

Abstract
As defined by the World Health Organization, an endocrine disruptor is an exogenous substance or mixture that alters function(s) of the endocrine system and consequently causes adverse health effects in an intact organism, its progeny, or (sub)populations. Traditional experimental testing regimens to identify toxicants that induce endocrine disruption can be expensive and time-consuming. Computational modeling has emerged as a promising and cost-effective alternative method for screening and prioritizing potentially endocrine active compounds. The efficient identification of suitable chemical descriptors and machine learning algorithms, including deep learning, is a considerable challenge for computational toxicology studies. Here, we sought to apply classic machine learning algorithms and deep learning approaches to a panel of over 7,500 compounds tested against 18 Toxicity Forecaster (ToxCast) assays related to nuclear estrogen receptor (ERα and ERβ) activity. Three binary fingerprints (Extended Connectivity FingerPrints, Functional Connectivity FingerPrints, and Molecular ACCess System) were used as chemical descriptors in this study. Each descriptor was combined with four machine learning, and two deep learning (normal and multitask neural networks) approaches to construct models for all 18 ER assays. The resulting model performance was evaluated using the area under the receiving operating curve (AUC) values obtained from a five-fold cross-validation procedure. The results showed that individual models have AUC values that range from 0.56 to 0.86. External validation was conducted using two additional sets of compounds (n=592 and n=966) with established interactions with nuclear ER demonstrated through experimentation. An agonist, antagonist, or binding score was determined for each compound by averaging its predicted probabilities in relevant assay models as an external validation, yielding AUC values ranging from 0.63 to 0.91. The results suggest that multitask neural networks offer advantages when modeling mechanistically-related endpoints. Consensus predictions based on the average values of individual models remain the best modeling strategy for computational toxicity evaluations.
Estrogen receptors (ERs) play essential roles in cell differentiation1, reproductive function2–4, and morphogenesis4. ERs exist in two major subclasses: those that act via a classical genomic mechanism of transcriptional regulation (nuclear ERα and ERβ) and those that act via nongenomic mechanisms (estrogen-related receptors and membrane-bound G-protein coupled ERs)5. Nuclear ERα has a large binding pocket, which allows for nonspecific ER binding by compounds that are estrogen-like6. In the classical genomic mechanism, nuclear ERα or ERβ binds to an estrogenic compound. This ligand binding triggers a conformational change and activates the receptor1,4,7. Two activated nuclear ERs then can dimerize, bind to the estrogen response element (ERE) promoter region on the cell’s DNA, and recruit cofactors required for transcription1,7. The resulting increased production of mRNA can trigger cell proliferation downstream7. This cell proliferation has been linked to adverse effects such as uterine and breast cancers4,8. Therefore, screening new compounds (e.g., drugs as well as commercial and personal care products) for undesired nuclear ER interactions early in development may be valuable.
Traditional experimental testing to identify toxicants relies on costly and time-consuming in vivo animal testing, which is impractical to efficiently assess the toxicity potential of the tens of thousands of registered compounds that require screening9. Computational modeling and in vitro high-throughput screening (HTS) assays are promising alternative methods for toxicity evaluation. However, traditional computational methods such as quantitative structure-activity relationship (QSAR) models often have limitations when they were developed by using small datasets. QSAR models trained with datasets of insufficient size are limited by narrow coverage of chemical space10, activity cliffs11, and overfitting12, which in turn reduce their utility for predicting more complex chemical modes of action.
Over the past 20 years, deep learning emerged as an integral field of machine learning, especially with regards to the processing of big data13. Deep learning has advanced many fields, including voice and image recognition, language processing, and bioinformatics14. Most current deep learning studies employ biologically-inspired deep neural networks (DNNs)15. Both classic QSAR models and DNNs usually undergo training to predict a single activity (e.g., a single toxicity endpoint). However, many toxicologically-relevant modes of action require complex biological pathway perturbations to elicit an adverse biological effect, and consequently, the evaluation of the overall potential of a compound to exert an adverse outcome requires the prediction of multiple biological endpoints in a comprehensive manner. Multitask learning allows for the development of models that can simultaneously predict multiple activities and is a potential solution to this challenge. The application of a multitask learning approach can improve the ability of a model developed for related endpoints to generalize to new compounds due to information sharing during model development, thereby increasing prediction accuracy on new compounds. Successful modeling efforts using both normal and multitask deep learning demonstrate the potential for this technique to improve drug discovery16–19 and toxicology20,21. However, currently, no universal criteria for the selection of machine versus deep learning methods exist22–26.
The development of in vitro testing protocols using robots27 rather than humans allows for the rapid generation of data through HTS programs, advancing computational modeling into a big data era28–33. One of the first significant HTS programs in toxicology was the Environmental Protection Agency (EPA) Toxicity Forecaster (ToxCast) initiative, which used an extensive battery of HTS assays to screen over 1,000 compounds34,35. The success of ToxCast led to the development of the Toxicity in the 21st Century (Tox21) collaboration of the EPA, Food and Drug Administration (FDA), National Center for Advancing Translational Sciences (NCATS), and National Toxicology Program (NTP), which has a goal of testing approximately 10,000 compounds in HTS assays36–38. The direct result of these HTS efforts is the generation of large datasets that researchers can use in computational toxicity modeling studies.
The availability of big data in public repositories brings urgent needs for researchers to create innovative computational models that can overcome the limitations associated with models based on small datasets. The application of non-animal models for toxicity evaluation using computational toxicology is becoming feasible with newly developed algorithms and modeling strategies39–44. Recently, Browne et al.42 and Judson et al.43 described models trained using a subset of 18 ToxCast and Tox21 in vitro assays that are mechanistically relevant to the ER pathway. However, despite the success of these models, they require experimental concentration-response data, which makes them inapplicable to new, untested compounds for which only structural information is available. Our goal was to address these limitations by evaluating machine learning and deep learning approaches for their ability to predict compound activity using models based upon mechanistically related suites of assays. In this study, we assessed the applicability of traditional machine learning algorithms and deep learning approaches, including multitask learning with DNNs, to model these 18 mechanistic in vitro assays addressing ER pathway perturbations. The consensus predictions from averaging the predicted probabilities in relevant assays showed advantages compared to individual models, including multitask learning models. The agonist, antagonist, or binding score was determined for new compounds based on consensus predictions and compared to their known experimental in vitro and in vivo toxicities. The results from this study suggest that a lack of universal criteria for chemical descriptor and algorithm selection for computational toxicology modeling continues to exist, and consensus predictions will still be the best strategy for computational chemical toxicity evaluation purposes.
Materials and Methods
ER HTS Assay Dataset
The toxicity dataset used for modeling is the output of 18 high-throughput in vitro assays from the ToxCast and Tox21 programs (Table 1)42,43. In total, the ToxCast and Tox21 programs tested 8,589 compounds against these 18 assays. However, the chemical fingerprints calculated in this study are two-dimensional, which exclude the differences between stereoisomers and cannot deal with inorganic compounds. Therefore, the chemical structures needed further curation before modeling. The CASE Ultra v1.8.0.0 DataKurator tool was used to accomplish this chemical structure standardization. All salts and mixtures were separated into their constituent parts, and the largest organic fraction was kept. Compounds with duplicate structures but different activities in the same assays were evaluated, and the compound with the most active responses across all assays was retained. Compounds with missing/inconclusive results in all 18 assays were removed from the dataset.
Table 1.
