

Abstract
Admixed populations are routinely excluded from genomic studies due to concerns over population structure. Here, we present a statistical framework and software package, Tractor, to facilitate inclusion of admixed individuals in association studies by leveraging local ancestry. We test Tractor with simulated and empirical 2-way admixed African-European cohorts. Tractor generates accurate ancestry-specific effect size estimates and p values, can boost GWAS power, and improves the resolution of association signals. Using a local ancestry aware regression model, we replicate known hits for blood lipids, discover novel hits missed by standard GWAS, and localize signals closer to putative causal variants.
Introduction
Admixed groups, whose genomes contain more than one ancestral population, make up more than a third of the US populace, with the population becoming increasingly mixed over time1. Many common, heritable diseases including prostate cancer2-5, asthma6-9, and certain cardiovascular disorders10,11 are enriched in admixed populations, which include African American and Hispanic/Latino individuals. However, only a minute proportion of association studies address the genetic architecture of complex traits in these groups12,13; admixed individuals are systematically removed from many large-scale collections. This is due in large part to the lack of methods and pipelines to effectively account for their ancestry such that population substructure can infiltrate analyses and bias results if they are run with standard analysis of homogeneous individuals14-21. Efforts to collect genetic data alongside medically-relevant phenotypes are beginning to focus more on diverse groups containing higher amounts of admixture22-27, motivating the development of scalable methods to allow well-calibrated statistical genomic work on these populations. If not addressed, this inability to analyze admixed people will limit the clinical utility of large-scale data-collection efforts for minorities, exacerbating existing health disparities28-32.
In GWAS, the specific concern regarding including admixed participants is false positive hits due to alleles being at different frequencies across populations. Most studies currently attempt to control for this by using Principal Components (PCs) in a linear or linear mixed model framework. However, PCs capture broader admixture fractions, and individuals’ local ancestry makeup may differ between case and control cohorts even if their global fractions are identical. Even including PCs as covariates, then, still leaves open the possibility for false positive associations, as well as absorbing power.
Studying diverse populations in gene discovery efforts not only reduces disparities but also benefits genetic analysis for individuals of all ancestries. A notable example of this is multi-ancestry fine-mapping, which can reduce the variant credible set by leveraging populations’ differing linkage disequilibrium (LD) patterns33-38. This is particularly helpful in populations of African descent, where LD blocks are the shortest and individuals have nearly a million more variants per person than individuals outside of the continent39. We note that, in admixed populations, we can utilize LD patterns from multiple ancestries as well as disrupted LD blocks within each, offering a more refined linkage landscape with which to localize GWAS signal.
Here, we have developed a scalable framework that allows for the incorporation of admixed individuals into large-scale genomics efforts by using local ancestry inference (LAI). Our framework, distributed as a software package named Tractor, generates ancestry dosages at each site from LAI calls, extracts painted haplotype segments to correct population structure at the genotype level, and runs a local ancestry-aware regression model, producing ancestry-specific effect size estimates and p values. Admixed individuals can thereby be analyzed in a well-calibrated manner, either in a cohort alone or alongside homogenous groups. Through testing in simulations and with empirical data on continuous and case/control phenotypes with differing levels of polygenicity, we demonstrate that Tractor produces accurate results in admixed cohorts and boosts GWAS power across many genetic contexts. We further demonstrate improvements in association signal localization from accounting for the ancestral backbone on which alleles fall. These efforts fill a gap in existing resources and will improve our understanding of complex diseases across diverse populations.
Incorporating local ancestry into variant identification for admixed populations has been discussed previously40,41,50,42-49, particularly with regard to ‘admixture mapping,’ whereby researchers associate an increase of a given ancestry at a locus with increased risk of a disease that is known to be stratified in prevalence across ancestries51-55. Admixture mapping has proven successful in diseases which are highly stratified, such as asthma and cardiovascular phenotypes56-61. However, this strategy cannot be employed for phenotypes which are observed at equivalent rates across groups and can be subject to false positives from local ancestry increases that are unrelated to the phenotype of interest. We build upon this important work by modeling the local ancestry dosage for each person at each variant in a way that accounts for differences in minor allele frequency (MAF) across populations, controlling for demography without an increased false positive risk. Tractor thereby generates accurate ancestry-specific summary statistics, which admixture mapping and traditional GWAS both cannot, and can be utilized for phenotypes which are seen at similar or different rates across populations.
The statistical model built into Tractor for binary phenotypes tests each SNP for an association with the phenotype using the logistic regression model:
where X1 is the number of haplotypes of the index ancestry present at that locus for each individual, X2 is the number of copies of the risk allele coming from the first ancestry, X3 is the number of copies coming from the second ancestry, and X4 to Xk are other covariates such as age, sex, an estimate of global ancestry, etc. The significance of the risk allele is evaluated with a likelihood ratio test comparing the full model to a model fit without the risk allele, thus allowing estimation of the aggregated effects in the presence of effect size heterogeneity. The (two degree of freedom) model presented here is for a 2-way admixed scenario but can be readily scaled to an arbitrary number of ancestries with the addition of terms.
Results
LAI has high accuracy for African Americans
We ran LAI using RFmixv2, a discriminative approach which estimates local ancestry using conditional random fields parameterized with random forests62. RFmix can run on multi-way admixed populations, outperforms other local ancestry inference methods for minority populations, and leverages the ancestry components in admixed reference panel individuals, important when there is a lack of homogenous reference panels as is often the case for understudied groups63,64. As Tractor relies heavily on LAI calls, we quantified RFmix’s accuracy using a simulated 2-way admixed cohort created from African American (AA) individuals from the Psychiatric Genomics Consortium PTSD working group (PGC-PTSD). LAI was highly accurate in a realistic demographic model for AA individuals65-67, assigning the correct ancestry ~98% of the time (Methods, Supplementary Table 1).
To ensure that Tractor performed well across demographic models, we varied admixture fractions and pulse timings. Specifically, we varied the pulse of admixture in time to 3 generations and 20 generations ago and changed the admixture fractions to 30/70% and 50/50% EUR and AFR ancestry, respectively (Extended Data Figure 1). We also checked the ancestry-specific accuracy in the realistic demographic scenario to assess if there was a bias in calling dependent on ancestry. Across all demographic models and ancestries, site-wise LAI calls were similarly accurate, with the correct call being obtained ~98% of the time (Supplementary Table 1). While we refer solely to continental level ancestry here, we appreciate the high level of diversity and admixture within the continents and particularly in Africa. As reference panels for diverse groups grow in size, we will have increased ability to examine ever more geographically refined groups.
Recovering haplotypes disrupted by statistical phasing
While errors in statistical phasing can lead to errors in LAI, we found that iterating between LAI and statistical phasing improved the accuracy of both. Errors in statistical phasing are a major concern68,69, but few methods to recover disrupted haplotypes exist. Phasing errors result in incorrect switches of the chromosome strand on which the haplotype is assigned, which artificially reduces tract lengths and increases the overall tract counts. This phenomenon could also be misinterpreted as recombination events. Taking advantage of the ability to visualize tracts offered by admixed individuals, we are able to consistently correct switch errors and recover disrupted haplotypes, making tract distributions significantly more realistic (Extended Data Figure 2, Supplementary Table 2, Supplementary Information).
Evaluating the landscape of GWAS power gains from Tractor
To quantify the potential increase in power from the inclusion of local ancestry, we simulated individuals’ likelihoods of being cases as a function of AFR admixture fraction, the risk allele dosage, and the ancestral background of each allele. This initial framework can be thought of as modeling a risk allele with a true effect only in the AFR background, and/or as modeling a tagging variant having a marginal effect size estimated in only one ancestry. This latter case may arise for multiple reasons due to genomic differences across ancestries, such as a mutation being monomorphic in EUR but variable in AFR (probable as individuals from Africa contain almost a million more variants than other populations70), differing demographies resulting in LD structure variation that affects causal variant tagging, and MAF differences. Therefore, our model quantifies both Tractor’s ability to estimate and leverage power from ancestral differences in true effect sizes, as well as for differing marginal effect sizes of tagging SNPs, as may be systematically expected across the genome. Our simulation framework also incorporates the clinically observed phenomenon of disease prevalence differing as a function of ancestry. We then ran association tests and compared the power across the odds ratio spectrum under the traditional GWAS and Tractor models.
Compared to the traditional model, we observe a significant gain in power using the Tractor framework with comparable improvements across sample sizes and disease prevalences (Figure 2A). We ran similar simulations varying the effect size difference, absolute MAF, MAF difference across ancestries, and admixture fractions. To summarize, Tractor is most powerful when there is heterogeneity in the apparent effect size for a variant across ancestries (Figure 2, Extended Data Figures 3,4). As described above, such heterogeneity may be a consequence of a variant truly having different effects in different populations (e.g., in the context of gene-environment interactions) or may arise from differences in the indirect association evidence of the variant (i.e., the contribution to the estimated effect size from tagging other causal genetic variants). The biggest power gain within the case of effect size heterogeneity comes if an allelic effect is present in the smaller fraction ancestry only (Figure 2D). For example, using a realistic AA demographic history, if we model an allele with an effect only in the EUR background (~20% of the sample), analyzing the tracts without LAI information will have essentially no power to detect an association due to the higher noise relative to signal from uninformative tracts. However, Tractor is able to recover the effective sample size and power analyzing just the effect haplotypes, i.e. the EUR segments alone.
