

Summary
Nervous system injury and disease have broad effects on the functional connectivity of the nervous system, but how injury signals are spread across neural circuits remains unclear. We explored how axotomy changes the physiology of severed axons and adjacent uninjured “bystander” neurons in a simple in vivo nerve preparation. Within hours after injury, we observed suppression of axon transport in all axons, whether injured or not, and decreased mechano- and chemosensory signal transduction in uninjured bystander neurons. Unexpectedly, we found the axon death molecule dSarm, but not its NAD+ hydrolase activity, was required cell-autonomously for these early changes in neuronal cell biology in bystander neurons, as were the voltage-gated calcium channel Cacophony (Cac) and the MAP kinase signaling cascade. Bystander neurons functionally recovered at later time points, while severed axons degenerated via dSarm/Axundead signaling, and independently of Cac/MAP kinase. Interestingly, suppression of bystander neuron function required Draper/MEGF10 signaling in glia, indicating glial cells spread injury signals and actively suppress bystander neuron function. Our work identifies a new role for dSarm and glia in suppression of bystander neuron function after injury, and defines two genetically and temporally separable phases of dSarm signaling in the injured nervous system.
In Brief
Hsu et al. demonstrate that uninjured bystander neurons temporarily suppress their physiology in response to adjacent severed axons via cell-autonomous Cac/dSarm/MAPK signaling, independent of dSarm NADase activity. Injury signals are spread to bystander neurons via Draper-mediated signaling in glia. They propose a two-phase model for dSarm signaling in injured nerves.
Graphical Abstract
Introduction
Nervous system injury or neurodegenerative disease can lead to profound alterations in neural circuit function. The precise cellular basis is poorly defined in any context, but disruption of circuit signaling is generally thought to occur as a result of a loss of physical connectivity between damaged neurons. Indeed, axon and synapse degeneration are among the best correlates of functional loss in patients with a variety of brain injuries or neurological diseases (Burke and O’Malley, 2013; Hill et al., 2016; Kasahara et al., 2010; Kneynsberg et al., 2017) But whether, and the extent to which, an injured or diseased neuron might also alter the functional properties of neighboring healthy “bystander” neurons (i.e. those not damaged or expressing disease-associated molecules) is an important and open question. If the physiology of bystander neurons is radically altered by their damaged neighbors, this would force us to reconsider the simple loss-of-physical-connectivity model as the appropriate explanation for functional loss in neural circuits after trauma.
It is well documented that bystander neurons can change their physiology in response to their neighbors being injured (Meyer and Ringkamp, 2008). For instance, mouse L5 spinal nerve transection results in the degeneration of distal L5 afferents in sciatic nerve alongside intact L4 C fiber afferents. Within one day after L5 lesion, L4 C fibers develop spontaneous activity that lasts for at least a week and appears to mediate injury-induced pain and hyperalgesia behaviors (Wu et al., 2001). Bystander effects have also been observed in the central nervous system (CNS). In a mouse model of mild TBI, one day after injury pyramidal neurons with severed axons and intact bystander neurons both exhibited injury-induced changes in action potential firing and afterhyperpolarization. Injured neurons failed to recover while bystander neurons ultimately exhibited a return to normal firing properties (Greer et al., 2012). How injured neurons or surrounding glia signal to bystander neurons, or how bystander neurons receive this signal, is not known, but the similar electrophysiological changes observed in axotomized and intact dorsal root ganglion neurons have been proposed to be associated with Wallerian degeneration (Chao et al., 2003).
Recent work has begun to illuminate the mechanisms by which damaged axons autonomously drive their own degeneration during Wallerian degeneration. A forward genetic screen in Drosophila identified the sterile α/Armadillo/Toll-interleukin receptor homology domain (dSarm) molecule as essential for axon auto-destruction—loss of dSarm completely blocked Wallerian degeneration (Osterloh et al., 2012). All known dSarm pro-degenerative function requires the BTB and BACK domain molecule Axundead (Axed), another powerful regulator of axon degeneration (Neukomm et al., 2017). dSarm function in axon degeneration after injury is conserved in mouse: Sarm1−/− mutants block Wallerian degeneration (Gerdts et al., 2013; Osterloh et al., 2012), and loss of Sarml also suppresses axon degeneration in mouse models of TBI (Henninger et al., 2016) and peripheral neuropathy (Geisler et al., 2019; Turkiew et al., 2017). Sarm1 inhibition is thus an exciting potential approach for blocking axon loss and neuroinflammation in human disease (Wang et al., 2018).
dSarm/Sarm1 has been studied primarily in the nervous system as a positive regulator of axonal degeneration. In mammals, axotomy leads to the depletion of the labile NAD+ biosynthetic enzyme Nmnat2 (Gilley and Coleman, 2010) and a decrease in NAD+ in severed axons (Wang et al., 2005). Nmnat2 loss somehow activates Sarm1 (Gilley et al., 2015), which is proposed to lead to further NAD+ depletion and metabolic catastrophe in the severed axon (Gerdts et al., 2015) through a Sarm1 intrinsic NAD+ hydrolase activity (Essuman et al., 2017). The Sarm1 NAD+ hydrolase activity appears to be activated directly by the NAD+ precursor, NMN, presumably through allosteric conformational changes in Sarm1 upon NMN binding (Bratkowski et al., 2020; Di Stefano et al., 2014; Zhao et al., 2019). This NAD+ depletion model has been proposed as the primary mechanism by which Sarm1 drives axon loss, and to explain the mechanistic basis of protection by several other neuroprotective molecules (Gerdts et al., 2016). For instance, the slow Wallerian degeneration molecule (Wlds), which includes the highly stable NAD+ biosynthetic enzyme Nmnat1, is thought to protect axons by substituting for the labile Nmnat2 molecule, thereby reducing NMN levels and avoiding NAD+ depletion (Gilley and Coleman, 2010; Lunn et al., 1989; Wang et al., 2005). Similarly, the protective effects of loss of the E3 ubiquitin ligase Highwire/Phr1 is thought to result from blockade of its direct role in degrading Nmnat2, such that in hiw/phr1 mutants Nmnat2 is stabilized and continues to maintain NAD+ levels (Babetto et al., 2013; Xiong et al., 2012).
Elegant genetic studies in C. elegans demonstrated that TIR-1 (the worm homolog of dSarm/Sarm1) is part of a signaling cascade downstream of the voltage gated calcium channel UNC −36 and CamK-II, and signals via the MAP kinase (MAPK) signaling cascade (Chuang and Bargmann, 2005). Based on this work, MAPK signaling was examined for roles in Wallerian degeneration, but met with mixed results. Changes in MAPK signaling (i.e. phosphorylation of MAPK pathway members) were found in axons within 15-30 minutes after axotomy, were Sarm1-dependent and suppressed by Nmnat overexpression (Yang et al., 2015), and partial suppression of axon degeneration was observed after simultaneous blockade of multiple MAPK components (Walker et al., 2017; Yang et al., 2015). But how MAPK signaling modulates axon degeneration, particularly in the context of Sarm1 signaling, remains controversial as one study proposed MAPK signals downstream of Sarm1 (Yang et al., 2015), while another argued Sarm1 was upstream of MAPK signaling (Walker et al., 2017), and the neuroprotective phenotypes resulting from MAPK blockade do not approach levels afforded by loss of Sarm1 in vivo.
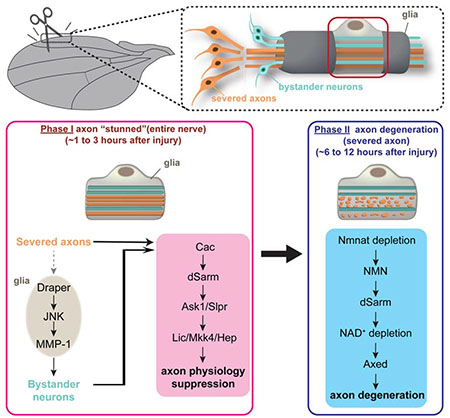
In this study, we used a partial nerve injury model to examine early changes in the physiology of severed axons and neighboring uninjured bystander neurons. We show that axotomy of even a small subset of neurons leads to inhibition of cargo transport in all axons within the nerve, and suppression of sensory signal transduction in bystander neurons. Surprisingly, this early blockade of axon transport and sensory signal transduction required dSarm, in both severed and uninjured bystander neurons, where it signaled via the conserved UNC-36/MAPK signaling pathway. Early suppression of axon transport and bystander neuron function did not require dSarm NAD+ hydrolase function or Axed, was not modulated by NMN, nor was it induced by depletion of dNmnat. This suggests it is mechanistically different from later events in axon death where dSarm drives axon degeneration with Axed. Intriguingly, we found that this early spreading of injury signals to bystander neurons required the Draper receptor in surrounding glia, indicating that glial cells actively signal to inhibit the function of bystander neurons in vivo. Our work identifies new roles for dSarm and glia in modifying neurophysiology early after injury, assigns the NAD+ hydrolase function exclusively to later axon degenerative events, and reveals a new role for UNC-36/MAPK signaling in promoting these dSarm-dependent changes early after an injury has occurred in the nervous system. We propose that two temporally and genetically separable phases of dSarm signaling exist that mediate these distinct injury-induced changes in neurophysiology and axon degeneration.
Results
Partial nerve injury blocks vesicle trafficking in both severed and intact axons
We wished to examine immediate injury-induced changes in neurophysiology in severed axons along with neighboring intact neurons. To accomplish this, we used a partial axotomy model in the adult Drosophila wing. The L1 wing vein nerve houses ~290 sensory neurons whose cell bodies are aligned along the anterior wing margin, with all axons projecting medially and synapsing in the thoracic ganglion (Fang et al., 2012; Neukomm et al., 2014). Sensory axons are ensheathed by glial cells along their length (Neukomm et al., 2014). Transecting the wing nerve at its midpoint severs the axons of approximately half of the sensory neurons (Figure 1A); those with cell bodies distal to the injury site are axotomized, while those with cell bodies proximal to the injury site remain intact and survive for weeks after injury. Severed axons undergo Wallerian degeneration ~8-10 hours after axotomy and neuronal debris is cleared by surrounding glial cells within 5-7 days (Neukomm et al., 2014).
Figure 1: Nerve injury blocks vesicle trafficking in both severed and intact axons. (See also Figure S1.).
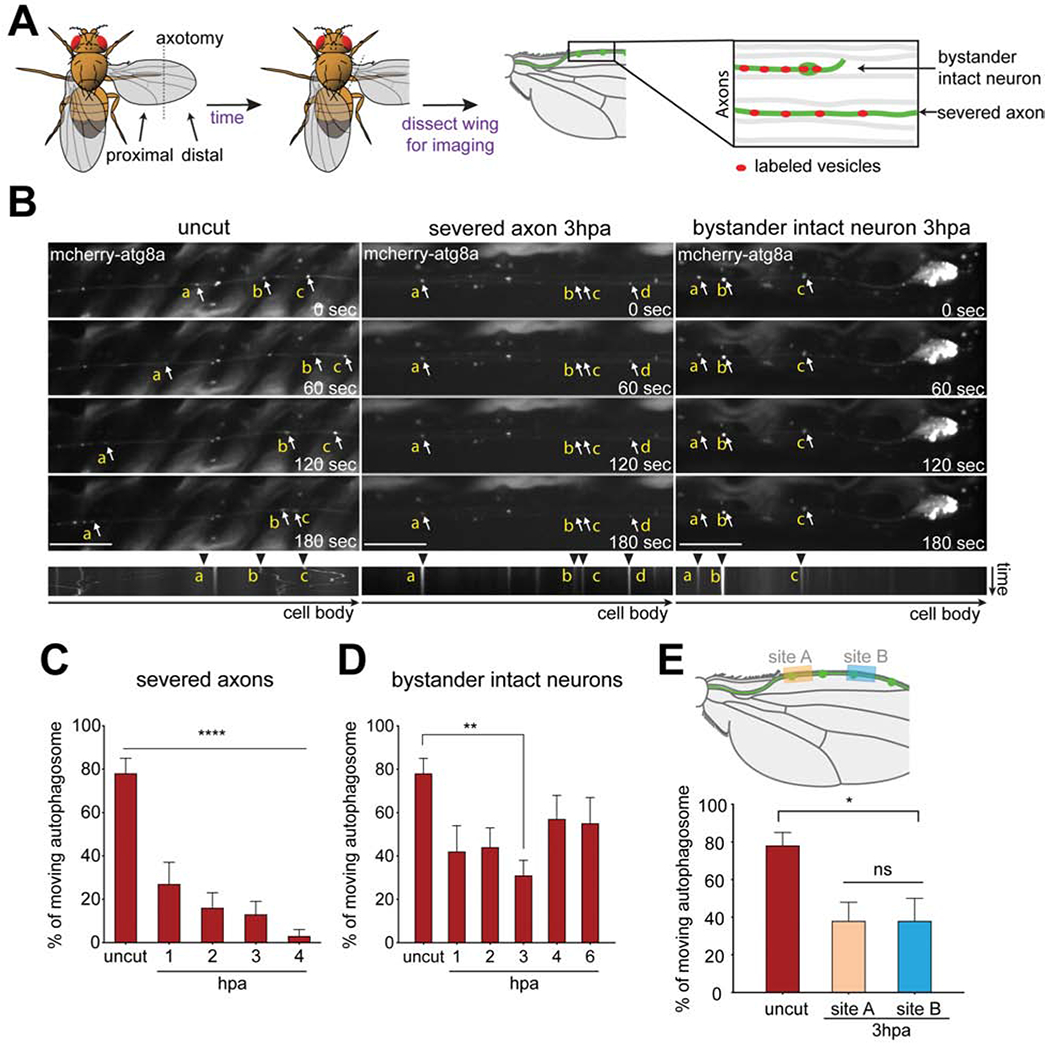
(A) Schematic of injury and vesicle transport live imaging assay in Drosophila wings. Adult fly wings were axotomized, animals were incubated, after which wings were dissected and imaged. Individual clones were labeled using MARCM (green), which allowed for clone-specific expression of markers; severed and intact axons were identified by tracing to cell bodies; unlabeled neurons (gray). (B) Representative time series and kymographs of mCherry-atg8a (arrows) transports in axons of uncut (left column), severed axons 3 hours post axotomy (middle column), and intact neurons 3 hours post axotomy (hpa) (right column). Autophagosomes (arrowheads) were highly motile in uncut axons but exhibited reduced movement in severed and intact axons by hours 3hpa. The individual autophagosome puncta were labeled with yellow lower-case alphabet in the montage images. Scale bar,10 μm. Time is total 3 minutes. (C, D) Quantification of moving autophagosomes over time in (C) severed axons or (D) proximal intact axons. Ordinary one-way ANOVA with Sidak multiple comparisons test. (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, n = 10 axons of each, Error bar = S.E.M.). (E) Upper panel: Schematic showing distal wing nerve injury and sites where intact axons were subsequently imaged (site A and B) 3hpa. Lower panel: Quantification of autophagosome movement. Ordinary one-way ANOVA with Sidak multiple comparisons test. (ns = not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, n = 10 axons of each, Error bar = S.E.M.).
