

Abstract
Aim:
Macrophage migration inhibitory factor (MIF) is a pro-inflammatory cytokine, which has been shown to promote disease severity in cystic fibrosis.
Methods:
In this study, aerosolized drug-loaded nanoparticles containing SCD-19, an inhibitor of MIF's tautomerase enzymatic activity, were developed and characterized.
Results:
The aerosolized nanoparticles had an optimal droplet size distribution for deep lung deposition, with a high degree of biocompatibility and significant cellular uptake.
Conclusion:
For the first time, we have developed an aerosolized nano-formulation against MIF's enzymatic activity that achieved a significant reduction in the inflammatory response of macrophages, and inhibited Pseudomonas aeruginosa biofilm formation on airway epithelial cells. This represents a potential novel adjunctive therapy for the treatment of P. aeruginosa infection in cystic fibrosis.
Keywords: : aerosolized drug delivery, biofilm formation, chronic infection, cystic fibrosis, inflammatory disease, macrophage migration inhibitory factor (MIF), nanodrug delivery system, Pseudomonas aeruginosa, PLGA NPs, pulmonary disease
Graphical abstract
Cystic fibrosis (CF) is a life limiting disease which impacts multiple organ systems. In CF, the lunges provide a microenvironment, which is conducive to the development of recurrent Pseudoomonas aeruginosa infections, which ultimately results in the acquisition of a chronic respiratory infection [1,2]. Chronic P. aeruginosa infection, which is driven by P. aeruginosa biofilm formation, results in the development of chronic inflammation within the lungs which drives progressive lung damage, resulting in respiratory failure and mortality [3,4]. As such, anti-inflammatory drugs have been identified as a promising treatment modality in CF [5]. Macrophage migration inhibitory factor (MIF) is a pro-inflammatory cytokine, which has been shown to be a key driver in both acute and chronic inflammatory diseases [6]. There is now a critical mass of published work implicating MIF in promoting a more aggressive phenotype in respiratory diseases, including CF [7–10]. MIF possesses ketol-enol tautomerase enzymatic activity [11], which has been shown to be responsible for many of MIF's unique biological activities [12]. SCD-19 is a novel small-molecular-weight inhibitor which targets this tautomerase activity, and has been shown to have significant anti-inflammatory and anticancer activity in vivo [12]. In this study, it is our unifying hypothesis that the development of an aerosolized nanodrug delivery system loaded with SCD-19 would allow for the inhibition of P. aeruginosa induced inflammation in CF, while enabling maximal delivery to the lungs and minimizing systemic toxicity. In CF, a selection of treatments are administered via the inhaled/nebulized route [2]. Previously, it has been shown that 75% of nanotherapeutics administered via injection systemically dissipate; ultimately resulting in poor drug delivery to the lung for the treatment of respiratory diseases. This results in not only loss of treatment efficiency, but also enhances the risk for systemic drug side-effects. In contrast, more than 80% of the drug payload nanoparticles (NPs) can be delivered to the lung when using an inhaled delivery strategy [13]. Local pulmonary delivery has significant advantages over traditional systemic delivery systems. Primarily, local delivery allows higher drug concentrations to be delivered to the target organ promoting a rapid onset of action. There is less associated systemic toxicity and an improvement in patient adherence to treatment [14,15]. Furthermore, targeting the lung has organ-specific advantages such a significant surface area for absorption efficiency and the removal of first-pass metabolism. Aerosol-mediated nanodrug delivery to the lungs can combine the advantages of both inhalation therapy and nanodrug delivery methods, providing an effective organ-specific treatment for a variety of lung diseases [15].
Poly lactic-co-glycolic acid (PLGA) is a linear copolymer which has been approved by the US FDA for drug delivery, due to its high bio-safety and low immunogenicity [16,17]. Moreover, PLGA-based drug delivery systems are biodegradable, thus therapeutic agents can be released in a controlled manner by taking the advantages of biodegradation kinetics of the polymers, which can limit toxicity and prolong the circulation of therapeutic agents [18]. Therefore, we wished to develop and characterize aerosolized PLGA NPs, which were loaded with SCD-19. Furthermore, we examined the efficacy of nano-packaged SCD-19 as an inhibitor of P. aeruginosa induced macrophage inflammatory responses and biofilm formation on airway epithelial cells in the context of CF.
Materials & methods
Nanoparticle synthesis
NPs were synthesized using a single emulsion-solvent evaporation technique. To synthesize PLGA-MNP-SCD-19, Oleic acid-stabilized Fe3O4 NPs (magnetic NPs [MNPs]) 40 μl solution (25 mg/ml in CH2Cl2) was added to Acetone:dichloromethane (4 ml, 1:2 w/v) at room temperature (this step was skipped in the synthesis of PLGA-SCD-19 NPs). PLGA and SCD-19 were added with 1:1 PLGA (100 mg) and SCD-19 (10 mg) to the mixture. The mixture was then stirred and 2% (w/v) poly (vinyl alcohol) (PVA, 8 ml), was added slowly dropwise. The emulsion formed was homogenized using a probe sonicator (Sonics Vibra-Cell™ Ultrasonic) (70% amplitude, 30 sec) and poured into 50 ml 0.2% PVA. The resulting emulsion was stirred with an overhead stirrer (Lab egg, IKA® RW 11) for 16 h to allow the organic solvents to evaporate. NPs were then recovered by centrifugation (21,000 × g, 10 min, 4°C), washed three-times with endotoxin free Millipore water (50 ml), resuspended in saline (0.9% NaCl; 1 ml) and stored at 4°C until use. To synthesis blank NPs (PLGA NPs), the same protocol was employed without adding MNPs and SCD-19.
Limulus amebocyte lysate assay
Endotoxin levels were quantified before and after endotoxin removal using a Pierce limulus amebocyte lysate (LAL) chromogenic endotoxin quantitation assay kit (Thermo Scientific). Briefly, the standard solutions were prepared according to the manufacturer's specification. The NPs (50 μl) were mixed with LAL reagent (50 μl) in a prewarmed 96-well plate and incubated for 10 min at 37°C followed by the addition of 50 μl substrate solution provided by the kit. Finally, the stop reagent 50 μl (acetic acid, 25% (v/v) in Millipore water) was added into each well. Absorbance (OD) was measured at 405 nm using an UV-spectrometer (SpectraMax i3x). The developed color intensity is proportional to the amount of endotoxin present in the sample and the standard curve was employed to determine the endotoxin concentration.
Optimization of NPs for Aerogen® vibrating mesh nebulizer
A serial dilution of the PLGA-SCD-19 NPs (8, 16 and 40 mg/ml) was prepared either in saline and dH2O. A vibrating mesh nebulizer (Aerogen Solo, Aerogen Ltd) was used to nebulize the samples. To nebulize the NPs, one ml of each concentration was loaded into the nebulizer before the nebulizer was placed on the 50 ml tube and sealed with parafilm. Following nebulization, the tube was shaken vigorously for 30 seconds to detach NPs that may have attached to the walls of the tube. Finally, the nebulized samples were collected by centrifugation at 2000 × g, 5 min, 4°C. To assess the efficiency of nebulization of each concentration of the NPs, the time required to nebulize 1 ml of NPs and the concentration of the NPs collected following nebulization were examined. Sample concentration was measured by Varian Cary 50 UV-VIS in a quartz cuvette with 1 cm path length and 1 ml capacity. The size distribution of the NPs was investigated by a dynamic light scattering (DLS; Malvern Instruments Ltd).
Transmission electron microscopy
The NPs (20 μl) were diluted in 10 ml of Millipore water and 100 μl of this solution was deposited on a 100-mesh copper grid covered with amorphous carbon film (Electron Microscopy Sciences). Samples were incubated for 15 min under ventilation to allow for complete drying. Samples were then examined using a JEOL 2100 LaB Transmission Electron Microscope (JEOL Ltd) at 5 kV. The average particle size was calculated based on the measurements of 100 randomly chosen particles.
Scanning electron microscopy
The nanospheres were prepared by sprinkling 30 μl of NPs onto silicon wafers and allowing them to dry. This was then gently tapped on a piece of clean aluminum foil on a work bench to remove excess nanosphere before being loaded into a Sputter Coater machine (Cressington Scientific) where they were coated in platinum for 2 min at 13 mA and An acceleration voltage of 10 keV. Then, the samples were analyzed by a Zeiss NEON 40 EsB Cross Beam scanning electron microscopy.
NP tracking analysis
Measurement of NP hydrodynamic swelling size was carried out using the NP tracking analysis (NTA) technique using a NanoSight device equipped with a 50 mW 532 nm laser (Malvern Instruments Ltd). NTA software version 3.1 was used. NPs were dispersed in ultrapure, 20 nm filtered dH2O and loaded into the NanoSight machine. Following the selection of the appropriate threshold, an automated script was run, and the positioning and motion of individual NPs was tracked. The analysis of each sample was conducted in batch mode, enabling a mean hydrodynamic size could be calculated.
Dynamic light scattering
The particle size distribution and polydispersity index (PDI) of the NPs was investigated by dynamic light scattering (DLS) on a Zetasizer Nano ZS90 instrument (Malvern). To produce a suitable scattering intensity, the samples were diluted to a suitable concentration by diluting 1:500. A sample volume of 1 ml was used in 10 mm path length cuvettes (Sarstedt). The size distribution profile and PDI of the NPs were determined by using a 659 nm/100 mW laser at an angle of 90° in 10 mm diameter cells at 25°C. Measurements were taken in triplicate and the data are the average of all three runs.
Drug loading, drug encapsulation & yield efficiency
NPs were transferred into screw cap micro tubes and incubated for 1 min in Liquid Nitrogen to snap-freeze. All samples were then placed into the 4.5 Liter Freeze Dry System (Labconco) at 0.02 mbar for 72 h and maintained at a temperature of -80 ± 5°C. NPs (2 mg) were dissolved in 200 μl of 70% DMSO followed by 10 min vigorous vortex. The solution was measured with a validated spectrophotometric method (Spectra Max i3x) at 330 nm. The concentration of loaded SCD-19 was assessed based on the standard curve that was generated from different known SCD-19 concentrations. Each experiment was repeated three-times. Drug loading (% DL), yield efficiency (%YE), and encapsulation efficiency (%EE) were calculated using the following equations [19]. The same yield efficiency (%YE) was achieved for the blank NPs (PLGA NPs) (Supplementary Table 1).
Drug release
PLGA-SCD-19 NPs (3 ml) which contained 100 μM SCD-19 were injected into a Slide-A-Lyzer MINI dialysis cassette of 3.5K MWCO pore size (Thermo Fisher Scientific). The dialysis cassette was then immersed in a Millipore water bath (20 l) and kept under shaking (150 rpm). To assess the amount of SCD-19 remaining inside particles at predetermined intervals (0, 2, 4, 6, 24, 48 and 72 h), 180 μl was collected from inside the dialysis cassette. Samples were then adjusted to 500 μl by adding DMSO and vortexed vigorously for 5 min to dissolve the NPs. The concentration of SCD-19 in each collected sample was assessed by a Varian Cary 50 UV–Vis (Agilent Technologies) in a quartz cuvette with 1 cm path length and 0.5 ml capacity for the 330 nm peak. The samples were measured in triplicate. The SCD-19 release percentage was obtained according to;
Where Dt and D0 indicate the amount of SCD-19 released from the PLGA NPs suspension at certain intervals and the total amount of drug in the PLGA NPs suspension, respectively.
