

Abstract
The neocortex is composed of layers. Whether layers constitute an essential framework for the formation of functional circuits is not well understood. We investigated the brain-wide input connectivity of vasoactive intestinal polypeptide (VIP) expressing neurons in the reeler mouse. This mutant is characterized by a migration deficit of cortical neurons so that no layers are formed. Still, neurons retain their properties and reeler mice show little cognitive impairment. We focused on VIP neurons because they are known to receive strong long-range inputs and have a typical laminar bias toward upper layers. In reeler, these neurons are more dispersed across the cortex. We mapped the brain-wide inputs of VIP neurons in barrel cortex of wild-type and reeler mice with rabies virus tracing. Innervation by subcortical inputs was not altered in reeler, in contrast to the cortical circuitry. Numbers of long-range ipsilateral cortical inputs were reduced in reeler, while contralateral inputs were strongly increased. Reeler mice had more callosal projection neurons. Hence, the corpus callosum was larger in reeler as shown by structural imaging. We argue that, in the absence of cortical layers, circuits with subcortical structures are maintained but cortical neurons establish a different network that largely preserves cognitive functions.
Keywords: barrel cortex, corpus callosum, rabies tracing, reelin, VIP neurons
Introduction
The neurons of the mammalian neocortex are stacked in layers (L). This organization is considered as an essential scaffolding principle that evolution could expand to generate increasingly sophisticated cognitive abilities (Shepherd and Rowe 2017). Layers are seen as units of processing, and their input–output connectivity defines the flow of information within the cortex (Miller 2011; Feldmeyer 2012). However, the notion of layers structuring the flow of cortical processing might be misleading. Cortical computations require foremost a functional neuronal circuit between defined cell types (Harris and Shepherd 2015; Guy and Staiger 2017). Because neurons show high specificity in targeting other neurons, there seem to be strict rules of connectivity (Kasthuri et al. 2015; Motta et al. 2019). These wiring rules might run in the framework of layers but perhaps are not determined by it. Whether the organization of layers constitutes an indispensable feature for the formation of cortical circuits is still unclear.
Therefore, we asked the question if correct laminar position is linked to a neuron’s capacity to receive inputs and become properly embedded in brain-wide circuits. A promising model system to study the significance of layers is the reeler mouse (Guy and Staiger 2017). Homozygous mice lack the protein reelin, which is secreted by Cajal–Retzius cells during development, to orchestrate the migration of neurons (Lee and D’Arcangelo 2016). In consequence, the formation of layers is strongly compromised. In the somatosensory cortex, neurons destined to become part of a certain layer are almost uniformly dispersed across the cortex (Dekimoto et al. 2010; Wagener et al. 2010; Boyle et al. 2011). However, this perturbation seems to have little effect on properties of neurons and formation of functional circuits. Neurons retain their molecular fate as well as their electrophysiological properties (Silva et al. 1991; Wagener et al. 2010; Guy et al. 2016). The structure of synaptic boutons remains virtually unchanged (Prume et al. 2018, 2019). Areal boundaries within the cortex are preserved (Boyle et al. 2011). Moreover, whisker stimulation of reeler mice leads to the same hemodynamic responses in the cortex and activates the same cell types indicating a preservation of circuit modules comparable, if not identical, to a layered cortex (Guy et al. 2015; Wagener et al. 2016). For these reasons, the reeler mouse seems well suited to study functional brain circuits of defined cell types in the absence of layers. However, the global absence of reelin could have direct effects on neuron and circuit maturation on top of the effects caused by cellular dispersion (Bock and May 2016). This confounder needs to be kept in mind when using the reeler mouse model to study changes in response to aberrant layering.
We decided to focus on the connectivity of vasoactive intestinal polypeptide (VIP) expressing neurons. They constitute a major subgroup of GABAergic inhibitory neurons (Tremblay et al. 2016). We chose this type of neuron for several reasons. First, VIP neurons have a very distinct laminar distribution with about 60% occurring in LII/III, 20% in LIV, and only very few cells in deep layers (Prönneke et al. 2015). So they are heavily biased toward upper layers. Second, VIP neurons have been suggested as major integrators of long-range input. They receive more inputs per cell than other inhibitory cell types (Wall et al. 2016). They are most strongly activated by cortical long-range input compared with pyramidal and somatostatin- and parvalbumin-expressing neurons (Lee et al. 2013; Zhang et al. 2014). Because VIP cells strongly inhibit somatostatin cells (Walker et al. 2016), which themselves inhibit pyramidal cells (Zhou et al. 2020), it has been proposed that VIP neurons become activated by long-range input during active states, disinhibit pyramidal cells, and thereby open a precisely timed window for integration and plasticity at excitatory synapses (Pfeffer et al. 2013; Fu et al. 2014; Williams and Holtmaat 2019). Third, the input connectivity of VIP neurons is already well characterized across many cortical areas (Wall et al. 2016; Zhang et al. 2016; Ährlund-Richter et al. 2019; Sun et al. 2019). Finally, with the VIP-Cre line, we can access these cells with high specificity having no contamination from other cell types (Taniguchi et al. 2011; Prönneke et al. 2015).
We first confirmed that VIP neurons lose their laminar bias in reeler mice, establishing this mouse line as a valid model system to study the circuitry of VIP cells in the absence of layers. Then, we generated maps of brain-wide long-range input to VIP neurons in the barrel cortex of wild-type (WT) and reeler mice, using rabies virus tracing. While we found that subcortical inputs innervated malpositioned VIP cells to the same extent as in a layered cortex, the balance of cortical inputs was fundamentally different. Ipsilateral cortical input was reduced, and contralateral cortical input was increased compared with WT. Reeler mice had more callosal projection neurons (CPNs) and hence a larger corpus callosum (CC). We thoroughly discuss if these changes in connectivity result as a consequence of cellular dispersion or the absence of reelin’s influence on dendritic, axonal, and synaptic maturation. Especially in the light of evidence that basic cognitive functions in the reeler mouse show little decline (Salinger et al. 2003; Wagener et al. 2010; Pielecka-Fortuna et al. 2015; Imai et al. 2017), we are inclined to the possibility that the different circuit arrangement seen in reeler constitutes an adaption to retain the necessary cognitive abilities without the presence of layers.
Material and Methods
Experimental Animals
We crossed the reeler line (B6C3Fe a/a-Relnrl/J, The Jackson Laboratory) with the VIP-Cre line (VIPtm1(cre) Zjh, The Jackson Laboratory) to breed VIP-Cre/reeler mice heterozygous for reelin mutation and homozygous for Cre. These animals were crossed to generate VIP-Cre/reeler mice homozygous for reelin knockout. WT littermates or animals from the VIP-Cre line were used for comparison in tracing experiments. For control experiments to check the quality of our Cre-dependent constructs, we used C57BL/6J WT mice (The Jackson Laboratory). For tracing experiments not requiring Cre expression and for imaging, we used WT and homozygous littermate pairs of the reeler line.
To visualize the population of VIP cells, we crossed the VIP-Cre/reeler line with the Ai9 tdTomato reporter line (B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J, The Jackson Laboratory) to achieve tdTomato expression in VIP neurons.
Mice were housed in standard cages in a 12-h light/dark cycle and with ad libitum access to food and water. All experimental procedures were performed in accordance with German laws on animal research. All tracing experiments were performed with 12- to 20-week-old mice of either gender.
Viral Constructs
pAAV-DIO-TVA66T-EGFP-oG was generated based on the backbone of pAAV-hSyn-oG-EGFP-TVA-WPRE-hGpA (gift from Euiseok Kim, Salk Institute). The sequence for oG-EGFP was extracted using PCR with the primers F1-CTATACGAAGTTATGGTACCTTAGAGCCG; R1-GGATCCGGAGCTACTAACTTCAGC. The sequence for TVA66T was extracted from pAAV-CAG-FLEX-TC66T (Miyamichi et al. 2013; Addgene #48331) using PCR with primers F1-AGTAGCTCCGGATCCCCCACCCCCCTTGGATGC; R1-CGGTAACGTGACCGGTAACGG. pAAV backbone was cut with restriction enzymes KpnI-HF and AgeI-HF to open the backbone, and oG-EGFP fragment was reinserted together with the TVA66T fragment using infusion cloning with an In-Fusion HD cloning kit (Takara bio). The plasmid was packaged into AAV8 by the Salk Viral Vector Core, from where it is available.
For RV tracing experiments, 100–200 nl of AAV8-DIO-TVA66T-EGFP-oG was injected at a titer of 1.6 × 1013 IU/ml. RV-SADΔG-mCherry (EnvA) was injected at the same location 14 days later at a titer of 1 × 107 IU/ml. Animals were sacrificed 7 days later. For non-transsynaptic tracing experiments, 150 nl of AAV-retrograde-hSyn-EGFP (Addgene, #50465) at a titer of 2–4 × 1012 IU/ml was used. Animals were sacrificed after 14 days.
Surgery and Viral Injection
For viral tracing experiments, mice underwent intrinsic signal optical imaging to localize the whisker C2-related cortical column. The injection pipette was inserted at this location in WT and reeler animals. The surgery was performed as in the study of Hafner et al. (2019).
For sedation and analgesia, mice were injected intraperitoneally with 10 μg xylazine (Xylariem, Ecuphar) and 0.065 μg buprenorphine (Temgesic, Indivior UK Limited) per gram of body weight diluted in sterile saline. Anesthesia was induced with 3% isoflurane (vol/vol) and maintained between 0.5% and 1% throughout the entire surgical procedure (Harvard Apparatus). Mice were mounted on a custom-built frame with rigid earbars. A mixture of 2 μg of bupivacaine/lidocaine (Astra Zeneca) per gram of body weight was injected subcutaneously under the scalp for local anesthesia. A heating pad was used to maintain body temperature at 37 °C (ATC 1000, World Precision Instruments). Subsequently, a small incision was made in the scalp to expose the right hemisphere of the skull. The bone over the somatosensory area was thinned to transparency with a dental drill (OS-40, Osada Electric Company). Then, the location of the C2 whisker-related column was determined and mapped on the blood vessel pattern as described below. The bone above the target area was removed with a syringe tip. A glass pipette cut to 20-μm tip diameter (Drummond Scientific Co) was front-filled with AAV helper virus. The pipette holder was attached to a micromanipulator (Luigs & Neumann). The pipette was inserted at the target location into the brain in an approximately 45° angle, orthogonal to the curvature of the cortex. AAV was pressure-injected with a syringe at 3 depths (750, 500, and 250 μm below pia). To reduce backflow, the needle was left in place at each depth for at least 3 min. The scalp was sutured and the mouse received a subcutaneous injection of 5 μg Carprofen (Pfizer) per gram of body weight for prolonged pain relief. Fourteen days later, the mouse was injected with RV-mCherry without prior imaging. The injection was guided based on the blood vessel pattern and landmarks from the previous surgery.
Intrinsic Signal Optical Imaging
This procedure was performed as described earlier (Guy et al. 2015; Hafner et al. 2019). Briefly, whisker C2 on the left muzzle was stimulated with a piezoelectric actuator at 5 Hz. Red light was shone on the exposed cortex above the right barrelfield and its reflectance was measured with a CCD camera. Changes in reflectance were averaged across 30 trials. The blood vessel pattern and the intrinsic signal were overlaid to guide the subsequent injection.
Fixation and Tissue Sectioning
Mice were sacrificed by injecting an overdose of ketamine (100 μg/g; Medistar) and perfused transcardially with ice-cold 10% sucrose solution followed by 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer. The brain was postfixed in 4% PFA for 4 h. A vibratome (VT 1200 S, Leica) was used to section the brain into 100-μm-thick coronal sections rostral and caudal to barrel cortex, while barrel cortex was sectioned at 50-μm-thick intervals. Sections spanning the barrel cortex were subjected to immunohistochemistry, while all other sections were stained for 4′,6-diamidino-2-phenylindole (DAPI) only.
