

Abstract
Checkpoint blocking antibodies that interfere in the PD-1/PD-L1 axis provide effective cancer immunotherapy for tumors that are immune inflamed or induced to become “hot”. It has also been demonstrated that a small molecule inhibitor of the signaling hub kinase GSK3 can interfere in the PD-1/PD-L1 axis in T-cells by suppressing PD-1 expression. This provides an alternative approach to intervening in the PD-1/PD-L1 axis to provide cancer immunotherapy. In this communication, we demonstrate the remote loading of GSK3 inhibitor AZD1080 into the porous interior of mesoporous silica nanoparticles coated with a lipid bilayer (a.k.a. silicasomes). In a MC38 colon cancer model, intravenous injection (IV) of silicasome-encapsulated AZD1080 significantly improved biodistribution and drug delivery to the tumor site. The improved drug delivery was accompanied by cytotoxic MC38 tumor cell killing by perforin-releasing CD8+ T-cells, exhibiting reduced PD-1 expression. IV injection of encapsulated AZD1080 also resulted in significant tumor shrinkage in other syngeneic mouse tumor models, including another colorectal tumor (CT26), as well as pancreas (KPC) and lung (LLC) cancer models. Not only was the therapeutic efficacy of encapsulated AZD1080 similar or better than anti-PD-1 antibody, but the treatment was devoid of treatment toxicity. These results provide proof-of-principal demonstration of the feasibility of using encapsulated delivery of a GSK3 inhibitor to provide cancer immunotherapy, with the possibility to be used as a monotherapy or in combination with chemotherapy or other immunomodulatory agents.
Keywords: immune checkpoint, silicasome, nano drug delivery, GSK3 inhibitor, PD-1/PD-L1 axis, solid tumor
Graphical Abstract
1. Introduction
Despite considerable advances in the past decade, cancer remains a significant global health burden and a major cause of mortality. While there has been a major paradigm shift in cancer treatment through the introduction of immune checkpoint blocking antibodies, only some cancers respond and even among those only ~20% of patients are responsive to checkpoint blockade [1]. These response differences reflect the “hot” or “cold” immune status of the tumor microenvironment (TME), which refers to the presence or absence of cytotoxic T-lymphocytes in the TME. In addition, there are a number of immune escape mechanisms that contribute to the heterogeneity of the immune landscape in different tumor types. This includes the role of immune checkpoint pathways, such as CTLA-4 or the PD-1/PD-L1 axis, the marginalization or exclusion of T-cells from the cancer site, or the recruitment of immunosuppressive cellular elements such as myeloid derived suppressor cells to the tumor stroma [2-4]. There could also be impediments in antibody biodistribution to the tumor site as a result of the relatively large molecular weight of immunoglobulins or Fc-mediated binding interactions [5], in addition to significant immune-related adverse events associated with antibody therapy [6]. To date, the development of ‘generic’ biologics has been hindered by quality control issues and difficulty in demonstrating bio-similarity [7]. A key question therefore becomes whether there are alternatives to checkpoint receptor blocking antibodies or alternative ways to interfere in immune checkpoint pathways.
One potential solution to these challenges would be to develop small molecule inhibitors (SMI) of the immune checkpoint pathways, e.g., the PD-1/PD-L1 axis. PD-L1 is a transmembrane protein expressed on multiple cells including antigen presenting cells (APC), tumor cells, and stromal cells in the TME [8]. Its binding partner, the transmembrane protein PD-1, is predominantly expressed on exhausted antigen-specific T-cells and pro-B cells. PD-L1 binding to PD-1 expressed on CD8+ T-cells is capable of interfering in signal transduction by the T-cell antigen receptor (TCR) and consequent tumor cell killing (Figure 1a) [8]. However, with the advent of anti-PD-1 or anti-PD-L1 antibodies, it has become possible to interfere in the immune suppressive effects of the PD-1/PD-L1 axis, thereby restoring T-cell receptor (TCR) signal transduction and the ability for activated T-cells to commence cytotoxic tumor cell killing [9]. While some work has been performed on macrocyclic peptides and other compounds capable of interfering in the binding of PD-1 to PD-L1, a significant recent advance has been the demonstration that inhibition of glycogen synthase kinase 3 (GSK3) can inhibit the expression of PD-1 in T-cells [10]. GSK3, a signaling hub protein at the intersection of several key intracellular signaling pathways, including TCR signal transduction, is a well-established drug target for which more than 20 compounds have been developed that are capable of interfering in its serine-threonine kinase activity [11, 12]. More specifically, the use of the compound SB415286 to inhibit GSK3 activity was found to increase the expression of the transcriptional regulator T-bet, which can interfere in PD-1 expression in T-cells (Figure 1b) [10, 13, 14]. This pharmacological interference in PD-1 expression can reverse the inhibition of exhausted cytotoxic T-cell responses in the mouse B16 melanoma cancer model, with the same level of efficacy as anti-PD-1 antibodies [14]. Thus, SMIs of GSK3 have the potential to be used as surrogates for replacing or augmenting the effect of immune checkpoint blocking antibodies, in addition to their use in other applications such as Alzheimer’s disease, diabetes and certain cancers [15].
Figure 1.

Interference in PD-1 expression by small molecule GSK3 inhibitors. (a) Illustration of the PD-1/PD-L1 signaling axis, which suppresses cytotoxic T-cell tumor cell killing by inhibiting signal transduction by the T-cell antigen receptor (TCR) complex. PD-1 is expressed on “exhausted” T-cells, leading to the recruitment of the SHP2 phosphatase, which interferes in recruitment of signaling components to tyrosine-based motifs in post-TCR signaling complexes. This prevents the release of cytolytic granules. Constitutionally active GSK3 is responsible for preventing the transcriptional activation of the T-bet promoter (Tbx21). (b) Introduction of a GSK3 inhibitor (e.g., by a nanocarrier) allows restoration of T-bet expression, leading to transcriptional interference of the PD-1 promoter (Pdcd1) complex. The disappearance of PD-1 from the cell surface restores TCR signal transduction, allowing tumor cell killing by cytotoxic T-cells. In this sense, the transcriptional suppression of PD-1 exerts the same effect as blocking of the interaction of PD-1 with its ligand by antibodies.
However, while potentially promising for interfering in the PD-1/PD-L1 axis, SMIs of GSK3 face their own challenges from a drug development perspective [12, 16]. This includes possible issues of systemic or off-target toxicity resulting from the “pleiotropic effects” and involvement of the α- or β-isoforms of this kinase in multiple signaling pathways, with the possibility that GSK3 inhibition could lead to potentially serious adverse effects. Indeed, toxicity concerns in preclinical and phase I trials resulted in the abandonment of a GSK3 inhibitor to treat Alzheimer’s disease [12, 16]. Moreover, from the perspective of cancer treatment, GSK3 inhibitors may need to be combined with other drugs [17-19], requiring harmonization of their pharmacokinetics (PK) and the possibility that adverse drug/drug interactions may occur.
Some of the therapeutic shortcomings of GSK3 inhibitors can be overcome through encapsulated drug delivery, with the additional benefit of improved biodistribution and PK profiles. In this regard, we have previously developed a lipid bilayer coated mesoporous silica nanoparticle (MSNP) platform that morphologically resembles a liposome, yet results in improved drug loading capacity, reduced leakage, and improved safety [20-23]. This multifunctional carrier has also been labeled as a “silicasome”, a scalable platform that can be synthesized in large quantities (e.g., 120 g batch sizes) [23] and capable of delivering up to ~8% of the total injected drug dose to the site of multiple tumor types in animal models [20-22]. Moreover, silicasomes are biodegradable and can be administered to mice as multiple doses of up to 100 mg/kg [23] or a single high dose of 1000 mg/kg (unpublished). In order to provide proof-of-principal testing of the utility of a silicasome carrier to interfere in PD-1 expression through GSK3i delivery, we developed a custom-designed carrier to encapsulate a potent GSK3 inhibitor, AZD1080. This inhibitor was chosen based on medicinal chemistry criteria for high remote loading capacity [24]. We demonstrate that intravenous (IV) injection of the silicasome-AZD1080 carrier could significantly reduce tumor growth, with the comparable efficacy as anti-PD1 in two colorectal (MC38 and CT26) models, a pancreatic cancer (KPC), and a lung (LLC) cancer. The response was accompanied by enhanced tumor cell killing that was mediated by cytotoxic T-cells showing decreased PD-1 expression. These findings provide proof-of principal demonstration of the utility of a nano-encapsulated GSK3 inhibitor for cancer immunotherapy, with the promise to be used as a monotherapy or in combination with other drug components.
2. Materials and Methods
2.1. Materials
All GSK3 inhibitors were purchased from Cayman Chemical (Ann Arbor, MI). The following antibodies for flow cytometry were purchased from BioLegend (San Diego, CA): BV510 anti-CD45 (cat 103137), AF647 anti-CD8a (cat 100727), BV785 anti-NK1.1 (cat 108749), BV711 anti-CD107a (cat 121631), BV421 anti-CD279 (cat 135217), and PE anti-granzyme B antibody (cat 372207). Chemical reagents for MSNP synthesis were purchased from Sigma Aldrich (St. Louis, MO), as described previously [23]. 18:0 DSPC (cat 850365), 18:0 PEG2000 PE (cat 880120), and cholesterol (cat 700100) for silicasome synthesis were purchased from Avanti Polar Lipids (Alabaster, AL).
2.2. Use of medicinal chemistry criteria to identify GSK3 inhibitors for carrier remote loading
Commercially available GSK3 inhibitors were analyzed using MarvinSketch software (ChemAxon, Budapest, Hungary) to evaluate the chemical properties of the compounds listed in Supplemental Figure 1. This includes calculating the partitioning (cLogP) and solubility coefficients (logS) at pH 7.4. IC50 values were obtained from Cayman Chemical and Selleck Chemicals.
2.3. Silicasome preparation for encapsulation of a trapping agent
Mesoporous silica nanoparticles (MSNPs) were synthesized as a large batch, as previously described [23]. Briefly, this involves the addition of 0.9 L of 25 wt% cetyltrimethylammonium chloride (CTAC) in water to 17.1 L pure water in a beaker, stirred at 85 °C. 72 g triethanolamine was added, followed by 600 mL tetraethyl orthosilicate (TEOS). After stirring for 4 hours and cooling to room temperature, the bare MSNPs were precipitated with ethanol and CTAC was removed by washing in acidic ethanol, with sonication. MSNPs at 80 mg/mL in ethanol were centrifuged at 21,000 x g for 15 minutes to pellet the nanoparticles. After removal of the ethanol supernatant, the MSNP pellet was resuspended in 123 mM ammonium sulfate in water by bath sonication. 40 mg of MSNP was resuspended in 1 mL of 123 mM ammonium sulfate. Materials for the lipid bilayer were dissolved in ethanol to provide a molar ratio of 60:40:3 for 1,2-distearoylsn-glycero-3-phosphocholine (DPSC), cholesterol, and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (PE-PEG2000). Altogether, this amounted to 120 mg lipid (3x the MSNP mass), which was dissolved in 240 μL ethanol at 65 °C. The aqueous MSNP suspension was rapidly added to the lipid solution, followed by dilution with 5 mL of 123 mM ammonium sulfate. The crude lipid/MSNP mixture was probe sonicated at 40% intensity, using two rounds of pulsing (each for 10 seconds, 5 seconds pause, 5 minutes pulsing). The silicasome/liposome mixture was centrifuged for 5 minutes at 5,000 x g to pellet large aggregates. The supernatant was collected and centrifuged at 21,000 x g for 15 minutes to pellet the silicasomes. After washing and repeat of the centrifugation step, silicasomes were resuspended in 0.9% NaCl solution.
2.4. Comparative analysis of drug loading capacity and efficiency of GSK3 inhibitor silicasomes
Four GSK3 inhibitor-laden silicasome formulations were analyzed to assess encapsulation efficiency and loading capacity. 100 μL of the silicasomes suspension at 10 mg/mL in 0.9% NaCl (1 mg silicasomes) was added to AZD1080, AZD2858, LY2090314, and 1-azakenpaullone solutions, which were prepared by adding 50, 100 or 200 μg of each inhibitor to 900 μL water. These drug mass quantities provided feed weight percentages of 5, 10, and 20%, in comparison to the silicasome mass. Following execution of the remote loading procedure as detailed above, the crude solutions were used to assess absorbance in a glass bottom 96-well plate (Total Drug Absorbance). Silicasomes were centrifuged at 21,000 x g for 15 minutes to pellet the particles. After removal of the supernatant, the silicasomes were re-suspended in a fresh 0.9% NaCl solution. This was repeated three times to remove any unloaded inhibitor. The silicasome solution was then sampled and absorbance was measured to determine Loaded Drug Absorbance. Concentration-matched unloaded ‘blank’ silicasomes also had absorbance measured (Silicasome Blank Absorbance). Encapsulation efficiency was calculated by the following formula:
All measurements were taken using a SpectraMax M5 plate reader (Molecular Devices). All solutions were diluted 1/10 in 0.9% NaCl solution prior to absorbance reads. The absorbances used for each compound were: 418 nm for AZD1080, 383 nm for AZD2858, 213 nm for LY2090314, and 334 nm for 1-azakenpaullone.
2.5. Physicochemical and release characterization of AZD1080-laden silicasomes
Bare MSNPs were characterized by transmission electron microscopy (JEOL 1200-EX). Bare and AZD1080-laden silicasomes were characterized by cryogenic transmission electron microscopy (cryoTEM, TF20 FEI Tecnai-G2). Silicasomes were prepared at 100 μg/mL in PBS and were analyzed for hydrodynamic diameter and zeta potential, using dynamic light scattering in a benchtop Zetasizer (Brookhaven).
Release of AZD1080 from silicasomes was analyzed abiotically at pH 7.4 and 6.0. Fetal bovine serum (FBS) was diluted to 50% in either PBS at pH 7.4 or citrate buffer in pH 6.0 citrate buffer, followed by pH calibration, using HCl and a pH meter. 1 mg of a sAZD1080 suspension was added to 10 mL of a buffered solution containing 50% FBS and was placed on a shaker at 37 °C. At each timepoint tested, sAZD1080 was pelleted by centrifugation at 10,000 x g for 15 minutes. A fresh buffer solution was added and AZD1080 concentration was determined by analyzing the absorbance via SpectraMax M5 plate reader (Molecular Devices) at 418 nm. AZD1080 remaining in the silicasomes was analyzed at pH 7.4 after 0.5, 4, 8, 16, and 24 hours. Similar analysis under pH 6.0 conditions was carried out after 0.5, 3, 8, 16, 22 s, and 24 hours (n = 4 for each pH).
2.6. Real time RT-qPCR in cultured primary murine T-cells
Wells of a 96-well tissue culture plate were coated with anti-mouse CD3 antibody (Ultra-LEAF, Biolegend). 7.5 μL of 1 mg/mL antibody solution was diluted in 1 mL of 1xPBS, before 50 μL of the dilution was added to each plate well for 2 hours at 37 °C. The wells were thoroughly washed 3x with 1x PBS.
T-cells were extracted from C57BL/6 mouse spleens, using a standard procedure with minor modifications [25]. Two mouse spleens were mechanically disrupted and the released cells were passed through a 70 μm filter. Red blood cells (RBC) were lysed using RBC lysis buffer (eBioscience) at 4 °C for 5 minutes. 10 mL of 1x PBS was added to stop the lysis process, after which cells were pelleted at 300 x g for 5 minutes. Cells were resuspended in 1 mL of Mojosort buffer, and negative T-cell selection proceeded as per the manufacturer’s instructions (BioLegend, MojoSort Mouse CD3 T-cell Isolation Kit). Briefly, splenocytes were incubated with an antibody cocktail that provide negative selections (biotin anti-Gr-1, biotin anti-B220, biotin anti-CD49b, biotin anti-CD19, biotin anti-CD11b, biotin anti-CD24, biotin anti-TER-119). Following the addition of streptavidin magnetic nanobeads, the cell suspension was incubated within the Mojosort magnet for 5 minutes to aggregate and capture non-T-cells. The still-suspended T-cells were decanted from the test tube and washed. After cell counting and pelleting at 300 x g for 5 min, cells were resuspended in RPMI + 10% FBS + penicillin/streptomycin (100 U) + glutamax (1x) to a concentration of 1.2 x 106 cells/mL. 200 μL cells (~2.4 x 105 cells) were added to each well of the anti-CD3 coated plate. 10 μL of each of the inhibitor compounds, solubilized in 10% DMSO/PBS, were added to each well to reach a final concentration of 10 μM. 10 μL of 10% DMSO without any inhibitor was used as a control. Cells were incubated in the anti-CD3 coated plates together with the inhibitors for 72 hours. All treatments were performed in quadruplicate, and the experiment was reproduced twice.
Cells were collected and RNA was extracted using Direct-zol RNA MiniPrep Plus kits (Zymo), as per the manufacturer’s instructions. Extracted RNA was converted into cDNA using Superscript III First-Strand Synthesis System kits (Invitrogen). cDNA from treated T-cells was analyzed by RT-PCR using a LightCycler 480 instrument (Roche) and the LightCycler 480 SYBR Green I Master kit (Roche), as per manufacturer’s instructions. cDNA was analyzed using forward and reverse primers for Pdcd1, Tbx21, and Gapdh as a control, as described previously [10, 14]. This included use of the following primers: tbet-fw – 5’-GATCGTCCTGCAGTCTCTCC-3’, tbet-rv – 5’-AACTGTGTTCCCGAGGTGTC-3’, pdcd1-fw – 5’-CCGCCTTCTGTAATGGTTTGA-3’, pdcd1-rv – 5’-GGGCAGCTGTATGATCTGGAA-3’, gapdh-fw – 5’-CAACAGCAACTCCCACTCTTC-3’, gapdh-rv – 5’-GGTCCAGGGTTTCTTACTCCTT-3’ (ThermoFisher). Pdcd1 and Tbx21 expression were normalized in relation to Gapdh expression.
2.7. MTS viability assay
MC38, CT26, LLC, and KPC cell lines were grown in DMEM supplemented with 1x pen/strep, 10% FBS, 10 mM HEPES, 1x nonessential amino acids, 1x Glutamax, and 1x sodium pyruvate. Splenic T-cells were harvested from murine spleens as described in section 2.6. Cells were seeded overnight into the wells of a 96 well plate at a density of 10,000 cells per well. AZD1080 was solubilized at 1 mg/mL in water and was added to cell culture medium at concentrations of 100, 10, 1, 0.1, and 0.01 μg/mL. Cells were incubated for 24 hours and were washed twice with 1xPBS. 100 μL of cell culture media was added to cells, along with 20 μL of CellTiter 96 Aqueous One Solution Cell Proliferation Assay MTS stock solution (Promega). Cells were incubated for 1 hour at 37 °C and were analyzed for formazan development using a SpectraMax M5 plate reader at 490 nm absorbance wavelength. Background was subtracted (cell free media + MTS solution) and readings were normalized according to the viability of untreated cells (regarded as exhibiting 100% viability).
2.8. Animals
Animal care was carried out according to the “Principles of Laboratory Animal Care” by the National Society for Medical Research (USA). The experimental protocol was approved by Animal Research Committee at University of California, Los Angeles. All mice (female) were purchased from Charles River at an age of 6-8 weeks old.
2.9. Maximum tolerated dose study
The maximum tolerated dose of AZD1080 was determined using a protocol from the National Cancer Institute [26]. C57BL/6 mice were injected with AZD1080 solutions via tail vein (100 μL) and weights were recorded daily for 14 days. AZD1080 was solubilized in 30% PEG400, 0.5% Tween-80, 5% polyethylene, and 64.5% 1xPBS. The injected amounts of AZD1080 were 5, 7.5, 11.25, 16.87, or 25.31 mg/kg per mouse, n = 4 mice per group.
2.10. HPLC and IVIS analysis to assess AZD1080 biodistribution
1,1'-Dioctadecyl-3,3,3',3'-Tetramethylindodicarbocyanine (DiD)-labeled fluorescent sAZD1080 silicasomes were generated as described in section 2.3, with minor modification. 1 mol% DiD, a lipophilic carbocyanine dye (ThermoFisher Scientific), was added to the lipid solution prior to coating of MSNP particles, which were fabricated as described above.
MC38 cells, stably transfected with a luciferase vector, were inoculated into the right flank of C57BL/6 mice, using 1.2x106 cells in 40% Matrigel 1x PBS solution [23]. We used four mice per group. Tumors were allowed to grow to a size of ~300 mm3 prior to the administration of a single dose of either sAZD1080 (5 mg/kg/mouse AZD1080; IV) or free AZD1080 (5 mg/kg/mouse AZD1080; IP) in carrier solution (30% PEG400, 0.5% Tween-80, 5% propylene glycol, 64.5% 1x PBS). Tumors, livers, spleens, kidneys, lungs, hearts, and blood were harvested 24 or 48 hours after administration. Organs from free AZD1080-treated mice were snap frozen in liquid nitrogen and stored at −80 °C. Organs from sAZD1080-treated mice were placed on ice, then imaged in an IVIS Lumina II ex vivo imaging system (PerkinElmer), which used AlexaFluor 633 excitation and emission wavelengths of 633 and 660, respectively. Organ radiant efficiency was calculated using the Living Image software (PerkinElmer). Following IVIS imaging, organs were snap frozen as described for the organs from mice treated with free-AZD1080.
Organs were thawed, weighed, and a 100 μg portion of each organ was obtained for homogenization, using bead milling in a Bead Mill 4 Homogenizer (Fisher Scientific) at a level 4 homogenization setting for 2 rounds of 30 seconds each. For organs weighing less than 100 μg, the entire organ was used. To obtain serum, blood was centrifuged at 4,000 rpm for 10 minutes, following which 100 μL of the serum supernatant was treated similarly as for 100 μg of the homogenized tumor samples. Homogenized samples were doped with 3.3 μg of AZD2858 as an internal standard for extraction efficiency. The small molecule compounds were extracted from the homogenate using an equal volume of dichloromethane (DCM), with vigorous vortexing. After phase separation, the DCM layer was removed into a conical tube. This process was repeated three times, with pooling of the DCM extractions. DCM was removed under vacuum, and the extracted AZD1080 was resuspended in 300 μL methanol for High Performance Liquid Chromatography (HPLC) analysis. HPLC was performed using a Brownlee SPP 2.7 μm C18 column, 4.6 x 150 mm, a flow rate of 0.75 mL/min, injection volume of 10 μL, 416 nm absorbance wavelength, and a temperature of 30 °C. The mobile phase was HPLC grade methanol:15 mM NH4OAc buffer (pH 7.2) (60:40 ratio, isocratic). Concentrations of AZD1080 and AZD2858 were calculated by determining the area under the curve, using a calibration curve that was established by known concentrations of the two compounds. Percent injected dose (%ID) was calculated using the formula:
2.11. Use of the MC38 syngeneic colon cancer model to assess the effect of GSK3 inhibitors
MC38 cells, stably transfected with a luciferase vector, were inoculated into the right flank of C57BL/6 mice, using 0.7x106 cells in 40% Matrigel 1x PBS solution. Tumors were allowed to grow to a size of ~50 mm3 prior to the initiation of treatment, approximately 14 days after initial inoculation. Mice with outlier tumor sizes (either too small or too large) were excluded from further analysis. The remaining animals were randomly assigned to the different treatment groups. Mice were treated with three injections, set three days apart, and were sacrificed 4 days after the final treatment administration. Mice were either treated with saline (IV), aPD-1 (4 mg/kg/mouse; IP), sAZD1080 (5 mg/kg/mouse AZD1080; IV), free AZD1080 in vehicle solution (30% PEG400, 0.5% Tween-80, 5% propylene glycol, 64.5% 1x PBS) (5 mg/kg/mouse AZD1080; IP), or free SB415286 in vehicle solution (8 mg/kg/mouse; IP). Two separate experiments were performed, the first with n = 6 mice per group and the second with n = 9 mice per group.
2.12. Flow Cytometry
Tumors were harvested from MC38 mice after sacrifice. Tumors larger than 100 mm3 were divided in two, one half was preserved for histology and the other for conducting flow cytometry. To prepare samples for flow cytometry, tumor chunks of 2-3 mm3 were incubated in an enzyme cocktail (1 mg/mL collagenase type IV, 2000 U DNase type IV, 0.1 mg/mL hyaluronidase type V in 1xHBSS) for 1 hour, and the digests passed through a 70 μm nylon filter. Cells were pelleted and washed in HBSS.
Cells were incubated in Zombie NIR dye (BioLegend) for 10 minutes, and then treated with the following antibodies against T-cell surface antigens: BV510, anti-CD45 (1:80); AF647, anti-CD8a (1:400); BV785, anti-NK1.1 (1:300); BV711, anti-CD107a (1:50); and BV421, anti-CD279 (1:40) for 15 minutes. Cells were diluted in cell staining buffer, pelleted by centrifuging at 350 x g for 5 minutes, and washed twice. Cell pellets were resuspended in 1x permeabilization buffer (Biolegend), before incubation with PE-labeled anti-granzyme B antibody (1:40) for 20 minutes. After washing and resuspension in cell staining buffer, flow cytometric analysis was performed in a BD Fortessa flow cytometer. Single-stained cells and single-color beads were used for construction of a compensation matrix, which was applied to flow cytometry data prior to gating and analysis. Analysis was performed using the online flow cytometry software, Cytobank [27].
2.13. Tissue Histology
The histology preparation and staining of tumor tissue and mouse organs was performed in the Pathology Core Facility of the UCLA Jonsson Comprehensive Cancer Center. Tissues were harvested at the time of mouse sacrifice, fixed in 10% neutral-buffered formalin for 24 hours, then gradually dehydrated in 25-75% ethanol solutions over 3 days. In the UCLA histology core facility, tissues were embedded in paraffin, sectioned at 4 μm thickness, and stained by anti-CD8, anti-cleaved caspase 3, anti-perforin, anti-PD-1, or haemotoxylin and eosin (H&E). Slides were digitally scanned for analysis using Aperio ImageScope software (Leica Biosystems, Buffalo Grove, IL). Four fields of view were analyzed in each tumor, counting the number of positively stained cells within each field at 20x magnification. The cell counts were then averaged across the fields and divided by 0.15 mm2 to derive the average number of cells per mm2.The investigator was blinded with respect to the animal groups to avoid bias during counting. Fields of view were chosen to avoid the tumor periphery and processing artifacts.
2.14. Blood Chemistry Panel
At the time of sacrifice, blood was obtained by cardiac puncture from MC38 tumor growing animals and collected into Greiner Bio-One MiniCollect Capillary Blood collection system tubes. Tubes were centrifuged as per manufacturer’s instructions to separate serum from blood cells. The serum was frozen at −20 °C and was subsequently analyzed with a comprehensive serum chemistry panel, developed by IDEXX Laboratories (Westbrook, ME) for use in the UCLA Division of Laboratory Animal Medicine (DLAM).
2.15. Assessment of the Impact of AZD1080 in Additional Syngeneic Cancer Models
Three other subcutaneous cancer models were examined: KPC, CT26, and Lewis Lung Carcinoma (LLC). While CT26 and LLC cells were obtained from commercial sources, the immortalized KPC cell line was derived from a spontaneous primary tumor growing in a transgenic KrasLSL-G12D/+; Trp53LSL-R172H/+; Pdx-1-Cre mouse [22]. KPC cells were subcutaneously injected in syngeneic B6/129 mice, CT26 cells into syngeneic BALB/c mice, and LLC cells into syngeneic C57BL/6 mice. Each animal received 1x106 cells in 40% Matrigel in PBS into the right flank. Tumors were allowed to grow to a size of ~100 mm3 prior to the initiation of treatment, typically 10-11 days after inoculation. Animals with outlier tumor sizes (either too small or too large) were excluded from further study, and the remaining mice were randomly assigned to the treatment groups. Mice were treated with three injections, delivered three days apart, and were sacrificed 4 days after administration of the final treatment. Mice were either treated with saline IV, aPD-1 (4 mg/kg/mouse; IP), or sAZD1080 (5 mg/kg/mouse AZD1080; IV). For the KPC model, the number of animals per group were: 10 for saline, 8 for aPD1, and 7 for sAZD1080. For the CT26 model, the n values were: 9 for saline, 6 for aPD1, and 6 for sAZD1080. For the LLC model, the n values were: 9 for saline, 7 for aPD1, and 7 for sAZD1080.
2.16. Statistics
Comparative analysis of the differences between groups was performed using Brown-Forsythe ANOVA and Dunnett’s T3 multiple comparisons test (GraphPad Prism 9.0.0) unless otherwise stated in the figure caption. Values were expressed as mean ± standard deviation, unless otherwise stated within the figure caption. For all statistical analyses, p < 0.05 was considered statistically significant.
3. Results
3.1. Screening selection of commercially available GSK3 inhibitors to produce silicasome carriers
We have established an effective drug loading approach to remote load weak-basic molecules into the silicasome carrier, making use of a protonating agent (Figure 2a) [23, 28]. Ammonium sulfate is one example of an FDA-approved loading agent that, upon entrapment in the porous interior of a MSNP by a lipid bilayer, is capable of generating a proton gradient according to the equation: (NH4)2SO4 ↔ 2NH4+ + SO42− ↔ 2NH3 + 2H+ + SO42− [24]. The released H+ ions are capable of protonating weak-basic compounds of amphiphilic nature, capable of diffusing across the lipid bilayer. Protonation of the amphiphilic compounds renders them hydrophilic, preventing their back diffusion across the lipid bilayer. Moreover, in the protonated state, the active pharmaceutical ingredients (API) are capable of interacting with SO42− ions, leading to the formation of drug precipitates in the carrier pores. The success of the loading technique has been demonstrated in the development of the doxorubicin carrier, Doxil®, which has also received FDA approval [29].
Figure 2.

Remote loading considerations based on the chemical properties of GSK3 inhibitors. (a) Illustration of remote loading by the silicasome carrier, requiring the generation of a proton gradient for drug import across the lipid bilayer. (b) Scheme demonstrating the chemical criteria to assess the remote loading capacity of commercially available GSK3 inhibitors (Supplemental Figure 1). These criteria include compound, molecular weight (MW), partition coefficient (cLogP), isoelectric point (pI) and solubility coefficient (LogS) at pH 7.4 (c) Table of relevant chemical properties of commercially available GSK3 inhibitors, arranged according to IC50 values, chemical abstracts service number (CAS No), and MW, as provided by chemical vendors. The compounds highlighted in bold fulfill the prediction criteria for chemical characteristics amenable to remote loading. These compounds were acquired to conduct remote loading studies, in addition to the GSK3 inhibitor, SB415286, that was previously used to assess the impact on PD-1 expression [14]. The IC50 values, as displayed, are provided by the chemical manufacturers.
Using medicinal chemistry criteria and analysis of quantitative structure-activity relationships (QSAR), it has been possible to screen drug libraries for chemical properties that allow proton gradient remote loading [24, 30]. While classically these studies have been performed in liposomes, we hypothesized that the same principles apply to lipid membrane-bound nanoparticles such as the silicasome (Figure 2a and 2b). Drugs that are expected to qualify for remote loading generally exhibit the following chemical properties: (i) small molecular weight (< 1 kDa) and geometric diameter (2~3 nm) to allow the compounds to pass through the lipid bilayer into the porous interior; (ii) an isoelectric point between 7 < pI < 11 to allow lipid bilayer transit before entrapment; (iii) high water solubility coefficient (logS) in the range of 1-10 mg/mL at physiological pH (7.4); and (iv) amphipathic nature, allowing the compound to pass through the lipid bilayer. The latter property can be evaluated by using of a partition coefficient (clogP). Compounds with clogP values > 5 are difficult to solubilize in water and have an increased propensity to partition into the lipid bilayer, while low logP values (< 0) are representative of hydrophilic compounds that will be less amenable to passing through the lipid membrane. These selection criteria were applied to 17 commercially available GSK3 inhibitors (Figure 2c), the chemical structures of which are shown in Supplemental Figure 1. Among these, two compounds provided good matches based on all the listed criteria (AZD2858 and AZD1080), while two others were moderate matches (LY2090314 and 1-Azakenpaullone) (Figure 2c). All four were purchased for experimental validation of their remote loading efficiency.
3.2. Remote loading of GSK3 inhibitors and silicasome characterization
The overall scheme for the synthesis of silicasome carriers to deliver GSK3 inhibitors is outlined in Figure 3. Briefly, a large batch (i.e. 120g) synthesis of bare MSNPs was carried out as previously described, yielding ~70 nm particles (Figure 3a) [23]. 20 mg/mL bare particles were soaked in a 123 mM ammonium sulfate solution to generate a pH of 5 inside the porous interior (Figure 2a) [31]. 20 mg of soaked-in MSNPs were then used together with an ethanol suspended lipid solution to apply a lipid bilayer by a flow through sonication (Figure 3b). This ethanol solution was comprised of 60 mg of a mixture of DSPC:Cholesterol:PE-PEG2000 at molar ratios of 6:4:0.3. Following the removal of the non-encapsulated trapping agent, AZD2858, AZD1080, LY2090314, and 1-Azakenpaullone were dissolved in 0.9% saline and incubated with the ammonium sulfate carrier to assess the loading capacity in response to different feed weight percentages (Supplemental Figure 2a). Whereas 1-Azakenpaullone encapsulation efficiency was comparatively low, AZD1080, AZD2858, and LY2090314 could be loaded with moderate efficiency. AZD2858 showed poor solubility at feed weight percentages ≥ 20% (w/w%, drug/particle). In contrast, AZD1080 showed good aqueous solubility and displayed a good encapsulation efficiency of 73% at 5% feed weight; this allowed a maximum loading capacity of 8.1 w/w% (Figure 3c). AZD1080 remote loading did not appreciably impact the carrier size or morphology, as demonstrated by cryogenic transmission electron microscopy (cryoTEM) (Figure 3d and Supplemental Figure 3). However, there was a slight increase in the hydrodynamic diameter of AZD1080-silicasomes (151.5 nm vs 160.9 nm) by dynamic light scattering (DLS). AZD1080 is released from the silicasome particles over time, amounting to ~5% cumulative release over 24 hours in 50% fetal bovine serum (FBS) / 50% PBS at 37 °C (Supplemental Figure 4). Release of AZD1080 from silicasomes is considerably faster in a citrate acidified buffer (pH 6) (containing 50% FBS), leading to ~24% release over 24 hours. Based on these performance characteristics, we opted to use the fully characterized AZD1080-silicasomes to conduct further in vitro and in vivo studies looking at the effect of the carrier on PD-1 expression.
Figure 3.

Characterization of the silicasome carrier for delivery of a GSK3 inhibitor. (a) Representative images to explain large batch synthesis of bare MSNPs. A 20L sol-gel synthesis of ~70 nm bare MSNP was prepared as previously described, yielding a 120 g batch of high-quality particles [22]. (b) Illustration of the subsequent synthesis procedure to obtain lipid-coated MSNPs (silicasomes) for soaking in the trapping agent, ammonium sulfate. (c) Table of silicasome physical characteristics, encapsulation efficiency, and loading capacity for AZD1080. DLS, zeta potential, and encapsulation data are representative of 3 samples (SD = standard deviation). (d) CryoTEM micrograph of silicasomes after loading of GSK3 inhibitor, AZD1080. Scale bar = 100 nm. A schematic illustration of the AZD1080 laden silicasome appears on the right-hand side.
3.3. T-cell screening to assess PD-1 inhibition by free and encapsulated GSK3 inhibitors
In order to assess the ability of AZD1080 to inhibit PD-1 expression in vitro, we utilized an RT-qPCR assay, developed by Taylor et al [14] to demonstrate the inhibitory effect of SB415286 on Pdcd1 expression and stimulatory effect on Tbx21 expression in anti-CD3 activated T-cells (Figure 4a). Compared to a ~2.2-fold reduction in Pdcd1 mRNA expression by SB415286, the comparable decreases by free AZD1080 and silicasome-encapsulated AZD1080 (sAZD1080) were ~6- and ~3.7-fold, respectively (Figure 4b). Compared to free drug, sAZD1080 generated a lesser but significant cellular response, likely due to slower but sustained drug release under tissue culture conditions. These results were the inverse of Tbx21 mRNA expression in response to the same stimuli (Figure 4c). These data are in agreement with the findings of Taylor et al, demonstrating that an SMI, AZD1080, is capable of inhibiting Pdcd1 mRNA expression in free or encapsulated form in T-cells.
Figure 4.

In vitro assay to show GSK3 inhibition of Pdcd1 expression. (a) Experimental procedure for assessing anti-CD3 induced Pdcd1 expression and inhibition by GSK3 inhibitors. The expression of (b) Pdcd1 and (c) Tbx21 was determined by qRT-PCR and reported as log2-fold change. The negative control was comprised of unstimulated cells, while cells treated with immobilized anti-CD3 antibody only (“aCD3 Only”) represents the positive control. Significance was determined using 2-way ANOVA, * p < 0.05 and ** p < 0.01. Error bars = SD, n = 4.
3.4. AZD1080 impacts on cancer cell viability
GSK3 exerts pleiotropic effects in cancer cells, which could lead to complicated and even paradoxical treatment outcomes [32]. For instance, GSK3 is an inhibitor of the Wnt/β-catenin signaling pathway, which may result in growth promotion or anti-apoptotic effects under some circumstances [33], while GSK3 inhibition can enhance the development of apoptosis in other cancer cell lines [34]. In order to determine whether AZD1080 impacts the viability of the MC38, CT26, LLC, and KPC cancer cells used in this study, a cell viability study was undertaken following 24-hour exposure to AZD1080 (Supplemental Figure 5). Use of an MTS assay demonstrated that only the LLC cell line showed decreased viability at a high dose (100 μg/mL) of AZD1080, while slightly enhancing the viability of MC38 and CT26 cells at 10 and 100 μg/mL (Supplemental Figure 5c). No significant changes were seen in KPC cells (Supplemental Figure 5d). AZD1080 failed to show a significant impact on the viability of primary T-cells isolated from murine spleens, except at 100 μg/mL, which is unlikely to be relevant to in vivo experimentation (Supplemental Figure 5e). Moreover, there was even a slight increase in cell viability over the dose range 0.01 to 1 μg/mL.
3.5. AZD1080 delivery by silicasome leads to efficient intratumoral drug delivery in MC38 tumors
In order to establish a safe working concentration for the performance of murine experiments, we performed a maximum tolerated dose study, which was performed by a one-time injection of 5-25.3 mg/kg AZD1080 and following animal survival, body weight, behavioral changes in posture over 10 days (Supplemental Figure 6). No toxicity was observed, which prompted us to consider the lowest dose of 5 mg/kg, which was considered efficacious and safer in the performance of a rat study to treat an Alzheimer’s disease process [35]. The free drug had to be administered intraperitoneal (IP) because of the vascular sclerosing effects of the solvents (e.g., 30% PEG400, 0.5% Tween-80, 5% propylene glycol) to keep free AZD1080 in solution [36], whereas the encapsulated drug could be injected into the tail vein repetitively. The ability to maintain vascular access for repetitive IV injection, constitutes an additional reason for considering the use of a nanocarrier for ease of encapsulated delivery.
In order to compare intratumoral drug biodistribution between free and encapsulated AZD1080, an in vivo experiment was performed in the MC38 colon cancer model (Figure 5a). We have previously demonstrated that the silicasome nanocarrier can significantly improve the delivery of chemotherapeutic agents at the tumor site, compared to free drug [23]. After establishment of subcutaneous tumors in a size range of ~300 mm3, mice were injected IV with 5 mg/kg of a fluorescently-labeled sAZD1080 nanocarrier, with a comparable dose of free AZD1080 being administered IP. Fluorescent labeling was performed by incorporating the lipophilic dye, 1,1'-Dioctadecyl-3,3,3',3'-Tetramethylindodicarbocyanine (DiD), into the lipid bilayer of sAZD1080 nanoparticles Animals were sacrificed after 24 and 48 hours, followed by harvesting of the tumors, livers, spleens, kidneys, lungs, and hearts and blood withdrawal. The explanted organs from sAZD1080-treated mice were used to obtain IVIS images, using an AlexaFluor 633 filter set (633 nm excitation, 660 nm emission) lipophilic dye (633 nm excitation, 660 nm emission) capable of incorporation into lipid bilayers was used to generate fluorescently-labeled sAZD1080 nanoparticles, which accumulated in various organs in vivo (Figure 5b). Quantification of the total radiant efficiency (Figure 5c and 5d) also demonstrated that nearly all the signal is confined to the tumors, livers, and spleens. The organs were subsequently used to extract for AZD1080 by an organic solvent, followed by HPLC analysis, in which the AZD1080 concentration was expressed as μg of drug per g of organ (Figure 5e and 5f). This demonstrated that the mean concentration of 6.07 μg AZD1080/g tumor tissue (95% CI: 3.43-8.71 μg/g) accomplished during encapsulated delivery was significantly higher (p < 0.0001) than the tumor concentration 0.46 μg/g (95% CI: 0.08-0.85 μg/g) for free drug after 24 hours. Moreover, this trend was sustained with further enhancement by 48 hours (p < 0.0001). This represents ~13-fold and ~18-fold enhancement of encapsulated AZD1080 delivery over free drug at 24 and 48 hours, respectively. When the tumor drug concentration was expressed as a percentage of the injected dose (%ID), the respective biodistribution values for sAZD1080 and free drug were 4.84 ± 1.45% and 0.32 ± 0.19% at 24 hours (Supplemental Figure 7a). This amounted to 9.18 ± 2.96 and 0.24 ± 0.18 %ID for sAZD1080 vs free AZD1080 by 48 hours (Supplemental Figure 7b). Silicasome delivery also allowed a significant increase in the splenic concentrations of AZD1080 compared to free drug at both timepoints (p < 0.0001). While there was no significant difference between free and encapsulated AZD1080 concentrations in the liver at 24 hours, encapsulated delivery that lead to a significant difference by 48 hours (p < 0.05). These findings are compatible with the established role of the mononuclear phagocyte system in nanocarrier sequestration from the circulation [37]. The only organ in which free AZD1080 administration resulted in a significant increase over sAZD1080 was in the kidneys after 24 hours (p < 0.0001) (Figure 5e). This is also compatible with the potential renal toxicity of AZD1080, with the clear implication that encapsulated delivery protects against this possibility because of stable drug retention and pattern of biodistribution by the silicasomes. In addition to the tissue biodistribution studies, encapsulated AZD1080 delivery also significantly increased the serum levels of the drug by 24 hour compared to free drug (p < 0.0001) (Figure 5e). This difference disappeared by 48 hours, which is in agreement with previous drug studies with the silicasome [23].
Figure 5.

In vivo biodistribution of AZD1080. (a) Illustration of the experimental scheme. MC38 tumor-bearing mice were injected with DiD-labeled sAZD1080, or free AZD1080 and organs were harvested 24 and 48 hours after drug administration. The organs were used for quantitative assessment of sAZD1080 fluorescence (633 nm excitation), followed by homogenization and AZD1080 extraction by an organic solvent. (b) IVIS imaging of explanted organs from DiD-sAZD1080 treated mice (633 nm excitation, 660 nm emission), including quantification of fluorescence intensity (c) 24 hours and (d) 48 hours after administration. n = 4 mice per treatment. Tu = tumor, Li = liver, Sp = spleen, Ki = kidneys, Lu = lungs, He = heart, Bl = blood. HPLC quantification of organ AZD1080 concentration at (e) 24 hours and (f) 48 hours. Significance was determined using 1-way ANOVA, * p < 0.05, ****, p < 0.0001 n = 4.
3.6. sAZD1080 inhibits tumor growth by impacting cytotoxic T-cell killing in a syngeneic MC38 cancer model
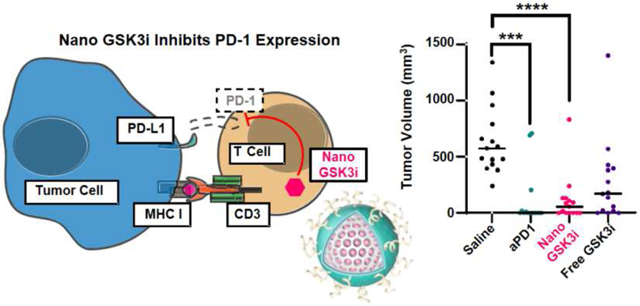
The first assessment of sAZD1080 efficacy was carried out in a MC38 colon cancer model, which is regarded as highly responsive to anti-PD1 monoclonal antibody treatment (Table 1). Following the establishment of subcutaneous tumor growth, treatment with free AZD1080, sAZD1080, anti-PD-1 antibody, SB415286, or saline commenced 10 days after inoculation. Animals treated with sAZD1080 received IV injection of 5 mg/kg (corresponding to a particle dose of ~62 mg/kg). Controls included animals receiving IP doses of 5, 8 and 4 mg/kg, respectively, of free AZD1080, SB415286 and anti-PD-1. Treatment was repeated every three days for a total of three administrations. Mice were sacrificed four days after the final treatment. The study was executed in two separate experiments, using animal group sizes of n=6 and n=9, respectively. Although each experiment yielded significant data in its own right, there were no significant differences between the experiments in terms of the tumor sizes, prompting us to combine the data for pooled analysis, which will be used in subsequent data presentation (Supplemental Figure 8). The first series of analyses documented the tumor growth curves (Figure 6a), impact on the final tumor volumes (Figure 6b), and the tumor weights (Figure 6c) at the time of sacrifice. These data demonstrate that all the treatment modalities yielded significant tumor growth inhibition, except for free AZD1080. The most robust tumor volume reduction was obtained with sAZD1080 (Figure 6b), while the biggest tumor weight decrease was obtained with anti-PD1 (Figure 6c). Inspection of the cumulative growth curves (spaghetti plots) for each animal demonstrate intra-group variances, including tumor disappearance in some animals (Figure 6d). Thus, while all the tumors in the saline group expanded exponentially, the number of “tumor-free” animals were 1/15 during treatment with SB415286, 3/15 for free AZD1080, 6/15 for sAZD1080, and 10/15 for anti-PD-1 treatment.
Table 1.
Syngeneic tumor models in this study.
| Name | Type | Syngeneic Mouse |
Method of Generation |
Mutational Load * |
aPD-1 Monotherapy Responsiveness *** |
Major Oncogenes *** |
sAZD1080 Responsiveness **** |
|---|---|---|---|---|---|---|---|
| MC38 | Colon Carcinoma | C57BL/6 | Chemical (DMH) | ~3,400 | Responsive | Alk, Braf, Erbb4, Jak2, Jak3, Pten, Ptpn11, Stat3, Trp53 | Responsive |
| KPC | Pancreatic Ductal Adenocarcinoma | B6/129 | Genetic | 2 ** | Poor | Kras, Trp53 ** | Moderate |
| CT26 | Colon Carcinoma | BALB/C | Chemical (NMU) | ~3,300 | Moderate | Brca2, Cdk4, Erbb3, Fgfr1, Kras, Pdgfra | Moderate |
| LLC (LL/2) | Lung Carcinoma | C57BL/6 | Spontaneous | ~2,300 | Poor | Alk, Apc, Cdk4, Csf1r, Erbb3, Flt1, Flt3, Jak2, Jak3, Met | Moderate |
Mosely et al. Cancer Immunol. Res. 2017
Lee et al. Curr Protoc Pharmacol 2016
Charles River Syngeneic Model Immunotherapy Responsiveness Data
This study
Figure 6.

Comparison of the effects of free and encapsulated GSK3 inhibitors and anti-PD1 on MC38 tumor growth and the effect on cytotoxic T-cell responses. Animal treatment is discussed in the method section. (a) Tumor volume growth curves starting from the time of inoculation up to sacrifice. The green arrows represent days on which treatment was administered. Error bars = SEM. n = 15 in each group (pooled data from 2 experiments). (b) Final tumor volumes, n = 15 per treatment. (c) Final weights for each tumor, with the bars representing the mean values for each group (n = 15). (d) Spaghetti plots to show individual tumor growth curves (tumor volume) for the duration of the experiment. Fractions displayed in each graph represent the number of mice (out of 15 animals) with no detectable tumor at the time of sacrifice. Statistical analyses were performed by Brown-Forsythe ANOVA and Dunnett’s T3 multiple comparisons test, * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001, NS = not significant.
In order to address the treatment’s impact on anti-tumor immunity, we asked whether sAZD1080 could alter the T-cell responses that are generally used to assess the therapeutic efficacy of immune checkpoint blocking antibodies. These include CD8+ T-cell abundance, the level of PD-1 expression on this subset, cytotoxic cancer cell death (via activated caspase 3 expression) and perforin release. Perforin is an indicator of cytotoxic T-cell maturation and acts as a pore-forming protein that leads to cancer cell killing [38, 39]. First, cell suspensions from tumor digests were used to assess the number of CD8+/CD107a+/granzyme B+ lymphocytes by flow cytometry [14, 38]. This demonstrated a significant increase in the number of activated CD8+ T-cells in all treatment groups except for free AZD1080 (Figure 7a). In order to assess the status of PD-1 expression on CD8+ T-cells, flow cytometry was used to assess the % of CD8+/ PD-1+ cells in the tumor digests [14, 39]. This demonstrated that while anti-PD-1, SB415286 or free AZD1080 had no impact on this cell subset, sAZD1080 administration could significantly suppress the % of CD8+/PD-1+ cells (Figure 7b). It is important to mention that due to the higher rate of tumor disappearance in the anti-PD1 treated group, fewer tumors were available for flow cytometry analysis, explaining why it was not possible to achieve statistical significance.
Figure 7.

Flow cytometry analysis of tumor-infiltrating CD8+ T-cells: (a) activated cytotoxic T-cells, i.e. CD107a+ Gzmb+ CD3+ CD8+ cells; (b) PD-1 staining intensity. Group sizes: n = 10 for saline and SB415286 treatments, n = 9 for free AZD1080 or sAZD1080 treatment, and n = 3 for anti-PD-1 (the reduced animal number for the latter group was due to disappearance or extremely reduced size of most of the tumors in this group). Statistical analysis was performed by Brown-Forsythe ANOVA and Dunnett’s T3 multiple comparisons test. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001, NS = not significant.
The flow cytometry data were supported by immunohistochemistry (IHC) staining of embedded tumor sections to display the presence of CD8+ T-cells, perforin and cleaved caspase 3 (Figure 8a). Low magnification images appear in Supplemental Figure 9. The analysis was further assisted by using imaging software for data quantification (Figure 8b-d). Treatment with sAZD1080 and anti-PD-1 resulted in the doubling of the number of CD8+ cells per mm2, amounting to a statistically significant increase (Figure 8b). Perforin expression was also significantly increased by the same treatment groups (Figure 8c). Moreover, these therapies increased the rate of tumor cell death as reflected by increased cleaved caspase 3 (CC3) staining (Figure 8d). No statistically significant change in CC3 expression was observed in mice receiving the free GSK3 inhibitors. All considered, the flow and IHC data strongly support the involvement of cytotoxic T-cell activity in decreasing tumor volume (Figure 7). As discussed previously, AZD1080 does not have a significant direct effect on cancer cell toxicity at relevant concentrations for these in vivo experiments, as demonstrated in Supplemental Figure 5.
Figure 8.

Immunohistochemical analysis of MC38 subcutaneous tumors, obtained from the efficacy study in Fig. 6. (a) Representative images of tumor sections stained with anti-CD8, anti-perforin, or anti-cleaved-caspase 3 antibodies. Scale bar (lower right corner) = 100 μm; all images were reviewed under the same magnification. Quantitative image analysis showing: (b) CD8+ cells per mm2, (c) perforin -positive cells per mm2, and (d) cleaved caspase 3+ (CC3) cells per mm2. The bars represent the mean values for each group. Statistical analysis by 2-way Brown-Forsythe ANOVA and Dunnett’s T3 multiple comparisons test, n = 6 for saline and free AZD1080 treatments, 5 for silicasome AZD1080 and anti-PD-1 treatments, and 8 for SB415286 treatment. * p < 0.05, ** p < 0.01.
3.7. Safety of the sAZD1080 treatment platform
We have previously demonstrated the intrinsic safety of the silicasome platform in pancreatic cancer therapy, including the role of the supported lipid bilayer in preventing the premature release and toxicity of irinotecan [21, 22, 28]. While there is sparse literature on the use of AZD1080 in animals, it was necessary to discontinue this agent in a phase I clinical trial in humans due to nephrotoxicity [16]. Our safety analysis in mice failed to show any changes in animal weight or evidence of organ toxicity during treatment with sAZD1080 (Supplemental Figure 10). This includes the absence of significant effects on serum chemistry, though free AZD1080 could be seen to depress the albumin/globulin ratio, a finding that is of unknown significance (Supplemental Figure 11a). Interestingly, both encapsulated and free AZD1080 administration was also associated with reductions in total bilirubin and blood urea nitrogen (BUN) levels, which do not reflect toxicity. Principal component analysis indicated that free AZD1080 was responsible for more variation in serum chemistry than sAZD1080 or other treatments (Supplemental Figure 11b). Finally, H&E staining of a number of organs collected at the time of sacrifice failed to show any histological evidence of tissue damage, including histological analysis of the kidney (Supplemental Figure 12). All considered, no evidence of toxicity was observed for SAZD1080 use.
3.8. The favorable immunotherapeutic effects of sAZD1080 also applies to syngeneic models for pancreatic, lung and colon cancer
While the MC38 model is highly responsive to the administration of immune checkpoint blocking antibodies, most animal and human cancers are either unresponsive or only partially responsive to this form of therapy [40-42]. To obtain a more comprehensive picture of the efficacy of SAZD1080 versus anti-PD-1 treatment, three additional syngeneic cancer models were investigated, namely a second colon cancer (CT26) model, a Lewis lung cancer (LLC) and a Kras-derived pancreatic cancer (KPC). These animal tumor models differ with respect to the cancer cell type, tumor origin, mutational load, oncogene expression, and tumor immune escape mechanisms (Table 1). Moreover, in contrast to the characterization of MC38 as a tumor type that is “highly responsive” to anti-PD-1, the responsiveness of CT26 to the same treatment is considered as “moderate”, with KPC and LLC classified as “poorly responsive”.
The first comparison of SAZD1080 to anti-PD-1 was carried out in CT26, a colon model that was derived from a chemically-induced cancer that exhibits a high mutational load (~3,300 neoantigens) [42]. In contrast to MC38, CT26 is considered moderately responsive to anti-PD-1 therapy [40, 42]. Systemic administration of above therapeutic agents, used at similar doses and treatment intervals as in the MC38 model, demonstrated differences in tumor growth inhibition (Figure 9a). Thus, while SAZD1080 reduced tumor growth by ~58%, which was significantly different from saline (p = 0.0425), tumor growth reduction of ~21% by anti-PD-1 treatment was not statistically significant (p = 0.3656). There was no statistical significance in the final tumor volume size of SAZD1080 vs. anti-PD-1 (p = 0.2349). In contrast to the robustness of the immunotherapy response in MC38, none of treatments resulted in tumor disappearance in CT26.
Figure 9.

Tumor growth inhibition by PD-1/PD-L1 axis inhibition in three additional syngeneic tumor models in mice. Tumor volume shrinkage in response to treatment with anti-PD-1 antibody or silicasome AZD1080 for: (a) CT colon cancer model, (LLC lung cancer model, and (c) KPC pancreatic cancer model. Animal treatment is discussed in the methods section. For each model, tumor volume growth curves are shown starting from time of inoculation to time of sacrifice. Green arrows represent days of treatment administration. Error bars = SEM. The spaghetti plots are used to show the growth curve for every animal, in addition to showing shrinkage of the final tumor volumes for each model. The error bars represent the mean values in each group. Statistical analysis by Brown-Forsythe ANOVA and Dunnett’s T3 multiple comparisons test. For CT26, n = 9 for saline treatment and n =6 for silicasome AZD1080 and anti-PD-1 treatments. For LLC, n = 9 for saline and 7 for silicasome AZD1080 and anti-PD-1 treatments. For KPC, n = 10 for saline, 7 for silicasome AZD1080, and 8 for anti-PD-1 treatments. * p < 0.05.
The next investigation was into the LLC model. These tumor cells are derived from a spontaneously developing lung cancer and also exhibit a high mutational load (~2,300 neoantigens) [42]. This model is considered poorly responsive to anti-PD-1 monotherapy [43]. The experimentation in LLC demonstrated good responses to both sAZD1080 and anti-PD-1, with statistically significant differences in tumor growth inhibition and final tumor volume (compared to saline) (Figure 9b). Final tumor volume was reduced by ~60% by sAZD1080 treatment (p = 0.0250) and ~57% (p = 0.0277) by anti-PD-1.
The last comparison of sAZD1080 with anti-PD-1 was carried out in a pancreatic ductal adenocarcinoma (PDAC) model, known for its complex TME and treatment resistance to multiple treatment modalities [44, 45]. The KPC model was established using a cell line derived from a transgenic KrasLSL-G12D/+; Trp53LSL-R172H/+; Pdx-1-Cre animal [46]. Similar to human PDAC, KPC has a low mutational load and is poorly responsive to immunotherapy [45], including anti-PD-1 monotherapy [47, 48]. Systemic administration of sAZD1080 led to significant tumor growth inhibition (p = 0.0258), with a ~66% reduction in tumor volume compared to saline (Figure 9c). However, while anti-PD-1 treatment reduced the tumor volume by ~55%, the growth inhibition in KPC did not achieve statistical significance (p = 0.1126).
All considered, our data demonstrate that encapsulated sAZD1080 delivery can induce significant growth inhibition in four different cancers, including in animal models where anti-PD-1 treatment did not make a significant impact. In order to demonstrate whether, similar to MC38, the responses to sAZD1080 in the rest of the tumors are mediated by cytotoxic T-cells, IHC analysis was performed to assess CD8 recruitment and tumor cell death (CC3 staining) (Figure 10a). This demonstrated that sAZD1080 treatment resulted in a significant increase in the number of CD8+ T-cells for all the cancers (Figure 10a). While similar responsiveness was seen for MC38 and KPC during anti-PD-1 treatment, this therapy did not result in significant cytotoxic T-cell recruitment in CT26 and LLC models (Figure 10a). These results were also compatible with the increase in CC3 staining. Thus, while sAZD1080 treatment resulted in a significant increase in tumor cell death in all tumor types, anti-PD1 did not impact CC3 staining in the KPC and CT26 models (Figure 10b). Moreover, the response to sAZD1080 in CT26 was significantly higher than the anti-PD-1 treatment group (p < 0.05). All considered, these data support the notion that encapsulated delivery of the GSK3 inhibitor AZD1080, induces significant tumor growth inhibition by increasing the abundance and tumor cell-killing effects of CD8+ T-cells in heterogeneous cancer models.
Figure 10.

Histological comparison of the immune responses across the syngeneic cancer models. Immunohistochemistry was performed using anti-CD8 or anti-cleaved caspase 3 antibodies on tumor sections from mice treated with saline, silicasome AZD1080 (sAZD1080) or anti-PD-1 antibody (aPD1). Quantification of positively stained cells was performed for (a) anti-CD8+ cells per mm2, or (b) anti-cleaved caspase 3+ cells per mm2. The bars represent the mean value for each group. The group sizes for CT26 were: n = 9 for saline treatment, n=6 for sAZD1080, as well as anti-PD-1. Group sizes for LLC: n = 9 for saline, n =7 for sAZD1080, as well as for anti-PD-1 treatments. Group sizes for KPC: n = 10 for saline, n =7 for sAZD1080, and n = 8 for anti-PD-1. The MC38 data were derived from the experiment shown in Figure 8. N values were higher for saline treatment groups due to previously recognized increased variance in tumor sizes. Statistical analyses were performed by 2-way Brown-Forsythe ANOVA and Dunnett’s T3 multiple comparisons test. * p < 0.05, ** p < 0.01 compared to saline control, and † p < 0.05 compared to anti-PD-1 treatment.
4. Discussion
In this study, we demonstrate the utility of a silicasome carrier to deliver the GSK3 inhibitor AZD1080 to the tumor site of multiple syngeneic animal cancer models. AZD1080 was selected from a panel of GSK3 inhibitors by medicinal chemistry criteria for predicting remote loading into silicasomes using a proton gradient. AZD1080 was found to inhibit Pdcd1 expression in murine T-cells in free as well as encapsulated drug form. Following the establishment of an MC38 colon cancer model in immunocompetent C57BL/6 mice, we could demonstrate that systemic administration of sAZD1080 was associated with a significant reduction of tumor growth, resulting 6 of 15 mice becoming tumor-free. This response outcome was comparable to the tumor-inhibiting effects of anti-PD-1 antibody. In contrast, free AZD1080 had no significant effect on tumor growth inhibition compared to the negative control. The assessment of intratumoral AZD1080 concentrations showed a 13~18-fold increase in drug delivery to the MC38 tumor site by silicasome compared to free drug delivery. Encapsulated delivery also increased biodistribution of AZD1080 to the spleen, while decreasing the renal content of AZD1080. Flow cytometric analysis further demonstrated that sAZD1080 administration could significantly increase the number of activated CD8+ T-cells in the tumor microenvironment. Moreover, IHC analysis revealed a significant increase in perforin, granzyme B and CC3 staining at the tumor site, comparable to the effect of anti-PD-1. In contrast, free GSK3 inhibitors did not significantly impact any of these biomarkers reflecting CD8+ T-cell activity. Additional assessment of sAZD1080 in a second colon cancer (CT26), a lung cancer (LLC) and a pancreatic cancer (KPC) model confirmed the carrier’s significant tumor growth inhibitory effects as a result of cytotoxic T-cell recruitment and induction of lytic tumor cell death. These results are compatible with a principal immunological mechanism rather than other possible mechanisms of action of sAZD1080. All considered, sAZD1080 was capable of generating roughly equivalent rates of tumor growth inhibition as anti-PD-1 antibody for all tumors tested. This demonstrates the potential utility of an encapsulated small molecule inhibitor of GSK3 as an alternative treatment modality for immune checkpoint inhibition.
The PD-1/PD-L1 axis serves as an important immune suppressive pathway that is amenable to immune checkpoint therapy [49]. Newly generated cytotoxic T-cells are capable of accomplishing tumor cell killing during TCR engagement, which leads to the release of perforin and granzyme B from cytotoxic granules [50]. There are number of adjunct mechanisms, however, that can constrain cytotoxic killing, including the acquisition of PD-1 expression that leads to a functional state of T-cell exhaustion [51]. Mechanistically, this involves the expression of an immunoreceptor tyrosine-based switch motif by PD-1, which leads to the recruitment of the Src homology region 2 domain of SHP-2, a phosphatase that strips away phosphate groups from post-TCR signaling molecules such as CD3ζ, ZAP70, Akt, and ERK [8]. The accompanying interference in TCR signal transduction results in reduced signal transduction and gene expression pathways engaged in tumor cell killing by cytotoxic T cells. In the process, the gene expression program of the T cells switch towards an “exhaustion program”, which further hinders effective tumor cell killing [52]. Against this background, Taylor et al. discovered that siRNA knockdown or small molecule inhibition of GSK3 in murine CD8+ T-cells was capable of reducing the cell surface expression of PD-1, in addition to boosting cytolytic killing of a lymphoma [13]. Moreover, it was demonstrated that the GSK3 inhibitor increases transcriptional activation of the Tbx21 promoter, leading to increased T-bet expression in CTLs [10]. Chromatin immunoprecipitation assays further confirmed that T-bet binding to the Pdcd1 promoter, with the ability to suppress the transcriptional expression of PD-1. Thus, the use of GSK3 inhibitors provides an additional approach to restoring the cytotoxic activity of exhausted T-cells, in addition to the availability of checkpoint blocking antibodies [8, 53]. Here it is also important to mention that to be effective in preventing T-cells from becoming exhausted, anti-PD-1 antibodies must remain bound to T-cells but that tumor-associated macrophages can rapidly remove the antibodies from T-cells, with the possibility of inducing anti-PD1 mAb resistance [54]. It is possible that the use of encapsulated GSK3 inhibitors may overcome this problem.
While inhibition of PD-1/PD-L1 is an attractive therapeutic target, the use of monoclonal antibodies (mAbs) for blocking the PD-1/PD-L1 has some detracting features. mAbs are large bio-reactive proteins and interact with a number of other biomolecules, proteins, and cell surfaces after systemic administration, including FcRn and Fcγ receptors [5]. These interactions and the hydrodynamic size of the antibodies can constrain the PK/PD profiles relative to small molecules [55, 56]. Moreover, even fully humanized mAbs are potentially immunogenic (sometimes toxic) and can result in the production of anti-IgG antibodies that could lead to rapid antibody clearance [6, 57, 58]. mAbs are also expensive and complex to manufacture. While SMIs capable of directly blocking avidity interactions of PD-1 with PD-L1 have had some success in preclinical studies [59], small molecule inhibitors of GSK3 have not as yet achieved FDA approval for advancement to the clinic [11, 16]. Moreover, even for the most-advanced GSK3 inhibitor, LY2090314 (a Phase 2 candidate), the clinical trial was abandoned due to the poor PK and drug bioavailability (clinical trial ID: NCT01214603, NCT01632306) [60]. Another concern in the use of SMI for GSK3 include off-target effects due to the pleiotropic involvement of this signaling hub kinase [61]. For instance, in one preclinical study, GSK3β disruption led to embryonic lethality in mice, generating a phenotype similar to the disruption of the IKKβ gene in the NF-κB pathway [62]. Moreover, the favorable characteristic of AZD1080 for crossing the blood-brain barrier to treat Alzheimer’s disease [35] was offset by the development of nephrotoxicity in a phase I clinical trial [16]. It is notable that in our study that administration of free AZD1080 resulted in significantly higher renal concentrations of the drug compared to silicasome delivery. Apparently, most of the drug reaching the tumor site or RES organs do not reenter the systemic circulation to make it to the kidney. This is also compatible with the retention of relatively high AZD1080 concentrations at the sites of silicasomes, distribution, compared to rapid decline in blood levels after 24 hours. All considered, from the perspective of treatment safety, the encapsulation of GSK3 inhibitors by a nanocarrier reduces the systemic biodistribution of the free drug, in exchange for increased drug delivery to the tumor site, where the action counts. In this regard, the high drug loading capacity, improved circulatory stability, and excellent biodistribution of silicasomes to the tumor site offer several different advantages to improve the treatment efficacy and safety of AZD1080.
GSK3 interfaces with a number of signaling pathways in cells and is overexpressed in a number of cancers [63]. It is possible, therefore, that inhibition of GSK3 could directly impact tumor cell proliferation or apoptosis, thereby directly impacting cancer growth [32, 34, 64, 65]. As one example, the Wnt/β-catenin signaling pathway plays a role in cancer cell proliferation and survival; GSK3 can interfere in this effect by phosphorylating β-catenin, which leads to its degradation [33]. In contrast, it has been demonstrated in a number of cancers expressing a Kras mutation that GSK3 inhibition can result in apoptosis and/or reduced proliferation of cancer cells [34]. In spite of these direct effects on tumor cells, most of our findings point toward the effect of the GSK3 inhibitors on the function of T-cells as the predominant mechanism of tumor growth suppression. This includes the in vitro observation that the free drugs have little or no cytotoxic effects on the tumor cells that were used in this study. Moreover, there is a demonstrated effect on increased intratumoral presence of perforin-producing cytotoxic T-cells, which exhibited reduced surface expression of PD-1. As with most nanocarriers, it is currently difficult to show the exact intratumoral distribution of silicasomes after entrance to the tumor site, other than previous demonstrations that MSNPs incorporating a gold core can be observed in the tumor stroma and in cancer cells within KPC tumors [22]. How exactly the T-cells are targeted in addition to the contribution of macrophages in the stroma is not known. The prevailing literature indicates that there is negligible nanocarrier uptake by T-cells in the absence of targeting ligands [66-70]. This suggests that the most likely site of drug release is in the tumor stroma, leading to uptake of the GSK3 inhibitor by T-cells from that locality. A recent study has demonstrated that GSK3 inhibition can inhibit the expression of Lag3, another inhibitory immune checkpoint [71]. This may further help to explain why sAZD1080 treatment was more consistently effective at inhibiting tumor growth, compared to anti-PD-1 treatment, and warrants further investigation.
The ability of the AZD1080-silicasome to inhibit growth in four syngeneic mouse cancer models by interfering in PD-1 expression illustrates its utility as a possible substitute for antibody-based checkpoint monotherapy under specified circumstances. While sAZD1080 and anti-PD-1 monotherapy were effective in the MC38 colon and LLC lung cancer models, only sAZD1080 (but not anti-PD-1) could inhibit CT26 and KPC tumor growth. The KPC model, which carries a point mutation in p53 gene (TP53R172H) and a point mutation in the KRAS gene (KRASG12D), is generally recognized as poorly responsive to treatment by immune checkpoint blocking antibodies, similar to the findings in analogous human tumors [46]. While there are several reasons for poor responsiveness, the dysplastic PDAC stroma plays an important role in promoting drug resistance through restricted vascular access or drug catabolism [72]. In spite of these challenges, we demonstrate that the silicasome is as effective for sAZD1080 delivery as was previously shown for the delivery of irinotecan, gemcitabine and paclitaxel to the PDAC site [20, 23, 28]. This includes the ability to deliver up to 6% of the injected drug dose to the KPC tumor site, in addition to the possibility to further improve transport by a transcytosis-inducing cyclic iRGD peptide [20-23, 28].
Anti-PD-1 antibodies (Pembrolizumab, Nivolumab) have been approved as immunotherapies for use in solid tumors such as gastric cancer, hepatocellular carcinoma, head and neck squamous cell carcinoma, urothelial carcinoma, cervical cancer, non-small cell lung cancer, and broadly for unresectable solid tumors with microsatellite instability (MSI-H) or defects in DNA mismatch repair [73]. It is also conceivable, based on our data, that sAZD1080 could serve as effective monotherapy for these cancers. In addition, there is a growing trend for combining immune checkpoint blockade with chemotherapy, radiotherapy, or other targeted therapies. Clinicaltrials.gov currently lists ~870 active clinical studies involving anti-PD-1 or anti-PD-L1 treatment, 60% of which is premised on combination therapy. Anti-PD1 antibodies have already received FDA approval for combination therapy with paclitaxel or oxaliplatin to treat melanoma and biliary tract cancer, pemetrexed to treat non-small cell lung cancer, and axitinib to treat renal cell carcinoma [9, 74-77]. Moreover, the heterogeneous immune landscape across multiple cancer types holds the key to the development of additional treatment combinations premised on the “hot” or “cold” immune status of the tumor, or a number of immune escape pathways operating in the TME. One type of intervention is to convert “cold” into “hot” tumors through the induction of immunogenic cell death (ICD) by chemotherapy, radiotherapy, or photodynamic therapy [78-80]. It is possible to propagate these immunogenic responses through the use of immune checkpoint interference. One strategy would be to design a “2-in-1” nanocarrier that co-encapsulates ICD-inducing chemo agents (i.e. doxorubicin, oxaliplatin) with AZD1080. This is accomplishable through the selection of weak-basic, amphiphilic drugs to achieve remote loading. Another option is to select an ICD-inducing chemotherapeutic agent such as paclitaxel [81] for incorporation into a silicasome lipid bilayer, allowing for AZD1080 remote loading into the porous interior. In this regard, we have previously shown the utility of silicasomes for co-delivery of paclitaxel and gemcitabine in PDAC [82]. An alternative strategy would be to develop a lipid conjugated prodrug that allows the GSK3 inhibitor to be incorporated into the lipid bilayer, allowing an ICD-inducing chemotherapeutic agent to be remotely imported into the porous interior.
While sAZD1080 has been shown to be highly effective in 4 syngeneic cancer models, a limitation of this study is that tumor growth was established by subcutaneous implantation which yield tumors that are less representative of human disease. This limitation may be addressed in future studies utilizing orthotopic models [83-85]. In this study we focused primarily on T cells and the impact of nano-enabled GSK3 inhibition on T cell biology. There are a number of other cells in the tumor microenvironment, some of which may also be affected by GSK3 inhibition [71] – the extent to which these cells are impacted by silicasome delivery of a GSK3 inhibitor is an interesting avenue for potential research.
5. Conclusions
In summary, silicasome-assisted delivery of a GSK3 inhibitor, AZD1080, was quite effective for cancer immunotherapy in four different syngeneic mouse models, based on pharmacological inhibition of PD-1 expression in T-cells. Silicasome delivery outperformed systemic administration of free AZD1080 in terms of intratumoral drug concentrations and the triggering of cytotoxic T-cell killing. The use of a nano-enabled approach for interfering in the PD-1/PD-L1 axis through the delivery of a small molecule inhibitor provides a promising new strategy for cancer immunotherapy.
Supplementary Material
6. Acknowledgments
This study was funded by U.S. Public Health Service Grant 1U01CA198846 and 1R01CA247666-01A1. We thank the UCLA Division of Laboratory Animal Services, UCLA Translational Pathology Core, UCLA Preclinical Imaging Technology Center, and UCLA Flow Cytometry Core.
Footnotes
Conflict of Interests
The authors declare the following competing financial interest(s): Andre E. Nel and Huan Meng are co-founders, board members and equity holders in Westwood Bioscience Inc. Andre E. Nel and Huan Meng are co-founders and equity holders in NAMMI therapeutics.
The remaining authors declared no conflict of interest.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data Availability
The processed data required to reproduce these findings are available to download from https://data.mendeley.com/datasets/5cs6vtdrk6/1. The raw data required to reproduce these findings can be shared upon request.
References
- [1].Haslam A, Prasad V, Estimation of the Percentage of US Patients With Cancer Who Are Eligible for and Respond to Checkpoint Inhibitor Immunotherapy Drugs, JAMA Netw Open 2(5) (2019) e192535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su MJ, Melms JC, Leeson R, Kanodia A, Mei S, Lin JR, Wang S, Rabasha B, Liu D, Zhang G, Margolais C, Ashenberg O, Ott PA, Buchbinder EI, Haq R, Hodi FS, Boland GM, Sullivan RJ, Frederick DT, Miao B, Moll T, Flaherty KT, Herlyn M, Jenkins RW, Thummalapalli R, Kowalczyk MS, Canadas I, Schilling B, Cartwright ANR, Luoma AM, Malu S, Hwu P, Bernatchez C, Forget MA, Barbie DA, Shalek AK, Tirosh I, Sorger PK, Wucherpfennig K, Van Allen EM, Schadendorf D, Johnson BE, Rotem A, Rozenblatt-Rosen O, Garraway LA, Yoon CH, Izar B, Regev A, A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade, Cell 175(4) (2018) 984–997 e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Umansky V, Blattner C, Gebhardt C, Utikal J, The Role of Myeloid-Derived Suppressor Cells (MDSC) in Cancer Progression, Vaccines (Basel) 4(4) (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Marin-Acevedo JA, Dholaria B, Soyano AE, Knutson KL, Chumsri S, Lou Y, Next generation of immune checkpoint therapy in cancer: new developments and challenges, J Hematol Oncol 11(1) (2018) 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Zhao L, Ren TH, Wang DD, Clinical pharmacology considerations in biologics development, Acta Pharmacol Sin 33(11) (2012) 1339–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Descotes J, Immunotoxicity of monoclonal antibodies, MAbs 1(2) (2009) 104–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Dolan C, Opportunities and challenges in biosimilar uptake in oncology, Am J Manag Care 24(11 Suppl) (2018) S237–S243. [PubMed] [Google Scholar]
- [8].Mizuno R, Sugiura D, Shimizu K, Maruhashi T, Watada M, Okazaki IM, Okazaki T, PD-1 Primarily Targets TCR Signal in the Inhibition of Functional T Cell Activation, Front Immunol 10 (2019) 630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Seidel JA, Otsuka A, Kabashima K, Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations, Front Oncol 8 (2018) 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Taylor A, Harker JA, Chanthong K, Stevenson PG, Zuniga EI, Rudd CE, Glycogen Synthase Kinase 3 Inactivation Drives T-bet-Mediated Downregulation of Co-receptor PD-1 to Enhance CD8(+) Cytolytic T Cell Responses, Immunity 44(2) (2016) 274–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Saraswati AP, Ali Hussaini SM, Krishna NH, Babu BN, Kamal A, Glycogen synthase kinase-3 and its inhibitors: Potential target for various therapeutic conditions, Eur J Med Chem 144 (2018) 843–858. [DOI] [PubMed] [Google Scholar]
- [12].Declercq LD, Vandenberghe R, Van Laere K, Verbruggen A, Bormans G, Drug Development in Alzheimer's Disease: The Contribution of PET and SPECT, Front Pharmacol 7 (2016) 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Taylor A, Rudd CE, Glycogen Synthase Kinase 3 Inactivation Compensates for the Lack of CD28 in the Priming of CD8(+) Cytotoxic T-Cells: Implications for anti-PD-1 Immunotherapy, Front Immunol 8 (2017) 1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Taylor A, Rothstein D, Rudd CE, Small-Molecule Inhibition of PD-1 Transcription Is an Effective Alternative to Antibody Blockade in Cancer Therapy, Cancer Res 78(3) (2018) 706–717. [DOI] [PubMed] [Google Scholar]
- [15].Maqbool M, Hoda N, GSK3 Inhibitors in the Therapeutic Development of Diabetes, Cancer and Neurodegeneration: Past, Present and Future, Current Pharmaceutical Design 23(29) (2017) 4332–4350. [DOI] [PubMed] [Google Scholar]
- [16].Eldar-Finkelman H, Martinez A, GSK-3 Inhibitors: Preclinical and Clinical Focus on CNS, Front Mol Neurosci 4 (2011) 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ugolkov A, Gaisina I, Zhang JS, Billadeau DD, White K, Kozikowski A, Jain S, Cristofanilli M, Giles F, O'Halloran T, Cryns VL, Mazar AP, GSK-3 inhibition overcomes chemoresistance in human breast cancer, Cancer Lett 380(2) (2016) 384–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ding L, Madamsetty VS, Kiers S, Alekhina O, Ugolkov A, Dube J, Zhang Y, Zhang JS, Wang E, Dutta SK, Schmitt DM, Giles FJ, Kozikowski AP, Mazar AP, Mukhopadhyay D, Billadeau DD, Glycogen Synthase Kinase-3 Inhibition Sensitizes Pancreatic Cancer Cells to Chemotherapy by Abrogating the TopBP1/ATR-Mediated DNA Damage Response, Clin Cancer Res 25(21) (2019) 6452–6462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].O'Flaherty L, Shnyder SD, Cooper PA, Cross SJ, Wakefield JG, Pardo OE, Seckl MJ, Tavare JM, Tumor growth suppression using a combination of taxol-based therapy and GSK3 inhibition in non-small cell lung cancer, PLoS One 14(4) (2019) e0214610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Meng H, Wang M, Liu H, Liu X, Situ A, Wu B, Ji Z, Chang CH, Nel AE, Use of a lipid-coated mesoporous silica nanoparticle platform for synergistic gemcitabine and paclitaxel delivery to human pancreatic cancer in mice, ACS Nano 9(4) (2015) 3540–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lu J, Liu X, Liao YP, Salazar F, Sun B, Jiang W, Chang CH, Jiang J, Wang X, Wu AM, Meng H, Nel AE, Nano-enabled pancreas cancer immunotherapy using immunogenic cell death and reversing immunosuppression, Nat Commun 8(1) (2017) 1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Liu X, Lin P, Perrett I, Lin J, Liao YP, Chang CH, Jiang J, Wu N, Donahue T, Wainberg Z, Nel AE, Meng H, Tumor-penetrating peptide enhances transcytosis of silicasome-based chemotherapy for pancreatic cancer, J Clin Invest 127(5) (2017) 2007–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Liu X, Jiang J, Chan R, Ji Y, Lu J, Liao YP, Okene M, Lin J, Lin P, Chang CH, Wang X, Tang I, Zheng E, Qiu W, Wainberg ZA, Nel AE, Meng H, Improved Efficacy and Reduced Toxicity Using a Custom-Designed Irinotecan-Delivering Silicasome for Orthotopic Colon Cancer, ACS Nano 13(1) (2019) 38–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Cern A, Golbraikh A, Sedykh A, Tropsha A, Barenholz Y, Goldblum A, Quantitative structure-property relationship modeling of remote liposome loading of drugs, J Control Release 160(2) (2012) 147–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lim JF, Berger H, Su IH, Isolation and Activation of Murine Lymphocytes, J Vis Exp (116) (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Aston WJ, Hope DE, Nowak AK, Robinson BW, Lake RA, Lesterhuis WJ, A systematic investigation of the maximum tolerated dose of cytotoxic chemotherapy with and without supportive care in mice, BMC Cancer 17(1) (2017) 684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Chen TJ, Kotecha N, Cytobank: providing an analytics platform for community cytometry data analysis and collaboration, Curr Top Microbiol Immunol 377 (2014) 127–57. [DOI] [PubMed] [Google Scholar]
- [28].Liu X, Situ A, Kang Y, Villabroza KR, Liao Y, Chang CH, Donahue T, Nel AE, Meng H, Irinotecan Delivery by Lipid-Coated Mesoporous Silica Nanoparticles Shows Improved Efficacy and Safety over Liposomes for Pancreatic Cancer, ACS Nano 10(2) (2016) 2702–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Haran G, Cohen R, Bar LK, Barenholz Y, Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases, Biochim Biophys Acta 1151(2) (1993) 201–15. [DOI] [PubMed] [Google Scholar]
- [30].Cern A, Barenholz Y, Tropsha A, Goldblum A, Computer-aided design of liposomal drugs: In silico prediction and experimental validation of drug candidates for liposomal remote loading, J Control Release 173 (2014) 125–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Bolotin EM, Cohen R, Bar LK, Emanuel N, Ninio S, Barenholz Y, Lasic DD, Ammonium Sulfate Gradients for Efficient and Stable Remote Loading of Amphipathic Weak Bases into Liposomes and Ligandoliposomes, Journal of Liposome Research 4(1) (1994) 455–479. [Google Scholar]
- [32].Mancinelli R, Carpino G, Petrungaro S, Mammola CL, Tomaipitinca L, Filippini A, Facchiano A, Ziparo E, Giampietri C, Multifaceted Roles of GSK-3 in Cancer and Autophagy-Related Diseases, Oxid Med Cell Longev 2017 (2017) 4629495–4629495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Krishnamurthy N, Kurzrock R, Targeting the Wnt/beta-catenin pathway in cancer: Update on effectors and inhibitors, Cancer Treat Rev 62 (2018) 50–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kazi A, Xiang S, Yang H, Delitto D, Trevino J, Jiang RHY, Ayaz M, Lawrence HR, Kennedy P, Sebti SM, GSK3 suppression upregulates β-catenin and c-Myc to abrogate KRas-dependent tumors, Nature Communications 9(1) (2018) 5154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Georgievska B, Sandin J, Doherty J, Mortberg A, Neelissen J, Andersson A, Gruber S, Nilsson Y, Schott P, Arvidsson PI, Hellberg S, Osswald G, Berg S, Falting J, Bhat RV, AZD1080, a novel GSK3 inhibitor, rescues synaptic plasticity deficits in rodent brain and exhibits peripheral target engagement in humans, J Neurochem 125(3) (2013) 446–56. [DOI] [PubMed] [Google Scholar]
- [36].Thackaberry EA, Wang X, Schweiger M, Messick K, Valle N, Dean B, Sambrone A, Bowman T, Xie M, Solvent-based formulations for intravenous mouse pharmacokinetic studies: tolerability and recommended solvent dose limits, Xenobiotica 44(3) (2014) 235–241. [DOI] [PubMed] [Google Scholar]
- [37].Gustafson HH, Holt-Casper D, Grainger DW, Ghandehari H, Nanoparticle Uptake: The Phagocyte Problem, Nano Today 10(4) (2015) 487–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Aktas E, Kucuksezer UC, Bilgic S, Erten G, Deniz G, Relationship between CD107a expression and cytotoxic activity, Cellular Immunology 254(2) (2009) 149–154. [DOI] [PubMed] [Google Scholar]
- [39].Kansy BA, Concha-Benavente F, Srivastava RM, Jie HB, Shayan G, Lei Y, Moskovitz J, Moy J, Li J, Brandau S, Lang S, Schmitt NC, Freeman GJ, Gooding WE, Clump DA, Ferris RL, PD-1 Status in CD8(+) T Cells Associates with Survival and Anti-PD-1 Therapeutic Outcomes in Head and Neck Cancer, Cancer Res 77(22) (2017) 6353–6364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Larimer BM, Bloch E, Nesti S, Austin EE, Wehrenberg-Klee E, Boland G, Mahmood U, The Effectiveness of Checkpoint Inhibitor Combinations and Administration Timing Can Be Measured by Granzyme B PET Imaging, Clin Cancer Res 25(4) (2019) 1196–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Homet Moreno B, Zaretsky JM, Garcia-Diaz A, Tsoi J, Parisi G, Robert L, Meeth K, Ndoye A, Bosenberg M, Weeraratna AT, Graeber TG, Comin-Anduix B, Hu-Lieskovan S, Ribas A, Response to Programmed Cell Death-1 Blockade in a Murine Melanoma Syngeneic Model Requires Costimulation, CD4, and CD8 T Cells, Cancer immunology research 4(10) (2016) 845–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Mosely SI, Prime JE, Sainson RC, Koopmann JO, Wang DY, Greenawalt DM, Ahdesmaki MJ, Leyland R, Mullins S, Pacelli L, Marcus D, Anderton J, Watkins A, Coates Ulrichsen J, Brohawn P, Higgs BW, McCourt M, Jones H, Harper JA, Morrow M, Valge-Archer V, Stewart R, Dovedi SJ, Wilkinson RW, Rational Selection of Syngeneic Preclinical Tumor Models for Immunotherapeutic Drug Discovery, Cancer Immunol Res 5(1) (2017) 29–41. [DOI] [PubMed] [Google Scholar]
- [43].Li HY, McSharry M, Bullock B, Nguyen TT, Kwak J, Poczobutt JM, Sippel TR, Heasley LE, Weiser-Evans MC, Clambey ET, Nemenoff RA, The Tumor Microenvironment Regulates Sensitivity of Murine Lung Tumors to PD-1/PD-L1 Antibody Blockade, Cancer Immunol Res 5(9) (2017) 767–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Gopinathan A, Morton JP, Jodrell DI, Sansom OJ, GEMMs as preclinical models for testing pancreatic cancer therapies, Disease Models & Mechanisms 8(10) (2015) 1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lee JW, Komar CA, Bengsch F, Graham K, Beatty GL, Genetically Engineered Mouse Models of Pancreatic Cancer: The KPC Model (LSL-Kras(G12D/+) ;LSL-Trp53(R172H/+) ;Pdx-1-Cre), Its Variants, and Their Application in Immuno-oncology Drug Discovery, Curr Protoc Pharmacol 73 (2016) 14 39 1–14 39 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA, Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice, Cancer Cell 7(5) (2005) 469–83. [DOI] [PubMed] [Google Scholar]
- [47].Kabacaoglu D, Ciecielski KJ, Ruess DA, Algul H, Immune Checkpoint Inhibition for Pancreatic Ductal Adenocarcinoma: Current Limitations and Future Options, Front Immunol 9 (2018) 1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Steele CW, Karim SA, Leach JDG, Bailey P, Upstill-Goddard R, Rishi L, Foth M, Bryson S, McDaid K, Wilson Z, Eberlein C, Candido JB, Clarke M, Nixon C, Connelly J, Jamieson N, Carter CR, Balkwill F, Chang DK, Evans TRJ, Strathdee D, Biankin AV, Nibbs RJB, Barry ST, Sansom OJ, Morton JP, CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma, Cancer Cell 29(6) (2016) 832–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Zitvogel L, Kroemer G, Targeting PD-1/PD-L1 interactions for cancer immunotherapy, Oncoimmunology 1(8) (2012) 1223–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Isaaz S, Baetz K, Olsen K, Podack E, Griffiths GM, Serial killing by cytotoxic T lymphocytes: T cell receptor triggers degranulation, re-filling of the lytic granules and secretion of lytic proteins via a non-granule pathway, Eur J Immunol 25(4) (1995) 1071–9. [DOI] [PubMed] [Google Scholar]
- [51].Jiang Y, Li Y, Zhu B, T-cell exhaustion in the tumor microenvironment, Cell Death Dis 6 (2015) e1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Simon S, Labarriere N, PD-1 expression on tumor-specific T cells: Friend or foe for immunotherapy?, Oncoimmunology 7(1) (2017) e1364828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Wang S, He Z, Wang X, Li H, Liu XS, Antigen presentation and tumor immunogenicity in cancer immunotherapy response prediction, Elife 8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Arlauckas SP, Garris CS, Kohler RH, Kitaoka M, Cuccarese MF, Yang KS, Miller MA, Carlson JC, Freeman GJ, Anthony RM, Weissleder R, Pittet MJ, In vivo imaging reveals a tumor-associated macrophage-mediated resistance pathway in anti-PD-1 therapy, Sci Transl Med 9(389) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Castelli MS, McGonigle P, Hornby PJ, The pharmacology and therapeutic applications of monoclonal antibodies, Pharmacol Res Perspect 7(6) (2019) e00535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Lobo ED, Hansen RJ, Balthasar JP, Antibody pharmacokinetics and pharmacodynamics, J Pharm Sci 93(11) (2004) 2645–68. [DOI] [PubMed] [Google Scholar]
- [57].Garces S, Demengeot J, The Immunogenicity of Biologic Therapies, Curr Probl Dermatol 53 (2018) 37–48. [DOI] [PubMed] [Google Scholar]
- [58].Ovacik M, Lin K, Tutorial on Monoclonal Antibody Pharmacokinetics and Its Considerations in Early Development, Clin Transl Sci 11(6) (2018) 540–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Ganesan A, Ahmed M, Okoye I, Arutyunova E, Babu D, Turnbull WL, Kundu JK, Shields J, Agopsowicz KC, Xu L, Tabana Y, Srivastava N, Zhang G, Moon TC, Belovodskiy A, Hena M, Kandadai AS, Hosseini SN, Hitt M, Walker J, Smylie M, West FG, Siraki AG, Lemieux MJ, Elahi S, Nieman JA, Tyrrell DL, Houghton M, Barakat K, Comprehensive in vitro characterization of PD-L1 small molecule inhibitors, Sci Rep 9(1) (2019) 12392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Rizzieri DA, Cooley S, Odenike O, Moonan L, Chow KH, Jackson K, Wang X, Brail L, Borthakur G, An open-label phase 2 study of glycogen synthase kinase-3 inhibitor LY2090314 in patients with acute leukemia, Leuk Lymphoma 57(8) (2016) 1800–6. [DOI] [PubMed] [Google Scholar]
- [61].Hu S, Begum AN, Jones MR, Oh MS, Beech WK, Beech BH, Yang F, Chen P, Ubeda OJ, Kim PC, Davies P, Ma Q, Cole GM, Frautschy SA, GSK3 inhibitors show benefits in an Alzheimer's disease (AD) model of neurodegeneration but adverse effects in control animals, Neurobiol Dis 33(2) (2009) 193–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR, Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation, Nature 406(6791) (2000) 86–90. [DOI] [PubMed] [Google Scholar]
- [63].Zeng J, Liu D, Qiu Z, Huang Y, Chen B, Wang L, Xu H, Huang N, Liu L, Li W, GSK3β Overexpression Indicates Poor Prognosis and Its Inhibition Reduces Cell Proliferation and Survival of Non-Small Cell Lung Cancer Cells, PLOS ONE 9(3) (2014) e91231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Leung N, Turbide C, Balachandra B, Marcus V, Beauchemin N, Intestinal tumor progression is promoted by decreased apoptosis and dysregulated Wnt signaling in Ceacam1−/− mice, Oncogene 27(36) (2008) 4943–4953. [DOI] [PubMed] [Google Scholar]
- [65].Yuan G, Zhang B, Yang S, Jin L, Datta A, Bae S, Chen X, Datta PK, Novel role of STRAP in progression and metastasis of colorectal cancer through Wnt/β-catenin signaling, Oncotarget 7(13) (2016) 16023–16037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Irvine DJ, Hanson MC, Rakhra K, Tokatlian T, Synthetic Nanoparticles for Vaccines and Immunotherapy, Chemical Reviews 115(19) (2015) 11109–11146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Ramishetti S, Kedmi R, Goldsmith M, Leonard F, Sprague AG, Godin B, Gozin M, Cullis PR, Dykxhoorn DM, Peer D, Systemic Gene Silencing in Primary T Lymphocytes Using Targeted Lipid Nanoparticles, ACS Nano 9(7) (2015) 6706–6716. [DOI] [PubMed] [Google Scholar]
- [68].Beltran-Sastre V, Navarro E, Measuring activity of endocytosis-regulating factors in T-lymphocytes by flow cytometry, Cytotechnology 67(3) (2015) 551–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Yi S, Allen SD, Liu Y-G, Ouyang BZ, Li X, Augsornworawat P, Thorp EB, Scott EA, Tailoring Nanostructure Morphology for Enhanced Targeting of Dendritic Cells in Atherosclerosis, ACS Nano 10(12) (2016) 11290–11303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Allen SD, Liu Y-G, Bobbala S, Cai L, Hecker PI, Temel R, Scott EA, Polymersomes scalably fabricated via flash nanoprecipitation are non-toxic in non-human primates and associate with leukocytes in the spleen and kidney following intravenous administration, Nano Research 11(10) (2018) 5689–5703. [Google Scholar]
- [71].Rudd CE, Chanthong K, Taylor A, Small Molecule Inhibition of GSK-3 Specifically Inhibits the Transcription of Inhibitory Co-receptor LAG-3 for Enhanced Anti-tumor Immunity, Cell Rep 30(7) (2020) 2075–2082 e4. [DOI] [PubMed] [Google Scholar]
- [72].Neesse A, Michl P, Frese KK, Feig C, Cook N, Jacobetz MA, Lolkema MP, Buchholz M, Olive KP, Gress TM, Tuveson DA, Stromal biology and therapy in pancreatic cancer, Gut 60(6) (2011) 861–8. [DOI] [PubMed] [Google Scholar]
- [73].Wu X, Gu Z, Chen Y, Chen B, Chen W, Weng L, Liu X, Application of PD-1 Blockade in Cancer Immunotherapy, Comput Struct Biotechnol J 17 (2019) 661–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Sun D, Ma J, Wang J, Han C, Qian Y, Chen G, Li X, Zhang J, Cui P, Du W, Wu Z, Chen S, Zheng X, Yue Z, Song J, Gao C, Zhao X, Cai S, Hu Y, Anti-PD-1 therapy combined with chemotherapy in patients with advanced biliary tract cancer, Cancer Immunol Immunother 68(9) (2019) 1527–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Burtness B, Harrington KJ, Greil R, Soulieres D, Tahara M, de Castro G Jr., Psyrri A, Baste N, Neupane P, Bratland A, Fuereder T, Hughes BGM, Mesia R, Ngamphaiboon N, Rordorf T, Wan Ishak WZ, Hong RL, Gonzalez Mendoza R, Roy A, Zhang Y, Gumuscu B, Cheng JD, Jin F, Rischin D, K.-. Investigators, Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): a randomised, open-label, phase 3 study, Lancet 394(10212) (2019) 1915–1928. [DOI] [PubMed] [Google Scholar]
- [76].Langer CJ, Gadgeel SM, Borghaei H, Papadimitrakopoulou VA, Patnaik A, Powell SF, Gentzler RD, Martins RG, Stevenson JP, Jalal SI, Panwalkar A, Yang JC-H, Gubens M, Sequist LV, Awad MM, Fiore J, Ge Y, Raftopoulos H, Gandhi L, Carboplatin and pemetrexed with or without pembrolizumab for advanced, non-squamous non-small-cell lung cancer: a randomised, phase 2 cohort of the open-label KEYNOTE-021 study, The Lancet Oncology 17(11) (2016) 1497–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Atkins MB, Plimack ER, Puzanov I, Fishman MN, McDermott DF, Cho DC, Vaishampayan U, George S, Olencki TE, Tarazi JC, Rosbrook B, Fernandez KC, Lechuga M, Choueiri TK, Axitinib in combination with pembrolizumab in patients with advanced renal cell cancer: a non-randomised, open-label, dose-finding, and dose-expansion phase 1b trial, Lancet Oncol 19(3) (2018) 405–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Bezu L, Gomes-de-Silva LC, Dewitte H, Breckpot K, Fucikova J, Spisek R, Galluzzi L, Kepp O, Kroemer G, Combinatorial strategies for the induction of immunogenic cell death, Front Immunol 6 (2015) 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Rapoport BL, Anderson R, Realizing the Clinical Potential of Immunogenic Cell Death in Cancer Chemotherapy and Radiotherapy, Int J Mol Sci 20(4) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Golden EB, Apetoh L, Radiotherapy and immunogenic cell death, Semin Radiat Oncol 25(1) (2015) 11–7. [DOI] [PubMed] [Google Scholar]
- [81].Wang YJ, Fletcher R, Yu J, Zhang L, Immunogenic effects of chemotherapy-induced tumor cell death, Genes Dis 5(3) (2018) 194–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Von Hoff DD, Ramanathan RK, Borad MJ, Laheru DA, Smith LS, Wood TE, Korn RL, Desai N, Trieu V, Iglesias JL, Zhang H, Soon-Shiong P, Shi T, Rajeshkumar NV, Maitra A, Hidalgo M, Gemcitabine plus nab-paclitaxel is an active regimen in patients with advanced pancreatic cancer: a phase I/II trial, J Clin Oncol 29(34) (2011) 4548–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Hage C, Hoves S, Ashoff M, Schandl V, Hort S, Rieder N, Heichinger C, Berrera M, Ries CH, Kiessling F, Poschinger T, Characterizing responsive and refractory orthotopic mouse models of hepatocellular carcinoma in cancer immunotherapy, PLoS One 14(7) (2019) e0219517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Zhao X, Li L, Starr TK, Subramanian S, Tumor location impacts immune response in mouse models of colon cancer, Oncotarget 8(33) (2017) 54775–54787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Song W, Tiruthani K, Wang Y, Shen L, Hu M, Dorosheva O, Qiu K, Kinghorn KA, Liu R, Huang L, Trapping of Lipopolysaccharide to Promote Immunotherapy against Colorectal Cancer and Attenuate Liver Metastasis, Adv Mater 30(52) (2018) e1805007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The processed data required to reproduce these findings are available to download from https://data.mendeley.com/datasets/5cs6vtdrk6/1. The raw data required to reproduce these findings can be shared upon request.