

Abstract
Bardet–Biedl Syndrome (BBS) is a pleiotropic genetic disease caused by the dysfunction of primary cilia. The immune system of patients with ciliopathies has not been investigated. However, there are multiple indications that the impairment of the processes typically associated with cilia may have influence on the hematopoietic compartment and immunity. In this study, we analyze clinical data of BBS patients and corresponding mouse models carrying mutations in Bbs4 or Bbs18. We find that BBS patients have a higher prevalence of certain autoimmune diseases. Both BBS patients and animal models have altered red blood cell and platelet compartments, as well as elevated white blood cell levels. Some of the hematopoietic system alterations are associated with BBS‐induced obesity. Moreover, we observe that the development and homeostasis of B cells in mice is regulated by the transport complex BBSome, whose dysfunction is a common cause of BBS. The BBSome limits canonical WNT signaling and increases CXCL12 levels in bone marrow stromal cells. Taken together, our study reveals a connection between a ciliopathy and dysregulated immune and hematopoietic systems.
Keywords: Bardet–Biedl Syndrome, ciliopathy, CXCL12, immunity, obesity
Subject Categories: Haematology, Immunology, Molecular Biology of Disease
Bardet‐Biedl Syndrome is a pleiotropic genetic disease caused by the dysfunction of primary cilia. BBS is associated with altered hematopoiesis and homeostasis of the immune system in patients and mouse models.

Introduction
Bardet–Biedl Syndrome (BBS) is a recessive genetic disorder caused by complete or partial loss‐of‐function mutations in any of 24 genes known to date (BBS1–23 (Lindstrand et al, 2016; Forsythe et al, 2018; Wormser et al, 2019), NPHP1 (Lindstrand et al, 2014)). BBS belongs to a group of ciliopathies, i.e., disorders caused by defective formation and/or function of primary cilia. Eight BBS proteins (BBS1, BBS2, BBS4, BBS5, BBS7, BBS8, BBS9, and BBS18) form a transport complex called the BBSome, which sorts selected cargoes into and out of the cilium (Berbari et al, 2008; Jin et al, 2010; Wei et al, 2012; Klink et al, 2017). Other commonly mutated BBS genes (ARL6/BBS3, MKKS/BBS6, BBS10, and BBS12) assist the assembly or function of the BBSome (Jin et al, 2010; Zhang et al, 2012a). The BBSome acts as an adaptor connecting ciliary cargoes to intraflagellar transport (IFT) machinery (Liu & Lechtreck, 2018; Ye et al, 2018).
Bardet–Biedl Syndrome is a pleiotropic disease whose primary diagnostic features are rod‐cone dystrophy, polydactyly, obesity, learning difficulties, hypogonadism, and renal anomalies (Beales et al, 1999). The immune system of patients with ciliopathies including BBS has not been studied in detail. An exception in this respect is a case report of three BBS patients suffering from autoimmune diseases in a cohort of 15 studied BBS patients (Halac & Herzog, 2012). The possible connection between ciliopathies and the immune system has not been addressed most likely because immune cells do not form primary cilia (Wheatley et al, 1996; Plotnikova et al, 2009). However, there are several lines of evidence suggesting that the BBS affects the function of the immune system.
First, the immunological synapse formed between T cells and antigen‐presenting cells exhibits a striking analogy to the primary cilium (Finetti et al, 2011; de la Roche et al, 2016). The formation of these structures involves the reorganization of cortical actin and the centrosome polarization. In the same vein, some components of the IFT machinery have been shown to participate in the organization of the immunological synapse to promote T‐cell activation (Yuan et al, 2014; Vivar et al, 2016). In particular, it has been shown that vesicles containing key T‐cell signaling molecules, the TCR/CD3 complex and LAT, are transported toward the immunological synapse by IFT proteins (Finetti et al, 2009; Finetti et al, 2014). The BBSome may regulate T‐cell activation by transporting signaling proteins into and/or out of the immunological synapse. Depending on the character of these putative cargoes and the direction of the transport, the BBSome could be a positive or a negative regulator of T‐cell signaling.
Second, the BBSome is required for Sonic hedgehog (SHH) signaling (Zhang et al, 2011; Zhang et al, 2012b; Goetz et al, 2017). The SHH signaling pathway regulates multiple processes in the organism including T‐cell development and differentiation (Shah et al, 2004; El Abdaloussi et al, 2006; Crompton et al, 2007; Rowbotham et al, 2008; Drakopoulou et al, 2010; Furmanski et al, 2013). Key components of the SHH signaling pathway, SMO, IHH, GLI1, and PTCH2, are upregulated in effector cytotoxic T cells and transported toward the immunological synapse in vesicles (de la Roche et al, 2013). Moreover, Smo KO/KO T cells show reduced cytotoxicity associated with defects in the polarized release of cytotoxic granules (de la Roche et al, 2013).
Third, the BBSome regulates trafficking of the leptin receptor (Guo et al, 2016). Leptin is a signaling molecule which acts as a pro‐inflammatory cytokine (Procaccini et al, 2012; La Cava, 2017). In particular, leptin signaling inhibits the proliferation of regulatory T cells (De Rosa et al, 2007) and promotes the proliferation of effector T cells and their polarization toward Th1 helper cells (Martin‐Romero et al, 2000). Moreover, T cells deficient in the leptin receptor show impaired differentiation into Th17 helper T cells in mice (Reis et al, 2015), indicating a T‐cell intrinsic role of leptin signaling.
Fourth, the BBSome tunes WNT signaling by promoting the non‐canonical pathway and suppressing the canonical WNT signaling (Ross et al, 2005; Gerdes et al, 2007). Since WNT signaling is an important regulator of hematopoiesis (Staal et al, 2016; Richter et al, 2017), the BBSome deficiency might lead to defective formation of blood cells including leukocytes.
Fifth, one of the major symptoms of BBS is obesity, which is believed to undermine the immune tolerance (Johnson et al, 2012). Obesity induces production of pro‐inflammatory cytokines, such as TNF (Park et al, 2005) and IL‐6 (Moro et al, 2010). These may predispose individuals to the development of autoimmune diseases (Gremese et al, 2014; Granata et al, 2017; Owczarczyk‐Saczonek & Placek, 2017; Singh et al, 2017; Wang & He, 2018). Thus, the BBSome may have an extrinsic role in the immune system via inducing obesity.
In this study, we addressed the intrinsic and extrinsic roles of the BBSome in the immune system by investigating BBS patients and mouse models of the BBSome deficiency. We found that BBS patients show elevated prevalence of particular autoimmune diseases. Moreover, our data revealed dysregulated homeostasis of blood cells both in BBS mouse models and in BBS patients.
Results
Autoimmune diseases are more prevalent in BBS patients
In this work, we studied the potential role of the BBSome in the immune system. Initially, we analyzed two cohorts of BBS patients from the CRIBBS NIH registry and from the Guy's Hospital of Guy's and St Thomas' NHS Foundation Trust, London, or Great Ormond Street Hospital, London. We found out that certain autoimmune and inflammatory diseases, such as type I diabetes, Hashimoto's thyroiditis, rheumatoid arthritis, and inflammatory bowel diseases, are more prevalent in BBS patients than in the overall population (Table 1). The co‐occurence of multiple autoimmune diseases was relatively small (Appendix Tables S1 and S2). The prevalence of autoimmunity does not seem to be restricted to any causative gene (Appendix Tables S3 and S4). Most frequent causative genes in our cohort were BBS1 and BBS10. Patients with mutations in BBS10 showed slightly higher prevalence of autoimmunity than patients with mutations in BBS1, which is in line with typically higher disease severity in the former group (Niederlova et al, 2019). However, the sample size was too small to draw any conclusions. Overall, these findings suggested that the BBSome has an intrinsic or extrinsic role in the immune system, particularly in the immune tolerance.
Table 1.
Autoimmune diseases in BBS patients.
| CRIBBS registry | ||||||
|---|---|---|---|---|---|---|
| Cases/total | % Prevalence in BBS patients | % Normal prevalence | Fold difference in prevalence | Odds ratio | P‐value | |
| Arthritis | 17/172 | 9.88 | 0.86 | 11.49 | 12.64 | 2.73e‐13 |
| Type 1 diabetes mellitus | 6/217 | 2.76 | 0.48 | 5.76 | 5.9 | 6.99e‐4 |
| Hashimoto's thyroiditis | 6/235 | 2.55 | 0.79 | 3.23 | 3.29 | 0.01 |
| Ulcerative colitis | 2/255 | 0.78 | 0.03 | 26.14 | 26.34 | 2.77e‐3 |
| Celiac disease | 2/255 | 0.78 | 0.75 | 1.05 | 1.05 | 0.72 |
| At least one autoimmune condition CRIBBS | 29 a /255 | 11.37 | 4.93 | 2.31 | 2.47 | 3.16e‐5 |
| Great Ormond Street Hospital & Guy's Hospital cohort | ||||||
|---|---|---|---|---|---|---|
| Cases/total | % Prevalence in BBS patients | % Normal prevalence | Fold change prevalence | Odds ratio | P‐value | |
| Hypothyroidism | 22/198 | 11.11 | 5.30 | 2.10 | 2.23 | 1.15e‐3 |
| Celiac disease | 4/198 | 2.02 | 0.75 | 2.69 | 2.73 | 0.06 |
| Inflammatory bowel diseases | 3/198 | 1.51 | 0.40 | 3.79 | 3.83 | 0.05 |
| Type 1 diabetes mellitus | 3/198 | 1.51 | 0.48 | 3.16 | 3.19 | 0.07 |
| Psoriasis | 2/198 | 1.01 | 0.20 | 5.05 | 5.09 | 0.06 |
| Other diseases with one occurrence (Immune thrombocytopenic purpura, Henoch‐Schönlein purpura, Sarcoidosis, Vitiligo) | 4/198 | |||||
| At least one autoimmune condition UK | 33 b /198 | 16.67 | 4.93 | 3.38 | 3.86 | 9.76e‐10 |
The table shows the fold difference in the prevalence (prevalence in BBS patients/general prevalence) and the odds ratio of autoimmune diseases in the CRIBBS cohort of 255 BBS patients (upper part) and in the cohort of 198 BBS patients from Great Ormond Street Hospital and Guy's Hospital in London (lower part). Normal prevalence of autoimmune diseases was adopted from (Hayter & Cook, 2012; Martel‐Pelletier et al, 2016; Taylor et al, 2018). Statistical significance was calculated using exact binomial test.
Total count is not equal to sum of all the patients with autoimmune diseases because of the co‐occurrence of more than one disease in 4 patients. Co‐occurring diseases are listed in Appendix Table S1.
Total count is not equal to sum of all the patients with autoimmune diseases because of the co‐occurrence of more than one disease in 5 patients. Co‐occurring diseases are listed in Appendix Table S2.
BBSome subunit genes are expressed in mouse lymphoid tissues
In the next step, we employed mouse models to study the putative role of the BBSome in the immune and hematopoietic systems in controlled experiments. First, we tested if the BBSome subunits are expressed in murine lymphoid tissues. We detected the expression of all eight BBSome subunits in the spleen, lymph nodes, and isolated T cells on the mRNA level (Fig 1A). The expression levels of Bbs2, Bbs4, Bbs9, and Bbs18 in the lymphoid tissues were comparable to the brain and the kidney, two organs where the BBSome plays a major role (ODea et al, 1996; Davis et al, 2007; Keppler‐Noreuil et al, 2011; Putoux et al, 2012). The other four subunits (Bbs1, Bbs5, Bbs7, and Bbs8) were expressed in the lymphoid tissues at 10‐ to 50‐fold lower levels than in the brain and the kidney. Moreover, we detected BBS4 protein in isolated T and B cells (Fig 1B). Altogether, all the BBSome subunits are variably expressed in lymphocytes and lymphocyte‐rich tissue, despite of the fact that hematopoietic cells are commonly considered as non‐ciliated cells. This suggested that the BBSome as a whole or some individual BBSome subunits might have an intrinsic role in lymphocytes.
Figure 1. Basic characterization of Bbs4‐deficient mouse models.
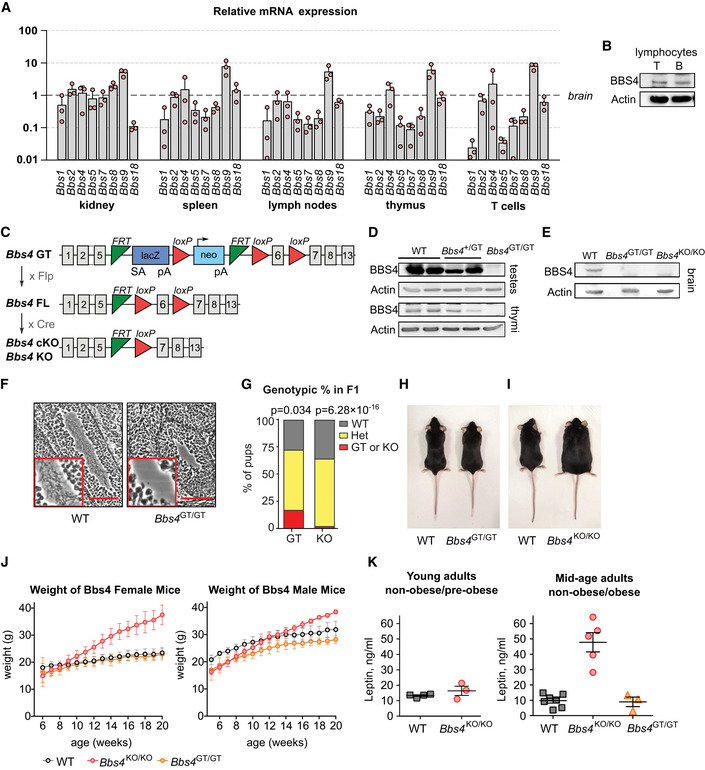
- Relative expression of the BBSome subunits in the indicated organs and T cells measured by qPCR. C T values of the BBS genes were normalized to the geometric mean of the C T values of reference genes Gapdh, Tubb2a, and Eef1a1. The expression levels are normalized to those of brain (= 1). Mean + SD. Three biological replicates (the result of each biological replicate is a median of three technical replicates).
- Immunoblot analysis of BBS4 expression in T and B lymphocytes isolated from the lymph nodes and spleen of C57Bl/6 mouse. β‐actin staining served as a loading control. The identical immunoblot is shown in Fig EV4A. A representative experiment out of three biological replicates in total is shown.
- Schematic representation of mouse models of Bbs4 deficiency used in this study. Bbs4 GT, gene trap allele interrupting the Bbs4 gene. Bbs4 FL, allele with floxed Bbs4 exon 6. Bbs4 KO and cKO alleles with deleted exon 6.