Figure 2. GWAS power gains across sample sizes, ancestral MAF differences, admixture proportions, and effect size differences.
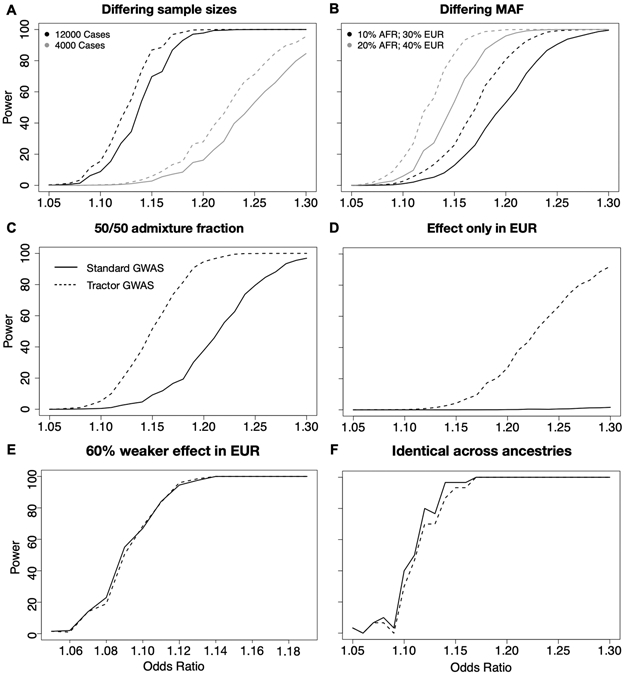
In all scenarios shown, dashed lines correspond to the power from the Tractor model incorporating local ancestry, solid lines are for a traditional GWAS model. In all panels we modeled a 10% disease prevalence. Unless otherwise noted, we used the parameters for a realistic demographic scenario for AA individuals: 80% AFR ancestry, an effect present only in the AFR genetic background, 12k cases and 30k controls, and 20% MAF. (A) There are similar gains in GWAS power when using the Tractor LAI-aware model across samples sizes of 4,000 (grey) and 12,000 (black) cases with 2x controls. (B) When there is a MAF difference between ancestries, the boost in power is even more pronounced. Gains vary across the allele frequency spectrum: black=MAF 10% AFR, 30% EUR; grey=MAF 20% AFR, 40% EUR. (C) Gains become more pronounced when the admixture fractions are modified to 50/50. (D) Dramatic gains are obtained when the effect is switched to instead only be present on the less common EUR background. (E) The threshold for heterogeneity in ancestral effect sizes required to observe gains in Tractor power over traditional GWAS assuming 20% MAF in both ancestries and an effect that is stronger in AFR with varying difference to the EUR effect. (F) There is a small loss in power from incorporating local ancestry into the GWAS model when all parameters are identical across ancestries.
We assessed the required level of heterogeneity in effect sizes required for Tractor to benefit from power gains over standard GWAS. We note that the amount of heterogeneity required depends heavily on other parameters, in particular on the relative admixture fraction of the ancestry containing a larger effect. This is because the inflation of variance and bias of the effect estimate will vary based on not only the difference in means, but the relative sizes of groups. However, all parameters being equal, unless group effects are substantially different the penalty of additional model complexity can be more severe than the penalty of a biased estimate of effect and higher variance estimate found in a regular model. In our AA simulation framework with equivalent MAF and an effect present in both ancestries but stronger in AFR, Tractor would require an effect difference of more than ~60% to benefit from effect size heterogeneity (Figure 2E, Extended Data Figure 3E-F).
Notably, however, under scenarios where there is no expected benefit from incorporating local ancestry (that is, when all features are equivalent across ancestries), the power loss is minimal, reflecting the penalty for adding an additional model parameter (Extended Data Figures 3,4). In no case does Tractor dramatically underperform compared to the traditional GWAS model. Additionally, Tractor reliably estimates ancestry-specific effects whereas in a regular GWAS the effect estimate generated would be a weighted average of these. Therefore, even when failing to demonstrate elevated power in variant identification, the Tractor model may provide more power in downstream applications where correct effect estimates are required.
We additionally benchmarked Tractor against asaMap50. Instead of local ancestry, asaMap is based on a mixture model, where the mixture components are the phenotype distributions corresponding to the given ancestries of the tested SNP and the mixture weights are the probabilities of these ancestries. asaMap thereby generates ancestry-specific effect sizes without local ancestry, making it a useful point of comparison. We also tested the impact of variability in LAI accuracy on Tractor, modeling perfect, realistic (98%), and a lower bound of 90% LAI accuracy. All Tractor models reached higher statistical power than both standard GWAS and asaMap (Extended Data Figure 5). As expected, decreasing LAI accuracy reduced power for Tractor, however even at the lowest accuracy tested, Tractor outperformed these other methods.
Tractor accurately estimates ancestry-specific effects
To ensure that Tractor generates reliable ancestry-specific effect sizes, we checked the effect size estimated as compared to that modeled across a range of absolute and ancestrally differing effect sizes using the simulation framework described above. Across all genomic models, Tractor accurately estimated the ancestry-specific effect size (Figure 3).
Figure 3. Tractor accurately estimates ancestry-specific effect sizes.

Boxplots show the effect size estimated by Tractor as compared to that modeled in the simulation across a range of effect sizes where the center is the median, the bounds of box represent the first quantile to third quantile, and whiskers are 1.5 * IQR. The lines indicate the simulated values for each ancestry. Blue represents effects in AFR, red in EUR. The models presented all include 1000 simulation replicates with 12k cases, 30k controls at 10% disease prevalence in a realistic AA population with an admixture ratio of 80/20 AFR/EUR. Unless otherwise noted, the risk allele MAF was set at 20% in both ancestries. (A) The initial simulation framework of an effect only in AFR. (B) An effect only in AFR with differing minor allele frequencies across ancestries: AFR being 10% and EUR 30%. (C) An effect in both ancestries, with a 30% weaker effect modeled in the EUR. (D) Effect only in EUR.
No increase in false positive rate with the Tractor model
We quantified the type I error rate of the Tractor model by simulating a variant with no effect in either ancestry and counting the spurious significant associations identified in a simulated AA population with 10% disease prevalence given α = 0.05. Across our tests, we observe no statistically significant difference in false positive rate between Tractor and the expected type I error rate of 5% (Supplementary Figure 1). In addition, we calculated the genomic inflation factor, λGC, of null simulated phenotypes across GWAS permutations and confirmed no significant inflation using the Tractor GWAS model (Supplementary Figure 2). Therefore, there does not appear to be an elevation in false positive rates with the Tractor framework, suggesting that the observed power increases result from improved detection of true biological signal.
Tractor replicates known loci and identifies novel hits
To ensure that our Tractor joint-analysis GWAS model also performs well on empirical data, we ran the method on well characterized blood lipid phenotypes previously demonstrated to have ancestry-specific effects: Total Cholesterol and Low-Density Lipoprotein Cholesterol. We constructed a pseudo-cohort of 4,309 two-way African-European admixed individuals from the UK Biobank (UKB) with blood panel phenotype data to serve as our sample. To ensure LAI was unbiased across regions of the genome, we examined its genomic distribution. Local ancestry was relatively evenly distributed and was proportional to global fractions, being 97.5% correlated with admixture proportions (Supplementary Figure 3).
Tractor GWAS replicated known associations for blood lipids in this cohort43,71-73, reaching the standard genome-wide significance level of 5×10−8 at previous top associations, including in genes PCSK9, LDLR, and APOE (Figure 4, Supplementary Figure 4). In some cases, Tractor improved the observed top hit significance. We describe Total Cholesterol in detail here; see Supplemental Information for further description of validation with LDL Cholesterol.
Figure 4. Tractor GWAS replicates established hits for Total Cholesterol in admixed African-European individuals and identifies new ancestry-specific loci.

QQ and Manhattan plots for Total Cholesterol using (A) the standard GWAS model compared to Tractor joint-analysis results for the (B) AFR and (C) EUR backgrounds. The traditional genome-wide significance threshold of 5e-08 is shown as the red dashed line.
Our model also identified additional hits in these admixed individuals missed by standard GWAS (Figure 4). For example, we identify an association present only on the AFR background on chr1 (rs12740374, p=3.46x10−8). This locus has previously been shown to affect blood lipid levels, metabolic syndrome, and coronary heart disease risk in independent AA cohorts71,74-79, and was determined to be the causal variant for affecting LDL Cholesterol in a multi-ancestry fine-mapping study80. Had we not deconvolved ancestral tracts for our GWAS, we would have missed this site with a demonstrated effect in the phenotype and population of interest.
We additionally identify a novel peak on chr15 that only reached significance in the AFR tracts. The lead SNP (rs12594517, p= 1.915x10-8) lies in an intergenic area and is uncharacterized. The closest gene neighboring it is MEIS2, lying ~70kb upstream, followed by C15orf41. While the precise role and mechanism this locus plays in affecting blood lipids remains unclear, MEIS2 has previously been found to be associated to body mass index and waist circumference and C15orf41 was a significant hit in a previous GWAS of cholesterol81,82. Though further follow-up is needed to clarify any direct relationship to Total Cholesterol, this association highlights the utility of Tractor to identify signals that would be undetectable in admixed cohorts without accounting for local ancestry.
