

Abstract
Clinical laboratory tests are a critical component of the continuum of care. We evaluate the genetic basis of 35 blood and urine laboratory measurements in the UK Biobank (n=363,228 individuals). We identify 1,857 loci associated with at least one trait, containing 3,374 fine-mapped associations, and additional sets of large-effect (> 0.1 sd) protein-altering, HLA, and copy-number variant associations. Through Mendelian Randomization analysis, we discover 51 causal relationships, including previously known agonistic effects of urate on gout and cystatin C on stroke. Finally, we develop polygenic risk scores for each biomarker and built ‘multi-PRS’ models for diseases using 35 PRSs simultaneously, which improved chronic kidney disease, type 2 diabetes, gout, and alcoholic cirrhosis genetic risk stratification in an independent dataset (FinnGen; n=135,500) relative to single-disease PRSs. Together, our results delineate the genetic basis of biomarkers, their causal influences on diseases, and improve genetic risk stratification for common diseases.
Introduction
Serum and urine biomarkers are frequently measured to diagnose and monitor chronic disease conditions. Understanding the genetic predisposition to particular biomarker states, and the factors that confound them, may have implications for disease treatment. While the genetics of some biomarkers have been extensively studied, most notably lipids1,2,3, glycemic traits4–6, and measurements of kidney function7–9, the genetic basis of most biomarkers has not been queried in large population-scale datasets.
To this end, UK Biobank (UKB) has performed laboratory testing of >30 commonly measured biomarkers in serum and urine on a cohort of >480,000 individuals with extensive phenotype and genome-wide genotype data, including the unrelated individuals in this study (Supplementary Figure 1)10.
Here, we 1) performed a systematic analysis of the genetic architecture and detailed fine-mapping of biomarker-associated loci in 363,228 individuals including protein-altering, protein-truncating (PTV), non-coding, human leukocyte antigen (HLA), and copy number variants; 2) built phenome-wide associations for implicated genetic variants; 3) evaluated causal relationships between biomarkers and 40 medically relevant phenotypes; and 4) constructed polygenic prediction models (Figure 1).
Figure 1. Schematic overview of the study.
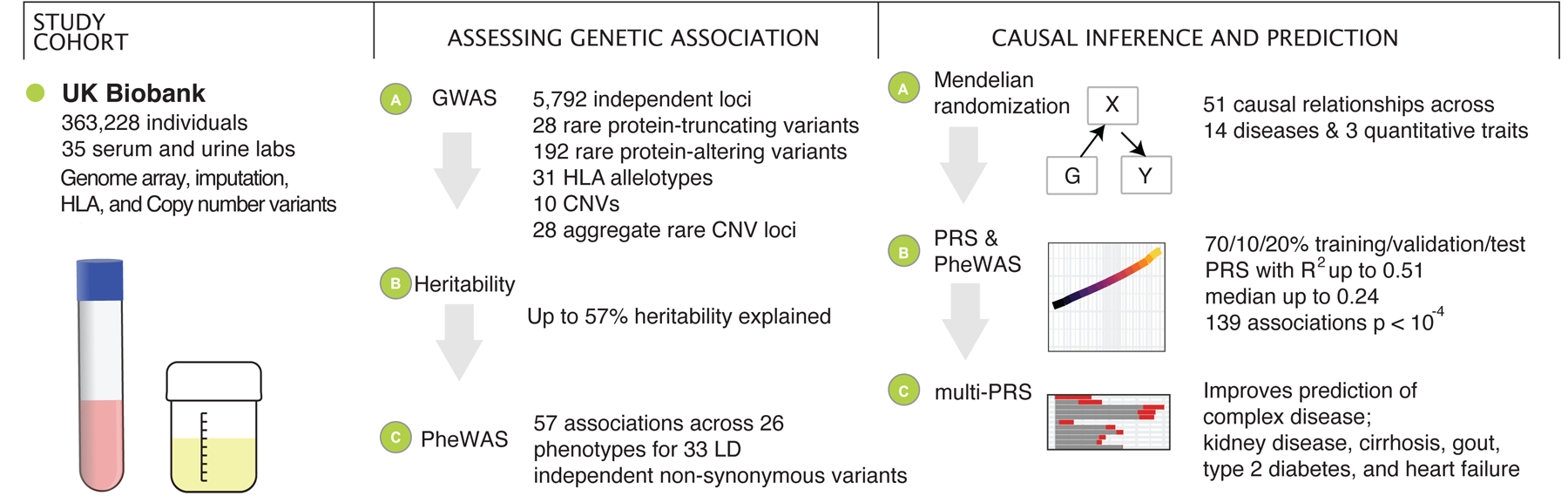
We prepared a dataset of 35 serum and urine biomarkers from 363,228 individuals in UK Biobank. We analyzed the genetic basis of these biomarkers, assessed their relationship to medically relevant phenotypes, and generated predictive models of disease outcomes from genome-wide data.
Results
Biomarker phenotype distributions
We first examined the consistency of the biomarker measurements10,11. After adjusting for statin usage (Supplementary Table 1a–c), we fit a regression model with multiple covariates (see Methods). For each biomarker, we measured the proportion of phenotypic variance explained by these covariates; this ranged from 1.7% (Rheumatoid factor) to 90% (Testosterone) depending on the biomarker (Supplementary Figure 2a–c, Supplementary Table 2). We evaluated body mass index as a confounder in associations, and there were minimal differences in genetic effects under this model (Supplementary Tables 3–4). Taking all the 35 lab phenotypes together, we recover several previously estimated phenotype correlations (Figure 2)12,13.
Figure 2. Genetics of 35 biomarkers. (top left inset).
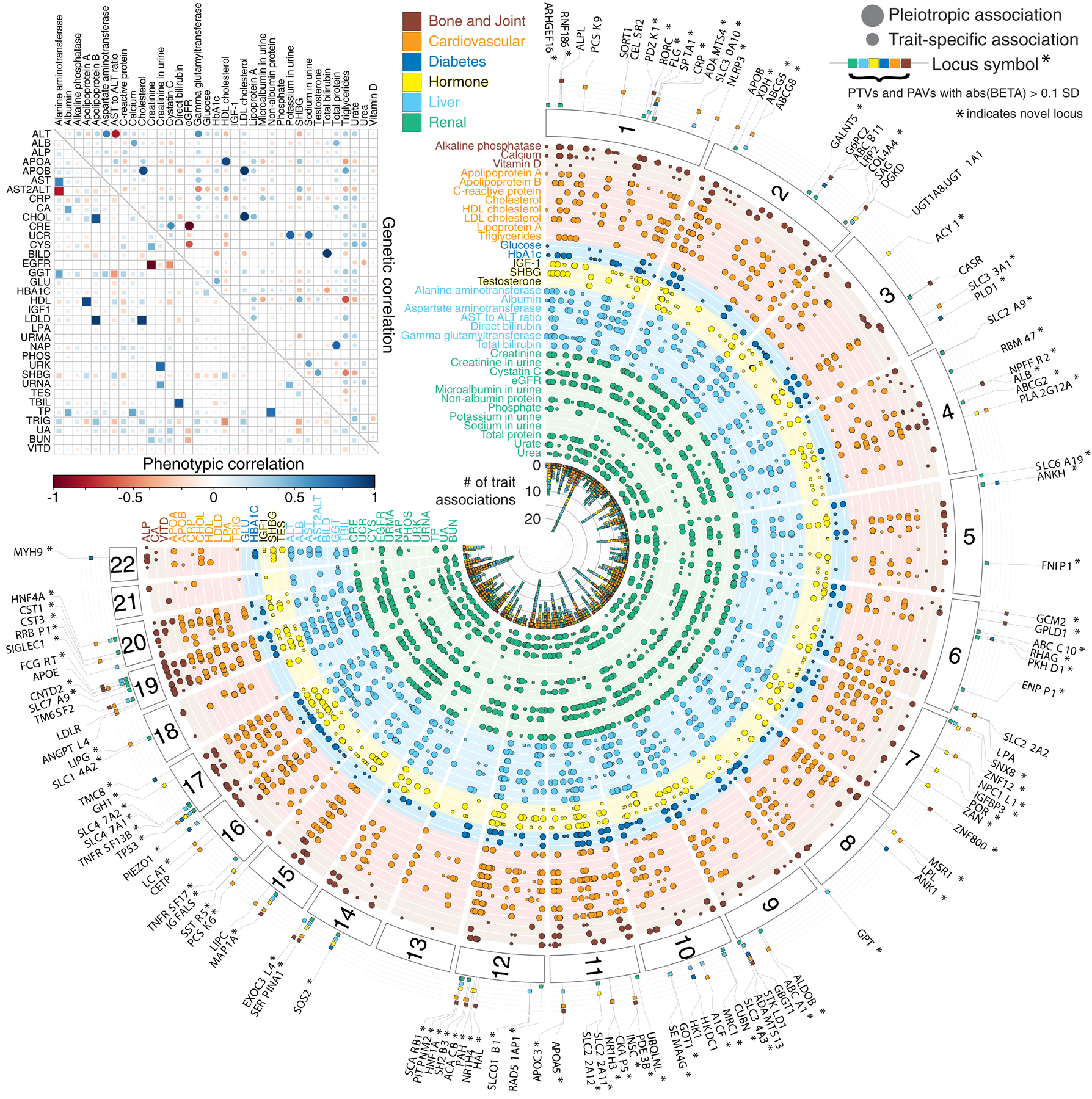
Correlation of phenotypic (lower triangular matrix) and genetic (upper triangular matrix) effects plot between the 35 lab phenotypes, estimated using LD Score regression. The absolute heritability estimates with standard errors are in Supplementary Table 11a. (main panel) Fuji plot of lab phenotypes across the six categories provided by UK Biobank and genetic variant associations shown for LD independent variants with meta-analysis p < 5 × 10−9. Large-effect protein-truncating and protein-altering variants (labeled when abs(beta) >= 0.1 standard deviation [SD]) annotated with the category of association displayed (colored fill boxes) and highlighted if the loci were not previously reported in the comparison studies (Methods). Pleiotropic association and trait-specific association are shown by different sized circles. The p-values were from two-sided tests and were not corrected for multiple hypothesis testing.
Genetics of biomarkers
We performed association analysis between directly genotyped and imputed autosomal genetic variants, copy number variations (CNVs), and HLA allelotypes and 35 biomarkers in the unrelated individuals in UK Biobank across 5 population groups (N = 318,953 for White British, 23,582 for non-British White, 6,019 for African, 7,338 for South Asian, and 1,082 for East Asian) followed by meta-analysis of all but East Asian populations (N in meta-analysis = 355,891, Methods, Figure 2, Supplementary Figure 3). We stratified the genetic variants into three bins: 1) protein-truncating (27,816), 2) protein-altering (87,430), and 3) synonymous and non-coding variants (minor allele frequency [MAF] > 0.1% and INFO score > 0.3, imputed variants present in Haplotype Reference Consortium [HRC], 9,444,56114) (Figure 2). Comparison of effect sizes estimated across 42 other previously published study cohorts for 25 of the biomarkers showed overall high agreement (Supplementary Figure 4, Supplementary Table 5). This was true when comparing to previous studies of lipids1,2,15,16, glycemic traits17,18, kidney function tests19,20, liver function tests17, and other biomarker measurements21,22.