Stability of the NPs in synthetic lung fluids
To investigate the colloidal stability of NPs in lung artificial fluids, 0.5 mg/ml PLGA-SCD-19 NPs that contained 100 μM SCD-19 and 0.5 mg/ml PLGA NPs (control) were dispersed in saline, artificial lung fluid and artificial lysosomal fluid (Supplementary Table 2). The size and PDI of the NPs were investigated after 48 h using a Zetasizer Nano ZS90 instrument (Malvern Instruments Ltd).
Nebulizer efficiency assay
A serial dilution of PLGA-SCD-19 NPs (8, 16 & 40 mg/ml) was prepared in saline and dH2O. An autoclavable vibrating mesh nebulizer (Aerogen Solo, Aerogen, Galway, Ireland) was used to nebulize the samples. To avoid clogging the vibrating mesh, the samples were run from low (0.06 mg/ml) to high (40 mg/ml) concentration. Before loading each sample, 1 ml of saline was nebulized to remove the NPs left in the nebulizer. One millilitre of each concentration was loaded into the nebulizer then, the nebulizer was placed on the 50 ml tube and sealed with parafilm. The nebulized samples were collected via centrifuge at 2000 × g for 5 min at 4°C. To assess the efficiency of nebulization of each concentration of NPs, the time required to nebulize 1 ml of NPs was examined. The volume of NPs collected in the tube following nebulization was also measured. Furthermore, the concentration of NPs in the tube after nebulization was quantified. The size distribution of the NPs was investigated by a dynamic light scattering (DLS) (Malvern Instruments Ltd., Amesbury, UK), and compared with the sample before nebulization. The volume of delivered sample was also determined by Thermo fisher micropipette. Sample concentration was measured by Varian Cary 50 UV-VIS in a quartz cuvette with 1 cm path length and 1 ml capacity.
Laser diffraction
The Volume median diameter (VMD) and geometric standard deviation (GSD) of the aerosolized PLGA-SCD-19 (16 mg/ml) were determined by using a Malvern Spraytec particle size analyzer with RT Sizer software (version 5.60). The NPs were nebulized using a vibrating mesh nebulizer (Aerogen, Galway, Ireland). Nebulizers were connected to the inhalation cell and aspiration was carried past the laser by means of a vacuum pump set to 20 l/min, ensuring laminar flow and reducing artificial droplet size growth through collision with other droplets. The data acquisition rate was set to 500 Hz, that is, 500 individual readings per second were taken characterizing the droplet size distribution. All experiments were performed at room temperature and ambient relative humidity (40–60%).
Cascade impactor
To determine the relative amount of deposition of the NPs in the different regions of the lung and Mass Median Aerodynamic Diameter (MMAD), the PLGA-SCD-19 NPs were nebulized by a vibrating mesh nebulizer (Aerogen Solo, Aerogen, Galway, Ireland) in to a Next Generation Impactor (NGI) (Copley Scientific Ltd., Nottingham, UK) that was operated at 15 l/min at room temperature. The droplet size cut-off points were set at 14.1, 8.61, 5.39, 3.3, 2.08, 1.36, 0.98 and 0.70 μm, respectively, for each of the eight stages of the cascade impactor. After nebulization of PLGA-SCD-19 (1 ml) (16 mg/ml), each plate of the impactor and the throat were rinsed with 5 and 10 ml of PBS (Gibco™, Glasgow, UK), respectively. The amount of SCD-19 recovered from each stage filter was determined with the use of a spectrophotometer (Molecular Devices, California, US) by measuring the absorbance at 330 nm and interpolation on a standard curve of SCD-19 concentrations. The Fine Particle Fraction (FPF), FPF5 μm, equivalent to the respiratory fraction to target lung and the fine particle fraction, FPF2 μm, equivalent to alveolar fraction to target alveoli were calculated as the percentage of aerosol particles <5 μm and <2 μm, respectively (ISO 27427), relative to the total emitted dose by the Next Generation Impactor.
Human breathing simulator & human mechanical ventilator
To simulate a spontaneously breathing adult, a breathing simulator (ASL5000, Ingmar Medical, PA, USA) was used. The breathing simulator was programmed to simulate the recorded human breathing patterns with adult setting (inhalation: exhalation ratio 1:3, 15 breaths/min and tidal volume 500 ml). A vibrating mesh nebulizer (Aerogen, Galway, Ireland) was placed on the system. To nebulize the NPs and at the end of the system a bacterial/viral filter (Respirgard II 303, Baxter, Ireland) was also placed to collect the nebulized NPs.
To simulate a mechanically ventilated adult, a Maquet SERVO-i mechanical ventilator (Getinge, Sweden) was used. Breathing parameters were inhalation: exhalation ratio 1:1, 15 breaths/min and tidal volume 500 ml. Active humidification was supplied to the limb circuit by a hot pot humidifier (Fisher & Paykel, Auckland, New Zealand). The bacterial/viral filter (Respirgard II 303, Baxter, Ireland) was placed before the humidifier while the nebulizer was placed at the limb circuit.
For both breathing simulator and mechanical ventilator, a nominal dose of 1 ml (16 mg/ml) PLGA-SCD-19 NPs was nebulized in each test run. The amount of NPs captured in the filter was considered the lung dose. The filter was removed upon completion of the run time, eluted 10 ml of PBS (Gibco™, Glasgow, UK), and the washings were tested for SCD-19 with an UV spectrophotometry (Biochro, Cambridge, UK) and interpolation on a standard curve at 330 nm. All test iterations were run in triplicate.
Routine bacterial culture
Bacterial cultures were grown in LB broth at 37°C with shaking for 16 h. Lab strain P. aeruginosa PA01 was purchased from ATCC. Clinical isolates were provided by Dr. Julie Renwick (Department of Clinical Microbiology, Trinity College Dublin). In order to generate a standard curve of OD600 vs CFU/ml, serial dilutions of overnight cultures of PA01 were prepared, the OD600 measured and samples subsequently plated on LB agar plates. A standard curve was generated that yielded the following formula which was then used to calculate respective MOI of experiments Equation 1 P. aeruginosa CFU/ml equation:
| (Eq. 1) |
Epithelial cell culture
Cells were maintained in a humidified atmosphere containing 5% CO2 at 37°C. 16HBE14o- and CFBE41o- cells were kindly donated by Dr Catherine Greene (RCSI, Clinical Microbiology, Beaumount Hospital, Dublin, Ireland). 16HBE14o - cells are an SV40-transformed human bronchial epithelial cell line and CFBE41o- cells are human ΔF508 homozygote bronchial epithelial cells. Both cell lines were maintained in a 37°C, humidified CO2 incubator. 16HBE14o- and CFBE41o- cells were maintained in minimal essential medium (MEM) supplemented with 10% fetal bovine serum (FBS), 1% L-glutamine, 100 U/ml penicillin and streptomycin. A549 cells were obtained from ATCC®. A549 cells were maintained in DMEM supplemented with 10% FBS and 100 U/ml pen/strep. Cells were passaged every 2–3 days by removing medium and adding 5 ml of 0.05% Trypsin-EDTA for 5 min at 37°C. Trypsin was neutralized by adding medium before cells were collected and spun for 5 min in at 1200 RPM.
Biofilm formation on airway epithelial cells
Methods adapted from Anderson et al. [20]. Epithelial cells were plated at 2 × 105/ml in 500 μl of medium in a 24 well plate. Cells were cultured under normal conditions for 5 days to enable monolayer formation, with media changes every 2 days. On day 5, media was removed, and cells were washed twice with warm PBS. Cells were inoculated with PA01 at an MOI of 30:1 (PAO1:Cells) for 1 h using antibiotic free DMEM. Inoculum was removed and cells were washed with warm PBS. A total of 500 μl of antibiotic free DMEM was added to each well and cells were incubated for a further 3 h under normal conditions. Supernatants were removed and the cells were washed twice with warm PBS. Cells were lysed with 500 μl of 0.1% Triton X-100 (Thermo, #28314) for 10 min. Cell lysates were collected and serially diluted using PBS before plating on agar plates for CFU/ml enumeration. Where recombinant MIF (rMIF) or SCD-19 were added, cells were pretreated for 30 min and treatments were maintained for the duration of the experiment. For experiments involving PLGA-SCD-19 NPs, cells were pre-treated for 24 h.
Macrophage cell culture
Bone marrow cells were collected from the hind legs of mice and differentiated for 7 days in DMEM supplemented with 10% heat-inactivated fetal bovine serum, 100 U/ml penicillin/streptomycin and 20% L929 conditioned medium. Differentiated cells were plated for treatment the following day. Cells were stimulated with the respective stimulants and treatments for the indicated times. RAW264.7 cells were maintained in DMEM supplemented with 10% heat-inactivated fetal bovine serum, 100 U/ml penicillin/streptomycin. Differentiated cells were plated for treatment the following day. Cells were stimulated with the respective stimulants and treatments for the indicated times.
RAW 264.7 cell stimulations
RAW 264.7 cells were stimulated with Heat Killed Pseudomonas aeruginosa (HKPA) or planktonic P. aeruginosa filtrate (PPF) for 24 h. Treatment with SCD-19 was given at the same time as HKPA or PPF, respectively, for 24 h. For experiments involving nano packaged SCD-19, cells were pre-treated for 24 h.
Analysis of P. aeruginosa killing by macrophages
Cells were seeded at a density of 1 × 105 cells per well in a 96 well plate before infection the next day. Overnight cultures of PA01 were centrifuged at 4500 RPM for 5 min at room temperature to collect the bacterial pellet. The pellet was resuspended in antibiotic free DMEM and the OD600 measured, using medium only as a blank. The number of CFU/ml was calculated using the following equation; CFU/ml = (2 × 109)(x) - (5 × 107), where x = OD600. Macrophages were infected at a multiplicity if infection (MOI) of 10 CFU: 1 macrophage for 4 h. In experiments involving 4-OI, this was added at the time of inoculation. Following this, supernatant was removed and placed into a fresh 96 well plate. Macrophages were then lysed using 20 μl of 0.1% Triton X for 15 min. The supernatant was then added back to the corresponding well and mixed with the cell lysate. This was then serially diluted and subsequently plated on LB agar plates for CFU/ml analysis.
Enzyme-linked immunosorbent assay
Supernatants were collected and centrifuged at 3000 × g to remove dead cells and cell debris and stored at -80°C. Supernatants were diluted when necessary to be in the optimal optical range and analyzed by enzyme-linked immunosorbent assay (ELISA) using the DuoSet ELISA Development Systems© according to manufacturer's instructions. Optical densities were measured using a Spectramax i3X microplate spectrophotometer at 490 nm. Final cytokine concentrations were calculated using a standard curve.
Cellular uptake of NPs
Perls' Prussian blue stain, which reveals the presence of iron, was used on the A549 cells to visualize the cellular uptake of PLGA-MNP-SCD-19 NPs. A549 cells were seeded at a density of 2 × 104 cells/well in the 8-well chambered cell culture slides and after overnight incubation the cells were exposed to different concentrations of PLGA-MNP-SCD-19 NPs. Following this, cells were washed three times with pre-warmed PBS and fixed using 4% paraformaldehyde (Sigma-Aldrich). The chamber was removed and slides were allowed to air dry. Equal parts of 4% hydrochloric acid and 4% potassium ferrocyanide were prepared (potassium ferrocyanide staining solution) immediately prior to use and slides were immersed in this solution for 20 min. The slides were then dipped in dH2O three times and counterstained with 0.5% Neutral Red (Sigma-Aldrich) for 2 min to stain the nucleus of the cells. Slides were rinsed a further two times with dH2O and allowed to air dry. Finally, the slides were cover slipped with D.P.X (Sigma-Aldrich). Imaging was completed 24 h post mounting using the Nikon E800W microscope with 20× and 40× objectives. The images were acquired using the micropublisher 3.3RTV color camera (Photometrics).
