

Abstract
Objective
The aim of this study is to confirm the hepatocellular protective functions of apigenin and the molecular mechanism on liver fibrosis in mice.
Methods
Carbon tetrachloride (CCl4) and bile duct ligature (BDL) mouse fibrosis models were used to investigate the effects of apigenin on liver fibrosis. Sixty-six male C57 mice were randomly divided into eight groups, including the vehicle group, CCl4 group, CCl4+L-apigenin (20 mg/kg) group, CCl4+H-apigenin (40 mg/kg) group, sham group, BDL group, BDL+L-apigenin(20 mg/kg) group, and BDL+H-apigenin(40 mg/kg) group. Serum liver enzymes (ALT and AST), proteins associated with autophagy, and indicators linked with the TGF-β1/Smad3 and p38/PPARα pathways were detected using qRT-PCR, immunohistochemical staining, and western blotting.
Results
Our findings confirmed that apigenin could decrease the levels of ALT and AST, suppress the generation of ECM, inhibit the activation of HSCs, regulate the balance of MMP2 and TIMP1, reduce the expression of autophagy-linked protein, and restrain the TGF-β1/Smad3 and p38/PPARα pathways.
Conclusion
Apigenin could alleviate liver fibrosis by inhibiting hepatic stellate cell activation and autophagy via TGF-β1/Smad3 and p38/PPARα pathways.
1. Introduction
Liver fibrosis is a chronic pathological change caused by a variety of reasons, such as chronic infection by hepatotropic viruses, excess alcohol consumption, nonalcoholic fatty liver disease, autoimmune liver diseases, and hereditary disease, and is a necessary stage for the development of many liver diseases to liver cirrhosis and even liver cancer [1, 2]. Liver fibrosis is a wound healing response characterized by excessive deposition of extracellular matrix (ECM). The possible treatments for liver fibrosis including curing the primary disease, reducing inflammation and immune response, inhibiting stellate cell activation, and increasing the degradation of scar matrix had been generally accepted [3, 4]. Although liver transplantation is the most efficient therapy, there are great limitations because of huge cost of treatment and shortage of liver donor available for transplantation [1, 5, 6]. Therefore, revealing the molecular mechanism of liver fibrosis and finding key drug targets are an important issue that needs to be solved urgently.
The progression of fibrosis is a complex process which involves nonparenchymal hepatocytes, parenchymal hepatocytes, and infiltrating immune cells. The activation of inflammation mediators and profibrotic genes caused by cell death in both nonparenchymal and infiltrating immune cells thereby trigger the fibrosis process. Hepatic stellate cells (HSCs) are the most powerful fibrogenic effector cells and are also considered as the initial process during liver fibrosis [4, 7–10]. The activation of HSCs by several cellular events including immune/inflammatory injury as well as molecular regulation especially transforming growth factor-β1 (TGF-β1) will contribute to the excessive accumulation of ECM which promotes liver fibrosis [11]. It had been reported that suppressing the activation HSCs and expression of TGF-β1 could reduce the levels of myofibroblast markers, increase the ratio of MMPs/TIMPs, and decrease Smad2/Smad3 associated collagen production which further attenuated liver fibrosis [11–15].
Autophagy is a self-selective mode of cell death, which can remove necrotic cells to maintain organ homeostasis [12]. Results have shown that autophagy could provide energy for the activation of HSCs by stimulating the metabolism of lipid droplets [16]. At the same time, many literatures have confirmed that inhibiting the autophagy of HSCs can play a positive role in liver protection [17–20]. So, inhibition of autophagy which could significantly reduce activation of HSCs can attenuate liver fibrosis [21, 22].
Apigenin is a kind of dietary flavonoid extracted mainly from celery, parsley, thyme, chamomile, and onions [23]. Recently, apigenin has reported many pharmacological effects including anticancer [24–28], anti-inflammation [29–32], antifibrosis [33–36], and so on. Zhang et al. confirmed apigenin could downregulate the miR34a expression to suppress mouse peritoneal fibrosis [35]. Jiao et al. demonstrated that apigenin could inhibit fibroblast proliferation and reduce epidural fibrosis by suppressing the Wnt3a/β-catenin signaling pathway [34]. However, whether apigenin has the antihepatic fibrosis effect and the specific molecular mechanism of this effect are still unclear and need to be explored.
Carbon tetrachloride (CCl4) and bile duct ligature (BDL) mouse models are extremely practical models to investigate the underlying molecular mechanisms of liver fibrosis, which have been widely applied to the establishment of liver fibrosis [15, 37]. Therefore, this study is aimed at exploring the antihepatic fibrosis effect and the specific molecular mechanism of apigenin using the CCl4 and BDL models. We hypothesized that apigenin could alleviate liver fibrosis by inhibiting hepatic stellate cell activation and autophagy via TGF-β1/Smad3 and p38 MAPK/PPARα pathways.
2. Materials and Methods
2.1. Drugs and Reagents
Apigenin (HPLC ≥ 98% CAS:520-36-5) was purchased from Shanghai Yuanye Bio-Technology Co., Ltd. (Shanghai, China). When used, it is dissolved into dimethyl sulfoxide (DMSO) at 2 mg/ml and 4 mg/ml concentrations. Carbon tetrachloride (CCl4) was purchased from China Sinopharm International Corporation (Shanghai, China). Alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were tested by microplate test kits purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). Quantitative real-time (qRT) PCR kits were purchased from TaKaRa (Dalian, China). The primers were obtained from Generay (Shanghai, China). Detailed information of the primary antibodies used in this study are listed in Table 1. Dulbecco's Modified Eagle Medium (DMEM) and foetal bovine serum (FBS) were purchased from HyClone (GE Healthcare). Apigenin was dissolved in DMSO (<0.1% [v/v]) for in vitro treatment.
Table 1.
The primary antibodies used for western blotting and immunohistochemistry in the study.
| Antibody | Species | Targeted species | Supplier | Catalogue number |
|---|---|---|---|---|
| β-Actin | M | H, M, R | CST | 3700 |
| IL-1β | Rbt | M | CST | 12507 |
| α-SMA | M | H, M, R | Abcam | ab7817 |
| Collagen 1 | Rbt | H, M, R | Abcam | ab34710 |
| MMP2 | Rbt | H, M, R | PT | 10373-2-AP |
| TIMP1 | Rbt | H, M, R | PT | 10753-1-AP |
| p62 | Rbt | H, M, R | PT | 55274-1-AP |
| LC3 | Rbt | H, M, R | PT | 14600-1-AP |
| Beclin-1 | Rbt | H, M, R | PT | 11306-1-AP |
| TGF-β1 | Rbt | H, M, R | PT | 21898-1-AP |
| Smad3 | Rbt | H, M, R | Abcam | ab40854 |
| p-Smad3 | Rbt | H, M | Abcam | ab52903 |
| p38 MAPK | Rbt | H, M, R | Zenbio | 200782 |
| p-p38 MAPK | Rbt | H, M, R | CST | 4511 |
| PPARα | Rbt | H, M, R | PT | 15540-1-AP |
Abbreviations: H: human; M: mouse; Rbt: rabbit; R: rat; CST: Cell Signaling Technology (Danvers, MA, USA); PT: Proteintech (Chicago, IL, USA).
2.2. Cell Culture and CCK8 Assay
The human immortal LX2 cell line was cultured in high glucose DMEM with 10% FBS, 100 U/mL of penicillin, and 100 g/mL of streptomycin. The apparent logarithmic phase cells were seeded in 96-well plates for 48 hours, then apigenin was added at concentrations of 10, 20, 30, 40, 50, 60, 70, or 80 μM for 24 hours, and the cytotoxicity analysis was performed. Cell viability was then measured with the CCK8 assay according to the manufacturer's protocol. All the experiments were performed in triplicate.
2.3. BrdU Assay
Proliferation of the cells was evaluated using the BrdU Cell Proliferation ELISA Kit (ab126556, Abcam, Cambridge, MA) according to the manufacturer's instruction. Briefly, cells were cultured in 96-well plates and exposed to apigenin (20, 40, and 60 μM) for 24 hours. Subsequently, 10 μM BrdU was added to each well, and samples were incubated for 12 h at 37°C. BrdU signaling was determined by measuring the absorbance at 450 nm.
