

Abstract

SARS-CoV-2, the etiologic agent of the COVID-19 pandemic, emerged as the cause of a global crisis. Rapid and reliable clinical diagnosis is essential for effectively controlling transmission. The gold standard assay for SARS-CoV-2 identification is the highly sensitive real-time quantitative polymerase chain reaction (RT-qPCR); however, this assay depends on specialized reagents and may suffer from false results. Thus, additional assays based on different approaches could be beneficial. Here, we present a novel method for SARS-CoV-2 identification based on mass spectrometry. The approach we implemented combines a multistep procedure for the rational down-selection of a set of reliable markers out of all optional in silico derived tryptic peptides in viral proteins, followed by monitoring of peptides derived from tryptic digests of purified proteins, cell-cultured SARS-CoV-2, and nasopharyngeal (NP) swab matrix spiked with the virus. The marker selection was based on specificity to SARS-CoV-2 and on analytical parameters including sensitivity, linearity, and reproducibility. The final assay is based on six unique and specific peptide markers for SARS-CoV-2 identification. The simple and rapid (2.5 h) protocol we developed consists of virus heat inactivation and denaturation, tryptic digestion, and identification of the selected markers by liquid chromatography coupled to high-resolution mass spectrometry (LC-MS/MS). The developed assay enabled the identification of 104 PFU/mL SARS-CoV-2 spiked into buffer. Finally, the assay was successfully applied to 16 clinical samples diagnosed by RT-qPCR, achieving 94% concordance with the current gold standard assay. To conclude, the novel MS-based assay described here is specific, rapid, simple, and is believed to provide a complementary assay to the RT-qPCR method.
1. Introduction
Human coronavirus disease 2019 (COVID-19) initially emerged in China toward the end of 2019 and rapidly spread globally causing serious global public health concern.1,2 As of September 10, 2020, more than 25 million COVID-19 cases have been confirmed worldwide with approximately 1 million deaths (https://covid19.who.int). The disease has a high human-to-human transmission rate,3 making reliable diagnosis critical for disease control.
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2),4 a novel strain of coronaviruses (CoVs), is the etiologic agent of COVID-19. CoVs are linear, single-stranded positive-sense RNA viruses belonging to the Coronavirinae subfamily in the Coronaviridae family.5 They have a crownlike appearance under electron microscopy due to the presence of spike glycoproteins on their envelope.6,7 SARS-CoV-2 is classified as a betacoronavirus, similar to the two other highly virulent human CoV (hCoV) pathogens: SARS-CoV and Middle East respiratory syndrome (MERS-CoV).6,7 In addition, four common hCoVs (OC43, HKU1, 229E, NL63) are responsible for mild respiratory tract infections,8 making it seven hCOVs that have been identified to date. The full-length genome sequence of SARS-CoV-2 is 29 903 bp (GenBank no. MN908947).9 Notably, SARS-CoV-2 shares the highest nucleotide sequence identity (88%) with two bat-derived SARS-like coronaviruses, but is more distant from SARS and MERS, with ∼79 and ∼50% identity, respectively.9
The SARS-CoV-2 genome comprises 10 genes, which ultimately produce 26 proteins,9−11 most of which are functional proteins, including proteases, RNA polymerase, and associated factors needed to copy the genome, a proofreading exonuclease, and several other nonstructural proteins. The remaining genes predominantly encode the four structural components of the virus:11 (1) the trimeric outer spike glycoprotein (S) that binds the mammalian cellular angiotensin-converting enzyme 2 (ACE2) receptor to enable cell entry, (2) a nucleoprotein (N), that directly binds to viral RNA providing stability;12 (3) an envelope protein (E) which has both structural and functional aspects; and (4) a membrane protein (M) that forms the outer layer of the virus. These four structural proteins are the most abundant proteins in the virus, estimated to exist at ∼1000 and ∼300 copies per virion for the N and S proteins, respectively.11
A fast, convenient, and reliable diagnosis of patients suspected to be infected with SARS-CoV-2 is urgently needed for both appropriate treatment and isolation of confirmed cases, with the aim of preventing further spread of the infection. Currently, the majority of testing is done on respiratory samples, usually NP swabs, using nucleic acid-based tests,13 mainly quantitative real-time polymerase chain reaction (RT-qPCR).10 This relatively rapid assay is highly sensitive and therefore is recommended by the World Health Organization (WHO) and considered as the gold standard assay for SARS-CoV-2 identification. However, due to the high sensitivity of these nucleic acid-based methods (limit of detection is estimated as 1–10 PFU/mL14,15), they are prone to false-positive results.10,16 On the other hand, nucleic acid tests are susceptible inhibition by substances in clinical samples, leading to possible false-negative results.16−18 Thus, in some cases, a positive result might immediately follow repeated negative tests.16 Importantly, nucleic acid-based tests depend on the availability of specific reagents for RNA extraction and the RT-qPCR procedure.19 The global nature of the pandemic has already caused reagent shortages.20,21 The urgent need to implement and rapidly expand testing has led to the development of serological assays22 such as enzyme-linked immunosorbent assays (ELISA), lateral flow (LAF), chemiluminescence immunoassay (CLIA), and immunofluorescence assay (IFA). These assays are based on the detection of antibodies that are part of the immune response to the virus;10,22 yet, for early detection of infection, direct identification of the virus is essential. Thus, an alternative or complementary assay for SARS-CoV-2 diagnosis is urgently needed.
Mass spectrometry (MS)-based approaches for targeted and untargeted protein identification have become essential tools in clinical laboratories. During the SARS outbreak at 2003, the matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) technique was applied successfully for the characterization of SARS S and N proteins.23 Very recently, a few reports suggested identifying SARS-CoV-2 based on the LC-MS/MS analysis of its tryptic peptides. Marker selection was based mainly on a proteomics approach, starting directly from specimens with high viral loads and searching against the UniProt SARS-CoV-2 proteins database. Although Gouveia et al.24 listed candidate peptides derived from the virus N, S, or M proteins, the applicability of these markers has not been confirmed in clinical samples. A “proof-of-concept” assay for the detection of SARS-CoV-2 from gargle solution samples was demonstrated using acetone precipitation followed by tryptic digestion and a targeted MS analysis.25 This approach identified peptides originating from SARS-CoV-2 N protein, thus enabling virus detection in two of three gargle samples. Another recently published approach for SARS-CoV-2 detection in nasal swab specimens is based on the combination between MALDI-TOF MS and machine learning analysis.26
Here, we demonstrate the development and application of a rapid, simple, and specific MS-based SARS-CoV-2 identification assay. The assay was developed based on our novel approach for viral marker selection, starting from a bioinformatics in silico analysis, through evaluation of purified viral proteins, cultured virus, and negative NP clinical samples spiked with virus. The applicability of this novel assay for SARS-CoV-2 identification in clinical samples is demonstrated by screening 16 predetermined positive or negative NP swabs collected from human patients. The proposed novel assay is in very strong concordance with the well-established nucleic acid-based test and may serve as a complementary approach.