Immunohistochemistry
Barrel cortex sections were washed in TRIS buffer (TB) 1 × 15 min, TRIS-buffered saline (TBS) 1 × 15 min, and TBS + 0.5% Triton X-100 (TBST) 2 × 15 min, all at pH 7.6. For blocking, sections were incubated 90 min at room temperature in 0.25% bovine serum albumin/10% goat serum/TBST (Jackson ImmunoResearch). For primary antibody labeling, sections were incubated 48–72 h at 4 °C with (1) chicken anti-GFP (Aves; Cat#GFP-1020, RRID:AB_10000240) diluted 1:500 and (2) mouse anti-RFP (Rockland; Cat#200–301-379S, RRID:AB_2611064) diluted 1:2000. Sections were washed 4 × 15 min with TBST. For secondary antibody labeling, sections were incubated 4 h at room temperature with (1) Alexa Fluor 488-conjugated goat anti-chicken IgG (Molecular Probes; Cat#A11039) and (2) Alexa Fluor 568-conjugated goat anti-mouse IgG2a (Molecular Probes; Cat#A21134) diluted 1:500 in TBST. In some cases, sections were stained in addition with the primary antibody guinea pig anti-vGluT2 (Millipore; Cat#AB2251, RRID: AB_1587626) diluted 1:2000 and the secondary antibody Alexa Fluor 633-conjugated goat anti-guinea pig (Molecular probes; Cat#A21105) diluted 1:500. After washing 2 × 15 min with TBST and 1 × 15 min with TBS, sections were stained with DAPI, diluted 1:1000 in TBS. After several washes in TB, sections were mounted in Aqua-Poly-Mount (Polysciences).
Fluorescent In Situ Hybridization
Sections of the VIPCre/Reeler/tdTomato mouse were stained with a riboprobe against VIP mRNA. VIP riboprobe was generated as described earlier (Prönneke et al. 2015) using the following primers, FP: CCTGGCA TTCCTGATACTCTTC; RP: ATTCTCTGATTTCAGCTCTGCC (527 bp; Allen Brain Atlas Riboprobe ID: RP_070116_02_E09). The staining procedure was performed exactly as in the study of Prönneke et al. (2015). The tdTomato signal was amplified after the in situ hybridization using the immunohistochemistry protocol described above but without Triton X-100. Primary antibody staining with rabbit anti-RFP (Rockland; Cat#600–401-379, RRID:AB_2209751) was followed by Alexa Fluor 594-conjugated goat anti-rabbit (Molecular probes; Cat#A11037) as the secondary antibody.
Image Acquisition and Processing
Images were acquired on an inverted epifluorescence slide-scanning microscope (Axio Observer, Zeiss) with a ×10 objective (NA = 0.3) or ×25 objective (NA = 0.8). For overview images, only one plane was imaged; for the injection sites, stacks were acquired. Tiles were stitched after imaging and stacks were deconvoluted in ZEN blue software (Zeiss) to reduce out-of-focus light. Confocal acquisitions were taken on a LSM 880 (Zeiss) with a ×25 objective operated by ZEN black software (Zeiss).
Magnetic Resonance Imaging Data Acquisition and Preprocessing
Ex vivo brain samples (n = 3 per group) were imaged in a high-field 9.4 Tesla MR system, using a mouse brain 4-channel coil array (Bruker BioSpin MRI GmbH). The scanning protocol included magnetization transfer (MT)-weighted images and diffusion-weighted images. For MT, a 3D Fast Low Angle Shot (FLASH) sequence was used to acquire 3 datasets: MT-weighted, proton density-weighted, and T1-weighted (repetition time 15 ms, echo time 3.2 ms, flip angles [5°, 5°, 25°], 10 averages, voxel size 125 μm × 125 μm × 125 μm). These datasets were used to estimate MT saturation (MTsat) according to the method described by Helms et al. (2008). Diffusion-weighted images were acquired using a Stejskal–Tanner pulsed gradient spin-echo sequence (repetition time 2000 ms, echo time 23.2 ms, 20 slices, 3 averages, voxel size 125 × 125 × 500 μm3, b values 3000 and 6000 s/mm2, 30 directions each, 5 b0 images). The diffusion data were preprocessed through denoising (Veraart et al. 2016), correction for eddy current distortions (Andersson and Sotiropoulos 2016), motion correction (Avants et al. 2011), and bias field correction (Tustison et al. 2010). A diffusion tensor model (Basser et al. 1994) was fitted to the preprocessed data and fractional anisotropy (FA) was derived (Garyfallidis et al. 2014). An RGB color-coded FA map was also computed. To ensure that all images were centered at a common origin and oriented in the same way, we registered the individual mouse brains to the digital template of the Allen Mouse Brain Common Coordinate Framework version 3 (Wang et al. 2020). The registration employed 6 degrees of freedom (3 translations and 3 rotations) to maximize the mutual information between each individual brain and the template (Avants et al. 2011). Since this constitutes a rigid body transform, the original volumes of the structures were preserved.
CC and Isocortex Segmentation
To measure the cross-sectional area of the CC at its midline crossing in a consistent way across animals, we followed a multistep process involving the computed maps for MTsat and RGB FA. The MTsat maps, which exhibit good gray/white matter contrast, were used to generate white matter masks through simple thresholding (voxels with MTsat > 0.006 were classified as white matter). We subsequently restricted the white matter masks to the 5 middle sagittal slices (left-to-right thickness 0.625 mm) and further cropped them along the rostrocaudal (5 mm) and the dorsoventral (3.125 mm) directions—thus confining them within a thin midline slab that contained the entire CC crossing. This slab also included several other white matter tracts, some of which (e.g., dorsal fornix) run adjacent to the CC and are challenging to separate based on MTsat alone. We addressed this issue by incorporating information about the orientation of white matter tracts. This was available in the form of the RGB FA map, which is a color-coded representation of the principal diffusion direction at each voxel (Pajevic and Pierpaoli 1999). Using ITK-SNAP (Yushkevich et al. 2006), we overlaid the RGB FA map on the MTsat image, and segmented the CC manually, excluding voxels with principal diffusion directions other than left to right. In reeler mutants, this step proved especially useful for excluding a midline tract with a rostrocaudal orientation (cingulum), running dorsal to the CC. Using the final CC mask, we computed its midline cross-sectional area by multiplying the number of CC voxels on the midsagittal slice with the 2D area of each voxel (0.125 × 0.125 mm2).
The isocortical volume was derived from MTsat images, with the help of the Allen Brain Atlas. First, we pooled all isocortical regions of the atlas into a single mask and overlaid it on each individual MTsat image using ITK-SNAP. Then, we manually edited the mask for each brain, until it covered the entire cortical thickness, without protruding into extracortical areas. The final isocortical volume was computed by multiplying the number of voxels in the edited mask with the 3D volume of each voxel (0.125 × 0.125 × 0.125 mm3).
Quantification and Statistical Analysis
Mapping of RV-labeled input cells was done by overlaying the tissue section with the corresponding section of the Allen Brain Atlas (http://mouse.brain-map.org/experiment/thumbnails/100048576?image_type=atlasand). Labeled cells on all sections spanning from bregma +3 to −4.5 mm were counted manually in Neurolucida (MBF Bioscience). Double-labeled starter cells were manually counted the same way on all sections on which they could be found. Cell counts in an area were either normalized by the total number of starter cells (input magnitude) or by the total number of input cells (input fraction). Cell counts were exported with Neurolucida Explorer to Excel. R software (www.R-project.org) was used to sort data and perform statistical tests with custom-written scripts.
For pairwise comparisons between WT and reeler mice, data were first tested for normality (Shapiro–Wilk test) and equal variance (Bartlett test). For normally distributed data with equal variances, we used the Student’s t-test. For non-normally distributed data, we used the Wilcoxon rank-sum test. For normally distributed data with unequal variances, we used the Welch’s t-test. For multiple comparisons, we used the alpha-error adjustment by Holm. All values are given as mean ± standard deviation (SD).
For quantification of contralateral input labeled with AAV-retro-EGFP, we manually counted all EGFP-positive cells in contralateral barrel cortex on sections from bregma −0.8 to −2.1 mm as well as all cells in a volume from pia to white matter with a 200 μm diameter around the injection site. Contralateral neurons were then divided by the number of cells at the injection site to normalize for different labeling intensities.
For quantification of VIP neurons in VIP-Cre/tdTomato mice, neurons were manually counted on six 40-μm-thick sections per mouse on a patch of barrel cortex that spanned from pia to white matter and was 1000 μm wide.
For a layer-independent analysis of neuronal distribution in the cortex, we divided the space between the pia and the white matter border into 20 bins of equal size. We counted the number of labeled cells in each of these bins and normalized them by the total count of cells in all bins. To test for differences in the neuronal distribution between WT and reeler, we performed pairwise comparisons between genotypes for values in each bin with the same tests as indicated above.
Graphs were produced using Origin software (Origin Lab). Adobe Illustrator and InDesign CS6 were used for arrangement of pictures.
Results
VIP Neurons Are Uniformly Distributed across the Cortex in Reeler Mice
It is known from previous studies in reeler mice that parvalbumin- (Boyle et al. 2011) and somatostatin-positive (Yabut et al. 2007) inhibitory neurons lose their typical laminar distribution, but VIP neurons have not been investigated yet. We generated VIP-Cre/tdTomato/reeler mice to visualize VIP neurons (Fig. 1A,A’). First, to check if Cre-expressing tdTomato cells are VIP expressing, we stained sections of reeler barrel cortex against VIP-RNA with in situ hybridization. We found that 99.4 ± 0.8% of tdTomato cells expressed VIP (n = 3 mice, 3 sections each; Supplementary Fig. S1A,B). This is the same level of specificity demonstrated for the VIP-Cre/tdTomato/WT mouse (Prönneke et al. 2015). While in WT, VIP neurons showed their typical bias toward the upper layers, in reeler mice, they appeared homogenously distributed throughout the thickness of the cortex (Fig. 1B,B’,C).
Figure 1.
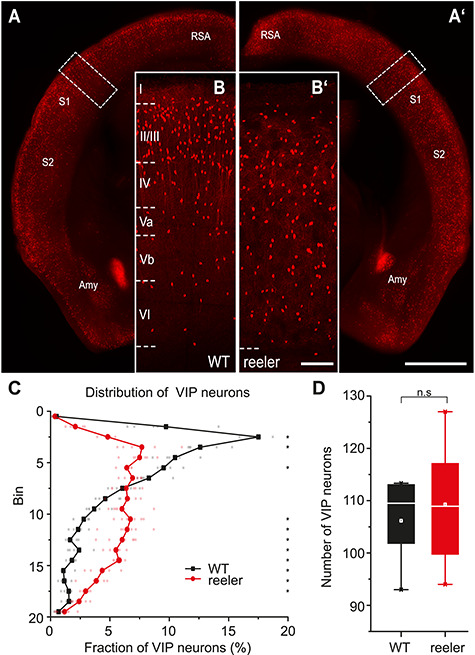
Distribution of VIP cells is different between WT and reeler mice. (A, A’) Coronal sections at the level of the barrel cortex of WT and reeler mice in which VIP neurons are labeled with tdTomato. The areal/nuclear locations of VIP expression remained the same in WT and reeler (scale bar: 1000 μm; Amy, amygdala; RSA, retrosplenial agranular cortex; S1/S2, primary/secondary somatosensory cortex). (B, B′) Close-up of barrel cortex area in WT and reeler mice (insert in A, A’). In WT, VIP neurons showed a stronger bias toward upper layers (II–IV). In reeler, VIP neurons were uniformly dispersed across the cortical thickness (scale bar: 100 μm). (C) Distribution of VIP neurons across the cortical depth. In WT, they showed a prominent peak in the upper layers. In reeler, they were fairly uniformly distributed, with few neurons close to pia and white matter (n = 5 WT mice, 6 reeler mice; 6 sections for each mouse; symbols connected by lines show average; small, transparent symbols show individual animals; asterisks highlight significantly different fractions per bin between genotypes with P < 0.05). (D) Number of VIP neurons counted on 40-μm-thick sections in an area of barrel cortex spanning from pia to white matter and being 1000 μm wide (n = 5 WT mice, 6 reeler mice; 6 sections each mouse; box plot: white line = median; white dot = mean). Counts were almost the same in WT and reeler (P > 0.05).