2.4. Animals
66 six-week-old male C57 mice (22-26 g) were obtained from Shanghai SLAC Laboratory Animal (Shanghai, China) and housed in a standard animal laboratory with free access to food and water. All experimental procedures involving mice were approved by the Animal Care and Use Committee of Shanghai Tongji University. Handling and care of mice conformed to the National Institutes of Health Guidelines.
2.5. Establishment of Mouse Liver Fibrosis Models
We established two different mouse liver fibrosis models. To create the CCl4-induced liver fibrosis model, mice were injected with 10% CCl4 (1.0 mL/kg, diluted in peanut oil) intraperitoneally three times a week for 8 weeks. In the bile duct ligation- (BDL-) induced liver fibrosis model, all mice were fasted for 12 h and anesthetized intraperitoneally by 1.25% pentobarbital sodium salt (40 mg/kg). After opening the abdomen via the linea alba, the bile duct was exposed and isolated over a certain length. Two surgical knots were tied in the isolated bile duct, which was then cut between the knots. The abdomen was then closed.
2.6. Experimental Design
2.6.1. Preliminary Study
In order to verify whether the apigenin dose (20 mg/kg and 40 mg/kg) could cause damage to the structure and function of the liver and other internal organs, we designed a preliminary experiment. The eighteen mice were randomly divided into the following 3 groups.
Normal control (NC) (n = 6): no treatment
Vehicle group (n = 6): mice were injected intraperitoneally with DMSO three times a week
API (40 mg/kg) group (n = 6): apigenin (40 mg/kg) was given to mice by intragastric administration three times a week.
2.6.2. Formal Experiment
In the CCl4-induced liver fibrosis model, 24 mice were randomly divided into the following 4 groups.
Vehicle group (n = 6): mice were injected intraperitoneally with DMSO three times a week for 8 weeks
CCl4 group (n = 6): mice were injected with CCl4 intraperitoneally three times a week for 8 weeks
CCl4+L-API group (n = 6): mice were injected with CCl4 intraperitoneally and gavaged with 20 mg/kg apigenin three times a week for 8 weeks
CCl4+H-API group (n = 6): mice were injected with CCl4 intraperitoneally and gavaged with 40 mg/kg apigenin three times a week for 8 weeks
In the BDL-induced liver fibrosis model, 24 mice were randomly divided into the following 4 groups.
Sham group (n = 6): all mice underwent laparotomy without BDL
BDL group (n = 6): all mice underwent BDL surgery
BDL+L-API group (n = 6): all mice were gavaged with 20 mg/kg apigenin once a day for 14 days after BDL
BDL+H-API group (n = 6): all mice were gavaged with 40 mg/kg apigenin once a day for 14 days after BDL
Vehicle and sham groups were used as controls in both models. At the end of the experiment, blood samples and liver tissues were collected with diethyl ether anesthesia. Serum was acquired by centrifugation (4,500 rpm, 4°C, 10 min) and kept at −80°C. Liver tissues were stored at −80°C.
2.7. Serum Biochemical Analysis
The blood sample collected from the mouse orbit was placed at 4°C for 5 hours. And then, the serum sample was separated from the blood by centrifuging at 4,600 × g at 4°C for 10 minutes. Serum levels of ALT and AST were detected by microplate test kits.
2.8. Histopathology
A part of the fresh left liver lobe was excised and then fixed in 4% paraformaldehyde for 24 h. The tissues were dehydrated with ethanol and embedded in paraffin. Next, the liver tissues were cut into 3 μm thick sections and stained with hematoxylin and eosin (H&E) to determine the severity of injury.
2.9. Reverse Transcription PCR (RT-PCR) and Quantitative Real-Time PCR (qRT-PCR)
The total RNA was extracted from 100 mg liver tissue by TRIzol (Thermo Fisher Scientific, Waltham, MA, USA). Then, the purified RNA was reverse-transcribed into cDNA. The levels of mRNA were determined by SYBR Premix EX Taq through a 7900HT fast PCR system (Applied Biosystems, Foster City, CA, USA). The primers used for qRT-PCR are listed in Table 2.
Table 2.
Oligonucleotide sequences of primers used for qRT-PCR.
| Gene name | Forward (5′-3′) | Reverse (5′-3′) |
|---|---|---|
| β-Actin | GTGACGTTGACATCCGTAAAGA | GCCGGACTCATCGTACTCC |
| IL-1β | GAAATGCCACCTTTTGACAGTG | TGGATGCTCTCATCAGGACAG |
| Collagen 1 | CAATGGCACGGCTGTGTGCG | AGCACTCGCCCTCCCGTCTT |
| α-SMA | CCCAGACATCAGGGAGTAATGG | TCTATCGGATACTTCAGCGTCA |
| MMP2 | GGACAAGTGGTCCGCGTAAA | CCGACCGTTGAACAGGAAGG |
| TIMP1 | CGAGACCACCTTATACCAGCG | ATGACTGGGGTGTAGGCGTA |
| p62 | GAGGCACCCCGAAACATGG | ACTTATAGCGAGTTCCCACCA |
| LC3 | TTATAGAGCGATACAAGGGGGAG | CGCCGTCTGATTATCTTGATGAG |
| Beclin-1 | ATGGAGGGGTCTAAGGCGTC | TGGGCTGTGGTAAGTAATGGA |
| TGF-β1 | CCACCTGCAAGACCATCGAC | CTGGCGAGCCTTAGTTTGGAC |
| PPARα | AACATCGAGTGTCGAATATGTGG | CCGAATAGTTCGCCGAAAGAA |
Abbreviation: qRT-PCR: quantitative real-time PCR.
2.10. Immunohistochemistry (IHC)
Paraffin sections were baked in a 60°C oven for 1 hour and then dewaxed and rehydrated. Antigen was placed into a citrate buffer, which was then heated to 95°C for 10 minutes and cooled to room temperature. Next, the sections were covered in 3% hydrogen peroxide for 20 minutes to block endogenous peroxidase activity, and then 5% BSA was added to block nonspecific binding for 15 minutes (both at room temperature). Slices were then incubated overnight at 4°C with the following antibodies: anti-IL-1β, anti-α-SMA, anti-Col, anti-LC3, anti-Beclin-1, anti-p62-, anti-TGF-β1-, anti-p-Smad3-, anti-p-38-, and anti-PPARα (all 1 : 200). Then, the primary antibodies in the liver sections were incubated with secondary antibodies using a diaminobenzidine (DAB) kit. Final evaluations were performed with Image-Pro Plus software 6.0 to calculate the mean of integrated optical densities (MIOD = sum IOD/sum area) of the positive staining area.
2.11. Western Blotting
Firstly, liver tissues were ground (100 mg) into powder in liquid nitrogen, and then the powder was homogenized in RIPA lysis containing phenylmethanesulfonyl fluoride (PMSF) and protease inhibitors (PI). The protein concentrations were detected using the bicinchoninic acid method before being mixed with a 6x loading buffer and boiled at 100°C for 10 minutes. Secondly, protein samples were electrophoresed by 10% or 12.5% SDS-PAGE and transferred onto polyvinylidene fluoride or nitrocellulose membranes. Next, membranes were blocked with 5% skimmed milk for at least 1 hour and subsequently incubated overnight at 4°C with the primary antibodies (Table 1). Thirdly, the membranes were incubated with anti-rabbit or anti-mouse secondary antibodies after washing thrice with PBST (1% Tween diluted in PBS). Finally, the expression of protein was measured by an Odyssey two-color infrared laser imaging system (LI-COR Biosciences, Lincoln, NE, USA).
2.12. Statistical Analysis
Experimental data which was repeated at least three times was presented as mean ± SD (n = 6; ∗P < 0.05 for CCl4 (BDL) vs. control; #P < 0.05 for CCl4 (BDL)+API (20 mg/kg) vs. CCl4 (BDL); +P < 0.05 for CCl4 (BDL)+API (40 mg/kg) vs. CCl4 (BDL); !P < 0.05 for CCl4 (BDL)+API (40 mg/kg) vs. CCl4 (BDL)+API(20 mg/kg)). One-way ANOVA using the Student–Newman–Keuls method was used to compare statistical differences among three or four groups using SPSS version 20.0 software (IBM, Armonk, NY, USA). P < 0.05 was regarded as statistically significant.