Confocal microscopy
The uptake of PLGA-MNP-SCD-19 NPs and control MNPs were visualized using confocal microscopy. A549 cells were seeded at a density 2 × 104 cells/well in a the 8-well chambered cell culture slide (FalconTM, MA, USA), treated with MNPs (100 μg/ml), PLGA-MNP-SCD-19 NPs (100 μg/ml based on MNPs concentration) for 24 h. Cells were thoroughly washed twice in PBS (Gibco™, Glasgow, UK) and fixed using 4% Paraformaldehyde (Sigma-Aldrich) for 10 min, followed by further washing with PBS. Cells were then stained using fluorescent dyes. Briefly, cells were stained with 100 μl of 10 μg/ml Hoechst 33342 (Invitrogen, CA, USA) and Rhodamine phalloidin (Invitrogen, California, USA) for 2 h at room temperature to stain nuclei and F-actin filaments respectively. Cells were then washed three times with PBS before a coverslip was mounted with a transparent mounting medium (VECTASHIELD, Vector Laboratories Inc., USA) and sealed. Cellular internalization of PLGA-MNP-SCD-19 NPs and MNPs was evaluated using Laser Scanning Confocal Microscopy (LSCM). LSCM imaging and analysis was carried out with equipped with a Zeiss LSM 5 software (Carl Zeiss Microscopy GmbH, Jena, Germany). MNPs were imaged in reflectance mode at λexc = 561 nm.
Colorimetric ferrozine-based assay
The colorimetric ferrozine-based assay allows the quantification of iron in cultured cells. A549 cells seeded in 24-well plates were incubated with different concentrations of MNP-PLGA-SCD-19 NPs (50, 100 and 200 μg/ml based on MNPs concentration) and intracellular accumulation of the NPs were quantified after 24 h and 48 h of incubation respectively. Briefly, the cells were lysed with 0.5 ml of 50 mM NaOH (Sigma-Aldrich, UK) per well and incubated for 2 h at room temperature on a shaker. The lysed cells were then mixed with 500 μl of 10 mM HCl, and 500 μl of fresh iron-releasing reagent (a freshly mixed solution of 1:1 volumes of 1.4 M HCl and 4.5% (w/v) KMnO4 in dH2O). The mixture were incubated for 2 h at 60°C within a fume hood and then incubated for 1 h at room temperature. Finally, 150 μl of iron-detection reagent were added to each tube. After 30 min incubation in room temperature, the absorbance was measured at 570 nm (SpectraMax i3x, Molecular Devices, CA, USA). The iron content was calculated by comparing the absorbance of the sample a standard curve.
Statistical analysis
Statistical analyses were carried out using GraphPad Instat Software (GraphPad Software Inc. CA, USA). For datasets with only two experimental groups with equal standard deviations, unpaired T-tests were used to compare datasets. Statistical significance was recorded at p < 0.05. One-way analysis of variance (ANOVA) was used to test for statistical significance between experimental groups of three or more where there is only one independent variable. Two-way ANOVA was used was used to test for statistical significance (two tailed analysis) between experimental groups of three or more where there are two independent variables. Multiple comparisons between groups were then assessed using the Tukey–Kramer post-hoc test (for parametric analysis) or Dunn's post-hoc test (for non-parametric analysis). Statistical significance was recorded at p < 0.05. N = 3, representative of three independent experiments.
Results
Scanning electron microscopy, transmission electron microscopy & NTA analysis
The morphology of both PLGA-SCD-19 and PLGA-MNP-SCD-19 NPs was examined by Scanning electron microscopy (SEM). In these experiments, iron oxide NPs - magnetic NPs (MNPs) - was as a contrast agent. SEM images revealed that both PLGA-SCD-19 and PLGA-MNP-SCD-19 NPs possessed spherical shape and smooth morphology without any aggregation (Figure 1A & B). TEM was also employed to determine the size range and sample uniformity of the NPs (Figure 1C & D). The average size estimated for both PLGA-SCD-19 and PLGA-MNP-SCD-19 from the transmission electron microscopy (TEM) images was 180.0 ± 25.0 nm. NTA analysis confirmed that the NPs had a uniform size distribution (Figure 1E & F). The average hydrodynamic size of the samples as measured by NTA were within the nanometric range. Both PLGA-SCD-19 and PLGA-MNP-SCD-19 NPs in aqueous solution remained monodispersed without any aggregation with an associated modest increase in size of 200.0 ± 20.0 nm compared with 225 ± 25 nm, respectively.
Figure 1. . Electron microscopy images, size distribution, stability and optimization of the nanopaticles for Aerogen vibrating mesh nebulizer.
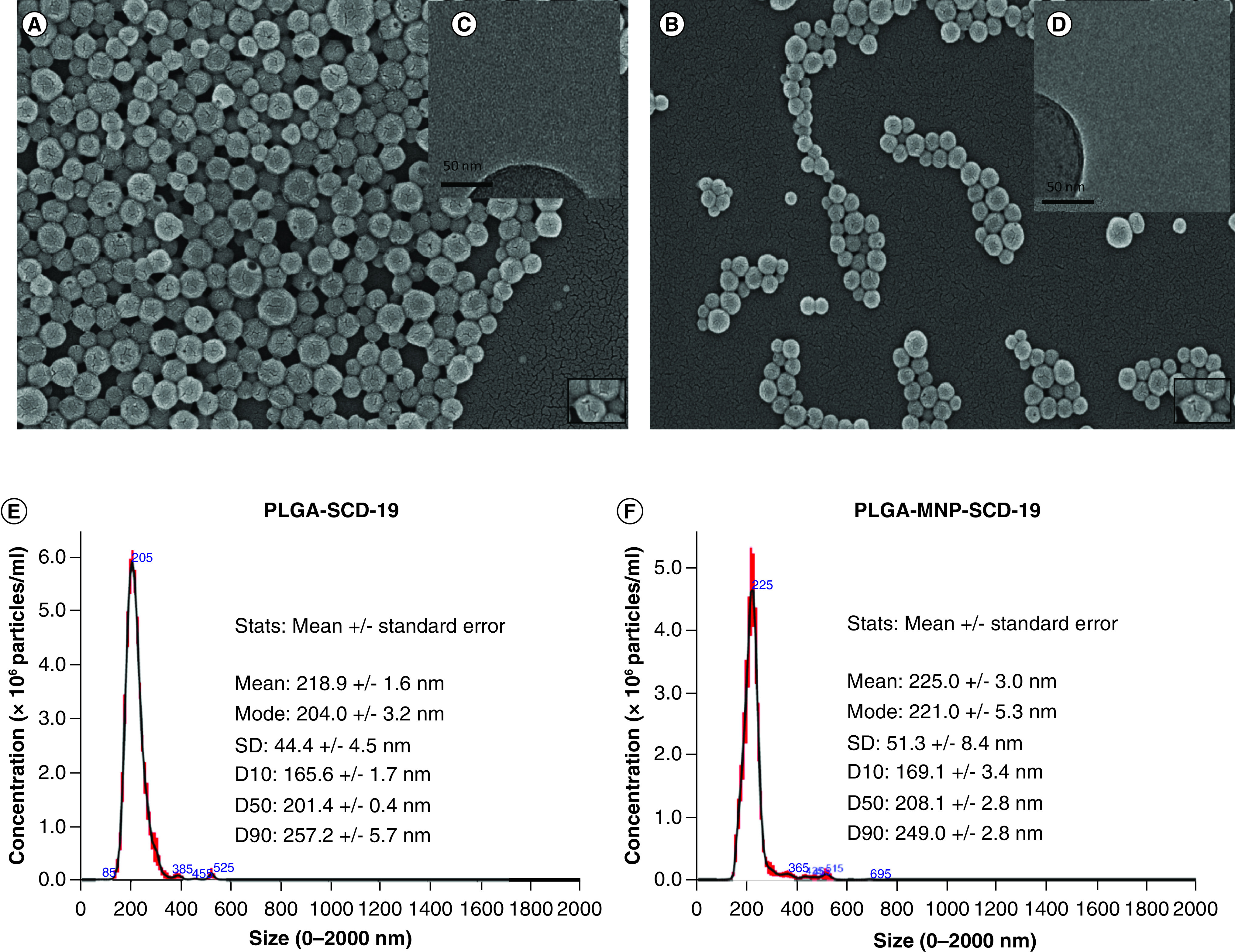
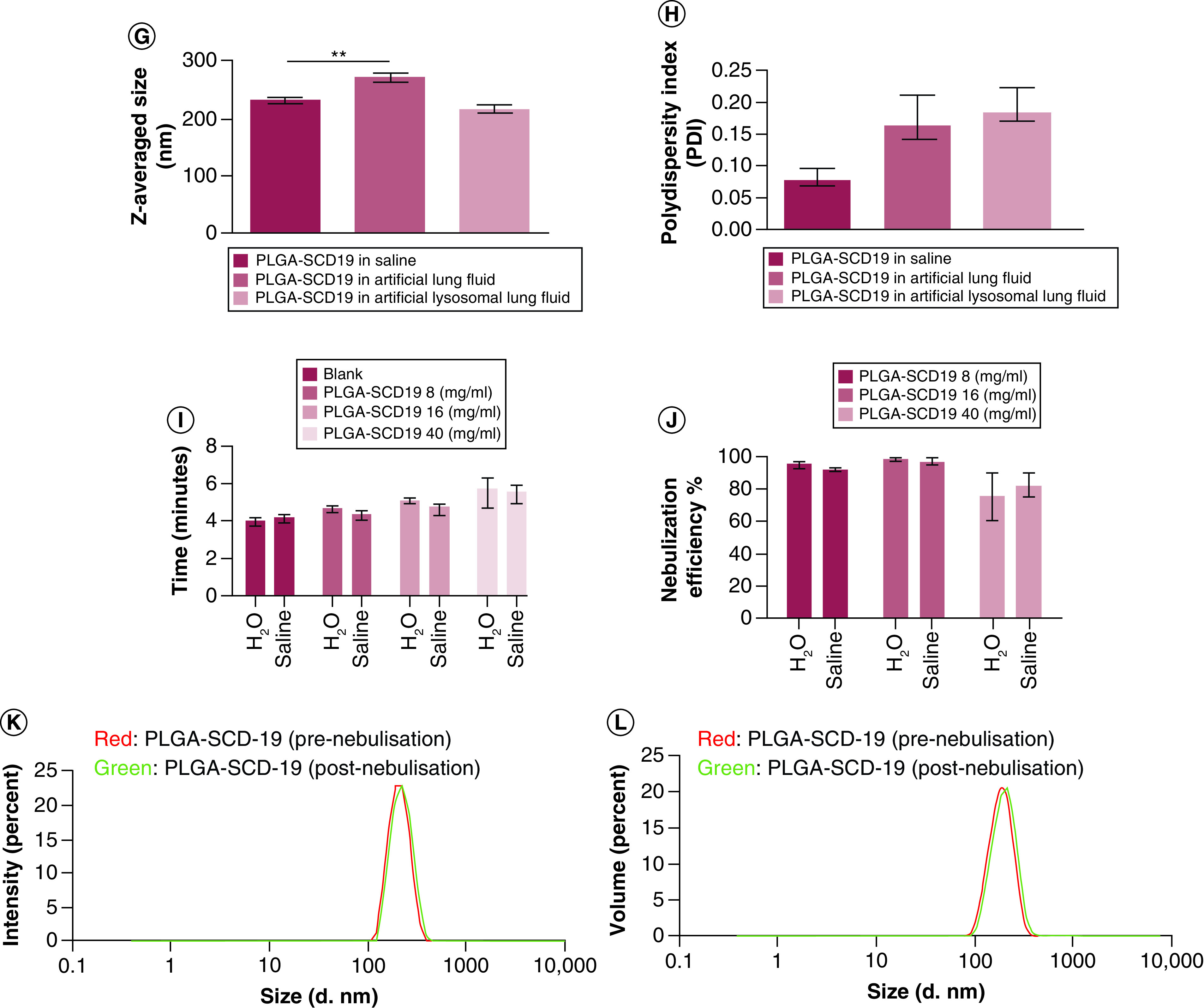
(A) Scanning electron microscopy (SEM) image and (B) TEM image of PLGA-SCD-19 NPs. (C) SEM image and (D) TEM image of PLGA-MNP-SCD-19 NPs in which the encapsulated MNPs can be seen in PLGA NPs (higher dark contrast). Scale bars correspond to 200 nm for SEM images and 50 nm for (TEM image. (E) Size distribution of PLGA SCD-19 NPs (F) Size distribution of PLGA-MNP-SCD-19 NPs. (G & H) The stability of PLGA-SCD-19 NPs in artificial lung fluids and saline (control; 0.9% (v/v). To analyze the size distribution and PDI, a one-way analysis of variance with Dunnett (compare all vs control) test and nonparametric one-way analysis of variance were used, respectively, to test for statistical significance. (I) The nebulization time for sample (1 ml) delivery was measured. (J) The nebulizer efficiency to deliver NPs was analyzed after comparing the concentration of SCD-19 before and after nebulization in the samples by a UV-Vis machine. Blank = saline or dH2O as the vehicle controls. (K) The size of the NPs was measured before and after nebulization using dynamic light scattering technique. (L) The impact of nebulization on the volume of PLGA-SCD-19 NPs (before and after nebulization). Data are represented as the mean ± SEM (n = 3).
