

Summary
Hutchinson-Gilford progeria syndrome (HGPS) is typically caused by a dominant-negative C•G-to-T•A mutation (c.1824 C>T, G608G) in LMNA, the nuclear lamin A gene. This mutation causes RNA mis-splicing that produces progerin, a toxic protein that induces rapid aging and shortens lifespan to ~14 years1–4. Adenine base editors (ABEs) perform targeted A•T-to-G•C base pair conversion with minimal byproducts and without requiring double-strand DNA breaks or donor DNA templates5,6. Here, we describe the use of an ABE to directly correct the pathogenic HGPS mutation in cultured progeria patient-derived fibroblasts and in a mouse model of HGPS. Lentiviral delivery of ABE to patient-derived fibroblasts results in ~90% correction of the pathogenic allele, mitigation of RNA mis-splicing, reduced progerin levels, and correction of nuclear abnormalities. Unbiased off-target DNA and RNA analysis did not detect off-target editing activity in treated patient-derived fibroblasts. In transgenic mice homozygous for the human LMNA c.1824 C>T allele, a single retro-orbital injection of adeno-associated virus 9 (AAV9) encoding the ABE resulted in substantial, durable correction of the pathogenic mutation (~20-60% across various organs 6 months post-injection), restoration of normal RNA splicing, and reduction of progerin protein. In vivo base editing rescued vascular pathology, preserving vascular smooth muscle cell counts and preventing adventitial fibrosis. A single ABE AAV9 injection at P14 improved animal vitality and greatly extended median lifespan from 215 to 510 days. These findings support the potential of in vivo base editing to treat HGPS, and other genetic diseases, by directly correcting the root cause of disease.
Hutchinson-Gilford progeria syndrome (HGPS, progeria) is a rare genetic disease characterized by accelerated aging4. In over 90% of HGPS patients, the disease is caused by a single de novo point mutation (c.1824 C>T; p.G608G) in the lamin A (LMNA) gene1,2. This mutation potentiates a cryptic splice site in exon 11, leading to a mis-splicing event that results in the loss of 50 amino acids from the lamin A protein (Fig. 1a)1,2. This truncated protein, known as progerin, lacks a proteolytic cleavage site for ZMPSTE24/FACE1, which cleaves the farnesylated C-terminus of wild-type pre-lamin A1. Progerin protein impairs nuclear structure and function, culminating in premature senescence and cell death3,7,8. The pathogenic mutation is dominant negative, so a single copy of the allele is sufficient to cause progeria3. Cardiovascular disease, characterized by premature atherosclerosis, loss of vascular smooth muscle cells (VSMCs), and vascular stiffening, is the predominant cause of death in children with progeria, who have an average lifespan of ~14 years3,7–11. While strategies for treating progeria such as global inhibition of protein farnesylation3,12,13 offer benefits to patients, no strategy has yet been reported to directly reverse the mutation that causes HGPS14–17.
Figure 1. ABE-mediated correction of the LMNA c.1824 C>T mutation in progeria patient-derived cell lines.
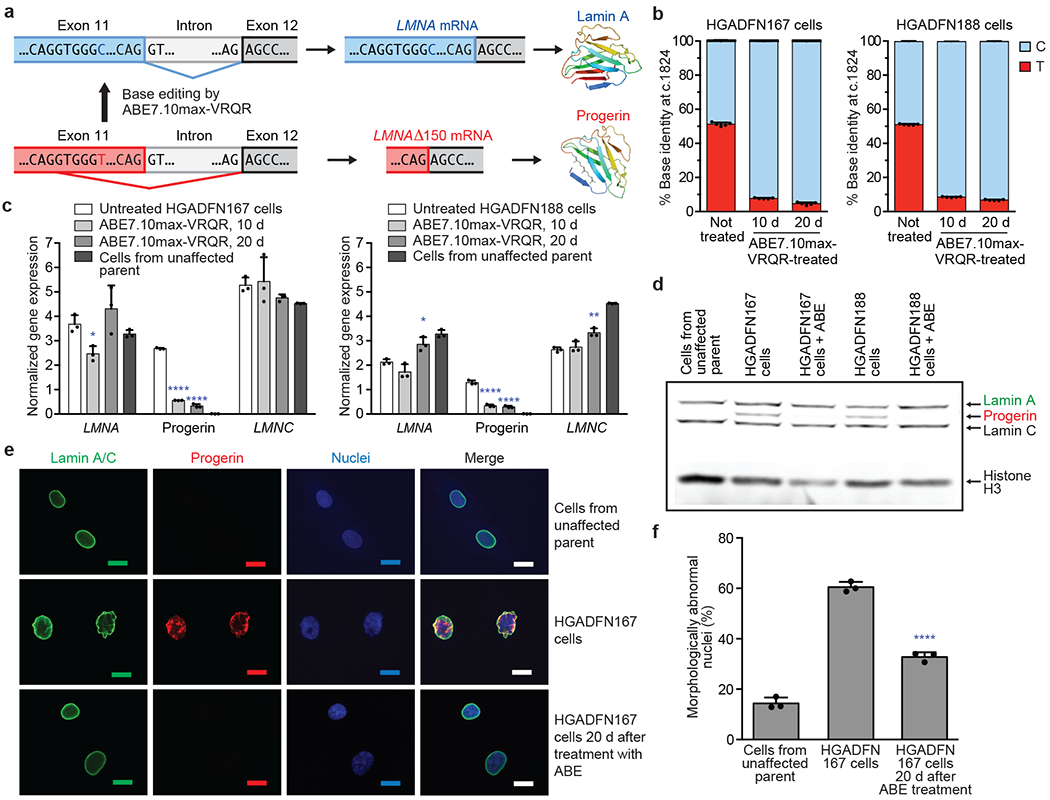
(a) The LMNA c.1824 C>T mutation potentiates a cryptic splice site in exon 11 of the LMNA gene, resulting in the pathogenic progerin protein. (b) LMNA c.1824 nucleotide identity in HGADFN167 and HGADFN188 patient-derived cells untreated or treated with ABE7.10max-VRQR lentivirus after 10 or 20 days. Values and error bars reflect mean±SD of five technical replicates. (c) Quantification by digital droplet PCR (ddPCR) of LMNA, progerin, and LMNC (a normal alternative splice form) transcripts in untreated cells, cells 10 or 20 days after ABE lentivirus treatment, and cells from an unaffected parent. Gene expression levels were normalized to transferrin receptor (TRFC) expression levels. Data from the unaffected parent is shown in both graphs for ease of comparison. Values and error bars reflect mean±SD of three technical replicates. (d) Western blot of cells from an unaffected parent, HGADFN167 cells, or HGADFN188 cells untreated or 20 days after ABE lentiviral treatment using the JOL2 antibody specific for human lamin A, progerin, and lamin C. Complete blots with molecular weight markers are available in Supplementary Figure 1. Additional replicates are provided in Extended Data Fig. 1. (e) Nuclear morphology of cells stained with a lamin A-specific antibody, with a progerin-specific antibody, or with DAPI. Scale bar=20 μm. Additional replicates were not performed. (f) Frequency of morphologically abnormal nuclei in samples of cells shown in (e). Values and error bars reflect mean±SD from three counts of independent images from the experiment in (e). *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 by Student’s unpaired two-sided t-test.
The dominant negative function of progerin poses challenges for treatment of HGPS by gene augmentation or gene disruption strategies. Overexpression of wild-type LMNA does not rescue cellular phenotypes18. While CRISPR-Cas9 nuclease-mediated gene disruption of the pathogenic allele has been reported to improve phenotypes in mouse models of progeria15–17, the resulting diversity of uncharacterized insertion and deletion (indel) products at the target locus, together with the risk of disrupting the wild-type LMNA allele that differs only at a single base pair from the pathogenic allele19,20, impede clinical translation of gene disruption strategies to treat progeria.
Base editors are genome editing agents that directly convert targeted base pairs without making double-strand DNA breaks6. Cytosine base editors (CBEs) convert C•G to T•A, while adenine base editors (ABEs) convert A•T to G•C5. Base editors function in many mitotic and post-mitotic cell types and in a wide array of organisms6. ABEs use a laboratory-evolved deoxyadenosine deaminase to convert adenine to inosine (which base pairs like guanine) within a small window of ~4-5 nucleotides at a Cas protein-specified locus, and induce the cell to replace the complementary thymine with cytosine by nicking and stimulating repair of the non-edited strand5,6.
Here, we report ABE-mediated correction of the LMNA c.1824 C>T mutation in HGPS patient-derived fibroblasts and in a mouse model carrying two genomically integrated copies of the human LMNA c.1824 C>T progeria allele21. In cultured patient-derived cells, we observed efficient (~90%) genomic DNA correction that ameliorates pathogenic LMNA transcript mis-splicing, reduces progerin protein production, and restores normal nuclear morphology. When delivered into a mouse model of human progeria by single retro-orbital injection of therapeutically relevant doses of AAV9 encoding the ABE and single guide RNA (sgRNA), the ABE corrected the human LMNA c.1824 C>T allele in various tissues at the DNA, RNA, and protein levels. P14-treated animals showed remarkable improvement in vascular disease compared to saline-injected controls, with aortic VSMC counts and adventitial fibrosis indistinguishable from those of wild-type mice, as well as reduced numbers of progerin-positive VSMCs and increased lamin A/C-positive VSMCs. The median lifespan of ABE-treated progeria mice was up to 2.4-fold longer than that of saline-injected controls. These findings suggest a potential therapeutic strategy for HGPS that directly corrects the causative mutation in vivo and inform applications of base editors to treat other genetic diseases.
Results
ABE correction of the progeria mutation in patient-derived cells
To position the pathogenic human LMNA c.1824 C>T mutation within the activity window of an ABE (positions 4-7, where the PAM is positions 21-23 for ABE7.10max5), we chose a target site with an NGA protospacer-adjacent motif (PAM) that places the mutation at protospacer position 6. To target this PAM, we used ABE7.10max-VRQR22, which combines an optimized ABE7.10 variant22 with an engineered SpCas9-VRQR variant23 that targets NGA PAMs.
We tested the ability of ABE7.10max-VRQR to correct the mutation in two progeria patient-derived primary fibroblast cell lines, HGADFN167 and HGADFN188 (Methods), using a lentivirus to deliver ABE7.10max-VRQR and the sgRNA targeting the LMNA c.1824 C>T mutation. After lentiviral transduction and puromycin selection of HGADFN167 and HGADFN188 cells, we observed 84% and 85% correction of the pathogenic mutation at 10 days, and 87% and 91% correction at 20 days, respectively (Fig. 1b). A low frequency (1.1-2.2%) of bystander editing was observed at the A•T at protospacer position 10, which results in Val690Ala (Extended Data Fig. 1a). Indel frequencies were minimal (≤0.15%) for both cell lines (Extended Data Fig. 1b). These results indicate that an ABE can efficiently correct the HGPS mutation to wild-type with few editing byproducts at the target locus.
Consistent with genomic correction of LMNA c.1824 C>T, we observed 8.1-fold and 4.4-fold reduction of mis-spliced LMNA mRNA in ABE lentivirus-transduced HGADFN167 and HGADFN188 cells, respectively, 20 days post-treatment compared to untreated cells (Fig. 1c). ABE treatment also reduced progerin protein levels 6.1- and 15-fold, respectively, relative to untreated cells and modestly increased lamin A abundance (Fig. 1d). Nuclear morphology improved among ABE-treated cells, with 1.8-fold fewer abnormal nuclei compared to untreated cells (Fig. 1e–f and Extended Data Fig. 1c–e). Together, these results establish that base editing to correct the LMNA c.1824 C>T mutation in HGPS patient-derived cells rescues the molecular and phenotypic consequences of the mutation.
Off-target DNA and RNA editing analysis in patient-derived cells
Canonical ABE7.10 editors can induce Cas-dependent off-target DNA editing and transient, low-level Cas-independent off-target RNA editing6. Cas-independent off-target DNA editing by ABE7.10 has been reported to be minimal or undetectable6. To identify candidate Cas-dependent off-target DNA editing sites associated with the sgRNA and Cas9-VRQR variant used to correct the progeria mutation, we performed CIRCLE-seq24 on genomic DNA from HGADFN167 and HGADFN188 cells treated in vitro with Cas9-VRQR nuclease and the LMNA-targeting sgRNA (Extended Data Fig. 2). We performed targeted sequencing of genomic DNA at the top 32 CIRCLE-seq identified candidate off-target loci in HGADFN167 and HGADFN188 cells 20 days after ABE lentivirus transduction. We observed no detectable (>0.1%) off-target DNA editing at the 32 tested candidate off-target loci in either cell line, despite 87–91% on-target editing (Fig. 2a).
Figure 2. Off-target DNA and RNA editing analysis and gene expression changes upon ABE7.10max-VRQR treatment of progeria patient-derived fibroblasts.
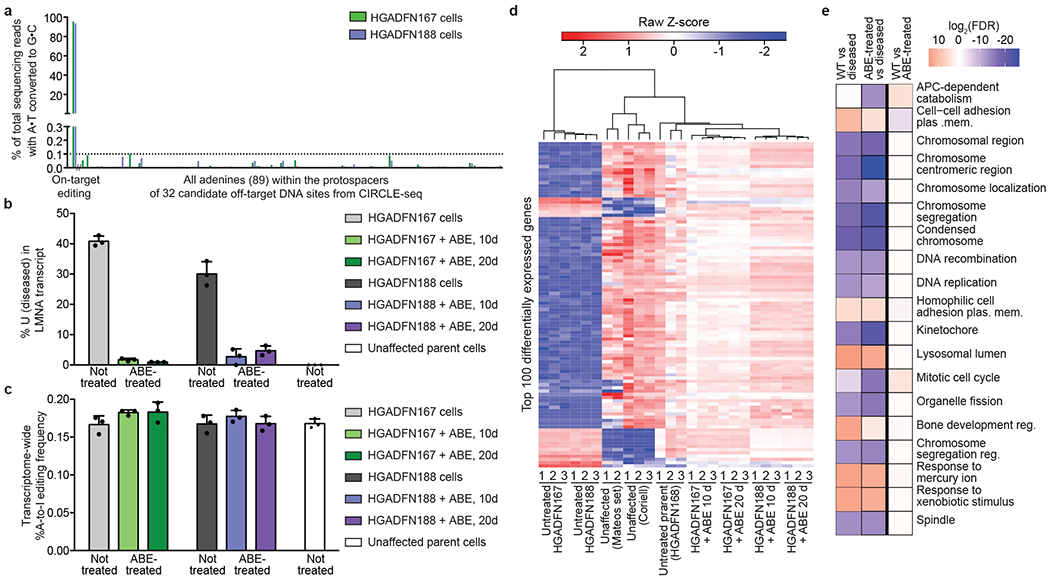
(a) DNA sequencing for the top 32 CIRCLE-seq-identified24 candidate off-target loci from HGADFN167 and HGADFN188 progeria patient-derived cells 20 days post-treatment with lentiviral ABE. (b) Uncorrected LMNA transcript frequency by RNA-seq in unaffected parental cells, untreated patient-derived cells, and ABE lentivirus-treated cells 10 or 20 days post-treatment. Values and error bars reflect mean±SD of three technical replicates. (c) Transcriptome-wide cellular A-to-I RNA editing levels in unaffected parental cells, untreated patient-derived cells, and in ABE lentivirus-treated cells 10 or 20 days post-treatment. Values and error bars reflect mean±SD of three technical replicates. (d) Heatmap of Z-scores for the top 100 differently expressed genes between unaffected control fibroblasts (Coriell and the Mateos dataset; see Methods) and untreated or lentiviral ABE-treated progeria patient-derived cells. Expression Z-scores across each gene are scaled so mean expression=0 and SD=1. Samples and genes are ordered by hierarchical clustering. Progeria patient-derived cells treated with lentiviral ABE for 10 and 20 days cluster with unaffected fibroblasts. (e) Gene ontology molecular function analysis45 of differentially-expressed genes. The 19 most significantly enriched gene sets in the Broad Institute molecular signatures database were identified between differentially-expressed genes in WT cells (Mateos, Coriell, and unaffected parent), disease cells (untreated HGADFN167 and HGADFN188), and treated cells (lentiviral ABE-treated HGADFN 167 and 188 at 10 and 20 days). A heat map of log2 FDR values for these 19 gene sets is shown, with overexpressed gene sets in red and underexpressed gene sets in blue.
To assess off-target RNA editing, we performed transcriptome-wide RNA-seq on ABE lentivirus-treated or untreated HGADFN167 and HGADFN188 cells, measuring the frequency of adenine-to-inosine RNA deamination (which naturally occurs throughout the transcriptome from endogenous cellular deaminases)25. The on-target nucleotide within the LMNA transcript was efficiently (>80%) corrected from U to C in ABE-treated cells (Fig. 2b). The average frequency and distribution of A-to-I conversion in the transcriptome of ABE-treated cells was similar to those of untreated cells (Fig. 2c, Extended Data Fig. 1h). Notably, ABE treatment of HGPS patient-derived cells restored the transcriptome to a state resembling that of cells from an unaffected parent (Fig. 2d,e). These results collectively show that treatment of cells with the LMNA-targeting sgRNA and ABE7.10max-VRQR did not result in detected off-target DNA or RNA editing using the above analysis methods, despite high levels of on-target editing.
In vivo ABE delivery in a mouse model of progeria
Encouraged by these findings, we applied base editing in vivo to correct a mouse model of progeria. We used C57BL/6 mice homozygous for a transgene that includes the complete human LMNA c.1824 C>T allele (C57BL/6-tg(LMNA*G608G)HClns/J, previously used as heterozygous mice21); these homozygous mice hereafter are referred to as “progeria mice”. Phenotypically, this model recapitulates hallmark HGPS symptoms including VSMC defects, hair loss, lack of subcutaneous fat, musculoskeletal abnormalities, and shortened lifespan3,4,21. Given the diverse tissues affected by progeria, we sought systemic in vivo delivery of the ABE and sgRNA characterized above.