2. Results and Discussion
2.1. Methodology for Assay Development
The main target protein documented until now for MS-based detection of SARS-CoV-2 was the N protein, since it is known to be the most abundant protein in the virus.25,27,28 While N protein units link together in a long spiral, wrapping and coiling the RNA, the S protein is found in the outer layer of the virus, possibly presenting a more accessible target for tryptic digestion. In addition, S protein has a significantly higher molecular weight (Mw) than N protein (∼139 vs ∼42 kDa, respectively), resulting in a larger number of tryptic peptides and hence potential markers; therefore, we have added the S protein to our analysis. Lately, Gouveia et al. reported on 14 peptides from the N, S, and M proteins that could be used as potential markers for SARS-CoV-2 diagnosis,20 reinforcing the importance of marker selection from S protein, which presented the lowest diversity in terms of sequence variants.20
The selection of a set of SARS-CoV-2-specific markers described in this study was based on a targeted approach, in which a rational, multistep procedure was applied on all possible tryptic peptides derived from N and S SARS-CoV-2 proteins. An in silico tryptic digestion of the N and S protein sequences revealed a set of potential tryptic peptides markers. Theoretical considerations, followed by experimental evaluations, in both purified proteins and SARS-CoV-2 spiked samples, based on LC-MS/MS analysis, resulted in six unique markers determined for SARS-CoV-2-specific identification, three of which are novel (to the best of our knowledge). The down-selection and evaluation steps toward the determination of the six specific markers are schematically illustrated in Figure 1 and further detailed in the following sections.
Figure 1.

Methodology flowchart for SARS-CoV-2 marker selection. The first part was performed computationally, including in silico tryptic digest followed by length restriction (6–14 aa) and specificity consideration (removal of peptides that appear in human pathogen databases). The second part is LC-MS analysis of tryptic peptides derived from purified proteins, cell-cultured SARS-CoV-2, negative NP sample spiked with SARS-CoV-2, and positive NP samples. Evaluating parameters as sensitivity, reproducibility, and linearity helped reduce the number of potential markers.
2.2. Criteria for Markers Selection
Rational selection of SARS-CoV-2-specific peptide markers, whose identification provides unambiguous proof for the presence of virus, is the most important step in developing a targeted MS method. Four criteria for marker selection were defined: (a) selectivity—the clinical specimens are NP swabs; therefore, to avoid false-positive results, selected markers should not be shared by other organisms, particularly by human respiratory system pathogens. To verify the selectivity of markers, potential markers should also be tested against a wide range of existing sequences in available databases. (b) Sensitivity—an essential condition for good sensitivity in LC-MS-based analysis is high electrospray ionization (ESI) efficiency, which is expressed by MS signal response of the potential markers. This can be altered by matrix effects; therefore, it is important to evaluate the sensitivity of markers derived from purified proteins, cell-cultured virus, and virus spiked to the relevant clinical matrix. (c) Chemical stability—the selected peptide markers should be chemically stable. Therefore, the markers’ sequence should exclude amino acids such as methionine or cysteine, which are prone to oxidation. (d) Reproducibility and linearity—reliable markers, derived from purified proteins and from the entire SARS-CoV-2 virus, should exhibit high values of linearity and precision, to ensure constant tryptic digest efficiency, minimum missed cleavage, no adsorption to reaction vessels and/or LC-MS system, as well as minimum mutations.
2.3. In Silico Tryptic Digestion and Sequence Analysis
In silico tryptic digestion of SARS-CoV-2 N and S protein sequences revealed 57 and 102 peptides, respectively. The length of the tryptic peptides selected for further evaluation was limited to 6–14 amino acids (aa) to increase the probability of marker specificity (aa≥6) and minimize peptide loss due to adsorption (aa≤14). Under these restrictions, 16 and 36 predicted tryptic peptides derived from N and S proteins, respectively, remained. To exclude the possibility that these peptides might be present in nonrelevant human clinical samples, we conducted a sequence similarity search of the 52 predicted tryptic peptides against a list of clinically relevant human pathogens (www.kariusdx.com/pathogenlist/3.6), as well as against the comprehensive NCBI nr database. This analysis revealed 14 peptides that are identical to other human pathogens’ proteins and were therefore excluded from further evaluation. All other peptides may have an identical sequence in nonrelevant organisms, nonhuman coronaviruses proteins, or SARS proteins (Figure S1, nonhighlighted sequences). Obviously, these organisms are not expected to be present in SARS-CoV-2-suspected human samples; thus, these findings do not impose a limitation with regard to candidate markers. A total of 38 peptides (29 and 9 originating from S and N proteins, respectively) were therefore selected for further evaluation as specific markers of SARS-CoV-2.
2.4. Evaluating Purified Protein Tryptic Digestion Efficiency and Analytical Performance
To evaluate the actual efficiency of tryptic digestion process, recombinant SARS-CoV-2-purified S or N proteins were used. A rapid, simple, and efficient tryptic digestion process was performed, similar to the procedure used in our study of protein toxins digestion described recently.29 Samples were preheated (95 °C, 10 min) to inactivate the virus and denature viral proteins (improving subsequent digestion efficiency), after which direct tryptic digestion of the purified proteins (50 °C, 120 min) was performed. The samples containing 10 ppm purified S or N proteins (1 μg protein in 100 μL buffer) were digested for 120 min in triplicate and analyzed by LC coupled to a high-resolution Q Exactive Plus Orbitrap MS/MS instrument. Of a possible 38 predicted peptides, 33 (25 S-derived, 8 N-derived) with the same m/z as the in silico tryptic peptides were identified according to accurate mass and amino acid sequences using LC-MS/MS analysis, demonstrating high efficiency of the digestion process.
To characterize the potential markers, buffer solutions were spiked with four concentrations (0.01, 0.1, 1, and 10 ppm) of purified S or N proteins. The samples were trypsin-digested and analyzed by LC-MS. Three parameters, signal intensity, precision, and linearity, were used for the evaluation of a marker’s performance and as indications of markers’ sensitivity, chemical and physical (nonadsorption) stability, and tryptic digestion process efficiency and reproducibility. The values obtained are presented in Table 3 (and detailed in Tables S1 and S2 for N and S proteins, respectively). The intensities obtained for N-derived peptides were ∼10-fold higher compared to those observed for the same concentrations of S-derived peptides. Tables S1 and S2 present LC-MS-based analytical performance of SARS-CoV-2 tryptic peptides. Five out of eight tryptic peptides derived from N protein (peptides numbers 3, 4, 5, 6, and 7) and 8 out of 25 peptides derived from S protein (1, 3, 6, 11, 13, 18, 21, 25) exhibited relative high sensitivity with good precision (<30%) and linearity in the range of 10–10 000 ppb.
Table 3. Analytical Parameters for the Six Selected Tryptic Peptides Derived from SARS-CoV-2-Purified Proteins and Cell-Cultured Virus Spiked to Buffer with or without Acetone Precipitation.
| intensity (a.u) | precision (%) | linearity range | ||
|---|---|---|---|---|
| purified proteins | N | 1.4 × l07–2.0 × 107 | 22–28 | 10 ppb–10 ppm |
| S | 9.9 × 105–2.5 × 106 | 7–28 | 10 ppba–10 ppm | |
| cell-cultured SARS-CoV-2 | N | 1.4 × 105–2.1 × 105 | 9 | 104–106 PFU/mL |
| S | 1.6. × 105–2.8 × 105 | 5–24 | 104–106 PFU/mL | |
| acetone precipitation | N | 6.8 × 105–1.9 × 106 | 12–30 | 103*–106 PFU/mL |
| S | 3.5 × 105–6.3 × 105 | 16–30 | 104–106 PFU/mL |
For the best performance.
