

Abstract
Lysine succinylation (Ksucc) is an evolutionarily conserved and widespread post‐translational modification. Histone acetyltransferase 1 (HAT1) is a type B histone acetyltransferase, regulating the acetylation of both histone and non‐histone proteins. However, the role of HAT1 in succinylation modulation remains unclear. Here, we employ a quantitative proteomics approach to study succinylation in HepG2 cancer cells and find that HAT1 modulates lysine succinylation on various proteins including histones and non‐histones. HAT1 succinylates histone H3 on K122, contributing to epigenetic regulation and gene expression in cancer cells. Moreover, HAT1 catalyzes the succinylation of PGAM1 on K99, resulting in its increased enzymatic activity and the stimulation of glycolytic flux in cancer cells. Clinically, HAT1 is significantly elevated in liver cancer, pancreatic cancer, and cholangiocarcinoma tissues. Functionally, HAT1 succinyltransferase activity and the succinylation of PGAM1 by HAT1 play critical roles in promoting tumor progression in vitro and in vivo. Thus, we conclude that HAT1 is a succinyltransferase for histones and non‐histones in tumorigenesis.
Keywords: epigenetic regulation, glycolysis, HAT1, succinylation, tumorigenesis
Subject Categories: Cancer; Post-translational Modifications, Proteolysis & Proteomics
Histone acetyltransferase 1 is a succinyltransferase for histone H3 and PGAM1, thereby contributing to epigenetic regulation and glycolysis to promote tumor growth.
Introduction
Protein post‐translational modifications (PTMs) serve as the critical cellular mechanisms to extend the genetic code, participating in various biological processes (Walsh, Garneau‐Tsodikova et al, 2005; Witze, Old et al, 2007; Zhang, Tan et al, 2011). As an essential residue for protein structure and function, lysine modification significantly contributes to the complex PTM networks (Andrews, Strahl et al, 2016). Lysines can be modified by various PTMs, such as acetylation, methylation, phosphorylation, and ubiquitination (Patel & Wang, 2013). The development of non‐restrictive sequence alignment technology provides the possibility to identify novel PTMs. (Bandeira, Tsur et al, 2007; Chen, Chen et al, 2009; Carrico, Meyer et al, 2018; Zhao, Zhang et al, 2018). Histone acetyltransferase 1 (HAT1) is the sole known example of type B histone acetyltransferase which is responsible for acetylation of newly synthesized histones, and the acetylation pattern on newly synthesized histones is crucial for nucleosome assembly in host cells (Shahbazian & Grunstein, 2007), (Parthun, 2007; Parthun, 2013). HAT1 is able to regulate the acetylation on both histones and non‐histones. As a histone acetyltransferase, HAT1 modulates the acetylation of multiple specific histone sites containing H4K5, H4K12, H3K9, H3K18, and H3K27 (Shahbazian & Grunstein, 2007; Nagarajan, Ge et al, 2013). In addition, HAT1 acetylates transcriptional regulator PLZF to modulate the NF‐κB response (Sadler, Suliman et al, 2015). Our previous study also revealed that HAT1‐mediated acetylation contributed to the assembly and epigenetic regulation of hepatitis B virus covalently closed circular DNA minichromosome (Yang, Feng et al, 2019). However, whether HAT1 is able to modulate other PTMs of histones and non‐histones has not been reported.
Recently, lysine succinylation (Ksucc), an evolutionarily conserved and widespread post‐translational modification like lysine acetylation (Kac), has been identified and investigated (Zhang et al, 2011; Weinert, Scholz et al, 2013). Critically, the writers that are responsible for catalyzing the succinylation and the significance of succinylation in tumorigenesis have not been well documented. Accumulated evidence suggests that the succinylation is modulated by acetyltransferase and deacetylases (Zhao et al, 2018). Lysine acetyltransferase 2A (KAT2A) identified as a histone succinyltransferase is responsible for the succinylation of histone H3K79 for the regulation of gene expression (Wang, Guo et al, 2017). It has been reported that SIRT7, serving as a histone desuccinylase, is an eraser of succinylation of histone H3K122 for modulating DNA damage response and genome stability (Li, Shi et al, 2016). However, the writers for succinylation of histone H3K122 have not been reported. For the succinylation of non‐histones, significant progress has been made in identification of deacylation enzymes, called desuccinylation or the eraser of succinylation, in the past several years. The classically annotated HDACs were shown to have activities not only for acetylation but also for other acylations, such as succinylation (Du, Zhou et al, 2011). SIRT5‐mediated lysine desuccinylation impacts diverse metabolic pathways (Park, Chen et al, 2013; Rardin, He et al, 2013). However, there are no ideas for the succinyltransferases (writers) for both histones and non‐histones. Meanwhile, previous investigation of succinylation mainly focuses on the physiology, while the significance of succinylation in tumorigenesis is elusive. Notably, the role of HAT1 in modulation of succinylation remains unclear.
It has been well‐identified that the metabolism remodeling is crucial for the maintenance of cancer cells (Hanahan & Weinberg, 2011; Ward & Thompson, 2012). The enhanced glucose uptake and lactate production are the general features during the metabolism alterations (Liberti & Locasale, 2016). The process is well‐known as the Warburg Effect, a process termed aerobic glycolysis (DeNicola & Cantley, 2015; Liberti & Locasale, 2016). The glycolysis is mainly performed by 10 key enzymes, including hexokinase, phosphoglucose isomerase (GPI), phosphofructokinase (PFK), aldolase (ALDO), triose phosphate isomerase (TPI), glyceraldehyde 3‐phosphate dehydrogenase (GAPDH), phosphoglycerate kinase (PGK), phosphoglyceromutase (PGAM), enolase, and pyruvate kinase (PKM) (Lunt & Vander Heiden, 2011; Huang, Tang et al, 2018). For this reason, it is not surprising that many glycolytic enzymes are commonly overexpressed in tumors (Lunt & Vander Heiden, 2011). Phosphoglyceromutase 1 (PGAM1) catalyzes the conversion of 3‐phosphoglycerate (3‐PG) into 2‐phosphoglycerate (2‐PG) during glycolysis (Hitosugi, Zhou et al, 2012; Hitosugi, Zhou et al, 2013; Oslund, Su et al, 2017). It has been reported that PGAM1 is a glycolytic switch for controlling biosynthesis (Chaneton & Gottlieb, 2012). Protein PTMs, such as phosphorylation and lysine acetylation, are known to swiftly modulate the activities of key glycolytic enzymes in response to acute nutritional signals (Hitosugi et al, 2013; Chang, Su et al, 2015; Liu & Shyh‐Chang, 2017). Variations in succinylation levels occur during physiological conditions and may serve as a signal inducing the differential succinylation and altered function or properties of a subset of cellular proteins. However, whether lysine succinylation of key glycolytic enzymes, such as PGAM1, contributes to the glycolysis in tumorigenesis is poorly understood.
In the present study, we are interested in the role of HAT1 in the modulation of succinylation in tumorigenesis. Surprisingly, we identified that HAT1 was a novel succinyltransferase to modulate the succinylation of histones and non‐histones. HAT1 succinylated histone H3 on K122, conferring to gene expression in cancer epigenetics. For non‐histone, HAT1 catalyzed the succinylation of PGAM1 on K99 to increase its enzyme activity, promoting glycolysis in cancer cells. Functionally, HAT1‐mediated succinylation contributed to tumorigenesis. Thus, our finding provides new insight into the mechanism by which a histone acetyltransferase HAT1 modulates succinylation in cancer development.
Results
HAT1 is able to modulate succinylation on multiple proteins
The Ksucc is a widespread PTM like Kac, raising the possibility that certain acetyltransferase may catalyze the succinylation on the targeted proteins (Weinert et al, 2013; Wang et al, 2017). HAT1 is an acetyltransferase, which is able to regulate the acetylation of histones and non‐histones (Shahbazian & Grunstein, 2007; Sadler et al, 2015). Here, we hypothesized that HAT1 might be able to modulate the succinylation as well. To answer the question, we generated HAT1‐knockout HepG2 cell lines by CRISPR/Cas9 system. Our data showed that the depletion of HAT1 in #3, #20, and #34 clonal cells were identified by Western blot analysis and Sanger sequencing from 48 isolated clonal cells (Appendix Fig S1A and B). To demonstrate our model is effective, we repeated some key assays in two independent clones including Clone #3 and Clone #20, sometimes termed Clone 1 and Clone 2. And if not marked, Clone #3 (Clone 1) cells were used for the experiments. Furthermore, we sought to determine whether the alterations of HAT1 expression could change the Ksucc levels by immunoblotting with the indicated pan‐anti‐Ksucc or pan‐anti‐Kac antibody in the cells. Consistent with the well‐known effect of HAT1 on acetylation, we found that the Kac levels were decreased upon the deletion of HAT1 as a positive control. Surprisingly, the depletion of HAT1 substantially reduced the levels of Ksucc on a number of proteins in HepG2 cells (Fig 1A and Appendix Fig S2A), but not the levels of succinyl‐CoA in the system (Appendix Fig S2B), suggesting that the HAT1 may regulate succinylation of various proteins. And, HAT1 knockout failed to affect the levels of other acylations, such as butyrylation, crotonylation, and propionylation, in the cells (Appendix Fig S2C).
Figure 1. HAT1 is able to modulate succinylation on multiple proteins.
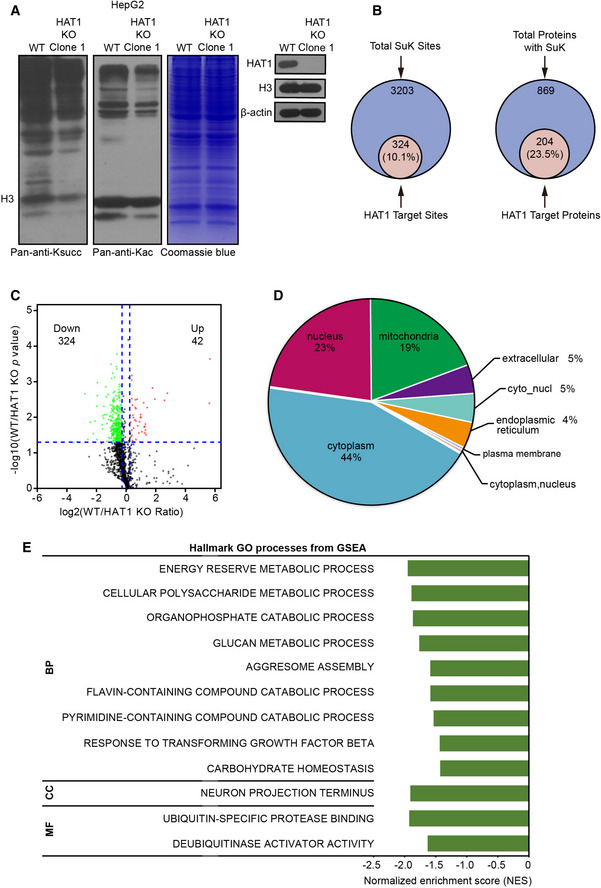
- Total cell lysates from wild‐type (WT) and HAT1 knockout (KO) Clone 1 HepG2 cells were analyzed for the levels of Ksucc and Kac by Western blot analysis (left). The expression levels of HAT1, Histone H3 (H3), and beta‐actin (β‐actin) were measured by Western blot analysis in the cells (right).