- Immunoblot analysis of BBS4 expression in testes and thymi lysates of Bbs4 +/+ (WT), Bbs4 GT/+, and Bbs4 GT/GT mice. β‐actin staining serves as a loading control. A representative experiment out of three biological replicates in total is shown.
- Immunoblot analysis of BBS4 expression in the brain lysates of Bbs4 +/+, Bbs4 GT/GT, and Bbs4 KO/KO mice. β‐actin staining served as a loading control. A representative experiment out of two biological replicates is shown.
- Hematoxylin and eosin staining of sections of seminiferous tubules from ~ 4 weeks old Bbs4 +/+ (n = 2 mice) and Bbs4 GT/GT (n = 4) males. Scale bar, 100 µm. Representative images are shown.
- Genotypic ratio of Bbs4 +/+, Bbs4 GT/+ or Bbs4 KO/+ (Het), and Bbs4 GT/GT (GT) or Bbs4 KO/KO (KO) offspring at weaning from mating of Bbs4 GT/+ (n = 145 pups) or Bbs4 +/KO (n = 168 pups) parents. Binomial test was used for statistical comparison of the observed distribution to the expected Mendelian ratio.
- Bbs4 +/+ and Bbs4 GT/GT female littermates at 20 weeks of age. Representative litter out of seven in total.
- Bbs4 +/+ and Bbs4 KO/KO female littermates at 20 weeks of age. Representative litter out of five in total.
- Growth curves of Bbs4‐deficient mice, mean ± SD is shown. Females: Bbs4 +/+ (n = 12 mice), Bbs4 GT/GT (n = 12), Bbs4 KO/KO (n = 6). Males: Bbs4 +/+ (n = 6 mice), Bbs4 GT/GT (n = 7), Bbs4 KO/KO (n = 5).
- Leptin concentration in blood plasma taken from young adult (7–8 weeks) or mid‐age (14–20 weeks) mice. Young adult mice: Bbs4 +/+ (n = 4 mice), Bbs4 KO/KO (n = 3), analyzed in two independent experiments. Mid‐age adult mice: Bbs4 +/+ (n = 7 mice), Bbs4 KO/KO (n = 5), Bbs4 GT/GT (n = 3), analyzed in 4 independent experiments. Kruskal–Wallis tests were used for the statistical analysis. Mean ± SEM.
Source data are available online for this figure.
Mouse models for studying the role of the BBSome in the immune system
Our next step was to obtain a mouse model of BBS. We decided to use the Bbs4‐deficient mouse for the following reasons: (i) BBS4 is an essential part of the BBSome (Klink et al, 2017), (ii) Bbs4 KO/KO mouse has been shown to have a relatively severe phenotype in comparison to other BBSome‐deficient mice (Rahmouni et al, 2008; Guo et al, 2011), (iii) Bbs4 had a relatively high expression in lymphoid tissues (Fig 1A and B). In the following experiments, we used mice with an interrupted Bbs4 gene with a gene trap (GT allele) cassette, mice with deleted Bbs4 exon 6 (KO allele), and mice with Bbs4 exon 6 flanked with LoxP sites (FL allele) enabling the conditional deletion of this exon (cKO; Fig 1C).
BBS4 protein was not detectable in the testes, thymi and brain from the Bbs4 GT/GT mice (Fig 1D and E). As expected, the Bbs4 deficiency lead to the absence of sperm flagella in testes of 30‐days‐old Bbs4 GT/GT males (Fig 1F). However, we did not observe two previously reported features of BBS mouse models in Bbs4 GT/GT mice. Although mating of Bbs4 +/GT heterozygotes resulted to 17% Bbs4 GT/GT pups at weaning, only 2% Bbs4 KO/KO pups were produced in mating of Bbs4 +/KO heterozygotes (Fig 1G). This suggests that Bbs4 KO/KO, but not Bbs4 GT/GT, mice suffer from frequent pre‐weaning lethality. Moreover, Bbs4 KO/KO mice, but not Bbs4 GT/GT, developed obesity (Fig 1H–J). As expected, adult Bbs4 KO/KO mice, suffering from obesity, had elevated levels of leptin in blood plasma in comparison to non‐obese Bbs4 GT/GT mice, and pre‐obese young Bbs4 KO/KO mice (Fig 1K). Because leptin has been proposed to act as a pro‐inflammatory molecule, it might have an impact on the immune system of obese BBS mice.
To explain the phenotypic difference between Bbs4 GT/GT and Bbs4 KO/KO mice, we analyzed the respective Bbs4 transcripts via RT–PCR. Although transcript of Bbs4 KO allele lacked the exon 6 as expected, we detected a transcript containing all exons 5–11 in Bbs4 GT/GT mice (Fig EV1A). Unexpectedly, the RNA polymerase II is able to read through two transcription termination sites in the gene trap cassette in Bbs4 GT/GT tissues to generate Bbs4 mRNA at ~ 10% of the wild‐type (WT) levels (Fig EV1B). However, the PCR product of Bbs4 GT mRNA was longer than the respective amplicon of the WT allele (Fig EV1A and B). Sequencing revealed that Bbs4 GT allele is mis‐spliced and includes a short insert originating from the gene trap cassette, which induces a frame shift in the open reading frame (Fig EV1C). Expression of this Bbs4 GT cDNA in HEK293T cells resulted in a very tiny production of truncated BBS4 (Fig EV1D), possibly via an alternative transcription/translation start. It is plausible that low levels of this truncated BBS4 are present in Bbs4 GT/GT tissues as well. Moreover, we could detect a small amount (~ 1% of WT levels) of properly spliced Bbs4 mRNA in Bbs4 GT/GT brain (Fig EV1E). Overall, these data indicate that Bbs4 KO is a null allele, whereas Bbs4 GT is a hypomorphic allele.
Figure EV1. Analysis of the hypomorphic nature of the Bbs4 GT allele.
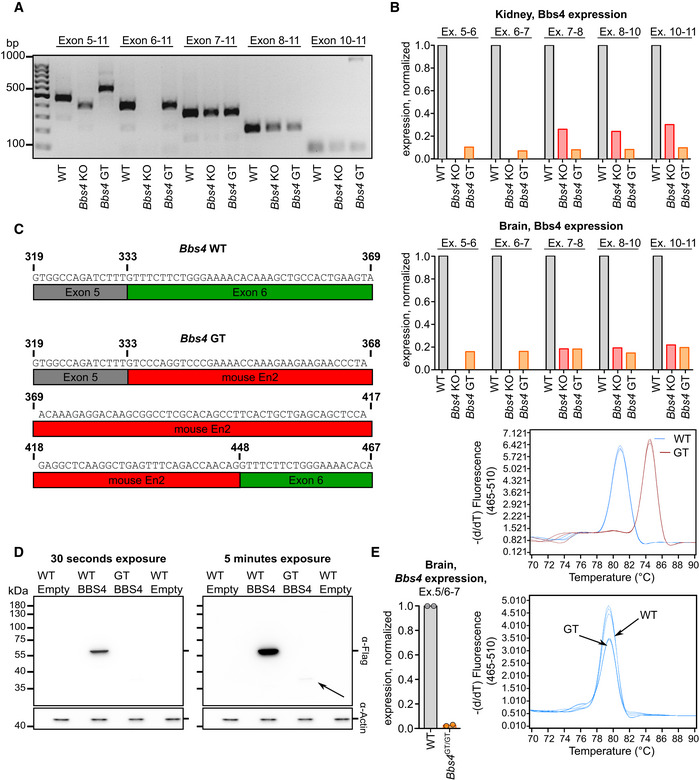
- The exon inclusion in Bbs4 transcript in the brain of Bbs4 +/+ (WT), Bbs4 GT/GT, and Bbs4 KO/KO mice was analyzed by end‐point RT–PCR with primers specific to exons 5, 6, 7, 8, or 10 (forward) and exon 11 (reverse). A single mouse of each genotype was analyzed.
- The relative expression of Bbs4 exon 5–6, exon 6–7, exon 7–8, exon 8–10, exon 10–11 in the brain and kidney of Bbs4 +/+ (n = 2 mice), Bbs4 GT/GT (n = 1), and Bbs4 KO/KO (n = 1) mice was analyzed using RT–qPCR. Melting curve analysis for the amplicon representing exon 5–6 is shown for Bbs4 +/+ and Bbs4 GT/GT mice. The average from two animals is shown for Bbs4 +/+.
- Results of Sanger sequencing of the amplicon representing Bbs4 exon 5–6 of the Bbs4 GT/GT mice. The sequence shown in red originates from the Bbs4 GT cassette (En2 exon) and is absent in Bbs4 WT RNA.
- C‐terminally FLAG‐tagged full‐length Bbs4 WT and Bbs4 GT cDNA was transiently expressed in HEK293T cells. The expression of BBS4 was detected by anti‐FLAG antibody via immunoblotting. Re‐probing the membrane for actin served as a loading control. The same actin staining (30 s exposure) is shown for both exposures of anti‐FLAG staining. The arrow indicates a weak band with low apparent molecular weight which was present only in cells transfected with Bbs4 GT cDNA. Representative experiment out of two biological replicates is shown.
- The expression of properly spliced Bbs4 (primers annealed to exon 5/6 boundary and exon 7) in the brains of Bbs4 +/+ and Bbs4 GT/GT mice was analyzed by RT–qPCR. The melting curve analysis of the amplicon is shown. The experiment shows two technical replicates (the tissue was divided into halves before RNA isolation) using a single animal of each strain.
Source data are available online for this figure.
Altered B‐cell compartment in Bbs4 deficient mice
To analyze the role of the BBSome in the formation of adaptive immune cells, we analyzed the development and homeostasis of T and B cells in Bbs4‐deficient mice. We did not observe any major alterations in the T‐cell compartment in Bbs4 KO/KO and Bbs4 GT/GT mice (Fig EV2A–F). The only significant differences were decreased percentage of T cells among the splenocytes (Fig EV2C) and decreased percentage of CD44+ cells among splenic CD8+ T cells of the Bbs4 KO/KO mice (Fig EV2F).
Figure EV2. T‐cell compartment is mildly affected in Bbs4 KO/KO mice and unaffected in Bbs4 GT/GT .
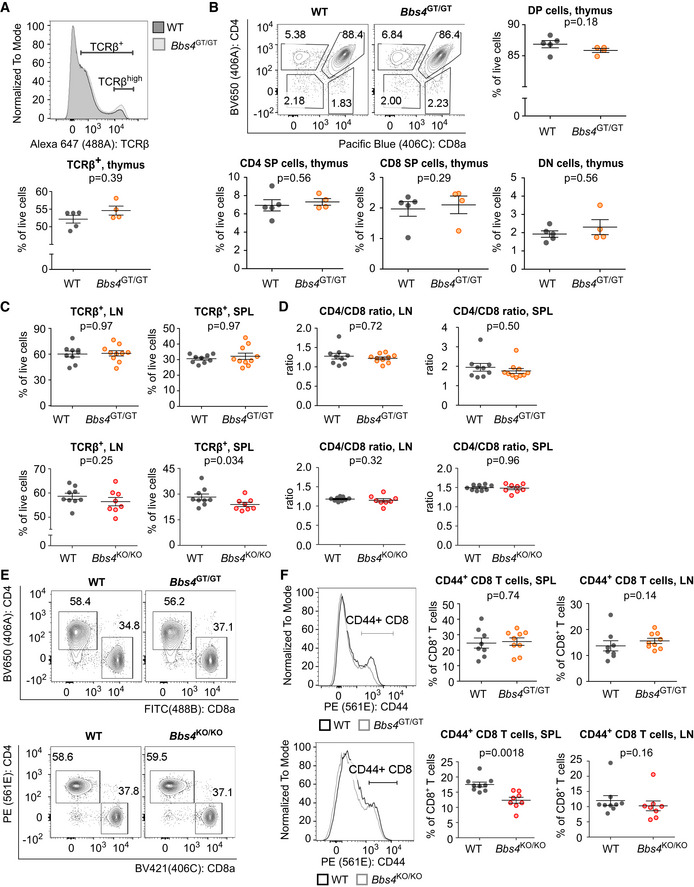
-
A, BCells isolated from thymi of 6 weeks old Bbs4 +/+ (WT) (n = 5 mice) and Bbs4 GT/GT (n = 4) littermates were analyzed by flow cytometry in three independent experiments. Representative staining is shown. (B) Thymic cell populations: double‐positive (DP) (CD4+ CD8+), CD4 single‐positive (SP) (CD4+, CD8−), CD8 SP (CD4− CD8+), double‐negative (DN) (CD4− CD8−) cells.
-
C, DCells isolated from lymph nodes (LN) and spleens (SPL) of Bbs4 +/+ (n = 9 mice) and Bbs4 GT/GT (n = 10) littermates, or Bbs4 +/+ (n = 9) and Bbs4 KO/KO (n = 8) littermates were analyzed by flow cytometry, and percentage of TCRβ+, or CD8+, CD4+ cells was determined. Seven independent experiments for Bbs4 GT/GT, six independent experiments for Bbs4 KO/KO. Mean ± SEM. Statistical significance was calculated using two‐tailed Mann–Whitney test.
-
ERepresentative experiment showing CD4+ and CD8+ T‐cell populations in spleens of Bbs4 GT/GT, Bbs4 KO/KO, and their WT littermates.
-
FCells isolated from lymph nodes (LN) and spleens (SPL) of Bbs4 +/+ (n = 8 mice) and Bbs4 GT/GT (n = 9) littermates, or Bbs4 +/+ (n = 9) and Bbs4 KO/KO (n = 8) littermates were analyzed by flow cytometry, and percentage of CD8+ CD44+ cells was determined. Six independent experiments were performed for each strain. Representative staining of T cells from spleen is shown. Mean ± SEM. Statistical significance was calculated using two‐tailed Mann–Whitney test.
Source data are available online for this figure.
We found an alteration of the B‐cell development and/or homeostasis in Bbs4‐deficient mice. The total numbers of bone marrow B220+ B‐cell lineage cells were not significantly altered in Bbs4 KO/KO or Bbs4 GT/GT mice (Fig EV3A). However, the ratio of B220high and B220low cells in the bone marrow was shifted toward less mature B220low cells in Bbs4 KO/KO mice and, to a lesser extent, in Bbs4 GT/GT mice (Fig 2A). Both Bbs4 KO/KO and Bbs4 GT/GT mice had higher percentage of IgD− IgM− B‐cell precursors than controls (Fig 2B). A deeper analysis of this population showed that Bbs4‐deficiency results in the accumulation of pre‐B cells, which is the developmental stage when the pre‐B‐cell receptor selection occurs (Fig 2C).
Figure EV3. Bbs4 deficiency in mice leads to B‐cell compartment alterations.