We live imaged axon transport in the L1 wing vein as a first step toward determining how axon biology changes in response to nerve injury. Sensory neurons were sparsely labeled using the mosaic analysis with a repressible cell marker (MARCM) approach (Lee and Luo, 1999; Neukomm et al., 2014), which allowed for the GAL4/UAS-dependent labeling of single cell clones with membrane tethered GFP or RFP. After surgical removal of the distal half of the wing, we directly compared the phenotypes of severed axons alongside remaining intact neighboring neurons (Figure 1A). For examination of changes in axon transport, we labeled four different types of axon cargoes: autophagosomes (UAS-mCherry-Atg8a), lysosomes (UAS-GFP-lamp), synaptic vesicles (UAS-syt-GFP), and mitochondria (UAS-mito-GFP). In uninjured wings, autophagosomes were transported in anterograde and retrograde directions along axons, with almost ~80% of the total population exhibiting movement (both anterograde and retrograde) during the 3-minute observation window (Figures 1B, C, Video S1).
In severed axons, the number of motile autophagosomes decreased to ~25% within 1 hour, and continued to decrease at each hour timepoint until it was <4% at 4 hrs after injury (Figures 1B, C, Video S2). To our surprise, we found that the percent of moving autophagosomes also dropped significantly in the axons of neighboring intact bystander neurons (~40% by 1 hour after injury; Figures 1B, D, Video S3), and partially recovered by 6 hours after axotomy. We observed a similar time course for suppression of axon transport of lysosomes (Figures S1A, B) and synaptic vesicles (Figures S1C, D), with a near full blockade of transport in severed axons, and partial suppression in bystander neurons. We found mitochondria were largely stationary in adult post-mitotic neurons, which is consistent with work showing that as axons mature mitochondria become more stable (Lewis et al., 2016), so they were not examined further.
Does wing injury or nerve injury drive changes in bystander axon physiology? To answer this question, we lesioned the wing from the posterior margin anteriorly through the wing, but left the L1 nerve intact. We found no change in axon transport in response to this surgery, suggesting that lesion of L1 nerve itself was required to induce bystander effects (Figure S2). Finally, we sought to determine whether the proximity of neuronal cell bodies of bystander neurons to the injury site affected the severity of axon transport blockade, or whether blockade of axon transport was uniform throughout the wing nerve. We lesioned the L1 wing nerve distally, removing only ~20 neuronal cell bodies from the tip of the wing, and then imaged the transport of autophagosomes in intact bystander neurons at 3 hrs after injury at two sites along the wing nerve, one distant from (Site A) and one closer to (Site B) the injury site (Figure 1E). We found that regardless of the distance of the cell body from the lesion site, or even the reduced number of severed axons in the more distally injured nerve preparation (~20 in the distal lesion, versus ~110 with midpoint lesions), the suppression of autophagosome transport was similar in bystander neurons and approximated that observed when we lesioned the midpoint of the wing. These data indicate that nerve injury efficiently suppresses axon transport throughout the L1 wing nerve within 3 hrs after injury, even with relatively small injuries (~20 neurons out of 290 total). Because the axons of uninjured cells also exhibited this phenotype, we conclude that bystander neurons can detect nerve injuries even at a distance, and respond by suppression of axon transport.
dSarm is required for the blockade of vesicle trafficking in both severed and intact axons after injury independent of its NADase activity
Approximately 8-10 hours after axotomy of the L1 wing vein, axon death is driven cell autonomously by dSarm in severed axons (Neukomm et al., 2014). While dSarm is primarily thought to drive axon death at these later stages, we used MARCM technology to assay dsarm mutant clones to determine whether dSarm was also required for this early suppression of axon transport. We generated sparse MARCM clones of dsarm null alleles (dsarm896), which lead to the production of ~8-10 homozygous mutant, GFP+ clones being present in a field of (~280) otherwise wild type neurons in the L1 nerve. Under MARCM conditions, we can therefore assay directly for cell-autonomous requirements for dsarm in GFP+ clones. We found normal rates of axon transport of autophagosomes (Figure 2A, B, and F, Video S4) and lysosomes (Figure S3) in axons in dsarm896 null mutant clones before injury. However, in contrast to wild type clones, dsarm896 null clones exhibited normal rates of axon transport of autophagosomes in severed axons for at least 6 hours after axotomy (Figures 2A, F, Video S5). This was confirmed using markers for lysosomes (Figure S3), and this could be rescued by a bacterial artificial chromosome (BAC) that covers dsarm locus to restore endogenous expression of dsarm (Figure 2F). We conclude that dSarm is required within the first few hours after axotomy for blockade of axon transport in the distal stumps of severed axons.
Figure 2: dSarm is required for the blockade of vesicle trafficking in both severed and intact axons after injury independent to its NADase activity. (See also Figure S2–S3.).
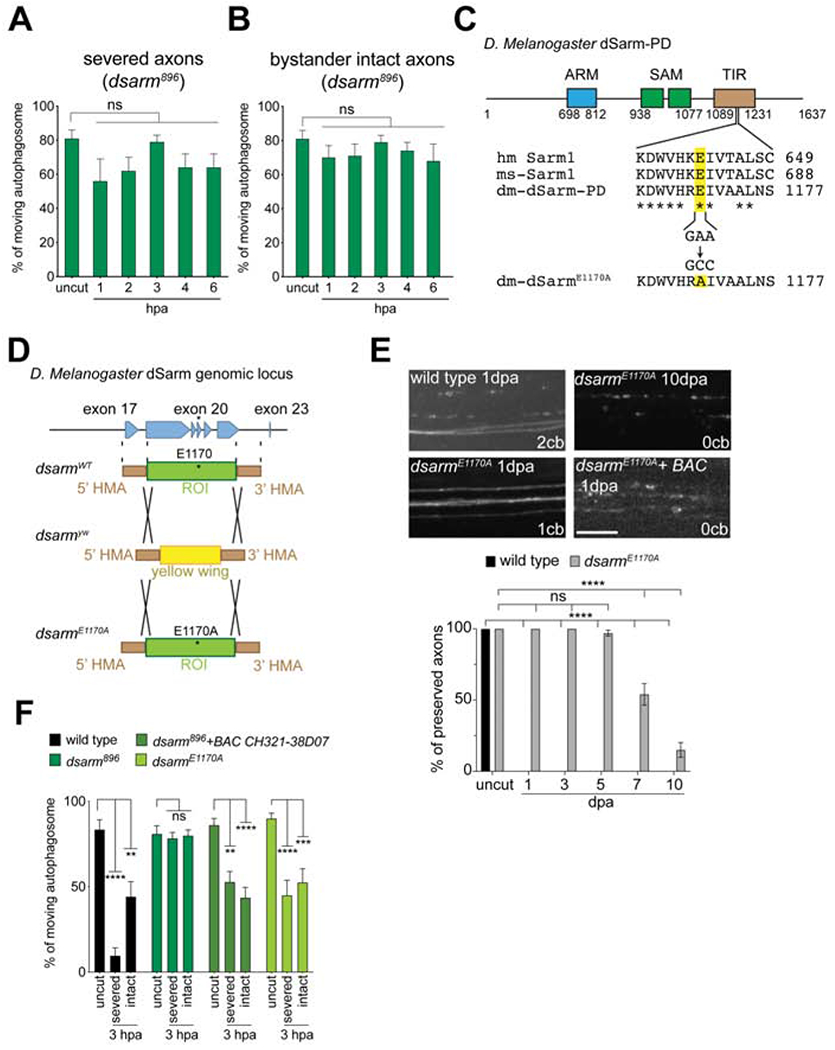
(A, B) Quantification of autophagosome trafficking over the first 6 hrs after injury in dsarm mutant axons. One-way ANOVA with Sidak multiple comparison. (hpa = hours post axotomy, ns = not significant, n = 10 axons of each, Error bar = S.E.M.). (C) Schematic indicates the conserved glutamic acid position in human Sarm1 (hm-Sarm1), mouse Sarm1 (ms-Sarm1), and fly dSarm-PD isoform (dm-dSarm-PD). The nucleotides GAA were mutated to GCC, which encodes alanine, to generate E1170A mutation (dm-dSarmE1170A). (D) Schematic shows the strategy of fly genome cassette swapping. The region of interest (ROI) across exon 18-22, flanking with 5’ and 3’ homology arm (HMA) in wild-type dsarm fly genome (dsarmWT), was replaced with yellow wing cassette by Crispr/Cas9 genome editing to generate dsarmyw mutant. And then the yellow wing cassette were replaced again by the ROI contained E1170A mutation to generate dsarmE1170A mutant (also see Star Method). (E) Images and quantification of axon preservation after axotomy as previously described (Neukomm et al., 2014). dpa. Days post axotomy. cb. Cell body. BAC is BAC CH321-38D07. Two-way ANOVA with Sidak multiple comparisons test. (scale bar = 5 μm, ns = not significant, ****p < 0.0001, n = 10 animals of each, Error bar = S.E.M.). (F) Quantification of autophagosome movement in dsarm mutants (dsarm896, dsarmE1170A, and dsarm896+BAC CH321-38D07) 3hpa, compared to uninjured wings (uncut). Two-way ANOVA with Sidak multiple comparison. (For all, ns = not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, n = 13-20 axons of each, Error bar = S.E.M.).
We next examined axon transport in adjacent uninjured dsarm null mutant MARCM clones and found, unexpectedly, that transport of autophagosomes was also maintained at normal levels in dsarm mutant bystander neurons (Figures 2B, F). This was confirmed by examination of lysosome transport (Figure S3), and was also rescued by introduction of the BAC clone covering the dsarm locus (Figure 2F). To further confirm this represented a cell autonomous role for dSarm in bystander neurons, we performed injuries such that all MARCM clones remained intact (i.e. all were bystanders), and found that while control clones again suppressed axon transport, dsarm mutant MARCM clones maintained normal axon transport (Figure S1E). These observations indicate that dSarm is required cell-autonomously in bystander neurons to detect nerve injury and suppress axon transport. To our knowledge, this is the first description of a cell autonomous role for dSarm/Sarm1 after nerve injury in changing the physiology of uninjured neurons.
Sarm1-dependent axon degeneration requires a recently identified NAD+ hydrolase activity in the Sarm1 TIR domain that is conserved in Drosophila (Essuman et al., 2017). We wished to determine whether dSarm-mediated axon transport blockade also required this NAD+ hydrolase function. dSarm contains a conserved glutamic acid (E1170, equivalent to human Sarm1 E642), which is critical for NAD+ hydrolase activity (Figure 2C) (Essuman et al., 2017). We generated an NADase-dead dSarm mutant allele (dsarmE1170A) by substituting the glutamic acid at position 1170 to alanine using Cas9-mediated genome editing (Li-Kroeger et al., 2018) (Figure 2D, also see Star Methods). Consistent with this leading to an NADase-dead dSarm molecule, severed axons were strongly delayed in their degeneration in dsarmE1170A mutant MARCM clones (Figure 2E). However, in contrast to dsarm null mutants where axons survive for >30 days (Osterloh et al., 2012), dsarmE1170A mutant axons remained intact only for ~ 5 dpa (Figure 2E). These data suggest that the NAD+ hydrolase function might not fully account for dSarm pro-degenerative function, however, we cannot exclude the possibility that the E1170A mutant protein retains low level NAD+ hydrolase activity. Interestingly, we found that dsarmE1170A mutant MARCM clones exhibited a suppression of axon transport similar to dsarm896 nulls alleles in both severed axons and bystander neurons. These data indicate that although the NADase activity of dSarm is important for axon degeneration, it is dispensable for the early suppression of axon transport after injury (Figure 2F).
Axed is not required for the blockade of vesicle trafficking in both severed and intact axons after injury
Loss of Axed suppresses axon degeneration at levels equivalent to loss of dSarm (Neukomm et al., 2017). To determine whether dSarm functions to suppress axon transport through Axed, we assayed axon transport in axed null mutant MARCM clones before and after nerve injury. We found normal rates of axon transport for autophagosomes (Figure 3A) and lysosomes (Figure S3) before axon injury in axed null clones. However, in both severed and bystander neurons, we found that loss of axed failed to maintain axon transport after nerve injury (Figures 3A–C), with axon transport being suppressed within 1 hour after axotomy (Figure 3C). This was confirmed with a second null allele of axed (Figure 3A), and similar results were found with lysosomes (Figure S3). We conclude that Axed is not required for nerve injury-induced blockade of axon transport, despite its critical role at later stages in executing dSarm-dependent axon degeneration (Neukomm et al., 2017).
Figure 3: Axed is not required for the blockade of vesicle trafficking in both severed and intact axons after injury. (See also Figure S3.).
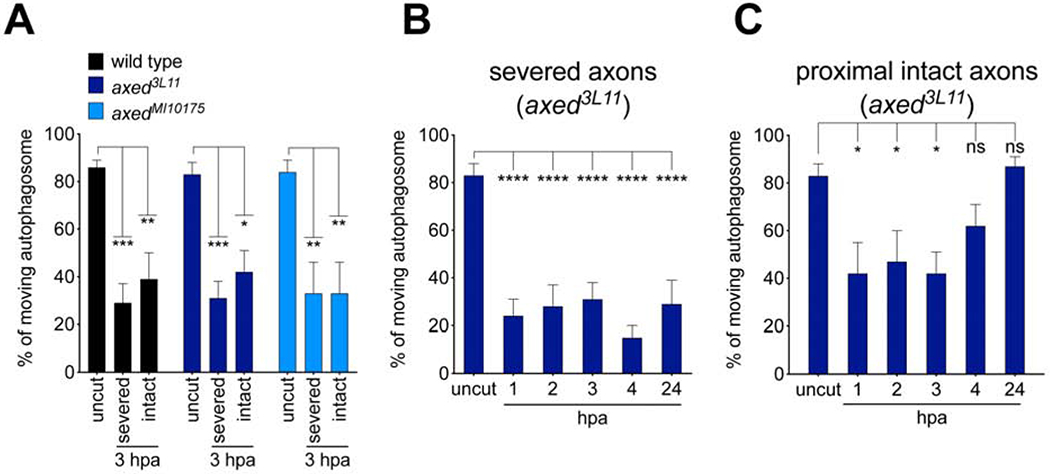
(A) Quantification of autophagosome movement null mutants of axed (axed3L11 and axedMI1017S) 3hpa compared uncut controls. Two-way ANOVA with Sidak multiple comparison. (B, C) Quantification of autophagosome trafficking over the first 6 hrs after injury in axed mutant axons. Ordinary one-way ANOVA with Sidak multiple comparisons test. (For all, ns = not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, n = 10 axons of each, Error bar = S.E.M.).
