

Abstract
Mitochondrial diseases (MD) are a heterogeneous group of disorders resulting from genetic mutations in nuclear or mitochondrial DNA (mtDNA) genes encoding for mitochondrial proteins 1,2. MD cause pathologies with severe tissue damage and ultimately death 3,4. There are no cures for MD and current treatments are only palliative 5–7. Here we show that tetracyclines improve fitness of cultured MD cells and ameliorate disease in a mouse model of Leigh syndrome. To identify small molecules that prevent cellular damage and death under nutrient stress conditions, we conduct a chemical high-throughput screen with cells carrying human MD mutations and discover a series of antibiotics that maintain survival of various MD cells. We go on to show that a sub-library of tetracycline analogs, including doxycycline, rescues cell death and inflammatory signatures in mutant cells through partial and selective inhibition of mitochondrial translation, resulting in an ATF4-independent mitohormetic response. Doxycycline treatment strongly promotes fitness and survival of Ndufs4−/− mice, a pre-clinical Leigh syndrome mouse model 8. A proteomic analysis of brain tissue reveals that doxycycline treatment largely prevents neuronal death and the accumulation of neuroimmune and inflammatory proteins in Ndufs4−/− mice, indicating a potential causality of these proteins in the brain pathology. Our findings suggest that tetracyclines deserve further evaluation as a potential drugs for the treatment of MD.
MD cause diverse pathologies including neurodegeneration, cardiac and muscle myopathies, liver and kidney failures, deafness, and ultimately death 3,9. Since there are currently no effective therapies for MD, we aimed to identify small molecules that ameliorate the detrimental phenotypes and death associated with mitochondrial mutations. As a cellular model for MD, we used human MELAS homoplasmic mutant cybrid cells (A3243G tRNALeu(UUR)) 10 that exhibit complete reduction in respiratory complex function (Extended Data Fig. 1a) and rely heavily on glycolysis for survival and proliferation 11. These mutant cells die in glucose restriction conditions from apoptosis (Extended Data Fig. 1b), a phenotype that can be exploited for screening readouts (Fig. 1a). We designed a high-throughput chemical screen platform using MELAS cybrid cells scoring viability after 48 hours in low-glucose culture medium (2.5 mM) (Fig. 1a). Of a library of 4,937 known bioactive compounds screened, we identified 184 hits (z-score>1.96, 97.5% confidence interval) that promoted cell survival (Fig. 1b). The main classes of compounds included tetracycline antibiotics, p53 activators, and HSP90 and mTOR inhibitors (Fig. 1c). mTOR inhibitors improve lifespan in multiple models of MD 12–15, validating our screening platform for moving compounds towards in vivo testing. Top hits from the screen re-tested in a non-high-throughput 7 day low-glucose cell survival assay (Fig. 1d). Among the top scored compounds, tetracyclines such as doxycycline promoted the highest survival and proliferation in MELAS cells (Fig. 1d). These effects were not exclusive to MELAS cybrids as other mitochondrial mutant cells such as Rieske (complex III) KO mouse fibroblasts 16 and ND1 A3796G and ND6 G14459A (complex I) 17 homoplasmic mutant cybrid cells were rescued from cell death by doxycycline treatment in glucose restriction conditions (Fig. 1e–i). Notably, the pro-survival effects of doxycycline were independent of shifts in mutant mtDNA levels (Extended Data Fig. 1c) in cybrids or respiratory chain complex levels (Extended Data Fig. 1d–f). Doxycycline also improved survival in wild-type cells treated with piericidin (complex I inhibitor) or antimycin (complex III inhibitor) during glucose deprivation (Fig. 1j). These results indicate that tetracycline antibiotics promote cell survival under nutrient stress in different cellular models of MDs.
Fig. 1. Small molecule screen identifies antibiotics as suppressors of cell death in cellular models of human mitochondrial disease.
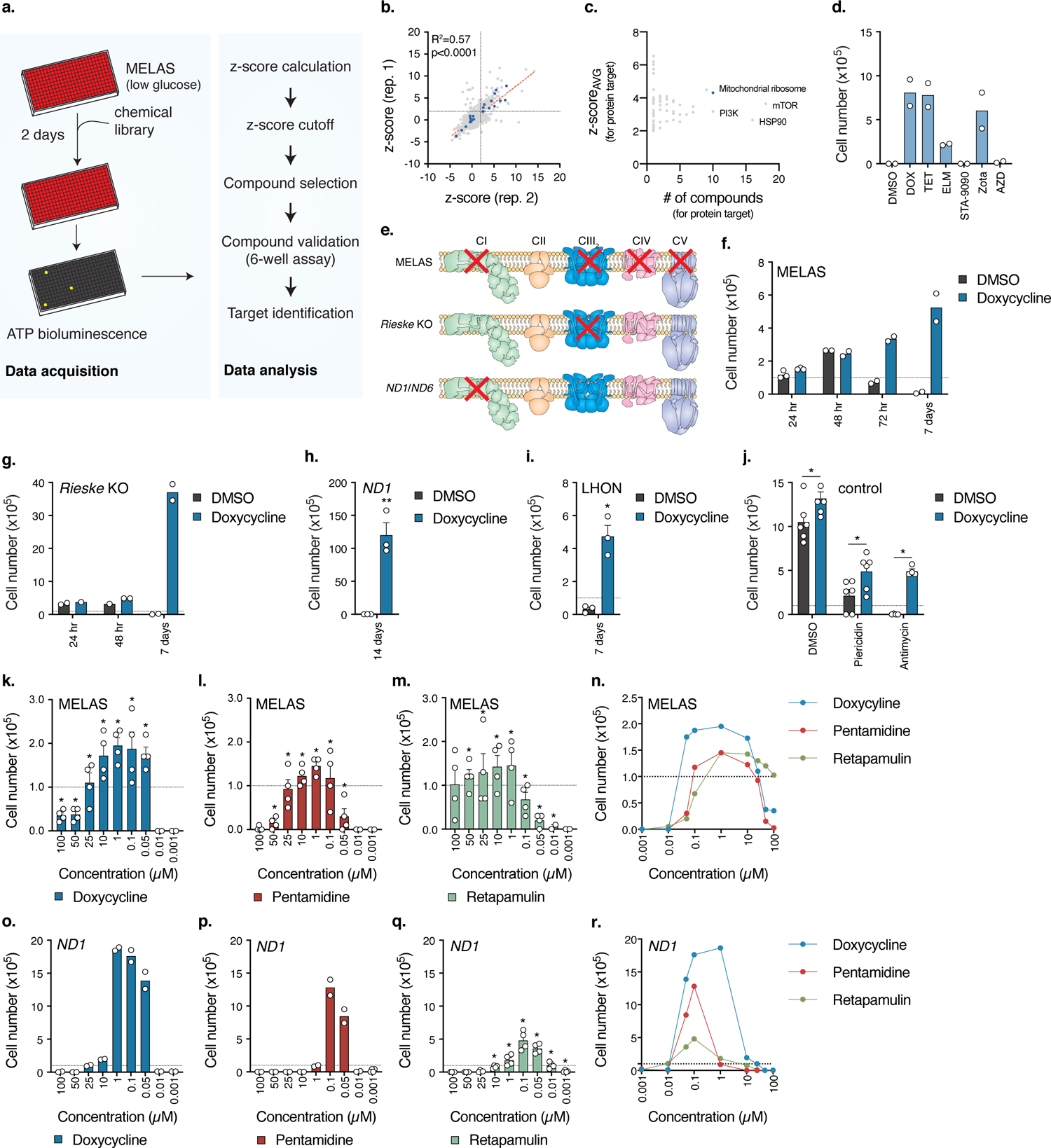
a. Schematic of the high-throughput small molecule screen to identify compounds that increase survival of MELAS cybrid cells. b. Plot of z-scores for each compound from two replicates. Antibiotics targeting the mitochondrial ribosome are highlighted in blue, R2 = 0.5704 (p<0.0001, Pearson). c. Plot of average z-scores for a particular protein against the number of compounds for that target. For compounds with multiple annotated target proteins, the compound z-score was incorporated into each protein target z-scoreAVG. d. 7 day low-glucose survival assay of MELAS cybrid cells treated with 3.3 μM compound (n=2 biologically independent samples). e. Comparison of electron transfer chain defects between the different cell lines used in the studies. MELAS, ND1, and ND6 (LHON) are human cybrid cell lines and Rieske is a mouse fibroblast cell line. Adapted from Rhee, H.W. et al.58 Reprinted with permission from AAAS. f–g. 7 day low-glucose survival assay of MELAS cybrid or Rieske cells treated with doxycycline (MELAS, n=2–3; Rieske, n=2 biologically independent samples). h. 14 day galactose survival assay of ND1 cybrid cells treated with doxycycline (n=3 biologically independent samples). i. 7 day galactose survival of LHON cybrid cells treated with doxycycline (n=3 biologically independent samples). j. Cell survival of U2OS control cells treated with 1 nM piericidin or 5 nM antimycin with 1 μM doxycycline in galactose media (n=2–3 biologically independent samples over n=2 independent experiments). k–m. 48 hour low-glucose survival assay of MELAS cybrid cells (n=2 biologically independent samples over n=2 independent experiments). n. Overlay of Fig. 1k–m. o–q. 7 day galactose survival assay of ND1 cybrid cells titrated with 3 structurally distinct antibiotics (Doxycycline, n=2; Pentamidine, n=2; Retapamulin, n=4 biologically independent samples). r. Overlay of Fig. 1o–q. Survival assays were initially seeded with 1.0×105 cells with media changed every 24 hours for low-glucose assays and 48 hours for galactose assays. Data are presented as mean values +/− s.e.m. error bars, Student’s t-test with a two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli, with Q = 5%, * q<0.05.
In addition to tetracyclines, the antiparasitic agent pentamidine and the antibiotic retapamulin also scored positive in our screen and retested at dose in MELAS and ND1 cybrid cells (Fig. 1k–r). This data suggested that mitochondrial translation might be a relevant target: while doxycycline and pentamidine are known to inhibit eukaryotic mitochondrial translation 18–20, retapamulin inhibits prokaryotic translation and could potentially inhibit mitochondrial translation 21. To examine this, we measured rates of mitochondrial translation with 35S-methionine/cysteine protein labelling using different concentrations of these antibiotics. Consistent with cell survival effects between 100 nM and 1 μM (Fig. 1k–r and Extended Data Fig. 2a), antibiotics did not reduce mitochondrial translation at 10 nM, partially reduced it between 100 nM and 1μM, and strongly suppressed it above 10 μM in ND1 cybrid cells (Figs. 1k–r and 2b,c). This sensitivity also occurred in MELAS cybrid cells, despite the mitochondrial tRNALeu mutation (Extended Data Fig. 2b). Thus, cell survival from antibiotics followed a mitohormetic distribution 22 (Figs. 1n,r and 2c), suggesting that partial or attenuated mitochondrial translation caused by these antibiotics promotes a protective cellular response.
Fig. 2. Antibiotic-mediated mitochondrial translation attenuation is required to promote cell survival in human mitochondrial disease mutant cells.
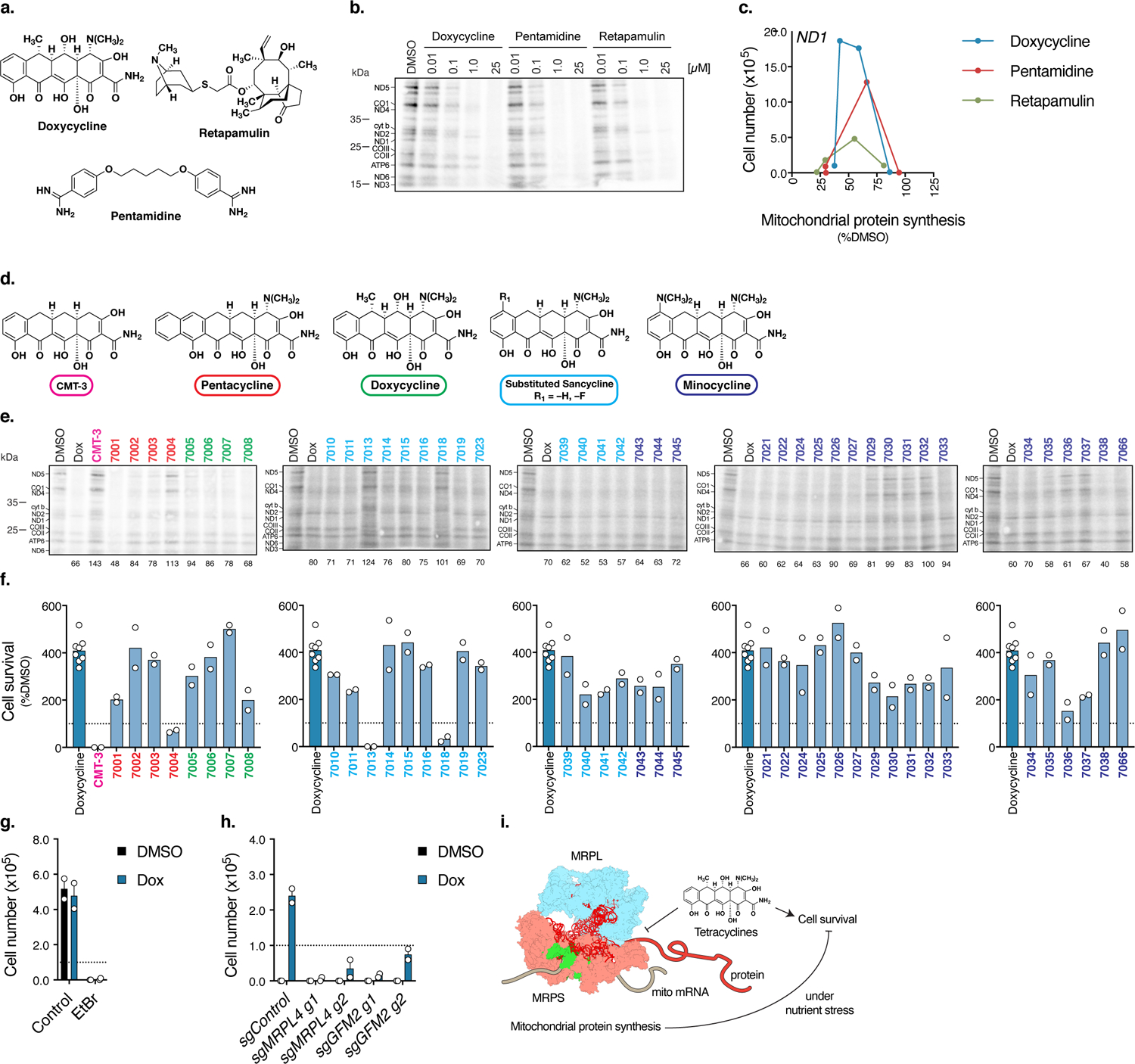
a. Chemical structures of doxycycline, retapamulin, and pentamidine. b. 35S-labelled cysteine and methionine pulse in ND1 cybrid cells treated with doxycycline, pentamidine, or retapamulin at 10 nM, 100 nM, 1 μM, or 25 μM with a 48 hour pre-treatment and 1 hour pulse (n=2 experiments). c. Mitochondrial protein synthesis and cell survival in ND1 cybrid cells (Data from Fig. 1o–q and quantification of Fig. 2b). d. Parent chemical structures for tetracycline analogs used in the screen. e. 35S-labelled cysteine and methionine pulse in ND1 cybrid cells pretreated with 1 μM tetracycline compounds for 48 hours in glucose conditions prior to the 1 hour pulse in the presence of compound (n=2 experiments). f. 4 day galactose survival assay of ND1 cybrid cells treated with 1 μM tetracycline analogs (n=2 biologically independent samples). g. 48 hour galactose survival assay of ND1 cybrid cells propagated in ethidium bromide (EtBr) (50 ng/mL for 3 weeks) and treated with doxycycline (n=2 biologically independent samples). h. 48 hour galactose survival assay of sgControl, sgMRPL4, and sgGFM2 ND1 cybrid cells treated with doxycycline (n=2 biologically independent samples). i. Tetracyclines antibiotics attenuate mitochondrial translation and promote cell survival of mitochondrial disease mutant cells under nutrient stress conditions. Ribosome model adapted from PDB:5AJ4 59. MRPL, mitochondrial ribosome protein, large; MRPS, mitochondrial ribosome protein, small. Survival assays were initially seeded with 1.0×105 cells with media changed every 48 hours. Data are presented as mean values +/− s.e.m. error bars, Student’s t-test with a two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli, with Q = 5%, * q<0.05.