MNP: Magnetic nanoparticle; NP: nanoparticle; PLGA: Poly lactic-co-glycolic acid.
Drug loading efficiency, drug content & yield efficiency
The concentration of SCD-19 that was successfully encapsulated into PLGA NPs was 3.7 ± 0.7 mg or 15.0 ± 0.1 mM per batch, indicating that the loading efficiency was 63.0 ± 13.0%. In addition, the optimized formulation presented satisfactory yield efficiency (66.0 ± 4.0%), which is the weight of the raw materials (PLGA and SCD-19) used in the preparation and the final weight of the produced PLGA-SCD-19 NPs. (Table S1)).
Stability of PLGA-SCD-19 NPs in artificial lung fluids
The size and PDI of the NPs were determined by using a DLS technique. The NPs were significantly larger when incubated in artificial lung fluids (Supplementary Table 2), compared with those incubated in saline (p < 0.01). However, no significant change in average NP size was detected for those incubated with the artificial lysosomal fluid. The average size of PLGA-SCD-19 NPs after incubation for 48 h in saline, artificial lung fluid and artificial lysosomal fluid was 234 ± 10.4 nm, 271 ± 22.8 nm and 215 ± 21.9 nm, respectively (Figure 1G and Supplementary Table 3).
The PDI value of PLGA-SCD-19 NPs incubated with artificial lung fluid and artificial lysosomal fluid, respectively, was increased compared to those incubated with saline, although this was not significant (p = 0.15). The average PDI value as determined by DLS for the NPs incubated with saline, artificial lung fluid and artificial lysosomal fluid was 0.07 ± 0.05, 0.18 ± 0.1 and 0.16 ± 0.14, respectively (Figure 1H and Supplementary Table 3).
Optimization of NPs for the Aerogen vibrating mesh nebulizer
The time required to nebulize 1 ml of two different nanoformulation concentrations (8 mg/ml and 16 mg/ml) was comparable to that observed for blank samples (dH2O and 0.9% (w/v) saline). However, a clogging issue on the vibrating mesh from the high concentration sample (40 mg/ml) was observed after each run and extra washing procedures were required to clean the vibrating mesh. Due to this clogging issue, the time required to nebulize 40 mg/ml was notably higher than controls (Figure 1I). The concentration of PLGA-SCD-19 NPs (mass of SCD-19) before and after nebulization was examined to assess the nebulizer drug delivery efficiency. Maximum delivery efficiency of almost 100% was achieved for the 8 mg/ml and 14 mg/ml samples (Figure 1J). The samples with the highest concentration (40 mg/ml), dispersed in either dH2O or 0.9% (w/v) saline, resulted in approximately 80% delivery efficiency.
To examine the impact of the Aerogen nebulizer on the size and volume of PLGA-SCD-19 NPs following nebulization, nebulized NPs were collected and the particle size and volume of the NPs examined by DLS. The particle size and volume of the NPs remained similar to the initial nanoformulation before nebulization and the Aerogen vibrating mesh nebulizer had no detectable impact on the NPs (Figure 1K & L).
Surface charge & drug release profile
The zeta potential from PLGA-SCD-19 NPs as determined by using dynamic light scattering (DLS) was −26.0 ± 5.4 mV. In vitro, PLGA-SCD-19 NPs released approximately 30% of their contents within 24 h, followed by a plateau release for two days (Supplementary Figure 1).
Cytotoxicity & biocompatibility
Lactate dehydrogenase assays were performed on different cell lines to quantify cytotoxicity. No significant cytotoxicity was observed at concentrations of 1 mM or less. At 2 mM concentrations after 48 h incubation, cytotoxicity of 18% was observed in A549 cells. For other cell types, the assessed cytotoxicity level at this timepoint was <10% (See Supplementary Figures).
High content screening (HCS) was employed to determine the changes in cellular morphology. A549 cellular morphology and nuclei area remained unchanged following treatment both PLGA-SCD-19 and PLGA-MNP-SCD-19 NPs respectively (Supplementary Figure 2B & D). Furthermore, cells that were treated with toxic CdSe quantum dots as a positive control demonstrated significantly decreased cell and nuclei area (Supplementary Figure 2C & D).
Internalization of NPs in alveolar epithelial cells
Internalization of NPs by A549 cells was examined using laser scanning confocal microscopy. Significant uptake of the NPs was observed after 24 h. There were no notable differences between uncoated MNPs (control) and PLGA-MNP-SCD-19 in terms of cellular uptake and internalization. After cellular uptake, no detectable cellular deformation or cell necrosis was observed (Figure 2).
Figure 2. . Internalization of poly lactic-co-glycolic acid-magnetic nanoparticles-SCD-19 nanoparticles by A549 cells.

The confocal images were acquired after cell exposure topoly lactic-co-glycolic acid-magnetic nanoparticles-SCD-19 and uncoated magnetic nanoparticles for 24 h. The cells were stained with Hoechst 33342 and Rhodamine phalloidin to stain nuclei (blue) and actin filaments (red), respectively. (A) The A549 cells exposed to PLGA-MNP-SCD-19 (100 μg/ml). (B) The positive control was only treated by uncoated magnetic nanoparticles (100 μg/ml). Scale bar 10 μm.
MNP: Magnetic nanoparticle; NP: nanoparticle; PLGA: Poly lactic-co-glycolic acid.
To assess cellular uptake, A549 cells were incubated with three different concentrations of PLGA-MNP-SCD-19 NPs (50, 100, 200 μg/ml) at 24 h and 48 h time-points. (Figure 3 A). The NPs accumulated only in the cytoplasm close to nuclei and did not penetrate into the nuclei after 48 h of incubation (Figure 3 B). To determine the intracellular NPs content, the ferrozine colorimetric assay technique was performed. The internalization of the NPs demonstrated a linear correlation with the NP concentration. Saturation of cellular uptake was observed at 24 h (Figure 3 C).
Figure 3. . Cellular uptake of poly lactic-co-glycolic acid-magnetic nanoparticles-SCD-19 by A549 cells.
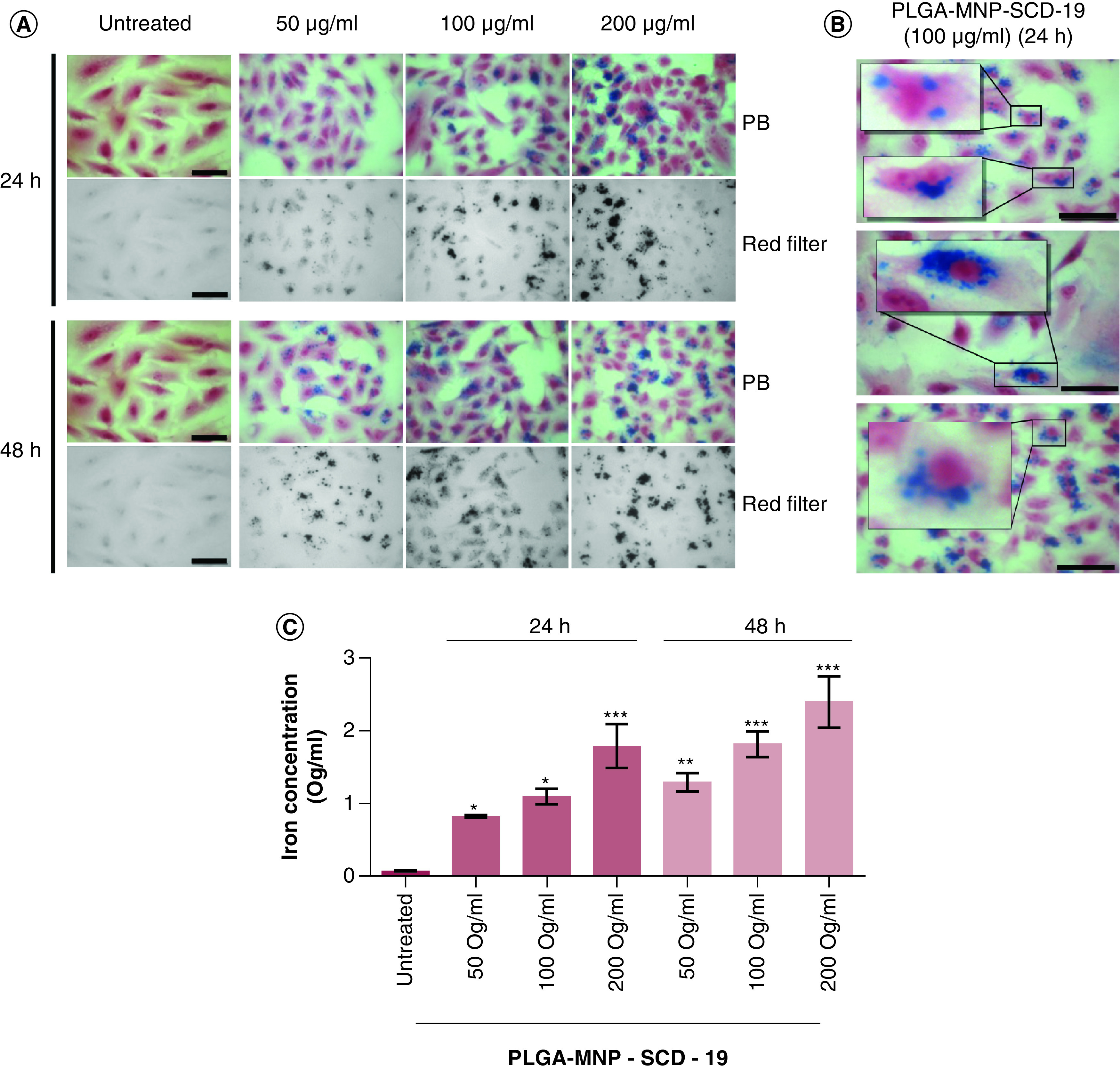
(A) Cells incubated with three different concentrations of the poly lactic-co-glycolic acid-magnetic nanoparticles-SCD-19 nanoparticles (NPs) (50, 100, 200 μg/ml) at two time points (24 and 48 h). The MNPs were stained with Perls Prussian blue and Neutral Red (0.5% [v/v]) to stain nuclei and cytoplasm, respectively (B) Representative micrographs with selected area of interest (magnification 40×) showing the uptake and accumulation pattern of the NPs inside the cytosol and around the nucleus of A549 cell at 100 ug/ml. Microscopy image of the NPs. Scale bar 10 μm. (C) Intracellular iron content quantification by ferrozine assay. A one-way analysis of variance (ANOVA) with Dunnett (compare all vs control [untreated]) was used to test for statistical significance. The results are presented as mean ± SEM and is expressed in μg/ml.