Estrogen Receptor Toxicity Forecaster (ToxCast) Agonism, Antagonism, and Binding Assays
| Assay ID | Assay Endpoint Name | Assay Source | Organism | Gene Name | Timepoint (min) | Biological Process Target | Assay Design Type | Cell Line |
|---|---|---|---|---|---|---|---|---|
| A1 | NVS_NR_bER | NovaScreen | Bovine | ERα | 1080 | Receptor binding | Radioligand binding | NA |
| A2 | NVS_NR_hER | NovaScreen | Human | ERα | 1080 | Receptor binding | Radioligand binding | NA |
| A3 | NVS_NR_mERa | NovaScreen | Mouse | ERα | 1080 | Receptor binding | Radioligand binding | NA |
| A4 | OT_ER_ERaERa_0480 | Odyssey Thera | Human | ERα | 480 | Protein stabilization | Protein fragment complementation assay | HEK293T |
| A5 | OT_ER_ERaERa_1440 | Odyssey Thera | Human | ERα | 1440 | Protein stabilization | Protein fragment complementation assay | HEK293T |
| A6 | OT_ER_ERaERb_0480 | Odyssey Thera | Human | ERα, ERβ | 480 | Protein stabilization | Protein fragment complementation assay | HEK293T |
| A7 | OT_ER_ERaERb_1440 | Odyssey Thera | Human | ERα, ERβ | 1440 | Protein stabilization | Protein fragment complementation assay | HEK293T |
| A8 | OT_ER_ERbERb_0480 | Odyssey Thera | Human | ERβ | 480 | Protein stabilization | Protein fragment complementation assay | HEK293T |
| A9 | OT_ER_ERbERb_1440 | Odyssey Thera | Human | ERβ | 1440 | Protein stabilization | Protein fragment complementation assay | HEK293T |
| A10 | OT_ERa_EREGFP_0120 | Odyssey Thera | Human | ERα | 120 | Regulation of gene expression | Fluorescent protein induction | HeLa |
| A11 | OT_ERa_EREGFP_0480 | Odyssey Thera | Human | ERα | 480 | Regulation of gene expression | Fluorescent protein induction | HeLa |
| A12 | ATG_ERa_TRANS_up | Attagene, Inc. | Human | ERα | 1440 | Regulation of transcription factor activity | mRNA induction | HepG2 |
| A13 | ATG_ERE_CIS_up | Attagene, Inc. | Human | ERα | 1440 | Regulation of transcription factor activity | mRNA induction | HepG2 |
| A14 | TOX21_ERa_BLA_Agonist_ratio | Tox21 | Human | ERα | 1440 | Regulation of transcription factor activity | Beta lactamase induction | HEK293T |
| A15 | TOX21_ERa_LUC_BG1_Agonist | Tox21 | Human | ERα | 1320 | Regulation of transcription factor activity | Luciferase induction | BG1 |
| A16 | ACEA_T47D_80hr_Positive | ACEA Biosciences, Inc. | Human | ERα | 1920 | Cell proliferation | Real-time cell-growth kinetics | T47D |
| A17 | TOX21_ERa_BLA_Antagonist_ratio | Tox21 | Human | ERα | 1440 | Regulation of transcription factor activity | Beta lactamase induction | HEK293T |
| A18 | TOX21_ERa_LUC_BG1_Antagonist | Tox21 | Human | ERα | 1320 | Regulation of transcription factor activity | Luciferase induction | BG1 |
The final dataset used for modeling in this study consisted of 7,576 unique compounds, each of which showed conclusive active or inactive test results in at least one of the 18 nuclear ER-related in vitro assays (Supplementary Table SI). Inconclusive results were treated as missing data for modeling purposes. Each chemical was assigned an activity vector consisting of 18 active, inactive, or missing/inconclusive results for all assays.
Chemical Descriptors
Three types of two-dimensional binary chemical fingerprints, Molecular ACCess System (MACCS), Extended Connectivity FingerPrint (ECFP), and Functional Connectivity FingerPrint (FCFP) descriptors, were generated for all compounds in Python v3.6.2 using the cheminformatics package RDKit v2017.09.1 (http://rdkit.org/). MACCS descriptors are a set of 167 fingerprints based on chemical substructures widely used in cheminformatics modeling45. ECFP and FCFP descriptors are substructure fingerprints calculated using a modified version of the Morgan algorithm (i.e., by evaluating the environment surrounding particular atoms in a molecule using a specified bond radius)46. FCFP descriptors can represent functional group information about a molecule rather than a specific substructure, whereas ECFP descriptors can represent specific chemical information about a molecule. For example, FCFP descriptors detect the presence of an aryl halide rather than the specific presence of chlorine bonded to a benzene ring that ECFP descriptors detect. In this study, 1,024 ECFP and FCFP descriptors were calculated for all compounds using a bond radius of 3.
QSAR Model Development
Four machine learning (ML) algorithms were used to develop QSAR models for each ToxCast assay endpoint: Bernoulli Naïve Bayes (BNB), k-Nearest Neighbors (kNN), Random Forest (RF), and Support Vector Machines (SVM). In this study, all four ML algorithms were implemented in Python v3.6.2 using scikit-learn v0.19.0 (http://scikit-learn.org/)47. Briefly, BNB models apply Bayes’ theorem to datasets with binary features by “naively” assuming that features are independent of one another48. kNN models learn and predict a compound based on the activities of its k nearest neighbors calculated by a subspace similarity search49. RF models are ensemble models that construct a series of decision trees using a random selection of features and training set compounds50. RF models ultimately produce an average of the output from each decision tree to prevent overfitting. SVM models represent training compounds in the descriptor space and attempt to locate the optimal hyperplane that separates active and inactive compounds51. The ML algorithms were tuned to identify the optimal input parameters for model performance, as described previously23. Briefly, hyperparameters, or any other parameters set before model training, were optimized using an exhaustive grid-search algorithm23. Each machine learning algorithm was fit to the ER HTS training data using each possible set of hyperparameters to identify the best performing model. The model with the best combination of hyperparameters was retained and then used for the prediction of the test set.
Both normal and multitask DNNs were implemented in Python v3.6.2 using keras v2.1.2 (http://keras.org) and TensorFlow v1.4.0 (https://www.tensorflow.org/). DNNs consist of an input layer that contains information about the features of the data, such as chemical fingerprints, used to train the model, and an output layer, which is a prediction for the activity of interest15. A series of “dense” layers connect the input and output layers, such that every node in each layer shares a weighted connection with every node in the previous and next layers. These weighted connections undergo optimization in the model training process. All DNNs in this study were implemented with three hidden layers of width equal to the number of fingerprints in the input layer (i.e., 167 for MACCS descriptors and 1,024 for ECFP and FCFP descriptors). Before model training, the weights between the neurons of each layer were randomly initiated using the He normal method52. These weights were optimized during training to achieve the minimum binary cross-entropy. To this end, the following standard deep learning methods were implemented: stochastic gradient descent (SGD) optimization53 (learning rate = 0.01, Nesterov momentum54 = 0.9), Rectified Linear Unit (ReLU) hidden layer activation55, and automatic learning rate reduction56 (90% reduction upon 50 consecutive epochs with no loss improvement, minimum = 0.0001). Dropout57 (rate = 0.5) and L258 (β = 0.001) regularizations and early stopping59 (upon 200 epochs with no loss improvement) were implemented to avoid overfitting. The model output layer used a sigmoid activation function60 so that the predicted result was interpretable as a probability.
Model performance was evaluated using the area under the receiver operating curve (ROC) metric (AUC). Each model developed in this study computes a probability that a tested compound will be active in a given bioassay. Tested compounds are classified as active when they exceed a determined probability threshold. The ROC curve for model performance is a plot of the true positive rate (TPR, Equation 1) against the false positive rate (FPR, Equation 2) using various probability thresholds for the classification of active compounds61. The area under this plotted curve (AUC) is interpretable as a measure of the likelihood of a model to distinguish active compounds from inactive compounds correctly. An AUC of 0.5 represents a random model performance as the baseline. The AUC is a suitable metric for this study due to the highly imbalanced nature of the assay data used to train the models. In modeling studies using imbalanced datasets (e.g., HTS assay data), the default probability threshold of 0.5 is not always appropriate62. Using the AUC as an evaluation method takes this consideration into account by evaluating model performance at several different probability thresholds.
| [1] |
| [2] |
External Validation
The developed models can be used to predict new compounds to prove their predictivity. To this end, external validation was performed using two datasets: the Collaborative Estrogen Receptor Activity Prediction Project (CERAPP) in vitro agonist, antagonist, and binding datasets63 and the Estrogenic Activity Database (EADB) in vivo rodent uterotrophic dataset64. Before model validation, the CASE Ultra v1.8.0.0 DataKurator tool was used to prepare the structures of new compounds as previously described. Only the new compounds not existing in the training dataset were kept. The final curated CERAPP in vitro agonist, antagonist, and binding validation sets contained 368, 264, and 569 compounds, respectively (Supplementary Table SII). The final curated EADB in vivo rodent uterotrophic agonist validation set contained 966 compounds (Supplementary Table SIII).