2.5. Evaluation of Potential Markers in Cell-Cultured SARS-CoV-2
To evaluate the applicability of the method for direct identification of the entire virus, the assay was performed on samples containing SARS-CoV-2 viruses cultured in Vero E6 cells. Buffer samples were spiked with virus at final concentrations of 103–106 PFU/mL, followed by tryptic digestion and LC-MS/MS analysis, as described above. Results with high-concentration samples (106 PFU/mL) demonstrated high efficiency of the tryptic digestion process, enabling identification of almost all tryptic peptides that were detected in the purified SARS-CoV-2 proteins (23/25 for S-derived peptides and all N-derived peptides; Tables S1 and S2).
The analytical parameters of viral peptides derived from cell-cultured SARS-CoV-2 proteins (peak intensities, precision values, and linearity) were calculated, and the results are presented in Table 3 (and detailed in Tables S1 and S2). Considering that the amount of S protein in SARS-CoV-2 is 3 times lower than N protein11 and that S protein tryptic digestion efficiency is 10 times lower as well,23 we would expect significantly lower intensities of S tryptic peptides compared to N tryptic peptides, when tryptic digests of the virus are used. However, as shown in Tables 3, S1, and S2, similar marker intensities were obtained from virus tryptic digestion of both proteins. This observation may indicate greater accessibility of the S protein to tryptic digestion. Three out of five N markers (3, 4, and 6), and five out of eight S markers (6, 11, 13, 18, and 21), showed a strong potential for serving as markers according to their analytical parameters, including relative high sensitivity, reproducibility (<30%), and linearity in the range of 104–106 PFU/mL (Tables S1 and S2). Figure 2 demonstrates the performance of a representative marker (N protein, marker number 3) obtained from tryptic digest of the purified protein and from cell-cultured virus.
Figure 2.

Example of tryptic digest-derived marker behavior (AYNVTQAFGR, N protein, marker no. 3). (A) Extracted-ion chromatogram (EIC) of m/z 563.7856 from a full-scan LC-MS run of 10 ppm N protein (left). Linearity of a tryptic peptide derived from tryptic digestion of N protein in the range of 10 ppb–10 ppm (right). (B) Extracted-ion chromatogram (EIC) of m/z 563.7856 from a full-scan LC-MS run of 106 PFU/mL SARS-CoV-2 (left). Linearity of a tryptic peptide derived from tryptic digestion of SARS-CoV-2 in the range of 104–106 PFU/mL (right). (C) Extracted-ion chromatogram (EIC) of m/z 563.7856 from a full-scan LC-MS run of 106 SARS-CoV-2 after precipitation (left). Linearity of a tryptic peptide derived from tryptic digestion of SARS-CoV-2 in the range of 103–106 PFU/mL.
To determine the incubation time required for viral digestion, buffer samples containing 106 PFU/mL SARS-CoV-2 were digested for increasing durations (0, 10, 30, 60, 120, and 240 min). The kinetic profiles for tryptic digest of both proteins, N and S were performed by LC-MS analysis of the above eight markers. After 120 min of tryptic digestion, the reaction was still found to be ongoing, although the sensitivity improvement after 240 min was <40%. On the other hand, shortening the digestion time to 30 min reduced the sensitivity by 60%. The representative marker intensities derived from N and S proteins (Nos. 3 and 6, respectively) at specific time points are presented at Figure S2. Based on these results, the optimal digestion time for SARS-CoV-2 was determined to be 120 min.
2.6. Assay Performance in Spiked NP Samples
To examine the assay performance in NP swab samples and the relevant (and challenging) clinical matrix, known negative specimens (n = 10) were spiked with SARS-CoV-2 to achieve virus concentrations of 105 and 106 PFU/mL. Samples were subjected to tryptic digestion and compared with the parallel spiked buffer sample. The matrix background significantly interfered with two peptides, one derived from N protein (Table S1, peptide number 4) and the other from S protein (18), and were therefore excluded from further evaluation. The other six potential markers (N protein markers 3 and 6, S protein markers 6, 11, 13, and 21) were detected, yet suffered from decreased signal intensities, compared to buffer-spiked samples. To avoid this sensitivity decrease, whether as a result of matrix effect on ESI or a lower efficiency of the tryptic digestion process, several dilution compositions of NP specimens with ammonium bicarbonate buffer were spiked with 106 PFU/mL SARS-CoV-2 and compared to the same final concentrations spiked directly into buffer. The minimum necessary dilution to maintain the sensitivity obtained in buffer-spiked samples was 3:2 (buffer:sample). Figure 3 illustrates the sample dilution effect on markers’ signal intensities. By diluting the samples prior to tryptic digestion, all six markers were successfully observed, having signal intensities similar to spiked buffer and high precision (<30%) in replicates with different concentrations of virus; these were therefore selected for further evaluation in clinical samples. Table 1 presents the selected markers and their detected MS/MS fragments using purified proteins. The fragments that were detected in negative NP samples, spiked with SARS-CoV-2, are highlighted. The greater the complexity of the matrix and the lower the virus concentration in it, the smaller the number of detectable fragments. Retention time stability is of great importance especially in complex matrices that do not undergo sample pretreatment. Retention time variations were evaluated for the six markers, derived from different concentrations of N and S pure proteins, SARS-CoV-2 spiked to buffer, and negative NP samples, which were injected at different days (Table S4). Low run-to-run variations, <±0.05 min, were observed indicating the chromatographic stability of the analytical method.
Figure 3.

SARS-CoV-2 spiked to buffer or to NP-negative samples. SARS-CoV-2 was spiked to 50 mM ammonium bicarbonate buffer and to NP samples as is or diluted with 50 mM ammonium bicarbonate (2:3 respectively) to give 106 PFU/mL. A reduction in peak intensities was observed in nondiluted matrix, while similar peak intensities in buffer and diluted matrix were observed monitoring our six selected markers.
Table 1. Rationally Selected Markers for the Identification of SARS-CoV-2a.
| marker | protein | m/z (multiple charge) | retention time (min) | MS/MS fragments |
|---|---|---|---|---|
| AYNVTQAFGR | N | 563.7856 (M + 2H)/2 | 3.5 | 175.1039(y1), 207.1134(a2), 232.1253, 235.1083(b2)*, 321.1562(13), 349.1512(b3), 379.1937(y3), 420.2247(a3), 450.2308(y4), 578.2894(y5), 679.3371(y6), 748.3630(b7), 778.4055 (y7), 892.4484(y8) |
| GFYAEGSR | N | 443.7063 (M + 2H)/2 | 2.9 | 120.0813(imm Phe), 136.0762(imm Tyr), 205.0977(b2)*, 319.1730(y3), 368.1610(b3), 439.1982(b4), 448.2156(y4)*,519.2527(y5)*, 682.3160(y6)* |
| SFIEDLLFNK | S | 613.3268 (M + 2H)/2 | 4.7 | 86.0970(imm lle), 120.0813(imm phe), 147.1134(y1), 207.1134(a2)*, 235.1083(b2)*, 261.1563(y2), 348.1923(b3), 408.2247(y3), 521,3087(y4), 592.2618(b5), 634.3928(y5), 749.4197(y6), 878.4623(y7), 991.5464(y8) |
| FLPFQQFGR | S | 570.3035 (M + 2H)/2 | 4.2 | 86.0970(imm lle), 101.0715(imm Gln), 120.0813(imm phe) 175.1195(y1), 232.1410(y2), 233.1654(a2)*, 261.1603(b2)*, 358.2131(b3), 379.2094(y3), 505.2815(b4), 507.2680(y4), 635.3265(y5), 782.3949(y6), 879.447(y7)*, 992.5317(y8) |
| FQTLLALHR | S | 366.8854 (M + 3H)/3, 549.8246 (M + 2H)/2 | 3.6 | 86.0970(imm Leu), 120.0813(imm phe), 156.0773(y1-NH3), 175.1039(y1), 276.1348(b2), 312.1628(Y2), 377.1825(b3), 425.2468(Y3), 496.2839(Y4)*, 609.3680(Y5), 722.4521(Y6), 823.4998(Y7) |
| HTPINLVR | S | 475.2825 (M + 2H)/2 | 3.0 | 86.0970(imm Leu), 110.0718(imm His), 138.0667(b2), 175.1195(y1), 158.0930(Z1), 211.1195(a2), 221.1039(b2-H2O), 239.114(b2), 274.1879(y2), 387.2720(y3), 449.2513(b4), 501.3149(y4), 563.2942(b5), 614.3989(y5), 676.3782(b6), 711.4517(y6)*, 775.4467(b7), 812.4994(y7)* |
All markers do not exist in human pathogen databases, including the four human coronavirus OC43, NL63, HKU1, and 229E. The list of MS/MS diagnostic fragments was obtained from tryptic digest of N and S proteins spiked to buffer (10 ppm). The fragments indicated in bold were also found in negative spiked samples with 105 PFU/mL SARS-CoV-2. The fragments with asterisks were only found in negative samples spiked with 106 PFU/mL. Mass accuracy of parent ion <2 ppm. Mass accuracy of daughters ions <5 ppm.