Tractor is also able to refine the location of signals closer to the causal variant than is possible using standard GWAS procedures. Total Cholesterol was previously mapped to an intron in the gene DOCK6 in AA cohorts71, a finding we replicate at the suggestive significance threshold with standard GWAS. Tractor identified a lead DOCK6 SNP 20kb downstream in the AFR and a meta-analysis of hits from deconvolved AFR and EUR tracts. This new lead SNP (rs2278426) spans DOCK6 as well as ANGPTL8, where it is a missense mutation (NC_000019.9:g.[11350488C>T]) that is predicted to be possibly damaging and deleterious by polyphen and SIFT, respectively83,84 (Discussion, Figure 5). ANGPTL8, also known as lipasin, has been shown to regulate plasma lipid levels in mice by inhibiting the enzyme lipoprotein lipase85-88. In humans, ANGPTL8 levels correlate with metabolic phenotypes including type 2 diabetes and obesity89-92 and HDL Cholesterol levels across diverse populations have been demonstrated to better correlate with ANGPTL8 than DOCK693. Altogether ANGPTL8 appears to be a more promising candidate, highlighting how leveraging diverse populations allows for improved identification of risk variants. Notably, asaMap improved localization compared to standard GWAS, but was unable to identify the putative causal variant (Supplementary Figure 5).
Figure 5. Tractor better localizes a top hit for Total Cholesterol.

Runs on UKB admixed individuals with (A) standard GWAS model, (B) AFR-specific GWAS with Tractor, and (C) a meta-analysis of GWAS runs on deconvolved EUR and AFR tracts. Both Tractor runs pinpoint a lead SNP ~20kb downstream of the intronic standard GWAS top hit in DOCK6 spanning a better candidate gene, ANGPTL8. No significant signal was seen in the EUR segments. In all plots, point size is proportional to the number of samples included for that test, and color indicates r2 to the named lead SNP. The recombination rate is shown as a blue line generated from the AFR superpopulation of the 1000 Genomes Project in B, or the EUR superpopulation for other panels.
To assess whether our ability to identify rs2278426 was due to a true or marginal effect size difference driven by MAF or LD differences across ancestries, we attempted validating its association with fine-mapping in 345,235 white, British UKB individuals and 135,808 Japanese individuals from Biobank Japan94. rs2278426 was successfully fine-mapped to a 95% credible set in both populations, with maximum posterior inclusion probability of 0.993 in Biobank Japan. This variant occurs at 26% frequency in gnomAD East Asian individuals, 18% in African, and 4% in non-Finnish Europeans95. We estimated the frequencies in our admixed empirical cohort to be ~22% in the AFR and ~4% in the EUR. To assess the role of LD on localization improvement, we created within-sample LD heatmaps for this DOCK6 region of interest from the full dataset, AFR and EUR tracts, finding broadly similar patterns (Supplementary Figure 6). These trends suggest that localization improvement was driven by higher power in non-European populations to identify the causal variant rather than LD differences directly. Though below the traditional genome-wide significance level, this locus highlights the improved ability to localize GWAS signal thanks to leveraging additional ancestral breakpoints in admixed genomes. See Supplemental Information for demonstration of signal localization for LDLC in the canonical gene PCSK9 (Supplementary Figure 7).
Discussion
Despite recent advances in understanding the genetics of complex diseases, major limitations remain in our knowledge of the architecture of such disorders in minority and admixed populations. Here, we present an analytic framework and gene discovery method distributed as a scalable software package named Tractor, which allows admixed samples to be appropriately analyzed alone or alongside homogenous cohorts in statistical genomics efforts. We test our framework in a simulation model designed to emulate AA cohorts. We then apply it to empirical data from admixed African-descent individuals of the UKB. We observe a gain in power to detect risk loci across sample sizes, demographic models, and disease prevalences using Tractor, particularly when true or marginal effect sizes are heterogeneous across populations. Our approach incorporates a local ancestry aware GWAS method that generates ancestry-specific estimates, which admixture mapping and standard GWAS cannot, and which can be extremely helpful in downstream efforts such as constructing genetic risk scores for understudied populations. Tractor also gives increased precision in localizing GWAS signal closer to the causal variant in recently admixed groups. This reduces the credible set of SNPs and aids in the prioritization of variants for subsequent functional testing.
The Tractor pipeline requires several inputs, most importantly accurate local ancestry calls. Users should ensure good LAI performance in their target cohorts. A major determinant of accurate LAI calls is a comprehensive and well-matched reference panel47,96. Existing reference panels are more plentiful for Eurasian populations than for other groups, underscoring the need to expand sequencing efforts in diverse global populations. An additional consideration is to ensure high quality imputation if Tractor is to be run on imputed genotype array data. A final consideration is to ensure consistent phenotyping across ancestry groups, as is standard in multi-ancestry GWAS. We note that we have thoroughly tested Tractor here in the two-way admixture scenario reflecting AA demographic history. The analytic infrastructure can currently also run on three-way admixed populations, and our statistical model can scale to an arbitrary number of ancestries. Future work will test power and optimize the code for multi-way admixed models.
We evaluated the landscape of when Tractor does and does not add GWAS discovery power using simulated data modeled after AA cohorts (Figure 2, Extended Data Figures 3-5). Modifying several interacting parameters (absolute and difference in effect size, absolute and difference in MAF, sample size, disease prevalence, and admixture proportions), power gains from Tractor are generally most dramatic when there is a large effect size difference between ancestries. Gains from observed effect size differences are heightened when coupled with highly differing MAF across groups. The most extreme examples of such a case are when a there is an effect only on one haplotype background or an allele only present in one ancestry. Another interacting feature is the overall admixture proportions and whether the stronger effect allele is on the more frequent or more rare ancestry background. Tractor power gains are most dramatic when a variant with an effect in only one ancestry falls on the ancestry that is less common in the dataset. In such an instance in a standard GWAS setting, noise from the uninformative majority haplotypes would result in extremely low power to detect the locus (Figure 2D). Power can be recovered, however, by analyzing genotypes on ancestry-specific haplotypes, thus controlling for population structure as well as identifying risk variants that would otherwise be undetectable.
Conversely, we find that it is generally not necessary to include local ancestry in a GWAS model when there is no difference in the estimated effect size between ancestry groups. Notably, we are referring here to detection of both marginal as well as true effects. By a true effect, we mean an effect at a causal mutation directly impacting the phenotype. By a marginal effect, we imply the estimated effect at a variant tagging a causal variant; these marginal effects will represent the majority of results from a standard GWAS. Differences in indirect association can come from features such as ancestry-specific variation or different patterns of LD. Both true and marginal effect sizes are useful in post-GWAS efforts such as the construction of genetic risk scores and heritability estimation. There is evidence suggesting that in most cases (with some notable exceptions8,71,97), the effects of causal variants are similar across ancestries33,38,98-103. However, the marginal effect sizes of tag SNPs are routinely expected to differ across ancestries due to differences in ancestral MAF and the LD patterns resulting from each ancestral population’s demographic history104,105. Therefore, the most powerful use case for Tractor – when there are effect size differences across ancestries – should impact many more sites across the genome than just variants which have a true biological difference in effect across ancestry groups. Said differently, we clarify that Tractor benefits from power gains to detect the marginal beta in addition to the rarer case of variants with true effects only on one haplotypic backbone.
Tractor is also expected to benefit from increased power when functionally important (and likely rare) alleles only present in one population are missed by genotyping or imputation. In such situations the common alleles in LD, despite being shared across populations, would be associated with the phenotype as a function of which haplotypic background they are found on and thus would have a haplotype-specific effect. Another relevant scenario is the presence of LD in regions where there are ancestry-specific markers intermingled with shared ones. This would affect univariate scan results such that considering the haplotypic background on which alleles fall would particularly aid in localizing signal through improved marginal beta estimates, even with consistent causal effects in both ancestries. To expand on this idea, assuming a shared causal variant across ancestries, even in genomic regions with all variants present in both groups, one could hone in on the causal variant by minimizing the difference between betas across ancestries. If one assumes the beta at the causal variant is the same in both populations, then optimizing for reduced beta difference across groups using ancestry-specific effect size estimates can help in fine-mapping signal.
To summarize, the primary benefits we observed from Tractor are power gains from leveraging ancestral genomic differences, accurate estimation of ancestry-specific effect sizes, and improved GWAS signal resolution. At admixed cohort sample sizes of the scale we tested (4-12k cases), 10 PCs adequately controlled for false positives using standard GWAS procedures. With increasing sample sizes, however, Tractor’s fine-scale correction of population structure is expected to better control false positive associations than PCs alone, in addition to improving GWAS signal.
In empirical data, Tractor was able to replicate established GWAS hits, discover new ones, and aid in the localization of GWAS signal. We replicated known hits for blood lipids in ~4300 admixed African-European individuals from the UKB and demonstrate an improved ability to identify putative causal SNPs fine-mapped in another diverse collection, Biobank Japan94 (Figures 4,5, Supplementary Figures 4,7). We additionally identify novel hits using our local ancestry aware method that would have been missed using standard GWAS.