- Quantitative proteomics of lysine (K) succinylation was performed in HepG2 and HAT1 knockout HepG2 cells. Graphs show the overlap of the total number of succinylated sites (left) and proteins (right) identified with those targeted by HAT1 (> 1.2‐fold decrease and P < 0.05, Student's t‐test).
- Volcano plot depicting the relative fold change of Ksucc levels in the succinylation quantitative proteomics of WT versus HAT1 KO cells. The vertical blue, dashed lines represent the threshold of 1.2‐fold, and the horizontal blue, dashed lines represents the threshold of P = 0.05. Student's t‐test.
- The diagram showing the cellular distribution of succinylated proteins targeted by HAT1 identified in the quantitative succinyl‐proteome.
- GSEA analysis showing the GO processes for the succinylated proteins targeted by HAT1 in the succinylation quantitative proteomic. BP, biological process; CC, cellular component; MF, molecular function.
To better understand the landscape of HAT1‐mediated succinylation in tumor cells, we quantified Ksucc in wild‐type (WT) and HAT1 knockout (KO) HepG2 cells using a succinylation quantitative proteomic by dimethyl‐labeling succinylation enrichment with pan‐anti‐Ksucc antibody and high‐resolution liquid chromatography‐mass spectrometry (Appendix Fig S3A and B). Importantly, our data revealed that the levels of Ksucc of 324 sites on 204 proteins were down‐regulated by the depletion of HAT1 with varied numbers of modification sites on each substrate and specific motif (Fig 1B and C, and Appendix Fig S3C and D), suggesting that HAT1 is able to modulate the succinylation of multiple proteins. Interestingly, we found that the subcellular distribution of succinylation proteins targeted by HAT1 was widespread in the subcellular compartment (Fig 1D). Gene set enrichment analysis (GSEA), Kyoto Encyclopedia of Genes and Genomes (KEGG), and Gene ontology protein domain analysis revealed that the HAT1‐targeted succinylation proteins displayed important roles in a series of cellular processes (Fig 1E and Appendix Fig S4). Thus, we conclude that HAT1 is able to modulate the succinylation of various proteins.
HAT1 is a novel histone succinyltransferase
Given that the modification of histones played critical roles in the epigenetic regulation and chromatin remodeling and widely affected the gene expression in tumorigenesis (Brien, Valerio et al, 2016; Chen, Li et al, 2017), then we focused on the investigation of HAT1 in modulation of histone succinylation. Interestingly, HAT1 knockout significantly reduced the levels of histone H3 succinylation in HepG2 cells, which was more obvious than those of histone H4 succinylation (Fig 2A and B, and Appendix Fig S5A and B). To validate the effect of HAT1 on histone succinylation, we established a HAT1‐depleted pancreatic cancer PANC1 cell line and a cholangiocarcinoma HuCCT1 cell line. Three shRNAs targeting HAT1 mRNA were designed, among which shHAT1‐3 showed the strongest knockdown efficiency and was used in the following experiments in PANC1 and HuCCT1 cells (Appendix Fig S5C). As expected, we observed the similar results in the cells (Appendix Fig S5D and E), suggesting that HAT1 may be involved in the modulation of histone H3 succinylation.
Figure 2. HAT1 is a novel histone succinyltransferase.
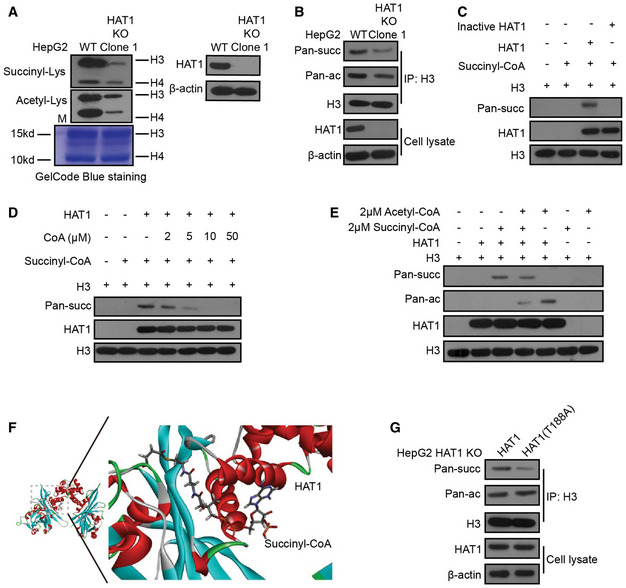
- Histones were extracted from HepG2 (WT) and HAT1 knockout (KO) Clone 1 HepG2 cells. The total histone succinylation and acetylation levels were analyzed by Western blot analysis. Total histone levels were measured by GelCode Blue staining. The efficiency of HAT1 knockout was measured by Western blot analysis.
- Histone H3 was immunoprecipitated from HepG2 cells with or without endogenous HAT1 depletion (by knockout). The levels of succinylation, acetylation, histone H3, HAT1, and β‐actin were analyzed by Western blot analysis in the cells.
- HAT1‐catalyzed histone H3 succinylation was determined by mixing purified HAT1, histone H3, and succinyl‐CoA in the in vitro succinylation assays. Heat‐inactivated HAT1 was used as a negative control. Western blot analysis was performed with the indicated antibodies.
- HAT1‐catalyzed histone H3 succinylation was analyzed by mixing purified HAT1, histone H3, and succinyl‐CoA (2 μM) with or without the addition of the indicated concentrations of CoA. Western blot analysis was performed with indicated antibodies.
- HAT1‐mediated histone H3 succinylation was assessed by mixing purified HAT1, histone H3, and succinyl‐CoA (2 μM) with or without acetyl‐CoA (2 μM). Western blot analysis was performed with the indicated antibodies.
- Diagram illustrating the catalytic pocket of HAT1 bound to succinyl‐CoA. Structures of HAT1 (PDB ID 2P0W) and succinyl‐CoA (PDB ID 5TRL) were used for the modeling by Discovery Studio.
- Histone H3 was immunoprecipitated from HAT1 KO HepG2 cells that were either reconstituted with wild‐type HAT1 or mutant HAT1 (T188A). The levels of succinylation, acetylation, histone H3, HAT1, and β‐actin were tested by Western blot analysis in these cells.
Based on that HAT1 bound to acetyl‐CoA and acted as a histone acetyltransferase (Parthun, 2007; Shahbazian & Grunstein, 2007; Nagarajan et al, 2013), we further tested whether HAT1 directly transferred the succinyl group of succinyl‐CoA to histone H3. The in vitro succinylation assays showed that wild‐type His‐HAT1, but not heat‐inactivated His‐HAT1, could succinylate histone H3 (Fig 2C), suggesting that HAT1 directly catalyzes the succinylation of histone H3. Notably, HAT1‐mediated histone H3 succinylation was inhibited by CoA at high doses (Fig 2D), implying that CoA is able to compete with succinyl‐CoA to bind HAT1, and the CoA group in succinyl‐CoA is involved in its interaction with HAT1. Then, we investigated the effect of HAT1 on succinylation and acetylation of histone H3 in vitro. Interestingly, we observed that the succinyl‐CoA markedly inhibited the HAT1‐mediated histone H3 acetylation, but the acetyl‐CoA only caused a smaller decrease in HAT1‐mediated histone H3 succinylation when the purified HAT1 was mixed with the purified histone H3 in the presence of equal amounts of succinyl‐CoA and acetyl‐CoA (Fig 2E), implying that HAT1‐mediated histone H3 succinylation is moderately affected by acetyl‐CoA. Meanwhile, in vitro acylation assays revealed that HAT1 directly catalyzed the succinylation of histone H3, but not the propionylation, butyrulation, and crotonylation of histone H3 (Appendix Fig S5F).
To further validate the effect HAT1 on succinylation, we analyzed the interaction of HAT1 with succinyl‐CoA by bioinformatics using Discovery Studio. The structures were analyzed by molecular replacement with the known structure of the HAT1‐acetyl‐CoA complex (Protein Data Bank ID: 2P0W). Interestingly, we observed that several amino acid residues of HAT1 were responsible for the interaction of HAT1 with succinyl‐CoA, among which the T188 was one the most crucial sites (Fig 2F and Appendix Fig S6A and B). Then, we measured the velocity of enzyme‐catalyzed reaction at infinite concentration of substrate (V max) and Michaelis constant of the modification of histone H3 by HAT1 in the presence of succinyl‐CoA or acetyl‐CoA (Suganuma & Workman, 2011). Our data revealed that the K m of HAT1 was lower toward succinyl‐CoA than that toward acetyl‐CoA (Appendix Fig S6C). Moreover, HAT1 succinylated histone H3 showed a higher velocity than HAT1 acetylated histone H3 (Appendix Fig S6D), suggesting that HAT1 may have a higher binding affinity for succinyl‐CoA than acetyl‐CoA. Next, we validated that the reconstituted expression of HAT1 (T188A) mutant reduced histone H3 succinylation, but not histone H3 acetylation, relative to the reconstituted expression of wild‐type HAT1 in HepG2, PANC1, and HuCCT1 cells with depleted endogenous HAT1 (Fig 2G and Appendix Fig S6E and F), suggesting that the site of T188 on HAT1 is critical for HAT1‐mediated succinylation. Meanwhile, HAT1 knockout and HAT1 (T188A) mutant failed to affect the levels of propionylation, butyrulation, and crotonylation of histone H3 in the cells (Appendix Fig S6G). Taken together, we conclude that HAT1 is a novel histone succinyltransferase.