2.7. Markers’ Intraspecies Universality and Specificity
The sequence conservation of the six selected peptide markers, two derived from N protein: AYNVTQAFGR (peptide no. 3) and GFYAEGSR (6), and four derived from S protein: SFIEDLLFNK (6), FLPFQQFGR (11), FQTLLALHR (13), and HTPINLVR (21), among different known strains of SARS-CoV-2 is a major concern, in light of the accumulating knowledge of mutations occurring during the spread of the virus. Intraspecies conservation was therefore verified against all known variants of the virus (56 940 complete genomes with high coverage, as of September 2020, downloaded from the GISAID database (https://gisaid.org)). The S and N proteins were retrieved from the downloaded genomes by translated BLAST against the corresponding proteins in the reference Wuhan strain. Each of the six markers was analyzed for sequence similarity against its parental protein. This analysis revealed an extremely high degree of sequence conservation, with identical sequence in >99.8% of known variants for 4/6 markers and >97.5% for the remaining two markers (see Table S3 for FQTLLALHR and HTPINLVR peptides). These results indicate that a diagnostic array based on the combination of six markers originating from distinct regions in two different proteins will provide a targeted assay for specific detection of SARS-CoV-2, which might transcend the slight genetic drift that may occur in future strains.
The uniqueness of the selected markers was verified in silico against the four common hCoVs (OC43, HKU1, 229E, NL63), revealing no identical markers in any of them, highlighting our assay’s specificity. To experimentally verify the assay’s specificity, the protocol was applied in buffer samples spiked with a high concentration (107 PFU/mL) of hCoV (OC43). As expected, none of the SARS-CoV-2-specific markers was observed.
2.8. Clinical Samples Analysis
The next step was to evaluate our assay in a real-life setting using actual clinical NP samples, previously diagnosed by RT-PCR. In complex matrices, such as NP specimens, unexpected interference, that differ in nature from sample to sample, may occur. Using a set of six markers increases the probability of proper identification. Each of the selected markers is unique for SARS-CoV-2 so that identification of a single marker is sufficient to provide a reliable basis for virus identification. As defined by the EU and the SANCO guide, the following analytical criteria are required for SARS-CoV-2 identification: a specific marker with proper chromatographic retention time, accurate mass (<2 ppm), and at least two MS/MS fragments.30,31 Using these criteria, our new assay enables the identification of SARS-CoV-2 at concentrations as low as 104 PFU/mL of cell-cultured virus (spiked into buffer). This LOD was evaluated according to the lowest concentration at which a chromatographic peak with at least two MS-MS fragments ions could be detected using mass tolerance of 5 ppm (Figure S3).
To evaluate further the performances and specificity of the assay in relevant clinical specimens, NP swab samples previously tested negative (n = 10) or positive (n = 6) by the “gold standard” RT-qPCR were inactivated at 95 °C for 10 min. From each specimen, 40 μL was diluted with 60 μL of buffer and subjected to tryptic digestion for 2 h at 50 °C. The reaction was ended by adding formic acid at a final concentration of 1%, and followed by LC-MS/MS analysis. Figure 4 demonstrates the identification of a representative SARS-CoV-2-specific marker (FQTLLALHR) in a positive NP swab specimen as well as in SARS-CoV-2 spiked into buffer; this marker is absent from a negative NP sample. In a similar way, 5/6 NP swab specimens, which were previously tested positive by RT-qPCR with Ct values in the range of 19–25, were found to contain the virus using our new MS analysis method. In these five specimens, the virus concentrations were estimated by comparison NP matrix spiked with known concentrations of SARS-CoV-2. These samples were found to contain 5 × 104–1 × 106 PFU/mL. For one sample, a mismatch was observed between the results of RT-qPCR and MS methods, probably due to a false-negative result of the MS method. However, there is a possibility that this mismatch was caused by a false-positive result of the RT-qPCR assay. None of the markers was found in the RT-qPCR-negative samples analyzed. Table 2 summarizes the results of marker identification in NP swab samples using LC-MS/MS analysis. The retention time values of the detected markers were in the estimated range (<±0.05 min, Table S4). MS-MS fragments of marker precursor ions were in accordance with those observed in negative NP spiked samples (Table 1). Notably, a substantial diversity of the identified markers was observed in these clinical samples, probably due to a variety of matrix interferences between the different samples. This emphasizes the importance of the selection of an array of six specific markers, to increase the identification probability in unexpectedly challenging matrices.
Figure 4.