Three new parameters were created to evaluate a chemical’s potential to act as a nuclear ER agonist, antagonist, or binder based on its predicted activity in relevant assays: agonist score (SAg, Equation 3), antagonist score (SAnt, Equation 4), and binding score (SB, Equation 5). In these equations, P(Ai) is the probability for a predicted compound to be active in Assay i. The 18 total assays contain 16 agonism assays (A1-A16), 13 antagonism assays (A1-A11, A17, and A18), and 11 binding assays (A1 – A11). These three parameters integrate relevant models of ER agonism, antagonism, and binding to evaluate new compounds for their toxicity potential at nuclear ERs. The performance of models during external validation was evaluated using ROC curve plots and AUC calculations, as previously described for the cross-validation procedure.
| [3] |
| [4] |
| [5] |
Results
Dataset
Figure 1 shows a summary of the 7,576 unique compounds tested against at least one of the 18 ToxCast and Tox21 nuclear ER-related in vitro assays. HTS assay data usually contain missing and inconclusive data points, and the results are biased (i.e., more inactive than active)28,29. In total, these compounds consist of over 53,000 total conclusively active or inactive assay hit calls, indicating that missing/inconclusive results exist in the dataset. The results show a diverse number of conclusive activities per compound, ranging from 2 to 18 hit calls in these assays (Figure 1A). Only 476 compounds showed conclusive results for all 18 assays, representing 6.3% of the full dataset. The low active response ratio across all assays (i.e., active ratio ranges from 1:16 to 1:3) compared to inactive responses reflects the nature of HTS results for chemical toxicity testing28,29. Furthermore, no individual assay has conclusive results for all 7,576 compounds. Instead, the size of each assay dataset ranges from 883 to 7,263 compounds, depending on the assay nature (Table 1, Figure 1B). For example, NVS_NR_bER (A1, 1,004 compounds), NVS_NR_hER (A2, 1,076 compounds), and NVS_NR_mERa (A3, 883 compounds) show the lowest number of tested compounds, and they are NovaScreen assays. TOX21_ERa_BLA_Agonist_ratio (A14), TOX21_ERa_LUC_BG1_Agonist (A15), TOX21_ERa_BLA_Antagonist_ratio (A17), and TOX21_ERa_LUC_BG1_Antagonist (A18) are Tox21 assays that each consist of 7,263 compounds with conclusive results, representing the richest individual assay datasets. Therefore, these 18 assay datasets represent a large range of data size and chemical diversity, which are suitable for modeling studies to evaluate the machine learning algorithms.
Figure 1.
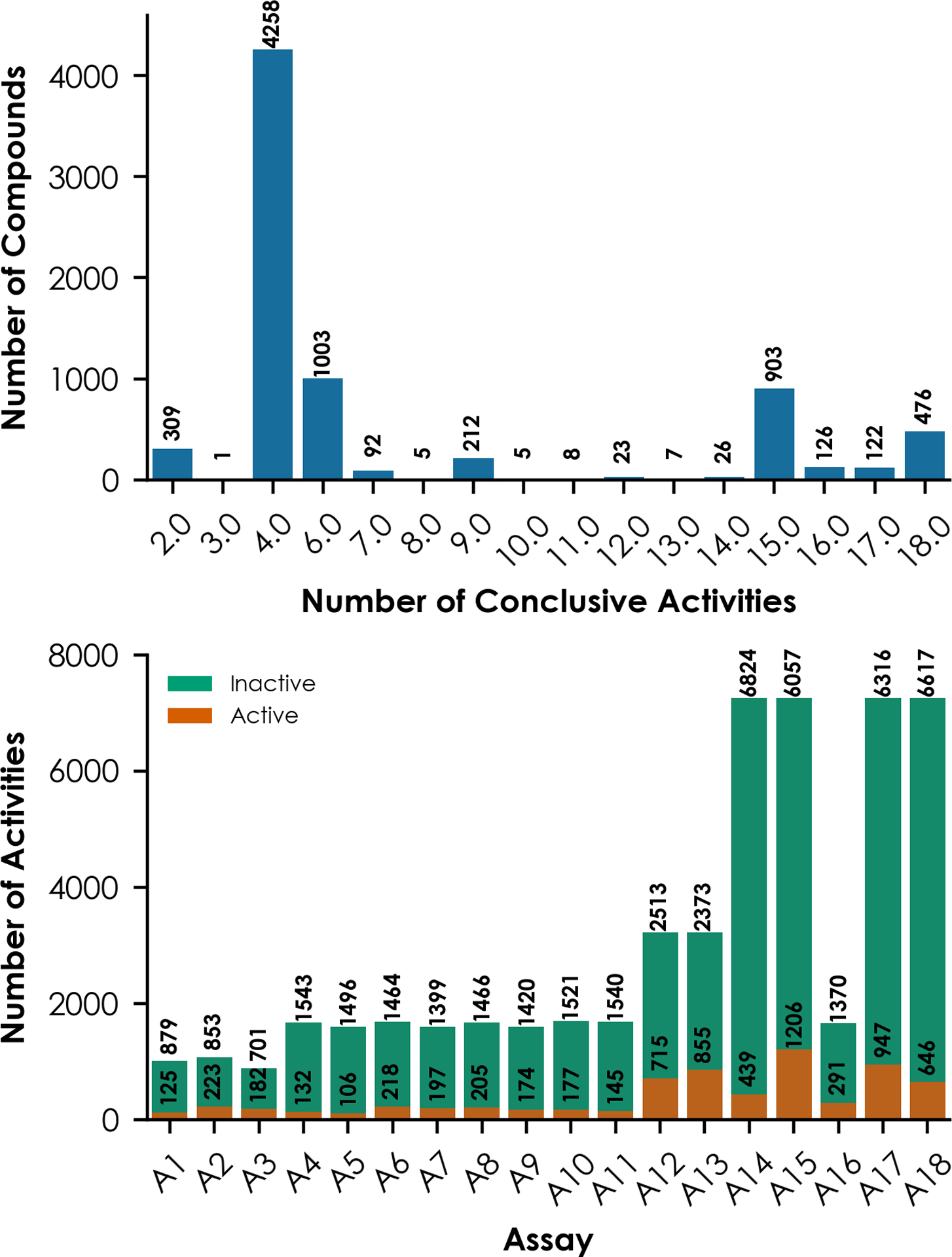
Distributions of (A) compounds in the ToxCast and Tox21 dataset (n=7,576) by the number of conclusive active or inactive results per compound and (B) individual assay datasets (n=18) by the number of active and inactive compounds.
The data used in this study also show a bias toward inactive responses. Out of the full dataset, only six of these compounds showed active results across all 18 assays: Bisphenol AF (CAS 1478-61-1), 2-ethylhexyl 4-hydroxybenzoate (CAS 5153-25-3), 4-tert-octylphenol (CAS 140-66-9), diethylstilbestrol (CAS 56-53-1), 4-cumylphenol (CAS 599-64-4), and hexestrol (CAS 84-16-2). These six compounds show uterotrophic activity in at least one guideline-like study65. By comparison, 4,698 compounds show only inactive results in one or more of these 18 assays, representing a majority (62.0%) of all compounds. The individual assay datasets reveal a similar trend, with small ratios of active versus inactive results. For example, ATG_ERE_CIS_up (A13), which is an mRNA induction assay, has the highest active ratio of approximately 1:3. Compared to this assay, TOX21_ERa_BLA_Agonist_ratio (A14), which is a beta-lactamase induction assay, has the lowest active ratio of approximately 1:16. Some previous studies showed that downsampling to remove some inactive compounds from training datasets was beneficial to the resulted QSAR models66,67. However, in this study, the full dataset was retained to preserve an ample chemical space for the prediction of new compounds.
QSAR Model Development
Four machine learning (BNB, kNN, RF, and SVM) and two DNN algorithms were paired with ECFP, FCFP, and MACCS descriptors individually to develop 18 models for each ER assay (Figure 2). Simpler algorithms, such as logistic regression, were not used in this study since previous studies have shown the advantages of advanced machine learning algorithms23,68. Therefore, in total, 273 models (216 ML models, 54 normal DNN models, and 3 multitask DNN models) were developed for all of the ER assay data. In 2007, the Organization for Economic Co-Operation and Development (OECD) published a guidance document on the validation of QSAR models developed for risk assessment purposes69. The guidelines set forth by this document require that models undergo statistical evaluation for goodness-of-fit, robustness, and predictivity, including model cross-validation69. Cross-validation procedures that leave compounds out during each iteration provide reliable model evaluations70. In this study, all models were evaluated using a five-fold cross-validation procedure, with 20% of the dataset left out for prediction purposes during each iteration. Each assay dataset was randomly split into five equal subsets maintaining the original proportion of active and inactive responses. In this procedure, four subsets (80% of the total compounds) were combined as a training set, and the remaining 20% was used as a test set. This procedure was repeated five times, such that each compound was used in a test set one time. The six resulting models for each assay-descriptor combination were averaged to give a consensus prediction, as described in previous publications66,71–73.
Figure 2.