HAT1 catalyzes histone H3K122 succinylation for epigenetic regulation
Next, we examined the histone succinylation sites catalyzed by HAT1 in detail. Interestingly, succinylation quantitative proteomic analysis demonstrated 45 succinylation sites on 5 histones, in which K36 of histone H3 was firstly identified (Appendix Fig S7A). Moreover, the histone succinylation sites targeted by HAT1 were listed by further analysis (Appendix Fig S7B). It has been reported that SIRT7, serving as an eraser, is able to catalyze the desuccinylase of histone H3 at the site of K122 (Li et al, 2016). However, the writers for succinylation of histone H3K122 have not been identified. Hence, we supposed that HAT1 might serve as the writer of histone H3K122 succinylation. As expected, we validated that the depletion of HAT1 remarkably reduced the levels of histone H3K122 succinylation and H3K27 acetylation, but not histone H3K122 acetylation, in HepG2, PANC1, and HuCCT1 cells, in which HAT1‐mediated histone H3K27 acetylation was used as a positive control (Fig 3A and Appendix Fig S7C and D). Importantly, reconstituted expression of the HAT1 (T188A) mutant, which failed to affect histone H3K27 acetylation, reduced histone H3K122 succinylation, but not histone H3K122 acetylation, compared with reconstituted expression of wild‐type HAT1 in the cells depleted endogenous HAT1 (Fig 3A and Appendix Fig S7C and E), suggesting that HAT1 is required for the succinylation of histone H3K122. Then, we asked whether HAT1 directly catalyzed the succinylation of histone H3 on K122. Interestingly, the in vitro succinylation assays showed that the wild‐type His‐HAT1, but not heat‐inactivated His‐HAT1, could succinylate histone H3K122 (Fig 3B), while the histone H3 (K122R) mutant could not be succinylated (Fig 3B). Interestingly, CoA could inhibit the HAT1‐mediated H3K122 succinylation at high doses in vitro (Fig 3C), suggesting that HAT1 can directly catalyze the succinylation of H3K122. We further observed that the succinyl‐CoA markedly inhibited HAT1‐mediated histone H3K27 acetylation, but the acetyl‐CoA only caused a smaller decrease in HAT1‐mediated histone H3K122 succinylation when the purified HAT1 was mixed with the purified histone H3 in the presence of equal amounts of succinyl‐CoA and acetyl‐CoA (Appendix Fig S7F), suggesting that HAT1‐mediated histone H3K122 succinylation is moderately affected by acetyl‐CoA. Next, we explored the function of HAT1‐mediated histone H3K122 succinylation. To assess the chromatin occupancy of histone H3K122 succinylation and its correlation with HAT1 at a high resolution, we performed chromatin immunoprecipitation‐sequencing (ChIP‐seq) assays. Surprisingly, we identified 145, 499 H3K122 succinylation‐enriched peaks and 1387 HAT1‐binding‐enriched peaks, among which 609 peaks were common and widely distributed in the genome of HepG2 cells (Appendix Fig S7G). The ChIP‐seq peaks for H3K122 succinylation (2.95%, 4292/145, 499) and HAT1‐binding (4.48%, 62/1387) were occupied in the gene promoter regions of HepG2 cells (Appendix Fig S7H and S8A), suggesting that HAT1‐mediated H3K122 succinylation may be required for the epigenetic regulation and gene expression. As expected, we validated that HAT1 depletion significantly reduced H3K122 succinylation, but not H3K122 acetylation, in the promoter region of representative gene including CREBBP, BPTF, and RPTOR. However, the re‐expression of the HAT1 (T188A) mutant in the cells depleted endogenous HAT1 failed to re‐constitute this effect compared with re‐expression of wild‐type HAT1 in the cells (Fig 3D and Appendix Fig S9A and B). Consistently, the mRNA levels of these genes were inhibited by the depletion of HAT1. However, the reconstituted expression of the HAT1 (T188A) mutant failed to rescue this inhibition compared with reconstituted expression of wild‐type HAT1 in the cells depleted endogenous HAT1 (Fig 3E and Appendix Fig S9C and D), supporting that HAT1 contributes to the modulation of gene expression by succinylation of histone H3K122. However, KAT2A failed to affect histone H3K122 succinylation, and HAT1 could not modulate histone H3K79 succinylation (Appendix Fig S9E), implying that HAT1 and KAT2A may independently work in modulation of histone succinylation. Overall, we conclude that HAT1 catalyzes histone H3K122 succinylation for epigenetic regulation and gene expression (Fig 3F).
Figure 3. HAT1 catalyzes histone H3K122 succinylation for epigenetic regulation.
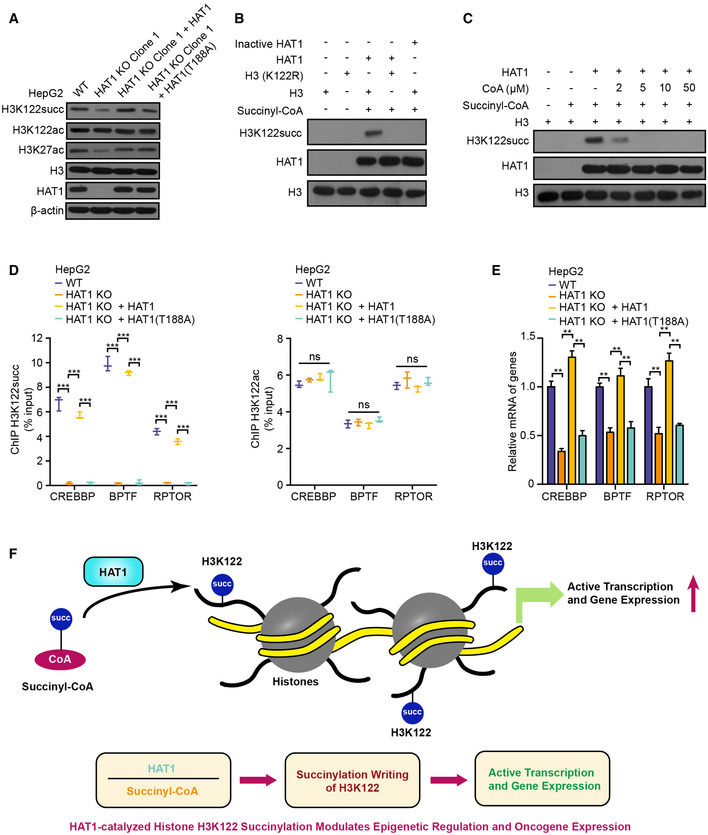
- The levels of histone H3K122 succinylation, histone H3K122 acetylation, histone H3K27 acetylation, histone H3, HAT1, and β‐actin were examined by Western blot analysis in HepG2, HAT1 knockout (KO) Clone 1 HepG2 cells, and in HAT1 KO cells reconstituted with either HAT1 or mutant HAT1 (T188A).
- HAT1‐catalyzed histone H3K122 succinylation was analyzed by mixing purified HAT1, histone H3 or histone H3 mutant (K122R), and succinyl‐CoA in the in vitro succinylation assays. Heat‐inactivated HAT1 was used as a negative control. Western blot analysis was performed with indicated antibodies.
- HAT1‐catalyzed histone H3K122 succinylation was analyzed by mixing purified HAT1, histone H3, and succinyl‐CoA (2 μM) with or without the addition of the indicated concentrations of CoA. Western blot analysis was performed with indicated antibodies.
- The effect of HAT1 on histone H3K122 succinylation and histone H3K122 acetylation at the indicated gene promoters was determined by ChIP assays with anti‐H3K122 succinylation antibody and anti‐H3K122 acetylation antibody and followed by quantitative PCR with primers of the promoter regions of CREBBP, BPTF, and RPTOR in WT HepG2 cells, HAT1 KO HepG2 cells, and HAT1 KO HepG2 cells reconstituted with either wild‐type HAT1 or mutant HAT1 (T188A). N = 3 biological replicates. Data are presented as mean ± SD. Student's t‐test, ***P < 0.001; ns, no significance.
- The mRNA levels of CREBBP, BPTF, and RPTOR were measured by RT–qPCR in HepG2 cells, HAT1 KO HepG2 cells, and HAT1 KO HepG2 cells reconstituted with either wild‐type HAT1 or mutant HAT1 (T188A). N = 3 biological replicates. Data are presented as mean ± SD. Student's t‐test, **P < 0.01.
- A model illustrating how HAT1‐catalyzed histone H3K122 succinylation modulates epigenetic regulation and gene expression.
HAT1‐mediated non‐histone succinylation specifically targets glycolysis
In addition to histone succinylation, the succinylation of non‐histone is also widely identified, which affects various cellular processes such as metabolic pathway (Park et al, 2013; Weinert et al, 2013). Previous studies reported that SIRT5 served as an eraser to modulate the lysine desuccinylation and impact diverse metabolic pathways (Du et al, 2011; Park et al, 2013; Rardin et al, 2013; Nishida, Rardin et al, 2015). However, up to now the writers of non‐histone succinylation have not been well documented. Accordingly, we further explored the effect of HAT1 on non‐histone succinylation. We performed the KEGG pathway enrichment analysis and interaction analysis on the potential HAT1‐targeted non‐histones identified in the succinylation quantitative proteomic. Interestingly, our data showed that HAT1 specifically mediated the succinylation of proteins involving glycolysis, including 7 of 10 key enzymes of glycolysis such as GPI, TPI, GAPDH, PGK, PGAM, enolase, and PKM (Fig 4A–C and Appendix Fig S10). Moreover, we validated that the depletion of HAT1 significantly reduced the relative levels of glucose consumption, lactate production, and intracellular abundance of many key metabolites related to glycolysis, such as pyruvate, ATP and NADH in the HepG2, PANC1, and HuCCT1 cells (Fig 4D–F, and Appendix Fig S11A–F). Interestingly, reconstituted expression of HAT1 (T188A) mutant in the cells depleted endogenous HAT1 also reduced these markers compared with reconstituted expression of wild‐type HAT1 (Fig 4D–F and Appendix Fig S11A–F), suggesting that HAT1‐mediated succinylation promotes glycolysis. Thus, we conclude that HAT1‐mediated non‐histone succinylation specifically targets glycolysis.
Figure 4. HAT1‐mediated non‐histone succinylation specifically targets glycolysis.
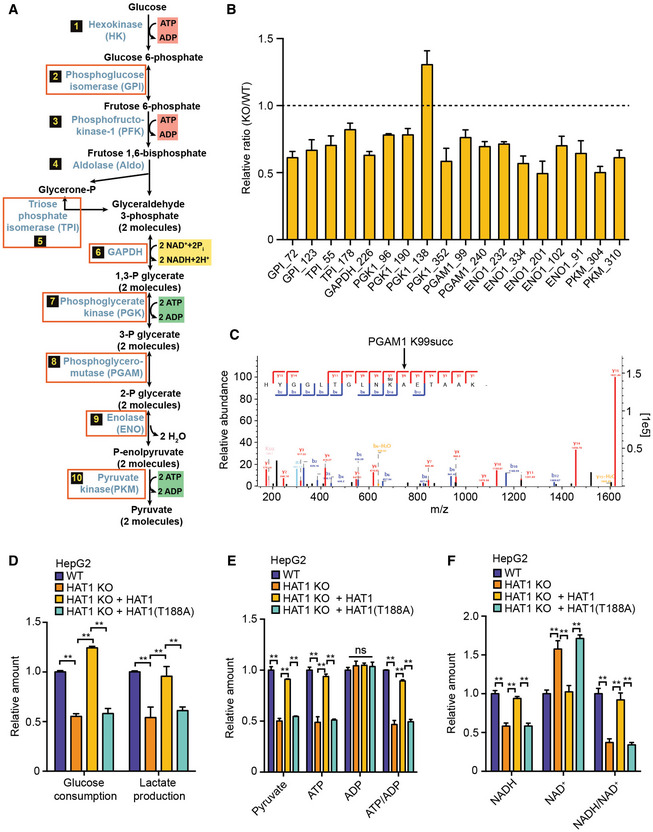
-
ADiagram showing 7 of 10 key glycolytic enzymes that were identified as HAT1 succinylation targets (red boxes) in the succinylation quantitative proteomics.
-
BBar graphs depicting the relative ratio of succinylation in HAT1 KO versus WT of the 7 key enzymes in the glycolysis pathway depicted in (A). N = 3 biological replicates. Data are presented as mean ± SD.
-
CThe diagram showed the MS/MS spectra of PGAM1 K99succ peptide for its identification and quantification. The b and y represented the fragment ions of the N and C termini in peptide backbone, respectively. m/z, mass‐to‐charge ratio.
-
DThe decrease of glucose and increase of lactate in culture medium was measured by ELISA assays in HepG2 cells, HAT1 KO HepG2 cells, and HAT1 KO HepG2 cells reconstituted with either wild‐type HAT1 or mutant HAT1 (T188A). N = 3 biological replicates. Data are presented as mean ± SD. Student's t‐test, **P < 0.01.