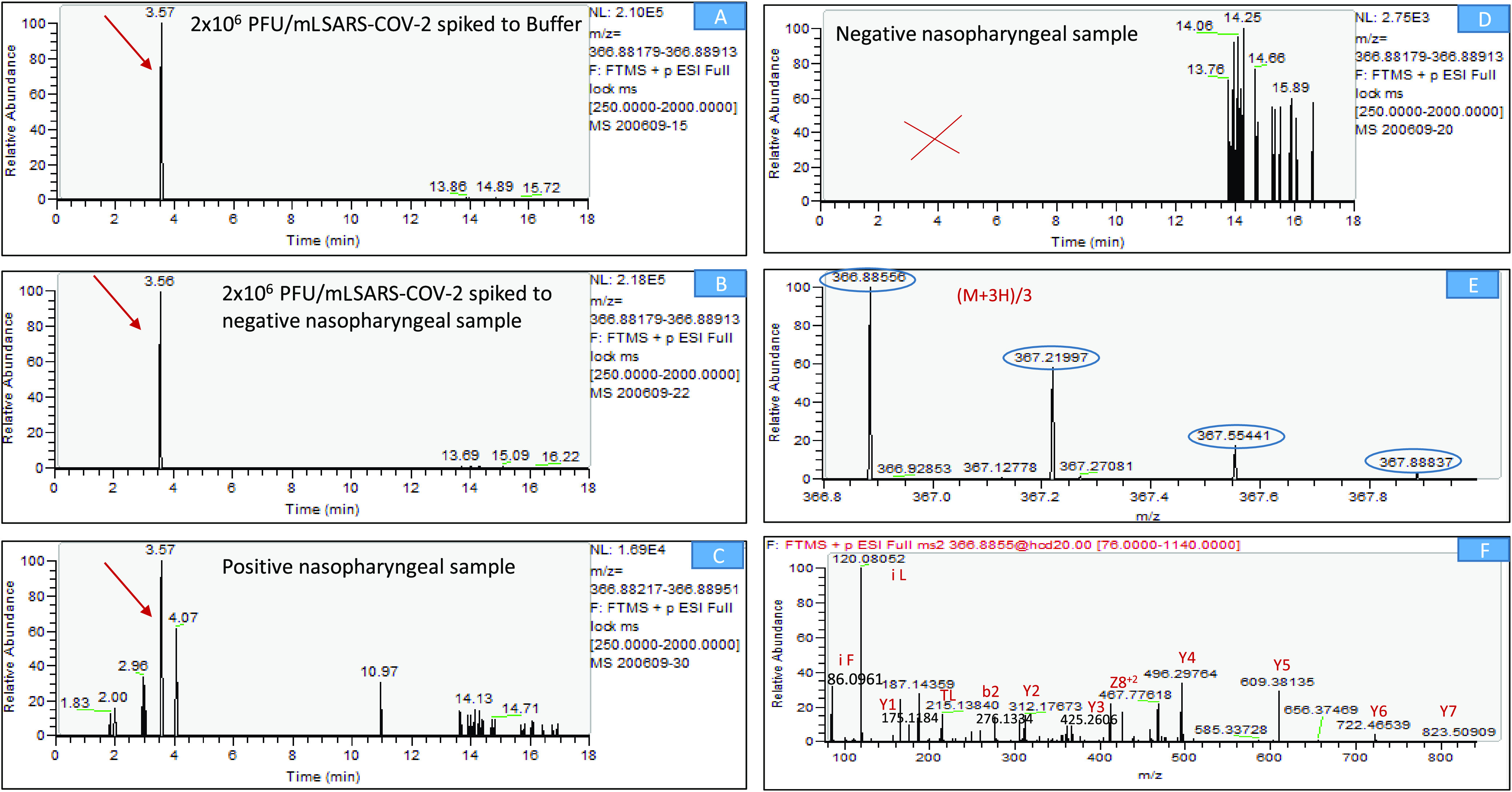
LC-MS/MS (Orbitrap) identification of SARS-CoV-2-specific marker (FQTLLALHR, S protein, peptide no. 13). (A) Extracted-ion chromatogram (EIC) of m/z 366.8854 from a full-scan LC-MS run of 106 PFU/mL SARS-CoV-2 spiked to buffer. (B) Extracted-ion chromatogram (EIC) of m/z 366.8854 from a full-scan LC-MS run of 106 PFU/mL SARS-CoV-2 spiked to negative NP swab. (C) Extracted-ion chromatogram (EIC) of m/z 366.8854 from a full-scan LC-MS run of positive NP swab. (D) Extracted-ion chromatogram (EIC) of m/z 366.8854 from a full-scan LC-MS run of negative NP swab. (E) Mass spectrum of the specific marker FQTLLALHR (parent ion, (M + 3H)/3, at m/z 366.8856, chromatographic peak at 3.5 min) derived from spiked protein. (F) Marker fragmentation spectrum (MS-MS of 366.8854). Y—Ions that result from the cleavage of the C–N bonds (amide bond) of a peptide backbone with the C-terminal fragments retaining the charge. b—Ions that result from the cleavage of the C–N (amide bond) bonds of a peptide backbone with the N-terminal fragments retaining the charge. Z—Ions that result from the cleavage of the N–C bonds of a peptide backbone with the C-terminal fragments retaining the charge. i—Immonium ion is an internal fragment formed by a combination of a type (C–C bond) and y type (C–N bond) cleavage.
Table 2. SARS-CoV-2 Markers Detected in NP Swab Samples by LC-MS/MS (Orbitrap) Analysisa.
| samples | PCR (Ct) | markers detected m/z (multiple charge) | r.t (min) | MS-MS fragments |
|---|---|---|---|---|
| 1 | +(19.6) | 563.7356 (M + 2H)/2 | 3.55 | 207.113, 235.108 |
| 443.7063 (M + 2H)/2 | 2.89 | 120.081, 136.076, 448.215, 519.253, 682.316 | ||
| 570.3035 (M + 2H)/2 | 4.19 | 86.097, 120.081, 175.119, 233.165, 261.160, 879.447 | ||
| 366.8354, (M + 2H)/2 | 3.57 | 86.097, 120.081, 496.284 | ||
| 475.2825 (M + 2H)/2 | 3.01 | 86.097, 110.071, 175.119, 239.114, 711.451 | ||
| 2 | +(21.1) | 613.3268 (M + 2H)/2 | 4.74 | 86.097, 120.081, 235.108 |
| 3 | +(22.3) | 570.3035 (M + 2H)/2 | 4.17 | 86.097, 101.071,120.081, 175.119 |
| 4 | +(24.6) | - | ||
| 5 | +(19.1) | 613.3268 (M + 2H)/2 | 4.70 | 86.097, 120.081 |
| 366.8854 (M + 2H)/2 | 3.58 | 86.097, 120.081, 496.284 | ||
| 6 | +(18.7) | 613.3268 (M + 2H)/2 | 4.68 | 86.097, 120.081, 147.113, 207.113,235.108, 261.156 |
| 443.7063 (M + 2H)/2 | 2.85 | 120.081, 136.076 | ||
| 7–16 | –(>40) |
Mass accuracy of the parent ion <2 ppm. Mass accuracy of the fragments ions <5 ppm. Retention time accuracy <±0.05 min (see Table S4).
2.9. Sample Processing for Sensitivity Improvement
The sensitivity of RT-qPCR assay is estimated to be 1–10 PFU/mL at Ct ≤ 40,14,15 which is significantly greater than that shown in our assay (∼104 PFU/mL). To enable effective COVID-19 diagnosis, the assay’s sensitivity needs to be improved; therefore, several virus enrichment processes were evaluated. Increasing the initial sample volume to 500 μL, ultracentrifugation at 20 000g for 20 min and pellet resuspension with 100 μL of buffer did not yield significant improvement. In contrast, acetone precipitation of the virus was found to increase sensitivity. Buffer samples spiked with known concentrations of SARS-CoV-2 were precipitated prior to tryptic digestion by adding acetone to 500 μL of sample (1:1) followed by freezing (−20 °C, 60 min), allowing sedimentation of the proteins. The samples were then centrifuged (20 000g, 20 min), supernatant was removed, and the pellet was resuspended in 100 μL of buffer and subjected to the tryptic digestion. Using this protocol, the sample was essentially concentrated by an order of magnitude, starting with 500 μL instead of 40 μL in the simple standard procedure. Under these conditions, we achieved an increase in sensitivity of up to 10-fold for markers derived from the N protein and up to 3-fold for markers derived from the S protein (Figure 2C, Tables S1 and S2). The difference in sensitivity enhancement between N and S protein markers was found to result from acetone traces (<10%), which apparently reduced tryptic digestion of the S protein more than of the N protein. Buffer samples spiked with virus concentrations of 103–106 PFU/mL and prepared with the acetone precipitation step were evaluated for marker performance based on analytical parameters (intensities, precision, and linearity). The results, presented in Table 3 (and Tables S1 and S2), illustrate the potential of improving the assay sensitivity, even recognizing that the precipitation procedure affects the assay duration and complexity.