3. Result
3.1. Effects of Apigenin on Liver and LX2 Cells
The human immortal HSC cell line (LX2 cells) was used in this study to investigate the effect of apigenin on HSCs. The CCK8 assay was used to measure the toxicity of apigenin in LX2 cells (Figure 1(a)). Apigenin decreased the viability of LX2 cells in a dose-dependent manner, and the half-maximal inhibitory concentration (IC50) was 28.80 μM. At the same time, the BrdU incorporation assay was performed to explore the effect of apigenin on cell proliferation. As shown in Figure 1(b), apigenin could reduce the proportion of proliferating cells in a dose-dependent manner. Besides, in the preliminary experiment, 12 mice were injected with vehicle (DMSO) or gavaged with 40 mg/kg apigenin to explore security of drug and solvent used in this study. As shown in Figure 1(c), there was no hepatocellular injury or structural damage compared with the NC group. The results of ALT, AST, and western blotting shown in Figures 1(b) and 1(d) could also verify no statistically significant differences between the vehicle, apigenin, and NC groups. So, we got the conclusion that the apigenin could inhibit proliferation and decrease the viability of LX2 cells, but has no harmful effects on the liver tissues.
Figure 1.
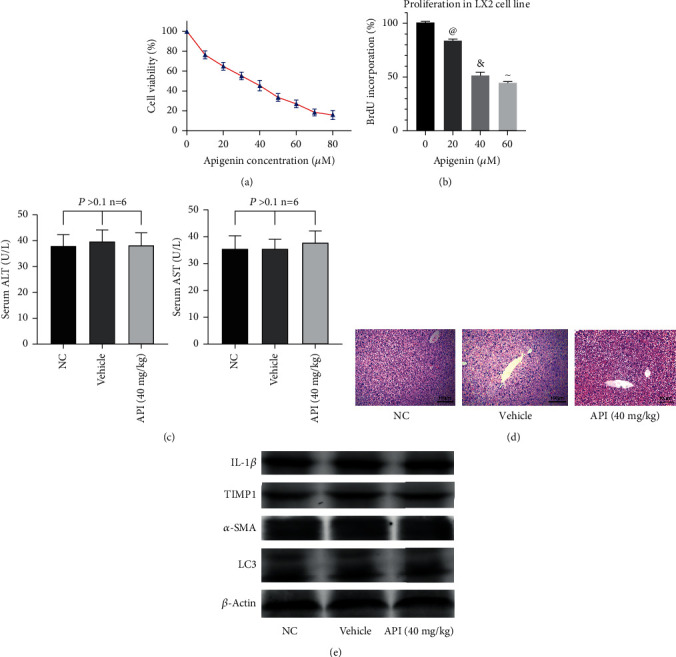
Effects of apigenin on liver and LX2 cells. Notes: (a) the CCK8 assay was used to determine the effects of apigenin on the viability of LX2 cells. (b) Cells were treated with the indicated concentrations of apigenin for 24 hours, and the degree of apigenin to inhibit cell proliferation was measured using BrdU Cell Proliferation ELISA Kit (@P < 0.05 for 20 μM apigenin vs. 0 μM apigenin; &P < 0.05 for 40 μM apigenin vs. 20 μM apigenin; ~P < 0.05 for 60 μM apigenin vs. 40 μM apigenin). (c) The levels of serum ALT and AST are presented as mean ± SD. One-way ANOVA indicated that there was no significant difference among the three groups (n = 6; P > 0.1). (d) Representative H&E-stained hepatic sections were examined under light microscopy and imaged at a 200x magnification. (e) Western blot analysis of IL-1β, TIMP1, α-SMA, and LC3 protein levels.
3.2. Apigenin Protects the Liver against Fibrosis Induced by CCl4 and BDL in Mice
The levels of serum ALT and AST are important indicators of liver parenchymal damage. So, we detected the levels of ALT and AST in the serum to explore the extent of liver parenchymal damage in both fibrosis models. We could see it clearly from the Figures 2(a) and 2(b) that ALT and AST elevated dramatically in model groups compared with vehicle and sham groups. However, we also noticed apigenin groups could reverse the increase induced by CCl4 and BDL surgery in a dose-dependent manner. Next, HE and Masson staining were used to evaluate the pathological changes of liver tissues. HE staining showed that the morphology and structure of mouse liver cells in the control groups were normal, with normal arrangement, normal hepatic lobules and portal area, and no inflammatory cell exudation. Compared with the control groups, the disordered arrangement of liver cells, the damaged normal structure, the exudation of many inflammatory cells, and the proliferation of collagen fibers were significantly observed in the CCl4 and BDL groups. When apigenin was given at the same time, the disordered arrangement of liver cells was significantly reduced, the structure of portal area was almost normalized, fibrous tissue hyperplasia and inflammatory cell infiltration were significantly decreased, and the morphological structure was close to normal liver tissue (Figures 2(c) and 2(d)). The results of Masson staining could further confirm the protective effect on liver fibrosis of apigenin. The above results showed that apigenin had an obviously protective effect on CCl4- and BDL-induced liver fibrosis in mice.
Figure 2.
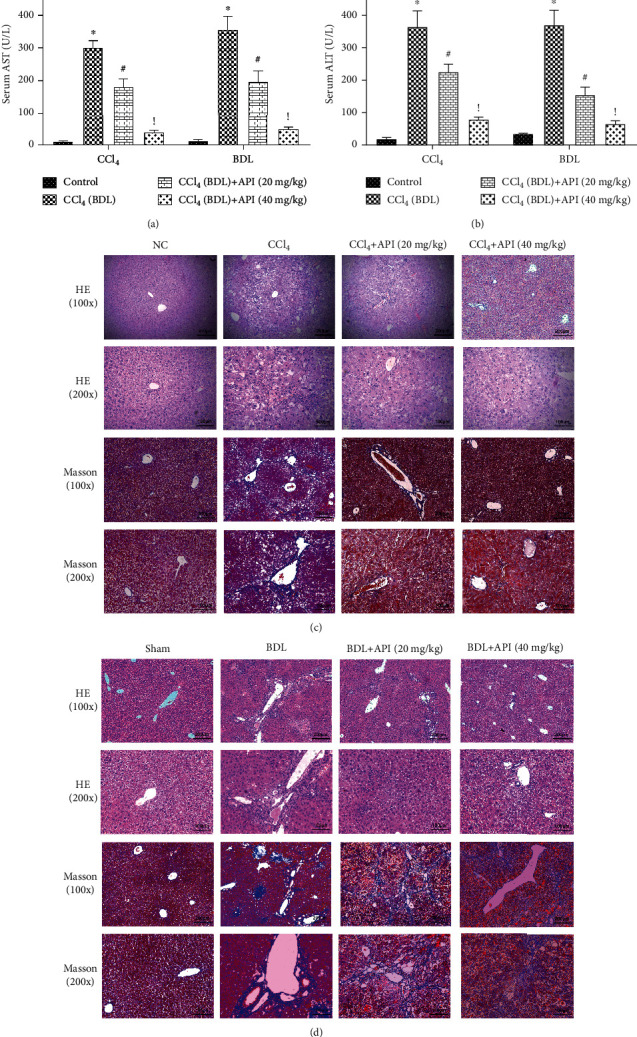
Apigenin protects the liver against fibrosis induced by CCl4 and BDL in mice. Notes: (a, b) the levels of serum ALT and AST are presented as mean ± SD (n = 6; ∗P < 0.05 for CCl4 (BDL) group vs. control group; #P < 0.05 for CCl4 (BDL)+apigenin (20 mg/kg) group vs. CCl4 (BDL) group; !P < 0.05 for CCl4 (BDL)+apigenin (40 mg/kg) group vs. CCl4 (BDL)+apigenin (20 mg/kg) group). (c, d) Representative H&E- and Masson-stained hepatic sections were examined under light microscopy and imaged at 200x and 100x magnifications.
3.3. Apigenin Restrained the Activation of HSC and Regulated the Balance of TIMP1 and MMP2
α-SMA was an important indicator of HSC activation, and collagen 1 was the main component of ECM, which were often used as important indicators to test the degree of liver fibrosis. In order to further prove the effect of apigenin on mouse liver fibrosis, mRNA and protein expression of collagen 1, α-SMA, and IL-1β in the mouse liver tissues were measured by real-time PCR, western blotting, and IHC. The results showed that compared with the vehicle or sham control groups, mRNA and protein expression of collagen 1, α-SMA, and IL-1β in the model group were significantly increased, but their expressions were decreased after apigenin treatment (Figures 3(a)–3(c)). The synthesis and degradation of hepatic ECM are regulated by matrix metalloproteinases (MMPs) and matrix metalloproteinase inhibitors (TIMPs). Injury factors can lead to the activation of HSC, resulting in the imbalance of MMPS/TIMPs; therefore, we measured the levels of TIMP1 and MMP2 in liver tissues. The results indicated that the expression of TIMP1 increased obviously in the CCl4 and BDL groups, and this trend could be inhibited by apigenin treatment. On the contrary, MMP2 decreased in the fibrosis model groups but increased in the apigenin groups. In general, the above experimental results showed that apigenin could restrain the activation of HSC and regulated the balance of TIMP1 and MMP2 to relieve liver fibrosis in mice.
Figure 3.
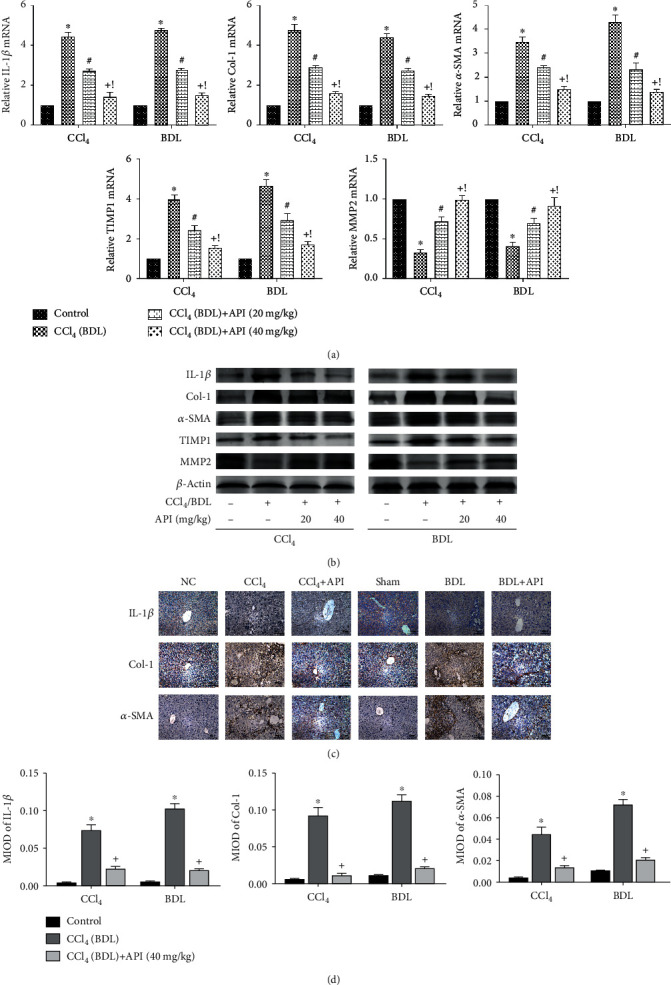
Apigenin restrained the activation of HSC and regulated the balance of TIMP1 and MMP2. Notes: (a) relative IL-1β, Col-1, α-SMA, TIMP1, and MMP2 mRNA levels were determined by qRT-PCR. (b) Western blot analysis of IL-1β, Col-1, α-SMA, TIMP1, and MMP2 protein levels. (c) IL-1β, Col-1, and α-SMA protein expressions in liver tissues are shown by immunohistochemical staining (200x). (d) Final evaluations were made using Image-Pro Plus 6.0 software to calculate the MIOD of the positive staining area. Data are presented as mean ± SD (n = 6; ∗P < 0.05 for CCl4 (BDL) group vs. control group; #P < 0.05 for CCl4 (BDL)+apigenin (20 mg/kg) group vs. CCl4 (BDL) group; +P < 0.05 for CCl4 (BDL)+apigenin (40 mg/kg) group vs. CCl4 (BDL) group; !P < 0.05 for CCl4 (BDL)+apigenin (40 mg/kg) group vs. CCl4 (BDL)+apigenin (20 mg/kg) group). Abbreviation: MIOD: mean of integrate optical density.
3.4. Apigenin Alleviated Autophagy during Liver Fibrosis
Beclin-1, LC3, and p62, which are autophagy signature proteins, were analyzed by qRT-PCR, IHC, and western blotting to explore the protective effect of apigenin. As demonstrated in Figures 4(a)–4(d) and S1, the expressions of Beclin-1 and LC3II/LC3I augmented obviously, while p62 decreased drastically, in the CCl4 and BDL groups. However, apigenin groups could ameliorate these changes in a dose-dependent manner. The above results suggested that apigenin could alleviate autophagy during liver fibrosis.
Figure 4.
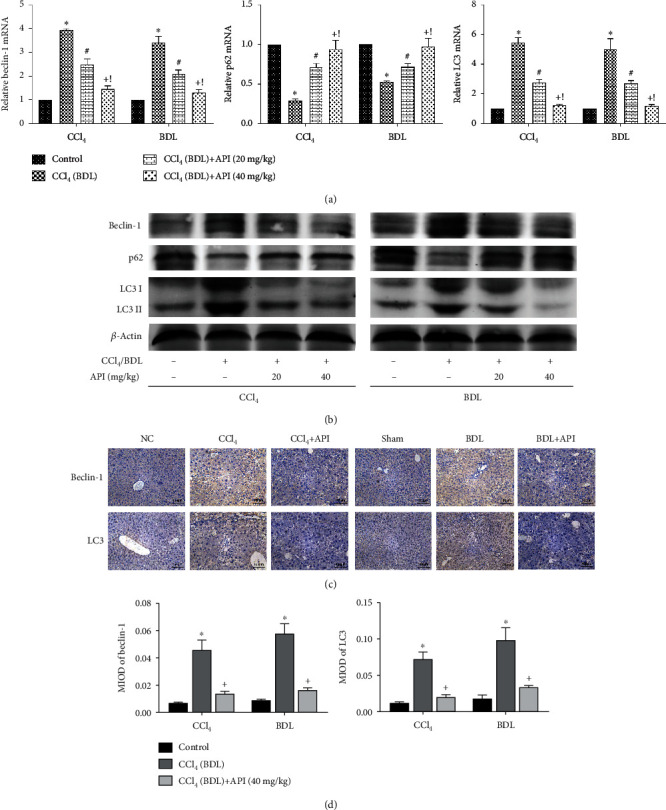
Apigenin alleviated autophagy during liver fibrosis. Notes: (a) relative Beclin-1, p62, and LC3 mRNA levels were determined by qRT-PCR. (b) Western blot analysis of Beclin-1, p62, and LC3. (c) Beclin-1 and LC3 protein expressions in liver tissues are shown by immunohistochemical staining (200x). (d) Final evaluations were made using Image-Pro Plus 6.0 software to calculate the MIOD of the positive staining area. Data are presented as mean ± SD (n = 6; ∗P < 0.05 for CCl4 (BDL) group vs. control group; #P < 0.05 for CCl4 (BDL)+apigenin (20 mg/kg) group vs. CCl4 (BDL) group; +P < 0.05 for CCl4 (BDL)+apigenin (40 mg/kg) group vs. CCl4 (BDL) group; !P < 0.05 for CCl4 (BDL)+apigenin (40 mg/kg) group vs. CCl4 (BDL)+apigenin (20 mg/kg) group). Abbreviation: MIOD: mean of integrate optical density.
3.5. Apigenin Could Relieve Hepatic Fibrosis Induced by CCl4 and BDL via Downregulating TGF-β1/Smad3 and p38/PPARα Pathways
TGF-β1 is a pluripotent cytokine that is involved in inflammatory infiltration, cell growth, apoptosis, differentiation, and other processes in fibrosis. The Smad protein family is the downstream molecule of TGF-β1. Therefore, we evaluated the expressions of the TGF-β1/Smad3 pathway. The results of qRT-PCR, IHC, and western blotting in Figures 5(a)–5(d) illustrated that CCl4 and BDL surgery could significantly active the TGF-β1/Smad3 pathway, but apigenin treatment could reverse this activation. It means that the protective effects of apigenin were associated with restraining the TGF-β1/Smad3 pathway. Next, we measured the levels of p38 and PPARα which was also a downstream molecule of TGF-β1. In our results, we found that liver fibrosis induced by CCl4 and BDL surgery could lead to phosphorylation of p38, which further inhibited PPARα. In apigenin treatment groups, p-p38 was dramatically downregulated and PPARα increased obviously. Therefore, we can draw the conclusion that apigenin could relieve hepatic fibrosis induced by CCl4 and BDL via downregulating the TGF-β1/Smad3 and p38/PPARα pathways.
Figure 5.
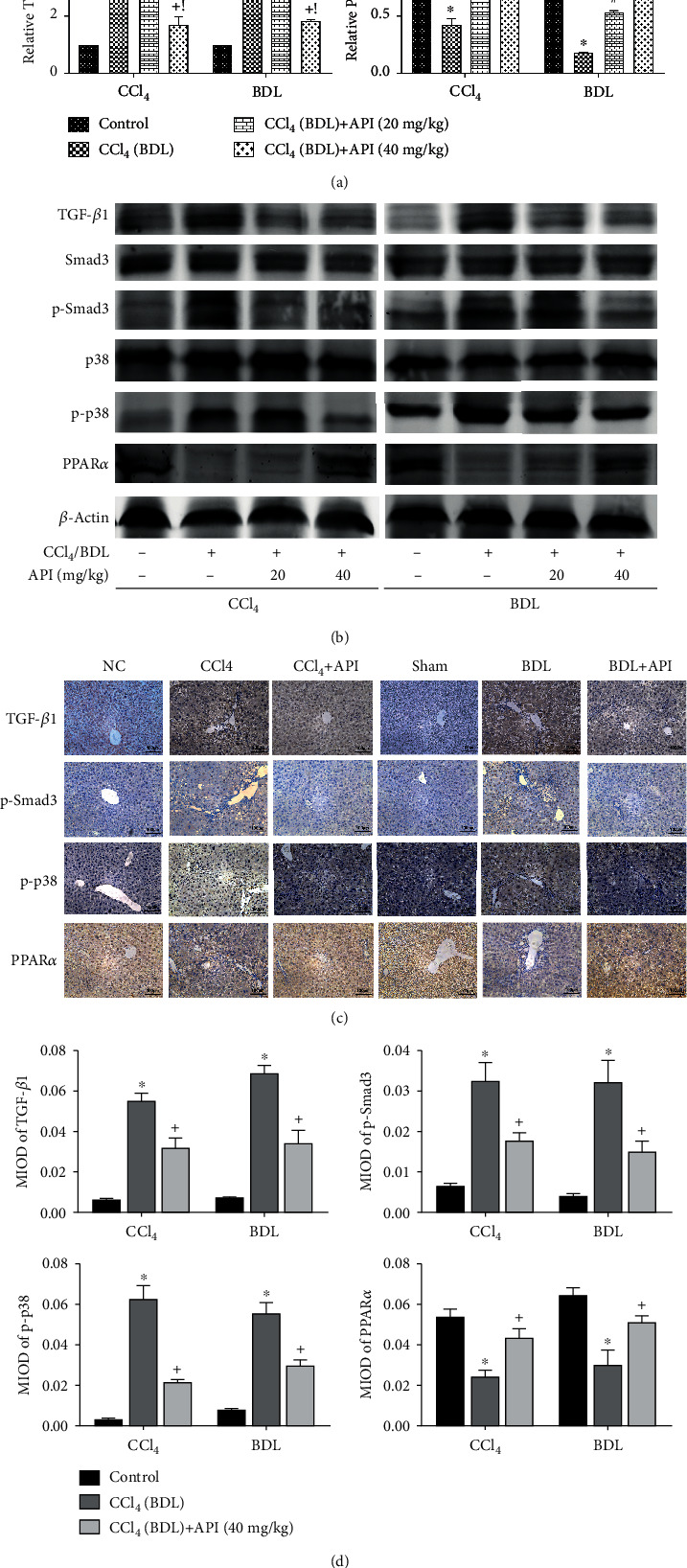
Apigenin could relieve hepatic fibrosis induced by CCl4 and BDL via downregulating TGF-β1/Smad3 and p38/PPARα pathways. Notes: (a) relative TGF-β1 and PPARα mRNA levels were determined by qRT-PCR. (b) Western blot analysis of TGF-β1, Smad3, p-Smad3, p38, p-p38, and PPARα. (c) TGF-β1, p-Smad3, p-p38, and PPARα protein expressions in liver tissues are shown by immunohistochemical staining (200x). (d) Final evaluations were made using Image-Pro Plus 6.0 software to calculate the MIOD of the positive staining area. Data are presented as mean ± SD (n = 6; ∗P < 0.05 for CCl4 (BDL) group vs. control group; #P < 0.05 for CCl4 (BDL)+apigenin (20 mg/kg) group vs. CCl4 (BDL) group; +P < 0.05 for CCl4 (BDL)+apigenin (40 mg/kg) group vs. CCl4 (BDL) group; !P < 0.05 for CCl4 (BDL)+apigenin (40 mg/kg) group vs. CCl4 (BDL)+apigenin (20 mg/kg) group). Abbreviation: MIOD: mean of integrate optical density.
4. Discussion
Liver fibrosis is a chronic wounding-healing response with a long-time liver injury [4, 38]. Although there are little symptoms at the beginning of liver fibrosis, the risk of mortality increases significantly once liver fibrosis progresses to cirrhosis and even hepatocellular carcinoma [2]. More than 30,000 deaths per year caused by cirrhosis and 1,000 deaths per year occurred related to liver cancer in the United States are enough to warn us that halting and reversing the progression of fibrosis is currently an effective way to reduce mortality rather than only relying on highly limited liver transplants [39].
Apigenin is a kind of dietary flavonoid extracted mainly from celery, parsley, thyme, chamomile, and onions [23]. It had been reported that apigenin is of great effect in antifibrosis [33–36] and liver protection [26, 40–42]. Mirzoeva et al. demonstrated that apigenin could reduce TGF-β-induced VEGF production and suppress prostate carcinogenesis by regulating the Smad2/3 and Src/Fak/Akt pathways. Apigenin is also reported to inhibit metastasis and angiogenesis by the p38 MAPK pathway [43].These indicate that apigenin may become an efficient drug to prevent liver fibrosis, and the molecular mechanism might be closely related to the TGF-β and p38 MAPK pathway. Therefore, in our study, CCl4- and BDL-induced liver fibrosis models are used to explore the effects of apigenin and the specific molecular mechanism. Our results of HE and Masson staining confirmed that apigenin could improve liver fibrosis in a dose-dependent manner.
The first step to try to stop and reverse liver fibrosis is to explore the molecular mechanisms of this disease. The formation of liver fibrosis is a complex pathophysiological process involving many cells, molecules, and signaling pathways. The accumulation of ECM is regarded as the important character, and the activation of HSCs is considered as the initial process of liver fibrosis [2, 7, 10]. HSCs are one of the mesenchymal cells which account for one-third of the nonparenchymal cells in the liver and 15% of the total number of liver cells [11]. In normal conditions, HSC is at a quiescent condition and could store vitamin A and triglycerides in the cytoplasm [44]. However, when the liver suffers from acute or chronic injury, HSCs are activated and differentiated into myofibroblasts, which have a strong ability of proliferation, migration, and secretion. Activated HSCs are the main cells to produce ECM, and a large amount of ECM is continuously deposited in the Disse space. In addition, the main components of ECM also change from type IV collagen to type I collagen [45], resulting in the increase of density and hardness of ECM, and accumulated ECM also becomes the liver fibrosis tissue microenvironment containing α-SMA, TGF-β1, chemokines such as PDGF, hepatocyte growth factor (HGF), fibroblast growth factor (FGF), epidermal growth factor (EGF), and VEGF [46]. The synthesis and degradation of liver ECM is regulated by the combination of matrix metalloproteinases (MMPs) and tissue matrix metalloproteinase inhibitors (TIMPs). Under normal conditions, MMPs and TIMPs can be synthesized by hepatocytes and various mesenchymal cells and play a key role in maintaining the dynamic balance between ECM synthesis and degradation in normal liver tissues through complex regulatory mechanisms [12, 13, 47]. In our study, we explored the function of apigenin in the activation of HSCs and the levels of ECM. Our results illustrated that apigenin could suppress the activation of HSCs and decrease ECM by increasing the ratio of MMP2/TIMP1.
TGF-β is generally considered to be the strongest fibrogenic factor. The activation of the TGF-β1/Smad3 signaling pathway plays an important role in liver fibrosis [48]. Smad3 is phosphorylated into p-Smad3 which could promote the transcription of type 1 and type 3 collagen after the activation of TGF-β1 [49]. In addition, TGF-β can also activate the p38 MAPK signaling pathway to promote the transcription of collagen which is the main ingredient of ECM [50]. Besides, large amounts of literature have confirmed that inhibiting the TGF-β1/Smad pathway could efficiently reduce the injury of liver fibrosis [4, 15, 47, 51–53]. In our study, we proved that TGF-β1 and Smad3 expressed much more in fibrosis model groups than in control groups, and at the same time, apigenin groups obviously reduced the expression of TGF-β1, Smad3, and the other related proteins. Thus, we concluded that the protective effect of apigenin was closely related to the inhibition of the TGF-β1/Smad3 pathway.
p38 MAPK belongs to the family of MAPKs that affects a variety of intracellular responses including cell-cycle regulation, inflammation, cell death, and tumorigenesis [54]. p38 MAPK could be phosphorylated by many extracellular stimulants through a classic MAPK pathway, and phosphorylated p38 (p-p38) could further regulate many substrates that include transcription factors, peroxisome proliferator-activated receptors (PPARs), and so on [54, 55]. The study of Liu et al. demonstrated that p38 MAPK activated by TGF-β1 could exert a positive effect on liver fibrosis [12]. In addition, Lu et al. illustrated that the inhibition of p-p38 MAPK could increase the expression of PPARα to protect liver from concanavalin A-induced injury [55]. PPARs which belong to the subfamily of the nuclear receptor superfamily containing PPARα, PPAR β/δ, and PPARγ have many biological functions such as liver protection, antitumor, antiasthma, antidiabetes, and antineuropathic pain [56–62]. It was reported that PPARα could reverse fibrosis by reducing lipid peroxides and inhibiting the activation of HSCs and Kupffer cells (KCs) [63, 64]. So, in our study, we detected the expressions of p-p38 and PPARα and proved that apigenin could inhibit the phosphorylation of p38 which further increased PPARα to protect the liver from fibrosis.
Autophagy is a self-selective mode of cell death which contributes a lot to the basic liver functions [65]. Hernandez-Gea and Friedman demonstrated that autophagy could provide energy for the activation of HSCs by stimulating metabolism of lipid droplets [16]. However, inappropriate autophagy activity may aggravate damage in hepatic injury such as liver fibrosis [66]. The conclusion of Li et al. proved that suppressing autophagy could alleviate liver fibrosis [52]. Autophagy is closely related to the TGF-β1/Smad3 pathway, which could increase the expression of Bechin1 and LC3 and decrease the generation of p62 [67]. In addition, the inhibition of autophagy via the p38/PPARα pathway could exert positive effects in liver injury [55]. Our current results confirmed that apigenin could ameliorate liver fibrosis by inhibiting autophagy via the TGF-β1/Smad3 and p38/PPARα pathways.
In general, our study illustrated the liver-protective effect of apigenin in CCl4- and BDL-induced liver fibrosis models. Apigenin could inhibit the activation of HSCs which promote the accumulation of ECM and the secretion of many fibrogenic factors such as α-SMA and collagen 1. In addition, the TGF-β1/Smad3 and p38/PPARα pathways are proven to be the main signaling pathways through which apigenin exerts its function (Figure 6). Therefore, apigenin may be a new clinical option for the treatment of fibrosis; however, more drug safety and clinical trials need to be accomplished before clinical applications.
Figure 6.
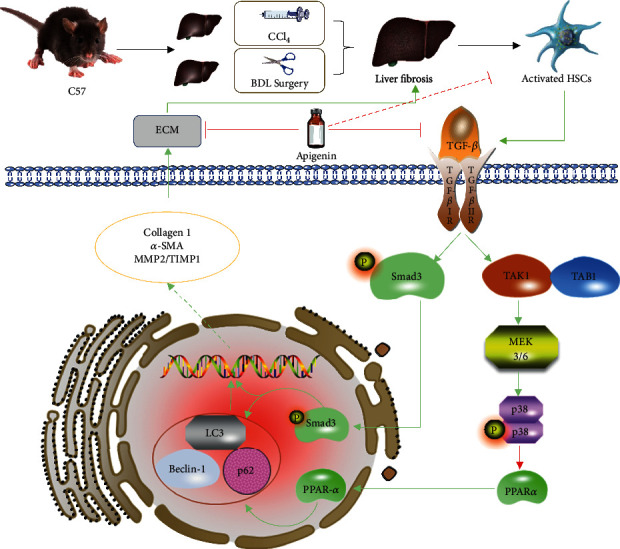
Probable mechanisms of apigenin against liver fibrosis. Notes: apigenin inhibits the production of TGF-β1 in HSCs. The decrease of TGF-β1 results in the reduced activation of HSCs and downregulation of the TGF-β1/Smad3 and p38/PPARα signaling pathway, including decreased ECM production and inhibition of autophagy. Abbreviations: BDL: bile duct ligation; CCl4: carbon tetrachloride; ECM: extracellular matrix; HSCs: hepatic stellate cells.
5. Conclusion
Our study illustrated the liver-protective effect of apigenin in CCl4- and BDL-induced liver fibrosis models. Inhibiting the TGF-β1/Smad3 and p38/PPARα pathways, reducing autophagy, and decreasing ECM formation are the major mechanism of the antifibrotic effects of apigenin.
Acknowledgments
This research was funded by the National Natural Science Foundation of China (grant number: 81670472), the Yangfan Plan of the Shanghai Science and Technology Commission (grant number: 20YF1443300), the National Natural Science Foundation of Shanghai (grant number: 19ZR1447700), the Health System Innovation Plan of Shanghai Putuo District Science and Technology Committee (grant numbers: PTKWWS2018001 and PTKWWS201903), and the WBN Liver Disease Research Fund of China Hepatitis Prevention Foundation (grant number: CFHPC2019031).
Data Availability
The data used to support the findings of this study are available from the corresponding author upon request.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Authors' Contributions
Jie Ji, Qiang Yu, Weiqi Dai, Liwei Wu, and Jiao Feng conducted the experiments. Jie Ji, Yuanyuan Zheng, and Yan Li analyzed the data. Jie Ji wrote the manuscript. Chuanyong Guo provided the reagents and materials.
Supplementary Materials
supplementary Figure S1: the quantitative data of western blotting of relative LC3II/LC3I ratio.
References
- 1.Parola M., Pinzani M. Liver fibrosis: pathophysiology, pathogenetic targets and clinical issues. Molecular Aspects of Medicine. 2019;65:37–55. doi: 10.1016/j.mam.2018.09.002. [DOI] [PubMed] [Google Scholar]
- 2.Hernandez-Gea V., Friedman S. L. Pathogenesis of liver fibrosis. Annual Review of Pathology. 2011;6(1):425–456. doi: 10.1146/annurev-pathol-011110-130246. [DOI] [PubMed] [Google Scholar]
- 3.Friedman S. L. Liver fibrosis - from bench to bedside. Journal of Hepatology. 2003;38:38–53. doi: 10.1016/S0168-8278(02)00429-4. [DOI] [PubMed] [Google Scholar]
- 4.Feng J., Chen K., Xia Y., et al. Salidroside ameliorates autophagy and activation of hepatic stellate cells in mice via NF-κB and TGF-β1/Smad3 pathways. Drug Design, Development and Therapy. 2018;Volume 12:1837–1853. doi: 10.2147/DDDT.S162950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eom Y. W., Shim K. Y., Baik S. K. Mesenchymal stem cell therapy for liver fibrosis. The Korean Journal of Internal Medicine. 2015;30(5):580–589. doi: 10.3904/kjim.2015.30.5.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Czaja A. J. Hepatic inflammation and progressive liver fibrosis in chronic liver disease. World Journal of Gastroenterology. 2014;20(10):2515–2532. doi: 10.3748/wjg.v20.i10.2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trautwein C., Friedman S. L., Schuppan D., Pinzani M. Hepatic fibrosis: concept to treatment. Journal of Hepatology. 2015;62(1):S15–S24. doi: 10.1016/j.jhep.2015.02.039. [DOI] [PubMed] [Google Scholar]
- 8.Lee Y. A., Wallace M. C., Friedman S. L. Pathobiology of liver fibrosis: a translational success story. Gut. 2015;64(5):830–841. doi: 10.1136/gutjnl-2014-306842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pinzani M., Macias-Barragan J. Update on the pathophysiology of liver fibrosis. Expert Review of Gastroenterology & Hepatology. 2014;4(4):459–472. doi: 10.1586/egh.10.47. [DOI] [PubMed] [Google Scholar]
- 10.Bataller R., Brenner D. A. Liver fibrosis. Journal of Clinical Investigation. 2005;115(2):209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Higashi T., Friedman S. L., Hoshida Y. Hepatic stellate cells as key target in liver fibrosis. Advanced Drug Delivery Reviews. 2017;121:27–42. doi: 10.1016/j.addr.2017.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu N., Feng J., Lu X., et al. Isorhamnetin inhibits liver fibrosis by reducing autophagy and inhibiting extracellular matrix formation via the Tgf-Beta1/Smad3 and Tgf-Beta1/P38 Mapk pathways. Mediators of Inflammation. 2019;2019 doi: 10.1155/2019/6175091.6175091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hemmann S., Graf J., Roderfeld M., Roeb E. Expression of Mmps and Timps in liver fibrosis - a systematic review with special emphasis on anti-fibrotic strategies. Journal of Hepatology. 2007;46(5):955–975. doi: 10.1016/j.jhep.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 14.Shen M., Chen K., Lu J., et al. Protective Effect of Astaxanthin on Liver Fibrosis through Modulation of TGF-β1 Expression and Autophagy. Mediators of Inflammation. 2014;2014 doi: 10.1155/2014/954502.954502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu L., Zhang Q., Mo W., et al. Quercetin prevents hepatic fibrosis by inhibiting hepatic stellate cell activation and reducing autophagy via the TGF-β1/Smads and PI3K/Akt pathways. Scientific Reports. 2017;7(1):p. 9289. doi: 10.1038/s41598-017-09673-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hernandez-Gea V., Friedman S. L. Autophagy fuels tissue fibrogenesis. Autophagy. 2014;8(5):849–850. doi: 10.4161/auto.19947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ji J., Wu L., Feng J., et al. Cafestol preconditioning attenuates apoptosis and autophagy during hepatic ischemia-reperfusion injury by inhibiting Erk/Pparγ pathway. International Immunopharmacology. 2020;84 doi: 10.1016/j.intimp.2020.106529. [DOI] [PubMed] [Google Scholar]
- 18.Yu Q., Wu L., Liu T., et al. Protective effects of levo-tetrahydropalmatine on hepatic ischemia/reperfusion injury are mediated by inhibition of the ERK/NF-κB pathway. International Immunopharmacology. 2019;70:435–445. doi: 10.1016/j.intimp.2019.02.024. [DOI] [PubMed] [Google Scholar]
- 19.Deng J., Feng J., Liu T., et al. Beraprost sodium preconditioning prevents inflammation, apoptosis, and autophagy during hepatic ischemia-reperfusion injury in mice via the P38 and Jnk pathways. Drug Design, Development and Therapy. 2018;Volume 12:4067–4082. doi: 10.2147/DDDT.S182292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu L., Zhang Q., Dai W., et al. Quercetin Pretreatment Attenuates Hepatic Ischemia Reperfusion-Induced Apoptosis and Autophagy by Inhibiting ERK/NF-κB Pathway. Gastroenterology Research and Practice. 2017;2017 doi: 10.1155/2017/9724217.9724217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hernández-Gea V., Ghiassi-Nejad Z., Rozenfeld R., et al. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology. 2012;142(4):938–946. doi: 10.1053/j.gastro.2011.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thoen L. F. R., Guimarães E. L. M., Dollé L., et al. A role for autophagy during hepatic stellate cell activation. Journal of Hepatology. 2011;55(6):1353–1360. doi: 10.1016/j.jhep.2011.07.010. [DOI] [PubMed] [Google Scholar]
- 23.Madunic J., Madunic I. V., Gajski G., Popic J., Garaj-Vrhovac V. Apigenin: a dietary flavonoid with diverse anticancer properties. Cancer Letters. 2018;413:11–22. doi: 10.1016/j.canlet.2017.10.041. [DOI] [PubMed] [Google Scholar]
- 24.Bauer D., Mazzio E., Soliman K. F. A. Whole transcriptomic analysis of apigenin on Tnfα immuno-activated Mda-Mb-231 breast cancer cells. Cancer Genomics Proteomics. 2019;16(6):421–431. doi: 10.21873/cgp.20146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang J., Pi C., Wang G. Inhibition of Pi3k/Akt/Mtor pathway by apigenin induces apoptosis and autophagy in hepatocellular carcinoma cells. Biomedicine & Pharmacotherapy. 2018;103:699–707. doi: 10.1016/j.biopha.2018.04.072. [DOI] [PubMed] [Google Scholar]
- 26.Bhattacharya S., Mondal L., Mukherjee B., et al. Apigenin loaded nanoparticle delayed development of hepatocellular carcinoma in rats. Nanomedicine. 2018;14(6):1905–1917. doi: 10.1016/j.nano.2018.05.011. [DOI] [PubMed] [Google Scholar]
- 27.Shao H., Jing K., Mahmoud E., Huang H., Fang X., Yu C. Apigenin sensitizes colon cancer cells to antitumor activity of Abt-263. Molecular Cancer Therapeutics. 2013;12(12):2640–2650. doi: 10.1158/1535-7163.MCT-13-0066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lefort É. C., Blay J. Apigenin and its impact on gastrointestinal cancers. Molecular Nutrition & Food Research. 2013;57(1):126–144. doi: 10.1002/mnfr.201200424. [DOI] [PubMed] [Google Scholar]
- 29.Chen P., Huo X., Liu W., Li K., Sun Z., Tian J. Apigenin exhibits anti-inflammatory effects in Lps-stimulated Bv2 microglia through activating Gsk3β/Nrf2 signaling pathway. Immunopharmacology and Immunotoxicology. 2020;42(1):9–16. doi: 10.1080/08923973.2019.1688345. [DOI] [PubMed] [Google Scholar]
- 30.Zhou Q., Cheng K. W., Gong J., Li E. T. S., Wang M. Apigenin and its methylglyoxal-adduct inhibit advanced glycation end products- induced oxidative stress and inflammation in endothelial cells. Biochemical Pharmacology. 2019;166:231–241. doi: 10.1016/j.bcp.2019.05.027. [DOI] [PubMed] [Google Scholar]
- 31.Mirzoeva S., Tong X., Bridgeman B. B., Plebanek M. P., Volpert O. V. Apigenin inhibits Uvb-induced skin carcinogenesis: the role of thrombospondin-1 as an anti-inflammatory factor. Neoplasia. 2018;20(9):930–942. doi: 10.1016/j.neo.2018.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li F., Lang F., Zhang H., et al. Apigenin alleviates endotoxin-induced myocardial toxicity by modulating inflammation, oxidative stress, and autophagy. Oxidative Medicine and Cellular Longevity. 2017;2017:10. doi: 10.1155/2017/2302896.2302896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang H., Guo B., Lin S., Chang P., Tao K. Apigenin inhibits growth and migration of fibroblasts by suppressing Fak signaling. Aging (Albany NY) 2019;11(11):3668–3678. doi: 10.18632/aging.102006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiao R., Chen H., Wan Q., et al. Apigenin inhibits fibroblast proliferation and reduces epidural fibrosis by regulating Wnt3a/Β-catenin signaling pathway. Journal of Orthopaedic Surgery and Research. 2019;14(1):p. 258. doi: 10.1186/s13018-019-1305-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y., Sun Q., Li X., et al. Apigenin suppresses mouse peritoneal fibrosis by down-regulating Mir34a expression. Biomedicine & Pharmacotherapy. 2018;106:373–380. doi: 10.1016/j.biopha.2018.06.138. [DOI] [PubMed] [Google Scholar]
- 36.Wei X., Gao P., Pu Y., et al. Activation of Trpv4 by dietary apigenin antagonizes renal fibrosis in deoxycorticosterone acetate (Doca)-salt-induced hypertension. Clinical Science (London, England) 2017;131(7):567–581. doi: 10.1042/CS20160780. [DOI] [PubMed] [Google Scholar]
- 37.Yanguas S. C., Cogliati B., Willebrords J., et al. Experimental models of liver fibrosis. Archives of Toxicology. 2016;90(5):1025–1048. doi: 10.1007/s00204-015-1543-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bottcher K., Pinzani M. Pathophysiology of liver fibrosis and the methodological barriers to the development of anti-fibrogenic agents. Advanced Drug Delivery Reviews. 2017;121:3–8. doi: 10.1016/j.addr.2017.05.016. [DOI] [PubMed] [Google Scholar]
- 39.El-Serag H. B., Rudolph K. L. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132(7):2557–2576. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- 40.Yue S., Xue N., Li H., Huang B., Chen Z., Wang X. Hepatoprotective effect of apigenin against liver injury via the non-canonical Nf-Κb pathway in vivo and in vitro. Inflammation. 2020;43(5):1634–1648. doi: 10.1007/s10753-020-01238-5. [DOI] [PubMed] [Google Scholar]
- 41.Wang F., Liu J. C., Zhou R. J., et al. Apigenin protects against alcohol-induced liver injury in mice by regulating hepatic Cyp2e1-mediated oxidative stress and Pparα-mediated lipogenic gene expression. Chemico-Biological Interactions. 2017;275:171–177. doi: 10.1016/j.cbi.2017.08.006. [DOI] [PubMed] [Google Scholar]
- 42.Feng X., Yu W., Li X., et al. Apigenin, a modulator of Pparγ, attenuates Hfd-induced Nafld by regulating hepatocyte lipid metabolism and oxidative stress via Nrf2 activation. Biochemical Pharmacology. 2017;136:136–149. doi: 10.1016/j.bcp.2017.04.014. [DOI] [PubMed] [Google Scholar]
- 43.Peng Q., Deng Z., Pan H., Gu L., Liu O., Tang Z. Mitogen-activated protein kinase signaling pathway in oral cancer. Oncology Letters. 2018;15(2):1379–1388. doi: 10.3892/ol.2017.7491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shirakami Y., Lee S. A., Clugston R. D., Blaner W. S. Hepatic metabolism of retinoids and disease associations. Biochimica et Biophysica Acta. 2012;1821(1):124–136. doi: 10.1016/j.bbalip.2011.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsuchida T., Friedman S. L. Mechanisms of hepatic stellate cell activation. Nature Reviews. Gastroenterology & Hepatology. 2017;14(7):397–411. doi: 10.1038/nrgastro.2017.38. [DOI] [PubMed] [Google Scholar]
- 46.Friedman S. L., Sheppard D., Duffield J. S., Violette S. Therapy for fibrotic diseases: nearing the starting line. Science Translational Medicine. 2013;5(167):p. 167sr1. doi: 10.1126/scitranslmed.3004700. [DOI] [PubMed] [Google Scholar]
- 47.Liu T., Xu L., Wang C., et al. Alleviation of hepatic fibrosis and autophagy via inhibition of transforming growth factor-β1/Smads pathway through Shikonin. Journal of Gastroenterology and Hepatology. 2019;34(1):263–276. doi: 10.1111/jgh.14299. [DOI] [PubMed] [Google Scholar]
- 48.Meng X. M., Nikolic-Paterson D. J., Lan H. Y. TGF-β: the master regulator of fibrosis. Nature Reviews. Nephrology. 2016;12(6):325–338. doi: 10.1038/nrneph.2016.48. [DOI] [PubMed] [Google Scholar]
- 49.Macias M. J., Martin-Malpartida P., Massague J. Structural determinants of Smad function in TGF-β signaling. Trends in Biochemical Sciences. 2015;40(6):296–308. doi: 10.1016/j.tibs.2015.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsukada S., Westwick J. K., Ikejima K., Sato N., Rippe R. A. Smad and P38 Mapk signaling pathways independently regulate α1(I) collagen gene expression in unstimulated and transforming growth factor-β-stimulated hepatic stellate cells. The Journal of Biological Chemistry. 2005;280(11):10055–10064. doi: 10.1074/jbc.M409381200. [DOI] [PubMed] [Google Scholar]
- 51.Xu S., Mao Y., Wu J., et al. TGF-β/Smad and Jak/Stat pathways are involved in the anti-fibrotic effects of propylene glycol alginate sodium sulphate on hepatic fibrosis. Journal of Cellular and Molecular Medicine. 2020;24(9):5224–5237. doi: 10.1111/jcmm.15175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li J., Chen K., Li S., et al. Protective effect of fucoidan from Fucus vesiculosus on liver fibrosis via the Tgf-beta1/Smad pathway-mediated inhibition of extracellular matrix and autophagy. Drug Design, Development and Therapy. 2016;10:619–630. doi: 10.2147/DDDT.S98740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xia Y., Li J., Chen K., Feng J., Guo C. Bergenin attenuates hepatic fibrosis by regulating autophagy mediated by the Ppar-Γ/Tgf-Β pathway. PPAR Research. 2020;2020:13. doi: 10.1155/2020/6694214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Coulthard L. R., White D. E., Jones D. L., McDermott M. F., Burchill S. A. p38MAPK: stress responses from molecular mechanisms to therapeutics. Trends in Molecular Medicine. 2009;15(8):369–379. doi: 10.1016/j.molmed.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lu X., Liu T., Chen K., et al. Isorhamnetin: a hepatoprotective flavonoid inhibits apoptosis and autophagy via P38/Ppar-a pathway in mice. Biomedicine & Pharmacotherapy. 2018;103:800–811. doi: 10.1016/j.biopha.2018.04.016. [DOI] [PubMed] [Google Scholar]
- 56.Du X., Wu M., Tian D., et al. Microrna-21 contributes to acute liver injury in Lps-induced sepsis mice by inhibiting Pparα expression. PPAR Research. 2020;2020:7. doi: 10.1155/2020/6633022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kytikova O. Y., Perelman J. M., Novgorodtseva T. P., et al. Peroxisome proliferator-activated receptors as a therapeutic target in asthma. PPAR Research. 2020;2020:18. doi: 10.1155/2020/8906968.8906968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang P. L., Wang J. S., Cheng X. M., et al. Ppar-Γ ligand inhibits nasopharyngeal carcinoma cell proliferation and metastasis by regulating E2f2. PPAR Research. 2019;2019:9. doi: 10.1155/2019/8679271.8679271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Holm L. J., Monsted M. O., Haupt-Jorgensen M., Buschard K. Ppars and the development of type 1 diabetes. PPAR Research. 2020;2020 doi: 10.1155/2020/6198628.6198628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Blitek A., Szymanska M. Expression and role of peroxisome proliferator-activated receptors in the porcine early placenta trophoblast. Domestic animal endocrinology. 2019;67:42–53. doi: 10.1016/j.domaniend.2018.12.001. [DOI] [PubMed] [Google Scholar]
- 61.Morinishi T., Tokuhara Y., Ohsaki H., Ibuki E., Kadota K., Hirakawa E. Activation and expression of peroxisome proliferator-activated receptor alpha are associated with tumorigenesis in colorectal carcinoma. PPAR Research. 2019;2019 doi: 10.1155/2019/7486727.7486727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Alsalem M., Haddad M., Aldossary S. A., et al. Effects of dual peroxisome proliferator-activated receptors alpha and gamma activation in two rat models of neuropathic pain. PPAR Research. 2019;2019 doi: 10.1155/2019/2630232.2630232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ip E., Farrell G., Hall P., Robertson G., Leclercq I. Administration of the potent Pparalpha agonist, Wy-14,643, reverses nutritional fibrosis and steatohepatitis in mice. Hepatology. 2004;39(5):1286–1296. doi: 10.1002/hep.20170. [DOI] [PubMed] [Google Scholar]
- 64.Pawlak M., Lefebvre P., Staels B. Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. Journal of Hepatology. 2015;62(3):720–733. doi: 10.1016/j.jhep.2014.10.039. [DOI] [PubMed] [Google Scholar]
- 65.Ueno T., Komatsu M. Autophagy in the liver: functions in health and disease. Nature reviews. Gastroenterology & hepatology. 2017;14(3):170–184. doi: 10.1038/nrgastro.2016.185. [DOI] [PubMed] [Google Scholar]
- 66.Mallat A., Lodder J., Teixeira-Clerc F., Moreau R., Codogno P., Lotersztajn S. Autophagy: a multifaceted partner in liver fibrosis. BioMed Research International. 2014;2014:7. doi: 10.1155/2014/869390.869390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kiyono K., Suzuki H. I., Matsuyama H., et al. Autophagy is activated by Tgf-Beta and potentiates Tgf-Beta-mediated growth inhibition in human hepatocellular carcinoma cells. Cancer Research. 2009;69(23):8844–8852. doi: 10.1158/0008-5472.CAN-08-4401. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supplementary Figure S1: the quantitative data of western blotting of relative LC3II/LC3I ratio.
Data Availability Statement
The data used to support the findings of this study are available from the corresponding author upon request.