We adjusted the nominal association p-values for multiple hypothesis testing and identified over 10,000 significant associations (Bonferroni-corrected meta-analysis p < 5 × 10−9 for assayed and imputed variants; Bonferroni-corrected p < 1 × 10−6 for non-rare [MAF > 0.1%] CNVs and CNV burden test for 23,598 genes; and Benjamini-Yekutieli [BY] adjusted p < 0.05 for HLA alleles, Methods, Supplementary Figure 5, Supplementary Tables 6–10). Linkage disequilibrium (LD) Score intercepts for single-variant association results were between 0.999 and 1.137 for all 35 phenotypes, consistent with anthropometric traits in UKB and suggesting that population structure in our analysis is well-controlled23 (Supplementary Table 11a).
Global and local heritability of biomarkers
To characterize the heritability of the 35 biomarkers we first applied LD Score regression24 to stratify heritability into 10 tissue types and 53 general genomic features (e.g. coding variants and regulatory variants) and further applied the Heritability Estimator from Summary Statistics (HESS)25,26. We found that both LD Score regression and HESS indicate common SNPs explain substantial heritability of some but not all biomarkers (0.6% [Lipoprotein A, also referred to as lipoprotein(a)] to 23.9% [IGF-1] using LD Score regression and 3.2% [Microalbumin in urine] to 57% [Total bilirubin] using HESS across the studied continuous phenotypes, Supplementary Tables 11a,b). Estimates were lower in LD Score regression than HESS for traits with lower polygenicity (e.g. Lipoprotein A, h2ldsc = 0.6% and h2HESS = 24%), as LD Score regression estimates polygenic heritability24. We compared the polygenicity of all 35 biomarkers by computing the fraction of total SNP heritability attributable to loci by the top 1% of SNPs. We found that three biomarkers have more than 50% of the SNP heritability explained by the top 1% of loci (Lipoprotein A 67.7%, total bilirubin 60.9%, and direct bilirubin 57.5%) while the remaining 32 phenotypes show patterns of moderate to high polygenicity (Supplementary Table 11b).
Associated variants prioritize therapeutic targets
We found 58 (43 rare, MAF < 1%, and 55 not reported in comparison study, Methods) PTV associations and 1,323 (306 rare, 1,079 not reported in comparison studies) protein-altering variant (PAV) associations outside the major histocompatibility complex (MHC) region (hg19 chr6:25,477,797–36,448,354; meta-analyzed p < 5 × 10−9). We found 19 non-MHC PTV associations (17 rare [MAF < 1%]) with large estimated biomarker-lowering effects (>0.1 sd) and 26 (24 rare) with biomarker-raising effects (>0.1 sd) across 31 (27 rare) PTVs and at least one biomarker phenotype, where the same PTV may have both increasing and decreasing associations across different biomarkers (Figure 2, Supplementary Table 6). Similarly, there were 240 (161 rare) and 182 (125 rare) non-MHC PAV associations with large estimated lowering and raising effects (>0.1 sd) across 241 (179 rare) PAVs and at least one biomarker phenotype, respectively (Figure 2, Supplementary Table 7). To assess whether the variants associated with biomarkers impact medically relevant phenotypes, we performed a phenome-wide association analysis (PheWAS) across 166 traits in UK Biobank, compared our findings with previously published literature, and sought independent replication in the FinnGen R2 cohort (Supplementary Tables 12–13, Methods). We found 57 phenotype associations (33 and 24 for increasing and decreasing disease risk, respectively) across 26 medically relevant phenotypes for 2 PTVs and 31 PAVs (p < 1 × 10−7), of which 31 associations were previously reported and 26 were novel (Supplementary Tables 13a, Methods).
For eight cardiovascular biomarkers (Supplementary Table 4a), we identified a stop-gain variant in PDE3B with documented protection against high cholesterol and a range of effects on increasing HDL cholesterol and Apolipoprotein A (0.40, 0.27 sd) and decreasing triglycerides and Apolipoprotein B (0.43, 0.27 sd)2,27; a stop gain variant in ANGPTL8, where we replicated a previously-reported effect on HDL cholesterol (0.06 sd in our dataset) and discovered a triglyceride-lowering effect (0.06 sd)28; two PTVs in LPA with lowering effects on Lipoprotein A levels (0.37, 0.42 sd), of which one is known to be associated with decreased risk of coronary artery disease (p = 3 × 10−11; OR = 0.89 [95% CI 0.86, 0.92])29; a 0.2% MAF missense allele in ACACB associated with LDL, triglyceride, ApoB, and alkaline phosphatase30; two independent missense alleles in PLA2G12A with increasing effects on triglycerides, sex-hormone binding globulin (SHBG), and testosterone, and lowering effects on HDL cholesterol, ApoA, and HbA1c levels (Supplementary Table 6); a splice region variant in CPT1A, with lowering effects on triglycerides; and a missense variant in PCSK6 with ApoB- and LDL-lowering effects (Supplementary Table 7).
For seven liver biomarkers (Supplementary Table 4a), we found a 0.05% MAF inframe deletion in GOT1 with a lowering effect on aspartate aminotransferase (2.6 sd); a 0.1% MAF missense allele in SLC30A10 with increasing effects on alanine and aspartate aminotransferases; four missense alleles in GPT with alanine aminotransferase lowering effects; a missense variant in ABCB4 with increasing effect on alanine aminotransferase and increased risk of gallstones in UK Biobank (p = 1.2 × 10−8, OR = 1.38 [95% CI: 1.23, 1.38]); an allelic series of 3 missense variants in SERPINA1 with pleiotropic increasing effects on albumin, aspartate aminotransferase, direct bilirubin, and gamma glutamyltransferase, and lowering effects on AST to ALT ratio, with one of these missense alleles associated with increased risk of gallstones (p = 8.1 × 10−17, OR = 1.36 [95% CI: 1.27, 1.47]) and cholecystitis (p = 1.6 × 10−8, OR = 1.26 [95% CI: 1.16, 1.37]) in UK Biobank; and two missense alleles in DGKD, with raising and lowering effects, respectively, on direct and total bilirubin (Supplementary Tables 7, 13a).
For 12 renal biomarkers (Supplementary Table 4a), we found a PTV in COL4A4 associated with an increasing effect on microalbumin in urine (0.77 sd) and an increased risk of kidney disease (p = 6.7 × 10−13, OR = 6.9 [95% CI: 4.06, 11.6]) in UK Biobank, which is defined using a combination of hospital in-patient record (ICD-10 codes: Q60 [Renal agenesis and other reduction defects of kidney], and its sub-concepts) and self-report kidney diseases (coded as 1405 [other renal/kidney problem] in UK Biobank)31; a frame-shift variant in SLC22A2 with strong lowering effects on eGFR (0.52 sd) and increasing effect on creatinine (0.52 sd); a stop-gain variant in SLC22A11 with raising effects on urate (0.14 sd; Supplementary Tables 6, 13a); a 0.1% rare missense allele in SLC34A3 with strong eGFR and phosphate lowering and serum creatinine, Cystatin C, and urea raising effects; missense alleles in SLC6A19, LRP2, ALDOB, and SLC7A9, and two missense variants in SLC25A45, all associated with creatinine lowering and eGFR raising, among other examples (Supplementary Table 7). Notably, the majority of these genes are known to have high expression levels in renal tissue32.
For three bone and joint biomarkers (Supplementary Table 4a), we found an allelic series of two frame-shift variants and a missense variant in GPLD133, in addition to an allelic series of missense variants in ALPL. Similarly, we found an allelic series in CASR that is associated with both calcium increasing and lowering effects (Supplementary Tables 6, 7).
For glucose and HbA1C (biomarkers for diabetes, Supplementary Table 4a), we found a known missense variant association in ANKH (−0.11 and −0.17 sd for glucose and HbA1C, respectively), which we also replicated the documented protective effects to diabetes (p = 1.2 × 10−8, OR = 0.66 [95% CI: 0.57, 0.76]). We also found a splice-donor variant in RHAG that is strongly associated with lower HbA1c (0.80 sd) and allelic series containing 4 missense variants each in G6PC2 and TMC8 (Figure 2, Supplementary Tables 6, 7).
For three hormone biomarkers (Supplementary Table 4a), we found a PTV in ADH1C, MSR1, and NUBP2 affecting serum IGF-1 levels, and an allelic series including the hepatocyte growth factor genes HGFAC, HGF, and HNF4A with effects on SHBG. Among those, we identified novel associations with HNF4A alleles: a missense variant with MAF 0.02% was associated with increased risk for diabetic eye disorders (p = 3.1 × 10−8, OR = 9.60 [95% CI: 4.30, 21.4]) and diabetes (p = 4.7 × 10−8, OR = 3.8 [95% CI: 2.34, 6.09]) and another missense variant (MAF 3.1%) was associated with increased risk for cholecystitis (p = 2.2 × 10−13, OR = 1.27 [95% CI: 1.22, 1.38] in UK Biobank, and also replicated in FinnGen R2, p = 2.9 × 10−17, OR = 1.46 [95% CI: 1.34, 1.60]) (Figure 2, Supplementary Tables 6, 7, 13a).
These results suggest that the genetic underpinning of biomarker levels could aid in prioritizing and better understanding the mechanisms of disease-associated variants.
CNVs and HLA allelotypes influencing biomarkers
Copy number variations (CNV) constitute a significant fraction of all base pair differences between individuals. We found 13 unique associations across 10 individual CNVs (Bonferroni p < 1 × 10−6, MAF > 0.01%, Supplementary Table 10a)34. We performed aggregate rare (MAF < 0.1%) CNV burden tests, pooling CNVs in each gene, for 23,598 genes. We found 29 gene-level associations (Bonferroni p < 1 × 10−6; Supplementary Table 10a) including a burden of rare CNVs overlapping HNF1B associated with serum urea, eGFR, creatinine, and cystatin C (the least significant p = 8.8 × 10−13) estimated to have large effects (beta = 0.77, −0.90, 0.93, 0.98 sd, respectively; Supplementary Figure 6a). Previous studies have associated mutations in HNF1B with maturity onset diabetes of the young (MODY5) and altered kidney function35.The rare CNVs overlapping HNF1B were associated with chronic kidney disease (p = 1 × 10−7; OR = 4.94, SE = 0.30; Supplementary Figure 6a)36,37 in a diabetes-dependent fashion (Supplementary Table 10b). We found a rare duplication in the CST3 gene associated with increased levels of cystatin C, the protein it encodes, of opposite effect to a rare PTV at the same locus (Supplementary Figure 6b). These results highlight the value of CNV analysis with potentially large effects on lab measurements.
To identify HLA allelotype associations that are not driven by pervasive LD structure in the HLA region, we applied Bayesian model averaging (Methods) to the significant allelotype-trait pairs (BY adjusted p-value < 0.05). We found 58 associations across 28 biomarker traits and allelotypes (Supplementary Table 9).
Fine-mapping of common associated variants
To nominate potentially causal variants at loci with common (MAF > 1%) variant associations, we performed fine-mapping analysis. Specifically, focusing on the White British summary statistics, we applied FINEMAP38,39. From over 9,000 biomarker-associated loci, we identified 27,853 distinct signals in 5,363 regions across 35 traits. In the identified credible sets, 17,696 signals were fine-mapped to 50 or fewer variants with posterior probability of including the causal variant > 0.99; at 2,547 biomarker-associated loci, we resolved the signal to a single nominated causal variant (Figure 3a, Supplementary Table 14a). Moreover, we identified 3,374 unique trait-variant associations with a posterior probability > 0.99 of being the causal variant (Figure 3b, Supplementary Table 14a). These explain between 0% (urine potassium) and 48% (Lipoprotein A) of the residual trait variance (Supplementary Table 14b).
Figure 3. Summary of fine-mapped associations across 35 biomarker traits.
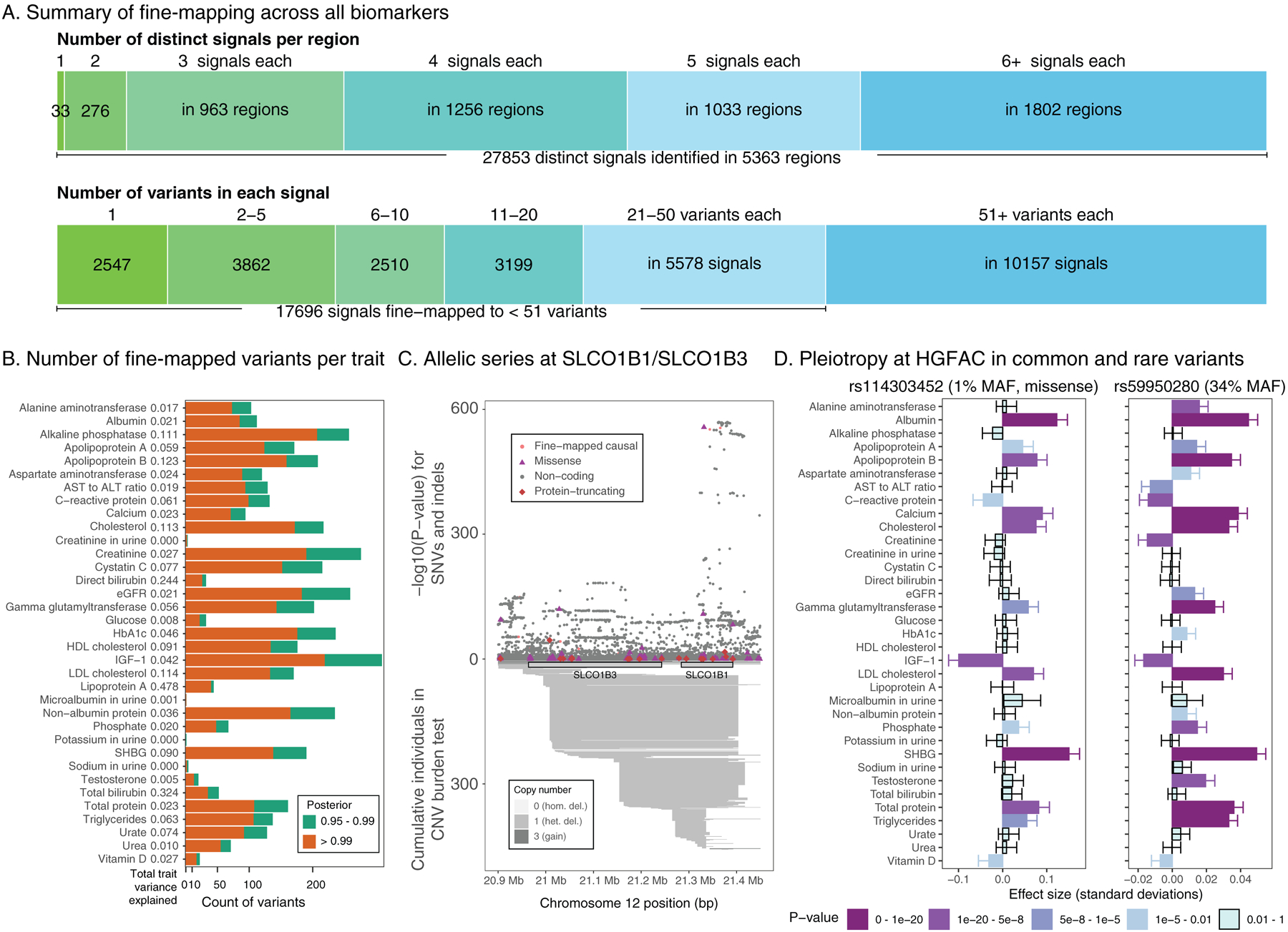
(a) FINEMAP analysis summary. (top) The number of identified distinct association signals (color gradient from green to blue) in each region with at least one genome-wide significant (UK Biobank meta-analysis p < 5 × 10−9) association and the number of regions are shown, such as a single signal at 33 regions and two to forty signals at 5,330 regions across 35 traits. (bottom) The number of identified candidate causal variants in the credible set with >= 99% posterior probability (color gradient from green to blue) and the number of signals are shown, such as 2,547 signals were mapped to a single variant in the credible set across 35 traits. (b) Breakdown of the number of fine-mapped associations with posterior probability greater than 0.95 or 0.99 across all biomarkers. Orange, posterior greater than 0.99, green, posterior between 0.95 and 0.99. The total variance explained for each trait is shown and in Supplementary Table 14b. (c) Allelic series showing combined missense, non-coding, and rare copy number variants at the SLCO1B1/SLCO1B3 on total bilirubin levels. Copy number variants annotated below axis and SNPs and short indels annotated above the axis. (d) Pleiotropic effects of fine-mapped rare coding (rs114303452, left) and common non-coding (rs59950280, right) variants at the HGFAC locus. Darker colors of purple indicate more significant associations. The p-values were from two-sided tests and were not corrected for multiple hypothesis testing. The error bars represent standard deviations.
Glycemic trait fine-mapping
We discovered fine-mapped associations for glycemic traits, including multiple variants at the TGFB1/AXL locus; rare missense variants in PFN1 and GYPC (previously implicated in a small GWAS of Mexican Americans)40; an intronic variant at the cytokine receptor IL6R; a downstream variant at VEGFA41; a missense variant at HFE, the gene responsible for hemochromatosis42,43; and an intronic variant at CD33 and 3’UTR variant at CD36 (Supplementary Table 14a). CD36 encodes for a well-studied fatty acid receptor and biomarker for type 2 diabetes (T2D)44,45, and CD33 levels are known to be perturbed in T2D cases46.
Allelic series at the SLCO1B locus
We discovered several alleles implicated in the genetic control of bilirubin levels at the SLCO1B locus (Figure 3c). We find several heterozygous deletion events, and single-nucleotide variants that we fine-mapped to two main signals: a missense variant in SLCO1B1 (rs34671512, marginal beta = −0.11 sd, p = 1.25 × 10−95) and a non-coding association in an intron of SLCO1B3 (rs11045598, marginal beta = 0.076 sd, p = 1.31 × 10−139). Despite two PTVs (one in SLCO1B1 and one in SLCO1B3) at the locus, neither had a conditionally independent effect on bilirubin levels. The diversity of variant types at this critical bilirubin and drug transporter suggest that large-effect loci can harbor variants with multiple independent genetic mechanisms contributing to their trait associations.
HGFAC pleiotropy
We scanned for loci with large effects across multiple biomarkers. The most prominent of these is HGFAC, the gene encoding hepatocyte growth factor activator. At this locus, we discovered two independent fine-mapped variants, rs114303452 (a missense variant with MAF = 1%) and rs59950280 (a non-coding variant with MAF = 34%). These two variants show significant associations with a number of diverse biomarker traits, including lipids, IGF-1, albumin, and calcium (Figure 3d). In addition, rs114303452 has been previously associated with serum HGF levels47, supporting the role of HGFAC in control of a number of other serum biomarkers through regulation of hepatocyte growth factor.
Targeted phenome-wide association analysis
We conducted PheWAS of the fine-mapped imputed variants across 166 UK Biobank phenotypes and identified 14 and 263 coding and non-coding associations, of which 109 were not previously reported in literature (p < 10−7, Supplementary Table 12, 13b–c, Methods). For example, a common (MAF = 33%) intronic variant in DPEP1 has protective effects against skin cancers (OR = 0.88 [95% CI: 0.86, 0.90], 0.81 [0.77, 0.84], and 0.89 [0.87, 0.91] for skin cancer, malignant melanoma, and non-melanoma skin cancer, respectively), with replication in FinnGen R2 (p = 3.1 × 10−5, OR = 0.81 [95% CI: 0.74, 0.90] for malignant neoplasm of skin). An allelic series of two intronic variants in ABCG2 identified with increasing and lowering urate level associations that have risk-increasing (p = 2.8 × 10−67, OR = 1.38 [95% CI: 1.33, 1.44]) and protective (OR = 0.72 [95% CI: 0.69, 0.74]) associations with gout, respectively. Both of these associations with gout are also replicated in FinnGen R2 (p = 6.3 × 10−6, OR = 1.25 [95% CI: 1.13, 1.37] and p = 8.4 × 10−5, OR = 0.84 [95% CI: 0.78, 0.92]). Those two variants (r2 = 0.47) have low linkage with a known common protein-altering variant in ABCG2 (r2 = 0.22 and 0.11 in UKB White British for rs2231142 [Q141K]) which contributes to risk of gout48. These results indicate that variants with effects on biomarkers may have pleiotropic effects across medically relevant phenotypes.
Causal inference
Given the relevance of several of the biomarkers studied to disease conditions we estimated causal effects of biomarker levels on 40 medically relevant phenotypes (including 32 diseases; Supplementary Table 15) using two-sample Mendelian Randomization with the genome-wide significant variants for each biomarker as instrumental variables49–52 (see Methods). We identified 51 significant causal relationships at FDR of 5% (Figure 4a, Supplementary Table 16). Many of these and their causal effects are well-described. We found genetic evidence supporting the protective effect of sex hormone binding globulin on diabetes (0.7 OR/SD) consistent with existing reports53–54, of ApoA on fasting glucose (0.84 OR/SD, FDR-adjusted p = 0.02)55–57, as well as an increasing effect of alanine aminotransferase levels on diabetes (1.53 OR/SD, FDR-adjusted p = 0.0018)58,59. There was a consistent effect of cystatin C on stroke (1.2 OR/SD, FDR-adjusted p = 8.7×10−4 for any stroke and 1.21 OR/SD, FDR-adjusted p = 2.8×10−3 for ischemic stroke)60,61. Finally, both HDL and ApoA were associated with increased risk of age-related macular degeneration62, as was cystatin C63–65.
Figure 4. Causal inference, transferability of polygenic risk scores, and complex trait association in polygenic risk tails.
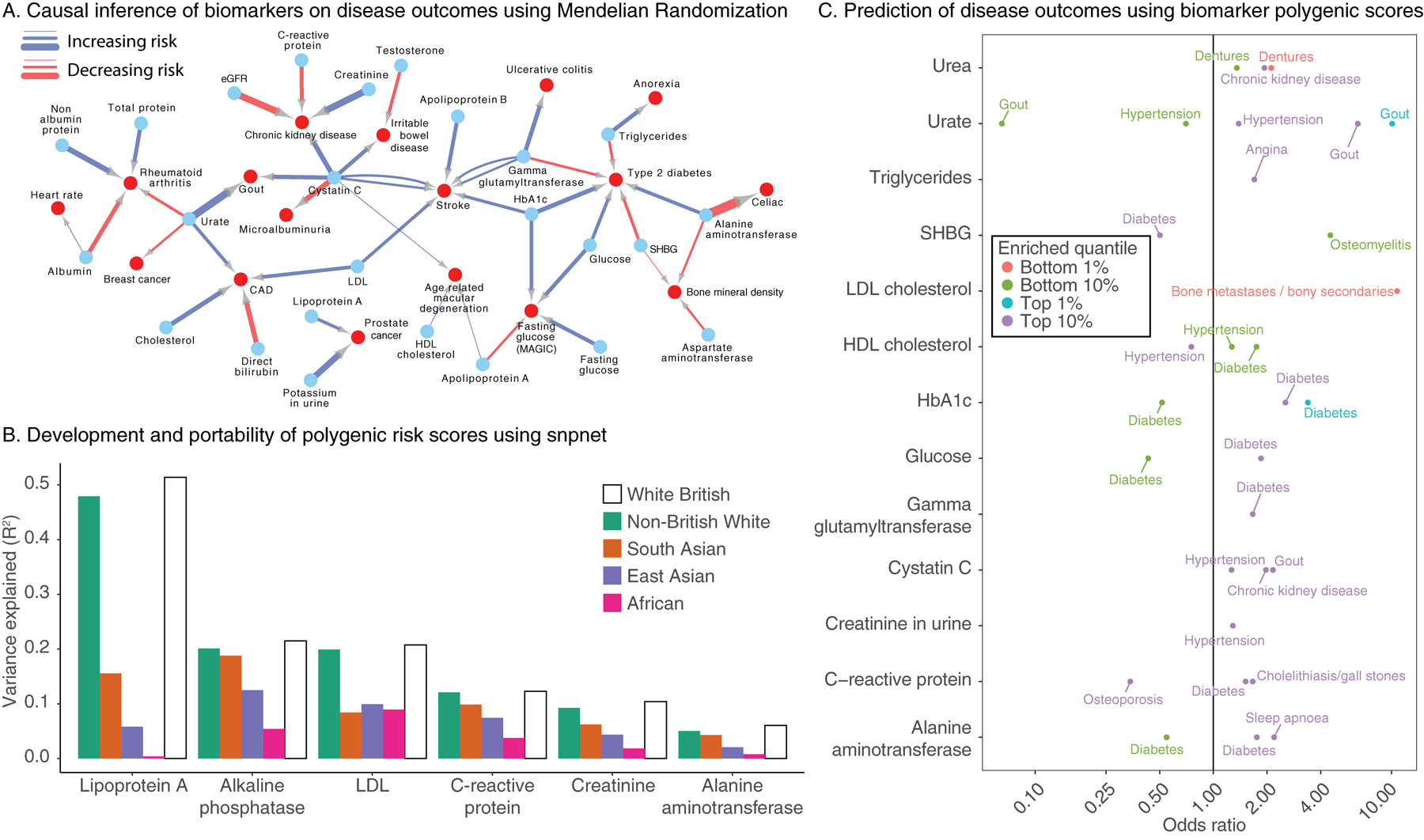
(a) Mendelian Randomization estimates causal links between biomarkers (blue nodes) and selected complex traits (red nodes). Association arrows are drawn based on effect direction (red decreasing, blue increasing). Associations were adjusted for FDR 5% cutoff across all tests (Methods, Supplementary Table 16). Edge width is proportional to the absolute causal effect size (log odds per standard deviation). (b) Summary of prediction accuracy of the snpnet polygenic scores across traits, evaluated on a held-out test set in White British as well as other 4 populations in UK Biobank. (c) (x-axis) Biomarker polygenic risk scores for the top 1%, top 10%, bottom 1%, and bottom 10% of individuals and their association to different diseases in UK Biobank, represented as the odds ratio of the disease in this group relative to the 40–60% quantiles. Traits without rows did not have any outcomes with FDR-adjusted significant associations.
We estimated a causal protective effect of testosterone on inflammatory bowel disease (0.70 OR/SD, FDR-adjusted p = 3.86×10−3)66,67, and a protective effect of urate on breast cancer risk (0.87 OR/SD, FDR-adjusted p = 0.033)68.
Polygenic prediction of biomarkers
The vast size of the UK Biobank cohort affords the opportunity to build predictive polygenic risk models of biomarkers from genotype data alone69. We constructed PRS for all 35 biomarkers using batch screening iterative lasso (BASIL) implemented in the R snpnet package70,71. Specifically, we split the White British individuals into 70% training, 10% validation (to identify the optimal sparsity parameter), and 20% test sets and evaluated the predictive performance (R2) in the held-out test set (n = 63,818) as well as in 4 populations in UK Biobank (see Methods). We found the mean predictive performance relative to the White British test set for these 4 populations were 93%, 70%, 51%, and 24%, respectively, suggesting these polygenic models have limited generalizability across populations (Figure 4b, Supplementary Figure 7, Supplementary Table 17)72. As an external validation, we found that the PRSs had high portability to self-identified white individuals from the MESA cohort (Supplementary Table 18)73.
Multiple regression with PRSs
We hypothesized that the 35 biomarker PRSs may improve the prediction of higher-level traits and diseases in combination with the PRS for the trait or disease itself. To this end, we constructed multi-PRS models for traits by using multiple regression to predict the trait or disease from a) its own PRS, b) the PRSs for each of the 35 biomarkers, and c) the covariates age, sex, and principal components (Methods).
We selected disease endpoints for multi-PRSs analysis by considering the enrichment of disease prevalence at the tails of the distribution of the single-trait biomarker PRSs (Figure 4c, Supplementary Table 19, Supplementary Figure 8). We focused on traits with three or more associated biomarkers (Supplementary Figure 9), as we reasoned these would benefit most from the combination of multiple biomarker PRSs.
For chronic kidney disease, the multi-PRS stratified individuals according to disease status better than the snpnet PRS (Figure 5a–b, Supplementary Table 20). In contrast, the myocardial infarction snpnet PRS was equally stratifying as compared to the multi-PRS, with both explaining a substantial portion of trait heritability (AUC 0.58–0.59; Figure 5c). This trend held after including additional existing polygenic scores for type 2 diabetes and myocardial infarction as well (AUC 0.594 and 0.611 respectively; Supplementary Figure 10, Supplementary Tables 20–23). This suggests that the genetic basis of myocardial infarction, as previously reported74, already captures the majority of the genetic component of serum lipids and other biomarkers. Similar weak effects of biomarkers were estimated in hypertension, angina, and gallstones, while alcoholic cirrhosis, gout, type 2 diabetes, and heart failure were better predicted with multi-PRS models (Figure 5c, Supplementary Table 20). Improved predictions relied on relevant and variable biomarkers across these traits (Supplementary Figure 11), including eGFR, creatinine, cystatin C, and bilirubin for CKD; creatinine, bilirubin, total and LDL cholesterol, cystatin C, and eGFR for heart failure; and bilirubin, GGT, eGFR, HDL cholesterol, and IGF-1 for alcoholic cirrhosis.
Figure 5. Multiple regression with biomarker polygenic scores improve prevalent and incident disease prediction.
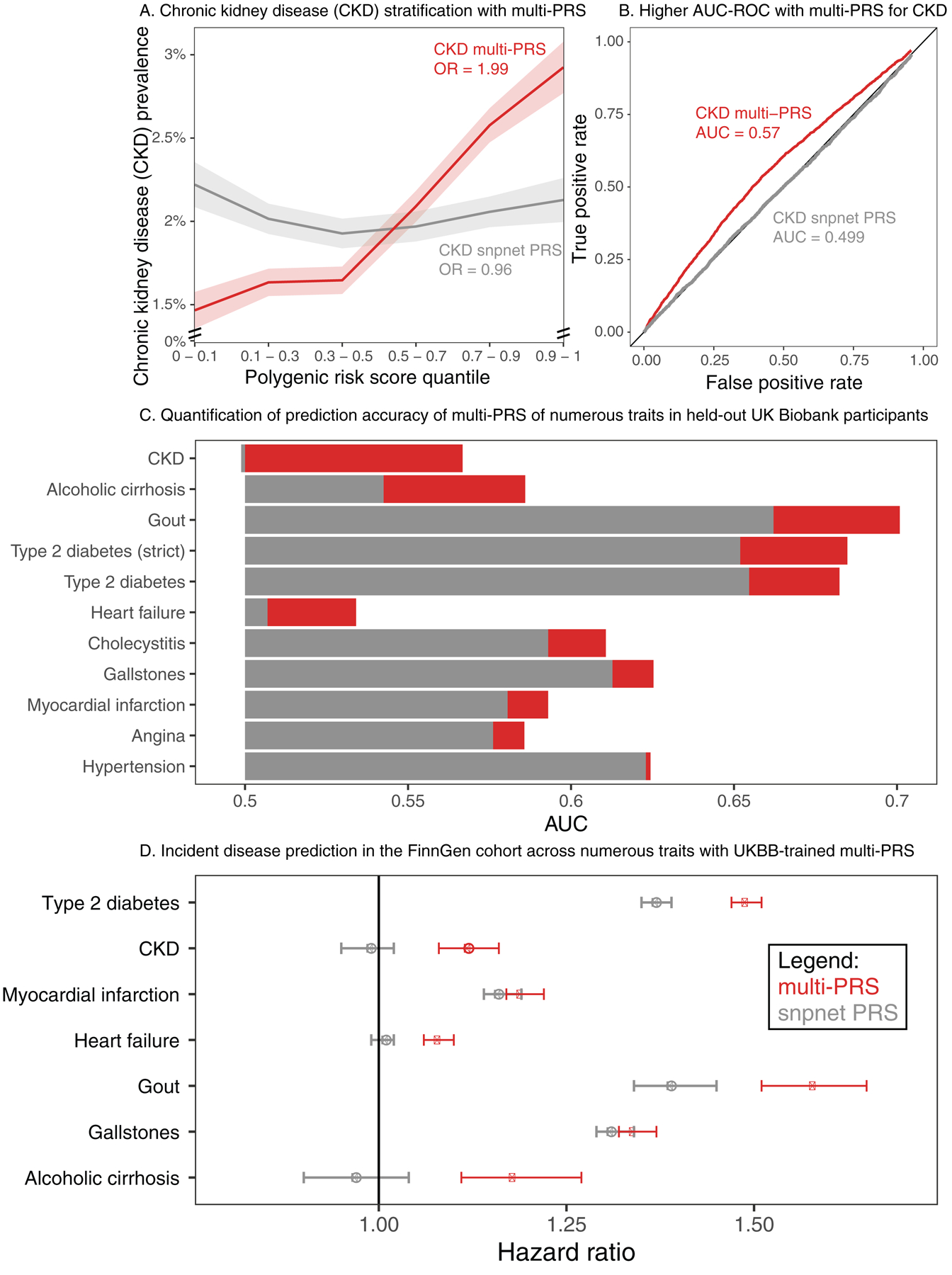
(a) (x-axis) quantiles of polygenic risk score, spaced to linearly represent the mean of the corresponding bin of scores. (y-axis) Prevalence of chronic kidney disease (n = 2,780 cases and n = 89,409 total, defined by verbal questionnaire and hospital in-patient record ICD code data) within each quantile bin of the polygenic risk score. Error bars represent the standard error around each measurement, and individuals evaluated are held-out European-ancestry individuals in UK Biobank. (b) ROC curve with AUC for chronic kidney disease, comparing the snpnet-derived polygenic score to a multi-PRS model trained across biomarkers as well. Individuals evaluated are held -out European-ancestry individuals in UK Biobank. (c) AUC-ROC estimates for prediction of 10 disease outcomes in a held-out test set of the UK Biobank. Diabetes was run using both a strict definition (excluding from control individuals with HbA1c < 39) and the complete sample (Methods). (d) Hazard ratios for the incidence of type 2 diabetes (n = 17,519), chronic kidney disease (n=3,058), myocardial infarction (n=7,913), heart failure (n = 13,965), gout (n = 1,936), gallstones (n = 11,629), and cirrhosis (n=845) in FinnGen using the standard single-disease PRS trained on UK Biobank using snpnet versus the multi-PRS including both biomarker PRSs and the trait PRS. The strict definition of type 2 diabetes is shown. Error bars represent 95% confidence intervals and points represent mean hazard ratio estimates.
Encouraged by these findings, we evaluated the potential of these improved polygenic scores in identifying disease cases by applying both trait-specific PRS and combined PRS in an independent replication cohort, FinnGen (R3, n = 135,500, Supplementary Tables 24–26). Here, we found evidence that the combination of PRS increased the effect size in chronic kidney disease (hazard ratio (HR) = 0.99, p = 0.46 for snpnet PRS and hazard ratio = 1.12, p = 2.09 × 10−10 for multi-PRS, Figure 5d, Supplementary Figure 12, Supplementary Table 24), type 2 diabetes (HR = 1.37, p < 2 × 10−16 for snpnet PRS and HR = 1.49 for multi-PRS), gout (HR = 1.39, p < 2 × 10−16 for snpnet PRS and HR = 1.58 for multi-PRS), heart failure (HR = 1.01, p = 0.38 for snpnet PRS and HR = 1.08, p < 2 × 10−16 for multi-PRS), and alcoholic cirrhosis (HR = 0.97, p = 0.35 for snpnet PRS and HR = 1.18, p = 1.04 × 10−6 for multi-PRS, Supplementary Table 24). Similar results to UK Biobank were found in models including existing polygenic scores (Supplementary Table 26, Supplementary Figure 13), with the integrated type 2 diabetes model, including both pre-existing PRS and biomarker PRS, resulting in 1.67 HR change per standard deviation. This suggests that multiple regression of polygenic risk for biomarkers might capture multiple underlying disease states and/or underlying causes, and that these multiple states are predictive of disease.
Discussion
Using data from 35 biomarkers in ~363,000 UKB samples, we provide an assessment of genetic associations with biomarker levels, the relevance of these associations in disease phenotypes, and their utility in risk stratification.
Protein-altering variants that modify biomarker levels and disease risk can provide in vivo validation of therapeutic targets75,76. Here, we found multiple protein-altering variants that directly implicate genes associated with the studied biomarkers, and we hypothesize that some of these genes may provide potential therapeutic targets.
To assess the translatability of our findings, we built predictive models aggregating trait PRS with those of the biomarkers, improving the predictive accuracy of multiple disease outcomes both overall and especially at the extremes of genetic risk. Given that biomarker values are already routinely collected in structured data formats, we anticipate that the multi-PRS methods could inform clinical practice in the coming years, as a larger fraction of the population is genotyped and sequenced.
In addition to the discovery of multiple individual loci and candidate causal variants, we can also draw some general conclusions across the traits evaluated with our multi-PRS models. Traits and diseases were predicted best when they had individually predictive biomarkers and a complex etiology (e.g. chronic kidney disease), but underpowered genetic studies. We believe that a large number of disease cases is typically most useful in developing well-powered models, as it helps both with the baseline polygenic score and fitting of the multi-PRS components. Further exploration of the conditions where multi-PRS models perform particularly well is an area of future study.
Numerous limitations to this work are present. We assigned individuals to ancestry groups based on self-reported ethnicity categories and the top two principal components of the genotype matrix. We included many technical covariates in order to reduce bias in the measurements of the biomarkers, but doing so has the potential to reduce power. We fine-map based on imputed genotypes and summary statistics, and both reduce power to detect true causal variants. In addition, the large and complex linkage present at some loci, including notably the LPA locus, might result in spurious fine-mapped and rare coding variant associations, though conditional analyses (e.g. of a rare coding variant in SLC22A2) are inconclusive (Supplementary Figure 14). Similarly, causal inference using individual-level data77 can increase power and reduce bias, and we recommend it for future studies. Lastly, we anticipate that including other genetic risk scores will fit well into the multi-PRS framework to further improve prediction of common complex disease.
The genome-wide resource made available with this study, including the association summary statistics, fine-mapped regions, and polygenic prediction models (Data availability)78, provides a starting point for causal mapping of genetic variants affecting the 35 biomarkers and their relevance to medical phenotypes. These results highlight the benefits of direct measurements of biomarkers in population cohorts for interpreting the genetic basis of biomarkers and improved prediction of multiple common diseases.
Online Methods
Genotype and phenotype data in UK Biobank
We used genotype datasets from the UK Biobank (release version 2 for the directly genotyped variants and the imputed HLA allelotype datasets and release version 3 for the imputed genotype dataset), the copy number variation dataset34, and the hg19 human genome reference for all analyses in the study10. To minimize the variability due to population structure in our dataset, we restricted our analyses to unrelated individuals based on the following four criteria reported by the UK Biobank in the sample QC file, “ukb_sqc_v2.txt”: 1) used to compute principal components (“used_in_pca_calculation” column); 2) not marked as outliers for heterozygosity and missing rates (“het_missing_outliers” column); 3) do not show putative sex chromosome aneuploidy (“putative_sex_chromo- some_aneuploidy” column); and 4) have at most 10 putative third-degree relatives (“excess_relatives” column).
Additionally, we used the “in_white_British_ancestry_subset” column in the sample QC file as a part of the population definition as shown below.
We used a combination of self-reported ethnicity (UK Biobank field ID: 21000) and principal component analysis and analyzed 5 subpopulations in the study: self-identified White British (n = 337,151 individuals), African (6,498), East Asian (1,772), South Asian (7,962), and self-identified non-British White (24,909). We first used the genotype principal components (PCs) of the genotyped variants from the UK Biobank and defined thresholds on PC1 and PC2 and further refine the population definition (described in Supplementary Note). We subsequently focused on a subset of individuals with non-missing values for covariates and biomarkers as described below.
Variant annotation and quality control
Detailed information on variant annotation and quality control is described in Supplementary Note.
Biomarker phenotype definition
Phenotype and covariate quality control excluded rheumatoid factor and estradiol from further analyses, and fasting glucose (available for 17,439 self-reported fasting individuals) was used as a phenotype-level quality control for the glucose measurements -- throughout the text, “glucose” refers to glucose levels adjusted for fasting time rather than the GWAS among only fasting individuals (self-report of more than 7 and less than 24 hours of fasting, n = 17,439) unless otherwise noted. We focused on 32 biomarkers for genetic analysis and also defined three derived phenotypes, estimated glomerular filtration rate (eGFR), non-albumin protein, and AST to ALT ratio, for a total of 35 biomarkers (Supplementary Table 4a). The eGFR measure is an indicator of renal function and is defined by the CKD-EPI equation79. We defined non-albumin protein as the difference between the total protein and albumin. Then after applying covariate correction (see Covariate correction below, Supplementary Table 4b), we additionally defined the AST to ALT ratio as the difference of the (log-transformed) estimates for aspartate aminotransferase and alanine aminotransferase.
Statin identification and LDL adjustment
Statin identification and LDL adjustment is described in Supplementary Note.
Covariate correction
Covariate adjustment is described in Supplementary Note.
Definition of type 2 diabetes
We used the definition of type 2 diabetes from our previous paper, including removal of type 1 diabetes cases from both cases and controls80. We use the terminology from Eastwood et al. throughout this description81. Type 2 diabetes was assigned case status for “probable type 2 diabetes” and “possible type 2 diabetes” and control status for “type 2 diabetes unlikely”; in addition, individuals with “probable type 1 diabetes,” “possible type 1 diabetes,” or “ probable gestational diabetes” were excluded. Finally, for the “strict” type 2 diabetes definition, we removed controls with HbA1c ≥ 39 mmol/mol.
Genome-wide association analysis
We performed genome-wide association analyses using the following four datasets: 1) The directly genotyped variants on the array (for protein-truncating and protein-altering variants); 2) The imputed variants (version 3); 3) The imputed HLA alleles; and 4) The copy number variations (CNVs) and gene-level aggregated CNV burden34. All the p-values from the association analyses are from two-sided tests. Detailed description of the association analysis is described in Supplementary Note.
Meta-analysis
Using the GWAS summary statistics for four analyzed populations (White British, Non-British White, South Asian, and African; East Asian GWAS were excluded) in UK Biobank, we performed inverse-variance weighted meta-analysis using METAL (version 2011-03-25) and included heterogeneity of effects analysis.
For the summary statistics from the meta-analysis, we checked whether the A1 and A2 alleles match with the alternate and reference allele in GRCh37/hg19 reference genome (fasta file) using bedtools getfasta subcommand82, and canonicalized our association summary statistics such that we always report the effect size with respect to the alternate allele in the reference genome.
Derivation of independent loci
Once we ran the GWAS, full summary statistics were clumped to r2 > 0.1 using the following clump command:
plink1.9 --bfile <1000G Phase 3 European plink file> --clump <summary statistics> --clump-p1 1e-6 --clump-p2 1e-4 --clump-r2 0.1 --clump-kb 10000 --clump-field P --clump-snp-field ID
Then, to avoid calling very large signals as multiple associations, these were further filtered such that any SNPs within 0.1cM of each other (as annotated by 1000 Genomes) were considered part of the same association signal, with the cM annotation derived from the 1000G Phase 3 European samples (n = 489)24 -- variants within 0.1cM were chose to only have the minimum p-value.
In order to report independent signals, we ran the following plink command:
plink1.9 --bfile <1000G Phase 3 European plink file> --extract <all unique hit SNPs, n = 6269> --indep 50 5 2
And counted the number of independent SNPs it reported.
Comparison of effect sizes with published studies
Full summary statistics from comparison studies (PMID in Supplementary Table 5) were downloaded and overlapped with our GWAS summary statistics using the munging framework from LD Score regression to align alleles (modified to additionally report the unnormalized beta). The observed correlation coefficients and linear effect regression coefficients across variants with p < 1 × 10−6 in either study (subthreshold) or p < 5 × 10−8 in our study (GWAS hits) are listed in Supplementary Table 5. Using the same set of comparison studies, we also checked whether the protein-truncating and protein-altering associations were previously reported for a given trait by calling the association reported if the p-value of the variant is less than 1 × 10−6 in any comparison study for a given trait.
Fine-mapping biomarker-associated regions
Independent loci were defined by clumping White British GWAS summary statistics (see section “Derivation of independent loci”). For each putative SNP, we defined distance-independent regions by collating all variants in linkage disequilibrium with the following plink command:
plink1.9 --clump-p1 1e-3 --clump-p2 1e-3 --clump-r2 0.0001 --clump-kb 10000 --clump-field P-value --clump-snp-field MarkerName
In this way, we defined the individual loci contributing to the fine-mapping. We identified putative causal SNPs in each locus by using the FINEMAP software version 1.3 and 1.439. The output from FINEMAP is (1) a list of potential causal configurations together with their posterior probabilities and Bayes factors, (2) for each SNP, the posterior probability, and Bayes factor of being causal, and (3) credible sets for each identified causal signal. We applied FINEMAP with its default settings while allowing for a maximum of forty causal SNPs and by using pairwise correlations between SNPs computed from the original GWAS genotype data as previously recommended38.
We ran fine mapping on all associations with more than one variant for which the most significantly associated variant had a p-value less than 1 × 10−3. We filtered regions based on the unique variant ID (in MFI file from UK Biobank) to those regions for which at least one of the variants in the region was annotated as an association lead SNP in our analysis (p < 5 × 10−9).
Heritability estimates
Heritability analysis is described in Supplementary Note.
Targeted phenome-wide association analysis
We curated a list of 166 medically relevant phenotypes from previously-reported binary phenotypes in Global Biobank Engine (GBE)27,31,76. Specifically, we selected phenotypes with at least 700 cases in white British grouping and removed phenotypes that were likely to be duplicated (Supplementary Table 12). Those phenotypes include non-cancer disease-outcome endpoints derived from a combination of the ICD codes from hospital inpatient records as well as self-reported disease ascertainment status31, family history phenotype (UK Biobank data category 100034), cancer phenotypes derived from a combination of the UK cancer registry data and questionnaire data76, and additional set of medically relevant phenotypes derived from the following data fields in UK Biobank: 2247, 2463, 2834, 3591, 6148, 6149, 6152, 6153, 20126, 20406, 20483, and 21068. For example, the chronic kidney disease phenotype is defined based on the combination of self-reported kidney disease (coded as “1192” in UKB Data coding ID 6) and ICD-10 code (N17 [“Acute kidney failure”], N18 [“Chronic kidney disease (CKD)”], N19 [“Unspecified kidney failure”], and its sub-concepts) from hospital inpatient data (Supplementary Figure 8a), which is visualized with R UpSetR package version 1.4.0. The data source of the phenotype definitions is described in “Source of the phenotype” column in Supplementary Table 12.
After performing LD-pruning using PLINK with “--indep 50 5 2” as previously described27,76, we prioritized (A) the 632 LD-independent protein-altering or protein-truncating variants outside of the MHC region that showed the significant associations (p < 5×10−9) on the genotyping array, as well as (B) 43 non-synonymous and (C) 2,442 synonymous or non-coding variants with significant associations (p < 5×10−9) from the imputation dataset (Supplementary Tables 13 a–c). We applied the PheWAS analysis for those variants with a p-value threshold of p < 1×10−7.
For the resulting associations, we checked the NHGRI-EBI GWAS catalog to see whether they are already reported in the previous studies83. Specifically, we identified the LD proxy (r2 > .9) of the PheWAS target variants and manually inspected the reported associations for those variants. For associations with no supporting prior studies, we additionally queried Open Target Genetics and manually assessed the novelty of the associations84. In addition, we also checked the FinnGen study (Freeze R2, http://r2.finngen.fi/) and asked whether the PheWAS target variants and its LD proxy have similar associations. Those PheWAS results and the reference to the prior association reports are summarized in Supplementary Tables 13 a–c.
For the CNV PheWAS, we queried summary statistics from previous CNV association tests for the 173 traits of interest34. Results for a burden of HNF1B copy number variation are in (Supplementary Figure 6a), along with the corresponding meta-analyzed summary statistics for biomarker traits described in this work.
Correlation of genetic effects across relevant phenotypes
We used LD Score regression in genetic correlation mode to estimate genetic correlation effects between biomarkers and the 166 medically relevant phenotypes used in the PheWAS analysis. The exact arguments were:
ldsc.py --rg <traits> --ref-ld-chr ldsc/1000G.EUR.QC/ --w-ld-chr ldsc/weights_hm3_no_hla/weights.
Causal inference
For the final results, all lead variants with p < 5 × 10−8 were kept for the Mendelian Randomization analyses. All MR calculations were done using TwoSampleMR, which was also used to perform trait munging85.
We used the Rücker model-selection framework for causal inference as follows50–52,86. For each exposure-outcome pair, we started with a simple fixed-effects inverse variance weighted (FE-IVW) MR analysis and computed the model’s significance and the Q statistic for heterogeneity. If the significance of Q was <0.01 then we used it as evidence for heterogeneity and switched to a mixed-effects IVW (ME-IVW) model instead. We then computed an MR-Egger model and compared it to the IVW selected model. Let Qivw be the Q-statistic of the IVW model and Qe be the Q statistic of the MR-Egger model. We computed the significance of the difference Qe − Qivw using a χ12 distribution and switched to the MR-Egger model if the result was significant (p<0.01). The significance of all selected models was adjusted using a BY FDR correction at 5%87. Network visualization of the results was done using Cytoscape version 3.7 and 3.888.
Polygenic prediction within and across populations
To construct polygenic risk scores for each of the traits, we applied the batch screening iterative lasso (BASIL) algorithm implemented in the R snpnet package. This method is capable of finding the exact solution for L1-penalized multivariate regression (lasso) on an ultra-high dimensional large dataset through an iterative procedure built on top of the R glmnet package70,71,89. Because this method considers all of the genetic variants available in the input dataset and performs variable selection and multivariate regression fit simultaneously, it is suitable for the polygenic risk prediction from a large-scale dataset.
We randomly split the White British individuals into training (70%, n = 223,327 with non-missing phenotype for at least one biomarker trait), validation (10%, n = 31,929), and test (20%, n = 63,818) sets and used both training and validation sets to fit multivariate Lasso regression models. The validation set is used to find the optimal penalization (sparsity) parameter with respect to the predictive performance (R2). To maximize the performance of polygenic prediction, we combined the directly genotyped variants, the imputed HLA allelotypes, and the CNV dataset with PLINK version 1.9, and used it as the input genotype dataset consists of 1,080,968 variants. For each biomarker phenotype, we applied the R snpnet package for the log-transformed and covariate-adjusted phenotypes and regression coefficients, BETAs71.
Using the beta values from multivariate Lasso regression, we computed the polygenic risk score for each individual with PLINK2 --score subcommand90. To evaluate the performance of the models, we computed R2 values for log-transformed phenotypes using individuals in the held out White British test set (n = 63,818), as well as self-identified non-British white (n = 23,595), African (n = 6,021), South Asian (n = 7,341), and East Asian (n = 1,082) populations. To assess the incremental predictive performance compared to the covariates, we evaluated the R2 values for the risk score computed from the covariate (defined as the difference between the log-transformed phenotype value and log-transformed and covariate-adjusted phenotype values) as well as the combined risk score (the sum of the covariate score and genotype PRS, Supplementary Table 17a). Polygenic score accuracy was generally independent of residualization strategy (Supplementary Table 17b).
For the evaluation of multi-PRS models, we also trained snpnet PRS models for disease outcomes using the R snpnet package in the same way as in the biomarker phenotypes, except that we used the binomial family for logistic regression and AUC as the criterion to select the sparsity parameter.
Evaluation of snpnet PRS models with MESA cohort is described in Supplementary Note.
Single-trait biomarker PRS-PheWAS
We started by enumerating all our 166 high-confidence traits which were replicated between ICD codes and self-reported, cancer, family history, and manually curated traits31,76 described in the PheWAS analysis above. For each of the 35 biomarkers, we used R’s fisher.test implementation of the Fisher’s Exact test between the 40–60 percentile and the top and bottom 1%ile and 1–10%ile of PRS in the union of the unrelated non-British White and held-out test set of unrelated White British individuals. We then corrected for multiple hypotheses using a Bonferroni-adjusted q-value less than 5% within each biomarker and reported the enrichment as the odds ratio estimate from Fisher’s exact test.
Models for multi-PRS prediction of disease outcomes
In order to perform out-of-sample validation, we trained L1-regularized logistic regression models with glmnet using just the 35 biomarker PRSs and the snpnet PRS for the trait of interest as predictors70. Results were evaluated using the area under the receiver operating characteristic curve (AUC-ROC) in the union of the held-out test set of self-identified White British individuals and all unrelated, self-identified non-British White individuals for which the corresponding phenotype was available (as used in the cross-population testing; see above). We also performed the lasso regression additionally including age, sex, genotyping array, and the top ten global principal components of the genotyping matrix as covariates for each outcome (referred to as “Age/sex/PCs”) and additional information provided in Supplementary Note.
Finally, we derived versions of the multi-PRS model with these covariates and relevant pre-existing polygenic scores for gallstones91, type 2 diabetes92,93, and heart attack94,95 in the model as well (Supplementary Table 20). We refer to models trained just on covariates and trait polygenic scores as “Baseline” models and those which additionally include the 35 biomarker PRSs as “multi-PRS” models throughout the manuscript.
Evaluation of multi-PRS prediction in an external cohort
The FinnGen Data Freeze 3 comprised 135,300 Finnish participants, with phenotypes derived from International Classification of Diseases (8th, 9th, and 10th revision) diagnosis codes obtained from national registries, including the national Finnish hospital discharge and cause-of-death registries as a part of the FinnGen project (Supplementary Table 24).
FinnGen samples were genotyped with Illumina and Affymetrix arrays (Thermo Fisher Scientific, Santa Clara, CA, USA). Genotype imputation was carried out by using the population-specific SISu v3 imputation reference panel with Beagle 4.1 (version 08Jun17.d8b, https://faculty.washington.edu/browning/beagle/b4_1.html) as described in the following protocol: dx.doi.org/10.17504/protocols.io.nmndc5e. Post-imputation quality control involved excluding variants with INFO score < 0.6.
We estimated a full weighting matrix for each SNP from the corresponding coefficients of the regression model, then applied the per-SNP weighted model to individuals in FinnGen. To assess the risk for first disease events, hazard ratios, and 95% confidence intervals per SD increment were estimated with Cox proportional hazards models after evaluation of the proportionality assumption. For the comparison, on type 2 diabetes and myocardial infarction, with models including the existing polygenic scores, scores were standardized within the population before being applied using the weights of standardized PRSs (Supplementary Table 22–23) in order to capture the differences in SNP sets used across the scores. An R script to perform these analyses, which takes as input the raw PRS for each outcome of interest, is available in the code repository. With age as the time scale, the survival models were stratified by sex and adjusted for batch, and the first ten principal components of ancestry calculated within Finns.
Statistics
For computational and statistical analysis, we used Jupyter notebook with Python (3.6 and 2.7) and R kernels (http://jupyter.org/), R (version 3.5.2 and 3.4.0), R studio (3.5.2), R tidyverse package version 1.3.0, and Stata version 15. Software and packages used for specific analysis are listed in the corresponding subsection above. The p-values are computed from two-sided tests, unless otherwise specified.
Supplementary Material
Acknowledgments
This research has been conducted using the UK Biobank Resource under Application Number 24983, “Generating effective therapeutic hypotheses from genomic and hospital linkage data” (http://www.ukbiobank.ac.uk/wp-content/uploads/2017/06/24983-Dr-Manuel-Rivas.pdf). Based on the information provided in Protocol 44532 the Stanford IRB has determined that the research does not involve human subjects as defined in 45 CFR 46.102(f) or 21 CFR 50.3(g). All participants of UK Biobank provided written informed consent (more information is available at https://www.ukbiobank.ac.uk/2018/02/gdpr/). For FinnGen, all patients and control subjects provided informed consent, a biobank research consent, based on the Finnish Biobank Act. Recruitment protocols followed the biobank protocols approved by Valvira, the National Supervisory Authority for Welfare and Health. The Ethical Review Board of the Hospital District of Helsinki and Uusimaa approved the FinnGen study protocol Nr HUS/990/2017. All DNA samples and data in this study were pseudonymized. Statin adjustment analyses were further conducted via UK Biobank application 7089 using a protocol approved by the Partners HealthCare Institutional Review Board. We thank all the participants in the UK Biobank and FinnGen studies. We thank Andrew Paterson and members of the Rivas, Pritchard, and Bejerano labs for their feedback. The Genotype-Tissue Expression (GTEx) Project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The data used for the analyses described in this manuscript were obtained from the GTEx Portal on 10/19/2020. This work was supported by National Human Genome Research Institute (NHGRI) of the National Institutes of Health (NIH) under awards R01HG010140 (M.A.R.), R01EB001988-21 (T.H.) and R01HG008140 (J.K.P.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Some of the computing for this project was performed on the Sherlock cluster at Stanford University. We would like to thank Stanford University and the Stanford Research Computing Center for providing computational resources and support that contributed to these research results. N.S.A. is supported by the Department of Defense through a National Defense Science and Engineering Grant and by a Stanford Graduate Fellowship. Y.T. is supported by a Funai Overseas Scholarship from the Funai Foundation for Information Technology and the Stanford University School of Medicine. N.M. is supported by Academy of Finland (#331671). H.M.O is supported by Academy of Finland (#309643). F.R. is supported by a National Heart, Lung, and Blood Institute grant (1K01HL144607). FinnGen is supported by Abbvie, Astra Zeneca, Biogen, Celgene, Genentech, GSK, Merck, Pfizer, and Sanofi. M.A.R. is in part supported by National Human Genome Research Institute (NHGRI) of the National Institutes of Health (NIH) under award R01HG010140 (M.A.R.), and a National Institute of Health center for Multi- and Trans-ethnic Mapping of Mendelian and Complex Diseases grant (5U01 HG009080). The land upon which some of this work was performed is the ancestral and unceded land of the Muwekma Ohlone, and we pay our respects to their Elders past and present.
Footnotes
Competing financial interests
The Board of Trustees of the Leland Stanford Junior University filed U.S. Provisional Application “Methods for diagnosis of polygenic diseases and phenotypes from genetic variation” (Serial No. 62/852,738) describing this work. J.K.P., M.A.R., N.S.-A., and Y.T. are designated as inventors of the patent. M.A.R is on the SAB of 54Gene and Computational Advisory Board for Goldfinch Bio and has advised BioMarin, Third Rock Ventures, MazeTx and Related Sciences. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Data availability
Summary-level data generated in this work are available at NIH’s instance of figshare (the meta-analyzed GWAS summary statistics [https://doi.org/10.35092/yhjc.12355382], the fine-mapped associations [https://doi.org/10.35092/yhjc.12344351], the snpnet polygenic risk score coefficients [https://doi.org/10.35092/yhjc.12298838], and the multi-PRS weights [https://doi.org/10.35092/yhjc.12355424], please see Supplementary Note for details)78. Other data are displayed in the Global Biobank Engine (https://biobankengine.stanford.edu).
Code Availability
Analysis scripts and notebooks are available on GitHub at https://github.com/rivas-lab/biomarkers/.
References for main text
- 1.Willer CJ et al. Discovery and refinement of loci associated with lipid levels. Nat. Genet 45, 1274–1283 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klarin D et al. Genetics of blood lipids among ~300,000 multi-ethnic participants of the Million Veteran Program. Nat. Genet 50, 1514 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu DJ et al. Exome-wide association study of plasma lipids in >300,000 individuals. Nat. Genet 49, 1758–1766 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wheeler E et al. Impact of common genetic determinants of Hemoglobin A1c on type 2 diabetes risk and diagnosis in ancestrally diverse populations: A transethnic genome-wide meta-analysis. PLoS Med. 14, e1002383–e1002383 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spracklen CN et al. Identification and functional analysis of glycemic trait loci in the China Health and Nutrition Survey. PLoS Genet. 14, e1007275 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scott RA et al. Large-scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nat. Genet 44, 991–1005 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Graham SE et al. Sex-specific and pleiotropic effects underlying kidney function identified from GWAS meta-analysis. Nat. Commun 10, 1847 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wuttke M et al. A catalog of genetic loci associated with kidney function from analyses of a million individuals. Nat. Genet 51, 957 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Okada Y et al. Meta-analysis identifies multiple loci associated with kidney function-related traits in east Asian populations. Nat. Genet 44, 904–909 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bycroft C et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fry Daniel, Almond Rachael, Stewart Moffat Mark Gordon & Parmesher Singh. UK Biobank Biomarker Project: Companion Document to Accompany Serum Biomarker Data. UK Biobank Document Showcase http://biobank.ctsu.ox.ac.uk/showcase/docs/serum_biochemistry.pdf (2019). [Google Scholar]
- 12.Kathiresan S et al. A genome-wide association study for blood lipid phenotypes in the Framingham Heart Study. BMC Medical Genetics vol. 8 S17 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Snell-Bergeon JK et al. Evaluation of urinary biomarkers for coronary artery disease, diabetes, and diabetic kidney disease. Diabetes Technol. Ther 11, 1–9 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McCarthy S et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet 48, 1279–1283 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu X et al. Exome chip meta-analysis identifies novel loci and East Asian-specific coding variants that contribute to lipid levels and coronary artery disease. Nat. Genet 49, 1722–1730 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kettunen J et al. Genome-wide study for circulating metabolites identifies 62 loci and reveals novel systemic effects of LPA. Nat. Commun 7, 11122 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kanai M et al. Genetic analysis of quantitative traits in the Japanese population links cell types to complex human diseases. Nat. Genet 50, 390–400 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Horikoshi M et al. Discovery and Fine-Mapping of Glycaemic and Obesity-Related Trait Loci Using High-Density Imputation. PLoS Genet. 11, e1005230 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Teumer A et al. Genome-wide Association Studies Identify Genetic Loci Associated With Albuminuria in Diabetes. Diabetes 65, 803–817 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gorski M et al. 1000 Genomes-based meta-analysis identifies 10 novel loci for kidney function. Sci. Rep 7, 45040 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang X et al. Genome-wide association study in 79,366 European-ancestry individuals informs the genetic architecture of 25-hydroxyvitamin D levels. Nat. Commun 9, 260 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Köttgen A et al. Genome-wide association analyses identify 18 new loci associated with serum urate concentrations. Nat. Genet 45, 145 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Devlin B & Roeder K Genomic Control for Association Studies. Biometrics vol. 55 997–1004 (1999). [DOI] [PubMed] [Google Scholar]
- 24.Bulik-Sullivan BK et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet 47, 291–295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi H, Mancuso N, Spendlove S & Pasaniuc B Local Genetic Correlation Gives Insights into the Shared Genetic Architecture of Complex Traits. Am. J. Hum. Genet 101, 737–751 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shi H, Kichaev G & Pasaniuc B Contrasting the Genetic Architecture of 30 Complex Traits from Summary Association Data. Am. J. Hum. Genet 99, 139–153 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tanigawa Y et al. Components of genetic associations across 2,138 phenotypes in the UK Biobank highlight adipocyte biology. Nat. Commun 10, 4064 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peloso GM et al. Association of low-frequency and rare coding-sequence variants with blood lipids and coronary heart disease in 56,000 whites and blacks. Am. J. Hum. Genet 94, 223–232 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van der Harst P & Verweij N Identification of 64 Novel Genetic Loci Provides an Expanded View on the Genetic Architecture of Coronary Artery Disease. Circ. Res 122, 433–443 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Myocardial Infarction Genetics and CARDIoGRAM Exome Consortia Investigators et al. Coding Variation in ANGPTL4, LPL, and SVEP1 and the Risk of Coronary Disease. N. Engl. J. Med 374, 1134–1144 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DeBoever C et al. Assessing Digital Phenotyping to Enhance Genetic Studies of Human Diseases. Am. J. Hum. Genet 106, 611–622 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.GTEx Consortium. GTEx Portal https://www.gtexportal.org/home/ (2020).
- 33.McComb RB, Bowers GN & Posen S Alkaline Phosphatase. (Springer, 1979). [Google Scholar]
- 34.Aguirre M, Rivas MA & Priest J Phenome-wide Burden of Copy-Number Variation in the UK Biobank. Am. J. Hum. Genet 105, 373–383 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Horikawa Y et al. Mutation in hepatocyte nuclear factor-1 beta gene (TCF2) associated with MODY. Nat. Genet 17, 384–385 (1997). [DOI] [PubMed] [Google Scholar]
- 36.Iwasaki N et al. Liver and kidney function in Japanese patients with maturity-onset diabetes of the young. Diabetes Care 21, 2144–2148 (1998). [DOI] [PubMed] [Google Scholar]
- 37.Nishigori H et al. Frameshift mutation, A263fsinsGG, in the hepatocyte nuclear factor-1beta gene associated with diabetes and renal dysfunction. Diabetes 47, 1354–1355 (1998). [DOI] [PubMed] [Google Scholar]
- 38.Benner C et al. Prospects of Fine-Mapping Trait-Associated Genomic Regions by Using Summary Statistics from Genome-wide Association Studies. Am. J. Hum. Genet 101, 539–551 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Benner C et al. FINEMAP: efficient variable selection using summary data from genome-wide association studies. Bioinformatics 32, 1493–1501 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hayes MG et al. Identification of type 2 diabetes genes in Mexican Americans through genome-wide association studies. Diabetes 56, 3033–3044 (2007). [DOI] [PubMed] [Google Scholar]
- 41.Abhary S et al. Common sequence variation in the VEGFA gene predicts risk of diabetic retinopathy. Invest. Ophthalmol. Vis. Sci 50, 5552–5558 (2009). [DOI] [PubMed] [Google Scholar]
- 42.Barton JC & Acton RT Diabetes in Hemochromatosis. J Diabetes Res 2017, 9826930 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Raju K & Venkataramappa SM Primary Hemochromatosis Presenting as Type 2 Diabetes Mellitus: A Case Report with Review of Literature. Int J Appl Basic Med Res 8, 57–60 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marks JD et al. Pressure indices of myocardial oxygen consumption during pulsatile ventricular assistance. ASAIO Trans. 35, 436–439 (1989). [DOI] [PubMed] [Google Scholar]
- 45.Alkhatatbeh MJ, Enjeti AK, Acharya S, Thorne RF & Lincz LF The origin of circulating CD36 in type 2 diabetes. Nutr. Diabetes 3, e59 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gonzalez Y et al. High glucose concentrations induce TNF-α production through the down-regulation of CD33 in primary human monocytes. BMC Immunol. 13, 19 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Larson NB et al. Trans-ethnic meta-analysis identifies common and rare variants associated with hepatocyte growth factor levels in the Multi-Ethnic Study of Atherosclerosis (MESA). Ann. Hum. Genet 79, 264–274 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Woodward OM et al. Identification of a urate transporter, ABCG2, with a common functional polymorphism causing gout. Proc. Natl. Acad. Sci. U. S. A 106, 10338–10342 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Verbanck M, Chen C-Y, Neale B & Do R Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet 50, 693–698 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bowden J et al. Improving the visualization, interpretation and analysis of two-sample summary data Mendelian randomization via the Radial plot and Radial regression. Int. J. Epidemiol 47, 2100 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rücker G, Schwarzer G, Carpenter JR, Binder H & Schumacher M Treatment-effect estimates adjusted for small-study effects via a limit meta-analysis. Biostatistics 12, 122–142 (2011). [DOI] [PubMed] [Google Scholar]
- 52.Bowden J et al. A framework for the investigation of pleiotropy in two-sample summary data Mendelian randomization. Stat. Med 36, 1783–1802 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Curhan GC, Willett WC, Rimm EB & Stampfer MJ A prospective study of dietary calcium and other nutrients and the risk of symptomatic kidney stones. N. Engl. J. Med 328, 833–838 (1993). [DOI] [PubMed] [Google Scholar]
- 54.Ruth KS et al. Using human genetics to understand the disease impacts of testosterone in men and women. Nat. Med 26, 252–258 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang F et al. Apolipoprotein A-IV improves glucose homeostasis by enhancing insulin secretion. Proc. Natl. Acad. Sci. U. S. A 109, 9641–9646 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pietzsch J, Julius U, Nitzsche S & Hanefeld M In vivo evidence for increased apolipoprotein A-I catabolism in subjects with impaired glucose tolerance. Diabetes 47, 1928–1934 (1998). [DOI] [PubMed] [Google Scholar]
- 57.Zhang P, Gao J, Pu C & Zhang Y Apolipoprotein status in type 2 diabetes mellitus and its complications (Review). Mol. Med. Rep 16, 9279–9286 (2017). [DOI] [PubMed] [Google Scholar]
- 58.Vozarova B et al. High alanine aminotransferase is associated with decreased hepatic insulin sensitivity and predicts the development of type 2 diabetes. Diabetes 51, 1889–1895 (2002). [DOI] [PubMed] [Google Scholar]
- 59.De Silva NMG et al. Liver Function and Risk of Type 2 Diabetes: Bidirectional Mendelian Randomization Study. Diabetes 68, 1681–1691 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huang G-X, Ji X-M, Ding Y-C & Huang H-Y Association between serum cystatin C levels and the severity or potential risk factors of acute ischemic stroke. Neurol. Res 38, 518–523 (2016). [DOI] [PubMed] [Google Scholar]
- 61.van der Laan SW et al. Cystatin C and Cardiovascular Disease: A Mendelian Randomization Study. J. Am. Coll. Cardiol 68, 934–945 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Colijn JM et al. Increased High-Density Lipoprotein Levels Associated with Age-Related Macular Degeneration: Evidence from the EYE-RISK and European Eye Epidemiology Consortia. Ophthalmology 126, 393–406 (2019). [DOI] [PubMed] [Google Scholar]
- 63.Kay P et al. Age-related changes of cystatin C expression and polarized secretion by retinal pigment epithelium: potential age-related macular degeneration links. Invest. Ophthalmol. Vis. Sci 55, 926–934 (2014). [DOI] [PubMed] [Google Scholar]
- 64.Klein R, Knudtson MD, Lee KE & Klein BEK Serum cystatin C level, kidney disease markers, and incidence of age-related macular degeneration: the Beaver Dam Eye Study. Arch. Ophthalmol 127, 193–199 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zurdel J, Finckh U, Menzer G, Nitsch RM & Richard G CST3 genotype associated with exudative age related macular degeneration. Br. J. Ophthalmol 86, 214–219 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Khalili H et al. Endogenous levels of circulating androgens and risk of Crohn’s disease and ulcerative colitis among women: a nested case-control study from the nurses’ health study cohorts. Inflamm. Bowel Dis 21, 1378–1385 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nasser M et al. Testosterone therapy in men with Crohn’s disease improves the clinical course of the disease: data from long-term observational registry study. Horm. Mol. Biol. Clin. Investig 22, 111–117 (2015). [DOI] [PubMed] [Google Scholar]
- 68.Kühn T et al. Albumin, bilirubin, uric acid and cancer risk: results from a prospective population-based study. Br. J. Cancer 117, 1572–1579 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wray NR, Kemper KE, Hayes BJ, Goddard ME & Visscher PM Complex Trait Prediction from Genome Data: Contrasting EBV in Livestock to PRS in Humans. Genetics vol. 211 1131–1141 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tibshirani R Regression Shrinkage and Selection Via the Lasso. Journal of the Royal Statistical Society: Series B (Methodological) vol. 58 267–288 (1996). [Google Scholar]
- 71.Qian J et al. A fast and scalable framework for large-scale and ultrahigh-dimensional sparse regression with application to the UK Biobank. PLoS Genet. 16, e1009141 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Martin AR et al. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat. Genet 51, 584–591 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bild DE et al. Multi-Ethnic Study of Atherosclerosis: objectives and design. Am. J. Epidemiol 156, 871–881 (2002). [DOI] [PubMed] [Google Scholar]
- 74.Inouye M et al. Genomic Risk Prediction of Coronary Artery Disease in 480,000 Adults: Implications for Primary Prevention. J. Am. Coll. Cardiol 72, 1883–1893 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cohen JC, Boerwinkle E, Mosley TH, Jr & Hobbs, H. H. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N. Engl. J. Med 354, 1264–1272 (2006). [DOI] [PubMed] [Google Scholar]
- 76.DeBoever C et al. Medical relevance of protein-truncating variants across 337,205 individuals in the UK Biobank study. Nat. Commun 9, 1612 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Amar D, Ashley E & Rivas MA Constraint-based analysis for causal discovery in population-based biobanks. bioRxiv 566133 (2019) doi: 10.1101/566133. [DOI] [Google Scholar]
- 78.Tanigawa Y, Sinnott-Armstrong N, Benner C & Rivas MA Datasets described in ‘Genetics of 35 blood and urine biomarkers in the UK Biobank’. (2020) doi: 10.35092/yhjc.c.5043872.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Levey AS et al. A new equation to estimate glomerular filtration rate. Ann. Intern. Med 150, 604–612 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wainberg M et al. Homogeneity in the association of body mass index with type 2 diabetes across the UK Biobank: A Mendelian randomization study. PLoS Med. 16, e1002982 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Eastwood SV et al. Algorithms for the Capture and Adjudication of Prevalent and Incident Diabetes in UK Biobank. PLoS One 11, e0162388 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Quinlan AR & Hall IM BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Buniello A et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 47, D1005–D1012 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Carvalho-Silva D et al. Open Targets Platform: new developments and updates two years on. Nucleic Acids Res. 47, D1056–D1065 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hemani G et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Slob EAW, Groenen PJF, Thurik AR & Rietveld CA A note on the use of Egger regression in Mendelian randomization studies. International journal of epidemiology vol. 46 2094–2097 (2017). [DOI] [PubMed] [Google Scholar]
- 87.Benjamini Y & Yekutieli D The Control of the False Discovery Rate in Multiple Testing under Dependency. Ann. Stat 29, 1165–1188 (2001). [Google Scholar]
- 88.Shannon P et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Friedman J, Hastie T & Tibshirani R Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw 33, 1–22 (2010). [PMC free article] [PubMed] [Google Scholar]
- 90.Chang CC et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Joshi AD et al. Four Susceptibility Loci for Gallstone Disease Identified in a Meta-analysis of Genome-Wide Association Studies. Gastroenterology 151, 351–363.e28 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mahajan A et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat. Genet 50, 1505–1513 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Läll K, Mägi R, Morris A, Metspalu A & Fischer K Personalized risk prediction for type 2 diabetes: the potential of genetic risk scores. Genet. Med 19, 322–329 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Abraham G et al. Genomic prediction of coronary heart disease. Eur. Heart J 37, 3267–3278 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Khera AV et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet 50, 1219–1224 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.