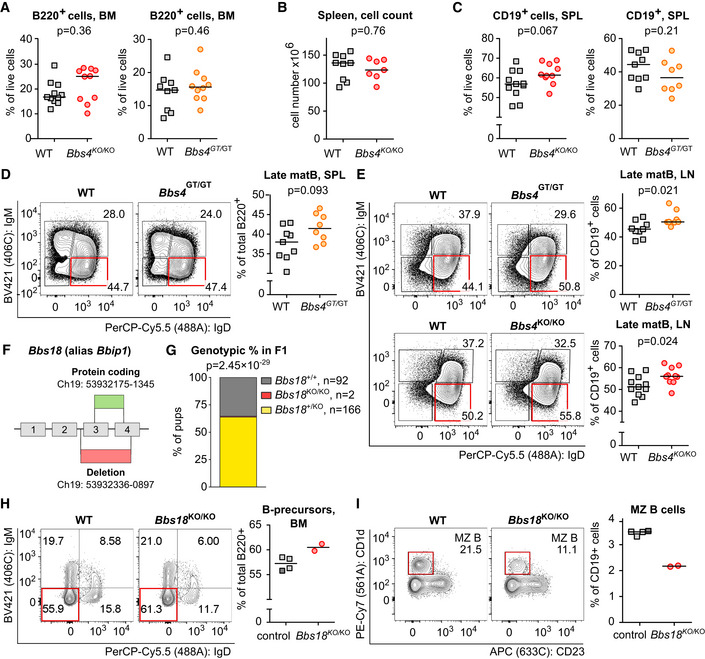
-
APercentage of B220+ cells in the bone marrow (BM) of Bbs4 GT/GT (n = 10 mice), Bbs4 KO/KO (n = 10) and their Bbs4 +/+ (WT) littermates (n = 9, n = 11, respectively) was determined by flow cytometry. For Bbs4 GT strain, a representative experiment out of six in total is shown. For Bbs4 KO strain, a representative experiment out of eight in total is shown. Statistical significance was calculated using two‐tailed Mann–Whitney test. Medians are shown.
-
BSplenocyte count in Bbs4 +/+ (n = 9 mice) and Bbs4 KO/KO (n = 7) mice. Medians are shown. Statistical significance was calculated using two‐tailed Mann–Whitney test.
-
CPercentage of B cells (CD19+) in the spleen (SPL) of Bbs4 GT/GT (n = 8 mice), Bbs4 KO/KO (n = 10) and their Bbs4 +/+ littermates (n = 9 and n = 11, respectively) was determined. Six independent experiments for Bbs4 GT/GT and eight experiments for Bbs4 KO/KO were performed. Statistical significance was calculated using two‐tailed Mann–Whitney test. Medians are shown.
-
DPercentage of splenic (SPL) late mature (IgM− IgD+) B cells (Late matB) among viable CD19+ cells in Bbs4 +/+ (n = 9 mice) and Bbs4 GT/GT (n = 8) mice. Representative experiment out of six in total is shown. Statistical significance was calculated using two‐tailed Mann–Whitney test. Medians are shown.
-
EPercentage of late mature (IgM− IgD+) B cells (Late matB) among viable CD19+ cells in lymph nodes (LN) from Bbs4 GT/GT (n = 8 mice) and Bbs4 +/+ littermates (n = 9), or Bbs4 KO/KO (n = 10) mice and Bbs4 +/+ controls (n = 11). Representative experiments are shown. Statistical significance was calculated using two‐tailed Mann–Whitney test. Medians are shown.
-
FSchematic representation of the Bbs18 KO mouse model. Bbs18 reference sequence: ENSMUSG00000084957 (Ensembl database).
-
GGenotypic ratio of Bbs18 +/+, Bbs18 +/KO, or Bbs18 KO/KO at weaning from mating of Bbs18 +/ KO parents. Binomial test was used for statistical comparison of the observed distribution to the expected Mendelian ratio.
-
H, IComparison of Bbs18 KO/KO mice (n = 2 mice) mice with pooled Bbs18 +/+ (n = 1, dark gray square) and Bbs18 +/KO (n = 3, light gray square) controls. All six mice were littermates and were analyzed side by side in a single experiment. (H) Percentage of B‐cell precursors (IgM− IgD−) in the bone marrow (BM). Gated on viable B220+ cells. Medians are shown. (I) Percentage of splenic MZ B cells (CD23− CD1d+) in Bbs18 KO/KO mice and their control littermates was determined. Gated on viable CD19+, IgD− IgM+, CD138− cells. Medians are shown.
Source data are available online for this figure.
Figure 2. B‐cell compartment is moderately affected in Bbs4‐deficient mice.
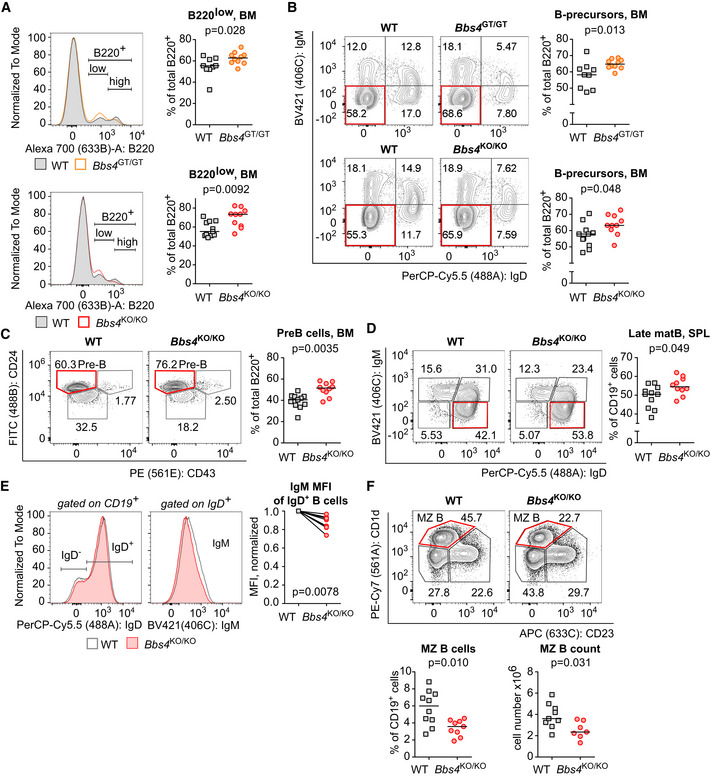
Cells isolated from the bone marrow (BM) (A–C) and spleens (SPL) (D–F) of 18‐ to 25‐week‐old Bbs4 +/+ (WT), Bbs4 GT/GT, and Bbs4 KO/KO mice were analyzed by flow cytometry. Number of analyzed mice (n) is indicated. Six (Bbs4 GT/GT strain and Bbs4 +/+ controls) or eight (Bbs4 KO/KO strain and Bbs4 +/+ controls) independent experiments were performed.
- Percentage of B220low positive cells in the bone marrow. Up: Bbs4 +/+ n = 9, Bbs4 GT/GT n = 10. Down: Bbs4 +/+ (n = 11), Bbs4 KO/KO (n = 10). Statistical significance was calculated using two‐tailed Mann–Whitney test. Medians are shown.
- Percentage of B‐cell precursors (IgM− IgD−) in the bone marrow. Gated on viable B220+ cells. Up: Bbs4 +/+ (n = 9), Bbs4 GT/GT (n = 10). Down: Bbs4 +/+ (n = 11), Bbs4 KO/KO (n = 10). Statistical significance was calculated using two‐tailed Mann–Whitney test. Medians are shown.
- Percentage of pre‐B cells (CD43− CD24high) in the bone marrow of Bbs4 +/+ (n = 11) and Bbs4 KO/KO mice (n = 10). Gated on viable B220+ IgM− IgD− cells. Statistical significance was calculated using two‐tailed Mann–Whitney test. Medians are shown.
- Percentage of late mature (IgM− IgD+) B cells in the spleen of Bbs4 +/+ (n = 11) and Bbs4 KO/KO mice (n = 10) Gated on viable CD19+ cells. Statistical significance was calculated using two‐tailed Mann–Whitney test. Medians are shown.
- An alternative analysis of the experiment shown in (D). Geometric mean fluorescence intensity (MFI) of BV421 (IgM) on IgD+ B cells was determined. Mean of MFI values for each genotype per experiment was quantified, and obtained values were normalized to Bbs4 +/+ (= 1) for each experiment. Two‐tailed one sample Wilcoxon signed rank test was used for the statistical analysis.
- Percentage of splenic marginal zone (MZ) B cells (CD23− CD1d+) in Bbs4 +/+ controls (n = 10) and Bbs4 KO/KO mice (n = 9) was determined. Gated on viable CD19+, IgD− IgM+, CD138− cells. Statistical significance was calculated using two‐tailed Mann–Whitney test. Medians are shown.
Source data are available online for this figure.
We observed slightly increased numbers of B cells in the spleen of Bbs4 KO/KO, but not Bbs4 GT/GT mice (Fig EV3B and C). In the spleen and lymph nodes, Bbs4 KO/KO and Bbs4 GT/GT mice showed larger late mature B‐cell population (IgD+ IgM−) than WT mice (Figs 2D and E, and EV3D and E). In addition, Bbs4 KO/KO showed ~ 2‐fold lower percentage of splenic marginal zone (MZ) B cells (Fig 2F).
To address whether the phenotype of Bbs4 KO/KO is indeed caused by the BBSome dysfunction, we generated a second mouse model of BBS based on the genetic disruption of another BBSome subunit, BBS18 (alias BBIP1; Fig EV3F). Unfortunately, we observed strong pre‐weaning lethality resulting in only two adult Bbs18 KO/KO animals overall (Fig EV3G). The comparison of these Bbs18 KO/KO animals to their WT and Bbs18 +/KO littermates showed very similar phenotype to Bbs4 KO/KO mice, i.e., increased B‐cell precursors in the bone marrow (Fig EV3H) and decreased MZ B cells in the spleen (Fig EV3I). These data indicated that the alterations of the B‐cell lineage are not unique to the deficiency in BBS4, but are likely a common feature of the BBSome disruption.
Overall, these data show that BBSome deficiency results in an abnormal development of B cells in the bone marrow, a slightly increased percentage of late mature splenic B cells and a reduction of splenic MZ B cells in the periphery. The developmental alteration was observed also in non‐obese Bbs4 GT/GT mice, indicating that this phenotype is largely obesity‐independent.
Bbs4 deficiency does not intrinsically influence antigen‐specific T‐cell and B‐cell responses
As we observed an alteration of B‐cell homeostasis in Bbs4‐deficient mice, we decided to investigate how it affects the response of the adaptive immune system. First, we activated monoclonal B cells specific for 4‐hydroxy‐3‐nitrophenyl acetyl (NP) from B‐cell receptor transgenic B1–8 mice (Sonoda et al, 1997) and Bbs4 GT/GT B1–8 mice using NP‐labeled cells. In this setup, we monitored the upregulation of the activation marker CD69 (Fig 3A and B). Moreover, we stimulated B cells from Bbs4 KO/KO, Bbs4 +/KO, and Bbs4 +/+ mice with plate bound anti‐IgM antibody (Fig 3C). In these assays, we did not observe any role of Bbs4 deficiency in the antigenic B‐cell response.
Figure 3. BBS4 is not required for T‐cell and B‐cell antigenic responses.
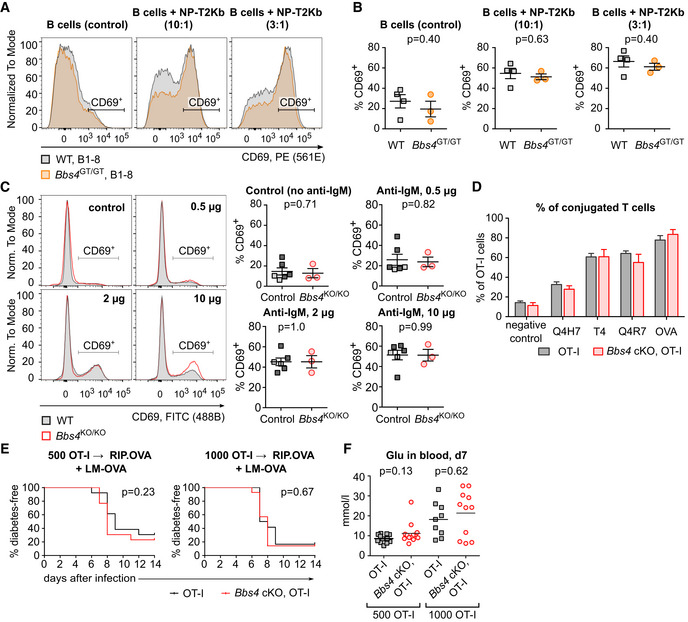
-
A, BSplenocytes isolated from Bbs4 +/+ (n = 4 mice) and Bbs4 GT/GT (n = 3) B‐cell transgenic B1–8 littermates were activated with 4‐hydroxy‐3‐nitrophenylacetic acid succinimide ester‐loaded T2‐Kb cells in three independent experiments. Percentage of activated B cells (gated as CD69+ B220+ IgLλ+ viable cells) in the samples without T2‐Kb (negative control), and in the samples with T2‐Kb added in ratios 1:10 or 1:3 was determined by flow cytometry. (A) Representative mice are shown. (B) Mean ± SEM. Statistical significance was calculated using two‐tailed Mann–Whitney test.
-
CB cells from 18‐ to 20‐week‐old Bbs4 KO/KO (red circle, n = 3 mice), and control mice (Bbs4 +/+, dark gray square, n = 4, or Bbs4 +/KO, light gray square, n = 2) were activated with F(ab')2‐goat anti‐mouse IgM (Mu chain). Percentage of activated B cells (gated as CD69+ CD19+ viable cells) was determined by flow cytometry. Representative experiment out of three biological replicates is shown. Mean ± SEM. Statistical significance was calculated using two‐tailed Mann–Whitney test.
-
DCFSE‐loaded T cells isolated from lymph nodes of Bbs4 FL/FL OT‐I Rag2 KO/KO (OT‐I) and CD4‐Cre Bbs4 FL/FL OT‐I Rag2 KO/KO (Bbs4 cKO, OT‐I) littermates were incubated with DDAO‐labeled WT splenocytes loaded with OVA peptide or with the indicated altered peptide ligands for 20 min. Percentage of T cells conjugated with the APCs was determined by flow cytometry. Four biological replicates were performed. Mean + SEM. Statistical significance was calculated using two‐tailed Mann–Whitney test, P > 0.05 for all peptides.