-
E, FThe relative amounts of indicated metabolites were quantified by ELISA assays in HepG2 cells, HAT1 KO HepG2 cells, and HAT1 KO HepG2 cells reconstituted with either wild‐type HAT1 or mutant HAT1 (T188A). N = 3 biological replicates. Data are presented as mean ± SD. Student's t‐test, **P < 0.01; ns, no significance.
HAT1 succinylates PGAM1 to enhance glycolysis
Protein PTMs, such as phosphorylation and lysine acetylation, are known to swiftly modulate the activities of key glycolytic enzymes in response to acute nutritional signals (Hitosugi et al, 2013; Chang et al, 2015; Liu & Shyh‐Chang, 2017). Next, we further investigated whether the modulation of glycolysis by HAT1 is due to the succinylation of glycolytic enzymes. Firstly, we validated that the succinylation of PGAM1, ENO1, and PKM was inhibited by depletion of HAT1 in the HepG2, PANC1, and HuCCT1 cells, and re‐expression of the HAT1 (T188A) mutant in the cells depleted endogenous HAT1 failed to rescue the succinylation of PGAM1, ENO1, and PKM compared with reconstituted expression of wild‐type HAT1 (Fig 5A and Appendix Fig S12A–C), suggesting that HAT1 is responsible for the succinylation of PGAM1, ENO1, and PKM. Then, our data showed that the HAT1‐mediated succinylation remarkably affected the enzyme activity of PGAM1 among three enzymes in HepG2 cells (Appendix Fig S12D). Hence, we selected PGAM1 for further investigation. The interaction of HAT1 with PGAM1 was validated by immunoprecipitation (Fig 5A and Appendix Fig S12A and B). Meanwhile, the acetylation of PGAM1 was observed in the cells, but not propionylation, butyrulation, and crotonylation of PGAM1. But, HAT1 knockout and HAT1 (T188A) mutant failed to affect the acetylation of PGAM1 in the cells (Appendix Fig S12E). Moreover, since we observed that HAT1 could catalyze histone H3K122 succinylation for epigenetic regulation and gene expression (Fig 3A), we concerned whether PGAM1 was transcriptionally regulated by HAT1‐mediated histone H3 succinylation. ChIP assays showed that H3K122 succinylation and H3K122 acetylation were undetectable on the promoter of PGAM1 in the cells (Appendix Fig S12F). Consistently, HAT1 knockout and re‐expression of the HAT1 (T188A) mutant failed to modulate the mRNA expression of PGAM1 in the cells (Appendix Fig S12G), suggesting that HAT1 modulates the succinylation of PGAM1 at post‐translational level, but not at transcriptional level mediated by histone H3K122 succinylation. The depletion of HAT1 significantly reduced the relative intracellular PGAM1 activity and the levels of 2‐PG, which was the metabolite directly modulated by PGAM1, and increased the levels of 3‐PG in HepG2, PANC1, and HuCCT1 cells (Fig 5B and C, and Appendix Fig S13A and B). However, re‐expression of the HAT1 (T188A) mutant in the cells depleted endogenous HAT1 failed to rescue this effect compared with re‐expression of wild‐type HAT1 (Fig 5B and C, and Appendix Fig S13A and B), suggesting that HAT1 modulates enzyme activity of PGAM1 by succinylation.
Figure 5. HAT1 succinylates PGAM1 to enhance glycolysis.
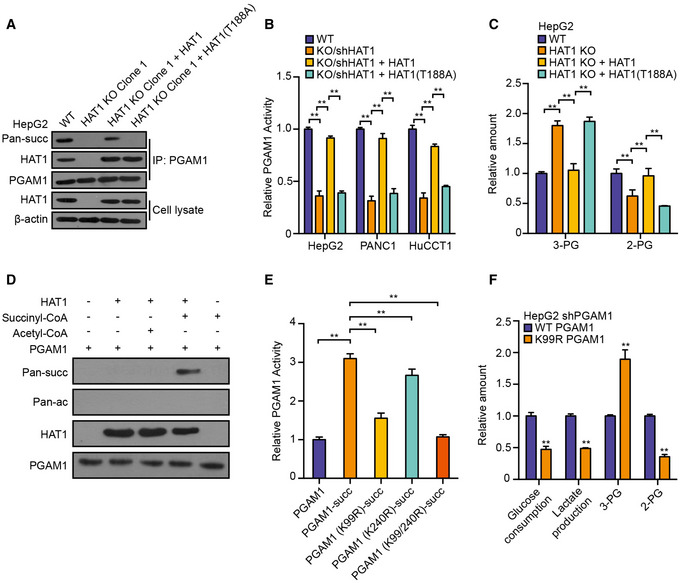
- PGAM1 was immunoprecipitated from HepG2, HAT1 KO Clone 1 HepG2, and HAT1 KO Clone 1 HepG2 cells reconstituted with either wild‐type HAT1 or mutant HAT1 (T188A). The levels of succinylation, PGAM1, HAT1, and β‐actin were analyzed by Western blot analysis in the cells.
- Relative PGAM1 activity was measured by PGAM1 activity assays in the indicated tumor cells. N = 3 biological replicates. Data are presented as mean ± SD. Student's t‐test, **P < 0.01.
- The decrease of 2‐PG and increase of 3‐PG were measured by ELISA assays in HepG2, HAT1 KO HepG2, and HAT1 KO HepG2 cells reconstituted with either wild‐type HAT1 or mutant HAT1 (T188A). N = 3 biological replicates. Data are presented as mean ± SD. Student's t‐test, **P < 0.01.
- HAT1‐catalyzed PGAM1 succinylation was analyzed by mixing purified HAT1, PGAM1, and succinyl‐CoA/acetyl‐CoA in the in vitro succinylation assays. Western blot analysis was performed with indicated antibodies.
- Purified HAT1, PGAM1 or PGAM1 mutant, and succinyl‐CoA were mixed in the in vitro succinylation assays and relative PGAM1 activity was measured by PGAM1 activity assays in vitro. N = 3 biological replicates. Data are presented as mean ± SD. Student's t‐test, **P < 0.01.
- The decrease of glucose and increase of lactate in the culture medium and the relative amounts of 2‐PG and 3‐PG were analyzed by ELISA assays in HepG2 cells depleted endogenous PGAM1 and reconstituted expression of PGAM1 or PGAM1 mutant (K99R). N = 3 biological replicates. Data are presented as mean ± SD. Student's t‐test, **P < 0.01.
To explore whether HAT1 directly catalyzed the succinylation of PGAM1, we performed the in vitro succinylation assays. Our data showed that wild‐type His‐HAT1 was able to succinylate PGAM1, but not acetylated PGAM1 (Fig 5D). Meanwhile, the in vitro acylation assays revealed that HAT1 directly catalyzed the succinylation of PGAM1, but not the propionylation, butyrulation, and crotonylation of PGAM1 (Appendix Fig S13C–E). Succinylation quantitative proteomic showed that HAT1 could modulate the succinylation of PGAM1 on K99 and K240. Importantly, we revealed that the succinylation of PGAM1 mutant (K99R) was significantly reduced relative to the succinylation of PGAM1 mutant (K240R) when the purified HAT1 was mixed with the purified PGAM1, PGAM1 mutant (K99R), PGAM1 mutant (K240R), and PGAM1 mutant (K99R/K240R) in the presence of succinyl‐CoA (Appendix Fig S13F), suggesting that PGAM1 K99 is the key succinylation site. Consistently, the findings of in vitro PGAM1 activity were similar to the data of in vitro succinylation assays (Fig 5E and Appendix Fig S14A and B), suggesting that HAT1 modulates PGAM1 activity by catalyzing its succinylation. Functionally, we observed that the mutant of PGAM1 (K99R) inhibited the glucose consumption, lactate production, and the levels of 2‐PG and increased the levels of 3‐PG in the HepG2, PANC1, and HuCCT1 cells (Fig 5F and Appendix Fig S14C–E), suggesting that the succinylation of PGAM1 may contribute to the glycolysis. Taken together, we conclude that HAT1 contributes to the glycolysis through modulation of PGAM1 succinylation, but not acetylation, in tumor cells.
HAT1‐mediated succinylation displays critical roles in tumor growth
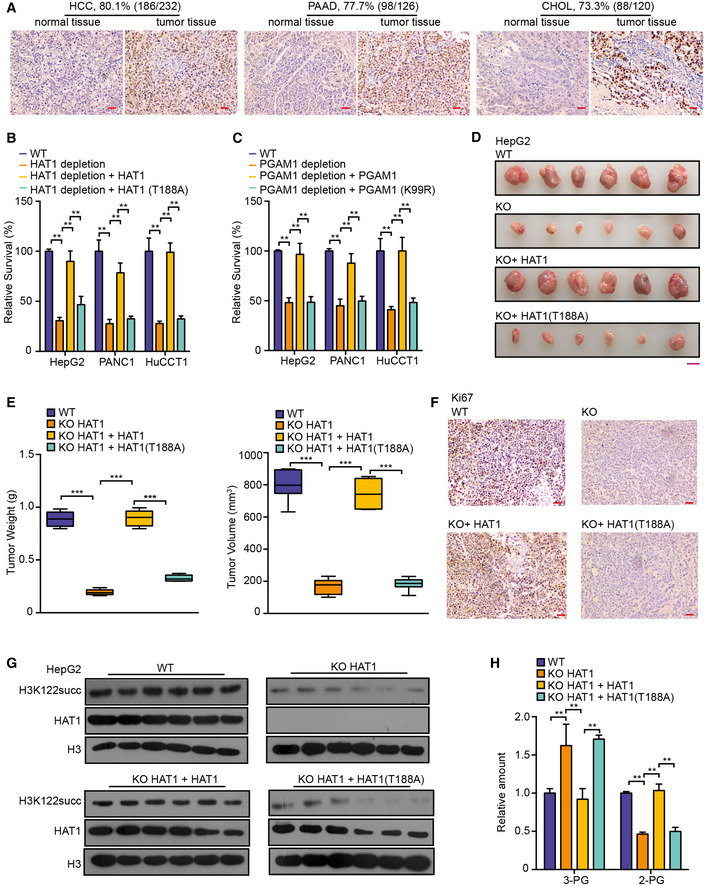
Based on the extensive influence and precise regulation of HAT1‐mediated succinylation in tumor cells, we further explored the roles of HAT1‐mediated succinylation in tumorigenesis. Firstly, we examined the clinical significance of HAT1 expression. Strikingly, immunohistochemistry showed that HAT1 was positive in 80.1% (186/232) clinical liver cancer tissues, 77.7% (98/126) clinical pancreatic cancer tissues, and 73.3% (88/120) clinical cholangiocarcinoma tissues (Fig 6A), suggesting that HAT1 broadly expresses in cancers. Functionally, MTS assays showed that the depletion of HAT1 remarkably reduced the proliferation of liver cancer HepG2 cells, pancreatic cancer PANC1 cells, and cholangiocarcinoma HuCCT1 cells (Fig 6B). Moreover, re‐expression of HAT1 (T188A) mutant failed to rescue the effect relative to reconstituted expression of wild‐type HAT1 in the cells depleted endogenous HAT1 (Fig 6B), suggesting that HAT1‐mediated succinylation may contribute to the proliferation of tumor cells in vitro. Moreover, MTS assays revealed that the depletion of PGAM1 remarkably reduced the proliferation in above cells, and re‐expression of the PGAM1 (K99R) mutant failed to rescue the effect compared with reconstituted expression of wild‐type PGAM1 in the cells depleted endogenous PGAM1 (Fig 6C), suggesting that the succinylation of PGAM1 may enhance the proliferation of tumor cells in vitro. Strikingly, nude mice tumorigenicity experiments displayed that the depletion of HAT1 significantly inhibited the growth of HepG2 and PANC1 cells, in which the expression of Ki67 and succinylation of H3K122, and the levels of glycolytic markers were decreased as well in the tumor tissues (Fig 6D–H. and Appendix Fig S15A–E). Importantly, reconstituted expression of the HAT1 (T188A) mutant HAT1 failed to work compared with the reconstituted expression of wild‐type HAT1 in the cells depleted endogenous HAT1 (Fig 6D–H. and Appendix Fig S15A–E), suggesting that the HAT1‐mediated succinylation may play crucial roles in promotion of tumor growth in vivo. In addition, nude mice tumorigenicity experiments showed that the reconstituted expression of the PGAM1 (K99R) mutant failed to rescue the growth of PANC1 cells compared with reconstituted expression of wild‐type PGAM1 in the cells depleted endogenous PGAM1 (Appendix Fig S16A–E), suggesting that the succinylation of PGAM1 may contribute to the growth of pancreatic cancer as well. Thus, we conclude that HAT1‐mediated succinylation is required for tumor growth.