3. Conclusions
In the study presented here, a simple, rapid, and specific high-resolution MS-based assay was developed for the identification of SARS-CoV-2. Six unique markers, three of which are novel to the best of our knowledge, derived from the N and the S viral proteins were selected for COVID-19 diagnosis. This panel of markers was established using a rational down-selection process, which comprised in silico considerations and technical-based filtering criteria, followed by experimental evaluation using the HR-MS technology and implementation of the developed assay with clinical samples. We consider the sample preparation protocol to be straightforward, consisting of sample heating for virus deactivation and denaturation followed by 2 h of tryptic digestion and 20 min of LC-MS/MS analysis. The duration of the entire process is approximately 2.5 h.
This new assay enabled the identification of 104 PFU/mL SARS-CoV-2 in buffer with the promise of a 1 order of magnitude improvement in sensitivity by introducing a virus precipitation step prior to tryptic digestion. The method was successfully applied in 5/6 NP swab specimens that were previously tested as positive by RT-qPCR. NP samples with Ct values <25 were selected, assumed to be detectable based on the assay’s sensitivity in spiked buffer samples. An estimated viral load detection, based on comparison to known spiked concentrations of SARS-CoV-2 into virus-negative NP specimens, reached 5 × 104–1 × 106 PFU/mL. The assay specificity, which is essential for avoiding false-positive results, was verified in NP samples determined to be negative (n = 10), as well as in samples containing the closely related (but nonvirulent) hCoV OC43.
Granted, the sensitivity of a nucleic acid-based approach (1–10 PFU/mL) is higher than that achieved in the assay presented herein (104 PFU/mL). However, there is no certainty that the RT-qPCR test sensitivity is indeed clinically required, and it may possibly lead to false-positive results.10,16 The alternative diagnostic approach presented here is likely to serve as a complementary assay for RT-qPCR, which can suffer from reagent shortages or false results in typical COVID-19 patients. Further significant improvement in sensitivity, while reducing background noise, may be achieved by preconcentration and purification steps using immunomagnetic beads designed for SARS-CoV-2 capture and by developing a sensitive MRM-based LC-MS/MS analytical method for the target markers. Such approaches are currently under development in our laboratory.
4. Materials and Methods
4.1. Reagents
All solvents and chemicals used in the LC-MS/MS analysis were of LC-MS grade. Water (Cat. Number 232141B1), acetonitrile (Cat. Number 120410100), and formic acid (99% purity, Cat. Number 691413) were purchased from Bio Lab. Ammonium bicarbonate (NH4HCO3, Cat. Number A6141-500G), and octyl-β-d-glucopyranoside (OG, Cat. Number O8001-1G) were acquired from Sigma-Aldrich. Phosphate-buffered saline (PBS, pH 7.4, Cat. Number 02-023-1A) and grade-modified trypsin (Cat. Number V5111) were purchased from Biological Industries.
4.2. Expression of SARS-CoV-2 S and N Recombinant Proteins
Recombinant SARS-CoV-2 spike glycoprotein, expressed as recently described,32 was kindly provided by Dr. Ohad Mazor Lab (IIBR, Israel). A stabilized soluble version of the spike protein (based on GenPept: QHD43416 ORF amino acids 1–1207) was designed to include proline substitutions at positions 986 and 987, and disruptive replacement of the furin cleavage site RRAR (position 682-685) with GSAS. Protein expression was carried out using ExpiCHO system (Thermo Scientific).
Nucleocapsid phosphoprotein (N) sequences of SARS-CoV-2 (GenBank accession: YP_009724397.2) were codon-optimized for Escherichia coli, synthesized, and cloned in the expression vector pET-28a (+) by twist bioscience (Twist Bioscience). The resulting plasmid DNA was maintained in the T7 Express lysY Competent E. coli strain (New England Biolabs, France). The bacterial culture was induced for protein expression by 0.1 mM IPTG and incubated at 15 °C overnight. A cell pellet was harvested from 250 mL of the bacterial culture by centrifugation (10 000g) at 4 °C for 10 min. Bugbaster master mix lysis buffer (25 mL, Merck Millipore) was added to the harvested cell pellet and harvested according to the manufacturer’s protocol. Cell lysis mix was harvested by centrifugation (10 000g) at 4 °C for 10 min, and 25 mL of urea (8 M) was added to the pellet. The N protein was purified using HIS–SelectNickel affinity gel (Sigma-Aldrich) according to the manufacturer’s protocol.
4.3. Cell Lines and Viruses
African green monkey kidney clone E6 cells (Vero E6, ATCC CRL-1586) were grown in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS), MEM nonessential amino acids (NEAA), 2 mM l-glutamine, 100 units/mL penicillin, 0.1 mg/mL streptomycin, and 12.5 units/mL Nystatin (P/S/N) (Biological Industries, Israel). The cells were cultured at 37 °C in a 5% CO2 in 95% air atmosphere.
SARS-CoV-2 (GISAID accession EPI_ISL_406862) was kindly provided by Bundeswehr Institute of Microbiology, Munich, Germany. Virus stocks were propagated (four passages) and titered on Vero E6 cells. All virus handling and work were conducted in a BSL3 facility in accordance with the biosafety guidelines of the Israel Institute for Biological Research (IIBR).
To determine viral titers, Vero E6 cells were seeded in 12-well plates (5 × 105 cells/well) and grown overnight in DMEM containing 10% FBS, MEM nonessential amino acids, 2 nM l-glutamine, and P/S/N (Biological Industries, Israel). Serial dilutions of SARS-CoV-2 were prepared in MEM containing 2% FCS with NEAA, glutamine, and P/S/N and used to infect Vero E6 monolayers in triplicate (200 μL/well). Plates were incubated for 1 h at 37 °C to allow viral adsorption. Then, 2 mL/well of overlay (MEM containing 2% FBS and 0.4% Tragacanth; Merck, Israel) was added to each well and the plates were incubated at 37 °C, 5% CO2 for 48 h. The medium was aspirated, and the cells were fixed and stained with crystal violet solution (Biological Industries, Israel). The number of plaques in each well was determined, and SARS-CoV-2 titer was calculated.
4.4. Clinical Samples
NP swabs from suspected COVID-19 patients were transported to our lab in viral transport medium (Biological Industries). Samples were kept at 4 °C until use and randomly selected for this study. Clinical samples processing was done by vortexing the swabs vigorously for 30 s to release the virus into solution. For MS preparation, the samples were diluted with ammonium bicarbonate buffer in a ratio of 4:6 (40 μL of sample with 60 μL of buffer).
The RT-qPCR assay was performed according to WHO instructions, as previously described,14 using the SensiFAST Probe Lo-ROX One-Step kit (Bioline). Primers and probe for SARS-CoV-2 E gene were taken from the Berlin protocol.33
4.5. Tryptic Digestion
Tryptic digestion was conducted as previously described.29 In brief, a total volume of 100 μL of samples (SARS-CoV-2-purified N or S proteins, buffer-spiked viruses, or clinical NP swabs) was heated for denaturation (95 °C, 10 min). After 2 min cooling, 2 μL of sequencing-grade-modified trypsin (0.5 μg/μL) was added (final concentration, 1 μg/100 μL) to the sample tubes in the presence of 0.2% octyl-β-glucopyranoside (OG), followed by 2 h of incubation at 50 °C with continuous rotating (600 rpm). The tryptic digestion was stopped by adding 10 μL of 10% formic acid (final concentration, 1%), followed by 2 min of centrifugation (14 000 rpm). The resulting supernatants were transferred to LC-MS analysis vials.