To visualize the differences in the cell distribution, we divided the cortex into 20 equal-sized bins and plotted the fraction of VIP neurons in each bin (Fig. 1C). We performed pairwise comparisons for each bin between the fraction of VIP neurons in both genotypes (Supplementary Table S1). In reeler animals, there was a lower proportion of VIP cells in the upper part of the cortex and a higher proportion in the middle and lower part of the cortex compared with WT (Fig. 1C). Therefore, VIP neurons in reeler lost their typical bias seen in WT and had a rather uniform distribution.
We counted the absolute number of VIP neurons in barrel cortex on 40-μm-thick sections of WT and reeler mice and found it to be almost the same (Fig. 1D; WT: n = 5 mice, 6 sections each, mean = 106 ± 8.7; WT: n = 6 mice, 6 sections each, mean = 109 ± 11.9; two-sample t-test, t = −0.49, P > 0.05). In sum, VIP neurons in the reeler mouse retain their VIP expression and appear in equal numbers as in WT but show no laminar bias. Hereafter, we used the VIP-Cre/reeler mouse to specifically target VIP neurons and investigate if their altered distribution affects their ability to integrate long-range inputs.
Rabies Virus Tracing for Mapping of Brain-Wide Inputs to VIP Neurons
We used Cre-dependent rabies virus tracing to map the monosynaptic long-range inputs to VIP cells in barrel cortex of WT and reeler mice (Wickersham et al. 2007; Wall et al. 2016; Fig. 2). We injected AAV8-DIO-TVA66T-EGFP-oG (hereafter AAV-TVA66T-EGFP-oG) into the cortex of VIP-Cre mice having WT or reeler genotypes. This led to the expression of 3 proteins: the cell surface receptor TVA (here we used a mutated version, TVA66T; Miyamichi et al. 2013), the optimized version of the rabies glycoprotein (oG; Kim et al. 2016), and EGFP (Fig. 2A). Modified rabies virus RV-SADΔG-mCherry (EnvA) (hereafter RV-mCherry; Fig. 2A) was injected 2 weeks later at the same location. Because it is coated with the TVA-ligand EnvA, it can only transduce cells presenting TVA on their surface. Furthermore, RV-mCherry is G-deleted, so that it needs the glycoprotein provided in trans from the AAV to spread to presynaptic neurons. Its mCherry expression labels these presynaptic neurons red. The starter cells appear yellow due to the mixture of EGFP and mCherry (Fig. 2B). This two-virus system allows the visualization of brain-wide monosynaptic inputs to unequivocally identifiable starter cells.
Figure 2.
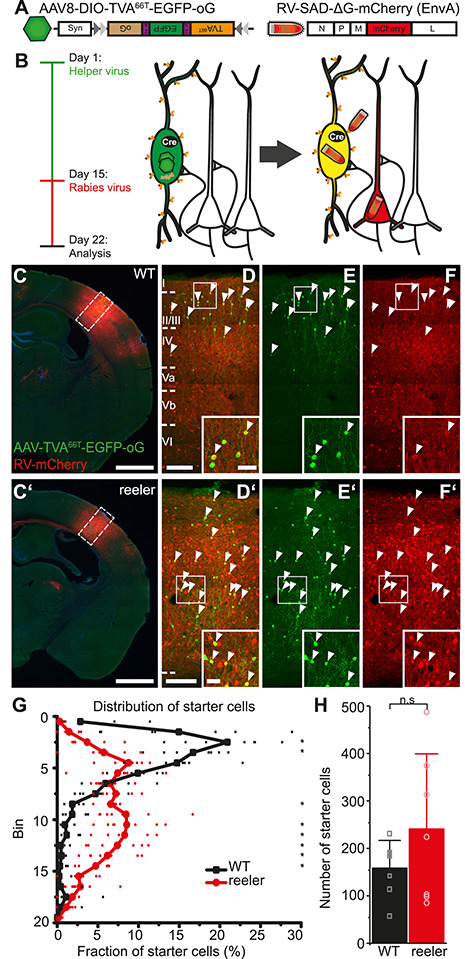
RV tracing in VIP-Cre mice is based on a different distribution and number of VIP starter cells in WT and reeler mice. (A) Viral constructs for RV tracing. TVA66T is a mutated version of TVA, to which EnvA in the RV envelope has a reduced affinity. This ensures that low-level Cre-independent TVA expression does not permit RV entry into cells. oG is the optimized rabies glycoprotein necessary for transsynaptic spread. (B) Injection of Cre-dependent helper AAV on day 1 induces high expression of TVA66T and oG only in VIP cells. RV-mCherry pseudotyped with TVA-ligand EnvA is injected 14 days later to infect VIP neurons and spread from there to first-order presynaptic neurons using oG. Seven days later, starter VIP cells appear yellow due to the mixture of fluorophores; presynaptic neurons have solely mCherry. (C, C’) Coronal sections through an injection site in the barrel cortex of WT and reeler mice (scale bar: 1000 μm). (D/D’–F/F’) Inserts in C, C’. Cells marked by white arrowheads are double-labeled starter cells that have been co-transduced by AAV-TVA66T-EGFP-oG and RV-mCherry. Inserts at the bottom show some of these cells in higher resolution. Exclusively RV-mCherry-positive cells represent local inputs, presynaptic to the starter cells, forming an extremely dense network of cell bodies and neuropil at the injection site (scale bar overview: 100 μm; scale bar insert: 20 μm). (G) Distribution of starter cells across the cortical depth. Cortical thickness was divided into 20 equal-sized bins. The proportion of starter cells in each bin was plotted. While in WT starter cells were predominantly in the upper third, starter cells in reeler were much more dispersed (n = 7 per group; symbols connected by lines show average; small, transparent symbols show individual animals; asterisks highlight significantly different fractions per bin between genotypes with P < 0.05). (H) Number of starter cells in each genotype. (n = 7 per group; mean ± SD, symbols show individual animals; P > 0.05).
To check if our viruses are specific, we performed control experiments in BL6 animals without Cre. Injection of RV-mCherry alone did not result in any labeling, showing that TVA is required for RV entry into cells (Supplementary Fig. S2A). When we injected AAV-TVA66T-EGFP-oG and RV-mCherry as in tracing experiments, we did not detect any RV-mCherry labeling, neither at the injection site nor in the thalamus (Supplementary Fig. S2B). A few EGFP-positive cells most likely resulted from a minimal Cre-independent expression of the AAV construct. While RV requires very little TVA to enter cells, which can result in Cre-independent leak expression, TVA66T has lower affinity to RV and thus needs to be expressed in much higher levels (Miyamichi et al. 2013). Therefore, this receptor abolishes leak expression and each mCherry-positive neuron can be surely considered an input neuron.
Injections Were Centered on the Whisker C2 Representation
In the barrel cortex, each whisker is represented by a cortical column. Although the reeler barrel cortex has no laminar organization, it retains a somatotopic organization such that adjacent whiskers are represented by adjacent modules (Guy et al. 2015). We localized the cortical whisker C2 representation to guide our injections using intrinsic signal optical imaging (Grinvald et al. 1986; Guy et al. 2015). Repetitive single-whisker C2 stimulation elicited hemodynamic responses, which appeared similar among WT and reeler mice (Supplementary Fig. S3A, A’). The overlay of the signal with the blood vessel pattern on the cortical surface (Supplementary Fig. S3B, B’) provided a map to accurately target the whisker-related cortical module with an injection (Hafner et al. 2019). As a proof of principle, we injected AAV-TVA66T-EGFP-oG and RV-mCherry centered on C2 in a WT animal and sectioned the cortex tangentially. We could confirm our target specificity as the highest density of inputs was present within C2 (Supplementary Fig. S3C). In this way, we could target VIP neurons that belong to the same functional modules in WT and reeler mice to have optimal comparability between genotypes.
VIP Starter Cells in Reeler Did Not Show a “laminar” bias
VIP-Cre and VIP-Cre/reeler mice (n = 7 per group) were injected with AAV-TVA66T-EGFP-oG and 2 weeks later with RV-mCherry into the right barrel cortex. Cells double labeled with EGFP/mCherry were considered starter cells and counted on each section where they appeared (Fig. 2C–F). To visualize the distribution of starter cells in the two genotypes, we again divided the cortex into 20 equal-sized bins and calculated the proportion of starter cells in each bin relative to the total number of starter cells (Fig. 2G). Pairwise comparisons between genotypes in each bin (Supplementary Table S2) highlighted very similar differences as in the general distribution of VIP neurons (Figs 1C and 2G). Thus, the starter cell distribution matched the general distribution of VIP cells in both genotypes. In WT, there was a clear bias of starter cells toward the upper third of the cortex. In reeler, the distribution of starter cells was broader with the majority of cells in the middle part of the cortex. The mean number of starter cells was higher in reeler, but this difference was not significant (Fig. 2H; mean WT: 158 ± 58.3; mean reeler: 241 ± 158.1; Wilcoxon rank sum test, W = 30, P > 0.05). The potential confounder of an imperfectly matched number of starter cells between genotypes is addressed below.
VIP Cells Received Input from the Same Areas in WT and Reeler
To address the question if the absence of layers impacts the capacity of VIP cells to receive proper long-range inputs, we manually counted all RV-labeled cells in the entire brain but omitted cells in the barrel cortex itself because of their extreme abundance. Therefore, local connectivity of VIP neurons was not assessed. Each coronal section was overlaid with the corresponding atlas section of the Allen Brain atlas. RV-mCherry-positive cells were assigned to an area, based on the outlines of the atlas and the cytoarchitectonic features discernable with nuclear stain.
VIP neurons in reeler mice received input from the very same areas as WT mice with no exception. Examples for areas in which we found transynaptically labeled cells are presented in Figure 3. Secondary somatosensory cortex contributed the highest number of input cells in WT and reeler (Fig. 3B,B’). In some areas, morphological differences of input cells became apparent, for example, in motor cortex where somata in reeler appeared larger (Fig. 3A,A’). Quantitative differences were easily noticeable in contralateral barrel cortex, which contained more cells in reeler (Fig. 3C,C’), and in primary auditory cortex, which contained fewer cells in reeler compared with WT (Fig. 3D,D’). Thalamic inputs appeared very similar (Fig. 3E,E’). In visual cortex, the distribution of projection neurons was clearly different with inputs in WT located at the LIII/IV border but being more dispersed in reeler (Fig. 3F,F’). From these observations, we suspected that the absence of layers could affect most strongly the magnitude and the proportion of cortical inputs to VIP cells.
Figure 3.