Figure 6. HAT1‐mediated succinylation displays critical roles in tumor growth.
-
AHAT1 levels were assessed by immunohistochemical staining in clinical tissues of liver cancer patients, pancreatic cancer patients, and cholangiocarcinoma patients. Scale bar = 100 μm
-
BThe effect of HAT1‐mediated succinylation on cancer cell proliferation was measured by MTS assays in the indicated cells. N = 3 biological replicates. Data are presented as mean ± SD. Student's t‐test, **P < 0.01.
-
CThe effect of PGAM1 succinylation on cancer cell proliferation was measured by MTS assays in the indicated cells. N = 3 biological replicates. Data are presented as mean ± SD. Student's t‐test, **P < 0.01.
-
D–HHepG2 cells, HAT1 KO HepG2 cells, and HAT1 KO HepG2 cells reconstituted with either wild‐type HAT1 or mutant HAT1 (T188A) were subcutaneously injected into athymic nude mice (N = 6). (D, E) Photographs showing the tumors from the nude mice. Tumor volumes and average tumor weight were calculated. N = 6 biological replicates. Scale bar = 10 mm. In the boxplots, boxes extend from the 25th to 75th percentiles (inter‐quartile range (IQR)), central band represents the median, and whiskers indicate the lowest and highest data within 1.5 × IQR from the lower and upper quartiles, respectively. Student's t‐test, ***P < 0.001. (F) Immunohistochemical analysis of the indicated tumor sections was performed with an anti‐Ki67 antibody. The images represent the results of six tissue slides. Scale bar = 100 μm. (G) Western blot analysis was performed with the indicated antibodies. Representative images of triplicate experiments are shown. (H) The relative amounts of 3‐PG and 2‐PG were determined by ELISA assays in the tumor tissues. N = 6 biological replicates. Data are presented as mean ± SD. Student's t‐test, **P < 0.01.
Discussion
PTM is one of the most wide and precise regulation mechanisms, playing important roles in the cellular physiological and pathological processes (Walsh et al, 2005). The reversible succinylation of lysine has been identified with largely unknown prevalence and biological functions (Zhang et al, 2011; Weinert et al, 2013). HAT1 as an acetyltransferase is able to regulate the acetylation of histones and non‐histones (Parthun, 2007; Shahbazian & Grunstein, 2007; Sadler et al, 2015). Our previous study also identified that HAT1‐mediated histone acetylation modulated the assembly and epigenetic regulation of hepatitis B virus covalently closed circular DNA minichromosome (Yang et al, 2019). In this study, we try to identify the novel succinylation function of HAT1 on both histones and non‐histones in tumorigenesis.
Succinylation is a widespread PTM like acetylation (Zhang et al, 2011; Weinert et al, 2013). KAT2A is a histone H3 succinyltransferase (Wang et al, 2017), which is responsible for the succinylation of histone H3K79. In this study, we found that the depletion of HAT1 in HepG2 cells substantially reduced Ksucc levels on a number of proteins including histones and non‐histones. Meanwhile, HAT1 knockout failed to affect the levels of other acylations, including propionylation, butyrylation, and crotonylation, in the HepG2 cells. It suggests that succinylation may be one of the new PTMs modulated by HAT1. To better understand the landscape of HAT1‐mediated succinylation in tumor cells, we quantified Ksucc in WT (wild‐type) and HAT1 KO (knockout) HepG2 cells using a succinylation quantitative proteomic in conjunction with immunoaffinity enrichment with pan‐anti‐Ksucc antibody and mass spectrometric analyses. Strikingly, our data revealed that the levels of Ksucc of 324 sites of 204 proteins were down‐regulated by depletion of HAT1 with varied numbers of modification sites on each substrate and specific motif. It suggests that HAT1 is essential for the succinylation of these proteins. In addition, we found that the subcellular distribution of the succinylated proteins targeted by HAT1 was widespread. It suggests that HAT1‐mediated succinylation may be involved in multiple cellular processes. Previous study of succinylation mainly focused on the function of physiology (Zhang et al, 2011; Weinert et al, 2013). In this study, GSEA, KEGG, GO, and protein domain analysis showed that the HAT1‐targeted succinylation was significantly enriched in the multiple cellular pathways. It suggests that HAT1‐mediated succinylation may globally affect the cellular physiological and pathological processes.
The PTM of histones plays critical roles in epigenetic regulation and chromatin remodeling, which widely affects gene expression (Brien et al, 2016; Chen et al, 2017). For succinylation of histones, KAT2A is the only one histone succinyltransferase until now, which is responsible for the succinylation of histone H3K79 (Wang et al, 2017). In this study, we discovered that HAT1 was able to catalyze the succinylation of histone H3. It suggests that HAT1 is a novel histone H3 succinyltransferase. It has been reported that SIRT7, serving as a histone desuccinylase, is an eraser for desuccinylation of H3K122 for modulating DNA damage response and genome stability (Li et al, 2016). However, the writers for succinylation of histone H3K122 are unknown. Strikingly, here we found that HAT1 could catalyze histone H3K122 succinylation, which contributed to the epigenetic regulation and gene expression, such as CREBBP, BPTF, and RPTOR in tumor cells. It suggests that HAT1 is a novel writer of histone succinylation, and HAT1‐mediated histone succinylation has wide implication in cancer epigenetics. Accordingly, we were interested in the relationship between the two writers such as KAT2A and HAT1 in histone succinylation. However, we observed that KAT2A failed to affect histone H3K122 succinylation, and HAT1 could not modulate histone H3K79 succinylation as well. It suggests that HAT1 and KAT2A independently work in the modulation of histone succinylation.
In addition to histones, the succinylation of multiple non‐histone substrates was identified (Zhang et al, 2011; Weinert et al, 2013). For succinylation of non‐histones, significant progress has been made in identification of deacylation enzymes, called desuccinylation or the eraser of succinylation, in the past several years (Zhao et al, 2018). However, there are no ideas about the succinyltransferases (writers) for non‐histones. Notably, the succinyltransferases catalyzing the succinylation on both histones and non‐histones have not been identified until now. Interestingly, in this study we reported that HAT1 specifically mediated the non‐histone succinylation involving glycolysis. It suggests that HAT1‐mediated succinylation may contribute to glycolysis. Functionally, our data suggest that HAT1‐mediated succinylation promotes glycolysis. Specifically, we found that HAT1 was required for the glycolysis through succinylation of PGAM1, but not its acetylation, in tumor cells. It suggests that the HAT1‐mediated PGAM1 succinylation is critical for glycolysis in tumorigenesis.
Basically, the study for succinylation still remains at an early stage, and the function of succinylation in diseases, especially in tumors, is unclear (Zhang et al, 2011; Weinert et al, 2013; Wang et al, 2017). It has been reported that KAT2A is able to promote the tumor growth of neuroglioma cells by modulation of histone H3K79 succinylation (Wang et al, 2017). According to the critical role of HAT1‐mediated succinylation in epigenetic regulation and glycolysis, here we were interested in the role of HAT1‐mediated succinylation in tumorigenesis. Clinically, immunohistochemistry showed that the expression of HAT1 was significantly elevated in liver cancer tissues, pancreatic cancer tissues, and cholangiocarcinoma tissues. It suggests that HAT1 may display important roles in tumors. Next, we further explore the function of HAT1‐mediated succinylation in vitro and in vivo. Our data suggest that HAT1‐mediated succinylation is required for the tumor growth including liver cancer and pancreatic cancer. Thus, we conclude that HAT1 displays a novel role in the modulation of succinylation in tumorigenesis. Our finding provides new insights into the mechanism by which a histone acetyltransferase HAT1 modulates succinylation in cancer progression.
Overall, we summarize a model for the succinylation function of HAT1 in tumorigenesis (Appendix Fig S17). As a writer, HAT1 affects Ksucc levels on various proteins including histones and non‐histones, which is involved in multiple cellular pathways of physiology and pathology. For histones, HAT1 is a novel histone succinyltransferase and enhances the epigenetic regulation and gene expression profiling by catalyzing histone H3K122 succinylation. For non‐histones, HAT1 facilitates glycolysis through catalyzing the succinylation of PGAM1 on K99 in cancer cells. Functionally, HAT1‐mediated succinylation is essential for the tumorigenesis.
Materials and Methods
Establishment of stable depletion cell lines
HAT1 knockout HepG2 cell lines were established by using CRISPR/Cas9 system previously. Briefly, multiple sgRNAs for HAT1 DNA target were screened from the BlueHeron (https://benchling.com). The sgRNAs were designed based on the target site sequence (21 bp) and needed to be flanked on the 3' end by a 3 bp NGG PAM sequence. DNA oligonucleotides, which harbored variable 21‐nt sequences for Cas9 targeting, were annealed to generate short double‐strand DNA and inserted into BbsI‐digested plasmid pX330 according to previous study (Ran, Hsu et al, 2013; Maddalo, Manchado et al, 2014). To assess the efficiency generated by Cas9/sgRNA, the cell lysate was subjected to Western blot analysis.
Three shRNAs targeting HAT1 were designed, among which shHAT1‐3 showed the strongest knockdown efficiency in 293T cells and was used in the following experiments. Three shRNAs targeting PGAM1 mRNA were designed, among which shPGAM1‐2 showed the strongest knockdown efficiency in 293T cells and was used in the following experiments in HepG2, PANC1, and HuCCT1 cells. All targeted sequences were listed in Appendix Table S1. The stably depletion of HAT1 or PGAM1 in the cells was generated by using lentiviruses containing control shRNA vector or shRNA constructs expressing shRNA specifically against human HAT1, and lentiviral particles were then prepared to stably infected PANC1 and HuCCT1 cells. The deletion of endogenous HAT1 was confirmed by Western blot analysis.
Cell lines and cell culture
Human hepatoma cell line HepG2, human pancreatic cancer cell line PANC1, and 293T cell line were maintained in Dulbecco's modified Eagle’s medium (DMEM) (Gibco, Grand Island, NY, USA). Human cholangiocarcinoma cell line HuCCT1 was cultured in Roswell Park Memorial Institute 1640 medium (RPMI 1640) (Gibco, Grand Island, NY, USA). All of the cell lines were cultured in DMEM or RPMI 1640 medium supplemented with 10% FBS, 100 U/ml penicillin, and 100 mg/ml streptomycin in 5% CO2 at 37°C. The cells were cultured in a 6‐well, 24‐well, or 96‐well plate for 12 h and then were transfected with plasmid or shRNAs. The transfections were performed using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA).
Dimethyl‐labeling succinylation quantitative proteomics and analysis
The dimethyl‐labeling succinylation quantitative proteomics in HepG2 and HAT1 knockout HepG2 cells were performed by Jingjie PTM BioLab Co. Ltd (China). Briefly, sample was sonicated three times on ice using a high intensity ultrasonic processor in lysis buffer (8 M urea, 1% Protease Inhibitor Cocktail. Note: For PTM experiments, inhibitors were also added to the lysis buffer, e.g. 3 μM TSA and 50 mM NAM). The remaining debris was removed by centrifugation at 12,000 g at 4°C for 10 min. Finally, the supernatant was collected, and the protein concentration was determined with BCA kit according to the manufacturer’s instructions. For digestion, the protein solution was reduced with 5 mM dithiothreitol for 30 min at 56°C and alkylated with 11 mM iodoacetamide for 15 min at room temperature in darkness. The protein sample was then diluted by adding 100 mM TEAB to urea concentration less than 2 M. Finally, trypsin was added at 1:50 trypsin‐to‐protein mass ratio for the first digestion overnight and 1:100 trypsin‐to‐protein mass ratio for a second 4‐h digestion (Liang, Xie et al, 2020).
The trypsin‐digested peptide was desalted with Strata X C18 (Phenomenex, USA) and vacuum‐dried. The peptide was solubilized with 0.5 M TEAB, and the peptides were labeled according to the dimethyl‐labeling kit instructions. CH2O was labeled for control group, and treatment group was labeled with CD2O. The simple operation is showed as follows: The labeling reagent was mixed with the peptide and incubated for 2 h at room temperature, and the labeled peptides were mixed, desalted, and vacuum‐dried. The tryptic peptides were fractionated into fractions by high pH reverse‐phase HPLC using Thermo Betasil C18 column (5 μm particles, 10 mm ID, 250 mm length). Briefly, peptides were first separated with a gradient of 8–32% acetonitrile (pH 9.0) over 60 min into 60 fractions. Then, the peptides were combined into 4 fractions and dried by vacuum centrifuging.
The peptide was dissolved in IP buffer (100 mM NaCl, 1 mM EDTA, 50 mM Tris–HCl, 0.5% NP‐40, pH 8.0), and the supernatant was transferred to the prewashed succinylated resin (Resin antibody product number PTM402, Jingjie PTM BioLab Co. Ltd., China), was placed on a rotary shaker at 4°C, gently shaken, and incubated overnight. After the incubation, the resin was washed 4 times with IP buffer solution and twice with deionized water. Finally, the resin‐bound peptide was eluted with 0.1% trifluoroacetic acid eluate and eluted three times in total. The eluate was collected and vacuum‐dried and drained. After draining, the salt was removed according to the C18 ZipTips (Millipore, USA) instructions, vacuum‐dried, and drained for liquid mass analysis. The protocol of HPLC/MS/MS analysis was conducted as previously described (Rardin et al, 2013; Wang et al, 2017; Huang et al, 2018). The enriched peptides obtained above were dissolved in 0.1% formic acid in water and loaded onto a reversed‐phase microcapillary column (10 cm length with 75 mm inner diameter) packed in‐house with Reprosil 100 C18 resin (3 mm particle size, Dr. Maisch GmbH, Beim Bruckle, Germany). The loaded samples were separated using a gradient of 5–80% HPLC buffer B (0.1% formic acid in 90% acetonitrile, v/v) in buffer A (0.1% formic acid in water, v/v) at a flow rate of 200 nl/min over 60 min by an EASY‐nLC 1000 UPLC (Thermo Fisher Scientific, Waltham, MA, USA). The samples were analyzed by a Q ExactiveTM hybrid quadrupole‐orbitrap mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). A data‐dependent procedure that alternated between one full mass scan followed by the top 15 most intense precursor ions was applied with a 25‐s dynamic exclusion. Intact peptides were detected with a resolution of 70,000, and the tandem mass spectra were acquired with a mass resolution of 17,500 at 27% normalized collision energy.
The protocol of database search and data filter criteria analysis was performed as previously described (Rardin et al, 2013; Wang et al, 2017; Huang et al, 2018). The acquired MS/MS data were searched by MaxQuant with integrated Andromeda search engine (v.1.3.0.5) (Cox and Mann, 2008; Cox et al, 2009). All the data were searched against UniProt Human protein database (88,277 entries, http://www.uniprot.org). Trypsin was specified as cleavage enzyme allowing a maximum of 2 missing cleavages. Cysteine carbamidomethylation was specified as fixed modification. The lysine succinylation was specified as variable modifications. FDR thresholds for protein, peptide, and modification site were specified at 1%. The following peptides were considered as false positives and removed from our list: peptides identified from reverse or contaminant protein sequences, peptides with score below 40, site localization probability below 0.75, Ksucc sites on peptide C terminus unless the peptide C‐terminal was also the corresponding protein C‐terminal. To ensure that the Ksucc level change in the cells is not caused by the protein level change, we quantified the protein expression levels. Briefly, the proteolytic peptides obtained in previous step were separated on preparative HPLC into 20 fractions or separated into 12 fractions with SDS–PAGE. These samples were analyzed using the same procedures for the Ksucc peptides quantification. Then, all the ratios of quantified Ksucc peptides were normalized by the ratios of their corresponding protein expression levels.
DNA constructs and mutagenesis
The oligos of HAT1, PGAM1, and KAT2A shRNAs were synthesized by Genscript (Nanjing, P.R. China). The shRNAs of HAT1, PGAM1 and KAT2A were cloned into pLKO.1 TRC. The CDS region of HAT1, I186 mutant (I to E) HAT1, T188 mutant (T to A) HAT1, S190 mutant (S to A) HAT1, M241 mutant (M to K) HAT1, K249 mutant (K to R) HAT1, G251 mutant (G to A) HAT1, G253 mutant (G to A) HAT1, A254 mutant (A to E) HAT1, E276 mutant (E to A) HAT1, S279 mutant (S to A) HAT1, S281 mutant (S to A) HAT1, and F288 mutant (F to A) HAT1 were constructed into the plasmid of pCMV‐3Tag‐1A. The CDS region of K122 mutant (K to R) H3, K99 mutant (K to R) PGAM1, K240 mutant (K to R) PGAM1, and K99/K240 mutant (K to R) PGAM1 were synthesized and constructed into the plasmid of pET‐30a (+) by Genscript (Nanjing, P.R. China). The CDS region of PGAM1, K99 mutant (K to R) PGAM1, K240 mutant (K to R) PGAM1, and K99/K240 mutant (K to R) PGAM1 was synthesized and constructed into the plasmid of pCMV‐3Tag‐1A by Genscript (Nanjing, P.R. China). The K99 mutant (K to E) PGAM1 was constructed into the plasmid of pCMV‐3Tag‐1A. All primers are listed in Appendix Table S2.
Western blot analysis
Total protein lysates were extracted from hepatoma cells with RIPA buffer. Protein concentrations were measured using the BCA protein Quantification Kit (YEASEN, China), and 20–50 μg protein extracts were subjected to SDS–PAGE. Then, proteins were transferred to a PVDF membrane, blocked with 5% non‐fat milk, and incubated with first antibodies for 1 h at R.T or 12 h at 4°C. After incubation with secondary antibody against mouse (1:10,000) or rabbit (1: 10,000) for 1 h at 37°C, the membrane was visualized by ECL Western Blot Detection Kit (GE Healthcare, Waukesha, WI). All antibodies are listed in Appendix Table S3.
Histone extraction
The protocol of histone extraction was performed as previously described (Wang et al, 2017). Cells were collected and washed twice with ice‐cold PBS and then resuspended in triton extraction buffer (TEB: PBS containing 0.5% Triton X‐100 (v/v), 2 mM phenylmethylsulfonyl fluoride (PMSF), 0.02% (w/v) NaN3) at a cell density of 107 cells per ml. Cells were then lysed on ice for 10 min with gentle stirring and centrifuged at 1000 g for 10 min at 4°C. The supernatant was removed and discarded. The cells were washed with TEB and centrifuged as before, and then, the pellet was resuspended in 0.2 N HCl at a cell density of 4 × 107 cells per ml. Histones were acid extracted overnight at 4°C. Then, samples were centrifuged at 2,000 rpm for 10 min at 4°C. The supernatant was removed, and protein content determined using the Bradford assay, and pH was neutralized by adding 10% (v/v) 2 M NaHCO3. Aliquots were stored at −20°C.
Expression and purification of recombinant proteins
The protocol of expression and purification of recombinant proteins was performed as previously described (Gao, Feng et al, 2017; Wang et al, 2017). The CDS region of K122 mutant (K to R) H3, K99 mutant (K to R) PGAM1, K240 mutant (K to R) PGAM1, and K99/K240 mutant (K to R) PGAM1 was synthesized and constructed into the plasmid of pET‐30a (+) by Genscript (Nanjing, P.R. China). And then, the plasmids were expressed in BL21 strain of Escherichia coli. The cultures grew at 37°C to an optical density (at a wavelength of 600 nm) of about 0.6 and were then treated with 0.5 mM isopropyl β‐d‐1‐thiogalactopyranoside (IPTG) at 18°C. Cell pellets were collected, resuspended in BugBuster lysis buffer (EMD Millipore) with a supplement of cocktail proteinase inhibitors and DNase I, and processed using sonication. Soluble His‐tagged proteins were purified as described previously (Gao et al, 2017). The protein purity was identified via gel staining with GelCode Blue Staining.
Immunoprecipitation assays
Co‐immunoprecipitation was measured using Pierce Co‐Immunoprecipitation Kit (Fisher Scientific‐Germany, Schwerte, Germany) according to the manufacturer’s instructions (Gao et al, 2017). All antibodies are listed in Appendix Table S3.
In vitro succinylation assays
The protocol of expression and purification of recombinant proteins was conducted as previously described (Wang et al, 2017). To analyze HAT1‐catalyzed histone H3 succinylation, we incubated purified wild‐type His‐histone H3 (Sino Biological inc., China) or His‐histone H3 (K122R) with purified full‐length active HAT1 (Fitzgerald, USA) in the presence of HAT buffer (50 mM Tris–HCl, pH 8.0, 50 mM KCl, 5% glycerol, 0.1 mM EDTA, 1 mM dithiothreitol, 1 mM PMSF, 10 mM sodiumbutyrate), and 2 μM succinyl‐CoA (SIGMA, USA), or CoA (SIGMA, USA), at 37°C for 10 min. Histone H3 succinylation was determined by using immunoblotting analysis.