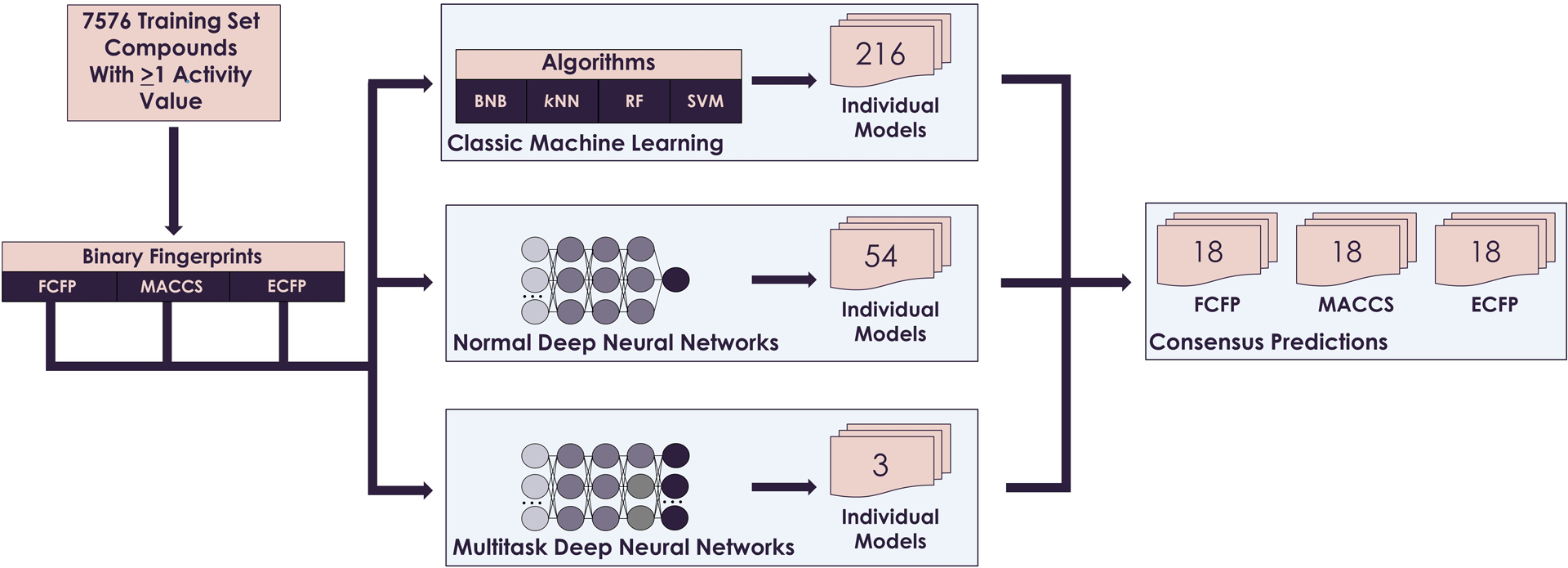
Consensus QSAR modeling workflow used in this study.
Table 2 shows the five-fold cross-validation results for each model. The AUC values for all the resulted models ranged between 0.562–0.870. The highest AUC value ranged between 0.645–0.870 for each assay, indicating that at least one descriptor-algorithm combination yielded a satisfactory model for each endpoint. OT_ER_ERaERb_0480 (A6) had the best performing models, with AUC values ranging between 0.609–0.870. Compared to this assay, TOX21_ERa_LUC_BG1_Agonist (A15) and ACEA_T47D_80hr_Positive (A16) consistently had lower performing models with AUC values ranging between 0.562–0.660 and 0.562–0.645, respectively. In previous studies, QSAR model performance was high when modeling simple endpoints (e.g., physical-chemical properties) but became lower for complex biological activities (e.g., cellular responses)29. A15 and A16 are nuclear ER agonism assays that represent protein production induced by ER-mediated transcriptional activation74 and the resulting cell proliferation75,76 (Table 1). Among the biological processes represented by these 18 assays, transcriptional activation and cell proliferation represent the farthest downstream processes in the classical genomic ER signaling pathway43, which may be the reason that they are the most difficult to model.
Table 2.
Performance of Individual Models for 18 ToxCast and Tox21 ER Assays Using a Five-Fold Cross-Validation
| Algorithms | Descriptors | AUC | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A1 | A2 | A3 | A4 | A5 | A6 | A7 | A8 | A9 | A10 | A11 | A12 | A13 | A14 | A15 | A16 | A17 | A18 | ||
| BNB | MACCS | 0.732 | 0.702 | 0.664 | 0.803 | 0.764 | 0.788 | 0.705 | 0.770 | 0.723 | 0.688 | 0.672 | 0.716 | 0.670 | 0.698 | 0.618 | 0.597 | 0.685 | 0.716 |
| FCFP6 | 0.723 | 0.725 | 0.727 | 0.819 | 0.764 | 0.829 | 0.749 | 0.820 | 0.749 | 0.740 | 0.720 | 0.742 | 0.687 | 0.724 | 0.645 | 0.645 | 0.725 | 0.746 | |
| ECFP6 | 0.722 | 0.704 | 0.723 | 0.828 | 0.763 | 0.824 | 0.705 | 0.800 | 0.725 | 0.688 | 0.692 | 0.735 | 0.682 | 0.730 | 0.643 | 0.632 | 0.722 | 0.736 | |
| kNN | MACCS | 0.625 | 0.649 | 0.639 | 0.681 | 0.676 | 0.729 | 0.634 | 0.693 | 0.651 | 0.707 | 0.682 | 0.686 | 0.659 | 0.712 | 0.616 | 0.601 | 0.654 | 0.636 |
| FCFP6 | 0.597 | 0.597 | 0.596 | 0.639 | 0.643 | 0.650 | 0.614 | 0.641 | 0.627 | 0.603 | 0.616 | 0.622 | 0.622 | 0.650 | 0.592 | 0.588 | 0.615 | 0.605 | |
| ECFP6 | 0.593 | 0.600 | 0.610 | 0.626 | 0.642 | 0.609 | 0.576 | 0.599 | 0.597 | 0.590 | 0.573 | 0.618 | 0.587 | 0.644 | 0.562 | 0.578 | 0.601 | 0.599 | |
| RF | MACCS | 0.740 | 0.687 | 0.689 | 0.843 | 0.814 | 0.848 | 0.733 | 0.827 | 0.736 | 0.743 | 0.714 | 0.750 | 0.704 | 0.762 | 0.658 | 0.620 | 0.799 | 0.818 |
| FCFP6 | 0.730 | 0.723 | 0.707 | 0.796 | 0.735 | 0.837 | 0.708 | 0.812 | 0.743 | 0.751 | 0.696 | 0.748 | 0.683 | 0.733 | 0.642 | 0.635 | 0.748 | 0.747 | |
| ECFP6 | 0.742 | 0.685 | 0.726 | 0.805 | 0.783 | 0.843 | 0.716 | 0.809 | 0.715 | 0.677 | 0.729 | 0.740 | 0.689 | 0.740 | 0.646 | 0.617 | 0.745 | 0.726 | |
| SVM | MACCS | 0.737 | 0.717 | 0.679 | 0.845 | 0.795 | 0.864 | 0.712 | 0.819 | 0.715 | 0.759 | 0.737 | 0.770 | 0.712 | 0.782 | 0.652 | 0.622 | 0.819 | 0.827 |
| FCFP6 | 0.713 | 0.677 | 0.701 | 0.822 | 0.736 | 0.827 | 0.735 | 0.818 | 0.733 | 0.768 | 0.709 | 0.742 | 0.698 | 0.744 | 0.639 | 0.626 | 0.794 | 0.789 | |
| ECFP6 | 0.706 | 0.697 | 0.713 | 0.827 | 0.748 | 0.810 | 0.667 | 0.792 | 0.683 | 0.684 | 0.664 | 0.756 | 0.697 | 0.785 | 0.641 | 0.613 | 0.802 | 0.798 | |
| Normal DNN | MACCS | 0.695 | 0.690 | 0.679 | 0.827 | 0.771 | 0.855 | 0.659 | 0.751 | 0.723 | 0.737 | 0.699 | 0.724 | 0.674 | 0.777 | 0.637 | 0.596 | 0.798 | 0.790 |
| FCFP6 | 0.687 | 0.656 | 0.673 | 0.780 | 0.689 | 0.738 | 0.658 | 0.770 | 0.725 | 0.662 | 0.661 | 0.675 | 0.631 | 0.648 | 0.609 | 0.562 | 0.649 | 0.641 | |
| ECFP6 | 0.708 | 0.682 | 0.672 | 0.811 | 0.752 | 0.661 | 0.605 | 0.701 | 0.667 | 0.588 | 0.643 | 0.696 | 0.624 | 0.590 | 0.574 | 0.592 | 0.678 | 0.674 | |
| Multitask DNN | MACCS | 0.707 | 0.705 | 0.700 | 0.853 | 0.752 | 0.849 | 0.743 | 0.822 | 0.733 | 0.775 | 0.746 | 0.761 | 0.699 | 0.781 | 0.647 | 0.635 | 0.815 | 0.818 |
| FCFP6 | 0.709 | 0.685 | 0.677 | 0.810 | 0.732 | 0.818 | 0.755 | 0.790 | 0.751 | 0.726 | 0.720 | 0.709 | 0.647 | 0.724 | 0.625 | 0.618 | 0.748 | 0.722 | |
| ECFP6 | 0.691 | 0.677 | 0.664 | 0.810 | 0.705 | 0.791 | 0.694 | 0.776 | 0.686 | 0.679 | 0.674 | 0.723 | 0.650 | 0.735 | 0.614 | 0.626 | 0.775 | 0.739 | |
| Consensus | MACCS | 0.749 | 0.729 | 0.703 | 0.852 | 0.796 | 0.870 | 0.718 | 0.819 | 0.739 | 0.749 | 0.728 | 0.764 | 0.718 | 0.785 | 0.660 | 0.634 | 0.824 | 0.830 |
| FCFP6 | 0.741 | 0.703 | 0.731 | 0.809 | 0.742 | 0.829 | 0.742 | 0.827 | 0.750 | 0.782 | 0.726 | 0.752 | 0.700 | 0.745 | 0.644 | 0.638 | 0.779 | 0.784 | |
| ECFP6 | 0.725 | 0.707 | 0.728 | 0.833 | 0.770 | 0.798 | 0.700 | 0.798 | 0.713 | 0.686 | 0.710 | 0.754 | 0.697 | 0.743 | 0.639 | 0.642 | 0.781 | 0.784 | |
Notably, no algorithm can outperform the others across all of the 18 assay endpoints and three descriptor sets (Table 2). However, compared to normal DNNs, multitask DNNs had better predictivity for 16 out of 18, 18 out of 18, and 13 out of 18 assay endpoints using MACCS, FCFP, and ECFP descriptors, respectively (Table 2), indicating the advantage of using multitask learning to model these mechanistically-related endpoints. The three consensus models showed better or similar results compared to all other algorithms. For example, when using MACCS descriptors, the five-fold cross-validation results of the consensus model achieve AUC values as high as 0.870, representing the best performance for 10 out of 18 assay endpoints (55.5%) compared to individual models. When using the FCFP descriptors, the consensus model achieves AUC values as high as 0.829, representing the best performance for 8 out of 18 assay endpoints (44.4%) compared to individual models. When using the ECFP descriptors, the consensus model achieves AUC values as high as 0.833, representing the best performance for 5 out of 18 assay endpoints (27.8%) compared to individual models. No individual model shows better performance than the consensus model across all 18 assay endpoints.