To investigate structure-activity relationships (SAR) within the tetracycline scaffold, we used chemical synthesis to introduce structural diversity at positions C5, C5a, C6, C7, and C9, generating a library of 45 fully synthetic tetracycline analogs including substituted sancyclines, minocyclines, doxycyclines, and pentacyclines, as well as the semisynthetic analog known as CMT-3 23–26. The compounds were screened in cell survival and mitochondrial protein synthesis assays. Fig. 2e,f show that a number of the novel tetracycline analogs both promoted cell survival in ND1 cybrids when cultured under conditions of nutrient stress and inhibited mitochondrial translation. Tetracycline analogs that most strongly rescued cell death in ND1 cybrids include various C5- or C5a-substituted minocycline analogs (e.g.: 7026, 7038, and 7066), pentacyclines (e.g.: 7002, 7003), and certain (e.g.: 7007, 7014, 7015, and 7019), but not all, C6-aryl-substituted tetracyclines (e.g.: 7004, 7013, and 7018) (Fig. 2e,f and Supplementary Table 1). Among the C6-aryl analogs, those that did not promote cell survival also did not inhibit mitochondrial translation, whereas all those that rescued cells did inhibit mitochondrial translation to varying levels (Fig. 2e,f). These data suggest that mitochondrial translation attenuation is beneficial to the survival of ND1 mutant cells. Though small, the data set makes clear that the SAR for cell rescue within the tetracycline class is far from narrow, but suggests instead that there is broad opportunity for further molecular optimization.
To clarify the dependence on mitochondrial translation inhibition for cell survival, we targeted components of the mitochondrial translation machinery. First, we treated ND1 cybrid cells with ethidium bromide to deplete mtDNA and thus mitochondrial-encoded rRNA 27. Doxycycline did not rescue ND1 cybrid cells without mtDNA (Fig. 2g). Furthermore, ND1 mutant CRISPR KO cells depleted of MRPL4, a mitochondrial ribosome subunit, or GFM2, a mitochondrial translation factor, were not rescued under nutrient-stress conditions with doxycycline treatment (Fig. 2h and Extended Data Fig. 2c,d). Mitochondrial translation inhibition initiates an integrated stress response mediated by ATF4 28. We confirmed that MELAS cells treated with doxycycline induced ATF4 and p-eIF2α levels (Extended Data Fig. 3a), however, cell survival was independent of ATF4 (Extended Data Fig. 3b–d). Previous studies implicate different protein targets for doxycycline including PARP-1 29, PAR-1 30, and MMPs 31. Consistent with the requirement of mitochondrial translation attenuation, inhibitors and/or genetic deletion of these factors were not sufficient to promote survival in MELAS or ND1 cells (Extended Data Fig. 3e–l).
MD mutant cells activate inflammatory responses, particularly under nutrient stress conditions32–36 (Extended Data Fig. 4a). Doxycycline and tetracycline analogs suppressed inflammatory gene expression in MELAS or ND1 cybrid cells (Extended Data Fig. 4b,c) in a manner that correlated with mitochondrial translation inhibition. To further explore this relationship and its connection to metabolism, we determined the metabolic response of ND1 cells treated with analogs with (7066 and doxycycline) or without (7004, 7013, and CMT-3) effects on mitochondrial translation and cell survival (Supplementary Table 2). There were 15 metabolites that differentiated these groups (p<0.05, Student’s t-test, Bonferroni) in processes such as glycolysis, pentose phosphate pathway, and nucleotide synthesis (Extended Data Fig. 5a). A subset of these metabolites (10/15) increased in ND1 cells propagated in galactose media from 24 hours to 72 hours and correlated with inflammatory gene expression (Extended Data Fig. 5b,c and Supplementary Table 3). NADPH redox changes were apparent at 24 hours with doxycycline (Extended Data Fig. 5c), suggesting normalization of cellular redox may be the initial anti-inflammatory pro-survival event consistent with our previous findings 37. Interestingly, suppression of inflammation with the p38 inhibitor SB203580 replicated aspects of the doxycycline metabolic signature (Extended Data Fig. 6a,b and Supplementary Table 4). The NADP+/NADPH ratio decreased with SB203580 and decreased further with doxycycline (Extended Data Fig. 6c,d), arguing a fraction of doxycycline effects on redox occurs independently of inflammatory signaling. Together, this data supports a model where partial or attenuated mitochondrial translation inhibition normalizes cellular redox, reduces inflammatory gene expression, and increases survival in MD mutant cells (Fig. 2i).
Given the ability of tetracyclines to rescue several in vitro models of MD, we investigated whether doxycycline shows efficacy in a mouse model of a human MD. Mice lacking the mitochondrial complex I protein NDUFS4 closely recapitulate Leigh Syndrome, the most common pediatric manifestation of MD38. These mice are generally healthy until ~P35, at which point they begin to lose weight and become ataxic and lethargic. Without treatment, the mice meet the criteria for humane euthanasia at ~P58. Following weaning at ~P21, Ndufs4−/− (KO) mice along with their wild-type and heterozygous littermate controls (referred to collectively as “WT”) were randomly assigned to 5000 ppm, 8000 ppm doxycycline, or control diet groups. With either diet, there was accumulation of doxycycline in brain and liver of wild-type and KO animals at concentrations consistent with other feeding studies 39 (Extended Data 7 a,b). Doxycycline ameliorated the decline in body weight that occurred in KO mice around P35 (Fig. 3a). Doxycycline-treated KO mice lived significantly longer than untreated controls (P<0.0001 for both treatment groups). Median survival was 100 and 107 days, for the 5000 ppm and 8000 ppm groups respectively, with a maximum lifespan of 341 days (Fig. 3b). KO mice exhibit a progressive neurological decline that leads to decreased motor function and activity 8. A rotarod test was used at P30, P40, and P50 to assess motor function in doxycycline-treated KO and WT mice. Doxycycline significantly improved rotarod performance in KO mice at P40 and P50 without affecting WT performance (Fig. 3c–e). These results indicate that doxycycline treatment increases survival and fitness in a pre-clinical mouse model of MD.
Fig. 3. Doxycycline significantly increases the lifespan and fitness of a mouse model of complex I deficiency.
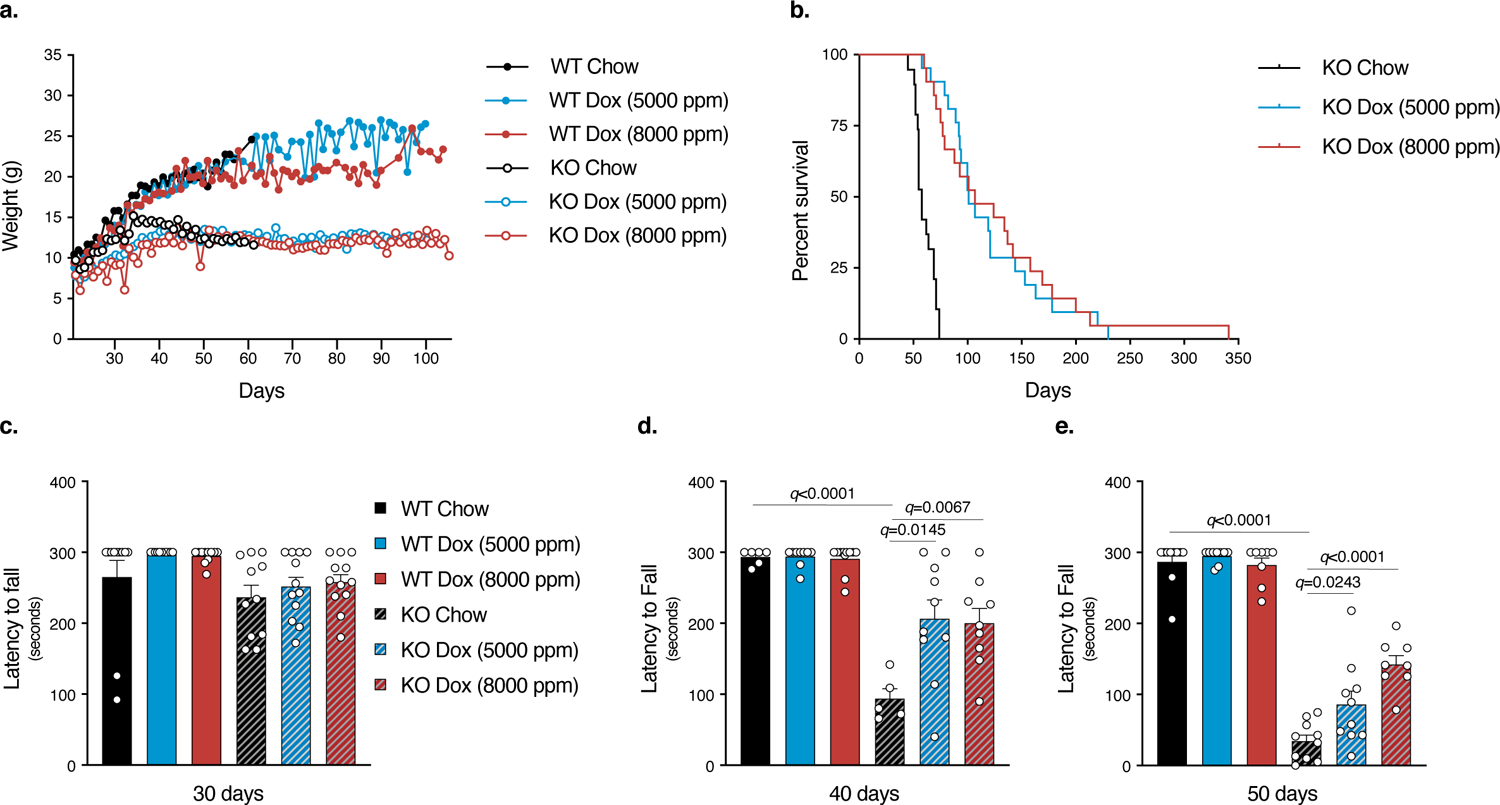
a. Body weight of Ndufs4−/− KO and WT mice over time (n=10 WT and KO Chow, n=8 WT and KO Dox 5000 ppm, n=10 KO Dox 8000 ppm, and n=7 WT Dox 8000 ppm mice). b. Survival of Ndufs4−/− mice is significantly increased with dietary doxycycline supplementation at 5000 ppm and 8000 ppm (n=19 Chow, n=21 Dox 5000 ppm, and n=21 Dox 8000 ppm mice) (p<0.0001, log rank test for both treatments compared to chow control). c–e. Rotarod performance of KO and WT mice at 30, 40, and 50 days. Untreated KO mice show a progressive decline in performance which is partially prevented with doxycycline treatment. Maximum time of 300 seconds with 3 attempts per mouse per time point. The maximum time is represented (at least n=5 mice were tested for each treatment at each time point). Data are presented as mean values +/− s.e.m. error bars, Student’s t-test with a two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli, with Q = 5%, * q<0.05.
Given the profound phenotypic improvement of the doxycycline-treated mice, we tested whether doxycycline may correct the underlying neuropathology observed in the Ndufs4−/− mouse model. KO mice developed bilateral lesions in the vestibular nuclei and the olfactory bulb. These lesions were marked by microglial activation evidenced by elevated Iba-1 expression (Fig. 4a). Immunohistochemistry analysis show that Iba-1 levels were significantly lower in the vestibular nucleus and olfactory bulb in the doxycycline-treated KO mice compared to untreated KO mice (q<0.05) (Fig. 4a,b). To identify how doxycycline prevents this neuropathology in KO mice, we performed isobaric quantitative-based mass spectrometry analysis 40 using whole brain tissues from WT, KO, and doxycycline-treated KO mice at P55 (Supplementary Table 5). As predicted, mitochondrial complex I proteins were robustly decreased in KO mice compared to WT controls and were not rescued by doxycycline (Extended Data Fig. 7c). Gene ontology identified “immune process” and “innate immune system” as the most significantly altered pathways by doxycycline treatment (Fig. 4c). Proteins associated with neuroimmune and inflammation, including markers of microglial and astrocyte activation (LEG3, CD180, GFAP, S100A4), complement components (C1QA, C1QB), and the interferon response (IFIT1 and IFIT3) 41–43, were significantly upregulated in KO mice compared to WT, and largely suppressed by doxycycline (Fig. 4d–f). Doxycycline treatment also upregulated proteins associated with anti-inflammatory responses (CLINT1, OTUD4, APOA4)44–46 (Supplementary Table 5). To further investigate these protective effects of doxycycline, we performed targeted metabolomics using brains of P55 WT and KO mice and compared them to ND1 cybrid cells under galactose conditions with and without doxycycline treatment (Supplementary Tables 6 and 7). Doxycycline changed a subset of metabolites that were altered in KO mice and ND1 cybrid cells (Extended Data Fig. 7d,e) and increased metabolomic signatures associated with attenuation of immune, inflammatory, and oxidative stress responses such as NADPH/glutathione redox, polyamine biosynthesis, and itaconic acid 47–50 (Fig. 4g–i and Extended Data Fig. 7e–g). Since oxidative stress is considered the main pathological cause of MDs, these metabolic changes are consistent with suppression of the disease phenotype 47–49,51–53. Doxycycline also increased glutamine and glutamate levels in Ndufs4 KO mice and ND1 cybrid cells (Fig. 4g–i), similar to rapamycin in brains of Ndufs4 KO mice 54. These findings suggest that mitochondrial dysfunction and associated pathology in the brains of KO mice leads to an innate immune and inflammatory response, which is strongly suppressed with doxycycline treatment.