Cacophony and the TIR-1-like-MAPK signaling pathway promotes blockade of vesicle trafficking after nerve injury
C. elegans TIR-1 (worm dSarm/Sarm1) signals downstream of the voltage-gated calcium channel (VGCC) UNC-36, and activates a conserved MAP kinase cascade (Figure 4A) (Chuang and Bargmann, 2005). In injured mouse DRG axons, the MAPKK MKK4, and JNKs, are phosphorylated within 15 min in a Sarm1-dependent fashion (Yang et al., 2015), arguing for an early role for Sarm1 in injury-induced changes in axon biology. To determine whether dSarm might signal through a VGCC/dSarm/MAPK-like cascade early in axonal injury signaling, we first visualized axon transport in MARCM mutant clones for cacophony, which encodes the Drosophila ortholog of UNC-36. We found that transport of autophagosomes was unchanged after injury in MARCM clones of cacK, similar to dsarm null mutants, (Figure 4B). Likewise transport of synaptic vesicles (Figure S3A) was unaffected in cacK neurons in both severed axons and bystander neuron. Similar results were found with a second strong loss of function allele of cac, cacF (Figures 4B, Figure S4A), which is a channel conductance mutant (Tian et al., 2015), arguing for a role for ion conductance through Cacophony in promoting suppression of axon transport.
Figure 4: Cacophony and the TIR-1-like-MAPK signaling pathway promotes blockade of vesicle trafficking after nerve injury. (See also Figures S4–S6.).
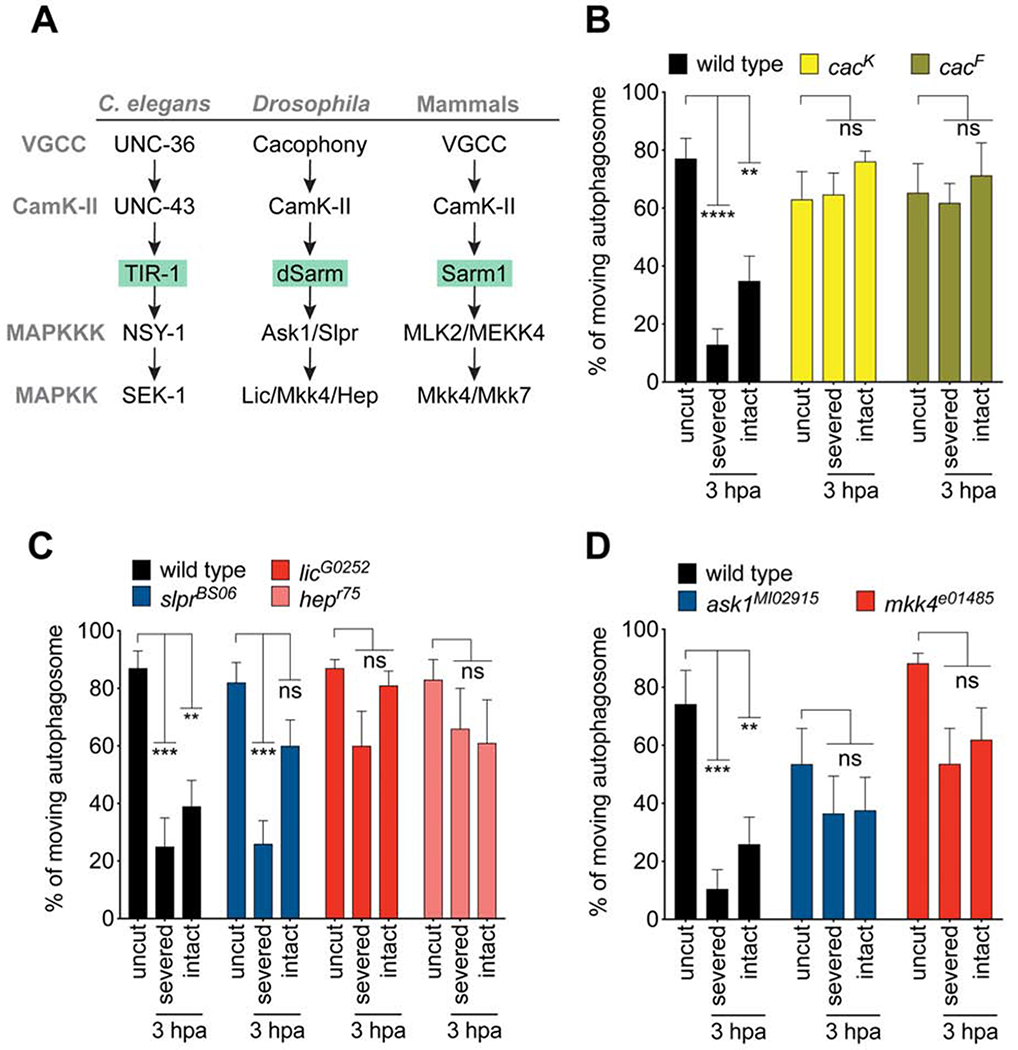
(A) The Tir-1/MAPK signaling pathway and conservation in flies and mammals. (B) Quantification of autophagosome movement in two cac mutants (cacK and cacF) in severed or proximal intact axons 3hpa compared uncut controls. (For all, Two-way ANOVA with Sidak multiple comparisons test. ns = not significant, **p < 0.01, ***p < 0.001, ****p < 0.0001, n = 10 axons of each, Error bar = S.E.M.). (C) Quantification of autophagosome movement in mkk4 and askl mutants (mkk4e01485, and askMI02915) in both severed axons or proximal intact axons in injured wings (3hpa) compared to that in uninjured wings (uncut). (D) Quantification of autophagosome movement in slpr, lic, and hep mutants (slprBS06, licG0252, and hepr75) in both severed axons or proximal intact axons in injured wings (3hpa) compared to that in uninjured wings (uncut).
We next assayed for roles of the MAPKKKs Ask1 and Slpr, and the MAPKKs Lic, Mkk4, and Hep. We found that MAPK signaling components were required for potent suppression of axon transport after axotomy in severed and intact bystander neurons (Figures 4A, C, and D; Figures S4B, C), although their phenotypes did not appear as strong null alleles of dsarm or cac. MAPKs often function in a partially redundant fashion, as do Mkk4 and Mkk7 in axon degeneration (Yang et al., 2015). We therefore tested the double RNAi knockdowns of mkk4 and hep using two different mkk4 RNAi lines, but found results comparable to the single loss of function backgrounds (Figure S4D), suggesting that in this case they do not act redundantly. Importantly, despite their clear roles in early blockade of axon transport, even strong loss-of-function alleles of cac or MAPK signaling molecules failed to suppress axon degeneration at later stages at any level (Figure S5). We conclude that dSarm signals with the VGCC Cac and at least partially through the MAPK signaling cascade within the first few hours after axonal injury to suppress axon transport, while loss of Cac or MAPK components does not suppress later phases of axon degeneration.
The involvement of the VGCC Cac led us to test whether initial calcium influx after axotomy was altered by manipulation of Cac or dSarm, and might thereby be suppressing blockade of axon transport. We sparsely labeled control and cac mutant MARCM clones with GCaMP6, laser-ablated a subset of axons in the L1 wing nerve, and then live imaged GCaMP6s fluorescence in clones following ablation. We found strong GCaMP signal induction in the cell bodies of severed axons and bystander neurons across all the genotypes we tested within seconds of injury (Figure S6). We found no differences in the maximum amplitude or time to maximum amplitude in either severed or bystander neurons when we compared controls to dsarm or axed mutants (Figure S6A–C). However, we did observe a significant decrease in maximum amplitude and increase in time to maximum amplitude for GCaMP6s signals in injured neurons lacking Cac, although this change was small (Figure S6A–C). We conclude that while Cacophony may partially contribute to initial calcium influxes after axon injury, it plays a relatively minor role in total Ca2+ influx immediately after axotomy despite its potent role in suppression of axon transport within the following 1-2 hours.
Degradation of NMN does not suppress axon transport blockade, and dNmnat is required for the bystander response
Nmnat loss, NAD+ depletion, and activation of Sarml by NMN to drive further NAD+ depletion, are proposed to be key steps in activating the dSarm/Sarm1-dependent explosive axon destruction during Wallerian degeneration. We wished to determine whether these events were also required for early suppression of axon transport in severed and bystander neurons. Overexpression of E. coli NMN deamidase (NMNd), which degrades NMN, has been shown to partially protect axons from degeneration after axotomy (Figure 5A) (Di Stefano et al., 2014; Loreto et al., 2014). We generated transgenic Drosophila where we could express NMNd under the control of Gal4-UAS system. We found that overexpression of NMNd in neurons led to strong suppression of axon degeneration for 3-5 days after axotomy (Figure 5B), arguing that NMN is also an activator of dSarm-dependent axon degeneration in Drosophila. We next assayed whether expression of NMNd could suppress the axotomy-induced blockade of axon transport. We found that NMNd expression failed to sustain the transport of both autophagosome and synaptic vesicles in both injured axons and bystander neurons 3 hours after axotomy (Figure 5C, S7A). These data argue against a role for NMN in activating dSarm in the context of axon transport blockade.
Figure 5: Suppression of axon transport is not caused by the accumulation of NMN, or the depletion of dNmnat. (See also Figures S7.).
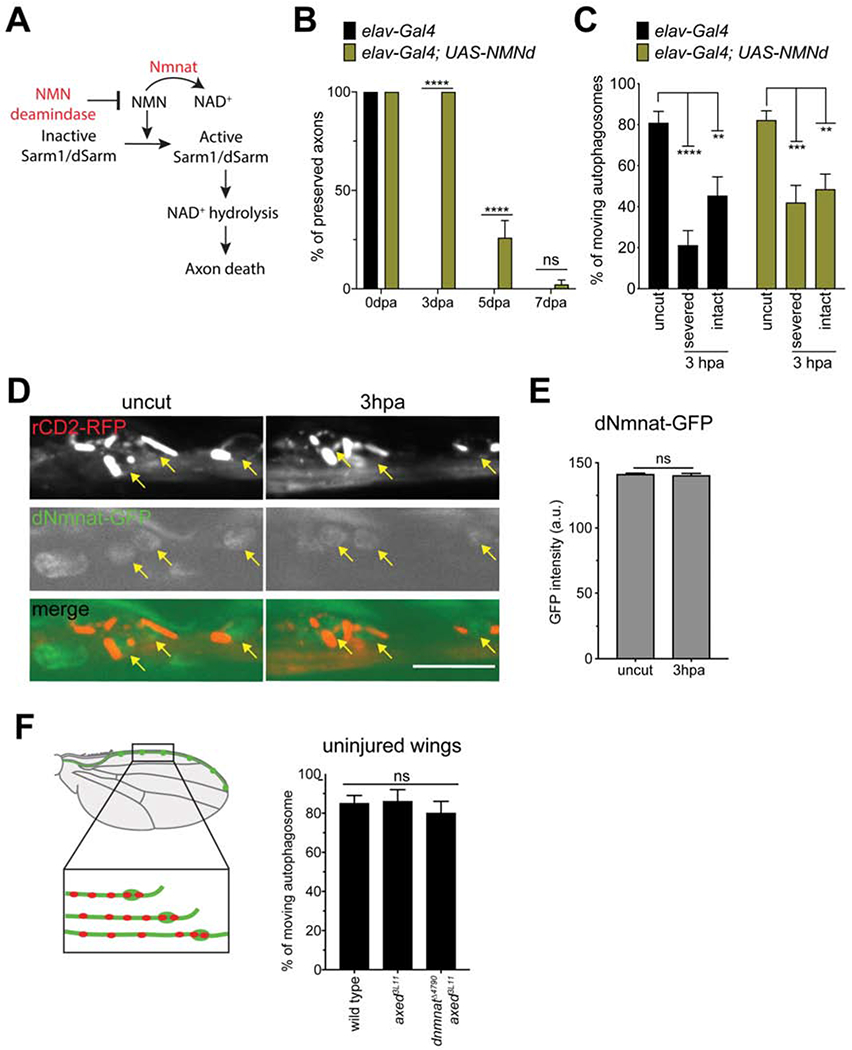
(A) Diagram of NMN induced Sarm1 NAD+ hydrolase activity. (B) Quantification of axon preservation after axotomy as previously described (Neukomm et al., 2014). Dpa, days post axotomy. Two-way ANOVA with Sidak multiple comparisons test. (ns = not significant, ****p < 0.0001, n = 25 animals of each, Error bar = S.E.M.). (C) Severed and intact axons expressing NMN deamidase (NMNd) showed decrease of axon transport after axonal injury 3hpa compared to that in uninjured wings. Two-way ANOVA with Sidak multiple comparisons test. (ns = not significant, **p < 0.01, ***p < 0.001, ****p < 0.0001, n = 10~20 axons of each, Error bar = S.E.M.). (D) Representative images of nSyb-lexA; 13xLexAop-rCD2-RFP labeling the neurons (top panels), dNmnat-GFP-dNmnatWT labeling the endogenous dNmnat (middle panels). The dNmnat-GFP-dNmnatWT in neurons are distinguished by the merged images (bottom panels). Arrows points the neurons and dNmnat-GFP expression. There is no visible difference of dNmnat-GFP expression between uncut and 3hpa images. Scale bar, 10 μm. (E) The GFP intensity of dNmnat-GFP-dNmnatWT showed no difference between uninjured and injured wings. Unpaired two-tailed t-test. (ns = not significant, n = 20 animals, Error bar = S.E.M.). (F) Schematic indicates the neuronal clones in uninjured wings were labeled and imaged for axon transport quantification. The autophagosomes transport in axed3111; dnmnat14790 axons is comparable to wild-type axons and axed3L11 axons in uninjured wings. One-way ANOVA with Tukey multiple comparisons test. (ns = not significant, n = 10 animals, Error bar = S.E.M.).
Depletion of dNmnat in intact neurons also causes spontaneous axon degeneration that can be suppressed by loss of dSarm or Axed (Gilley et al., 2015; Neukomm et al., 2017). Is dNmnat depleted in neurons within hours after nerve injury? To explore this possibility, we utilized a functional dNmnat allele that is endogenously tagged with GFP (Nmnat-GFP-NmnatWT) (Li-Kroeger et al., 2018) to visualize the level of dNmnat before and immediately after axotomy. To ensure this version of dNmnat was not hyper-stable and neuroprotective, as previously found when mouse Nmnat2 was GFP-tagged (Milde et al., 2013a), we assayed axon degeneration (Figure S7D) and injury-induced suppression of axon transport (Figure S7C), and both were normal, arguing this is a functional version of dNmnat. When we then examined dNmnat-GFP levels in partially injured nerves and controls, we found no discernible change of the dNmnat-GFP when we compared intact nerves to samples 3 hours after axotomy (Figures 5D, E). These data argue that dNmnat is not depleted within 3 hours after injury, when suppression of axon transport is strongest.
Is dNmnat loss sufficient to induce suppression of axon transport? There is only one Nmnat molecule encoded in the fly genome, dNmnat, and its loss causes spontaneous axon degeneration and cell death within 1 day after depletion (Fang et al., 2012; Neukomm et al., 2017; Xiong et al., 2012). To explore the potential role for dNmnat in injury-induced suppression of axon transport we generated dnmnat null clones in axed mutant backgrounds, where axon integrity is preserved despite the loss of dNmnat (Neukomm et al., 2017). In uninjured wings, we did not observe a blockade of axon transport in axed3L11, dnmnatΔ4790 double mutant clones, arguing that loss of dNmnat does not cause a spontaneous blockade of axon transport (Figure 5F). Unexpectedly, we instead found evidence supporting a positive role for dNmnat in suppressing axon transport early after injury. Specifically, while we observed a strong blockade of axon transport 3 hours after axotomy in controls (Figure S7B) and in axed single mutant clones (e.g. Figure 3), we did not find such blockade in axed, dnmnat null clones in either severed axons or uninjured bystanders (Figure S7B).
In summary, these data support the notion that early blockade of axon transport is not mediated by accumulation of NMN or early depletion of dNmnat. Rather, dNmnat plays a positive role in promoting suppression of axon transport in injured nerves. The latter observation is somewhat paradoxical, given that both loss- (dnmnat mutants) versus gain-of-fUnction (dNmnat and WldS overexpression, below) approaches have similar rather than opposing effects on bystander neuron responses (see Discussion).
Overexpression of Wlds or dNmnat, or mutation of hiw, rescue axon transport defects after nerve injury
WldS is thought to suppress axon degeneration by substituting for the axon survival factor Nmnat2 after its depletion (Gilley and Coleman, 2010). Our data above argue against a role for dNmnat depletion or NMN accumulation (which results from loss of Nmnat activity) in modulating axon transport immediately after injury. In the context of the Nmnat/NAD+ depletion model, we predicted that WldS overexpression or loss of Hiw/Phr1 (which normally degrades dNmnat) would not alter the early injury-induced blockade of axon transport. However, we found that expression of WldS or dNmnat, was indeed sufficient to suppress the axotomy-induced inhibition of axon transport in both severed axons and intact bystander neurons 3 hours after axotomy (Figure 6A, B). Likewise, we found axon transport was maintained 3 hours after axotomy in hiw mutant clones in both severed axons and intact bystander neurons (Figure 6C). We conclude that early blockade of axon transport in injured nerves is sensitive to WldS or dNmnat, and loss of Hiw/Phr1. These data, coupled with that above demonstrating a positive role for dNmnat in the early blockade of axon transport in injured nerves, are consistent with the notion that WldS/dNmant and Hiw act in this context by providing additional Nmnat activity, or enhancing dNmnat stability, respectively.
Figure 6: Overexpression of Wlds, or increasing the level of dNmnat, is sufficient to rescue axon transport defects after nerve injury.
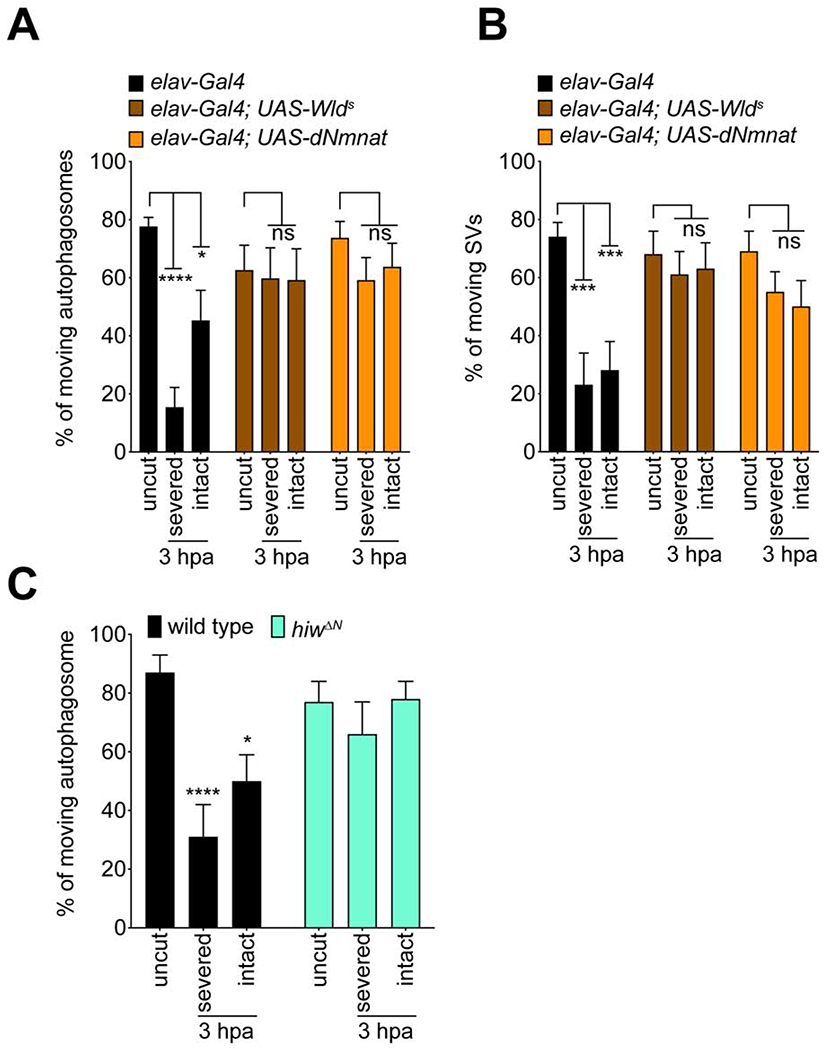
(A, B) Severed and intact axons expressing either WldS or dNmnat showed no changes in autophagosomes and synaptic vesicles (SV) transport after axonal injury 3hpa compared to that in uninjured wings. Two-way ANOVA with Sidak multiple comparisons test. (ns = not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, n = 10 axons of each, Error bar = S.E.M.). (C) Severed and intact axons of hiwΔN mutant clones showed comparable percentage of axon transport after axonal injury 3hpa with that in uninjured wings. Two-way ANOVA with Sidak multiple comparisons test. (ns = not significant, *p < 0.05, ****p < 0.0001, n = 10 axons of each, Error bar = S.E.M.)
Draper-dependent signaling functions in glia to suppress axon transport in the intact neurons
How are injury signals spread to bystander neurons? The Drosophila L1 wing vein houses ~290 sensory neurons that project into the thoracic ganglion. We examined the morphology of this nerve by TEM and found that individual sensory neuron axons were fully ensheathed by glial processes (Figures 7A–C), suggesting they are not in direct contact with one another. It is plausible that severed axons signal to glia, and then glia alert bystander neurons to alter their axonal physiology. Alternatively, glia could be damaged during the nerve injury and signal directly to neurons that an injury has occurred. To explore these possibilities, we assayed for blockade of axon transport in draper null mutants. Draper is a receptor required for glial phagocytic function (Freeman et al., 2003; MacDonald et al., 2006), as is its mammalian ortholog MEGF10 (Wu et al., 2009), but Draper/MEGF10 also plays additional non-phagocytic roles in cell-cell signaling (Kay et al., 2012; McPhee et al., 2010). We sparsely labeled neurons and autophagosomes with MARCM in control and whole animal draper null mutant backgrounds (drprΔ5) and assayed transport in severed axons and bystander neurons. We found that axon transport was suppressed in severed axons in draper mutants, similar to controls (Figure 7D). However, axon transport in intact bystander neurons was maintained in draper mutants (Figure 7D). To confirm this represented a role for Draper in glia, we knocked down draper with RNAi selectively in glia using the pan-glia Gal4 driver line repo-Gal4 and assayed axon transport. We found that glial depletion of draper was sufficient to maintain axon transport in bystander neurons, but not severed axons (Figure S8). These data demonstrate that glial Draper is required for active spreading of injury signals to bystander neurons, while suppression of axon transport in severed axons occur in a Draper-independent manner.
Figure 7: Draper-dependent signaling functions in glia to suppress axon transport in the intact neurons. (See also Figures S8.).
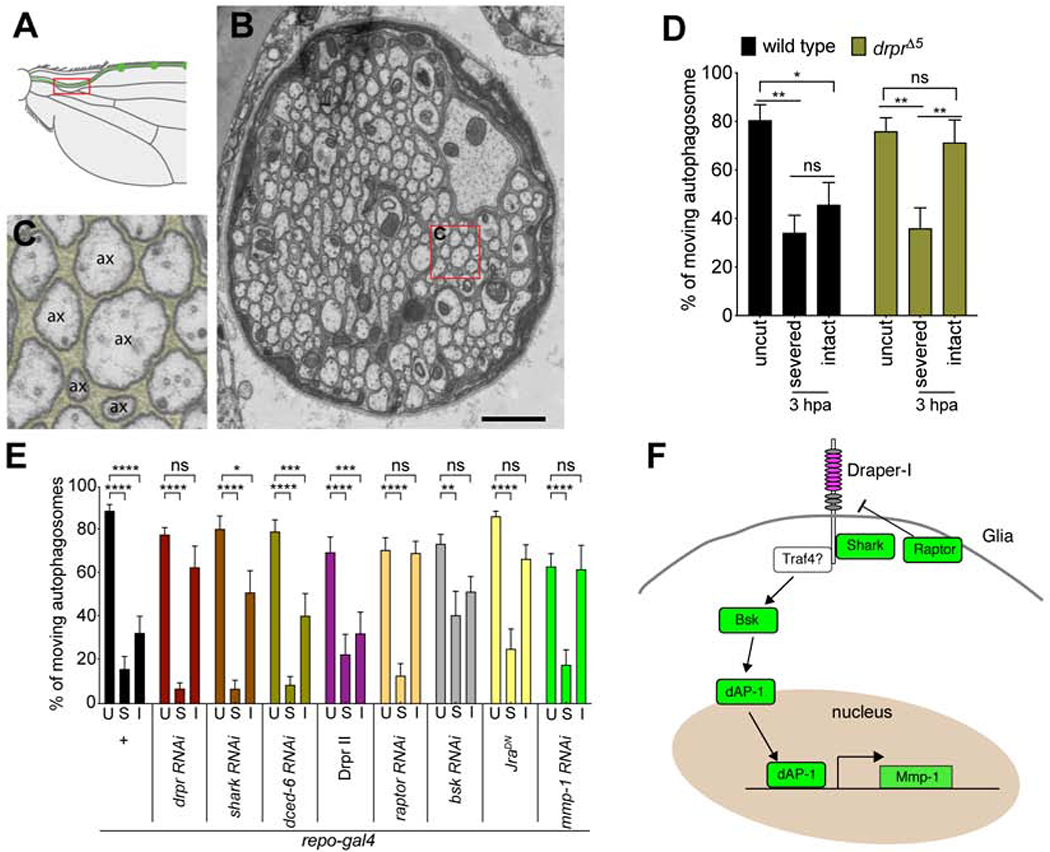
(A) Schematic shows that the region where transmission electron microscopy (TEM) image was taken (red rectangle). (B) TEM image of the nerve in L1 wing vein. Red rectangle indicates the selected region of (C). Scale bar = 1μm. (C) 5x magnified TEM image from the indicated region on (B). Glia are pseudocolored. ax = axon. (D) Severed axons, but not intact axons, showed decrease of axon transport after axonal injury 3hpa in drprΔ5 homozygote flies. Two-way ANOVA with Sidak multiple comparisons test. (ns = not significant, *p < 0.05, **p < 0.01, n = 10~20 axons of each, Error bar = S.E.M.). (E) Quantifications of autophagosome transport in glial-specific RNAi knockdown, or overexpression (repo-gal4, uas-transgenes) neurons of the genes indicated. Two-way ANOVA with Sidak multiple comparisons test. U = uncut, S = severed axons, I = intact bystander neurons (ns = not significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, n = 10~20 axons of each, Error bar = S.E.M.). (F) Schematic showed the model of Draper-dependent glial signaling pathway to suppress axon transport in bystander intact neurons.
Draper promotes glial engulfment through a well-defined signaling cascade (Awasaki et al., 2006; Doherty et al., 2009; Doherty et al., 2014; Logan et al., 2012; Lu et al., 2017; Purice et al., 2017). To more deeply explore the mechanism by which Draper spreads injury signals to bystander neurons, we performed glial knock-down of the majority of key molecules known to act downstream of Draper. Glial-specific depletion of the c-Jun kinase Basket partially phenocopied draper mutants, and led to a suppression of blockade of axon transport in bystander neurons, as did depletion of Shark (Figure 7E) and the overexpression of a dominant negative version of the transcriptional regulator Jra (the Drosophila c-Jun homolog) (Figure 7E). Overexpression of Draper-II, an ITIM-bearing isoform than can potently block Draper signaling in the context of engulfment via Csw (Shp1/2) (Logan et al., 2012), or dCed-6, an adaptor protein essential for Draper engulfment signaling, had no significant effect (Figure 7E). Glial knockdown of the mmp-1 gene, a transcriptional target of dAP-1 downstream of Draper (Purice et al., 2017) that is required for phagocytic function, also suppressed blockade of axon transport in bystander neurons at 3 hrs after axotomy (Figure 7E), as did glial depletion of Raptor, a key component of the mTORC1 complex that regulates basal Draper levels in glia (Doherty et al., 2014). These data further support the notion that a transcriptional cascade downstream of Draper, involving JNK, dAP-1, Mmp-1 and PI3K/Raptor signaling is required for glial suppression of bystander neuron function after partial nerve injury (Figure 7F).
Axon injury leads to a dSarm-dependent reversible suppression of sensory signaling in bystander neurons
We sought to determine whether bystander neurons exhibited changes in their functional properties beyond suppression of axon transport. We therefore examined the effects of L1 wing nerve injury on mechanosensory-driven behaviors. Stimulation of mechanosensory neurons in the wing (Figure 8A) with a gentle touch leads to a kicking response that is thought to represent a type of grooming behavior (Li et al., 2016). We stimulated mechanosensory bristles in control wings and wings lesioned at the midpoint of the L1 nerve and found that 3 hours after axotomy there was a significant reduction (~40%) in kicking responses in injured wings (Figure 8B, Videos S6 and 7). By 6 hours after axotomy responses had recovered to control levels (Figure 8B). We found this injury-induced decrease in mechanosensory function could not be suppressed by neuronal expression of NMN deamidase, but could be fully suppressed by neuronal expression of WldS or dNmnat (Figure 8C), or RNAi-mediated knockdown of dsarm in sensory neurons (Figure 8D). hiwΔN mutants showed defective kicking response even without any injury (Figure S9), possibly due to the changes of synaptic growth during development (Collins et al., 2006), so we were unable to test its potential role in suppressing mechanosensory behavior. These data indicate that axon death signaling molecules regulate these early, reversible changes in behavior mediated by intact mechanosensory neurons.
Figure 8: Axon injury leads to a dSarm-dependent reversible suppression of sensory signaling in bystander neurons. (See also Figure S9.).
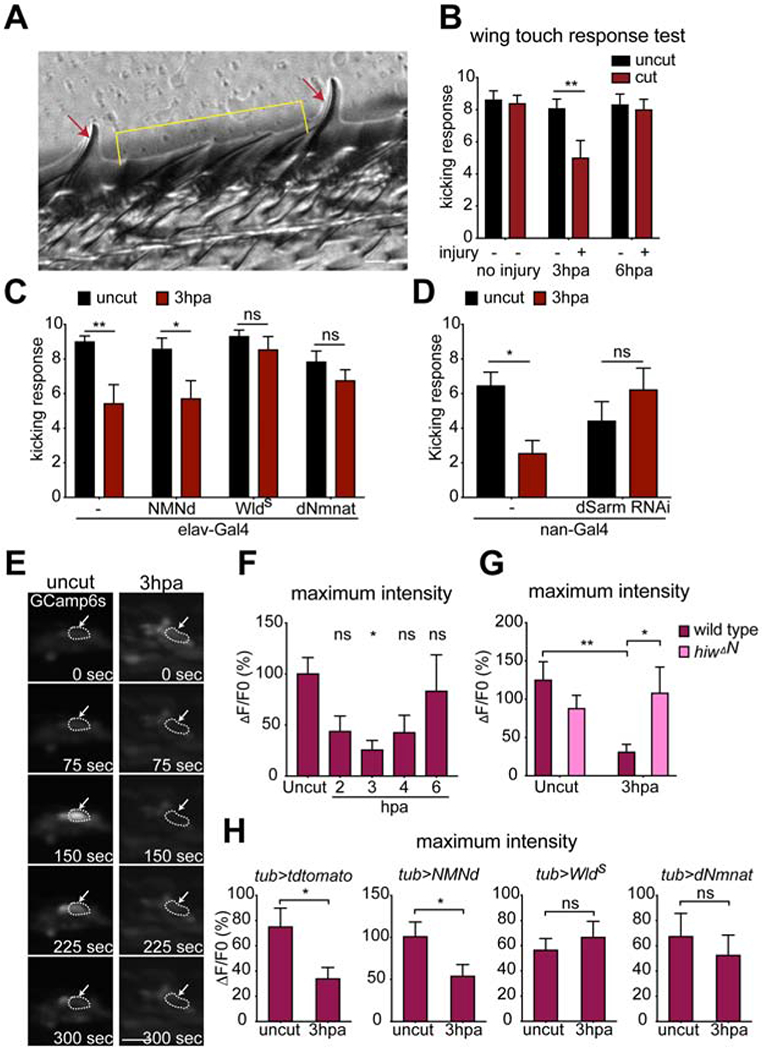
(A) Representative image showed slender bristles for chemosensation (red arrows), and stout bristles for mechanosensation (yellow bracket) on the fly wing margin. (B) Injury reduced the number of kicking responses upon bristle stimulation at 3 hours post axotomy (3hpa) of injured wings (left wings) in w1118 control animals, which recovered by 6 hpa. Two-way ANOVA with Sidak multiple comparisons test. (**p < 0.01, n = 13~14 animals of each, Error bar = S.E.M.). (C) Overexpression of NMNd, Wlds, dNmnat or driver only by pan-neuronal driver elav-Gal4. Two-way ANOVA with Sidak multiple comparisons test, (ns = not significant, *p < 0.05,**p < 0.01, n = 10~14 animals, Error bar = S.E.M.). (D) Knocking down of dsarm in Nan+ neurons resulted in normal kicking responses 3 hpa. Unpaired two-tailed t-test. (ns = not significant, *p < 0.05, n = 10~14 animals, Error bar = S.E.M.). (E) GCamp6s signals are increased in the wing chemosensilla of UAS-GCamp6s/+; tub-Gal4/+ flies in response to 200mM glucose application, but this response was suppressed by nerve injury (3 hpa), arrows point the representative chemosensillar. Scale bar, 10 μM. Dash line circled the chemosensilla with responses upon stimulation. (F) The average maximum intensity (maximum ΔF/F0%) of GCamp6s fluorescence stimulated by glucose was significantly reduced at 3hpa, but recovered by 6 hpa. F0 was defined as the average of fluorescent intensity of 100-120 seconds. Ordinary one-way ANOVA with Sidak multiple comparisons test, (ns = not significant, *p < 0.05, n = 10 animals, Error bar = S.E.M.). (G) In hiwΔN homozygous mutant animals, the average maximum GCamp6s intensity is comparable in the uninjured and injured wings. Two-way ANOVA with Sidak multiple comparisons test. (*p < 0.05, **p < 0.01, n = 10~18 animals of each, Error bar = S.E.M.). (H) Overexpression of tdTomato, NMNd, Wlds or dNmnat by tub-Gal4. Unpaired two-tailed t-test. (ns = not significant, n = 15 animals, Error bar = S.E.M.).
To directly assay the sensitivity of intact sensory neurons to stimulation we live imaged L1 wing vein chemosensory neuron activity using the Ca2+ indicator GCaMP6s (expressed pan neuronally with tub-Gal4) in response to bath application of 200mM glucose (Raad et al., 2016). Glucose exposure led to a robust activation of sugar-sensitive wing sensory neurons, which was potently suppressed within 3 hours after axotomy, but recovered by 6 hours after axotomy (Figures 8E, F, Videos S8 and 9). Injury-induced inhibition of sensory neuron activity was not modified by neuronal expression of NMN deamidase, but was suppressed by WldS, dNmnat (Figure 8H) or hiwΔN mutants (Figure 8G). Together these data demonstrate that nerve injury profoundly alters the physiological responsiveness of bystander neurons within 3 hours, and that this reversible modification of neurophysiology is modulated by dSarm, WldS/dNmnat and Hiw/Phr1.
DISCUSSION
The underlying cellular and molecular changes that drive neural circuit dysfunction after injury or in neurodegenerative disease remain poorly defined. The loss of physical connections between neurons, through synapse or axon degeneration, is thought to be a major contributor. Here we show that relatively small injuries can lead to the rapid and efficient spreading of injury signals across nerves that potently suppress axon transport throughout the nerve, and broadly inhibit neurophysiology in uninjured bystander neurons. Surprisingly, we found the same molecule required to drive explosive axon degeneration in severed axons at later stages, dSarm/Sarm1, is required for this early suppression of neuronal function, although the signaling mechanisms at each stage appear to be different. Our data support a model whereby early (i.e. 1-3 hrs after injury) dSarm signals with Cac and MAPK components, but independent of its NAD+ hydrolase activity, to suppress axon transport and neurophysiology, while at later stages (8-12 hrs) dSarm signals with Axed to promote explosive axon degeneration. This significantly expands the role for dSarm/Sarm1 in regulating nervous system responses to injury to include even uninjured bystander neurons. Furthermore, we discovered a critical role for glial cells, through Draper, in signaling to bystander neurons to inhibit their axon transport and neurophysiology. Together this work suggests that a significant amount of functional loss after neural trauma is not a result only of frank degeneration, but more subtle changes in neuronal function, and it occurs in uninjured neurons through glial spreading of injury signals.
Injury signals spread efficiently across the injured nerve through Cac/dSarm/MAPK signaling
Our data support the notion that widespread signaling occurs between cells in injured neural tissues immediately after injury, and that injury signals can radically alter neuronal function. Severing even a small number of axons led to a suppression of axon transport within hours in all axons in the adult wing nerve, even in uninjured bystander neurons. Beyond axon transport, local uninjured bystander sensory neurons also exhibited a disruption of mechano- and chemosensory signal transduction, which was partially reversible within a few hours. These observations suggest that beyond simple breakage of connectivity, a significant part of functional loss after brain injury or in neurodegenerative disease may also be occurring in healthy, intact neurons that have received function-suppressing signals from nearby damaged neurons.
Surprisingly, we found that dSarm is required cell autonomously in bystander neurons to alter axon transport and nerve function in response to injury, and that this role does not require its NAD+ hydrolase activity. Reception of this injury signal in the bystander neuron (and severed axons) requires the voltage gated calcium channel Cac and the MAPK signaling cascade, similar to Tir-1 signaling in C. elegans, but not Axed. Reciprocally, Cac and MAPK components are not required for Wallerian degeneration at later stages. Explosive axon degeneration requires dSarm, its NAD+ hydrolase activity, and Axed. Based on the timing of these different events (i.e. changes in neuronal function versus frank degeneration) with our genetic studies indicating they are separable, we propose a two-phase model for dSarm signaling in injured neural tissues: early dSarm-dependent changes in axon biology and neurophysiology that occur within hours after injury are mediated by the Cac/dSarm/MAPK signaling cascade (Phase I), while late stage axon degeneration is driven by dSarm signaling through Axed (Phase II). The existence of these temporally distinct phases of dSarm signaling likely explain previous results that seemed in conflict, where MAPK signaling was proposed to act both upstream (Yang et al., 2015) and downstream (Walker et al., 2017) of Sarm1 after axotomy. According to our model both of these assertions would be correct—with dSarm/Sarm1 acting upstream of MAPK early (Phase I) and independent but ultimately downstream of MAPK later to drive dSarm/Axed-dependent axon degeneration (Phase II).
To date dSarm/Sarm1 has been thought of primarily as a cell autonomous regulator of explosive axon degeneration, but our work shows that dSarm can also drive important changes in circuit function through altering neuronal cell biology and neurophysiology. That bystander neurons recover and remain viable also demonstrates that activation of dSarm after injury does not necessarily lead to axon death. We suspect that recovery occurs in large part because bystander neurons have not been severed, which is an extreme injury, and depends on their connection to the cell body, which is a source of axon survival factors like Nmnat2 (Milde et al., 2013b). Connection to the cell body may also explain why axon transport was less severely suppressed in the bystander neurons; additional transport factors can still be continuously supplied to the distal axon from the soma. Defining how dSarm activity is regulated in each of these contexts to interact with Cac/MAPKs versus Axed, and why the first phase does not require NAD+ hydrolase function, are key questions for the future.
Roles for axon death molecules in suppression of neuronal function in bystander neurons
A compelling case exists for the NAD+ depletion hypothesis for dSarm/Sarm1 function in axon degeneration (Essuman et al., 2017; Gerdts et al., 2015; Gerdts et al., 2016), although arguments have been made this dSarm/Sarm1 signaling is likely more complex (Neukomm et al., 2017). In this model, depletion of Nmnat2 via Hiw/Phr1 results in the accumulation of NMN, which functions as an activator of Sarm1, with Sarm1 NAD+ hydrolase activity driving metabolic catastrophe. We provide several lines of evidence that the above, newly-described early dSarm signaling events (i.e. suppression of axon transport and neurophysiology) are mechanistically distinct, but are nevertheless also regulated by some axon death-associated molecules. First, while NMN deamidase (NMNd) can suppress axon degeneration in flies and other species (Di Stefano et al., 2014), it cannot block early suppression of axon transport or changes in bystander neuron function. This argues that NMN is not a driving force for dSarm activation in the early phase. Second, although limited to tagged versions of dNmnat for our analysis, we observe no depletion of dNmnat within the time frame of 6 hours after injury. Previous studies in SCG or DRG cultures in vitro suggest Nmnat2 depletion takes 4-6 hours and NAD+ depletion begins ~ 2-3 hrs after axotomy (Gilley and Coleman, 2010; Wang et al., 2005), which is slightly later than we observe the bystander effect in vivo. And because full axon degeneration is prolonged in vivo compared to in vitro studies, the timing of Nmnat2 loss and NAD+ depletion is likely also prolonged in vivo, further suggesting this likely happens after cessation of axon transport. Third, Axed, which is genetically downstream of dSarm during axon degeneration (Neukomm et al., 2017), is not required for early suppression of nerve responses to injury in either severed or intact neurons, only later axon degenerative events in the severed axons. Finally, we show that while the NAD+ hydrolase function of dSarm is required in vivo for efficient axon degeneration, it is dispensable for early suppression of axon transport.
Despite these clear molecular and genetic differences between early and late phase signaling events, WldS or dNmant expression, or hiw mutants are capable of suppressing early changes in axon transport and neurophysiology, even in bystander neurons. This could be interpreted as evidence for similarity in signaling mechanisms at early and late stages of dSarm signaling (i.e. that they act by maintaining NAD+). However, we favor the alternative possibility that these data point to an important role for dNmnat in mediating early dSarm signaling events during suppression of bystander neuron function. Loss of Axed does not affect the bystander effect, and axon transport is suppressed. However, we found that loss of dNmnat in axed null backgrounds (which allows for preservation of neuronal integrity despite loss of dNmnat) blocked the ability of injury to induce the bystander effect. This result reveals a paradoxical, positive role for dNmnat in promoting the bystander effect early. We suspect dNmnat exerts this effect through modulating MAPK signaling, whose interactions are complex: loss of Nmant has been shown to suppress MAPK signaling (Zhou et al., 2016), while increased Nmnat activity can also potently block the activation of MAPK signaling within the first few hours after axotomy (Yang et al., 2015). We propose that dNmnat activity is required early for the bystander effect, and that dNmnat levels need to be precisely tuned for proper signaling at each phase.
Glial Draper signaling promotes suppression of bystander neuron function
Glial cells are well-positioned to rapidly spread signals to all axons in the wing nerve. Much like Remak bundles in mammals, the Drosophila L1 wing nerve has glial cells that appear to wrap axons individually, which would imply that axon-to-axon signals must pass through glia. Our observation that selective elimination of Draper signaling in glia is sufficient to inhibit the spreading of injury signals to bystander neurons is consistent with an axon→glia→bystander neuron signaling event, although it is also possible that glia are directly injured by the axotomy and signal to bystander neurons without input from the severed axons. Given the similarities in the response of severed axons and those of bystanders (i.e. both block axon transport on the same time scale), and the selective effects of Draper on the bystander neuron axons, we favor the former model rather than the latter.
Draper signals to bystander neurons through a transcriptional JNK/dAP-1 cascade, likely through activating MMP-1. Nerve injury also rapidly activates JNK/c-Jun signaling in mammalian Schwann cells, where JNK/c-Jun mediate most aspects of Schwann cell injury responses and reprogramming events (Parkinson et al., 2008). This conserved glial response likely occurs in Schwann cell-like wrapping glia present in the Drosophila L1 wing nerve, although it may be activated in the subperineurial glia, which can act in a partially redundant fashion with wrapping glia (Neukomm et al., 2014). The involvement of Mmp-1 is intriguing given its well-known role in neuroinflammatory responses to brain injury in mammals where it functions to break down the extracellular matrix and has been proposed to promote diffuse axon injury (Bell et al., 2012; Candelario-Jalil et al., 2011). Other key components of the Draper signaling pathway—dCed-6 in particular, which is required for Draper signaling in all other known contexts—were not required for suppression of bystander neuron neurophysiology.
How bystander neurons receive injury signals and respond has remained unclear, although injury or disease-induced effects on bystander neurons is well-documented (Ising and Heneka, 2018). In most cases this has been explored in the context of bystander neuron cell death driven by neuroinflammatory cells. For instance, release of C1q, IL-1a, TNF from microglia following brain injury drive the formation of neurotoxic astrocytes which can promote the death of neurons through release of yet-to-be-identified toxins (Liddelow et al., 2017). Bystander neuronal cell death is also driven by brain-infiltrating inflammatory monocytes in viral encephalitis, in a way that is mediated by calpains (Howe et al., 2016), which are also important regulators of axon degeneration. Secondary axon degeneration (i.e. that occurring in neurons not damaged by the initial injury) can be driven in a way that requires intracellular Ca2+ release through IP3Rs and ryanodine receptors (Orem et al., 2017). These represent extreme cases of bystander effects, where cells undergo apoptosis or their axons degeneration. Whether dSarm/Sarm1 is involved in these effects is an open question. The model we employ is likely most relevant to partial nerve injury, where non-autonomous changes in bystander neurons have been well-documented (Meyer and Ringkamp, 2008). Uninjured bystander neurons in mild TBI models are certainly altered physiologically, in a reversible way (Greer et al., 2012). The molecular basis of any of these signaling events remains unknown, but our study points to dSarm/Sarm1 as a candidate mediator. It is interesting to note that in contrast to control mice, which show significant behavioral defects for hours after mild TBI, Sarm1−/− animals exhibited almost immediate recovery (Henninger et al., 2016)—and this was at a time point long before diffuse axon injury is observed in TBI models. It is plausible that this early loss of function is mediated in part by the bystander effect.
In summary, our study defines two genetically separable phases of dSarm signaling, places dSarm/Sarm1 at the heart of neuronal injury signaling throughout neural tissues, identifies new signaling partners for dSarm, and expands its role to regulating the responses of uninjured neurons to local tissue injury.
STAR★METHODS
RESOURCE AVAILABILITY
Leading Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Marc R. Freeman (freemmar@ohsu.edu).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
Data and Code Availability
This study did not generate any unique datasets or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Flies (D. melanogaster) were kept on standard cornmeal agar supplemented with dry yeast at 25 C unless stated otherwise. For MARCM crosses, the following gender was used: X chromosome: females, and autosomes (chromosomes 2L, 2R, 3L and 3R): males & females, respectively. We did not observe any gender specific differences in clone numbers or axon death phenotype. The list of genotypes used in this study are listed in KEY RESOURCE TABLE.
KEY RESOURCE TABLE
| Chemicals, Peptides, and Recombinant Proteins | ||
| Halocarbon Oil 27 | Sigma | Cat#H8773 |
| D-Glucose | Sigma | Cat#G7021 |
| One Shot™ TOP 10 Chemically Competent E. coli | ThermoFisher | Cat#C404010 |
| Gibson assembly | NEB | Cat#E5510S |
| Experimental Models: D. melanogaster genotype: | ||
| 10xUAS-ivs-mCD8-gfp | Bloomington Drosophila Stock Center (BDSC) | RRID:BDSC_32189 |
| elav-Gal4 | BDSC | RRID:BDSC_458 |
| FRT19A | BDSC | RRID:BDSC_1709 |
| 5xUAS-wldS | Freeman lab | N/A |
| hepr75 | BDSC | RRID:BDSC_6761 |
| OK371-Gal4 | BDSC | RRID:BDSC_26160 |
| FRT40A | BDSC | RRID:BDSC_8212 |
| FRTG13 (FRT42B) | BDSC | RRID:BDSC_1956 |
| ase-FLP2e | (Neukomm et al., 2014), Freeman lab | N/A |
| FRT2A (FRT79D-F) | BDSC | RRID:BDSC_1997 |
| FRT82B | BDSC | RRID:BDSC_2035 |
| slprBS06 | BDSC | RRID:BDSC_58807 |
| licG0252 | BDSC | RRID:BDSC_11880 |
| ask1MI02915 | BDSC | RRID:BDSC_36163 |
| mkk4e01458 | BDSC | RRID:BDSC_17956 |
| dnmnatD4790-1 | BDSC | RRID:BDSC_39698 |
| Nmnat:GFP:NmnatWT | BDSC | RRID:BDSC_80087 |
| hiwΔN | BDSC | RRID:BDSC_51637 |
| drprΔ5 | (MacDonald et al., 2006) | N/A |
| nSyb-Gal4 | BDSC | RRID:BDSC_39698 |
| ase-FLP3b | (Neukomm et al., 2014), Freeman lab | N/A |
| 5xUAS-drprRNAi | (MacDonald et al., 2006) | N/A |
| 5xUAS-mcherry-atg8a | BDSC | RRID:BDSC_37750 |
| 5xUAS-gfp-lamp | BDSC | RRID:BDSC_42714 |
| 5xUAS-syt-gfp | BDSC | RRID:BDSC_6925 |
| 5xUAS-syt-gfp | BDSC | RRID:BDSC_6926 |
| 5xUAS-Gal4 | This paper | N/A |
| PBac[brp(FRT.Stop)V5-2A-LexA-VP16]VK00033 | BDSC | RRID:BDSC_56747 |
| 13xLexAop-ivs-myr-gfp | BDSC | RRID:BDSC_56747 |
| 13xLexAop2-rab3-mcherry | BDSC | RRID:BDSC_52252 |
| 13xLexAop2-mcherry-atg8a | This paper | N/A |
| 13xLexAop-rCD2-rfp10 | BDSC | RRID:BDSC_58755 |
| nSyb-LexA | BDSC | RRID:BDSC_52247 |
| 20xUAS-ivs-gcamp6s | BDSC | RRID:BDSC_42746 |
| tub-Gal4 | BDSC | RRID:BDSC_5138 |
| nan-Gal4 | BDSC | RRID:BDSC_24903 |
| 5xUAS-dsarm RNAi | BDSC | RRID:BDSC_63525 |
| hsFLPD5.fco | BDSC | RRID:BDSC_55815 |
| dsarm896 | (Osterloh et al., 2012) | N/A |
| dsarmE1170A | This paper | N/A |
| dsarmywg2+ | This paper | N/A |
| axed0011 | (Neukomm et al., 2017) | N/A |
| axedMI13270 (transposon insertion) | BDSC | RRID:BDSC_58041 |
| cack | BDSC | RRID:BDSC_57064 |
| cacF | BDSC | RRID:BDSC_67175 |
| nos-Cas9 | BDSC | RRID:BDSC_54591 |
| 5xUAS-NMNd | This paper | N/A |
| 5xUAS-mcherry-T2A-NMNd | This paper | N/A |
| BAC CH321-38D07 | BDSC | RRID:BDSC_90151 |
| UAD-JraJbz (JraDN) | BDSC | RRID:BDSC 7217 |
| shark RNAi | VDRC | 105706 |
| dced-6 RNAi | VDRC | 108101 |
| bsk RNAi | VDRC | 104569 |
| raptor RNAi | VDRC | 13112 |
| 5xUAS-draper II | (Logan et al., 2012) | N/A |
| mmp-1 RNAi | (Uhlirova and Bohmann, 2006) | N/A |
| repo-Gal4 | BDSC | RRID:BDSC_7415 |
| Oligonucleotides | ||
| Primer for pattB-5xUAS-mCherry-T2A-NMNd forward: AAACCCCTCGAGCAAAACATGACAGATAGCGAGCTGATGCAGC | This paper | N/A |
| Primer for pattB-5xUAS-NMNd forward: AAAAACCTCGAGCAAAACATGGTGAGCAAGGGCGAGG | This paper | N/A |
| Primer for both NMNd recombinant DNA reverse: CCCCCATCTAGATCAAGTGTTCTGGAGGAACTGTTGC | This paper | N/A |
| Primer for atg8a forward: GGCAGGAATTCTATGAAGTTCCAATACAAGGAGGAGC | This paper | N/A |
| Primer for atg8a reverse: CGAGCCGTCGACTTAGTTAATTTTGGCCATGCCGTAAAC | This paper | N/A |
| Primer for mCherry forward: GGAAGATCTATGGTGAGCAAGGGC | This paper | N/A |
| Primer for mCherry reverse: TTTGGATCCCTTGTACAGCTCGTCCATGCC | This paper | N/A |
| Primer for 5’HMA forward: GAGGACCTGAGACCGACCTGAGACGGTTCGCAAGGGCATCTTC | This paper | N/A |
| Primer for 5’HMA reverse: AGAGAGGGTCTCGATCCGACTGGGATATCGCCTTC | This paper | N/A |
| Primer for 3’HMA forward: ATATATGGTCTCTTTCCTCGGGGAGCCAGTTTG | This paper | N/A |
| Primer for 3’HMA reverse: GACTTGACGGGACGGCTCTAGATATAGTAAATATTTTCGTTTTAAGCCACACGGG | This paper | N/A |
| Primer for dSarm endogenous mutation second half forward: ACTCGTTTCGCAGGCCATCGTGGCTGCCCTGAACTCGAAC | This paper | N/A |
| Primer for dSarm endogenous mutation first half reverse: CAGCCACGATGGCCTGCGAAACGAGTATCCCCTTATGAG | This paper | N/A |
| Single-guide RNA upstream for deletion: TCGTCTGCGATTGCTGCGAC | This paper | N/A |
| Single-guide RNA downstream for deletion: GCATCTGCCTCGGTTCATCG | This paper | N/A |
| Single-guide RNA upstream for rescue: CAAGAAGGCGATATCCCAGT | This paper | N/A |
| Single-guide RNA downstream for rescue: CTGGTCAAACTGGCTCCCCG | This paper | N/A |
| Recombinant DNA | ||
| pattB-5xUAS-NMNd | This paper | N/A |
| pattB-5xUAS-mCherry-T2A-NMNd | This paper | N/A |
| pBH-5’HMA-ywing-3’HMA | This paper | N/A |
| pBH-5’HMA-dSarm(E1170A)-3’HMA | This paper | N/A |
| pattB-13xLexA-mCherry-atg8a | This paper | N/A |
| pCFD3-dU6:3gRNA | Addgene | Cat#49410 |
| pBH vector | Bellen lab | N/A |
| Software and Algorithms | ||
| Prism8 | GraphPad | N/A |
| FIJI | imageJ | N/A |
| Other: Tools | ||
| MicroPoint Scissors (5-mm cutting edge) | EMS | Cat#72933-04 |
| Tweezers (high precision, ultra-fine) | EMS | Cat#78520-5 |
METHOD DETAILS
Axotomy assays
Adult flies were anesthetized on a CO2 pad, and wings were cut with MicroPoint Scissors at the indicated location (Fig 1A and E, and Fig S2). Flies were returned to the vials and incubated at 25°C after injury. The remaining wings were left attached to the fly body for behavioral assays or dissected for imaging.
Confocal microscopy
Live imaging of Vesicle trafficking:
Intact, injured or crushed wings (1 to 5 days old flies) were dissected and mounted in Halocarbon oil 27 beneath a coverslip and imaged immediately. Images of vesicle trafficking were acquired on a spinning disc microscopy (Zeiss Examiner Z1) with a 63X oil-immersion objective at the location indicated (Fig. 1A). Time-lapse images were acquired with CCD camera (Hamamatsu Orca Flash 4.0) using Zen software (Zeiss) for a 3 minutes window. The wings were sitting in halocarbon oil no longer than 15 min before imaging. All the wings were imaged including the those with only labeled severed neurons or intact neurons.
Sensory transduction assay and calcium imaging:
The procedures for sugar application and imaging were adapted from a previous study (Raad et al., 2016). Intact or injured wings (3 to 5 days old flies) were dissected and mounted in a drop of water (~1.5ul) under a coverslip, and imaged immediately. Time-lapse images were acquired on a 3I spinning disc microscopy (Zeiss Imager X1) with a 63X oil-immersion objective, CCD camera (Hamamatsu Orca Flash 4.0) and by slide book software at 1 frame every 1 s for a 5 minutes duration. 5 μl of D-glucose solution (200 mM in water, Sigma cat.# G6152) was loaded at the edge of coverslip at indicated time point in real time when the time course images were recorded.
Laser ablation and calcium imaging:
Flies (3 to 7 days old) were anesthetized with CO2 and had the dorsal side of both wings and abdomen glued to a coverslip using a UV light-cured adhesive. Sparse GCaMP6s-expressing neuronal clones were generated using MARCM. Wing nerve ablation time-lapse images were taken on a 3I spinning disc confocal microscope with a Micropoint laser ablation system (430nm). An ablation region was selected by manually drawing a line across the wing nerve at the location for ablation to occur. All cell bodies analyzed were within 200 μm from the site of ablation.
Image of Nmnat-GFP-NmnatWT:
Injured and intact wings of 3 days old adult flies were dissected and mounted between slide and coverslip with a drop of Halocarbon oil 27, and imaged immediately. Images were acquired on a spinning disc microscopy (Zeiss Examiner Z1) with a 63X oil-immersion objective, and CCD camera (Hamamatsu Orca Flash 4.0).
Transmission electron microscopy
Wings were prepared for TEM using microwave-assisted tissue processing as follows. Wings were dissected from anesthetized 5 dpe female flies and immediately placed in fresh modified Karnofsky’s fixative (2% glutaraldehyde, 4% paraformaldehyde, 0.1M sodium cacodylate buffer, pH 7.4). Primary fixation, secondary fixation in 2% OsO4 and imidazole, en bloc uranyl acetate staining, dehydration, and resin infiltration of samples was carried out in a Pelco laboratory microwave following the procedure for zebrafish larvae described (Cunningham and Monk, 2018). After resin infiltration, wings were flat embedded between two sheets of Aclar plastic. After overnight polymerization at 60C, the Aclar was removed and flat-embedded wings were trimmed to access the ROI and remounted in fresh resin for sectioning on an Ultramicrotome (Leica). 70nm sections were collected on Formvar coated grids, counterstained with 5% uranyl acetate for 20 minutes, and Reynold’s lead citrate for 7 minutes before being imaged on an FEI Technai T12 operating at 80kV with an Advanced Microscopy Techniques camera.
Behavioral assay
The procedures of behavior test were adapted from previous study (Li et al., 2016). Adult female flies (3 to 7 days old) were anesthetized on ice and decapitated with micro forceps. Decapitated flies were allowed to recover on wet kimwipes for 10 min, and those flies able to stand up and respond to light touch on the body were used for behavioral tests. An eyelash was used as the probe to stimulate gentle touch and kicking responses by touching the wing margin bristles for 1-2 s. For each animal, the wing was touched 3 times. If the animal did not respond to all 3 touches, this test was counted as no response, if it responded to any of the 3 touches, it was counted as a positive response. Each wing (intact or injured) was touched for 10 times, with ~30 s interval between each touch. The number of kicking response was recorded. Videos were recorded by iPhone 7.
Image analysis and quantification
Vesicle movement:
Kymographs were created in Fiji imageJ for time-lapse confocal images using the multiple kymograph plugin. Lysosomes, autophagosomes, mitochondria, or synaptic vesicles were classified as stationary (no movement during the 3 min video), or moving (with any movement, including oscillation during the 3 min video). The percentage of moving or stationary vesicles in the imaged region was calculated, and similar genotypes and conditions (e.g. injured vs uninjured) were pooled. The n value represents the total number of axons scored. If too many cells were labeled in a wing for unique identification, we only included those axons were clearly identifiable as severed or intact (by connection to cell bodies or lack thereof).
Calcium imaging:
Time-Lapse confocal images were analyzed by using Fiji ImageJ. The average fluorescent intensity of GFP in three individual chemosensilla were measured in each imaged wing. The F0 was defined as the average fluorescent intensity in 20 seconds before glucose application (figure 2d). Chemosensilla were identified based on morphology, or the change of fluorescent intensity upon glucose application. In injured wings, if there was no increase of the fluorescent intensity in any cell bodies, the percentage of ΔF/F0 was count as 0%.
Laser ablation and calcium imaging:
Time-lapse confocal images were analyzed using Fiji (ImageJ). Cell body average fluorescence intensity was analyzed by manually drawing regions of interest and F0 was defined as the average fluorescence intensity for the ten seconds before ablation. Outliers for values of either max amplitude or the time to maximum amplitude were identified (ROUT method, Q=1%)
Quantification of Nmnat-GFP-NmnatWT fluorescent intensity:
Confocal images were analyzed by using Fiji ImageJ. The average fluorescent intensity of the GFP signal of 5 neuron cell bodies were quantified in each wing, and 20 wings were quantified for both injured or uninjured wings.
Generation of dSarmE1170A fly
The dSarmE1170A fly was generated following a protocol described previously (Li-Kroeger et al., 2018). A 3795 bp region containing the DNA sequence for Glutamic acid at position 1170 (dsarm-PD isoform) was selected for swapping to ywmg2+ (Figure 2D). To construct the donor DNA (pBH-5’HMA-p{yWing2+}-3’HMA), the homology arms were PCR amplified from genomic DNA of y1 M{w+mC=nos-Cas9.P}ZH-2A w* (Bloomington fly center) flies (Primers listed in Key Resource Table). The homology arm (HMA), pBH donor vector, and p{ywing2+} were combined by Golden Gate assemble using restriction enzyme BsaI (NEB #E1601S). The product was transformed into TOP10 Chemically Competent Cells (ThermoFisher #404010), and proceeded to DNA preparation. The entire homology arm sequences and ~200 bps of the adjacent cassette sequences were verified by sequencing.
sgRNA were cloned into the pCFD3-dU6:3gRNA plasmid using standard protocols (www.flvcrisoprdesign.org). Briefly, sense and antisense oligos containing the 20 bp guide target sequence were annealed, and then inserted between Bbsl sites in the plasmid, and proceeded to DNA preparation.
To generate the dsarmywg2+ flies, a mix of 25 ng/μl sgRNA and 150 ng/μl donor DNA were injected into y1 M{w+mC=nos-Cas9.P}ZH-2A w* (Bloomington fly center) flies by Bestgene. Injected animals were then crossed to y,w flies. The offspring were screened for a yellow+ wings and crossed to the appropriate balancer. We sequence-verified the entire homology arm sequence and ~200 bps of the adjacent cassettes.
To replace the ywing2+ cassette with the dsarmE1170A sequence, we first generated the donor DNA with the E1170A mutation (pBH-5’HMA-dSarm(E1170A)-3’HMA) (primers listed in Key Resource Table). In brief, the primers contained mutated nucleotides were used for DNA fragments amplification (5’HMA-dSarm(E1170A) first half and dSarm(E1170A) second half-3’HMA), and all DNA fragments and pBH vector were combined by Gibson assembly (NEB #E5510S). The sgRNA was generated as explained above using different 20 bp guide target sequences (Key Resource Table). A mix of 25 ng/μl sgRNA and 150 ng/μl donor DNA were injected into our y; dsarmywg2+/CyO flies by Bestgene, and we screened for the loss of the yellow+ wing of progeny. These flies were balanced, and the whole replaced region was sequenced to confirm no additional mutations were generated.
5xUAS-NMN demidase and 13xLexAop-atg8-mcherry DNA cloning and transgenesis
The NMN deamidase DNA fragment were cloned from E. Coli NMN deamidase cDNA (Liu et al., 2018) with the primer pairs listed in Key Resource Table, and inserted into 5xUAS vector (Phi31 integration, 5xUAS, w+ marker, attB). The mCherry and atg8a DNA fragments were cloned from the DNA of 5xUAS-mCherry-Atg8a flies, with primers listed in Key Resource Table, and combined with attB-13xLexAop vector (Phi31 integration, 13xLexAop, w+ marker, attB). Both plasmids were injected into y1 w67c23; P{CaryP}attP40 flies by Bestgene.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data acquisition and quantification were performed in a non-blinded fashion. Statistical analyses were performed using GraphPad Prism 8. Statistical details (definition of test, exact value of n (e.g., number of wings, number of animals, etc.), mean ± deviations, p values) are listed in Figure Legends and below. All statistical comparisons were conducted on data originating from 3 or more biologically independent experimental replicates.
Supplementary Material
Video S1. Autophagosomes are highly motile in the axons of uninjured wings. (Related to Figure 1)
Fluorescent labeled autophagosomes (ok371-Gal4, UAS-mcherry-atg8a) were monitored in the axon clones of uninjured wings, video is recorded for 3 minutes.
Video S2. The transport of autophagosomes is blocked in the severed axons after wing injury. (Related to Figure 1)
Fluorescent labeled autophagosomes (ok371-Gal4, UAS-mcherry-atg8a) were monitored in the severed axon clones at 3 hours after wing injury, video is recorded for 3 minutes.
Video S3. The transport of autophagosomes are blocked in the intact neurons after wing injury. (Related to Figure 1)
Fluorescent labeled autophagosomes (ok371-Gal4, UAS-mcherry-atg8a) were monitored in intact neurons, which is distinguished by the connection of cell body, at 3 hours after wing injury, video is recorded for 3 minutes.
Video S4. The transport of autophagosomes in dSarm896 clones in the uninjured wings is normal. (Related to Figure 2)
Fluorescent labeled autophagosomes (ok371-Gal4, UAS-mcherry-atg8a) were monitored in the intact dSarm896 axon clones of uninjured wings, video is recorded for 3 minutes.
Video S5. The transport of autophagosomes is maintained in dSarm896 clones of both severed axons and intact neurons after wing injury. (Related to Figure 2)
Fluorescent labeled autophagosomes (ok371-Gal4, UAS-mcherry-atg8a) were monitored in the intact dSarm896 axon clones of uninjured wings, video is recorded for 3 minutes.
Video S6. Mechanical stimulation on the uninjured wing margins can elicit kicking responses. (Related to Figure 8)
Video S7. Wing injury strongly reduced the kicking response stimulated by the mechanical stimulation of the wing. (Related to Figure 8)
Video S8. Glucose exposure leads to robust activation of wing sensory neurons. (Related to Figure 8)
200mM of glucose was applied at the indicated time point, the activity of wing sensory neurons in the uninjured wings was monitored by GCamp6s calcium sensor (tub-Gal4, 20xuas-ivs-gcamp6s). Video was recorded for 5 minutes.
Video S9. Glucose-stimulated wing sensory neuron activation is suppressed within 3 hours after wing axotomy. (Related to Figure 8)
200mM of glucose was applied at the indicated time point, the activity of wing sensory neurons in the wild type wings within 3 hours after injury was monitored by GCamp6s calcium sensor (tub-Gal4, 20xuas-ivs-gcamp6s). Video was recorded for 5 minutes.
Highlight.
Partial nerve injury broadly suppresses neurophysiology, even in uninjured bystander neurons
Cac/dSarm/MAPK signaling is required in bystander neurons to suppress neurophysiology
dSarm NADase activity is required for axon degeneration but not bystander effects
Glia spread injury signals to suppress bystander neurons through Draper signaling
ACKNOWLEDGEMENTS
We thank all the lab members of Freeman lab for the suggestions and discussions on this work and manuscript writing. We thank Dr. Richard Goodman in OHSU for sharing E. Coli NMNdeamidase cDNA. We thank Dr. Li-Kroeger in Dr. Hugo Bellen lab in Baylor College of Medicine for sharing pBH vector and helping on dsarmE1170A fly generation. We thank Dr. Lukas Neukomm in University of Lausanne, Switzerland, for sharing 5xUAS-Gal4 transgenic flies. We thank Dr. Kelly Monk for providing the microwave and assisting with EM fixation procedures, Jo Hill and Sue Aicher for technical support with TEM, and the P30 grant number OHSU Neuroscience Imaging Center grant NS061800. We thank Dr. Sean D. Speese for sharing the technology of imaging Ca2+ response after laser ablation on living animals. This work was supported by NIH RO1 NS059991 (to MRF) and OHSU.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
MRF is a co-founder of Nura Bio, whose goal is to block axon loss in disease.
REFERENCES
- Awasaki T, Tatsumi R, Takahashi K, Arai K, Nakanishi Y, Ueda R, and Ito K (2006). Essential role of the apoptotic cell engulfment genes draper and ced-6 in programmed axon pruning during Drosophila metamorphosis. Neuron 50, 855–867. [DOI] [PubMed] [Google Scholar]
- Babetto E, Beirowski B, Russler EV, Milbrandt J, and DiAntonio A (2013). The Phr1 ubiquitin ligase promotes injury-induced axon self-destruction. Cell Rep 3, 1422–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, Holtzman DM, Betsholtz C, Armulik A, Sallstrom J, et al. (2012). Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 485, 512–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bratkowski M, Xie T, Thayer DA, Lad S, Mathur P, Yang YS, Danko G, Burdett TC, Danao J, Cantor A, et al. (2020). Structural and Mechanistic Regulation of the Pro-degenerative NAD Hydrolase SARM1. Cell Rep 32, 107999. [DOI] [PubMed] [Google Scholar]
- Burke RE, and O’Malley K (2013). Axon degeneration in Parkinson’s disease. Exp Neurol 246, 72–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candelario-Jalil E, Thompson J, Taheri S, Grossetete M, Adair JC, Edmonds E, Prestopnik J, Wills J, and Rosenberg GA (2011). Matrix metalloproteinases are associated with increased blood-brain barrier opening in vascular cognitive impairment. Stroke 42, 1345–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang CF, and Bargmann CI (2005). A Toll-interleukin 1 repeat protein at the synapse specifies asymmetric odorant receptor expression via ASK1 MAPKKK signaling. Genes Dev 19, 270–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins CA, Wairkar YP, Johnson SL, and DiAntonio A (2006). Highwire restrains synaptic growth by attenuating a MAP kinase signal. Neuron 51, 57–69. [DOI] [PubMed] [Google Scholar]
- Cunningham RL, and Monk KR (2018). Transmission Electron Microscopy for Zebrafish Larvae and Adult Lateral Line Nerve. Methods Mol Biol 1739, 385–400. [DOI] [PubMed] [Google Scholar]
- Di Stefano M, Nascimento-Ferreira I, Orsomando G, Mori V, Gilley J, Brown R, Janeckova L, Vargas ME, Worrell LA, Loreto A, et al. (2014). A rise in NAD precursor nicotinamide mononucleotide (NMN) after injury promotes axon degeneration. Cell Death Differ 22, 731–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty J, Logan MA, Tasdemir OE, and Freeman MR (2009). Ensheathing glia function as phagocytes in the adult Drosophila brain. J Neurosci 29, 4768–4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty J, Sheehan AE, Bradshaw R, Fox AN, Lu TY, and Freeman MR (2014). PI3K signaling and Stat92E converge to modulate glial responsiveness to axonal injury. PLoS Biol 12, e1001985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essuman K, Summers DW, Sasaki Y, Mao X, DiAntonio A, and Milbrandt J (2017). The SARM1 Toll/Interleukin-1 Receptor Domain Possesses Intrinsic NAD(+) Cleavage Activity that Promotes Pathological Axonal Degeneration. Neuron 93, 1334–1343 e1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y, Soares L, Teng X, Geary M, and Bonini NM (2012). A novel Drosophila model of nerve injury reveals an essential role of Nmnat in maintaining axonal integrity. Curr Biol 22, 590–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman MR, Delrow J, Kim J, Johnson E, and Doe CQ (2003). Unwrapping glial biology: Gcm target genes regulating glial development, diversification, and function. Neuron 38, 567–580. [DOI] [PubMed] [Google Scholar]
- Geisler S, Huang SX, Strickland A, Doan RA, Summers DW, Mao X, Park J, DiAntonio A, and Milbrandt J (2019). Gene therapy targeting SARM1 blocks pathological axon degeneration in mice. J Exp Med 216, 294–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdts J, Brace EJ, Sasaki Y, DiAntonio A, and Milbrandt J (2015). SARM1 activation triggers axon degeneration locally via NAD(+) destruction. Science 348, 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdts J, Summers DW, Milbrandt J, and DiAntonio A (2016). Axon Self-Destruction: New Links among SARM1, MAPKs, and NAD+ Metabolism. Neuron 89, 449–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdts J, Summers DW, Sasaki Y, DiAntonio A, and Milbrandt J (2013). Sarm1-mediated axon degeneration requires both SAM and TIR interactions. J Neurosci 33, 13569–13580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilley J, and Coleman MP (2010). Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol 8, e1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilley J, Orsomando G, Nascimento-Ferreira I, and Coleman MP (2015). Absence of SARM1 rescues development and survival of NMNAT2-deficient axons. Cell Rep 10, 1974–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer JE, Povlishock JT, and Jacobs KM (2012). Electrophysiological abnormalities in both axotomized and nonaxotomized pyramidal neurons following mild traumatic brain injury. J Neurosci 32, 6682–6687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henninger N, Bouley J, Sikoglu EM, An J, Moore CM, King JA, Bowser R, Freeman MR, and Brown RH Jr. (2016). Attenuated traumatic axonal injury and improved functional outcome after traumatic brain injury in mice lacking Sarm1. Brain 139, 1094–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill CS, Coleman MP, and Menon DK (2016). Traumatic Axonal Injury: Mechanisms and Translational Opportunities. Trends Neurosci 39, 311–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe CL, LaFrance-Corey RG, Mirchia K, Sauer BM, McGovern RM, Reid JM, and Buenz EJ (2016). Neuroprotection mediated by inhibition of calpain during acute viral encephalitis. Sci Rep 6, 28699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ising C, and Heneka MT (2018). Functional and structural damage of neurons by innate immune mechanisms during neurodegeneration. Cell Death Dis 9, 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara M, Menon DK, Salmond CH, Outtrim JG, Taylor Tavares JV, Carpenter TA, Pickard JD, Sahakian BJ, and Stamatakis EA (2010). Altered functional connectivity in the motor network after traumatic brain injury. Neurology 75, 168–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay JN, Chu MW, and Sanes JR (2012). MEGF10 and MEGF11 mediate homotypic interactions required for mosaic spacing of retinal neurons. Nature 483, 465–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneynsberg A, Combs B, Christensen K, Morfini G, and Kanaan NM (2017). Axonal Degeneration in Tauopathies: Disease Relevance and Underlying Mechanisms. Front Neurosci 11, 572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T, and Luo L (1999). Mosaic analysis with a repressible cell marker for studies of gene function in neuronal morphogenesis. Neuron 22, 451–461. [DOI] [PubMed] [Google Scholar]
- Lewis TL Jr., Turi GF, Kwon SK, Losonczy A, and Polleux F (2016). Progressive Decrease of Mitochondrial Motility during Maturation of Cortical Axons In Vitro and In Vivo. Curr Biol 26, 2602–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhang W, Guo Z, Wu S, Jan LY, and Jan YN (2016). A Defensive Kicking Behavior in Response to Mechanical Stimuli Mediated by Drosophila Wing Margin Bristles. J Neurosci 36, 11275–11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li-Kroeger D, Kanca O, Lee PT, Cowan S, Lee MT, Jaiswal M, Salazar JL, He Y, Zuo Z, and Bellen HJ (2018). An expanded toolkit for gene tagging based on MiMIC and scarless CRISPR tagging in Drosophila. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Munch AE, Chung WS, Peterson TC, et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HW, Smith CB, Schmidt MS, Cambronne XA, Cohen MS, Migaud ME, Brenner C, and Goodman RH (2018). Pharmacological bypass of NAD(+) salvage pathway protects neurons from chemotherapy-induced degeneration. Proc Natl Acad Sci U S A 115, 10654–10659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan MA, Hackett R, Doherty J, Sheehan A, Speese SD, and Freeman MR (2012). Negative regulation of glial engulfment activity by Draper terminates glial responses to axon injury. Nat Neurosci 15, 722–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loreto A, Di Stefano M, Gering M, and Conforti L (2014). Wallerian Degeneration Is Executed by an NMN-SARM1-Dependent Late Ca(2+) Influx but Only Modestly Influenced by Mitochondria. Cell Rep 13, 2539–2552. [DOI] [PubMed] [Google Scholar]
- Lu TY, MacDonald JM, Neukomm LJ, Sheehan AE, Bradshaw R, Logan MA, and Freeman MR (2017). Axon degeneration induces glial responses through Draper-TRAF4-JNK signalling. Nat Commun 8, 14355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunn ER, Perry VH, Brown MC, Rosen H, and Gordon S (1989). Absence of Wallerian Degeneration does not Hinder Regeneration in Peripheral Nerve. Eur J Neurosci 1, 27–33. [DOI] [PubMed] [Google Scholar]
- MacDonald JM, Beach MG, Porpiglia E, Sheehan AE, Watts RJ, and Freeman MR (2006). The Drosophila cell corpse engulfment receptor Draper mediates glial clearance of severed axons. Neuron 50, 869–881. [DOI] [PubMed] [Google Scholar]
- McPhee CK, Logan MA, Freeman MR, and Baehrecke EH (2010). Activation of autophagy during cell death requires the engulfment receptor Draper. Nature 465, 1093–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer RA, and Ringkamp M (2008). A role for uninjured afferents in neuropathic pain. Sheng Li Xue Bao 60, 605–609. [PubMed] [Google Scholar]
- Milde S, Fox AN, Freeman MR, and Coleman MP (2013a). Deletions within its subcellular targeting domain enhance the axon protective capacity of Nmnat2 in vivo. Sci Rep 3, 2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milde S, Gilley J, and Coleman MP (2013b). Axonal trafficking of NMNAT2 and its roles in axon growth and survival in vivo. Bioarchitecture 3, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neukomm LJ, Burdett TC, Gonzalez MA, Zuchner S, and Freeman MR (2014). Rapid in vivo forward genetic approach for identifying axon death genes in Drosophila. Proc Natl Acad Sci U S A 111, 9965–9970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neukomm LJ, Burdett TC, Seeds AM, Hampel S, Coutinho-Budd JC, Farley JE, Wong J, Karadeniz YB, Osterloh JM, Sheehan AE, et al. (2017). Axon Death Pathways Converge on Axundead to Promote Functional and Structural Axon Disassembly. Neuron 95, 78–91 e75. [DOI] [PubMed] [Google Scholar]
- Orem BC, Pelisch N, Williams J, Nally JM, and Stirling DP (2017). Intracellular calcium release through IP3R or RyR contributes to secondary axonal degeneration. Neurobiol Dis 106, 235–243. [DOI] [PubMed] [Google Scholar]
- Osterloh JM, Yang J, Rooney TM, Fox AN, Adalbert R, Powell EH, Sheehan AE, Avery MA, Hackett R, Logan MA, et al. (2012). dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science 337, 481–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson DB, Bhaskaran A, Arthur-Farraj P, Noon LA, Woodhoo A, Lloyd AC, Feltri ML, Wrabetz L, Behrens A, Mirsky R, et al. (2008). c-Jun is a negative regulator of myelination. J Cell Biol 181, 625–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purice MD, Ray A, Munzel EJ, Pope BJ, Park DJ, Speese SD, and Logan MA (2017). A novel Drosophila injury model reveals severed axons are cleared through a Draper/MMP-1 signaling cascade. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raad H, Ferveur JF, Ledger N, Capovilla M, and Robichon A (2016). Functional Gustatory Role of Chemoreceptors in Drosophila Wings. Cell Rep 15, 1442–1454. [DOI] [PubMed] [Google Scholar]
- Tian X, Gala U, Zhang Y, Shang W, Nagarkar Jaiswal S, di Ronza A, Jaiswal M, Yamamoto S, Sandoval H, Duraine L et al. (2015). A voltage-gated calcium channel regulates lysosomal fusion with endosomes and autophagosomes and is required for neuronal homeostasis. PLoS Biol 13, e1002103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turkiew E, Falconer D, Reed N, and Hoke A (2017). Deletion of Sarm1 gene is neuroprotective in two models of peripheral neuropathy. J Peripher Nerv Syst 22, 162–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlirova M, and Bohmann D (2006). JNK- and Fos-regulated Mmp1 expression cooperates with Ras to induce invasive tumors in Drosophila. EMBO J 25, 5294–5304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker LJ, Summers DW, Sasaki Y, Brace EJ, Milbrandt J, and DiAntonio A (2017). MAPK signaling promotes axonal degeneration by speeding the turnover of the axonal maintenance factor NMNAT2. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Zhai Q, Chen Y, Lin E, Gu W, McBurney MW, and He Z (2005). A local mechanism mediates NAD-dependent protection of axon degeneration. J Cell Biol 170, 349–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Zhang S, Liu T, Wang H, Liu K, Wang Q, and Zeng W (2018). Sarm1/Myd88-5 Regulates Neuronal Intrinsic Immune Response to Traumatic Axonal Injuries. Cell Rep 23, 716–724. [DOI] [PubMed] [Google Scholar]
- Wu G, Ringkamp M, Hartke TV, Murinson BB, Campbell JN, Griffin JW, and Meyer RA (2001). Early onset of spontaneous activity in uninjured C-fiber nociceptors after injury to neighboring nerve fibers. J Neurosci 21, RC140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HH, Bellmunt E, Scheib JL, Venegas V, Burkert C, Reichardt LF, Zhou Z, Farinas I, and Carter BD (2009). Glial precursors clear sensory neuron corpses during development via Jedi-1, an engulfment receptor. Nat Neurosci 12, 1534–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X, Hao Y, Sun K, Li J, Li X, Mishra B, Soppina P, Wu C, Hume RI, and Collins CA (2012). The Highwire ubiquitin ligase promotes axonal degeneration by tuning levels of Nmnat protein. PLoS Biol 10, e1001440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Wu Z, Renier N, Simon DJ, Uryu K, Park DS, Greer PA, Tournier C, Davis RJ, and Tessier-Lavigne M (2015). Pathological axonal death through a MAPK cascade that triggers a local energy deficit. Cell 160, 161–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao ZY, Xie XJ, Li WH, Liu J, Chen Z, Zhang B, Li T, Li SL, Lu JG, Zhang L et al. (2019). A Cell-Permeant Mimetic of NMN Activates SARM1 to Produce Cyclic ADP-Ribose and Induce Non-apoptotic Cell Death. iScience 15, 452–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou RM, Shen Y, Yao J, Yang H, Shan K, Li XM, Jiang Q, and Yan B (2016). Nmnat 1: a Security Guard of Retinal Ganglion Cells (RGCs) in Response to High Glucose Stress. Cell Physiol Biochem 38, 2207–2218. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. Autophagosomes are highly motile in the axons of uninjured wings. (Related to Figure 1)
Fluorescent labeled autophagosomes (ok371-Gal4, UAS-mcherry-atg8a) were monitored in the axon clones of uninjured wings, video is recorded for 3 minutes.
Video S2. The transport of autophagosomes is blocked in the severed axons after wing injury. (Related to Figure 1)
Fluorescent labeled autophagosomes (ok371-Gal4, UAS-mcherry-atg8a) were monitored in the severed axon clones at 3 hours after wing injury, video is recorded for 3 minutes.
Video S3. The transport of autophagosomes are blocked in the intact neurons after wing injury. (Related to Figure 1)
Fluorescent labeled autophagosomes (ok371-Gal4, UAS-mcherry-atg8a) were monitored in intact neurons, which is distinguished by the connection of cell body, at 3 hours after wing injury, video is recorded for 3 minutes.
Video S4. The transport of autophagosomes in dSarm896 clones in the uninjured wings is normal. (Related to Figure 2)
Fluorescent labeled autophagosomes (ok371-Gal4, UAS-mcherry-atg8a) were monitored in the intact dSarm896 axon clones of uninjured wings, video is recorded for 3 minutes.
Video S5. The transport of autophagosomes is maintained in dSarm896 clones of both severed axons and intact neurons after wing injury. (Related to Figure 2)
Fluorescent labeled autophagosomes (ok371-Gal4, UAS-mcherry-atg8a) were monitored in the intact dSarm896 axon clones of uninjured wings, video is recorded for 3 minutes.
Video S6. Mechanical stimulation on the uninjured wing margins can elicit kicking responses. (Related to Figure 8)
Video S7. Wing injury strongly reduced the kicking response stimulated by the mechanical stimulation of the wing. (Related to Figure 8)
Video S8. Glucose exposure leads to robust activation of wing sensory neurons. (Related to Figure 8)
200mM of glucose was applied at the indicated time point, the activity of wing sensory neurons in the uninjured wings was monitored by GCamp6s calcium sensor (tub-Gal4, 20xuas-ivs-gcamp6s). Video was recorded for 5 minutes.
Video S9. Glucose-stimulated wing sensory neuron activation is suppressed within 3 hours after wing axotomy. (Related to Figure 8)
200mM of glucose was applied at the indicated time point, the activity of wing sensory neurons in the wild type wings within 3 hours after injury was monitored by GCamp6s calcium sensor (tub-Gal4, 20xuas-ivs-gcamp6s). Video was recorded for 5 minutes.
Data Availability Statement
This study did not generate any unique datasets or code.