To analyze the priority of HAT1‐catalyzed histone H3 succinylation and acetylation, we incubated purified wild‐type His‐histone H3 with purified full‐length active HAT1 in the presence of HAT buffer (50 mM Tris–HCl, pH 8.0, 50 mM KCl, 5% glycerol, 0.1 mM EDTA, 1 mM dithiothreitol, 1 mM PMSF, 10 mM sodiumbutyrate), and 2 μM succinyl‐CoA and acetyl‐CoA (SIGMA, USA), at 37°C for 10 min. Histone H3 succinylation was determined by using immunoblotting analysis.
To analyze HAT1‐catalyzed PGAM1 succinylation, we incubated purified wild‐type His‐PGAM1 (Proteintech, China), His‐PGAM1 (K99R), His‐PGAM1 (K240R), or His‐PGAM1 (K99/240R) with purified full‐length active HAT1 in the presence of HAT buffer (50 mM Tris–HCl, pH 8.0, 50 mM KCl, 5% glycerol, 0.1 mM EDTA, 1 mM dithiothreitol, 1 mM PMSF, 10 mM sodiumbutyrate), and 2 μM succinyl‐CoA or acetyl‐CoA, at 37°C for 10 min. The PGAM1 succinylation was determined by using immunoblotting analysis.
Steady‐state kinetics of HAT1 activity for histone H3 modifications
Immunoprecipitated full‐length Flag‐HAT1 from HepG2 cells was immobilized on anti‐Flag‐agarose beads and incubated with purified histone H3 (4 μM) in HAT buffer in the presence of 0.009 μM, 0.027 μM, 0.082 μM, 0.247 μM, 0.741 μM, 2.222 μM, 6.667 μM, or 20.000 μM of acetyl‐CoA or succinyl‐CoA at 25°C for 5 min. The reaction mixture was precipitated via centrifugation at 1,000 g for 5 min. The production of CoA in the supernatant was quantitatively analyzed using a modified protocol from the CoA Fluorometric Assay Kit (BioVision, USA). An identical reaction was set up for each acetyl‐CoA or succinyl‐CoA titration without HAT1 protein and used as the blank control. Data are presented as the means ± SD of three independent experiments.
Molecular docking analysis
The docking analysis of HAT1 with succinyl‐CoA was tested by Discovery Studio software according to the reported methods, with some modifications (Fu, Alashi et al, 2017; Tu, Liu et al, 2018). The structure of HAT1 (PDB ID 2P0W) and succinyl‐CoA (PDB ID 5TRL) was downloaded from RCSB Protein Data Bank. The docking of HAT1 with succinyl‐CoA was analysis by CDOCKER.
ChIP and ChIP‐seq assays
ChIP was performed using a SimpleChIP Enzymatic Chromatin IP Kit (Cell Signaling Technology, USA). Chromatin prepared from cells in a 15‐cm dish was used to determine total DNA input and was incubated overnight with specific antibodies or normal rabbit IgG. All primers are listed in Appendix Table S2. All antibodies are listed in Appendix Table S3. ChIP sequencing was performed at the ANOROAD (Beijing, China). In brief, a total of 5 × 107 cells were used for each ChIP assays. Enriched DNA fragments were subjected to library preparation and next‐generation sequencing with Annoroad Gene Technology using a Novaseq6000 (Illumina). Short reads were mapped to the Homo_sapiens.GRCh38.87.chr reference genome using Bowtie2 (Langmead & Salzberg, 2012), and ChIP peaks were called using model‐based analysis of ChIP‐seq 14 (MACS 14), with the input sample as the control (Zhang, Liu et al, 2008). Enrichment heat‐maps that surrounded the ChIP peaks were generated using seqMINER (Ye, Krebs et al, 2011), and signal plotting of individual genes was generated using the Integrated Genome Viewer (Robinson, Thorvaldsdottir et al, 2011).
RNA extraction, reverse‐transcription polymerase chain reaction (RT–PCR), and quantitative real‐time PCR (RT–qPCR)
Total RNAs were extracted from cells using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). First‐strand cDNA was synthesized as reported before. RT–qPCR was performed by a Bio‐Rad sequence detection system according to the manufacturer's instructions using double‐stranded DNA‐specific SYBR GreenPremix Ex TaqTM II Kit (TaKaRa, Ohtsu, Japan). Experiments were conducted in duplicate in three independent assays. Relative transcriptional folds were calculated as 2‐∆∆Ct. GAPDH was used as an internal control for normalization. All the primers used are listed in Appendix Table S2.
Analyses of metabolites
The protocol of analyses of metabolites was conducted as previously described (Hitosugi et al, 2012). The levels of glucose and lactate in the supernatant of the cell culture medium were measured by using the ELISA kit (Mlbio, China). The levels of succinyl‐CoA were measured by using the ELISA kit (Mlbio, China). To determine cellular concentration of pyruvate ATP, ADP, NADH, NAD+, 2‐PG, and 3‐PG, 100 μl of packed cell pellets was homogenized in 1.5 ml of hypotonic lysis buffer (20 mM HEPES (pH 7.0), 5 mM KCl, 1 mM MgCl2, 5 mM DTT, and protease inhibitor cocktail). The homogenates were centrifuged in a cold room at 4°C for 10 minutes at maximum speed, and the supernatants were applied to Amicon Ultra tubes with 10 KDa cutoff filter (Millipore, USA). The flow through containing the metabolites was used for the measurement by using the ELISA kit (Mlbio, China).
Activity assays for glycolytic enzymes
The protocol of activity assays for PGAM1 was performed by multiple enzymes coupled assay as previously described (Hitosugi et al, 2012; Song, Baek et al, 2018). PGAM1 enzyme mix containing 100 mM Tris–HCl, 100 mM KCl, 5 mM MgCl2, 1 mM ADP, 0.2 mM NADH, 5 mg/ml recombinant PGAM1, 0.5 units/ml enolase, 0.5 units/ml recombinant pyruvate kinase M1, and 0.1 units/ml recombinant LDH was prepared. 3‐PG was added last at the final concentration of 2 mM to initiate the reaction. The decrease in autofluorescence (ex: 340 nm, em: 460 nm) from oxidation of NADH was measured as PGAM1 activity. The enzyme activity of ENO1 was analyzed by using ENO1 Assay (Abcam ab117994, UK), and the enzyme activity of PKM was analyzed by using Pyruvate Kinase Assay Kit (Abcam ab83432, UK).
Cell viability assays
The cell viability was determined as previously described (Stehling, Vashisht et al, 2012) by using the MTS assay (Promega, USA). The MTS assay reagent was made by dissolving CellTiter 96R Aqueous MTS powder at a concentration of 2 mg/ml in 1 × PBS containing 300 μM phenazine ethosulfate. The MTS assay reagent was added to cells at a final concentration of 20%. Cells were incubated for 3 h at 37°C before absorbance of the sample was read at 490 nm. All absorbance values were averaged for control and treated samples.
Immunohistochemistry
Immunohistochemical (IHC) staining of samples was measured as previously reported (Zhang, Zhang et al, 2012; Cui, Xiao et al, 2015; Wang et al, 2017). The microarrays including 232 liver cancer tissues, 126 pancreatic cancer tissues, and 120 cholangiocarcinoma tissues were obtained from the Xi'an Aomei Biotechnology Co., Ltd. (Xi'an, China). Clinic pathological information about the patients was obtained from patient records and was summarized in Dataset EV1. These microarrays included duplicate core biopsies (1 mm in diameter) from fixed, paraffin‐embedded tumors. Mouse tumor samples were fixed, paraffin‐embedded, sectioned (5 μm), and stained with Mayer’s hematoxylin and eosin (BioGenex Laboratories). Slides were then mounted using universal mount (Research Genetics) and examined with a light microscope. The antibody of rabbit anti‐HAT1 and rabbit anti‐Ki‐67 was used (Appendix Table S3). Categorization of immunostaining intensity was performed by three independent observers. Written consents approving the use of their tissues for research purposes after operation were obtained from patients. The Institute Research Ethics Committee at the Nankai University approved the study protocol.
Analysis of tumorigenicity in nude mice
The protocol of analysis of tumorigenicity in nude mice was conducted as previously described (Zhang et al, 2012; Gao et al, 2017). HepG2 or PANC1 cells (5 × 106) with or without modified gene expression were subcutaneously injected into male 4‐week‐old athymic nude mice (in 100 μl of Dulbecco’s modified Eagle’s medium per mouse). Twenty‐four mice were randomly grouped in four groups (six per group) or twelve mice were randomly grouped in two groups (six per group) before tumor cell injection. Tumor growth was measured after 10 days from injection and then every 5 days. At 30 days after injection, mice were sacrificed, and tumors were weighed after necropsy. Tumor volume (V) was monitored by measuring the length (L) and width (W) with calipers and calculated with the formula × 0.5 (Verrier et al, 2018). The Institute Research Ethics Committee at the Nankai University approved the study protocol.
The succinylation site stoichiometry analysis of representative glycolytic enzymes
The succinylation site stoichiometry analysis of representative glycolytic enzymes was performed as previously described (Olsen, Vermeulen et al, 2010). The results were listed in Appendix Table S4. The calculate formula was briefly showed as follow:
Succinylpeptide HAT1 KO/WT = x.
Non‐Succinylpeptide HAT1 KO/WT = y.
Protein HAT1 KO/WT = z.
WT: proportion = z – y / x ‐ z = a.
HAT1 KO: proportion = x * (z ‐ y) / y * (x ‐ z) = b.
WT: succinylation site stoichiometry = a / (1 + a).
HAT1 KO: succinylation site stoichiometry = b / (1 + b).
Statistical analysis
Each experiment was repeated at least three times. Statistical significance was assessed by comparing mean values (± SD) using a Student t‐test for independent groups and was assumed for *P < 0.05; **P < 0.01; ***P < 0.001 unless specifically indicated.
Author contributions
XDZ and GY designed the experiments and interpreted the data. GY performed the experiments shown in Figs 1, 2, 3, 4, 5, 6, and Appendix Figs S1–S12. YY, HFY, JPW, and HLY performed the experiments shown in Figs 2, 3, 4, 5, and Appendix Figs S4–S10, and 11. LHL, YJW, and BNW performed and analyzed the succinylation quantitative proteomic. YG, MZ, ZXL, JYF, YNB, and LL performed the experiments shown in Figs 2 and 6, and Appendix Figs S4 and 11. XDZ and GY wrote the paper with input from all authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Dataset EV1
Review Process File
Acknowledgements
The study was supported in part by National Basic Research Program of China (973 Program, No. 2015CB553703) and National Natural Science Foundation of China (No. 31670769).
EMBO reports (2021) 22: e50967.