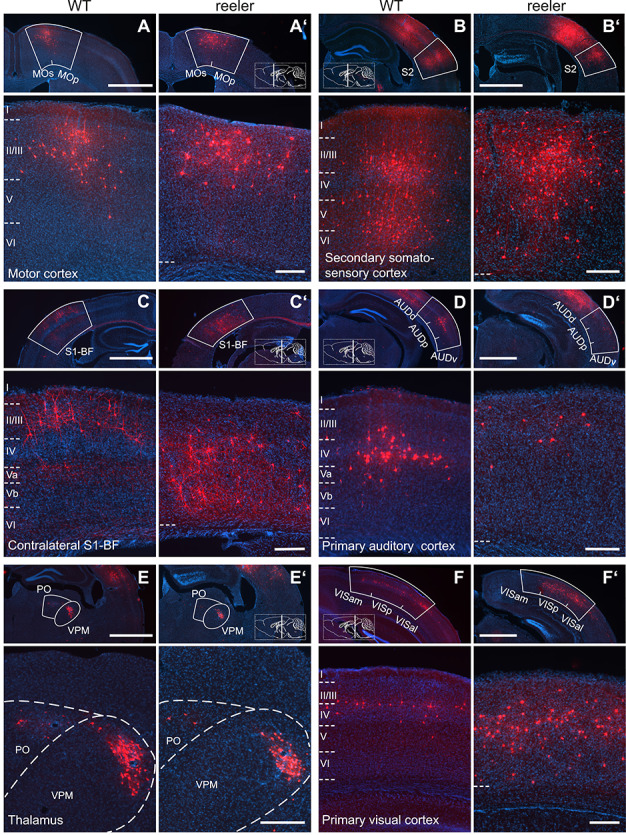
Long-range input to VIP cells in barrel cortex of WT and reeler mice. (A/A’–F/F’) Sections along the rostrocaudal extent of WT and reeler mice showing consistently labeled areas with presynaptic partners of VIP cells in barrel cortex. In the top overview panels, the white contour delineates the borders of the respective source area. Higher magnification close-ups are shown below. Section planes are indicated on the schematic sagittal brain section (scale bar overview: 1000 μm; scale bar close-up: 200 μm; AUDd/AUDp/AUDv, dorsal/primary/ventral auditory area; MOp/MOs, primary/secondary motor cortex; PO, posterior complex of the thalamus; S1-BF, primary somatosensory cortex, barrel field; S2, secondary somatosensory cortex; VISal/VISam/VISp, anterolateral/anteromedial/primary visual area; VPM, ventral posteromedial nucleus of the thalamus).
VIP Cells in Reeler Displayed an Imbalance between Ipsilateral and Contralateral Inputs
When analyzing quantitative differences of the inputs between genotypes, we first focused on more global categories of input (Fig. 4). We summed up all long-range inputs and then separated inputs from ipsilateral and contralateral cortical areas, from the thalamus, and from all subcortical areas. We normalized the inputs by dividing the cell count by the number of starter cells to calculate the input magnitude, which can be seen as a proxy for the strength of an input. We performed pairwise comparisons of these global input categories between WT and reeler (statistical values are listed in Supplementary Table S3). VIP neurons in reeler received overall fewer inputs per cell. This reduction was exclusively due to fewer inputs from the ipsilateral cortex in reeler, while they received more contralateral inputs per cell. There was no difference for thalamic or total subcortical input.
Figure 4.
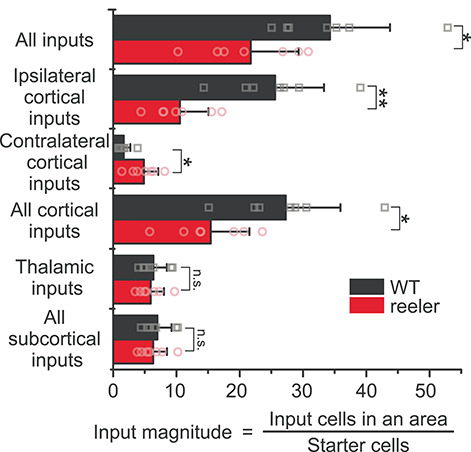
Input magnitude from global cortical and subcortical areas highlights a cortical phenotype. Histograms representing the input magnitude from summed-up cell counts of brain areas. Reeler mice received overall less input per cell, which was due to less input from the ipsilateral cortex. Input from the contralateral hemisphere was increased in reeler, whereas subcortical input remained unaffected (n = 7 per group; mean ± SD; symbols show individual animals; *P < 0.05, **P < 0.01, n.s. P > 0.05).
The normalization of inputs by starter cells could be misleading, if one presynaptic neuron synapses on multiple starter cells. This divergent connectivity would prompt that with a rising number of starter cells, the count of additionally labeled presynaptic cells would decrease. Hence, the higher the number of starter cells, the lower the ratio of starter cells to input cells (input magnitude). Because we had on average more starter cells in the reeler group, this might cause the smaller overall input magnitude in reeler. To investigate this possibility, we plotted the number of starter cells against the input magnitude for the two genotypes (Supplementary Fig. S2C). For both genotypes, the slope was close to zero, indicating no relationship between the number of starter cells and the input magnitude (WT: input magnitude = 0.002 * starter cells + 33.9, R2 = 0.2; reeler: input magnitude = −0.002 * starter cells + 22.2, R2 = −0.2). This result does not negate that tracing a higher number of starter cells will label more inputs but it implies that the normalization of inputs by starter cells is not biased by the total number of starter cells. Similarly, the higher number of starter cells in reeler cannot explain the lower input magnitude. Instead, the lower input magnitude seems to reflect an overall decreased innervation of VIP neurons in reeler.
In sum, VIP neurons in reeler mice are embedded in a different long-range cortical circuitry that contains fewer inputs from the ipsilateral and more inputs from the contralateral cortical hemisphere.
Auditory and Motor Cortices Showed the Strongest Reduction in Ipsilateral Input
Because each VIP neuron in reeler received on average less input, the subsequent analyses for differences among individual areas using the input magnitude would be inherently biased. Therefore, we employed another means of normalization: input fraction. It is the number of inputs in an area divided by the number of total inputs in the brain. It reflects how different inputs are balanced. We selected 41 consistently labeled areas that constituted the majority of inputs (97.6% in WT; 98.8% in reeler). We calculated the input fraction for each area and made pairwise comparisons between genotypes (Fig. 5; statistical values are listed in Supplementary Table S4). For motor, somatosensory body region, auditory, and visual cortices, we also summed up counts in their individual subareas to calculate a total input fraction. VIP neurons in reeler mice received a notably lower fraction from almost all ipsilateral cortical areas. The strongest reduction was seen for primary auditory cortex. The input fraction for motor cortex was also significantly reduced. The contralateral barrel cortex as well as the contralateral secondary somatosensory cortex contributed a significantly higher fraction of inputs in reeler. Subcortical input fraction was fairly similar, and only the ventral posterolateral nucleus of the thalamus constituted a higher fraction of inputs in reeler. This analysis of the fraction of inputs validates that the proportion of inputs from the ipsilateral hemisphere is reduced and from the contralateral homotopic area increased.
Figure 5.
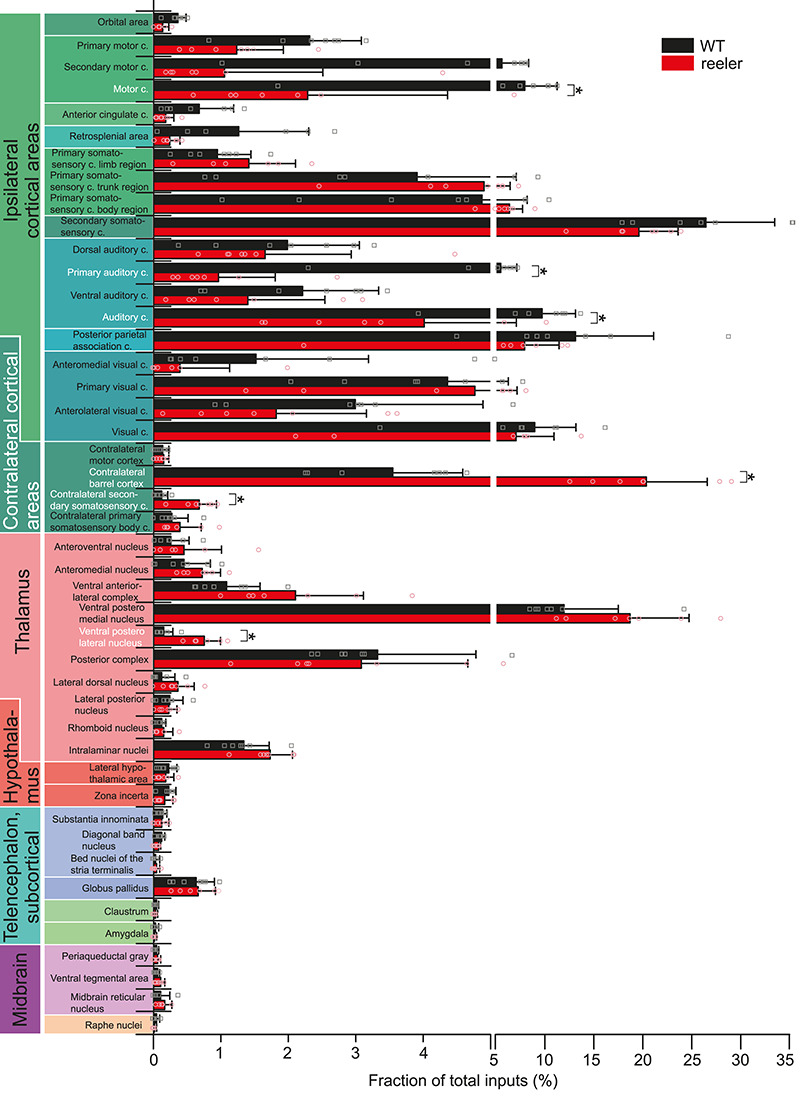
Comparative analysis of the input fraction from individual brain areas. Mean proportion of RV-labeled cells in 41 individual areas normalized against the total number of inputs in the whole brain for the two genotypes. For motor cortex, primary somatosensory cortex body region, auditory cortex, and visual cortex, the summated proportions of the individual subareas are shown as well. Pairwise comparisons were carried out to assess differences in input fraction for individual areas (n = 7 per group; mean ± SD; symbols show individual animals; *P < 0.05; c, cortex).
Reeler Mice Had a Larger CC
The fact that VIP cells in reeler received more input from the contralateral somatosensory cortex prompted the question if there are more callosal projection neurons (CPNs) in reeler. We injected the retrograde tracer AAV-retro-EGFP (Tervo et al. 2016) into the right barrel cortex of WT and reeler mice and counted the CPNs in the other hemisphere (Supplementary Fig. S4A, A’). We normalized the count of CPNs by the number of labeled cells in a given volume at the injection site. In reeler, there was about a 3-fold increase in the average number of CPNs (Supplementary Fig. S4C; n = 4 per group, mean ± SD: WT, 1.2 ± 1.4; reeler, 3.6 ± 2.7). Of note, in WT, our virus injections did not transduce neurons uniformly across layers with more neurons being labeled in upper than lower layers, while in reeler the transduction was fairly uniform across the cortical thickness (Supplementary Table S5). This could indicate that we also transduced fewer callosal fibers reaching deeper layers in WT and hence underestimate the number of CPNs. Still, the increase in CPNs we observed in reeler with AAV-retro-EGFP injections was the same as for contralateral inputs to VIP cells detected by rabies virus tracing. Moreover, the distribution of CPNs was very similar to the contralateral inputs to VIP cells, being more biased toward the lower part of the cortex (Supplementary Fig. S4C, Fig. 7F).
Figure 7.
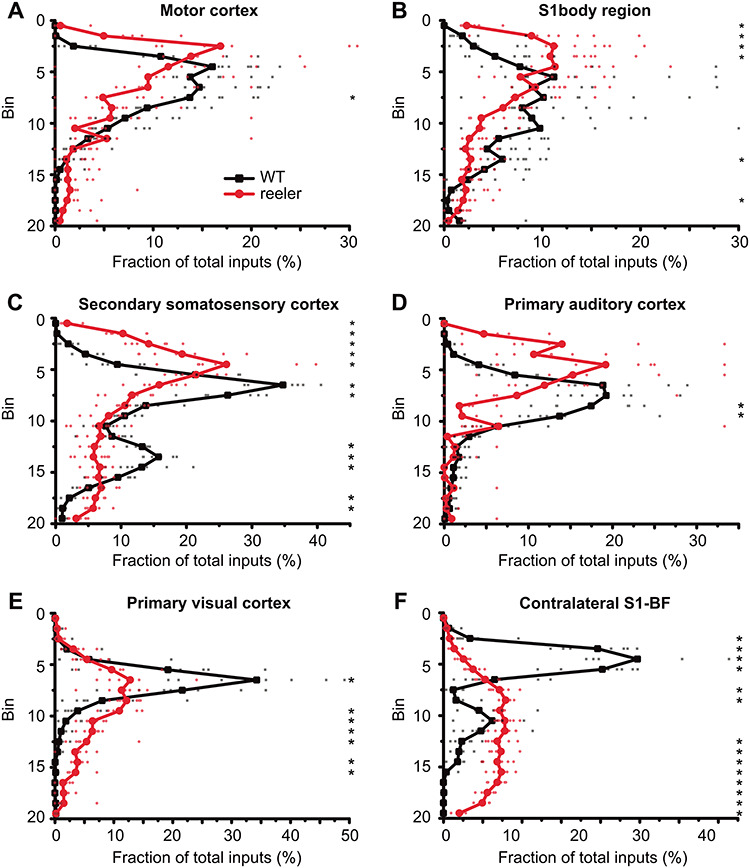
Distribution of projection neurons in cortical input areas. (A–F) The thickness of the cortex was divided into 20 equal-sized bins. We plotted the fraction of inputs in a bin normalized against the total inputs of VIP cells from this area. In ipsilateral areas, the distribution of projection neurons had one peak that was usually shifted more superficially in reeler, except for visual cortex where the peak was just broader. The distribution of contralateral projection neurons from the barrel cortex was very different, with neurons in WT being mostly located in the upper third, while in reeler predominantly in the lower two thirds of the cortex (n = 7 per group; symbols connected by lines show average; small, transparent symbols show individual animals; asterisks highlight significantly different fractions per bin between genotypes with P < 0.05; S1, primary somatosensory cortex; BF, barrel field).