We recently developed an in vivo base editor delivery strategy using adeno-associated virus (AAV)26, an FDA-approved delivery modality. This approach uses trans-splicing inteins to reconstitute full-length base editor in cells from a pair of AAVs, each expressing one half of the base editor (Fig. 3a)26,27. We adapted this system to deliver ABE7.10max-VRQR and the LMNA c.1824-targeting sgRNA. We chose the AAV9 capsid for its broad tissue tropism, clinical validation, and ability to transduce progeria-relevant tissues including heart and muscle28,29.
Figure 3. Pathogenic DNA, RNA, and protein correction from a single in vivo ABE-AAV9 injection of a mouse model of human progeria.
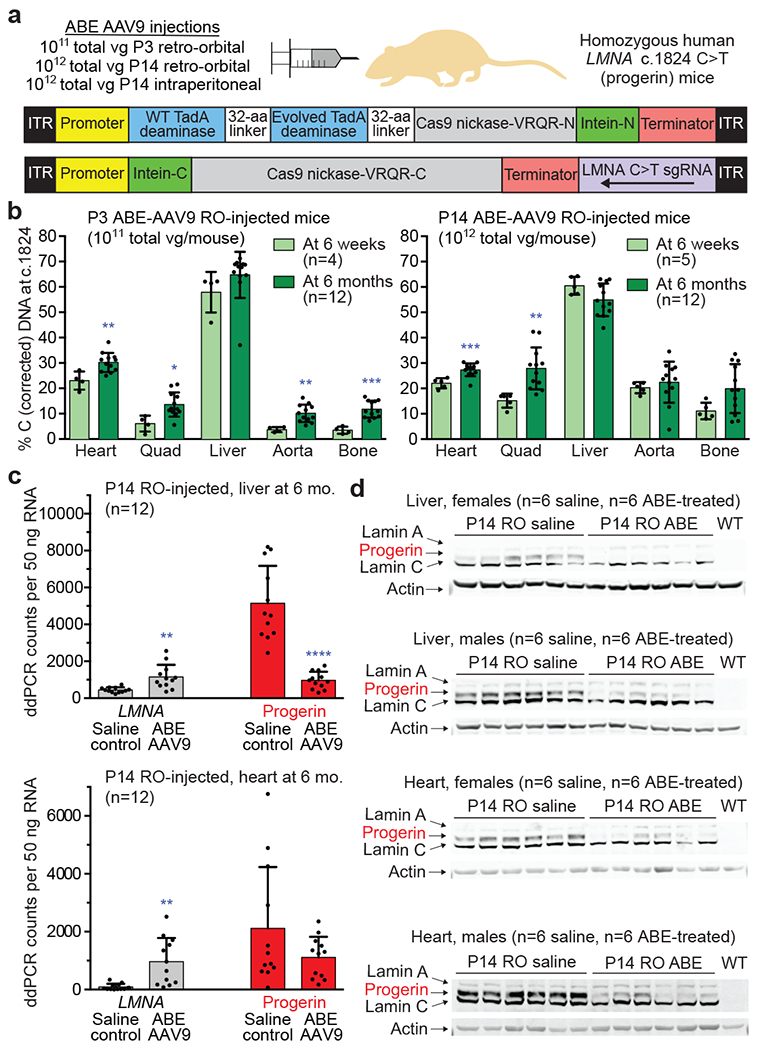
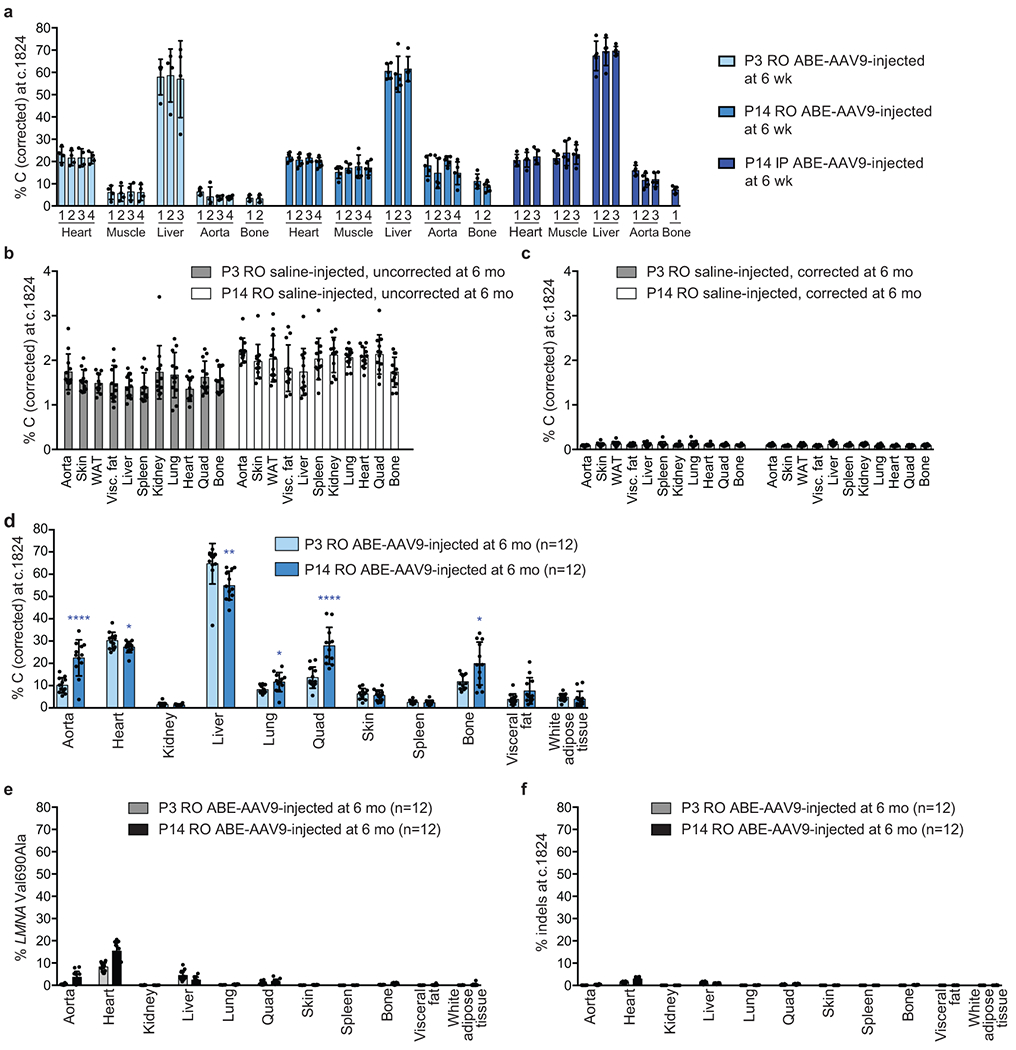
(a) Dual AAV9 encoding split-intein ABE7.10max-VRQR base editor halves26 and the LMNA-targeting sgRNA were injected into progeria mice. P3 retro-orbital (RO) injections (5×1010 of each AAV vg, 1×1011 vg total), P14 RO injections (5×1011 vg of each AAV vg, 1×1012 vg total), or P14 intraperitoneal (IP) injections (5×1011 vg of each AAV, 1×1012 vg total) were administered. (b) DNA editing efficiencies correcting LMNA c.1824 from T (pathogenic) to C (wild-type) for P3 RO-injected mice (left) or P14 RO-injected mice (right) in 6-week- or 6-month-old mice. Editing in P14 IP-injected mice is in Extended Data Fig. 3a. (c) ddPCR counts for human LMNA (grey bars) and progerin (red bars) RNA transcript abundance in P14 RO saline- or ABE-AAV9-injected mice in liver and heart. See Extended Data Fig. 4 for additional data. (d) Western blot analysis of human lamin A, progerin, and lamin C proteins in liver and heart of P14 RO saline- or ABE-AAV9-injected mice. Each lane shows tissue from a different mouse. WT is a C57BL/6 mouse lacking the transgene, showing that the antibody is specific to human lamin proteins. See Extended Data Fig. 5 for additional data. Values and error bars represent mean±SD for the indicated number of biological replicates. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 by Student’s unpaired two-sided t-test.
To compare the effects of injection route and timing on in vivo editing, we performed retro-orbital (RO) injection of P3 (3 day-old) progeria mice (n=4), RO injection of P14 (2 week-old) mice (n=5), and intraperitoneal (IP) injection of P14 mice (n=5). P3 injections used 5×1010 viral genomes (vg) of each AAV for a total of 1×1011 vg per mouse. Both P14 injections used 5×1011 vg of each AAV for a total injection of 1×1012 vg per mouse. 6-week-old mice were sacrificed and editing was evaluated in various tissues (Fig. 3, Extended Data Fig. 3a–c).
At 6 weeks of age, P14 RO injection resulted in the highest aorta and bone editing efficiencies among the tested injection routes (Fig. 3b, Extended Data Fig. 3a). Editing efficiencies in bulk heart tissue (excluding aorta) were similar for the three injection routes. P14 injections generally achieved higher base editing efficiencies that P3 injections, possibly due to the 10-fold higher AAV dose that could be injected into P14 mice or increased expression of AAV9 receptors in the older mice26,30,31. Together, these data reveal that a single in vivo injection of AAV encoding base editors results in modest to high levels of correction (10-60%) of the causative LMNA point mutation in various organs.
Treatment of progeria mice for 6-month tissue and longevity analyses
We performed long-term animal studies using both P3 and P14 retro-orbital AAV injections to assess the relationship between long-term in vivo outcomes and editing efficiencies in disease-relevant tissues such as the aorta, where editing levels in P14-injected mice were 3.8-fold that of P3-injected animals at 6 weeks of age. We retro-orbitally injected 24 progeria mice at P3 with 1011 total vg of dual AAV9, and 24 mice at P14 with 1012 total vg as before. As controls, 24 mice at P3 and 24 mice P14 were injected retro-orbitally with saline. All animal groups were gender-balanced. At 6 months of age, when untreated progeria mice typically show phenotypic decline but are not yet at the end of their lifespan, 24 P3-injected mice and 24 P14-injected mice (half AAV9-treated, half saline controls) were sacrificed and analyzed for DNA base editing efficiency, LMNA RNA splicing, human progerin and lamin protein levels, and tissue histology. We placed the remaining 24 P3-injected mice and 24 P14-injected mice (half AAV9-treated, half saline controls) in a longevity study to assess animal lifespan.
DNA, RNA, and protein analysis at 6 months of age
Analysis of the DNA base editing outcomes in 6-month-old mice revealed important differences between the P3- and P14-injected cohorts. Both cohorts showed increases in DNA editing efficiency in several tissues compared to the 6-week time point (Fig. 3b). For example, editing in aorta editing rose from 4.5±2.5% to 10±3.4% in P3-injected mice, and from 17±5.2% to 23±8.1% in P14-treated mice. Modest editing was observed in the lung, skin, visceral fat, and white adipose tissue (WAT), while minimal editing was observed in kidney or spleen (Extended Data Fig. 3d). Bystander Val690Ala editing and indels varied by tissue but were generally observed at low frequencies compared to on-target editing (Extended Data Fig. 3e–f). These results suggest that base editing may continue in vivo from 6 weeks to 6 months of age, consistent with the known persistence of AAV in mammals32,33, or that edited cells may have a survival advantage over uncorrected cells in some organs, increasing the prevalence of edited alleles over time.
Editing efficiencies at 6 months in most tissues remained higher in the P14-injected cohort than the P3-injected cohort, including by 2.2-fold in the aorta, 2.1-fold in skeletal muscle, 1.7-fold in bone, and 1.4-fold in lung (Extended Data Fig. 3d). These results indicate that injecting mice with 1012 total vg at P14 results in higher levels of LMNA correction 6 months after treatment, compared to mice injected with 1011 total vg at P3.
Next, we quantified the effect of in vivo ABE treatment on transcript abundance and protein levels for progerin and human lamin A in 6-month-old mice. Base editing led to decreases in progerin transcript abundance (Fig. 3c, Extended Data Fig. 4) that were sometimes larger than DNA correction levels; for example, in P14 WAT we observed 31% reduction in progerin mRNA levels despite only 4.0±3.6% DNA correction. These findings suggest that corrected cells may be more transcriptionally active than uncorrected cells or that cells with higher transcriptional activity may be more efficiently edited in vivo. Finally, we noted increases in the abundance of correctly spliced LMNA transcripts among ABE-treated mice in a variety of tissues compared with saline-injected controls (Extended Data Fig. 4).
We evaluated progerin and lamin A protein levels in 6-month-old mice by western blot. P14-injected ABE-treated mice showed robust reduction of progerin protein in the liver (87±14% reduction), heart excluding aorta (86±9.1% reduction), and aorta (49±19% reduction) compared to saline-injected controls (Fig. 3d, Extended Data Fig. 5). P3-injected ABE-treated mice showed similar reductions in progerin protein levels (Extended Data Fig. 5). Progerin protein levels were generally reduced more than progerin mRNA levels in the same organ, suggesting that corrected cells may be translationally more active than uncorrected cells. Taken together, these findings indicate that in vivo ABE-mediated correction of the pathogenic human LMNA c.1824 C>T allele can reduce progerin RNA and protein levels in multiple clinically relevant tissues.
ABE treatment improves histopathology in aorta and adipose tissue
To probe the physiological consequences of base editing the pathogenic LMNA c.1824 C>T allele, we performed histological analysis of aorta and adipose tissue. Progeria patients exhibit loss of VSMCs in aortic vessel walls and periadventitial thickening that together contribute to aortic stiffening and impairment of cardiac function7–11. We observed these hallmark vascular features of progeria in saline-injected control mice (Fig. 4a)21. Aortas from 6-month-old P3-injected ABE-treated mice showed 3.3-fold higher average VSMC counts per cross-sectional area compared with saline-injected controls, but similar (1.2-fold lower) adventitial thickness (Fig. 4a–b, Extended Data Figs. 6–7). Remarkably, P14 injection of ABE completely rescued both aortic VSMC counts (11-fold increase) and adventitial thickness (5.5-fold decrease) compared to saline-injected controls, such that P14-injected ABE-treated mice were indistinguishable from wild-type mice in these two parameters (Fig. 4a–b, Extended Data Figs. 6–7).
Figure 4. Aortic histopathology and lifespan of progeria mice following a single in vivo ABE-AAV9 injection.
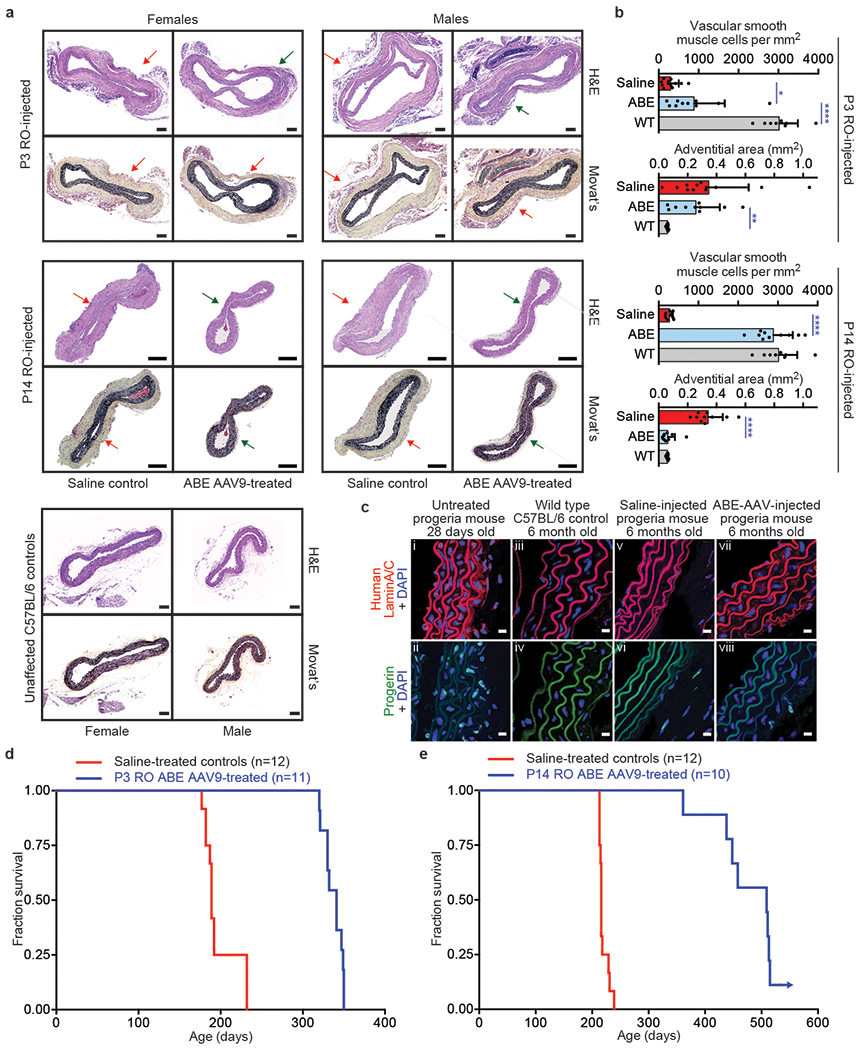
(a) Representative aorta cross-sections from 6-month-old mice showing vascular smooth muscle cell (VSMC) nuclei and adventitia in saline- or ABE-AAV9-treated mice injected at P3 or P14. Upper images were stained with hematoxylin and eosin (H&E); lower images were stained with Movat’s pentachrome. Red arrows emphasize decreased VSMC counts and adventitial fibrosis; green arrows indicate preserved VSMC counts and less adventitial fibrosis. P3 and C57BL/6 scale bar=100 μm, P14 scale bar=200 μm. Additional replicates are shown in Extended Data Figs. 6 and 7. (b) Quantification of VSMC nuclei counts and adventitia area in mouse cohorts. Values and error bars reflect mean±SD of n=12 (P3 saline), n=10 (P3 ABE-AAV9, P14 saline, and P14 ABE-AAV9), or n=8 (WT) replicates. Data from WT samples are shown in both graphs for ease of comparison. Replicates analyzed are provided in Extended Data Figs. 6 and 7. (c) Representative fixed aortas stained for human lamin A/C + DAPI and progerin + DAPI for untreated progeria mice at P28 (i and ii), wild-type C57BL/6 mice at 6 months (iii and iv), saline-injected progeria mice at 6 months (v and vi), and ABE-treated progeria mice at 6 months (vii and viii). Autofluorescent elastin fibers in the tunica media appear as wavy lines. Scale bar=10 μm. Additional replicates are shown in in Extended Data Fig. 8. (d) Kaplan-Meier curve for P3 RO saline- and ABE-AAV9-injected progeria mice. Median lifespans: P3 saline-injected mice=189 days, P3 ABE-AAV9-injected mice=337 days (1.8-fold longer, p<0.0001). (e) Kaplan-Meier curve for P14 RO saline- and ABE-AAV9-injected progeria mice. Median lifespans: P14 saline-injected mice=215 days, P14 ABE-AAV9-injected mice=510 days (2.4-fold longer, p<0.0001). *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 by Student’s unpaired two-sided t-test for (b). Mantel-Cox test for (d) and (e).