MNP: Magnetic nanoparticle; NP: nanoparticle; PLGA: Poly lactic-co-glycolic acid.
Regional aerosol deposition of the NPs
The VMD of NPs was 4.4 ± 0.33 μm, as characterized by laser diffraction. This methodology characterizes the diameter of the aerosol droplets, and the mass median aerodynamic diameter (MMAD) of the nebulized PLGA-SCD-19 NPs as determined by a Next Generation Impactor (NGI) was 2.2 ± 0.4 μm (Supplementary Table 4). Over the course of the experiment, the aerosolized NPs are separated into different size fractions, on the basis of their aerodynamic mass. Quantification of deposited NPs on each size fraction collection plates showed that most of NPs have deposited in the stages of 3, 4, 5, 6 and 7, indicating that the vast majority of the aerosolized PLGA-SCD-19 NPs had a droplet size range between approximately 1 to 5 μm. The highest deposition rate was detected in the stages of 4 and 5, which had almost half of the deposited nanoformulation. Also, approximately 4% of the NPs deposited in throat region and <2% of the nanoformulation had a droplet size greater than 14 μm (Figure 4) (Figure S3)). A difference in the recorded aerosol characteristics for these two regulator mandated methods is to be expected, considering the fundamentally different approaches to how droplet sizes are characterized.
Figure 4. . Quantification of the deposited poly lactic-co-glycolic acid-SCD-19 nanoparticles.
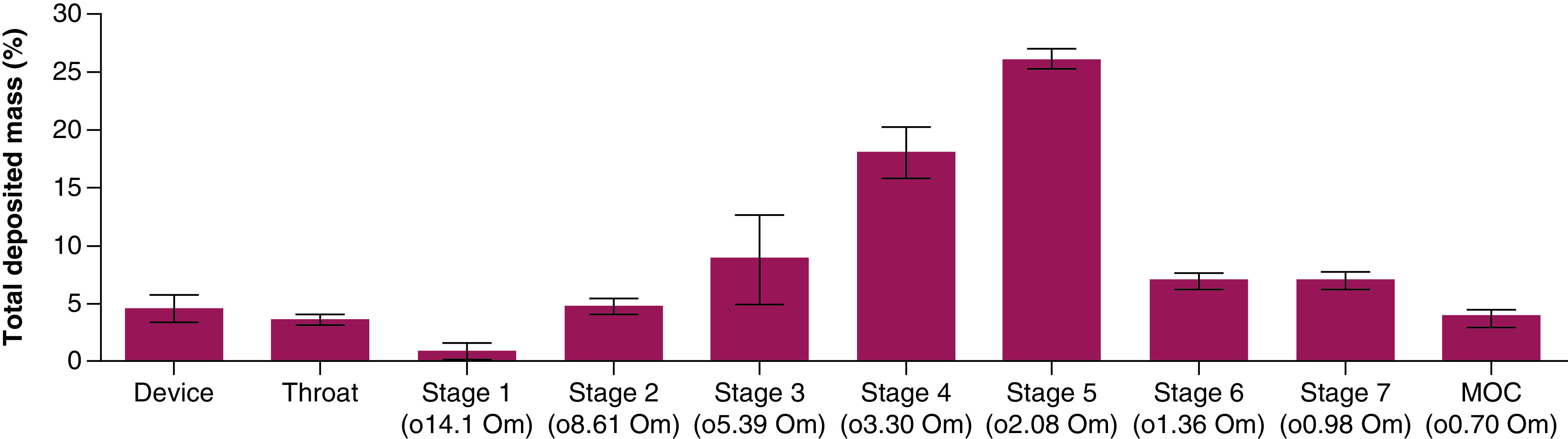
Mass distribution of nebulized nanoparticles using a Next Generation Impactor, Model 170. Data were expressed as the percentage of total drug deposited on all stages of the impactor including the throat and was represented by the mean ± standard deviation. The data are represented as mass percentage of nominal dose (n = 3).
Drug delivery efficiency
To assess the in vitro drug delivery efficiency PLGA-SCD-19 NPs were aerosolized during simulated patient interventions in which aerosols are often administered; mechanical ventilation and spontaneous breathing. The delivery time for all the samples was in the standard range (i.e., less than 4.5 min). During mechanical ventilation, the recorded lung dose was 23% (standard deviation 0.88%) of the nominal dose placed in the nebulizer. During simulated spontaneous breathing, the recorded lung dose was 14.1% (standard deviation again recorded at 0.88%) (Supplementary Table 5).
SCD-19 & PLGA SCD-19 NPs modulate HKPA planktonic PPF induced TNF-α & RANTES protein production
The inflammatory response of RAW 264.7 macrophages to HKPA stimulation, a TLR2 and TLR5 agonist, was investigated (Figure 5). A significant induction of TNF-α and RANTES protein production was observed at 24 h following HKPA stimulation (Figure 5 A–D). Our previous work has described the inhibition of LPS driven cytokine production and inflammation by SCD-19 and the inhibition of P. aeruginosa mediated lung inflammation in vivo [12]. Here, we investigated the effect of SCD-19 on HKPA induced TNF-α and RANTES protein production by RAW 264.7 cells. SCD-19 significantly inhibited HKPA induced TNF-α and RANTES protein production respectively following 24 h treatment (Figure 5 A–B). These data demonstrate for the first time the regulation of the immune response to HKPA by SCD-19.
Figure 5. . Effects of SCD-19 and poly lactic-co-glycolic acid SCD-19 nanoparticles on heat killed Pseudomonas aeruginosa and P. aeruginosa filtrate induced TNF-α and RANTES protein production.
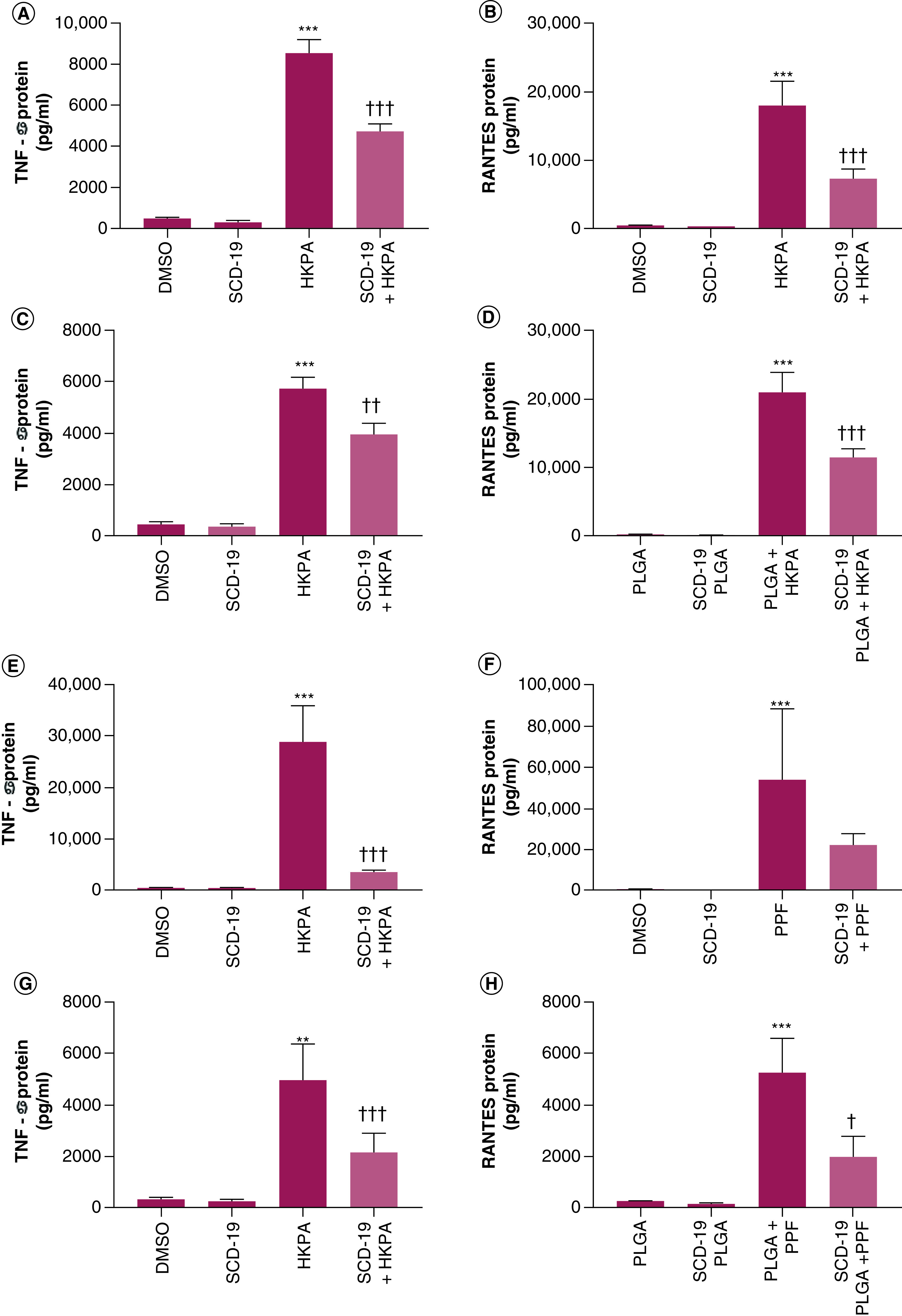
SCD-19 (100 μM) inhibits Heat Killed Pseudomonas aeruginosa (HKPA) mediated (A) TNF-α and (B) RANTES protein production. Nanopackaged SCD-19 inhibits HKPA mediated (C) TNF-α and (D) RANTES protein production. SCD-19 inhibits P. aeruginosa filtrate (PPF) mediated (E) TNF-α but not (F) RANTES protein production. poly lactic-co-glycolic acid-SCD-19 (PLGA-SCD-19) nanoparticles (100 μM) inhibits PPF mediated (G) TNF-α and (H) RANTES protein production. Protein expression was measured by ELISA. Data are presented as mean ± SEM of 3 independent experiments with 3 technical replicates. A One-way ANOVA with a Tukey's multiple comparison post-hoc test was used to test for statistical differences. (A & B) ***p < 0.001; DMSO compared with HKPA. ††† p < 0.001; HKPA compared with SCD-19 + HKPA. (C & D) ***p < 0.001; PLGA compared with PLGA + HKPA. †††p < 0.001; PLGA + HKPA compared with PLGA SCD-19 + HKPA. (E, F) ***p < 0.001; DMSO compared with PPF. †††p < 0.001; PPF compared with SCD-19 + PPF. (G & H) **p < 0.05; ***p < 0.001; PLGA compared with PLGA + PPF. †p < 0.01, †††p < 0.001; PLGA + PPF compared with PLGA-SCD-19 nanoparticles + PPF.
In this article, the formulation and characterization of PLGA-SCD-19 NPs has been detailed (Figures 1–4). Here, the ability of PLGA-SCD-19 NPs to inhibit the induction of TNF-α and RANTES protein production by RAW 264.7 cells in response to HKPA stimulation was investigated. The results demonstrated that PLGA-SCD-19 NPs significantly inhibited TNF-α and RANTES protein production respectively following 24 h of pretreatment with PLGA SCD-19 NPs and subsequent 24 h treatment with HPKA and PLGA SCD-19 NPs (Figure 5C–D). These data demonstrate for the first time, the modulation of the inflammatory response to HKPA using SCD-19 loaded NPs targeting MIF.
Planktonic P. aeruginosa filtrate (PPF) contains a variety of diffusible products including LPS, quorum sensing molecules and bacterial DNA [21]. PPF induced significant TNF-α and RANTES protein production in RAW 264.7 cells following 24 h of stimulation (Figure 5E & F). Furthermore, SCD-19 significantly inhibited PPF induced TNF-α protein production, but not RANTES protein production respectively following 24-h treatment. (Figure 5E & F) These data demonstrate for the first time the regulation of the immune response to PPF by SCD-19. Subsequently, the ability of PLGA-SCD-19 NPs to inhibit TNF-α and RANTES protein production in RAW 264.7 cells was examined. PLGA-SCD-19 NPs significantly inhibited TNF-α (Figure 5 G) and RANTES (Figure 5 H) protein production following 24 h of pretreatment with PLGA-SCD-19 NPs and subsequent 24 h treatment with PPF and PLGA-SCD-19 NPs. These results demonstrate for the first time, the modulation of TNF-α and RANTES protein production in response to secreted P. aeruginosa products using SCD-19 loaded NPs targeting MIF.
MIF inhibits macrophage mediated killing of P. aeruginosa
We have previously reported a correlation between circulating MIF levels in CF patients and P. aeruginosa infection [10,22]. Furthermore, MIF has been successfully targeted using various inhibitors in vivo in models of P. aeruginosa induced sepsis, ocular keratitis and chronic lung infection resulting in improved outcomes compared with untreated mice [9,23,24]. Here, the effects of MIF on macrophage mediated killing of P. aeruginosa were examined using bone marrow derived macrophages (BMDMs) (Figure 6). Wild type BMDMs, which express MIF, exhibited significantly impaired killing of P. aeruginosa compared with MIF deficient macrophages (Figure 6A). Furthermore, enhanced macrophage mediated killing of P. aeruginosa was observed in SCD-19 treated macrophages compared with those treated with DMSO (Figure 6B). These data show for the first time that expression of MIF impairs the bactericidal capacity of macrophages against P. aeruginosa. Moreover, we also show for the first time that SCD-19 enhances macrophage mediated killing of P. aeruginosa.
Figure 6. . MIF inhibits macrophage mediated killing of P. aeruginosa.

(A) Wild-type bone marrow derived macrophages (BMDMs) exhibit decreases bactericidal capacity against P. aeruginosa infection compared with BMDMs derived from MIF-/- mice. (B) Treatment of wild type BMDMs with SCD-19 (100 μM) enhances P. aeruginosa killing. Data are presented as mean ± SEM of three independent experiments with three technical replicates. A two-tailed unpaired t-test was used to test for statistical significance of differences. *p < 0.05; **p < 0.01.
MIF is a valid target for the inhibition of P. aeruginosa attachment to human airway epithelial cells
Previously, we have demonstrated that MIF enhances P. aeruginosa biofilm formation on abiotic surfaces, and that SCD-19 represents an effective therapeutic in vivo [9]. Here, we examine the effect of MIF on PA01 attachment to airway epithelial cells, and the effects of SCD-19 on this (Figure 7). rMIF significantly enhanced the attachment of PA01 to A549 airway epithelial cell monolayers (Figure 7A). Furthermore, SCD-19 significantly inhibited PA01 attachment to A549 cell monolayers (Figure 7B). Subsequently, the effect of PLGA-SCD-19 NPs on PA01 attachment to A549 monolayers was investigated. PLGA-SCD-19 NPs significantly inhibited PA01 attachment to A549 monolayers compared with blank NPs (PLGA NPs) (Figure 7C). Next, the effects of PLGA-SCD-19 NPs on PA01 attachment to both healthy and CF airway epithelial cell monolayers were examined. PLGA-SCD-19 NPs significantly inhibited attachment of PA01 to both healthy (Figure 7D) and CF (Figure 7E) airway cell monolayers respectively after 24 h of pre-treatment and subsequent 4 h of infection. These data illustrate for the first time that MIF enhances early biofilm formation on human airway epithelial cells and also that SCD-19 inhibits the attachment of P. aeruginosa to host epithelial cells.
Figure 7. . MIF is a valid target for the inhibition of PA01 attachment to human airway epithelial cells.
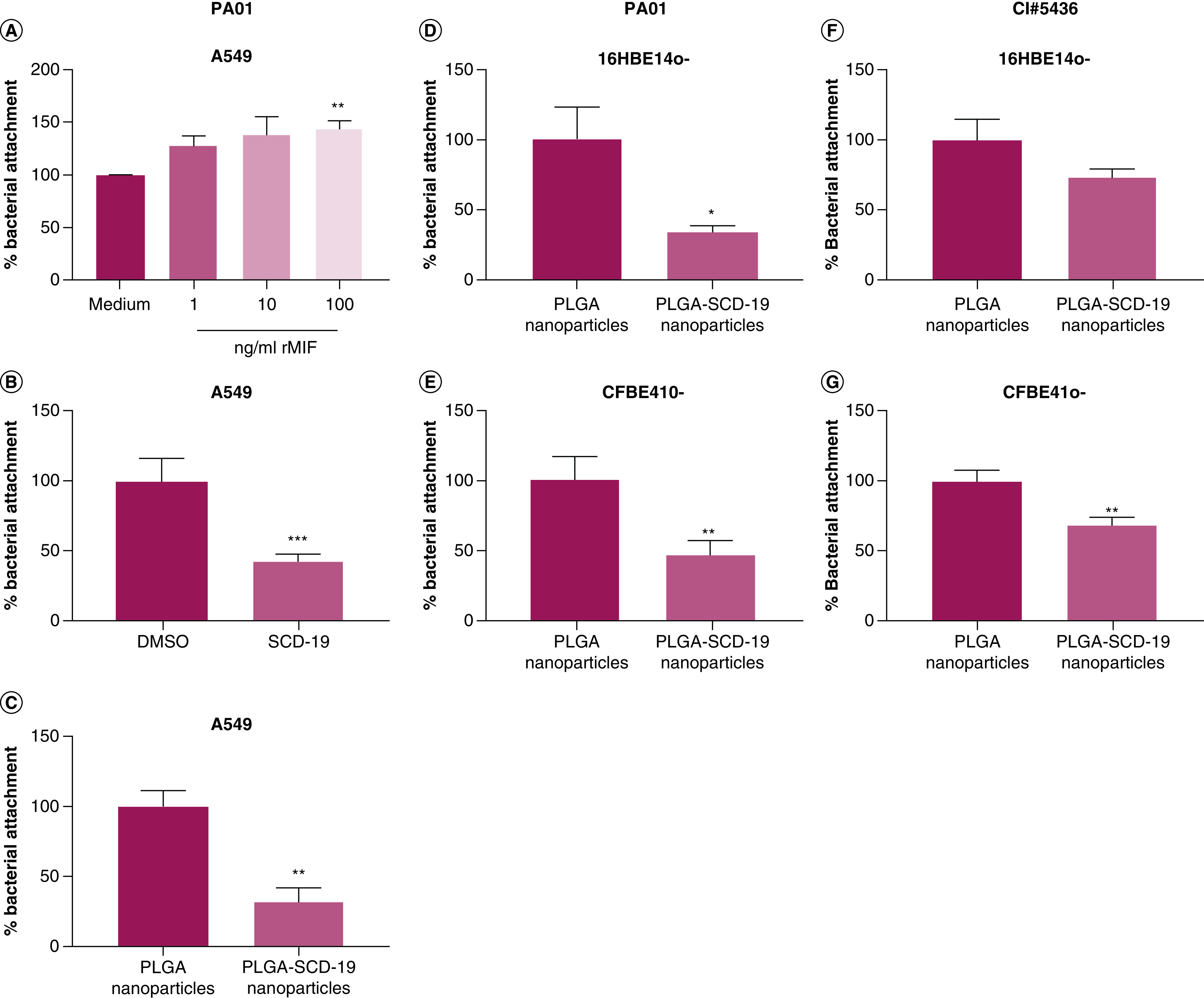
(A) Recombinant migration inhibitory factor enhances attachment of PA01 to A549 epithelial cells. (B) SCD-19 (100 μM) significantly decreases attachment of PA01 to A549 epithelial cells. (C) SCD-19 PLGA nanoparticles (NPs) (100 μM) inhibits attachment of PA01 to A549 epithelial cells. (D) PLGA-SCD-19 NPs (100 μM) inhibits attachment of PA01 to 16HBE14o- epithelial cells. (E) PLGA SCD-19 NPs (100 μM) inhibits attachment of PA01 to CFBE410- epithelial cells. PLGA-SCD-19 NPs (100 μM) inhibit attachment of clinically isolated P. aeruginosa to (F) 16HBE14o- and (G) CFBE410- epithelial cell monolayers. Data are presented as mean ± SEM of three replicates from three independent experiments. (A) A One-way ANOVA with a Tukey's multiple comparison post-hoc test was used to test for statistical differences; **p < 0.01 medium compared with 100 ng/ml migration inhibitory factor. (B–F) A two-tailed unpaired t test was used to test for statistical significance; *p < 0.05; **p < 0.01; ***p < 0.001.
PLGA: Poly lactic-co-glycolic acid.
As our data demonstrated the inhibition of PA01 attachment to airway epithelial cell monolayers using PLGA-SCD-19 NPs (Figure 7), we next examined their efficacy with a strain of P. aeruginosa (CI#5436), which was isolated from a CF patient. The growth rate of CI#5436 was similar to PA01, however CI#5436 was more resistant to treatment of tobramycin, meropenem and piperacillin/tazobactam than PA01 (See Supplementary Figures). Treatment of 16HBE14o- cell monolayers with PLGA-SCD-19 NPs inhibited CI#5436 attachment following 4 h of culture compared with cells treated with blank NPs (PLGA NPs) (vehicle), although this was not significant (Figure 7F). Furthermore, treatment of CFBE41o- cell monolayers with PLGA-SCD-19 NPs significantly inhibited CI#5436 attachment following 4 h of culture compared with cells treated with blank NPs (vehicle) (Figure 7G). This data illustrates, for the first time, the use of a nano-packaged compound which targets MIF to inhibit bacterial attachment of clinically isolated P. aeruginosa to airway epithelial cells.
Discussion
This study describes the synthesis and biological efficacy in in vitro systems of aerosolized NPs as part of a nanodrug delivery system targeting the enzymatic activity of MIF and its ability to modulate P. aeruginosa mediated inflammation, a key pathogen in cystic fibrosis (CF). The development of a nebulized nanodrug delivery system has distinct advantages over alternative modalities of administration including targeted organ drug delivery, improved drug stability and limitations in systemic toxicity [15]. One key advantage of PLGA NPs is that they enhance the solubility of therapeutic compounds in water [25]. To date, an estimated 40% of commercially available drugs and 90% of drugs in development are poorly water-soluble [15,26]. Solubility of therapeutic agents is a key factor which is required to achieve the desired drug concentrations in vivo to achieve the optimal pharmacological response [27]. As with most drugs in development, SCD-19 is poorly water-soluble, hence this study focus on developing PLGA-SCD-19 NPs as an inhaled therapeutic option in lung diseases.
In CF, enhanced MIF activity has been previously shown to be associated with a more aggressive clinical phenotype. Specifically, it has been shown to be associated with increased P. aeruginosa induced lung inflammation and earlier P. aeruginosa colonization in CF. Moreover, in vitro studies demonstrate accelerated biofilm formation and antibiotic resistance in MIF treated P. aeruginosa isolates [9,10,22,24]. Furthermore, SCD-19 has been shown to promote bacterial clearance and inhibit inflammation within the lung in a model of chronic P. aeruginosa infection [9]. Chronic P. aeruginosa infection drives progressive lung injury in CF [1,28,29]. As such, there is a significant clinical unmet need for treatments that are both anti-inflammatory and antibacterial for CF patients. The development of a nebulized pulmonary nano-drug strategy targeting CF would allow higher doses of selected anti-inflammatory agents to be delivered to the lung compared with alternative administration routes. Other distinct advantages would be patient acceptability and the ability for home-administration and the consequent potential reduction in hospital healthcare costs [15].
In order to address this unmet clinical need, in this study, a novel aerosolized delivery system was developed and validated for the nanopackaged inhibitor targeting MIF. The nanocarrier PLGA was selected based on published work showing it to be bio-degradable, targetable, biocompatible and detectable [30]. It has been reported that the ideal drug-to-polymer mass ratio to achieve maximum drug loading efficiency and enhance drug release profile is 1:10 [31,32], hence we utilized PLGA (100 mg) and SCD-19 (10 mg) to prepare PLGA-SCD-19 NPs. The drug content was 5.7% which is above-average compared with nondrug delivery systems [33].
In examining cellular internalization of the NPs we found the average size for both PLGA-SCD-19 NPs and PLGA-MNP-SCD-19 NPs from the TEM and SEM images was 180.0 ± 25.0 nm. These findings were in accordance with previous published works where PLGA NPs synthesized by the oil/water technique resulted in a hydrodynamic radius of the range 100.0 to 250.0 nm [34,35].
The size of nanodrug carriers can affect drug release and cellular internalization, two key features of NPs [36]. Larger sized particles present slower release rates because of smaller surface area to volume ratio compared with smaller sized nanodrug carriers [37]. In the present study, in vitro drug release behavior of PLGA-SCD-19 NPs showed a high initial burst release in the first 24 h, which is possibly due to the percentage of drug that is over or just under the surface of the NPs, followed by a plateau release over two days. It has previously been shown that the degradation of PLGA nanodrug carriers, characteristically is rapid in the initial stage, approximately 30% in the first 24 h, followed by a plateau which results in an extended and sustained drug release profile [30].
In the context of NP stability, recent studies have suggested that PDI values smaller than 0.3 are desired to ensure monodisperse PLGA nanoformulations [38,39]. The Zeta potential of the nanoformulation, in this study, was -26.5 ± 5.4 mV. Zeta potential in NPs has been highlighted as a key factor for the stability of particle solution/suspension [40,41]. It has been reported that zeta potential value greater than +25 mV or less than -25 mV results in the high stability of nanoformulation [42]. Moreover, previous studies indicated that negatively charged NPs have more chance for cellular uptake, whereas, NPs with positive surface charge can increase pulmonary side effects along with a transient systemic toxicity mainly on white blood cells [41].
The PDI value for all samples incubated in lung synthetic fluids were less than 0.2, showing an acceptable monodisperse size distribution and indicating that the NPs are stable under physiological conditions.
Providing an endotoxin free nanodrug delivery system is a crucial issue that must be taken into careful consideration in the context of nanosafety. Endotoxin contamination in NPs promotes the activation of innate immune responses [43]. The endotoxin concentration of PLGA-SCD-19 NPs synthesized in this study was subpicogram level which satisfies current FDA regulations [44].
Vibrating mesh devices are among the most efficient that have been developed and employed for inhalation therapy [45,46]. Vibrating mesh devices are practical, portable, quiet and small in size. They also have improved drug delivery efficiency and minimal residual volume aligned with shorter treatment times (3–10 min). Their capacity to generate high concentration fine-particle fractions increases drug delivery to the lung peripheries [46,47]. Aerosol droplet size has been shown to play a vital role in evading the physiological barriers of the pulmonary system, maximizing delivery and targeting the therapeutics to the appropriate respiratory tract [48]. The VMD of the aerosolized PLGA-SCD-19 NPs was measured to be 4.4 ± 0.33 μm with a GSD of 1.5 ± 0.2 μm. This midpoint droplet size is recognized as the ideal particle size distribution for deep lung deposition [49]. For the same devices, the FPF of the aerosolized nanoformulation revealed that at least 60% of the particles are perfect size range with an MMAD of 2.2 ± 0.4 μm, predicting targeting and achieving deep deposition within the respiratory tract. Moreover, the results of the in vitro regional aerosol deposition measurement predict that in excess of 70% of the aerosolized PLGA-SCD-19 NPs will be deposited within the alveolar region.
A recent review of NP aerosolized systems in clinical trials has shown that whole lung drug deposition via mechanical ventilators is 1% to 16% [50], whereas in this study the data revealed values of 23 and 14% drug deposition in the mechanical ventilator and human breathing simulator systems. We suggest that these improved values are secondary to a high degree of nanoformulation uniformity without aggregation, high stability of NPs and the optimized nanoformulation concentration used in the systems.
Previously published work supports the hypothesis that the gram-negative bacterium, P. aeruginosa utilizes human MIF for its survival advantage. Building on this initial work, we evaluated the effects of targeting MIF's enzymatic activity in the context of P. aeruginosa mediated inflammation and also P. aeruginosa biofilm formation. LPS induced-MIF secretion has previously been shown to be regulated by MIF [10,51]. In this study, it was demonstrated that both free and nano-packaged SCD-19 significantly attenuated both HKPA) and PPF induced pro-inflammatory cytokine production in macrophages.
It has previously been reported that human MIF has the capacity to enhanced P. aeruginosa biofilm formation [9]. Bacterial cellular attachment represents an important initial step in biofilm formation. In this present study, it is demonstrated for the first time that MIF promotes biofilm formation of both lab strain and clinically isolated P. aeruginosa on both healthy and CF airway epithelial cell monolayers respectively. Furthermore, it is demonstrated that both free and nano-packaged SCD-19 significantly inhibits P. aeruginosa biofilm formation on these cell monolayers. Finally, a role for MIF in the promotion of impaired macrophage mediated P. aeruginosa clearance is described. MIF knock-out cells demonstrated enhanced P. aeruginosa killing compared with wild type cells, while BMDMs treated with SCD-19 demonstrated enhanced P. aeruginosa killing compared to those treated with DMSO vehicle. This is in line with observations that promotes P. aeruginosa colonization in CF patients, and also that SCD-19 promotes P. aeruginosa clearance in a murine model of chronic P. aeruginosa infection [9,10,23].
Conclusion
Overall, the results of this study provide persuasive evidence that targeting MIF's unique enzymatic activity represents a valid therapeutic strategy for the treatment of P. aeruginosa infection in the context of CF. Furthermore, the characterization and biological studies of encapsulated SCD-19 within a PLGA nanocarrier, provides evidence for a lung specific delivery system that would be, on the one hand, clinically effective while minimizing systemic toxicity. This is the first study to investigate the synthesis and characterization of an aerosolized nanodrug delivery system targeting the enzymatic active site of MIF in order to treat facets of pulmonary disease. The packaging of SCD-19 in PLGA NPs overcomes the issue of poor water-solubility, enabling the therapy to be delivered using water or saline as a vehicle. These NPs have been optimized for nebulization using the Aerogen® nebulizer. Furthermore, the NPs exhibited minimum toxicity, high levels of cellular uptake and high levels of stability. They also showed the optimal size for deep lung deposition, with the majority of nebulized NPs reaching the deep lung in experiments utilizing a human breathing simulator. We believe that the development of encapsulated SCD-19 within a PLGA nanocarrier given in an inhaled form, represents a potential valid therapeutic therapy for the treatment of P. aeruginosa lung infections.
Summary points.
Macrophage migration inhibitory factor (MIF) has been associated with earlier P. aeruginosa colonization, enhanced biofilm formation, increased disease severity and increased decline in lung function in cystic fibrosis (CF) patients.
Targeted inhibition of MIF's unique tautomerase enzymatic activity has been shown to significantly decrease inflammation and enhance bacterial clearance in an in vivo model of chronic respiratory P. aeruginosa infection.
For the first time, we have developed an aerosolized nanodrug delivery system targeting the enzymatic activity of MIF.
These nanoparticles (NPs) have low levels of toxicity, high levels of cellular uptake and are stable in synthetic lung fluids. Furthermore, these NPs are the optimal size for deep lung deposition, which maximizes drug delivery efficiency.
In vitro, these aerosolized NPs significantly reduce P. aeruginosa induced inflammation.
The aerosolized NPs significantly decrease biofilm formation of lab strain and clinically isolated P. aeruginosa on healthy and CF bronchial epithelial cell monolayers.
Overall, these NPs represent a novel potential adjunctive therapy for the treatment of chronic P. aeruginosa infection in CF.
Supplementary Material
Footnotes
Supplementary data
To view the supplementary data that accompany this paper please visit the journal website at: www.futuremedicine.com/doi/suppl/10.2217/nnm-2020-0344
Financial & competing interests disclosure
This work was supported by grant funding obtained from Science Foundation Ireland (SFI) and the Irish Lung Foundation (MD, AO'N, CO'R, AT, LM, AmcE, SCD) and from the NIH (grant AI83256-06 (GAO'T, SSW)). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as: • of interest; •• of considerable interest
- 1.Folkesson A, Jelsbak L, Yang L et al. Adaptation of Pseudomonas aeruginosa to the cystic fibrosis airway: an evolutionary perspective. Nat. Rev. Microbiol. 10, 841 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Rey MM, Bonk MP, Hadjiliadis D Cystic fibrosis: emerging understanding and therapies. Annu. Rev. Med. 70, 197–210 (2019). [DOI] [PubMed] [Google Scholar]; •• Provides an up to date review of the current understand in of cystic fibrosis (CF) disease and pathogenesis.
- 3.Elborn JS Cystic fibrosis. Lancet (London, England) 388(10059), 2519–2531 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Moreau-Marquis S, Stanton BA, O'toole GA Pseudomonas aeruginosa biofilm formation in the cystic fibrosis airway. Pulmon. Pharmacol. Ther. 21(4), 595–599 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chmiel JF, Konstan MW, Elborn JS Antibiotic and anti-inflammatory therapies for cystic fibrosis.. Cold Spring Harb. Perspect. Med. 3(10), a009779–a009779 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kang I, Bucala R The immunobiology of MIF: function, genetics and prospects for precision medicine. Nat. Rev. Rheumatol. 15(7), 427–437 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Donnelly SC, Haslett C, Reid PT et al. Regulatory role for macrophage migration inhibitory factor in acute respiratory distress syndrome. Nat. Med. 3(3), 320–323 (1997). [DOI] [PubMed] [Google Scholar]
- 8.Hams E, Armstrong ME, Barlow JL et al. IL-25 and type 2 innate lymphoid cells induce pulmonary fibrosis. Proc. Natl Acad. Sci. U SA 111(1), 367–372 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tynan A, Mawhinney L, Armstrong ME et al. Macrophage migration inhibitory factor enhances Pseudomonas aeruginosa biofilm formation, potentially contributing to cystic fibrosis pathogenesis. FASEB J. 31(11), 5102–5110 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Indicates the targeting of migration inhibitory factor's (MIF's) tautomerase activity as a valid adjunctive therapy in combating bacterial infections.
- 10.Adamali H, Armstrong ME, Mclaughlin AM et al. Macrophage migration inhibitory factor enzymatic activity, lung inflammation, and cystic fibrosis. Am. J. Respir. Crit. Care Med. 186(2), 162–169 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Indicates the describes the role of MIF tautomerase activity in P. aeruginosa induced lung inflammation in CF.
- 11.Rosengren E, Bucala R, Aman P et al. The immunoregulatory mediator macrophage migration inhibitory factor (MIF) catalyzes a tautomerization reaction. Mol. Med. 2(1), 143–149 (1996). [PMC free article] [PubMed] [Google Scholar]
- 12.Mawhinney L, Armstrong ME, O'reilly C et al. Macrophage migration inhibitory factor (MIF) enzymatic activity and lung cancer. Mol. Med. 20(1), 729–735 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Highlights the role of MIF's enzyme activity in its physiological activity. Also, this study showed that targeting the tautomerase enzymatic activity of MIF, represents a promising approach for lung cancer treatment.
- 13.Moreno-Sastre M, Pastor M, Salomon CJ, Esquisabel A, Pedraz JL Pulmonary drug delivery: a review on nanocarriers for antibacterial chemotherapy. J. Antimicrob. Chemother. 70(11), 2945–2955 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Kuzmov A, Minko T Nanotechnology approaches for inhalation treatment of lung diseases. J. Control. Release 219, 500–518 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Doroudian M, Macloughlin R, Poynton F, Prina-Mello A, Donnelly SC Nanotechnology based therapeutics for lung disease. Thorax 74(10), 965–976 (2019). [DOI] [PubMed] [Google Scholar]; •• Comprehensive review discussing recent progress and development in clinical applications of nanotechnology in respiratory diseases. This paper covers a wide range of attempts in clinical trials employing nanotechnology for therapeutic agent delivery, vaccination and detection for the treatment of lung diseases.
- 16.Hudson D, Margaritis A Biopolymer nanoparticle production for controlled release of biopharmaceuticals. Crit. Rev. Biotechnol. 34(2), 161–179 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Ungaro F, D'Angelo I, Miro A, La Rotonda MI, Quaglia F Engineered PLGA nano- and micro-carriers for pulmonary delivery: challenges and promises. J. Pharm. Pharmacol. 64(9), 1217–1235 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Kamaly N, Yameen B, Wu J, Farokhzad OC Degradable controlled-release polymers and polymeric nanoparticles: mechanisms of controlling drug release. Chem. Rev. 116(4), 2602–2663 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; • An illustrative review of biocompatible and degradable polymeric nanoparticles (NPs) and their mechanism for drug release.
- 19.Olivieri C, Bargagli E, Inghilleri S, Campo I, Cintorino M, Rottoli P Macrophage migration inhibitory factor in lung tissue of idiopathic pulmonary fibrosis patients. Exp. Lung Res. 42(5), 263–266 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Anderson GG, Moreau-Marquis S, Stanton BA, O'Toole GA In vitro analysis of tobramycin-treated pseudomonas aeruginosa biofilms on cystic fibrosis-derived airway epithelial cells. Infect. Immun. 76(4), 1423–1433 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chekabab SM, Silverman RJ, Lafayette SL, Luo Y, Rousseau S, Nguyen D Staphylococcus aureus inhibits IL-8 responses induced by pseudomonas aeruginosa in airway epithelial cells. PLoS One 10(9), e0137753 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Plant BJ, Gallagher CG, Bucala R et al. Cystic fibrosis, disease severity, and a macrophage migration inhibitory factor polymorphism. Am. J. Respir. Crit. Care Med. 172(11), 1412–1415 (2005). [DOI] [PubMed] [Google Scholar]
- 23.Bozza M, Satoskar AR, Lin G et al. Targeted disruption of migration inhibitory factor gene reveals its critical role in sepsis. J. Exp. Med. 189(2), 341–346 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gadjeva M, Nagashima J, Zaidi T, Mitchell RA, Pier GB Inhibition of macrophage migration inhibitory factor ameliorates ocular pseudomonas aeruginosa-induced keratitis. PLoS Pathogens 6(3), e1000826 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma X, Williams RO Polymeric nanomedicines for poorly soluble drugs in oral delivery systems: an update. Int. J. Pharm. Investig. 48(1), 61–75 (2018). [Google Scholar]
- 26.Loftsson T, Brewster ME Pharmaceutical applications of cyclodextrins: basic science and product development. J. Pharm. Pharmacol. 62(11), 1607–1621 (2010). [DOI] [PubMed] [Google Scholar]
- 27.Savjani KT, Gajjar AK, Savjani JK Drug solubility: importance and enhancement techniques. ISRN Pharm. 2012, 195727–195727 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Burns JL, Gibson RL, Mcnamara S et al. Longitudinal assessment of Pseudomonas aeruginosa in young children with cystic fibrosis. J. Infect. Diseases 183(3), 444–452 (2001). [DOI] [PubMed] [Google Scholar]
- 29.Malhotra S, Hayes D Jr, Wozniak DJ Cystic fibrosis and pseudomonas aeruginosa: the host–microbe interface. Clin. Microbiol. Rev. 32(3), e00138–18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Makadia HK, Siegel SJ Poly lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers (Basel) 3(3), 1377–1397 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jelvehgari M, Soltani S, Milani P, Jalali MB Fabrication and in vitro evaluation of Ketotifen Fumarate-loaded PLGA nanoparticles as a sustained delivery system. Iran. J. Pharm. Res. 16(1), 22–34 (2017). [PMC free article] [PubMed] [Google Scholar]
- 32.Madani F, Esnaashari SS, Mujokoro B, Dorkoosh F, Khosravani M, Adabi M Investigation of effective parameters on size of paclitaxel loaded PLGA nanoparticles. Adv. Pharm. Bull. 8(1), 77–84 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Park K Facing the truth about nanotechnology in drug delivery. ACS Nano 7(9), 7442–7447 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramalho MJ, Pereira MC Preparation and characterization of polymeric nanoparticles: an interdisciplinary experiment. J. Chem. Educ. 93(8), 1446–1451 (2016). [Google Scholar]
- 35.Ling Y, Wei K, Luo Y, Gao X, Zhong S Dual docetaxel/superparamagnetic iron oxide loaded nanoparticles for both targeting magnetic resonance imaging and cancer therapy. Biomaterials 32(29), 7139–7150 (2011). [DOI] [PubMed] [Google Scholar]
- 36.Hsu MN, Luo R, Kwek KZ, Por YC, Zhang Y, Chen CH Sustained release of hydrophobic drugs by the microfluidic assembly of multistage microgel/poly (lactic-co-glycolic acid) nanoparticle composites. Biomicrofluidics 9(5), 052601 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kohane DS Microparticles and nanoparticles for drug delivery. Biotechnol. Bioeng. 96(2), 203–209 (2007). [DOI] [PubMed] [Google Scholar]
- 38.Fernández E, Santos-Carballal B, De Santi C et al. Biopolymer-based nanoparticles for cystic fibrosis lung gene therapy studies. Materials (Basel) 11(1), 122 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lopalco A, Ali H, Denora N, Rytting E Oxcarbazepine-loaded polymeric nanoparticles: development and permeability studies across in vitro models of the blood-brain barrier and human placental trophoblast. Int. J. Nanomedicine 10, 1985–1996 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sabbah M, Esposito M, Pierro P, Giosafatto C, Mariniello L, Porta R Insight into zeta potential measurements in biopolymer film preparation. J. Biotechnol. Biomater. 6, e126 (2016). [Google Scholar]
- 41.Honary S, Zahir F Effect of zeta potential on the properties of nano-drug delivery systems-a review (Part 1). Trop. J. Pharm. Res. 12(2), 255–264 (2013). [Google Scholar]
- 42.Kesarkar R, Yeole M, Dalvi B, Sharon M, Chowdhary A Simplistic approach towards synthesis of highly stable and biocompatible L-cysteine capped gold nanosphere intermediate for drug conjugation. Int. J. Pharm. Sci. Rev. Res. 31, 143–146 (2015). [Google Scholar]
- 43.Li Y, Fujita M, Boraschi D Endotoxin contamination in nanomaterials leads to the misinterpretation of immunosafety results. Front. Immunol 8, 472–472 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guidance for Industry: Pyrogen and Endotoxins Testing: Questions and Answers. www.fda.gov/regulatory-information/search-fda-guidance-documents/guidance-industry-pyrogen-and-endotoxins-testing-questions-and-answers
- 45.Zhou Q, Leung SSY, Tang P, Parumasivam T, Loh ZH, Chan H-K Inhaled formulations and pulmonary drug delivery systems for respiratory infections. Adv. Drug Deliv. Rev 85, 83–99 (2015). [DOI] [PubMed] [Google Scholar]
- 46.Pritchard JN, Hatley RH, Denyer J, Hollen DV Mesh nebulizers have become the first choice for new nebulized pharmaceutical drug developments. Ther. Deliv. 9(2), 121–136 (2018). [DOI] [PubMed] [Google Scholar]
- 47.Waldrep JC, Dhand R Advanced nebulizer designs employing vibrating mesh/aperture plate technologies for aerosol generation. Curr. Drug Deliv. 5(2), 114–119 (2008). [DOI] [PubMed] [Google Scholar]
- 48.Labiris NR, Dolovich MB Pulmonary drug delivery. Part I: physiological factors affecting therapeutic effectiveness of aerosolized medications. Br. J. Clin. Pharmacol. 56(6), 588–599 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Patton JS, Byron PR Inhaling medicines: delivering drugs to the body through the lungs. Nat. Rev. Drug Discov. 6(1), 67–74 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Dugernier J, Ehrmann S, Sottiaux T et al. Aerosol delivery during invasive mechanical ventilation: a systematic review. Crit. Care (London) 21(1), 264–264 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Roger T, David J, Glauser MP, Calandra T MIF regulates innate immune responses through modulation of Toll-like receptor 4. Nature 414(6866), 920–924 (2001). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.