Portions of the Tractor pipeline may be helpful for working with admixed cohorts in use cases beyond GWAS. For example, recovering long-range haplotypes and correcting for population structure is key in evolutionary genomic studies for analyses such as genome-wide scans of selection106-108. Within medical genetics, accounting for the ancestral background of alleles will be valuable in studies of rarer variants which are more population specific109,110.
In sum, Tractor allows users to account for genotype-level ancestry in a precise manner, enabling the well-calibrated inclusion of admixed individuals in large-scale gene discovery efforts. This approach provides a number of benefits over traditional GWAS, including the production of accurate ancestry-specific summary statistics, improved localization of GWAS signals, and power boosts in many genetic contexts. This infrastructure is designed as a series of steps to be flexible and easily ported into other statistical genomics activities. Tractor advances the existing methodologies for studying the genetics of complex disorders in admixed populations.
Online Methods
QC and LAI Pipeline
The core feature of the Tractor framework relies on accounting for fine-scale population structure as informed by local ancestry (i.e. ancestral chromosome painting). Tractor then uses this information to (i) correct for individuals’ ancestral dosage at all variant sites, (ii) recover long-range tracts in admixed individuals; and (iii) extract the tracts and ancestry dosage counts from each ancestry component for use in ancestry-specific association tests. We have tested and built this framework around LAI calls from RFmix_v262, and have built an automated pipeline (https://github.com/eatkinson/Post-QC) to perform post-genotyping QC, data harmonization, phasing, and LA inference to consistently prepare the data for downstream analysis (Supplementary Information)1,2.
In all tests, we ran RFmix with 1 EM iteration and a window size of 0.2 cM with the HapMap combined recombination map3 to inform switch locations. The -n 5 flag (terminal node size for random forest trees) was included to account for an unequal number of reference individuals per reference population. We used the --reanalyze-reference flag, which recalculates admixture in the reference samples for improved ability to distinguish ancestries. This is especially important when the reference samples are themselves admixed. As a reference panel, we used AFR and EUR individuals from the 1000G reference panel. Painted karyogram plots were produced using a modified version of publicly available code (https://github.com/armartin/ancestry_pipeline). We have optimized this pipeline under the two-way admixed AA demographic model. Tractor additionally supports 3-way admixture calls with an expanded set of scripts.
LAI Accuracy
We validated that LAI was performing well in the AA use case. To do this, we generated a truth dataset by simulating individuals with known phase and LA from empirical data. Our simulation reference panel consisted of haplotypes from homogenous PGC-PTSD individuals who had ≥95% EUR or AFR ancestry as inferred by SNPweights4. We simulated admixture between these reference individuals with admix-simu5 using a realistic AA demographic model of 1 pulse of admixture 9 generations ago with 84% contribution from Africa and 16% from Europe. The resultant population mixes amongst itself until the present day, copying haplotypes from the previous generation with break points informed by the recombination map. This retains the LD structure and genetic variation present in real genomic data and ensures that the truth dataset resembles cohort data as closely as possible. We then ran LAI and calculated LAI accuracy as how often the ancestry call was correct in the simulated truth data. The overall proportion of the genome estimated by RFmix in the realistic scenarios was within the range of expectations given the simulation model of 16% European, 84% African ancestry (15.1% and 84.9%, respectively).
Correcting Switch Errors using Local Ancestry
Despite LAI calling ancestry dosage accurately, frequent chromosomal switches were visible in painted karyograms (Figure 1), which we determined were due to phasing errors. It is important to retain complete tracts, as spurious breakpoints will reduce the accuracy of haplotype-based tests. Tractor detects and fixes phase switches, which we define as a swap of ancestry across a chromosome within a 1 cM window at a region with heterozygous ancestry dosage, using local ancestry. This level of information is accessible due to the admixture; no local ancestry switches would be visible if a cohort were homogenous.
Figure 1. Painted karyograms of a simulated AA individual showing local EUR (red) and AFR (blue) ancestral tracts across data treatments.

The top panel shows the truth results for an example individual in our simulated AA cohort. The same person after statistical phasing is shown in the second row – note the disruption of long haplotypes resulting from phasing switch errors. The third panel illustrates our recovery of tracts broken by switch errors in phasing. The bottom panel shows the smoothing and further improvement of tracts acquired through an additional round of LAI. The same section of chr13 showing an example tract at higher resolution is pictured on the right to highlight tract recovery.
To ensure that correcting phase switch errors improved results compared to the truth expectations, we modeled the expected distributions of EUR tract within AA individuals using a Poisson process with rate equal to the number of generations ago when the pulse of admixture occurred. The waiting time until a recombination event disrupts a tract is expected to follow this distribution, with a slight shortening of tracts proportional to the percent admixture due to the inability to visualize tract switches that occur across regions of the same ancestry. In this way we could quantify the probability of observing the observed number of tracts after a particular data treatment given the truth expectations (See Supplementary Information, Supplementary Table 2, Extended Data Figures 1,2).
GWAS power simulations incorporating local ancestry
We assessed the improvements in GWAS power from using Tractor through simulations. We formulated our simulation framework on the suggestions of Skotte et al. (2019). Power calculations were based on a simulation framework that initially models an AA population assuming a bi-allelic disease risk allele with a 20% overall MAF and an additive effect in the AFR genetic background but not in the EUR. Specifically, the overall admixture proportions were drawn from a beta distribution with shape parameters 7.76 and 2.17, the fitted parameters to this distribution for AFR ancestry proportions observed in the PGC-PTSD Freeze 2 AA cohorts. The genotype of each copy of the allele was drawn from a binomial distribution with the probability of having the minor allele set to the MAF. We simulated a disease phenotype with individuals’ risk drawn from a binomial distribution assuming a 10% disease prevalence. Risk of developing the phenotype was modified on a log-additive scale according to the admixture proportions and the presence of the minor allele on an AFR background using a logit model. In this model, the probability of disease was set to −2.19 + log of allelic risk effect size*number of copies of the minor allele coming from an AFR ancestral background + 0.5*AFR admixture proportion. −2.19 was chosen as it represents a 10% probability of disease given no AFR admixture or copies of the minor allele from either ancestral background. The 0.5 value in 0.5*AFR proportion was set in order to induce stratification in the simulated population, as is observed in empirical data. In other words, all of our simulations modeled increasing disease prevalence with admixture fractions, reflective of clinical observation. With this simulation design, individuals with higher AFR ancestry proportions are more likely to be cases whereas those with higher EUR ancestry proportions are more likely to be controls. Subjects’ disease status was then drawn from a binomial distribution with the probability parameterized to their individual disease risk according to the logit model. Cases and controls were sampled at random from the simulated population at a 2.5:1 control to case ratio, the approximate ratio of controls to cases in PGC-PTSD freeze 2.
Under each simulation, we fit three logistic regression models of disease status that included: M1) admixture only, M2) number of copies of the risk allele only, and M3) admixture + number of copies of the risk allele on a EUR background + number of copies on an AFR background. M1 serves as a null comparison to evaluate the significance of including the SNP as a predictor. M2 is equivalent to standard GWAS procedures. M3 models our Tractor method. In all cases we included the first 10 PCs to account for population structure, as is common practice in current GWAS on admixed cohorts. The significance of M2 and M3 are evaluated by likelihood ratio tests comparing them to M1. For each 100 simulations at a given effect size and sample size, for both M2 and M3 we estimated power as the proportion of the time that the likelihood ratio test was significant (p < 5e-8). We performed 1000 rounds of simulation with this model at each level of allelic effect size ranging from Odds Ratio (OR) 1.05 to 1.3 and case sample size N=4000 and 12000. It should be noted that the 2-way Tractor model is by nature a 2 degree of freedom test. Adding additional ancestries/terms into the model could reduce power gains, due to the additional penalty.
We additionally benchmarked Tractor against asaMap under our primary models assessing the effect of differing sample sizes, MAF differences, admixture fractions, and relative ancestry-specific effects (Figure S5). The specific asaMap model test that we assessed in these runs is equivalent to the M1 vs M5 comparison described in Skotte et al. (2019), namely an effect in either population. A comparison of results from different asaMap models for the DOCK6 region of interest can be seen in Supplementary Figure 5.
We also built in LAI uncertainty into the Tractor model in these comparisons to quantify the effect of LAI accuracy on Tractor power. For this, we modeled perfect, realistic - 98%, and a lower tolerable bound of 90% LAI accuracy (Extended Data Figure 5). In all cases, 1000 replicates were run under a model of 12k cases, 30k controls, 10% disease prevalence, and a realistic 80/20 AFR/EUR admixture ratio using 10 PCs as covariates.
Characterizing the landscape of Tractor power gains
To evaluate Tractor power gains, we ran similar sets of simulations varying effect size differences across ancestries, MAF differences, admixture fractions, and disease prevalence (Figures 2, Extended Data Figures 3, 4). Additional detail about these runs can be found in Supplementary Information.
Varying effect size across populations:
To examine the effect of modifying the effect sizes, we introduced an effect on EUR haplotypes as well, rather than just on AFR. All these simulations assumed 80% admixture, 10% disease prevalence, and 20% MAF in both groups. We modeled cases across the OR spectrum where there was an effect of equal size in both ancestries, a 30% larger effect size on the EUR background, an effect size 30% larger on the AFR haplotype, and an effect size only in the EUR.