Progerin-induced VSMC death is a key driver of mortality in progeria patients7,11. The improvements in aortic pathology in ABE-treated animals prompted us to examine progerin and human lamin A/C protein levels in aortic VSMCs. Sections of aorta from saline- and P14-injected ABE-treated mice were fixed and stained with antibodies specific to human lamin A/C and human progerin (Fig. 4c, Extended Data Fig. 8). As expected given their young age, VSMC nuclei from 4-week-old progeria animals stained robustly for both proteins (Fig. 4c, i and ii). No staining for human lamin A/C or progerin was observed in wild-type C57BL/6J aortic VSMCs, demonstrating antibody specificity (Fig. 4c, iii and iv). Consistent with VSMC counts, aortas from 6-month-old saline-injected progeria mice contained virtually no VSMCs (Fig. 4c, v and vi). In contrast, ABE-treated progeria mice maintained much higher numbers of VSMCs that expressed human lamin A/C with minimal progerin (Fig. 4c, vii and viii). These observations suggest that ~25% DNA base editing in the aorta rescues two key progeria vascular defects—VSMC loss and periadventitial fibrosis–while preserving lamin A/C expression and reducing progerin abundance in VSMCs.
Lipodystrophy (reduced subcutaneous body fat) is a clinical feature of progeria patients.4 ABE treatment modestly rescued the loss of the hypodermal fat layer in both P3-and P14-injected mice relative to saline-injected controls (Extended Data Figs. 9–10). Both ABE-treated and saline-injected mice exhibit moderate dermal hypoplasia compared to wild-type C57BL/6 mice (Extended Data Figs. 9–10).
ABE treatment greatly increases lifespan in progeria mice
We conducted longevity studies on 24 P3-injected and 24 P14-injected progeria mice (half ABE-AAV9-injected, half saline-injected, gender-balanced). Over the 1.5-year duration of the longevity studies, 3 of 48 mice were excluded following health issues deemed unrelated to progeria or treatment (Supplementary Note 1).
Body weights were maintained in both ABE-treated cohorts (Extended Data Fig. 11). Median survival of P3 saline-injected mice (n=12) was 189 days, while median survival of P3 ABE-treated mice (n=11) was 337 days (1.8-fold longer, p<0.0001 by Mantel-Cox test) (Fig. 4d). Median survival for P14 saline-injected mice (n=12) was 215 days, while median survival of P14 ABE-treated mice (n=10) is 510 days (2.4-fold longer, p<0.0001 by Mantel-Cox test) (Fig. 4e), which corresponds approximately to the beginning of old age in wild-type C57BL/6 mice34. P14-injected mice exhibit remarkably normal behavior and vitality at ages well beyond the lifespan of saline-injected animals (Supplementary Videos 1–5). Complete longevity study animal data is in Supplementary Table 1.
Among the nine deceased P14-injected ABE-treated mice (one is still in good apparent health as of this writing), necropsy revealed gastrointestinal necrosis in one, liver tumors in five, and no apparent abnormalities in three (Supplementary Table 1). To probe the potential origins of the liver tumors, we performed whole-genome sequencing of liver tissue samples from two P14 saline-injected mice and from livers and liver tumors of three P14 AAV9 ABE-treated mice that showed liver tumors. Samples from all AAV-injected mice, but none from the saline-injected mice, showed evidence of rare AAV integration in genomic regions where AAV integration has been previously associated with liver tumors in mice (Abr, Alb, BC057079/Focad, and Ksr1)35 (Supplementary Data 1). Although samples from liver tumors showed similar numbers of AAV integration events as samples from non-tumor liver tissue, previous observations of AAV integration at these regions in liver tumors from AAV-injected mice35 suggest that AAV integration may have contributed to liver tumor formation. We note that AAV-associated liver tumors, while documented in mice, have thus far not been reported in human patients treated with therapeutic AAV vectors36.
The fraction of A•T-to-G•C point mutations among all point mutations detected by whole-genome sequencing was not significantly different among saline-injected mouse liver tissue, ABE-treated mouse liver tissue, and ABE-treated mouse tumor tissue (FDR-adjusted p=0.28–0.50), suggesting no apparent change in the relative genome-wide abundance of A•T-to-G•C point mutations from ABE treatment (Extended Data Fig. 12a–b, Supplementary Data 2). We also searched for A•T-to-G•C mutations and indels in the exons, introns, and ATAC-seq-defined regulatory regions within 100 kb of 84 genes recurrently mutated in liver cancer (see Methods), and found no evident patterns distinguishing saline-injected mouse liver tissue, ABE-treated mouse liver tissue, or ABE-treated mouse tumor tissue (Extended Data Fig. 12c, Supplementary Data 3). These data collectively suggest no apparent role of base editing in liver tumor formation. Finally, we note that the progeria mice used in this study contain two copies of human RAB25 downstream of LMNA, and overexpression of RAB25 promotes human hepatocellular carcinoma37, raising the possibility that expression of transgenic RAB25 in these mice (confirmed by RT-qPCR in Extended Data Fig. 12d) may contribute to liver tumorigenesis that manifests only once lifespan is greatly extended. Additional studies are needed to understand the potential roles of uncorrected LMNA c.1824 C>T alleles, RAB25, AAV transduction, ABE treatment, and mouse age in the long-term health of these animals.
Discussion
Correcting pathogenic alleles that cause devastating diseases is a longstanding challenge in therapeutic science. Base editing provides an opportunity to directly correct point mutations that drive many disorders without requiring double-strand DNA breaks. Here we demonstrate direct correction of the mutation that underlies the majority of HGPS cases using an ABE. In patient-derived cells, base editing efficiently corrects the pathogenic allele, substantially reduces RNA mis-splicing, decreases progerin protein abundance, and rescues nuclear morphology abnormalities. Characterization of off-target DNA and RNA editing suggests a low degree of off-target editing in patient-derived cells despite ~90% on-target editing.
Systemic single-dose in vivo injection of dual AAV9 encoding the ABE and sgRNA into a mouse model of human progeria resulted in durable correction of the pathogenic allele, amelioration of RNA mis-splicing, reduction of progerin protein in various tissues, and greatly improved aortic health, fully rescuing VSMC counts and adventitial fibrosis in progeria mice treated with ABE at P14, which in the course of normal mouse maturation corresponds to approximately year 5-6 in humans34. These findings are particularly encouraging because aortic pathology is a major determinant of morbidity and mortality in children with progeria4,7,8.
These results also establish that single injections of an ABE packaged in a clinically-relevant AAV capsid can greatly extend the lifespan of a progeria animal model, with median lifespan increasing in P14-treated mice from 215 to 510 days, approaching old age in wild-type C57BL/6 mice34. The striking improvements in aortic pathology, lifespan, and animal vitality among P14-injected mice long after a single ABE injection collectively suggest that this strategy has the potential to restore cellular and tissue functions in progeria patients.
Although single administration with possible transient immunosuppression may help mitigate potential immunological responses to editing agents, efforts to translate these findings into patients must closely monitor immune responses to treatment. AAV integration and liver tumors observed in some of the longest-lived mice, consistent with previous reports of AAV integration-induced liver tumors in mice35,38–41, further highlights the importance of vector and dose optimization as well as post-treatment monitoring, even though liver tumors have not yet been observed in human patients treated with AAV vectors36.
In some tissues modest DNA editing resulted in disproportionately large benefits at the RNA, protein, or tissue levels. For example, ~25% editing in the aorta of P14-injected mice resulted in 11-fold higher VSMC counts, a 5-fold decrease in adventitia fibrosis, and a lack of observed progerin-positive VSMC nuclei. The outsized benefits of DNA editing suggest that edited cells may contribute disproportionately to the health of tissues in this animal model. Additional studies are needed to understand the molecular basis of this phenomenon.
A number of additional studies may further advance base editing treatments for progeria toward clinical application. ABE variants with much higher editing activity than ABE7.10max were recently reported42,43. These variants may further increase editing efficiency and phenotypic rescue, or may reduce required dosage. The timing of treatment may also need to be further optimized for best results, taking into account the time to diagnosis. Finally, ABE editing has the potential to synergize with emerging progeria treatments3,14–16 including farnesyltransferase inhibitors (FTIs)3, other small-molecule drugs14, or antisense oligonucleotides targeting the mutant LMNA allele.18,44
Methods
Cell culture
HGADFN167 and HGADFN188 cells (Progeria Research Foundation) were maintained in antibiotic-free DMEM (Thermo Fisher 10569044) supplemented with 20% (v/v) fetal bovine serum (Thermo Fisher), at 37 °C with 5% CO2. HEK239T/17 (ATCC CRL-11268) cells were maintained in antibiotic-free DMEM (Thermo Fisher 10569044) supplemented with 10% (v/v) fetal bovine serum (Thermo Fisher), at 37 °C with 5% CO2.
Lentiviral vector cloning
The ABEmax-VRQR gene was inserted into the lentiCRISPRv2 backbone (Addgene 52961) via restriction cloning. Backbone plasmid was digested using AgeI and BamHI according to the manufacturer’s protocol. ABEmax-VRQR was amplified from (Addgene 119811) using the primers LWK901 and LWK902. Gibson assembly was performed using a 3:1 molar ratio of insert to vector backbone according to the manufacturer’s protocol. The LMNA c.1824 C→T targeted sgRNA was installed by digesting the cloned backbone with BsmBI and gel extracting the resulting cleaved backbone. DNA oligonucleotides encoding the sgRNA were ordered to match the corresponding overhangs generated by BsmbI digestion (top oligo: caccggtccAcccacctgggctcc, bottom oligo: aaacggagcccaggtgggTggacc). Oligonucleotides were annealed and phosphorylated using T4-PNK according to the manufacturer’s instructions and ligated into the digested backbone as previously described22.
LWK901: 5’-TTTGCCGCCAGAACACAGGACCGGTGCCACCATGAAACGGACAGCCGACG
LWK902: 5’-GGGAAAAGTTGGTGGCCCCGGATCCGACTTTCCTCTTCTTCTTGGGCTCG
AAV vector cloning
N-terminal virus was as we previously reported26. C-terminal virus required the installation of the VRQR mutations (D1135V, G1218R, R1335Q, T1337R) as well as the sgRNA sequence targeting LMNA c.1824 C→T. The VRQR mutations were installed by Gibson assembly and ligation of the ABEmax-VRQR C-terminus. The sgRNA targeting LMNA c.1824 C→T was installed by BsmBI digestion of the backbone as described for the lentiviral backbone and ligation of the same top and bottom oligonucleotides into the cut vector.
To generate the C-terminal AAV genome encoding Npu-ABEmax(VRQR) and the sgRNA targeting LMNA G608G, we first subcloned Npu-ABEmax(VRQR) in a mammalian (CMV) expression plasmid by deleting the UGI domains from an Npu-BE4max(VRQR) intermediate. We then amplified by PCR the Npu-ABEmax(VRQR) gene using primers 877(fwd)/670(rev) and cloned by Gibson assembly into AgeI/BglII-cut AAV plasmid. In a subsequent cloning step, annealed oligonucleotides encoding the sgRNA targeting LMNA G608G were ligated into BsmBI-cut plasmid.
877(fwd): 5’- TCACTTTTTTTCAGGTTGGACCGGTGCCACCATGAAACGGACAGCCGACGG
670(rev): 5’- AATCCAGAGGTTGATTATCAGATCTTAGACTTTCCTCTTCTTCTTGGGCTCGAATTCGC
Lentiviral production
HEK239T/17 (ATCC CRL-11268) cells were maintained in antibiotic free DMEM (Thermo Fisher 10569044) supplemented with 10% (v/v) fetal bovine serum (Thermo Fisher), at 37 °C with 5% CO2. Cells were verified to be free of mycoplasma by ATCC upon purchase. On day 1, cells were split 1:3 from rapidly dividing HEK293T/17 flasks that had been split 1:10 three days prior. The following day, media was changed on cells and cells were transfected using FuGENE HD according to manufacturer’s protocol. Transfection mix included 9 μg of transfer vector (the packaging genome of interest), 9 μg of psPAX2 (encoding the viral packaging proteins), and 6 μg of pVSV-G (encoding the VSV-G envelope protein). Transfection mix was then supplemented with 70 μL of room temperature equilibrated FuGENE and brought to a final volume of 1500 μL per flask with Opti-mem. Two days after transfection, media was collected and spun at 3000 g for 15 minutes to remove remaining cells. Centrifuged supernatant was passed through a 0.45-μm PVDF filter to eliminate all non-viral debris. Supernatant was either transferred directly to target cells.
The human non-targeting control sgRNA sequence was used from a previous study46. Oligos containing these non-targeting sgRNAs with 5’ overhang BsmBI digestion sites were synthesized by Integrated DNA Technologies, Inc. The oligos were first annealed and inserted into the lentiCRISPR v2 plasmids (a gift from Feng Zhang, Addgene plasmid #52961) as described previously47. The fragments containing these non-targeting sgRNAs were digested from the recombinant lentiCRISPR v2 plasmids by restriction enzymes KpnI and NheI. These fragments were then ligated into the ABEmax7.10 backbone, which was extracted from the digests of KpnI and NheI. The sequences of the recombinant plasmids were confirmed by Sanger sequencing. Control sgRNA oligo sequences:
ctrl sgRNA F CACCGGCCTGCCCTAAACCCCGGAA
ctrl sgRNA R AAACTTCCGGGGTTTAGGGCAGGCC
HGADFN167 and HGADFN188 lentiviral transduction
Lentivirus was generated as described above. HGADFN167 and HGADFN188 cells were cultured in 75-cm flasks in antibiotic-free DMEM supplemented with 20% FBS. Supernatant was removed from cells and media was replaced with 15-mL filtered lentiviral media per plate, supplemented with 5 mL of regular DMEM. Cells were grown for 3 days prior to media change with DMEM +20% FBS including 2 μg/mL puromycin to select for cells expressing full-length editor. Cells were maintained in selective media for 10 and 20 days post-infection prior to harvesting genomic DNA, RNA, and protein while also isolating cells at 20 days of age for histological analysis.
HGADFN167 and HGADFN188 genomic DNA isolation
Genomic DNA for DNA sequencing analysis was isolated first trypsinizing cells and centrifugation of one 15-cm dish per cell line per time point. Trypsinized cells were resuspended in media and spun gently at 100 g for 10 minutes. Cell pellets were resuspended in 200 μL of lysis buffer (10 mM Tris-HCl, pH 7.5, 0.05% SDS, 25 μg/mL proteinase K (NEB)). Lysing cells were incubated at 37 °C for 1 hour. Proteinase K was inactivated by 30-minute incubation at 80 °C.
HGADFN167 and HGADFN188 RNA extraction and ddPCR
Total RNA from the cell-lines was extracted with Trizol (Life Technologies) and purified using the RNeasy mini kit (Qiagen) per the manufacturer’s instructions. The total RNA yield was determined using the NanoDrop 2000 spectrophotometer. 1 μg of total RNA per condition was converted to cDNA using iScript Select cDNA Synthesis Kit (Bio-Rad). PCR cycling conditions consisted of an initial enzyme activation step for 10 minutes at 95 °C, followed by 40 cycles of 94 °C for 30 seconds and 59 °C for 30 seconds with a 2 °C/second ramp rate, and a 10-minute enzyme deactivation step at 98 °C for 10 minutes. Each reaction was duplexed with the Human TFRC PrimePCR Probe Assay (assayID #qHsaCIP0033292, HEX) and performed in triplicate. Upon completion of reactions, samples were analyzed on a QX200 droplet reader (BioRad) to obtain expression levels relative to murine Hprt and transcript-specific copy number, then further analyzed using Excel software.
Cell-line qPCR analysis
Quantitative RT-PCR was performed in triplicate using SYBR Green Supermix (Bio-Rad) on CFX96 real-time system (C1000 thermal Cycler; Bio-Rad). The relative mRNA level of LMNA and progerin is normalized to GAPDH, a housekeeping gene used as an internal control. Primers for GAPDH were obtained from OriGENE Technologies. For amplifying LNNA transcript, the forward PCR primer was 5’-GCAACAAGTCCAATGAGGACCA-3’ and the reverse primer was 5’-CATGATGCTGCAGTTCTGGGGGCTCTGGAT-3’; for progerin mRNA amplification, the sequence of the forward PCR primer was 5’-GCAACAAGTCCAATGAGGACCA-3’ and the reverse primer was 5’-CATGATGCTGCAGTTCTGGGGGCTCTGGAC-3’.
HGADFN167 and HGADFN188 RNA-seq analysis
Total RNA was applied to Oligo-dT(25) Dynabeads (Thermofisher) to enrich for polyadenylated transcripts. Stranded RNA-seq libraries were generated using the PrepX mRNA 48 kit (Takara) on the Apollo 324 automated liquid handling system (Wafergen) followed by barcoding and amplification (12 cycles). Following PCR and bead cleanup with AmpureXP beads (Beckman Coulter), libraries were visualized on a 4200 TapeStation system (Agilent) and quantified using a Library Quantification Kit (KAPA Biosystems). Libraries were sequenced on a NextSeq high-throughput flowcell (Illumina) as 75 bp paired-end reads. All raw fastq files generated are available from the NCBI SRA under BioProject PRJNA627465.
Fastq files were generated with bcl2fastq2 version 2.20 and trimmed using TrimGalore version 0.6.2 to remove low-quality bases, unpaired sequences, and adaptor sequences. Trimmed reads were aligned to Homo sapiens genome assembly GRCh38 with a custom Cas9-ABEmax gene entry by initially aligning with STAR (version 2.7.3a) to identify splice junction followed by an additional STAR alignment including the splice junctions identified in the first STAR alignment (2-STAR pass).
To calculate the average % of A-to-I editing amongst adenosines sequenced in transcriptome-wide sequencing analysis, we used REDItools v1.3 to quantify the % editing in each sample. We removed all nucleotides except adenosines from our analysis and then removed all adenosines with a read coverage less than 10 to avoid errors due to low sampling; additionally, we removed positions with a mapping or read quality score below 25. Next, we calculated the number of adenosines converted to inosine in each sample and divided this by the total number of adenosines in our dataset after filtering to obtain a percentage of adenosines edited to inosine in the transcriptome. The standard error of the mean (SEM) was calculated by comparison of 3 biological replicates. Significance was calculated using the student’s unpaired t-test.
Transcriptome correction analysis in progeria cell lines
Treated HGADFN167 and HGADFN188 cells at 10-day and 20-day time points were used for analysis. Untreated HGADFN167 and HGADFN188 cells were also used. Untreated unaffected donor cells (AG03257) were obtained from Coriell Cell Bank and were also used for analysis. Additional unaffected cells were also pulled from a previous study and used for analysis48.
Sorted bam files generated in the 2-STAR pass alignment described above where supplied to RSEM version 1.3.1 to count transcripts. The limma-voom R package was used to normalize gene expression levels and perform differential expression analysis.
HGADFN167 and HGADFN188 protein isolation and Western Blot analysis
HGPS fibroblasts were prepared by collecting cells with Trypsin, washing briefly in cold PBS and then lysing in RIPA lysis buffer (150 mM NaCl, 50 mM Tris-HCL pH8, SDS 1%, Sodium Deoxycholic Acid 0.5%, NP-40 1%) for 30 minutes. After high speed centrifugation at 4 °C for 15 minutes, protein was quantified using standard BCA. Lysates were diluted in LDS sample buffer and heated at 70 °C for 10 minutes and separated by SDS-PAGE on 4-12% Bis-Tris gels (Nupage, Invitrogen). Protein was transferred to a nitrocellulose membrane using a Mini Gel Tank for 90 minutes at 30 volts. Membranes were blocked with Intercept TBS Blocking Buffer (LI-COR) and primary antibodies were incubated overnight in Intercept buffer. Membranes were then washed 3 times for 10 minutes in TBS-T (0.1% tween) and incubated with IRDye conjugated secondary antibodies at 1:10000 dilution (LI-COR). The following antibodies were used for cell and tissue blotting: anti-Lamin A/C, which recognizes human Lamin A/C and Progerin (Abcam #40567, Jol2 Clone, 1:200); anti-beta Actin (Abcam #8227, 1:1000); anti-Histone 3 (Cell Signaling Technology 4499, 1:2000). Goat anti-rabbit secondary (IRDye 680LT) or goat anti-mouse secondary (IRDye 800cw) were used at 1:10000 dilution.
Western blot densitometry was performed to quantify relative protein abundance for in vivo samples. The relative abundance of Lamin A or progerin protein in saline- or ABE-treated samples was quantified by normalizing densitometry values for each band to its corresponding beta-actin band.
For Extended Data Figures 1e and 1g, whole cell lysates were prepared by dissolving cell pellets in Laemmli Sample Buffer containing 5% 2-mercaptoethanol (Bio-Rad). Subsequently, protein samples were resolved on 10% SDS-PAGE gels and transferred onto nitrocellulose membranes (Bio-Rad). Antibodies used in this study were lamin A/C (MAB3211; Millipore; 1:500 dilution), progerin (Collins, custom; 1:500 dilution), and GAPDH (SC-47724, Santa Cruz; 1:5000 dilution). Secondary antibodies used for these blots were Anti-mouse (SC-516102, Santa cruz) and anti-rabbit (211-035-109, Jackson ImmunoResearch), both at 1:5000 dilutions.
HGADFN167 and HGADFN188 immunocytochemistry
For immunocytochemistry, cells were fixed in 4% paraformaldehyde (PFA) for 15 min. The cells were then blocked with 4% BSA serum in tris-buffered saline (TBS) for 1 hour. Subsequently, the cells were incubated with the primary antibodies lamin A/C (MAB3211; Millipore; 1:250 dilution), progerin49 (Collins, custom; 1:250 dilution) in 4% BSA in TBS (0.3% Triton-X) overnight at 4°C. After three washes with TBS, cells were incubated in 1% BSA in TBS containing secondary antibodies and DAPI (Vector Laboratories). Secondary antibodies used were Alexa Fluor 594 donkey anti-rabbit IgG (Invitrogen), and Alexa Fluor 488 donkey anti-mouse IgG (Invitrogen) (both at 1:1000 dilutions). Images were acquired with Zeiss AX10 microscope equipped with a SPOT PURSUIT camera. Greater than 150 nuclei were analyzed per condition where nuclei were assigned by visual inspection into abnormal or normal bins based on nuclear blebbing phenotype.
Cell line high-throughput sequencing and data analysis
Genomic DNA was amplified by qPCR using Phusion Hot Start II DNA polymerase with use of SYBR gold for quantification. Human LMNA-specific primers listed at the end of this section which include Illumina Miseq adapter sequences (bolded). 3% DMSO was added to all gDNA PCR reactions. To minimize PCR bias, reactions were stopped during the exponential amplification phase. 1 μL of the unpurified gDNA PCR was used as a template for subsequent barcoding PCR (10 cycles, annealing temperature 61 °C). Pooled barcoding PCR products were gel-extracted (Min-elute columns, Qiagen) and quantified by qPCR (KAPA KK4824) or Qubit dsDNA HS assay kit (Thermo Fisher). Sequencing of pooled amplicons was performed using an Illumina MiSeq according to the manufacturer’s instructions. Oligonucleotide sequences for the primers used are provided below.
Initial de-multiplexing and fastq generation were performed by bcl2fastq2 running on BaseSpace (Illumina) with the following flags: –ignore-missing-bcls –ignore-missing-filter –ignore-missing-positions –ignore-missing-controls –auto-set-to-zero-barcode-mismatches –find-adapters-with-sliding-window –adapter-stringency 0.9 –mask-short-adapter-reads 35 –minimum-trimmed-read-length 35. Alignment of fastq files and quantification of editing frequency was performed by CRISPResso2 in batch mode with a window width of 10 nucleotides.
Forward: 5’-ACACTCTTTCCCTACACGACGCTCTTCCGATCTNNNNACCCCGCTGAGTACAACC
Reverse: 5’-TGGAGTTCAGACGTGTGCTCTTCCGATCTNNNNGCAGAAGAGCCAGAGGAGAT
Cas9-VRQR nuclease purification
Cas9-VRQR nuclease was cloned into a pET42b plasmid with a 6xHis tag, eliminating the normal glutathione-s-transferase fusion. BL21 Star DE3 chemically competent E. coli cells (Invitrogen) were transformed with the plasmid and picked into 2xYT+25 μg/mL kanamycin for overnight growth at 37 °C. The next day, 1 L of pre-warmed 2xYT+25 μg/mL kanamycin was inoculated at OD600=0.03 and shaken at 37 °C for about 3 hours until OD600 reached 0.8. Culture was cold shocked in an ice-water slurry for one hour, following which protein expression was induced by the addition of 1 mM IPTG. Culture was shaken at 16 °C for 16 hours to express protein. Cells were pelleted at 6000 g for 20 minutes and stored at −80 °C. The next day, cells were resuspended in 30 mL cold lysis buffer (1 M NaCl, 100 mM Tris-HCl pH 7.0, 5 mM TCEP, 20% glycerol, with 3 tablets of cOmplete, EDTA-free protease inhibitor cocktail (Millipore Sigma, Cat. No. 4693132001). Cells were lysed by sonification at 4 °C for a total treatment of 7.5 minutes, providing time to cool after every 3 seconds of treatment. Cell lysate was clarified for 20 minutes using a 20,000 g centrifugation at 4 °C. Supernatant was collected and added to 1.5 mL of Ni-NTA resin slurry (G Bioscience, Cat. No. 786-940, prewashed once with lysis buffer). Protein-bound resin was washed twice with 12 mL of lysis buffer in a gravity column. Protein was eluted in 3 mL of elution buffer (200 mM imidazole, 500 mM NaCl, 100 mM Tris-HCl pH 7.0, 5 mM TCEP, 20% glycerol). Eluted protein was diluted in 40 mL of low-salt buffer (100 mM Tris-HCl, pH 7.0, 5 mM TCEP, 20% glycerol) just before loading into a 50-mL Akta Superloop for ion exchange purification on the Akta Pure25 FPLC. Ion exchange chromatography was conducted on a 5-mL GE Healthcare HiTrap SP HP pre-packed column. After washing the column with 15 mL low salt buffer, the diluted protein was flowed through the column to bind. The column was washed in 15 mL of low salt buffer before being subjected to an increasing gradient to a maximum of 80% high salt buffer (1 M NaCl, 100 mM Tris-HCl, pH 7.0, 5 mM TCEP, 20% glycerol) over the course of 50 mL, at a flow rate of 5 mL per minute. 1-mL fractions were collected during this ramp to high salt buffer. Peaks were assessed by SDS-PAGE to identify fractions containing the desired protein, which were pooled and concentrated using an Amicon Ultra 15-mL centrifugal filter (100 kDa cutoff). SDS-PAGE stained with InstantBlue (Expedion, SKU ISB1L) was used to visualize the purity after each step (Extended Data Figure 2c). Concentrated protein was quantified using a BCA assay (ThermoFisher, Cat. No. 23227); the final concentration was 68.6 μM.
CIRCLE-seq sample preparation and off target analysis
Genomic DNA was isolated from both HGADFN167 and HGADFN188 cell lines using a Gentra Puregene Tissue Kit (Qiagen). CIRCLE-seq was performed as previously described24. Cas9-VRQR was complexed with a synthetic sgRNA (Synthego) containing phosphorothioate linkage and 2’MeO modification at the first and last three nucleotides, and this complex was used to treat circularized DNA. PCR amplification before sequencing was conducted using PhusionU polymerase. PCR product was gel-purified and quantified by QuBit dsDNA high sensitivity assay (Invitrogen) before loading onto an Illumina MiSeq. Data was processed using the CIRCLE-Seq analysis pipeline (https://github.com/tsailabSJ/circleseq) with parameters: “read_threshold: 4; window_size: 3; mapq_threshold: 50; start_threshold: 1; gap_threshold: 3; mismatch_threshold: 6; merged_analysis: True”. The human reference genome GRCh37 was used for alignment. The twenty off-target genomic loci that yielded the greatest read counts for each sample were chosen for more detailed analysis. Loci tied for the highest read counts were included, and seven loci were shared in the top 20 list from each cell line, thus primers were designed to amplify 35 sites in total. Of these, PCR product was successfully obtained for 32 sites. These PCR products of edited and unedited cells were sequencing using an Illumina MiSeq. For successfully amplified loci, amplicons were aligned using Crispresso2 as with other amplicon sequencing performed in this manuscript, however, these samples were stringently quality filtered with a flag for q=30 to ensure SNP calling was only performed on high-quality reads. The resulting data is in Supplementary Data 4.
AAV production
AAV production was performed as previously described26. Briefly, HEK293T/17 cells were maintained in DMEM/10% FBS without antibiotic in 150mm dishes (Thermo Fisher 157150) and passaged every 2-3 days. Cells for production were split 1:3 and allowed to grow near 100% confluent before PEI transfection the following day. 5.7 μg AAV genome, 11.4 μg pHelper (Clontech), and 22.8 μg rep-cap plasmid were transfected per plate. 1 day after transfection, media was exchanged for DMEM/5% FBS. 3 days after transfection, cells were scraped with a rubber cell scraper (Corning), pelleted by centrifugation for 10 minutes at 2000g, resuspended in 500 μL hypertonic lysis buffer per plate (40 mM Tris base, 500 mM NaCl, 2 mM MgCl2 with 100 U/mL Salt Active Nuclease (Arcticzymes 70910-202)), and incubated at 37 °C for 1 h to lyse cells. Media was decanted, combined with a 5x solution of 40% PEG / 2.5 M NaCl (final concentration 8% PEG / 500 mM NaCl), incubated on ice for 2 hours to facilitate PEG precipitation, and centrifuged at >3000 g for 40 minutes. The supernatant was discarded and the pellet resuspended in 500 μL lysis buffer per plate and added to the cell lysate. Incubation at 37 °C was continued for 30 minutes. Crude lysates were either incubated at 4 °C overnight or directly used for ultracentrifugation.
Cell lysates were gently clarified by centrifugation at 2000 g for 10 minutes and added to Beckman Quick-seal tubes via 16 g ×5” disposable needles (Air-Tite N165). A discontinuous iodixanol gradient was formed by sequentially floating layers: 9 mL 15% iodixanol in 500 mM NaCl and 1× PBS-MK (1× PBS plus 1 mM MgCl2 and 2.5 mM KCl), 6 mL 25% iodixanol in 1× PBS-MK, and 5 mL each of 40% and 60% iodixanol in 1× PBS-MK. 1 μg/mL final Phenol red was added to the 15, 25, and 60% layers to facilitate identification. Ultracentrifugation was performed using a Ti 70 rotor at 58600rpm for 2:15 (h:mm) at 18°C. Following ultracentrifugation, roughly 4 mL of solution was withdrawn from the 40%-60% iodixanol interface via an 18-gauge needle, dialyzed with PBS containing 0.001% F-68, and ultrafiltered via 100kD MWCO columns. The concentrated viral solution was sterile-filtered using a 0.22-μm filter, quantified via qPCR (Clontech AAVpro Titration Kit v.2), and stored at 4 °C until use.
Timing of 6-month tissue and longevity analysis
The 6-month time point was chosen as a time when untreated homozygous LMNA c.1824 C>T mice typically show phenotypic decline but are not yet at the end of their median lifespan (7.0 months, females; 7.3 months, males).
In vivo sample high-throughput sequencing
Genomic DNA was isolated by standard protocol using Extraction, Tissue Preparation, and Neutralization Solution (Sigma-Aldrich) from 5- to 10-mg tissue samples. Isolated DNA was amplified as described for the genomic DNA samples using the same PCR1 and PCR2 primers. Libraries were prepared, diluted, and sequenced on an Illumina MiSeq as previously described26.
In order to guarantee that only reads belonging to the Human LMNA gene were included for downstream analysis first, UCHIME version 4.2 was used to remove PCR generated chimeras. Sequences were further filtered by removing reads containing at least two sequence motifs unique to mouse LMNA. Code is available at https://github.com/CwilsonBroad/Koblan_2020_In-Vivo-Adenine-Base-Editing-Corrects-Hutchinson-Gilford-Progeria-Syndrome. Resulting reads where aligned and quantified of editing frequency by CRISPResso2 in batch mode with a window width of 10 nucleotides.
RNA isolation and ddPCR from mouse tissues
Murine tissues were collected into Trizol reagent (ThermoFisher), homogenized and immediately flash-frozen until ready for total RNA isolation. RNA was subsequently digested for 20 minutes at 37 °C with recombinant DNase I (ThermoFisher), then analyzed for integrity and concentration on an Agilent nucleic acid bioanalyzer (Agilent Technologies). Synthesis of cDNA used 1 μg of RNA, which was reverse transcribed using the iScript cDNA Synthesis kit (BioRad) according to the manufacturer’s protocol. For each transcript assay, droplets were generated using 50 ng of cDNA, 900 nM primers, 250 nM probes in 1x ddPCR Supermix for Probes (BioRad) on a QX200 Droplet Generator, followed by PCR amplification. The sequence of primers and probes used are listed in the appended table. PCR cycling conditions consisted of an initial enzyme activation step for 10 minutes at 95 °C, followed by 40 cycles of 94 °C for 30 seconds and 59 °C for 30 seconds with a 2 °C/second ramp rate, and a 10 minute enzyme deactivation step at 98 °C for 10 minutes. Each reaction was duplexed with the Mouse Hprt PrimePCR Probe Assay (assay ID #qMmuCEP0054164, HEX, BioRad) and performed in triplicate. Upon completion of reactions, samples were read on a QX200 droplet reader (BioRad) to obtain expression levels relative to murine Hprt and transcript-specific copy number, then further analyzed using Excel software.
| Species / Transcript | Oligo Name | Primer / Probe Sequence |
|---|---|---|
| human LMNA | hLMNA-F | CCCAGGTGGGCGGAC |
| hLMNA-R | AGGAGCGGGTGACCAGATT | |
| hLMNA-FAM | 56-FAM-CAGCTACCGCAGTGTGGGGG-IABkFQ | |
| human Progerin | hPROG-F | CTGTGCGGGACCTGCG |
| hPROG-R | AAGCCTCCACCCCCACC | |
| hPROG-FAM | 56-FAM-AGGAGCCCAAGCCCCCAGAACT-IABkFQ | |
| human LMNC | hLMNC-F | GTGGAAGGCACAGAACACCT |
| hLMNC-R | CATTCTTTAATGAAAAGATTTTTGG | |
| hLMNC-FAM | 56-FAM-CAGTGACTGTGGTTGAGGACGACG-IABkFQ | |
Protein isolation and western blotting from mouse tissues
After sacrifice, tissues from treated control or ABE-treated mice were flash frozen and stored in liquid nitrogen until the time of protein extraction. To extract proteins, 10-30 mg of frozen tissue was first pulverized in temperature resistant tubes (Covaris, tissueTube #520001) on a liquid nitrogen bath. This frozen tissue powder was resuspended in RIPA lysis buffer (see above), moved to a 2-mL collection tube, and homogenized for 30 seconds at 25 Hz with a 5-mm stainless steel bead in a TissueLyser II (Qiagen). Samples were then centrifuged for 5 seconds on table-top microcentrifuge and incubated on ice for 45 minutes while rocking. After incubation, lysates were clarified by centrifugation at 21,000 g for 15 minutes at 4 °C. The supernatant was transferred to a fresh tube and protein quantified using standard BCA assay. Tissue lysates were prepared identically to the cell line isolated samples and separated by SDS-PAGE (25 μg protein loaded for heart/liver; 10 μg for skeletal muscle; 7.5 μg for aorta, 7.5 μg for cells) on 4-12% Bis-Tris gels (Nupage, Invitrogen). Tissue western blots were performed using the same method as cell-line western blots.
Tissue histology
Tissues were fixed in 2% paraformaldehyde for 24 hours prior to dehydration with graded alcohols and embedding in paraffin. Cross-sections (4-μm thick) were cut and mounted on charged slides and visualized by haematoxylin and eosin staining. For aorta and skin sections, additional staining was performed with Movat’s pentachrome (CVPath Inc.) or Masson’s trichrome (Histoserv Inc.), respectively. Images were captured on an Axioscan imaging system (Zeiss) at 20x magnification and processed using ZenBlue 2.0 software. Images were further processed for VSMC counts and adventitial area assessment using Photoshop CC software (21.2.3).
Immunofluorescence histochemistry
Immunohistostaining was performed following the protocol previously described7 with modifications, and by using mouse monoclonal anti-lamin A/C (MABT538, clone 2E8.2, Millipore Sigma; 1:75 dilution) antibody or rabbit polyclonal anti-progerin antibody (Collins, custom; 1:75 dilution). Briefly, ascending aorta sections were dewaxed and rehydrated, and the antigens were retrieved by heating in EDTA buffer (1 mM, pH 8.0) for 2 min in a pressure cooker. Tissue sections were blocked in TBS buffer containing 10% donkey serum and 1% BSA, and then incubated with a Mouse-on-Mouse blocking reagent (Vector Laboratories) to reduce endogenous mouse antibody binding. Slides were incubated with the above primary antibody overnight at 4 °C. After washing thoroughly in TBS, the sections were then incubated with donkey anti-mouse Alexa Fluor 594-conjugated or donkey anti-rabbit Alexa Fluor 488-conjugated secondary antibodies (ThermoFisher Scientific; 1:3000 dilutions). All tissue sections were mounted in DAPI-containing medium (Vector Laboratories). Fluorescence images were captured by a confocal microscope system (Zeiss LMS 880) with 40x water lens.
Whole-genome sequencing of isolated tumor samples
Tumors were isolated from mouse livers following standard necropsy of deceased animals. Following tissue isolation, genomic DNA was isolated using the QIAamp DNA Mini Kit (Qiagen) according to the manufacturer’s recommendations. The resulting isolated genomic DNA was sheared to a mean size of 300 bps using a Covaris S220 sonicator (Covaris, USA). An Illumina sequencing library was prepared from the sheared DNA using the Apollo 324 automated liquid handler (WaferGen, USA) and the PrepX DNA library kit (Takara Bio, USA). This step included DNA end repairing, A-tailing, adaptor ligation, barcoding and 5 cycles of PCR amplification. After examination on a TapeStation 4200 system (Agilent, USA) with a high-sensitivity DNA 1000 ScreenTape (Agilent, USA) for size distribution, and library concentration quantification by Qubit fluorometer (Invitrogen, CA), the resulting libraries were pooled and sequenced on an Illumina Novaseq using an S4 flow cell as paired-end 150 bp reads, along with 6-bp index reads, following the manufacturer’s protocol (Illumina, USA).
Quality control of sequence reads
We assessed sequence quality of the paired-end reads with FastQC (v0.10.0, http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) for each of 16 whole-genome samples (WGS). Sequencing of a second tumor sample from mouse 9550 failed, and the sample was excluded from analysis. Singleton reads were not included in the FastQC analysis, and they were excluded from alignment (see below) and downstream variant calling. We used MultiQC50 (v1.8) to summarize the FastQC results. No outlier sample was detected in the evaluation of mean base quality and mapping quality across the samples.
Sequence alignment and variant calling
We assembled a comprehensive reference genome including following components: 1) the mouse genome GRCm38; 2) AAV9 C terminal contig; 3) AAV9 N terminal contig; 3) cloning vector pBACe3.6 (GenBank U80929.2)51; and RP11-702H12, a human LMNA BAC sequence present in the transgenic mouse model52. Each of the four non-mouse components was integrated in the reference genome as a separate contig. The alignment program ‘bwa mem’53 (v0.7.17) was used to align the sequence reads to the combined reference genome with “-M” option, and remaining parameters are set to default. On average, 1,542,162,680 sequence reads (>=548,801,830) per sample were generated, of which 99.58% mapped to the genome as primary alignments. For sequencing statistics, see the second tab of Supplementary Data 1.
Identification of AAV9 integration in mouse genomes
We implemented a two-step process to identify AAV9 integration sites in mice genomes. First, we used “samtools view” (v0.1.18)54 to extract any reads that map to AAV9 C- or N-terminal contigs. We excluded following reads: 1) mapping quality score < 30; 2) failed Illumina platform/vendor quality check; or 3) duplicates. Next, we anchored the reads mapped to AAV9 contigs and searched the ones whose paired read mapped to the mouse genome. These read pairs represented sequencing fragments that cover potential integration sites of AAV9 contigs and mouse chromosomes.
We annotated the mouse genomic DNA reads identified around the integration sites with genes defined in gencode vM24 (https://www.gencodegenes.org/pages/gencode.html) using “bedtools intersect”55. In order to capture integration sites that were near, but not within, a gene body, we extended both ends of each mouse gene by 2 kb of flanking sequence for the purposes of this annotation. These steps resulted in a list of AAV9 contigs that are integrated into the mouse genome. The resulting data is in Supplementary Data 1.
SNV calling from WGS data
Aligned reads were mapped to the reference genome as described above. Duplicates were removed using sambamba56 and GATK was applied57. Base quality score recalibration, indel realignment, and SNP and indel discovery and genotyping of all 15 samples were performed simultaneously using standard hard filtering parameters according to GATK Best Practices recommendations58,59. SNP and indels were annotated using ANNOVAR60. In order to compare the fraction of all SNVs that are A•T-to-G•C between different tissue samples, generalized linear models were fitted and FDR adjusted p-values were reported on three datasets: 1) three independent liver tumor samples and two independent saline-injected samples; 2) four independent normal liver samples and two saline-injected samples and 3) seven liver tumor samples and six normal liver samples from four independent mice with mouse identity as a covariate. The resulting data is summarized in Supplementary Data 2, with the full dataset available in Supplementary Data 5.
Analysis of liver cancer-associated genomic loci
Cancer-associated single nucleotide variants (SNVs) and insertion/deletions were obtained from the Cosmic Cancer Mutation Census (CMC; https://cancer.sanger.ac.uk/cmc/home)61. This list was first filtered to identify recurrent or cancer associated mutations. These were defined by matching at least one of the following three criteria:
Having >1% prevalence in at least one tumor type as measured by whole genome sequencing (WGS)
Scoring as likely pathogenic or pathogenic based on the ClinVar clinical significance criteria
Having a dn/ds diseases score with a significant q-value (q-value < 0.05) in at least one tumor type as defined by COSMIC analysis of TCGA whole exome data
These criteria resulted in 19,986 high-confidence human cancer-associated mutations spanning 6,614 genes and 171 liver cancer-associated mutations spanning 84 genes. High-confidence human liver cancer-associated mutations included those affecting many well-known genes implicated in liver cancer such as HNF1A, CTNNB1 (β-catenin), and IL6ST (Interleukin 6 Signal Transducer). Note that this analysis did not include copy number or structural variants.
Whole-genome sequencing GATK ANNOVAR analysis output from mouse liver and tumor samples (see above) was first filtered for variants not found in both saline-treated mouse livers, and then further filtered to only include insertion/deletions and events associated with adenine base editing (A>G and T>C mutations).
These mutations were then classified based on the following:
Falling within the coding region or affecting the splicing of a gene with known high confidence cancer associated mutations (denoted as CODING).
Mutations not in the first category that reside in an active cis-regulatory region within 100 kb of a gene with known high-confidence cancer-associated mutations (denoted as REGULATORY). Active cis-regulatory regions were defined using ATAC-seq (a measure of open and active regulatory chromatin)62 in postnatal mouse livers from a processed dataset obtained from the Mouse ENCODE project (ENCFF168WUC – IDR threshold narrow peak bed output from replicate analysis in the experiment ENCSR6090HJ). This definition resulted in 24,758 ATAC-seq defined mouse liver cis-regulatory regions within 100 kb of high-confidence cancer-associated genes and 526 cis-regulatory regions associated with liver cancer-specific genes.
The resulting data is in Supplementary Data 3.
RAB25 transcript detection
RNA was isolated from liver samples as described above and reverse transcribed using the iScript cDNA Synthesis kit (BioRad) according to the manufacturer’s protocol. PCR was carried out using gene-specific primers for mouse Gapdh (Biorad; 10031231), mouse Actb (Biorad; 10031237), and human RAB25 (Biorad; 10031234) with the iQ multiplex powermix (Biorad). PCR products were analyzed by gel electrophoresis on a 2% agarose gel.
Mice
Mice were housed in barrier facilities with a 12-h light/dark cycle at both National Institutes of Health (NIH) and Vanderbilt University Medical Center (VUMC). Genotyping was performed using standard PCR methods with primers: TTGGACCAAACAAGTACATATCA (Common Forward); CCAATGATAGTGACAGGTATACGG (Wild Type Reverse); CTGACATTCTAGTGGAGGGAGA (Mutant Reverse). Body weights were measured and recorded during health observation twice per week. Mice were injected with split AAV constructs at postnatal day 3 (P3, retro-orbital, 1011 viral genomes total in 10 microliter total volume) or postnatal day 14 (P14, retro-orbital and intraperitoneal, 1012 viral genomes total in 100 microliter total volume). All retro-orbital injections were performed at NIH, while intraperitoneal injections were done at VUMC. Full pathological examination included the ascending aorta, descending aorta, carotid artery, abdominal aorta, external ear, femur, skin, liver, spleen, kidney, heart, skeletal muscle, visceral adipose tissue and subcutaneous adipose tissue. One cohort of P3 and P14 animals were euthanized at 6 weeks of age and individual tissues harvested for DNA sequencing analysis. Another cohort of P3 and P14 injected mice were euthanized at 6 months of age and individual tissues harvested for DNA, RNA, protein and histological analysis. A separate cohort was followed for longevity. All animal use complied with the Animal Care and Use Committee guidelines under protocol G-03–05 (NIH) and M1800126 (VUMC).
A linear regression of the t-distribution of longevity history of the homozygous mice in the colony with mouse sex as a covariate using 12 male/female treated mice vs 12 male/female control mice suggests a significant difference in longevity at 42.0 days with 80% power, or 48.6 days with 90% power. The Mantel-Cox test was used for longevity statistics.
Extended Data
Extended Data Figure 1. Additional characterization of progeria patient-derived cells treated with ABE7.10max-VRQR.
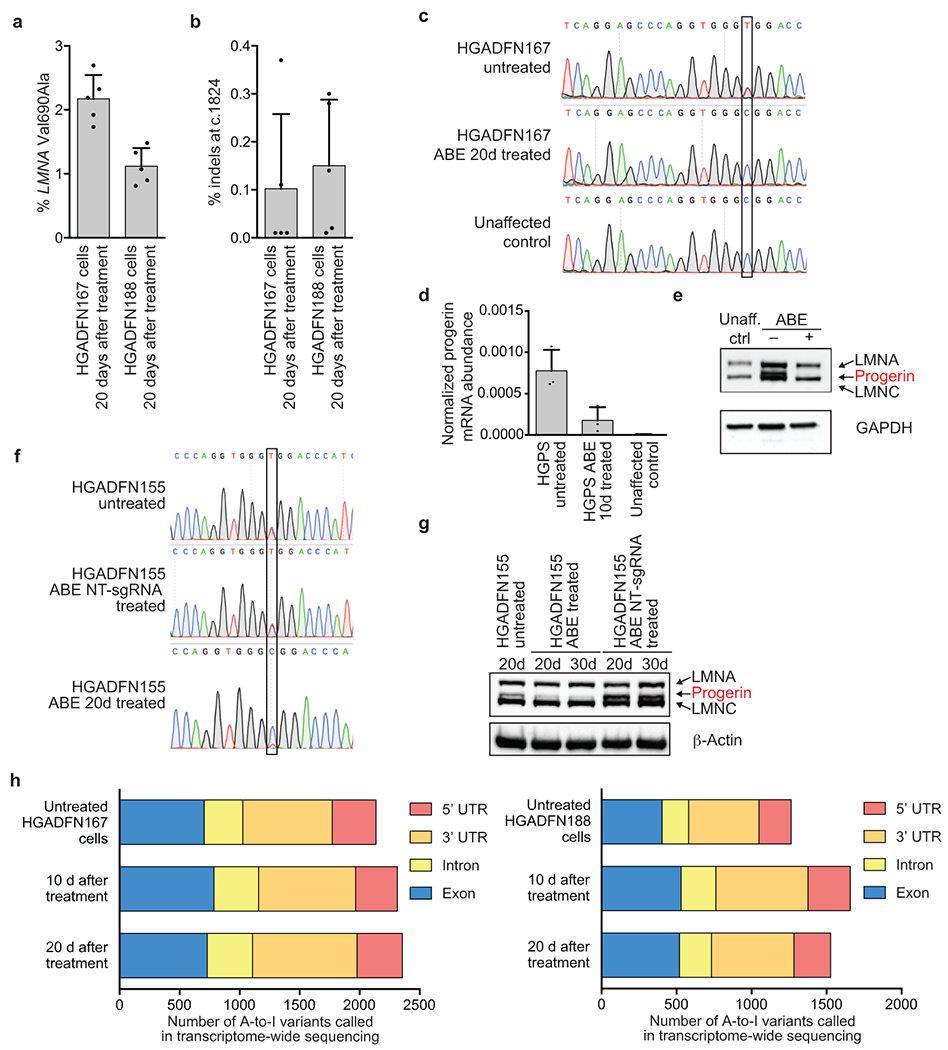
(a) Bystander Val690Ala editing in progeria patient-derived HGADFN167 and HGADFN188 cells 20 days after treatment with lentiviral ABE7.10max-VRQR. (b) Indel formation frequency at the c.1824 target locus in HGADFN167 and HGADFN188 cells 20 days after treatment with lentiviral ABE7.10max-VRQR. Values and error bars represent mean±SD for n=5 technical replicates (individual points) for (a) and (b). (c) Sanger DNA sequencing traces of untreated HGADFN167 cells, 20-day treated HGADFN167 cells, and unaffected control cells. The target nucleotide is boxed. (d) qPCR-normalized progerin mRNA abundance in cells described in c. Values and error bars represent mean±SD for n=3 biological replicates. (e) Western blot analysis of HGADFN167 cells described in c. LMNA, progerin, and LMNC protein are all stained on the gel, A GAPDH loading control is shown below. An additional replicate is provided in Fig. 1d. Unaff. ctrl, control cells from an unaffected parent. (f) Sanger DNA sequencing traces of untreated, non-targeting (NT) sgRNA treated, and ABE treated HGADFN155 fibroblasts at 20d and 30d time-points to ensure NT-sgRNA did not lead to DNA editing. (g) Western blot analysis of cells described in f. LMNA, progerin, and LMNC proteins are all stained on the gel, A β-actin loading control is shown below. Expected molecular weights: lamin A, 74 kDa; progerin, 69 kDa; lamin C, 65 kDa. Complete blots are available in Supplementary Figure 1. Additional replication was not performed. (h) HGADFN167 (left) and HGADFN188 (right) cell lines untreated or treated with lentiviral ABE7.10max-VRQR after 10 or 20 days show similar relative distributions of A-to-I single-nucleotide variants (SNVs) in their transcriptomes compared with the hg38 human genome reference sequence. On average, 36±3.6% of SNPs in these samples occur with ~100% frequency, suggesting they arise from genomic sequence variations; however, we cannot explicitly exclude them from consideration since no whole genome sequence is available for these cell lines. Raw counts of 100% edited SNPs per sample are: Untreated HGADFN167 cells (849), HGADFN167 10 d after treatment (883), HGADFN167 20 d after treatment (871), Untreated HGADFN188 cells (488), HGADFN188 10 d after treatment (501), HGADFN188 20 d after treatment (510).
Extended Data Figure 2. CIRCLE-seq analysis of HGADFN167 and HGADFN188 cells using Cas9-VRQR and the progeria-targeting sgRNA.
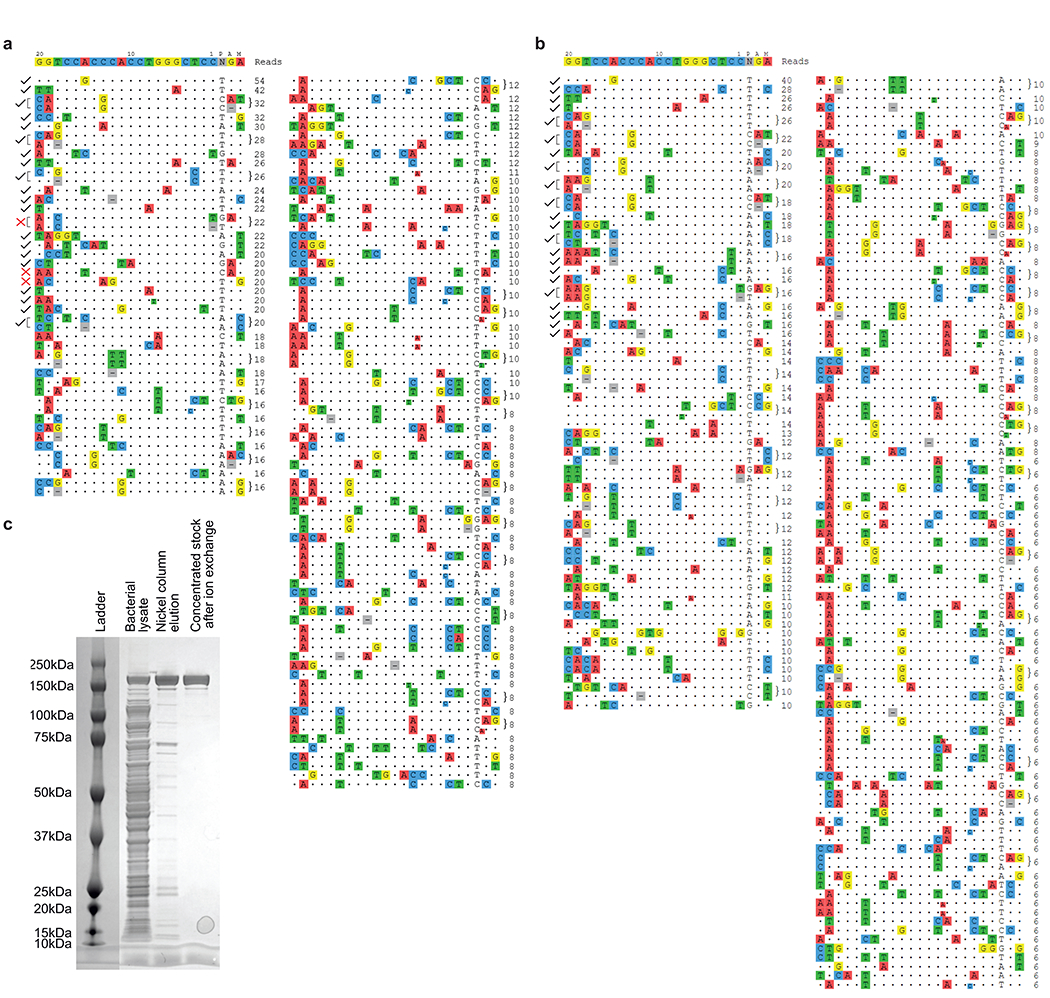
CIRCLE-seq read counts for Cas9-VRQR nuclease-treated genomic DNA from HGADFN167 (a) and HGADFN188 (b) cell lines. Targeted amplicon sequencing was used to assess the off-target base editing for 36 noted total loci distributed across both cell lines. DNA at 32 of 35 loci amplified efficiently from both cell lines (denoted by black check marks), DNA at 3 loci failed to amplify (denoted by red X marks). Complete CIRCLE-seq data is provided in Supplementary Data 4. (c) SDS-PAGE gel stained with InstantBlue to follow protein purification of Cas9-VRQR. 0.5 μL of clarified lysate, 0.25 μL of nickel column elution, or 0.1 μL of the concentrated protein stock following His-tag purification and ion exchange chromatography were added to 5 μL of NuPAGE loading buffer. Samples were denatured at 98 °C for 5 minutes before loading onto the 4-12% acrylamide gel. Precision Plus Protein Kaleidoscope pre-stained Ladder (Bio-Rad) was used as reference. The desired Cas9-VRQR has a predicted molecular weight of 161.9 kDa. Additional replication was not performed.
Extended Data Figure 3. DNA on-target editing, bystander editing, and indel efficiencies across tissues from in vivo injection route optimization experiments.
(a) Dual AAV9 encoding split-intein ABE7.10max-VRQR base editor halves and the LMNA-targeting sgRNA were injected into homozygous human LMNA c.1824 C>T mice. P3 retro-orbital (RO) injections (5×1010 of each AAV vg, 1×1011 vg total), P14 RO injections (5×1011 of each AAV vg, 1×1012 vg total), and P14 intraperitoneal (IP) injections (5×1011 of each AAV vg, 1×1012 vg total) were tested. At 6-weeks of age, mice were harvested and heart, muscle, liver, aorta, and bone were isolated for sequencing analysis. Tissues were sub-sectioned for sequencing analysis to ensure sub-sections did not show differences in editing efficiencies for downstream analyses. Each bar represents a different tissue subsection. DNA editing efficiencies correcting LMNA c.1824 from T (pathogenic) to C (wild-type) for P3 RO-injected mice (left, n=4), P14 RO-injected mice (middle, n=5), and P14 RO-injected mice (right, n=5) at 6 weeks of age are shown for five disease-relevant tissues. Values and error bars represent mean±SD. (b) Apparent LMNA c.1824 T (pathogenic) to C (wild-type) mutations from tissue samples of saline-injected P3 RO (left) and P14 RO (right) control mice at 6 months of age show background signal due to amplicon crossover during PCR between the human diseased allele and the wild-type mouse allele, which share 90% overall sequence identity within the amplified region. Similar crossover levels were observed across 11 tissues in both P3 RO and P14 RO saline-injected mice. Values and error bars represent mean±SD for n=12 mice (6 male, 6 female). (c) Computational filtering of same sequencing reads shown in (b) after removing any reads containing any mouse-specific sequence variations, analyzing only reads containing exclusively human sequence. The script used to remove mouse-containing sequencing reads is in Supplementary Note 3 and is described in the methods. WAT white adipose tissue. (d) DNA editing for P3- and P14-injected mice at 6 months of age across 11 tissues. Each point represents a biological replicate of a tissue harvested from a unique mouse (n=12 for each group). (e) Val690➔Ala bystander editing frequency across eleven tissues for P3 RO and P14 RO ABE-treated mice at 6 months of age (n=12 for each group). (f) Indel frequencies at the c.1824 target locus across 11 tissues for P3 RO and P14 RO ABE-treated mice at 6 months of age. Values and error bars represent mean±SD for the indicated number of biological replicates. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 by Student’s unpaired two-sided t-test.
Extended Data Figure 4. Quantification of LMNA and progerin transcript abundance by ddPCR in P3- and P14-injected mice.
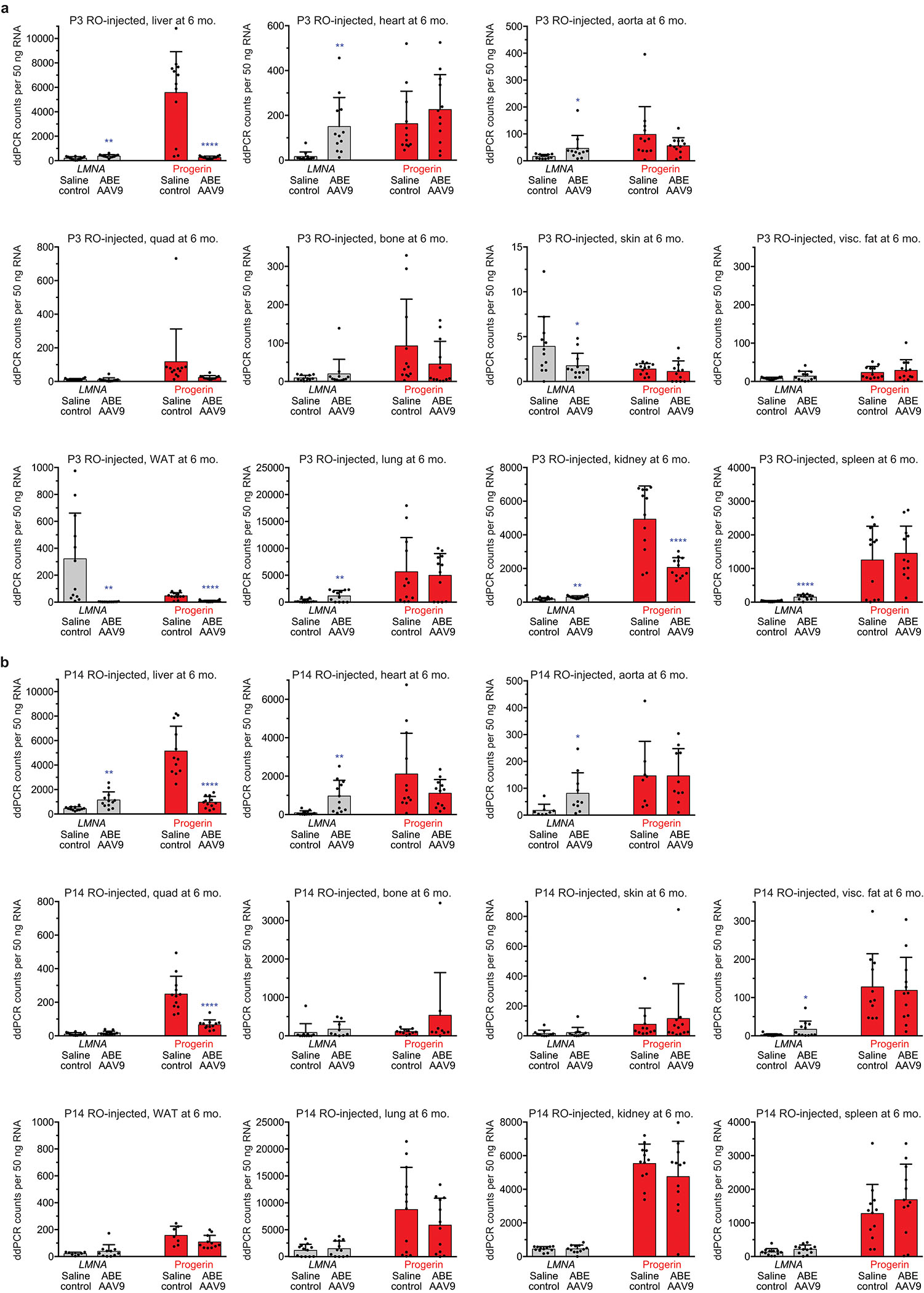
(a) ddPCR counts for LMNA (grey bars) and progerin (red bars) RNA transcript abundance in P3 RO saline- and ABE-AAV9-injected mice. Values and error bars represent mean±SD for n=12 mice. (b) ddPCR counts for LMNA (grey bars) and progerin (red bars) RNA transcript abundance in P14 RO saline- and ABE-AAV9-injected mice. Values and error bars represent mean±SD for n=12 biological replicates for all samples except for saline-injected mouse skin (n=11), WAT (n=7), visceral fat (n=11), tibia (n=11), aorta (n=8); and ABE-AAV9-injected mouse WAT (n=11), tibia (n=9), and aorta (n=10). WAT white adipose tissue. Visc. fat, Visceral fat. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 by Student’s unpaired two-sided t-test for (a) and (b). Liver and heart values are reproduced from Fig. 3c for ease of comparison.
Extended Data Figure 5. Quantification of western blots.
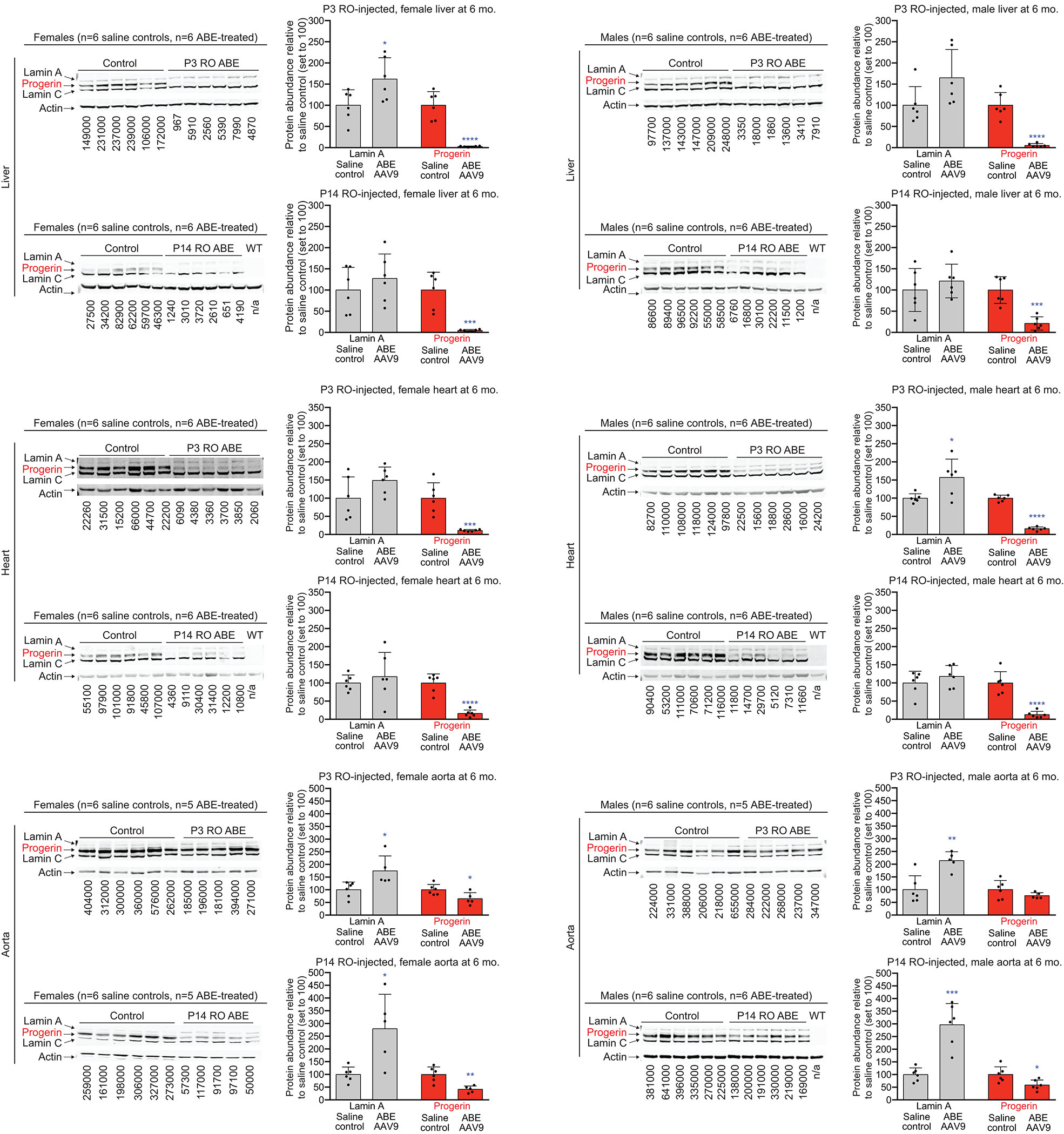
Liver, heart, and aorta tissue western blots for P3-injected (top half of each tissue set) and P14-injected mice (bottom half of each tissue set) were quantified by western blot. Samples from females appear in the left column, and samples from males are in the right column. Each lane represents the tissue type specified on the left taken from a different mouse. Control mice were treated with saline instead of ABE-AAV9. WT indicates C57BL/6 mouse lacking the transgene, showing that the antibody is specific to human lamin proteins and progerin. The abundance of lamin A or progerin protein relative to β-actin in saline- or ABE-treated mouse tissues was quantified by normalizing the fluorescence signal from the secondary antibody for each band (800 nm for progerin and lamins, and 680 nm for actin; see Methods). The normalized protein abundance relative to saline-treated samples (set to 100) is shown in the bar graphs. Control mice were treated identically to the corresponding ABE-treated mice except injected with saline instead of ABE-AAV9. Raw fluorescent signal for progerin protein measured at the 800nm wavelength (using licor,IRDye labeled antibody) displayed under each lane. Values and error bars represent mean±SD for n=5 or 6 biological replicates, as indicated. The n for each sample type is listed in each figure panel. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 by Student’s unpaired two-sided t-test. Expected molecular weights: lamin A, 74 kDa; progerin, 69 kDa; lamin C, 65 kDa. Liver and heart blots are reproduced from Fig. 3c for ease of comparison. Complete blots are available in Supplementary Figure 1.
Extended Data Figure 6. P3 RO saline- and ABE AAV9-injected male and female mouse aortic histology assessed by H&E and Movat’s staining.
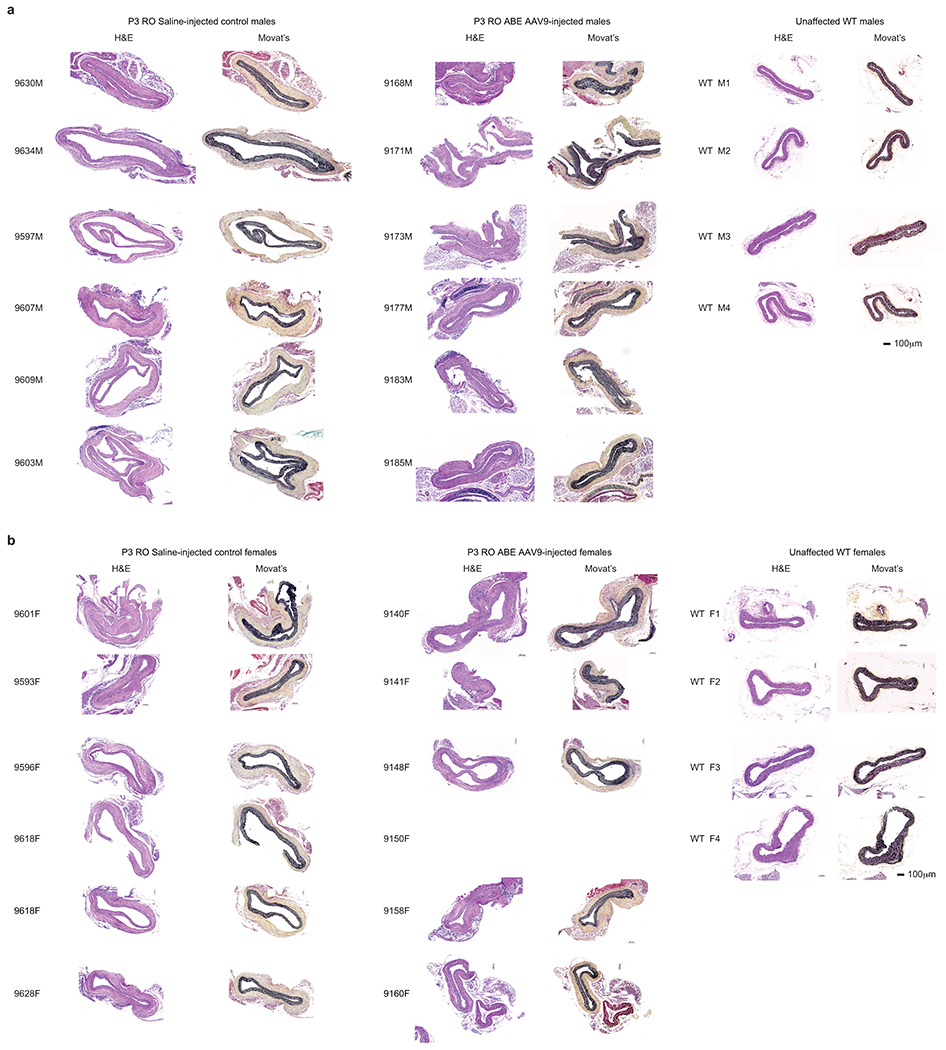
(a) Representative aorta cross-sections for P3 RO saline- or ABE-AAV9-injected males at 6 months of age. Left images were stained with hematoxylin and eosin (H&E); right images were stained with Movat’s stain. (b) Representative aorta cross-sections for P3 RO saline- or ABE-AAV9-treated females at 6 months of age. Left images were stained with hematoxylin and eosin (H&E); right images were stained with Movat’s pentachrome. Unaffected WT are wild-type C57BL/6 mice. WT M2, 9609M, 9177M, WT F3, 9628F, and 9148F are replicated from the main figure for ease of comparison. These sections each represent replicates from different mice.
Extended Data Figure 7. P14 RO saline- and ABE AAV9-injected male and female mouse aortic histology assessed by H&E and Movat’s staining.
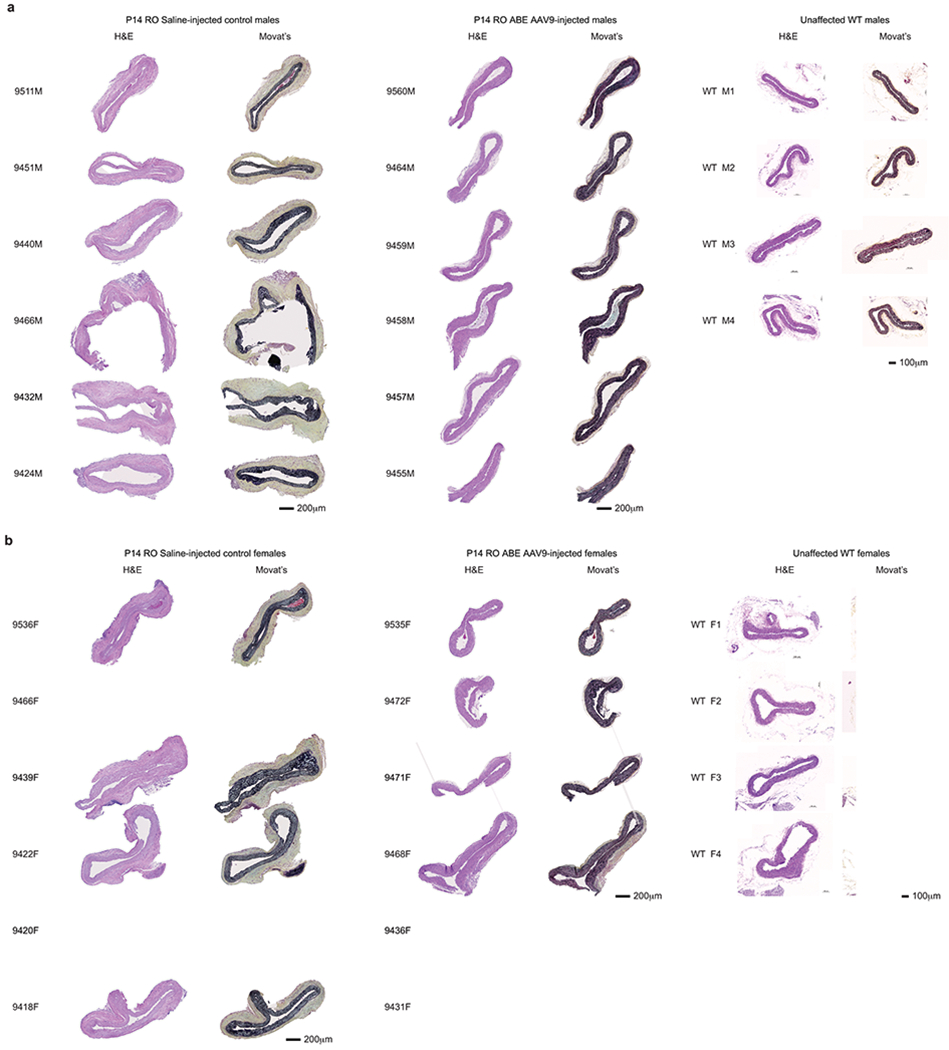
(a) Representative aorta cross-sections for P14 RO saline- or ABE-AAV9-injected males at 6 months of age. Left images were stained with hematoxylin and eosin (H&E); right images were stained with Movat’s stain. (b) Representative aorta cross-sections for P14 RO saline- or ABE-AAV9-treated females at 6 months of age. Left images were stained with hematoxylin and eosin (H&E); right images were stained with Movat’s pentachrome. Unaffected WT are wild-type C57BL/6 mice, reproduced from Extended Data Fig. 9a,b for ease of comparison. Images from the following mice are reproduced from Fig. 4 for ease of comparison: WT M2, 9440M, 9459M, WT F3, 9536F, and 9535F. These sections each represent replicates from different mice.
Extended Data Figure 8. C57BL/6 control, untreated homozygous human LMNA c.1824 C>T mice, P14 RO saline- and ABE AAV9-injected male and female aorta immunofluorescence staining.
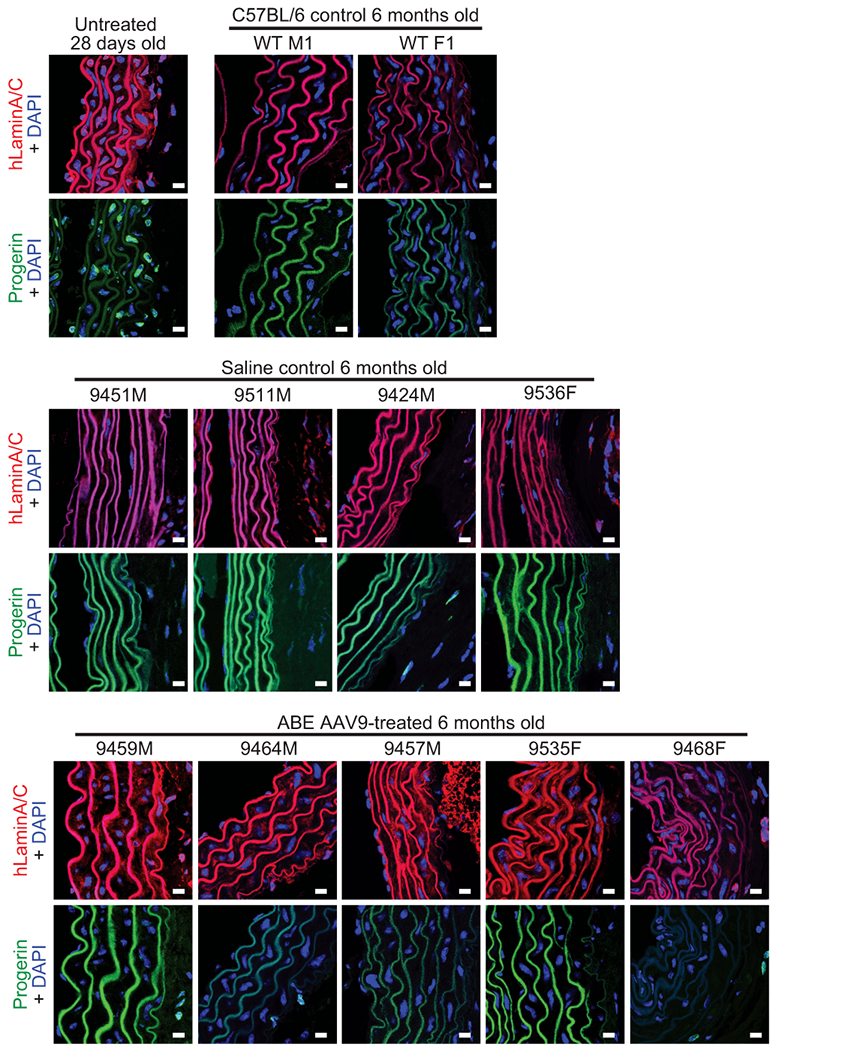
Immunofluorescence staining of C57BL/6 (n=2), untreated P28 homozygous human LMNA c.1824 C>T (n=1), saline-treated homozygous human LMNA c.1824 C>T (n=4), and ABE-treated homozygous human LMNA c.1824 C>T (n=5) mouse aortas stained for human lamin A/C + DAPI or for progerin + DAPI. Scale bar=10 μm. Images from untreated 28 day-old, WT M1, 9424M, and 9464M are replicated from Fig. 4 for ease of comparison.
Extended Data Figure 9. P3 RO saline- and ABE AAV9-injected male and female mouse skin histology assessed by H&E and Masson staining.

(a) Representative skin cross-sections for P3 RO saline-injected (left), ABE-AAV9-injected (middle), and wild-type C57BL/6 males at 6 months of age. Left images were stained with hematoxylin and eosin (H&E); right images were stained with Masson stain. (b) Representative skin cross-sections for P3 RO saline-injected (left), ABE-AAV9-injected (middle), and wild-type C57BL/6 females at 6 months of age. Left images were stained with hematoxylin and eosin (H&E); right images were stained with Masson stain. Unaffected WT are wild-type C57BL/6 mice. These sections each represent replicates from different mice.
Extended Data Figure 10. P14 RO saline- and ABE AAV9-injected male and female skin histology assessed by H&E and Masson staining.
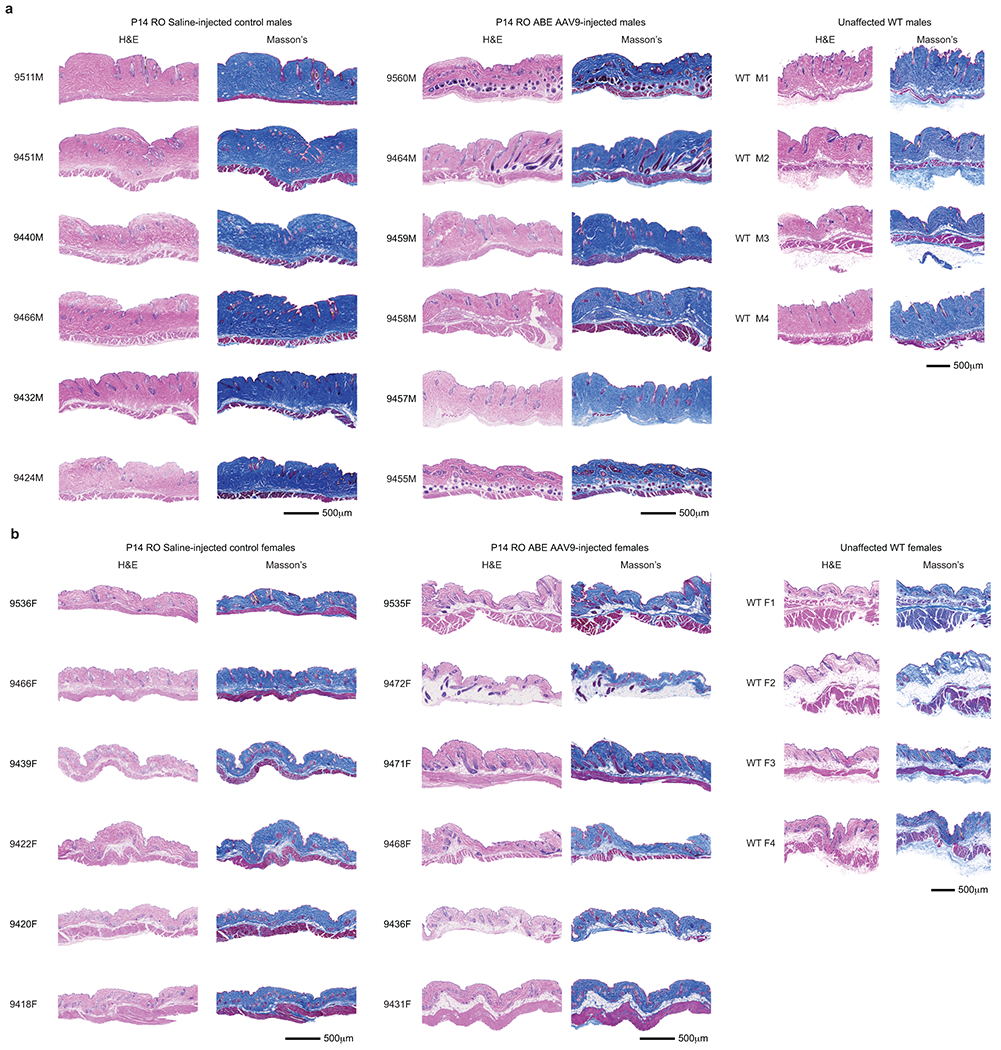
(a) Representative skin cross-sections for P14 RO saline-injected (left), ABE-AAV9-treated (middle), and wild-type C57BL/6 males at 6 months of age. Left images were stained with hematoxylin and eosin (H&E); right images were stained with Masson stain. (b) Representative skin cross-sections for P14 RO saline-injected (left), ABE-AAV9-treated (middle), and wild-type C57BL/6 females at 6 months of age. Left images were stained with hematoxylin and eosin (H&E); right images were stained with Masson stain. Unaffected WT are wild-type C57BL/6 mice, reproduced from Extended Data Figure 12a,b for ease of comparison. These sections each represent replicates from different mice.
Extended Data Figure 11. P3 RO and P14 RO saline- and ABE-AAV9-injected mouse weights.
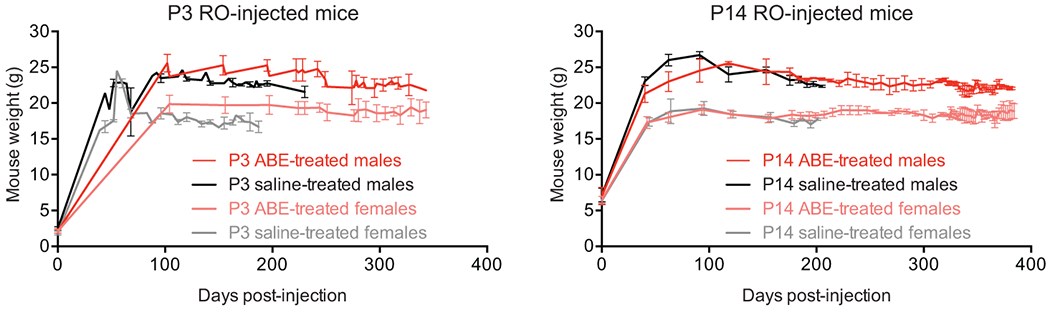
Weights of homozygous human LMNA c.1824 C>T mice taken across animal lifespans for cohorts of P3 RO (left) and P14 RO (right) saline- and ABE-AAV9-injected cohorts. Mouse weights are shown by gender. The X-axis shows days post-injection, rather than age. Values and error bars represent mean±SD for the number of surviving mice at each time point, complete data can be accessed in Supplementary Data 5.
Extended Data Figure 12. Whole-genome sequencing analysis of single-nucleotide variants and indels from mouse tissue samples, and quantification of RAB25 transcript levels in mouse tissue samples.
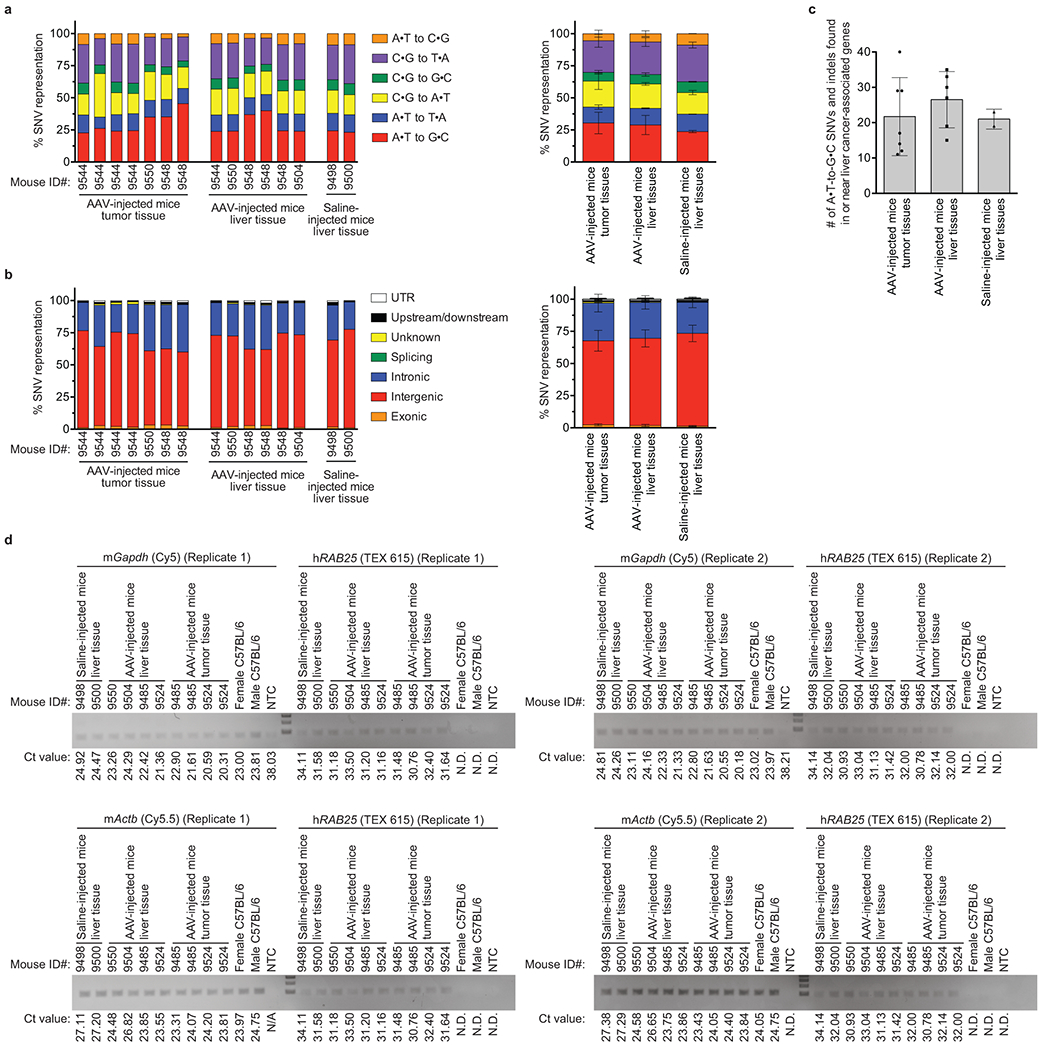
(a) Distribution of all possible single-nucleotide variant (SNV) types in non-tumor liver tissue and liver tumor tissue samples isolated from ABE AAV9-injected and saline-injected mice. Values from individual tissue samples are shown on the left. Aggregated values from all AAV-injected mouse tumor tissue samples, all AAV-injected mouse liver tissue samples, and all saline-injected mouse liver tissue samples are shown on the right, where values represent the mean of each sample type and error bars reflect the standard deviation with each tissue section treated as a different sample: AAV-injected tumor tissue (n=7), AAV-injected liver tissue (n=6), and saline-injected liver tissue (n=2). (b) Genomic classification of A•T-to-G•C SNVs. Values from individual tissue samples are shown on the left. Aggregated values from AAV-injected mouse tumor tissue samples, AAV-injected mouse liver tissue samples, and saline-injected mouse liver tissue samples from all tissue types are shown on the right, where values represent the mean of each sample type and error bars reflect the standard deviation with each tissue section treated as a different sample: AAV-injected tumor tissue (n=7), AAV-injected liver tissue (n=6), saline-injected liver tissue (n=2). (c) A•T-to-G•C SNVs and indels found in or near genes that are recurrently mutated in human liver cancers62,63 including introns, exons, and at ATAC-seq-defined cis-regulatory regions within 100-kb of each gene’s transcription start site, in AAV-injected mouse tumor tissue samples, AAV-injected mouse liver tissue samples, and saline-injected mouse liver tissue samples. Values represent the mean of individual tissue samples and error bars represent standard deviation. Individual data points are shown for each sample. The complete list of SNVs from ANNOVAR analysis is provided as Supplementary Data 6. Summary statistics for SNV calls are in Supplementary Data 2. (d) RNA isolated from mouse liver tissue samples was reverse transcribed and amplified with primer sets specific to mouse Gapdh (detected with Cy5), mouse Actb (detected with Cy5.5), and human RAB25 (detected with TEX 615). Ct values were determined by quantitative PCR and are shown below each lane. N.D. = not detected.
Supplementary Material
Acknowledgements
This work was supported by U.S. NIH U01AI142756, UG3AI150551, UG3TR002636, RM1HG009490, R01EB022376, R35GM118062, Z01HG200305, R01HL146654, R01HL126784, and HHMI. L.W.K. acknowledges an NSF GRFP fellowship. C.W. acknowledges a Damon Runyon Cancer Research Foundation fellowship (DRG-2343-18). G.A.N. acknowledges a Helen Hay Whitney postdoctoral fellowship. L.B.G., L.W.K. and D.R.L. acknowledge Progeria Research Foundation support.
Footnotes
The authors declare competing financial interests: D.R.L. is a co-founder of Beam Therapeutics, Prime Medicine, Pairwise Plants, and Editas Medicine, companies that use genome editing.
Online Content
Methods, along with any additional Extended Data display items, are available in the online version of the paper; references unique to these sections appear only in the online paper.
Data Availability
DNA sequencing files can be accessed using PRJNA627465. Raw data is available in Supplementary Data 1, 2, 3, 5, and 6.
Supplementary Information is available in the online version of the paper.
References
- 1.Eriksson M et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 423, 293–298, doi: 10.1038/nature01629 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Sandre-Giovannoli A et al. Lamin a truncation in Hutchinson-Gilford progeria. Science 300, 2055, doi: 10.1126/science.1084125 (2003). [DOI] [PubMed] [Google Scholar]
- 3.Gordon LB et al. Impact of farnesylation inhibitors on survival in Hutchinson-Gilford progeria syndrome. Circulation 130, 27–34, doi: 10.1161/CIRCULATIONAHA.113.008285 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gordon LB, Brown WT & Collins FS Hutchinson-Gilford progeria syndrome. GeneReviews (2019). [Google Scholar]
- 5.Gaudelli NM et al. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 551, 464–471, doi: 10.1038/nature24644 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anzalone AV, Koblan LW, and Liu DR . Genome Editing with CRISPR-Cas Nucleases, Base Editors, Transposases, and Prime Editors Nature Biotechnology 38, 824–844 (2020). [DOI] [PubMed] [Google Scholar]
- 7.Olive M et al. Cardiovascular pathology in Hutchinson-Gilford progeria: correlation with the vascular pathology of aging. Arterioscler Thromb Vasc Biol 30, 2301–2309, doi: 10.1161/ATVBAHA.110.209460 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gerhard-Herman M et al. Mechanisms of premature vascular aging in children with Hutchinson-Gilford progeria syndrome. Hypertension 59, 92–97, doi: 10.1161/HYPERTENSIONAHA.111.180919 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rivera-Torres J et al. Cardiac electrical defects in progeroid mice and Hutchinson-Gilford progeria syndrome patients with nuclear lamina alterations. Proc Natl Acad Sci U S A 113, E7250–E7259, doi: 10.1073/pnas.1603754113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prakash A et al. Cardiac Abnormalities in Patients With Hutchinson-Gilford Progeria Syndrome. JAMA Cardiol 3, 326–334, doi: 10.1001/jamacardio.2017.5235 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stehbens WE, Wakefield SJ, Gilbert-Barness E, Olson RE & Ackerman J Histological and ultrastructural features of atherosclerosis in progeria. Cardiovasc Pathol 8, 29–39, doi: 10.1016/s1054-8807(98)00023-4 (1999). [DOI] [PubMed] [Google Scholar]
- 12.Gordon LB et al. Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A 109, 16666–16671, doi: 10.1073/pnas.1202529109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Capell BC & Collins FS Human laminopathies: nuclei gone genetically awry. Nat Rev Genet 7, 940–952, doi: 10.1038/nrg1906 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Lai W-F & Wong W-T Progress and trends in the development of therapies for Hutchinson–Gilford progeria syndrome. Aging Cell 19, e13175, doi: 10.1111/acel.13175 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beyret E et al. Single-dose CRISPR-Cas9 therapy extends lifespan of mice with Hutchinson-Gilford progeria syndrome. Nat Med 25, 419–422, doi: 10.1038/s41591-019-0343-4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Santiago-Fernandez O et al. Development of a CRISPR/Cas9-based therapy for Hutchinson-Gilford progeria syndrome. Nat Med 25, 423–426, doi: 10.1038/s41591-018-0338-6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suzuki K et al. Precise in vivo genome editing via single homology arm donor mediated intron-targeting gene integration for genetic disease correction. Cell Res 29, 804–819, doi: 10.1038/s41422-019-0213-0 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scaffidi P & Misteli T Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat Med 11, 440–445, doi: 10.1038/nm1204 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang F & Doudna JA CRISPR-Cas9 Structures and Mechanisms. Annu Rev Biophys 46, 505–529, doi: 10.1146/annurev-biophys-062215-010822 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Pattanayak V et al. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol 31, 839–843, doi: 10.1038/nbt.2673 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Varga R et al. Progressive vascular smooth muscle cell defects in a mouse model of Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A 103, 3250–3255, doi: 10.1073/pnas.0600012103 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koblan LW et al. Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat Biotechnol 36, 843–846, doi: 10.1038/nbt.4172 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kleinstiver BP, Pattanayak V, Prew MS & Nature T-SQ High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsai SQ et al. CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat Methods 14, 607–614, doi: 10.1038/nmeth.4278 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eisenberg E & Levanon EY A-to-I RNA editing - immune protector and transcriptome diversifier. Nat Rev Genet 19, 473–490, doi: 10.1038/s41576-018-0006-1 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Levy JM et al. Cytosine and adenine base editing of the brain, liver, retina, heart and skeletal muscle of mice via adeno-associated viruses. Nat Biomed Eng 4, 97–110, doi: 10.1038/s41551-019-0501-5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Villiger L et al. Treatment of a metabolic liver disease by in vivo genome base editing in adult mice. Nat Med 24, 1519–1525, doi: 10.1038/s41591-018-0209-1 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Zincarelli C, Soltys S, Rengo G & Rabinowitz JE Analysis of AAV serotypes 1–9 mediated gene expression and tropism in mice after systemic injection. Mol Ther 16, 1073–1080, doi: 10.1038/mt.2008.76 (2008). [DOI] [PubMed] [Google Scholar]
- 29.Inagaki K, Fuess S, Storm TA & Therapy G-GA Robust systemic transduction with AAV9 vectors in mice: efficient global cardiac gene transfer superior to that of AAV8. Molecular Therapy (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bostick B, Ghosh A, Yue Y, Long C & Duan D Systemic AAV-9 transduction in mice is influenced by animal age but not by the route of administration. Gene Ther 14, 1605–1609, doi: 10.1038/sj.gt.3303029 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Wang L, Wang H, Bell P, McMenamin D & Wilson JM Hepatic gene transfer in neonatal mice by adeno-associated virus serotype 8 vector. Hum Gene Ther 23, 533–539, doi: 10.1089/hum.2011.183 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kessler PD et al. Gene delivery to skeletal muscle results in sustained expression and systemic delivery of a therapeutic protein. Proc Natl Acad Sci U S A 93, 14082–14087, doi: 10.1073/pnas.93.24.14082 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nathwani AC et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N Engl J Med 371, 1994–2004, doi: 10.1056/NEJMoa1407309 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hagan C When Are Mice Considered Old? (2017). [Google Scholar]
- 35.Chandler RJ, Sands MS & Venditti CP Recombinant Adeno-Associated Viral Integration and Genotoxicity: Insights from Animal Models. Hum. Gene Ther. 28, 314–322, doi: 10.1089/hum.2017.009 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nault J-C et al. Wild-type AAV Insertions in Hepatocellular Carcinoma Do Not Inform Debate Over Genotoxicity Risk of Vectorized AAV. Molecular Therapy 24, 660–661, doi: 10.1038/mt.2016.47 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Geng D, Zhao W, Feng Y & Liu J Overexpression of Rab25 promotes hepatocellular carcinoma cell proliferation and invasion. Tumor Biology (2016). [DOI] [PubMed] [Google Scholar]
- 38.Donsante A et al. Observed incidence of tumorigenesis in long-term rodent studies of rAAV vectors. Gene Therapy 8, 1343–1346, doi: 10.1038/sj.gt.3301541 (2001). [DOI] [PubMed] [Google Scholar]
- 39.Donsante A et al. AAV Vector Integration Sites in Mouse Hepatocellular Carcinoma. Science 317, 477–477, doi: 10.1126/science.1142658 (2007). [DOI] [PubMed] [Google Scholar]
- 40.Embury JE, Charron CC, Poirier AE & Zori A Long term portal vein administration of AAV-WPRE vector results in increased incidence of neoplastic disease and hepatic pathology. Molecular Therapy (2006). [Google Scholar]
- 41.Sands MS AAV-mediated liver-directed gene therapy. Adeno-Associated Virus (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Richter MF, Zhao KZ, Eton E, Lapinaite A, Newby GA, Thuronyi BW., Wilson C, Koblan LW, Zeng J, Bauer DE, Doudna JA, and Liu DR. Phage-Assisted Evolution of an Adenine Base Editor with Enhanced Cas Domain Compatibility and Activity. . Nature Biotechnology (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gaudelli NM et al. Directed evolution of adenine base editors with increased activity and therapeutic application. Nat Biotechnol, doi: 10.1038/s41587-020-0491-6 (2020). [DOI] [PubMed] [Google Scholar]
- 44.Osorio FG et al. Splicing-directed therapy in a new mouse model of human accelerated aging. Sci Transl Med 3, 106ra107, doi: 10.1126/scitranslmed.3002847 (2011). [DOI] [PubMed] [Google Scholar]
- 45.Wu D & Smyth GK Camera: a competitive gene set test accounting for inter-gene correlation. Nucleic Acids Res 40, e133, doi: 10.1093/nar/gks461 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Doench JG et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nature Biotechnology 34, 184–191, doi: 10.1038/nbt.3437 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sanjana NE, Shalem O & Zhang F Improved vectors and genome-wide libraries for CRISPR screening. Nature Methods 11, 783–784, doi: 10.1038/nmeth.3047 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mateos J, Fafian-Labora J & one M-L-M Next-Generation Sequencing and Quantitative Proteomics of Hutchinson-Gilford progeria syndrome-derived cells point to a role of nucleotide metabolism in premature aging. PloS one (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cao K et al. Rapamycin reverses cellular phenotypes and enhances mutant protein clearance in Hutchinson-Gilford progeria syndrome cells. Sci Transl Med 3, 89ra58, doi: 10.1126/scitranslmed.3002346 (2011). [DOI] [PubMed] [Google Scholar]
- 50.Ewels P, Magnusson M, Lundin S & Kaller M MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048, doi: 10.1093/bioinformatics/btw354 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frengen E et al. A modular, positive selection bacterial artificial chromosome vector with multiple cloning sites. Genomics 58, 250–253, doi:DOI 10.1006/geno.1998.5693 (1999). [DOI] [PubMed] [Google Scholar]
- 52.DuBose AJ et al. Use of microarray hybrid capture and next-generation sequencing to identify the anatomy of a transgene. Nucleic Acids Res 41, doi:ARTN e70 10.1093/nar/gks1463 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li H & Durbin R Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760, doi: 10.1093/bioinformatics/btp324 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li H et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079, doi: 10.1093/bioinformatics/btp352 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Quinlan AR & Hall IM BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842, doi: 10.1093/bioinformatics/btq033 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tarasov A, Vilella AJ, Cuppen E, Nijman IJ & Prins P Sambamba: fast processing of NGS alignment formats. Bioinformatics 31, 2032–2034, doi: 10.1093/bioinformatics/btv098 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McKenna A et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303, doi: 10.1101/gr.107524.110 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.DePristo MA et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature genetics 43, 491–498 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.der Auwera VGA et al. From FastQ data to high-confidence variant calls: the genome analysis toolkit best practices pipeline. Current protocols in bioinformatics 43, doi:doi: 10.1002/0471250953.bi1110s43. (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang K, Li M & Hakonarson H ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Research 38, e164–e164, doi: 10.1093/nar/gkq603 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sondka Z et al. The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers. Nature reviews. Cancer 18, 696–705, doi: 10.1038/s41568-018-0060-1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Buenrostro JD, Giresi PG, Zaba LC, Chang HY & Greenleaf WJ Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature methods, 1213–1218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sondka Z et al. The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers. Nature reviews cancer 18, 696–705 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.