytoskeleton proteins, together with extracellular matrix, cell adhesion/tight junction, and autophagy genes (e.g., cathepsins H and S) in the upregulated pan-neurodegenerative signature could be explained by an increase in cell motility and phagocytosis, since microglia are actively surveilling the neuropil and removing synapses and dead neurons (Fuhrmann et al., 2010), likely in concert with astrocytes (Damisah et al., 2020).
At the pathway level, the pan-neurodegenerative signature was defined by an upregulation of pro-inflammatory (including Toll-like receptor, TNF, and NFκB signaling) and phagocytic pathways, and a downregulation of mitochondrial oxidative phosphorylation. Intriguingly, lysosomal acidification (exemplified by ATP6V1G2 in the pan-neurodegenerative gene signature) and ubiquitin-proteasome pathways were also downregulated. This finding is noteworthy as aging itself may be associated with the existence of a metastable sub-proteome comprised of proteins whose physiologic concentration exceeds their solubility and are, therefore, prone to aggregation (Ciryam et al., 2016; Walther et al., 2017). Thus, our results suggest that the clearance pathways for these aggregated proteins may also be impaired in neurodegenerative diseases; experimental data on all three diseases support this conclusion (Gao et al., 2017; Scrivo et al., 2018; Vilchez et al., 2014). Furthermore, both autophagy and proteasome-mediated protein degradation require high levels of ATP, which may be in short supply in neurons due to the failure of energy metabolism (suggested by the downregulation of energy metabolism genes). Besides being an energy source, at high concentrations ATP may act as a biological hydro-trope, keeping proteins soluble or dissolving previously formed protein aggregates (Patel et al., 2017); therefore, an ATP deficit may favor further protein aggregation.
In addition, our meta-analysis enabled us to evaluate the functional gains and losses characteristic of each disease. Among the most distinct gains in AD were cholesterol transport – which has also been implicated in AD pathogenesis by genome-wide association studies (Kunkle et al., 2019) – and extracellular matrix organization. Similarly, protein chaperone activity and response to oxidative stress were upregulated in LBD and ALS-FTD, respectively. Conversely, the main functional losses were GABAergic and glutamatergic neurotransmission and synaptic plasticity in AD, catecholaminergic and cholinergic neurotransmission in LBD, and microtubule organization and axonal transport in ALS-FTD.
Importantly, while a shift in cell-type proportions associated with neuron loss could have partially contributed to the observed findings, our analyses aimed at correcting for this shift reinforce the conclusion that this pan-neurodegenerative signature is related to a dysfunction of surviving neurons rather than neuronal loss. Several other lines of evidence support this conclusion. First, recent single nuclei RNA-seq studies from human postmortem AD and control brains have shown that most DEGs assigned to nuclei from excitatory and inhibitory neurons are downregulated (Mathys et al., 2019). Second, a failure of energy metabolism, specifically glucose utilization, can be seen in disease-affected CNS regions of AD, LBD and ALS-FTD patients with [18F]-deoxy-glucose (FDG)-PET imaging, even after co-registering with MRI and correcting for severity of atrophy, and FDG-PET scans are helpful for the clinical diagnosis of AD (McKhann et al., 2011), DLB (McKeith et al., 2017) and FTD (Rascovsky et al., 2011). Third, a recent CSF and brain proteomic study in a large cohort of control and AD subjects has reported a downregulation of neuronal and mitochondrial proteins in AD as well as an upregulation of astrocyte and microglial proteins, suggesting that our results derived from transcriptomics hold true at the proteomics level (Johnson et al., 2020). Finally, although the 18 kDa translocator protein (TSPO) is not only expressed by microglia (Gui et al., 2020), an increased uptake of TSPO radiotracers in disease-relevant CNS areas has been shown for AD (Fan et al., 2015), LBD (Fan et al., 2015; Gerhard et al., 2006) and ALS-FTD (Alshikho et al., 2018), supporting microglial activation.
Some limitations of our study should be acknowledged. Combining datasets from different CNS regions obviates the regional heterogeneity in gene expression of the brain (Sjöstedt et al., 2020), however we discarded datasets from regions generally spared by the neurodegenerative process and assumed that the progression of the proteinopathy through the affected neural networks would attenuate those regional differences. Further, bulk RNA data analyses are inherently affected by shifts in cell-type proportion and cannot reliably account for transcripts that are upregulated by one cell type and downregulated by another. While our correction for neuron-predominant gene expression yielded similar results, future meta-analyses of existing single-nuclei RNA-seq studies are required to resolve these outstanding questions. Finally, as we filtered for duplicate probe mappings, spliced isoform-specific signal may have been omitted from our analyses. Nonetheless, our study has several strengths. We conducted a systematic review of microarray transcriptomic datasets in two different repositories following PRISMA guidelines to minimize selection bias. We rigorously meta-analyzed a large number of datasets of disease-affected CNS regions from three neurodegenerative diseases, totaling 1293 control and 1307 diseased samples (to our knowledge, the largest transcriptomics meta-analysis across AD, LBD, and ALS-FTD to date), and overlapped the three resulting meta-analytic signatures to obtain a robust, common neurodegenerative signature shared by all three.
In summary, our meta-analysis of AD, LBD, and ALS-FTD microarray transcriptomic studies identified neuroinflammation, together with a failure in neuronal energy metabolism and protein degradation, as the substrates underlying neurodegeneration. These results imply a gain of function in microglia and loss of function in neurons. Additional single-cell and single-nuclei RNA-seq studies in postmortem specimens from patients with various neurodegenerative diseases, as well as healthy subjects across the lifespan, are needed to confirm these findings and contrast them with normal aging.
Supplementary Material
Acknowledgements
The authors would like to thank Lori Chibnik, Rosemary J. Jackson, Townley Chisholm, and Alison Hobbie for useful discussions. The graphical abstract was created using BioRender.com.
Funding
This work was supported by the Alzheimer’s Association (AACF-17-524184 to AS-P), the National Institute of Aging (K08AG064039 to AS-P and P30AG062421 to BTH and SD), the Rainwater Charitable Foundation (to BTH), and a MassLife Sciences MassCATS award (to BTH and SD). The funding sources had no role in study design; data collection, analysis and interpretation; or manuscript preparation.
Footnotes
Declaration of Competing Interest
The authors declare no competing financial interests.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.nbd.2020.105225.
References
- Alshikho MJ, Zürcher NR, Loggia ML, Cernasov P, Reynolds B, Pijanowski O, Chonde DB, Izquierdo Garcia D, Mainero C, Catana C, Chan J, Babu S, Paganoni S, Hooker JM, Atassi N, 2018. Integrated magnetic resonance imaging and [11 C]-PBR28 positron emission tomographic imaging in amyotrophic lateral sclerosis. Ann. Neurol 83, 1186–1197. 10.1002/ana.25251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G, 2000. Gene ontology: tool for the unification of biology. Nat. Genet 25, 25–29. 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brion JP, Flament-Durand J, Dustin P, 1986. Alzheimer’s disease and tau proteins. Lancet Lond. Engl 2, 1098 10.1016/s0140-6736(86)90495-2. [DOI] [PubMed] [Google Scholar]
- Carvalho BS, Irizarry RA, 2010. A framework for oligonucleotide microarray preprocessing. Bioinformatics 26, 2363–2367. 10.1093/bioinformatics/btq431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciryam P, Kundra R, Freer R, Morimoto RI, Dobson CM, Vendruscolo M, 2016. A transcriptional signature of Alzheimer’s disease is associated with a metastable subproteome at risk for aggregation. Proc. Natl. Acad. Sci. U. S. A 113, 4753–4758. 10.1073/pnas.1516604113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damisah EC, Hill RA, Rai A, Chen F, Rothlin CV, Ghosh S, Grutzendler J, 2020. Astrocytes and microglia play orchestrated roles and respect phagocytic territories during neuronal corpse removal in vivo. Sci. Adv 6 10.1126/sciadv.aba3239 eaba3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Li Z, Noori A, Hyman BT, Serrano-Pozo A, 2020. Meta-analysis of mouse transcriptomic studies supports a context-dependent astrocyte reaction in acute CNS injury versus neurodegeneration. J. Neuroinflammation 17, 227 10.1186/s12974-020-01898-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrenberger PF, Fernando FS, Kashefi SN, Bonnert TP, Seilhean D, Nait-Oumesmar B, Schmitt A, Gebicke-Haerter PJ, Falkai P, Grünblatt E, Palkovits M, Arzberger T, Kretzschmar H, Dexter DT, Reynolds R, 2015. Common mechanisms in neurodegeneration and neuroinflammation: A BrainNet Europe gene expression microarray study. J. Neural Transm. Vienna Austria 1996 122, 1055–1068. 10.1007/s00702-014-1293-0. [DOI] [PubMed] [Google Scholar]
- Fan Z, Aman Y, Ahmed I, Chetelat G, Landeau B, Ray Chaudhuri K, Brooks DJ, Edison P, 2015. Influence of microglial activation on neuronal function in Alzheimer’s and Parkinson’s disease dementia. Alzheimers Dement. J. Alzheimers Assoc 11, 608–621 e7. 10.1016/j.jalz.2014.06.016. [DOI] [PubMed] [Google Scholar]
- Fuhrmann M, Bittner T, Jung CKE, Burgold S, Page RM, Mitteregger G, Haass C, LaFerla FM, Kretzschmar H, Herms J, 2010. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nat. Neurosci 13, 411–413. 10.1038/nn.2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F-B, Almeida S, Lopez-Gonzalez R, 2017. Dysregulated molecular pathways in amyotrophic lateral sclerosis-frontotemporal dementia spectrum disorder. EMBO J. 36, 2931–2950. 10.15252/embj.201797568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhard A, Pavese N, Hotton G, Turkheimer F, Es M, Hammers A, Eggert K, Oertel W, Banati RB, Brooks DJ, 2006. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol. Dis 21, 404–412. 10.1016/j.nbd.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Glenner GG, Wong CW, 1984. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun 120, 885–890. 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- Grundke-Iqbal I, Iqbal K, Quinlan M, Tung YC, Zaidi MS, Wisniewski HM, 1986. Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J. Biol. Chem 261, 6084–6089. [PubMed] [Google Scholar]
- Gui Y, Marks JD, Das S, Hyman BT, Serrano-Pozo A, 2020. Characterization of the 18 kDa translocator protein (TSPO) expression in post-mortem normal and Alzheimer’s disease brains. Brain Pathol. Zurich Switz 30, 151–164. 10.1111/bpa.12763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP, 2003. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostat. Oxf. Engl 4, 249–264. 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- Jarosz DF, Khurana V, 2017. Specification of physiologic and disease states by distinct proteins and protein conformations. Cell 171, 1001–1014. 10.1016/j.cell.2017.10.047. [DOI] [PubMed] [Google Scholar]
- Jassal B, Matthews L, Viteri G, Gong C, Lorente P, Fabregat A, Sidiropoulos K, Cook J, Gillespie M, Haw R, Loney F, May B, Milacic M, Rothfels K, Sevilla C, Shamovsky V, Shorser S, Varusai T, Weiser J, Wu G, Stein L, Hermjakob H, D’Eustachio P, 2020. The reactome pathway knowledge base. Nucleic Acids Res. 48, D498–D503. 10.1093/nar/gkz1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson ECB, Dammer EB, Duong DM, Ping L, Zhou M, Yin L, Higginbotham LA, Guajardo A, White B, Troncoso JC, Thambisetty M, Montine TJ, Lee EB, Trojanowski JQ, Beach TG, Reiman EM, Haroutunian V, Wang M, Schadt E, Zhang B, Dickson DW, Ertekin-Taner N, Golde TE, Petyuk VA, De Jager PL, Bennett DA, Wingo TS, Rangaraju S, Hajjar I, Shulman JM, Lah JJ, Levey AI, Seyfried NT, 2020. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat. Med 26, 769–780. 10.1038/s41591-020-0815-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffmann A, Huber W, 2010. Microarray data quality control improves the detection of differentially expressed genes. Genomics 95, 138–142. 10.1016/j.ygeno.2010.01.003. [DOI] [PubMed] [Google Scholar]
- Kauffmann A, Gentleman R, Huber W, 2009. arrayQualityMetrics - A bioconductor package for quality assessment of microarray data. Bioinformatics 25, 415–416. 10.1093/bioinformatics/btn647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly J, Moyeed R, Carroll C, Albani D, Li X, 2019. Gene expression meta-analysis of Parkinson’s disease and its relationship with Alzheimer’s disease. Mol. Brain 12, 16 10.1186/s13041-019-0436-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, Boland A, Vronskaya M, van der Lee SJ, Amlie-Wolf A, Bellenguez C, Frizatti A, Chouraki V, Martin ER, Sleegers K, Badarinarayan N, Jakobsdottir J, Hamilton-Nelson KL, Moreno-Grau S, Olaso R, Raybould R, Chen Y, Kuzma AB, Hiltunen M, Morgan T, Ahmad S, Vardarajan BN, Epelbaum J, Hoffmann P, Boada M, Beecham GW, Garnier J-G, Harold D, Fitzpatrick AL, Valladares O, Moutet M-L, Gerrish A, Smith AV, Qu L, Bacq D, Denning N, Jian X, Zhao Y, Del Zompo M, Fox NC, Choi S-H, Mateo I, Hughes JT, Adams HH, Malamon J, Sanchez-Garcia F, Patel Y, Brody JA, Dombroski BA, Naranjo MCD, Daniilidou M, Eiriksdottir G, Mukherjee S, Wallon D, Uphill J, Aspelund T, Cantwell LB, Garzia F, Galimberti D, Hofer E, Butkiewicz M, Fin B, Scarpini E, Sarnowski C, Bush WS, Meslage S, Kornhuber J, White CC, Song Y, Barber RC, Engelborghs S, Sordon S, Voijnovic D, Adams PM, Vandenberghe R, Mayhaus M, Cupples LA, Albert MS, De Deyn PP, Gu W, Himali JJ, Beekly D, Squassina A, Hartmann AM, Orellana A, Blacker D, Rodriguez-Rodriguez E, Lovestone S, Garcia ME, Doody RS, Munoz-Fernadez C, Sussams R, Lin H, Fairchild TJ, Benito YA, Holmes C, Karamujić-Čomić H, Frosch MP, Thonberg H, Maier W, Roshchupkin G, Ghetti B, Giedraitis V, Kawalia A, Li S, Huebinger RM, Kilander L, Moebus S, Herńandez I, Kamboh MI, Brundin R, Turton J, Yang Q, Katz MJ, Concari L, Lord J, Beiser AS, Keene CD, Helisalmi S, Kloszewska I, Kukull WA, Koivisto AM, Lynch A, Tarraga L, Larson EB, Haapasalo A, Lawlor B, Mosley TH, Lipton RB, Solfrizzi V, Gill M, Longstreth WT, Montine TJ, Frisardi V, Diez-Fairen M, Rivadeneira F, Petersen RC, Deramecourt V, Alvarez I, Salani F, Ciaramella A, Boerwinkle E, Reiman EM, Fievet N, Rotter JI, Reisch JS, Hanon O, Cupidi C, Andre Uitterlinden AG, Royall DR, Dufouil C, Maletta RG, de Rojas I, Sano M, Brice A, Cecchetti R, George-Hyslop PS, Ritchie K, Tsolaki M, Tsuang DW, Dubois B, Craig D, Wu C-K, Soininen H, Avramidou D, Albin RL, Fratiglioni L, Germanou A, Apostolova LG, Keller L, Koutroumani M, Arnold SE, Panza F, Gkatzima O, Asthana S, Hannequin D, Whitehead P, Atwood CS, Caffarra P, Hampel H, Quintela I, Carracedo Á, Lannfelt L, Rubinsztein DC, Barnes LL, Pasquier F, Frölich L, Barral S, McGuinness B, Beach TG, Johnston JA, Becker JT, Passmore P, Bigio EH, Schott JM, Bird TD, Warren JD, Boeve BF, Lupton MK, Bowen JD, Proitsi P, Boxer A, Powell JF, Burke JR, Kauwe JSK, Burns JM, Mancuso M, Buxbaum JD, Bonuccelli U, Cairns NJ, McQuillin A, Cao C, Livingston G, Carlson CS, Bass NJ, Carlsson CM, Hardy J, Carney RM, Bras J, Carrasquillo MM, Guerreiro R, Allen M, Chui HC, Fisher E, Masullo C, Crocco EA, DeCarli C, Bisceglio G, Dick M, Ma L, Duara R, Graff-Radford NR, Evans DA, Hodges A, Faber KM, Scherer M, Fallon KB, Riemenschneider M, Fardo DW, Heun R, Farlow MR, Kölsch H, Ferris S, Leber M, Foroud TM, Heuser I, Galasko DR, Giegling I, Gearing M, Hüll M, Geschwind DH, Gilbert JR, Morris J, Green RC, Mayo K, Growdon JH, Feulner T, Hamilton RL, Harrell LE, Drichel D, Honig LS, Cushion TD, Huentelman MJ, Hollingworth P, Hulette CM, Hyman BT, Marshall R, Jarvik GP, Meggy A, Abner E, Menzies GE, Jin L-W, Leonenko G, Real LM, Jun GR, Baldwin CT, Grozeva D, Karydas A, Russo G, Kaye JA, Kim R, Jessen F, Kowall NW, Vellas B, Kramer JH, Vardy E, LaFerla FM, Jöckel KH, Lah JJ, Dichgans M, Leverenz JB, Mann D, Levey AI, Pickering-Brown S, Lieberman AP, Klopp N, Lunetta KL, Wichmann H-E, Lyketsos CG, Morgan K, Marson DC, Brown K, Martiniuk F, Medway C, Mash DC, Nöthen MM, Masliah E, Hooper NM, McCormick WC, Daniele A, McCurry SM, Bayer A, McDavid AN, Gallacher J, McKee AC, van den Bussche H, Mesulam M, Brayne C, Miller BL, Riedel-Heller S, Miller CA, Miller JW, Al-Chalabi A, Morris JC, Shaw CE, Myers AJ, Wiltfang J, O’Bryant S, Olichney JM, Alvarez V, Parisi JE, Singleton AB, Paulson HL, Collinge J, Perry WR, Mead S, Peskind E, Cribbs DH, Rossor M, Pierce A, Ryan NS, Poon WW, Nacmias B, Potter H, Sorbi S, Quinn JF, Sacchinelli E, Raj A, Spalletta G, Raskind M, Caltagirone C, Bossù P, Orfei MD, Reisberg B, Clarke R, Reitz C, Smith AD, Ringman JM, Warden D, Roberson ED, Wilcock G, Rogaeva E, Bruni AC, Rosen HJ, Gallo M, Rosenberg RN, Ben-Shlomo Y, Sager MA, Mecocci P, Saykin AJ, Pastor P, Cuccaro ML, Vance JM, Schneider JA, Schneider LS, Slifer S, Seeley WW, Smith AG, Sonnen JA, Spina S, Stern RA, Swerdlow RH, Tang M, Tanzi RE, Trojanowski JQ, Troncoso JC, Van Deerlin VM, Van Eldik LJ, Vinters HV, Vonsattel JP, Weintraub S, Welsh-Bohmer KA, Wilhelmsen KC, Williamson J, Wingo TS, Woltjer RL, Wright CB, Yu C-E, Yu L, Saba Y, Pilotto A, Bullido MJ, Peters O, Crane PK, Bennett D, Bosco P, Coto E, Boccardi V, De Jager PL, Lleo A, Warner N, Lopez OL, Ingelsson M, Deloukas P, Cruchaga C, Graff C, Gwilliam R, Fornage M, Goate AM, Sanchez-Juan P, Kehoe PG, Amin N, Ertekin-Taner N, Berr C, Debette S, Love S, Launer LJ, Younkin SG, Dartigues J-F, Corcoran C, Ikram MA, Dickson DW, Nicolas G, Campion D, Tschanz J, Schmidt H, Hakonarson H, Clarimon J, Munger R, Schmidt R, Farrer LA, Van Broeckhoven C, O’Donovan M, DeStefano AL, Jones L, Haines JL, Deleuze J-F, Owen MJ, Gudnason V, Mayeux R, Escott-Price V, Psaty BM, Ramirez A, Wang L-S, Ruiz A, van Duijn CM, Holmans PA, Seshadri S, Williams J, Amouyel P, Schellenberg GD, Lambert J-C, Pericak-Vance MA, Alzheimer Disease Genetics Consortium (ADGC), European Alzheimer’s Disease Initiative (EADI), Cohorts for Heart and Aging Research in Genomic Epidemiology Consortium (CHARGE), Genetic and Environmental Risk in AD/Defining Genetic, Polygenic and Environmental Risk for Alzheimer’s Disease Consortium (GERAD/PERADES), 2019. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet 51, 414–430. 10.1038/s41588-019-0358-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski TJ, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, Valdmanis P, Rouleau GA, Hosler BA, Cortelli P, de Jong PJ, Yoshinaga Y, Haines JL, Pericak-Vance MA, Yan J, Ticozzi N, Siddique T, McKenna-Yasek D, Sapp PC, Horvitz HR, Landers JE, Brown RH, 2009. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323, 1205–1208. 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- Labadorf A, Choi SH, Myers RH, 2018. Evidence for a pan-neurodegenerative disease response in Huntington’s and Parkinson’s disease expression profiles. Front. Mol. Neurosci 10 10.3389/fnmol.2017.00430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek JT, Storey JD, 2007. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 3, e161 10.1371/journal.pgen.0030161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD, 2012. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883. 10.1093/bioinformatics/bts034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MD, Burns TC, Morgan AA, Khatri P, 2014. Integrated multi-cohort transcriptional meta-analysis of neurodegenerative diseases. Acta Neuropathol. Commun 2, 93 10.1186/s40478-014-0093-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberati A, Altman DG, Tetzlaff J, Mulrow C, Gøtzsche PC, Ioannidis JPA, Clarke M, Devereaux PJ, Kleijnen J, Moher D, 2009. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate healthcare interventions: Explanation and elaboration. The BMJ 339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP, 2011. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740. 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipták T, 1958. On the combination of independent tests. Magy. Tud Akad Mat Kut. Int. Kozl 3, 171–197. [Google Scholar]
- López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G, 2013. The hallmarks of aging. Cell 153, 1194–1217. 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maglott D, Ostell J, Pruitt KD, Tatusova T, 2007. Entrez gene: Gene-centered information at NCBI. Nucleic Acids Res. 35, D26–D31. 10.1093/nar/gkl993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X, Martorell AJ, Ransohoff RM, Hafler BP, Bennett DA, Kellis M, Tsai L-H, 2019. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337. 10.1038/s41586-019-1195-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeith IG, Boeve BF, Dickson DW, Halliday G, Taylor J-P, Weintraub D, Aarsland D, Galvin J, Attems J, Ballard CG, Bayston A, Beach TG, Blanc F, Bohnen N, Bonanni L, Bras J, Brundin P, Burn D, Chen-Plotkin A, Duda JE, El-Agnaf O, Feldman H, Ferman TJ, Ffytche D, Fujishiro H, Galasko D, Goldman JG, Gomperts SN, Graff-Radford NR, Honig LS, Iranzo A, Kantarci K, Kaufer D, Kukull W, Lee VMY, Leverenz JB, Lewis S, Lippa C, Lunde A, Masellis M, Masliah E, McLean P, Mollenhauer B, Montine TJ, Moreno E, Mori E, Murray M, O’Brien JT, Orimo S, Postuma RB, Ramaswamy S, Ross OA, Salmon DP, Singleton A, Taylor A, Thomas A, Tiraboschi P, Toledo JB, Trojanowski JQ, Tsuang D, Walker Z, Yamada M, Kosaka K, 2017. Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology 89, 88–100. 10.1212/WNL.0000000000004058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R, Mohs RC, Morris JC, Rossor MN, Scheltens P, Carrillo MC, Thies B, Weintraub S, Phelps CH, 2011. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. J. Alzheimers Assoc 7, 263–269. 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori K, Weng S-M, Arzberger T, May S, Rentzsch K, Kremmer E, Schmid B, Kretzschmar HA, Cruts M, Van Broeckhoven C, Haass C, Edbauer D, 2013. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339, 1335–1338. 10.1126/science.1232927. [DOI] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM-Y, 2006. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133. 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Oerton E, Bender A, 2017. Concordance analysis of microarray studies identifies representative gene expression changes in Parkinson’s disease: A comparison of 33 human and animal studies. BMC Neurol. 17, 58 10.1186/s12883-017-0838-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A, Malinovska L, Saha S, Wang J, Alberti S, Krishnan Y, Hyman AA, 2017. ATP as a biological hydrotrope. Science 356, 753–756. 10.1126/science.aaf6846. [DOI] [PubMed] [Google Scholar]
- Patel H, Dobson RJB, Newhouse SJ, 2019. A meta-analysis of Alzheimer’s disease brain transcriptomic data. J. Alzheimers Dis 68, 1635–1656. 10.3233/JAD-181085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phipson B, Lee S, Majewski IJ, Alexander WS, Smyth GK, 2016. Robust hyperparameter estimation protects against hypervariable genes and improves power to detect differential expression. Ann. Appl. Stat 10, 946–963. 10.1214/16-AOAS920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rascovsky K, Hodges JR, Knopman D, Mendez MF, Kramer JH, Neuhaus J, van Swieten JC, Seelaar H, Dopper EGP, Onyike CU, Hillis AE, Josephs KA, Boeve BF, Kertesz A, Seeley WW, Rankin KP, Johnson JK, Gorno-Tempini M-L, Rosen H, Prioleau-Latham CE, Lee A, Kipps CM, Lillo P, Piguet O, Rohrer JD, Rossor MN, Warren JD, Fox NC, Galasko D, Salmon DP, Black SE, Mesulam M, Weintraub S, Dickerson BC, Diehl-Schmid J, Pasquier F, Deramecourt V, Lebert F, Pijnenburg Y, Chow TW, Manes F, Grafman J, Cappa SF, Freedman M, Grossman M, Miller BL, 2011. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain J. Neurol 134, 2456–2477. 10.1093/brain/awr179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK, 2015. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scrivo A, Bourdenx M, Pampliega O, Cuervo AM, 2018. Selective autophagy as a potential therapeutic target for neurodegenerative disorders. Lancet Neurol. 17, 802–815. 10.1016/S1474-4422(18)30238-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjöstedt E, Zhong W, Fagerberg L, Karlsson M, Mitsios N, Adori C, Oksvold P, Edfors F, Limiszewska A, Hikmet F, Huang J, Du Y, Lin L, Dong Z, Yang L, Liu X, Jiang H, Xu X, Wang J, Yang H, Bolund L, Mardinoglu A, Zhang C, von Feilitzen K, Lindskog C, Pontén F, Luo Y, Hökfelt T, Uhlén M, Mulder J, 2020. An atlas of the protein-coding genes in the human, pig, and mouse brain. Science 367 10.1126/science.aay5947. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M, 1997. Alpha-synuclein in Lewy bodies. Nature 388, 839–840. 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP, 2005. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A 102, 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Kuehn H, Gould J, Tamayo P, Mesirov JP, 2007. GSEA-P: A desktop application for gene set enrichment analysis. Bioinform. Oxf. Engl 23, 3251–3253. 10.1093/bioinformatics/btm369. [DOI] [PubMed] [Google Scholar]
- Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, Jensen LJ, von Mering C, 2019. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613. 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Gene Ontology Consortium, 2019. The gene ontology resource: 20 years and still GOing strong. Nucleic Acids Res. 47, D330–D338. 10.1093/nar/gky1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth-Petroczy A, Palmedo P, Ingraham J, Hopf TA, Berger B, Sander C, Marks DS, 2016. Structured states of disordered proteins from genomic sequences. Cell 167, 158–170 e12. 10.1016/j.cell.2016.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilchez D, Saez I, Dillin A, 2014. The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat. Commun 5, 5659 10.1038/ncomms6659. [DOI] [PubMed] [Google Scholar]
- Walsh CJ, Hu P, Batt J, Santos CCD, 2015. Microarray meta-analysis and cross-platform normalization: Integrative genomics for robust biomarker discovery. Microarrays Basel Switz. 4, 389–406. 10.3390/microarrays4030389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther DM, Kasturi P, Zheng M, Pinkert S, Vecchi G, Ciryam P, Morimoto RI, Dobson CM, Vendruscolo M, Mann M, Hartl FU, 2017. Widespread proteome remodeling and aggregation in aging C. elegans. Cell 168, 944 10.1016/j.cell.2016.12.041. [DOI] [PubMed] [Google Scholar]
- Wan Y-W, Al-Ouran R, Mangleburg CG, Perumal TM, Lee TV, Allison K, Swarup V, Funk CC, Gaiteri C, Allen M, Wang M, Neuner SM, Kaczorowski CC, Philip VM, Howell GR, Martini-Stoica H, Zheng H, Mei H, Zhong X, Kim JW, Dawson VL, Dawson TM, Pao P-C, Tsai L-H, Haure-Mirande J-V, Ehrlich ME, Chakrabarty P, Levites Y, Wang X, Dammer EB, Srivastava G, Mukherjee S, Sieberts SK, Omberg L, Dang KD, Eddy JA, Snyder P, Chae Y, Amberkar S, Wei W, Hide W, Preuss C, Ergun A, Ebert PJ, Airey DC, Mostafavi S, Yu L, Klein H-U, Accelerating Medicines Partnership-Alzheimer’s Disease Consortium, Carter GW, Collier DA, Golde TE, Levey AI, Bennett DA, Estrada K, Townsend TM, Zhang B, Schadt E, De Jager PL, Price ND, Ertekin-Taner N, Liu Z, Shulman JM, Mangravite LM, Logsdon BA, 2020. Meta-analysis of the Alzheimer’s disease human brain transcriptome and functional dissection in mouse models. Cell Rep. 32, 107908 10.1016/j.celrep.2020.107908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Lin Y, Song C, Sibille E, Tseng GC, 2012. Detecting disease-associated genes with confounding variable adjustment and the impact on genomic meta-analysis: With application to major depressive disorder. BMC Bioinform. 13, 52 10.1186/1471-2105-13-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Zhang Y, Wang M, Song W-M, Shen Q, McKenzie A, Choi I, Zhou X, Pan P-Y, Yue Z, Zhang B, 2019. The landscape of multiscale transcriptomic networks and key regulators in Parkinson’s disease. Nat. Commun 10, 5234 10.1038/s41467-019-13144-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesseling H, Mair W, Kumar M, Schlaffner CN, Tang S, Beerepoot P, Fatou B, Guise AJ, Cheng L, Takeda S, Muntel J, Rotunno MS, Dujardin S, Davies P, Kosik KS, Miller BL, Berretta S, Hedreen JC, Grinberg LT, Seeley WW, Hyman BT, Steen H, Steen JA, 2020. Tau PTM profiles identify patient heterogeneity and stages of Alzheimer’s disease. Cell. 10.1016/j.cell.2020.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaykin DV, 2011. Optimally weighted Z-test is a powerful method for combining probabilities in meta-analysis. J. Evol. Biol 24, 1836–1841. 10.1111/j.1420-9101.2011.02297.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, Vogel H, Steinberg GK, Edwards MSB, Li G, Duncan JA, Cheshier SH, Shuer LM, Chang EF, Grant GA, Gephart MGH, Barres BA, 2016. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 89, 37–53. 10.1016/j.neuron.2015.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B, Liao Z, Locascio JJ, Lesniak KA, Roderick SS, Watt ML, Eklund AC, Zhang-James Y, Kim PD, Hauser MA, Grünblatt E, Moran LB, Mandel SA, Riederer P, Miller RM, Federoff HJ, Wüllner U, Papapetropoulos S, Youdim MB, Cantuti-Castelvetri I, Young AB, Vance JM, Davis RL, Hedreen JC, Adler CH, Beach TG, Graeber MB, Middleton FA, Rochet J-C, Scherzer CR, Global PD Gene Expression (GPEX) Consortium, 2010. PGC-1α, a potential therapeutic target for early intervention in Parkinson’s disease. Sci. Transl. Med 2, 52–73. 10.1126/scitranslmed.3001059. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.