External Validations
External validation is necessary to prove the predictivity of the resulted QSAR models. An external validation procedure was conducted using two new datasets: the in vitro CERAPP dataset consisting of 368 new agonists, 264 new antagonists, and 569 new binders, and the in vivo EADB uterotrophic dataset consisting of 966 new agonists. Before performing external validation, compounds that were also included in the model training set were removed from both datasets, resulting in 569 and 966 unique compounds that were not tested in the ToxCast and Tox21 ER HTS assays and are new to the developed models. Since each assay is only relevant to a specific target of a binding mechanism, using the parameters SAg, SAnt, and SB, which were defined to integrate all relevant models, can estimate the estrogenic activities of new compounds more reliably compared to using a single QSAR model for the external compounds (Equations 3–5). For example, the SB parameter represents the likelihood of a compound to be an in vitro ER binder (Equation 5). This parameter includes 11 assays (A1 to A11) that represent receptor binding77–80, receptor dimerization81–83, and DNA binding83 (Table 1). The SAg parameter (Equation 3) represents the likelihood of a compound to be an in vitro ER agonist and includes five additional assays (A12 to A16) that represent RNA transcription84, protein production74, and cell proliferation75,76. The SAnt parameter (Equation 4) includes all assays used to calculate SB and two extra assays (A17 and A18) that represent transcriptional suppression74.
Table 3 shows the results of these external validations. The AUC values of the prediction results using the SAg parameter for the new agonists in the CERAPP and EADB datasets ranged from 0.732–0.906 and 0.640–0.802, respectively. The highest performing models for the CERAPP dataset were RF models regardless of the descriptors used. The combination of normal DNNs with FCFP descriptors showed the best performance for the EADB dataset. The AUC values of the prediction results using the SAnt parameter for the new antagonists in the CERAPP dataset ranged from 0.711–0.869. The highest performing model for this dataset used multitask DNNs with FCFP descriptors and achieved an AUC value of 0.869. The AUC values of the prediction of new binders in the CERAPP dataset using the SB parameter ranged from 0.622–0.754. The highest performing model for the CERAPP dataset is the combination of normal DNNs with MACCS descriptors. Although the consensus model does not show the best performance in the external predictions, its prediction accuracy is similar to the best performing model in the four datasets (Table 3).
Table 3.
External Validation of ER Agonists, Antagonists, and Binders
| Algorithms | Descriptors | AUC | |||
|---|---|---|---|---|---|
| CERAPP in vitro
Agonists |
CERAPP in vitro Antagonists | CERAPP in vitro Binders | EADB in vivo Uterotrophic | ||
| BNB | MACCS | 0.859 | 0.731 | 0.684 | 0.640 |
| FCFP6 | 0.799 | 0.815 | 0.715 | 0.757 | |
| ECFP6 | 0.780 | 0.831 | 0.702 | 0.686 | |
| kNN | MACCS | 0.796 | 0.768 | 0.688 | 0.729 |
| FCFP6 | 0.732 | 0.711 | 0.622 | 0.751 | |
| ECFP6 | 0.736 | 0.786 | 0.626 | 0.684 | |
| RF | MACCS | 0.901 | 0.759 | 0.713 | 0.756 |
| FCFP6 | 0.884 | 0.747 | 0.703 | 0.726 | |
| ECFP6 | 0.906 | 0.706 | 0.707 | 0.747 | |
| SVM | MACCS | 0.887 | 0.820 | 0.739 | 0.770 |
| FCFP6 | 0.829 | 0.830 | 0.667 | 0.765 | |
| ECFP6 | 0.829 | 0.849 | 0.670 | 0.790 | |
| Normal DNN | MACCS | 0.879 | 0.860 | 0.754 | 0.767 |
| FCFP6 | 0.794 | 0.780 | 0.691 | 0.802 | |
| ECFP6 | 0.801 | 0.733 | 0.681 | 0.724 | |
| Multitask DNN | MACCS | 0.866 | 0.749 | 0.698 | 0.720 |
| FCFP6 | 0.822 | 0.869 | 0.672 | 0.787 | |
| ECFP6 | 0.821 | 0.751 | 0.736 | 0.757 | |
| Consensus | MACCS | 0.889 | 0.828 | 0.726 | 0.766 |
| FCFP6 | 0.826 | 0.817 | 0.704 | 0.784 | |
| ECFP6 | 0.823 | 0.831 | 0.726 | 0.738 | |
Discussion
Computational methods offer potential advantages for rapid early screening of compounds for possible estrogenic and antiestrogenic effects. In 2015, the US EPA published a computational model that incorporated concentration-response data from 18 quantitative HTS (qHTS) assays from the ToxCast and Tox21 programs42,43. The success of this model to predict in vivo uterotrophic activity led to the acceptance of its results as an alternative to rodent uterotrophic testing85. However, this model requires experimental concentration-response data for evaluating compounds and cannot be applied to new compounds that did not yet undergo testing in these assays. Further, not all of the included assays are readily available to be applied. This issue was solved in the current study by developing machine learning and deep learning models to predict the ER activity of new compounds directly from chemical structure. Multitask deep learning outperformed normal deep learning for the prediction of in vitro activity in almost all cases across the 18 ToxCast and Tox21 assays. None of the six algorithms used for modeling could consistently outperform all others across the 18 assays, regardless of the descriptors used. Consensus modeling is, therefore, still the most suitable and robust modeling approach. These advantages are evident in this study, with consensus models yielding the highest AUC for 11 of the 18 total assays across all descriptor-algorithm combinations (61%, Table 2). The combination of all descriptor-algorithm sets to generate one consensus prediction instead of selecting an algorithm that is specific to a descriptor set is still the best strategy for future model development.
The SAg, SAnt, and SB parameters used for the prediction of the in vitro agonist, antagonist, and binding activities of external validation datasets are also based on the concept of consensus modeling (Equations 3–5). Each of these parameters incorporates predictions using assays that represent between three and six different biological processes relevant to the activity of interest. For example, the SAg parameter includes 16 assays related to nuclear ER agonism, which represent six biological processes: receptor binding, receptor dimerization, DNA binding, RNA transcription, protein production, and cell proliferation (Table 1). Furthermore, these assays represent four general types of technology: radioligand, fluorescence, bioluminescence, and electrical impedance42,43 (Table 1). By incorporating assays that represent a variety of technologies, the results are more reliable because technology-specific artifacts will affect fewer probabilities.
The predictivity of new compounds, especially toxic compounds, can be explained by revealing their nearest neighbor compounds. For example, 6α-hydroxyestradiol (CAS 1229-24-9) was classified as a binder and strong agonist in the CERAPP dataset63. This compound is an estrogenic product from the liver metabolism of the prominent endogenous estrogen estradiol (E2)86. 6α-hydroxyestradiol showed both the highest SB score (SB = 0.882) and the highest SAg score (SAg = 0.879) among all new compounds using the consensus models. 6α-hydroxyestradiol was predicted to be active in all binding-related assays (A1 to A11) and all agonism-specific assays (A12 to A16). Its nearest neighbor in the training set was alfatradiol (CAS 57-91-0), a stereoisomer of E2 that behaves as a nuclear ER agonist in both in vitro63 and in vivo65 assays. Alfatradiol also showed active responses in all binding and agonist assays used to train the models in this study. Among the EADB in vivo uterotrophic agonists, mestilbol (CAS 18839-90-2) showed the highest SAg score (SAg = 0.870). Mestilbol is a synthetic monomethyl ether derivative of diethylstilbestrol (CAS 56-53-1), which is its nearest neighbor in the training set. Diethylstilbestrol (DES) is a well-known synthetic nonsteroidal estrogen that was previously prescribed to pregnant women to prevent miscarriages87. DES is a known strong agonist of the ER that showed uterotrophic activity in several independent guideline-like studies65. Another external compound, pipendoxifene (CAS 198480-55-6), was classified as an ER antagonist in the CERAPP dataset52 and was predicted correctly. Pipendoxifene is an investigational drug currently undergoing clinical trials as a selective ER modulator (SERM)88. Pipendoxifene is under development to treat ER-positive breast cancers as well as osteoporosis89. Pipendoxifene showed mixed (either active or inactive) results in binding assay model predictions but was predicted as an antagonist in the specific assays (A17 and A18). Among these assays, this compound’s two nearest neighbors were raloxifene hydrochloride (CAS 82640-04-8) and bazedoxifene acetate (CAS 198481-33-3), which are FDA-approved SERMs for the treatment of osteoporosis89,90. Clinical trials of these compounds indicated ER antagonist activity in breast and uterine tissue89,90.
The predictive accuracy of this study can be improved by implementing applicability domains. The QSAR models were based on chemical structures and therefore are most reliable when predicting new compounds that are chemically and structurally similar to compounds in the training dataset. A common method to implement a QSAR model applicability domain is only to predict compounds that are within a certain similarity threshold with their nearest neighbor in the training set91,92. Figure 3 shows the effect of only predicting compounds within a Jaccard similarity of 0.8, 0.4, or 0.3 using models with MACCS, FCFP, or ECFP descriptors, respectively, on the five-fold cross-validation and external validation results. For external validation, new compounds were predicted if the SAg, SAnt, and SB parameters can be calculated with at least half of their constituent assay models (Equations 3–5). Using these thresholds allows for 42% to 83% coverage of the external predictions. Implementing these applicability domains enhanced the cross-validation performance of all the algorithms, including consensus predictions, for the 18 ER assays (Figure 3A, 3C, and 3E). The average AUC value for each algorithm improved from 0.600–0.759 to 0.617–0.800 using the applicability domains (i.e., Jaccard similarity 0.8 for MACCS, 0.3 for ECFP, and 0.4 for FCFP descriptors). The use of the applicability domains also enhanced most external predictions (Figure 3B, 3D, and 3F). For CERAPP compounds, the AUC values improve from 0.622–0.906 to 0.696–0.923 using the applicability domain. However, for the EADB compounds, implementing the applicability domain does not improve the results significantly (Figure 3B, 3D, and 3F). Although the SAg, SAnt, and SB parameters as currently calculated show good predictivity (Table 3), utilizing applicability domains and reducing the weight of binding assays in the calculations is expected to enhance the results further. Defining the applicability domain is also one of the principles for validation of QSAR use for regulatory purposes, and thus is a prudent consideration if the ultimate purpose of the QSAR model is to make a regulatory decision93.
Figure 3.
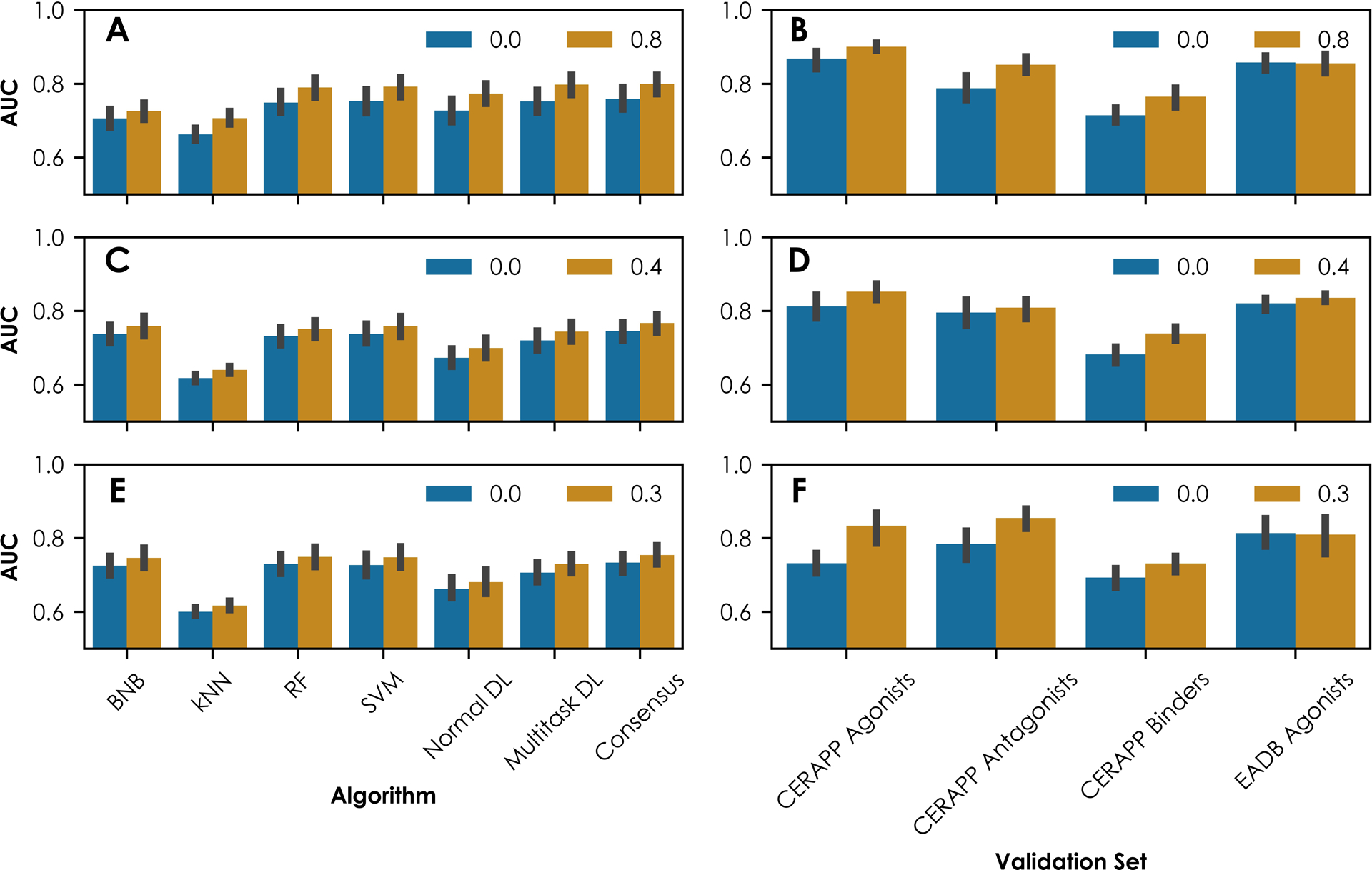
Predictivity of individual and consensus QSAR models using MACCS descriptors for (A) cross-validation and (B) external validation with a chemical similarity threshold of 0.8, using FCFP descriptors for (C) cross-validation and (D) external validation with a chemical similarity threshold of 0.4, and using ECFP descriptors for (E) cross-validation and (F) external validation with a chemical similarity threshold of 0.3. All AUC values are reported as the mean value ± standard deviation.
In this study, 7,576 compounds that were tested in ToxCast and Tox21 assays related to nuclear ER agonism, antagonism, and binding were used for exhaustive modeling using classic machine learning, normal deep learning, and multitask deep learning approaches. To this end, 273 individual QSAR models were developed for 18 assay datasets related to nuclear ER activity. QSAR models developed using multitask deep learning outperformed models developed with normal deep learning (i.e., trained for a single endpoint) in almost all endpoints. However, no individual algorithm can consistently outperform all others across the 18 endpoints. The consensus models generated by averaging the predictions of the individual models had similar or higher predictivity than the individual models. Three parameters were defined to incorporate predictions from models that represent mechanistically-relevant assays to predict a compound’s likelihood of behaving like a nuclear ER agonist, antagonist, or binder. External validation based on these parameters showed reliable predictivity for new compounds that did not undergo experimental testing in the 18 assays. The results of this study demonstrate the advantages of multitask deep learning for the QSAR modeling of mechanistically-related assay endpoints. Furthermore, consensus modeling remains the most reliable strategy for QSAR modeling in the current big data era, as no algorithm or chemical descriptor set is universally better than others are.
Supplementary Material
Acknowledgments
This project was partially supported by the National Institute of Environmental Health Sciences (Grant numbers R01ES029275, R01ES031080, R15ES023148, and P30ES005022) and an ExxonMobil research grant for Rutgers University.
Footnotes
Conflict of Interest
The authors declare that they have no conflict of interest.
References
- 1.Hall JM, Couse JF, Korach KS. The Multifaceted Mechanisms of Estradiol and Estrogen Receptor Signaling. J Biol Chem 2001;276:36869–36872. [DOI] [PubMed] [Google Scholar]
- 2.Eddy EM, Washburn TF, Bunch DO, Goulding EH, Gladen BC, Lubahn DB, et al. Targeted Disruption of the Estrogen Receptor Gene in Male Mice Causes Alteration of Spermatogenesis and Infertility. Endocrinology 1996;137:4796–4805. [DOI] [PubMed] [Google Scholar]
- 3.Lubahn DB, Moyer JS, Golding TS, Couse JF, Korach KS, Smithies O. Alteration of reproductive function but not prenatal sexual development after insertional disruption of the mouse estrogen receptor gene. Proc Natl Acad Sci U S A 1993;90:11162–11166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J, et al. Estrogen Receptors: How Do They Signal and What Are Their Targets. Physiol Rev 2007;87:905–931. [DOI] [PubMed] [Google Scholar]
- 5.Prossnitz ER, Arterburn JB. International Union of Basic and Clinical Pharmacology. XCVII. G Protein-Coupled Estrogen Receptor and Its Pharmacologic Modulators. Pharmacol Rev 2015;67:505–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engström O, et al. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 1997;389:753–758. [DOI] [PubMed] [Google Scholar]
- 7.Björnström L, Sjöberg M. Mechanisms of estrogen receptor signaling: Convergence of genomic and nongenomic actions on target genes. Mol Endocrinol 2005;19:833–842. [DOI] [PubMed] [Google Scholar]
- 8.De Coster S, van Larebeke N. Endocrine-disrupting chemicals: Associated disorders and mechanisms of action. J Environ Public Health 2012;2012:713696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meigs L, Smirnova L, Rovida C, Leist M, Hartung T. Animal testing and its Alternatives – the Most Important Omics is Economics. ALTEX 2018; 35:275–305. [DOI] [PubMed] [Google Scholar]
- 10.Stouch TR, Kenyon JR, Johnson SR, Chen X-Q, Doweyko A, Li Y. In silico ADME/Tox: Why models fail. J Comput Aided Mol Des 2003;17:83–92. [DOI] [PubMed] [Google Scholar]
- 11.Maggiora GM. On outliers and activity cliffs - Why QSAR often disappoints. J Chem Inf Model 2006;46:1535. [DOI] [PubMed] [Google Scholar]
- 12.Dearden JC, Cronin MTD, Kaiser KLE. How not to develop a quantitative structure-activity or structure-property relationship (QSAR/QSPR). SAR QSAR Environ Res 2009;20:241–266. [DOI] [PubMed] [Google Scholar]
- 13.Najafabadi MM, Villanustre F, Khoshgoftaar TM, Seliya N, Wald R, Muharemagic E. Deep learning applications and challenges in big data analytics. J Big Data 2015;2:1. [Google Scholar]
- 14.LeCun Y, Bengio Y, Hinton G. Deep learning. Nature 2015;521:436–444. [DOI] [PubMed] [Google Scholar]
- 15.Schmidhuber J Deep Learning in Neural Networks: An Overview. Neural Netw 2015;61:85–117. [DOI] [PubMed] [Google Scholar]
- 16.Ramsundar B, Liu B, Wu Z, Verras A, Tudor M, Sheridan RP, et al. Is Multitask Deep Learning Practical for Pharma? J Chem Inf Model 2017;57,2068–2076. [DOI] [PubMed] [Google Scholar]
- 17.Xu Y, Ma J, Liaw A, Sheridan RP, Svetnik V. Demystifying Multitask Deep Neural Networks for Quantitative Structure–Activity Relationships. J Chem Inf Model 2017;57:2490–2504. [DOI] [PubMed] [Google Scholar]
- 18.Dahl GE, Jaitly N, Salakhutdinov R. Multi-task Neural Networks for QSAR Predictions. arXiv 2014;1406.1231. [Google Scholar]
- 19.Simões RS, Maltarollo VG, Oliveira PR, Honorio KM. Transfer and multi-task learning in QSAR modeling: Advances and challenges. Front Pharmacol 2018;9:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mayr A, Klambauer G, Unterthiner T, Hochreiter S. DeepTox: Toxicity Prediction using Deep Learning. Front Environ Sci 2015;3:80. [Google Scholar]
- 21.Wenzel J, Matter H, Schmidt F. Predictive Multitask Deep Neural Network Models for ADME-Tox Properties: Learning from Large Data Sets. J Chem Inf Model 2019;59:1253–1268. [DOI] [PubMed] [Google Scholar]
- 22.Byvatov E, Fechner U, Sadowski J, Schneider G. Comparison of Support Vector Machine and Artificial Neural Network Systems for Drug/Nondrug Classification. J Chem Inf Comput Sci 2003;43:1882–1889. [DOI] [PubMed] [Google Scholar]
- 23.Korotcov A, Tkachenko V, Russo DP, Ekins S. Comparison of Deep Learning with Multiple Machine Learning Methods and Metrics Using Diverse Drug Discovery Data Sets. Mol Pharm 2017;14:4462–4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koutsoukas A, Monaghan KJ, Li X, Huan J. Deep-learning: Investigating deep neural networks hyper-parameters and comparison of performance to shallow methods for modeling bioactivity data. J Cheminform 2017;9:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Russo DP, Zorn KM, Clark AM, Zhu H, Ekins S. Comparing Multiple Machine Learning Algorithms and Metrics for Estrogen Receptor Binding Prediction. Mol Pharm 2018;15:4361–4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou Y, Cahya S, Combs SA, Nicolaou CA, Wang J, Desai PV, et al. Exploring Tunable Hyperparameters for Deep Neural Networks with Industrial ADME Data Sets. J Chem Inf Model 2019;59:1005–1016. [DOI] [PubMed] [Google Scholar]
- 27.Attene-Ramos MS, Miller N, Huang R, Michael S, Itkin M, Kavlock RJ, et al. The Tox21 robotic platform for the assessment of environmental chemicals – from vision to reality. Drug Discov Today 2013;18:716–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ciallella HL, Zhu H. Advancing Computational Toxicology in the Big Data Era by Artificial Intelligence: Data-Driven and Mechanism-Driven Modeling for Chemical Toxicity. Chem Res Toxicol 2019;32:536–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu H Big Data and Artificial Intelligence Modeling for Drug Discovery. Annu Rev Pharmacol Toxicol 2020;60:573–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu H, Zhang J, Kim MT, Boison A, Sedykh A, Moran K. Big data in chemical toxicity research: The use of high-throughput screening assays to identify potential toxicants. Chem Res Toxicol 2014;27:1643–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao L and Zhu H. Big Data in Computational Toxicology: Challenges and Opportunities. In: Ekins S, editor. Computational Toxicology: Risk Assessment for Chemicals. Hoboken, NJ: John Wiley & Sons, 2018. p. 291–312. [Google Scholar]
- 32.Luechtefeld T, Rowlands C, Hartung T. Big-data and machine learning to revamp computational toxicology and its use in risk assessment. Toxicol Res (Camb) 2018;7:732–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang L, Tan J, Han D, Zhu H. From machine learning to deep learning: progress in machine intelligence for rational drug discovery. Drug Discov Today 2017;22:1680–1685. [DOI] [PubMed] [Google Scholar]
- 34.Dix DJ, Houck KA, Martin MT, Richard AM, Setzer RW, Kavlock RJ. The ToxCast Program for Prioritizing Toxicity Testing of Environmental Chemicals. Toxicol Sci 2007;95:5–12. [DOI] [PubMed] [Google Scholar]
- 35.Judson RS, Houck KA, Kavlock RJ, Knudsen TB, Martin MT, Mortensen HM, et al. In Vitro Screening of Environmental Chemicals for Targeted Testing Prioritization: The ToxCast Project. Environ Health Perspect 2010;118:485–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shukla SJ, Huang R, Austin CP, Xia M. The future of toxicity testing: A focus on in vitro methods using a quantitative high-throughput screening platform. Drug Discov Today 2010;15:997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thomas RS, Paules RS, Simeonov A, Fitzpatrick SC, Crofton KM, Casey WM, et al. The US Federal Tox21 Program: A Strategic and Operational Plan for Continued Leadership. ALTEX 2018;35:163–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hsu C-W, Huang R, Attene-Ramos MS, Austin CP, Simeonov A, Xia M. Advances in high-throughput screening technology for toxicology. Int J Risk Assessment and Management 2017;20:109–135. [Google Scholar]
- 39.Russo DP, Strickland J, Karmaus AL, Wang W, Shende S, Hartung T, et al. Nonanimal models for acute toxicity evaluations: Applying data-driven profiling and read-across. Environ Health Perspect 2019;127:47001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao L, Russo DP, Wang W, Aleksunes LM, Zhu H. Mechanism-Driven Read-Across of Chemical Hepatotoxicants Based on Chemical Structures and Biological Data. Toxicol Sci 2020;174:178–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luechtefeld T, Marsh D, Rowlands C, Hartung T. Machine Learning of Toxicological Big Data Enables Read-Across Structure Activity Relationships (RASAR) Outperforming Animal Test Reproducibility. Toxicol Sci 2018;165:198–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Browne P, Judson RS, Casey WM, Kleinstreuer NC, Thomas RS. Screening Chemicals for Estrogen Receptor Bioactivity Using a Computational Model. Environ Sci Technol 2015;49:8804–8814. [DOI] [PubMed] [Google Scholar]
- 43.Judson RS, Magpantay FM, Chickarmane V, Haskell C, Tania N, Taylor J, et al. Integrated model of chemical perturbations of a biological pathway using 18 in vitro high-throughput screening assays for the estrogen receptor. Toxicol Sci 2015;148:137–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kleinstreuer NC, Ceger P, Watt ED, Martin M, Houck K, Browne P, et al. Development and Validation of a Computational Model for Androgen Receptor Activity. Chem Res Toxicol 2017;30:946–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leach AR and Gillet VJ. Introduction to Chemoinformatics. Dordrecht, The Netherlands: Springer, 2007. [Google Scholar]
- 46.Rogers D, Hahn M. Extended-Connectivity Fingerprints. J Chem Inf Model 2010;50:742–754. [DOI] [PubMed] [Google Scholar]
- 47.Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, et al. Scikit-learn: Machine Learning in Python. J Mach Learn Res 2011;12:2825–2830. [Google Scholar]
- 48.Manning CD, Raghavan P, Schuetze H. The Bernoulli model. In: Introduction to Information Retrieval. New York, NY: Cambridge University Press, 2009. p. 234–265. [Google Scholar]
- 49.Cover TM, Hart PE. Nearest neighbor pattern classification. IEEE Trans Inf Theory 1967;13:21–27. [Google Scholar]
- 50.Breiman L Random Forests. Mach Learn 2001;45:5–32. [Google Scholar]
- 51.Vapnik VN. Methods of Pattern Recognition. In: The Nature of Statistical Learning Theory. New York: Springer Science+Business Media, 2000. p. 123–170. [Google Scholar]
- 52.He K, Zhang X, Ren S, Sun J. Delving Deep into Rectifiers: Surpassing Human-Level Performance on ImageNet Classification. In: Proceedings of the IEEE International Conference on Computer Vision (ICCV). 2015. p. 1026–1034. [Google Scholar]
- 53.Bottou L Large-Scale Machine Learning with Stochastic Gradient Descent. In: 19th International Conference on Computational Statistics. 2010. p. 177–186. [Google Scholar]
- 54.Sutskever I, Martens J, Dahl G, Hinton G. On the importance of initialization and momentum in deep learning. In: Proceedings of the 30th International Conference on Machine Learning. Atlanta, Georgia: 2013. p. 1139–1147. [Google Scholar]
- 55.Nair V, Hinton GE. Rectified Linear Units Improve Restricted Boltzmann Machines. In: Proceedings of the 27th International Conference on Machine Learning. Haifa, Israel: 2010. p. 807–814. [Google Scholar]
- 56.Goodfellow I, Bengio Y, Courville A. Challenges in Neural Network Optimization. In: Learning Deep. Cambridge, MA: The MIT Press, 2016. p. 279–290. [Google Scholar]
- 57.Srivastava N, Hinton G, Krizhevsky A, Sutskever I, Salakhutdinov R. Dropout: A Simple Way to Prevent Neural Networks from Overfitting. J Mach Learn Res 2014;15:1929–1958. [Google Scholar]
- 58.Ng AY. Feature selection, L1 vs. L2 regularization, and rotational invariance. In: Proceedings of the 21st International Conference on Machine Learning. Banff, Canada: 2004. p. 78. [Google Scholar]
- 59.Li M, Soltanolkotabi M, Oymak S. Gradient Descent with Early Stopping is Provably Robust to Label Noise for Overparameterized Neural Networks. In: Proceedings of the 23rd International Conference on Artificial Intelligence and Statistics (AISTATS) 2020. Palermo, Italy: 2020. p. 4313–4324. [Google Scholar]
- 60.Han J, Moraga C. The influence of the sigmoid function parameters on the speed of backpropagation learning. In: Mira J and Sandoval F, editors. International Workshop on Artificial Neural Networks: From Natural to Artificial Neural Computation. Springer, Berlin, Heidelberg: Malaga-Torremolinos, Spain, 1995. p. 195–201. [Google Scholar]
- 61.Fawcett T An introduction to ROC analysis. Pattern Recognit Lett 2006;27:861–874. [Google Scholar]
- 62.Zakharov AV, Peach ML, Sitzmann M, Nicklaus MC. QSAR modeling of imbalanced high-throughput screening data in PubChem. J Chem Inf Model 2014;54:705–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mansouri K, Abdelaziz A, Rybacka A, Roncaglioni A, Tropsha A, Varnek A, et al. CERAPP: Collaborative Estrogen Receptor Activity Prediction Project. Environ Health Perspect 2016; 124:1023–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shen J, Xu L, Fang H, Richard AM, Bray JD, Judson RS, et al. EADB: An estrogenic activity database for assessing potential endocrine activity. Toxicol Sci 2013;135:277–291. [DOI] [PubMed] [Google Scholar]
- 65.Kleinstreuer NC, Ceger PC, Allen DG, Strickland J, Chang X, Hamm JT, et al. A Curated Database of Rodent Uterotrophic Bioactivity. Environ Health Perspect 2016;124:556–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ribay K, Kim MT, Wang W, Pinolini D, Zhu H. Predictive Modeling of Estrogen Receptor Binding Agents Using Advanced Cheminformatics Tools and Massive Public Data. Front Environ Sci 2016;4:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang L, Fourches D, Sedykh A, Zhu H, Golbraikh A, Ekins S, et al. Discovery of novel antimalarial compounds enabled by QSAR-based virtual screening. J Chem Inf Model 2013;53:475–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang J, Deng F, Zeng F, Shanahan AJ, Li WV, Zhang L Predicting long-term multicategory cause of death in patients with prostate cancer: random forest versus multinomial model. Am J Cancer Res 2020;10:1344–1355. [PMC free article] [PubMed] [Google Scholar]
- 69.Organisation for Economic Co-operation and Development. Guidance Document on the Validation of (Quantitative)Structure-Activity Relationship [(Q)SAR] Models. OECD Environ Heal Saf Publ Ser Test Assess 2007;69:1–154. [Google Scholar]
- 70.Tropsha A, Gramatica P, Gombar VK. The importance of being earnest: Validation is the absolute essential for successful application and interpretation of QSPR models. QSAR Comb Sci 2003;22:69–77. [Google Scholar]
- 71.Kim MT, Sedykh A, Chakravarti SK, Saiakhov RD, Zhu H. Critical Evaluation of Human Oral Bioavailability for Pharmaceutical Drugs by Using Various Cheminformatics Approaches. Pharm Res 2014;31:1002–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang W, Kim MT, Sedykh A, Zhu H. Developing Enhanced Blood-Brain Barrier Permeability Models: Integrating External Bio-Assay Data in QSAR Modeling. Pharm Res 2015;32:3055–3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Solimeo R, Zhang J, Kim M, Sedykh A, Zhu H. Predicting chemical ocular toxicity using a combinatorial QSAR approach. Chem Res Toxicol 2012;25:2763–2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Huang R, Sakamuru S, Martin MT, Reif DM, Judson RS, Houck KA, et al. Profiling of the Tox21 10K compound library for agonists and antagonists of the estrogen receptor alpha signaling pathway. Sci Rep 2014;4:5664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rotroff DM, Dix DJ, Houck KA, Kavlock RJ, