

SARS-CoV-2 infection elicits a distinct host response in nasal swabs and blood that can be used to diagnose COVID-19.
Abstract
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which causes coronavirus disease-19 (COVID-19), has emerged as the cause of a global pandemic. We used RNA sequencing to analyze 286 nasopharyngeal (NP) swab and 53 whole-blood (WB) samples from 333 patients with COVID-19 and controls. Overall, a muted immune response was observed in COVID-19 relative to other infections (influenza, other seasonal coronaviruses, and bacterial sepsis), with paradoxical down-regulation of several key differentially expressed genes. Hospitalized patients and outpatients exhibited up-regulation of interferon-associated pathways, although heightened and more robust inflammatory responses were observed in hospitalized patients with more clinically severe illness. Two-layer machine learning–based host classifiers consisting of complete (>1000 genes), medium (<100), and small (<20) gene biomarker panels identified COVID-19 disease with 85.1–86.5% accuracy when benchmarked using an independent test set. SARS-CoV-2 infection has a distinct biosignature that differs between NP swabs and WB and can be leveraged for COVID-19 diagnosis.
INTRODUCTION
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the cause of coronavirus disease-19 (COVID-19), emerged in December 2019 and has resulted in more than 56 million cases and more than 1.3 million deaths globally as of mid-November 2020 (1). Although the majority of patients with COVID-19 are asymptomatic or have mild symptoms, approximately 16 to 19% of patients develop acute respiratory failure, and 0.4 to 11.1% die from the disease (2–7). The exact mechanisms underlying the development of severe disease remain unclear, although cytokine storm and dysregulated cellular immune responses are thought to play important roles (8, 9). Currently, diagnostic testing relies on reverse transcription quantitative polymerase chain reaction (RT-PCR), which can yield false-negative results as viral loads in patients may be low and fluctuate substantially during the course of the illness (10–12). Host response–based testing may be useful as a complementary tool for differential diagnosis of SARS-CoV-2 and other respiratory viral or bacterial infections (13–16).
Here, we apply transcriptome profiling to evaluate and compare host responses among patients with COVID-19, other viral and nonviral acute respiratory illnesses (ARIs) from nasopharyngeal (NP) swab samples, and with COVID-19, influenza, and bacterial sepsis from whole-blood (WB) samples. Host response data are also compared between outpatients with mild COVID-19 disease and hospitalized patients with severe COVID-19, including intensive care unit (ICU) patients requiring mechanical ventilation. Several studies have previously demonstrated that gene expression profiles using NP swabs and/or WB can identify patients with viral or bacterial infections (17–23). We therefore used the host response data to generate a classifier for differential diagnosis of SARS-CoV-2 infection.
RESULTS
Population characteristics and sequencing metrics
A total of 380 remnant NP swab samples from 351 individuals (138 SARS-CoV-2–positive patients, 213 SARS-CoV-2–negative patients, including 88 with documented influenza or seasonal coronavirus infection, and 11 donor controls) and 53 WB samples from 53 individuals (7 SARS-CoV-2–positive patients, 26 SARS-CoV-2–negative patients with influenza or bacterial sepsis, and 20 donor controls) were collected for RNA sequencing (RNA-seq) analysis (Fig. 1A). Of the 351 NP samples, 286 remnant NP swab samples from 286 individuals (137 SARS-CoV-2–positive patients and 149 SARS-CoV-2–negative patients) and all the WB samples were initially used to evaluate the host response. To ensure more balanced numbers across the categories for subsequent classifier generation, we included an additional 65 NP samples, composed of 1 SARS-CoV-2–positive sample and 64 other acute viral respiratory illness samples.
Fig. 1. Overview of sample collection and metatranscriptomic analysis.
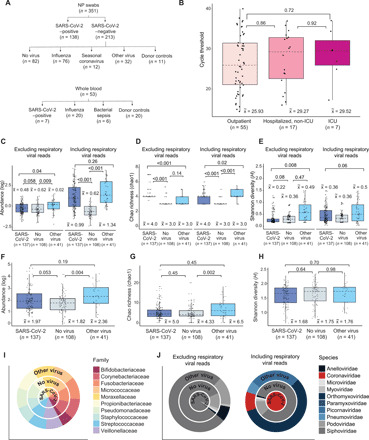
(A) Flowchart of NP swab and WB sample collection. (B) Box and whisker plots of RT-PCR cycle threshold (Ct) values of SARS-CoV-2–positive individuals who are outpatients (n = 55) were compared with those who are hospitalized, non-ICU (n = 17), or in the ICU (n = 7). There was no difference in viral load, inversely related to the Ct value, regardless of disease severity [P = 0.89 by analysis of variance (ANOVA)]. (C to E) Box and whisker plots of abundance (C), Chao richness (D), and Shannon diversity (E) of the viral metatranscriptome in patients with SARS-CoV-2 (COVID-19) (n = 137), respiratory viruses (“Other virus”) (n = 41), and without respiratory viruses (“No virus”) (n = 108), stratified by the inclusion (“Including respiratory viral reads”) or exclusion (“Exclusion respiratory viral reads”) of respiratory viral reads. (F to H) Box and whisker plots of abundance (F), Chao richness (G), and Shannon diversity (H) of the bacterial metatranscriptome. (I) Distribution of viral families in each group, expressed as log10-normalized RPM. (J) Distribution of the top 10 bacterial families in each group. For box and whisker plots, the median is represented by a dotted line, boxes represent the first to third quartiles, whiskers represent the minimum and maximum values, and jitters represent the distribution of the population. For (C) to (H), statistical analysis was conducted by Kruskal-Wallis test, followed by the Nemenyi test for post hoc analysis.
Clinical history was available from 177 of 340 (52.1%) patients with NP swabs, and for all 33 patients with WB samples (tables S1 and S2). Clinical history was unavailable for the remaining 163 NP samples collected from non–University of California, San Francisco (UCSF) institutional partners in outreach settings or from the California Department of Public Health (CPDH). Only demographic information was available from the 20 WB control donors (table S3).
Among patients with COVID-19, there was a median of 5 ± 11 days (range, 0 to 65 days) between symptom onset and NP sample collection, and a median of 9 ± 29 days (range, 6 to 72 days) between symptom onset and WB sample collection. Six patients with COVID-19 had paired NP swabs and WB available for comparison. As a surrogate indicator for disease severity, patients with COVID-19 were also stratified according to the highest level of care received (outpatient, hospitalized but not requiring intensive care, and ICU admission). The median age for patients with COVID-19 was 49 years versus 44 years for non–COVID-19 patients, with proportionally fewer women in the COVID-19 group (P = 0.0021) (table S1). Patients with COVID-19 were more likely to have fever (P < 0.0001), chills (P = 0.003), malaise (P = 0.0009), and anosmia (P = 0.0002) than non–COVID-19 patients with ARI (table S1). Hypertension and hyperlipidemia were significantly associated with patients with COVID-19 (P = 0.0406 and P = 0.0128). The presence of fever (P = 0.004) and cough (P = 0.0008) appeared to correlate with high viral loads as indicated by low cycle threshold (Ct) values by PCR (<18). In contrast, viral loads in more severely ill hospitalized patients, including patients in the ICU, were not significantly different from those in outpatients (P = 0.72) (Fig. 1B).
A total of 22.0 billion and 3.4 billion raw reads were sequenced from 351 NP swab and 53 WB samples, respectively. For the NP swab samples, the median human transcriptome coverage achieved was 53.5 ± 17.8% (range, 0.69 to 84.7%), corresponding to 14,037 of 26,486 genes from the University of California, Santa Cruz (UCSC) Genome Browser Database (24). A median of 29.6 ± 87.0 million reads (range, 0.061 to 604 million reads) were generated for each sample (fig. S1, A and B). Of these NP swab samples, 286 were sequenced initially and used to evaluate the host response and metatranscriptome, from 19 billion raw sequencing reads, with a median transcriptome coverage of 58.5 ± 15.1% (range, 4.4 to 84.7%), generated from a median of 28.8 ± 96.1 million reads (range, 0.45 to 604 million reads). For the WB samples, the median coverage achieved was 37.5 ± 1 6.2% (range, 20.8 to 89.2%), generated from a median of 30.8 ± 41.7 million reads (range, 16.5 to 182 million reads) (fig. S1, C and D).
All 351 samples (138 SARS-CoV-2 positive, 93 nonviral ARI, and 120 viral ARI) were then used to generate a machine learning–based classifier. Samples from each of the three disease groups were randomly but proportionally assigned into a training set (80%) or independent test set (20%). There was no statistical difference between transcriptome coverage and raw read counts in the training set (P = 0.09) nor in the test set (P = 0.11) (table S4).
Viral coinfections in patients with SARS-CoV-2
Of 286 NP swab samples, 137 (47.9%) were SARS-CoV-2 positive and 108 (37.8%) were negative for any respiratory virus (including NP swab samples from the 11 donor controls). A respiratory virus was identified by metatranscriptome analysis in 41 cases (14.3%), including 27 patients with previously confirmed influenza or seasonal coronavirus infection by RT-PCR testing. These respiratory viruses included seasonal coronavirus, influenza virus, human rhinovirus, human parainfluenza virus, and human metapneumovirus (fig. S2). Coinfections were identified in 10 of 137 (7.3%) SARS-CoV-2–positive and 4 of 41 (9.76%) SARS-CoV-2–negative individuals (P = 0.61), while 2 of 137 SARS-CoV-2–positive (1.5%) and 2 of 41 SARS-CoV-2–negative (4.88%) individuals were infected by three viruses (P = 0.20) (table S5). These triply infected individuals had additional infections from human rhinovirus (multiple genotypes) and human metapneumovirus.
Analysis of WB samples identified anelloviruses and human herpesvirus 6B in SARS-CoV-2–positive individuals (but no SARS-CoV-2 reads), and hepatitis B virus, HIV, and anelloviruses in patients with influenza (table S6). The absence of SARS-CoV-2 viremia is consistent with the results from other published studies showing that viremia is rare in acutely infected individuals (25).
Impact of SARS-CoV-2 infection on the NP metatranscriptome
We next investigated the effect of SARS-CoV-2 infection on the NP viral and bacterial metatranscriptome. We compared the virome of SARS-CoV-2–positive individuals (COVID, n = 137) to SARS-CoV-2–negative individuals either with another respiratory virus (seasonal coronavirus, influenza, human rhinovirus, and human metapneumovirus) detected by sequencing (“Other virus”; n = 41) or with no virus detected (“No virus”; n = 108) (Fig. 1, C to E). Additional detected respiratory viruses included all four seasonal coronaviruses (229E, HKU1, NL63, and OC43), influenza virus, human rhinovirus, human parainfluenzavirus 2, and human metapneumovirus. Relative abundance (P < 0.001) and richness (Chao Richness Score) (P < 0.001) of respiratory viruses, as calculated from the viral sequencing reads, were higher in patients with SARS-CoV-2 and patients infected with other respiratory viruses than in patients without respiratory viral infection. In comparison to patients infected with another respiratory virus, patients with SARS-CoV-2 had no difference in abundance (P = 0.26) and a decrease in richness (P = 0.02) (“Including respiratory viral reads,” Fig. 1, C and D). There was no difference in diversity in any population (P = 0.06) (“Including respiratory viral reads,” Fig. 1E). If respiratory viral reads are excluded (“Excluding respiratory viral reads,” Fig. 1, C to E), patients with SARS-CoV-2 infection showed no difference in abundance (P = 0.06) or diversity (P = 0.08) but revealed an increase in richness (P < 0.001) relative to individuals without a respiratory virus. In comparison to patients infected with another respiratory virus, patients with SARS-CoV-2 had decreased abundance (P = 0.04) and diversity (P = 0.008) but increased richness (P < 0.001) in their viral metatranscriptome.
There was no difference in relative abundance, richness, or alpha diversity of the bacterial metatranscriptome in SARS-CoV-2–positive individuals compared with those without a virus or with another respiratory virus (Fig. 1, F to H). Furthermore, infections from SARS-CoV-2 or other respiratory viruses did not appear to affect the overall distribution of families in the bacterial metatranscriptome (Fig. 1I). On the basis of the relative distribution of viral families found in the nasopharynx, patients with SARS-CoV-2 had an increase in the proportion of Siphoviridae (95%; Fig. 1J) compared with those infected with another respiratory virus (90%) or without a respiratory virus identified (86%). These findings are consistent with a study evaluating the microbiome using NP swabs in patients with mild SARS-CoV-2 infections (26).
Comparison of cell types and proportions between SARS-CoV-2 and other infections
Cell type and proportion analyses of NP swabs and WB were performed using the Multi-Subject Single Cell (MuSiC) deconvolution algorithm (figs. S3 and S4) (27). SARS-CoV-2–positive patients had increased ciliated epithelial cells relative to influenza (P = 0.03) and seasonal coronavirus (P = 0.02), increased neutrophils relative to nonviral ARIs (P < 0.0001), and increased eosinophils relative to donor samples (P = 0.008) and nonviral ARIs (P < 0.0001). SARS-CoV-2–positive patients had decreased fibroblasts relative to influenza (P = 0.008) and seasonal coronaviruses (P = 0.01), and decreased macrophages relative to influenza (P = 0.02) and other respiratory viruses (P = 0.04). Endothelial cells and other cells (mast, myeloid, basal, plasma, and glandular epithelial cells) were also lower in SARS-CoV-2 relative to influenza (P = 0.02) and other viruses (P = 0.04). Influenza had increased fibroblasts (P < 0.0001), macrophages (P < 0.03), and neutrophils (P < 0.0001), but decreased ciliated epithelial cells (P = 0.02), endothelial cells (P = 0.03), and other cells (P = 0.03) relative to nonviral ARIs. Seasonal coronaviruses had increased neutrophils (P < 0.0001), fibroblasts (P < 0.0001), and other cells (P = 0.04), but decreased ciliated epithelial cells (P < 0.0001) compared with nonviral ARIs. There was no difference in the proportion of cell types among different levels of severity of SARS-CoV-2 infection (fig. S3B).
When looking at cell proportions in WB, there was an increase in basophils and smooth muscle cells in SARS-CoV-2 relative to influenza (P = 0.007 and P = 0.003, respectively), sepsis (P = 0.008 and P = 0.008, respectively), and donor controls (P = 0.0002 and P = 0.001, respectively) (fig. S4). There were also increased bone marrow progenitor cells (P = 0.002) and platelets (P = 0.004) in SARS-CoV-2 relative to influenza and decreased CD8+ T cells (P = 0.004) and erythrocytes (P = 0.004) relative to donor controls. Compared with sepsis, SARS-CoV-2 had decreased neutrophils (P = 0.03) and increased platelets (P = 0.002).
NP swab transcriptome analysis
Pathway analysis of differentially expressed genes (DEGs) in NP swabs from patients with COVID-19 relative to uninfected donor controls showed prominent activation of genes related to interferon (IFN) signaling and IFN-stimulating genes (ISGs) (including IFI6, IFIT1–3, and ISG15), but inhibition of interleukin-6 (IL-6) and IL-8 signaling genes (including IRAK1 and MAP2K7) (Fig. 2A). Patterns of activation and inhibition associated with COVID-19 were markedly different from those associated with influenza or other viral infections (Figs. 2A and 3, A to F). In particular, patients with COVID-19 showed activation of pathways involved primarily in cell death and survival, and both activation and inhibition of pathways associated with organismal injury and survival and inflammatory response (Fig. 3, A to C). Relative to donor controls, influenza and other viral respiratory infections shared IFN signaling activation pathways in common with COVID-19 (Figs. 2A, 3D, and 6A, and tables S7 and S8). However, other immune response pathways that were activated by influenza and other viral infections, such as acute phase, B cell receptor, and Toll-like receptor signaling (including genes IRAK1, MAPK12, and MAP2K7), and chemokine signaling (including IL-6 and IL-8) were inhibited in COVID-19.
Fig. 2. Comparison of canonical pathways predicted to be involved in COVID-19.
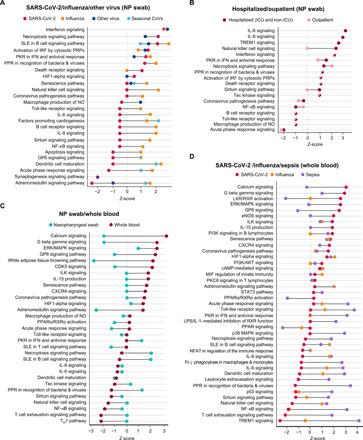
(A) Comparison of NP swab pathways in COVID-19, influenza, other respiratory viruses (human metapneumovirus, human rhinovirus, and human parainfluenza 2), and seasonal coronaviruses. (B) Comparison of NP swab pathways in COVID-19 outpatients and hospitalized patients (non-ICU and ICU). (C) Comparison of NP swab and WB pathways in COVID-19–positive patients. (D) Comparison of WB pathways in COVID-19, influenza, and bacterial sepsis. Pathway prediction is determined by z-score; a positive value denotes up-regulation, and a negative value denotes down-regulation. All pathways and z-scores were calculated relative to NP swab and WB donor controls.
Fig. 3. Heatmap of pathways predicted to be involved in COVID-19 and other respiratory viruses from NP swabs.
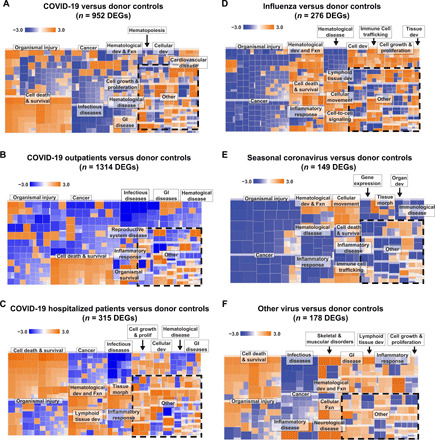
Comparison of pathways for (A) all patients with COVID-19 compared with donor controls, (B) outpatients compared with donor controls, and (C) hospitalized (non-ICU and ICU) patients compared with donor controls. (D) Influenza compared with donor samples. (E) Seasonal coronaviruses (HCoV HKU-1, HCoV NL-63, HCoV OC43, and HCoV 229E) compared with donor samples. (F) Other respiratory viruses (human metapneumovirus and human rhinovirus) compared with donor samples. Dev, development; Fxn, function; GI, gastrointestinal; morph, morphology.
Fig. 6. Venn diagrams of DEGs.
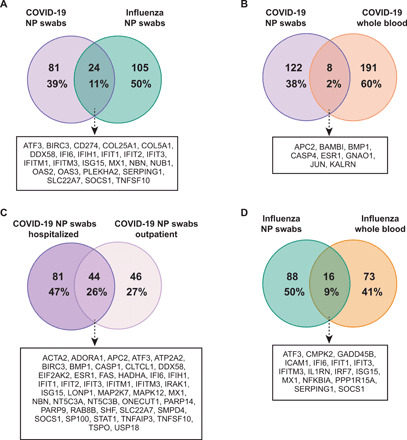
(A) Comparison of NP swab DEGs in patients with SARS-CoV-2 and influenza. (B) Comparison of NP swab and WB DEGs in patients with SARS-CoV-2. (C) Comparison of NP swab DEGs in COVID-19 outpatients and hospitalized patients. (D) Comparison of NP swab and WB DEGs in patients with influenza. The DEGs are calculated relative to donor controls.
Hierarchical clustering of DEGs generated from pairwise comparisons of the NP swab transcriptome in patients with SARS-CoV-2 infection and other viral ARI relative to individuals with nonviral ARI revealed three distinct gene groups (Fig. 5A and table S9). Group A (n = 35; including IFIT2, IFI6, and OAS2) was enriched in immune signaling genes and was up-regulated in SARS-CoV-2 infections but not other viral and nonviral ARIs. Group B consisted mostly of genes related to cell metabolism, signaling, and transport, as well as many uncharacterized genes (n = 41; including SOX3, CLCN1, and CCL2), and was increased in viral infections other than SARS-CoV-2, particularly influenza and seasonal coronavirus, compared with nonviral ARIs. Group C (n = 24; including COX15, FLI-1, and POLD1) was enriched in immune signaling, cell signaling, and cellular metabolism genes and was increased in viral infection, including from SARS-CoV-2.
Fig. 5. Hierarchically clustered heatmaps of DEGs.
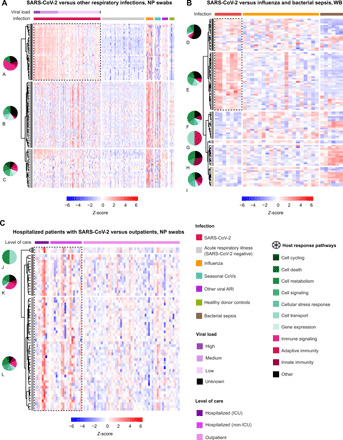
(A) NP swab DEGs from patients with infection by SARS-CoV-2 compared with influenza, seasonal coronaviruses, and other respiratory viruses, or virus-negative patients. Samples are stratified by diagnosis (“Infection”) and highest level of care (“Level of care” including outpatient, hospitalized, non-ICU, and ICU). (B) WB DEGs from patients with infection by SARS-CoV-2 compared with influenza, bacterial, and bacterial sepsis. Samples are stratified by diagnosis (“Infection”). (C) Comparison of DEGs among outpatients and hospitalized patients admitted (“ICU”) or not admitted (“non-ICU”) to the ICU. For (A), DEGs with Bonferroni-corrected P value <0.001 were included. For (B) and (C), DEGs with Bonferroni-corrected P value <0.01 were included. All noncoding genes were removed. For (A), DEGs are calculated relative to individuals with nonviral ARIs, and the top 100 DEGs are included; for (B), DEGs are calculated relative to donor controls, and the top 100 DEGs are included; for (C), DEGs are calculated on the basis of pairwise comparisons between outpatients and hospitalized patients, and all DEGs are included. Up-regulated genes are colored in red, and down-regulated genes are colored in blue. The distribution of predicted pathways (“Host response pathways”) is shown by pie graphs above each group in the heatmap; cellular functions are indicated in shades of green, immune functions are indicated in shades of red, and other pathways are indicated in black. DEG groups (A to L) were identified on the basis of hierarchical clustering with complete linkage, after exclusion of noncoding genes. The lettering is presented in alphabetical order, starting with A in the top left corner. Dotted outlines highlight key DEG groups, which are distinctly up-regulated in SARS-CoV-2 infection compared with other disease states.
Differential NP host responses in COVID-19 hospitalized patients versus outpatients
Hospitalized patients with COVID-19, including those requiring intensive care, had overlapping but heightened inflammatory responses compared with outpatients, relative to donor controls (Figs. 2B and 3, B and C), with up-regulation of DEGs implicated in innate antiviral immunity, such as triggering receptor expressed on myeloid cells 1 (TREM1) signaling and proinflammatory cytokines related to interleukin-6 (IL-6) and interleukin-8 (IL-8) signaling, including CXCL2, CXCL8, and IL6R relative to uninfected donor controls (Fig. 2B). In a direct comparison between hospitalized patients and outpatients with COVID-19, there was increased activation of pathways involved in hematological development and function, cellular movement, immune cell trafficking, inflammatory responses, and cell-to-cell signaling (Fig. 4A).
Fig. 4. Heatmaps of NP swab and WB pathways.
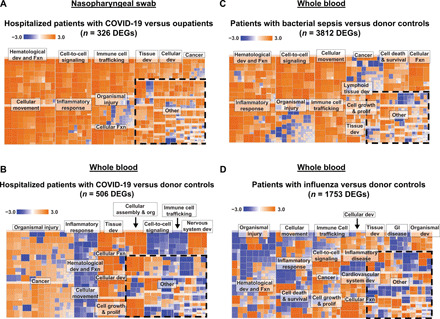
(A) Hospitalized patients compared with outpatients with COVID-19 using NP swabs. Comparison of WB pathways in hospitalized patients with COVID-19 compared with (B) donor controls, (C) bacterial sepsis, and (D) influenza.
Hierarchical clustering of DEGs based on direct comparison between outpatients versus hospitalized patients with COVID-19 revealed three distinct groups (Fig. 5C and table S10). The groups consisted of genes related to cell signaling, cellular metabolism, immune signaling, and innate immunity (group L) (n = 52; including IL1R1, IL6R, and CXCL2); cellular metabolism, immune signaling, and innate immunity (group M) (n = 13; including CXCL1, CXCL8, and VEGFA); and cellular metabolism and transport (group N) (n = 2; including SAT1 and FTH1). Genes from all three groups had increased overall expression in hospitalized patients relative to outpatients. Relative to donor controls, 26% (44 of 171) of DEGs were shared between outpatients and hospitalized patients (Fig. 6C and table S11), of which 21 of 44 (48%) were related to IFN signaling and innate immunity, including IFIT1, IFIT3, ISG15, EIF2AK2, and MAPK2K7.
WB transcriptome analysis
Pathway analysis of WB from patients with COVID-19, all of whom were hospitalized, compared with patients with influenza or bacterial sepsis showed notable inhibition of genes in multiple pathways associated with immune cell signaling and antiviral IFN responses, particularly genes in the nuclear factor κB and TREM1 signaling pathways (IL-1B,TLR1, TLR4, and TLR6), as well as natural killer cell signaling pathways (FCGR2A, FCGR3A, and FCGR3B) (Fig. 2D and tables S12 and S13). Up-regulated pathways in COVID-19 were primarily related to cell signaling [extracellular signal–regulated kinase (ERK)/mitogen-activated protein kinase (MAPK) and GP6 signaling], tissue development, cellular function and proliferation, and organismal injury and included only a few immune pathways, such as phosphatidylinositol 3-kinase (PI3K) signaling in B lymphocytes, CXCR4 signaling, and IL-15 production (Figs. 2D and 4, B to D). In contrast, bacterial sepsis was characterized by generalized up-regulation of immune-mediated pathways as well as multiple additional pathways associated with hematological development and other cellular functions (Figs. 2D and 3C). Hierarchical clustering of DEGs among patients with COVID-19, influenza, or bacterial sepsis based on comparisons to donor controls revealed six distinct groups (Fig. 5B and table S14). Groups D (n = 20) and E (n = 36) were up-regulated in COVID-19 and were primarily composed of genes related to cell death, cell metabolism, cell signaling, and multiple additional pathways, including DUSP8, CCR3, STX1A, and HBEGF. Groups H (n = 13) and I (n = 12) were up-regulated in bacterial sepsis and were enriched in genes related to innate immunity, immune signaling, cell signaling, and cell metabolism, including TLR8, DDIT4, IFIT1, and MMP9. Influenza showed mild up-regulation of all pathways.
Comparison of COVID-19 host responses between NP swabs and WB
COVID-19 host responses in NP swabs and WB shared common pathways related to antiviral response, innate immunity, ISG signaling (e.g., IL-6 and IL-8), and dendritic cell maturation. However, the directionality of signaling was discordant between NP swabs and WB for multiple additional immune-related pathways, including acute phase response signaling (z-score of −1.30 for NP swabs versus 0.33 for WB), IL-15 signaling (z-score of 0 versus 1.89), CXCR4 signaling (z-score 0 versus 1.63), natural killer cell signaling (z-score 0 versus −1.63), T helper 1 pathway (z-score 0 versus −2.24), and B cell receptor signaling (z-score 2.11 versus −0.5) (Fig. 2C). Very few DEGs (≤3%) were shared between NP swabs and WB from patients with COVID-19 (Fig. 6B and figs. S5, A and B), suggesting that the host response was localized and body site specific. In contrast, heightened IFN responses in both NP swabs and WB were observed for influenza (Fig. 2, A and D), consistent with a systemic immune and inflammatory response. Notably, among the 16 DEGs shared between NP swabs and WB from patients with influenza, the majority of those genes (11 of 16, 69%) were related to innate immunity and IFN signaling (Fig. 6D).
Host response classifier
As transcriptome analysis had revealed distinct patterns of gene expression in patients with COVID-19 (Figs. 2A and 5A), we hypothesized that we would be able to construct a classifier that accurately discriminates between SARS-CoV-2 infection and other viral or nonviral ARIs from NP swabs. After randomly partitioning 20% of samples into an independent test cohort, we developed a two-layer classifier that first differentiates between SARS-CoV-2–positive cases and SARS-CoV-2–negative cases for which no pathogen was identified (layer 1), followed by a second layer that differentiates SARS-CoV-2 from microbiologically confirmed viral ARIs, including influenza and seasonal coronavirus infections, among others (layer 2) (Fig. 7A and table S15). The initial set of DEGs for each classifier was selected using a Bonferroni-corrected P value of <0.001 for both layers. Using read counts corresponding only to the DEGs identified from training set samples, we generated optimal binary classifiers using fivefold cross-validation to evaluate the performance of 13 candidate classification models in differentiating between SARS-CoV-2 infection and nonviral ARIs (layer 1) (table S16) and between SARS-CoV-2 infection and other viral ARIs (layer 2). Samples assigned to SARS-CoV-2 by both classifiers were designated positive for SARS-CoV-2 infection. The cutoff for the prediction score of each classifier was determined by generating receiver operating characteristic (ROC) curves for the training data and comparing a cutoff based on Youden’s index, an arbitrary 0.5 cutoff, and a manually selected threshold that prioritized specificity (“high-specificity threshold”) (tables S17 and S18). After review of the training set results, we elected to use the manually selected high-specificity threshold.
Fig. 7. Diagnostic classifier for COVID-19.
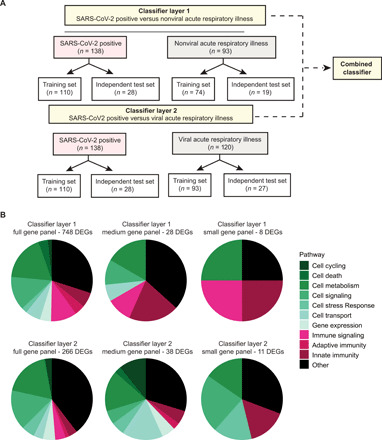
(A) Overview of classifier design and distribution of samples for training and test sets in layers 1 and 2. (B) Pie graphs showing the distribution of predicted pathways (“Host response pathways”) represented by the genes within the full gene panel (left), medium gene panel (middle), or the small gene panel (right).
The layer 1 classifier, generated using a training set of 110 SARS-CoV-2–positive and 74 nonviral ARI samples, contained 748 DEGs, consisting of genes associated with both cell processes and immune signaling (Fig. 7B, top left, and table S19). This classifier had a sensitivity of 97.3%, specificity of 97.3%, and an area under the ROC curve (AUC) of 0.993 at a threshold of 0.4515 (fig. S6A and table S15). The layer 2 classifier, generated using a training set of the same 110 SARS-CoV-2–positive and 93 viral ARI samples, contained 266 DEGs with a smaller proportion of immune signaling genes than in the layer 1 classifier (Fig. 7B, bottom left, and table S20). This classifier had a sensitivity of 95.5%, specificity of 98.9%, and AUC of 0.999 at a threshold of 0.6066 (fig. S6D and table S15). On the basis of the training set data, the full 1014-gene two-layer classifier (containing a full complement of 1014 genes) had an overall sensitivity of 95.5%, specificity of 98.2%, and AUC of 0.999 (Fig. 8A and table S15).
Fig. 8. Performance characteristics of the two-layered (combined layers 1 and layer 2) classifier for full, medium, and small gene panels.
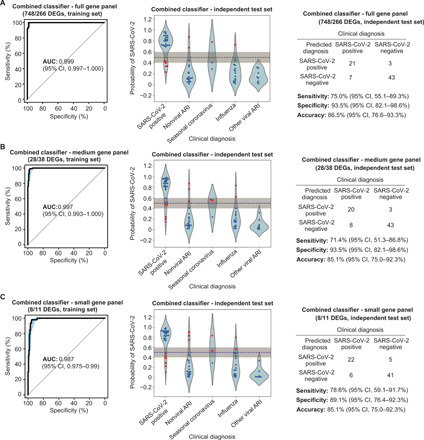
(A to C) Training set ROC curve (left) and test set violin plot (middle) and confusion matrix (right) for the two-layer classifier, using either the full gene panel (A), medium gene panel (B), or the small gene panel (C).
The performance of the two-layer classifier was then evaluated using an independent test set that included NP swab samples from 28 SARS-CoV-2–positive, 19 nonviral ARI, and 27 viral ARI patients (Fig. 7A and table S15). The layer 1 classifier had 82.1% sensitivity, 89.5% specificity (fig. S7A and table S15), and AUC of 0.944, while the layer 2 classifier yielded 92.9% sensitivity, 96.3% specificity (fig. S7D and table S15), and AUC of 0.991. On the basis of test set data, the full 1014-gene two-layer classifier had an overall sensitivity of 75.0% [95% confidence interval (CI), 55.0 to 89.0%], specificity of 93.5% (95% CI, 82.1 to 98.6%), and AUC of 0.933 (range, 0.879 to 987), yielding an overall accuracy of 86.5% (Fig. 8A).
Because panels containing a smaller number of genes would be more practical to translate into a clinical assay, we used lasso regression analysis to find an optimal set of genes for a medium two-layer classifier with an a priori specification of no more than 100 genes (tables S21 and S22). The medium classifier consisted of 29 genes for layer 1 and 38 genes for layer 2 (Fig. 7B, middle, and tables S23 and S24). On the basis of the training set, the medium 67-gene two-layer classifier had a sensitivity of 88.2%, specificity of 97.6%, and AUC of 0.997 (fig. S6 and table S15). When applied to the test set, the medium two-layer classifier had a sensitivity of 71.4% (95% CI, 51.3 to 86.8%), specificity of 93.5% (95% CI, 82.1 to 98.6%), AUC of 0.922 (range, 0.863 to 0.982), and 85.1% overall accuracy (Fig. 8B). We then explored narrowing the number of genes to <20 total by iteratively removing one gene at a time from the 29 genes for layer 1 (table S21) and 37 genes for layer 2 (table S22). Maximum performance was identified for a small two-layer classifier consisting of 19 genes, 8 genes for layer 1, and 11 genes for layer 2 (Fig. 7B and tables S25 and S26). On the basis of the training set, the small 19-gene two-layer classifier had a sensitivity of 94.6%, specificity of 94.6%, and AUC of 0.984 for layer 1 (fig. S6 and table S15). When applied to the test set, the small two-layer classifier had a sensitivity of 78.6% (95% CI, 76.5 to 99.1%), specificity of 89.1% (95% CI, 59.1 to 91.7%), AUC of 0.906 (range, 0.837 to 0.974), and 85.1% accuracy (Fig. 8C).
There was >50% overlap in the misclassified patients among all three classifiers, suggesting internal consistency between them (table S27). No obvious clinical factors, including days between symptom onset and sample collection (fig. S8), appeared to be associated with classifier performance.
DISCUSSION
Here, we use RNA-seq to characterize the differential host responses to SARS-CoV-2 infection in 286 NP swab and 53 WB samples from 333 individuals. Both NP swabs and WB from patients with COVID-19 show distinct patterns of activation or inhibition relative to other infections (influenza, seasonal coronaviruses, and bacterial sepsis) and to each other. SARS-CoV-2 infection was found to activate IFN-mediated antiviral pathways and paradoxically inhibit multiple additional immune and inflammatory pathways, resulting in an overall dysregulated immune response. Although overall DEGs and pathways were similar between outpatients and hospitalized patients with COVID-19, the magnitude of host response was found to increase with clinical severity of disease. We also demonstrated that diagnostic two-layer host response classifiers based on RNA-seq data can discriminate SARS-CoV-2 infection from other viral and nonviral ARIs from NP swab samples with an accuracy of 85.1 to 86.5%.
Viral metatranscriptome analyses show that coinfections of SARS-CoV-2 with other respiratory viruses are uncommon and occur at similar frequencies among COVID-19 and non–COVID-19 cases. They also reveal a decrease in abundance and diversity in SARS-CoV-2 infection relative to nonviral ARI, with displacement of the normal viral flora in the nasopharynx (i.e., anelloviruses and bacteriophages). The degree of perturbation of the virome appears to be less for SARS-CoV-2 than for other respiratory viral infections. In contrast, viral infection, whether caused by SARS-CoV-2 or another respiratory virus, did not appear to markedly alter the bacterial metatranscriptome of the nasopharynx.
IFN-mediated antiviral responses and chemokine expression are critical to host defense against viral infection (28, 29). Notably, the specific patterns of activation or inhibition of these pathways in COVID-19 are distinct from those associated with influenza or other respiratory viruses. Overall, the host response in patients with COVID-19 shows increased expression of genes involved in IFN responses and ISGs (30–32) but inhibition of multiple other immune-mediated pathways including IL-6 and IL-8 signaling. Our finding of IL-6 down-regulation in patients with COVID-19 is consistent with prior published reports describing the host response to SARS-CoV-2 in bronchoalveolar lavage and peripheral blood mononuclear cell samples (33, 34). Although other reports describe increased circulating plasma IL-6 levels in COVID-19 (35–37), this may be due to a negative feedback loop driving IL-6 gene down-regulation. Our data also support the hypothesis that tissue-associated neutrophils may be an important contributor to severe COVID-19, as suggested by the increased relative neutrophil counts estimated in NP swab samples by cell deconvolution analysis and induction of CXCL2 and CXCL8 cytokines, which are associated with neutrophil chemotaxis (38, 39).
Of note, ciliated epithelial cells appear to be major contributors to the host transcriptome in NP swab samples versus white blood cells in WB. Differing cell types and proportions may thus explain the lack of overlap in shared DEGs and pathways between NP swabs and WB. Notably, there are no IFN-associated DEGs or pathways shared between NP swabs and WB from patients with COVID-19. In contrast, activation of IFN-associated pathways in both the upper airway and blood of patients with influenza suggests a global, more systemic host response relative to COVID-19. Although angiotensin converting enzyme 2 (ACE2) has been shown to be the cellular receptor for entry of SARS-CoV-2 and has been described as an ISG (40, 41), we did not find ACE2 to be up-regulated in patients with COVID-19, whether from NP swab or WB samples.
Our findings of a distinct host response biosignature in patients with COVID-19 and an augmented response in the setting of more severe illness underscore the potential diagnostic utility of host response–based classifiers for SARS-CoV-2 infection. Here, we present a 19-gene diagnostic classifier with >85% overall accuracy (~80% sensitivity and ~90% specificity). The size of the classifier is compatible with existing multiplex diagnostic platforms (42, 43). A host response–based test may be particularly useful as a complementary diagnostic tool for SARS-CoV-2 infection. Here, we also identify a panel of DEGs associated with more severe COVID-19 disease. No correlation is generally observed between viral load and severity of disease (Fig. 1B) (44, 45), and a robust biomarker for disease severity is not yet clinically available. Validation with additional longitudinal samples will be needed to determine the utility of a separate host response–based classifier in predicting clinical severity and outcomes.
Although PCR has been shown to have excellent sensitivity and specificity overall for the detection of SARS-CoV-2 and other respiratory viruses, host gene expression classifiers may eventually play a complementary role in the diagnosis of COVID-19 disease. Studies published to date have reported host classifiers that can distinguish between bacterial and viral illness (16, 20–22). Here, we present a diagnostic host classifier from NP swabs that can distinguish among SARS-CoV-2 and other viral and bacterial respiratory infections. Future efforts will focus on evaluating the utility of this classifier for diagnosis of SARS-CoV-2 infection in presymptomatic or asymptomatic individuals during the incubation period, 38% of whom are still PCR negative at time of symptom onset (11, 46), and in generating classifiers to evaluate and predict COVID-19 severity.
MATERIALS AND METHODS
Ethics
This study was approved by the institutional review board (IRB) at the UCSF (IRB number 10-02598) and as a no-subject contact study with waiver of consent. Samples from the CDPH were deidentified and deemed not research or exempt by the Committee for the Protection of Human Subjects (project number 2020-30) issued under the California Health and Human Services Agency’s Federal Wide Assurance #00000681 with the Office of Human Research Protections. Remnant NP swab and WB samples after clinical testing were collected for RNA-seq analysis, and review of the patient electronic medical records was performed, with the data presented in aggregate.
NP swab sample collection
The study population consisted of patients with available remnant NP samples collected in universal transport media (UTM) or DNA/RNA Shield (Zymo Research) from the clinical laboratories at the UCSF (n = 316). Samples from patients who were positive or negative by SARS-CoV-2 real-time RT-PCR testing or were positive by respiratory virus panel PCR on NP swabs were collected from 20 September 2014 to 30 April 2020 (Fig. 1A). Patients who tested negative by SARS-CoV-2 RT-PCR were selected randomly. In addition, RNA extracts from patients who had tested positive by SARS-CoV-2 RT-PCR (n = 4) and UTM from patients with seasonal coronavirus or influenza were provided by the CDPH (Richmond, CA) (n = 20). NP swabs from donor controls were obtained from asymptomatic volunteers at the UCSF (n = 11).
WB sample collection
Remnant WB from patients with COVID-19 was collected from the clinical laboratories at the UCSF from 8 March 2020 to 13 April 2020 (n = 7) (Fig. 1A). Remnant WB from patients with influenza (n = 20) and sepsis (n = 6) were collected from 7 March 2018 to 15 November 2018. Additional donor controls were obtained from volunteers at the UCSF (n = 20).
Nucleic acid extraction
All NP swab samples obtained at the UCSF were pretreated with a 1:1 ratio of DNA/RNA Shield (Zymo Research) before extraction. An input volume of 200 μl of NP swab sample was used for all extraction methods performed at the UCSF and eluted in 100 μl. NP swab samples obtained from the CDPH were extracted using the easyMag instrument (bioMérieux) according to the manufacturer’s instructions with an input volume of 300 μl and elution volume of 110 μl, except for 4 seasonal coronavirus and 3 influenza samples, which were extracted using the Mag-Bind Viral DNA/RNA 96 kit (Omega Bio-Tek) on a KingFisher Flex (Thermo Fisher Scientific) instrument according to the manufacturer’s instructions. For NP swab samples collected at the UCSF, 297 were extracted using the Mag-Bind Viral DNA/RNA 96 kit (Omega Bio-Tek) on the KingFisher Flex (Thermo Fisher Scientific), and 30 samples were extracted using the EZ1 Advanced XL (Qiagen) according to the manufacturer’s instructions.
All WB samples (300 μl) were pretreated with a 2:1 ratio of DNA/RNA Shield (Zymo Research) and extracted using Direct-zol RNA MiniPrep kit (Zymo Research) according to the manufacturer’s instructions. Samples were on-column deoxyribonuclease (DNase) treated with DNase I (Zymo Research) and eluted in 30 μl. Extracted material was stored at −80°C.
Library preparation and sequencing
Extracted RNA from NP swab samples (25 μl) was treated with a nuclease cocktail of TURBO DNase (Thermo Fisher Scientific) and Baseline Zero DNase (Ambion) for 30 min at 37°C and purified using AMPure XP beads (Beckman Coulter) on the epMotion 5075 (Eppendorf). Purified RNA (7 μl) was used for library preparation using the SMART-Seq Stranded kit (Takara Bio) and purified using AMPure XP beads (Beckman Coulter) on the epMotion 5073 (Eppendorf). Libraries were quantified using the Qubit dsDNA HS Assay (Thermo Fisher Scientific) on the Qubit Flex (Thermo Fisher Scientific).
WB sample libraries were prepared using 9 μl of total RNA and TruSeq Total RNA with Ribo-Zero Globin (Illumina) and spiked with 1 μl of ERCC RNA Spike-In Mix (Thermo Fisher Scientific). Libraries were purified using AMPure XP beads (Beckman Coulter) and quantified using the Qubit dsDNA HS Assay (Thermo Fisher Scientific) on the Qubit Flex (Thermo Fisher Scientific).
NP swab and WB sample libraries were sequenced on the NovaSeq 6000 (Illumina) using 150–base pair paired-end sequencing at the UCSF Center for Advanced Technology. Included in each sequencing run were negative controls (nuclease-free water) to monitor for laboratory and reagent contamination and a Human Reference RNA Standard (Agilent) to monitor for sequencing efficiency.
Metatranscriptomic analysis
Metatranscriptomic next-generation sequencing (mNGS) data from all samples were analyzed for viral nucleic acids using SURPI+ (v1.0.7-build.4), a bioinformatics pipeline for pathogen detection and discovery from metatranscriptomic data, modified to incorporate enhanced filtering and classification algorithms (47, 48). The Scalable Nucleotide Alignment Program (SNAP) nucleotide aligner was run using an edit distance of 16 against the National Center for Biotechnology Information (NCBI) nucleotide database (March 2019, with inclusion of the SARS-CoV-2 WuHan-Hu-1 genome accession number NC_045512) filtered to retain only viral, bacterial, fungal, and parasitic reads, enabling detection of reads from microorganisms with ≥90% identity to reference sequences in the database. The preestablished criterion for viral detection by SNAP was the presence of reads mapping to at least three nonoverlapping regions of the viral genome (47). Diversity metrics, including the Chao Richness Score and Shannon diversity index, were calculated in R (version 4.00) (49) using the vegan package (version 2.5.3), and figures were produced using the ggplot2 package (50).
Transcriptome analysis
Following sequencing of sample libraries, quality control was performed on the fastq files to ensure that sequencing reads met preestablished cutoffs for number of reads and quality using FastQC (version 0.11.8) (51) and MultiQC (version 1.8) (52). Quality filtering and adapter trimming were performed using BBduk tools (version 38.76, https://sourceforge.net/projects/bbmap). Remaining reads were aligned to the ENSEMBL GRCh38 human reference genome assembly (release 33) using STAR (version 2.7.0f) (53), and gene frequencies were counted using featureCounts (version 2.0.0) within the Subread package (54). Comparative analysis of DEGs was performed using a generalized linear model implemented in the edgeR Bioconductor package (version 3.30.3) (55) using a Benjamini-Hochberg–corrected P value of <0.01.
Hierarchical clustering of DEGs was performed in R (version 4.0.0) using the ComplexHeatmap and pheatmap package (49). Figures were produced using the ggplot2 package (50). For NP and WB, the top 100 DEGs sorted by P value with Bonferroni-corrected P values of <0.001and <0.01, respectively, were included. For the comparison between hospitalized and outpatients, all the DEGs with a Bonferroni-corrected P value of <0.01 were included. Clustering was performed on the basis of Euclidean distance with complete linkage, after exclusion of noncoding genes.
Signaling pathway analyses and heatmaps were generated using the Ingenuity Pathway Analysis (IPA) software (Qiagen) (56). The molecule activity predictor tool of IPA was used to predict gene up- or down-regulation and pathway activation or inhibition. The enrichment score P value was used to evaluate the significance of the overlap between predicted and observed genes, while the z-score was used to assess the match between observed and predicted regulation or down-regulation.
Classifiers were developed using scikit-learn (version 1.2.2) (57) in Python. A total of 13 different classifier models, including Linear Support Vector Machine, Linear Discriminant Analysis, and Deep Neural Network, were trained in parallel using a cross-validation approach. Candidate classifier models included a Linear Support Vector Machine, Linear Discriminant Analysis, and a Deep Neural Network, all within the scikit-learn package. The performance of each model was evaluated on the basis of the average score achieved across five cross-validation iterations; these average scores were then compared to select the best-performing model (table S16). Reduced gene panels were selected using Lasso (58), and a customized reverse search across the resulting feature set was performed. This search iteratively removed the remaining gene with the lowest significance as measured by its Lasso coefficient, performed classifier training, and reported sensitivity, specificity, and accuracy across the training set. These results were then manually reviewed to balance each of them with a priority placed on specificity and number of genes. ROC curves were generated using pROC package in R (59).
Statistical analysis
To identify potentially important clinical predictors for COVID-19 score among RT-PCR–positive patients, linear regression models were used to check the association of each clinical variable with the transformed COVID-19 score while controlling for demographics (age, gender, and race/ethnicity). A stepwise procedure was then used to determine what clinical variables would be selected when all the variables were included in the model while controlling for demographics. Variables with a P value less than 0.15 from those models were further examined for their association with transformed COVID-19 score in one model together while controlling for demographics. In this exploratory analysis, we did not adjust P values for multiple comparisons to avoid missing potentially important variables.
Ct values were categorized as low (Ct <18), moderate (Ct ≥18 and ≤25), and high (Ct >25). The association of demographics and clinical variables with RT-PCR (positive versus negative), diagnosis (COVID-19, influenza, or bacterial sepsis), and viral load (low, medium, or high) was examined by Fisher’s exact test (values <5) or χ2 test (values >5) for categorical variables and two-sample t test or analysis of variance (ANOVA) for age, respectively. The association of demographics and clinical variables with Ct values was assessed with Wilcoxon rank sum test for variables with two categories or Kruskal-Wallis test for variables with more than two categories. The tetrachoric or polychoric correlation was estimated for the correlation between binary RT-PCR and binary or ordinal symptoms and outcome. The point-biserial correlation was estimated for the correlation between binary symptoms and continuous Ct values. For mNGS analysis, comparisons of virome or bacterial metatranscriptome abundance, richness, and alpha diversity between groups were analyzed using the Kruskal-Wallis test, followed by the Nemenyi test for post hoc analysis.
Comparisons of diagnosis and disease severity for cell types were conducted using the Kruskal-Wallis test, followed by Dunn’s test for pairwise multiple comparisons for post hoc analysis of viral diagnosis (fig. S3A, 16 comparisons), patient severity (fig. S3B, 4 comparisons), and WB diagnosis (fig. S4, 7 comparisons). All statistical tests were calculated as two sided at the 0.05 significance level.
Acknowledgments
We thank the staff at the UCSF Center for Advanced Technology for sequencing transcriptome libraries using the Illumina NovaSeq 6000. Funding: This work was funded by NIH grants R01HL105704 (C.Y.C.), R33AI29077 (C.Y.C.), R38HL143581 (G.M.G. and S.P.B.), R01CA249054 (C.E.M.), and R01NS076465 (C.E.M.). This work was also funded, in part, by the Charles and Helen Schwab Foundation (C.Y.C.), the Abbott Laboratories (C.Y.C.), the Pershing Square Sohn Cancer Research Alliance (C.E.M.), the Citadel Foundation (C.E.M.), and the WorldQuant Foundation (C.E.M.). These funders had no role in study design, data collection and analysis, writing of the manuscript, or decision to publish. Author contributions: C.Y.C. conceived, designed, and supervised the study. D.L.N. and A.C.G. coordinated the study. D.L.N., A.C.G., Y.A.S, V.S., and C.Y.C. analyzed the data, designed the figures, and wrote and edited the manuscript. G.M.G. performed chart review and designed the figures. A.S.-G. and C.S.-S.M. performed experiments and contributed to the collection of clinical specimens. A.G., R.C., C.S.C., C.-Y.P., H.G., D.W., and S.M. contributed to the collection of clinical specimens. N.A., N.M.N., D.N.N., A.G.L., J.B., C.A.W., A.S.Gl., S.P.B., L.M.H., and K.T. performed chart review. S.F. and T.L. provided bioinformatics analysis. J.C. performed biostatistical analysis. C.M., J.F., and C.E.M. performed the deconvolution analysis. All authors read the manuscript and agree to its contents. Competing interests: C.Y.C. is the director of the UCSF-Abbott Viral Diagnostics and Discovery Center (VDDC) and receives research support funding from the Abbott Laboratories Inc. D.L.N. receives research support funding from Cepheid Inc. C.Y.C., D.L.N., A.C.G., Y.A.S., and V.S. are inventors on a pending patent application related to the SARS-CoV-2 classifier entitled “Analysis of Host Gene Expression for Diagnosis of Severe Acute Respiratory Syndrome Coronavirus 2 Infection” (docket no. 64366-30026.00). The other authors declare that they have no competing interests. Patent filed by C.Y.C., D.L.N., A.C.G., V.S., and Y.A.S. Name of the organization filing the patent: UCSF. Current patent status: Submitted (Provisional Patent Application). Date of Filing: 10 December 2020. U.S. Patent Application Serial No. 62/591,660. Current patent status: Submitted (Provisional Patent Application). Data and materials availability: Metagenomic sequencing data (FASTQ files) after removal of human genomic reads have been deposited into the NCBI Sequence Read Archive (SRA) (accession number PRJNA691419, under umbrella project PRJNA234047). RNA-seq data (FASTQ files and count tables) have been deposited into the NCBI Gene Expression Omnibus (GEO) biorepository (accession number GSE163151). All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials or available for download from the Chiu lab Zenodo repository at DOI: 10.5281/zenodo.4283753. R scripts for computational and visualization tasks are available from the authors upon request.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/6/eabe5984/DC1
REFERENCES AND NOTES
- 1.Dong E., Du H., Gardner L., An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 20, 533–534 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abdollahi E., Champredon D., Langley J. M., Galvani A. P., Moghadas S. M., Temporal estimates of case-fatality rate for COVID-19 outbreaks in Canada and the United States. CMAJ 192, E666–E670 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guan W.-J., Ni Z.-Y., Hu Y., Liang W.-H., Ou C.-Q., He J.-X., Liu L., Shan H., Lei C.-L., Hui D. S. C., Du B., Li L.-J., Zeng G., Yuen K.-Y., Chen R.-C., Tang C.-L., Wang T., Chen P.-Y., Xiang J., Li S.-Y., Wang J.-L., Liang Z.-J., Peng Y.-X., Wei L., Liu Y., Hu Y.-H., Peng P., Wang J.-M., Liu J.-Y., Chen Z., Li G., Zheng Z.-J., Qiu S.-Q., Luo J., Ye C.-J., Zhu S.-Y., Zhong N.-S.; China Medical Treatment Expert Group for Covid-19 , Clinical characteristics of coronavirus disease 2019 in China. N. Engl. J. Med. 382, 1708–1720 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Russell T. W., Hellewell J., Jarvis C. I., van Zandvoort K., Abbott S., Ratnayake R.; Cmmid Covid-Working Group, Flasche S., Eggo R. M., Edmunds W. J., Kucharski A. J., Estimating the infection and case fatality ratio for coronavirus disease (COVID-19) using age-adjusted data from the outbreak on the Diamond Princess cruise ship, February 2020. Euro Surveill. 25, 2000256 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Verity R., Okell L. C., Dorigatti I., Winskill P., Whittaker C., Imai N., Cuomo-Dannenburg G., Thompson H., Walker P. G. T., Fu H., Dighe A., Griffin J. T., Baguelin M., Bhatia S., Boonyasiri A., Cori A., Cucunuba Z., FitzJohn R., Gaythorpe K., Green W., Hamlet A., Hinsley W., Laydon D., Nedjati-Gilani G., Riley S., van Elsland S., Volz E., Wang H., Wang Y., Xi X., Donnelly C. A., Ghani A. C., Ferguson N. M., Estimates of the severity of coronavirus disease 2019: A model-based analysis. Lancet Infect. Dis. 20, 669–677 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu Z., McGoogan J. M., Characteristics of and important lessons from the Coronavirus disease 2019 (COVID-19) outbreak in China: Summary of a report of 72314 cases from the Chinese Center for Disease Control and Prevention. JAMA 323, 1239–1242 (2020). [DOI] [PubMed] [Google Scholar]
- 7.Yang X., Yu Y., Xu J., Shu H., Xia J., Liu H., Wu Y., Zhang L., Yu Z., Fang M., Yu T., Wang Y., Pan S., Zou X., Yuan S., Shang Y., Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: A single-centered, retrospective, observational study. Lancet Respir. Med. 8, 475–481 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu C., Chen X., Cai Y., Xia J., Zhou X., Xu S., Huang H., Zhang L., Zhou X., Du C., Zhang Y., Song J., Wang S., Chao Y., Yang Z., Xu J., Zhou X., Chen D., Xiong W., Xu L., Zhou F., Jiang J., Bai C., Zheng J., Song Y., Risk factors associated with acute respiratory distress syndrome and death in patients with Coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern. Med. 180, 934–943 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou Z., Ren L., Zhang L., Zhong J., Xiao Y., Jia Z., Guo L., Yang J., Wang C., Jiang S., Yang D., Zhang G., Li H., Chen F., Xu Y., Chen M., Gao Z., Yang J., Dong J., Liu B., Zhang X., Wang W., He K., Jin Q., Li M., Wang J., Heightened innate immune responses in the respiratory tract of COVID-19 patients. Cell Host Microbe 27, 883–890.e2 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pan Y., Zhang D., Yang P., Poon L. L. M., Wang Q., Viral load of SARS-CoV-2 in clinical samples. Lancet Infect. Dis. 20, 411–412 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wolfel R., Corman V. M., Guggemos W., Seilmaier M., Zange S., Muller M. A., Niemeyer D., Jones T. C., Vollmar P., Rothe C., Hoelscher M., Bleicker T., Brunink S., Schneider J., Ehmann R., Zwirglmaier K., Drosten C., Wendtner C., Virological assessment of hospitalized patients with COVID-2019. Nature 581, 465–469 (2020). [DOI] [PubMed] [Google Scholar]
- 12.Zou L., Ruan F., Huang M., Liang L., Huang H., Hong Z., Yu J., Kang M., Song Y., Xia J., Guo Q., Song T., He J., Yen H. L., Peiris M., Wu J., SARS-CoV-2 viral load in upper respiratory specimens of infected patients. N. Engl. J. Med. 382, 1177–1179 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen M., Carlson D., Zaas A., Woods C. W., Ginsburg G. S., Hero A. III, Lucas J., Carin L., Detection of viruses via statistical gene expression analysis. IEEE Trans. Biomed. Eng. 58, 468–479 (2011). [DOI] [PubMed] [Google Scholar]
- 14.Huang Y., Zaas A. K., Rao A., Dobigeon N., Woolf P. J., Veldman T., Oien N. C., McClain M. T., Varkey J. B., Nicholson B., Carin L., Kingsmore S., Woods C. W., Ginsburg G. S., Hero A. O. III, Temporal dynamics of host molecular responses differentiate symptomatic and asymptomatic influenza a infection. PLOS Genet. 7, e1002234 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zaas A. K., Burke T., Chen M., McClain M., Nicholson B., Veldman T., Tsalik E. L., Fowler V., Rivers E. P., Otero R., Kingsmore S. F., Voora D., Lucas J., Hero A. O., Carin L., Woods C. W., Ginsburg G. S., A host-based RT-PCR gene expression signature to identify acute respiratory viral infection. Sci. Transl. Med. 5, 203ra126 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zaas A. K., Chen M., Varkey J., Veldman T., Hero A. O. III, Lucas J., Huang Y., Turner R., Gilbert A., Lambkin-Williams R., Øien N. C., Nicholson B., Kingsmore S., Carin L., Woods C. W., Ginsburg G. S., Gene expression signatures diagnose influenza and other symptomatic respiratory viral infections in humans. Cell Host Microbe 6, 207–217 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andres-Terre M., McGuire H. M., Pouliot Y., Bongen E., Sweeney T. E., Tato C. M., Khatri P., Integrated, multi-cohort analysis identifies conserved transcriptional signatures across multiple respiratory viruses. Immunity 43, 1199–1211 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bouquet J., Soloski M. J., Swei A., Cheadle C., Federman S., Billaud J. N., Rebman A. W., Kabre B., Halpert R., Boorgula M., Aucott J. N., Chiu C. Y., Longitudinal transcriptome analysis reveals a sustained differential gene expression signature in patients treated for acute lyme disease. MBio 7, e00100-16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Landry M. L., Foxman E. F., Antiviral response in the nasopharynx identifies patients with respiratory virus infection. J. Infect. Dis. 217, 897–905 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suarez N. M., Bunsow E., Falsey A. R., Walsh E. E., Mejias A., Ramilo O., Superiority of transcriptional profiling over procalcitonin for distinguishing bacterial from viral lower respiratory tract infections in hospitalized adults. J. Infect. Dis. 212, 213–222 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sweeney T. E., Wong H. R., Khatri P., Robust classification of bacterial and viral infections via integrated host gene expression diagnostics. Sci. Transl. Med. 8, 346ra391 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsalik E. L., Henao R., Nichols M., Burke T., Ko E. R., McClain M. T., Hudson L. L., Mazur A., Freeman D. H., Veldman T., Langley R. J., Quackenbush E. B., Glickman S. W., Cairns C. B., Jaehne A. K., Rivers E. P., Otero R. M., Zaas A. K., Kingsmore S. F., Lucas J., Fowler V. G. Jr., Carin L., Ginsburg G. S., Woods C. W., Host gene expression classifiers diagnose acute respiratory illness etiology. Sci. Transl. Med. 8, 322ra311 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Woods C. W., McClain M. T., Chen M., Zaas A. K., Nicholson B. P., Varkey J., Veldman T., Kingsmore S. F., Huang Y., Lambkin-Williams R., Gilbert A. G., Hero A. O. III, Ramsburg E., Glickman S., Lucas J. E., Carin L., Ginsburg G. S., A host transcriptional signature for presymptomatic detection of infection in humans exposed to influenza H1N1 or H3N2. PLOS ONE 8, e52198 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rosenbloom K. R., Armstrong J., Barber G. P., Casper J., Clawson H., Diekhans M., Dreszer T. R., Fujita P. A., Guruvadoo L., Haeussler M., Harte R. A., Heitner S., Hickey G., Hinrichs A. S., Hubley R., Karolchik D., Learned K., Lee B. T., Li C. H., Miga K. H., Nguyen N., Paten B., Raney B. J., Smit A. F., Speir M. L., Zweig A. S., Haussler D., Kuhn R. M., Kent W. J., The UCSC Genome Browser database: 2015 update. Nucleic Acids Res. 43, D670–D681 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang W., Xu Y., Gao R., Lu R., Han K., Wu G., Tan W., Detection of SARS-CoV-2 in different types of clinical specimens. JAMA 323, 1843–1844 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Maio F., Posteraro B., Ponziani F. R., Cattani P., Gasbarrini A., Sanguinetti M., Nasopharyngeal microbiota profiling of SARS-CoV-2 infected patients. Biol. Proced. Online 22, 18 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang X., Park J., Susztak K., Zhang N. R., Li M., Bulk tissue cell type deconvolution with multi-subject single-cell expression reference. Nat. Commun. 10, 380 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McNab F., Mayer-Barber K., Sher A., Wack A., O’Garra A., Type I interferons in infectious disease. Nat. Rev. Immunol. 15, 87–103 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salazar-Mather T. P., Hokeness K. L., Cytokine and chemokine networks: Pathways to antiviral defense. Curr. Top. Microbiol. Immunol. 303, 29–46 (2006). [DOI] [PubMed] [Google Scholar]
- 30.Bost P., Giladi A., Liu Y., Bendjelal Y., Xu G., David E., Blecher-Gonen R., Cohen M., Medaglia C., Li H., Deczkowska A., Zhang S., Schwikowski B., Zhang Z., Amit I., Host-viral infection maps reveal signatures of severe COVID-19 patients. Cell 181, 1475–1488.e12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chua R. L., Lukassen S., Trump S., Hennig B. P., Wendisch D., Pott F., Debnath O., Thurmann L., Kurth F., Volker M. T., Kazmierski J., Timmermann B., Twardziok S., Schneider S., Machleidt F., Muller-Redetzky H., Maier M., Krannich A., Schmidt S., Balzer F., Liebig J., Loske J., Suttorp N., Eils J., Ishaque N., Liebert U. G., von Kalle C., Hocke A., Witzenrath M., Goffinet C., Drosten C., Laudi S., Lehmann I., Conrad C., Sander L. E., Eils R., COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 38, 970–979 (2020). [DOI] [PubMed] [Google Scholar]
- 32.Liao M., Liu Y., Yuan J., Wen Y., Xu G., Zhao J., Cheng L., Li J., Wang X., Wang F., Liu L., Amit I., Zhang S., Zhang Z., Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. 26, 842–844 (2020). [DOI] [PubMed] [Google Scholar]
- 33.Wilk A. J., Rustagi A., Zhao N. Q., Roque J., Martinez-Colon G. J., McKechnie J. L., Ivison G. T., Ranganath T., Vergara R., Hollis T., Simpson L. J., Grant P., Subramanian A., Rogers A. J., Blish C. A., A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat. Med. 26, 1070–1076 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xiong Y., Liu Y., Cao L., Wang D., Guo M., Jiang A., Guo D., Hu W., Yang J., Tang Z., Wu H., Lin Y., Zhang M., Zhang Q., Shi M., Liu Y., Zhou Y., Lan K., Chen Y., Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg. Microbes Infect. 9, 761–770 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blanco-Melo D., Nilsson-Payant B. E., Liu W. C., Uhl S., Hoagland D., Moller R., Jordan T. X., Oishi K., Panis M., Sachs D., Wang T. T., Schwartz R. E., Lim J. K., Albrecht R. A., tenOever B. R., Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 181, 1036–1045.e9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen X., Zhao B., Qu Y., Chen Y., Xiong J., Feng Y., Men D., Huang Q., Liu Y., Yang B., Ding J., Li F., Detectable serum severe acute respiratory syndrome Coronavirus 2 viral load (RNAemia) is closely correlated with drastically elevated interleukin 6 level in critically Ill patients with Coronavirus disease 2019. Clin. Infect. Dis. 71, 1937–1942 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gao Y., Li T., Han M., Li X., Wu D., Xu Y., Zhu Y., Liu Y., Wang X., Wang L., Diagnostic utility of clinical laboratory data determinations for patients with the severe COVID-19. J. Med. Virol. 92, 791–796 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barnes B. J., Adrover J. M., Baxter-Stoltzfus A., Borczuk A., Cools-Lartigue J., Crawford J. M., Dassler-Plenker J., Guerci P., Huynh C., Knight J. S., Loda M., Looney M. R., McAllister F., Rayes R., Renaud S., Rousseau S., Salvatore S., Schwartz R. E., Spicer J. D., Yost C. C., Weber A., Zuo Y., Egeblad M., Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J. Exp. Med. 217, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang D., Hu B., Hu C., Zhu F., Liu X., Zhang J., Wang B., Xiang H., Cheng Z., Xiong Y., Zhao Y., Li Y., Wang X., Peng Z., Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA 323, 1061–1069 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hoffmann M., Kleine-Weber H., Schroeder S., Kruger N., Herrler T., Erichsen S., Schiergens T. S., Herrler G., Wu N.-H., Nitsche A., Muller M. A., Drosten C., Pöhlmann S., SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280.e8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ziegler C. G. K., Allon S. J., Nyquist S. K., Mbano I. M., Miao V. N., Tzouanas C. N., Cao Y., Yousif A. S., Bals J., Hauser B. M., Feldman J., Muus C., Wadsworth M. H. II, Kazer S. W., Hughes T. K., Doran B., Gatter G. J., Vukovic M., Taliaferro F., Mead B. E., Guo Z., Wang J. P., Gras D., Plaisant M., Ansari M., Angelidis I., Adler H., Sucre J. M. S., Taylor C. J., Lin B., Waghray A., Mitsialis V., Dwyer D. F., Buchheit K. M., Boyce J. A., Barrett N. A., Laidlaw T. M., Carroll S. L., Colonna L., Tkachev V., Peterson C. W., Yu A., Zheng H. B., Gideon H. P., Winchell C. G., Lin P. L., Bingle C. D., Snapper S. B., Kropski J. A., Theis F. J., Schiller H. B., Zaragosi L. E., Barbry P., Leslie A., Kiem H. P., Flynn J. L., Fortune S. M., Berger B., Finberg R. W., Kean L. S., Garber M., Schmidt A. G., Lingwood D., Shalek A. K., Ordovas-Montanes J.; HCA Lung Biological Network. Electronic address: Lung-network@humancellatlas.org; HCA Lung Biological Network , SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell 181, 1016–1035.e19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dueck M. E., Lin R., Zayac A., Gallagher S., Chao A. K., Jiang L., Datwani S. S., Hung P., Stieglitz E., Precision cancer monitoring using a novel, fully integrated, microfluidic array partitioning digital PCR platform. Sci. Rep. 9, 19606 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Popowitch E. B., O’Neill S. S., Miller M. B., Comparison of the biofire filmarray RP, Genmark eSensor RVP, Luminex xTAG RVPv1, and Luminex xTAG RVP fast multiplex assays for detection of respiratory viruses. J. Clin. Microbiol. 51, 1528–1533 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.He X., Lau E. H. Y., Wu P., Deng X., Wang J., Hao X., Lau Y. C., Wong J. Y., Guan Y., Tan X., Mo X., Chen Y., Liao B., Chen W., Hu F., Zhang Q., Zhong M., Wu Y., Zhao L., Zhang F., Cowling B. J., Li F., Leung G. M., Temporal dynamics in viral shedding and transmissibility of COVID-19. Nat. Med. 26, 672–675 (2020). [DOI] [PubMed] [Google Scholar]
- 45.Liu Y., Yan L. M., Wan L., Xiang T. X., Le A., Liu J. M., Peiris M., Poon L. L. M., Zhang W., Viral dynamics in mild and severe cases of COVID-19. Lancet Infect. Dis. 20, 656–657 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kucirka L. M., Lauer S. A., Laeyendecker O., Boon D., Lessler J., Variation in false-negative rate of reverse transcriptase polymerase chain reaction-based SARS-CoV-2 tests by time since exposure. Ann. Intern. Med. 173, 262–267 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miller S., Naccache S. N., Samayoa E., Messacar K., Arevalo S., Federman S., Stryke D., Pham E., Fung B., Bolosky W. J., Ingebrigtsen D., Lorizio W., Paff S. M., Leake J. A., Pesano R., DeBiasi R., Dominguez S., Chiu C. Y., Laboratory validation of a clinical metagenomic sequencing assay for pathogen detection in cerebrospinal fluid. Genome Res. 29, 831–842 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Naccache S. N., Federman S., Veeraraghavan N., Zaharia M., Lee D., Samayoa E., Bouquet J., Greninger A. L., Luk K. C., Enge B., Wadford D. A., Messenger S. L., Genrich G. L., Pellegrino K., Grard G., Leroy E., Schneider B. S., Fair J. N., Martínez M. A., Isa P., Crump J. A., DeRisi J. L., Sittler T., Hackett J. Jr., Miller S., Chiu C. Y., A cloud-compatible bioinformatics pipeline for ultrarapid pathogen identification from next-generation sequencing of clinical samples. Genome Res. 24, 1180–1192 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.R Core Team, A Language and Environmental for Statistical Computing (R Foundation for Statistical Computing, Vienna, Austria, 2018); www.R-project.org/.
- 50.H. Wickham, Use R!,. (Springer International Publishing, Springer, Cham, 2016), pp. 1 online resource. [Google Scholar]
- 51.Wingett S. W., Andrews S., FastQ Screen: A tool for multi-genome mapping and quality control. F1000Res 7, 1338 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ewels P., Magnusson M., Lundin S., Kaller M., MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dobin A., Gingeras T. R., Mapping RNA-seq Reads with STAR. Curr. Protoc. Bioinformatics 51, 11.14.1–11.14.19 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liao Y., Smyth G. K., Shi W., featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 55.Robinson M. D., McCarthy D. J., Smyth G. K., edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kramer A., Green J., Pollard J. Jr., Tugendreich S., Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 30, 523–530 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pedregosa F., Varoquaux G., Gramfort A., Michel V., Thirion B., Grisel O., Blondel M., Prettenhofer P., Weiss R., Dubourg V., Vanderplas J., Passos A., Cournapeau D., Brucher M., Perrot M., Duchesnay E., Scikit-learn: Machine learning in python. J. Mach. Learn. Res. 12, 2825–2830 (2011). [Google Scholar]
- 58.T. Hastie, R. Tibshirani, M. Wainwright, Statistical Learning with Sparsity: The Lasso and Generalizations (Monographs on statistics and applied probability, CRC Press, Taylor & Francis Group, Boca Raton, 2015), pp. xv, 351 pages. [Google Scholar]
- 59.Robin X., Turck N., Hainard A., Tiberti N., Lisacek F., Sanchez J. C., Muller M., pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics 12, 77 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/6/eabe5984/DC1