

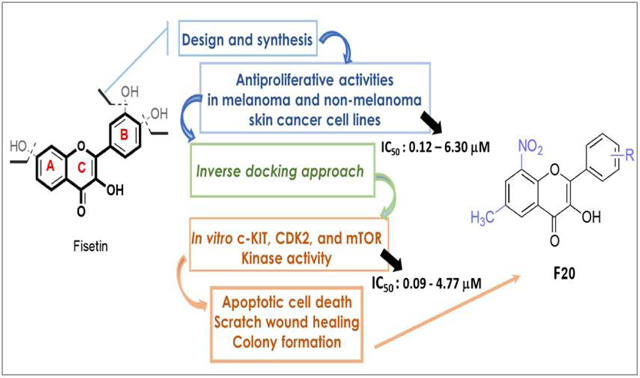
Abstract
Due to hurdles, including resistance, adverse effects, and poor bioavailability, among others linked with existing therapies, there is an urgent unmet need to devise new, safe, and more effective treatment modalities for skin cancers. Herein, a series of flavonol-based derivatives of fisetin, a plant-based flavonoid identified as an anti-tumorigenic agent targeting the mammalian targets of rapamycin (mTOR)-regulated pathways, were synthesized and fully characterized. New potential inhibitors of receptor tyrosine kinases (c-KITs), cyclin-dependent kinase-2 (CDK2), and mTOR, representing attractive therapeutic targets for melanoma and non-melanoma skin cancers (NMSCs) treatment, were identified using inverse-docking, in vitro kinase activity and various cell-based anticancer screening assays. Eleven compounds exhibited significant inhibitory activities greater than the parent molecule against four human skin cancer cell lines, including melanoma (A375 and SK-Mel-28) and NMSCs (A431 and UWBCC1), with IC50 values ranging from 0.12 to < 15 μM. Seven compounds were identified as potentially potent single, dual or multi-kinase c-KITs, CDK2, and mTOR kinase inhibitors after inverse-docking and screening against twelve known cancer targets, followed by kinase activity profiling. Moreover, the potent compound F20, and the multi-kinase F9 and F17 targeted compounds, markedly decreased scratch wound closure, colony formation, and heightened expression levels of key cancer-promoting pathway molecular targets c-Kit, CDK2, and mTOR. In addition, these compounds downregulated Bcl-2 levels and upregulate Bax and cleaved caspase-3/7/8 and PARP levels, thus inducing apoptosis of A375 and A431 cells in a dose-dependent manner. Overall, compounds F20, F9 and F17, were identified as promising c-Kit, CDK2 and mTOR inhibitors, worthy of further investigation as therapeutics, or as adjuvants to standard therapies for the control of melanoma and NMSCs.
Keywords: Anticancer activity, apoptosis, flavonols, melanoma, non-melanoma skin cancer, fisetin-analogs, inverse docking, kinase activity
Graphical Abstract
1. Introduction
Hyper-proliferation and compromised cell death machinery are universal features of cancer cells, as these impairments functionally underlie the resistance mounted to apoptotic pointers resulting from diverse therapeutic regimens (1–3). In spite of recent advances in cancer treatment, achieving durable complete responses for systemic metastatic cancer patients is challenging, as cancer cells, such as melanoma, evade eradication due to phenotypic complexity and the activation of cancer cells’ pro-survival machinery, which thwarts apoptotic stimuli and enhances tumor development and progression (4,5). Skin cancers constitute the highest-incidence among all forms of cancers in the United States (6). There are two predominant classifications, melanoma and non-melanoma skin cancers (NMSCs), the latter including basal cell carcinoma (BCC) and squamous cell carcinoma (SCC). Melanoma is the most aggressive culprit of the skin cancers, with a rising incidence of new cases exceeding 7.7% in the U. S. (6,7), in association with a high number of deaths, while BCC and SCC are the most prevalent forms of NMSCs, with over 5.3 million yearly cases in the US (6,8,9). Amongst other skin-cancer related diseases, the prognosis of malignant melanoma alone remains dismal despite recent advances in therapeutic strategies (6,10–12). In this context, to overcome the urgent treatment hurdles associated with increased cancer progression and mortality, the identification of novel, more effective, safe, and (ideally) low-cost synthetic analogs of natural bioactive compounds, with known anticancer health-benefits, represents a suitable alternative (13–16). Flavonoids are attractive chemotypes for drug discovery due to their molecular diversity. These compounds belong to a large family of secondary metabolites from plants, comprised of about 12 classes, harbouring a very characteristic phenylbenzopyran core, built around a C6-C3-C6 arrangement, with C6 being a benzene ring (17,18). Major structurally related classes from this family of compounds include flavones, flavonols, flavans, flavanones, flavanols, chalcones and dihydrochalcones, isoflavones, aurones, anthocyanins and anthocyanidins, and catechins, differing from each other primarily by the degree of oxidation of the C ring (19). Flavonoids are widely recognized to exhibit various pharmacological effects, including antioxidative, anticancer, cardiovascular ion channel modulator, antiprotozoan and antimalarial (13,14,17). One group member, namely 3´,4´,7-trihydroxyflavonol, also known as fisetin (F0, Fig. 1), is well known to possess a range of biological and pharmacological activities, including anti-cancer properties (24–32). Fisetin has shown efficacy in multiple in vitro and in vivo preclinical animal models of human cancers, including melanoma, colon and prostate cancers (14,25,29,33–46). Fisetin’s induction of cytotoxicity in metastatic human melanoma cells appears to involve the activation of both extrinsic and intrinsic apoptotic pathways, at least in part arising from the inhibition or beneficial modulation of kinases involved in melanoma growth and progression (28–35). However, one of the major limitations to fisetin’s clinical efficacy has been associated with the presence of the 7-hydroxyl substituent on ring A (47), which is prone to a rapid extensive first-pass biotransformation, resulting in biologically less-active and/or less-bioavailable metabolites upon oral administration (47). Moreover, the catechol moiety (ring B) is also susceptible to rapid oxidation, generating a reactive ortho-quinone metabolite (48–51). Due to fisetin’s high susceptibility to first-pass elimination, exacerbated by its relatively poor aqueous solubility resulting in a limited dissolution absorption rate, this molecule suffers from compromised oral bioavailability that reduces its therapeutic efficacy (43,44,46). Moreover, fisetin’s three hydroxyl moieties may be responsible for sub-optimal lipophilicity for transdermal or gut wall absorption and access to intracellular targets (47). Therefore, it was deemed imperative to investigate anticancer activity of the fisetin analogs, having improved lipophilicity, and in which the above-listed impediments deriving mainly from the presence of the 7-hydroxy group on ring A and the 3’,4’-dihydroxy moiety of ring B, are corrected. To achieve that objective, 24 fisetin-based derivatives (Figure 1) bearing a wide range of substituents at different positions of ring A and B of the 2-phenyl-4H-chromenone scaffold were synthesized, and their antitumor potentials were investigated. All the synthesized compounds were first tested for their cytotoxicity and antiproliferative efficacy against four human skin tumor cell lines, including two melanoma and two non-melanoma lines. In comparison, two normal primary and non-cancerous skin cell lines were used as controls. The most active compounds were further subjected to an inverse docking protocol to identify putative molecular targets from among a selection of prospectively and commonly dysregulated ones in melanoma and NMSCs. Finally, by employing in-vitro enzymatic, cell culture, immunocytochemical, and Western-blot assessment assays, the most potent fisetin analogs were evaluated against the identified target proteins for their inhibitory potential and mechanistic effects.
Figure 1.
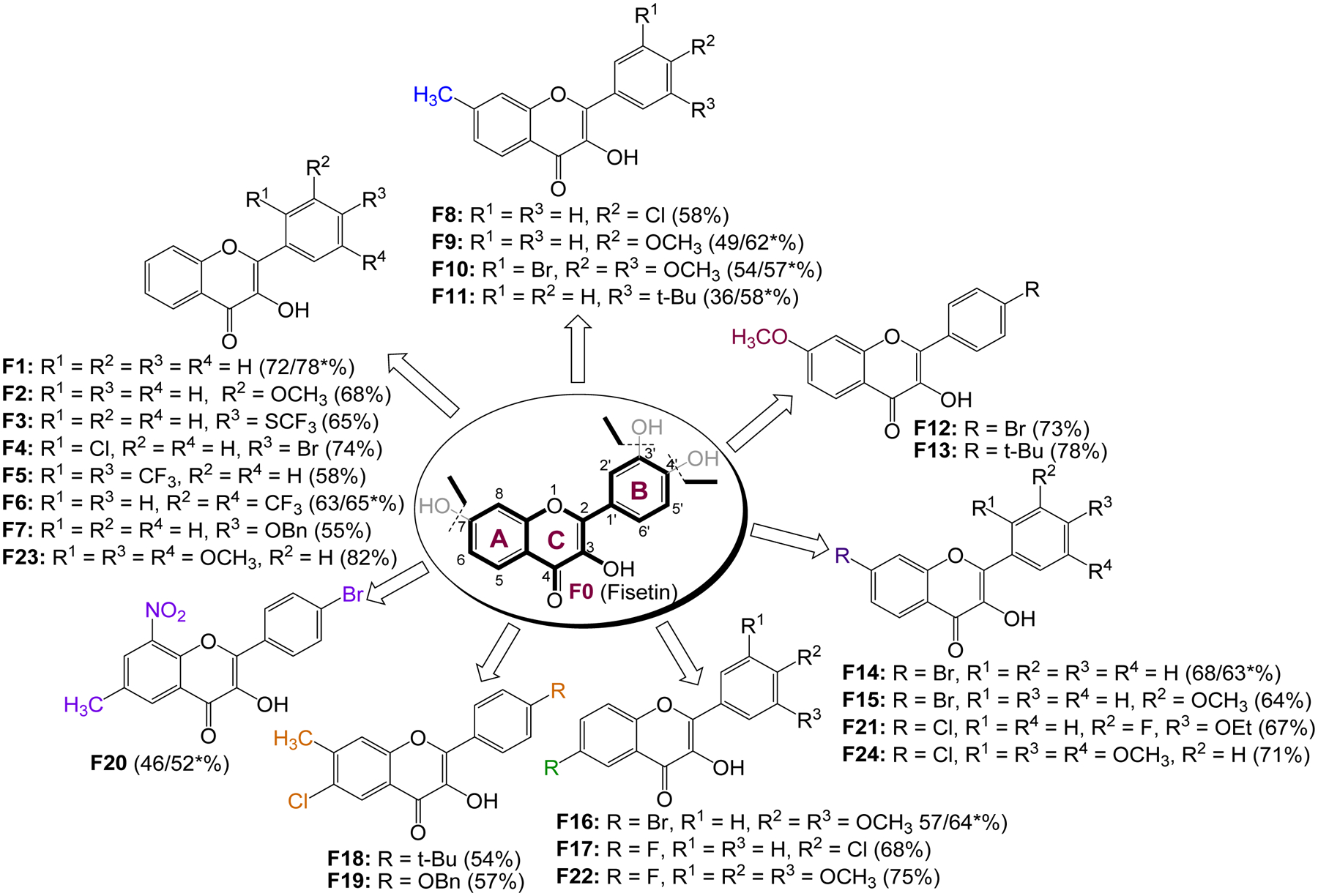
List of fisetin analogs designed and synthesized (*% yields from the one-pot reactions).
2. Results and Discussion
2.1. Chemistry
The overarching goal for this project was to synthesize and investigate the anticancer/anti-proliferative activities of fisetin (F0) analogs lacking both the 7-hydroxy group on ring A as well as the 3’,4’-dihydroxy moiety on ring B. In the process, it was also set out to investigate the electronic and steric effects of various groups at different positions of the 3-hydroxy-2-phenyl-4H-chromen-4-one skeleton on the cytotoxic activity of the resulting analogs, with a primary goal of modifying their physicochemical characteristics, and eventually improve their pharmacokinetic and pharmacodynamic properties (52, 53). Balancing the lipophilicity of the resulting analogs was also an important parameter in this design, as it has been shown that replacing the hydroxy groups on the flavonol moiety with substituents that are less susceptible to oxidation, increases the bioavailability and reduces the metabolic lability (54). As such, a number of substituents, including alkyl, alkoxy, halogen, trifluoromethyl, and nitro groups were introduced at the 6, 7, and/or 8 positions on the A-ring, as well as at positions 2’, 3’, 4’, and/or 5’ on the ring B of the core flavonol, resulting in an array of 24 analogs as illustrated in Figure 1.
The synthesis of flavonols is well defined and documented (55–61). The target compounds listed in Figure 1 were synthesized via a two-step (two-pot) reaction sequence proceeding through the corresponding 2’-hydroxychalcone intermediate, followed by an oxidative cyclization to produce the expected flavonol derivatives. To enable a good control over the reaction conditions, a microwave-assisted approach was utilized to prepare the chalcone intermediates. Microwave-assisted reactions are often much faster, cleaner, and thus more likely to produce higher yields than conventional heating procedures (62,63). In fact, a microwave-assisted Claisen-Schmidt condensation (64) of suitably substituted 2’-hydroxyacetophenones with benzaldehyde derivatives bearing the appropriate substituents at the desired positions, afforded the corresponding 2’-hydroxychalcones (Scheme 1), which were isolated in good to excellent yields, and characterized by TLC and GC-MS analyses. It should be mentioned that all the chalcone intermediates were synthesized using microwave irradiations, except those used in the preparation of compounds F3, F7, and F19, which were prepared following a previously reported procedure (65).
Scheme 1.
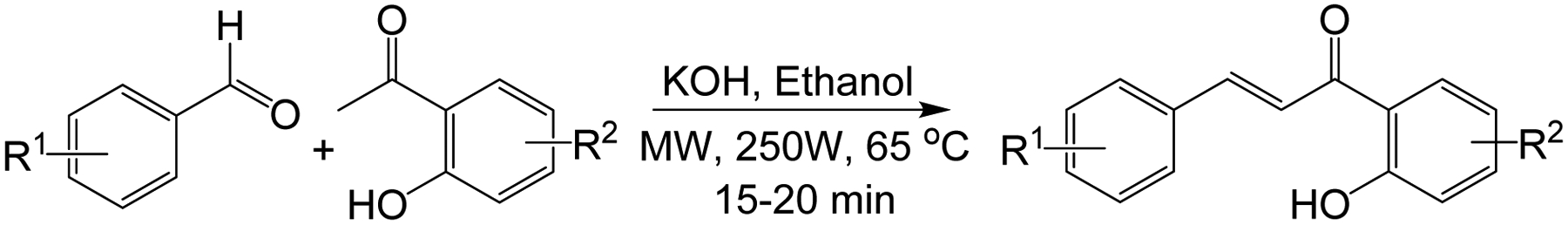
Microwave assisted synthesis of 2’-hydroxychalcones
These 2‘-hydroxychalcone intermediates were subsequently converted into flavonols via a base-promoted cyclization, using an aqueous solution of hydrogen peroxide in the presence of KOH (Scheme 2), following Algar-Flynn-Oyamada (AFO) reaction conditions (66, 67) as a starting point. However, under these initial conditions, the reaction failed to produce a good yield at low temperature, prompting a quick optimization through the variation of reaction parameters including temperature, solvent, and the equivalents of H2O2 and KOH. The best yield was obtained in methanol, using 1.5 equivalents of KOH and 6 equivalent of 30% aqueous H2O2 at room temperature. These reaction conditions appeared to tolerate a number of substituents on both rings A and B, and produced isolated yields of between 36 – 78% (Scheme 2).
Scheme 2.
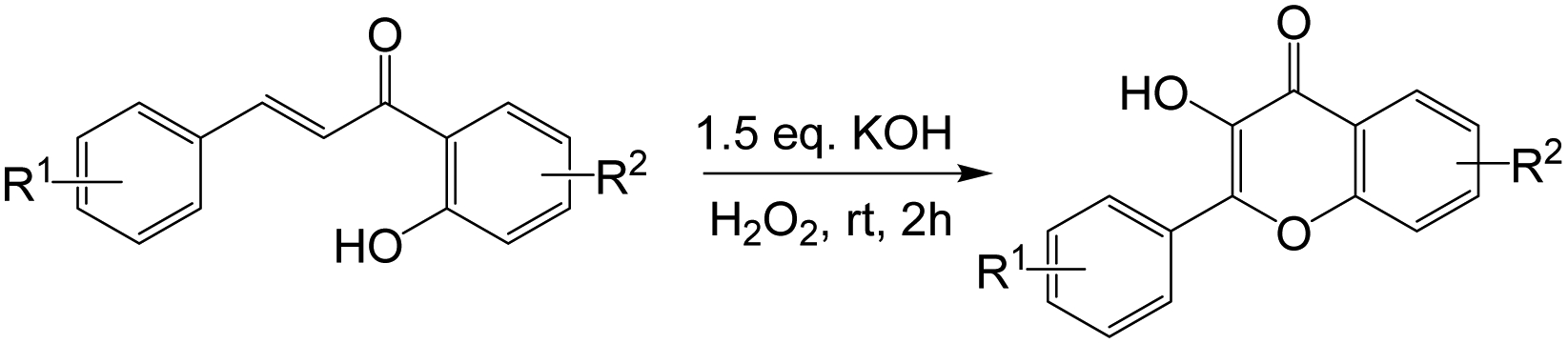
Synthesis of substituted flavonols via oxidative cyclization of 2’-hydroxychalcones at room temperature.
It should be mentioned that a microwave-based, one-pot (two-step) sequential addition approach through which the chalcone is oxidatively cyclized in situ was also explored (Scheme 3). Even though reaction times were much shorter, and the yields comparable to those obtained with the two-pot approach, the number of unidentified side products increased, resulting in complex mixtures that complicated the isolation of the desired product. As such, this latter approach was used only in the preparation of a few compounds, which could not be as efficiently prepared using the two-pot approach. In fact, with the exception of F14, the others compounds, namely F1, F6, F9–11, and F16 were produced in relatively higher yields with the one-pot approach (Figure 1). All of the final products were purified and fully characterized by a combination of spectroscopic and spectrometric techniques, including 1H-NMR, 13C-NMR, IR, GC-MS and HR-ESIMS, with the detailed 1H-NMR, 13C-NMR and HR-ESIMS spectra data provided as in supplementary dataset (68).
Scheme 3.
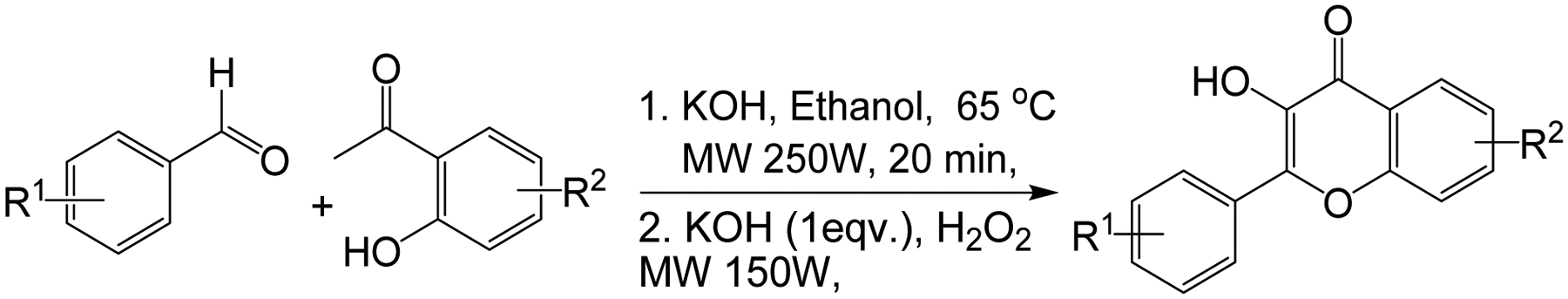
Microwave-assisted one-pot synthesis of substituted flavonol derivatives.
2.2. In vitro assessment of cytotoxic activity
The growth-suppressive and cytotoxic activities of all synthesized target compounds, (F1-F24), alongside fisetin (F0, the parent flavonol), and doxorubicin, co-assayed as positive controls, were investigated on human solid skin cancer cells: two melanoma (A375 and SK-Mel-28) and two NMSC (A431 and UWBCC1) cell lines, and for comparison, two normal control cells (melanocytes and HaCaT keratinocytes), using in vitro MTT and trypan blue dye exclusion assays. The screened anti-proliferative and cytotoxicity activities were assessed in terms of the concentration capable of reducing the measured viability of the cell population to 50% of maximal (expressed as IC50) for the above-listed six human skin-derived cell lines in culture, as described previously (38).
The obtained data (summarized in Table 1) indicates that several of the tested analogs exhibited variable degrees of anti-proliferative activities and potency against the human-skin-cancer cell lines as compared to the control lines. Eleven analogs (F1, F3, F6, F8, F9, F11, F14, F17, F19, F20 and F21) were more potent than fisetin, with the most potent compound F20, displaying the lowest IC50 values (0.12 and 0.20 μM against non-melanoma cell lines UWBCC1 and A431, respectively), which represents a 233- and 130-fold dose-advantage over fisetin, respectively. F20 was also more selective on cancer cells (UWBCC1 and A431) over normal (HaCaT) cells, with selectivity indexes of about 283 and 170, respectively, as compared to both fisetin (1.2) and doxorubicin (0.2). In melanoma cells, F20 displayed some level of potency against A375 cells (IC50 value of 6.3 μM), but was only marginally potent against SK-Mel-28 cells (IC50 value of 17 μM). F19, bearing a 6-Cl, 7-CH3 and 4’-OBn, respectively, also exhibited strong activity against both UWBCC1 and A431 cells (IC50s 1.1 and 1.5 μM, respectively), but with much lower selectivity indices (25 and 19, respectively).
Table 1.
Cytotoxicity of flavonol analogues F1-F24 plus parent fisetin against human melanoma (A375 and SK-Mel-28), and non-melanoma skin cancer (A431 and UWBCC1) cells relative to normal control HaCaT and Melanocytes.
| Compounds ID | Cell lines, IC50 values in μM, and (selectivity index)a | |||||
|---|---|---|---|---|---|---|
| Melanoma | Control | Non-Melanoma | Control | |||
| A375 | SK-Mel-28 | Melanocyte | A431 | UWBCC1 | HaCaT | |
| F1 | 14 ± 0.80 (1.9) | 21 ± 1.0 (1.2) | 27 ± 0.90 | 15 ± 0.04 (2.5) | 12 ± 0.80 (3.2) | 38 ± 0.02 |
| F2 | 28 ± 2.3 | ND | ND | 21 ± 16 | ND | ND |
| F3 | 14 ± 0.20 (2.2) | 5.2 ± 1.7 (6.0) | 31 ± 0.74 | 9.7 ± 2.3 (2.8) | ND | 27 ± 0.70 |
| F4 | >100 | ND | ND | 61 ± 30 | ND | ND |
| F5 | 95 ± 12 | ND | ND | >100 | ND | ND |
| F6 | 7.0 ± 0.32 (3.7) | 27 ± 4.1(10) | 26 ± 1.3 | 8.6 ± 0.50 (3.6) | 6.6 ± 0.79 (4.7) | 31 ± 1.9 |
| F7 | 88 ± 35 | ND | ND | >100 | ND | ND |
| F8 | 8.4 ± 0.4 (3.1) | 22 ± 3.0 (1.1) | 26 ± 3.8 | 8.3 ± 2.1 (2.7) | 6.7 ± 1.5 (3.3) | 22 ± 0.20 |
| F9 | 12 ± 0.9 (2.2) | 17 ± 1.0 (1.5) | 26 ± 1.6 | 9.2 ± 0.70 (2.9) | 9.4 ± 0.30 (2.9) | 27 ± 0.30 |
| F10 | 31 ± 13 | ND | ND | 36 ± 0.10 | 11 ± 5.0 | ND |
| F11 | 11 ± 2.0 (2.4) | 18 ± 2.0 (1.4) | 26 ± 1.1 | 4.7 ± 0.9 (4.7) | 9.7 ± 0.60 (2.3) | 25 ± 0.78 |
| F12 | >100 | ND | ND | >100 | ND | ND |
| F13 | 28 ± 24 | ND | ND | >100 | ND | ND |
| F14 | 15 ± 0.08 (1.4) | 22 ± 4.3 | 24 ± 0.84 | 5.3 ± 2.1 (4.5) | 3.3 ± 1.3 (7.3) | 24 ± 0.20 |
| F15 | >100 | ND | ND | >100 | ND | ND |
| 16 | 79.5 ± 9.0 | ND | ND | >100 | 40 ± 0.89 | ND |
| F17 | 13 ± 1.0 (2.5) | 25 ± 3.4 | 32 ± 0.72 | 10 ± 2.6 (3.8) | 8.1 ± 1.2 (4.7) | 38 ± 0.80 |
| F18 | >100 | ND | ND | 24 ± 5.6 | ND | ND |
| F19 | 13 ± 2.6 (1.7) | 19 ± 3.0 (1.2) | 27 ± 1.0 | 1.5 ± 0.30 (19) | 1.1 ± 0.40 (25) | 28 ± 0.31 |
| F20 | 6.3 ± 1.0 (5.6) | 17 ± 3.0 (2.1) | 35 ± 0.23 | 0.20 ± 0.10 (170) | 0.12 ± 0.11 (283) | 34 ± 0.60 |
| F21 | 8.6 ± 0.40 (3.2) | 12 ± 3.0 (2.3) | 27 ± 0.10 | 8.2 ± 0.20 (3.2) | ND | 26 ± 0.40 |
| F22 | 15 ± 1.9 (1.6) | ND | 24 ± 1.6 | 13 ± 2.6 (1.8) | 11 ± 2.0 (2.1) | 23 ± 1.9 |
| F23 | 13 ± 3.1 (1.7) | 19 ± 3.0 (1.6) | 22 ± 1.0 | 12 ± 1.9 (2.0) | 9.4 ± 1.4 (2.6) | 24 ± 1.4 |
| F24 | >100 | ND | ND | >100 | ND | ND |
| cFisetin (F0) | 21 ± 1.0 (1.6) | 29 ± 4 (1.1) | 33 ± 2.7 | 26 ± 1.0 (1.2) | 28 ± 0.20 (1.1) | 30 ± 1.1 |
| cDoxorubicin | 0.1±0.01 | 0.23±0.06 | ND | 0.37±0.02 (0.2) | ND | 0.07±0.01 |
The selectivity index was determined as the ratio of IC50 value in the respective normal cells (melanocytes or HaCaT keratinocytes) divided by the IC50 for the corresponding specified cancer (melanoma or non-melanoma) cell lines. IC50: compound concentration required to inhibit tumor cell proliferation (assessed as cell viability) by 50%.
Fisetin (the parent flavonoid) and doxorubicin (a well-known anticancer agent) were employed as positive controls. Data are expressed as the mean ± SD from the dose-response curves of at least five independent experiments performed in octuplicates. ND: not determined.
F6 (IC50s 8.6 and 6.6 μM), F8 (IC50s 8.3 and 6.7 μM), F9 (IC50s 9.2 and 9.4 μM), F11 (IC50s 4.7 and 9.7 μM) and F14 (IC50s 5.3 and 3.3 μM), which also displayed some potency against A431 and UWBCC1 cell lines, but the selectivity was generally poor. Although F6 (IC50 7.0 μM) and F8 (IC50 8.4 μM) also exhibited noteworthy potency against cells of the melanoma A375 line, they were essentially inactive against SK-Mel-28 cells. On the other hand, in melanoma cells, F9, F11, and F14 were either marginally active against A375 cells or totally inactive against SK-Mel-28 cells. It is worth noting that while both F6 (4’-CF3) and F8 (4’-Cl) bear an electron-withdrawing group at the 4’-position on ring B, F14 carries a Br at the position 7 of ring A, with F9 (4’-OCH3) and F11 (4’-t-Bu) bearing rather an electron-donating group at the 4’ position. Other analogs that displayed some activity against at least one of the non-melanoma cell lines include F3 (A431, IC50 9.7 μM), bearing a 4’-SCF3, F17 (UWBCC1, IC50 8.1 μM), bearing 6-F and 4’-Cl, and F23 (UWBCC1, IC50 9.4 μM), which carries electron-donating groups (OCH3) at the 2’, 4’ and 5’ positions. Among these, only F3 displayed some activity against SK-Mel-28 cells (IC50 of 5.2 μM). Therefore, the respective selective indexes in all cancer cell-lines (except for F6 in SKMEL-28) were greater than 1 (1.1–25), indicating a possible low toxicity profile when administered (though warranting further confirmation) as compared to doxorubicin.
In the light of the structure activity relationship (SAR), it was generally observed that compounds with a methoxy group at any position on the A or B ring or on both (F2, F10, F12, F13, F15, F16, F22, F23 and F24), were mostly marginally active or inactive against all the tested cell lines, except for F9. Though these observations are fairly consistent with previous reports on substituted flavonols (68–70), it is yet unclear how the position and the electronic profile of the different substituents actually affect the activity of these analogs against the tested melanoma or the NMSC cells. For example, compounds F18 and F19 both have the same substitution profile on ring A (6-Cl and 7-CH3) and differ merely by nature of the electron donating substituent (t-Bu and OBn) at position 4’, but displayed a difference in activity. On the other hand, F7, which bears a 4’-OBn like F19, but without 6-Cl and 7-CH3 substituents on ring A, was completely inactive, compared to F19, which was substantially active against the non-melanoma cells.
Given that no clear SAR emerged from this initial series of assays, more insight into the relationship between the structural patterns and the anticancer activities of this family of compounds was coveted and explored through an inverse-docking analysis platform. Other 3-hydroxychromen-4-one derivatives, with potent anticancer properties, have previously been identified as CDK2 inhibitors (69); and considering that various oncogenic pathways involve drug targets, including EGFR, cKit, Akt, MET, MEK1, VEGFR, MAPK, MTOR, PI3K, PIP5K1α, CDK2, and FGFR, prospectively enabling the design of active agents for the management of melanoma (14 for a review, 71), the eleven hits, obtained from the anti-proliferative assays, were computationally screened on the above-listed proteins.
Molecular targets involving these pathways are usually associated with the abnormal cell proliferation, malignant/carcinogenic transformation and cell-cycle aberrations observed in cancer progression (72–80). Nonetheless, the development of resistance by human cancer cells to established chemotherapeutic regimens is often associated with mutations affecting the apoptotic machinery or signalling pathways involving these processes (14). Moreover, combination therapies (“drug cocktails”) of agents with multiple and definite molecular mechanisms of action (MMOAs) are often or would be key to improving outcomes for patients, ideally by preventing the emergence of resistant cancerous cells and tumors, often at distant sites, which is particularly common and problematic with melanoma (72–80). Consequently, it is expected that computational screening via an inverse docking analysis of the eleven hits obtained from the above antiproliferative assays on the listed proteins could provide key parameters essential for future design of more potent and metabolically less-labile analogs of fisetin to carry forward to in vivo assays and other preclinical studies leading to clinical candidacy.
2.3. Inverse-docking analysis
All the eleven fisetin derivatives (F1, F3, F6, F8, F9, F11, F14, F17, F19, F20, and F21) identified via MTT assay, as potent anti-tumor molecules, were subjected to an exhaustive inverse docking protocol to identify the putative target for these hit molecules. In this technique, a molecule is attempted to dock to the ligand-binding domain of proteins associated with the potential activity. The term “inverse” is used because the method identifies proteins that can fit a specific ligand, rather than identifying ligands that fit within a specific protein (81). For the current inverse docking protocol, a pool of 12 proteins (EGFR, cKit, Akt, MET, MEK1, VEGFR, MAPK, MTOR, PI3K, PIP5K1α, CDK2 and FGFR), reported as key molecular targets for the management of melanoma, were utilized for the exhaustive docking analysis against the eleven hits obtained after MTT assay. The docking protocol was validated by re-docking the co-crystallized ligand within the catalytic domain of specific selected target protein. It was observed that the re-docked pose of co-crystallized ligand retained the binding conformation to the co-crystallized pose with the RMSD value of less than 1 Å. Key interactions reported in previous studies as essential for the inhibition of the selected proteins were considered as screening parameters along with the obtained docking score for each hit against corresponding proteins. For molecular docking, the 3D structures of the target proteins were selected from a data bank, using a cross-docking protocol with the available Protein Data Bank (PDB) number associated with each selected protein (82–85), also see supplemental dataset (68). The collected data indicated that in the case of EGFR, most of the analogs maintained the key H-bond interaction with Met769 as seen in PDB1M17 for the 4 anilinoquinazoline EGFR kinase inhibitor erlotinib (Table 2). However, the estimated interaction energies (based on Glide docking scores) for the interaction poses predicted to be optimal within the −7.41 to −6.05 Kcal/mol range, as compared to −8.41 Kcal/mol for fisetin. Against c-Kit, however, these molecules, when optimally docked per Glide scoring, maintained the required key interactions with Cys673 alone (F1, F3, F6, F11, F14 and F20) and/or Glu671 as seen for AZD3229 in PDB 6GQJ, and showed good docking scores (−11.04 to −8.54 Kcal/mol) in comparison to fisetin (−11.51 Kcal/mol). Thus, each of these 11 fisetin analogs studied are predicted to undergo stronger binding interactions with c-Kit than with the EGFR kinase domain (−11.04 to −8.54 Kcal/mol). The detailed results of docking scores for each of the fisetin analogs interacting with each of the proteins selected for inverse docking studies are summarized in Table 2, and the 3D interactions diagram of fisetin and its analogs with the 12 selected proteins are shown in Figure 2. Based on the collective analysis, CDK2, c-Kit and mTOR were predicted as the most likely targets for these analogs among the proteins screened. The inverse docking analysis with CDK2 revealed that most of the compounds bind with the catalytic domain in the same pose as fisetin, with the carbonyl on the flavone core moiety interacting as a hydrogen bond acceptor, forming a hydrogen bond with Leu83 backbone N-H. In addition, the 3-hydroxy substituent of the flavonol skeleton acts as a hydrogen bond donor, forming a hydrogen bond with the Glu81 side-chain carboxyl, while the 2-phenyl group (ring B) is predicted to consistently create π-π interactions with Phe80 throughout all of these analogs (Figure 2). Against c-Kit, these compounds are also predicted to adopt a binding poses consistent with that of fisetin, again with the flavone carbonyl interacting as a hydrogen bond acceptor, consistently forming a hydrogen bond with the backbone amide nitrogen of Cys673. The data also predict ring B to maintain a π-π interaction with Phe811 across these analogs. Pertaining to mTOR, it was found that ring B consistently maintains a π-π interaction with Tyr2225. Overall, the results of the computational evaluation revealed that analogs with a single and small substituent on ring B were able to easily maintain poses similar to that of fisetin with almost all of the target proteins, whereas a similar binding pose was not always maintained with analogs bearing multiple or bulky substituents on ring B.
Table 2.
Detailed results of inverse docking protocol studies for the selected flavonol analogs.
| S.No | Target Enzymes | F1 | F3 | F6 | F8 | F9 | F11 | F14 | F17 | F19 | F20 | Fisetin (F0) | Specific Co-crystallized ligand | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1. | EGFR | Docking Score | −7.07 | −7.17 | −6.25 | −7.31 | −7.31 | −6.05 | −7.19 | −7.41 | −7.41 | −7.13 | −8.46 | −8.96 |
| H-bond | Met769, Lys721 |
Met769 | Asp831 | Met769, Gln767 |
Met769, Gln767 |
Met769 | Met769 | Met769 | Met769 | Met769 | Met769, Lys721, Asp831 |
Met769 | ||
| 2. | c-Kit | Docking Score | −9.86 | −10.08 | −8.54 | −10.42 | −10.62 | −10.48 | −10.20 | −10.39 | −11.04 | −8.65 | −11.51 | −8.52 |
| H-bond | Cys673 | Cys673 | Cys673 | Glu671, Cys673 |
Glu671, Cys673 |
Cys673 | Cys673 | Glu671, Cys673 |
Glu671, Cys673, Asp810 |
Cys673 | Cys673, Asp810 |
Glu671, Cys673, Asp810 | ||
| 3. | Akt | Docking Score | −4.46 | −2.57 | −4.09 | −4.55 | −4.70 | −3.96 | −2.79 | −4.46 | −5.15 | −2.12 | −6.26 | −7.63 |
| H-bond | Glu17, Ile19, Arg23, Tyr18 |
Lys14, Arg86 |
Glu17, Ile19, Arg23, Tyr18 |
Glu17, Ile19, Arg23, Tyr18 |
Glu17, Ile19, Arg23, Tyr18 |
Glu17, Ile19, Arg23, Tyr18 |
Glu17, Ile19, Arg23, Tyr18 |
Glu17, Ile19, Arg23, Tyr18 |
Glu17, Ile19, Arg23, Asn53 |
Glu17, Ile19, Arg23, Tyr18 |
Lys14, Glu17, Ile19, Arg23, Tyr18 |
Lys14, Glu17, Ile19, Arg23, Tyr18 | ||
| 4. | MET | Docking Score | −7.86 | −8.45 | −7.81 | −8.05 | −7.78 | −8.16 | −7.72 | −8.08 | −8.25 | −8.22 | −8.18 | −9.26 |
| H-bond | Met1160 | Met1160 | Arg1208 | Met1160 | Met1160 | Met116 0 |
Met116 0 |
Met1160 | - | Met116 0 |
Met1160, Tyr1159 |
Met1160, Asp1222 | ||
| 5. | MEK1 | Docking Score | −5.61 | −5.29 | −5.56 | −6.05 | −6.28 | −6.20 | −5.57 | −6.09 | −6.03 | −4.85 | −8.45 | −9.12 |
| H-bond | - | - | Gln153 | - | Met146 | - | Gln153 | Ser150 | - | Ser194, Asp152 |
Glu144, Gln153, Ser194 |
Glu144, Met146, Lys192 | ||
| 6. | VEGFR | Docking Score | −8.04 | −8.84 | −9.12 | −6.05 | −6.37 | −9.11 | −8.92 | −8.52 | −8.76 | −4.89 | −6.81 | −10.15 |
| H-bond | Cys912 | Cys912 | Glu878, Asp1040 |
Ile1019, Arg1021 |
Ile1019, Arg1021 |
Cys912 | Cys912 | Cys912 | - | Arg1021 | Asp807, Glu878, Ile1019, Arg1021 |
Glu878, Cys912, Asp1040 | ||
| 7. | MAPK | Docking Score | −4.58 | −5.45 | −5.84 | −5.28 | −5.95 | −5.34 | −6.47 | −6.01 | −4.61 | −6.39 | −8.10 | −7.26 |
| H-bond | Lys114 | Asp111, Lys114 |
Met108 | Asp111, Lys114 |
Met108 | Lys114 | Met108 | Met108 | - | Met108 Lys114 |
Asp106, Met108, Asp111 |
Gln105, Asp106, Met108 | ||
| 8. | mTOR | Docking Score | −8.36 | −7.59 | −7.72 | −8.93 | −9.68 | −8.19 | −8.67 | −9.02 | −8.95 | −7.98 | −8.46 | - |
| H-bond | Gly2238 Val2240 |
Val2240 | - | Gly2238 Val2240 |
Gly2238 Val2240 |
Val224 0 |
Gly223 8 Val224 0 |
Val2240 | Val2240, Asp2357 |
Val224 0 |
Lys2187, Asp2195 |
- | ||
| 9. | PI3K | Docking Score | −8.11 | −8.65 | −7.73 | −7.86 | −8.82 | −8.96 | −7.16 | −9.49 | −8.44 | −8.53 | −9.79 | −10.41 |
| H-bond | Val851 | Val851 | Val851 | Val851 | Val851 | Val851 | Tyr836, Asp933 |
Val851 | Val851 | Val851 | Lys802, Tyr836, Val851 |
Val851, Gln859 | ||
| 10. | PIP5K1a | Docking Score | −6.20 | −5.00 | −4.45 | −6.32 | −6.39 | −4.58 | −6.21 | −6.59 | −4.21 | −4.33 | −7.49 | −8.94 |
| H-bond | Leu224 | Leu224 | Lys171 | Leu224 | Leu224 | Lys171 | Leu224 | Leu224 | Thr257 | Leu224 | Lys171, Leu224 |
Asn222, Leu224, Asp238 | ||
| 11. | FGFR | Docking Score | −7.11 | −7.55 | −6.98 | −7.36 | −7.78 | −6.68 | −6.82 | −7.13 | −7.43 | −7.55 | −7.91 | −8.38 |
| H-bond | Glu562, Ala564 |
Glu562, Ala564 |
Ala564 | Ala564 | Ala564 | Ala564 | Glu562, Ala564 |
Ala564 | Glu562, Ala564 |
Glu562, Ala564 |
Glu571, Ala564 |
Asp541, Ala564 | ||
| 12. | CDK2 | Docking Score | −8.42 | −8.47 | −7.19 | −8.96 | −9.14 | −7.78 | −8.62 | −8.81 | −6.93 | −8.39 | −9.97 | −7.22 |
| H-bond | Leu83, Glu81, |
Leu83 | Asp145, Thr14 |
Leu83, Glu81, |
Leu83, Glu81, Asp145 |
Leu83 | Leu83, Glu81, |
Leu83, Glu81, |
Phe80, Asp145 |
Leu83, Asp145 |
Leu83, Asp86, Asp145 |
Leu83, Asp145 | ||
pdb selected for mTOR (2NPU) did not have any co-crystallized ligand
Figure 2.
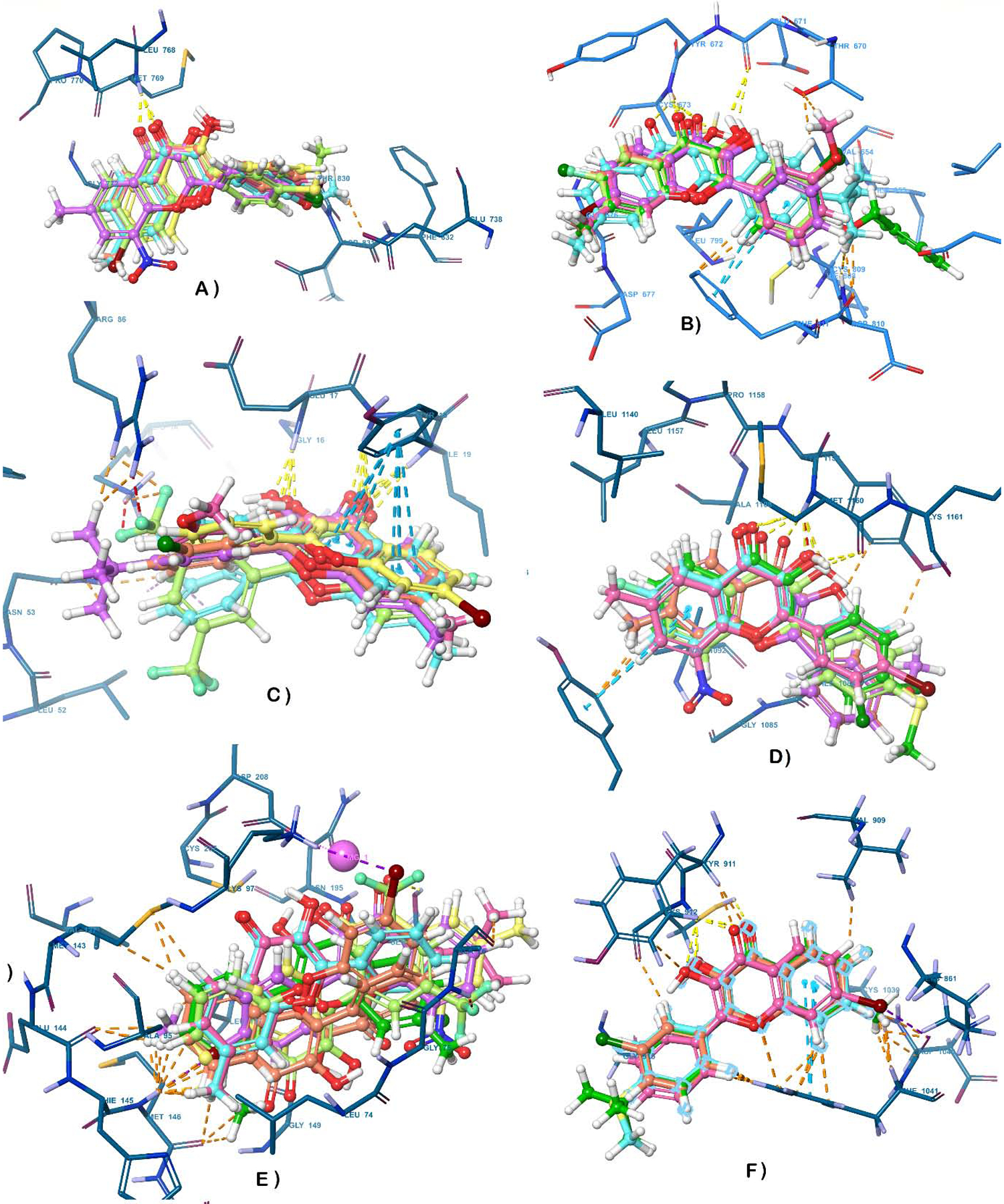
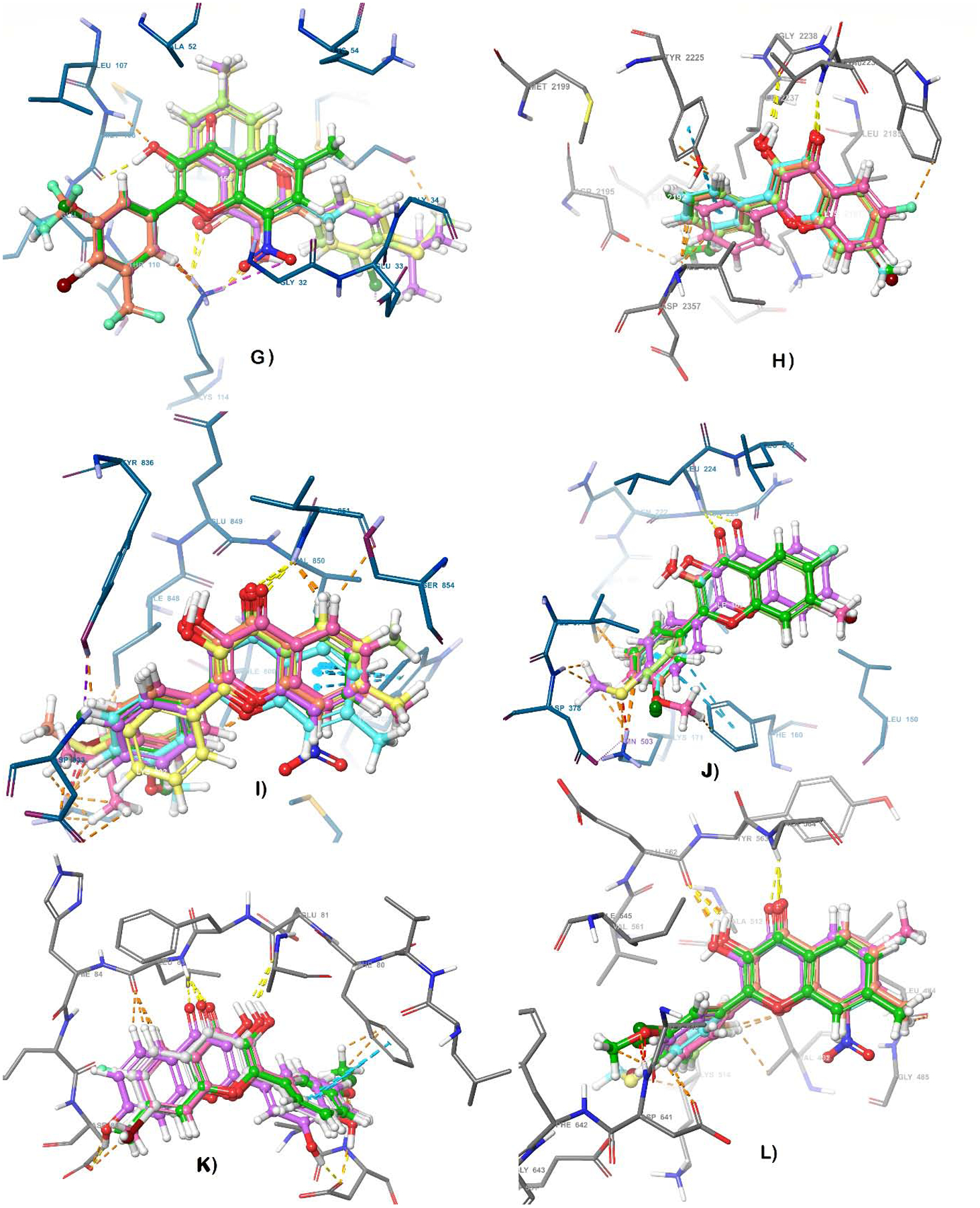
3-D interaction representations for the flavonol analogs superimposed over fisetin within the binding cavity of target enzymes a) EGFR b) cKIT c) Akt d) MET e) MEK1 f) VEGFR g) MAPK h) mTOR i) PI3K j) PIP5K1α k) CDK2 l) FGFR.
One interesting finding from in-silico studies was that F20 (the most potent compound from the MTT assay) was not predicted to conserve a similar binding pose and orientation as fisetin, which appears to be primarily due to the presence of a nitro group at position 8 on ring A, which forces the core scaffold out of the pocket in most of the cases. Nonetheless, the computed interaction energies for F20 predicted binding affinity comparable to fisetin’s for mTOR, CDK2, c-Kit and MET. It is also worth mentioning that although many compounds maintained key interactions within the catalytic domain of Akt, the obtained Glide docking scores were low, translating to predicted poor binding affinities with the protein, and thus, Akt was dropped from further studies. Also, although VEGFR and MET displayed better docking binding scores with most of the analogs than with fisetin itself, they were not predicted to maintain the key H-bonding interactions observed with fisetin. Based on these observations and taking into account the balance of both the binding energy (docking score) and essential H-bonds, only CDK2, c-KIT and mTOR were selected as possible molecular targets for further in-vitro enzymatic evaluation of the inhibitory potential of these compounds.
2.4. In-vitro enzymatic assay: c-KIT, CDK2, and mTOR kinase activity assays
With the aim of further elucidating the mechanism(s) of the observed anti-proliferative activities of these title analogs of continued interest, nine compounds (see below) selected from the initial cell-culture viability screens were further assessed for their inhibitory activities of kinases chosen based on the results of the inverse-docking studies, as discussed in the previous section. The four inverse-docking identified targets CDK2 (cyclin A, and cyclin E), c-Kit and mTOR are commonly associated with cancer progression (73–80) as already noted, and thus represent attractive targets for anticancer drug discovery. Consequently, F6, F8, F9, F11, F14, F17, F19, F20, and F21, as well as F0 were evaluated via an in vitro kinase-profiling platform from Reaction Biology Corporation. Rapamycin, staurosporine and PI-103 (at 1 and 10 μM), as well as the parent flavonol (fisetin, F0), which has been previously reported to inhibit mTOR/Akt and p70S6K1 as components of its observed anti-melanoma activity (28,29), were co-assayed as positive control drugs. For the initial screening, the activities were measured in a 10-dose IC50 mode for three-fold serial dilutions of the compounds at a starting concentration of 100 μM. The compounds were retested to exclude false positives whenever inhibition of a target kinase was seen to be pronounced, whereupon confirmed actives were further evaluated by kinase activity assay to derive IC50 values from response-vs.-concentration curves for c-Kit, CDK2/Cyclin A, CDK2/Cyclin E, and mTOR kinase inhibition, as displayed in detail in the supplementary report (68).
The obtained results summarized in Table 3 indicate that fisetin and several of the synthesized analogs exhibited potent inhibition of CDK2, c-Kit and mTOR kinase activities. In reference to fisetin used as a positive control, the respective IC50 values obtained for c-Kit, CDK2 (cyclin A, and cyclin E), and mTOR were determined to be 1.34, 2.62, 3.34 and 28.7 μM, respectively, indicating that the in vitro kinase assays reliably reproduced previous reports (28,29,38). Among the tested analogs, F20, showed the most potent kinase inhibitory activity against c-Kit (IC50 0.09 μM); F9 and F14 were comparably potent (IC50 0.12 and 0.14 μM, respectively); whereas, F8, F19, and F21 exhibited IC50 values of 0.54, 0.33, and 0.26 μM, respectively, and F17 (IC50 1.74 μM) displaying a lesser activity comparable to fisetin’s. Coincidentally, F20 also appears to be the most potent anti-proliferative and cytotoxic compound, though its molecular docking score did not predict increased binding potency vs. fisetin’s. Docking scores for F14 and F19 were similar to that of fisetin and predictive of their observed increased potencies.
Table 3.
IC50 values for c-Kit, CDK2, and mTOR inhibition by active compounds.
| Compound | c-Kit IC50 (μM) | CDK2 IC50 (μM) | mTOR/FRAP1 IC50 (μM) | |
|---|---|---|---|---|
| Cyclin A | Cyclin E | |||
| F0 | 1.34 | 2.62 | 3.34 | 28.69 |
| F6 | >100 | >100 | ND | ND |
| F8 | 0.54 | 7.16 | 17.12 | 23.93 |
| F9 | 0.12 | 2.95 | 1.78 | 1.42 |
| F11 | 101 | 32.46 | ND | ND |
| F14 | 0.13 | 6.64 | 5.38 | >100 |
| F17 | 1.74 | 4.57 | 5.70 | 11.45 |
| F19 | 0.33 | 12.74 | ND | 1.64 |
| F20 | 0.09 | 4.77 | >100 | ND |
| F21 | 0.26 | 30.10 | ND | ND |
With respect to the CDK2 kinase activities, five analogs (F8, F9, F14, F17, and F20) for CDK2/cyclin A, and three (F9, F14, and F17) for CDK2/cyclin E, showed IC50 values below 10 μM. For cyclin A inhibition, F9 was seen to be most potent (IC50 2.95 μM), with F8, F14, F17, and F20 exhibiting IC50 values of 7.12, 6.64, 4.57, and 4.77 μM, respectively. F9 was also the most potent inhibitor of cyclin E (IC50 1.78 μM), whereas compounds F14 and F17 displayed IC50 values of 5.38 and 5.70 μM, respectively (cf. Table 3). The detailed analysis of the kinase activities of these analogs are displayed in the supplementary report (68).
Compounds F9 and F19 emerged as the most potent inhibitors against mTOR, with IC50 values of 1.42 and 1.64 μM, respectively; analogs F8 and F17 showed mTOR kinase inhibitory activities (IC50 values of 23.9 and 11.5 μM, respectively) comparable to that of fisetin. However, analogs F11, F20 and F21 did not substantively inhibit cyclin E and mTOR kinase activity. Not all of the identified active antiproliferative analogs exhibited strong inhibitory effects against these kinases. This was particularly true for F6 and F11, with neither compound showing detectable kinase inhibitory activity against any of the tested targets (particularly surprising with respect to lack of c-Kit inhibition given the Glide docking scores), despite their noteworthy anti-proliferative and cytotoxic properties (see Table 1). These latter observations suggest the participation of some other yet-to-be-identified cellular targets and signalling pathways mediating the anti-proliferative activities of F6 and F11.
Taken together, the kinase inhibition data suggest that F8, F9, F17 and F19 may exert their antiproliferative activities at least in part through their various degrees of actions as multi-kinase (c-Kits, CDK2/cyclin A, CDK2/cyclin E, or mTOR) inhibitors (Table 3). Meanwhile, F20, the most-potent and selective anti-proliferative hit for cancerous vs. non-cancerous cells, may be acting primarily as a c-Kit kinase inhibitor, with some inhibitory effects on cyclin A. F14 displayed dual c-Kit/CDK2 kinase inhibition, while F19 is a potent dual c-Kit/mTOR inhibitor, with some effects on cyclin A. F21 seems to have a significant effect only on c-Kit among the kinases investigated. Reasonably, full-kinome screening would be needed to reach any further conclusions, and certainly, given the spectrum of biological activities of flavonols seen in the totality of the literature for this class of compounds (and for flavonoids more generally), other as-yet-to-be-identified non-kinase targets may likely contribute to the observed antiproliferative actions. Most importantly, it was noted that the multi-kinase activities observed in the current studies corresponded with the anti-cancer effects exhibited by these compounds on skin cancer cells, and that the kinase inhibition data for analogs F8, F9, F14, F17, F19, F20 and F21. However, noting both observations might provide valuable information regarding the anticancer mechanism(s) of actions of these compounds and for treatment of these types of cancers, further mechanistic and phenotypic functional studies were pursued.
2.5. Potent multi-kinase-inhibiting fisetin analogs modulate cellular biochemical parameters to induce apoptosis in A375 and A431 skin cancer cells.
To explore the mechanisms by which the synthesized flavonols induce cell death, compounds F8, F9, F14, F17, F19, F20, and F21 were selected based on their observed in vitro phenotypic efficacies (anti-proliferative activities) and target-level potencies(kinase(s) inhibition). Because the induction of apoptosis in cancer cells is a vital component for successful cancer therapy, as it is linked to the efficacy of the vast majority of anticancer agents, it was further investigated whether the above-listed chosen analogs could induce or promote apoptotic processes selectively in cancerous cells. Initially, their effects on nuclear morphology were assessed via 4′,6-diamidino-2-phenylindole (DAPI) staining, monitoring the effects and percentage of apoptotic cells by immunofluorescent microscopy analysis. We found that the nuclei of A375 and A431 cells treated with compounds F8, F9, F14, F17, F19, F20, and F21 exhibited highly shrunken/condensed and fragmented chromatin with punctuation, clearly distinguishing drug-treated apoptotic from the normally cobblestone-like appearances of non-apoptotic skin cancer cells (data not shown). As summarized (Table 4), the percentage of apoptotic cells was significantly increased by each of the seven best hit compounds at concentrations of ½ IC50 and IC50 in both A375 and A431 cells. Following this initial pro-apoptotic screening assay, only three among the seven tested analogs, namely F20, F9, and F17, were retained for further experiments, based on their aggregate anti-proliferative, kinase inhibitory and pro-apoptotic potencies.
Table 4.
Summary of percentage of apoptotic A375 and A431 cells as derived from relative numbers of DAPI-stained nuclei of cultured cells treated for 36 hours with selected flavonols at ½IC50 and IC50 antiproliferative concentrations (cf. Table 1).
| DAPI (+) apoptotic cells (%) | ||||||
|---|---|---|---|---|---|---|
| Cell line | A375 | A431 | ||||
| Compound ID | Control | ½IC50 | IC50 | Control | ½IC50 | IC50 |
| F8 | ND | 6.5 | 11.3 | 4.1 | 5.84 | 12.8 |
| F9 | 3.4 | 7.5 | 18.2 | 3.6 | 7.93 | 19.1 |
| F14 | 2.9 | 6.2 | 10.9 | 3.5 | 6.92 | ND |
| F17 | 3.9 | 6.9 | 17.7 | 3.8 | 8.81 | 18.4 |
| F19 | 3.3 | 5.6 | 13.3 | 3.6 | 6.71 | 14.2 |
| F20 | 3.0 | 8.2 | 20.4 | 4.2 | 14.6 | 27.7 |
| F21 | 3.2 | 5.4 | ND | 3.9 | 8.8 | 14.4 |
Apoptotic caspases belong to the family of cysteinyl aspartate specific proteases, often classified into sets including initiators (caspases 8, 9 and 10) and executioners (caspases 3, 6 and 7) (90,91). Employing both A375 and A431 cells, the modulatory effects of compounds F20, F9, and F17 treatment were examined on the intrinsic and extrinsic apoptosis signaling pathways, by probing the protein expression levels of a number of key apoptosis-related marker proteins using Western blot analysis. Thereto, expression levels of apoptotic markers including the pro- and cleaved (activated) forms of initiator (caspase 8) and executioner (caspase −3 and −7) caspases, and activation of Poly (ADP-ribose) polymerase (PARP) cleavage, were assessed. Treatment of A375 and A431 cells with F20, resulted in significant activation of apoptosis, as evidenced by the elevated protein expression levels of cleaved caspases −3, −7, and −8 (Figure 3), and PARP (Figure 4) in a concentration-dependent manner when matched with untreated controls. In-depth data displaying the effects of treatment with F9 and F17, indicating similar patterns on these cells, are presented in the supplementary dataset (68). The cleavage of caspase −3, −7, −8, and of PARP, was also present in UWBCC1 cells similarly treated with each of these three hit compounds (data not shown).
Figure 3. Potent flavonol F20 induces apoptosis through activation of the extrinsic and intrinsic apoptotic pathway caspases in melanoma and non-melanoma cancer cells.
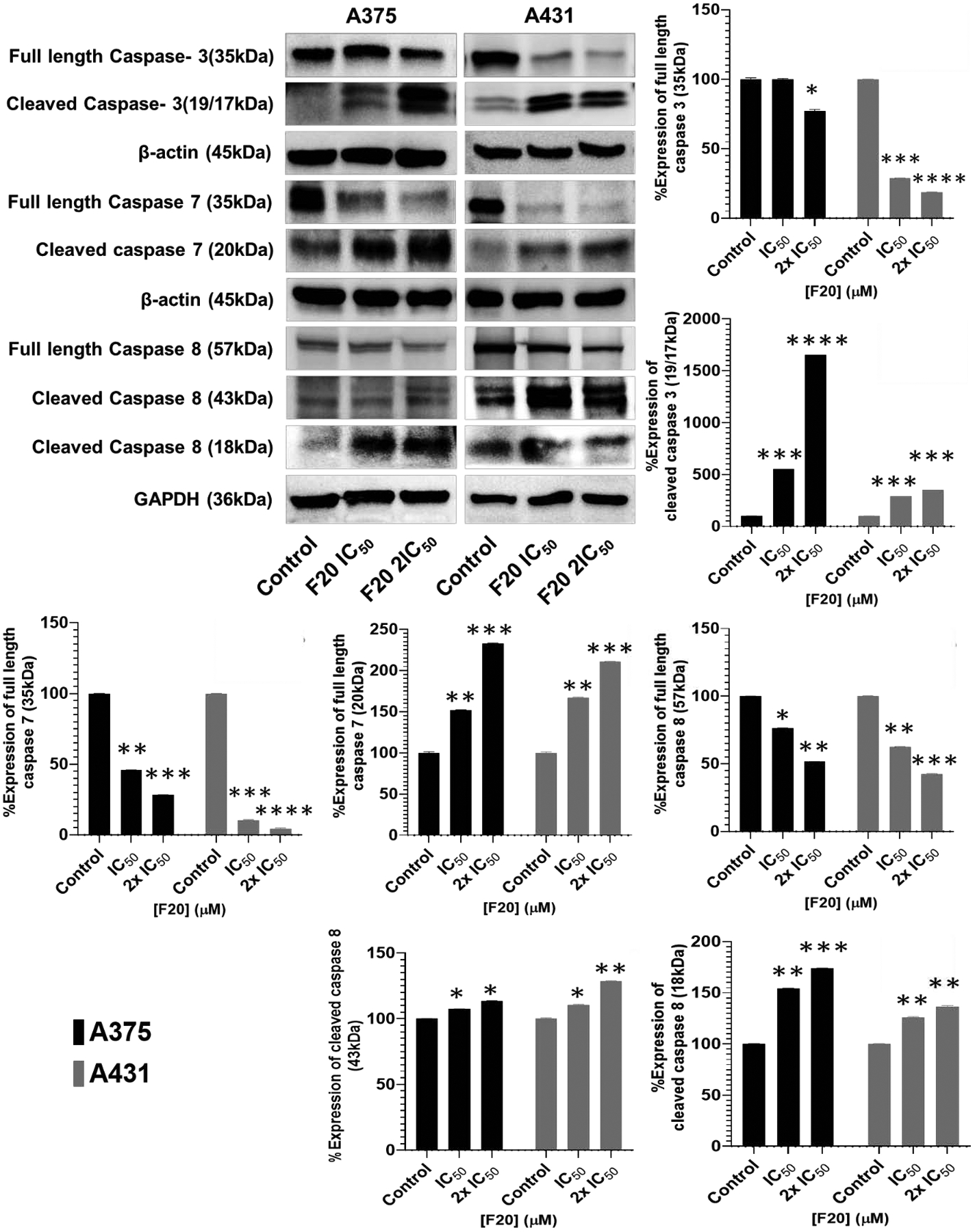
The blots show the effect of F20 at IC50 and 2×IC50 concentrations on the protein expression levels of markers of apoptosis, including pro- and-cleaved caspase−3, −7 and −8, components of the intrinsic apoptosis pathway, after 48-h treatment, comparing with untreated controls. A375 and A431 cells were incubated in the absence or presence of F20 (0, IC50, 2×IC50; 48 h), and whole-cell lysates were subjected to SDS-polyacrylamide gel electrophoresis. The Western blot data shown are representative immunoblots from three independent experiments with similar results. Equal protein loading was confirmed by re-probing for β-actin or GAPDH as loading control, and the actual protein levels were normalized to the loading control to obtain % Expression values shown on the bar graphs (mean ± SD of relative quantitative density values are plotted). The data are expressed as the percentage to which derivatives F20 suppressed or induced the protein expression levels of pro- and-cleaved caspases in A375 and A431 cells as compared with controls. Statistical significance was assessed using one-way ANOVA and Dunn’s multiple comparison tests, and p < 0.05 (*) was considered significant.
Figure 4. Potent flavonol F20 induces apoptosis through activation of mitochondrial pro-apoptotic pathways involving Bcl-2 family proteins and of PARP cleavage in melanoma and non-melanoma cancer cells.
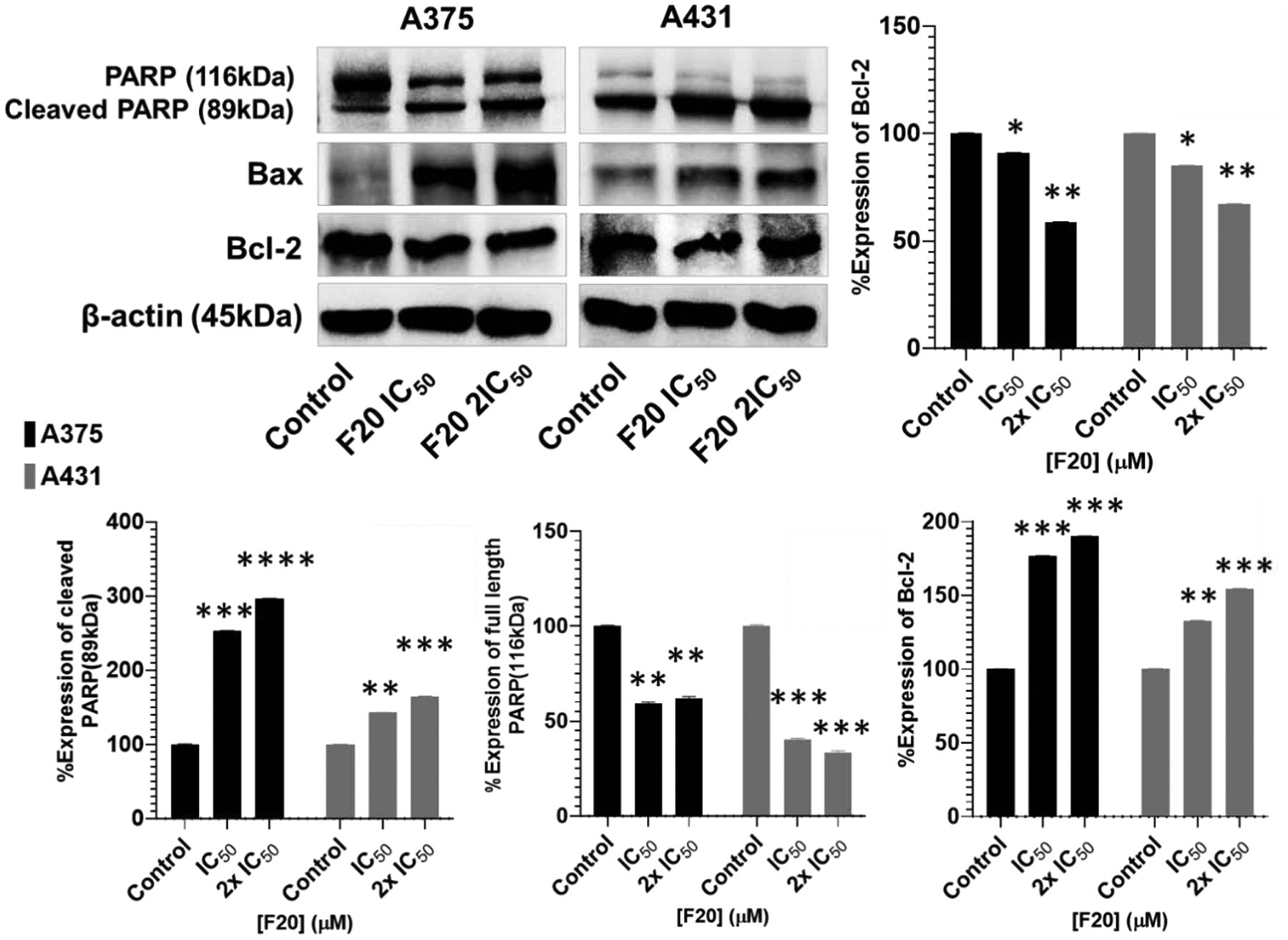
The Western blot data show the effects of F20 at IC50 and 2×IC50 concentrations for 48 h on the protein expression levels of intact and cleaved PARP (85 kDa), as well as Bcl-2 family of proteins (Bax and Bcl-2), components of the intrinsic apoptosis pathway, in A375 and A431 cells. A375 and A431 cells were incubated in the absence or presence of F20 (0, IC50, 2×IC50; 48 h), and whole-cell lysates were subjected to SDS-polyacrylamide gel electrophoresis. The blots shown are representative of immunoblots from three independent experiments with similar results. The data are expressed as the percentage suppression of the protein expression levels of Bcl-2 and cleaved PARP (116 kDa), and percentage increase in levels of cleaved PARP ((85 kDa)) and of Bax, in treated A375 and A431 cells as compared with controls. Equal protein loading was confirmed by re-probing for β-actin as loading control, and the actual protein levels were normalized to the loading control to obtain % Expression values shown on the bar graphs (mean ± SD of relative quantitative density values). Statistical significance was assessed using one-way ANOVA and Dunn’s multiple comparison tests, and p < 0.05 (*) was considered significant.
The expression level changes for Bcl-2-family proteins functioning as important regulator proteins in the mitochondrial pathways of apoptosis induction, namely Bax, (a pro-apoptotic protein) and Bcl-2 (an anti-apoptotic protein) (92), in A375 and A431 cells was studied upon treatment with compounds F9, F17 and F20. Besides the increased number of DAPI positively stained apoptotic cells, treatment of both A375 and A431 cells with F20 (Figure 4), significantly increased the expression level of the pro-apoptotic proteins, Bax, whereas the level of anti-apoptotic protein Bcl-2 was decreased (suppressed) after treatment as compared with the control. Similar results were obtained from in-depth analysis of these markers upon treatment with compounds F9 and F17, and are displayed in the supplementary report (68). Treatment with these compounds increased the ratio of Bax/Bcl-2 in a concentration-dependent manner (see Figure 4). Taken together, these data further support the hypothesis that compounds F20, F9 and F17 can induce the apoptosis of skin cancer cells, specifically cells of A375 and A431 lines, through activation of the intrinsic mitochondrial apoptotic signaling pathway, leading to cleavage of and caspases 3, 7 and 8 as well as of PARP activation.
2.6. Potent fisetin analogs inhibit proliferation and induce apoptosis through downregulation of the expression of CDK2, and suppression of phosphorylated c-Kit and downstream targets in A375 and A431 cells.
Aberrant cell proliferation and dysregulation of the cell cycle, hallmarks of malignant tumors, are dependent on the coordination of cyclins and cyclin-dependent kinases (CDKs), particularly the CDK2 which amongst others, crucially regulates G1/S and modulates S/G2 cell cycle progression (93). However, CDK2 activity exceeding a certain threshold has been suggested to worsen outcomes for “CDK2-low” cancers, thus predictive that inhibitors targeting CDK2 could represent promising anticancer strategy (93). Melanoma and squamous cell carcinoma represent the two such cancers, and together account for a major portion of skin cancer-related deaths, with the incidence and mortality rate having increased globally over the past few decades (94,95). Additionally, alterations in c-KIT, a proto-oncogene that encodes the corresponding transmembrane receptor tyrosine kinase, has been reported to be mutated and aberrantly expressed in melanomas and SCC (95 and for reviews see 94,96). Due to its important association with cell survival and differentiation, c-KIT inhibition has been therapeutically targeted in several advanced melanoma clinical trials using inhibitors, including imatinib, dasatinib, nilotinib, and sunitinib, all with problematic side-effects (94). Even so, the data from these trials and subsequent clinical experience provides endorsement of c-KIT as an opportune anti-cancer target, particularly when co-targeted with other pathways. Moreover, it has been documented that the downstream effectors of c-KIT driven melanoma are the MAPK/ERK and PI3K/AKT pathways (94,96). In this study, since most of these compounds, particularly F20, bind strongly c-KIT and/or CDK2 and inhibit their kinase activities, A375 and A431 cells were treated with various doses (0, IC50 and 2×IC50) of F9, F17, and F20 for 48 hours to evaluate their biological effects on CDK2, activated c-KIT and the downstream targets by Western blot analysis. As shown (Figure 5), compound F20 significantly suppress the protein expression levels of cyclin A2, cyclin E2 and CDK2 as compared to untreated control group, though somewhat disparately between the two cell lines. A similar trend was observed for F9 and F17 treated cells as detailed in supplementary report (68).
Figure 5: Flavonol analogs inhibit the protein expression levels of cyclin A/E, CDK2 and phosphorylated c-Kit and Stat3 in A375 melanoma and A431 non-melanoma cancer cells.
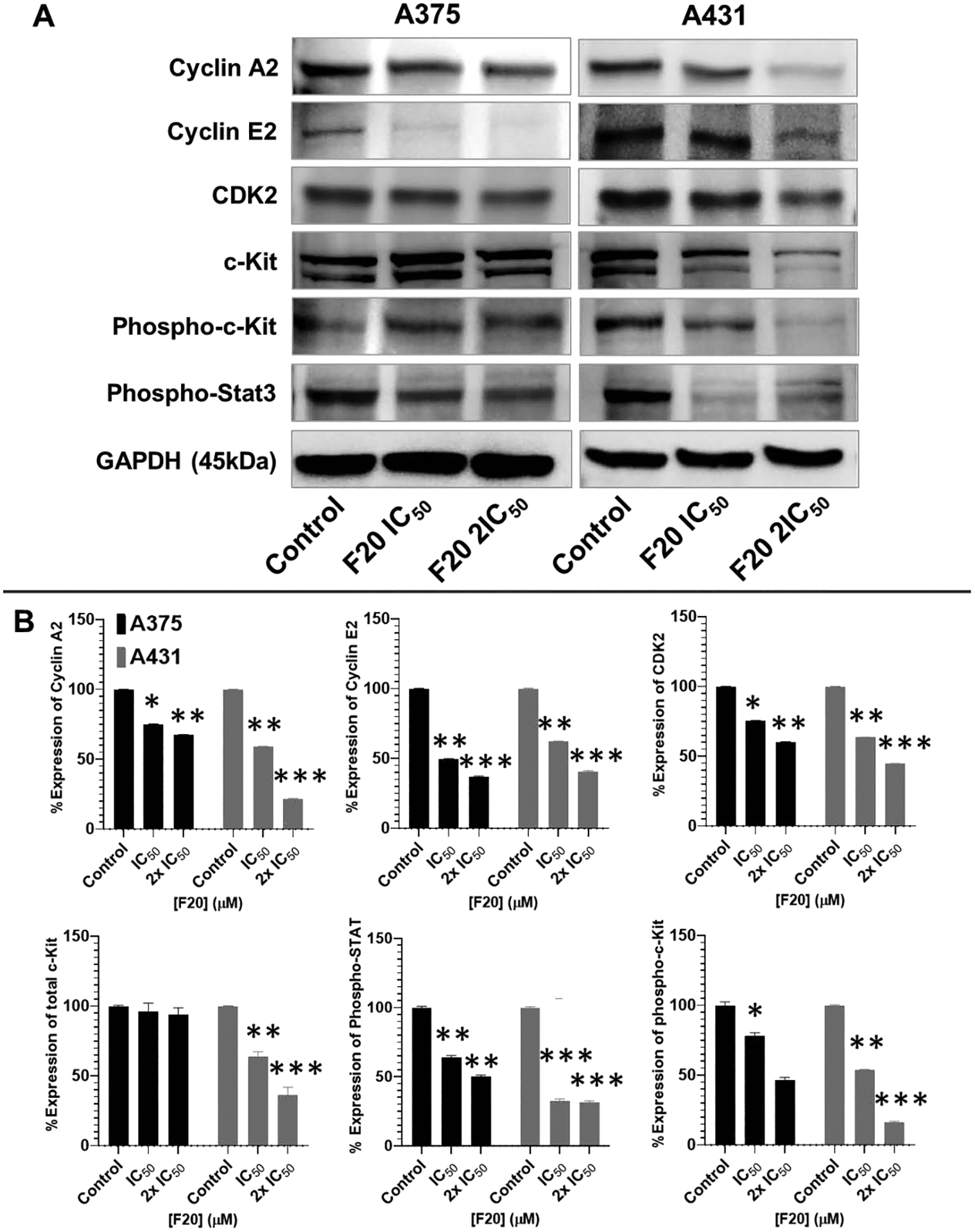
(A) Western blot data showing the effect of different concentrations of F20 on protein expression levels of cyclin A2, cyclin E2, CDK2, c-Kit, and phosphorylated c-Kit and Stat3 in 48 h-treated A375 (left panel) and A431 (right panel) cells. A375 and A431 cells were incubated with/without F20; 0, IC50, 2×IC50; μM, 48h), and western blotting performed as described in the method section. Equal protein loading was confirmed by reprobing for GAPDH as loading control, and protein levels were normalized to the loading control and expressed as percentage. The blot data shown are representative of immunoblots of three independent experiments with similar results. (B) The data expressed in the bar graphs represent mean ± SD of relative quantitative normalized density values in percentage with an internal loading control from three independent experiments. Analog F20 significantly suppressed the protein expression levels of these in A375 and A431 as compared with untreated control cells. Bar graphs, the statistical significance was determined using one-way ANOVA and Dunn’s multiple comparison test, and p < 0.05 (*) was considered significant.
Moreover, the protein expression levels of total c-Kit were not significantly altered by treatment with any of these drugs at indicated concentrations. In contrast, the protein expression levels of the phosphorylated form of c-Kit were significantly suppressed by F20 (Figure 5). Detailed results on the effects of F9 and F17, which showed similar activities, are presented in the supplementary report (68). Importantly, these compounds also modulated the downstream effector targets (signature) of c-Kit signalling, including those of the phosphorylated Stat3, PI3K/p90RSK/Akt/mTOR, and MAPK (ERK1/2) pathways (Figures 5 and 6) and, as detailed in the supplementary report (68). Western blot analysis for A375 and A431 cells treated with these compounds showed that along with activated c-Kit, phosphorylated p90RSK (Ser380), Akt (Ser473), ribosomal protein S6, and mTOR (Ser2448) were significantly suppressed when compared to untreated controls (Figure 5); see detailed results of F9 and F17 in the supplementary dataset (68). Similarly, these compounds notably suppressed the expression level of phosphorylated Stat3 (Tyr705) (see Figure 5 and supplementary dataset report (68)), and mechanistically downregulated the corresponding increases in protein expression levels of phosphorylated p44/42-MAPK (ERK1/2; Thr202/Try204) in A375 and A431 cancer cells (see Figure 6 and supplementary dataset report (68)). These results demonstrate that the selected compounds, particularly F20, effectively inhibit cell proliferation via targeting CDK2, c-Kit, and associated signalling pathways (see Figures 5–6 and supplementary dataset report (68)).
Figure 6: Potent flavonol analogs inhibit the protein expressions of activated c-Kit and downstream effectors levels, including phosphorylated mTOR/ ribosomal protein S6/ MAPK (ERK1/2) and p90RSK in A375 melanoma and A431 non-melanoma cells.
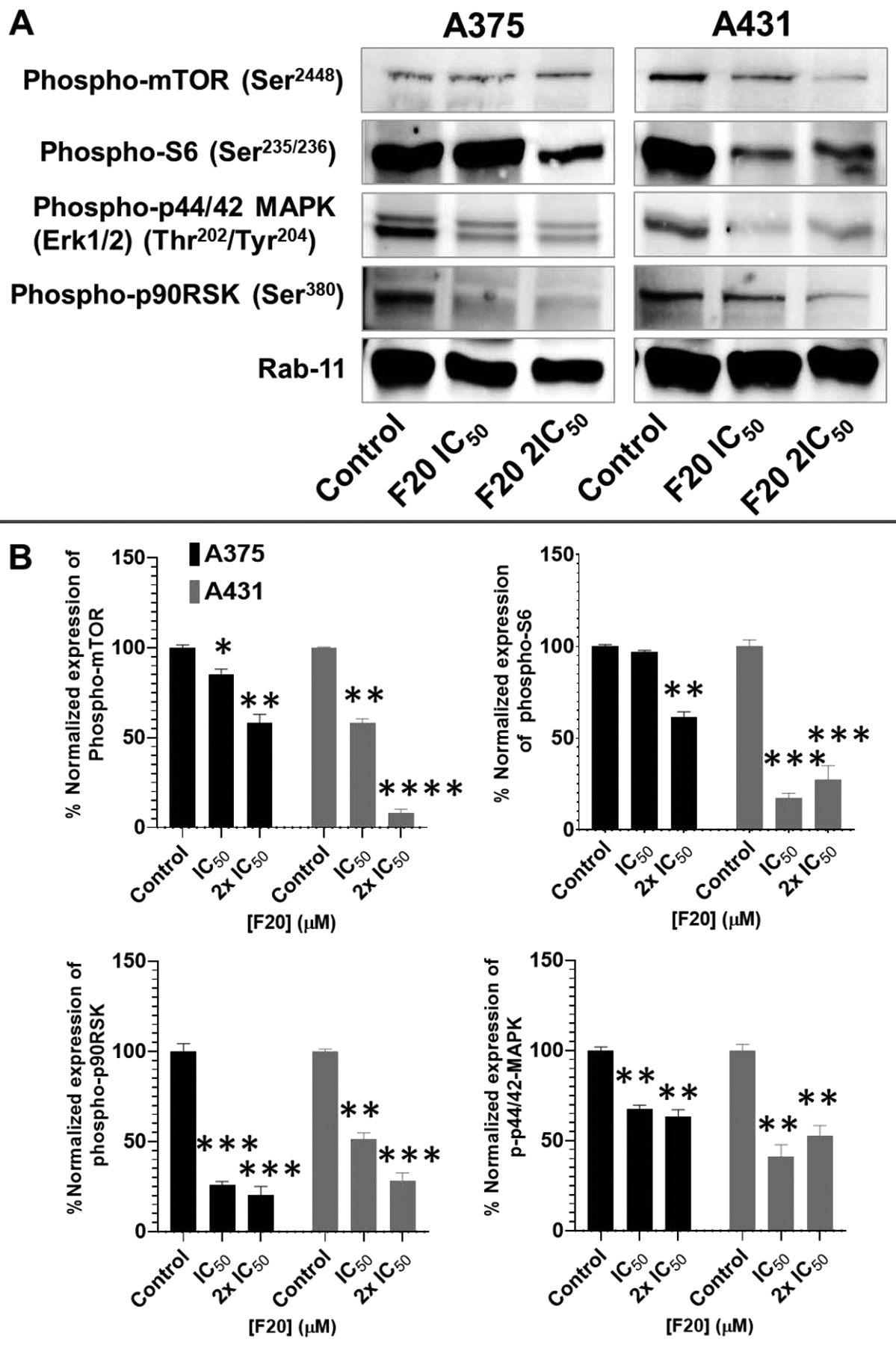
(A) Western blots displays are representatives of c-Kit downstream effectors the protein expression levels of phosphorylated mTOR, ribosomal protein S6, MAPK p44/42(ERK1/2) and p90RSK, in 48h treated A375 (left panel) and A431 (right panel) cells. A375/A431 cells were incubated with/without test compounds (F20; 0, IC50, 2×IC50; μM, 48h), and western blotting was carried out as described in the Methods section. Equal protein loading was confirmed by reprobing for Rab11 as loading control, and protein levels were normalized to the loading control and expressed as percentage. The data shown are representative of immunoblot of three independent experiments with similar results. (B) The data expressed in the bar graphs represent mean ± SD of relative quantitative normalized density values in percentage with an internal loading control. The analogs F20 significantly suppressed the protein expression levels of the phorphorylated form of the targets in A375 and A431 as compared with untreated control cells. Statistical significance was determined using one-way ANOVA and Dunn’s multiple comparison test, and p < 0.05 (*) was considered significant.
2.7. Favonol analogs inhibit Scratch Wound Healing and colony formation in 2D cultures of A375 and A431.
The selected flavonols of interest, F9, F17 and F20 were further investigated with respect to modulation of cell migration and were evaluated using scratch wound healing assay with plated aggressive melanoma (A375) and non-melanoma (A431) cells. Cells were plated and grown to confluence, scratch wounds were created as described in the Methods section 4.5.7, and were subsequently treated with or without different doses (½IC50, IC50, and 2×IC50) of the compounds, with a goal of analysing their impacts on the migration of cells into the wounded cell-free area after 48 h of scratch-wounding. Through imaging analysis, the invasion of cells into the wounded area or migration rates, were assessed by measuring the area of cell wound closure area into the initial cell-free areas, expressed as percentage of cell-free areas for comparisons. After 48 h of incubation, the test compounds (particularly F20) significantly decreased the cultured cells scratched wound area in a dose-dependent manner (Figure 7A–B). A similar trend was observed for compounds F9 and F17; detailed results are displayed in the supplementary dataset report) (68), which exhibited 28–72% inhibition of wound closure, as compared to untreated controls.
Figure 7: The most-potent analog, F20, significantly inhibited Scratch wound healing in 2D cultures of A375 and A431 in a dose-dependent manner.
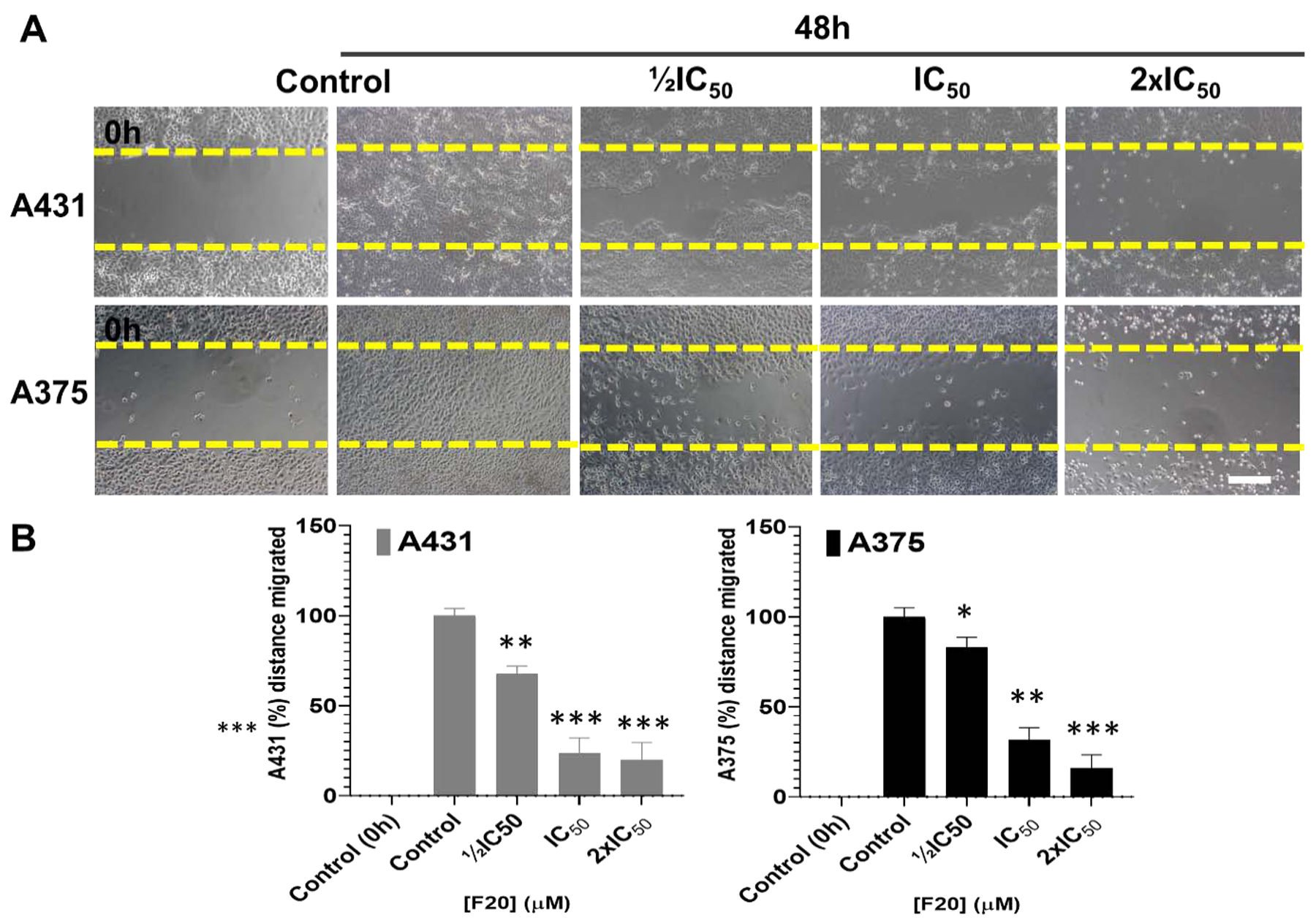
Effect of the flavonol analog F20 on the closure/migration into the initial cell-free scratch-wound areas compared to the percentage of control cell-free areas (A) (Upper panel) A431 and (lower panel) A375. (B) The data presented indicate significant dose-dependent decrease in cultured cells repopulating the scratch wound healing area in the presence of F20, as compared to untreated control cells, after 48 h of incubation. The data in the bar graphs represent mean ± SD of scratched-wound area values, expressed as percentage, compared to 0 h control and 48 h controls from three independent experiments. F20 significantly decreased wound healing area A431 (left panel) and A375 (right panel) as compared with untreated controls. Statistical significance was assessed using one-way ANOVA and Dunn’s multiple comparison test, and p < 0.05 (*) was considered significant.
To further assess the long-term effect of these selected analogs, including F9, F17, and F20, on the A375 and A431 cells growth inhibition, colony formation assays were performed in the presence or absence of different concentrations of the compounds (½IC50, IC50 and 2×IC50). As summarized in Figure 8A–B, flavonol F20, significantly reduced the percentage of colonies in a concentration-dependent manner as compared to respective control groups. The significant reduction in these parameters was similar upon treatment with F9 and F17, and the in-depth results are displayed in supplementary dataset (68). Taken together, these data from anti-proliferative/cytotoxic, scratch-wound healing, and colony formation assays, together with altered levels of an aggregation of protein markers of disease, indicate that the fisetin analogs prepared and investigated in these studies inhibit growth, migration and proliferation of cutaneous carcinoma cells.
Figure 8: Potent analog F20 significantly inhibits colony formation in 2D cultures of A375 and A431 cells in a dose-dependent manner.
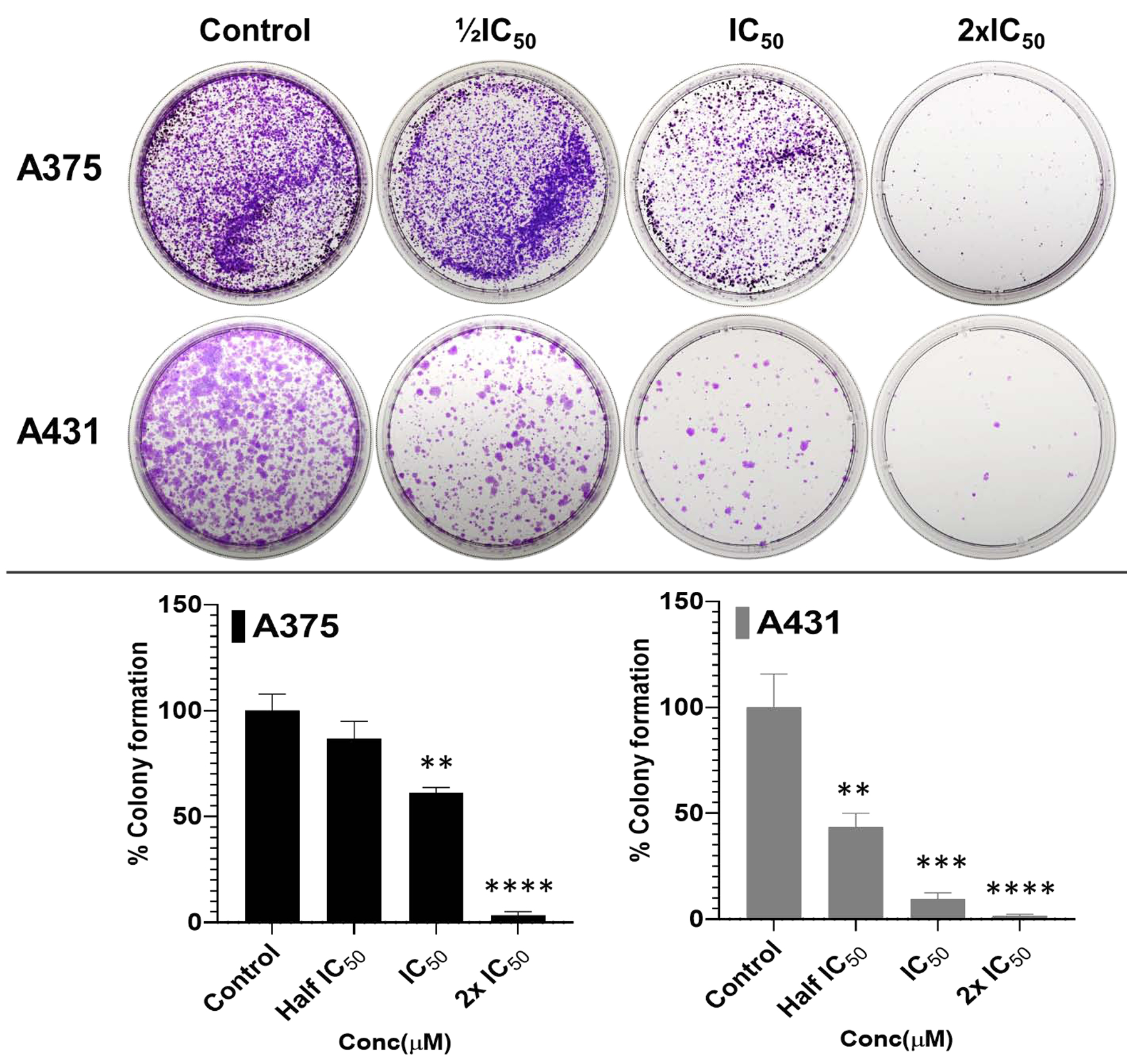
(A-B) Long-term effect of the most potent flavonol, F20, on clonogenic potential in A375, and A431 cells. Treatment with derivative F20 (½IC50, IC50 and 2×IC50) of the respective cell lines, significantly reduced/suppressed the percentage of colonies formed in a dose-dependent manner when compared to the respective control untreated cutaneous carcinoma cells. (B) The data expressed in the bar graphs represent mean ± SD of values in percentage control from three-independent experiments. Statistical significance was assessed using one-way ANOVA and Bonferoni’s multiple comparison test; p < 0.05 (*) was considered significant.
3. Conclusions
Growing evidence indicates that the potential high therapeutic index of fisetin is significantly reduced due to bioavailability, variable activity, toxicity and biotransformation into less active metabolites following oral administration in vivo. Therefore, new fisetin derivatives designed to overcome these issues may lead to the development of new compounds exhibiting high therapeutic indexes. In this study, the anticancer activity of 24 synthesized fisetin analogs were investigated, and the results indicate that some of the compounds displayed comparable to better anti-proliferative/cytotoxic properties compared to fisetin. A selected number of these analogs exhibited significant inhibition of the activation of deregulated disease molecular targets including specific kinases, activation of apoptosis, and suppression of colony formation and wound closure in a cell culture model of skin carcinogenesis. Therefore, these data indicate that the synthesized flavonol-analogs of fisetin lacking the 7-hydroxy group on ring A and the 3’,4’-dihydroxy groups on ring B, are capable of displaying potent anti-proliferative/cytotoxic properties up to 233-fold higher than the parent fisetin’s, with selectivity index of 283, over normal cell line as shown for compound F20. These fisetin derivatives likely mediate their actions through several parallel pathways, including direct and indirect effects on pro-survival and/or apoptotic pathways. Importantly, some of these analogs (i.e. F9, F17 and F20) exert their cytotoxicity against cancer cells by significantly inducing apoptosis markers; cleaved caspases −3, −7, −8, PARP, Bax/Bcl-2 ratio. In addition, they inhibited single or multi deregulated molecular cancer targets such as CDK2, phosphorylated c-Kit and its downstream effectors including Akt, mTOR, p70S6K, p90RSK, Stat3, and MAPK (ERK1/2), which are mostly hyper-activated in melanoma and NMSCs. Although extended cytotoxicity or in vivo studies are needed to assess whether the broad activity is specific and not connected to general cytotoxic effects, the current study provides information and an available model to develop chemotherapeutic agents with improved therapeutic index on the basis of phytochemical prototypes. However, the main takeaway from these studies is the fact that a number of derivatives lacking the aforementioned key impediments to fisetin’s clinical efficacy, have proven to display better potency against cells of both non-melanoma and melanoma lines when compared to fisetin. Finally, it should also be noted here that the effects of various substituents on lipophilicity, and thus, cell penetrability, might also matter and could perhaps be thought to confound the SAR studies to some degree; the equilibrative nature of these cell culture studies should have mostly averted that issue, but in that same vein, close attention was paid to any prospective constraints as might be imposed by solubility limits of compounds in the cell culture media. Taken together, the study data identify F20, as well as F9 and F17, as promising c-Kit, CDK2 and mTOR inhibitor prototypes that warrant further investigation as single agents or in combination with, or as adjuvants to standard therapies for the control of melanoma and NMSCs.
4. Materials and Experimental methods
4.1. Chemistry
4.1.1. Synthesis and Characterization of Flavonols
General information:
For the chemistry portion of this study, all solvents and other chemicals were purchased from Sigma-Aldrich, and TCI except for 2’-hydroxy-4’-methoxyacetophenone and m-anisaldehyde, which were obtained from Alfa Aesar. All the reagents were commercial grade, and were used without further purification unless otherwise indicated. Organic extracts were dried over anhydrous sodium sulfate, and solvents were removed under a reduced pressure on a Buchi Rotavapor equipped with a vacuum controller and vacuum pump. Reaction mixtures were monitored by TLC using silica gel 60 F254 plates, with a fluorescent indicator, and silica gel (60–120 mesh size) was used for column chromatography. NMR spectra were recorded on JEOL 400 MHz FT spectrometer in the indicated solvent. Chemical shifts are expressed in δ units (ppm) from tetramethylsilane (TMS) used as the internal standard for 1H NMR (400 MHz) and for 13C NMR (100 MHz). Infrared spectra (IR) were run on a SpectrumOne FT-ATR spectrophotometer (Perkin Elmer). Band position and absorption ranges are given in cm−1. GC-MS analysis carried out on an Agilent GC-MS (7890A – 5975C VL MSD) system. High Resolution Mass Spectrometry (HRMS) was performed on an Agilent 6230, and spectra recorded by electro-spray ionization (ESI) with a Q-TOF. Microwave-assisted reactions were performed on Discover S-Class (CEM) single mode microwave reactor, with the instrument settings controlled by PC-running Synergy 1.4 software. All experiments were carried out in microwave reaction vials using a stirring bar (length 20 mm, diameter 6 mm). Stirring, temperature, irradiation power, PowerMAX (in situ cooling during the microwave irradiation), ramp and hold times were set as indicated, and the temperature of the reaction was monitored by a built-in infrared sensor.
4.2. General synthetic procedures
4.2.1. Microwave Assisted Synthesis of Chalcones:
A 10 mL microwave reaction vial, equipped with a stir bar, charged with the substitued 2-hydroxyacetophenone (5 mmol), the appropriate aldehyde (5 mmol), ethanol (6 mL) and potassium hydroxide (1.5 eqv.), was stirred at room temperature. The vial was then sealed with Teflon-lined silicone cap and placed in the microwave reactor. Microwave irradiation of 250 W was used and the temperature ramped from 25 °C to 80 °C. Once it reached 80 °C in about 1 minute, the reaction was maintained for 20 minutes at the same temperature, then cooled to room temperature. The mixture was extracted with ethyl acetate and evaporated to get the crude product. The solid product mixture was recrystallized using acetonitrile to yield the product.
4.2.2. Room Temperature Synthesis of Flavonols from Chalcones:
To a solution of chalcone intermediate (1 mmol) in methanol (5 mL), 3M aqueous solution of KOH (1.4 mL, 3 mmol) was added and stirred for 10 minutes. Then a 30% aqueous solution of H2O2 (0.35 mL, 3 mmol) was added slowly under a continuous stirring for 2 hrs at room temperature. The resulting suspension was treated with 10% aqueous solution of HCl (5 mL), extracted with dichloromethane, dried with sodium sulfate, and evaporated. The products were purified by column chromatography (using hexane-ethyl acetate) or recrystallization (using 80:20 acetonitrile/ethanol solvent system).
4.2.3. Microwave Assisted One-Pot Synthesis of Flavonols:
A 10 mL microwave reaction vial, equipped with a stir bar, charged with substituted 2-hydroxyacetophenone (5 mmol), the appropriate aldehyde (5 mmol), ethanol (6 mL) and potassium hydroxide (1.5 equiv.) was stirred at room temperature. The vial was the sealed with Teflon-lined silicone cap and placed in the microwave reactor. Microwave irradiation of 250 W was used and the temperature ramped from 25 °C to 80 °C, then maintained for 20 min at the same temperature while stirring, and cooled to room temperature. A potassium hydroxide (0.45 mL, 1 mmol) solution in methanol and 30% solution of hydrogen peroxide (0.35 mL, 3 mmol) were added, and placed back into the microwave reactor. The mixture was stirred at 40 °C for 10 min and cooled to room temperature, extracted with ethyl acetate, dried over magnesium sulfate, and evaporated to get the crude product. The products were purified by column chromatography using hexane-ethyl acetate as eluent.
4.2.4. Characterization of the flavonols synthesized
3-Hydroxy-2-phenyl-4H-chromen-4-one (F1):
1H-NMR (400 MHz, CDCl3): 7.41 (t, 1H, J = 7.8 Hz, ArH), 7.46 (d, 1H, J = 7.3 Hz, ArH), 7.52 (t, 2H, J = 7.3 Hz, ArH), 7.57 (d, 2H, J = 8.7 Hz, ArH), 7.68 (t, 1H, J = 6.8 Hz, ArH), 8.24 (d, 2H, J = 8.2 Hz, ArH). 13C-NMR (100 MHz, CDCl3): 118.4, 120.7, 124.6, 125.5, 127.8, 128.7, 130.3, 131.1, 133.8, 138.5, 145.1, 155.5, 173.5. IR (KBr): 3258, 3153, 1619, 1607, 1562, 1481, 1471, 1415, 1211, 1129, 756, 705, 689 cm−1. HRMS (ESI) calcd for C15H10O3 [M + H]+ 239.07027; found, 239.0702.
3-Hydroxy-2-(3-methoxyphenyl)-4H-chromen-4-one (F2):
1H-NMR (400 MHz, CDCl3): 3.88 (s, 3H, OCH3), 7.09 (s, 1H, ArH), 7.01 (dd, 1H, J = 8.0 and 1.2 Hz, ArH), 7.24 (s, 1H, ArH), 7.39 – 7.45 (m, 2H, ArH), 7.57 (d, 1H, J = 8.2 Hz, ArH), 7.69 (t, 1H, J = 8.0 Hz, ArH), 7.80–7.85 (m, 2H, ArH), 8.22 (d, 1H, J = 6.8 Hz, ArH), 10.70 (brs, 1H, OH). 13C-NMR (100 MHz, CDCl3): 55.5, 113.4, 116.1, 118.3, 119.5, 120.5, 124.8, 125.5, 129.7, 130.9, 132.3, 133.9, 136.6, 155.4, 159.6, 162.2, 174.1. IR (KBr): 3252, 3098, 1621, 1605, 1563, 1482, 1473, 1288, 1204, 1168, 903, 774 cm−1. HRMS (ESI) calcd for C16H12O4 [M + H]+ 269.08084; found, 269.08060.
3-Hydroxy-2-(4-((trifluoromethyl)thio)phenyl)-4H-chromen-4-one (F3):
1H-NMR (400 MHz, CDCl3): 7.24 (brs, 1H, OH)7.42 (t, 1H, J = 7.3 Hz, ArH), 7.58 (d, 1H, J = 8.7 Hz, ArH), 7.72 (t, 1H, J = 7.1 Hz, ArH), 7.79 (d, 2H, J = 8.2 Hz, ArH), 8.24 (dd, 1H, J = 7.2 and 1.3 Hz, ArH), 8.30 (d, 2H, J = 8.7 Hz, ArH). 13C-NMR (100 MHz, CDCl3): 118.4, 120.6, 124.9, 125.6, 126.3, 128.6, 133.5, 134.2, 136.1, 139.2, 143.3, 155.5, 173.6. IR (KBr): 3243, 3012, 1622, 1606, 1495, 1478, 1422, 1286, 1213, 1127, 1100, 836, 758, 500 cm−1. HRMS (ESI) calcd for C16H9F3O3S [M + H]+ 339.02973; found, 339.02945.
2-(4-Bromo-2-chlorophenyl)-3-hydroxy-4H-chromen-4-one (F4):
1H-NMR (400 MHz, CDCl3): 6.58 (brs, 1H), 7.43 (t, 1H, J = 7.8 Hz, ArH), 7.50–7.54 (m, 4H, ArH), 7.72 (m, 1H, ArH), 8.26 (dd, 1H, J = 7.8 and 1.2 Hz, ArH). 13C-NMR (100 MHz, CDCl3): 118.2, 121.9, 124.2, 125.3, 129.4, 129.8, 132.5, 132.8, 133.4, 134.6, 139.5, 142.1, 155.3, 173.3. IR (KBr): 3261, 3019, 1625, 1611, 1468, 1292, 1215, 1130, 762, 664 cm−1. HRMS (ESI) calcd for C15H8BrClO3 [M + H]+ 350.94181; found, 350.94151.
2-(2,4-Bis(trifluoromethyl)phenyl)-3-hydroxy-4H-chromen-4-one (F5):
1H-NMR (400 MHz, CDCl3): 6.79 (brs, 1H, OH), 7.26–7.52 (m, 2H, ArH), 7.72–7.76 (m, 1H, ArH), 7.96–8.03 (m, 2H, ArH), 8.12 (s, 1H, ArH), 8.28 (dd, 1H, J = 8.4Hz and 1.6 Hz, ArH). 13C-NMR (100MHz, CDCl3): 118.4, 121.1, 125.0, 125.6, 128.6, 128.7, 132.8, 134.3, 138.8, 143.1, 155.7, 173.4.. IR (KBr): 3285, 2978, 1625, 1606, 1350, 1274, 1175, 1119, 904, 754, 661 cm−1. HRMS (ESI) calcd for C17H8F6O3 [M + H]+ 375.04504; found, 375.04475.
2-(3,5-Bis(trifluoromethyl)phenyl)-3-hydroxy-4H-chromen-4-one (F6):
1H-NMR (400 MHz, CDCl3): 7.46 (t, 1H, J = 8.0 Hz, ArH), 7.66 (d, 1H, J = 8.2 Hz, ArH), 7.77 (t, 1H, J = 6.8 Hz, ArH), 7.95 (s, 1H, ArH), 8.26 (d, 1H, J = 6.8 Hz, ArH), 8.72 (brs, 2H, ArH). 13C-NMR (100 MHz, CDCl3): 118.4, 120.6, 121.9, 123.3, 124.6, 125.2, 125.8, 127.6, 130.4, 132.0, 132.4, 133.3, 134.6, 139.6, 141.5, 155.5, 173.7. IR (KBr): 3215, 3187, 3023, 1617, 11451, 1368, 1281, 1126, 901, 754, 683 cm−1. HRMS (ESI) calcd for C17H8F6O3 [M + H]+ 375.04504; found, 375.04440.
2-(4-(Benzyloxy)phenyl)-3-hydroxy-4H-chromen-4-one (F7):
1H-NMR (400 MHz, CDCl3): 5.09 (s, 2H, ArCH2), 7.01–7.07 (m, 3H, ArH), 7.39–7.44 (m, 3H, ArH), 7.49–7.51 (m, 2H, ArH). 13C-NMR (100 MHz, CDCl3): 70.1, 73.6, 76.8, 77.1, 83.6, 115.1, 118.2, 122.1, 127.5, 128.1, 128.7, 129.0, 137.0, 159.6, 161.8. IR (KBr): 3188, 3086, 1619, 1603, 1565, 1478, 1282, 1165, 901, 773 cm−1. HRMS (ESI) calcd for C22H16O4 [M + H]+ 345.11214; found, 345.11155.
2-(3-Chlorophenyl)-3-hydroxy-7-methyl-4H-chromen-4-one (F8):
1H-NMR (400 MHz, CDCl3): 2.50 (s, 3H, CH3), 7.21 (dd, 1H, J = 8.0 and 1.2 Hz, ArH), 7.37–7.44 (m, 2H, ArH), 8.10 (d, 1H, J = 8.4 Hz, ArH), 8.13–8.15 (m, 1H, ArH), 8.20 (t, 1H, J = 2.0 Hz, ArH). 13C-NMR (100MHz, CDCl3): 22.1, 118.0, 118.3, 125.2, 125.9, 126.5, 127.4, 129.9, 130.0, 133.0, 134.8, 138.7, 145.6, 155.6, 173.4. IR (KBr): 3264, 2982, 2889, 1606, 1561, 1478, 1402, 1269, 1211, 1170, 1136, 775 cm−1. HRMS (ESI) calcd for C16H11ClO3 [M + H]+ 287.04695; found, 287.04639.
3-Hydroxy-2-(3-methoxyphenyl)-7-methyl-4H-chromen-4-one (F9):
1H-NMR (400 MHz, CDCl3): 2.50 (s, 3H, CH3), 3.88 (s, 3H, OCH3), 7.00 (dd, 1H, J = 8.4 and 2.8 Hz, ArH), 7.21 (d, 1H, J = 8.2 Hz, ArH), 7.37 (s, 1H, ArH), 7.42 (t, 1H, J = 7.8 Hz, ArH), 7.79–7.84 (m, 2H, ArH), 8.10 (d, 1H, J = 8.2 Hz, ArH). 13C-NMR (100 MHz, CDCl3): 22.1, 55.5, 113.1, 115.9, 118.0, 118.4, 120.2, 125.2, 126.4, 129.7, 132.5, 138.5, 144.4, 145.3, 155.6, 159.7, 173.4. IR (KBr): 3233, 2905, 1606, 1438, 1392, 1256, 1200, 1160, 1128, 1034, 868, 819, 776 cm−1. HRMS (ESI) calcd for C17H14O4 [M + H]+ 283.09649; found, 283.09629.
2-(2-Bromo-3,4-dimethoxyphenyl)-3-hydroxy-7-methyl-4H-chromen-4-one (F10):
1H-NMR (400 MHz, CDCl3): 2.49 (s, 3H, CH3), 3.89 (s, 3H, OCH3), 3.93 (s, 3H, OCH3), 7.09 (s, 1H, ArH), 7.16 (s, 1H, ArH), 7.24 (s, 1H, ArH), 7.32 (s, 1H, ArH), 8.14 (d, 1H, J = 7.76 Hz, ArH). 13C-NMR (100 MHz, CDCl3): 22.0, 56.3, 56.4, 113.8, 116.1, 118.2, 119.0, 125.3, 126.5, 138.4, 145.3, 145.9, 148.2, 151.0, 156.0, 173.3. IR (KBr): 3270, 2978, 1622, 1617, 1510, 1447, 1373, 1262, 1212, 1165, 1014, 869, 847, 773 cm−1. HRMS (ESI) calcd for C18H15BrO5 [M + H]+ 391.01756; found, 391.01684.
2-(4-(Tert-butyl)phenyl)-3-hydroxy-7-methyl-4H-chromen-4-one (F11):
1H-NMR (400 MHz, CDCl3): 1.36 (s, 9H, C(CH3)3), 2.49 (s, 3H, CH3), 7.21 (dd, 1H, J = 9.2 and 2.4 Hz, ArH), 7.36 (s, 1H, ArH), 7.54 (d, 2H, J = 8.7 Hz, ArH), 8.10 (d, 1H, J = 7.8 Hz, ArH), 8.16 (d, 2H, J = 8.2 Hz, ArH). 13C-NMR (100 MHz, CDCl3): 22.0, 31.2, 35.0, 117.9, 118.5, 125.2, 125.7, 125.4, 127.6, 128.4, 138.1, 145.1, 153.7, 155.6, 173.1. IR (KBr): 3284, 3094, 2928, 2856, 1611, 1596, 1473, 1425, 1248, 1165, 1098, 776 cm−1. HRMS (ESI) calcd for C20H20O3 [M + H]+ 309.14852; found, 309.14871.
2-(4-Bromophenyl)-3-hydroxy-7-methoxy-4H-chromen-4-one (F12):
1H-NMR (400 MHz, CDCl3): 3.89 (s, 3H), 6.89 (d, 1H, J = 2.2 Hz, ArH), 6.95 (dd, 1H, J = 9.2 and 2.4 Hz, ArH), 7.58 (d, 2H, J = 8.7 Hz, ArH), 8.06–8.08 (m, 3H, ArH). 13C-NMR (100 MHz, CDCl3): 55.9, 99.8, 114.6, 115.1, 124.3, 126.8, 129.0, 130.2, 131.8, 138.4, 143.2, 157.3, 164.5, 172.8. IR (KBr): 3237, 2983, 1614, 1595, 1446, 1286, 1202, 1167, 1124, 879, 831, 791 cm−1. C16H11BrO4 [M + H]+ 346.99135; found, 346.99113.
2-(4-(Tert-butyl)phenyl)-3-hydroxy-7-methoxy-4H-chromen-4-one (F13):
1H-NMR (400 MHz, CDCl3): 1.36 (s, 9H, C(CH3)3), 3.93 (s, 3H, OCH3), 6.94–6.97 (m, 2H, ArH), 7.54 (d, 1H, J = 8.2 Hz, ArH), 8.11–8.16 (m, 3H, ArH). 13C-NMR (100 MHz, CDCl3): 31.2, 35.0, 55.9, 99.9, 114.7, 115.0, 125.5, 125.7, 126.8, 127.4, 128.4, 137.9, 153.5, 157.4, 164.3, 172.7. IR (KBr): 3278, 3092, 2889, 1624, 1606, 1563, 1442, 1349, 1286, 1146, 887, 772, 687 cm−1. GC-MS m/z 324.3.
7-Bromo-3-hydroxy-2-phenyl-4H-chromen-4-one (F14):
1H-NMR (400 MHz, CDCl3): 7.46–7.54 (m, 4H, ArH), 7.78 (d, 1H, J = 1.2 Hz, ArH), 8.09 (d, 1H, J = 8.4 Hz, ArH), 8.20 (d, 2H, J = 7.2 Hz, ArH). 13C-NMR (100 MHz, CDCl3) 121.4, 126.9, 127.8, 128.2, 128.4, 128.5, 128.7, 130.5, 130.7, 138.8, 145.5, 155.4, 173.1. IR (KBr): 3268, 2922, 1622, 1603, 1451, 1398, 1268, 1230, 1209, 1130, 766, 732, 697 cm−1. HRMS (ESI) calcd for C15H9BrO3 [M + H]+ 316.98078; found, 316.98099.
7-Bromo-3-hydroxy-2-(3-methoxyphenyl)-4H-chromen-4-one (F15):
1H-NMR (400 MHz, CDCl3): 3.88 (s, 3H, OCH3), 7.00–7.02 (m, 2H, ArH), 7.42 (t, 1H, J = 8.2 Hz, ArH), 7.51 (dd, 1H, J = 8.8 and 1.8 Hz, ArH), 7.78 (t, 3H, J = 8.2 Hz, ArH), 8.08 (d, 1H, J = 8.6 Hz, ArH). 13C-NMR (100 MHz, CDCl3): 55.5, 113.1, 116.3, 119.5, 120.2, 121.4, 126.8, 128.2, 128.4, 129.8, 131.9, 138.7, 144.9, 155.3, 159.7, 173.0. IR (KBr): 3271, 2932, 1601, 1574, 1440, 1388, 1325, 1265, 1136, 896, 775 cm−1. HRMS (ESI) calcd for C16H11BrO4 [M + H]+ 346.99135; found, 346.99080.
6-Bromo-2-(3,4-dimethoxyphenyl)-3-hydroxy-4H-chromen-4-one (F16):
1H-NMR (400 MHz, CDCl3): 3.474 (s, 3H, OCH3), 3.478 (s, 3H, OCH3), 6.54 (d, 1H, J = 8.7 Hz, ArH), 7.08 (d, 1H, J = 8.7 Hz, ArH), 7.30 (dd, 1H, J = 9.2 and 2.7 Hz, ArH), 7.36 (d, 1H, J = 1.8 Hz, ArH), 7.42 (dd, 1H, J = 8.7 and 2.1 Hz, ArH), 7.81 (d, 1H, J = 2.7 Hz, ArH). 13C-NMR (100 MHz, CDCl3): 56.0, 56.1, 111.2, 111.3, 117.2, 120.9, 122.1, 123.9, 127.4, 136.1, 139.0, 146.4, 148.7, 150.7, 153.7, 172.1. IR (KBr): 3299, 3012, 2918, 1627, 1598, 1553, 1469, 1386, 1259, 1146, 1017, 809, 677 cm−1. HRMS (ESI) calcd for C17H13BrO5 [M + H]+ 377.00191; found, 377.00179.
2-(3-Chlorophenyl)-6-fluoro-3-hydroxy-4H-chromen-4-one (F17):
1H-NMR (400 MHz, CDCl3): 7.41–7.46 (m, 3H, ArH), 7.58 (dd, 1H, J = 9.1 and 4.1 Hz, ArH), 7.83 (dd, 1H, J = 8.2 and 3.2 Hz, ArH), 8.14 (m, 1H, ArH), 8.18 (t, 1H, J = 1.8 Hz, ArH). 13C-NMR (100 MHz, CDCl3): 109.9, 110.2, 120.5, 120.6, 122.5, 122.8, 126.0, 127.6, 129.9, 130.3, 132.5, 134.8, 138.6, 143.9, 151.7, 157.9, 171.3. IR (KBr): 3260, 3092, 1617, 1609, 1577, 1487, 1399, 1261, 1171, 1128, 1102, 887, 819, 792, 732, 686 cm−1. HRMS (ESI) calcd for C15H8ClFO3 [M + H]+ 291.02188; found, 291.02187.
2-(4-(Tert-butyl)phenyl)-6-chloro-3-hydroxy-7-methyl-4H-chromen-4-one (F18):
1H-NMR (400 MHz, CDCl3): 1.36 (s, 9H, C(CH3)3), 2.50 (s, 3H, CH3), 7.44 (s, 1H, ArH), 7.53 (d, 2H, J = 8.7 Hz, ArH), 8.12–8.16 (m, 3H, ArH). 13C-NMR (100 MHz, CDCl3): 21.0, 31.2, 35.0, 119.8, 120.0, 124.9, 125.7, 127.6, 128.1, 131.4, 138.2, 142.9, 145.5, 153.7, 153.9, 172.3. IR (KBr): 3225, 2966, 1598, 1459, 1432, 1381, 1304, 1230, 1202, 1106, 883, 840, 781 cm−1. HRMS (ESI) calcd for C20H19ClO3 [M + H]+ 343.1095; found, 343.1091.
2-(4-(Benzyloxy)phenyl)-6-chloro-3-hydroxy-7-methyl-4H-chromen-4-one (F19):
1H-NMR (400 MHz, CDCl3): 2.48 (s, 3H, CH3), 5.12 (s, 2H, ArCH2), 7.07 (d, 2H, J = 8.7 Hz, ArH), 7.39–7.43 (m, 6H, ArH), 8.15 (t, 3H, J = 8.7 Hz, ArH). 13C-NMR (100 MHz, CDCl3): 21.0, 70.1, 115.0, 119.8, 119.9, 123.6, 124.9, 127.6, 128.3, 128.7, 129.6, 131.3, 136.4, 137.6, 142.7, 145.4, 153.5, 160.4, 172.0. IR (KBr): 3226, 2892, 1599, 1483, 1384, 1202, 1096, 834, 779, 734, 696 cm−1. HRMS (ESI) calcd for C23H17ClO4 [M + H]+ 393.0888; found, 393.0881.
2-(4-Bromophenyl)-3-hydroxy-6-methyl-8-nitro-4H-chromen-4-one (F20):
1H-NMR (400 MHz, CDCl3): 1.22 (s, 3H, CH3), 7.13 (brs, 1H, OH), 7.65 (d, 2H, J = 8.2 Hz, ArH), 8.22 (d, 2H, J = 8.2 Hz, ArH), 8.26 (s, 1H, ArH), 8.31 (s, 1H, ArH). 13C-NMR (100 MHz, CDCl3): 20.6, 123.1, 124.5, 129.4, 129.7, 130.9, 131.1, 131.6, 133.9, 139.8, 172.0. IR (KBr): 3228, 3094, 1627, 1605, 1563, 1482, 1420, 151, 1288, 1213, 1132, 899, 776, 759, 690 cm−1. HRMS (ESI) calcd for C16H10BrNO5 [M + H]+ 375.9815; found, 375.9814.
7-Chloro-2-(4-ethoxy-3-fluorophenyl)-3-hydroxy-4H-chromen-4-one (F21):
1H-NMR (400 MHz, CDCl3): 1.50 (t, 3H, J = 6.8 Hz, CH3), 4.20 (q, 2H, J = 6.8 Hz, OCH2), 7.06 (t, 2H, J = 8.4 Hz, ArH), 7.37 (dd, 1H, J = 8.4 and = 1.6 Hz, ArH), 7.59 (d, 1H, J = 1.6 Hz, ArH), 7.98 (m, 2H, ArH), 8.15 (d, 1H, J = 8.4 Hz, ArH). 13C (100 MHz; CDCl3): 14.7, 64.8, 113.9, 115.4, 115.6, 118.1, 119.1, 123.2, 123.3, 124.4, 124.5, 125.6, 126.7, 138.0, 139.8, 144.1, 144.2, 148.7, 148.8, 150.9, 153.4, 155.1, 172.5. IR (KBr): 3234, 2919, 1605, 1518, 1452, 1282, 1108, 1040, 771 cm−1. HRMS (ESI) calcd for C17H13ClFO4 [M + H]+ 35.0481; found, 335.0477.
6-Fluoro-3-hydroxy-2-(3,4,5-trimethoxyphenyl)-4H-chromen-4-one (F22):
1H-NMR (400 MHz, CDCl3): 3.92 (s, 3H, OCH3), 3.94 (s, 6H, 2xOCH3), 7. 10 (brs, 1H, OH), 7.40–7.45 (m, 1H, ArH), 7.49 (s, 2H, ArH), 7.58–7.61 (m, 1H, ArH), 7.83–7.86 (m, 1H, ArH). 13C-NMR (100 MHz, CDCl3): 56.3, 61.1, 105.5, 109.9, 110.1, 120.4, 120.5, 122.1, 126.0, 138.0, 140.1, 145.3, 151.6, 153.3, 157.9, 160.3, 172.7. IR (KBr): 3252, 2953, 2889, 1606, 1560, 1490, 1391, 1261, 1169, 1126, 1098, 779, 665 cm−1. HRMS (ESI) calcd for C18H FO6 [M + H]+ 347.0925; found, 347.0924.
3-Hydroxy-2-(2,4,5-trimethoxyphenyl)-4H-chromen-4-one (F23):
1H-NMR (400 MHz, CDCl3): 3.87 (s, 3H, OCH3), 3.88 (s, 3H, OCH3), 3.96 (s, 3H, OCH3), 6.55 (brs, 1H, OH), 6.65 (s, 1H, ArH), 7.22 (s, 1H, ArH), 7.39 (t, 1H, J = 7.2 Hz, ArH), 7.51 (d, 1H, J = 8.4 Hz, ArH), 7.64–7.68 (m, 1H, ArH), 8.26 (d, 1H, J = 8.0 Hz, ArH). 13C (100 MHz; CDCl3): 56.1, 56.5, 57.0, 97.9, 110.8, 113.2, 118.29, 118.35, 121.3, 124.3, 125.40, 125.44, 133.2, 138.5, 143.1, 145.7, 151.9, 152.4, 155.8, 173.1. IR (KBr): 3284, 2922, 2876, 1606, 1566, 1469, 1386, 1282, 1210, 1163, 1105, 1029, 776 cm−1. HRMS (ESI) calcd for C18H17O6 [M + H]+ 329.1020; found, 329.1021.
7-Chloro-3-hydroxy-2-(2,4,5-trimethoxyphenyl)-4H-chromen-4-one (F24):
1H-NMR (400 MHz, CDCl3): 3.86 (s, 6H, 2xOCH3), 3.94 (s, 3H, OCH3), 6.63 (s, 1H, ArH), 7.08 (s, 1H, ArH), 7.33–7.35 (m, 1H, ArH), 7.52 (d, 1H, J = 1.6 Hz, ArH), 8.17 (d, 1H, J = 8.72 Hz, ArH). 13C-NMR (100 MHz, CDCl3): 56.2, 56.6, 57.1, 97.9, 110.5, 113.2, 118.4, 120.0, 125.4, 126.9, 138.8, 139.4, 143.2, 146.2, 152.1, 152.6, 155.9, 172.6. IR (KBr): 3288, 2921, 2886, 1603, 1513, 1441, 1272, 1211, 1163, 1036, 789 cm−1. HRMS (ESI) calcd for C18H15ClO6 [M + H]+ 363.063; found, 363.0626.
4.4. Inverse-docking analysis
All of the inverse docking analysis was performed on a personal Macbook installed with IOS operating system. For the docking analysis of all the selected proteins, “Glide” was used (85). Glide consisted of multiple modes for docking processing. For the present study, the XP mode of docking was used. Eleven flavonol hits obtained after MTT assay were sketched and cleaned by utilizing the “builder tool” option available in Maestro. The molecules were then optimized with OPLS3 force-field using the “Ligprep” module of Maestro at a pH range of 7 ± 2.0. For each selected target (Table 2), the co-crystallized structures available in the RCSB database were retrieved and subjected to a well-established cross docking protocol to select the optimum structure (see supplemental data) (81–85). All of the selected protein structures were then optimized using the “protein preparation wizard” module implemented in the Maestro program and the optimization included completion of bond order, addition of hydrogen atoms, construction of missing side chains and loops. Finally, the complexes were minimized using an OPLS3 force field to RMSD of 0.15 Å employing the “minimize” option of the protein preparation wizard.
4.5. Biological evaluation
4.5.1. Antibodies, chemicals and reagents
MTT dye (3-(4-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, 98% TLC) was purchased from Sigma-Aldrich Chemical Company (St. Louis, MO, USA). Dimethyl sulfoxide (DMSO) was purchased from MP Biomedicals (Irvine, California, USA). The antibodies for immunoblotting Bcl-2 (2876S), Bax(2772S), Caspase-3(9662S), mTOR, β-Actin, pS6 (Ser235/236), S6, TSC2, p4E-BP1 (Thr37/46), 4EBP1, Akt, pAkt (Ser473), phospho-mTOR(Ser2448) D9C2 (5536S), Phospho-p90RSK, Phospho-Akt, Phospho-p44/42 MAPK and Phospho-S6 Ribosomal Protein Detection Cocktail I (5301S), and horseradish peroxidase-conjugated (HRP) anti-mouse and anti-rabbit secondary antibodies were all obtained from Cell Signaling Technologies (Beverly, MA, USA). Antibody to peIF4E (Ser209) (76256) was from Abcam. Mini-protean precast Tris-Glycine Gels (TGX) were from Bio-Rad (Bio-Rad Laboratories Inc., Hercules, California, USA). The SuperSignal™ West Pico PLUS Chemiluminescent substrate detection system was from ThermoFisher Scientific (Waltham, Massachusetts, USA). A 2% (w/v) Aqueous Solution of Gentian Violet was from Ricca Chemical Company (Arlington, TX, USA). Dulbecco’s modified Eagle’s medium (DMEM) were from Corning (Corning, Manassas, VA, USA). Cascade BiologicsR PMA-free Human Melanocyte Growth Supplement (HMGS-2, # S0165), and medium 254 (# M-254-500) from Gibco™ Invitrogen Cell culture (ThermoFisher Scientific, Illinois, USA). RIPA buffer and Pierce BCA™ protein assay kit was purchased from ThermoFisher Scientific (Rockford, Illinois, USA). EpiLife® Medium with 60 μM calcium was from Life Technologies.
The quick coating solution (cAP-01) was purchased from Angio-Proteomie (Boston, MA, USA). The Dulbecco’s phosphate-buffered saline (DPBS) and phosphate buffer saline (PBS) 1X, defined trypsin inhibitor (DTI) 1X (R007-100), trypsin EDTA 0.25%, 1X(R25200-072), Trypsin neutralizer (TN) 1X (R-002-100), penicillin streptomycin (Pen Strep,15140-122) (PEST) 100X were purchased from Gibco, ThermoFisher Scientific (Rockford, IL, USA) and human keratinocyte growth supplement (HKGS) 100X (S-001-5) from Cascade Biologist. USDA approved Origin Fetal Bovine Serum (FBS) was from VWR Seradigm Life Science (Missouri, Texas, USA). The organic solvents including, ethanol (6183-10) and methanol (BDH 1135-4LP) were acquired from Macron Chemicals (Radnor, Pennsylvania, USA) and VWR (Missouri, Texas, USA) respectively.
4.5.2. In-vitro enzymatic assays
4.5.2.1. In-vitro CDK2, c-Kits, and mTOR kinase activity assays
The ability of the fesitin analogs to inhibit c-Kits, CDK2, and mTOR kinase enzymatic activity was assessed using an in vitro profiling platform, and performed at Reaction Biology Corporation (Malvern, PA, USA). Briefly, the specific kinase and substrate pairs alongside with their necessary cofactors were mixed in a reaction buffer consisting of (20 mM Hepes pH 7.5, 10 mM MgCl2, 1 mM EGTA, 0.02% Brij35, 0.02 mg/ml BSA, 0.1 mM Na3VO4, 2 mM DTT, 1% DMSO). Fisetin (F0), and 10 best inverse docking identified hit compounds, alongside respective positive activator molecules including rapamycin, staurosporine, and PI-103 at (1 and 10 μM) dissolved in DMSO were conveyed into their respective kinase reaction mixtures. The reaction was then initiated by the addition of 32P-ATP (specific activity 10 nCi/nl), and incubated for 2 h at rt, and the reactions later spotted onto P81 ion exchange filter paper (Whatman Inc., Piscataway, NJ), and the unbound phosphate was eliminated by extensively washing the filters in 0.75% phosphoric acid. Background derived from control reactions containing inactive enzyme was subtracted, and the residual kinase activity data expressed as percentage of the outstanding kinase activity in the test samples compared to vehicle (DMSO) reactions. Curve fitting and IC50 values were obtained using GraphPad Prism software version 8.1 (GraphPad Prism Inc., San Diego, CA, USA).
4.5.3. Cell lines and culture
Human epidermoid carcinoma A431, human melanoma A375 and SK-Mel-28, and Human immortalized keratinocytes (HaCaT) cell lines were purchased from American Type Culture Collection (ATCC; Manassas, VA, USA). The GFP-Expressing A431 (GFP-A431) and A375 (GFP-A375) cells were purchased from Angio-Proteomie (Boston, MA, USA). Primary normal human epidermal keratinocytes (NHEK) (86) and HEKa were procured from ThermoFisher Scientific (Waltham, Massachusetts, USA), respectively. Primary normal human melanocytes were kindly donated by Dr. I. Caroline Le Poole, from Loyola University Chicago (IL), USA, and were cultured in Medium 245 supplemented with HMGS-2, 1% antibiotic and antimycotic. All immortalized cell lines were grown in DMEM supplemented with FBS; 5% for A431 cells while A375 and HaCaT cells were grown in 10% FBS containing DMEM supplemented with 1% PEST (100X; 100U/ml penicillin, 100 μg/ml streptomycin). NHEK and HEKa were seeded unto collagen coated tissue culture flasks grown in low calcium EpiLife Medium supplemented with HKGS and 1% PEST. All cultured cell lines were maintained at 37°C in a 95% humidified atmosphere with 5% CO2. For all experiments in their logarithmic growth phase, the growth media were routinely changed every 2–3 days until cell reach desired confluence 65–80% prior to treatment. For subculturing, the cells were harvested after trypsin/EDTA treatment at 37 °C. For harvest, primary cells grown in serum-free medium were trypsinized, and trypsin was inhibited using DTI and/or TN. In A431 and A375 cells, the effect of trypsin was inhibited with two-fold volume of 10% FBS containing growth medium. A 10 mM stock solution of parent (fisetin) and each of the synthesized flavonol derivatives were prepared in DMSO, and further serially diluted in respective growth media during test treatment. Control cells were treated with equal volume of vehicle alone at a final concentration of 0.1% (v/v) DMSO, concentration that showed no effect on cell viability.
4.5.4. Cell growth and death by trypan blue dye exclusion assay
To estimate cell growth/viability, melanoma, non-melanoma alongside normal control cells were seeded (105 cells/well) in 6-well plates with in 2 mL of their respective FBS containing growth medium at 37°C in a 5% CO2 incubator overnight. Cells were refed with fresh medium, and treated with different doses of fisetin or various derivatives (0, 1, 10, or 25 μmol/L). After 24 or 48 hours of treatment, total cells were harvested by brief trypsinization, and washed with 1xPBS. Total cell number was determined by counting each sample in duplicate using a hemocytometer under an inverted microscope and by using analysis by an automated countess II cell counter from ThermoFisher Scientific (Waltham, Massachusetts, USA). Cell viability was determined using the trypan blue exclusion method. The data shown in this study are the mean of 3 independent experiments.
4.5.5. Evaluation of Cytotoxicity and cell viability by MTT assay
To estimate cell viability and cytotoxicity, proliferation of the different melanoma and non-melanoma cells and cytotoxicity of the different agents tested in diverse concentrations were assessed by using MTT assay. Seeded cells in 5–10% FBS/DMEM were incubated at 37°C in a 5% CO2 incubator and were re-fed with fresh medium after 24 hours. Briefly, cells were seeded in 200 μl of growth medium in a density of 5×103 cells per well on a 96-well plate and grown in their respective media in log growth phase until 70% confluent prior to treatment. The different cells, each in 8 to 10 repeats were treated with or without the different test compounds dissolved in DMSO (maximal incubation concentration of DMSO was 0.1%) at varied concentrations, and incubated as above for 48h, and the cells viability was measured by using MTT assay as previously described (38). The tested compounds were prepared according to their solubility in DMSO, at the incubation concentrations of two-fold-serial dilution with maximum concentration as indicated (1–80 μmol/L). The treatment was carried out in a humidified atmosphere with 5% CO2 at 37 °C, for 48 h. At harvest, the individual plates were vigorously shaken for 10–15 min to solubilize formazan crystals and absorbance was recorded at 562 nm on a SynergyLX, multi-mode microplate reader (BioTek Instruments Inc., Winooski, VT, USA). The effect of each agent on growth inhibition was assessed, as a percentage cell viability where DMSO-treated cells (untreated controls) was considered as 100% viable (87).
Cell proliferation inhibitory curves were constructed for each compound, plotting incubation concentrations vs. percentage of viability relative to untreated control. The standard toxicological parameter IC50 was calculated by a nonlinear regression analysis of the inhibitory curves using GraphPad Prism software version 9.1, (GraphPad Software, Inc., La Jolla, CA, USA).
4.5.6. Preparation of cellular protein lysates and western blotting/immunoblotting:
For cell lysates and immunoblot analyses, cell were plated in 10cm2 plates at 3 × 105 cells/well in respective media overnight. After cell monolayers were seeded overnight media were removed, and then cells were treated with or without appropriate test hit drug dilutions (< 0.1% DMSO final) and incubated for 48 h. Cells were harvested by aspirating media and washing the cells with cold 1xPBS (pH 7.4). The whole cell lysates were prepared with cell lysis RIPA buffer supplemented with protease and phosphatase inhibitors; 1mM PMSF (phenylmethylsulfonyl fluoride), 10mM RIPA buffer, 1mM Protease inhibitor cocktail (1 mM EDTA, 20 mM NaF, 0.5% NP-40, 1% Triton X-100) (Roche) and 0.5% Na3VO4 at pH 7.4. The cells were incubated for 15 minutes in added ice-cold lysis RIPA buffer lysates, prepared as described previously (38, 87). The protein concentration of the cleared lysates were determined using a BCA protein assay kit (Thermo Scientific, Pierce) according to the manufacturer’s protocol assayed using Eppendorf BioPhotometer D30, and were aliquoted and stored at −80 °C for further analysis as earlier described (38).
For Western blotting analyses, experiments were performed essentially as previously described (38), by denaturing in 2X Laemmli sample buffer and resolving equal amount of protein (approximately 20–40 μg of protein) loaded per lane on 8–12% sodium dodecyl sulfate-polyacrylamide gels (SDS-PAGE) or on 4–12% polyacrylamide ready mini-PROTEAN TGX gels electrophoresis to separate proteins based on molecular weight. After running, the protein separated were transferred onto nitrocellulose membrane using Trans-Blot Turbo Transfer Pack according to manufacturer’s instructions (Bio-Rad). Followed by washing, the membrane was blocked using 5% Bovine serum albumin (BSA) for phosphorylated-proteins, or 5–7% non-fat dry milk in 1% Tween-twenty in 1% Triton-X100-1xPBS (PBST) at room temperature for 45 minutes. Briefly, following blocking membranes were incubated with appropriate monoclonal or polyclonal primary antibody diluted at 1:250 to 1:1000 in the blocking buffer for 2 h at RT or overnight at 4°C, followed by two x10min washes. The blots were washed three times with PBS: 0.1% Tween 20 (PBST) and incubated for 2 h at room temperature (RT), on a shaker with 1:2000 diluted anti-mouse or anti-rabbit horseradish-peroxidase conjugated (HRP) secondary antibody obtained from Amersham Life Science Inc. (Arlington Height, IL, USA). The blots were then rewashed, exposed to enhanced chemiluminescence (ECL) (Amersham life science incorporation), plus west pico and subjected to autoradiography. The intensity of each band was measured using Bio-Rad ChemiDoc™ detection and imaging analysis software systems as earlier described (38). The blots were developed after 5 minutes of incubation with ECL and auto exposure. Equal loading of protein was confirmed by stripping the blotted membranes and re-probing with appropriate loading controls; for β-actin, GAPDH or vinculin, and the results displayed are representative of more than two independent experiments each performed in triplicate with comparable results. All primary antibodies were used at 1:250 to 1:1,000 dilutions except for β-actin, GAPDH, vinculin, which were at 1:2,000 dilution. Densitometric analyses of the visualized protein bands were performed using the BioRad digitized scientific software program Quantity One. Bands were scanned and processed with Adobe Photoshop CC 8.0 (Adobe Systems, San Jose, CA, USA). Linear range of band densities was ensured by performing multiple exposures and final data were analysed by one-way ANOVA.
4.5.7. Scratch wound healing assay
The cells (A431 and A375), were seeded in 24 well plates and cultured to confluence in DMEM complete media. Two parallel straight scratched wounds were made across the centre of the cell monolayer in each well using a sterile 200-μL Gilson pipette tip, followed by extensive washed. Their respective growth media with or without test compounds at varied concentrations were replenished and incubated for 24 – 48 h (Maximum of 0.1% DMSO containing medium served as control) as previously described (88). Briefly, phase contrast images were taken with a 10x, 20x objective lenses at regular intervals starting from 0 h when the wounds were created, to 24 h and 48h post-treatment using an Olympus IX51 phase-contrast microscope. Cell migration and wound healing was determined using ImageJ and calculated as a percentage of initial cell-free areas vs percentage of cell-free areas after treatment (88).
4.5.8. Colony formation assay
To investigate the effects of flavonol derivatives on clonogenic potential of A375 and A431cells, colony formation assay was performed as previously described (88). Briefly, cells were seeded in T-25 tissue culture flasks overnight, and 65% confluent log phased cells were treated with compounds F6, F9, F11, F19, F20 at various doses corresponding to 0, ½IC50, IC50 and 2xIC50 supplemented in respective growth media for 48h, with the exception of F6 and F9 which concentrations were 7.5, 15 and 30 μM in both cell lines (supplementary data (68)). After treatment and incubation period, cells were trypsinized, enumerated and then, low number of cells (~1000 cells) were seeded per 10-cm2 plate in fresh media, and cells were grown for 10–14 days with normal drug-free media. The media were replenished after every 2–3 days until control cell colonies were confluent prior to harvest as describe previously (88). As a vehicle control, control cells were treated with DMSO (≤ 0.1% v/v), and were maintained under standard cell culture conditions at 37°C and 5% CO2 in a humidified atmosphere. At harvest, cell colonies were stained with a cell fixative solution containing 0.5% Gentian violet staining agent in ≈ 64% (v/v) methanol and 4% paraformaldehyde in protocol as previously described (88). After staining, the plates were immersed three-consecutive times in running tap water and air-dried overnight. The photomicrographs of colonies were counted accordingly using a phase-contrast Olympus IX51 microscope at 10x magnification or using count PHICS analysis tool (89). Using the PHICS, the least counts for colonies was 22 for 10 cm plate and 21 for 6 well plate with a range of 0.1 to 1.4 mm.
4.5.9. Immunofluorescence analysis of GFP tagged cells for morphology and apoptosis:
Equal number of cells were seeded in T-25 flasks and treated with potent compounds in different concentrations for 36 hours. After that, the media was aspirated, and cells were washed with 1xPBS, and in PBS containing DAPI (4′,6-diamidino-2-phenylindole), the picture was taken using Olympus IX71 at 10x and 1.6×10x magnification.
4.5.10. Statistical analysis
Statistical analyses were performed using GraphPad Prism Software version 8.1 (GraphPad Prism Inc., San Diego, CA, U.S.A.). All quantitative data are presented as means ± standard error of the mean (SEM) or ± standard deviation (SD), from at least three independent experiments. One-way ANOVA, Student t-test or ANOVA with Bonferroni and/or turkey post hoc tests were used to determine the difference between two or more groups, and the differences were considered statistically significant when p-values < 0.05.
Supplementary Material
Highlights:
A series of flavonol derivatives of fisetin were synthesized, characterized, and evaluated as potential anti-skin-cancer agents.
Several of these compounds showed micromolar range anti-proliferative activity in vitro in melanoma (A375, SK-MEL-28) and non-melanoma (A431 and UWBCC-1) skin cancer cells.
Molecular docking and in-vitro enzymatic kinase assays identified CDK2, c-KIT, and mTOR kinase inhibitions, and validated the potencies of the selected hits as single, dual or multi-target inhibitors.
The potent analogs, F20, F9 and F17 significantly induced apoptosis and inhibited clonogenicity, wound healing and the activation of dysregulated cancer-related molecular targets CDK2, c-Kit and effectors including mTOR, S6K, p90RSK, Stat3, and MAPK (ERK1/2).
Acknowledgments
The authors thank, Professor I. Caroline Le Poole from Loyola University Chicago, Chicago, Illinois, USA for kindly providing primary human melanocytes. Kartik R. Roy, and Alexis D. Alexander for assistance with the biological assays. MS is also grateful to the Schrödinger Team for the opportunity to evaluate a trial of the latest version of software Maestro.
Funding
The research work reported in this publication was supported in part by a start-up fund from the University of Louisiana at Monroe (ULM) College of Pharmacy (to JCC), a ULM College of Pharmacy Faculty Research Seed grant 5CALHN-260615 (to JCC), LBRN Pilot Awards (to JCC and SM) from an Institutional Development Award (IDeA) Networks of Biomedical Research Excellence (INBRE), award from the National Institute of General Medical Sciences of the National Institutes of Health (NIGMS/NIH) grant number P2O GM103424-18 (to KGK), NIH NCI grants CA191550 and CA243167 (to VSS), and an LBRN-INBRE Administrative Supplement Award (to JCC) from NIH/NIGMS grant 3P20GM103424-18S1 (to KGK). MS thanks the Fondazione di Sardegna for its partial support.
Abbreviations
- BCA
bicinchoninic acid assay
- BSA
bovine serum albumin
- FBS
dialyzed fetal calf serum
- DMEM
dulbecco’s modified eagle’s medium
- DMSO
dimethyl sulfoxide
- DTI
defined trypsin inhibitor
- TN
trypsin neutralizing solution
- IC50
half maximal inhibitory concentration where 50% cells are dead
- EtOAc
ethyl acetate
- FBS
fetal bovine serum
- FDA
food and drug administration
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GC-MS
gas chromatography-mass spectrometry
- KOH
potassium hydroxide
- LC-MS
liquid chromatography-mass spectrometry
- MS
mass spectrometry
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide
- NMR
nuclear magnetic resonance
- SAR
structure activity relationship
- SDS
sodium dodecyl sulfate
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- TBS
tris buffered saline
- TLC
thin layer chromatography
- PBS
phosphate buffered saline
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Materials data Available: 1H- and 13C-NMR as well as mass spectra for the synthesized compounds, structures, tables; Table S1: List of PDB structures, schemes are available online at https://www.sciencedirect.com/journal/bioorganic-chemistry and in Data-In-Brief at https://www.journals.elsevier.com/data-in-brief supplementary report (68).
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Singh PK. Histone methyl transferases: A class of epigenetic opportunities to counter uncontrolled cell proliferation. Eur J Med Chem. 2019,166:351–368. [DOI] [PubMed] [Google Scholar]
- 2.Chan RJ, Teleni L, McDonald S, Kelly J, Mahony J, Ernst K, Patford K, Townsend J, Singh M, Yates P. Breast cancer nursing interventions and clinical effectiveness: a systematic review. BMJ Support Palliat Care. 2020. ahead of print (DOI: 10.1136/bmjspcare-2019-002120). [DOI] [PubMed] [Google Scholar]
- 3.Pasquali S, Hadjinicolaou AV, Chiarion SV, Rossi CR, Mocellin S. Systemic treatments for metastatic cutaneous melanoma. Cochrane database Syst Rev. 2018,2(2):CD011123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sun Y Tumor microenvironment and cancer therapy resistance. Cancer Lett. 2016, 380(1):205–215. [DOI] [PubMed] [Google Scholar]
- 5.Santomasso BD. Anticancer drugs and the nervous system. Continuum (Minneap Minn). 2020,26(3):732–764. [DOI] [PubMed] [Google Scholar]
- 6.Siegel RL, Miller KD, Jemal A. Cancer statistics-. CA Cancer J Clin. 2020,70(1):7–30. [DOI] [PubMed] [Google Scholar]
- 7.Rebecca VW, Sondak VK, Smalley KSM. A brief history of melanoma: from mummies to mutations. Melanoma Res. 2012,22(2):114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saida T Melanoma and non-melanoma skin cancers. Gan To Kagaku Ryoho. 2020,47(4):587. [PubMed] [Google Scholar]
- 9.Whiteman DC, Green AC, Olsen CM. The growing burden of invasive melanoma: projections of incidence rates and numbers of new cases in six susceptible populations through 2031. J Invest Dermatol. 2016,136(6):1161–1171. [DOI] [PubMed] [Google Scholar]
- 10.Dzwierzynski WW. Managing malignant melanoma. Plast Reconstr Surg. 2013,132(3):446e–460e. [DOI] [PubMed] [Google Scholar]
- 11.Hartman ML, Sztiller-Sikorska M, Gajos-Michniewicz A, Czyz M. Dissecting mechanisms of melanoma resistance to BRAF and MEK inhibitors revealed genetic and non-genetic patient- and drug-specific alterations and remarkable phenotypic plasticity. Cells. 2020,9(1):142(pp 1–28). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leonardi GC, Falzone L, Salemi R, Zanghì A, Spandidos DA, Mccubrey JA, Candido S, Libra M. Cutaneous melanoma: From pathogenesis to therapy (Review). Int J Oncol. 2018. April;52(4):1071–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fakhri S, Moradi SZ, Farzaei MH, Bishayee A. Modulation of dysregulated cancer metabolism by plant secondary metabolites: A mechanistic review. Semin Cancer Biol. 2020, ahead of print (DOI: 10.1016/j.semcancer.2020.02.007). [DOI] [PubMed] [Google Scholar]
- 14.Chamcheu JC, Roy T, Uddin MB, Banang-Mbeumi S, Chamcheu R-CN, Walker AL, et al. Role and therapeutic targeting of the PI3K/Akt/mTOR signaling pathway in skin cancer: A review of current status and future trends on natural and synthetic agents therapy. Cells. 2019,8(8):803 (pp 1–33). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alicea GM, Rebecca VW. Emerging strategies to treat rare and intractable subtypes of melanoma. Pigment Cell Melanoma Res. 2020, ahead of print (DOI: 10.1111/pcmr.12880) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garbe C, Amaral T, Peris K, Hauschild A, Arenberger P, Bastholt L, Bataille V, del Marmol V, Dréno B, Fargnoli MC, Grob J-J, Höller C, Kaufmann R, Lallas A, Lebbé C, Malvehy J, Middleton M, Moreno-Ramirez D, Pellacani G, Saiag P, Alexander J. Stratigos AJ, Vieira R, Zalaudek I, Alexander MM. Eggermont AMM. European consensus-based interdisciplinary guideline for melanoma. Part 2: Treatment - Update 2019. Eur J Cancer. 2020,126:159–177. [DOI] [PubMed] [Google Scholar]
- 17.Fotie J The antiprotozoan potential of flavonoids. Pharmacogn Rev. 2008;2(3):6. [Google Scholar]
- 18.Singh PK, Silakari O. The current status of O-heterocycles: A synthetic and medicinal overview. ChemMedChem. 2018,13(11):1071–1087. [DOI] [PubMed] [Google Scholar]
- 19.Salvamani S, Gunasekaran B, Shaharuddin NA, Ahmad SA, Shukor MY. Antiartherosclerotic effects of plant flavonoids. Biomed Res Int. 2014,480258 (pp 1–11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nagula RL, Wairkar S. Recent advances in topical delivery of flavonoids: A review. J Control Release. 2019,296:190–201. [DOI] [PubMed] [Google Scholar]
- 21.Bratkov VM, Shkondrov AM, Zdraveva PK, Krasteva IN. Flavonoids from the genus Astragalus: phytochemistry and biological activity. Pharmacogn Rev. 2016,10(19):11–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scholz EP, Zitron E, Katus HA, Karle CA. Cardiovascular ion channels as a molecular target of flavonoids. Cardiovascular Therapeutics 2010,28(4):46–52. [DOI] [PubMed] [Google Scholar]
- 23.Czaplinska M, Czepas J, Gwozdzinski K. Structure, antioxidative and anticancer properties of flavonoids. Postepy Biochemii 2012,58(3):235–244. [PubMed] [Google Scholar]
- 24.Khan N, Afaq F, Syed DN, Mukhtar H. Fisetin, a novel dietary flavonoid, causes apoptosis and cell cycle arrest in human prostate cancer LNCaP cells. Carcinogenesis. 2008,29(5):1049–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suh Y, Afaq F, Khan N, Johnson JJ, Khusro FH, Mukhtar H. Fisetin induces autophagic cell death through suppression of mTOR signaling pathway in prostate cancer cells. Carcinogenesis. 2010,31(8):1424–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Senthilkumar K, Arunkumar R, Elumalai P, Sharmila G, Gunadharini DN, Banudevi S, Krishnamoorthy G, Benson CS, Arunakaran J. Quercetin inhibits invasion, migration and signalling molecules involved in cell survival and proliferation of prostate cancer cell line (PC-3). Cell Biochem Funct. 2011,29(2):87–95. [DOI] [PubMed] [Google Scholar]
- 27.Aalinkeel R, Bindukumar B, Reynolds JL, Sykes DE, Mahajan SD, Chadha KC, Schwartz SA. The dietary bioflavonoid, quercetin, selectively induces apoptosis of prostate cancer cells by down-regulating the expression of heat shock protein 90. Prostate. 2008,68(16):1773–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Syed DN, Chamcheu J-C, Khan MI, Sechi M, Lall RK, Adhami VM, Mukhtar H. Fisetin inhibits human melanoma cell growth through direct binding to p70S6K and mTOR: findings from 3-D melanoma skin equivalents and computational modeling. Biochem Pharmacol. 2014,89(3):349–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sechi M, Lall RK, Afolabi SO, Singh A, Joshi DC, Chiu S-Y, Mukhtar H, Syed DN. Fisetin targets YB-1/RSK axis independent of its effect on ERK signaling: insights from in vitro and in vivo melanoma models. Sci Rep. 2018, 8(1):15726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Syed DN, Lall RK, Chamcheu JC, Haidar O, Mukhtar H. Involvement of ER stress and activation of apoptotic pathways in fisetin induced cytotoxicity in human melanoma. Arch Biochem Biophys. 2014,563:108–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Estevez-Sarmiento F, Said M, Brouard I, Leon F, Garcia C, Quintana J, Estévez F. 3’-Hydroxy-3,4’-dimethoxyflavone blocks tubulin polymerization and is a potent apoptotic inducer in human SK-MEL-1 melanoma cells. Bioorg Med Chem. 2017,25(21):6060–6070. [DOI] [PubMed] [Google Scholar]
- 32.Zheng Y, Pu W, Li J, Shen X, Zhou Q, Fan X, Yang S-Y, Yu Y, Chen Q, Wang C,Wu X, Peng Y. Discovery of a prenylated flavonol derivative as a Pin1 inhibitor to suppress hepatocellular carcinoma by modulating microRNA biogenesis. Chem Asian J. 2019,14(1):130–134. [DOI] [PubMed] [Google Scholar]
- 33.Pal HC, Hunt KM, Diamond A, Elmets CA, Afaq F. Phytochemicals for the management of melanoma. Mini Rev Med Chem. 2016,16(12):953–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pal HC, Diamond AC, Strickland LR, Kappes JC, Katiyar SK, Elmets CA, Athar M, Afaq F. Fisetin, a dietary flavonoid, augments the anti-invasive and anti-metastatic potential of sorafenib in melanoma. Oncotarget. 2016,7(2):1227–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pal HC, Baxter RD, Hunt KM, Agarwal J, Elmets CA, Athar M, Afaq F. Fisetin, a phytochemical, potentiates sorafenib-induced apoptosis and abrogates tumor growth in athymic nude mice implanted with BRAF-mutated melanoma cells. Oncotarget. 2015,6(29):28296–28311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khan N, Syed DN, Ahmad N, Mukhtar H. Fisetin: a dietary antioxidant for health promotion. Antioxid Redox Signal. 2013,19(2):151–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khan N, Afaq F, Khusro FH, Mustafa Adhami V, Suh Y, Mukhtar H. Dual inhibition of phosphatidylinositol 3-kinase/Akt and mammalian target of rapamycin signaling in human nonsmall cell lung cancer cells by a dietary flavonoid fisetin. Int J cancer. 2012,130(7):1695–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chamcheu JC, Esnault S, Adhami VM, Noll AL, Banang-Mbeumi S, Roy T, Singh SS, Huang S, Kousoulas KG, Mukhtar H. Fisetin, a 3,7,3’,4’-tetrahydroxyflavone inhibits the PI3K/Akt/mTOR and MAPK pathways and ameliorates psoriasis pathology in 2D and 3D organotypic human inflammatory skin models. Cells. 2019,8(9):1089 (pp 1–27). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maher P, Dargusch R, Ehren JL, Okada S, Sharma K, Schubert D. Fisetin lowers methylglyoxal dependent protein glycation and limits the complications of diabetes. PLoS One. 2011,6(6):e21226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ren Q, Guo F, Tao S, Huang R, Ma L, Fu P. Flavonoid fisetin alleviates kidney inflammation and apoptosis via inhibiting Src-mediated NF-kappaB p65 and MAPK signaling pathways in septic AKI mice. Biomed Pharmacother. 2020,122:109772. [DOI] [PubMed] [Google Scholar]
- 41.Syed DN, Adhami VM, Khan N, Khan MI, Mukhtar H. Exploring the molecular targets of dietary flavonoid fisetin in cancer. Semin Cancer Biol. 2016,40–41:130–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Varghese E, Samuel SM, Abotaleb M, Cheema S, Mamtani R, Buesselberg D. The “Yin and Yang” of natural compounds in anticancer therapy of triple-negative breast cancers. Cancers. 2018,10(10):346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kumar R, Kumar R, Khursheed R, Awasthi A, Khurana N, Singh SK, Khurana S, Sharma N, Gunjal P, Kaur J, Corrie L. Self-nanoemulsifying drug delivery system of fisetin: Formulation, optimization, characterization and cytotoxicity assessment. J Drug Deliv Sci Technol. 2019,54:101252. [Google Scholar]
- 44.Feng C, Yuan X, Chu K, Zhang H, Ji W, Rui M. Preparation and optimization of poly (lactic acid) nanoparticles loaded with fisetin to improve anti-cancer therapy. Int J Biol Macromol. 2019,125:700–710. [DOI] [PubMed] [Google Scholar]
- 45.Liu X-F, Long H-J, Miao X-Y, Liu G-L, Yao H-L. Fisetin inhibits liver cancer growth in a mouse model: relation to dopamine receptor. Oncol Rep 2017,38(1):53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen Y, Wu Q, Song L, He T, Li Y, Li L, Su W, Liu L, Qian Z, Gong C. Polymeric micelles encapsulating fisetin improve the therapeutic effect in colon cancer. ACS Appl Mater Interfaces. 2015,7(1):534–542. [DOI] [PubMed] [Google Scholar]
- 47.Shia C-S, Tsai S-Y, Kuo S-C, Hou Y-C, Chao P-DL. Metabolism and pharmacokinetics of 3,3’,4’,7-tetrahydroxyflavone (fisetin), 5-hydroxyflavone, and 7-hydroxyflavone and antihemolysis effects of fisetin and its serum metabolites. J Agric Food Chem. 2009,57(1):83–89. [DOI] [PubMed] [Google Scholar]
- 48.Osman A, Makris DP. Comparison of fisetin and quercetin oxidation with a cell-free extract of onion trimmings and peel, plant waste, containing peroxidase enzyme: a further insight into flavonol degradation mechanism. Int J Food Sci Tech. 2010,45(11):2265–2271. [Google Scholar]
- 49.Jin Y, Zhang W-Y, Meng Q-F, Li D-H, Garg S, Teng L-R, Wen J-Y. Forced degradation of flavonol glycosides extracted from Ginkgo biloba. Chem Res Chinese U. 2013,29(4): 667–670. [Google Scholar]
- 50.Kaizer J, Ganszky I, Speier G, Rockenbauer A, Korecz L, Giorgi M, Reglier M, Antonczak S. Cerium(IV)-mediated oxidation of flavonol with relevance to flavonol 2,4-dioxygenase. Direct evidence for spindelocalization in the flavonoxy radical. J Inorg Biochem. 2007,101(6):893–899. [DOI] [PubMed] [Google Scholar]
- 51.Szabados-Furjesi P, Pajtas D, Barta A, Csepanyi E, Kiss-Szikszai A, Tosaki A, Bak I. Synthesis, in vitro biological evaluation, and oxidative transformation of new flavonol derivatives: The possible role of the phenyl-N,N-dimethylamino group. Molecules. 2018, 23(12):3161(pp 1–15). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ghose AK, Viswanadhan VN, Wendoloski JJ. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J Comb Chem. 1999,1(1):55–68. [DOI] [PubMed] [Google Scholar]
- 53.Bocci G, Carosati E, Vayer P, Arrault A, Lozano S, Cruciani G. ADME-Space: a new tool for medicinal chemists to explore ADME properties. Sci Rep. 2017,7(1):6359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cai H, Sale S, Schmid R, Britton RG, Brown K, Steward WP, Gescher AJ. Flavones as colorectal cancer chemopreventive agents--phenol-o-methylation enhances efficacy. Cancer Prev Res (Phila). 2009,2(8):743–750. [DOI] [PubMed] [Google Scholar]
- 55.Parsons PJ, Penkett CS, Shell AJ. Tandem reactions in organic synthesis: novel strategies for natural product elaboration and the development of new synthetic methodology. Chem Rev. 1996,96(1):195–206. [DOI] [PubMed] [Google Scholar]
- 56.Domling A Recent developments in isocyanide based multicomponent reactions in applied chemistry. Chem Rev. 2006,106(1):17–89. [DOI] [PubMed] [Google Scholar]
- 57.Kraus GA, Gupta V, Kempema A. New approach to flavonols via base-mediated cyclization: total synthesis of 3,5,6,7-tetramethoxyflavone. Synlett. (2012),23(3):385–388. [Google Scholar]
- 58.Rao TS, Mathur HH, Trivedi GK. Reaction of chromous chloride with 3-nitroflavenes. A novel synthesis of flavonols. Tetrahedron Lett. 1984,25(48):5561–5562. [Google Scholar]
- 59.Rao TS, Singh AK, Trivedi GK. A novel photochemical method for the synthesis of flavonols. Heterocycles. 1984,22(6):1377–1382. [Google Scholar]
- 60.Deshpande SR, Mathur HH, Trivedi GK. A novel synthesis of flavonols. Synthesis. 1983,(10):835. [Google Scholar]
- 61.Looker JH, McMechan JH, Mader JW. An amine solvent modification of the Kostanecki-Robinson reaction. Application to the synthesis of flavonols. J Org Chem. 1978,43(12): 2344–2347 [Google Scholar]
- 62.Rahman AFMM, Ali R, Jahng Y, Kadi AA. A facile solvent free Claisen-Schmidt reaction: synthesis of alpha,alpha’-bis-(substituted-benzylidene)cycloalkanones and alpha,alpha’-bis-(substituted-alkylidene)cycloalkanones. Molecules. 2012,17(1):571–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rayar A, Veitia MS-I, Ferroud C. An efficient and selective microwave-assisted Claisen-Schmidt reaction for the synthesis of functionalized benzalacetones. Springerplus. 2015;4:221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rani A, Anand A, Kumar K, Kumar V. Recent developments in biological aspects of chalcones: the odyssey continues. Expert Opin Drug Discov. 2019,14(3):249–88. [DOI] [PubMed] [Google Scholar]
- 65.Li X, Chen G, Zhang X, Zhang Q, Zheng S, Wang G, et al. A new class of flavonol-based anti-prostate cancer agents: Design, synthesis, and evaluation in cell models. Bioorg Med Chem Lett. 2016,26(17):4241–4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Znati M, Bordes C, Forquet V, Lanteri P, Ben Jannet H, Bouajila J. Synthesis, molecular properties, anti-inflammatory and anticancer activities of novel 3-hydroxyflavone derivatives. Bioorg Chem. 2019,89:103009. [DOI] [PubMed] [Google Scholar]
- 67.Shen X, Zhou Q, Xiong W, Pu W, Zhang W, Zhang G, Wang C. Synthesis of 5-subsituted flavonols via the Algar-Flynn-Oyamada (AFO) reaction: The mechanistic implication. Tetrahedron 2017,73(32):4822–4829. [Google Scholar]
- 68.(Dataset) Roy T, Boateng ST, Banang-Mbeumi S, Singh PK, Basnet P, Chamcheu RN, et al. Identification of Kinase Inhibitors with Anti-Skin Cancer Activities via Data-Driven Evaluation of New Synthetic Fisetin Analogs. Data-In-Brief (in press), 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dias TA, Duarte CL, Lima CF, Proenca MF, Pereira-Wilson C. Superior anticancer activity of halogenated chalcones and flavonols over the natural flavonol quercetin. Eur J Med Chem. 2013,65:500–510. [DOI] [PubMed] [Google Scholar]
- 70.Lee J, Park T, Jeong S, Kim K-H, Hong C. 3-Hydroxychromones as cyclin-dependent kinase inhibitors: synthesis and biological evaluation. Bioorg Med Chem Lett. 2007,17(5):1284–1287. [DOI] [PubMed] [Google Scholar]
- 71.Chang YH, Jin HL,Tae SP, Jong HK, Sei HC, Sook KY, Hyun HC, Ho SS, Eunice EKK, Seong GR, Shin WJ, Dong MK. Cdk inhibitors having 3-hydroxychromen-4-one structure. WO2001083469A1, 2000. [Google Scholar]
- 72.Singh PK, Silakari O. Pharmacophore and molecular dynamics based activity profiling of natural products for kinases involved in lung cancer. J Mol Model. 2018,24(11):318. [DOI] [PubMed] [Google Scholar]
- 73.Liu Q, Wang J, Kang SA, Thoreen CC, Hur W, Ahmed T, Sabatini DM, Gray NS. Discovery of 9-(6-aminopyridin-3-yl)-1-(3-(trifluoromethyl)phenyl)benzo[h][1,6] naphthyridin-2(1H)-one (Torin2) as a potent, selective, and orally available mammalian target of rapamycin (mTOR) inhibitor for treatment of cancer. J Med Chem. 2011,54(5):1473–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Domingues B, Lopes JM, Soares P, Populo H. Melanoma treatment in review. ImmunoTargets Ther. 2018,7:35–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Naves LB, Dhand C, Venugopal JR, Rajamani L, Ramakrishna S, Almeida L. Nanotechnology for the treatment of melanoma skin cancer. Prog Biomater. 2017,6(1–2):13–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Furue M, Kadono T. Melanoma therapy: Check the checkpoints. J Dermatol. 2016,43(2):121–124. [DOI] [PubMed] [Google Scholar]
- 77.Ascierto PA, McArthur GA, Dreno B, Atkinson V, Liszkay G, Di Giacomo AM, Mandalà M, Demidov L, Stroyakovskiy D, Thomas, de la Cruz-Merino L, Dutriaux C, Garbe C, Yan Y, Wongchenko M, Chang I, Jessie J Hsu JJ, Koralek DO, Rooney I, Ribas A, Larkin J. Cobimetinib combined with vemurafenib in advanced BRAF(V600)-mutant melanoma (coBRIM): updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016,17(9):1248–1260. [DOI] [PubMed] [Google Scholar]
- 78.Patel H, Yacoub N, Mishra R, White A, Long Y, Alanazi S, Garrett JT. Current advances in the treatment of BRAF-mutant melanoma. Cancers. 2020,12(2):482 (pp 1–44). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lewis KD, Larkin J, Ribas A, Flaherty KT, McArthur GA, Ascierto PA, et al. Impact of depth of response on survival in patients treated with cobimetinib +/− vemurafenib: pooled analysis of BRIM-2, BRIM-3, BRIM-7 and coBRIM. Br J Cancer. 2019,121(7):522–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sullivan RJ, Hamid O, Gonzalez R, Infante JR, Patel MR, Hodi FS, Lewis KD, Tawbi HA, Hernandez G, Wongchenko MJ, Chang Y, Roberts L, Ballinger M, Yan Y, Cha E, Hwu P. Atezolizumab plus cobimetinib and vemurafenib in BRAF-mutated melanoma patients. Nat Med. 2019,25(6):929–935. [DOI] [PubMed] [Google Scholar]
- 81.Trojaniello C, Festino L, Vanella V, Ascierto PA. Encorafenib in combination with binimetinib for unresectable or metastatic melanoma with BRAF mutations. Expert Rev Clin Pharmacol. 2019,12(3):259–266. [DOI] [PubMed] [Google Scholar]
- 82.Singh PK, Negi A, Gupta PK, Chauhan M, Kumar R. Toxicophore exploration as a screening technology for drug design and discovery: techniques, scope and limitations. Arch Toxicol. 2016,90(8):1785–802. [DOI] [PubMed] [Google Scholar]
- 83.Kumar Singh P, Silakari O. In silico guided development of imine-based inhibitors for resistance-deriving kinases. J Biomol Struct Dyn. 2019,37(10):2593–2599. [DOI] [PubMed] [Google Scholar]
- 84.Mughal EU, Sadiq A, Ashraf J, Zafar MN, Sumrra SH, Tariq R, Mumtaz A, Javid A, Khan BA, Ali A, Javed CO. Flavonols and 4-thioflavonols as potential acetylcholinesterase and butyrylcholinesterase inhibitors: Synthesis, structure-activity relationship and molecular docking studies. Bioorg Chem. 2019;91:103124. [DOI] [PubMed] [Google Scholar]
- 85.Mughal EU, Javid A, Sadiq A, Murtaza S, Zafar MN, Khan BA, Sumrra SH, Tahir MN, Kanwal, Khan KM. Synthesis, structure-activity relationship and molecular docking studies of 3-O-flavonol glycosides as cholinesterase inhibitors. Bioorg Med Chem. 2018,26(12):3696–706. [DOI] [PubMed] [Google Scholar]
- 86.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004,47(7):1739–49. [DOI] [PubMed] [Google Scholar]
- 87.Chamcheu JC, Afaq F, Syed DN, Siddiqui IA, Adhami VM, Khan N, Singh S, Boylan BT, Wood GS, Mukhtar H. Delphinidin, a dietary antioxidant, induces human epidermal keratinocyte differentiation but not apoptosis: studies in submerged and three-dimensional epidermal equivalent models. Exp Dermatol. 2013,22(5):342–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sanna V, Chamcheu JC, Pala N, Mukhtar H, Sechi M, Siddiqui IA. Nanoencapsulation of natural triterpenoid celastrol for prostate cancer treatment. Int J Nanomedicine. 2015,10:6835–6846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chamcheu JC, Rady I, Chamcheu R-CN, Siddique AB, Bloch MB, Banang Mbeumi S, Babatunde AS, Uddin MB, Noubissi FK, Jurutka PW, Liu Y-Y, Spiegelman VS, Whitfield GK, El Sayed KA. Graviola (Annona muricata) exerts anti-proliferative, anti-clonogenic and pro-apoptotic effects in human non-melanoma skin cancer UW-BCC1 and A431 cells in vitro: involvement of hedgehog signaling. Int J Mol Sci. 2018,19(6):1791 (pp 1–24). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Brzozowska B, Galecki M, Tartas A, Ginter J, Kazmierczak U, Lundholm L. Freeware tool for analysing numbers and sizes of cell colonies. Radiat Environ Biophys. 2019,58(1):109–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Vickers JC, González-Páez EG, Wolan WD. Selective detection and inhibition of active caspase-3 in cells with optimized peptides. J Am Chem Soc. 2013, 28; 135(34):12869–76. [DOI] [PubMed] [Google Scholar]
- 92.Liu D, Tian Z, Yan Z, Wu L, Ma Y, Wang Q, Liu W, Zhou H, Yang C. Design, synthesis and evaluation of 1,2-benzisothiazol-3-one derivatives as potent caspase-3 inhibitors. Bioorg Med Chem. 2013;21(11):2960–7. [DOI] [PubMed] [Google Scholar]
- 93.Zhang S, Nie S, Huang D, FengY, Xie M. A novel polysaccharide from Ganoderma atrum exerts antitumor activity by activating mitochondria-mediated apoptotic pathway and boosting the immune system. J Agric Food Chem. 2014. February 19;62(7):1581–9. [DOI] [PubMed] [Google Scholar]
- 94.Liu H, Li Z, Huo S, Wei Q, Ge L. Induction of G0/G1 phase arrest and apoptosis by CRISPR/Cas9-mediated knockout of CDK2 in A375 melanocytes. Mol Clin Oncol. 2020. January;12(1):9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pham DDM, Guhan S, Tsao H. KIT and Melanoma: Biological Insights and Clinical Implications. Yonsei Med J. 2020. July;61(7):562–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Goto K, Takai T, Fukumoto T, Anan T, Kimura T, Ansai S, Oshitani Y, Murata Y, Sakuma T, Hirose T. CD117 (KIT) is a useful immunohistochemical marker for differentiating porocarcinoma from squamous cell carcinoma. J Cutan Pathol. 2016. March;43(3):219–26. [DOI] [PubMed] [Google Scholar]
- 97.Ponti G, Manfredini M, Greco S, Pellacani G, Depenni R, Tomasi A, Maccaferri M, Cascinu S. BRAF, NRAS and C-KIT Advanced Melanoma: Clinico-pathological Features, Targeted-Therapy Strategies and Survival. Anticancer Res. 2017. December;37(12):7043–7048. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.