If there are more CPNs in reeler, the CC should also contain more fibers and hence be larger. To provide additional evidence for an increased callosal connectivity, we performed magnetic resonance imaging to measure the dimensions of the CC. We performed a quantitative morphometric analysis on 3 WT–reeler littermate pairs. We segmented the CC based on structural information of MT maps (Fig. 6A,A’) and directional information of FA maps (Fig. 6C,C’). We measured the area of the CC on the mid-sagittal section, which was larger in reeler in every littermate pair (Fig. 6D). This effect was not generated by differences in cortical volume as the volume of the isocortex was fairly well matched between littermates, although in one pair the volume was slightly larger in the reeler mouse (Fig. 6E). Furthermore, we noticed on coronal sections that the CC was shaped differently in reeler. It did not have a typical U-shape in the medial area but was flattened out (Fig. 6B,B’). These experiments indicate that reeler mice have a larger CC because of a surplus of CPNs sending an axon to the other hemisphere.
Figure 6.
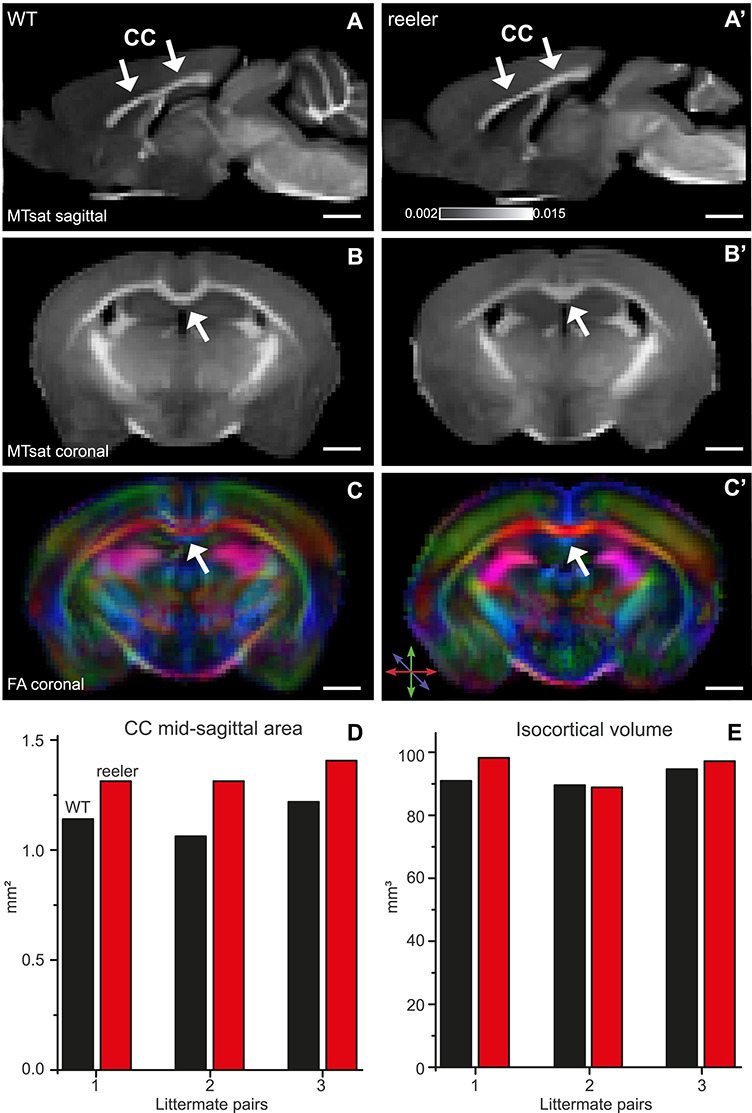
MRI reveals a larger and differently shaped CC (arrows) in reeler. (A, A’) Sagittal maps of MTsat of WT and reeler mice brain acquired ex vivo. These maps were used to determine the dimensions of the CC. (B, B’) Coronal MTsat map of WT versus reeler mice. At the midline, the CC showed a different geometry between genotypes. In reeler mice, the characteristic curvature of WT mice was absent so that the top of the CC appeared flattened. (C, C’) FA maps show the color-coded directionality of fibers. Because fibers of the CC run in the mediolateral direction, they could be distinguished from whiter matter bundles more dorsally or more ventrally that run in the rostrocaudal direction. (D) Midsagittal area of the CC of individual WT and reeler mice compared between littermate pairs. In each pair, the reeler mouse had a larger area, probably indicating that it is composed of a higher number of callosal fibers. (E) Total isocortical volume of individual WT and reeler mice compared between littermate pairs. The volume was almost the same, except for one pair in which the reeler mouse showed a slightly higher volume.
Projection Neurons in Reeler Showed Area-Specific Differences in Distribution
In reeler mice, the whole cortex shows alterations because of reelin deficiency. Therefore, the presynaptic projection neurons labeled in our study should show a markedly different distribution than in WT. To investigate the pattern of dispersion, we divided the cortex into 20 equal-sized bins and counted the proportion of presynaptic cells in each bin for ipsilateral motor, primary visual, primary auditory, somatosensory body (trunk + limbs), and secondary somatosensory cortices as well as for contralateral barrel cortex (Fig. 7). To quantify the differences between genotypes in each area, we performed pairwise comparisons for each bin (Supplementary Table S6). Projection neurons in reeler were never uniformly distributed across the cortex. Instead, for most ipsilateral areas, the neuronal distribution in reeler mice followed a similar shape as in WT but slightly shifted toward the pia (Fig. 7A–D). In visual cortex, however, the distribution looked notably different. In WT, most projection neurons were located around the LIII-IV border. This peak was smoothed out in reeler, indicating that the projection neurons were rather dispersed than inverted in their arrangement (Fig. 7E). The pattern of projection neurons in the contralateral barrel cortex was completely different (Fig. 7F). In WT, projection neurons were predominantly located in the upper third of the cortex, corresponding to LII/III. In reeler, projection neurons were predominantly located in the lower two-thirds of the cortex.
Discussion
We mapped the brain-wide long-range inputs to VIP neurons in barrel cortex of WT and reeler mice using retrograde RV tracing. We observed an overall smaller number of inputs to VIP cells in reeler. This reduction was due to fewer inputs from the ipsilateral cortical hemisphere. Contralateral cortical inputs, however, were much more numerous in reeler. There was no difference in the innervation by subcortical inputs. We will discuss possible mechanisms that could underlie this different cortical circuit architecture. We will hypothesize that the absence of layers necessitates an alternative connectivity to largely maintain sensory and cognitive abilities in the reeler mouse (Salinger et al. 2003; Pielecka-Fortuna et al. 2015).
Input to VIP Cells Originated from the Same Sources in WT and Reeler
We found that VIP cells in the barrel cortex of WT and reeler mice were innervated by the same sources in both genotypes. This is not surprising because the existence of the same types of cortical projection neurons has been confirmed in reeler, although they are in ectopic positions (Caviness 1976; Steindler and Colwell 1976; Yoshihara et al. 2010; Imai et al. 2012; Diodato et al. 2016; Wagener et al. 2016). Numerous tracing studies of cortical neurons in WT mice, including excitatory (DeNardo et al. 2015) and inhibitory (Wall et al. 2016) neurons in barrel cortex, demonstrated that all neuron classes in a cortical area receive input from the same sources. Therefore, the feature, which is considered to distinguish cell types and relates to potential functional implications, is the proportion of the input a given neuron type receives. In consequence, we expected to discover putative differences in the afferent connectivity to VIP neurons between genotypes specifically on a quantitative level, which has not been assessed for long-range projections in reeler yet.
Subcortical Fibers Target Ectopic VIP Cells to the Same Extent
When we looked at the proportion of inputs to VIP cells, we found profound differences between WT and reeler. VIP cells in reeler received less input per cell than in WT, which was exclusively due to a reduction in cortical inputs, while the subcortical inputs remained the same.
The thalamic input comprised the main source of subcortical input. The cells in the thalamus are -if at all- only subtly affected by the reelin mutation (Lambert de Rouvroit and Goffinet 1998; Wagener et al. 2010). Previous studies have shown that thalamic fibers from the VPM reach their malpositioned postsynaptic targets in the reeler cortex although they take an unusual trajectory, reaching LI before bouncing back to plunge down on the target cells (Molnár et al. 1998; Harsan et al. 2013; Wagener et al. 2016). Together with our results, these studies support the idea that fibers from properly developed subcortical structures find their ectopic targets in the mislaminated reeler cortex in approximately the same numbers. Therefore, neither the position of the VIP neurons nor reelin itself, as a molecular cue, seems to have an effect on the postsynaptic targeting.
Ipsilateral Long-Range Inputs Are Reduced
VIP cells in reeler barrel cortex received significantly fewer inputs from other ipsilateral cortical areas than in WT. Moreover, the extent to which the proportion of inputs in reeler was reduced varied considerably among the cortical areas. For example, the proportion of inputs from motor and auditory cortices was much more reduced than from visual or secondary somatosensory cortex. How could the global cellular dispersion influence connectivity in such a nuanced way? The pattern of developmental cellular malpositioning is not the same in each cortical area but varies along the rostrocaudal axis (Boyle et al. 2011; Mingo-Moreno et al., own unpublished observations). In motor cortex, cells are disorganized with elements of inversion of deep layer cells and normal migration of upper layer cells (Dekimoto et al. 2010; Boyle et al. 2011). In S1, cells are completely disorganized (Wagener et al. 2010). In visual cortex, cells acquire a “mirror-image type of architecture” centered around LIV cells (Boyle et al. 2011; Pielecka-Fortuna et al. 2015). Because each area seems to entail a unique distribution of cells, each area might comprise a unique environment. Considering that this cortical environment influences the development of projection neurons (Polleux et al. 2002), each area might vary in its capacity to induce connections with other areas.
Reeler Mice Have Increased Callosal Connectivity
In parallel to the reduction of ipsilateral cortical input, there was a massive increase of afferents from the contralateral hemisphere. We showed that in reeler there are more CPNs connecting the hemispheres of barrel cortex, and as a result, the CC is larger. Although previous studies have investigated the macroanatomy and trajectory of fiber bundles in the reeler mouse (Badea et al. 2007; Harsan et al. 2013), the enlargement of the CC has remained unnoticed. Similarly, earlier tracing studies have just confirmed the existence of callosal connections between homotopic areas but did not assess the numbers of CPNs (Caviness and Yorke 1976; Steindler and Colwell 1976; Imai et al. 2012).
Different Mechanisms Might Drive the Formation of Ipsilateral and Contralateral Projections
Because ipsilateral and contralateral long-range inputs showed such an opposing change in innervation intensity, we speculate that these two types of connections mature at different time points and based on different mechanisms. A sizeable fraction of ipsilateral projection neurons in reeler either fail to grow an axon or grow an axon but fail to find their destined postsynaptic partner. There is subtle evidence that ipsilateral projection neurons do not form excess projections that are pruned away but have a directed outgrowth and their connections are stably established by postnatal day (P) 7 (Klingler et al. 2018). On the contrary, during the development of callosal projections, a transiently higher number of callosal axons is reduced in an activity-dependent process of axonal elimination (reviewed in Innocenti and Price 2005). In the somatosensory system, CPNs invade the contralateral cortex by P5, reach their maximum density by P10, and then reduce again to reach a stable level of innervation by P20 (Fenlon et al. 2017; De Leon Reyes et al. 2019). Because the establishment of permanent callosal synapses is only finished in the third postnatal week, they might mature based on the network activity generated by already existing ipsilateral connections (Petreanu et al. 2007; Suárez et al. 2014). Perhaps in reeler, the pruning of callosal axons happens to a smaller degree than in WT to maintain a stable network in which the inputs of multiple afferent systems are balanced (Caviness and Rakic 1978). In this case, the excess callosal fibers would serve the purpose to compensate for the reduced innervation from ipsilateral areas.
Effects of Reelin on Neurite Outgrowth and Synaptogenesis Might Influence Neuronal Connectivity Directly
Reelin is a central player in the formation of circuits in several instances from early development into adulthood. Apart from its central function in controlling neuronal migration, it regulates growth of neurites, maturation of synapses, transmitter release, and synaptic plasticity (Bock and May 2016; Lee and D’Arcangelo 2016; Wasser and Herz 2017). For this reason, we cannot attribute the observed changes in the circuit of the adult reeler mouse solely to one particular effect of reelin. We would like to discuss our results in the light of possible mechanisms by which the absence of reelin could impact the formation of long-range connections and will argue that the observed effects are most likely caused by absent layering rather than other effects of reelin.
Role of Reelin in Dendritic Maturation
Reelin could affect the connectivity of neurons by influencing their input domain. The reduced number of cortical inputs to reeler VIP neurons could be explained by shorter dendritic length and/or reduced synaptic density. Reelin can directly promote dendritic growth (Niu et al. 2004; Jossin and Goffinet 2007). In reeler mice, dendrites of hippocampal (Niu et al. 2004, 2008) and LII/III cortical (Hoe et al. 2009) pyramidal neurons have shorter and less complex dendrites. However, spiny stellates in LIV of barrel cortex have virtually identical dendritic length as in WT (Guy et al. 2016) and neocortical GABAergic neurons labeled in the GIN mouse line (Ma et al. 2006) have even significantly longer dendrites in reeler (Yabut et al. 2007). Therefore, reelin absence does not necessarily go hand in hand with dendritic reduction. Instead, a direct effect of reelin on dendritic outgrowth might be cell type-specific. Reelin is also suspected to have a role in generation of synapses as its deficiency decreases the spine density of hippocampal neurons (Niu et al. 2008; Ventruti et al. 2011). In our case, these effects would need to be highly specific for ipsilateral cortical inputs because subcortical inputs were not decreased and contralateral inputs even increased. Therefore, alterations of VIP dendrites due to reelin absence might not be a key driver behind the changes in connectivity.
Role of Reelin in Axonal Maturation
Another possibility how the absence of reelin could affect the formation of circuits is via direct action on axonal outgrowth. During the early stages of axonal growth from entorhinal cortex into the hippocampus, axons are less numerous and have fewer branches, larger growth cones, and a lower bouton density in the reeler mouse (Borrell et al. 1999). This goes hand in hand with abnormal fiber trajectories and termination patterns (Del Rio et al. 1997; Borrell et al. 1999). These experiments led to the idea that reelin directly influences axonal growth (Ghosh 1997). However, this phenomenon is transient and the innervation pattern in the adult mouse is fairly similar between WT and reeler (Del Rio et al. 1997; Deller et al. 1999). It seems that not reelin but instead hyaluronan-associated cues play a pivotal role in the target-specific innervation of entorhinal fibers (Förster et al. 2001; Zhao et al. 2003). Considering a study showing that reelin does not impact axonal growth in vitro, too (Jossin and Goffinet 2001), the evidence so far does not suggest that reelin acts as a classical attracting or repelling signal for axons but rather as a modulator of growth cone stability (Leemhuis et al. 2010). Therefore, the absence of reelin most likely does not prevent axons from finding their targets but rather delays the process of axonal growth. Perhaps, signaling pathways that intersect with reelin signaling are upregulated as a compensatory mechanism to reinstate axonal outgrowth later in time. Because we did our tracing in adult mice, the effects of reelin absence on axonal outgrowth most likely have vanished at this point. Moreover, the effect of reelin would need to be highly specific for axons from a certain area. For example, we observed a strong reduction in inputs from auditory cortex but almost no reduction in inputs from visual cortex in reeler compared with WT.
Role of Reelin in Synaptic Maturation
Reelin can modulate synaptic plasticity both presynaptically by modulating transmitter release (Hellwig et al. 2011) and postsynaptically by changing channel composition (Groc et al. 2007; Ventruti et al. 2011). It influences synaptic strength measured by increased spine size when overexpressed (Pujadas et al. 2010; Bosch et al. 2016) and by increasing LTP (Weeber et al. 2002; Lane-Donovan et al. 2015). The effects of reelin on synaptic plasticity in the adult might not influence our tracing as RV is considered to jump synapses independent of synaptic activity (Ghanem and Conzelmann 2016). Nevertheless, the lack of reelin could influence the establishment of activity-dependent contacts. This might be especially relevant for the callosal fibers as we stated above.
Conditional KO of Reelin Could Filter Out Reelin’s Effects on Neurite Outgrowth
While a single secondary effect of reelin on maturation of neuronal processes is unlikely to underlie the changes in cortical connectivity, maybe their combination amounts to a substantial factor in the generation of reeler circuits. To pinpoint the influence of disturbed layering, it would be necessary to separate this component from the other effects of reelin. Once Cajal–Retzius cells wither, inhibitory neurons become the major source of reelin secretion in the cortex (Pohlkamp et al. 2013).
In our case, it would be elegant to switch off reelin expression after the layer formation is complete but before long-range connections, especially from the other hemisphere, are established. Then, we could investigate if reelin absence influences connectivity even in the presence of layers. Crossing the tamoxifen-inducible CreERT2 mouse line (Hayashi and McMahon 2002) with the Relnflox/flox mouse (Lane-Donovan et al. 2015) and injecting tamoxifen at P3 would allow to switch off reelin expression before callosal axons start to project to the other hemisphere (Wang et al. 2007). An alternative approach would be to delete reelin expression only from Cajal–Retzius cells. Then, layer formation would be disturbed but the postnatal effects of reelin would remain unaffected so that we can attribute patterns of connectivity solely to the absence of layers. Crossing the Frizzled10-Cre mouse line, which labels Cajal–Retzius cells (Zhao et al. 2006), with the Relnflox/flox mouse would restrict the reelin knockout to the Cajal–Retzius cells and leave reelin expression of other cortical neurons intact.
To perform the tracing as in our study, the reelin knockout line would need to be crossed to a VIP-Flp mouse confining Flp recombinase expression to VIP neurons. This would be complemented with a Flp-dependent AAV to induce gene expression for RV tracing and spread. Future efforts utilizing this approach would be able to isolate the different functions of reelin in maturation of brain-wide circuits.
An Alternative Circuit to Retain Cognitive Abilities
Despite the absence of layers, reeler mice display hardly any decline in cognitive abilities, have largely preserved sensory function, and only have a slight impairment of spatial memory and executive function (Salinger et al. 2003; Wagener et al. 2010; Pielecka-Fortuna et al. 2015; Imai et al. 2017). This is a strong indication that despite all the possible obstructions reelin absence can inflict on multiple levels of neuronal and circuit development, compensatory mechanisms take action to ensure the emergence of functional circuits. So far, the preservation of cognitive abilities has been attributed to the fact that in reeler neurons seem to retain not only their physiological properties but also their connectivity, implying the formation of similar or even the same circuits, despite the malposition of its parts (Guy and Staiger 2017). However, our map of brain-wide inputs to VIP neurons reveals that reeler mice have a different proportion of ipsilateral and contralateral inputs. We argue that this change in connectivity might emerge as a plastic adaption in pursuit of retaining global cognitive abilities like sensory perception but perhaps fails to realize more specialized and challenging domains of cognition. Because our connectivity map contains information on the number of connections, it allows to make very specific assumptions about behavioral impacts. For example, the auditory cortex fuels information about sound to the somatosensory cortex to be integrated into tactile processing (Lemus et al. 2010; Maruyama and Komai 2018). This input pathway comprises much fewer neurons in reeler. Behavioral experiments compelling reeler mice to integration of sound into tactile perception might be a starting point to study the functional significance of a diminished input. Similarly, the increased callosal connectivity might hint that reeler mice rely more on bilateral information during their sensory exploration. This could be tested perhaps in a variation of a corridor tracking task adapted for reeler to exclude the influence of motor deficits (Sofroniew et al. 2015).
Finally, we anticipate that the use of the reeler mouse (or related mouse models; see (Valiente and Marín 2010) as a model for neurological disorders will be accelerated by these findings, too. Reelin and its associated signaling pathway have been proposed as an important player in developmental disorders, such as lissencephaly, autism, or schizophrenia, which also show apparent callosal malformations (Fenlon and Richards 2015; Ishii et al. 2016; Romero et al. 2018). The reeler mouse has already been used to model these conditions and could help understanding mechanisms that lead to disorder-related connectivity changes, for example, in the CC.
Supplementary Material
Contributor Information
Georg Hafner, Institute for Neuroanatomy, University Medical Center, Georg-August-University Göttingen, 37075 Göttingen, Germany.
Julien Guy, Institute for Neuroanatomy, University Medical Center, Georg-August-University Göttingen, 37075 Göttingen, Germany.
Mirko Witte, Institute for Neuroanatomy, University Medical Center, Georg-August-University Göttingen, 37075 Göttingen, Germany.
Pavel Truschow, Institute for Neuroanatomy, University Medical Center, Georg-August-University Göttingen, 37075 Göttingen, Germany.
Alina Rüppel, Institute for Neuroanatomy, University Medical Center, Georg-August-University Göttingen, 37075 Göttingen, Germany.
Nikoloz Sirmpilatze, Functional Imaging Laboratory, German Primate Center, Leibniz Institute for Primate Research, 37077 Göttingen, Germany.
Rakshit Dadarwal, Functional Imaging Laboratory, German Primate Center, Leibniz Institute for Primate Research, 37077 Göttingen, Germany.
Susann Boretius, Functional Imaging Laboratory, German Primate Center, Leibniz Institute for Primate Research, 37077 Göttingen, Germany.
Jochen F Staiger, Institute for Neuroanatomy, University Medical Center, Georg-August-University Göttingen, 37075 Göttingen, Germany.
Notes
We thank Patricia Sprysch and Sandra Heinzl for excellent technical assistance. We also thank Christin Korb for help with cell counting of AAV-retro-EGFP-labeled cells. We are grateful to Karl-Klaus Conzelmann for kindly donating RV-SADΔG-mCherry (EnvA). We thank Michael Lingelbach and Edward Callaway for kindly donating pAAV-DIO-TVA66T-EGFP-oG. Conflict of Interest: None declared.
Funding
Deutsche Forschungsgemeinschaft via CRC 889 (Cellular mechanisms of sensory processing; TP C07 to J.F.S.) and STA 431/11-2.
Author Contributions
J.F.S., J.G., and G.H. conceived the study. G.H. conducted and analyzed all tracing experiments. J.G. and M.W. gave experimental advice. P.T. performed microscopic imaging. A.R. analyzed the VIPCre/reeler/tdTomato mouse line. N.S., R.D., and S.B. performed the MRI experiments. G.H., J.G., N.S., and J.F.S. wrote the manuscript.
References
- Ährlund-Richter S, Xuan Y, Anna van Lunteren J, Kim H, Ortiz C, Pollak Dorocic I, Meletis K, Carlén M. 2019. A whole-brain atlas of monosynaptic input targeting four different cell types in the medial prefrontal cortex of the mouse. Nat Neurosci. 22:657–668. [DOI] [PubMed] [Google Scholar]
- Andersson JLR, Sotiropoulos SN. 2016. An integrated approach to correction for off-resonance effects and subject movement in diffusion MR imaging. Neuroimage. 125:1063–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avants BB, Tustison NJ, Song G, Cook PA, Klein A, Gee JC. 2011. A reproducible evaluation of ANTs similarity metric performance in brain image registration. Neuroimage. 54:2033–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badea A, Nicholls PJ, Johnson GA, Wetsel WC. 2007. Neuroanatomical phenotypes in the reeler mouse. Neuroimage. 34:1363–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basser PJ, Mattiello J, LeBihan D. 1994. MR diffusion tensor spectroscopy and imaging. Biophys J. 66:259–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock HH, May P. 2016. Canonical and non-canonical reelin signaling. Front Cell Neurosci. 10:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrell V, Del Río JA, Alcántara S, Derer M, Martínez A, D’Arcangelo G, Nakajima K, Mikoshiba K, Derer P, Curran T et al. 1999. Reelin regulates the development and synaptogenesis of the layer-specific entorhino-hippocampal connections. J Neurosci. 19:1345–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch C, Muhaisen A, Pujadas L, Soriano E, Martínez A. 2016. Reelin exerts structural, biochemical and transcriptional regulation over presynaptic and postsynaptic elements in the adult hippocampus. Front Cell Neurosci. 10:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle MP, Bernard A, Thompson CL, Ng L, Boe A, Mortrud M, Hawrylycz MJ, Jones AR, Hevner RF, Lein ES. 2011. Cell-type-specific consequences of reelin deficiency in the mouse neocortex, hippocampus, and amygdala. J Comp Neurol. 519:2061–2089. [DOI] [PubMed] [Google Scholar]
- Caviness VS 1976. Patterns of cell and fiber distribution in the neocortex of the reeler mutant mouse. J Comp Neurol. 170:435–447. [DOI] [PubMed] [Google Scholar]
- Caviness VS, Rakic P. 1978. Mechanisms of cortical development: a view from mutations in mice. Annu Rev Neurosci. 1:297–326. [DOI] [PubMed] [Google Scholar]
- Caviness VS, Yorke CH. 1976. Interhemispheric neocortical connections of the corpus callosum in the reeler mutant mouse: a study based on anterograde and retrograde methods. J Comp Neurol. 170:449–460. [DOI] [PubMed] [Google Scholar]
- De Leon Reyes NS, Mederos S, Varela I, Weiss LA, Perea G, Galazo MJ, Nieto M. 2019. Transient callosal projections of L4 neurons are eliminated for the acquisition of local connectivity. Nat Commun. 10:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekimoto H, Terashima T, Katsuyama Y. 2010. Dispersion of the neurons expressing layer specific markers in the reeler brain. Dev Growth Differ. 52:181–193. [DOI] [PubMed] [Google Scholar]
- Del Rio JA, Heimrich B, Borell V, Förster E, Drakew A, Alcantara S, Nakajima K, Mijata T, Ogawa M, Mikoshiba K et al. 1997. A role for Cajal–Retzius cells and reelin in the development of hippocampal connections. Nature. 385:70–73. [DOI] [PubMed] [Google Scholar]
- Deller T, Drakew A, Frotscher M. 1999. Different primary target cells are important for fiber lamination in the fascia dentata: a lesson from reeler mutant mice. Exp Neurol. 156:239–253. [DOI] [PubMed] [Google Scholar]
- DeNardo LA, Berns DS, DeLoach K, Luo L. 2015. Connectivity of mouse somatosensory and prefrontal cortex examined with trans-synaptic tracing. Nat Neurosci. 18:1687–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diodato A, Ruinart De Brimont M, Yim YS, Derian N, Perrin S, Pouch J, Klatzmann D, Garel S, Choi GB, Fleischmann A. 2016. Molecular signatures of neural connectivity in the olfactory cortex. Nat Commun. 7:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmeyer D 2012. Excitatory neuronal connectivity in the barrel cortex. Front Neuroanat. 6:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenlon LR, Richards LJ. 2015. Contralateral targeting of the corpus callosum in normal and pathological brain function. Trends Neurosci. 38:264–272. [DOI] [PubMed] [Google Scholar]
- Fenlon LR, Suárez R, Richards LJ. 2017. The anatomy, organisation and development of contralateral callosal projections of the mouse somatosensory cortex. Brain Neurosci Adv. 1:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Förster E, Zhao S, Frotscher M. 2001. Hyaluronan-associated adhesive cues control fiber segregation in the hippocampus. Development. 128:3029–3039. [DOI] [PubMed] [Google Scholar]
- Fu Y, Tucciarone JM, Espinosa JS, Sheng N, Darcy DP, Nicoll RA, Huang ZJ, Stryker MP. 2014. A cortical circuit for gain control by behavioral state. Cell. 156:1139–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garyfallidis E, Brett M, Amirbekian B, Rokem A, van der Walt S, Descoteaux M, Nimmo-Smith I. 2014. Dipy, a library for the analysis of diffusion MRI data. Front Neuroinform. 8:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghanem A, Conzelmann KK. 2016. G gene-deficient single-round rabies viruses for neuronal circuit analysis. Virus Res. 216:41–54. [DOI] [PubMed] [Google Scholar]
- Ghosh A 1997. Axons follow reelin routes. Nature. 385:23–24. [DOI] [PubMed] [Google Scholar]
- Grinvald A, Lieke E, Frostig R, Gilbert C, Wiesel T. 1986. Functional architecture of cortex revealed by optical imaging of intrinsic signals. Nature. 324:361–364. [DOI] [PubMed] [Google Scholar]
- Groc L, Choquet D, Stephenson FA, Verrier D, Manzoni OJ, Chavis P. 2007. NMDA receptor surface trafficking and synaptic subunit composition are developmentally regulated by the extracellular matrix protein reelin. J Neurosci. 27:10165–10175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy J, Sachkova A, Möck M, Witte M, Wagener RJ, Staiger JF. 2016. Intracortical network effects preserve thalamocortical input efficacy in a cortex without layers. Cereb Cortex. 27:4851–4866. [DOI] [PubMed] [Google Scholar]
- Guy J, Staiger JF. 2017. The functioning of a cortex without layers. Front Neuroanat. 11:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy J, Wagener RJ, Möck M, Staiger JF. 2015. Persistence of functional sensory maps in the absence of cortical layers in the somatosensory cortex of reeler mice. Cereb Cortex. 25:2517–2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner G, Witte M, Guy J, Subhashini N, Fenno LE, Ramakrishnan C, Kim YS, Deisseroth K, Oberhuber M, Conzelmann K-K et al. 2019. Mapping brain-wide afferent inputs of parvalbumin- expressing GABAergic neurons in barrel cortex reveals local and long-range circuit motifs. Cell Rep. 28:3450–3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KD, Shepherd GMG. 2015. The neocortical circuit: themes and variations. Nat Neurosci. 18:170–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harsan L-A, Dávid C, Reisert M, Schnell S, Hennig J, von Elverfeldt D, Staiger JF. 2013. Mapping remodeling of thalamocortical projections in the living reeler mouse brain by diffusion tractography. Proc Natl Acad Sci U S A. 110:E1797–E1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi S, McMahon AP. 2002. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol. 244:305–318. [DOI] [PubMed] [Google Scholar]
- Hellwig S, Hack I, Kowalski J, Brunne B, Jarowyj J, Unger A, Bock HH, Junghans D, Frotscher M. 2011. Role for reelin in neurotransmitter release. J Neurosci. 31:2352–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helms G, Dathe H, Kallenberg K, Dechent P. 2008. High-resolution maps of magnetization transfer with inherent correction for RF inhomogeneity and T1 relaxation obtained from 3D FLASH MRI. Magn Reson Med. 60:1396–1407. [DOI] [PubMed] [Google Scholar]
- Hoe HS, Kea JL, Carney RSE, Lee J, Markova A, Lee JY, Howell BW, Hyman BT, Pak DTS, Bu G et al. 2009. Interaction of reelin with amyloid precursor protein promotes neurite outgrowth. J Neurosci. 29:7459–7473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai H, Shoji H, Ogata M, Kagawa Y, Owada Y, Miyakawa T, Sakimura K, Terashima T, Katsuyama Y. 2017. Dorsal forebrain-specific deficiency of reelin-Dab1 signal causes behavioral abnormalities related to psychiatric disorders. Cereb Cortex. 27:3485–3501. [DOI] [PubMed] [Google Scholar]
- Imai H, Yamamoto T, Katsuyama Y, Kikkawa S, Terashima T. 2012. Subcortically and callosally projecting neurons are distinct neuronal pools in the motor cortex of the reeler mouse. Kobe J Med Sci. 58:86–95. [PubMed] [Google Scholar]
- Innocenti GM, Price DJ. 2005. Exuberance in the development of cortical networks. Nat Rev Neurosci. 6:955–965. [DOI] [PubMed] [Google Scholar]
- Ishii K, Kubo K, Nakajima K. 2016. Reelin and neuropsychiatric disorders. Front Cell Neurosci. 10:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jossin Y, Goffinet AM. 2001. Reelin does not directly influence axonal growth. J Neurosci. 21:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jossin Y, Goffinet AM. 2007. Reelin signals through phosphatidylinositol 3-kinase and Akt to control cortical development and through mTor to regulate dendritic growth. Mol Cell Biol. 27:7113–7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasthuri N, Hayworth KJ, Berger DR, Schalek RL, Conchello JA, Knowles-Barley S, Lee D, Vázquez-Reina A, Kaynig V, Jones TR et al. 2015. Saturated reconstruction of a volume of neocortex. Cell. 162:648–661. [DOI] [PubMed] [Google Scholar]
- Kim EJ, Jacobs MW, Ito-cole T, Callaway EM. 2016. Improved monosynaptic neural circuit tracing using engineered rabies virus glycoproteins. Report Cell Rep. 15:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingler E, Prados J, Kebschull JM, Dayer A, Zador AM, Jabaudon D. 2018. Single-cell molecular connectomics of intracortically-projecting neurons. bioRxiv. 378760. doi: 10.1101/378760. [DOI] [Google Scholar]
- Lambert de Rouvroit C, Goffinet AM. 1998. The reeler mouse as a model of brain development. Adv Anat Embryol Cell Biol. 150:1–106. [PubMed] [Google Scholar]
- Lane-Donovan C, Philips GT, Wasser CR, Durakoglugil MS, Masiulis I, Upadhaya A, Pohlkamp T, Coskun C, Kotti T, Steller L et al. 2015. Reelin protects against amyloid β toxicity in vivo. Nat Commun. 8:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee GH, D’Arcangelo G. 2016. New insights into reelin-mediated signaling pathways. Front Cell Neurosci. 10:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Kruglikov I, Huang ZJ, Fishell G, Rudy B. 2013. A disinhibitory circuit mediates motor integration in the somatosensory cortex. Nat Neurosci. 16:1662–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leemhuis J, Bouché E, Frotscher M, Henle F, Hein L, Herz J, Meyer DK, Pichler M, Roth G, Schwan C et al. 2010. Reelin signals through apolipoprotein E receptor 2 and Cdc42 to increase growth cone motility and filopodia formation. J Neurosci. 30:14759–14772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemus L, Ndez AH, Luna R, Zainos A, Romo R. 2010. Do sensory cortices process more than one sensory modality during perceptual judgments. Neuron. 67:335–348. [DOI] [PubMed] [Google Scholar]
- Ma Y, Hu H, Berrebi AS, Mathers PH, Agmon A. 2006. Distinct subtypes of somatostatin-containing neocortical interneurons revealed in transgenic mice. J Neurosci. 26:5069–5082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama AT, Komai S. 2018. Auditory-induced response in the primary sensory cortex of rodents. PLoS One. 13:e0209266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RT 2011. Technical immunohistochemistry: achieving reliability and reproducibility of immunostains. Society for Applied Immunohistochemistry, 2001 Annual Meeting, p. 1–56.
- Miyamichi K, Yael S-F, Marvin S, Brandon CW, Liqun L, Mizrahi A. 2013. Dissecting local circuits: parvalbumin interneurons underlie broad feedback control of olfactory bulb output. Neuron. 80:1232–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnár Z, Adams R, Goffinet AM, Blakemore C. 1998. The role of the first postmitotic cortical cells in the development of thalamocortical innervation in the reeler mouse. J Neurosci. 18:5746–5765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motta A, Berning M, Boergens KM, Staffler B, Beining M, Loomba S, Schramm C, Hennig P, Wissler H, Helmstaedter M. 2019. Dense connectomic reconstruction in layer 4 of the somatosensory cortex. Science. (80):366. [DOI] [PubMed] [Google Scholar]
- Niu S, Renfro A, Quattrocchi CC, Sheldon M, D’Arcangelo G. 2004. Reelin promotes hippocampal dendrite development through the VLDLR/ApoER2-Dab1 pathway. Neuron. 41:71–84. [DOI] [PubMed] [Google Scholar]
- Niu S, Yabut O, D’Arcangelo G. 2008. The reelin signaling pathway promotes dendritic spine development in hippocampal neurons. J Neurosci. 28:10339–10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajevic S, Pierpaoli C. 1999. Color schemes to represent the orientation of anisotropic tissues from diffusion tensor data: application to white matter fiber tract mapping in the human brain. Magn Reson Med. 42:526–540. [PubMed] [Google Scholar]
- Petreanu L, Huber D, Sobczyk A, Svoboda K. 2007. Channelrhodopsin-2–assisted circuit mapping of long-range callosal projections. Nat Neurosci. 10:663–668. [DOI] [PubMed] [Google Scholar]
- Pfeffer CK, Xue M, He M, Huang ZJ, Scanziani M. 2013. Inhibition of inhibition in visual cortex: the logic of connections between molecularly distinct interneurons. Nat Neurosci. 16:1068–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pielecka-Fortuna J, Wagener RJ, Martens AK, Goetze B, Schmidt KF, Staiger JF, Löwel S. 2015. The disorganized visual cortex in reelin-deficient mice is functional and allows for enhanced plasticity. Brain Struct Funct. 220:3449–3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohlkamp T, Dávid C, Cauli B, Gallopin T, Bouché E, Karagiannis A, May P, Herz J, Frotscher M, Staiger JF et al. 2013. Characterization and distribution of reelin-positive interneuron subtypes in the rat barrel cortex. Cereb Cortex. 3046–3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polleux F, Dehay C, Goffinet A, Kennedy H. 2002. Pre- and post-mitotic events contribute to the progressive acquisition of area-specific connectional fate in the neocortex. Cereb Cortex. 11:1027–1039. [DOI] [PubMed] [Google Scholar]
- Prönneke A, Scheuer B, Wagener RJ, Möck M, Witte M, Staiger JF. 2015. Characterizing VIP neurons in the barrel cortex of VIPcre/tdTomato mice reveals layer-specific differences. Cereb Cortex. 25:4854–4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prume M, Rollenhagen A, Lübke JHR. 2018. Structural and synaptic organization of the adult reeler mouse somatosensory neocortex: a comparative fine-scale electron microscopic study of reeler with wild type mice. Front Neuroanat. 12:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prume M, Rollenhagen A, Yakoubi R, Sätzler K, Lübke JHR. 2019. Quantitative three-dimensional reconstructions of excitatory synaptic boutons in layer 5 of the adult human temporal lobe neocortex: a fine-scale electron microscopic analysis. Cereb Cortex. 29:2797–2814. [DOI] [PubMed] [Google Scholar]
- Pujadas L, Gruart A, Bosch C, Delgado L, Teixeira CM, Rossi D, De Lecea L, Martínez A, Delgado-García JM, Soriano E. 2010. Reelin regulates postnatal neurogenesis and enhances spine hypertrophy and long-term potentiation. J Neurosci. 30:4636–4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero DM, Bahi-Buisson N, Francis F. 2018. Genetics and mechanisms leading to human cortical malformations. Semin Cell Dev Biol. 76:33–75. [DOI] [PubMed] [Google Scholar]
- Salinger WL, Ladrow P, Wheeler C. 2003. Behavioral phenotype of the reeler mutant mouse: effects of Reln gene dosage and social isolation. Behav Neurosci. 117:1257–1275. [DOI] [PubMed] [Google Scholar]
- Shepherd GM, Rowe TB. 2017. Neocortical lamination: insights from neuron types and evolutionary precursors. Front Neuroanat. 11:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva LR, Gutnick MJ, Connors BW. 1991. Laminar distribution of neuronal membrane properties in neocortex of normal and reeler mouse. J Neurophysiol. 66:2034–2040. [DOI] [PubMed] [Google Scholar]
- Sofroniew NJ, Vlasov YA, Hires SA, Freeman J, Svoboda K. 2015. Neural coding in barrel cortex during whisker-guided locomotion. Elife. 4:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steindler DA, Colwell SA. 1976. Reeler mutant mouse: maintenance of appropriate and reciprocal connections in the cerebral cortex and thalamus. Brain Res. 105:386–393. [DOI] [PubMed] [Google Scholar]
- Suárez R, Fenlon LR, Marek R, Avitan L, Sah P, Goodhill GJ, Richards LJ. 2014. Balanced interhemispheric cortical activity is required for correct targeting of the corpus callosum. Neuron. 82:1289–1298. [DOI] [PubMed] [Google Scholar]
- Sun Q, Li X, Ren M, Zhao M, Zhong Q, Ren Y, Luo P, Ni H, Zhang X, Zhang C et al. 2019. A whole-brain map of long-range inputs to GABAergic interneurons in the mouse medial prefrontal cortex. Nat Neurosci. 22:1357–1370. [DOI] [PubMed] [Google Scholar]
- Taniguchi H, He M, Wu P, Kim S, Paik R, Sugino K, Kvitsiani D, Kvitsani D, Fu Y, Lu J et al. 2011. A resource of Cre driver lines for genetic targeting of GABAergic neurons in cerebral cortex. Neuron. 71:995–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tervo DGR, Hwang BY, Viswanathan S, Gaj T, Lavzin M, Ritola KD, Lindo S, Michael S, Kuleshova E, Ojala D et al. 2016. A designer AAV variant permits efficient retrograde access to projection neurons. Neuron. 92:372–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay R, Lee S, Rudy B. 2016. GABAergic interneurons in the neocortex: from cellular properties to circuits. Neuron. 91:260–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tustison NJ, Avants BB, Cook PA, Gee JC. 2010. N4ITK: improved N3 bias correction with robust B-spline approximation. IEEE Trans Med Imaging. 29:1310–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valiente M, Marín O. 2010. Neuronal migration mechanisms in development and disease. Curr Opin Neurobiol. 20:68–78. [DOI] [PubMed] [Google Scholar]
- Ventruti A, Kazdoba TM, Niu S, D’Arcangelo G. 2011. Reelin deficiency causes specific defects in the molecular composition of the synapses in the adult brain. Neuroscience. 189:32–42. [DOI] [PubMed] [Google Scholar]
- Veraart J, Novikov DS, Christiaens D, Ades-aron B, Sijbers J, Fieremans E. 2016. Denoising of diffusion MRI using random matrix theory. Neuroimage. 142:394–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagener RJ, Dávid C, Zhao S, C a H, Staiger JF. 2010. The somatosensory cortex of reeler mutant mice shows absent layering but intact formation and behavioral activation of columnar somatotopic maps. J Neurosci. 30:15700–15709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagener RJ, Witte M, Guy J, Mingo-Moreno N, Ku gler S, Staiger JF. 2016. Thalamocortical connections drive intracortical activation of functional columns in the mislaminated reeler somatosensory cortex. Cereb Cortex. 26:820–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker F, Möck M, Feyerabend M, Guy J, Wagener RJ, Schubert D, Staiger JF, Witte M. 2016. Parvalbumin- and vasoactive intestinal polypeptide-expressing neocortical interneurons impose differential inhibition on Martinotti cells. Nat Commun. 7:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall NR, La PMD, Sorokin JM, Taniguchi H, Huang ZJ, Callaway EM. 2016. Brain-wide maps of synaptic input to cortical interneurons. J Neurosci. 36:4000–4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C-L, Zhang L, Zhou Y, Zhou J, Yang X-J, Duan S-M, Xiong Z-Q, Ding Y-Q. 2007. Activity-dependent development of callosal projections in the somatosensory cortex. J Neurosci. 27:11334–11342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Ding SL, Li Y, Royall J, Feng D, Lesnar P, Graddis N, Naeemi M, Facer B, Ho A et al. 2020. The Allen Mouse Brain Common Coordinate Framework: a 3D Reference Atlas. Cell. 181:936–953.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasser CR, Herz J. 2017. Reelin: neurodevelopmental architect and homeostatic regulator of excitatory synapses. J Biol Chem. 292:1330–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeber EJ, Beffert U, Jones C, Christian JM, Förster E, David Sweatt J, Herz J. 2002. Reelin and apoE receptors cooperate to enhance hippocampal synaptic plasticity and learning. J Biol Chem. 277:39944–39952. [DOI] [PubMed] [Google Scholar]
- Wickersham IR, Lyon DC, Barnard RJO, Mori T, Conzelmann K, Young JAT, Callaway EM. 2007. Monosynaptic restriction of transsynaptic tracing from single, genetically targeted neurons. Neuron. 53:639–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams LE, Holtmaat A. 2019. Higher-order thalamocortical inputs gate synaptic long-term potentiation via disinhibition. Neuron. 101:1–12. [DOI] [PubMed] [Google Scholar]
- Yabut O, Renfro A, Niu S, Swann JW, Marín O, D’Arcangelo G. 2007. Abnormal laminar position and dendrite development of interneurons in the reeler forebrain. Brain Res. 1140:75–83. [DOI] [PubMed] [Google Scholar]
- Yoshihara Y, Setsu T, Katsuyama Y, Kikkawa S, Terashima T, Maeda K. 2010. Cortical layer V neurons in the auditory and visual cortices of normal, reeler, and yotari mice. Kobe J Med Sci. 56:50–59. [PubMed] [Google Scholar]
- Yushkevich PA, Piven J, Hazlett HC, Smith RG, Ho S, Gee JC, Gerig G. 2006. User-guided 3D active contour segmentation of anatomical structures: significantly improved efficiency and reliability. Neuroimage. 31:1116–1128. [DOI] [PubMed] [Google Scholar]
- Zhang S, Xu M, Chang W-C, Ma C, Hoang Do JP, Jeong D, Lei T, Fan JL, Dan Y. 2016. Organization of long-range inputs and outputs of frontal cortex for top-down control. Nat Neurosci. 19:1733–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Xu M, Kamigaki T, Hoang Do JP, Chang W-C, Jenvay S, Miyamichi K, Luo L, Dan Y. 2014. Long-range and local circuits for top-down modulation of visual cortex processing. Science. 345(80):660–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Guan W, Pleasure SJ. 2006. A transgenic marker mouse line labels Cajal-Retzius cells from the cortical hem and thalamocortical axons. Brain Res. 1077:48–53. [DOI] [PubMed] [Google Scholar]
- Zhao S, Förster E, Chai X, Frotscher M. 2003. Different signals control laminar specificity of commissural and entorhinal fibers to the dentate gyrus. J Neurosci. 23:7351–7357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Mansori I, Fischer T, Witte M, Staiger JF. 2020. Characterizing the morphology of somatostatin-expressing interneurons and their synaptic innervation pattern in the barrel cortex of the GIN mouse. J Comp Neurol. 528:244–260. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.