Data availability
The datasets (and computer code) produced in this study are available in the following databases:
References
- Andrews FH, Strahl BD, Kutateladze TG (2016) Insights into newly discovered marks and readers of epigenetic information. Nat Chem Biol 12: 662–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandeira N, Tsur D, Frank A, Pevzner PA (2007) Protein identification by spectral networks analysis. Proc Natl Acad Sci USA 104: 6140–6145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brien GL, Valerio DG, Armstrong SA (2016) Exploiting the epigenome to control cancer‐promoting gene‐expression programs. Cancer Cell 29: 464–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrico C, Meyer JG, He WJ, Gibson BW, Verdin E (2018) The mitochondrial acylome emerges: proteomics, regulation by sirtuins, and metabolic and disease implications. Cell Metab 27: 497–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaneton B, Gottlieb E (2012) PGAMgnam style: a glycolytic switch controls biosynthesis. Cancer Cell 22: 565–566 [DOI] [PubMed] [Google Scholar]
- Chang CM, Su H, Zhang DH, Wang YS, Shen QH, Liu B, Huang R, Zhou TH, Peng C, Wong CCL et al (2015) AMPK‐dependent phosphorylation of GAPDH triggers Sirt1 activation and is necessary for autophagy upon glucose starvation. Mol Cell 60: 930–940 [DOI] [PubMed] [Google Scholar]
- Chen Y, Chen W, Cobb MH, Zhao YM (2009) PTMap‐A sequence alignment software for unrestricted, accurate, and full‐spectrum identification of post‐translational modification sites. Proc Natl Acad Sci USA 106: 761–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Li S, Subramaniam S, Shyy JYJ, Chien S (2017) Epigenetic regulation: a new frontier for biomedical engineers. Annu Rev Biomed Eng 19: 195–219 [DOI] [PubMed] [Google Scholar]
- Cui M, Xiao ZL, Wang Y, Zheng MY, Song TQ, Cai XL, Sun BD, Ye LH, Zhang XD (2015) Long noncoding RNA HULC modulates abnormal lipid metabolism in hepatoma cells through an miR‐9‐mediated RXRA signaling pathway. Cancer Res 75: 846–857 [DOI] [PubMed] [Google Scholar]
- DeNicola GM, Cantley LC (2015) Cancer's fuel choice: new flavors for a picky eater. Mol Cell 60: 514–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Zhou Y, Su X, Yu JJ, Khan S, Jiang H, Kim J, Woo J, Kim JH, Choi BH et al (2011) Sirt5 is a NAD‐dependent protein lysine demalonylase and desuccinylase. Science 334: 806–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Alashi AM, Young JF, Therkildsen M, Aluko RE (2017) Enzyme inhibition kinetics and molecular interactions of patatin peptides with angiotensin I‐converting enzyme and renin. Int J Biol Macromol 101: 207–213 [DOI] [PubMed] [Google Scholar]
- Gao Y, Feng J, Yang G, Zhang S, Liu Y, Bu Y, Sun M, Zhao M, Chen F, Zhang W et al (2017) Hepatitis B virus X protein‐elevated MSL2 modulates hepatitis B virus covalently closed circular DNA by inducing degradation of APOBEC3B to enhance hepatocarcinogenesis. Hepatology 66: 1413–1429 [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144: 646–674 [DOI] [PubMed] [Google Scholar]
- Hitosugi T, Zhou L, Elf S, Fan J, Kang HB, Seo JH, Shan CL, Dai Q, Zhang L, Xie JX et al (2012) Phosphoglycerate mutase 1 coordinates glycolysis and biosynthesis to promote tumor growth. Cancer Cell 22: 585–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitosugi T, Zhou L, Fan J, Elf S, Zhang L, Xie JX, Wang Y, Gu TL, Aleckovic M, LeRoy G et al (2013) Tyr26 phosphorylation of PGAM1 provides a metabolic advantage to tumours by stabilizing the active conformation. Nat Commun 4: 1790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Tang S, Ji M, Tang Z, Shimada M, Liu X, Qi S, Locasale JW, Roeder RG, Zhao Y et al (2018) p300‐mediated lysine 2‐hydroxyisobutyrylation regulates glycolysis. Mol Cell 70: 984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL (2012) Fast gapped‐read alignment with Bowtie 2. Nat Methods 9: 357–U54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Shi L, Yang S, Yan R, Zhang D, Yang J, He L, Li W, Yi X, Sun L et al (2016) SIRT7 is a histone desuccinylase that functionally links to chromatin compaction and genome stability. Nat Commun 7: 12235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberti MV, Locasale JW (2016) The warburg effect: how does it benefit cancer cells? Trends Biochem Sci 41: 211–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu TM, Shyh‐Chang N (2017) SIRT2 and glycolytic enzyme acetylation in pluripotent stem cells. Nat Cell Biol 19: 412–414 [DOI] [PubMed] [Google Scholar]
- Lunt SY, Vander Heiden MG (2011) Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol 27: 441–464 [DOI] [PubMed] [Google Scholar]
- Maddalo D, Manchado E, Concepcion CP, Bonetti C, Vidigal JA, Han YC, Ogrodowski P, Crippa A, Rekhtman N, de Stanchina E et al (2014) In vivo engineering of oncogenic chromosomal rearrangements with the CRISPR/Cas9 system. Nature 516: 423–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan P, Ge Z, Sirbu B, Doughty C, Agudelo Garcia PA, Schlederer M, Annunziato AT, Cortez D, Kenner L, Parthun MR (2013) Histone acetyl transferase 1 is essential for mammalian development, genome stability, and the processing of newly synthesized histones H3 and H4. PLoS Genet 9: e1003518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida Y, Rardin MJ, Carrico C, He WJ, Sahu AK, Gut P, Najjar R, Fitch M, Hellerstein M, Gibson BW et al (2015) SIRT5 regulates both cytosolic and mitochondrial protein malonylation with glycolysis as a major target. Mol Cell 59: 321–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen JV, Vermeulen M, Santamaria A, Kumar C, Miller ML, Jensen LJ, Gnad F, Cox J, Jensen TS, Nigg EA et al (2010) Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci Signal 3: ra3 [DOI] [PubMed] [Google Scholar]
- Oslund RC, Su XY, Haugbro M, Kee JM, Esposito M, David Y, Wang BY, Ge E, Perlman DH, Kang YB et al (2017) Bisphosphoglycerate mutase controls serine pathway flux via 3‐phosphoglycerate. Nat Chem Biol 13: 1081‐+ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Chen Y, Tishkoff DX, Peng C, Tan M, Dai L, Xie Z, Zhang Y, Zwaans BM, Skinner ME et al (2013) SIRT5‐mediated lysine desuccinylation impacts diverse metabolic pathways. Mol Cell 50: 919–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parthun MR (2007) Hat1: the emerging cellular roles of a type B histone acetyltransferase. Oncogene 26: 5319–28 [DOI] [PubMed] [Google Scholar]
- Parthun MR (2013) Histone acetyltransferase 1: more than just an enzyme? Biochim Biophys Acta 1819: 256–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel DJ, Wang ZX (2013) Readout of Epigenetic Modifications. Annu Rev Biochem 82: 81‐118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR‐Cas9 system. Nat Protoc 8: 2281–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rardin MJ, He W, Nishida Y, Newman JC, Carrico C, Danielson SR, Guo A, Gut P, Sahu AK, Li B et al (2013) SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab 18: 920–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang S, Xie M, Tang J, Wang M, Zhang D, Hou S (2020) Proteomics reveals the effect of type I interferon on the pathogenicity of duck hepatitis A virus genotype 3 in Pekin ducks. Vet Microbiol 248: 108813 [DOI] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP (2011) Integrative genomics viewer. Nat Biotechnol 29: 24–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadler AJ, Suliman BA, Yu L, Yuan X, Wang D, Irving AT, Sarvestani ST, Banerjee A, Mansell AS, Liu JP et al (2015) The acetyltransferase HAT1 moderates the NF‐kappaB response by regulating the transcription factor PLZF. Nat Commun 6: 6795 [DOI] [PubMed] [Google Scholar]
- Shahbazian MD, Grunstein M (2007) Functions of site‐specific histone acetylation and deacetylation. Annu Rev Biochem 76: 75–100 [DOI] [PubMed] [Google Scholar]
- Song J, Baek IJ, Chun CH, Jin EJ (2018) Dysregulation of the NUDT7‐PGAM1 axis is responsible for chondrocyte death during osteoarthritis pathogenesis. Nat Commun 9: 3427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stehling O, Vashisht AA, Mascarenhas J, Jonsson ZO, Sharma T, Netz DJ, Pierik AJ, Wohlschlegel JA, Lill R (2012) MMS19 assembles iron‐sulfur proteins required for DNA metabolism and genomic integrity. Science 337: 195–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suganuma T, Workman JL (2011) Signals and combinatorial functions of histone modifications. Annu Rev Biochem 80: 473–499 [DOI] [PubMed] [Google Scholar]
- Tu ML, Liu HX, Zhang R, Chen H, Mao FJ, Cheng SZ, Lu WH, Du M (2018) Analysis and evaluation of the inhibitory mechanism of a novel angiotensin‐I‐converting enzyme inhibitory peptide derived from casein hydrolysate. J Agr Food Chem 66: 4139–4144 [DOI] [PubMed] [Google Scholar]
- Verrier ER, Yim SA, Heydmann L, El Saghire H, Bach C, Turon‐Lagot V, Mailly L, Durand SC, Lucifora J, Durantel D et al (2018) Hepatitis B virus evasion from cyclic guanosine monophosphate‐adenosine monophosphate synthase sensing in human hepatocytes. Hepatology 68: 1695–1709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh CT, Garneau‐Tsodikova S, Gatto GJ Jr (2005) Protein posttranslational modifications: the chemistry of proteome diversifications. Angew Chem Int Ed Engl 44: 7342–72 [DOI] [PubMed] [Google Scholar]
- Wang Y, Guo YR, Liu K, Yin Z, Liu R, Xia Y, Tan L, Yang P, Lee JH, Li XJ et al (2017) KAT2A coupled with the alpha‐KGDH complex acts as a histone H3 succinyltransferase. Nature 552: 273–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward PS, Thompson CB (2012) Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell 21: 297–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert BT, Scholz C, Wagner SA, Iesmantavicius V, Su D, Daniel JA, Choudhary C (2013) Lysine succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with acetylation. Cell Rep 4: 842–851 [DOI] [PubMed] [Google Scholar]
- Witze ES, Old WM, Resing KA, Ahn NG (2007) Mapping protein post‐translational modifications with mass spectrometry. Nat Methods 4: 798–806 [DOI] [PubMed] [Google Scholar]
- Yang G, Feng J, Liu Y, Zhao M, Yuan Y, Yuan H, Yun H, Sun M, Bu Y, Liu L et al (2019) HAT1 signaling confers to assembly and epigenetic regulation of HBV cccDNA minichromosome. Theranostics 9: 7345–7358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye T, Krebs AR, Choukrallah MA, Keime C, Plewniak F, Davidson I, Tora L (2011) seqMINER: an integrated ChIP‐seq data interpretation platform. Nucleic Acids Res 39: e35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Zhang JP, You XN, Liu Q, Du YM, Gao Y, Shan CL, Kong GY, Wang YL, Yang X et al (2012) Hepatitis B virus X protein modulates oncogene yes‐associated protein by CREB to promote growth of hepatoma cells. Hepatology 56: 2051–2059 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nussbaum C, Myers RM, Brown M, Li W et al (2008) Model‐based analysis of ChIP‐Seq (MACS). Genome Biol 9: R137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Tan M, Xie Z, Dai L, Chen Y, Zhao Y (2011) Identification of lysine succinylation as a new post‐translational modification. Nat Chem Biol 7: 58–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Zhang XR, Li HT (2018) Beyond histone acetylation‐writing and erasing histone acylations. Curr Opin Struc Biol 53: 169–177 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Dataset EV1
Review Process File
Data Availability Statement
The datasets (and computer code) produced in this study are available in the following databases: