

Abstract
Checkpoint blockade immunotherapy relies on the empowerment of the immune system to fight cancer. Why some patients fail to achieve durable clinical responses is not well understood, but unique individual factors such as diet, obesity, and related metabolic syndrome could play a role. The link between obesity and patient outcomes remains controversial and has been mired by conflicting reports and limited mechanistic insight. We addressed this in a C57BL/6 mouse model of diet-induced obesity using a western diet high in both fats and sugars. Obese mice bearing B16 melanoma or MC38 carcinoma tumors had impaired immune responses to immunotherapy and a reduced capacity to control tumor progression. Unexpectedly, these compromised therapeutic outcomes were independent of body mass and, instead, were directly attributed to dietary fructose. Melanoma tumors in mice on the high-fructose diet were resistant to immunotherapy and showed increased expression of the cytoprotective enzyme, heme oxygenase-1 (HO-1). This increase in HO-1 protein was recapitulated in human A375 melanoma cells exposed to fructose in culture. Induced expression of HO-1 shielded tumor cells from immune-mediated killing and was critical for resistance to checkpoint blockade immunotherapy, which could be overcome in vivo using a small-molecule inhibitor of HO-1. This study reveals dietary fructose as a driver of tumor immune evasion, identifying HO-1 expression as a mechanism of resistance and a promising molecular target for combination cancer immunotherapy.
Keywords: Obesity, T cells, immunotherapy, fructose, HO-1
Introduction
Immunotherapy is now a front-line treatment option for patients with cancer, but even with the aggressive combination regimen of anti-CTLA-4 and anti-PD-1, less than half of patients with melanoma experience durable remissions (1). Identifying the reasons for therapeutic success versus failure has become the focus of intense investigation and is critical for improving outcomes in a greater number of diverse cancer patients. Tumor infiltration by immune cells and the extent of PD-L1 expression have helped to predict and explain divergent outcomes in some cases, but other patient-specific factors likely influence outcomes too. For example, more than a third of Americans are now considered obese, a condition associated with impaired immune responses to infections and vaccines and a higher risk of cancer (2–5). This has led to the prediction that obese cancer patients will experience worse immunotherapy outcomes compared to leaner patients. Several studies have tested this hypothesis but came to different conclusions, ranging from obesity as a detrimental or neutral factor to even a benefit for some patients (6–10). Not surprisingly, reproducible mechanistic insight has also been lacking.
Most studies use body mass index (BMI) as a convenient way to assess human obesity, particularly in retrospective clinical analyses where height and weight data are the only accessible metrics. However, the simplicity of the BMI scale belies the considerable complexities of obesity, often associated with comorbidities like systemic inflammation, diabetes, liver dysfunction, metabolic syndrome and altered adipose tissue metabolism – none of which are captured by BMI. Thus, lack of consensus among clinical studies may stem from strict adherence to the BMI scale as the sole indicator of patient obesity.
The majority of obesity is diet-driven, and many nutritional components have the capacity to influence immunity and responses to therapy regardless of patient BMI. One example is fructose, a 5-carbon monosaccharide found naturally in fruit that also makes up half of the disaccharide sucrose (table sugar) when combined with glucose. Since 1970, daily per capita consumption of fructose in the United States is estimated to have increased by 26% and more than a 100-fold for high-fructose corn syrup (11), coinciding with increased obesity and an array of other human health conditions including cancer (12,13). Fructose is primarily metabolized by the liver and requires enzymatic pathways distinct from those involved in glycolysis (14). As a result, tumor cells utilizing fructose rather than glucose for energy assume an altered metabolic phenotype (15). In vivo studies have demonstrated that oral administration of high-fructose corn syrup to mice increases the development of intestinal tumors independent of obesity (16). But how dietary fructose impacts human cancer progression and the response to immunotherapy remains unresolved.
We investigated the hypothesis that diet-induced obesity compromises cancer immunotherapy outcomes. We first explored this in the B16 melanoma model using B6 mice on a western diet high in saturated fats and sugars, including fructose. As predicted, obese mice experienced erratic and typically worse outcomes after checkpoint blockade treatment, coinciding with dampened T-cell immune responses that were evident both systemically and within tumors. However, this inability to control tumor progression after immunotherapy was independent of body mass and instead was the result of high dietary fructose. Removal of fructose from the high-fat diet did not prevent obesity but completely restored the antitumor efficacy of immunotherapy. Despite our initial predictions, this restoration did not extend to T-cell function. Instead, we identified fructose as a driver of tumor immune evasion. Specifically, fructose induced a cytoprotective pathway involving expression of the known antiapoptotic enzyme heme oxygenase-1 (HO-1), endowing tumor cells with the ability to resist immune-mediated effector activity elicited during immunotherapy. Our study reveals fructose-mediated cytoprotection as a mechanism of immune evasion in mouse and human melanoma, and pinpoints the HO-1 pathway as a rational target in combination with immunotherapy strategies, particularly when resistance to checkpoint blockade is observed.
Materials and methods
Mice and diets
All mice were maintained under specific pathogen-free conditions and used in accordance with our animal use protocol approved by the Institutional Animal Care and Use Committee (IACUC) of the Department of Comparative Medicine, SLU School of Medicine. C57BL/6 (Stock No. 000664) and Pmel (Stock No. 005023) mice were purchased from The Jackson Laboratory. Both female and male mice were used throughout the studies. Mice were age- and sex-matched and between 4–6 months old when used for experiments and were assigned randomly to experimental groups. The western diet (WD) chow contained 40 kcal% from fat, 20 kcal% from fructose, and 2% cholesterol and was purchased from Research Diets (Cat# D17010103). The western diet with no added fructose (WDΔF) was the same formulation as D17010103 but without the added fructose (Cat# D19012301). Mice were placed on normal chow (NC), WD, or WDΔF upon weaning at approximately 4 weeks of age and maintained on the assigned diet for at least 10 weeks. The percent of lean and fat body mass was determined by nuclear magnetic resonance using a Bruker mini spec LF50. The mini spec acquired and analyzed time-domain nuclear magnetic resonance and provided body composition results for percentage of lean, fat, and fluid in the animal. Adiposity was then calculated from the ratio of fat mass to total body mass and reported as a percentage.
Tumor immunotherapy
The B16-F0 melanoma line was obtained from ATCC (Cat# CRL-6322). The MC38 carcinoma line was obtain from the Guangyong Peng lab (SLU). Both the B16 and MC38 lines were confirmed negative for mycoplasma on February 2, 2020 by PCR using a kit from ATCC (Cat# 30–1012K) according to the manufacturer’s protocol. Tumors were maintained in Dulbecco’s Modified Eagle medium (DMEM; ThermoFisher Cat# 11995–065) supplemented with 10% fetal bovine serum (Coring, Cat# 35–011-CV) and 1% Pen/Strep (Sigma Aldrich, Cat# P0781). Adherent cells were removed with 0.25% trypsin, and cells were passaged 3–5 times for each experiment. For all tumor studies, 1×106 cells were injected subcutaneously into both flanks of C57BL/6 mice. Tumors were established for 5–8 days mice prior to treatment with immune checkpoint blockade (ICB). Combination ICB consisted of anti-PD-1 (RMP1–14), anti-CTLA-4 (9D9), and anti-LAG-3 (C9B7W) from Bio × Cell (Cat# BE0146, BE0164, and BE0174, respectively) administered intraperitoneally at 5mg/kg on days 5, 10, and 15 for tumor growth studies, or on days 8, 11, and 14 for day 16 analysis of tumor-infiltrating lymphocytes (TILs). The HO-1 inhibitor, OB24 from TOCRIS (Cat# 6119), was administered intraperitoneally (IP) at 30mg/kg on days 3, 5, 7, 9, 11, and 13. Tumors were measured using digital calipers, and tumor volume was determined using the equation (L × W2)/2. In vivo leptin neutralization was accomplished by three 100 μg IP injections of recombinant mouse leptin-receptor Fc chimera (R&D Systems, Cat# 497-LR), as previously described (17). For TIL analysis, tumors were excised from mice and mechanically disrupted with the a sterile 3-mL syringe plunger and filtered through a 40uM strainer. All isolation steps were performed in complete DMEM. For B16 fructose pretreatment experiments, B16 tumor cells were cultured in 0g/L or 20g/L of fructose (Sigma Aldrich; Cat# F3510) in complete DMEM for three days, and 1×106 B16 tumor cells were injected subcutaneously into both flanks of C57BL/6 mice followed by ICB on days 1 and 3 with tumors measured every other day for 2 weeks.
Flow cytometry
Fluorochrome conjugated antibodies were purchased from Biolegend (anti-CD45 Cat# 103132, anti-CD4 Cat# 100546, anti-CD8 Cat# 100714, anti-NK1.1 Cat# 108710). Aqua fluorescent reactive dye (live/dead) was purchased from Invitrogen (Cat# L34966A), and Fc block was purchased from Biolegend (Cat# 101320) was performed. Flow cytometric analysis was performed on LSR II FACS analyzer (BD Biosciences) at the Saint Louis University Flow Cytometry Core Facility. Live lymphocytes were identified as staining negative for live/dead stain and positive for CD45. These were segregated into CD4+ and CD8+ T cells or CD4/8-negative NK1.1+ natural killer (NK) cells. CD4+ T cells were further segregated into conventional FoxP3-negative and Foxp3+ regulatory cells. Flow cytometry data was analyzed using FlowJo v.10 software (Tree Star Inc.).
Intracellular protein staining
Intracellular cytokine staining was performed using the Cytofix/Cytoperm Plus kit (BD Biosciences) per the manufacturer’s instructions. In brief, TILs were incubated with 10 ng/mL PMA (Sigma Aldrich; Cat# P8139) and 0.3 μg/mL Ionomycin (Sigma Aldrich; Cat# I0634) for 4 hours in the presence of GolgiPlug (BD Biosciences, 51–2301KZ) diluted 1:500 according to the manufacture’s protocol. Cells were first stained with live/dead dye and cell-surface markers as described above. Cells were then fixed, permeabilized, and stained with anti-IFNγ (Biolegend, Cat# 505808) and anti-TNF (Biolegend, Cat# 506324). Intracellular staining of cytoplasmic- and nuclear-associated proteins was performed using the eBioscience cellular perm kit per the manufacturer’s instructions. Briefly, TILs were processed and stained directly ex vivo with live/dead dye and cell-surface markers, including anti-PD-1 (Biolegend, Cat# 135231). Cells were fixed, permeabilized, and stained with antibodies specific for Foxp3 (eBioscience, FJK-16s) and granzyme B (Biolegend, Cat# 515403). Intracellular HO-1 was detected in a two-step staining process using anti-HO-1 (Cell Signaling Technology; E9H3A) and anti-rabbit-IgG linked to HRP (Cell Signaling Technology; Cat#7074S). Intracellular flow cytometry was analyzed using FlowJo v.10 software (Tree Star Inc.).
Microbiome analysis
Fresh fecal samples were collected one time each from individual C57BL/6 mice, snap frozen in dry ice, and stored at −80° C. Total genomic DNA was purified using the QIAamp DNA stool minikit (Qiagen, Germany) following manufacturer’s instructions. The concentration of extracted DNA samples was determined by UV absorbance at 260 nm, and purity from protein and RNA contamination by absorbance ratios of 260/280 nm and 260/230 nm. The relative abundance and identification of taxa-specific 16S rRNA genes was determined by quantitative PCR for individual bacterial taxa or species using primer sets described below (18,19). qPCR was performed using a 7500 Fast Real-Time PCR system on MicroAmp Fast Optical 96-well reaction plate (Applied Biosystems). Samples were prepared in 10μL reaction volumes composed of 10 ng genomic DNA, 500 nM primer, and 1X Powerup SYBR green PCR master mix (Applied Biosystems, Foster City, CA). Relative gene expression was quantified by 2−ΔΔCT.
| Actinobacteria | Act664F | TGTAGCGGTGGAATGCGC |
| Act941R | AATTAAGCCACATGCTCCGCT | |
| Bacteroidetes | Bac960F | GTTTAATTCGATGATACGCGAG |
| Bac1100R | TTAASCCGACACCTCACGG | |
| Bifidobacteria | BifidF | CGGGTGAGTAATGCGTGACC |
| BifidR | TGATAGGACGCGACCCCA | |
| Betaproteobacteria | Beta979F | AACGCGAAAAACCTTACCTACC |
| Beta1130R | TGCCCTTTCGTAGCAACTAGTG | |
| Saccharibacteria | Sac1031F | AAGAGAACTGTGCCTTCGG |
| Sac1218R | GCGTAAGGGAAATACTGACC | |
| Deferribacteres | Defer1115F | CTATTTCCAGTTGCTAACGG |
| Defer1265R | GAGHTGCTTCCCTCTGATTATG | |
| Delta/Gamma-proteobacteria | Gamma877F | GCTAACGCATTAAGTRYCCCG |
| Gamma1066R | GCCATGCRGCACCTGTCT | |
| Epsilonproteobacteria | Epsilon940F | TAGGCTTGACATTGATAGAATC |
| Epsilon1129R | CTTACGAAGGCAGTCTCCTTA | |
| Firmicutes | Firm934F | GGAGYATGTGGTTTAATTCGAAGCA |
| Firm1060R | AGCTGACGACAACCATGCAC | |
| Tenericutes | Ten662F | ATGTGTAGCGGTAAAATGCGTAA |
| Ten862R | CMTACTTGCGTACGTACTACT | |
| Verrucomicrobia | Ver1165F | TCAKGTCAGTATGGCCCTTAT |
| Ver1263R | CAGTTTTYAGGATTTCCTCCGCC |
Fecal transfer
Normal B6 mice were treated with antibiotic (enrofloxacin) at 100 mg/L in drinking water for 1 week and then put on normal food and water for 1 day. Fresh fecal material was collected from lean and obese mice and diluted in PBS at 15 mg/mL, and 100μL was administered to recipients by oral gavage on days −7 and −3 prior to establishing subcutaneous B16 tumors on day 0 followed by immunotherapy.
Serum chemistry analysis
Mouse blood samples were collected once per animal via the cheek and isolated serum was analyzed at Advanced Veterinary Lab (St. Louis, MO) using a Beckman Coulter AU480 system (Roche) for plasma lipids (total cholesterol, low-density lipoprotein, and high-density lipoprotein) and liver enzymes (ALT and AST) using reagents from Sekisui Diagnostics.
Histopathological assessment
Mouse organs were harvested and embedded in paraffin. Tissues were sectioned and slides created in the Saint Louis University microscopy core facility. Slides were stained for cleaved caspase-3 using a SignalStain® Apoptosis IHC detection kit (Cat.# 12692) from Cell Signaling Technology according to the manufacturer’s protocol.
Single-cell RNA sequencing
TILs were sorted to at least 99% purity based on CD45 staining using a BD FACS Aria III (BD Biosciences), and single-cell suspensions were loaded on a Chromium Single Cell Controller instrument (10x Genomics, Pleasanton, CA, USA) to generate single-cell gel beads in emulsion (GEMs). After sorting and processing, 8091 total CD45+ cells were analyzed (2129 from NC, 3189 from WD, and 2773 from WDΔF). Single-cell RNA libraries were prepared using the Chromium Single Cell 5′ Library & Gel Bead Kit (P/N 1000006, 10x Genomics). GEM-RT was performed in a Veriti 96-Well Thermal cycler (Applied Biosystems, 4375786): 53°C for 45 min, 85°C for 5 min; held at 4°C and stored at −20°C. The GEMs were then broken, and the single-strand cDNA was cleaned up with DynaBeads MyOne Silane Beads (Thermo Fisher Scientific; P/N 37002D). Barcoded, full length cDNA was amplified using the Veriti 96-Well Thermal Cycler: 98°C for 45 s; cycled 13 × : 98°C for 20 s, 67°C for 30 s, and 72°C for 1 min; 72 °C for 1 min; held at 4°C. Amplified cDNA product was cleaned up with the SPRIselect Reagent Kit (0.6 × SPRI; Beckman Coulter; P/N B23318). 5′ gene expression libraries were constructed using the reagents in the Chromium Single Cell 3′/5′ Library Construction kit (P/N 1000020). For 5′ gene expression library construction, these steps were followed: (1) fragmentation, end repair and A-tailing; (2) post fragmentation, end repair and A-tailing cleanup with SPRIselect; (3) adaptor ligation; (4) post ligation cleanup with SPRIselect; (5) sample index PCR and cleanup. Final QC and Illumina sequencing of the prepared libraries was performed on the NovaSeq6000 platform at the Washington University in St. Louis Genome Technology Access Center (GTAC). Raw data were processed through the CellRanger 3.0 pipeline (10x Genomics), and secondary clustering and differential expression analysis were conducted the open source R software package Seurat/R (20). Components for clustering were generated by canonical-correlation analysis. High-signal canonical correlates were determined in Seurat by ranked principal components based on the percent variance, explaining the most variance in comparison identity classes, and were aligned by dynamic time warping (DTW), and their dimensions were used for subsequent shared nearest neighbor clustering (SNN) and visualization by t-distributed stochastic neighbor embedding (t-SNE). Globally distinguishing genes for each cluster and comparison identity class were identified by calculating the normalized gene expression for the average single cell. Significant genes with at least a 2-fold change and corrected P value less than 0.01 were identified via Wilcoxon Rank-Sum Test with Bonferroni correction for multiple comparisons. Raw sequencing data files have been uploaded to the NCBI SRA database and can be accessed by referencing accession numbers SAMN15690252–7.
Western blot
Cultured melanoma cells or single-cell suspensions from whole ex vivo tumors were lysed with 1X lysis buffer from Cell Signaling (Cat# 9803) augmented with 1X protease and phosphatase inhibitors from Cell Signaling (Cat# 5872S) according to the manufacture’s protocol. B16 and A375 melanoma cells were cultured in 0–40 grams per liter of fructose (Sigma Aldrich; Cat# F3510) in complete DMEM for 3 days prior to lysis. Proteins were separated by SDS polyacrylamide gel electrophoresis (SDS-PAGE) on a pre-cast 4–12% Bis-Tris gel (Invitrogen Cat# NP0321BOX) and transferred onto nitrocellulose membrane (Invitrogen Cat# LC2001). HO-1 protein was detected by anti-mouse (Cat# 82206) or anti-human (Cat# 43966) antibodies from Cell Signaling, as was anti-human BACH1 (Cat# 4578). NRF2 was detected using a polyclonal anti-human antibody from Abcam (Cat# ab137550). All primary antibody were used at 1:1000 dilution of stock and detected with anti-rabbit IgG HRP-linked antibody from Cell Signaling (Cat# 7074) at 1:5000 dilution of stock, and beta-actin was detected with an anti-mouse/human cross-reactive antibody from Cell Signaling (Cat# 13E5) at 1:5000 dilution of stock. Western blots were visualized by chemiluminescence using ECL reagent from Cell Signaling (Cat# 6883S) according to the manufacturer’s protocol. Densitometry was performed using ImageJ software.
Apoptosis assay
Splenic T cells from naive Pmel mice were processed into a single-cell suspension at 1×106 c/mL, and stimulated with 4 μg/mL anti-CD3 (eBioscience; Cat# 14-0032-85), 4μg/mL anti-CD28 (Biolegend; Cat# 102102), and 20 U/mL recombinant human IL2 (National Cancer Institute Biological Resources Branch – Preclinical Biologics Repository) for 48 hours. B16 melanoma cells were pretreated with 10 μM Hemin (Sigma Aldrich, Cat# 51280) or 20 g/L of fructose with or without OB24 (10–30 μg/mL) for 24 hours. Splenocytes and B16 cells were then cocultured at a 10:1 effector to target ratio with or without fructose and OB24 for 72 hours. Tumor cell apoptosis was assessed by staining with pan-caspase (CaspGlow Invitrogen, Cat# 88-7003-4), and Annexin V (Invitrogen, Cat# A35110) according to the manufacturer’s protocol and analyzed by flow cytometry.
Statistics
Statistical analysis to compare multi-variable treatment groups was performed using a two-way ANOVA (Prism 7.0, GraphPad Software). Correlation between parameters was determined by calculating the Pearson’s value (r) and the corresponding P value for r using Prism 7.0. All error bars displayed within figures display the mean of distribution and represent the standard error of the mean (SEM) unless otherwise stated. Exact P values are indicated whenever possible.
Results
To test how diet-induced obesity influences cancer immunotherapy, four-week old B6 mice were fed a diet high in both saturated fat and sugars, modeling a western diet (WD) linked to obesity in people. After 12 weeks on this WD, both male and female mice recapitulated several comorbidities common in human obesity, including increased body mass and adiposity, high cholesterol, and fatty liver disease (Fig. 1A–E). Increases in body mass were not uniform, as mice on WD displayed a range of weights at 12 weeks. This is reflective of people who may have a poor diet but do not necessarily experience excessive weight gain, a phenomenon that is not fully captured by the standard body mass index (BMI) scale of obesity. For continuity between experiments, mice on this study were only considered obese after reaching 25% more mass than age- and sex-matched counterparts on normal chow (NC).
Figure 1. Mice on a western diet are resistant to immune checkpoint blockade (ICB).
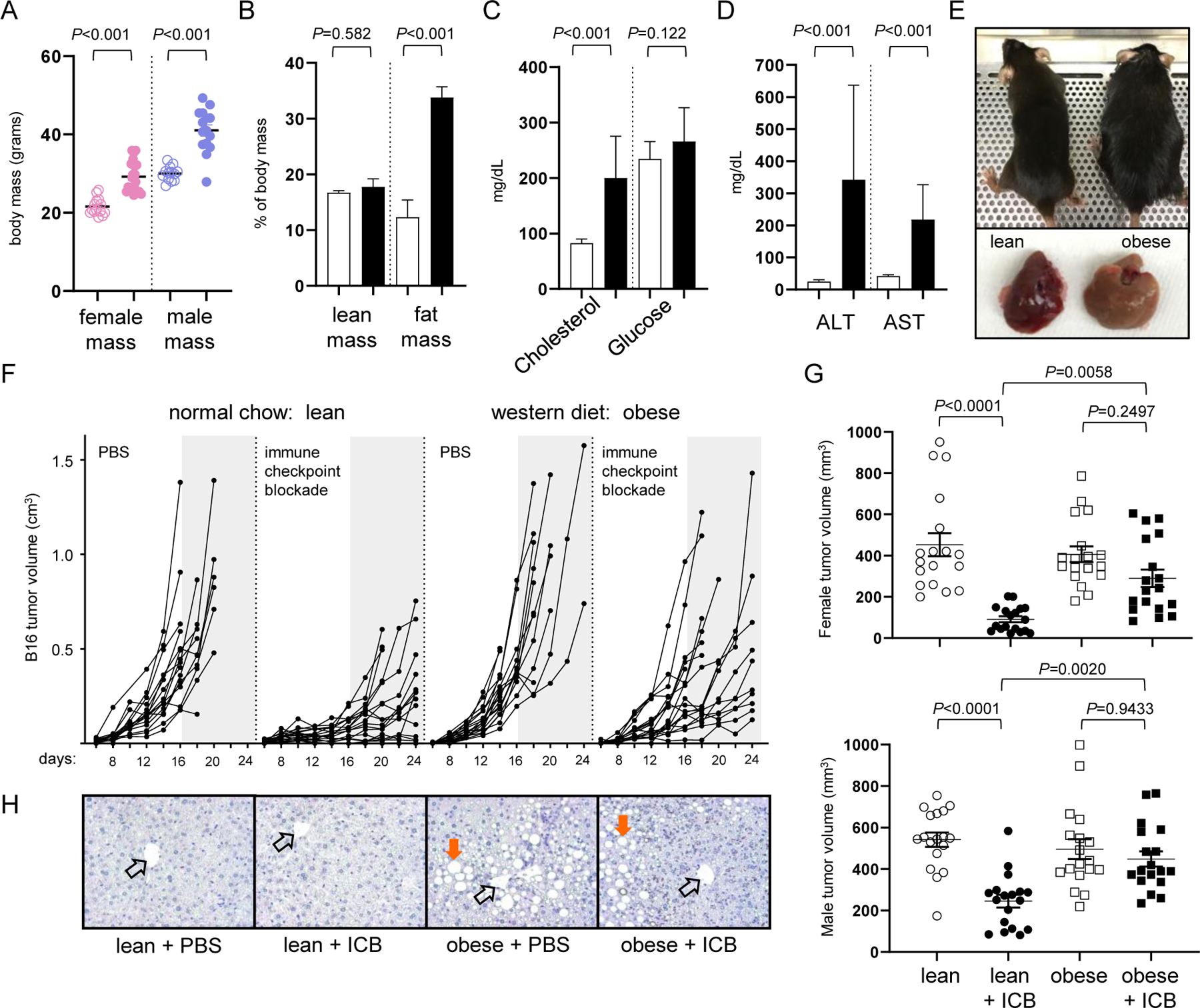
Male (n=30) and female (n=30) B6 mice were placed on normal chow (NC) or western diet (WD). After 12 weeks, (A) body mass, (B) adiposity, (C) serum cholesterol and glucose, and (D) liver enzymes were assessed, with open and closed bars representing data from NC and WD, respectively. (E) Representative images of mice on NC and WD with their corresponding liver. (F) Subcutaneous B16 melanoma tumor volume (cm3) over time for lean and obese mice treated with vehicle control (PBS) or ICB. Data are pooled from 2 independent experiments using an equal number of male and female mice (8 mice/treatment group). Grey shaded regions provide a common point of reference after day 16. (G) Day 16 melanoma tumor volumes in female (top panel) and male (bottom panel) recipients pooled from 3 separate studies each. Error bars represent SEM and exact P values were calculated by two-way ANOVA for the bracketed groups with each point representing data from an individual tumor (9 mice/treatment group). (H) Representative liver H&E stains with open arrows indicating central portal vein and orange arrows marking lipid deposits.
To determine if lean and obese mice responded differently to immunotherapy, bi-lateral subcutaneous B16 melanoma tumors were established in the flanks of mice on WD and age/sex-matched mice on NC. Recipients were treated with PBS or a combination immune checkpoint blockade (ICB; anti-PD-1/CTLA-4/LAG-3) on days 5, 10, and 15 after tumor inoculation. This triple-blockade regimen is superior to other ICB combinations for rescuing the function of tolerant CD8+ tumor-infiltrating lymphocytes (TILs) and providing a survival benefit to mice with disseminated leukemia or melanoma (21,22). Here, in the absence of immunotherapy, lean and obese mice experienced similar tumor growth despite differences in body weight (Fig. 1F). However, disparities in tumor growth were clearly evident in mice treated with ICB, which forestalled melanoma progression in lean mice but was less effective in obese mice resulting in overall larger tumor volumes (Fig. 1F). This impaired response to immunotherapy was quantified at day 16 across multiple pooled experiments, a time point when tumors were advanced but all recipients remained alive, and was observed in both female and male recipients (Fig. 1G). Although some variation in tumor sizes was apparent between the sexes, a diminished response to immunotherapy was evident in all obese recipients regardless of sex. Impaired therapeutic outcomes were not limited to the B16 melanoma model, as obese mice bearing an MC38 colon carcinoma also failed to benefit from ICB treatment relative to lean counterparts (Suppl. Fig. S1). These results implicated diet-induced obesity as a potential barrier to the success of cancer immunotherapy with checkpoint inhibitors. Although some studies support obesity as prognostic for poor immunotherapy outcomes (17,23), another concludes that obesity may be an advantage for both mice and people (10). Thus, consensus has been elusive, hinting that the influence of diet-induced obesity is more nuanced than previously appreciated.
In patients, treatment-related toxicity is an obstacle to the success of cancer immunotherapy and must be actively managed in clinical settings (24). There is evidence that obese mice are especially susceptible to immune-mediated toxicity (25), but this does not appear to be true for checkpoint blockade or other immune-modulating agents (10,17,23). In our study, combination ICB was well-tolerated in lean and obese mice, which were similarly active and maintained body weight throughout the course of treatment, indicative of low or no toxicity. Histopathological analysis also showed no evidence of treatment-related tissue damage in the gastrointestinal tract, liver, pancreas, or kidney regardless of body mass. Obese mice on WD mice had clear evidence of fatty liver disease (Fig. 1H) and high serum ALT/AST (Fig. 1D), but this was not exacerbated by immunotherapy. Thus, obesity does not appear to increase the risk of treatment-related toxicity in B6 mice receiving immune checkpoint inhibitors.
Poor therapeutic outcomes in obese mice on the WD could stem from impaired lymphocyte trafficking into tumors due to altered inflammation and chemokine gradients affected by excessive adipose tissue (2). Despite differences in tumor growth at day 16 (Fig. 1G), lean and obese mice had similar increases in CD8+ and CD4+ TIL frequencies after immunotherapy (Fig. 2A). Natural killer (NK) cells and Foxp3+CD4+ regulatory T cells (Tregs) were stable regardless of recipient weight or treatment (Fig. 2A). Therefore, disparities between lean and obese recipients could not be explained by impaired T-cell or NK-cell infiltration or expansion within tumors.
Figure 2. Mice on a western diet have functionally impaired TILs independent of body mass.
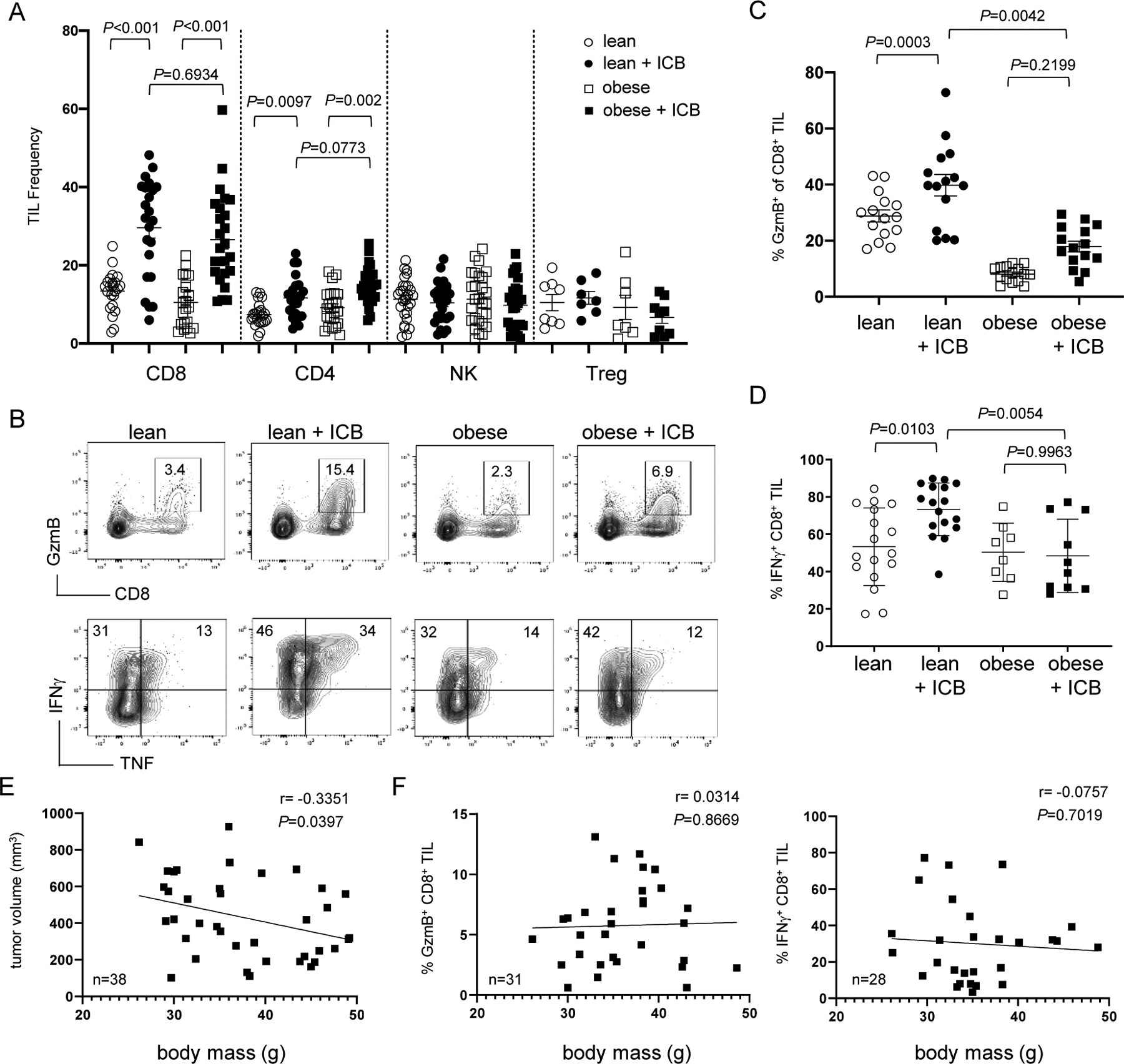
Mice on NC and WD were subcutaneously injected on each flank with 1×106 B16 tumor cells and treated with ICB on days 6, 10, and 14. On day 16, tumors were analyzed for (A) immune cell frequency by flow cytometry and graph shows data from 24 mice/treatment group (n=8/group for Tregs). (B) Representative flow plots show effector molecule expression on day 16. (C) The frequency of CD8+ TIL expressing GzmB was pooled from 4 independent experiments (15 mice/treatment group) using both male and female recipients. (D) The frequency of CD8+ TIL expressing IFNγ was pooled from 3 experiments (8–15 mice/treatment group). All error bars represent SEM and exact P values were calculated by two-way ANOVA for the bracketed groups. (E) Tumor volume and (F) CD8+ TIL granzyme B and IFNγ production were compared to the body mass of WD mice treated with ICB. Each square represents an individual mouse with inset lines representing the best fit for linear regression with the Pearson correlation coefficient (r) and exact P values indicated.
We next tested if CD8+ TILs in lean and obese mice had any functional differences that could account for disparate outcomes. Indeed, lean mice treated with ICB mounted a robust effector CD8+ T-cell response within tumors that included increased granzyme B (GzmB), IFNγ, and TNF protein expression (Fig. 2B–D), whereas these effector molecules were only modestly and inconsistently expressed in obese mice following ICB (Fig. 2B–D). Similar defects in IFNγ production were observed in CD4+ TILs but not NK cells (Suppl. Fig. S2), suggesting immune dysfunction in obese mice may be limited to T cells. The inability to effectively engage these requisite effector mechanisms during immunotherapy implied that obese mice on a WD were at least partially resistant to the immune modulatory effects of ICB.
We predicted that compromised T-cell responses in obese mice were attributed to increased body mass and adiposity. To test this, we leveraged the variations in weight gain among mice on WD and assessed if body mass correlated with outcomes. Tumor volumes only from mice on WD treated with ICB were directly compared to their corresponding host body masses, revealing a slight therapeutic advantage to mice with greater body mass (Fig. 2E). There was no correlation between body mass and CD8+ TIL effector responses in obese mice treated with immunotherapy (Fig. 2F). These unexpected results suggested that resistance to immunotherapy in obese hosts was independent of body mass or adiposity and may be attributable to other factors associated with the western diet.
To confirm that therapeutic outcomes were separate from obesity, we explored the possible role of obesity-associated leptin, which has been described as having immunosuppressive effects during immunotherapy (10,17), perhaps by increasing PD-1 expression on T cells. Although serum leptin concentrations were significantly elevated in obese mice, we observed no connection between leptin and PD-1 expression on T cells (Suppl. Fig. S3). Leptin neutralization had no impact on GzmB expression by CD8+ TILs or on immunotherapy outcomes in obese mice (Suppl. Fig. S3). We concluded that, despite its excess in obese hosts, leptin did not impair T-cell function nor influenced responses to immunotherapy. This is supported by several other studies in mice and humans reporting a range of leptin-induced effects, including that leptin may boost T-cell responses (26–29).
Mice on two different diets could also have divergent gut flora with the potential to influence responses to immunotherapy (18,30). Indeed, we observed clear differences in the abundance of many bacterial phyla in lean and obese mice on NC and WD, respectively (Suppl. Fig. S4). However, normal B6 recipients of fecal transfers from mice on WD responded equivalently well to ICB compared to identical B6 mice receiving fecal transfers from lean donors on NC (Suppl. Fig. S4). Thus, although the microbiome of lean and obese mice on NC and WD differed, this did not appear to significantly impact immunotherapy outcomes. This result illustrates that differences in microbiomes do not guarantee divergent immunotherapy outcomes. This is indirectly supported by clinical trial results, where diverse patients with varying tumor characteristics, geographical locations, and presumably different diets and gut flora can still experience durable immunotherapy responses (1,31–33).
Because poor therapeutic outcomes in obese mice could not be explained by increased body mass, adiposity, leptin concentration, or altered microbiome, we focused on the individual nutritional components of the western diet. In addition to being high in fat (40 kcal%), the WD was high in fructose, as both a monosaccharide and in disaccharide sucrose (55% fructose and 45% glucose). Fructose has been reported to increase gastrointestinal and pancreatic cancer cell growth and induce breast cancer metastases (16,34,35), suggesting dietary fructose can influence malignant disease progression but its impact immunotherapy outcomes has not been reported. To determine if dietary fructose played a role in shaping the success of ICB, B6 mice were provided NC, WD, or an identical western diet lacking the fructose monosaccharide component (WDΔF). After eight weeks, mice on either the WD or WDΔF had gained a significant weight compared to NC counterparts (Fig. 3A). At ten weeks, B16 tumors were established, and recipients were treated with ICB or PBS control as previously described. Obese mice on the WDΔF diet demonstrated a restored ability to control tumor growth following immunotherapy, whereas mice on WD containing fructose failed to respond to ICB (Fig. 3B). These diet-induced outcomes were not limited to the melanoma model, and were recapitulated in mice with MC38 tumors (Suppl. Fig. S5). Again, recipient body mass did not correlate with disease progression (Fig. 3C), implicating diet rather than obesity in dictating therapeutic outcomes.
Figure 3. Removal of dietary fructose restores responsiveness to cancer immunotherapy.
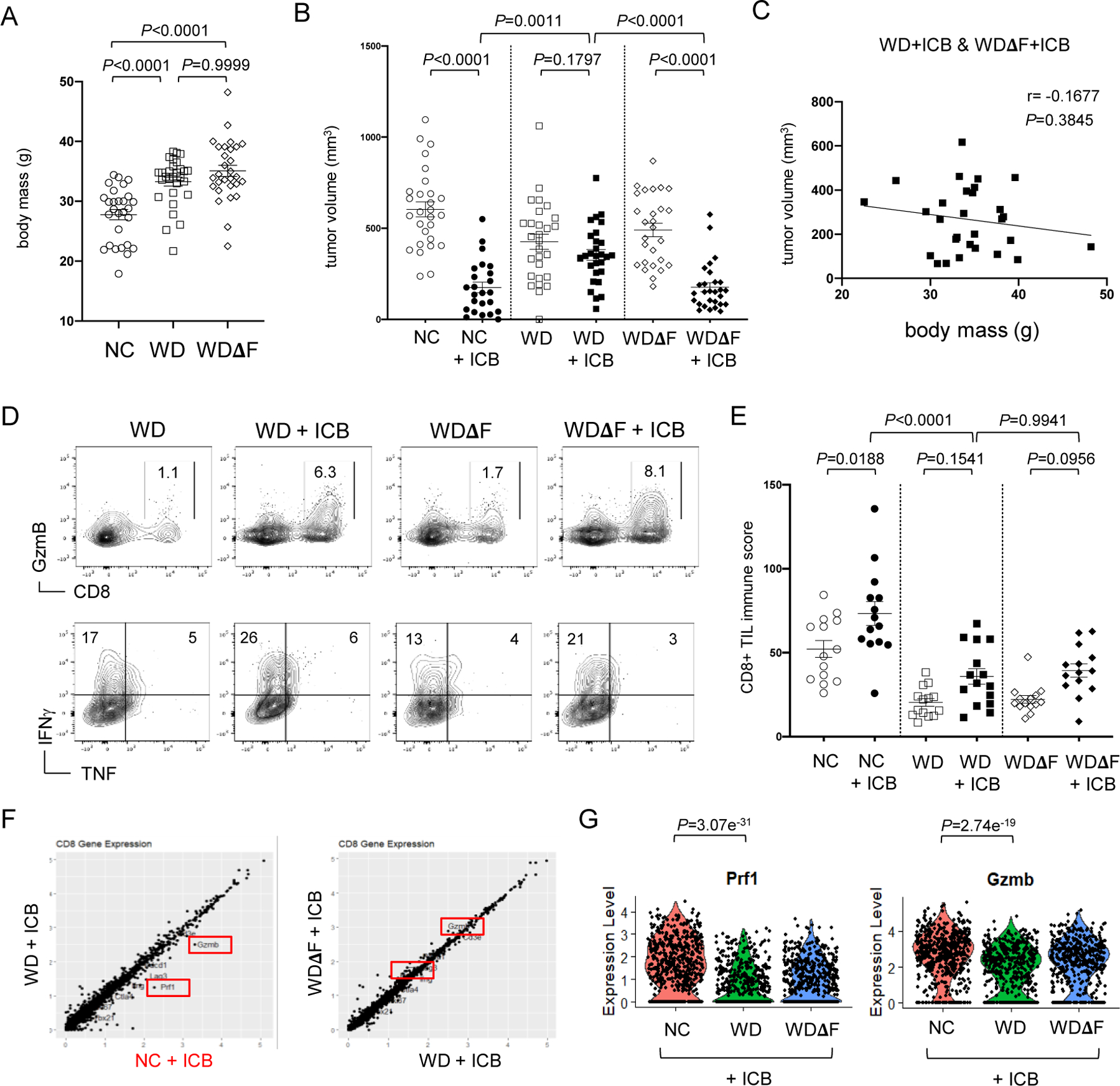
Male B6 mice on NC (n=28), WD (n=30) or WD without added fructose (WDΔF, n=29) were (A) weighed after 8 weeks on diet. At 10 weeks, bi-lateral subcutaneous B16 tumors were established and mice were treated with ICB on days 6, 10, and 14. (B) Tumors were measured on day 16 and data pooled from 3 independent experiments. Each point represents data from an individual tumor and error bars represent SEM with exact P values calculated by two-way ANOVA for the bracketed groups. (C) Tumor volume was determined at day 16 for mice on WD and WDΔF treated with ICB and compared with corresponding body mass. The inset line represents the best fit for linear regression with the Pearson correlation coefficient (r) and exact P values indicated. (D) CD8+ TIL function was assessed by flow cytometry and used to calculate an (E) immune score incorporating GzmB and IFNγ production and data was pooled from 3 separate experiments and error bars represent SEM. Single-cell RNA sequencing data showing (F) total gene expression by CD8+ TIL from individual mice on NC, WD, and WDΔF after ICB treatment. (G) Violin plots display expression of Prf1 and Gzmb genes with each dot representing data from an individual CD8+ TIL and exact P values indicated for the bracketed groups.
Because responses to immunotherapy were restored in mice on WDΔF, we expected that dietary fructose was impairing TIL effector function, which was presumably restored or maintained in the absence of fructose, but expression of the effector molecules GzmB, IFNγ, and TNF by CD8+ TILs was similar in mice on either WD or WDΔF (Fig. 3D, Suppl. Figs. S5-S6). To compare responses in mice across multiple independent experiments, a composite immune score was created by adding the percent of CD8+ TIL expressing GzmB to the percent of those expressing IFNγ. Mice on WD or WDΔF had relatively low immune scores after ICB compared to mice on NC (Fig. 3E). The data indicate that immune responses elicited during ICB were not improved in the absence of fructose despite better control of tumor growth. To confirm this unanticipated result, we mapped gene expression in TILs from mice on NC, WD, and WDΔF that had received ICB. Single-cell RNA sequencing of CD45+ TILs revealed few diet-related differences between recipients. However, Prf1 (Perforin) and Gzmb expression by CD8+ TILs (Fig. 3F) was significantly higher in mice on NC compared to identically treated obese mice regardless of fructose (Fig. 3G). Together with the protein expression data, these results demonstrated that improved immunotherapy outcomes upon the removal of fructose were not due to a boost in CD8+ TIL effector activity.
Because dietary fructose had no measurable effect on immune function, we focused on the tumor. Anecdotally, melanoma tumors from obese mice on either the WD or WDΔF were visibly darker in color compared to tumors from mice on NC (Fig. 4A). Although this difference presumably played no role in the response to immunotherapy, it highlights how changes in nutrition can alter tumor cell phenotype. We predicted that in order to affect therapeutic outcomes, fructose might also reduce the susceptibility of tumor cells to immune-mediated effector mechanisms.
Figure 4. Fructose induces HO-1 expression and protects melanoma tumors from CTLs.
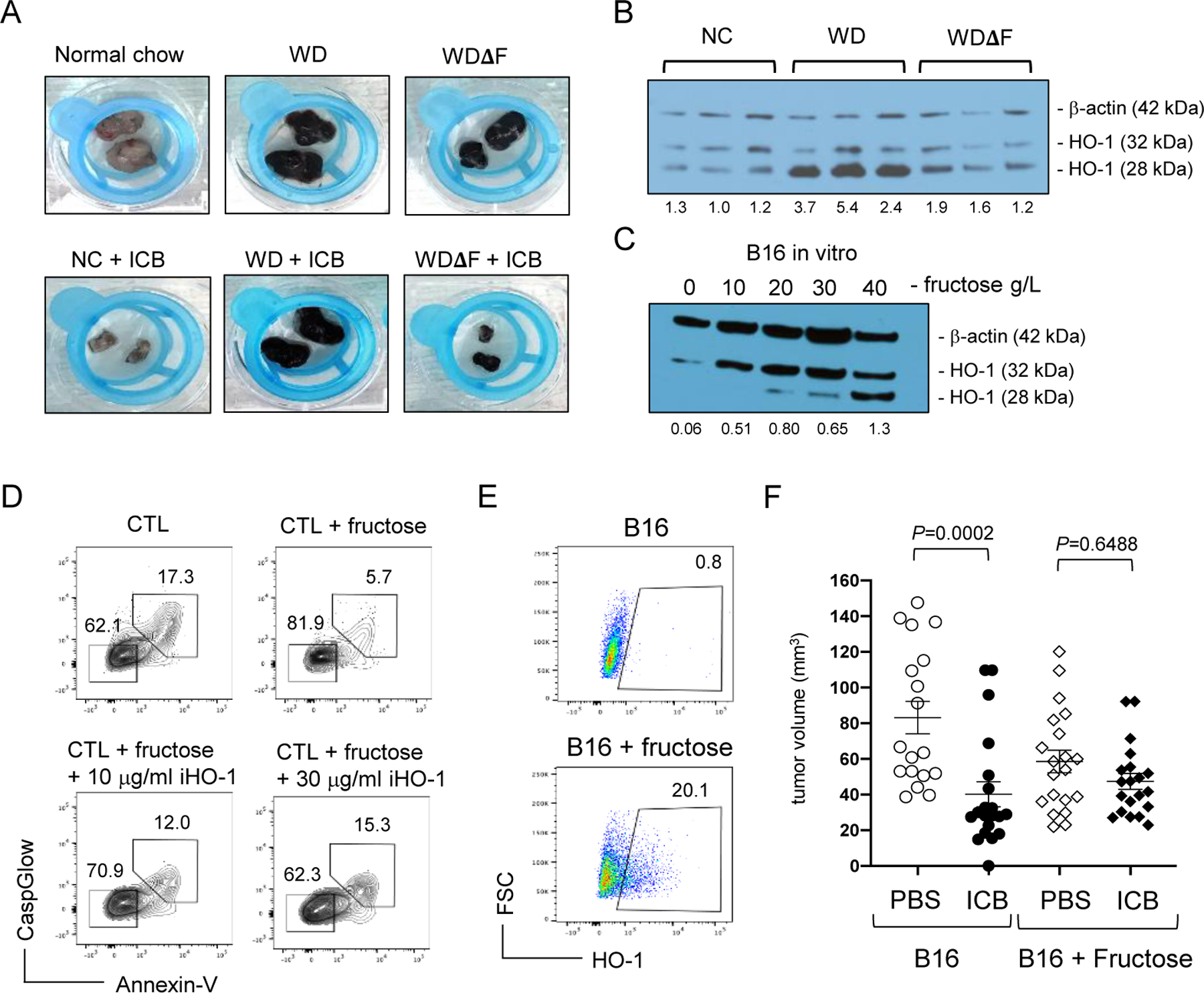
(A) Representative tumors from mice on the indicated diets with and without immunotherapy. (B) HO-1 and β-actin protein expression in B16 tumors from individual mice on NC, WD, and WDΔF, and in (C) B16 tumor cells cultured with fructose for 3 days at various concentrations. Densitometry results are shown below each band indicating the ratio of total HO-1 to β-actin. Data are representative of 3 independent experiments. (D) B16 cells were treated with 20g/L fructose in the presence or absence of OB24 and exposed to activated CTLs. Immune-mediated apoptosis was measured in B16 tumor cells by flow cytometric detection of CaspGlow and Annexin V. (E) B16 melanoma cells were cultured with 0g/L or 20g/L fructose for 3 days and assessed for HO-1 expression by flow cytometry. Inset numbers representing the percent of all B16 cells within the inscribed region. (F) Fructose-treated B16 cells were injected subcutaneously in normal male B6 mice and treated with ICB or PBS on days 1 and 3. Tumor volumes pooled from 3 separate experiments (9–10 mice/treatment group) measured at day 8 are displayed graphically (right) with each point representing data from an individual tumor. Error bars represent SEM and exact P values were calculated by 2-way ANOVA for the bracketed groups.
Dietary fructose has been reported to induce expression of the cytoprotective molecule, heme oxygenase-1 (HO-1) in hepatocytes and adipocytes (36–38), whereas fasting conversely reduces HO-1 expression in mouse tumors (39). HO-1 is induced under oxidative stress and catalyzes the breakdown of heme into free iron, biliverdin, and carbon monoxide, which have antioxidant, anti-apoptotic, and anti-inflammatory properties (40). HO-1 expression has also been identified as a poor prognostic indicator for a variety of human cancers and is associated with therapeutic resistance (41–43). To determine if high dietary fructose was associated with HO-1 expression in B16 melanoma, whole tumor tissue was analyzed from mice on NC, WD, and WDΔF at day 16, and HO-1 expression assessed by Western blot. Both the 28kDa and 32kDa forms of HO-1 were detected in all tumors but expression of the 28kDa form was increased in tumors from mice on the WD compared to NC or WDΔF (Fig. 4B). This finding has biological significance, as the truncated 28kDa form of HO-1 translocates to the nucleus and regulates genes involved in metabolism and cellular adaptation to oxidative stress (44,45). The 28kDa form of HO-1 was also more prominent in MC38 tumors from mice on the WD but was nearly absent in those from NC and WDΔF (Suppl. Fig. S5). To determine if fructose alone could induce HO-1 expression in melanoma cells directly, we cultured B16 tumor cells with varying concentrations of fructose. After 3 days, increased expression of both the 28kDa and 32kDa forms of HO-1 protein were observed (Fig. 4C). Fructose had minimal influence on tumor cell proliferation either in vitro or when grown in mice on the different diets, and these cells also showed little variation in surface expression of Fas (Suppl. Fig. S7), suggesting such processes are not major contributors to differential therapeutic outcomes among these recipient groups.
To directly test if fructose-induced HO-1 expression was cytoprotective, B16 tumor cells were pretreated with 20 g/L fructose and then cultured with cytolytic melanoma-reactive CD8+ Pmel cytotoxic T lymphocytes (CTLs) preactivated for 48 hours with anti-CD3/CD28. After 3 days, apoptotic B16 tumor cells were identified by detection of both active caspase and Annexin V binding. In the absence of fructose, approximately 17% of tumor cells were double-positive for these apoptosis markers (Fig. 4D), whereas pretreatment with fructose was at least partially protective and reduced the frequency of apoptotic cells to less than 6%. This effect was also evident in the viable double-negative cell population, which exceeded 81% of fructose-treated cells compared to only 62% in the absence of fructose. This resistance to apoptosis was dependent on HO-1, as treatment with the small-molecule inhibitor OB24 (iHO-1) restored tumor cell susceptibility to immune-mediated killing in a dose-dependent manner (Fig. 4D). Conversely, induction of HO-1 expression via treatment with 10μM Hemin protected B16 cells from CTL-mediated killing (Suppl. Fig. S8). The protective effect of fructose was recapitulated in vivo where B16 tumors pretreated with fructose displayed higher HO-1 expression (Fig. 4E) and demonstrated modest resistant to immune-mediated killing following recipient treatment with ICB (Fig. 4F). These data provide evidence that fructose induced cytoprotection through increased HO-1 expression, allowing melanoma tumor cells to resist immune-mediated killing elicited during ICB. These findings hold particular significance for obese individuals and others where immune responses may be diminished (46).
Extrapolation of these results predicted that targeting HO-1 during immunotherapy could weaken tumors and improve clinical outcomes. To test this intriguing possibility, B16 melanomas were established in mice on the high-fructose WD. Mice were treated with ICB and simultaneous inhibition of HO-1 (OB24) systemically. As before, mice on WD were resistant to immunotherapy, experiencing the same rate of tumor growth as control mice treated with PBS (Fig. 5A). Likewise, treatment with OB24 was ineffective as a monotherapy, but the combination of ICB and HO-1 inhibition significantly improved therapeutic outcomes (Fig. 5A–B). We repeated these experiments in obese mice bearing MC38 tumors and observed a similar trend but the impact of the combination therapy was far less pronounced (Suppl. Fig. S9). Specifically, whereas the combination treatment produced significantly smaller tumors than control mice (P=0.0173), they were not statistically different from those treated with ICB alone (P=0.1540). Our previous in vivo studies using MC38 suggested they are less responsive to ICB compared to B16 melanomas, and this is especially true in obese recipients (Suppl. Fig. S1, S5). This weak therapeutic effect indicated the likely presence of HO-1-independent resistance mechanisms operative in MC38 tumors. An important limitation here is that we currently have little insight into the in vivo distribution, efficacy, and specificity of this small-molecule inhibitor. Our data suggests that OB24 did not impact CD8+ TIL frequency or function (Suppl. Fig. S9), but it remains possible that the HO-1 inhibitor acts on other cells within the tumor microenvironment that could influence outcomes. Regardless, the clear therapeutic benefit in melanoma supports the predicted scenario in which disruption of fructose-induced HO-1 cytoprotection within tumor cells makes them more susceptible to antitumor immunity elicited in obese mice receiving ICB.
Figure 5. Inhibition of HO-1 overcomes immunotherapeutic resistance in melanoma.
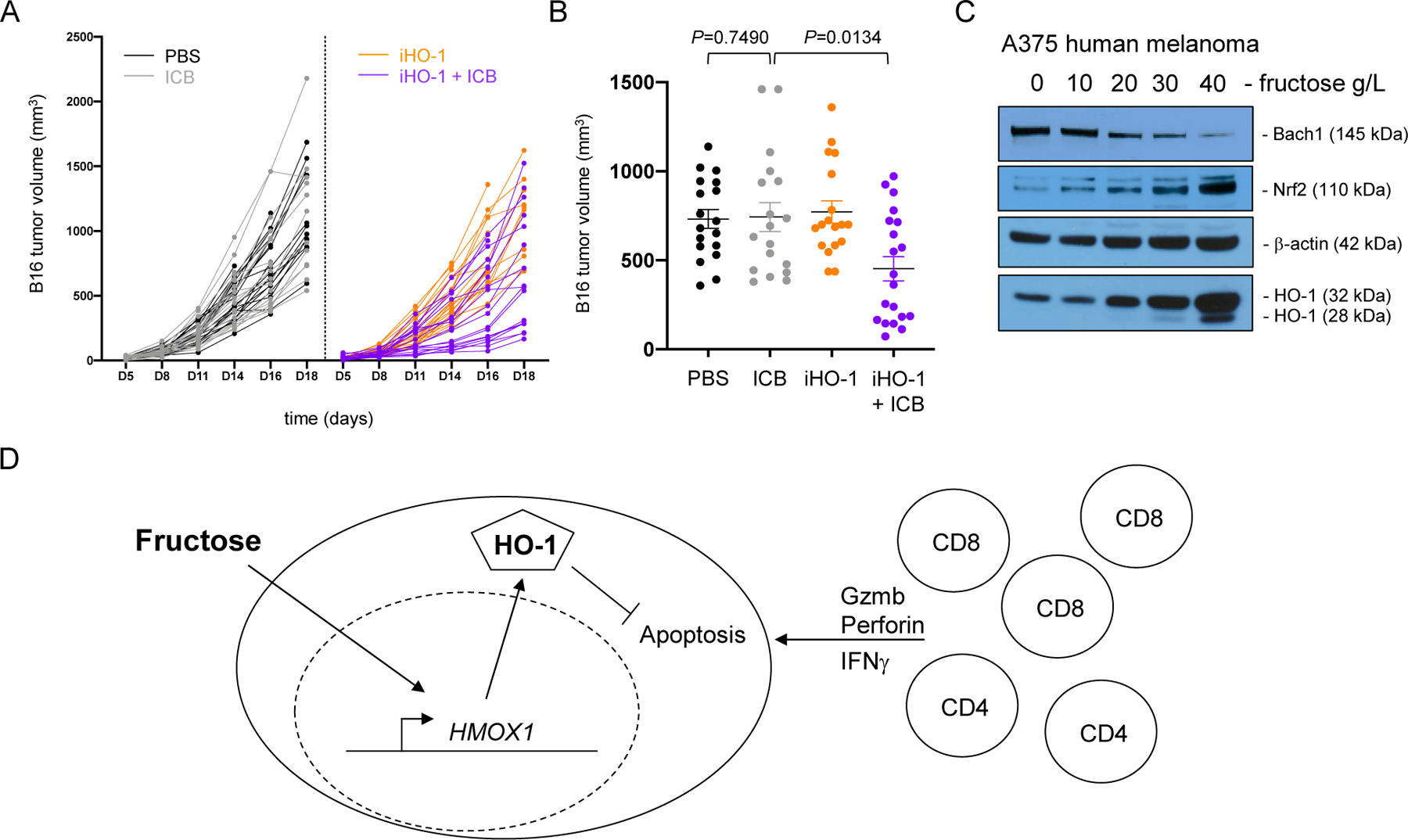
Subcutaneous B16 melanoma tumors were established in male B6 mice on western diet and treated as indicated (9–10 mice/treatment group). (A) Tumor volume (mm3) over time. (B) Day 16 tumor volumes for all four treatment groups. Error bars represent SEM and exact P values were calculated by two-way ANOVA for the bracketed groups with each point representing data from an individual tumor. Data are pooled from 2 independent experiments. (C) A375 human melanoma cells were cultured with the indicated concentration of fructose for 3 days. BACH1, NRF2, β-actin, and HO-1 protein expression was assessed by Western blot. Data are representative of 3 independent experiments. (D) Graphical abstract of working model depicting fructose inducing tumor cytoprotection and resistance to immune-mediated killing.
Our study revealed dietary fructose and subsequent HO-1 expression as part of an immune evasion mechanism in cancer. Whether such mechanisms are recapitulated in human melanoma is unknown. Expression of the HO-1 gene (HMOX1) is regulated by the balance between two transcription factors, Bach1 and Nrf2 (40). Under conditions of low oxidative stress, Bach1 protein is stable in both the cytoplasm and nucleus where it binds to the HMOX1 promotor and inhibits transcription. Oxidative stress leads to Bach1 degradation and simultaneous nuclear translocation of Nrf2 where it displaces Bach1 from the promotor and drives HMOX1 gene transcription (44). To determine if fructose induced this pathway in human tumors, we assessed expression of these molecules in the A375 human melanoma cell line. Here, fructose induced a dose-dependent loss of Bach1 in concert with the induction of Nrf2 and HO-1 (Fig. 5C), suggesting the cytoprotective influence of fructose is likely intact in human cancer.
Discussion
The significance of the HO-1 pathway, including Bach1 and Nrf2, in cancer has previously been recognized (41,47,48). Our study showed how this tumor-protective pathway could be engaged by dietary fructose, resulting in evasion of both CD4+ and CD8+ T cell effector mechanisms. This led us to the propose working model in Figure 5D, yet questions still remain. For example, what receptors are engaged by fructose to be absorbed by tumor cells, and what are the earliest signal transduction events that lead to HO-1 expression? The hexose transporter GLUT5 has high affinity for fructose and is the primary means of fructose uptake by intestinal epithelial cells (14). Expression of GLUT5 has been reported for several cancers to varying degrees including breast, lung, and colon and is often associated with more aggressive disease (34,49,50). We detected only low GLUT5 expression on B16 tumors in mice on western diet, perhaps indicating the involvement of other lower affinity transporters such as GLUT2, TXNIP or a combination (50,51). Regardless, although fructose uptake has been linked to altered tumor metabolism and disease progression, the signaling events necessary to promote tumor immune evasion mechanisms have not been described. We provide evidence that excessive dietary fructose induces cytoprotective HO-1 protein expression, endowing melanoma tumor cells with the ability to resist immune-mediated killing.
Our study fell short of identifying a mechanism behind the dampened immune responses observed in obese mice during immunotherapy, which was independent of fructose. Specifically, we observed reduced effector function by both CD4+ and CD8+ TILs in obese mice receiving immunotherapy regardless of dietary fructose. Immune defects associated with obesity have been described by others, but the variety of proposed mechanisms and conflicting data have unfortunately not produced a consensus (10,17,23,52,53). Two publications identify high levels of leptin in obese mice as a driver of immune suppression, with one study implicating leptin-induced PD-1 expression on T cells as the mechanism of action (10,17). We thoroughly explored this possibility and documented a clear association between serum leptin and host body mass. However, analysis of PD-1 expression on both peripheral and tumor-infiltrating T cells provided no evidence that leptin impacted surface protein expression. Moreover, successful neutralization of serum leptin did not improve T-cell immune responses or immunotherapy outcomes in obese mice. Clinical trial results have also not provided much clarity on this issue. Despite the well-documented relationship between serum leptin and human BMI (54), immunotherapy outcomes in cancer patients have not consistently aligned with this metric of obesity (6–9,46). Thus the question of how obesity influences cancer immunotherapy remains unanswered. Nevertheless, what our in vivo experiments demonstrated is that even when T-cell effector function is dampened, due to obesity or otherwise, such responses may still be sufficient to oppose tumor progression when immunotherapy is combined with inhibition of the HO-1 pathway.
In obese mice on high-fructose diet, immunotherapy alone was ineffective against tumors with high HO-1 expression but was effective at slowing progression of resistant melanomas when combined with HO-1 inhibition. This provides evidence that the cellular benefit of high HO-1 expression is protection and the ability to resist immune-mediated killing mechanisms. However, this same therapeutic strategy was not effective when applied to MC38 colon carcinomas, highlighting the need for improved understanding of HO-1 inhibition as a complement to immunotherapy. For example, systemic delivery of such an inhibitor likely has effects on other cell types including those within the tumor microenvironment. Whether those other cells contribute to differential therapeutic outcomes during HO-1 inhibition has not been investigated. There are also opportunities to improve and even customize such inhibitors. We chose OB24 as a proven and commercially available small-molecule inhibitor that worked well against B16 melanoma. However, several other small-molecules with similar structure to OB24 have been described and are predicted to inhibit HO-1 activity with variable efficacy (55), but these have not been tested in vivo or in combination with cancer immunotherapy. Our results provide clear justification for pursuing such studies and for subsequent development of optimized HO-1 inhibitors for therapeutic applications.
We have identified a relationship between fructose consumption and initiation of an anti-apoptotic cellular pathway involving HO-1 expression. This discovery raises important clinical questions with relevance to human cancer. Is the cytoprotective HO-1 pathway induced in normal or perhaps pre-cancerous cells throughout the body, potentially increasing the risk of malignancy? Is the HO-1 pathway a rational therapeutic target for human cancer? A myriad of clinical trials are investigating interventions that activate HO-1 to prevent tissue damage such as ischemia-reperfusion injury. However, strategies to inhibit HO-1 clinically are not as advanced and have been hampered by a lack of pharmaceutical grade reagents that are sufficiently specific for HO-1 and also potentially safe for use in people (56). Alternatively, targeting other pathway components such as Nrf2 is showing promise in animal tumor models (47,57). Our findings suggests that dietary interventions may also be successful in modulating the HO-1 pathway with the potential to improve immunotherapy outcomes. This is supported by at least one other report showing that fasting reduces HO-1 expression in murine tumors (39). The concept of targeting HO-1 in human cancer has only recently been proposed and more research is required to transform any of these approaches into realistic treatment options for cancer patients.
Supplementary Material
Acknowledgments
The authors thank Sherri Koehm and Joy Eslick (Saint Louis University) for technical assistance with flow cytometry and cell sorting. The authors also appreciate the financial support provided by grants from the Alvin J. Siteman Comprehensive Cancer Center (P30 CA091842) and from the National Institutes of Health National Cancer Institute (R01 CA238705) that supported this work.
Financial Support
Research reported in this manuscript was supported by grants from the Alvin J. Siteman Comprehensive Cancer Center (P30 CA091842) and from the National Institutes of Health National Cancer Institute (R01 CA238705) to R.M.T.
Footnotes
Conflict of Interest Statement
The authors declare no potential conflicts of interest.
References
- 1.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD, et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N Engl J Med 2019;381:1535–46 [DOI] [PubMed] [Google Scholar]
- 2.Kanneganti TD, Dixit VD. Immunological complications of obesity. Nat Immunol 2012;13:707–12 [DOI] [PubMed] [Google Scholar]
- 3.Tao W, Lagergren J. Clinical management of obese patients with cancer. Nat Rev Clin Oncol 2013;10:519–33 [DOI] [PubMed] [Google Scholar]
- 4.Sergentanis TN, Antoniadis AG, Gogas HJ, Antonopoulos CN, Adami HO, Ekbom A, et al. Obesity and risk of malignant melanoma: a meta-analysis of cohort and case-control studies. Eur J Cancer 2013;49:642–57 [DOI] [PubMed] [Google Scholar]
- 5.Green WD, Beck MA. Obesity altered T cell metabolism and the response to infection. Curr Opin Immunol 2017;46:1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Donnelly D, Bajaj S, Yu J, Hsu M, Balar A, Pavlick A, et al. The complex relationship between body mass index and response to immune checkpoint inhibition in metastatic melanoma patients. J Immunother Cancer 2019;7:222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferro M, Vartolomei MD, Russo GI, Cantiello F, Farhan ARA, Terracciano D, et al. An increased body mass index is associated with a worse prognosis in patients administered BCG immunotherapy for T1 bladder cancer. World J Urol 2018 [DOI] [PubMed] [Google Scholar]
- 8.Incio J, Ligibel JA, McManus DT, Suboj P, Jung K, Kawaguchi K, et al. Obesity promotes resistance to anti-VEGF therapy in breast cancer by up-regulating IL-6 and potentially FGF-2. Sci Transl Med 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McQuade JL, Daniel CR, Hess KR, Mak C, Wang DY, Rai RR, et al. Association of body-mass index and outcomes in patients with metastatic melanoma treated with targeted therapy, immunotherapy, or chemotherapy: a retrospective, multicohort analysis. Lancet Oncol 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Z, Aguilar EG, Luna JI, Dunai C, Khuat LT, Le CT, et al. Paradoxical effects of obesity on T cell function during tumor progression and PD-1 checkpoint blockade. Nat Med 2019;25:141–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elliott SS, Keim NL, Stern JS, Teff K, Havel PJ. Fructose, weight gain, and the insulin resistance syndrome. Am J Clin Nutr 2002;76:911–22 [DOI] [PubMed] [Google Scholar]
- 12.Makarem N, Bandera EV, Nicholson JM, Parekh N. Consumption of Sugars, Sugary Foods, and Sugary Beverages in Relation to Cancer Risk: A Systematic Review of Longitudinal Studies. Annu Rev Nutr 2018;38:17–39 [DOI] [PubMed] [Google Scholar]
- 13.Stanhope KL, Schwarz JM, Keim NL, Griffen SC, Bremer AA, Graham JL, et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J Clin Invest 2009;119:1322–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hannou SA, Haslam DE, McKeown NM, Herman MA. Fructose metabolism and metabolic disease. J Clin Invest 2018;128:545–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu H, Huang D, McArthur DL, Boros LG, Nissen N, Heaney AP. Fructose induces transketolase flux to promote pancreatic cancer growth. Cancer Res 2010;70:6368–76 [DOI] [PubMed] [Google Scholar]
- 16.Goncalves MD, Lu C, Tutnauer J, Hartman TE, Hwang SK, Murphy CJ, et al. High-fructose corn syrup enhances intestinal tumor growth in mice. Science 2019;363:1345–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murphy KA, James BR, Sjaastad FV, Kucaba TA, Kim H, Brincks EL, et al. Cutting Edge: Elevated Leptin during Diet-Induced Obesity Reduces the Efficacy of Tumor Immunotherapy. J Immunol 2018;201:1837–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015;350:1084–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang YW, Chen MK, Yang BY, Huang XJ, Zhang XR, He LQ, et al. Use of 16S rRNA Gene-Targeted Group-Specific Primers for Real-Time PCR Analysis of Predominant Bacteria in Mouse Feces. Appl Environ Microbiol 2015;81:6749–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 2018;36:411–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berrien-Elliott MM, Jackson SR, Meyer JM, Rouskey CJ, Nguyen TL, Yagita H, et al. Durable adoptive immunotherapy for leukemia produced by manipulation of multiple regulatory pathways of CD8+ T-cell tolerance. Cancer Res 2013;73:605–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuehm LM, Wolf K, Zahour J, DiPaolo RJ, Teague RM. Checkpoint blockade immunotherapy enhances the frequency and effector function of murine tumor-infiltrating T cells but does not alter TCRbeta diversity. Cancer Immunol Immunother 2019;68:1095–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.James BR, Tomanek-Chalkley A, Askeland EJ, Kucaba T, Griffith TS, Norian LA. Diet-induced obesity alters dendritic cell function in the presence and absence of tumor growth. J Immunol 2012;189:1311–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gangadhar TC, Vonderheide RH. Mitigating the toxic effects of anticancer immunotherapy. Nat Rev Clin Oncol 2014;11:91–9 [DOI] [PubMed] [Google Scholar]
- 25.Mirsoian A, Bouchlaka MN, Sckisel GD, Chen M, Pai CC, Maverakis E, et al. Adiposity induces lethal cytokine storm after systemic administration of stimulatory immunotherapy regimens in aged mice. J Exp Med 2014;211:2373–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dayakar A, Chandrasekaran S, Veronica J, Bharadwaja V, Maurya R. Leptin regulates Granzyme-A, PD-1 and CTLA-4 expression in T cell to control visceral leishmaniasis in BALB/c Mice. Sci Rep 2017;7:14664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lord GM, Matarese G, Howard JK, Baker RJ, Bloom SR, Lechler RI. Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature 1998;394:897–901 [DOI] [PubMed] [Google Scholar]
- 28.Naylor C, Petri WA Jr. Leptin Regulation of Immune Responses. Trends Mol Med 2016;22:88–98 [DOI] [PubMed] [Google Scholar]
- 29.Saucillo DC, Gerriets VA, Sheng J, Rathmell JC, Maciver NJ. Leptin metabolically licenses T cells for activation to link nutrition and immunity. J Immunol 2014;192:136–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vetizou M, Pitt JM, Daillere R, Lepage P, Waldschmitt N, Flament C, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015;350:1079–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med 2015;372:311–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Motzer RJ, Tannir NM, McDermott DF, Aren Frontera O, Melichar B, Choueiri TK, et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N Engl J Med 2018;378:1277–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tawbi HA, Forsyth PA, Algazi A, Hamid O, Hodi FS, Moschos SJ, et al. Combined Nivolumab and Ipilimumab in Melanoma Metastatic to the Brain. N Engl J Med 2018;379:722–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fan X, Liu H, Liu M, Wang Y, Qiu L, Cui Y. Increased utilization of fructose has a positive effect on the development of breast cancer. PeerJ 2017;5:e3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prado CM, Lieffers JR, McCargar LJ, Reiman T, Sawyer MB, Martin L, et al. Prevalence and clinical implications of sarcopenic obesity in patients with solid tumours of the respiratory and gastrointestinal tracts: a population-based study. Lancet Oncol 2008;9:629–35 [DOI] [PubMed] [Google Scholar]
- 36.Bekyarova G, Tzaneva M, Bratoeva K, Kotzev I, Radanova M. Heme-Oxygenase-1 Upregulated by S-Adenosylmethionine. Potential Protection against Non-Alcoholic Fatty Liver Induced by High Fructose Diet. Farmacia 2017;65:262–7 [Google Scholar]
- 37.Khitan Z, Harsh M, Sodhi K, Shapiro JI, Abraham NG. HO-1 Upregulation Attenuates Adipocyte Dysfunction, Obesity, and Isoprostane Levels in Mice Fed High Fructose Diets. J Nutr Metab 2014;2014:980547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sodhi K, Puri N, Favero G, Stevens S, Meadows C, Abraham NG, et al. Fructose Mediated Non-Alcoholic Fatty Liver Is Attenuated by HO-1-SIRT1 Module in Murine Hepatocytes and Mice Fed a High Fructose Diet. PLoS One 2015;10:e0128648. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 39.Di Biase S, Lee C, Brandhorst S, Manes B, Buono R, Cheng CW, et al. Fasting-Mimicking Diet Reduces HO-1 to Promote T Cell-Mediated Tumor Cytotoxicity. Cancer Cell 2016;30:136–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gozzelino R, Jeney V, Soares MP. Mechanisms of cell protection by heme oxygenase-1. Annu Rev Pharmacol Toxicol 2010;50:323–54 [DOI] [PubMed] [Google Scholar]
- 41.Alaoui-Jamali MA, Bismar TA, Gupta A, Szarek WA, Su J, Song W, et al. A novel experimental heme oxygenase-1-targeted therapy for hormone-refractory prostate cancer. Cancer Res 2009;69:8017–24 [DOI] [PubMed] [Google Scholar]
- 42.Berberat PO, Dambrauskas Z, Gulbinas A, Giese T, Giese N, Kunzli B, et al. Inhibition of heme oxygenase-1 increases responsiveness of pancreatic cancer cells to anticancer treatment. Clin Cancer Res 2005;11:3790–8 [DOI] [PubMed] [Google Scholar]
- 43.Fest S, Soldati R, Christiansen NM, Zenclussen ML, Kilz J, Berger E, et al. Targeting of heme oxygenase-1 as a novel immune regulator of neuroblastoma. Int J Cancer 2016;138:2030–42 [DOI] [PubMed] [Google Scholar]
- 44.Biswas C, Shah N, Muthu M, La P, Fernando AP, Sengupta S, et al. Nuclear heme oxygenase-1 (HO-1) modulates subcellular distribution and activation of Nrf2, impacting metabolic and anti-oxidant defenses. J Biol Chem 2014;289:26882–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin Q, Weis S, Yang G, Weng YH, Helston R, Rish K, et al. Heme oxygenase-1 protein localizes to the nucleus and activates transcription factors important in oxidative stress. J Biol Chem 2007;282:20621–33 [DOI] [PubMed] [Google Scholar]
- 46.Turbitt WJ, Buchta Rosean C, Weber KS, Norian LA. Obesity and CD8 T cell metabolism: Implications for anti-tumor immunity and cancer immunotherapy outcomes. Immunol Rev 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Galan-Cobo A, Sitthideatphaiboon P, Qu X, Poteete A, Pisegna MA, Tong P, et al. LKB1 and KEAP1/NRF2 Pathways Cooperatively Promote Metabolic Reprogramming with Enhanced Glutamine Dependence in KRAS-Mutant Lung Adenocarcinoma. Cancer Res 2019;79:3251–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee J, Yesilkanal AE, Wynne JP, Frankenberger C, Liu J, Yan J, et al. Effective breast cancer combination therapy targeting BACH1 and mitochondrial metabolism. Nature 2019;568:254–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bu P, Chen KY, Xiang K, Johnson C, Crown SB, Rakhilin N, et al. Aldolase B-Mediated Fructose Metabolism Drives Metabolic Reprogramming of Colon Cancer Liver Metastasis. Cell Metab 2018;27:1249–62 e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weng Y, Zhu J, Chen Z, Fu J, Zhang F. Fructose fuels lung adenocarcinoma through GLUT5. Cell Death Dis 2018;9:557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dotimas JR, Lee AW, Schmider AB, Carroll SH, Shah A, Bilen J, et al. Diabetes regulates fructose absorption through thioredoxin-interacting protein. Elife 2016;5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Michelet X, Dyck L, Hogan A, Loftus RM, Duquette D, Wei K, et al. Metabolic reprogramming of natural killer cells in obesity limits antitumor responses. Nat Immunol 2018;19:1330–40 [DOI] [PubMed] [Google Scholar]
- 53.Shirakawa K, Yan X, Shinmura K, Endo J, Kataoka M, Katsumata Y, et al. Obesity accelerates T cell senescence in murine visceral adipose tissue. J Clin Invest 2016;126:4626–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med 1996;334:292–5 [DOI] [PubMed] [Google Scholar]
- 55.Rahman MN, Vukomanovic D, Vlahakis JZ, Szarek WA, Nakatsu K, Jia Z. Structural Insights into Azole-based Inhibitors of Heme Oxygenase-1: Development of Selective Compounds for Therapeutic Applications. Curr Med Chem 2018;25:5803–21 [DOI] [PubMed] [Google Scholar]
- 56.Podkalicka P, Mucha O, Jozkowicz A, Dulak J, Loboda A. Heme oxygenase inhibition in cancers: possible tools and targets. Contemp Oncol (Pozn) 2018;22:23–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Evans JP, Winiarski BK, Sutton PA, Jones RP, Ressel L, Duckworth CA, et al. The Nrf2 inhibitor brusatol is a potent antitumour agent in an orthotopic mouse model of colorectal cancer. Oncotarget 2018;9:27104–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.