4.6. High-Resolution LC-MS/MS (Orbitrap)
LC-MS analysis was performed on an Agilent 1290 HPLC (Agilent Technologies, Palo Alto, CA) coupled to Q Exactive Plus Orbitrap MS/MS instrument (Thermo Fisher Scientific, Waltham, MA) equipped with a heated electrospray ionization source operated in positive mode. This high-resolution MS system enables the identification of N- and S protein-derived tryptic peptides according to their accurate mass and sequence determination using Full MS DIA acquisition mode. Chromatographic separations were performed on a Gemini C18 column (100 mm × 2.1 mm, 3 μm; Phenomenex, Le Pecq, France) kept at 40 °C. Mobile phases were 1% formic acid in H2O (A) and 1% formic acid in ACN:H2O (8:2 v/v, B). The LC gradient profile was 100% A linearly decreased to 5% A over 10 min and then held for 6 min and increased to 100% A over 0.1 min, held for another 4 min, for a total run time of 20 min. The flow rate was 0.3 mL/min, and the injection volume was 10 μL. The operating parameters were as follows: electrospray voltage, 1.25 kV; sheath gas flow rate, 45 (arbitrary units); auxiliary gas, 10 (arbitrary units); sweep gas, 2 (arbitrary units); auxiliary gas heater temperature, 400 °C; S-lens RF level, 55; and capillary temperature, 275 °C. The MS spectra were acquired with 140 000 mass resolution (at 200 m/z) from m/z 250–2000, Automatic gain control (AGC) target of 3 × 106, and maximum injection time (IT) of 100 ms. The MS/MS spectra were acquired using tryptic peptides inclusion list, 35 000 mass resolution (at 200 m/z), an isolation window of 1 m/z, and AGC target of 2 × 105. The collision gas was nitrogen, and the collision energy was set at 20 V. Data files will be provided upon request.
4.7. Bioinformatic Analysis
The protein sequences of the S and N proteins are derived from the Wuhan reference strain NC_045512.2, protein ID YP_009724390.1 and YP_009724397.2, respectively. Possible trypsin cleavage sites for the protein sequences were predicted with PeptideCutter (https://web.expasy.org/peptide_cutter/). The generated peptides were subjected to sequence similarity searches against the nr (nonredundant, https://www.ncbi.nlm.nih.gov/) database and against an in house constructed database of over 1000 clinically relevant human pathogens (https://kariusdx.com/pathogenlist/3.6), using BlastP.34 The algorithm parameters were optimized for short sequences (word_size = 2, gapopen = 9, gapxtend = 1, matrix = PAM30, threshold = 16). All other parameters were used with default values. Exact matches were considered as identity of 100% and query coverage of 100%. Self-hits (to SARS-CoV-2 sequences) were eliminated.
Acknowledgments
The authors thank Dr. Ohad Mazor, Dr. Itai Glinert, and Dr. Shmuel (Sandy) Livnat for their fruitful remarks and help in reviewing of this manuscript.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c04691.
S and N proteins sequences and their predicted tryptic peptides (Figure S1); SARS-CoV-2 kinetic profile (Figure S2); LC-MS/MS chromatogram and spectrum of a representative marker (Figure S3); LC-MS analysis of N protein markers (Table S1); LC-MS analysis of S protein markers (Table S2); universality of S3 and S4 markers (Table S3); and markers retention time stability (Table S4) (PDF)
Author Contributions
# O.S. and L.F. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Wu F.; Zhao S.; Yu B.; Chen Y. M.; Wang W.; Song Z. G.; Hu Y.; Tao Z. W.; Tian J. H.; Pei Y. Y.; Yuan M. L.; Zhang Y. L.; Dai F. H.; Liu Y.; Wang Q. M.; Zheng J. J.; Xu L.; Holmes E. C.; Zhang Y. Z. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P.; Yang X. L.; Wang X. G.; Hu B.; Zhang L.; Zhang W.; Si H. R.; Zhu Y.; Li B.; Huang C. L.; Chen H. D.; Chen J.; Luo Y.; Guo H.; Jiang R. D.; Liu M. Q.; Chen Y.; Shen X. R.; Wang X.; Zheng X. S.; Zhao K.; Chen Q. J.; Deng F.; Liu L. L.; Yan B.; Zhan F. X.; Wang Y. Y.; Xiao G. F.; Shi Z. L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y.; Shin W. I.; Pang Y. X.; Meng Y.; Lai J.; You C.; Zhao H.; Lester E.; Wu T.; Pang C. H. The First 75 Days of Novel Coronavirus (SARS-CoV-2) Outbreak: Recent Advances, Prevention, and Treatment. Int. J. Environ. Res. Public Health 2020, 17, 2323. 10.3390/ijerph17072323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5536–544.. 10.1038/s41564-020-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J.; Li F.; Shi Z. L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. 10.1038/s41579-018-0118-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Q.; Lin Q.; Jin S.; You L. Coronavirus 2019-nCoV: A brief perspective from the front line. J. Infect. 2020, 80, 373–377. 10.1016/j.jinf.2020.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G.; Fan Y.; Lai Y.; Han T.; Li Z.; Zhou P.; Pan P.; Wang W.; Hu D.; Liu X.; Zhang Q.; Wu J. Coronavirus infections and immune responses. J. Med. Virol. 2020, 92, 424–432. 10.1002/jmv.25685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su S.; Wong G.; Shi W.; Liu J.; Lai A. C. K.; Zhou J.; Liu W.; Bi Y.; Gao G. F. Epidemiology, Genetic Recombination, and Pathogenesis of Coronaviruses. Trends Microbiol. 2016, 24, 490–502. 10.1016/j.tim.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R.; Zhao X.; Li J.; Niu P.; Yang B.; Wu H.; Wang W.; Song H.; Huang B.; Zhu N.; Bi Y.; Ma X.; Zhan F.; Wang L.; Hu T.; Zhou H.; Hu Z.; Zhou W.; Zhao L.; Chen J.; Meng Y.; Wang J.; Lin Y.; Yuan J.; Xie Z.; Ma J.; Liu W. J.; Wang D.; Xu W.; Holmes E. C.; Gao G. F.; Wu G.; Chen W.; Shi W.; Tan W. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y.; Chang L.; Wang L. Laboratory testing of SARS-CoV, MERS-CoV, and SARS-CoV-2 (2019-nCoV): Current status, challenges, and countermeasures. Rev. Med. Virol. 2020, 30, e2106 10.1002/rmv.2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimoto F. K. The Proteins of Severe Acute Respiratory Syndrome Coronavirus-2 (SARS CoV-2 or n-COV19), the Cause of COVID-19. Protein J. 2020, 39, 198–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunewald M. E.; Fehr A. R.; Athmer J.; Perlman S. The coronavirus nucleocapsid protein is ADP-ribosylated. Virology 2018, 517, 62–68. 10.1016/j.virol.2017.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascarella G.; Strumia A.; Piliego C.; Bruno F.; Del Buono R.; Costa F.; Scarlata S.; Agrò F. E. COVID-19 diagnosis and management: a comprehensive review. J. Intern. Med. 2020, 288, 192–206. 