Varying absolute MAF:
We next fixed all other parameters and modified the absolute MAF of the simulated risk allele, with the relative difference in MAF between ancestries remaining constant. We changed our MAF from 20% to 10% and 40% under both the models of an effect only in the AFR background and with matching effect sizes between EUR and AFR.
MAF differences between groups:
To see if having a difference in the MAF between the two ancestral groups affected GWAS power, we varied the MAF in the EUR background to be 10, 20, and 30% while keeping the AFR MAF set to 20%.
False positive rate:
We quantified the false positive rate by simulating a variant with no effect and counting significant associations identified in a simulated realistic AA population given α = 0.05 across a range of absolute and relative differences in ancestral MAF. In all cases, 5000 replicates were run, individuals were 80/20% admixed AFR/EUR, the disease prevalence was set at 10%, and the sample size was modeled as 12k cases and 30k controls.
Selection of two-way admixed African-European individuals
To select individuals with 2-way admixture with European and West African ancestry (Supplementary Figure 3), we took a two-pronged approach. First, we combined genetic reference data from the 1000 Genomes Project and Human Genome Diversity Panel6, then harmonized meta-data according to consistent continental ancestries. We then ran PCA on unrelated individuals from the reference dataset. To partition individuals in the UKB based on their continental ancestry, we used the PC loadings from the reference dataset to project UK Biobank individuals into the same PC space. We trained a random forest classifier given continental ancestry meta-data based on the top 6 PCs from the reference training data. We applied this random forest to the projected UK Biobank PCA data and assigned AFR ancestries if the random forest probability was >50%, otherwise individuals were dropped from further analysis. To restrict to only two-way admixed West African-European ancestry individuals, we restricted to individuals with at least 12.5% European ancestry, at least 10% African ancestry, and who did not deviate more than 1 standard deviation from the AFR-EUR cline. This resulted in approximately 4300 individuals per blood lipid trait. Global ancestry fraction and ancestral allele frequency estimates were obtained from running ADMIXTURE7 with k=2 (which was the best fit k value to this dataset based on 5-fold cross-validation) on these individuals with 1000 Genomes Project EUR and AFR superpopulation individuals as reference data. To ensure there were no major areas of the genome where local ancestry inference was skewing significantly from the expected global fractions, we also assessed the cumulative local ancestry calls across the genome for the UKB admixed subset.
Software implementations
We developed separate scripts to deconvolve ancestry tracts and calculate haplotype dosages, correct phase switch errors, and run a Tractor GWAS to obtain ancestry-specific effect size estimates and p values. Pre-GWAS steps are available as independent python scripts to allow for maximum flexibility. To implement the joint modeling GWAS approach with the novel linear regression model described here, we have built a scalable pipeline in Hail8 which can be implemented locally or on the Google Cloud Platform9. Descriptions of the steps and an example Jupyter notebook10 demonstrating analytical steps and visualization of results of the Tractor joint-analysis GWAS are freely available on github (https://github.com/eatkinson/Tractor). The Hail implementation of Tractor GWAS generally runs on the order of minutes depending on the sample size and variant density. Alternatively, users can run a separate/meta-analysis GWAS version of Tractor (see Supplemental Information). This pipeline requires the initial processing steps to optionally correct phase switch errors and deconvolve ancestry tracts into their own VCF files. Next, GWAS can be run for the deconvolved files containing different ancestral components with the user’s preferred GWAS software, such as plink11. In this implementation, a standard GWAS model can be run on each ancestral component separately using the ancestry-specific VCF output by Tractor, which contains fully or partially missing data including only haplotypes from the ancestry in question. Results from the different ancestry runs could then be meta-analyzed to increase power by incorporating summary statistics from both populations, though we recommend preferentially using the joint-analysis method described in this manuscript to avoid any potential bias from combining multiple ancestral portions of the genome of the same individuals. This implementation is also compatible in large-scale collections where there are large numbers of homogenous individuals, for example many Europeans, but too limited a number of admixed individuals to be run in a GWAS alone. The EUR sections of the admixed cohorts could be analyzed alongside the homogenous European cohorts, making better use of the admixed samples even if other ancestry portions are not utilized, and increasing the effective sample size.
Empirical test of Tractor on blood lipid phenotypes
To ensure that Tractor replicated well-established associations, we ran standard GWAS, the Tractor joint-analysis model, and inverse variance weighted fixed effects meta-analysis of summary statistics from EUR and AFR deconvolved tracts on ~4300 admixed African-European individuals from the UKB on the biomarker blood lipid traits of Total Cholesterol and low-density lipoprotein cholesterol. We included covariates capturing global ancestry (PCs or global ancestry fraction), age, sex, and blood dilution factor in all runs. All simulation results for both Tractor and standard GWAS include the first 10 PCs to account for population stratification, as is currently standard practice in studies of admixed cohorts. To confirm good performance regardless of the global ancestry estimation strategy, we assessed meta-analysis performance using both metrics to capture global ancestry, namely PCs versus the AFR fraction as determined by ADMIXTURE, finding that these did not result in substantive differences. In the empirical joint-analysis framework, we used the measure of global AFR ancestry fraction to more directly capture global ancestry and avoid any potential collinearity with local ancestry from PCs. We generated QQ plots alongside each trait and compared the inflation of test statistics in each GWAS case by looking at the genomic inflation factor, λGC. We then compared results to those obtained from the same individuals using a standard GWAS approach. No individuals overlap between the previous study of interest, Natarajan et al. (2018), and the UKB individuals included here. As expected, near-identical results were obtained from the meta- and joint-approaches. Gene visualizations were produced with LocusZoom12, Manhattan and QQ plots with bokeh in Hail13.
We further investigated the pattern of LD in the DOCK6 region of interest to assess how much LD differences across ancestries aided in localization improvements with Tractor. For this we generated within-sample LD heatmaps for the region of interest (Supplementary Figure 6) in the full dataset, as well as within just the AFR and EUR tracts. Squared inter-variant allele count correlations were generated with PLINK11 for the region chr19:11325000-11360000. LD heatmaps were created with the LDheatmap package14 in R.
Statistical fine-mapping of top hits in independent cohorts
We conducted GWAS and statistical fine-mapping in two additional large-scale cohorts: 345,235 white British individuals from UKB and 135,808 Japanese individuals from BBJ. For the UKB white British, we used previously conducted fine-mapping results for Total Cholesterol (https://www.finucanelab.org/data). Briefly, we computed association statistics for the variants with INFO > 0.8, MAF > 0.01% (except for rare coding variants with MAC > 0), and HWE p-value > 1e-10 using BOLT-LMM15 with covariates including the top 10 PCs, sex, age, age2, sex * age, sex * age2, and blood dilution factor. We used FINEMAP v1.3.116,17 and susieR v0.8.1.052118 for fine-mapping using the GWAS summary statistics and in-sample dosage LD matrices computed by LDstore v2.0b. We defined regions based on 3 Mb window surrounding lead variants and merged them if overlapped. The maximum number of causal variants in a region was specified as 10. For BBJ, we additionally conducted fine-mapping using the same pipeline as we did for UKB. The GWAS summary statistics of Total Cholesterol was computed for the variants with Rsq > 0.7 and MAF > 0.01% using BOLT-LMM with the covariates including top 10 PCs, sex, age, age2, sex * age, sex * age2, and disease status (affected versus non-affected) for the 47 target diseases in the BBJ. The details about genotyping and imputation was extensively described previously19,20.
Assessment of the empirical p value threshold
To evaluate the appropriate p value threshold for Tractor associations, we estimated ancestry-specific empirical null p value distributions via permutation. Although the genome-wide significance threshold (p < 5 × 10−8) is widely adopted in the current literature, previous work has shown that different ancestry groups have different numbers of independent variants39. Here, we permuted a null continuous phenotype 1,000 times using the same admixed African-European individuals from UKB as in the Tractor cholesterol GWAS to assess the correct p value threshold for the admixed individuals in this study. We measured the minimum p values of associations (pmin) for each ancestry and derived an ancestry-specific empirical genome-wide significance threshold as the fifth percentile (α = 0.05) of pmin across permutations as previously described21. We calculated this percentile using the Harrell–Davis distribution-free quantile estimator22 and calculated the 95% confidence interval via bootstrapping. Based on the permutation results (Supplementary Figure 2), a study-wide significance threshold at a conservative level of p = 1 × 10−8 for both AFR- and EUR-specific associations was deemed appropriate for admixed African-European cohorts, consistent with the presence of additional recombination breakpoints present in such admixed populations. In addition, we calculated the genomic inflation factor, λGC, of null phenotypes across permutations and confirmed no significant inflation using the Tractor GWAS model (Supplementary Table 3).
Code Availability Statement
All code is freely available. The automated QC pipeline to prepare datasets for Tractor and run LAI is located at https://github.com/eatkinson/Post-QC. We freely provide Tractor code in python and Hail, as well as examples of implementation in a Jupyter notebook at https://github.com/eatkinson/Tractor alongside a detailed wiki. Specific scripts used to produce the simulated data and results is additionally freely provided at https://github.com/eatkinson/Tractor_ms_results.
Extended Data
Extended Data Fig. 1. Painted karyograms of a simulated AA individual showing EUR (red) and AFR (blue) ancestral tracts across demographic models.

