

Abstract
Background
Exclusive breastfeeding promotes beneficial modifications on the microbiota of cesarean born infants, but little is known about the role of specific breast milk components in this modulation. Women with an active FUT2 gene (called secretors) secrete α1–2 fucosylated human milk oligosaccharides (HMOs), which promote Bifidobacterium in the infant’s gut and may modulate the microbiota of cesarean born infants.
Objective
To compare the microbiota composition of cesarean and vaginally born infants breastfed by secretor mothers.
Methods
Maternal secretor status was determined by the occurrence of 4 different α1–2 fucosylated HMOs in breast milk by LC-MS. The fecal microbiota composition from cesarean and vaginally born infants was analyzed by 16S rRNA gene sequencing and qPCR, stratified by the maternal secretor status, and compared.
Results
Alpha and beta diversity were not significantly different in cesarean born, secretor-fed infants (CSe+) compared to vaginally born, secretor-fed infants (VSe+). There were no significant differences in the fecal relative abundance of Bifidobacterium between CSe+ and VSe+ infants, but the prevalence of the species B. longum was lower in CSe+. The fecal relative abundance of Bacteroides was also lower, while Akkermansia and Kluyvera were higher in CSe+ infants.
Conclusion
Cesarean and vaginally born infants fed with breast milk containing the α1–2 fucosylated HMOs fraction present similar amounts of Bifidobacterium in the feces, but differences are observed in other members of the microbiota.
Introduction
Disruptions in the natural assembly of the neonatal microbiota have been associated with increased risk of immune-mediated diseases [1,2] and excessive weight later in childhood [3–5]. The mode of birth is one of the major factors that significantly affect bacterial colonization and the development of the gut microbial community [6]. Vaginally born infants harbor a microbiota enriched with mutualistic bacteria such as Bifidobacterium, Escherichia, Bacteroides, and Parabacteroides. In contrast, cesarean born infants are depleted of these genera and are instead dominated by Enterococcus, Staphylococcus, Streptococcus, Klebsiella, Enterobacter, and Clostridium, which are commonly associated with the human skin and the hospital environment [6–10].
Previous studies demonstrated that exclusive breastfeeding can partially restore the disruptions caused by cesarean birth in the infant gut microbiota, without exploring the role of breast milk composition [11,12]. Liu et al. reported that cesarean born/exclusively breastfed infants share a more similar gut microbiota with vaginally born/exclusively breastfed than cesarean born/mixed-fed infants [12]. Recently, a study showed that 2’-fucosyllactose (2’FL) as an indicator of maternal secretor status, might be involved in the restoration promoted by exclusive breastfeeding on the gut microbiota of cesarean born infants [13]. So far, this is the only evidence on the role of a human milk component in repairing the changes caused by cesarean section on the infant gut microbiota.
Human milk oligosaccharides (HMOs) comprise the third largest solid fraction of human milk, after lactose and lipids, containing more than 150 different molecules with a total concentration between 5 and 20 g/L in mature human milk [14–16]. HMOs are not digested by the infant and reach the colon intact, where they act as prebiotics, antimicrobials, prevent pathogen binding, and promote the gut barrier function [15]. The composition and concentrations of HMOs are highly variable and influenced by several maternal characteristics, especially the secretor status, determined by the activity of the FUT2 gene [17–19]. FUT2 encodes the enzyme fucosyltransferase 2, which adds a fucose residue in an α1–2 linkage to the HMO chain [20,21]. Secretor women have an active FUT2 enzyme and produce high amounts of α1–2 fucosylated HMOs, such as 2’FL and lacto-N-fucopentaose I (LNFP I).
In contrast, due to a single nucleotide polymorphism in FUT2, non-secretors produce no or only small amounts of α1–2 fucosylated HMOs [13]. Breastfed infants from secretor mothers are colonized earlier by Bifidobacterium and present higher amounts of this genus in the feces than non-secretor-fed infants [22,23]. Furthermore, isolated Bifidobacterium from secretor-fed infants can consume 2’FL and distinct sets of Bifidobacterium dominate the microbial community in secretor-fed and non-secretor-fed infants [23].
Therefore, we hypothesized that besides the birth mode, the maternal secretor status (or the presence of α1–2 fucosylated HMOs in breast milk) also influences the fecal microbiota of exclusively breastfed infants. The purpose of this study was to compare the microbiota composition of cesarean and vaginally born infants breastfed by secretor mothers.
Materials and methods
Participants and sample collection
The participants of the study were a subset of mother-infant pairs enrolled in a cross-sectional, observational study whose aim was to identify maternal and infant factors associated with HMOs concentrations [19]. Mothers and their infants were included at one month postpartum (median: 34 days, 25th-75th percentile: 25–45 days postpartum). Data and samples were collected on the same day of enrollment in the study. Healthy full-term (gestational age ≥ 37 weeks), singleton, exclusively breastfed infants were included. Infants that received antibiotic treatment, probiotics, water, or any other food besides human milk were not enrolled. The inclusion criteria for the present study were the availability of a human milk sample from the mother and a stool sample from the infant. From the 78 pairs included in the original cohort, 54 provided both samples and were selected for this subset (S1 Fig).
Human milk samples (5 to 15 mL) were obtained by manual expression of the breast during the morning (8:30–12:00 a.m.) and stored at −20°C until HMOs analysis. Infant feces were collected from disposable diapers and transferred to a microfuge tube containing 1 mL of the ASL buffer from the QIAamp DNA Stool Mini Kit (Qiagen, Hilden, Germany) and stored at −20°C until DNA extraction. Human milk and infant feces samples were collected on the same day.
Ethics approval and consent to participate
This study was approved by the Ethics Committee of the Universidade Federal de São Paulo (protocol No. 419.162) and complied with the Declaration of Helsinki. All mothers received detailed oral and written information about the study and voluntarily agreed to participate. Written informed consent was obtained from each participant before the data and sample collection.
Maternal secretor status determination
Maternal secretor status was determined based on the presence of α1–2 fucosylated structures in the human milk sample by liquid chromatography-mass spectrometry (LC-MS), as previously described in detail [24]. Briefly, after fat and protein removal, the human milk sample was diluted and subjected to a reduction reaction with 0.25M sodium borohydride. The resulting extract containing the HMOs fraction was injected into the LC-MS system for HMOs analysis. Mothers whose human milk sample presented at least one of the α1–2 fucosylated structures 2’-FL, LNFP I, lacto-N-difucohexaose I (LNDFH I), and difucosyllacto-N-hexaose c (DFLNHc) were ascribed as secretors, and those that did not present any of those HMOs up to quantifiable amounts (0.039 μg/mL for 2’-FL, LNDFH I and DFLNHc and 0.156 μg/mL for LNFP I) were ascribed as non-secretors.
Fecal microbiota analysis
Whole genomic DNA was extracted from feces using the QIAamp DNA Stool Mini Kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions. Purified DNA was diluted to a final volume of 200 μL, and DNA quantification was performed using a spectrophotometer, model Denovix DS-11 (Denovix, Delaware, USA). All DNA samples were diluted to a final concentration of 20 ng/μL and stored at -20°C.
The overall fecal microbiota composition was analyzed by 16S rRNA gene sequencing. The bacterial 16S rRNA gene, hypervariable region V4, was amplified by PCR using V4 primers with specific Illumina adaptors [25]. Amplification of the target sequences was performed in two stages, following the instructions of the Illumina protocol (Illumina®, California, United States). After amplification, the samples were grouped in equimolar amounts, with a final concentration of 12 pM and sequenced using the 500-cycle Miseq V2 Kit (Illumina®, California, United States), including 20% PhiX as an internal control for low libraries diversity. Besides, all reagents and ultrapure water were used as blank samples, and no signal was detected in the sequencing cartridge, indicating that bacterial contamination was minimal during library preparation and sequencing.
The main bacterial genera and species were quantified by qPCR using specific primers (S2 Table). The qPCR reactions were conducted on the Rotor-gene Q (Qiagen) equipment, in duplicate, in a final volume of 10 μL. The qPCR conditions were as follows: initial denaturation at 95°C for 5 minutes, 40 cycles at 95°C for 10 s, and a final stretching step at 60°C for 15 s. The dissociation cycle of the products for the melting curve was 95°C for 1 minute and a step to perform the denaturation curve with a variation of 70°C to 95°C, with a gradual increase in temperature of 1°C/s.
The standard curve for the quantification was created by TopoTA cloning plasmids (Invitrogen, Carlsbad, CA, USA), containing the reference gene fragment for each bacterium, previously amplified by PCR. With the molecular mass of the plasmid and the size of the insert, it was possible to calculate the number of copies of the genes according to the formula: mass in Daltons (g/mol) = (double-strand size [ds] amplicon in base pairs [bp]) (330 Da × 2 nucleotides [nt] / bp). In this way, dividing the concentration in g/mol by the Avogadro constant, the number of molecules/g was obtained, which is equal to the number of copies/g of the gene. As a negative control, samples containing all reagents were used, except for DNA.
From this information, it was possible to determine the number of copies of each gene, providing a standard for the construction of a reference curve for quantification by qPCR. The results of qPCR were expressed as bacterial units/g of feces (U/g of feces). The detection limit was 1 cell/g for all organisms.
Bioinformatics
The generated fragments were joined and analyzed using the software QIIME version 1.9.1 [26] with each unique sequence assigned to an operational taxonomic unit (OTU) based on ≥ 97% similarity by the UCLUST algorithm, representing the smallest taxonomic entity and classified phylogenetically using the ribosomal sequences from the SILVA reference database, version 128 [27].
The resulting OTU table was filtered to remove singletons and any OTU with an abundance of less than 0.05% across all samples. The median (25th-75th percentiles) number of sequences found in the samples was 90158 (66398–153979), and the minimum number of sequences in a sample was 7044 sequences. Alpha diversity was analyzed using the number of observed OTUs, Chao1, and Shannon indexes, without rarefaction [28]. Beta diversity was calculated using weighted and unweighted UniFrac distances [29].
Statistical analysis
Statistical tests were performed to compare the microbiota composition of infants from secretor mothers, stratified in cesarean (CSe+, n = 27) and vaginally born (VSe+, n = 21). Non-secretor mothers and their infants were not included in the statistical analysis of the microbiota, but their data were described.
UniFrac distances were associated with birth mode using the method Adonis in QIIME [22]. Analysis of Covariance (ANCOVA) and non-parametric Analysis of Covariance (Ranked ANCOVA or Quade test [30]) were used to verify differences in α-diversity indices, relative abundances of the most abundant phyla and genera, and species absolute concentrations between CSe+ and VSe+ with age at inclusion as a covariate, according to the distribution of each variable. We used a free web program (www.masungur.com) to perform the ranked ANCOVA [31]. Chi-square test or Fisher’s exact test were used to compare the prevalence of bacterial genera and species between the groups. Clinical and demographic characteristics of the study population and the concentrations of α1–2 fucosylated HMOs were described using mean (SD), median (p25 –p75) or proportions and compared using Student’s t-test, Mann-Whitney rank-sum test, Chi-square test, or Fisher’s exact test according to the distribution and nature of the variables. Zero/non-detected levels were included in the comparisons of qPCR data and missing data were reported. Analyses were conducted with the software SigmaPlot version 12.0 (Systat Software, Inc., Chicago, IL) or R version 3.4.4 (The R Foundation for Statistical Computing, Vienna, Austria). The p-values for α-diversity indices, relative abundances of the most abundant phyla and genera, and species absolute concentrations were adjusted for multiple comparisons using the Benjamini and Hochberg method [32] to control the false discovery rate. Results were considered statistically significant when p < 0.05.
Results
Table 1 shows the characteristics of the study population according to the mode of birth and maternal secretor status. No significant differences were observed in clinical and demographic characteristics between the four groups (Table 1).
Table 1. Characteristics of the mothers and their exclusively breastfed infants according to the mode of birth and maternal secretor status.
| VSe+ (n = 21) | CSe+ (n = 27) | VSe- (n = 4) | CSe- (n = 2) | p | |
|---|---|---|---|---|---|
| Mothers | |||||
| Age, years (mean (SD)) | 31 (6) | 31 (7) | 31 (6) | 26 (2) | 0.738f |
| BMI, kg/m2 (median (p25 –p75)) | |||||
| Pre-gestational | 24.8 (22.4–27.9) | 23.6 (21.5–30.6) | 22.5 (21.3–22.7) | 22.5 (21.5–23.5) | 0.403g |
| At inclusion | 26.8 (24.4–29.0) | 25.8 (23.3–31.2) | 23.3 (22.1–24.8) | 25.2 (24.3–26.2) | 0.446g |
| Parity, n (median (p25 –p75)) | 2 (1–2) | 1 (1–2) | 2 (1–2) | 1 (1–2) | 0.535g |
| Allergic disease, n (%) | 6 (29) | 5 (19) | 0 (0) | 0 (0) | >0.498h |
| Education, n (%) a | |||||
| Elementary school | 2 (11) | 2 (8) | 1 (25) | 0 (0) | 1.000h |
| High school | 9 (47) | 6 (25) | 2 (50) | 2 (100) | >0.086h |
| Graduate | 5 (26) | 11 (46) | 1 (25) | 0 (0) | >0.221h |
| Postgraduate | 1 (5) | 5 (21) | 0 (0) | 0 (0) | >0.205h |
| Infants | |||||
| Gestational age at birth, weeks (mean (SD)) | 38.90 (1.10) | 38.97 (1.40) | 38.57 (1.41) | 38.78 (0.71) | 0.960f |
| Birth weight, g (mean (SD)) | 3097.50 (731.90) | 3179.77 (436.62) | 3232.50 (202.05) | 3097.50 (731.86) | 0.922f |
| Age at inclusion, days (mean (SD)) | 32.4 (10.7) | 40.0 (14.7) | 28.8 (5.9) | 33.5 (19.1) | 0.151f |
| Weight at inclusion, g (median (p25 –p75)) | 4177.50 (2690.00–5665.00) | 4250.00 (3677.50–5002.50) | 4192.50 (4037.50–4345.00) | 4177.50 (2690.00–5665.00) | 0.990g |
| Length at inclusion, g (mean (SD)) | 53.7 (2.4) | 54.0 (3.3) | 53.5 (1.3) | 52.8 (5.3) | 0.937f |
| Weight gain, g/day (mean (SD)) b | 28.49 (14.82) | 26.89 (11.98) | 32.35 (8.84) | 24.56 (26.95) | 0.864f |
| Sex, n (%) c | |||||
| Male | 8 (38) | 14 (54) | 2 (50) | 1 (50) | >0.380h |
| Female | 13 (62) | 12 (46) | 2 (50) | 1 (50) | >0.380h |
| Household | |||||
| Household with siblings, n (%) d | 10 (53) | 7 (33) | 3 (75) | 1 (50) | >0.270h |
| Household with pets, n (%) e | 7 (37) | 6 (30) | 2 (50) | 1 (50) | >0.580h |
VSe+: Vaginally born, secretor; CSe+: Cesarean born, secretor; VSe-: Vaginally born, non-secretor; CSe-: Cesarean born, non-secretor
a missing data from 2 mothers from group VSe+ and 3 mothers from group CSe+
b Weight gain = (weight at inclusion—birth weight)/age at inclusion
c missing data from 1 infant from group CSe+
d missing data from 2 infants from group VSe+ and 6 infants from group CSe+
e missing data from 2 mother-infant pairs from VSe+ and 7 pairs from group CSe+
f One-way ANOVA
g Kruskal-Wallis test
h Fisher’s Exact Test.
The median (25th-75th percentile) amount of LNFP I was significantly lower in the milk from mothers of cesarean born infants than in the milk from mothers of vaginally born infants (0.511 (0.269–0.962) g/L vs. 0.736 (0.444–1.564) g/L, respectively; p = 0.025). No significant difference was observed in 2’-FL or the other α1–2 fucosylated HMOs amounts between the groups (S1 Table).
The overall microbiota composition did not differ between CSe+ and VSe+ infants when comparing the unweighted (p = 0.75) and weighted (p = 0.87) UniFrac distances using the method Adonis (Fig 1A and 1B). The number of observed OTUs, the richness estimator (Chao1), and the diversity index (Shannon) also did not differ between CSe+ and VSe+ infants, as shown in Table 2.
Fig 1.
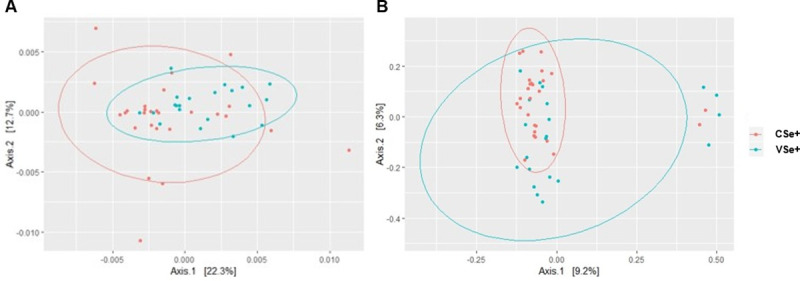
Principal coordinate analysis (PCoA) of the overall microbiota composition of exclusively breastfed infants from secretor mothers by birth mode using (A) unweighted and (B) weighted UniFrac distances. VSe+: vaginal, secretor; CSe+: cesarean, secretor.
Table 2. Fecal microbiota alpha diversity from vaginally and cesarean born, exclusively breastfed infants from secretor mothers.
| Index | VSe+ (n = 21) | CSe+ (n = 27) | p* |
|---|---|---|---|
| Observed OTUs | 595.381 ± 151.699 | 577.889 ± 214.780 | 0.841 |
| Chao 1 | 876.230 ± 169.751 | 866.394 ± 282.444 | 0.988 |
| Shannon | 1.871 ± 0.412 | 2.032 ± 0.483 | 0.394 |
Values are presented in mean ± SD; VSe+: Vaginally born, secretor; CSe+: Cesarean born, secretor.
*p-values were adjusted for age at inclusion using ANCOVA (analysis of covariance).
The relative abundance of the most abundant bacterial phyla was not different in CSe+ and VSe+ infants, except for Bacteroidetes and Verrucomicrobia (Table 3, Fig 2A). The relative abundances of the most abundant bacterial phyla and genera of cesarean born, non-secretor fed infants (CSe-, n = 2) and vaginally born, non-secretor fed infants (VSe-, n = 4) are presented in Fig 2 for descriptive purposes and were not included in the statistical analysis.
Table 3. The average relative abundance of the most abundant fecal bacterial groups from vaginally and cesarean born, exclusively breastfed infants from secretor mothers.
| Group | Average relative abundance (%) | p | |
|---|---|---|---|
| VSe+ | CSe+ | ||
| Phyla | |||
| Actinobacteria | 19.75 | 13.25 | 0.185a |
| Bacteroidetes | 13.81 | 1.45 | 0.003*b |
| Firmicutes | 28.43 | 35.36 | 0.465a |
| Proteobacteria | 37.88 | 48.73 | 0.094a |
| Verrucomicrobia | 0.00 | 1.08 | 0.030*b |
| Genera | |||
| Akkermansia | 0.00 | 1.18 | 0.030*b |
| Bacteroides | 13.94 | 1.32 | 0.006*b |
| Bifidobacterium | 17.43 | 13.48 | 0.342b |
| Clostridium sensu stricto 1 | 9.99 | 8.47 | 0.149b |
| Kluyvera | 13.13 | 33.82 | 0.001*b |
| Lactobacillus | 0.87 | 2.99 | 0.283b |
| Serratia | 19.78 | 13.91 | 0.950b |
| Streptococcus | 5.50 | 4.66 | 0.975b |
| Veillonella | 9.38 | 12.51 | 0.093b |
The most abundant taxa were those with an average relative abundance ≥ 1%; VSe+: Vaginally born, secretor; CSe+: Cesarean born, secretor. p-values were adjusted for multiple comparisons using the Benjamini and Hochberg method
a adjusted for age at inclusion using ANCOVA (analysis of covariance)
b adjusted for age at inclusion using a non-parametric ANCOVA (ranked analysis of covariance or Quade test)
*p < 0.05.
Fig 2.

Average relative abundances of the most abundant bacterial phyla (A) and genera (B) of the infant fecal microbiota according to the birth mode and maternal secretor phenotype. The taxa with an average relative abundance < 1% were removed and those with an average relative abundance ≥ 1% were set to 100%; VSe+: Vaginally born, secretor; CSe+: Cesarean born, secretor; VSe-: Vaginally born, non-secretor; CSe-: Cesarean born, non-secretor.
Bacteroidetes and its main genus Bacteroides (Fig 3H) were significantly lower, whereas Verrucomicrobia and its main genus Akkermansia (Fig 3I) were significantly higher in CSe+ infants. The prevalence of Verrucomicrobia was significantly higher in CSe+ infants when compared to the VSe+ (29.6% and 4.8%, respectively, Fisher’s exact test, p < 0.001). A high abundance of Proteobacteria was observed in both CSe+ (> 40%) and VSe+ (> 30%), of which Serratia and Kluyvera were the most abundant genera. While no differences were observed in Serratia abundance between the groups, CSe+ infants presented a significantly higher Kluyvera abundance (Table 3, Fig 3G). There were no significant differences in the average relative abundance of other bacterial genera between CSe+ and VSe+ (Fig 3A–3F).
Fig 3. The average relative abundance of the most abundant bacterial genera in infants from secretor mothers, by birth mode.

VSe+: Vaginally born (blue); CSe+: Cesarean born (yellow). The asterisks indicate statistical significance at p < 0.05.
In addition to the relative abundances of the main phyla and genera, there were no differences in the absolute amounts measured by qPCR of six Bifidobacterium species between CSe+ and VSe+, except for Bifidobacterium longum (Table 4). However, CSe+ group presented a lower (but not significant) prevalence of B. longum than VSe+ (22.2% vs. 52.4%, respectively; p = 0.062), and differences in absolute amounts between CSe+ and VSe+ were no longer significant when considering only the samples that presented a quantifiable amount of B. longum (6.00×105 cells/g vs. 6.66 ×105 cells/g, respectively; p = 0.725).
Table 4. Prevalence and absolute amounts of selected species obtained by qPCR from the feces of vaginally and cesarean born, exclusively breastfed infants from secretor mothers.
| Species | Prevalence, n (%) | Absolute amounts, cells/g (median (p25 –p75)) k | ||||
|---|---|---|---|---|---|---|
| VSe+ (n = 21) | CSe+ (n = 27) | p | VSe+ (n = 21) | CSe+ (n = 27) | p l | |
| Bifidobacterium infantis | 11 (52.4) | 13 (48.15) | 1.000 i | 4.00 ×104 (0–1.55 ×105) | 0 (0–1.08 ×105) | 0.950 |
| Bifidobacterium longum | 11 (52.4) | 6 (22.2) | 0.062 i | 2.94 ×104 (0–8.67 ×105) | 0 (0–0) | 0.009* |
| Bifidobacterium bifidum a | 20 (100.0) | 21 (100.0) | NA | 7.76 ×107 (4.51 ×106–3.34 ×108) | 3.65 ×107 (4.71 ×105–1.56 ×108) | 0.708 |
| Bifidobacterium breve b | 3 (14.3) | 6 (23.1) | 0.711 j | 0 (0–0) | 0 (0–8.70 ×102) | 0.342 |
| Bifidobacterium catenulatum c | 1 (4.8) | 1 (4.0) | 1.000 j | 0 (0–0) | 0 (0–0) | 0.903 |
| Bifidobacterium adolescentis d | 12 (60.0) | 14 (58.3) | 0.845 j | 2.70 ×104 (0–1.73 ×107) | 2.58 ×104 (0–2.39 ×106) | 0.769 |
| Bacteroides fragilis e | 7 (35.0) | 0 (0.0) | 0.003* j | 0 (0–2.48 ×106) | NA | NA |
| Clostridioides difficile f | 5 (25.0) | 9 (45.0) | 0.320 j | 0 (0–3.84 ×104) | 0 (0–1.16 ×104) | 0.229 |
| Escherichia coli g | 16 (80.0) | 14 (66.7) | 0.541 j | 1.86 ×106 (4.12 ×103–1.79 ×107) | 5.01 ×104 (0–5.15 ×106) | 0.820 |
| Methanobrevibacter smithii h | 6 (30.0) | 14 (61.0) | 0.086 j | 0 (0–1.04 ×104) | 2.30 × 103 (0–3.39 ×104) | 0.071 |
VSe+: Vaginally born, secretor; CSe+: Cesarean born, secretor; NA: Not applicable
a Missing data from 1 infant from VSe+ group and 6 infants from CSe+ group
b Missing data from 1 infant from group CSe+
c Missing data from 2 infants from group CSe+
d Missing data from 1 infant from VSe+ group and 3 infants from CSe+ group
e Missing data from 1 infant from VSe+ group and 5 infants from CSe+ group
f Missing data from 1 infant from VSe+ group and 7 infants from CSe+ group
g Missing data from 1 infant from VSe+ group and 6 infants from CSe+ group
h Missing data from 1 infant from VSe+ group and 4 infants from CSe+ group
i Chi-square test
j Fisher exact test
k Zero/non-detected levels were included as zero values in the analysis. The detection limit was 1 cell/g for all organisms
l p-values were adjusted for multiple comparisons using the Benjamini and Hochberg method and for age at inclusion using a non-parametric ANCOVA (ranked analysis of covariance or Quade test)
*p<0.05.
There were no significant differences in the prevalence and amounts of other bacterial species of clinical relevance such as Escherichia coli, Clostridioides difficile and Methanobrevibacter smithii (Table 4). The prevalence of Bacteroides fragilis was significantly lower in CSe+ than VSe+ infants (0.0% vs. 35.0%, respectively; p = 0.003), since it was not detected in any fecal sample from CSe+ infants, corroborating the 16S sequencing results.
Discussion
In this study, we compared the fecal microbiota of 48 full-term, healthy, exclusively breastfed infants from secretor mothers, according to the mode of birth. Cesarean born infants presented lower abundances of Bacteroides, lower prevalence of B. longum and higher abundances of Akkermansia and Kluyvera. The overall microbiota composition (alpha and beta diversity) was not different between cesarean and vaginally born infants breastfed by secretor mothers.
On the contrary to previous studies comparing the fecal microbiota of vaginally and cesarean born infants without considering the maternal secretor status [6,7], we did not observe differences in Bifidobacterium relative abundances between CSe+ and VSe+ infants. Besides the relative amounts obtained by 16S sequencing, we measured absolute numbers of some clinically relevant groups through qPCR, including six Bifidobacterium species, of which a difference between CSe+ and VSe+ infants was observed only in the concentration of B. longum. Previous studies reported that the presence of α1–2 fucosylated HMOs in breast milk (or maternal Se+ phenotype) affects Bifidobacterium establishment and abundance in the infant’s gut, being higher in secretor-fed infants [22,23]. This may explain the lack of difference in the relative abundances and counts of Bifidobacterium between CSe+ and VSe+ in our study. Bifidobacterium is essential for inhibiting the growth of pathogenic organisms, modulating mucosal barrier function, and promoting immunological and inflammatory responses in the infant’s gut [33,34]. The perturbance of Bifidobacterium establishment during the neonatal period may be involved in the development of immune diseases, such as eczema [35]. Bifidobacterium is also a component of the human milk microbiota [36], and its occurrence in the fecal microbiota of CSe+ infants observed in our study suggests that human milk may be an important source of Bifidobacterium besides the birth canal, contributing to the establishment of this genus in the infant gut microbiota.
Our results broadly support the findings of the recent and only previous published study, conducted in Finland, about the effects of maternal secretor phenotype on the microbiota of cesarean and vaginally born infants [13]. The authors reported that cesarean born infants from Se+ mothers presented a more modest deviation in the overall microbiota composition, compared to those of non-secretor mothers. Similar to our findings, the authors reported that CSe+ had significantly lower abundances of Bacteroidetes and Bacteroides and significantly higher abundances of Verrucomicrobia and Akkermansia muciniphila in their gut microbiota, compared to VSe+ infants. Considering that ethnicity and environmental factors are strongly associated with the microbial composition and function in healthy individuals [37], it is remarkable that similar associations were observed in such distinct populations as the Nordic and Brazilian. Differently, they found significantly lower Actinobacteria and Bifidobacterium and higher Firmicutes abundances in CSe+ compared to VSe+, which were not observed in our cohort [13].
The significantly higher abundance of Verrucomicrobia and its main genus Akkermansia observed only in CSe+ infants is a curious finding of both studies. Akkermansia is a mucin-degrading bacterium present in the human intestinal tract, which is involved in immune regulation and promotes the gut barrier function [38]. Lower abundance and prevalence of Akkermansia have been associated with obesity and related metabolic complications [39] and also with allergic diseases in children, such as atopy and asthma [40,41]. Akkermansia is also a component of the breast milk microbiota, and positive correlations between α1–2 fucosylated HMOs and Akkermansia muciniphila in human colostrum have been reported, as well as the ability of Akkermansia to degrade HMOs [42]. Interestingly, a study showed that CSe+ infants with high hereditary allergy risk, but who did not develop IgE allergic disease and IgE eczema at two years, consumed breast milk with higher levels of 2’-FL, suggesting a protective role of α1–2 fucosylated HMOs in the manifestation of atopy [43]. A possible explanation for this potential lower allergy risk in CSe+ infants is the higher abundance of Bifidobacterium and Akkermansia in the microbiota of CSe+ infants demonstrated in our study.
Some studies have reported impairment on Bacteroides transmission from mother to child by cesarean section [6–8,44], which remains absent from the microbiota at 12 months of age [7]. Interestingly, we also observed a significantly lower abundance of Bacteroides and the absence of B. fragilis in CSe+ compared to VSe+ infants, indicating that even exclusive breastfeeding and the α1–2 fucosylated HMOs cannot repair the disruption on Bacteroides transmission from mother to child caused by cesarean section. Cesarean birth has been associated with neurodevelopmental and psychiatric disorders, such as autism spectrum disorder and attention-deficit/hyperactivity disorder [45]. Curiously, Bacteroides is a producer of the neurotransmitter γ-aminobutyric acid (GABA)–whose misregulation has been linked to mental diseases–and low fecal Bacteroides levels were associated with brain signatures of depression [46]. In an experimental model, oral treatment with Bacteroides fragilis attenuated the behavioral symptoms of autism [47]. Besides neurological effects, an association among cesarean birth, delayed Bacteroidetes colonization, and reduced Th1 responses have also been described [44]. Further studies are needed to investigate the clinical consequences of Bacteroides deprivation at the neonatal period and early infancy.
We observed a high abundance of Proteobacteria in our cohort, of which Serratia and Kluyvera were the main genera. While Proteobacteria is one of the main phyla commonly reported in infant fecal samples [7,13], high abundances of Serratia and Kluyvera have not been reported in the literature. Serratia has been described as one of the dominant bacterial genera in human milk [48] and was reported to be highly abundant in breast milk from women with mastitis [49]. Kluyvera has been reported as a cause of pediatric infections [50,51]. Previous studies conducted in Brazil found a predominance of Escherichia in the fecal microbiota of healthy, vaginally born, one-month-old exclusively breastfed infants from low socioeconomic levels [52,53]. Besides Escherichia, the authors did not observe significant amounts of other members of the Proteobacteria phylum [52]. This difference to our study may be attributed to the middle-high socioeconomic level of our cohort, which possibly determines a different environmental exposure since the infants from these studies were also exclusively breastfed. However, the relative abundances of other principal members of the infant microbiota reported by these previous studies were very similar to the VSe+ group of our research, especially Bacteroides (~10%), Veillonella (~10%), and Streptococcus (~5%) [52].
Another novel finding of our study is the occurrence of Methanobrevibacter smithii on the microbiota of exclusively breastfed infants, independent of the birth mode. In previous studies, our group reported higher concentrations of M. smithii in the feces of school-aged children living near a landfill, from families of low socioeconomic level and poor sanitary housing conditions [54,55]. In the present study, we demonstrate that M. smithii is already present in the neonatal period in exclusively breastfed infants, but in lower levels than in school-aged children.
The limitations of the study include the absence of a bead-beating step during the DNA extraction, which may lead to an under-detection of Bifidobacterium. However, Bifidobacterium was still detected in high abundances, composing one of the main bacterial genera, both in VSe+ and CSe+ infants. The low prevalence of the non-secretor phenotype required a larger sample to compare the four groups according to the birth mode and maternal secretor status. Because of the cross-sectional design of the study, we were unable to ascertain whether the partial microbiota recovery in CSe+ infants is sustained or if other changes occur over time. Although the exposure to antibiotics was an exclusion criterion for the infants, we did not collect data on the maternal use of intra and postpartum antibiotics, which can be considered a limitation. Among the strengths of our study is our cohort, composed of healthy and exclusively breastfed infants, avoiding the bias on microbiota composition introduced by mixed feeding. Other strengths include the identification of the maternal secretor phenotype through the fucosylated HMOs profile and the use of high-throughput genetic sequencing complemented with the absolute quantification of the main bacterial genera and species by qPCR to profile the infant gut microbiota. Besides, we obtained a more balanced distribution of cesarean and vaginally born infants relative to previous studies.
In conclusion, our results show that there is no difference in the overall gut microbiota composition (alpha and beta diversity) and Bifidobacterium amounts between vaginally and cesarean born infants fed with breast milk containing the α1–2 fucosylated HMOs fraction. Differences between VSe+ and CSe+ infants were still observed in the abundances of the phyla Bacteroidetes and Verrucomicrobia, in the genera Bacteroides, Akkermansia, and Kluyvera and the species B. longum. Further studies are necessary to investigate the potential benefits of the HMOs on the microbiota of cesarean born infants.
Supporting information
CIAAM/UNIFESP, Breastfeeding Incentive and Support Center/Universidade Federal de São Paulo.
(PDF)
Values are presented as median (p25 –p75) unless otherwise indicated; * statistical comparisons between VSe+ and CSe+; 2’-FL: 2’-fucosyllactose; LNFP I: lacto-N-fucopentaose I; LNDFH I: lacto-N-difucohexaose I; DFLNHc: difucosyllacto-N-hexaose c; a breast milk from these groups presented levels of α1–2 fucosylated HMOs below the quantification limit; b quantification limit: 0.000039 g/L; c quantification limit: 0.000156 g/L; d Mann-Whitney rank sum test; e Student’s t test.
(PDF)
(PDF)
Acknowledgments
We are grateful to all the families who took part in this study for providing data and the human milk and fecal samples. We also thank Adriana Sañudo for the statistical assistance and the anonymous reviewers for their contribution to the improvement of the manuscript.
Data Availability
The dataset analyzed during the current study is available in the Sequence Read Archive repository, under accession number PRJNA615250 [https://www.ncbi.nlm.nih.gov/sra/PRJNA615250].
Funding Statement
KMT received funds from the National Council for Scientific and Technological Development (grant #142327/2013-1) and TBM received funds from the São Paulo Research Foundation (grant #2014/13514-5) to support this research. The URL for the National Council for Scientific and Technological Development is as follows: http://www.cnpq.br/. The URL for the São Paulo Research Foundation is as follows: http://www.fapesp.br/. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Thavagnanam S, Fleming J, Bromley A, Shields MD, Cardwell CR. A meta-analysis of the association between Caesarean section and childhood asthma. Clin Exp Allergy. 2008;38(4):629–33. 10.1111/j.1365-2222.2007.02780.x [DOI] [PubMed] [Google Scholar]
- 2.Sevelsted A, Stokholm J, Bonnelykke K, Bisgaard H. Cesarean Section and Chronic Immune Disorders. Pediatrics [Internet]. 2015. January 1;135(1):e92–8. Available from: http://pediatrics.aappublications.org/cgi/doi/10.1542/peds.2014-0596. [DOI] [PubMed] [Google Scholar]
- 3.Cai M, Loy SL, Tan KH, Godfrey KM, Gluckman PD, Chong Y-S, et al. Association of Elective and Emergency Cesarean Delivery With Early Childhood Overweight at 12 Months of Age. JAMA Netw Open [Internet]. 2018. November 21;1(7):e185025 Available from: http://jamanetworkopen.jamanetwork.com/article.aspx?doi=10.1001/jamanetworkopen.2018.5025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tun HM, Bridgman SL, Chari R, Field CJ, Guttman DS, Becker AB, et al. Roles of Birth Mode and Infant Gut Microbiota in Intergenerational Transmission of Overweight and Obesity From Mother to Offspring. JAMA Pediatr [Internet]. 2018. April 1;172(4):368 Available from: http://archpedi.jamanetwork.com/article.aspx?doi=10.1001/jamapediatrics.2017.5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sutharsan R, Mannan M, Doi SA, Mamun AA. Caesarean delivery and the risk of offspring overweight and obesity over the life course: a systematic review and bias-adjusted meta-analysis. Clin Obes [Internet]. 2015. December;5(6):293–301. Available from: 10.1111/cob.12114 [DOI] [PubMed] [Google Scholar]
- 6.Penders J, Thijs C, Vink C, Stelma FF, Snijders B, Kummeling I, et al. Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics [Internet]. 2006. August [cited 2013 Aug 1];118(2):511–21. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16882802. 10.1542/peds.2005-2824 [DOI] [PubMed] [Google Scholar]
- 7.Shao Y, Forster SC, Tsaliki E, Vervier K, Strang A, Simpson N, et al. Stunted microbiota and opportunistic pathogen colonization in caesarean-section birth. Nature [Internet]. 2019. September 18; Available from: 10.1016/j.jogc.2019.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bäckhed F, Roswall J, Peng Y, Feng Q, Jia H, Kovatcheva-Datchary P, et al. Dynamics and Stabilization of the Human Gut Microbiome during the First Year of Life. Cell Host Microbe [Internet]. 2015. May;17(5):690–703. Available from: http://linkinghub.elsevier.com/retrieve/pii/S1931312815001626. [DOI] [PubMed] [Google Scholar]
- 9.Stokholm J, Thorsen J, Chawes BL, Schjørring S, Krogfelt KA, Bønnelykke K, et al. Cesarean section changes neonatal gut colonization. J Allergy Clin Immunol [Internet]. 2016. September;138(3):881–889.e2. Available from: 10.1016/j.jaci.2016.01.028 [DOI] [PubMed] [Google Scholar]
- 10.Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci [Internet]. 2010. June 29;107(26):11971–5. Available from: http://www.pnas.org/cgi/doi/10.1073/pnas.1002601107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hill CJ, Lynch DB, Murphy K, Ulaszewska M, Jeffery IB, O’Shea CA, et al. Evolution of gut microbiota composition from birth to 24 weeks in the INFANTMET Cohort. Microbiome [Internet]. 2017. December 17;5(1):4 Available from: http://microbiomejournal.biomedcentral.com/articles/10.1186/s40168-016-0213-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu Y, Qin S, Song Y, Feng Y, Lv N, Xue Y, et al. The perturbation of infant gut microbiota caused by cesarean delivery is partially restored by exclusive breastfeeding. Front Microbiol. 2019;10(MAR):1–11. 10.3389/fmicb.2019.00598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Korpela K, Salonen A, Hickman B, Kunz C, Sprenger N, Kukkonen K, et al. Fucosylated oligosaccharides in mother’s milk alleviate the effects of caesarean birth on infant gut microbiota. Sci Rep [Internet]. 2018. December 13;8(1):13757 Available from: http://www.nature.com/articles/s41598-018-32037-6. 10.1038/s41598-018-32037-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elwakiel M, Hageman JA, Wang W, Szeto IM, van Goudoever JB, Hettinga KA, et al. Human Milk Oligosaccharides in Colostrum and Mature Milk of Chinese Mothers: Lewis Positive Secretor Subgroups. J Agric Food Chem [Internet]. 2018. July 11;66(27):7036–43. Available from: http://pubs.acs.org/doi/10.1021/acs.jafc.8b02021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bode L. Human milk oligosaccharides: every baby needs a sugar mama. Glycobiology [Internet]. 2012. September [cited 2013 May 27];22(9):1147–62. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22513036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thurl S, Munzert M, Boehm G, Matthews C, Stahl B. Systematic review of the concentrations of oligosaccharides in human milk. Nutr Rev [Internet]. 2017. November 1;75(11):920–33. Available from: http://academic.oup.com/nutritionreviews/article/doi/10.1093/nutrit/nux044/4558511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kunz C, Meyer C, Collado MC, Geiger L, García-Mantrana I, Bertua-Ríos B, et al. Influence of Gestational Age, Secretor, and Lewis Blood Group Status on the Oligosaccharide Content of Human Milk. J Pediatr Gastroenterol Nutr [Internet]. 2017. May;64(5):789–98. Available from: http://journals.lww.com/jpgn%0Ahttp://ovidsp.ovid.com/ovidweb.cgi?T=JS&PAGE=reference&D=emex&NEWS=N&AN=612076848. [DOI] [PubMed] [Google Scholar]
- 18.Azad MB, Robertson B, Atakora F, Becker AB, Subbarao P, Moraes TJ, et al. Human Milk Oligosaccharide Concentrations Are Associated with Multiple Fixed and Modifiable Maternal Characteristics, Environmental Factors, and Feeding Practices. J Nutr [Internet]. 2018. November 1;148(11):1733–42. Available from: https://academic.oup.com/jn/advance-article/doi/10.1093/jn/nxy175/5105883. [DOI] [PubMed] [Google Scholar]
- 19.Tonon KM, de Morais MB, Abrão ACFV, Miranda A, Morais TB. Maternal and Infant Factors Associated with Human Milk Oligosaccharides Concentrations According to Secretor and Lewis Phenotypes. Nutrients [Internet]. 2019. June 17;11(6):1358 Available from: 10.3390/nu11061358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kumazaki T, Yoshida A. Biochemical evidence that secretor gene, Se, is a structural gene encoding a specific fucosyltransferase. Proc Natl Acad Sci [Internet]. 1984. July 1;81(13):4193–7. Available from: http://www.pnas.org/cgi/doi/10.1073/pnas.81.13.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson PH, Watkins WM. Purification of the Lewis blood-group gene associated a-3/4-fucosyltransferase from human milk: an enzyme transferring fucose primarily to Type 1 and lactose-based oligosaccharide chains. Glycoconj J [Internet]. 1992. October;9(5):241–9. Available from: http://link.springer.com/10.1007/BF00731136. [DOI] [PubMed] [Google Scholar]
- 22.Smith-Brown P, Morrison M, Krause L, Davies PSW. Mothers Secretor Status Affects Development of Childrens Microbiota Composition and Function: A Pilot Study. PLoS One [Internet]. 2016;11(9):e0161211 Available from: 10.1371/journal.pone.0161211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lewis ZT, Totten SM, Smilowitz JT, Popovic M, Parker E, Lemay DG, et al. Maternal fucosyltransferase 2 status affects the gut bifidobacterial communities of breastfed infants. Microbiome [Internet]. 2015. December 10;3(1):13 Available from: http://www.microbiomejournal.com/content/3/1/13. 10.1186/s40168-015-0071-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tonon KM, Miranda A, Abrão ACFV, de Morais MB, Morais TB. Validation and application of a method for the simultaneous absolute quantification of 16 neutral and acidic human milk oligosaccharides by graphitized carbon liquid chromatography–electrospray ionization–mass spectrometry. Food Chem [Internet]. 2019; Available from: http://www.scopus.com/inward/record.url?eid=2-s2.0-85053204722&partnerID=MN8TOARS. 10.1016/j.foodchem.2018.09.036 [DOI] [PubMed] [Google Scholar]
- 25.Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. Development of a Dual-Index Sequencing Strategy and Curation Pipeline for Analyzing Amplicon Sequence Data on the MiSeq Illumina Sequencing Platform. Appl Environ Microbiol [Internet]. 2013. September 1;79(17):5112–20. Available from: 10.1128/AEM.01043-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods [Internet]. 2010. May 11;7(5):335–6. Available from: 10.1038/nmeth.f.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res [Internet]. 2012. November 27;41(D1):D590–6. Available from: 10.1093/nar/gks1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McMurdie PJ, Holmes S. Waste Not, Want Not: Why Rarefying Microbiome Data Is Inadmissible. McHardy AC, editor. PLoS Comput Biol [Internet]. 2014. April 3;10(4):e1003531 Available from: https://dx.plos.org/10.1371/journal.pcbi.1003531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lozupone C, Knight R. UniFrac: a New Phylogenetic Method for Comparing Microbial Communities. Appl Environ Microbiol [Internet]. 2005. December 1;71(12):8228–35. Available from: 10.1128/AEM.71.12.8228-8235.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cangür Ş, Sungur MA, Ankarali H. The Methods Used in Nonparametric Covariance Analysis. Duzce Tıp Fak Derg. 2018;20(1):1–6. [Google Scholar]
- 31.Cangür Ş, Sungur MA, Ankarali H. A web-based program for the Quade, Puri & Sen, and McSweeny & Porter ranked ANCOVA methods (post hoc Tukey-Kramer test) for one-factor covariance model with single-covariate [Internet]. [cited 2019 Jun 7]. Available from: http://www.masungur.com/nancova0.php. [Google Scholar]
- 32.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J R Stat Soc Ser B [Internet]. 1995. January;57(1):289–300. Available from: http://www.jstor.org/stable/2346101. [Google Scholar]
- 33.Turroni F, Duranti S, Bottacini F, Guglielmetti S, Van Sinderen D, Ventura M. Bifidobacterium bifidum as an example of a specialized human gut commensal. Front Microbiol [Internet]. 2014;5(August):1–8. Available from: http://journal.frontiersin.org/Journal/10.3389/fmicb.2014.00437/full. 10.3389/fmicb.2014.00437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hidalgo-Cantabrana C, Delgado S, Ruiz L, Ruas-Madiedo P, Sánchez B, Margolles A. Bifidobacteria and Their Health-Promoting Effects. Microbiol Spectr [Internet]. 2017. June 8;5(3):1–19. Available from: http://www.asmscience.org/content/journal/microbiolspec/10.1128/microbiolspec.BAD-0010-2016. 10.1128/microbiolspec.BAD-0010-2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hong P-Y, Lee BW, Aw M, Shek LPC, Yap GC, Chua KY, et al. Comparative Analysis of Fecal Microbiota in Infants with and without Eczema. Yang C-H, editor. PLoS One [Internet]. 2010. April 1;5(4):e9964 Available from: https://dx.plos.org/10.1371/journal.pone.0009964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murphy K, Curley D, O’Callaghan TF, O’Shea C-A, Dempsey EM, O’Toole PW, et al. The Composition of Human Milk and Infant Faecal Microbiota Over the First Three Months of Life: A Pilot Study. Sci Rep [Internet]. 2017. January 17;7(October 2016):40597 Available from: http://www.nature.com/articles/srep40597. 10.1038/srep40597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huttenhower C, Gevers D, Knight R, Abubucker S, Badger JH, Chinwalla AT, et al. Structure, function and diversity of the healthy human microbiome. Nature [Internet]. 2012. June 13;486(7402):207–14. Available from: 10.1038/nature11234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Geerlings S, Kostopoulos I, de Vos W, Belzer C. Akkermansia muciniphila in the Human Gastrointestinal Tract: When, Where, and How? Microorganisms [Internet]. 2018. July 23;6(3):75 Available from: http://www.mdpi.com/2076-2607/6/3/75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Derrien M, Belzer C, de Vos WM. Akkermansia muciniphila and its role in regulating host functions. Microb Pathog [Internet]. 2017. May;106:171–81. Available from: 10.1016/j.micpath.2016.02.005 [DOI] [PubMed] [Google Scholar]
- 40.Fujimura KE, Sitarik AR, Havstad S, Lin DL, Levan S, Fadrosh D, et al. Neonatal gut microbiota associates with childhood multisensitized atopy and T cell differentiation. Nat Med [Internet]. 2016;22(10):1187–91. Available from: 10.1038/nm.4176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Demirci M, Tokman HB, Uysal HK, Demiryas S, Karakullukcu A, Saribas S, et al. Reduced Akkermansia muciniphila and Faecalibacterium prausnitzii levels in the gut microbiota of children with allergic asthma. Allergol Immunopathol (Madr) [Internet]. 2019. July;47(4):365–71. Available from: 10.1016/j.aller.2018.12.009 [DOI] [PubMed] [Google Scholar]
- 42.Aakko J, Kumar H, Rautava S, Wise A, Autran C, Bode L, et al. Human milk oligosaccharide categories define the microbiota composition in human colostrum. Benef Microbes [Internet]. 2017. August 24;8(4):563–7. Available from: https://www.wageningenacademic.com/doi/10.3920/BM2016.0185. [DOI] [PubMed] [Google Scholar]
- 43.Sprenger N, Odenwald H, Kukkonen AK, Kuitunen M, Savilahti E, Kunz C. FUT2-dependent breast milk oligosaccharides and allergy at 2 and 5 years of age in infants with high hereditary allergy risk. Eur J Nutr [Internet]. 2017. April 24;56(3):1293–301. Available from: http://link.springer.com/10.1007/s00394-016-1180-6. 10.1007/s00394-016-1180-6 [DOI] [PubMed] [Google Scholar]
- 44.Jakobsson HE, Abrahamsson TR, Jenmalm MC, Harris K, Quince C, Jernberg C, et al. Decreased gut microbiota diversity, delayed Bacteroidetes colonisation and reduced Th1 responses in infants delivered by Caesarean section. Gut [Internet]. 2014. April;63(4):559–66. Available from: http://gut.bmj.com/lookup/doi/10.1136/gutjnl-2012-303249. [DOI] [PubMed] [Google Scholar]
- 45.Zhang T, Sidorchuk A, Sevilla-Cermeño L, Vilaplana-Pérez A, Chang Z, Larsson H, et al. Association of Cesarean Delivery With Risk of Neurodevelopmental and Psychiatric Disorders in the Offspring. JAMA Netw Open [Internet]. 2019. August 28;2(8):e1910236 Available from: https://jamanetwork.com/journals/jamanetworkopen/fullarticle/2749054. 10.1001/jamanetworkopen.2019.10236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Strandwitz P, Kim KH, Terekhova D, Liu JK, Sharma A, Levering J, et al. GABA-modulating bacteria of the human gut microbiota. Nat Microbiol [Internet]. 2019. March 10;4(3):396–403. Available from: http://www.nature.com/articles/s41564-018-0307-3. 10.1038/s41564-018-0307-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hsiao EY, McBride SW, Hsien S, Sharon G, Hyde ER, McCue T, et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell [Internet]. 2013;155(7):1451–63. Available from: 10.1016/j.cell.2013.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hunt KM, Foster JA, Forney LJ, Schütte UME, Beck DL, Abdo Z, et al. Characterization of the diversity and temporal stability of bacterial communities in human milk. PLoS One. 2011;6(6):1–8. 10.1371/journal.pone.0021313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Patel SH, Vaidya YH, Patel RJ, Pandit RJ, Joshi CG, Kunjadiya AP. Culture independent assessment of human milk microbial community in lactational mastitis. Sci Rep [Internet]. 2017. December 10;7(1):7804 Available from: http://www.nature.com/articles/s41598-017-08451-7. 10.1038/s41598-017-08451-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carter JE, Laurini JA, Mizell KN. Kluyvera infections in the pediatric population. Pediatr Infect Dis J [Internet]. 2008. September;27(9):839–41. Available from: http://content.wkhealth.com/linkback/openurl?sid=WKPTLP:landingpage&an=00006454-200809000-00015. 10.1097/INF.0b013e318170af5b [DOI] [PubMed] [Google Scholar]
- 51.Isozaki A, Shirai K, Mimura S, Takahashi M, Furushima W, Kawano Y. A case of urinary tract infection caused by Kluyvera ascorbata in an infant: Case report and review of the literature. J Infect Chemother. 2010;16(6):436–8. 10.1007/s10156-010-0067-3 [DOI] [PubMed] [Google Scholar]
- 52.Brandt K, Taddei C, Takagi E, Oliveira F, Duarte R, Irino I, et al. Establishment of the bacterial fecal community during the first month of life in Brazilian newborns. Clinics [Internet]. 2012. February 9 [cited 2013 Jul 31];67(2):113–23. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3275115/. 10.6061/clinics/2012(02)05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Talarico ST, Santos FE, Brandt KG, Martinez MB, Taddei CR. Anaerobic bacteria in the intestinal microbiota of Brazilian children. Clinics. 2017;72(3):154–60. 10.6061/clinics/2017(03)05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bezerra de Araujo Filho H, Silva Carmo-Rodrigues M, Santos Mello C, Cristina Fonseca Lahoz Melli L, Tahan S, Carlos Campos Pignatari A, et al. Children Living near a Sanitary Landfill Have Increased Breath Methane and Methanobrevibacter smithii in Their Intestinal Microbiota. Archaea [Internet]. 2014;2014:1–6. Available from: http://www.hindawi.com/journals/archaea/2014/576249/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mello CS, Carmo-Rodrigues MS, Filho HBA, Melli LCFL, Tahan S, Pignatari ACC, et al. Gut Microbiota Differences in Children From Distinct Socioeconomic Levels Living in the Same Urban Area in Brazil. J Pediatr Gastroenterol Nutr [Internet]. 2016. November;63(5):460–5. Available from: http://insights.ovid.com/crossref?an=00005176-201611000-00004. 10.1097/MPG.0000000000001186 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
CIAAM/UNIFESP, Breastfeeding Incentive and Support Center/Universidade Federal de São Paulo.
(PDF)
Values are presented as median (p25 –p75) unless otherwise indicated; * statistical comparisons between VSe+ and CSe+; 2’-FL: 2’-fucosyllactose; LNFP I: lacto-N-fucopentaose I; LNDFH I: lacto-N-difucohexaose I; DFLNHc: difucosyllacto-N-hexaose c; a breast milk from these groups presented levels of α1–2 fucosylated HMOs below the quantification limit; b quantification limit: 0.000039 g/L; c quantification limit: 0.000156 g/L; d Mann-Whitney rank sum test; e Student’s t test.
(PDF)
(PDF)
Data Availability Statement
The dataset analyzed during the current study is available in the Sequence Read Archive repository, under accession number PRJNA615250 [https://www.ncbi.nlm.nih.gov/sra/PRJNA615250].