10.1111/joim.13091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israeli O.; Beth-Din A.; Paran N.; Stein D.; Lazar S.; Weiss S.; Milrot E.; Atiya-Nasagi Y.; Yitzhaki S.; Laskar O.; Schuster O. Evaluating the efficacy of RT-qPCR SARS-CoV-2 direct approaches in comparison to RNA extraction. Int. J. Infect. Dis. 2020, 99, 352–354. 10.1016/j.ijid.2020.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won J.; Lee S.; Park M.; Kim T. Y.; Park M. G.; Choi B. Y.; Kim D.; Chang H.; Kim V. N.; Lee C. J. Development of a Laboratory-safe and Low-cost Detection Protocol for SARS-CoV-2 of the Coronavirus Disease 2019 (COVID-19). Exp. Neurobiol. 2020, 29, 107–119. 10.5607/en20009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C.; Ren L. Recent progress on the diagnosis of 2019 Novel Coronavirus. Transboundary Emerging Dis. 2020, 67, 1485–1491. 10.1111/tbed.13620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiseman J.; D’Amico T. A.; Zawadzka S.; Anyimadu H. False negative SARS-CoV-2 PCR - A case report and literature review. Respir. Med. Case Rep. 2020, 31, 101140 10.1016/j.rmcr.2020.101140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woloshin S.; Patel N.; Kesselheim A. S. False Negative Tests for SARS-CoV-2 Infection - Challenges and Implications. N. Engl. J. Med. 2020, 383, e38 10.1056/NEJMp2015897. [DOI] [PubMed] [Google Scholar]
- Younes N.; Al-Sadeq D. W.; Al-Jighefee H.; Younes S.; Al-Jamal O.; Daas H. I.; Yassine H. M.; Nasrallah G. K. Challenges in Laboratory Diagnosis of the Novel Coronavirus SARS-CoV-2. Viruses 2020, 12, 582. 10.3390/v12060582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fomsgaard A. S.; Rosenstierne M. W. An alternative workflow for molecular detection of SARS-CoV-2 - escape from the NA extraction kit-shortage, Copenhagen, Denmark, March 2020. Eurosurveillance 2020, 25, 2000398 10.2807/1560-7917.ES.2020.25.14.2000398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu A. W.; Chan W. M.; Ip J. D.; Yip C. C.; Chan J. F.; Yuen K. Y.; To K. K. Evaluation of simple nucleic acid extraction methods for the detection of SARS-CoV-2 in nasopharyngeal and saliva specimens during global shortage of extraction kits. J. Clin. Virol. 2020, 129, 104519 10.1016/j.jcv.2020.104519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z.; Yi Y.; Luo X.; Xiong N.; Liu Y.; Li S.; Sun R.; Wang Y.; Hu B.; Chen W.; Zhang Y.; Wang J.; Huang B.; Lin Y.; Yang J.; Cai W.; Wang X.; Cheng J.; Chen Z.; Sun K.; Pan W.; Zhan Z.; Chen L.; Ye F. Development and clinical application of a rapid IgM-IgG combined antibody test for SARS-CoV-2 infection diagnosis. J. Med. Virol. 2020, 92, 1518–1524. 10.1002/jmv.25727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krokhin O.; Li Y.; Andonov A.; Feldmann H.; Flick R.; Jones S.; Stroeher U.; Bastien N.; Dasuri K. V.; Cheng K.; Simonsen J. N.; Perreault H.; Wilkins J.; Ens W.; Plummer F.; Standing K. G. Mass spectrometric characterization of proteins from the SARS virus: a preliminary report. Mol. Cell. Proteomics 2003, 2, 346–356. 10.1074/mcp.M300048-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouveia D.; Grenga L.; Gaillard J. C.; Gallais F.; Bellanger L.; Pible O.; Armengaud J. Shortlisting SARS-CoV-2 Peptides for Targeted Studies from Experimental Data-Dependent Acquisition Tandem Mass Spectrometry Data. Proteomics 2020, 20, 2000107 10.1002/pmic.202000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihling C.; Tänzler D.; Hagemann S.; Kehlen A.; Hüttelmaier S.; Arlt C.; Sinz A. Mass Spectrometric Identification of SARS-CoV-2 Proteins from Gargle Solution Samples of COVID-19 Patients. J. Proteome Res. 2020, 19, 4389–4392. 10.1021/acs.jproteome.0c00280. [DOI] [PubMed] [Google Scholar]
- Nachtigall F. M.; Pereira A.; Trofymchuk O. S.; Santos L. S. Detection of SARS-CoV-2 in nasal swabs using MALDI-MS. Nat. Biotechnol. 2020, 38, 1168–1173. 10.1038/s41587-020-0644-7. [DOI] [PubMed] [Google Scholar]
- Nikolaev E.; Indeykina M.; Brzhozovskiy A.; Bugrova A.; Kononikhin A.; Starodubtseva N.; Petrotchenko E.; Kovalev G.; Borchers C.; Sukhikh G. Mass Spectrometric detection of SARS-CoV-2 virus in scrapings of the epithelium of the nasopharynx of infected patients via Nucleocapsid N protein. J. Proteome Res. 2020, 19, 4393–4397. 10.1021/acs.jproteome.0c00412. [DOI] [PubMed] [Google Scholar]
- Cardozo K.; Lebkuchen A.; Okai G.; Schuch R.; Viana L.; Olive A.; Lázari C.; Fraga A.; Granato C.; Carvalho V. Fast and low-cost detection of SARS-CoV-2 peptides by tandem mass spectrometry in clinical samples. Res. Square 2020, 10.21203/rs.3.rs-28883/v1. [DOI] [Google Scholar]
- Feldberg L.; Schuster O.; Elhanany E.; Laskar O.; Yitzhaki S.; Gura S. Rapid and sensitive identification of ricin in environmental samples based on lactamyl agarose beads using LC-MS/MS (MRM). J. Mass Spectrom. 2020, 55, e4482 10.1002/jms.4482. [DOI] [PubMed] [Google Scholar]
- Commission E. 2002/657/EC: Commission Decision of 12 August 2002 implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of results 2002.
- Tuija P.Method Validation and Quality Control Procedures for Pesticide Residues Analysis in Food and Feed. Document No. SANCO/10684/2009. SANCO 2009. [Google Scholar]
- Noy-Porat T.; Makdasi E.; Alcalay R.; Mechaly A.; Levy Y.; Bercovich-Kinori A.; Zauberman A.; Tamir H.; Yahalom-Ronen Y.; Israeli M.; Epstein E.; Achdout H.; Melamed S.; Chitlaru T.; Weiss S.; Peretz E.; Rosen O.; Paran N.; Yitzhaki S.; Shapira S. C.; Israely T.; Mazor O.; Rosenfeld R. A panel of human neutralizing mAbs targeting SARS-CoV-2 spike at multiple epitopes. Nat. Commun. 2020, 11, 4303 10.1038/s41467-020-18159-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corman V. M.; Landt O.; Kaiser M.; Molenkamp R.; Meijer A.; Chu D. K.; Bleicker T.; Brünink S.; Schneider J.; Schmidt M. L.; Mulders D. G.; Haagmans B. L.; van der Veer B.; van den Brink S.; Wijsman L.; Goderski G.; Romette J. L.; Ellis J.; Zambon M.; Peiris M.; Goossens H.; Reusken C.; Koopmans M. P.; Drosten C. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Eurosurveillance 2020, 25, 2000045 10.2807/1560-7917.ES.2020.25.3.2000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho C.; Coulouris G.; Avagyan V.; Ma N.; Papadopoulos J.; Bealer K.; Madden T. L. BLAST+: architecture and applications. BMC Bioinf. 2009, 10, 421. 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.