The first column shows the results for the demographic model of one pulse of admixture 3 generations ago, the middle column shows the realistic model of one pulse 9 generations ago, and the right column shows a pulse 20 generations ago. In all cases the model involved 84% AFR ancestry and 16% EUR. The rows show the results from treatments of the data across steps of the Tractor pipeline. The top row shows the truth results from our simulations. Painted karyograms after statistical phasing of this truth cohort is shown in the second row. The third row illustrates the recovery of tracts broken by switch errors in phasing obtained by unkinking. The bottom row shows the smoothing and further improvement of tracts acquired through an additional round of LAI.
Extended Data Fig. 2. Tractor recovers disrupted tracts, improving tract distributions.

The top row (A-C) shows the improvements to the distributions of the number of discrete EUR tracts observed in simulated AA individuals under demographic models of 1 pulse of admixture at 3, 9 (realistic for AA population history) and 20 generations ago. The bottom row (D,E) shows the results from different initial admixture fractions, of 70% and 50% AFR, respectively, at 9 generations since admixture. These can be compared to the inferred realistic demographic model shown in B. In all panels, the simulated truth dataset is shown in black, after statistical phasing in purple, immediately after tract recovery procedures is in orange, and after one additional round of LAI after tract recovery in yellow.
Extended Data Fig. 3. The contribution of absolute MAF and effect size to Tractor power.

All cases assume an 80/20 AFR/EUR admixture ratio, 10% disease prevalence, 12k cases/30k controls with an effect only in the AFR genetic background. In all panels, the solid line uses a traditional GWAS model while the dashed line is our LAI-incorporating Tractor model. (A,B): Equal effect in EUR and AFR with shifted absolute MAF. (C,D): effect only in AFR background. (A,C): MAF is set to 10% in both AFR and EUR. (B,D): MAF is set to 40% in both AFR and EUR. Panels E and F illustrate the heterogeneity in effect sizes required to observe gains in Tractor power over traditional GWAS assuming 20% MAF in both ancestries and an effect that is stronger in AFR with varying difference to the EUR effect.
Extended Data Fig. 4. The interaction of between-ancestry MAF differences and effect sizes on Tractor power.

In all cases, the grey solid line uses a traditional GWAS model while the black dashed line is our LAI-incorporating model, admixture proportions are 80/20 AFR/EUR, disease prevalence is 10%, and the AFR MAF is fixed at 20%. A and E model the same effect size between EUR and AFR while varying the EUR MAF. B,D,F model the case when there is no effect in the EUR background while varying EUR MAF. C models an effect size difference of 30% with the effect being stronger in the EUR background. For comparison, Figure 2F shows the same effect at matched 20% MAF.
Extended Data Fig. 5. The impact of LAI accuracy on Tractor’s performance as compared to standard GWAS and asaMap.

We modeled perfect accuracy, realistic accuracy as derived from simulations of our AA demographic model (98%), and a lower bound of 90% LAI accuracy. Black lines all indicate Tractor runs: the solid black line is Tractor’s performance with perfect LAI accuracy, the dashed line is at 98% accuracy, and the dotted line is at 90% accuracy. The red line represents the power obtained from standard GWAS, and the blue line for the asaMap model for the ancestry in which the effect was modeled (AFR for A,B, and C, and EUR for D). In all cases we included 10 PCs as covariates and 1000 replicates were run.
Supplementary Material
Acknowledgements
We thank the Psychiatric Genomics Consortium PTSD working group, P. Natarajan, S. Gagliano Taliun, and many other scientists within and beyond Boston for their intellectual contributions to this work. This project was supported by the National Institute of Mental Health (K01 MH121659 and T32 MH017119 to E.G.A.; K99MH117229 to A.R.M.; 2R01MH106595 to C.M.N. and K.C. K.). M.K. was supported by a Nakajima Foundation Fellowship and the Masason Foundation. M.L.S. was supported by Fundacao de Amparo a Pesquisa do Estado de Sao Paulo (#2018/09328-2). The BioBank Japan Project was supported by the Tailor-Made Medical Treatment Program of the Ministry of Education, Culture, Sports, Science, and Technology (MEXT), the Japan Agency for Medical Research and Development (AMED). This research has been conducted using the UK Biobank Resource under Application Number 31063.
Footnotes
Competing Interests statement
M.J.D. is a founder of Maze Therapeutics. A.R.M. serves as a consultant for 23andMe and is a member of the Precise.ly Scientific Advisory Board. B.M.N. is a member of the Deep Genomics Scientific Advisory Board and serves as a consultant for the Camp4 Therapeutics Corporation, Takeda Pharmaceutical and Biogen. The remaining authors declare no competing interests.
Data Availability Statement
All summary statistics described here for Total and LDL Cholesterol in ~4300 admixed UK Biobank individuals can be found at https://github.com/eatkinson/Tractor_ms_results and have been uploaded to the GWAS catalog under accession numbers GCST90012868-GCST90012873. The UK Biobank raw data can be obtained through a Data Access Application, available at https://www.ukbiobank.ac.uk. PGC-PTSD data can be obtained through a data access application at https://pgc-ptsd.com/data-samples/access-data/. Biobank Japan summary statistics are available at http://jenger.riken.jp/en/. The Thousand Genomes reference panel is available at: http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502/. The Human Genome Diversity Project dataset is available at https://www.internationalgenome.org/data-portal/data-collection/hgdp.
References
- 1.Parker K, Morin R, Horowitz Juliana Menasce & Rohal M Multiracial in America: Proud, Diverse and Growing in Numbers. (2015). [Google Scholar]
- 2.Bhardwaj A et al. Racial disparities in prostate cancer a molecular perspective. Front. Biosci 22, 4515 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grizzle WE et al. Self‐Identified African Americans and prostate cancer risk: West African genetic ancestry is associated with prostate cancer diagnosis and with higher Gleason sum on biopsy. Cancer Med. 8, 6915–6922 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duggan MA, Anderson WF, Altekruse S, Penberthy L & Sherman ME The Surveillance, Epidemiology, and End Results (SEER) Program and Pathology: Toward Strengthening the Critical Relationship. Am. J. Surg. Pathol 40, e94–e102 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Freedman ML et al. Admixture mapping identifies 8q24 as a prostate cancer risk locus in African-American men. Proc. Natl. Acad. Sci. U. S. A 103, 14068–14073 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bateman ED et al. Global strategy for asthma management and prevention: GINA executive summary. Eur. Respir. J 31, 143–78 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Daya M & Barnes KC African American ancestry contribution to asthma and atopic dermatitis. Ann. Allergy. Asthma Immunol 122, 456–462 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wyss AB et al. Multiethnic meta-analysis identifies ancestry-specific and cross-ancestry loci for pulmonary function. Nat. Commun 9, 2976 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Demenais F et al. Multiancestry association study identifies new asthma risk loci that colocalize with immune-cell enhancer marks. Nat. Genet 50, 42–50 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Benetos A & Aviv A Ancestry, Telomere Length, and Atherosclerosis Risk. Circ. Cardiovasc. Genet 10, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mozaffarian D et al. Heart Disease and Stroke Statistics—2015 Update. Circulation 131, (2015). [DOI] [PubMed] [Google Scholar]
- 12.Sirugo G, Williams SM & Tishkoff SA The Missing Diversity in Human Genetic Studies. Cell 177, 26–31 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Popejoy Alice B., Fullerton SM Genomics is falling. Nature 538, 161–164 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sul JH, Martin LS & Eskin E Population structure in genetic studies: Confounding factors and mixed models. PLOS Genet. 14, e1007309 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang H et al. Bootstrat: Population Informed Bootstrapping for Rare Variant Tests. bioRxiv 068999 (2016). doi: 10.1101/068999 [DOI] [Google Scholar]
- 16.Sohail M et al. Polygenic adaptation on height is overestimated due to uncorrected stratification in genome-wide association studies. Elife 8, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berg JJ et al. Reduced signal for polygenic adaptation of height in UK biobank. Elife 8, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lander ES & Schork NJ Genetic dissection of complex traits. Science (80-. ). 265, 2037–2048 (1994). [DOI] [PubMed] [Google Scholar]
- 19.Coram MA, Fang H, Candille SI, Assimes TL & Tang H Leveraging Multi-ethnic Evidence for Risk Assessment of Quantitative Traits in Minority Populations. Am. J. Hum. Genet 101, 218–226 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walters RK et al. Transancestral GWAS of alcohol dependence reveals common genetic underpinnings with psychiatric disorders. Nat. Neurosci 21, 1656–1669 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martin ER et al. Properties of global- and local-ancestry adjustments in genetic association tests in admixed populations. Genet. Epidemiol 42, 214–229 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stevenson A et al. Neuropsychiatric Genetics of African Populations-Psychosis (NeuroGAP-Psychosis): a case-control study protocol and GWAS in Ethiopia, Kenya, South Africa and Uganda. BMJ Open 9, e025469 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Consortium, T. H. Enabling the genomic revolution in Africa. Science 344, 1346–8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.TOPMed Whole Genome Sequencing Project. Freeze 5b, Phases 1 and 2. (2018). doi: 10.1155/2013/865181 [DOI] [Google Scholar]
- 25.Precision Medicine Initiative (PMI) Working Group. The precision medicine initiative cohort program – building a research foundation for 21st century medicine. Precis. Med. Initiat. Work. Gr. Rep. to Advis. Comm. to Dir. NIH September 17, 1–108 (2015). [Google Scholar]
- 26.Logue MW et al. The Psychiatric Genomics Consortium Posttraumatic Stress Disorder Workgroup: Posttraumatic Stress Disorder Enters the Age of Large-Scale Genomic Collaboration. Neuropsychopharmacology 40, 2287–97 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bien SA et al. The Future of Genomic Studies Must Be Globally Representative: Perspectives from PAGE. (2019). doi: 10.1146/annurev-genom-091416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martin AR et al. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat. Genet 51, 584–591 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peterson RE et al. Genome-wide Association Studies in Ancestrally Diverse Populations: Opportunities, Methods, Pitfalls, and Recommendations. Cell 179, 589–603 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hero JO, Zaslavsky AM & Blendon RJ The United States leads other nations in differences by income in perceptions of health and health care. Health Aff. 36, 1032–1040 (2017). [DOI] [PubMed] [Google Scholar]
- 31.Williams DR, Priest N & Anderson NB Understanding associations among race, socioeconomic status, and health: Patterns and prospects. Heal. Psychol 35, 407–411 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Agency for Healthcare Research & Quality. 2016 National Healthcare Quality and Disparities Report. (2017).
- 33.Li YR & Keating BJ Trans-ethnic genome-wide association studies: advantages and challenges of mapping in diverse populations. (2014). doi: 10.1186/s13073-014-0091-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spain SL & Barrett JC Strategies for fine-mapping complex traits. Hum. Mol. Genet 24, R111–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schaid DJ, Chen W & Larson NB From genome-wide associations to candidate causal variants by statistical fine-mapping. Nature Reviews Genetics 19, 491–504 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu Y et al. Trans-Ethnic Fine-Mapping of Lipid Loci Identifies Population-Specific Signals and Allelic Heterogeneity That Increases the Trait Variance Explained. PLoS Genet. 9, e1003379 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van de Bunt M et al. Evaluating the Performance of Fine-Mapping Strategies at Common Variant GWAS Loci. PLOS Genet. 11, e1005535 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mahajan A et al. Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat. Genet 46, 234–244 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Project, T. T. G. C. An integrated map of genetic variation from 1,092 human genomes. Nature 135, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang J & Stram DO The Role of Local Ancestry Adjustment in Association Studies Using Admixed Populations. Genet. Epidemiol 38, 502–515 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lachance J et al. Evolutionary history and adaptation from high-coverage whole-genome sequences of diverse African hunter-gatherers. Cell 150, 457–69 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tang H, Siegmund DO, Johnson NA, Romieu I & London SJ Joint testing of genotype and ancestry association in admixed families. Genet. Epidemiol 34, 783–791 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Coram MA et al. Genome-wide Characterization of Shared and Distinct Genetic Components that Influence Blood Lipid Levels in Ethnically Diverse Human Populations. Am. J. Hum. Genet 92, 904–916 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aschard H, Gusev A, Brown R & Pasaniuc B Leveraging local ancestry to detect gene-gene interactions in genome-wide data. BMC Genet. 16, 1–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zaitlen N, Pas B, Gur T, Ziv E & Halperin E ARTICLE Leveraging Genetic Variability across Populations for the Identification of Causal Variants. Am. J. Hum. Genet 86, 23–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pasaniuc B et al. Enhanced statistical tests for GWAS in admixed populations: assessment using African Americans from CARe and a Breast Cancer Consortium. PLoS Genet. 7, e1001371 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pasaniuc B et al. Analysis of Latino populations from GALA and MEC studies reveals genomic loci with biased local ancestry estimation. Bioinformatics 29, 1407–1415 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chimusa ER et al. Genome-wide association study of ancestry-specific TB risk in the South African coloured population. Hum. Mol. Genet 23, 796–809 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Smith EN et al. Genome-wide association study of bipolar disorder in European American and African American individuals. Mol. Psychiatry 14, 755–763 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Skotte L, Jørsboe E, Korneliussen TS, Moltke I & Albrechtsen A Ancestry‐specific association mapping in admixed populations. Genet. Epidemiol 43, 506–521 (2019). [DOI] [PubMed] [Google Scholar]
- 51.Shriner D Overview of admixture mapping. in Current Protocols in Human Genetics 1.23.1–1.23.8 (2013). doi: 10.1002/cphg.44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen M et al. Admixture mapping analysis in the context of GWAS with GAW18 data in BMC Proceedings 8, S3 (BioMed Central Ltd, 2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen W et al. A Generalized Sequential Bonferroni Procedure for GWAS in Admixed Populations Incorporating Admixture Mapping Information into Association Tests. Hum. Hered 79, 80–92 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hoggart CJ, Shriver MD, Kittles RA, Clayton DG & McKeigue PM Design and Analysis of Admixture Mapping Studies. Am. J. Hum. Genet 74, 965–978 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Patterson N et al. Methods for High-Density Admixture Mapping of Disease Genes. Am. J. Hum. Genet 74, 979–1000 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Spear ML et al. A genome-wide association and admixture mapping study of bronchodilator drug response in African Americans with asthma. Pharmacogenomics J. 19, 249–259 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gignoux CR et al. An admixture mapping meta-analysis implicates genetic variation at 18q21 with asthma susceptibility in Latinos. J. Allergy Clin. Immunol 143, 957–969 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shetty PB et al. Variants for HDL-C, LDL-C, and triglycerides identified from admixture mapping and fine-mapping analysis in African American families. Circ. Cardiovasc. Genet 8, 106–113 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shetty PB et al. Variants in CXADR and F2RL1 are associated with blood pressure and obesity in African-Americans in regions identified through admixture mapping. J. Hypertens 30, 1970–1976 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Reiner AP et al. Genome-wide association and population genetic analysis of c-reactive protein in african american and hispanic american women. Am. J. Hum. Genet 91, 502–512 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Florez JC et al. Strong Association of Socioeconomic Status and Genetic Ancestry in Latinos: Implications for Admixture Studies of Type 2 Diabetes in Racial Identities, Genetic Ancestry, and Health in South America: Argentina, Brazil, Colombia, and Uruguay (eds. Gibbon S, Santos RV & Sans M) 137–153 (Palgrave Macmillan; US, 2011). doi: 10.1057/9781137001702_7 [DOI] [Google Scholar]
- 62.Maples BK, Gravel S, Kenny EE & Bustamante CD RFMix: a discriminative modeling approach for rapid and robust local-ancestry inference. Am. J. Hum. Genet 93, 278–88 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Geza E et al. A comprehensive survey of models for dissecting local ancestry deconvolution in human genome. Brief. Bioinform 20, 1709–1724 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schubert R, Andaleon A & Wheeler HE Comparing local ancestry inference models in populations of two- and three-way admixture. Research Square (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tishkoff S.a et al. The genetic structure and history of Africans and African Americans. Science 324, 1035–44 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gravel S et al. Demographic history and rare allele sharing among human populations. Proc. Natl. Acad. Sci. U. S. A 108, 11983–8 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Martin AR et al. Human Demographic History Impacts Genetic Risk Prediction across Diverse Populations. (2017). doi: 10.1016/j.ajhg.2017.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Choi Y, Chan AP, Kirkness E, Telenti A & Schork NJ Comparison of phasing strategies for whole human genomes. PLOS Genet. 14, e1007308 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Andrés AM et al. Understanding the accuracy of statistical haplotype inference with sequence data of known phase. Genet. Epidemiol 31, 659–71 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Auton A et al. A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Natarajan P et al. Deep-coverage whole genome sequences and blood lipids among 16,324 individuals. Nat. Commun 9, 3391 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Musunuru K & Kathiresan S Genetics of Common, Complex Coronary Artery Disease. Cell 177, 132–145 (2019). [DOI] [PubMed] [Google Scholar]
- 73.Rotimi CN et al. The genomic landscape of African populations in health and disease. Hum. Mol. Genet 26, R225–R236 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Superko HR, Momary KM & Li Y Statins Personalized. Medical Clinics of North America 96, 123–139 (2012). [DOI] [PubMed] [Google Scholar]
- 75.Fu J et al. Unraveling the regulatory mechanisms underlying tissue-dependent genetic variation of gene expression. PLoS Genet. 8, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Avery CL et al. A phenomics-based strategy identifies loci on APOC1, BRAP, and PLCG1 associated with metabolic syndrome phenotype domains. PLoS Genet. 7, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lettre G et al. Genome-Wide association study of coronary heart disease and its risk factors in 8,090 african americans: The nhlbi CARe project. PLoS Genet. 7, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Talmud PJ et al. Gene-centric Association Signals for Lipids and Apolipoproteins Identified via the HumanCVD BeadChip. Am. J. Hum. Genet 85, 628–642 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sandhu MS et al. LDL-cholesterol concentrations: a genome-wide association study. Lancet 371, 483–491 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sanna S et al. Fine mapping of five loci associated with low-density lipoprotein cholesterol detects variants that double the explained heritability. PLoS Genet. 7, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fox CS et al. Genome-wide association to body mass index and waist circumference: the Framingham Heart Study 100K project. BMC Med. Genet 8, S18 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kathiresan S et al. A genome-wide association study for blood lipid phenotypes in the Framingham Heart Study. BMC Med. Genet 8, S17 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Adzhubei I, Jordan DM & Sunyaev SR Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. Chapter 7, Unit7.20 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ng PC & Henikoff S SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 31, 3812–4 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang R The ANGPTL3-4-8 model, a molecular mechanism for triglyceride trafficking. Open Biol. 6, 150272 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fu Z, Abou-Samra AB & Zhang R A lipasin/Angptl8 monoclonal antibody lowers mouse serum triglycerides involving increased postprandial activity of the cardiac lipoprotein lipase. Sci. Rep 5, 18502 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang R Lipasin, a novel nutritionally-regulated liver-enriched factor that regulates serum triglyceride levels. Biochem. Biophys. Res. Commun 424, 786–792 (2012). [DOI] [PubMed] [Google Scholar]
- 88.Siddiqa A et al. Visualizing the regulatory role of Angiopoietin-like protein 8 (ANGPTL8) in glucose and lipid metabolic pathways. Genomics 109, 408–418 (2017). [DOI] [PubMed] [Google Scholar]
- 89.Yamada H et al. Circulating betatrophin is elevated in patients with type 1 and type 2 diabetes. Endocr. J 62, 417–421 (2015). [DOI] [PubMed] [Google Scholar]
- 90.Espes D, Martinell M & Carlsson P-O Increased circulating betatrophin concentrations in patients with type 2 diabetes. Int. J. Endocrinol 2014, 323407 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hu H et al. Increased circulating levels of betatrophin in newly diagnosed type 2 diabetic patients. Diabetes Care 37, 2718–22 (2014). [DOI] [PubMed] [Google Scholar]
- 92.Fu Z et al. Elevated circulating lipasin/betatrophin in human type 2 diabetes and obesity. Sci. Rep 4, 5013 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cannon ME et al. Trans-ancestry Fine Mapping and Molecular Assays Identify Regulatory Variants at the ANGPTL8 HDL-C GWAS Locus. (2017). doi: 10.1534/g3.117.300088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kanai M et al. Genetic analysis of quantitative traits in the Japanese population links cell types to complex human diseases. Nat. Genet 50, 390–400 (2018). [DOI] [PubMed] [Google Scholar]
- 95.Karczewski KJ et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv 531210 (2019). doi: 10.1101/531210 [DOI] [Google Scholar]
- 96.Lin M et al. Population specific reference panels are crucial for the genetic analyses of Native Hawai’ians: an example of the CREBRF locus. bioRxiv 789073 (2019). doi: 10.1101/789073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ntzani EE, Liberopoulos G, Manolio TA & Ioannidis JPA Consistency of genome-wide associations across major ancestral groups. Hum. Genet 131, 1057–1071 (2012). [DOI] [PubMed] [Google Scholar]
- 98.Marigorta UM & Navarro A High trans-ethnic replicability of GWAS results implies common causal variants. PLoS Genet. 9, e1003566 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Waters K, Stram D, … Pl MH. & 2010, undefined. Consistent association of type 2 diabetes risk variants found in europeans in diverse racial and ethnic groups. ncbi.nlm.nih.govPaperpile [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lam M et al. Comparative genetic architectures of schizophrenia in East Asian and European populations. Nat. Genet (2019). doi: 10.1038/s41588-019-0512-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Liu J et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. nature.comPaperpile [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Carlson CS et al. Generalization and Dilution of Association Results from European GWAS in Populations of Non-European Ancestry: The PAGE Study. PLoS Biol. 11, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kuchenbaecker K et al. The transferability of lipid loci across African, Asian and European cohorts. Nat. Commun 10, 11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mägi R et al. Trans-ethnic meta-regression of genome-wide association studies accounting for ancestry increases power for discovery and improves fine-mapping resolution. Hum. Mol. Genet 26, 3639–3650 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wegmann D et al. Recombination rates in admixed individuals identified by ancestry-based inference. Nat. Genet 43, 847–853 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Atkinson EG et al. No Evidence for Recent Selection at FOXP2 among Diverse Human Populations. Cell 174, 1424–1435.e15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Deng L, Ruiz-Linares A, Xu S & Wang S Ancestry variation and footprints of natural selection along the genome in Latin American populations. Sci. Rep 6, 1–7 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jin W et al. Genome-wide detection of natural selection in African Americans pre- and post-admixture. Genome Res. 22, 519–527 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bomba L, Walter K & Soranzo N The impact of rare and low-frequency genetic variants in common disease. Genome Biol. 18, 77 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mathieson I & McVean G Differential confounding of rare and common variants in spatially structured populations. 44, 243–246 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Online Methods References
- 111.Van Rossum G, Drake FL Jr. Python reference manual. Centrum voor Wiskunde en Informatica; Amsterdam; 1995. [Google Scholar]
- 112.Gnu P Bash (3.2.48) [Unix shell program; ]. 2007. [Google Scholar]
- 113.International Hapmap Consortium T. The International HapMap Project. Nature 2003;426:789–96. 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 114.Chen CY, Pollack S, Hunter DJ, Hirschhorn JN, Kraft P, Price AL. Improved ancestry inference using weights from external reference panels. Bioinformatics 2013;29:1399–406. 10.1093/bioinformatics/btt144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Williams A. admix-simu: program to simulate admixture between multiple populations. 2016 doi: 10.5281/ZENODO.45517. . [DOI] [Google Scholar]
- 116.Cann HM, de Toma C, Cazes L, Legrand M-F, Morel V, Piouffre L, et al. A human genome diversity cell line panel. Science 2002;296:261–2. [DOI] [PubMed] [Google Scholar]
- 117.Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res 2009;19:1655–64. 10.1101/gr.094052.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.The Hail team. Hail. 2018. URL: https://github.com/hail-is/hail (Accessed 16 January 2019).
- 119.Google Cloud Platform Blog. Google Compute Engine launches, expanding Google’s cloud offerings. n.d. URL: https://cloudplatform.googleblog.com/2012/06/google-compute-engine-launches.html (Accessed 16 January 2019).
- 120.Kluyver T, Ragan-kelley B, Pérez F, Granger B, Bussonnier M, Frederic J, et al. Jupyter Notebooks—a publishing format for reproducible computational workflows. 2016. [Google Scholar]
- 121.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007;81:559–75. 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics 2010;26:2336–7. 10.1093/bioinformatics/btq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bokeh Development Team. Bokeh: Python library for interactive visualization. 2019. URL: https://bokeh.org/citation/ (Accessed 31 March 2020).
- 124.Shin J-H, Blay S, McNeney B, Graham J. LDheatmap: An R Function for Graphical Display of Pairwise Linkage Disequilibria between Single Nucleotide Polymorphisms. J Stat Softw 2006;16:. 10.18637/jss.v000.i00. [DOI] [Google Scholar]
- 125.Loh PR, Tucker G, Bulik-Sullivan BK, Vilhjálmsson BJ, Finucane HK, Salem RM, et al. Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nat Genet 2015;47:. 10.1038/ng.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Benner C, Spencer CCA, Havulinna AS, Salomaa V, Ripatti S, Pirinen M. FINEMAP: Efficient variable selection using summary data from genome-wide association studies. Bioinformatics 2016;32:1493–501. 10.1093/bioinformatics/btw018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Benner C, Havulinna A, Salomaa V, Ripatti S, Pirinen M. Refining fine-mapping: effect sizes and regional heritability. BioRxiv 2018:318618 10.1101/318618. [DOI] [Google Scholar]
- 128.Wang G, Sarkar A, Carbonetto P, Stephens M. A simple new approach to variable selection in regression, with application to genetic fine-mapping. BioRxiv 2019:501114 10.1101/501114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kanai M, Akiyama M, Takahashi A, Matoba N, Momozawa Y, Ikeda M, et al. Genetic analysis of quantitative traits in the Japanese population links cell types to complex human diseases. Nat Genet 2018;50:390–400. 10.1038/s41588-018-0047-6. [DOI] [PubMed] [Google Scholar]
- 130.Akiyama M, Ishigaki K, Sakaue S, Momozawa Y, Horikoshi M, Hirata M, et al. Characterizing rare and low-frequency height-associated variants in the Japanese population. Nat Commun 2019;10:1–11. 10.1038/s41467-019-12276-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kanai M, Tanaka T, Okada Y. Empirical estimation of genome-wide significance thresholds based on the 1000 Genomes Project data set. J Hum Genet 2016;61:861–6. 10.1038/jhg.2016.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.HARRELL FE, DAVIS CE. A new distribution-free quantile estimator. Biometrika 1982;69:635–40. 10.1093/biomet/69.3.635. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All summary statistics described here for Total and LDL Cholesterol in ~4300 admixed UK Biobank individuals can be found at https://github.com/eatkinson/Tractor_ms_results and have been uploaded to the GWAS catalog under accession numbers GCST90012868-GCST90012873. The UK Biobank raw data can be obtained through a Data Access Application, available at https://www.ukbiobank.ac.uk. PGC-PTSD data can be obtained through a data access application at https://pgc-ptsd.com/data-samples/access-data/. Biobank Japan summary statistics are available at http://jenger.riken.jp/en/. The Thousand Genomes reference panel is available at: http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502/. The Human Genome Diversity Project dataset is available at https://www.internationalgenome.org/data-portal/data-collection/hgdp.