-
E, F500 or 1,000 T cells isolated from lymph nodes of Bbs4 FL/FL OT‐I Rag2 KO/KO (OT‐I) and CD4‐Cre Bbs4 FL/FL OT‐I Rag2 KO/KO (Bbs4 cKO, OT‐I) littermates were adoptively transferred into RIP.OVA mice followed by infection with Listeria monocytogenes expressing ovalbumin (LM‐OVA) on the next day. (E) Glucose level in the urine of mice was monitored on a daily basis. The mouse was considered diabetic when it had urine glucose level ≥ 1,000 mg/dl for two consecutive days. Statistical significance was calculated by Log‐rank (Mantel‐Cox) test. (F) Glucose concentration in blood on day 7 post‐infection. 500 OT‐I ctrl (n = 13 mice), 500 OT‐I Bbs4 cKO (n = 13), 1,000 OT‐I ctrl (n = 9), 1,000 OT‐I Bbs4 cKO (n = 11), analyzed in four independent experiments, mean is shown. Statistical significance was calculated using two‐tailed Mann–Whitney test.
Source data are available online for this figure.
Based on the homology between the cilium and the immunological synapse (Finetti et al, 2009; Finetti et al, 2011; Stephen et al, 2018), we hypothesized that the BBSome might act as a positive or negative regulator of T‐cell responses. For this reason, we generated Bbs4 FL/FL Cd4‐Cre mouse line where Bbs4 deficiency was restricted to T cells (Fig EV4A). These mice did not show any obvious phenotype in the T‐cell compartment (Fig EV4B–E). To study the role of the BBSome in T‐cell antigenic responses, we crossed Bbs4 FL/FL Cd4‐Cre mice to TCR transgenic OT‐I Rag2 KO/KO mice. This mouse generates monoclonal T cells specific for Kb‐OVA, a model antigen originating from chicken ovalbumin. We did not observe any role of BBS4 in the conjugation of OT‐I T cells with antigen‐presenting cells loaded with OVA or its lower affinity variants (Fig 3D). Moreover, WT and Bbs4‐deficient OT‐I T cells showed the same ability to induce autoimmune diabetes upon a transfer into RIP.OVA mice expressing ovalbumin under the rat insulin promoter and subsequent priming by Listeria monocytogenes expressing ovalbumin (King et al, 2012; Palmer et al, 2016) (Fig 3E and F). This assay examines T cells for their ability to get primed by the OVA‐antigen, expand, infiltrate the pancreas, and kill the β‐cells. As the onset of diabetes caused by Bbs4‐deficient OT‐I T cells was not different from the control, we concluded that BBS4 does not play an important intrinsic role in any of indicated steps of the T‐cell‐mediated immune response.
Figure EV4. BBS4 has no intrinsic role in T‐cell development.
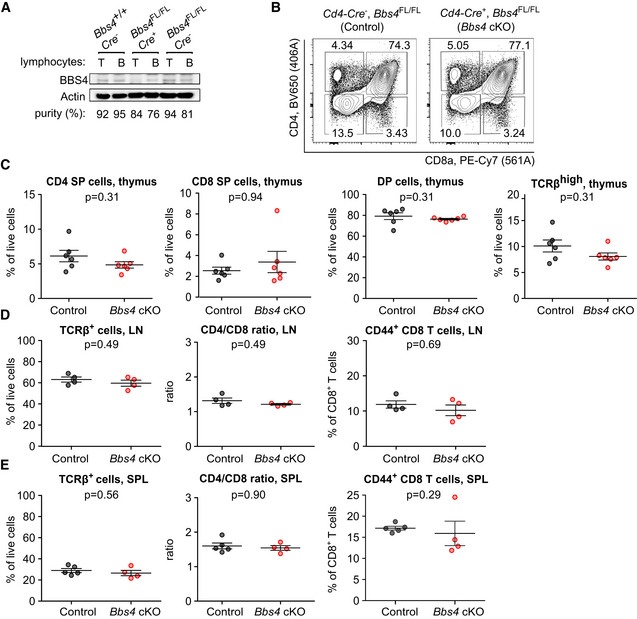
-
AThe immunoblot analysis of the BBS4 expression in enriched T or B cells taken from lymph nodes and spleens of Bbs4 +/+ Cd4‐Cre − , Bbs4 FL/FL Cd4‐Cre +, and Bbs4 FL/FL Cd4‐Cre − mice. The purity of the enriched populations is indicated. β‐actin staining served as a loading control. Part of the identical immunoblot is shown in Fig 1B. A representative experiment out of three biological replicates in total is shown.
-
B–ECells isolated from thymi (B, C), lymph nodes (LN) (D), and spleens (SPL) (E) of Bbs4 FL/FL Cd4‐Cre + (Bbs4 cKO) mice were analyzed by flow cytometry. Bbs4 +/+ Cd4‐Cre + and Bbs4 FL/FL Cd4‐Cre − mice were used as controls interchangeably. (B, C) Thymic cell populations: CD4 single‐positive (SP) (CD4+, CD8−), CD8 SP (CD4− CD8+), double‐positive (DP) (CD4+ CD8+), TCRβhigh cells. n = 6 mice per group, four independent experiments. Mean ± SEM, two‐tailed Mann–Whitney test. (D) T‐cell populations in lymph nodes, n = 4 mice per group, three independent experiments. Mean ± SEM, two‐tailed Mann–Whitney test. (E) T‐cell populations in spleens of control (n = 5 mice) and Bbs4 cKO (n = 4), analyzed in three independent experiments. Mean ± SEM, two‐tailed Mann–Whitney test.
Source data are available online for this figure.
The role of BBS4 in B‐cell homeostasis is not intrinsic
To investigate if the observed B‐cell developmental changes in Bbs4‐deficient mice are B‐cell intrinsic, we generated Bbs4 FL/FL Vav‐iCre mice with a specific deletion of Bbs4 in the hematopoietic and endothelial cells (Georgiades et al, 2002). We did not observe any evidence of altered B‐cell homeostasis in Vav‐iCre mice (Fig 4A–C). These results suggest that the B‐cell compartment in Bbs4 GT/GT and Bbs4 KO/KO mice is affected by factors extrinsic to the hematopoietic lineage.
Figure 4. The role of Bbs4 in B‐cell development is not intrinsic.
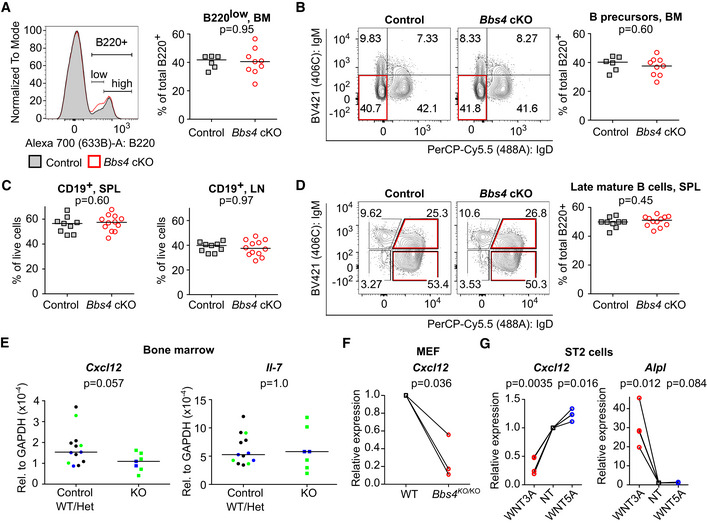
-
A–DCells isolated from bone marrow (BM) (A, B), lymph nodes (LN) and spleen (SPL) (C, D) of Bbs4 FL/FL (control) and Vav‐iCre Bbs4 FL/FL (cKO) mice were analyzed by flow cytometry. (A) Percentage of B220low in bone marrow. Representative experiment out of four in total is shown. Bbs4 FL/FL (n = 6 mice), Vav‐iCre Bbs4FL / FL (n = 9). (B) Percentage of B‐cell precursors (B220+, IgM− IgD−) in the bone marrow. Representative experiment out of four in total is shown. Bbs4 FL/FL (n = 6 mice), Vav‐iCre Bbs4FL / FL (n = 9). (C) Percentage of CD19+ B cells in spleens and lymph nodes of Vav‐iCre Bbs4 FL/FL (n = 12 mice) and Bbs4 FL/FL (n = 9) mice, six independent experiments. (D) Percentage of late mature B cells (CD19+, IgM−, IgD+) in spleens of Vav‐iCre Bbs4 FL/FL (n = 12 mice), and Bbs4 FL/FL mice (n = 9). Representative experiment out of six in total is shown. Statistical significance was calculated using two‐tailed Mann–Whitney test, P > 0.05 for all tests. Median is shown.
-
EExpression of Cxcl12 and Il‐7 in the bone marrow from Bbs4 +/+ Bbs18 +/+ (WT) mice (black circles, n = 7 mice), Bbs4 +/KO (Het, green circle, n = 3), Bbs18 +/KO (Het, blue circle, n = 3), Bbs4 KO/KO (green square, n = 5), and Bbs18 KO/KO (blue square, n = 2) was analyzed by RT–qPCR in five independent experiments. The expression was normalized to Gapdh. The statistical significance of the difference between the BBSome‐deficient mice (Bbs4 KO/KO and Bbs18 KO/KO) and controls were calculated using two‐tailed Mann–Whitney test. Median is shown.
-
FMEF cell lines were derived from a single Bbs4 +/+ (WT) or a single Bbs4 KO/KO embryos. The expression of Cxcl12 in these cell lines was analyzed by RT–qPCR. The expression was normalized to Gapdh and to Bbs4 +/+ MEFs (= 1) for each experiment. Three biological replicates. One sample t‐test was used for the statistical analysis.
-
GExpression of Cxcl12 and canonical WNT responsive gene Alpl in untreated (NT) ST2 cells, or ST2 cells treated with WNT3A or WNT5A was analyzed by RT–qPCR. The expression was normalized to Gapdh and untreated ST2 sample (= 1) for each experiment. Four biological replicates were performed. One sample t‐test was used for the statistical analysis.
Source data are available online for this figure.
The development of B cells is guided by the cytokine environment in the bone marrow niche with prominent roles of IL‐7 and CXCL12 (Zehentmeier & Pereira, 2019). We observed that the expression of Cxcl12, but not Il‐7, is decreased in the bone marrow of Bbs4 and Bbs18 KO mice (Fig 4E). In the next step, we generated mouse embryonic fibroblasts (MEF) from Bbs4 +/+ and Bbs4 KO/KO mice. Bbs4 KO/KO MEFs showed lower expression of Cxcl12 than Bbs4 +/+ MEFs (Fig 4F), indicating that the BBSome regulates Cxcl12 expression in mesenchymal cells. It has been shown previously that the BBSome inhibits canonical WNT signaling in vertebrates (Gerdes et al, 2007) and that the canonical WNT signaling downregulates Cxcl12 expression in bone marrow stroma‐derived ST2 cells (Tamura et al, 2011). Accordingly, canonical WNT signaling (triggered by WNT3A), but not non‐canonical WNT signaling (triggered by WNT5A), suppressed Cxcl12 expression in ST2 cells and induced the expression of canonical WNT responsive gene Alpl (Fig 4G). We produced Bbs4‐deficient ST2 cells using CRISPR/Cas9 (Fig EV5A, Appendix Table S6). Two out of four Bbs4 KO/KO ST2 cell clones showed significantly reduced expression of Cxcl12 (Fig EV5B). Overall, our data indicate that the BBSome deficiency amplifies canonical WNT signaling in bone marrow stromal cells leading to decreased production of CXCL12 and subsequent partial defects in B‐cell development.
Figure EV5. BBS4 regulates Cxcl12 expression in ST2 cells.
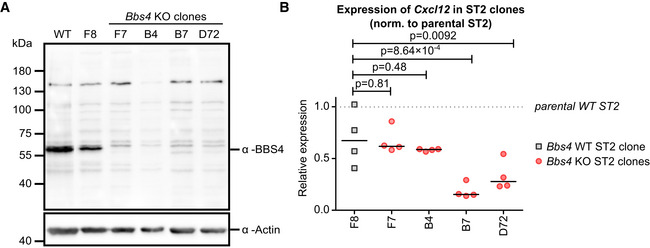
- The expression of BBS4 in the parental ST2 line and in five Bbs4 KO ST2 clones was analyzed by immunoblotting. The parental line and the clone F8 expresses BBS4, whereas the other clones lack BBS4. Re‐probing the membrane for actin served as a loading control. These results are in agreement with the results of the sequencing of the Bbs4 locus (Appendix Table S6). A representative experiment out of two biological replicates is shown.
- The expression of Cxcl12 in the parental ST2 line, in one Bbs4 WT clone (F8) and in four Bbs4 KO ST2 clones (indicated in red) was analyzed by RT–qPCR. The expression was normalized to Gapdh and to the parental ST2 line for each experiment (= 1, represented as a dotted line). The statistical significance was calculated with repeated measures ANOVA (P = 0.0007) with Dunnett's multiple comparison post‐test (indicated) for comparing each Bbs4 KO clone to clone F8. n = 4 biological replicates.
Source data are available online for this figure.
BBS‐induced obesity affects blood homeostasis
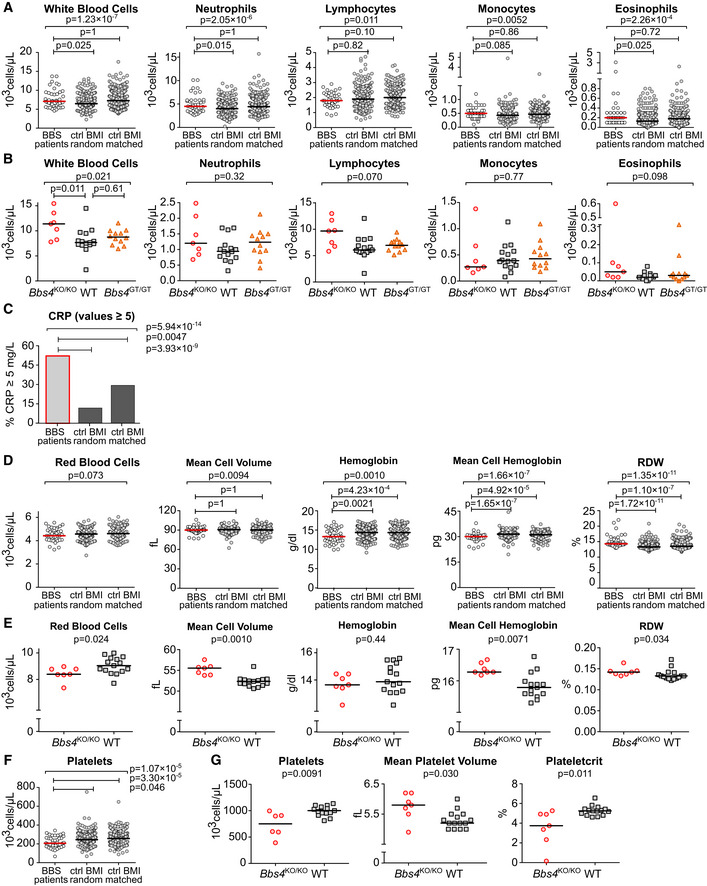
To investigate the possible factors predisposing BBS patients to the development of autoimmune diseases, we examined the blood test results of BBS patients. Intriguingly, immunity‐related parameters, such as counts of total white blood cells, neutrophils, and eosinophils, were increased in BBS patients, when compared to age and gender‐matched controls (BMI‐random controls) (Fig 5A). To address the role of obesity in BBS patients, we used an additional set of controls with body mass indexes (BMI) matching to those of BBS patients (BMI‐matched controls). We did not observe any difference when we compared the indicated leukocyte parameters between BBS patients and BMI‐matched controls.
Figure 5. Blood homeostasis is altered in BBS patients and Bbs4‐deficient mice.
-
A–G(A, C, D, F) Results of the blood tests of BBS patients from the Guy's Hospital and Great Ormond Street Hospital were extracted from the medical records and compared to two sets of healthy controls obtained from the UK Biobank. The BMI‐random controls were age‐ and gender‐matched to the set of BBS patients, with random BMIs. The BMI‐matched controls were matched for age, gender, and BMI. BBS patients n = 43 for parameters White blood cell count, Mean cell volume, Hemoglobin and Platelets; n = 41 for parameters Neutrophils, Lymphocytes, Monocytes, Eosinophils; n = 39 for parameters Red blood cells, Mean cell hemoglobin and Red cell distribution width (RDW). Both datasets of healthy controls were selected as 10‐fold larger than the set of BBS patients. Median is shown. Kruskal–Wallis test was used for the statistical analysis in (A, D and F). In (C), the percentage of patients or controls having CRP ≥ 5 were compared using Fisher's exact test with post hoc Sidak correction for multiple comparisons, BBS patients n = 42. (B, E, G) Indicated parameters were measured using the blood from 20–21 weeks old Bbs4 +/+ (WT) (n = 15), Bbs4 KO/KO (n = 7), and Bbs4 GT/GT (n = 12) mice. Kruskal–Wallis test with Dunn's multiple comparison post‐tests was used for the statistical analysis. Median is shown.
Source data are available online for this figure.
We analyzed the peripheral blood of Bbs4‐deficient mice to further explore the role of the BBSome and the potential involvement of obesity in blood cell homeostasis. In agreement with the analysis of the patients' blood tests, we did not observe major differences between WT and non‐obese Bbs4 GT/GT mice. However, obese Bbs4 KO/KO mice showed higher total white blood cell count than WT controls (Fig 5B), which corresponded with the data from patients as well (Fig 5A). These results indicate that obesity in BBS patients and in Bbs4‐deficient mice has an impact on the leukocyte homeostasis.
We found that elevated C‐reactive protein (CRP) was more frequent in BBS patients than in BMI‐matched and BMI‐random controls (Fig 5C), which indicates that obesity is not the only factor influencing the immune system of BBS patients. Notably, the homeostasis of red blood cells was altered in BBS patients as well as in the Bbs4 KO/KO mice, although at different levels (Fig 5D and E). BBS patients showed low overall hemoglobin levels caused by a mild decrease in the red blood cell count and low red blood cell hemoglobin (Fig 5D), indicating a possibility of a reduced oxygen transport capacity. Interestingly, the comparison of BBS patients with BMI‐matched controls showed that the alteration of the erythroid compartment was not caused by obesity. The mouse model of BBS showed a decreased number of red blood cells, which was compensated by enlarged red blood cell volume (Fig 5E).
In addition, we observed decreased platelet counts both in BBS patients and Bbs4 KO/KO mice (Fig 5F–G). The reduction of platelets in BBS patients was not obesity‐dependent. Furthermore, Bbs4 KO/KO mice showed higher mean platelet volume and lower plateletcrit than WT mice (Fig 5F–G), indicating enhanced removal of platelets in the periphery.
Altogether, our results suggest a role of the BBSome in the immune tolerance, hematopoiesis and/or blood homeostasis. Most of the effects seem to be extrinsic to the hematopoietic compartment as revealed by using tissue‐specific knock‐out mouse model and comparison of patients to BMI‐matched controls. However, altered CRP levels, red blood cell, and platelet homeostasis in BBS patients are obesity‐independent.
Discussion
In this study we focused on the putative connection between BBS and immune/hematopoietic defects. At first, we hypothesized that the immune system of BBS patients might be substantially influenced by obesity, which is a typical BBS symptom. Obesity can induce the state of low‐grade metabolic inflammation characterized by elevated TNFα, IL‐6, and CRP in blood (Visser et al, 1999; Tanaka et al, 2001; Bullo et al, 2003; Khaodhiar et al, 2004) and adipose tissue (Sindhu et al, 2015). Obesity is also considered as a risk factor for autoimmune disorders (Versini et al, 2014). Indeed, we have shown elevated prevalence of autoimmune disorders (namely T1DM, inflammatory bowel diseases, rheumatoid arthritis, and hypothyroidism) in two independent BBS cohorts. Obesity potentially contributes to the increased prevalence of autoimmune diseases in BBS patients, as obesity is associated with high prevalence of hypothyroidism, Hashimoto's thyroiditis, and rheumatoid arthritis (Song et al, 2019; Ohno et al, 2020). We also observed alteration of the white blood cell count in BBS patients and in a BBS mouse model. The comparison of BBS patients and non‐BBS obese controls, as well as the comparison of obese and non‐obese BBS mouse model, shows that this effect is caused by obesity.
One of the major players in obesity‐associated inflammation is leptin, an adipocyte‐derived hormone which acts as a pro‐inflammatory cytokine (Lord, 2006; Paz et al, 2012). The BBSome is proposed to transport the leptin receptor to the plasma membrane, which makes the BBSome‐deficient cells unresponsive to leptin (Guo et al, 2016). It has been shown that leptin signaling in the central nervous system regulates immune responses and prevents lethal overreaction of the immune system to sepsis in mice (Tschop et al, 2010). Thus, it is possible that defective leptin signaling in the nervous system directly contributes to high prevalence of autoimmunity in BBS patients.
Leptin also promotes T‐cell activation and proliferation (Martin‐Romero et al, 2000; Milner & Beck, 2012), suggesting a T‐cell intrinsic phenotype of the BBSome deficiency. Moreover, T cells repurpose a number of ciliary transport proteins for immunological synapse formation (Finetti et al, 2009; Vivar et al, 2016; Stephen et al, 2018). Last but not least, the BBSome is functionally connected to IFT20 and SHH signaling components (Wei et al, 2012; Williams et al, 2014; Nakayama & Katoh, 2018; Niederlova et al, 2019), which were shown to regulate early T‐cell development (El Abdaloussi et al, 2006; Michel et al, 2013; Yuan et al, 2014; Vivar et al, 2016). For all these reasons, we tested whether the BBSome plays a positive or negative intrinsic role in T‐cell biology. However, the development and function of T cells were largely unaffected in Bbs4 KO/KO mice and in Cd4‐Cre Bbs4 FL/FL mice.
Bardet–Biedl Syndrome patients exhibited additional defects in blood cell homeostasis, which cannot be solely attributed to obesity. First, BBS patients had elevated blood CRP levels, which might be a sign of ongoing inflammation. Second, BBS patients and Bbs4‐null mice exhibit low platelet counts. Furthermore, platelets from Bbs4 KO/KO mice had larger volume than WT controls. This indicates that a greater fraction of platelets in Bbs4KO/KO mice are immature. As these mice also have low platelet counts, their bone marrow is likely compensating for platelet loss on the periphery (Stasi, 2012). This state can be a result of immune‐mediated processes such as immune thrombocytopenia (Schmoeller et al, 2017; Norrasethada et al, 2019). Unfortunately, data concerning mean platelet volume are not available for BBS patients, but we can presume that the peripheral destruction of platelets is a cause of thrombocytopenia in BBS patients as well as in mice.
Our analysis of BBS mouse models revealed the role of the BBSome in B‐cell development. Bbs4 KO/KO, Bbs18 KO/KO, and non‐obese hypomorphic Bbs4 GT/GT mice showed an increased frequency of B‐cell precursors in the bone marrow, as well as low numbers of MZ B cells. These effects were not caused by BBSome deficiency in hematopoietic cells. Instead, BBSome‐deficient bone marrow stromal cells produce low levels of CXCL12, most likely as a consequence of enhanced canonical WNT signaling (Gerdes et al, 2007; Tamura et al, 2011). Our explanation that the BBSome regulates B‐cell development by increasing CXCL12 availability is supported by a previous study (Nie et al, 2004). This study showed that B‐cell‐specific deletion of CXCR4, the receptor for CXCL12, manifests with a phenotype resembling our BBS mouse models, i.e., increased frequency in immature IgM− IgD− B cells in the bone marrow and low numbers of MZ B cells in the spleen. Accordingly, deletion of WBP1L, a negative regulator of CXCR4 signaling, shows the opposite phenotype, i.e., decreased frequency of pre‐B cells in the bone marrow and increased frequency of MZ B cells in the spleen (Borna et al, 2020). The importance of ciliary signaling in non‐hematopoietic bone marrow cells for hematopoiesis should be addressed in further studies.
To our knowledge, this is the first study revealing the dysregulation of the immune and hematopoietic systems in patients suffering from a ciliopathy. Obesity, a common feature of ciliopathies such as BBS and Alström syndrome (Vaisse et al, 2017), leads to elevated concentration of white blood cells in BBS patients. However, BBS patients also exhibit elevated blood CRP levels and low platelet counts in an obesity‐independent manner. A detailed analysis of BBS mouse models revealed low CXCL12 production by BBSome‐deficient stromal cells resulting in defects in B‐cell development. Given the importance of the CXCL12‐CXCR4 axis in immunity and hematopoiesis, there might be additional effects of BBSome deficiency beyond our findings. Altogether, we identified BBS as a risk factor for autoimmune diseases.
Materials and Methods
Antibodies and reagents
Antibodies to the following antigens were used for flow cytometry: CD1d Pe‐Cy7 (1B1, #123524, Biolegend), CD4 BV650 (RM4‐5, #100545, Biolegend), CD8a PE‐Cy7 (53‐6.7, #1103610, SONY), CD8a FITC (53‐6.7, #100706, Biolegend), CD19 PE (6D5, #115508, Biolegend), CD23 APC (b3b4, #1108095, SONY), CD24 FITC (M1/69, #101806, Biolegend), CD43 PE (S7, #553271, BD PharMingen), CD44 PE (IM7, #103008, Biolegend), B220 Alexa Fluor 700 (RA3‐6B2, #103231, Biolegend), B220 FITC (RA3‐6B2, #103206, Biolegend), CD69 PE (H1.2F3, #104508, Biolegend), CD69 FITC (H1.2F3, #104506, Biolegend), IgM BV421 (rmm‐1, #2632585, SONY), IgD Per‐CP‐Cy5.5 (11‐26c.2a, #2628545, SONY), IgLλ APC (RML‐42, #407306, Biolegend), TCRβ APC (H57‐597, #109212, Biolegend).
Antibodies used for immunoblot analysis: BBS4 (rabbit, a kind gift from Prof. Maxence Nachury, UCSF, CA, USA, recognizing LQVGEALVWTKPVKDPKSKH peptide in exon 15 of human BBS4), β‐actin (mouse, #4967, Cell Signaling), anti‐FLAG (M2, mouse, F1804‐200UG, SIGMA), α‐mouse HRP, α‐rabbit HRP (both from Jackson ImmunoResearch).
Antibodies used for lymphocyte enrichment: biotinylated α‐TCRβ (H57‐597, #553169, BD PharMingen), α‐CD19 (6D5, #115503, Biolegend).
Antibody used for B‐cell activation: polyclonal F(ab')2‐Goat anti‐Mouse IgM (Mu chain), a kind gift from Dr. Tomas Brdicka (Institute of Molecular Genetics of the Czech Academy of Sciences in Prague, henceforth IMG).
Dyes and reagents: 4‐hydroxy‐3‐nitrophenylacetic acid succinimide ester (LGC, Biosearch Technologies), peptides OVA (SIINFEKL), Q4R7 (SIIRFERL), Q4H7 (SIIRFEHL), T4 (SIITFEKL) (Eurogentec or Peptides&Elephants), CFSE and DDAO cell tracker dyes (both Invitrogen), LIVE/DEAD near‐IR dye (Life Technologies), Hoechst 33258 (Life Technologies).
Mice
All mice were 5–25 weeks old and had C57Bl/6J background if not indicated otherwise. Mice were bred in specific‐pathogen‐free facility (IMG) (Flachs et al, 2014). Animal protocols were approved by the Czech Academy of Sciences, in accordance with the laws of the Czech Republic. Males and females were used for the experiments. If possible, age‐ and sex‐matched pairs of animals were used in the experimental groups. If possible, littermates were equally divided into the experimental groups. No randomization was performed since the experimental groups were based solely on the genotype of the mice. In case of the RIP.OVA mice used for the autoimmune diabetes model, mice were assigned to experimental groups randomly (defined by their ID numbers) prior to the visual contact between the experimenter and the mice. The experiments were not blinded since no subjective scoring method was used. For animal studies, minimal sample size was estimated using resource equation approach. As the number of mutant mice was limited due to pre‐weaning lethality, all the available mutant mice (and their WT littermates) were used for experiments. For the diabetes experiments, the number of mice was estimated based on our previous experience with this method.
B1–8 (Sonoda et al, 1997), RIP.OVA (Kurts et al, 1998), OT‐I Rag2 KO/KO (Palmer et al, 2016), Vav‐iCre (Shimshek et al, 2002; de Boer et al, 2003), Cd4‐Cre (Lee et al, 2001) strains were described previously. Mouse Bbs4 tm1a(EUCOMM)Hmgu allele with a gene trap cassette (abbreviated as Bbs4 GT) was obtained from KOMP (UC Davis, CA, USA) as frozen sperm and used for in vitro fertilization. Bbs4 tm1a(EUCOMM)Hmgu mice were crossed with a Flp‐deleter mouse strain, Gt(ROSA)26Sortm2(CAG–flpo,–EYFP)Ics (MGI: 5285396, Philippe Soriano), in order to obtain exon 6 flanked by LoxP sites for conditional or germ line Bbs4 ablation (Bbs4 tm1c(EUCOMM)Hmgu). In order to generate germ line knock‐out mice, the exon 6 was removed by a general Cre‐deleter strain, Gt(ROSA)26Sortm1(ACTB–cre,–EGFP)Ics (MGI: 5285392, Philippe Soriano), resulting in Bbs4 tm1d(EUCOMM)Hmgu (abbreviated as Bbs4 KO) as described previously (Skarnes et al, 2011). Bbs4 +/+ and Bbs4 GT/GT or Bbs4 KO/KO littermates were generated by intercrossing heterozygous animals.
Mice with a null‐mutation in Bbs18 (alias Bbip1) were generated in a C57BL/6N background using a CRISPR genome‐editing system. For this purpose, Cas9 protein and gene‐specific single guide (sg) RNAs (Integrated DNA Technologies, Coralville, IA, USA) were used for a zygote electroporation using a protocol described previously (Chen et al, 2016). sgRNA sequences with the PAM motif in bold (3′ end) were as follows:
sgRNA target 1: CTCTTCCCTGAAAATCGGTGAGG
sgRNA target 2: GGAATAACCAACTGGTCTTTAGG
The correct genome editing was confirmed by PCR amplification in the founder mouse with the primers listed in Appendix Table S5. Generation of Bbs18 KO mice was part of the International Mouse Phenotyping Consortium project (mouse ID 12685, Bbip1em1(IMPC)Ccpcz). The mutant allele was backcrossed for two generations to C57BL/6N mice and for one generation to C57BL/6J mice.
We did not confirm the efficacy of the Bbs4 deletion in Bbs4 FL/FL Vav‐iCre strain. However, this Vav‐iCre mouse line was proven as an efficient gene deleter in the hematopoietic bone marrow cells and splenocytes in our facility previously (Kardosova et al, 2018).
Cell culture
T2‐Kb is a H‐2Kb transgenic subline of the TAP1γ2‐deficient line (Alexander et al, 1989). The bone marrow‐derived stroma cell line, ST2, was kindly provided by Dr. Jana Balounova (IMG). The HEK293T cells were kindly provided by Dr. Tomas Brdicka (IMG). All cell lines were tested for Mycoplasma contamination by PCR on a regular basis. MEF cells were derived from E13.5 mouse embryos. After the removal of the placenta, yolk sac, head, liver, and hear, the tissue was trypsinized and homogenized. The adherent MEFs were genotyped and cultured. All cell lines were cultivated in DMEM supplemented with 10% fetal calf serum (FCS; Gibco), 100 U/ml penicillin (BB Pharma), 100 μg/ml streptomycin (Sigma‐Aldrich), and 40 μg/ml gentamicin (Sandoz).
Cloning, transfection, and generation of BBS4‐deficient cell lines
Bbs4 WT and GT ORFs were amplified from cDNA obtained from the kidney of respective mice. FLAG‐tag was fused to the Bbs4 WT and GT C‐terminally and cloned into pXJ41 vector (kindly provided by Dr. Tomas Brdicka, IMG) using EcoRI/XhoI restriction sites.
HEK293T cells at ~ 50% confluency were transfected with the Bbs4 WT or Bbs4 GT encoding pXJ41 plasmid using the following protocol. Thirty microgram of DNA plasmid was mixed with 75 μg polyethylenimine in 0.5 ml of 0.5% serum antibiotic‐free media for 10 min at room temperature. The mixture was then added onto cells in 3 ml of 0.5% serum and antibiotic‐free media. The media was replaced by DMEM with 10% serum and antibiotics after 3 h. Twenty‐four hours after transfection, the expression of BBS4 was analyzed by α‐FLAG antibody via immunoblotting.
BBS4‐deficient ST2 cell lines were generated using the CRISPR/Cas9 approach. sgRNA targeting mouse Bbs4 gene (NM_175325.3) was designed using the CHOPCHOP tool (Labun et al, 2019). sgRNA was cloned into pSpCas9(BB)‐2A‐GFP (PX458) vector kindly provided by Dr. Feng Zhang (Addgene plasmid #48138) (Ran et al, 2013). sgRNA sequences with the PAM motif in bold (3′ end) for mouse BBS4 are as follows:
Mouse BBS4 sgRNA#1: CATAATCCTTCCGGATATAGTGG
Mouse BBS4 sgRNA#2: GCACTGATTTTTCGCCTGGAAGG
ST2 cells were transfected with PX458 vector with the BBS4 sgRNA using Lipofectamine (Invitrogen). After 48 h, GFP positive cells were sorted as single cells in 96‐well plates using the 488nm laser on FACSAria IIu (BD Bioscience). Clones were analyzed for the expression of BBS4 proteins by immunoblotting and sequencing of DNA surrounding the sgRNA target site.
In vitro stimulation with WNT
Confluent ST2 cells were stimulated with 5 ng/ml recombinant mouse WNT‐3A (RD System) or 200 ng/ml WNT‐5A (Sigma), or left unstimulated in DMEM supplemented with 10% FCS (Gibco), 100 U/ml penicillin (BB Pharma), 100 μg/ml streptomycin (Sigma‐Aldrich), and 40 μg/ml gentamicin (Sandoz) for 48 h at 37°C/CO2 incubator. Total RNA was extracted from the cells, and Il‐7, Cxcl12, and Alpl mRNA expression level was determined by qRT–PCR.
RT–qPCR and end‐point PCR
Total RNA was extracted from tissues (kidney, brain, lymph nodes, spleen, bone marrow) and T cells from indicated mice, ST2 cells or mouse embryonic fibroblasts using Trizol LS (Invitrogen, # 10296010) and transcribed using RevertAid reverse transcriptase (Thermo Fisher, # EP0442) with oligo(dT)18 primers according to the manufacturer's instructions. cDNA was used for end‐point PCR or quantitative PCR using indicated primers (primer sequences are listed in the Appendix Table S5). Samples for quantitative PCR were measured by LightCycler 480 (Roche) in technical triplicates. Obtained C T values were normalized to indicated reference genes.
Enrichment of T and B lymphocytes
T and B lymphocytes for immunoblotting were enriched by positive selection using the Dynabeads Biotin Binder kit (Invitrogen, #11047), and biotinylated α‐TCRβ and α‐CD19 antibodies, respectively. B cells for B‐cell activation assay were enriched by negative selection using biotinylated α‐CD4 and α‐CD8 antibodies with the automated cell separator AutoMACS Pro system (Miltenyi Biotec).
Immunoblotting
For analysis of BBS4 expression, freshly isolated murine organs (testicles, thymi, brain), enriched lymphocytes, ST2 cells, or HEK293T cells were lysed and denatured in Laemmli buffer supplemented with 20mM dithiothreitol. The resulting lysates were separated on a polyacrylamide gel and transferred to nitrocellulose membrane using standard immunoblotting protocols. Membranes were probed with indicated antibodies followed by secondary anti‐rabbit or anti‐mouse HRP‐conjugated antibodies. Staining for β‐actin served as a loading control. The signal was visualized using chemiluminescence immunoblot imaging system Azure c300 (Azure Biosystems, Inc.).
Histological analysis
Testes isolated from 30‐day‐old male mice were collected, immediately dipped into Bouin solution, and fixed for 24 h at 4°C. Paraffin‐embedded tissue blocks were cut with a microtome (Leica RM2255), and the sections were stained with hematoxylin/eosin using standard technique. The images were taken using microscope system Axioplan 2 imaging (Zeiss) using 10×/0.50 NA objective.
Weighting of mice
Bodyweight of Bbs4 +/+, Bbs4 GT/GT, Bbs4 KO/KO mice was recorded weekly starting at 5 weeks of age. All the mice were kept in sex‐matched cages together with their littermates (≤ 6 per cage) and fed a standard chow diet ad libitum.
ELISA
Blood from Bbs4 KO/KO, Bbs4 GT/GT, and their age/sex‐matched controls was collected by submandibular bleeding (Golde et al, 2005) into EDTA‐coated tubes and centrifuged for 15 min at 1,000 g at 4°C in order to separate plasma. Obtained plasma samples were assayed immediately or stored at −80°C for later use. Leptin concentration was measured by mouse leptin ELISA Kit (Cloud‐Clone Corp., SEA084Mu) according to the manufacturer's instructions.
Flow cytometry
Live cells were stained with the above‐listed antibodies on ice. LIVE/DEAD near‐IR dye or Hoechst 33258 were used for discrimination of live and dead cells. Flow cytometry was carried out using an LSRII (BD Bioscience). Data were analyzed using FlowJo software (TreeStar).
B‐cell activation
For activation of nitrophenyl‐specific B cells, splenocytes isolated from B1–8 mice (Bbs4 +/+ and Bbs4 GT/GT) were mixed with 4‐hydroxy‐3‐nitrophenylacetic acid succinimide ester (NP‐Osu)‐loaded T2‐Kb cells at 1:10 or 1:3 ratios, and incubated in DMEM supplemented with 10% FCS (Gibco), 100 U/ml penicillin (BB Pharma), 100 μg/ml streptomycin (Sigma‐Aldrich), and 40 μg/ml gentamicin (Sandoz) for 6 h at 37°C/CO2 incubator.
For activation of Bbs4 KO/KO B cells, spleens and lymph nodes were isolated from Bbs4 KO/KO and control (Bbs4 +/KO or Bbs4 +/+) mice, and B cells were enriched by negative selection using automated cell separator AutoMACS. B cells were stimulated by indicated dose of plastic‐bounded F(ab')2‐Goat anti‐Mouse IgM (Mu chain) antibody in 96 well plate in DMEM supplemented with 10% FCS (Gibco), 100 U/ml penicillin (BB Pharma), 100 μg/ml streptomycin (Sigma‐Aldrich), and 40 μg/ml gentamicin (Sandoz) overnight at 37°C/CO2 incubator.
After incubation, cells were centrifuged (1,000 g, 2 min), resuspended in PBS/0.5% gelatin, stained with antibodies (CD19, B220, IgLλ, CD69) for 30 min on ice, and analyzed by flow cytometry.
T‐cell conjugation assay
T‐cell conjugation assay was performed as previously shown (Palmer et al, 2016). Briefly, OT‐I T cells from Bbs4 FL/FL Cd4‐Cre − or Bbs4 FL/FL Cd4‐Cre + littermates were stained with CFSE cell tracker dye, and splenocytes isolated from C57Bl/6 mice were stained with DDAO cell tracker dye. Splenocytes were loaded with OVA peptide or with indicated altered peptide ligands for 3 h in RPMI/10% FCS, mixed with OT‐I T cells at 2:1 ratio, and centrifuged (1,000 g, 1 min). After 20 min of co‐culture at 37°C/CO2 incubator, cells were fixed by adding formaldehyde (2% final, 35 min). Cells were centrifuged (1,000 g, 2 min), resuspended in PBS/0.5% gelatin, and analyzed by flow cytometry. The result of each of the four biological replicates was an average of technical duplicates.
Model of autoimmune diabetes
The model of autoimmune diabetes has been described previously (Drobek et al, 2018). Briefly, OT‐I cells from Bbs4 FL/FL Cd4‐Cre − or Bbs4 FL/FL Cd4‐Cre + sex‐matched littermates were adoptively transferred into a host RIP.OVA mice intravenously. On the following day, the host mice were immunized with 5,000 CFU of OVA expressing Listeria monocytogenes (Lm). Lm strain expressing OVA has been described previously (Chan et al, 2013). Level of glucose in the urine of RIP.OVA mice was monitored on a daily basis using test strips (GLUKOPHAN, Erba Lachema).
The animal was considered to suffer from diabetes when the concentration of glucose in the urine reached ≥ 1,000 mg/dl for two consecutive days. On day 7 post‐infection, blood glucose was measured using contour blood glucose meter (Bayer).
Blood analysis
Blood from 20–21 weeks old Bbs4 +/+, Bbs4 KO/KO, and Bbs4 GT/GT mice was collected by submandibular bleeding (Golde et al, 2005) into EDTA‐coated tubes and analyzed using BC5300 Vet Auto Hematology Analyzer (Mindray Bio‐Medical Electronics Co., Ltd.).
Analysis of the clinical data of BBS patients
Fully anonymized medical records of 255 BBS patients were obtained from the Clinical Registry Investigating BBS (CRIBBS) by the NIH through the National Center for Advancing Translational Sciences and the Office of Rare Diseases Research (https://grdr.hms.harvard.edu/transmart). Data were extracted from the following categories of the CRIBBS: Clinical data/ Physical findings/ Bone and joint information, Endocrine, Thyroid disorders, Digestive. Although the CRIBBS registry does not contain information about the specific type of arthritis, we assume that the arthritis reported in can be classified as rheumatoid arthritis due to the following facts: (i) the mean age of onset of arthritis in the CRIBBS cohort is 28.7 years, (ii) the mean age on onset of rheumatoid arthritis in general population is 20–29 years (Hayter & Cook, 2012), (iii) the typical onset of osteoarthritis is after 50 years (Martel‐Pelletier et al, 2016).
Medical records of BBS patients attending the BBS multidisciplinary clinic at Guy's Hospital of Guy's and St Thomas' NHS Foundation Trust, London, or Great Ormond Street Hospital, London, were studied in detail with focus on presence of any immune‐related phenotype. In addition to the manual control, the records were also automatically searched for the occurrence of the following terms: autoimm‐, immun‐, thyro‐, inflam‐, diabet‐, T1DM, ulcerative, crohn, IBD, rheuma‐, arthri‐, joints.
Data about the prevalence of autoimmune diseases in the two BBS cohorts were compared to normal prevalence of autoimmune diseases reported in the Autoimmune Registry or relevant publications in case of location‐specific prevalence of hypothyroidism and inflammatory bowel diseases (Stone et al, 2003; Hayter & Cook, 2012; Taylor et al, 2018). Statistical significance of the difference in the prevalence between the BBS patients and overall population was tested using two‐tailed exact binomial test in RStudio (function binom.test, R version: 3.6.2, RStudio version: 1.2.1335). The respective R‐script is deposited in Zenodo (https://doi.org/10.5281/zenodo.3733230).
Results of blood tests of BBS patients (total white blood cell count, leukocyte populations, hemoglobin, platelet counts, mean corpuscular volume, red blood cell count, hematocrit, red cell distribution width and mean corpuscular hemoglobin), their age ranges, genes with causative mutation and body mass indices (BMI) were retrospectively ascertained from medical records stored at the BBS multidisciplinary clinic at Guy's Hospital of Guy's and St Thomas' NHS Foundation Trust, London, or Great Ormond Street Hospital, London. Blood tests were performed during regular medical examination of the patients.
Two distinct sets of controls for the analyzed set of BBS patients were selected from the 14,750 participants of the UK Biobank project (ID: 40103) (Sudlow et al, 2015). First, we selected 10 controls for each patient matching by age range (categories 41–50, 51–60, 60+ years) and sex. These controls had random BMI and thus were used as BMI‐random controls. Second, we selected 10 controls for each patient matching by age range (categories 41–50, 51–60, 60+ years), sex, and BMI. These were used as BMI‐matched controls. For 34 of the 42 patients, we found controls with BMI difference ≤ 0.6 kg/m2. For the eight patients with extreme BMI values, that precise matching was not possible, so that we selected the best‐matching controls available for these cases. As the UK Biobank only includes participants older than 40 years, only BBS patients from this age group were included to this analysis.
All BBS patients gave informed consent or assent. The protocol for this study was approved by the Great Ormond Street Hospital Research Ethics Committee (Project Molecular Genetics of Human Birth Defects—mapping and gene identification, reference #08/H0713/82) and by the ethical committee of the IMG.
Statistical analyses
The statistical significance of the observed differences between experimental groups was calculated using frequentist statistics. Appropriate tests were applied for particular experiments, and P‐values were calculated using R or Prism 5.04 (GraphPad). The names of the tests are indicated in the Figure Legends. The P‐values are indicated in the Figures or in the Figure Legends. Two‐sided tests were used. We adjusted P‐values for multiple comparisons, when applicable. Whenever possible, we used non‐parametric tests. The only exceptions were RT–qPCR experiments with very low number of replicates, where non‐parametric tests were not applicable. It these cases we used one sample t‐test, assuming normal distribution of the data (which could not be statistically tested because of low number of replicates). We indicate the exact P‐value with the exception of some tests where the statistical software did not calculate the exact P‐value if it was very small (P < 0.0001).
Author contributions
MH and OS conceived the study and were in charge of the overall direction and planning. OT, VN, AD, AP, and OS performed the animal experiments. OT, AP, and MH performed experiments with cell lines. EF, KS, and PB collected the data of BBS patients from the Great Ormond Street Hospital and Guy's Hospital in London. VN filtered and analyzed the patients' data. PK, JP, and RS generated Bbs18KO / KO mouse. ZT contributed to the histological analysis of murine testes. OT, VN, and OS wrote the first draft of the manuscript. OT, VN, AP, MH, and OS wrote the final version of the manuscript with the contribution of all the other authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
We thank Ladislav Cupak for technical assistance and genotyping of mice and Dr. Alena Moudra for the assistance with Listeria infection. We are thankful to all colleagues who provided us reagents and cell lines. This study was supported by the Czech Science Foundation (17‐20613Y to MH), and the Charles University Grant Agency (1706119 to OT), and the National Institute for Health Research Biomedical Research Centre at Great Ormond Street Hospital for Children NHS Foundation Trust and University College London. The Group of Adaptive Immunity is supported by an EMBO Installation Grant (3259 to OS) and the Institute of Molecular Genetics of the Czech Academy of Sciences core funding (RVO 68378050). This research has been conducted using the UK Biobank Resource under application number #40103. OS is supported by the Purkyne Fellowship provided by the Czech Academy of Sciences. OT, VN, and AP are students partially supported by the Faculty of Science, Charles University, Prague. The graphical abstract was created with BioRender.com. The BBS18−/− mouse model was prepared in the Czech Centre for Phenogenomics supported by following grants: LM2015040, LM2018126, OP RDI CZ.1.05/2.1.00/19.0395, OP RDI BIOCEV CZ.1.05/1.1.00/02.0109 provided by the Czech Ministry of Education, Youth and Sports and the European Regional Development Fund.
Grantová Agentura České Republiky (GAČR) (17‐20613Y), Grantová Agentura, Univerzita Karlova (GA UK) (1706119), European Molecular Biology Organization (3259), Akademie Věd České Republiky (CAS) (RVO 68378050), Czech Academy of Sciences, Purkyne Fellowship, Charles University, Faculty of Science, Ministerstvo Školství, Mládeže a Tělovýchovy (MŠMT) (LM2015040, LM2018126, OP RDI CZ.1.05/2.1.00/19.0395, OP RDI BIOCEV CZ.1.05/1.1.00/02.0109)
EMBO reports (2021) 22: e50785.
See also: T Kanie & PK Jackson (February 2021)
Contributor Information
Martina Huranova, Email: martina.huranova@img.cas.cz.
Ondrej Stepanek, Email: ondrej.stepanek@img.cas.cz.
Data availability
No primary large datasets have been generated or deposited. R‐scripts used for the analysis of human patient data were deposited in Zenodo (https://doi.org/10.5281/zenodo.3733230).
References
- Alexander J, Payne JA, Murray R, Frelinger JA, Cresswell P (1989) Differential transport requirements of Hla and H‐2 class‐I glycoproteins. Immunogenetics 29: 380–388 [DOI] [PubMed] [Google Scholar]
- Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA (1999) New criteria for improved diagnosis of Bardet‐Biedl syndrome: results of a population survey. J Med Genet 36: 437–446 [PMC free article] [PubMed] [Google Scholar]
- Berbari NF, Lewis JS, Bishop GA, Askwith CC, Mykytyn K (2008) Bardet‐Biedl syndrome proteins are required for the localization of G protein‐coupled receptors to primary cilia. Proc Natl Acad Sci USA 105: 4242–4246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer J, Williams A, Skavdis G, Harker N, Coles M, Tolaini M, Norton T, Williams K, Roderick K, Potocnik AJ et al (2003) Transgenic mice with hematopoietic and lymphoid specific expression of Cre. Eur J Immunol 33: 314–325 [DOI] [PubMed] [Google Scholar]
- Borna S, Drobek A, Kralova J, Glatzova D, Splichalova I, Fabisik M, Pokorna J, Skopcova T, Angelisova P, Kanderova V et al (2020) Transmembrane adaptor protein WBP1L regulates CXCR4 signalling and murine haematopoiesis. J Cell Mol Med 24: 1980–1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullo M, Garcia‐Lorda P, Megias I, Salas‐Salvado J (2003) Systemic inflammation, adipose tissue tumor necrosis factor, and leptin expression. Obes Res 11: 525–531 [DOI] [PubMed] [Google Scholar]
- Chan SSM, Luben R, Olsen A, Tjonneland A, Kaaks R, Teucher B, Lindgren S, Grip O, Key T, Crowe FL et al (2013) Body mass index and the risk for Crohn's disease and ulcerative colitis: data from a European prospective cohort study (The IBD in EPIC Study). Am J Gastroenterol 108: 575–582 [DOI] [PubMed] [Google Scholar]
- Chen S, Lee B, Lee AYF, Modzelewski AJ, He L (2016) Highly efficient mouse genome editing by CRISPR ribonucleoprotein electroporation of zygotes. J Biol Chem 291: 14457–14467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton T, Outram SV, Hager‐Theodorides AL (2007) Sonic hedgehog signalling in T‐cell development and activation. Nat Rev Immunol 7: 726–735 [DOI] [PubMed] [Google Scholar]
- Davis RE, Swiderski RE, Rahmouni K, Nishimura DY, Mullins RF, Agassandian K, Philp AR, Searby CC, Andrews MP, Thompson S et al (2007) A knockin mouse model of the Bardet‐Biedl syndrome 1 M390R mutation has cilia defects, ventriculomegaly, retinopathy, and obesity. Proc Natl Acad Sci USA 104: 19422–19427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rosa V, Procaccini C, Cali G, Pirozzi G, Fontana S, Zappacosta S, La Cava A, Matarese G (2007) A key role of leptin in the control of regulatory T cell proliferation. Immunity 26: 241–255 [DOI] [PubMed] [Google Scholar]
- Drakopoulou E, Outram SV, Rowbotham NJ, Ross SE, Furmanski AL, Saldana JI, Hager‐Theodorides AL, Crompton T (2010) Non‐redundant role for the transcription factor Gli1 at multiple stages of thymocyte development. Cell Cycle 9: 4144–4152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drobek A, Moudra A, Mueller D, Huranova M, Horkova V, Pribikova M, Ivanek R, Oberle S, Zehn D, McCoy KD et al (2018) Strong homeostatic TCR signals induce formation of self‐tolerant virtual memory CD8 T cells. Embo J 37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Abdaloussi E, Graves S, Meng FY, Mandal M, Mashayekhi M, Aifantis I (2006) Hedgehog signaling controls thymocyte progenitor homeostasis and differentiation in the thymus. Nat Immunol 7: 418–426 [DOI] [PubMed] [Google Scholar]
- Finetti F, Paccani SR, Riparbelli MG, Giacomello E, Perinetti G, Pazour GJ, Rosenbaum JL, Baldari CT (2009) Intraflagellar transport is required for polarized recycling of the TCR/CD3 complex to the immune synapse. Nat Cell Biol 11: 1332–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finetti F, Paccani SR, Rosenbaum J, Baldari CT (2011) Intraflagellar transport: a new player at the immune synapse. Trends Immunol 32: 139–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finetti F, Patrussi L, Masi G, Onnis A, Galgano D, Lucherini OM, Pazour GJ, Baldari CT (2014) Specific recycling receptors are targeted to the immune synapse by the intraflagellar transport system. J Cell Sci 127: 1924–1937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flachs P, Bhattacharyya T, Mihola O, Pialek J, Forejt J, Trachtulec Z (2014) Prdm9 Incompatibility controls oligospermia and delayed fertility but no selfish transmission in mouse intersubspecific hybrids. PLoS One 9: e95806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsythe E, Kenny J, Bacchelli C, Beales PL (2018) Managing Bardet‐Biedl syndrome‐now and in the future. Front Pediatr 6: 23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furmanski AL, Saldana JI, Ono M, Sahni H, Paschalidis N, D'Acquisto F, Crompton T (2013) Tissue‐derived hedgehog proteins modulate Th differentiation and disease. J Immunol 190: 2641–2649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiades P, Ogilvy S, Duval H, Licence DR, Charnock‐Jones DS, Smith SK, Print CG (2002) VavCre transgenic mice: a tool for mutagenesis in hematopoietic and endothelial lineages. Genesis 34: 251–256 [DOI] [PubMed] [Google Scholar]
- Gerdes JM, Liu Y, Zaghloul NA, Leitch CC, Lawson SS, Kato M, Beachy PA, Beales PL, DeMartino GN, Fisher S et al (2007) Disruption of the basal body compromises proteasomal function and perturbs intracellular Wnt response. Nat Genet 39: 1350–1360 [DOI] [PubMed] [Google Scholar]
- Goetz SC, Bangs F, Barrington CL, Katsanis N, Anderson KV (2017) The Meckel syndrome‐associated protein MKS1 functionally interacts with components of the BBSome and IFT complexes to mediate ciliary trafficking and hedgehog signaling. PLoS One 12: e0173399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golde WT, Gollobin P, Rodriguez LL (2005) A rapid, simple, and humane method for submandibular bleeding of mice using a lancet. Lab Animal 34: 39–43 [DOI] [PubMed] [Google Scholar]
- Granata M, Skarmoutsou E, Trovato C, Rossi GA, Mazzarino MC, D'Amico F (2017) Obesity, Type 1 diabetes, and psoriasis: an autoimmune triple flip. Pathobiology 84: 71–79 [DOI] [PubMed] [Google Scholar]
- Gremese E, Tolusso B, Gigante MR, Ferraccioli G (2014) Obesity as a risk and severity factor in rheumatic diseases (autoimmune chronic inflammatory diseases). Front Immunol 5: 576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo DF, Beyer AM, Yang BL, Nishimura DY, Sheffield VC, Rahmouni K (2011) Inactivation of Bardet‐Biedl syndrome genes causes kidney defects. Am J Physiol‐Renal 300: F574–F580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo DF, Cui HX, Zhang QH, Morgan DA, Thedens DR, Nishimura D, Grobe JL, Sheffield VC, Rahmouni K (2016) The BBSome controls energy homeostasis by mediating the transport of the leptin receptor to the plasma membrane. Plos Genet 12: e1005890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halac U, Herzog D (2012) Bardet‐Biedl Syndrome, Crohn Disease, primary sclerosing cholangitis, and autoantibody positive thyroiditis: a case report and a review of a cohort of BBS patients. Case Rep Med 2012: 209827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayter SM, Cook MC (2012) Updated assessment of the prevalence, spectrum and case definition of autoimmune disease. Autoimmun Rev 11: 754–765 [DOI] [PubMed] [Google Scholar]
- Jin H, White SR, Shida T, Schulz S, Aguiar M, Gygi SP, Bazan JF, Nachury MV (2010) The conserved Bardet‐Biedl Syndrome proteins assemble a coat that traffics membrane proteins to cilia. Cell 141: 1208–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AR, Milner JJ, Makowski L (2012) The inflammation highway: metabolism accelerates inflammatory traffic in obesity. Immunol Rev 249: 218–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kardosova M, Zjablovskaja P, Danek P, Angelisova P, de Figueiredo‐Pontes LL, Welner RS, Brdicka T, Lee S, Tenen DG, Alberich‐Jorda M (2018) C/EBP gamma is dispensable for steady‐state and emergency granulopoiesis. Haematologica 103: E331–E335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keppler‐Noreuil KM, Blumhorst C, Sapp JC, Brinckman D, Johnston J, Nopoulos PC, Biesecker LG (2011) Brain tissue‐ and region‐specific abnormalities on volumetric MRI scans in 21 patients with Bardet‐Biedl syndrome (BBS). Bmc Med Genet 12: 101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaodhiar L, Ling PR, Blackburn GL, Bistrian BR (2004) Serum levels of interleukin‐6 and C‐reactive protein correlate with body mass index across the broad range of obesity. JPEN J Parenter Enteral Nutr 28: 410–415 [DOI] [PubMed] [Google Scholar]
- King CG, Koehli S, Hausmann B, Schmaler M, Zehn D, Palmer E (2012) T cell affinity regulates asymmetric division, effector cell differentiation, and tissue pathology. Immunity 37: 709–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klink BU, Zent E, Juneja P, Kuhlee A, Raunser S, Wittinghofer A (2017) A recombinant BBSome core complex and how it interacts with ciliary cargo. Elife 6: e27434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurts C, Miller JFAP, Subramaniam RM, Carbone FR, Heath WR (1998) Major histocompatibility complex class I‐restricted cross‐presentation is biased towards high dose antigens and those released during cellular destruction. J Exp Med 188: 409–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Cava A (2017) Leptin in inflammation and autoimmunity. Cytokine 98: 51–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labun K, Montague TG, Krause M, Cleuren YNT, Tjeldnes H, Valen E (2019) CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res 47: W171–W174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PP, Fitzpatrick DR, Beard C, Jessup HK, Lehar S, Makar KW, Perez‐Melgosa M, Sweetser MT, Schlissel MS, Nguyen S et al (2001) A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity 15: 763–774 [DOI] [PubMed] [Google Scholar]
- Lindstrand A, Davis EE, Carvalho CM, Pehlivan D, Willer JR, Tsai IC, Ramanathan S, Zuppan C, Sabo A, Muzny D et al (2014) Recurrent CNVs and SNVs at the NPHP1 locus contribute pathogenic alleles to Bardet‐Biedl syndrome. Am J Hum Genet 94: 745–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindstrand A, Frangakis S, Carvalho CMB, Richardson EB, McFadden Kelsey A, Willer Jason R, Pehlivan D, Liu P, Pediaditakis Igor L, Sabo A et al (2016) Copy‐number variation contributes to the mutational load of Bardet‐Biedl Syndrome. Am J Hum Genet 99: 318–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu PW, Lechtreck KF (2018) The Bardet‐Biedl syndrome protein complex is an adapter expanding the cargo range of intraflagellar transport trains for ciliary export. Proc Natl Acad Sci USA 115: E934–E943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord GM (2006) Leptin as a proinflammatory cytokine. Contrib Nephrol 151: 151–164 [DOI] [PubMed] [Google Scholar]
- Martel‐Pelletier J, Barr AJ, Cicuttini FM, Conaghan PG, Cooper C, Goldring MB, Goldring SR, Jones G, Teichtahl AJ, Pelletier JP (2016) Osteoarthritis. Nat Rev Dis Primers 2: 16072 [DOI] [PubMed] [Google Scholar]
- Martin‐Romero C, Santos‐Alvarez J, Goberna R, Sanchez‐Margalet V (2000) Human leptin enhances activation and proliferation of human circulating T lymphocytes. Cell Immunol 199: 15–24 [DOI] [PubMed] [Google Scholar]
- Michel KD, Uhmann A, Dressel R, van den Brandt J, Hahn H, Reichardt HM (2013) The Hedgehog receptor patched1 in T Cells is dispensable for adaptive immunity in mice. PLoS One 8: e61034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner JJ, Beck MA (2012) The impact of obesity on the immune response to infection. Proc Nutr Soc 71: 298–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, Furusawa J, Ohtani M, Fujii H, Koyasu S (2010) Innate production of T(H)2 cytokines by adipose tissue‐associated c‐Kit(+)Sca‐1(+) lymphoid cells. Nature 463: 540–544 [DOI] [PubMed] [Google Scholar]
- Nakayama K, Katoh Y (2018) Ciliary protein trafficking mediated by IFT and BBSome complexes with the aid of kinesin‐2 and dynein‐2 motors. J Biochem 163: 155–164 [DOI] [PubMed] [Google Scholar]
- Nie YC, Waite J, Brewer F, Sunshine MJ, Littman DR, Zou YR (2004) The role of CXCR4 in maintaining peripheral B cell compartments and humoral immunity. J Exp Med 200: 1145–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederlova V, Modrak M, Tsyklauri O, Huranova M, Stepanek O (2019) Meta‐analysis of genotype‐phenotype associations in Bardet‐Biedl syndrome uncovers differences among causative genes. Hum Mutat 40: 2068–2087 [DOI] [PubMed] [Google Scholar]
- Norrasethada L, Khumpoo W, Rattarittamrong E, Rattanathammethee T, Chai‐Adisaksopha C, Tantiworawit A (2019) The use of mean platelet volume for distinguishing the causes of thrombocytopenia in adult patients. Hematol Rep 11: 23–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Dea D, Parfrey PS, Harnett JD, Hefferton D, Cramer BC, Green J (1996) The importance of renal impairment in the natural history of Bardet‐Biedl syndrome. Am J Kidney Dis 27: 776–783 [DOI] [PubMed] [Google Scholar]
- Ohno T, Aune D, Heath AK (2020) Adiposity and the risk of rheumatoid arthritis: a systematic review and meta‐analysis of cohort studies. Sci Rep 10: 16006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owczarczyk‐Saczonek A, Placek W (2017) Compounds of psoriasis with obesity and overweight. Postep Hig Med Dosw 71: 761–772 [DOI] [PubMed] [Google Scholar]
- Palmer E, Drobek A, Stepanek O (2016) Opposing effects of actin signaling and LFA‐1 on establishing the affinity threshold for inducing effector T‐cell responses in mice. Eur J Immunol 46: 1887–1901 [DOI] [PubMed] [Google Scholar]
- Park HS, Park JY, Yu R (2005) Relationship of obesity and visceral adiposity with serum concentrations of CRP, TNF‐alpha and IL‐6. Diabetes Res Clin Pr 69: 29–35 [DOI] [PubMed] [Google Scholar]
- Paz G, Mastronardi C, Franco CB, Wang KB, Wong ML, Licinio J (2012) Leptin: molecular mechanisms, systemic pro‐inflammatory effects, and clinical implications. Arq Bras Endocrinol 56: 597–607 [DOI] [PubMed] [Google Scholar]
- Plotnikova OV, Pugacheva EN, Golemis EA (2009) Primary cilia and the cell cycle. Method Cell Biol 94: 137–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Procaccini C, Jirillo E, Matarese G (2012) Leptin as an immunomodulator. Mol Aspects Med 33: 35–45 [DOI] [PubMed] [Google Scholar]
- Putoux A, Attie‐Bitach T, Martinovic J, Gubler MC (2012) Phenotypic variability of Bardet‐Biedl syndrome: focusing on the kidney. Pediatr Nephrol 27: 7–15 [DOI] [PubMed] [Google Scholar]
- Rahmouni K, Fath MA, Seo S, Thedens DR, Berry CJ, Weiss R, Nishimura DY, Sheffield VC (2008) Leptin resistance contributes to obesity and hypertension in mouse models of Bardet‐Biedl syndrome. J Clin Invest 118: 1458–1467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR‐Cas9 system. Nat Protoc 8: 2281–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis BS, Lee K, Fanok MH, Mascaraque C, Amoury M, Cohn LB, Rogoz A, Danner OS, Moraes‐Vieira PM, Domingos AI et al (2015) Leptin receptor signaling in T cells is required for Th17 differentiation. J Immunol 194: 5253–5260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter J, Traver D, Willert K (2017) The role of Wnt signaling in hematopoietic stem cell development. Crit Rev Biochem Mol 52: 414–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Roche M, Ritter AT, Angus KL, Dinsmore C, Earnshaw CH, Reiter JF, Griffiths GM (2013) Hedgehog signaling controls T cell killing at the immunological synapse. Science 342: 1247–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Roche M, Asano Y, Griffiths GM (2016) Origins of the cytolytic synapse. Nat Rev Immunol 16: 421–432 [DOI] [PubMed] [Google Scholar]
- Ross AJ, May‐Simera H, Eichers ER, Kai M, Hill J, Jagger DJ, Leitch CC, Chapple JP, Munro PM, Fisher S et al (2005) Disruption of Bardet‐Biedl syndrome ciliary proteins perturbs planar cell polarity in vertebrates. Nat Genet 37: 1135–1140 [DOI] [PubMed] [Google Scholar]
- Rowbotham NJ, Furmanski AL, Hager‐Theodorides AL, Ross SE, Drakopoulou E, Koufaris C, Outram SV, Crompton T (2008) Repression of hedgehog signal transduction in T‐lineage cells increases TCR‐induced activation and proliferation. Cell Cycle 7: 904–908 [DOI] [PubMed] [Google Scholar]
- Schmoeller D, Picarelli MM, Munhoz TP, de Figueiredo CEP, Staub HL (2017) Mean platelet volume and immature platelet fraction in autoimmune disorders. Front Med‐Lausanne 4: 146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah DK, Hager‐Theodorides AL, Outram SV, Ross SE, Varas A, Crompton T (2004) Reduced thymocyte development in sonic hedgehog knockout embryos. J Immunol 172: 2296–2306 [DOI] [PubMed] [Google Scholar]
- Shimshek DR, Kim J, Hubner MR, Spergel DJ, Buchholz F, Casanova E, Stewart AF, Seeburg PH, Sprengel R (2002) Codon‐improved Cre recombinase (iCre) expression in the mouse. Genesis 32: 19–26 [DOI] [PubMed] [Google Scholar]
- Sindhu S, Thomas R, Shihab P, Sriraman D, Behbehani K, Ahmad R (2015) Obesity is a positive modulator of IL‐6R and IL‐6 expression in the subcutaneous adipose tissue: significance for metabolic inflammation. PLoS One 10: e0133494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S, Dulai PS, Zarrinpar A, Ramamoorthy S, Sandborn WJ (2017) Obesity in IBD: epidemiology, pathogenesis, disease course and treatment outcomes. Nat Rev Gastro Hepat 14: 110–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skarnes WC, Rosen B, West AP, Koutsourakis M, Bushell W, Iyer V, Mujica AO, Thomas M, Harrow J, Cox T et al (2011) A conditional knockout resource for the genome‐wide study of mouse gene function. Nature 474: 337–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song RH, Wang B, Yao QM, Li Q, Jia X, Zhang JA (2019) The impact of obesity on thyroid autoimmunity and dysfunction: a systematic review and meta‐analysis. Front Immunol 10: 2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoda E, PewznerJung Y, Schwers S, Taki S, Jung S, Eilat D, Rajewsky K (1997) B cell development under the condition of allelic inclusion. Immunity 6: 225–233 [DOI] [PubMed] [Google Scholar]
- Staal FJT, Chhatta A, Mikkers H (2016) Caught in a Wnt storm: complexities of Wnt signaling in hematopoiesis. Exp Hematol 44: 451–457 [DOI] [PubMed] [Google Scholar]
- Stasi R (2012) How to approach thrombocytopenia. Hematol‐Am Soc Hemat 2012: 191–197 [DOI] [PubMed] [Google Scholar]
- Stephen LA, ElMaghloob Y, McIlwraith MJ, Yelland T, Sanchez PC, Roda‐Navarro P, Ismail S (2018) The ciliary machinery is repurposed for T cell immune synapse trafficking of LCK. Dev Cell 47: 122–132.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone MA, Mayberry JF, Baker R (2003) Prevalence and management of inflammatory bowel disease: a cross‐sectional study from central England. Eur J Gastroenterol Hepatol 15: 1275–1280 [DOI] [PubMed] [Google Scholar]
- Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, Downey P, Elliott P, Green J, Landray M et al (2015) UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Medicine 12: e1001779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura M, Sato MM, Nashimoto M (2011) Regulation of CXCL12 expression by canonical Wnt signaling in bone marrow stromal cells. Int J Biochem Cell Biol 43: 760–767 [DOI] [PubMed] [Google Scholar]
- Tanaka S, Isoda F, Ishihara Y, Kimura M, Yamakawa T (2001) T lymphopaenia in relation to body mass index and TNF‐alpha in human obesity: adequate weight reduction can be corrective. Clin Endocrinol 54: 347–354 [PubMed] [Google Scholar]
- Taylor PN, Albrecht D, Scholz A, Gutierrez‐Buey G, Lazarus JH, Dayan CM, Okosieme OE (2018) Global epidemiology of hyperthyroidism and hypothyroidism. Nat Rev Endocrinol 14: 301–316 [DOI] [PubMed] [Google Scholar]
- Tschop J, Nogueiras R, Haas‐Lockie S, Kasten KR, Castaneda TR, Huber N, Guanciale K, Perez‐Tilve D, Habegger K, Ottaway N et al (2010) CNS leptin action modulates immune response and survival in sepsis. J Neurosci 30: 6036–6047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaisse C, Reiter JF, Berbari NF (2017) Cilia and obesity. Cold Spring Harbor Perspect Biol 9: a028217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Versini M, Jeandel PY, Rosenthal E, Shoenfeld Y (2014) Obesity in autoimmune diseases: not a passive bystander. Autoimmun Rev 13: 981–1000 [DOI] [PubMed] [Google Scholar]
- Visser M, Bouter LM, McQuillan GM, Wener MH, Harris TB (1999) Elevated C‐reactive protein levels in overweight and obese adults. JAMA 282: 2131–2135 [DOI] [PubMed] [Google Scholar]
- Vivar OI, Masi G, Carpier JM, Magalhaes JG, Galgano D, Pazour GJ, Amigorena S, Hivroz C, Baldari CT (2016) IFT20 controls LAT recruitment to the immune synapse and T‐cell activation in vivo . Proc Natl Acad Sci USA 113: 386–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang TT, He CQ (2018) Pro‐inflammatory cytokines: the link between obesity and osteoarthritis. Cytokine Growth Factor Rev 44: 38–50 [DOI] [PubMed] [Google Scholar]
- Wei Q, Zhang YX, Li YJ, Zhang Q, Ling K, Hu JH (2012) The BBSome controls IFT assembly and turnaround in cilia. Nat Cell Biol 14: 950–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheatley DN, Wang AM, Strugnell GE (1996) Expression of primary cilia in mammalian cells. Cell Biol Int 20: 73–81 [DOI] [PubMed] [Google Scholar]
- Williams CL, McIntyre JC, Norris SR, Jenkins PM, Zhang L, Pei QL, Verhey K, Martens JR (2014) Direct evidence for BBSome‐associated intraflagellar transport reveals distinct properties of native mammalian cilia. Nat Commun 5: 5813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wormser O, Gradstein L, Yogev Y, Perez Y, Kadir R, Goliand I, Sadka Y, El Riati S, Flusser H, Nachmias D et al (2019) SCAPER localizes to primary cilia and its mutation affects cilia length, causing Bardet‐Biedl syndrome. Eur J Hum Genet 27: 928–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye F, Nager AR, Nachury MV (2018) BBSome trains remove activated GPCRs from cilia by enabling passage through the transition zone. J Cell Biol 217: 1847–1868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan X, Garrett‐Sinha LA, Sarkar D, Yang S (2014) Deletion of IFT20 in early stage T lymphocyte differentiation inhibits the development of collagen‐induced arthritis. Bone Res 2: 14038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehentmeier S, Pereira JP (2019) Cell circuits and niches controlling B cell development. Immunol Rev 289: 142–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang QH, Nishimura D, Seo S, Vogel T, Morgan DA, Searby C, Bugge K, Stone EM, Rahmouni K, Sheffield VC (2011) Bardet‐Biedl syndrome 3 (Bbs3) knockout mouse model reveals common BBS‐associated phenotypes and Bbs3 unique phenotypes. Proc Natl Acad Sci USA 108: 20678–20683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Yu D, Seo S, Stone EM, Sheffield VC (2012a) Intrinsic protein‐protein interaction‐mediated and chaperonin‐assisted sequential assembly of stable bardet‐biedl syndrome protein complex, the BBSome. J Biol Chem 287: 20625–20635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang QH, Seo S, Bugge K, Stone EM, Sheffield VC (2012b) BBS proteins interact genetically with the IFT pathway to influence SHH‐related phenotypes. Hum Mol Genet 21: 1945–1953 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Data Availability Statement
No primary large datasets have been generated or deposited. R‐scripts used for the analysis of human patient data were deposited in Zenodo (https://doi.org/10.5281/zenodo.3733230).