Fig. 4. Doxycycline corrects neuroimmune and inflammatory proteins and increases metabolites that suppress oxidative stress in models of complex I deficiency.
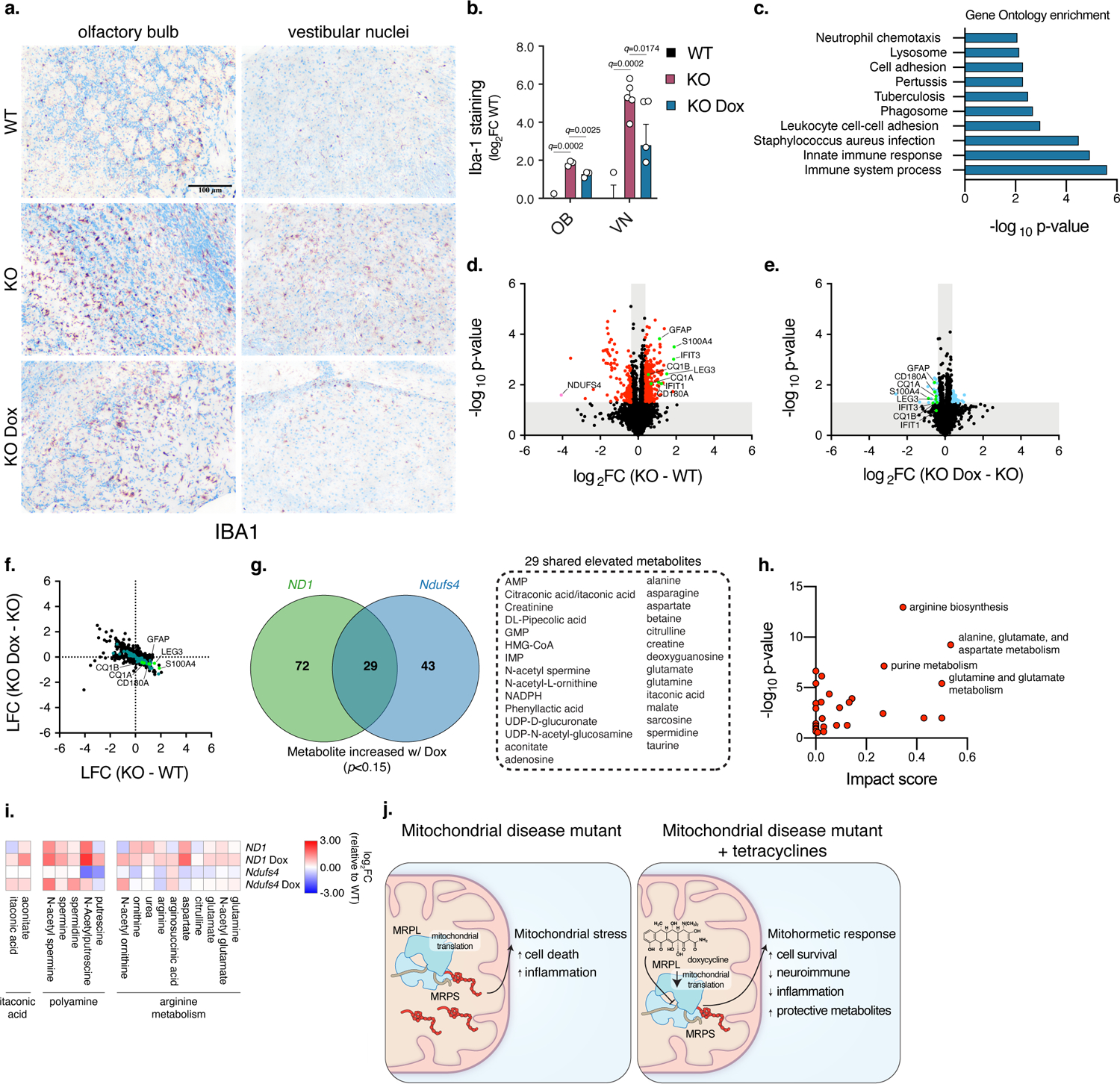
a–b. Representative images and quantification of Iba-1 staining in the olfactory bulb and vestibular nucleus regions of the brain at ~P55. Scale bar, 100 μm. (OB, n=3 mice for all groups; VN, n=3 WT, n=5 KO, and n=5 KO Dox 5000 ppm mice) (Student’s t-test with a two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli, with Q = 5%). c. Gene Ontology of proteomic data from doxycycline treated KO mice (Fisher’s Exact test). d. Volcano plot of proteomic changes in Ndufs4−/− mouse brains. Annotated dots indicate proteins associated with astrocyte activation, microglial activation, or interferon response (Student’s t-test, two-sided, unpaired). e. Volcano plot of proteomic changes in mouse brains of Ndufs4−/− mice treated with 5000 ppm doxycycline (Student’s t-test, two-sided, unpaired). f. Correlation plot of proteomic changes in KO mice (versus WT) compared to KO mice treated with doxycycline (versus KO). Points in green are significant changes in both comparisons (p<0.05, Student’s t-test, two-sided, unpaired). g. Venn diagrams of metabolites increased by doxycycline in ND1 cybrid cells or Ndufs4−/− mice (p<0.15, Student’s t-test, two-sided, unpaired). h. Pathway analysis of metabolomic changes from doxycycline treatment in ND1 cybrid cells and Ndufs4−/− mice. i. Heatmap of individual metabolites of specific pathways upregulated by doxycycline in ND1 cybrid cells and Ndufs4−/− mouse brains. j. Model of antibiotics causing a mitohormetic response. Data in Fig. 4b is presented as mean values +/− s.e.m. error bars.
The identification of antibiotics as potential drug-targeted therapies for MDs is highly paradoxical. Due to the close homology of the translational machinery in prokaryotes and human mitochondria, antibiotics designed to target the prokaryotic ribosome often induce dysregulation of both mitochondrial translation and organelle function in general 16. Antibiotic-dependent survival occurs at concentrations that reduce, but not block mitochondrial translation, supporting a mitohormesis model 22. Although the precise downstream mechanisms are unclear, our results suggest that antibiotic-selective mitochondrial translation attenuation suppresses cell death and neuroimmune and inflammatory processes (Fig. 4j). Despite this, we acknowledge that in vivo neuroimmune activation may be regulated independently of mitochondrial translation and instead, be sensitive to changes to the gut microbiome 55,56.
Although further studies are needed, our data suggest that tetracyclines might constitute a potential intervention to ameliorate symptoms for MD. Tetracyclines SAR studies could also be employed to identify improved analogs to treat MD. Doxycycline is yet another example, along with rapamycin and hypoxia 13,57, that complex I activity is not required to rescue pathologies associated with MD. Currently, there are no clinically effective therapies for mitochondrial disease, therefore, understanding the mechanisms downstream of antibiotic-dependent mitochondrial translation inhibition is valuable as a cell survival rescue process for mitochondrial disease mutations and future therapeutic strategies.
Methods:
Cell lines and media culture conditions.
MELAS cybrid cells, a gift of Carlos Moraes (University of Miami Medical School, Miami, Fl, USA) were generated through the fusion of a MELAS patient fibroblast harboring the A3243G point mutation in MT-TL1 with a U2Os based cell depleted of mitochondria 10. ND1 and ND6 cybrid cells, previously studied by our lab 17,33,37, were created by Rutger Vogel (Radboud University Medical Centre, Nijmegen, Gelderlan, Netherlands) by the same method described as the MELAS cybrid cells. In this case, ND1 and ND6 cybrid cells were made using patient fibroblasts carrying the A3796G mutation and ND6 G14459A, respectively. Control cybrids share the same U2Os nuclear background, but contain wild-type DNA. Rieske knockout fibroblasts were a gift of Carlos Moraes and generated from mouse lung fibroblasts containing a knock in allele with loxP sites flanking exon 2 of Rieske/ISP 16. High glucose experiments were performed using 25 mM glucose DMEM (Hyclone SH30022.01) supplemented with 10% FBS (GeminiBio 900–208), 1 mM sodium pyruvate (Gibco 11360070), Pen-Strep 100 U/mL (Gibco 15140148), 25 mM HEPES (Gibco 15630080), and 50 μg/mL uridine (Sigma-Aldrich U3003). For low glucose experiments, 2.5 mM glucose (Sigma G8270) and 22.5 mM galactose (Sigma-Aldrich G5388) were added to no glucose DMEM (Gibco 11966025) supplemented with 10% FBS, 1 mM sodium pyruvate, Pen-Strep 100 U/mL, 25 mM HEPES, and 50 μg/mL uridine. For galactose conditions, 25 mM galactose was added to no glucose DMEM supplemented with 10% FBS, 1 mM sodium pyruvate, Pen-Strep 100 U/mL, 25 mM HEPES Buffer, and 50 μg/mL uridine.
High-throughput chemical screen.
A high-throughput small molecule screen for cell survival in MELAS cybrid cells was performed in collaboration with the ICCB at Harvard Medical School. MELAS cybrids were seeded at a density of 1,500 cells per well in DMEM supplemented with 2.5 mM glucose, 22.5 mM galactose, 4 mM glutamine, 10% FBS, and 50 μg/mL uridine. For each 384-well plate, two columns were used as a positive controls containing cells grown in 25 mM glucose media. Chemical compounds were added by pin transfer at the time of seeding (final concentration of 3.3 μM). Under these conditions, untreated MELAS cells in 2.5 mM glucose die after 24 hours, while those in 25 mM glucose proliferate. The plates were measured for ATP luminescence (CellTiter-Glo, Promega, G7573) 48 hours after seeding as a readout of cell survival. After 48 hours, negative control wells were completely dead. In contrast, positive hits allowed the cells to survive with limited glucose. A total of 4,937 known bioactive compounds were screened from a diverse background. The screen was performed in technical duplicate so that each compound was added to two separate experimental wells on two separate plates. The z-score was calculated based on comparing an experimental well to the population mean on that plate. An average z-score was calculated for each compound and a cut-off of 1.96 (97.5% confidence interval) was used to determine positive hits.
Cell survival experiments.
MELAS cybrid cells and Rieske KO fibroblasts were seeded in 2.5 mM glucose with 22.5 mM galactose media at a density of 1.0×105 cells per well in 6-well plates. ND1 and ND6 mutant cybrid cells were seeded in 25 mM galactose media with 0 mM glucose at a density of 1.0×105 cells per well in 6-well plates. Stocks of doxycycline hyclate (Sigma, D9891), pentamidine isethionate (Sigma, 1504900), and retapamulin (Sigma, CDS023386) were dissolved in DMSO at 100 mM. Compounds were added to wells at the time of seeding at a final concentration indicated in the figures and figure legends (1 nM-100 μM). The p38 inhibitor, SB203580 (Cell Signaling, 5633S) was dissolved in DMSO and used at a final concentration of 25 μM as previously described 33. MELAS and Rieske KO cells were counted after 48 hours incubation, unless otherwise noted. ND1 and ND6 were maintained for the time point indicated in the figure legends with galactose (0 mM glucose) media. For long term survival experiments in low-glucose (e.g. MELAS/Rieske cells) or galactose (e.g. ND1/ND6 cells) media, fresh media and drug were replaced every 24 or 48 hours.
DNA Isolation.
DNA isolation protocol adapted from previous publications 60,61. MELAS, ND1, or control cybrid cells were seeded at a density of 1.0 × 105 cells per well in 25 mM glucose, 2.5 mM glucose, or galactose media and harvested after 24 hours. Cells were washed with PBS, pelleted, resuspended in 0.5 mL of 1x PBS + 0.5% SDS, mixed with 10 μL of 20 mg/mL Proteinase K, and incubated for 1 hour at 55°C. Following incubation, 0.5 mL phenol/chloroform was added and samples were vortexed for 1 minute and centrifuged at max speed for 5 minutes. The aqueous layer was then transferred to a new tube and 50 μL of 3 M Sodium Acetate (pH 5.2) was added, followed by 1.5 mL ice cold ethanol. DNA was precipitated overnight at −80°C. After thawing, DNA was pelleted at max speed for 30 minutes. The supernatant was carefully removed and the pellets were air dried for 10 minutes. The pellets were resuspended in 200 μL H2O and RNAse was added to a final concentration of 5 μg/mL and samples were incubated at 37°C for 30 minutes.
Heteroplasmy PCR.
PCR was performed from mtDNA using primers flanking ND1 or MT-TL1. PCR primer sequences were as follows: ND1 forward 5’-CCTAGGCCTCCTATTT-3’, ND1 reverse 5’-CGGCGTATTCGATGT-3’, MT-TL1 forward 5’-GGTTCGTTTGTTCAACGATT-3’, and MT-TL1 reverse 5’-TGCCATTGCGATTAGAATGG-3’. Each reaction contained 15 ng template DNA, 12.5 μL Q5 reaction buffer (New England Biolabs, M0491), 10 μM forward and reverse primers, and H2O up to 25 μL. The following protocol was performed: 98°C for 30 seconds, 98°C for 10 seconds, 57°C (ND1)/62°C (MT-TL1) for 30 seconds followed by 72°C for 15 seconds (with the last 3 steps repeated 29 times), 72°C for 2 minutes and hold at 10°C. A restriction digest was performed on the PCR products from MELAS, ND1, and control cybrids. MELAS and control cybrids were digested with Apal1 (New England Biolabs, R0114S) and ND1 and control cybrids were digested with Acil (New England Biolabs, R0551S). The reactions were incubated for 1 hour at 37°C and run on a 2% agarose gel.
Oxygen consumption measurements.
For oxygen consumption measurements for cells, 1.0×105 cells were seeded in an XFE-24 Seahorse plate (Seahorse Biosciences, 102340–100) and allowed to adhere for 24 hours at 37°C with 5% CO2. DMEM was then removed and cells were washed twice with pre-warmed unbuffered DMEM supplemented with 25 mM glucose, 1 mM sodium pyruvate, and 4 mM glutamine. After washing, 500 µl of identical buffer was added and the cells were then placed in a CO2 free incubator for 1 hour. The Seahorse 24 optical fluorescent analyzer cartridge was prepared by adding 4 μM oligomycin, 4 μM FCCP, and 1.5 μM rotenone/4 μM antimycin A to cartridge ports A, B, and C, respectively. Oxygen consumption rates (pmol/min) were measured for each treatment condition at 37°C using the Seahorse Bioanalyzer instrument with the following protocol: Mix: 1 minute, Wait: 3 minute, Measure: 3 minute.
Mitochondrial isolations and solubilizations for Blue native PAGE (BN-PAGE).
Mitochondrial isolations and BN-PAGE were performed as previously described 62,63. Frozen cell pellets were thawed on ice in hypotonic buffer (83 mM sucrose, 10 mM MOPS, 1X cOmplete EDTA-free Protease Inhibitor Cocktail (Roche, 4693159001)) and subsequently homogenized using a dounce homogenizer. After this, osmolarity was normalized using hypertonic sucrose buffer and the resulting solution was centrifuged at 1000xg for 5 minutes. The supernatant was then centrifuged for a second time to remove cell debris. Next, the supernatant containing mitochondria was centrifuged at 9,000xg for 10 minutes. The mitochondrial pellet was resuspended and washed twice in resuspension buffer (320 mM sucrose, 1 mM EDTA, 10 mM Tris pH 7.4, 1X protease inhibitor cocktail (Roche, 4693159001)). Protein content of each mitochondrial prep was quantified and mitochondria were divided into 100–200 μg aliquots that were flash frozen. Before running BN-PAGE, mitochondria were solubilized in NativePAGE 4x Sample Buffer (ThermoFisher Scientific, BN2003) at a 6 g/g digitonin/protein ratio for 20 minutes on ice. The insoluble portion was removed by centrifugation for 15 minutes at max speed. The soluble supernatant was then mixed with Coomassie G-250 (ThermoFisher Scientific, BN2004) prior to BN-PAGE.
Immunoblot.
Cells or tissue were harvested in RIPA buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA, 1% Triton X-100, 0.1% Sodium Deoxycholate, 0.1% SDS, 140 mM NaCl, 1X protease inhibitor cocktail, 1 mM PMSF) and proteins were quantified using the BCA assay (Pierce 23228). BN-PAGE analysis of 6 g/g digitonin/protein solubilized mitochondrial preparations was performed using 3–12% NativePAGE gels (ThermoFisher Scientific, BN1003BOX) according to manufacturer’s instructions. 20 µg of protein was run per well. BN-PAGE buffers included NativePAGE Running Buffer (20X) (ThermoFisher Scientific, BN2001) and NativePAGE Cathode Buffer Additive (20X) (ThermoFisher Scientific, BN2002). After electrophoresis, proteins and/or complexes were transferred to PVDF membranes and probed with specific antibodies. The following antibodies were used for SDS-PAGE and BN-PAGE: anti-SDHA (1:3000) (Abcam, ab14715), anti-MTCO1 (1:3000) (Abcam, ab14705), anti-NDUFA9 (1:1000) (Abcam, ab14713), anti-NDUFS2 (1:1000) (Abcam,192022), anti-UQCRB (1:1000) (Proteintech, 10756–1-AP), anti-ATP5A1 (1:1000) (Life Technologies, 459240), anti-ATF4 (1:1000) (Cell Signaling, 11815), anti-β-actin (1:2000) (Cell Signaling, 4967), p-eIF2α (1:1000) (Ser51) (Cell Signaling, 3398), total eIF2α (1:1000) (Cell Signaling, 5324), anti-CHOP (1:1000) (Cell Signaling, 2895), anti-β-Tubulin (1:5000) (Cell Signaling, 2146), anti-MRPL4 (1:1000) (Proteintech, 27484–1-AP), anti-GFM2 (1:1000) (Proteintech, 16941–1-AP), and anti-PAR1 (1:1000) (Abcam, ab32611).
Depletion of mtDNA with ethidium bromide.
Ethidium bromide (EtBr) solution (Biorad, 1610433) was diluted to 50 μg/mL with water and sterile filtered. ND1 cybrid cells cultured in glucose supplemented with 50 μg/mL uridine were treated with 50 ng/mL EtBr with media replaced daily as described previously 17,27. After 3 weeks of treatment, EtBr-treated and untreated ND1 cybrid cells were seeded at a density of 1.0×105 cells per well in galactose media with 1 μM doxycycline or DMSO and counted after 48 hours.
siRNA transfections.
siRNA oligo duplexes for ATF4 and F2R were purchased from Origene (ATF4, #SR319410 and F2R, #SR301496). The three siRNA guides were pooled and resuspended at a final concentration of 20 μM. For each infection, 6 μL Lipofectamine RNAi Max (Thermo Fisher Scientific, 13778100) was added to 100 μL Optimem media (Thermo Fisher Scientific, 31985088) followed by vortexing and left at room temperature for 5 minutes. In a separate tube 1 μL of the resuspended RNAi was added to 100 μL Optimem and vortexed. Both tubes were then combined and added directly to a 6-well plate. 1.0×105 cells in 2 mL of media were added to the well.
CRISPR editing experiments.
CRISPR-Cas9 editing was performed using the GeCKO system 64. Guides were cloned into either Lenti-CRISPR-V2-Puromycin (Addgene #98290). After cloning, HEK293T cells were reverse transfected in 6-well plates using PolyFect (Qiagen, 301105) following manufacturer’s instructions. Transfection reactions contained 600 ng of psPAX2 (Addgene plasmid #12260), 300 ng of pMD2 (Addgene, Plasmid #12259) and 900 ng of Lenti-CRISPR V2 with control guide RNAs. 24 hours after transfection, the media was replaced with fresh DMEM (3 mL per well). The next day, media was collected, filtered through a 0.45 µm filter and poured into ND1 cybrid cells grown from an overnight 6-well culture (1×105 seeding density). 24 hours after the infection, media was replaced with fresh media. Cells were selected 48 hours after infection with 0.5 μg/mL puromycin and selection continued for 2 weeks before cells were used for experiments. Guides for CRISPR-Cas9 editing can be found in Supplementary Table 8.
Measurement of mitochondrial translation.
ND1 mutant cybrid cells cultured in glucose for 48 hours in the presence of compound were washed with PBS and incubated for 30 min in 1 mL of labeling medium (1 mM sodium pyruvate in DMEM without methionine and cysteine) in the presence of compound. Emetine was added to a final concentration of 200 µg/mL, and cells were incubated for an additional 30 minutes before the addition of 50 µCi 35S-labeled methionine/cysteine mixture for 1 hour. Cells were recovered and washed twice in PBS before lysis and protein quantification. 5 μg of total proteins in a volume of 10 μL was loaded on a 4–12% SDS-PAGE. Cell treated with chloramphenicol (50 µg/mL) was used as a negative control to ensure the mitochondrial origin of the 35S signal. The protein identification associated with each band is proposed based on their relative abundance and molecular weight as determined by prior studies 65. Mitochondrial band intensities were quantified using ImageJ software 1.52 by plotting lanes and calculating area under the curve with a signal threshold set to a non-protein section of the blot.
Gene expression.
RNA was isolated with Trizol (Invitrogen, 15596–026) and a Zymo-Spin Direct-zol RNA Kit (Zymo Research, R2050). 1 μg of RNA was used to generate complementary DNA (cDNA) with a High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, 4368813) following the manufacturer’s protocol. For gene expression analysis, cDNA samples were mixed with Sybr Green qPCR mastermix (Applied Biosystems, 4309155) and were analyzed by a CFX 384 Real-Time system (Bio-Rad). All primers sequences can be found in Supplementary Table 8.
Animal care and lifespan determination.
C57BL/6J mice heterozygous for the Ndufs4 gene were purchased from the Jackson Laboratory (B6.129S4-Ndufs4tm1.1Rpa/J, Stock No. 027058) and crossed to generate Ndufs4−/− mice. Mice were housed in the Beth Israel Deaconess Medical Center Animal facility and kept on a 12 hour interval light-dark cycle. The facility was maintained at 20–22°C and 50% humidity. Pups were weaned at postnatal day 21 and genotyped immediately. Ndufs4−/− mice were randomly assigned to a treatment group along with 1–2 wild-type or heterozygous littermate controls (referred to collectively as “WT”). Males and females were included equally in all experiments and distributed equally amongst the treatment groups. Mice were given standard chow supplemented with 5000 ppm doxycycline, 8000 ppm doxycycline, or a re-pelleted control diet (Open Source Diets, Brunswick, NJ). Additional food and hydrogel were placed on the floor of the cage and replaced every 48 hours. The cages were inspected daily and mice were weighed every 48 hours to determine peak body weight. Mice were humanly euthanized with CO2 according to the IUACUC protocol when body weight declined by more than 20% of maximum (at that time, littermate controls were also euthanized). All animal studies were compliant with the IUACUC protocol approved by the Beth Israel Deaconess Medical Center Animal Facility.
Rotarod testing.
Motor function was assessed using a rotarod test at postnatal day 30, 40, and 50 (+/− 2 days). One day prior to the day 30 test, mice were acclimated to the rotarod. KO and WT littermate control mice were each placed on the rod and the latency to fall was measured for up to 5 minutes using an acceleration of 4 rpm/min and max speed of 40 rpm. Mice were considered to have fallen if they latched onto the rotarod for more than 10 seconds without walking. The test was performed three times and the maximum of the three attempts was reported. A minimum of five mice were tested for each time point and each group.
Histology.
KO and littermate control mice were euthanized with CO2 and the entire head was removed. The skin was removed from the head and a sagittal cut was made into the posterior cranium and the skull was lifted slightly away from the brain. The entire head was submerged into 10% formalin for 48 hours and then transferred to dilute formalin (1:10 dilution of formalin in PBS) for storage until processing. Samples were submitted to the Harvard Medical School Rodent Histopathology Core for paraffin embedding, parasagittal sectioning and H&E staining. Unstained sections were submitted to the Specialized Histopathology Core at Brigham and Women’s Hospital for immunohistochemistry. Antibody IBA-1 (Wako, 019–19741, polyclonal) was run at 1:500 dilution using the Leica Biosystems Refine Detection Kit with citrate antigen retrieval and Alexa 488 (Thermo Fisher Scientific, B40953). Sections were imaged at the Nikon Facility at Harvard Medical School using Nikon Ds-Fi1 cooled color digital microscope camera with Nikon Plan Apochromat 20x/0.75 NA objective. Additional sections were imaged at the Brigham and Women’s Specialized Histopathology Core using the Leica Aperio Versa 200 slide scanner. Staining quantification was performed using Image J software. Briefly, images were background subtracted (rolling ball radius, 200), colour deconvoluted, thresholded (Huang), and measured for integrated density of thresholded signal.
Metabolomics.
Brain tissue was collected from KO and littermate control mice ~55 days of age and stored at −80°C prior to metabolite extraction. Frozen tissues were powderized using liquid nitrogen and mortar and pestle. 800 μL of 60% cold methanol was used to resuspend 10 mg of powdered tissue. The samples were vortexed periodically for 10 minutes and returned to ice. 500 μL cold chloroform was added to each sample and vortexing continued for an additional 10 minutes. Samples were centrifuged at max speed for 10 minutes at 4°C. The top layer, which contains the polar metabolites, was removed and transferred to a new tube and dried down overnight using a speed vac. For cells, 1.0×105 ND1 or U2OS control cybrid cells were seeded in each well of a 6-well plate and cultured in galactose medium for 48 hours. Cells (3 wells/replicate) were harvested on dry ice with 0.8 mL pre-chilled 80% HPLC-grade methanol (Fluka Analytical). The cell mixture was incubated for 15 minutes on dry ice prior to centrifugation at 18,000xg for 10 minutes at 4°C. The supernatant was retained, and the remaining cell pellet was resuspended in 200 μL pre-chilled 80% methanol and centrifuged. Supernatants were combined and lyophilized using a SpeedVac (Thermo Fisher Scientific). Lyophilized samples, from both brains and cells, were resuspended in 20 μL HPLC quality water and subjected to metabolomics profiling using the AB/SCIEX 5500 QTRAP triple quadrupole instrument. Data analysis was performed using the GiTools software. p-values are from Metaboanalyst pathway enrichment and impact score is from pathway topology analysis.
Protein digestion and isobaric labelling for mass spectrometry analysis.
Brain tissues were extracted and snap-frozen in liquid nitrogen until further use. Brain tissues were lysed with 4 mL of SDS lysis buffer (2.0% SDS (w/v), 250 mM NaCl, 5 mM TCEP, EDTA-free protease inhibitor cocktail (Promega), and 100 mM HEPES, pH 8.5) using an Omni tissue homogenizer. Extracts were reduced at 57°C for 30 min and cysteine residues were alkylated with iodoacetamide (14 mM) in the dark at room temperature for 45 min. Extracts were purified by methanol–chloroform precipitation and pellets were washed with ice-cold methanol. Pellets were resuspended in 2 mL of 8 M urea (containing 50 mM HEPES, pH 8.5) and protein concentrations were measured by BCA assay (Thermo Fisher Scientific) before protease digestion. 100 µg of protein were diluted to 4 M urea and digested overnight with 4 µg LysC (Wako). Digests were diluted further to a 1.5 M urea concentration and 5 µg of trypsin (Promega) was added for 6 hours at 37°C. Digests were acidified with 50 µl of 20% formic acid and subsequently desalted by C18 solid-phase extraction (50 mg, Sep-Pak, Waters). Digested brain peptides were resuspended in 100 µl of 200 mM HEPES, pH 8.0. 10 μL of TMTpro reagents (Thermo Fisher Scientific) was added to each solution for 1 hour at room temperature (25°C). After incubating, the reaction was quenched by adding 4 μL of 5% (w/v) hydroxylamine. Labelled peptides were combined and subsequently desalted by C18 solid-phase extraction (50 mg, Sep-Pak, Waters) before basic pH reversed-phase separation.
Basic pH reverse-phase separation for mass spectrometry analysis.
Tandem mass tag (TMT)-labeled peptides were solubilized in 500 μL solution containing 5% acetonitrile in 10 mM ammonium bicarbonate, pH 8.0 and separated by an Agilent 300 Extend C18 column (5 mm particles, 4.6 mm inner diameter and 220 mm in length). An Agilent 1100 binary pump coupled with a photodiode array detector (Thermo Fisher Scientific) was used to separate the peptides. A 40-min linear gradient from 20% to 40% acetonitrile in 10 mM ammonium bicarbonate, pH 8 (flow rate of 0.6 ml/min) separated the peptide mixtures into a total of 96 fractions (30 sec). A total of 96 fractions was consolidated into 12 samples in a checkerboard fashion, acidified with 20 µl of 20% formic acid and vacuum dried to completion. Each sample was re-dissolved in 5% formic acid, 5% ACN, desalted via StageTips before liquid chromatograph–tandem mass spectrometry (LC–MS/MS) analysis.
LC–MS/MS analysis.
Data were collected using an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific, San Jose, CA) coupled with a Proxeon EASY-nLC 1200 LC pump (Thermo Fisher Scientific). Peptides were separated on a 100 μm inner diameter microcapillary column packed with 35 cm of Accucore C18 resin (2.6 μm, 100 Å, Thermo Fisher Scientific). Peptides were separated using a 3 hours gradient of 6–22% acetonitrile in 0.125% formic acid with a flow rate of ~400 nL/min. Each analysis used an MS3-based TMT method as described previously described 40.The data were acquired using a mass range of m/z 400 – 1400, resolution at 120,000, AGC target of 1 × 106, a maximum injection time 100 ms, dynamic exclusion of 180 seconds for the peptide measurements in the Orbitrap. Data dependent MS2 spectra were acquired in the ion trap with a normalized collision energy (NCE) set at 35%, AGC target set to 2.0 × 105 and a maximum injection time of 120 ms. MS3 scans were acquired in the Orbitrap with an HCD collision energy set to 45%, AGC target set to 1.5 × 105, maximum injection time of 200 ms, resolution at 50,000 and with a maximum synchronous precursor selection (SPS) precursors set to 10.
Mass spectrometry data processing and spectra assignment.
In-house developed software was used to convert acquired mass spectrometric data from the .RAW file to the mzXML format. Erroneous assignments of peptide ion charge state and monoisotopic m/z were also corrected by the internal software. SEQUEST algorithm was used to assign MS2 spectra by searching the data against a protein sequence database including Mouse Uniprot Database (downloaded June 2018) and known contaminants such as mouse albumin and human keratins. A forward (target) database component was followed by a decoy component including all listed protein sequences. Searches were performed using a 20 ppm precursor ion tolerance and requiring both peptide termini to be consistent with trypsin specificity. 16-plex TMT labels on lysine residues and peptide N termini (+304.2071 Da) were set as static modifications and oxidation of methionine residues (+15.99492 Da) as a variable modification. An MS2 spectra assignment false discovery rate (FDR) of less than 1% was implemented by applying the target-decoy database search strategy. Filtering was performed using a linear discrimination analysis method to create one combined filter parameter from the following peptide ion and MS2 spectra properties: XCorr and ΔCn, peptide ion mass accuracy, and peptide length. Linear discrimination scores were used to assign probabilities to each MS2 spectrum for being assigned correctly and these probabilities were further used to filter the data set with an MS2 spectra assignment FDR to obtain a protein identification FDR of less than 1%.
Determination of TMT reporter ion intensities.
For reporter ion quantification, a 0.003 m/z window centred on the theoretical m/z value of each reporter ion was monitored for ions, and the maximum intensity of the signal to the theoretical m/z value was recorded. Reporter ion intensities were normalized by multiplication with the ion accumulation time for each MS2 or MS3 spectrum and adjusted based on the overlap of isotopic envelopes of all reporter ions. Following extraction of the reporter ion signal, the isotopic impurities of the TMT reagent were corrected using the values specified by the manufacturer’s specification. Total signal-to-noise values for all peptides were summed for each TMT channel and all values were adjusted to account for variance and a total minimum signal-to-noise value of 200 was implemented.
Measurement of Doxycycline from Tissues.
Doxycycline extraction was modified from a previous protocol 66. Brain and liver from age P55 WT and KO mice treated with 5000 ppm and 8000 ppm was powderized using a mortar and pestle. 100 mg of tissue was homogenized using a bead better in 1 mL 0.02M oxalic acid buffer pH 4.0. For standard curves, brain or liver (1 g each) from untreated control mice were powderized and homogenized in oxalic acid buffer (10 mL). The sample was divided into 10× 1 mL samples to which doxycycline at known concentrations was added. These samples were processed identical to doxycycline-treated mouse samples. 50 μL of 2 μM doxycycline D3 standard (Cayman, 28528) was added to each sample and vortexed. Samples were centrifuged at max speed for 10 minutes and the supernatant was transferred to a 15 mL tube. The pellet was resuspended in 1 mL oxalic buffer for a second extraction which was pooled with the first following homogenization and centrifugation. 250 μL of 20% TCA was added to each sample, vortexed, and centrifuged at max speed for 15 minutes. SPE columns (Strata SPE, 8B-S100-ECH) were conditioned using 3 mL methanol followed by 3 mL HCL and then 3 mL H2O. Sample supernatants were transferred onto the conditioned cartridges and allowed to flow through. The cartridges were then washed with 3 mL oxalic acid buffer followed by 2 mL of water and then dried completely by opening the vacuum and letting air pass through the cartridges for 10 minutes. For elution, 10 mL tubes were placed below the vacuum manifold and 5 mL methanol was added to each cartridge. Samples were dried completely in a SpeedVac and submitted to the Harvard Center for Mass Spectrometry for processing. Samples were quantified on a QEplus mass spectrometer coupled to an Ultimate 3000 LC (Thermo Fisher Scientific). After resuspension in 50 μl of water, 10 μl was injected on a Luna C18 column (2mm x 150 mm, Phenomenex) maintained at 40°C. The mobile phases were A : water, 0.1% formic acid and B: Acetonitrile, 0.1% formic acid. The gradient was as follow : 5% B for 1 min, then to 60%B in 1 second, maintained at 60% B for 11 min, then to 90% B in 1 second, maintained at 90% for 3 min, followed by 5 min re-equilibration at 5% B. The flow rate was 0.2 mL min−1. Two standard curves were prepared by adding various concentration of doxycycline in blank liver or brain samples. The standard points were then extracted in the same way as the samples. Quantification was achieved using Tracefinder (Thermo Fisher Scientific), using the ratio of the area under the peak for Doxycycline and of the internal standard, using the accurate mass of the [M+H]+ ions.
Statistical Analysis and Reproducibility.
All measurements were taken from distinct biological samples. For RFLP mapping and SDS-PAGE or BN-PAGE western blots, n=1 experiments were performed unless otherwise noted in the figure legend. In general, for two experimental comparisons, a two-tailed unpaired Student’s t-test was used. For multiple comparisons to a control, we used Student’s t-test with a two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli, with Q = 5%. Statistical significance is represented by asterisks corresponding to *p/q < 0.05. GraphPad Prism software Version 8.4.0 was used to generate graphs and perform statistical analyses. Microsoft Excel for Mac Version 16.43 was used for analysis of small molecule screen data, metabolomics, and proteomics. Morpheus software from the Broad institute (https://software.broadinstitute.org/morpheus/) was used to generate heatmaps. DAVID Bioinformatics Resources 6.8 was used for gene ontology 67,68. Mitochondrial ribosomal structure in Fig. 2i was generated with UCSF ChimeraX 69.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1. MELAS cybrid cells exhibit deficient mitochondrial respiration and undergo apoptosis from nutrient stress.
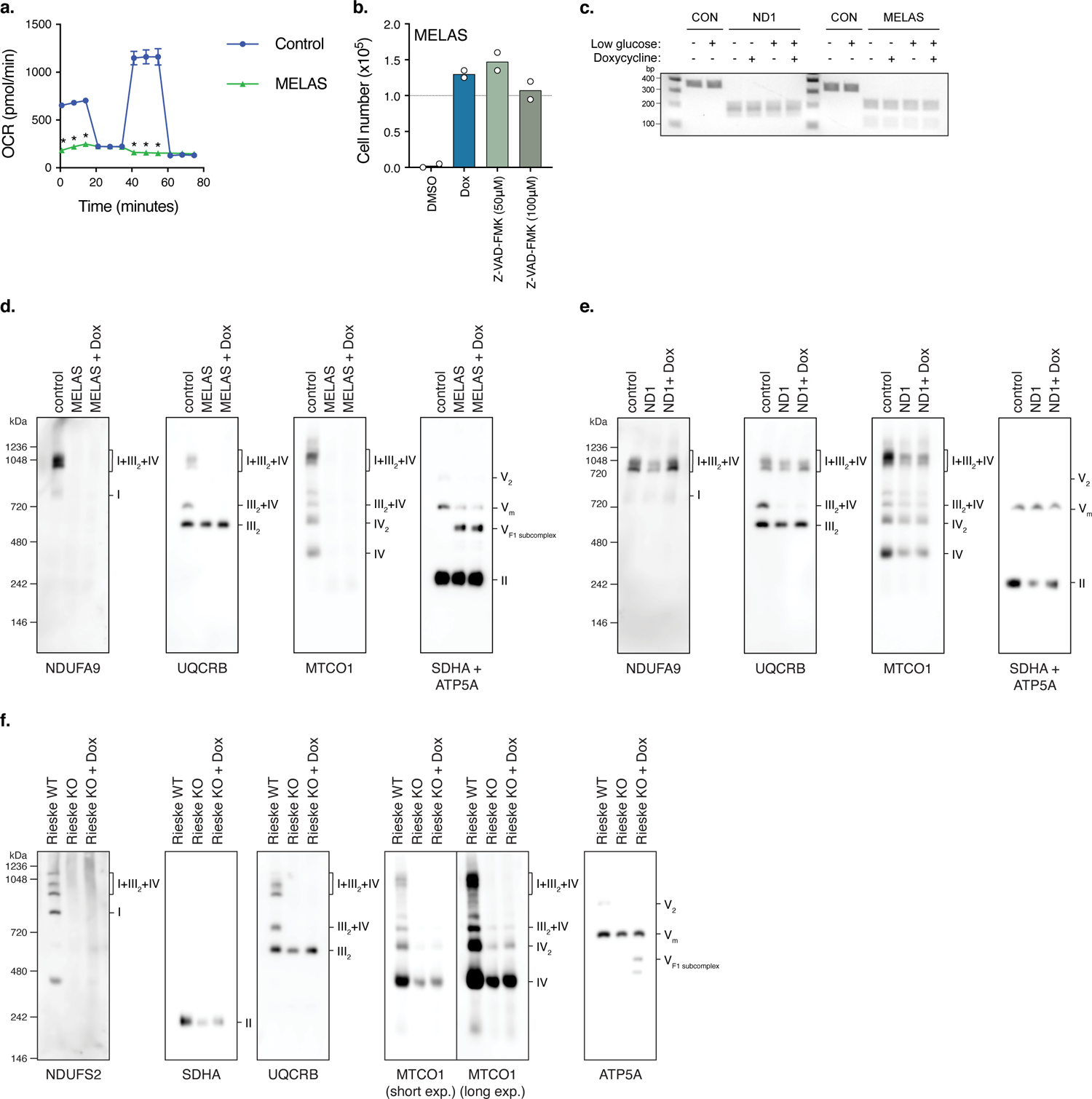
a. Oxygen Consumption Rates (OCR) measured in control and MELAS cybrid cells. Measurements 4–6 follow the injection of 4 μM oligomycin, measurements 7–9 follow the injection of 4 μM FCCP, and measurements 10–12 follow the injection of 1.5 μM rotenone/4 μM antimycin (n=5 biologically independent samples). b. 48 hour low-glucose survival assay of MELAS cybrid cells treated with the pan-caspase inhibitor Z-VAD-FMK (n=2 biologically independent samples). c. RFLP mapping of ND1 and MELAS mtDNA mutations in cybrids after 24 hour galactose or low-glucose conditions with 1 μM doxycycline. Expected band sizes are 193/159 bp for ND1 and 117/213 bp for MELAS mutations. d–f. BN-PAGE of isolated mitochondria from MELAS cybrids, ND1 cybrids, and Rieske KO fibroblasts treated with 1 μM doxycycline. MELAS cybrids were propagated 24 hours in low-glucose media, ND1 cybrids were propagated for 48 hours in galactose media, and Rieske KO fibroblasts were propagated for 48 hours in high-glucose media. Data are presented as mean values +/− s.e.m. error bars, Student’s t-test with a two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli, with Q = 5%, * q<0.05.
Extended Data Fig. 2. Doxycycline promotes cell survival through attenuation of mitochondrial translation.
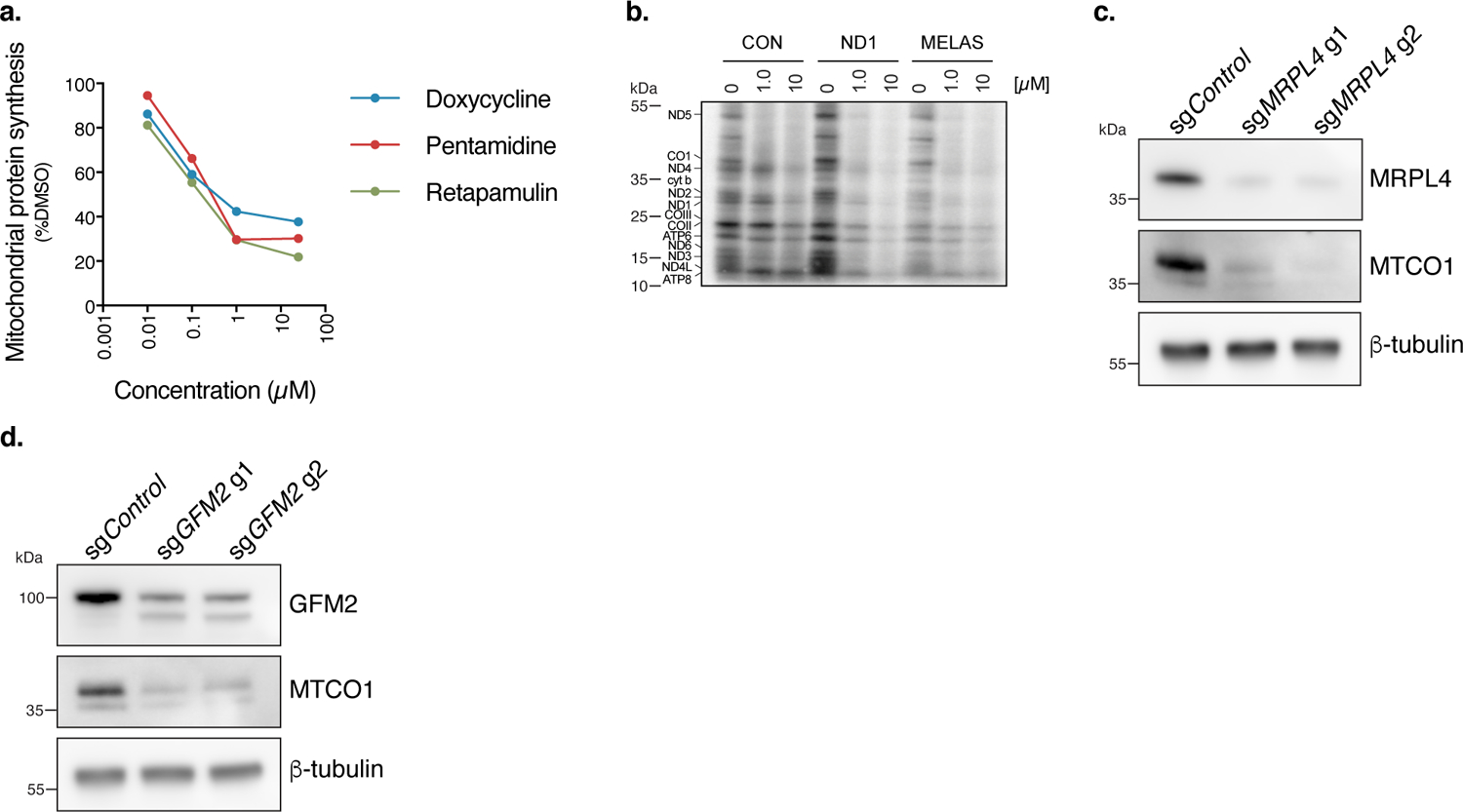
a. Mitochondrial protein synthesis (%DMSO) versus antibiotic concentration based on band quantification relative to DMSO from Fig. 2b (n=2 experiments). b. 35S-labelled cysteine and methionine pulse in cybrid cells treated with doxycycline at 1 μM or 10 μM with a 48 hour pre-treatment and 1 hour pulse (n=2 experiments). c. Western blot of MRPL4 in sgMRPL4 ND1 cybrid cells. d. Western blot of GFM2 in sgGFM2 ND1 cybrid cells.
Extended Data Fig. 3. Doxycycline does not promote cell survival in MELAS or ND1 cybrid cells through the integrated protein response (ATF4) or reported protein targets.
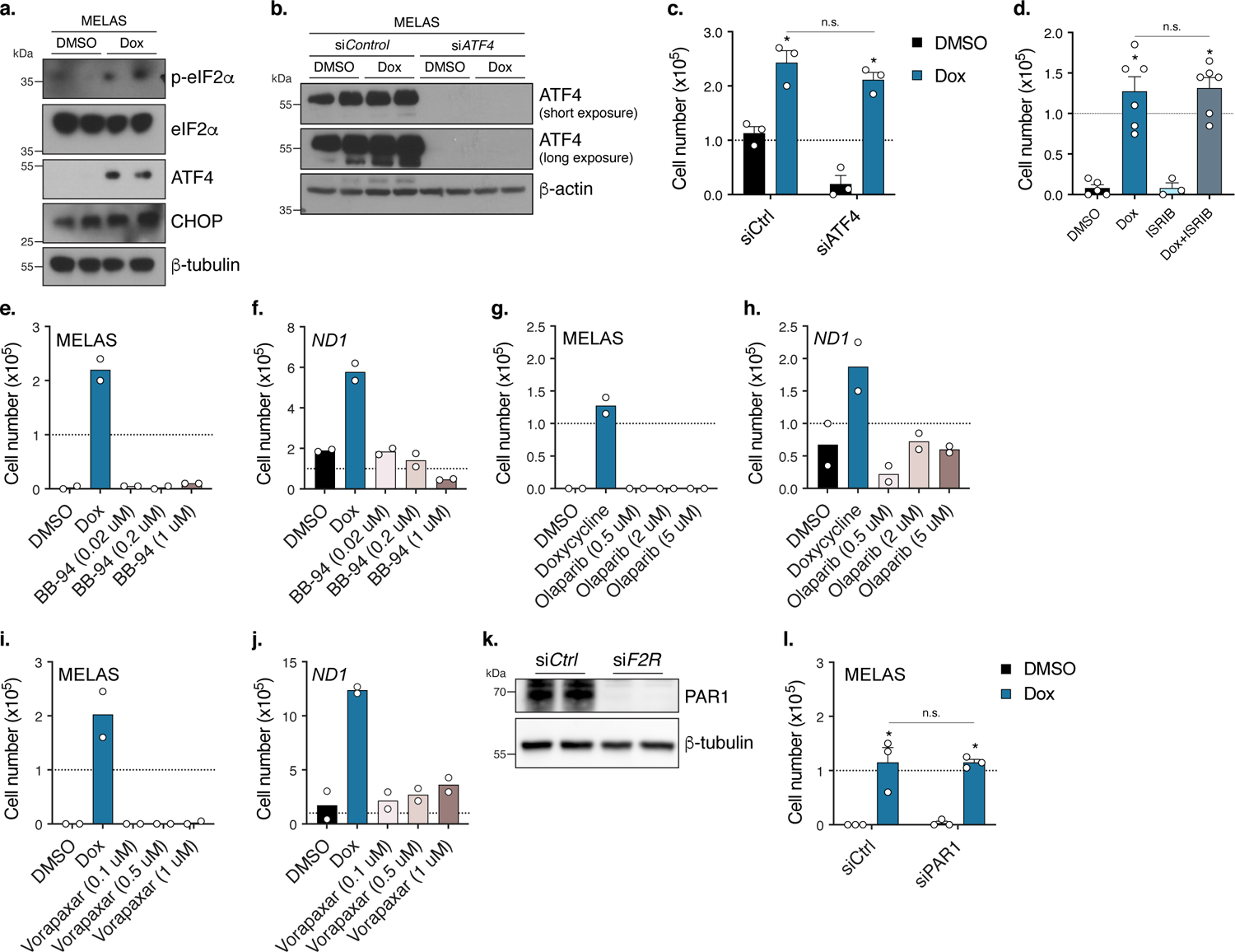
a. Western blot of integrated response proteins in MELAS cybrid cells treated with doxycycline for 24 hours (n=2 experiments). b. Western blot of ATF4 in siATF4 MELAS cybrid cells (n=2 experiments). c. 48 hour low-glucose survival of siATF4 MELAS cybrid cells treated with doxycycline (n=3 biologically independent samples). d. 48 hour low-glucose survival of MELAS cybrid cells treated with doxycycline with or without ISRIB (Integrated Stress Response Inhibitor) (n=3 biologically independent samples over n=2 independent experiments) e. 48 hour low-glucose survival of MELAS cybrid cells treated with pan matrix-metalloprotease (MMP) inhibitor BB-94 (n=2 biologically independent samples). f. 4 day galactose survival assay of ND1 cybrid cells treated with BB-94 (n=2 biologically independent samples). g. 48 hour low-glucose survival of MELAS cybrid cells treated with the PARP inhibitor Olaparib (n=2 biologically independent samples). h. 4 day galactose survival of ND1 cybrid cells treated with Olaparib (n=2 biologically independent samples). i. 48 hour low-glucose survival of MELAS cybrid cells treated with the PAR1 inhibitor Vorapaxar (n=2 biologically independent samples). j. 8 day galactose survival of ND1 cybrid cells treated with Vorapaxar (n=2 biologically independent samples). k. Western blot of PAR1 in siF2R MELAS cybrid cells (n=2 experiments). l. 48 hour low-glucose survival of MELAS cybrid cells depleted of PAR1 (siF2R) (n=3 biologically independent samples). Data are presented as mean values +/− s.e.m. error bars, Student’s t-test with a two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli, with Q = 5%, * q<0.05.
Extended Data Fig. 4. Extended Data 4. Mitochondrial mutant cells have basally elevated cytokines under nutrient stress conditions which are suppressed by doxycycline.
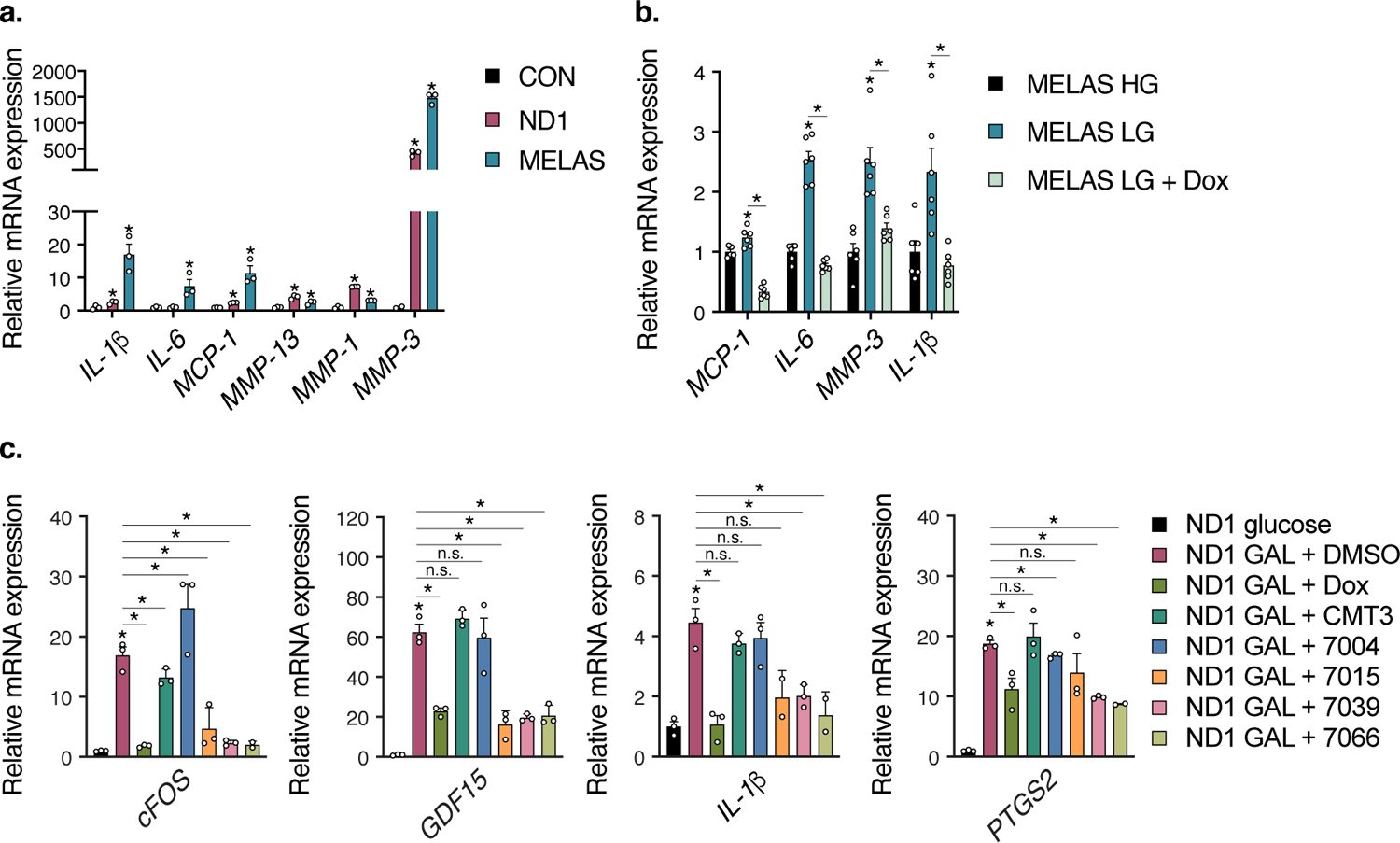
a. Gene expression of inflammatory cytokine panel under high-glucose conditions for control (CON), ND1, and MELAS cybrid cells (n=3 biologically independent samples). b. Gene expression of inflammatory cytokines in MELAS cybrid cells after 24 hour low-glucose (LG) conditions (n=6 biologically independent samples). c. Gene expression of inflammatory cytokines in ND1 cybrid cells after 48 hours galactose (GAL) conditions (n=3 biologically independent samples). Tetracycline analogs that rescue cell survival such as doxycycline, 7015, 7039, and 7066 suppress inflammatory gene expression. Data are presented as mean values +/− s.e.m. error bars, Student’s t-test with a two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli, with Q = 5%, * q<0.05.
Extended Data Fig. 5. Extended Data 5. Doxycycline suppresses inflammatory gene expression and metabolite levels in a time-dependent manner.
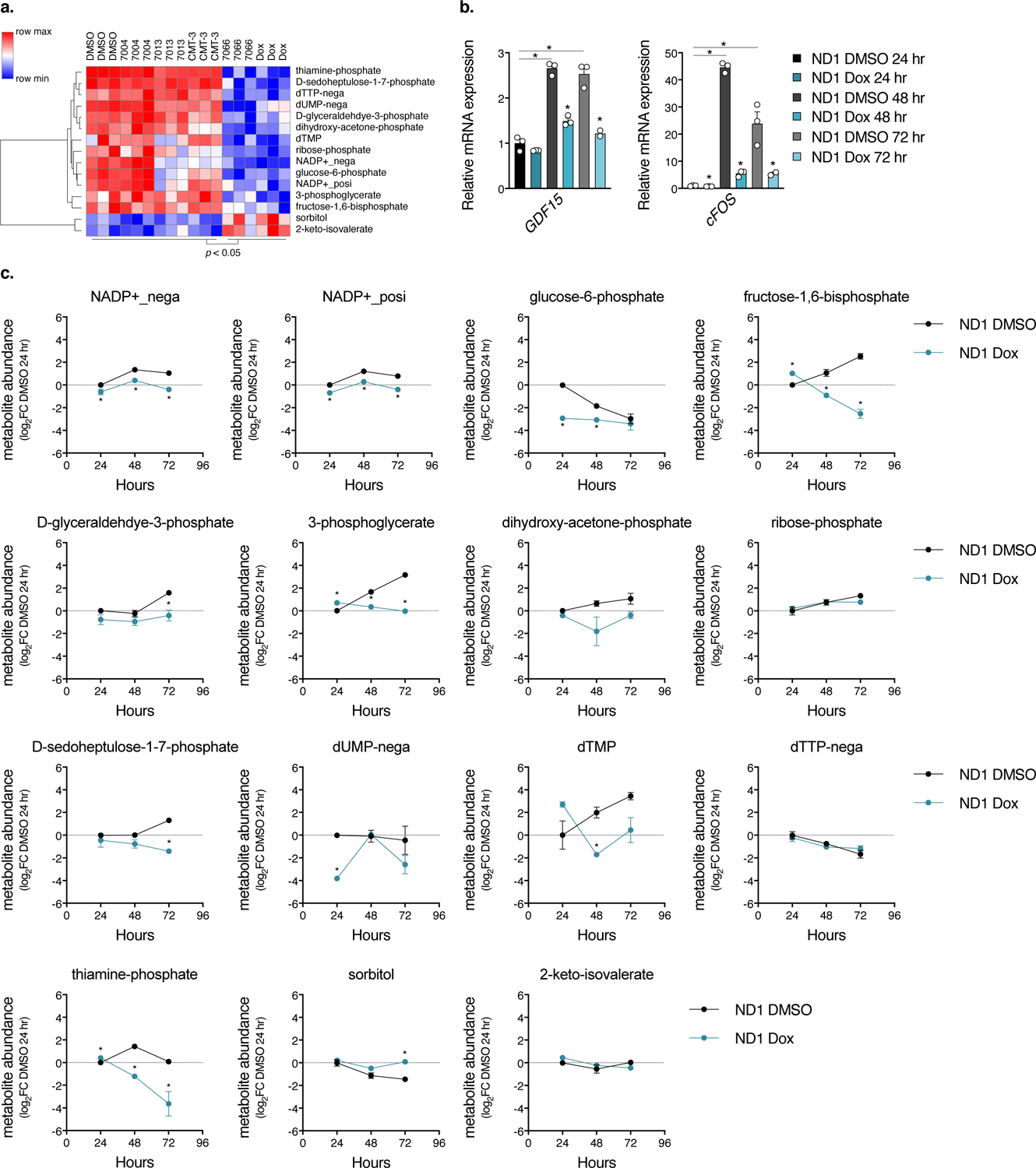
a. Heatmap of metabolites in ND1 cybrid cells that significantly change with doxycycline and 7066 compared to DMSO, 7004, 7013, and CMT-3 (n=3 biologically independent samples, p<0.05, Student’s t-test with Bonferroni’s correction). b. Time-dependent gene expression of inflammatory markers in ND1 cybrid cells under galactose conditions (n=3 biologically independent samples). c. Time-dependent metabolite changes in ND1 cybrid cells under galactose conditions (n=3 biologically independent samples). Data are presented as mean values +/− s.e.m. error bars, Student’s t-test with a two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli, with Q = 5%, * q<0.05.
Extended Data Fig. 6. Extended Data 6. p38 inhibition partially mimics the doxycycline anti-inflammatory metabolomic signature.
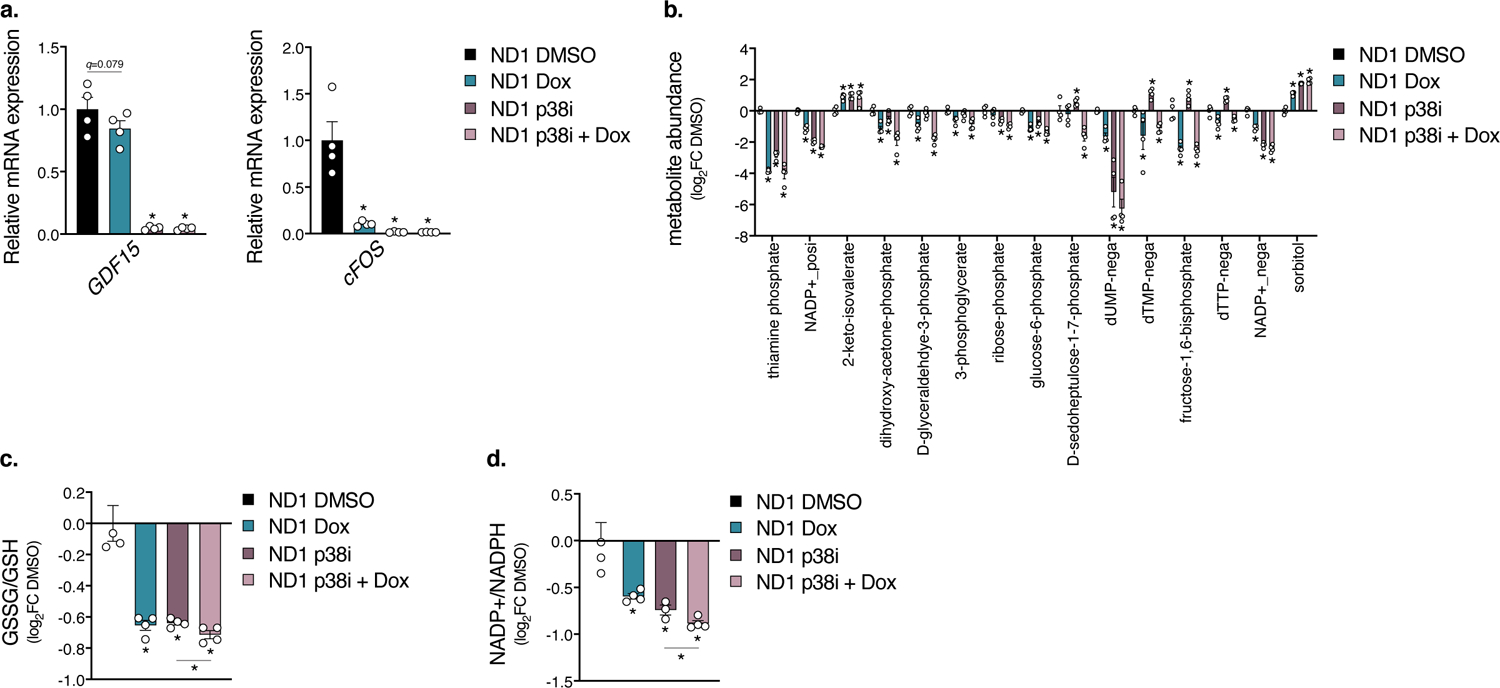
a. Gene expression of inflammatory markers in ND1 cybrid cells treated with doxycycline or p38i SB203580 after 48 hours galactose conditions (n=4 biologically independent samples). b. Metabolite changes in doxycycline signature in ND1 cybrid cells treated with doxycycline or SB203580 after 48 hours galactose conditions (n=4 biologically independent samples). c–d. Quantitation of GSSG/GSH and NADP+/NADPH ratio in ND1 cybrid cells with doxycycline or SB203580 treatment after 48 hours galactose conditions (n=4 biologically independent samples). Data are presented as mean values +/− s.e.m. error bars, Student’s t-test with a two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli, with Q = 5%, * q<0.05.
Extended Data Fig. 7. Extended Data 7. Doxycycline promotes metabolite redox homeostasis independent of complex I protein levels.
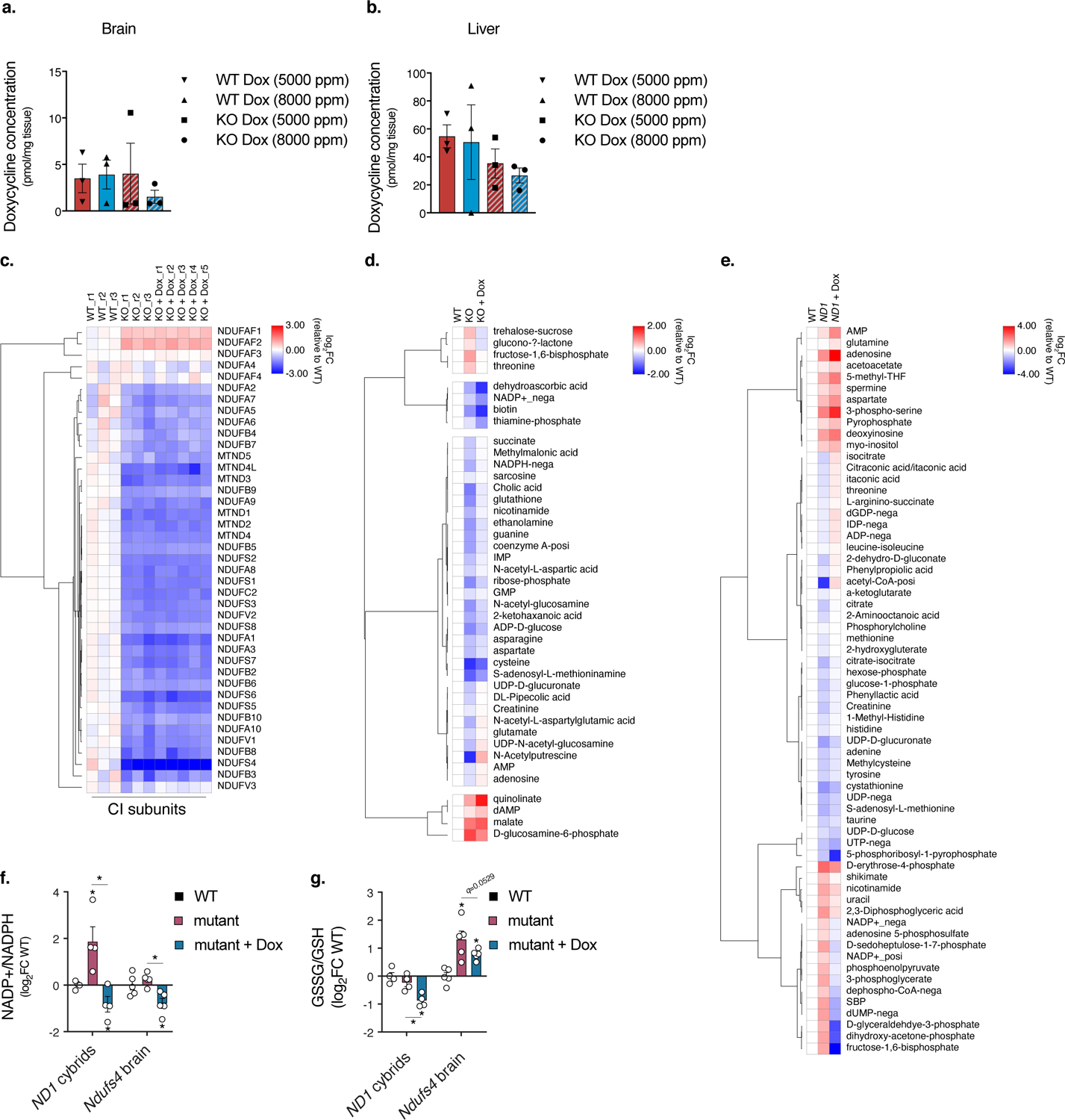
a. Quantitation of doxycycline in brains of wild-type and Ndufs4−/− (KO) animals fed 5000 or 8000 ppm doxycycline diets (n=3 mice per treatment group). b. Quantitation of doxycycline in livers of wild-type and Ndufs4−/− (KO) animals fed 5000 or 8000 ppm doxycycline diets (n=3 mice per treatment group). c. Heatmap of mitochondrial complex I subunits expressed in the mouse brains quantified through proteomics analysis (n=3 WT, n=3 KO, and n=5 KO Dox mice). d. Heatmap of metabolites altered in Ndufs4−/− (KO) brains (p<0.15, Student’s t-test, two-sided, unpaired) that are modulated by doxycycline (p<0.15, Student’s t-test, two-sided, unpaired) (n=5 mice). e. Heatmap of metabolites altered in ND1 cybrid cells (p<0.05, Student’s t-test, two-sided, unpaired) that significantly change with doxycycline (p<0.05, Student’s t-test, two-sided, unpaired) (n=4 biologically independent samples). f-g. Quantitation of NADP+/NADPH and GSSG/GSH ratios in ND1 cybrid cells or Ndufs4−/− (KO) brains with doxycycline treatment (cybrid cells, n=4 biologically independent samples; mice, n=5). Data are presented as mean values +/− s.e.m. error bars, Student’s t-test with a two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli, with Q = 5%, * q<0.05.
Supplementary Material
Supplementary Information:
Supplementary Table 1. Tetracycline analog library synthesized by the Myers Lab (Department of Chemistry and Chemical Biology, Harvard University)
Supplementary Table 2. Polar Metabolomics Analysis from ND1 cybrid cells +/− tetracycline analogs.
Supplementary Table 3. Polar Metabolomics Analysis from ND1 cybrid cells +/− doxycycline (Dox) at 24, 48, and 72 hours.
Supplementary Table 4. Polar Metabolomics Analysis from ND1 cybrid cells +/− doxycycline (Dox) and SB203580 (p38i).
Supplementary Table 5. Brain Proteomics from Ndufs4+/+ (WT), Ndufs4−/− (KO), and Ndufs4−/− with doxycycline treatment (DOX).
Supplementary Table 6. Polar Metabolomics Analysis from brains of Ndufs4+/+ (WT), Ndufs4−/− (KO) and Ndufs4−/− with doxycycline treatment (DOX) mice.
Supplementary Table 7. Polar Metabolomics Analysis from Control (WT) and ND1 cybrid cells +/− doxycycline (Dox).
Supplementary Table 8. RT-qPCR and CRISPR primer sequences.
Acknowledgments:
We thank members of the Puigserver Laboratory for helpful discussions regarding this project. We would like to thank A. Renner of the Myers lab for careful editing of the manuscript. We would like to thank C. Moraes (University of Miami Medical School) and J. Smeitink and R. Vogel (Radboud University Medical Centre, Nijmegen, Gelderlan, Netherlands) for providing us with the cell lines used in this study. Also, we would like to acknowledge the Nikon Imaging Center at Harvard Medical School for assistance with brightfield microscopy, the ICCB-Longwood Screening Facility at Harvard Medical School for facilitating our screening efforts, J.M. Asara and M. Yuan at the Beth Israel Deaconess Medical Center Mass Spectrometry Core for providing metabolomics profiling data, R. Bronson and the Rodent Histopathology Core at Harvard Medical School for careful sectioning and analysis of mouse sections and well as the Specialized Histopathology Core at Brigham and Women’s Hospital for immunohistochemistry. We would like to thank C. Vidoudez of the Harvard Center for Mass Spectrometry for analysis of doxycycline tissue concentrations. This work was supported by NIH RO1 DK089883–07 NIDDK and RO1 GM121452 NIGMS to P.P., NIH F30 DE028206–01A1 NIDCR to E.A.P., NIH F32 GM125243–01A1 NIGMS to C.F.B. and EMBO postdoctoral fellowship and MDA Development Grant to E.B., Human Frontier Science Program (LT-000033/2019-L) to P.L.M.
Footnotes
Competing interests: The authors declare no competing interests.
Data availability:
Datasets generated during this current study are included in the published article as supplementary tables or as publicly available datasets. Chemical screen data has been deposited in PubChem with accession dataset identifier 1508586 and proteomic data in PRIDE with accession dataset identifier PXD022860. Any additional data not included in this manuscript is available upon contact with the corresponding author upon request.
References:
- 1.Area-Gomez E & Schon EA Mitochondrial genetics and disease. J. Child Neurol 29, 1208–1215 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Wallace DC & Chalkia D Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb Perspect Biol 5, a021220 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.El-Hattab AW, Adesina AM, Jones J & Scaglia F MELAS syndrome: Clinical manifestations, pathogenesis, and treatment options. Mol. Genet. Metab 116, 4–12 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Gorman GS et al. Mitochondrial diseases. Nat Rev Dis Primers 2, 16080 (2016). [DOI] [PubMed] [Google Scholar]
- 5.El-Hattab AW, Zarante AM, Almannai M & Scaglia F Therapies for mitochondrial diseases and current clinical trials. Mol Genet Metab 122, 1–9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pfeffer G, Majamaa K, Turnbull DM, Thorburn D & Chinnery PF Treatment for mitochondrial disorders. Cochrane Database Syst Rev CD004426 (2012) doi: 10.1002/14651858.CD004426.pub3. [DOI] [PMC free article] [PubMed]
- 7.Russell OM, Gorman GS, Lightowlers RN & Turnbull DM Mitochondrial Diseases: Hope for the Future. Cell 181, 168–188 (2020). [DOI] [PubMed] [Google Scholar]
- 8.Kruse SE et al. Mice with mitochondrial complex I deficiency develop a fatal encephalomyopathy. Cell Metab 7, 312–320 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sproule DM & Kaufmann P Mitochondrial Encephalopathy, Lactic Acidosis, and Strokelike Episodes. Annals of the New York Academy of Sciences 1142, 133–158 (2008). [DOI] [PubMed] [Google Scholar]
- 10.Srivastava S et al. PGC-1α/β induced expression partially compensates for respiratory chain defects in cells from patients with mitochondrial disorders. Hum Mol Genet 18, 1805–1812 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pallotti F et al. Biochemical analysis of respiratory function in cybrid cell lines harbouring mitochondrial DNA mutations. Biochem J 384, 287–293 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khan NA et al. mTORC1 Regulates Mitochondrial Integrated Stress Response and Mitochondrial Myopathy Progression. Cell Metabolism 26, 419–428.e5 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Johnson SC et al. mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science 342, 1524–1528 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Civiletto G et al. Rapamycin rescues mitochondrial myopathy via coordinated activation of autophagy and lysosomal biogenesis. EMBO Mol Med 10, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu AK, Datta S, McMackin MZ & Cortopassi GA Rescue of cell death and inflammation of a mouse model of complex 1-mediated vision loss by repurposed drug molecules. Hum. Mol. Genet 26, 4929–4936 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diaz F, Enríquez JA & Moraes CT Cells Lacking Rieske Iron-Sulfur Protein Have a Reactive Oxygen Species-Associated Decrease in Respiratory Complexes I and IV. Mol Cell Biol 32, 415–429 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barrow JJ et al. Bromodomain Inhibitors Correct Bioenergetic Deficiency Caused by Mitochondrial Disease Complex I Mutations. Mol. Cell 64, 163–175 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moullan N et al. Tetracyclines disturb mitochondrial function across eukaryotic models: a call for caution in biomedical research. Cell Rep 10, 1681–1691 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun T & Zhang Y Pentamidine binds to tRNA through non-specific hydrophobic interactions and inhibits aminoacylation and translation. Nucleic Acids Res 36, 1654–1664 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chukwudi CU rRNA Binding Sites and the Molecular Mechanism of Action of the Tetracyclines. Antimicrob Agents Chemother 60, 4433–4441 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meydan S et al. Retapamulin-Assisted Ribosome Profiling Reveals the Alternative Bacterial Proteome. Molecular Cell 74, 481–493.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yun J & Finkel T Mitohormesis. Cell Metab 19, 757–766 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Charest MG, Lerner CD, Brubaker JD, Siegel DR & Myers AG A convergent enantioselective route to structurally diverse 6-deoxytetracycline antibiotics. Science 308, 395–398 (2005). [DOI] [PubMed] [Google Scholar]
- 24.Sun C et al. A Robust Platform for the Synthesis of New Tetracycline Antibiotics. J. Am. Chem. Soc 130, 17913–17927 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wright PM & Myers AG Methodological advances permit the stereocontrolled construction of diverse fully synthetic tetracyclines containing an all-carbon quaternary center at position C5a. Tetrahedron 67, 9853–9869 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Golub LM, McNamara TF, D’Angelo G, Greenwald RA & Ramamurthy NS A non-antibacterial chemically-modified tetracycline inhibits mammalian collagenase activity. J. Dent. Res 66, 1310–1314 (1987). [DOI] [PubMed] [Google Scholar]
- 27.Leibowitz RD The effect of ethidium bromide on mitochondrial DNA synthesis and mitochondrial DNA structure in HeLa cells. J. Cell Biol 51, 116–122 (1971). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quirós PM et al. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J Cell Biol 216, 2027–2045 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alano CC, Kauppinen TM, Valls AV & Swanson RA Minocycline inhibits poly(ADP-ribose) polymerase-1 at nanomolar concentrations. Proc. Natl. Acad. Sci. U.S.A 103, 9685–9690 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhong W et al. Doxycycline directly targets PAR1 to suppress tumor progression. Oncotarget 8, 16829–16842 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Golub LM et al. Tetracyclines inhibit tissue collagenase activity. A new mechanism in the treatment of periodontal disease. J. Periodont. Res 19, 651–655 (1984). [DOI] [PubMed] [Google Scholar]
- 32.Jin Z, Wei W, Yang M, Du Y & Wan Y Mitochondrial Complex I Activity Suppresses Inflammation and Enhances Bone Resorption by Tipping the Balance of Macrophage-Osteoclast Polarization. Cell Metab 20, 483–498 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Soustek MS et al. Inhibition of the ER stress IRE1α inflammatory pathway protects against cell death in mitochondrial complex I mutant cells. Cell Death & Disease 9, 658 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lorenzl S, Albers DS, Narr S, Chirichigno J & Beal MF Expression of MMP-2, MMP-9, and MMP-1 and their endogenous counterregulators TIMP-1 and TIMP-2 in postmortem brain tissue of Parkinson’s disease. Exp. Neurol 178, 13–20 (2002). [DOI] [PubMed] [Google Scholar]
- 35.Montero R et al. GDF-15 Is Elevated in Children with Mitochondrial Diseases and Is Induced by Mitochondrial Dysfunction. PLoS One 11, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sliter DA et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 561, 258–262 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 37.Balsa E et al. Defective NADPH production in mitochondrial disease complex I causes inflammation and cell death. Nat Commun 11, 2714 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lake NJ, Compton AG, Rahman S & Thorburn DR Leigh syndrome: One disorder, more than 75 monogenic causes. Ann. Neurol 79, 190–203 (2016). [DOI] [PubMed] [Google Scholar]
- 39.Gengenbacher M et al. Tissue Distribution of Doxycycline in Animal Models of Tuberculosis. Antimicrobial Agents and Chemotherapy 64, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McAlister GC et al. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem 86, 7150–7158 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stephan AH, Barres BA & Stevens B The complement system: an unexpected role in synaptic pruning during development and disease. Annu. Rev. Neurosci 35, 369–389 (2012). [DOI] [PubMed] [Google Scholar]
- 42.Torres-Odio S et al. Progression of pathology in PINK1-deficient mouse brain from splicing via ubiquitination, ER stress, and mitophagy changes to neuroinflammation. J Neuroinflammation 14, 154 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Benninger F, Glat MJ, Offen D & Steiner I Glial fibrillary acidic protein as a marker of astrocytic activation in the cerebrospinal fluid of patients with amyotrophic lateral sclerosis. J Clin Neurosci 26, 75–78 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Liuyu T et al. Induction of OTUD4 by viral infection promotes antiviral responses through deubiquitinating and stabilizing MAVS. Cell Res 29, 67–79 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dodd ME et al. The ENTH domain protein Clint1 is required for epidermal homeostasis in zebrafish. Development 136, 2591–2600 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Y et al. Effect of ApoA4 on SERPINA3 mediated by nuclear receptors NR4A1 and NR1D1 in hepatocytes. Biochem. Biophys. Res. Commun 487, 327–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lampropoulou V et al. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab 24, 158–166 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pegg AE Functions of Polyamines in Mammals. J. Biol. Chem 291, 14904–14912 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ying W NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid. Redox Signal 10, 179–206 (2008). [DOI] [PubMed] [Google Scholar]
- 50.Medina CB et al. Metabolites released from apoptotic cells act as tissue messengers. Nature 580, 130–135 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Madeo F, Eisenberg T, Pietrocola F & Kroemer G Spermidine in health and disease. Science 359, (2018). [DOI] [PubMed] [Google Scholar]
- 52.Holmström KM & Finkel T Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol 15, 411–421 (2014). [DOI] [PubMed] [Google Scholar]
- 53.Suomalainen A & Battersby BJ Mitochondrial diseases: the contribution of organelle stress responses to pathology. Nat. Rev. Mol. Cell Biol 19, 77–92 (2018). [DOI] [PubMed] [Google Scholar]
- 54.Johnson SC et al. Regional metabolic signatures in the Ndufs4(KO) mouse brain implicate defective glutamate/α-ketoglutarate metabolism in mitochondrial disease. Molecular Genetics and Metabolism 130, 118–132 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cerovic M, Forloni G & Balducci C Neuroinflammation and the Gut Microbiota: Possible Alternative Therapeutic Targets to Counteract Alzheimer’s Disease? Front Aging Neurosci 11, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sampson TR et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 167, 1469–1480.e12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ferrari M et al. Hypoxia treatment reverses neurodegenerative disease in a mouse model of Leigh syndrome. Proc. Natl. Acad. Sci. U.S.A 114, E4241–E4250 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rhee H-W et al. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 339, 1328–1331 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bank RPD RCSB PDB - 5AJ4: Structure of the 55S mammalian mitoribosome https://www.rcsb.org/structure/5AJ4.
- 60.Teves SS & Henikoff S Transcription-generated torsional stress destabilizes nucleosomes. Nat Struct Mol Biol 21, 88–94 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Belaghzal H, Dekker J & Gibcus JH Hi-C 2.0: An optimized Hi-C procedure for high-resolution genome-wide mapping of chromosome conformation. Methods 123, 56–65 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jha P, Wang X & Auwerx J Analysis of Mitochondrial Respiratory Chain Supercomplexes Using Blue Native Polyacrylamide Gel Electrophoresis (BN-PAGE). Curr Protoc Mouse Biol 6, 1–14 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Balsa E et al. ER and Nutrient Stress Promote Assembly of Respiratory Chain Supercomplexes through the PERK-eIF2α Axis. Molecular Cell 74, 877–890.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shalem O et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sasarman F & Shoubridge EA Radioactive labeling of mitochondrial translation products in cultured cells. Methods Mol. Biol 837, 207–217 (2012). [DOI] [PubMed] [Google Scholar]
- 66.Gajda A, Posyniak A & Tomczyk G LC-MS/MS analysis of doxycycline residues in chicken tissues after oral administration. Bulletin of the Veterinary Institute in Pulawy 58, 573–579 (2014). [Google Scholar]
- 67.Huang DW, Sherman BT & Lempicki RA Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4, 44–57 (2009). [DOI] [PubMed] [Google Scholar]
- 68.Huang DW, Sherman BT & Lempicki RA Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37, 1–13 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pettersen EF et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci (2020) doi: 10.1002/pro.3943. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information:
Supplementary Table 1. Tetracycline analog library synthesized by the Myers Lab (Department of Chemistry and Chemical Biology, Harvard University)
Supplementary Table 2. Polar Metabolomics Analysis from ND1 cybrid cells +/− tetracycline analogs.
Supplementary Table 3. Polar Metabolomics Analysis from ND1 cybrid cells +/− doxycycline (Dox) at 24, 48, and 72 hours.
Supplementary Table 4. Polar Metabolomics Analysis from ND1 cybrid cells +/− doxycycline (Dox) and SB203580 (p38i).
Supplementary Table 5. Brain Proteomics from Ndufs4+/+ (WT), Ndufs4−/− (KO), and Ndufs4−/− with doxycycline treatment (DOX).
Supplementary Table 6. Polar Metabolomics Analysis from brains of Ndufs4+/+ (WT), Ndufs4−/− (KO) and Ndufs4−/− with doxycycline treatment (DOX) mice.
Supplementary Table 7. Polar Metabolomics Analysis from Control (WT) and ND1 cybrid cells +/− doxycycline (Dox).
Supplementary Table 8. RT-qPCR and CRISPR primer sequences.
Data Availability Statement
Datasets generated during this current study are included in the published article as supplementary tables or as publicly available datasets. Chemical screen data has been deposited in PubChem with accession dataset identifier 1508586 and proteomic data in PRIDE with accession dataset identifier PXD022860. Any additional data not included in this manuscript is available upon contact with the corresponding author upon request.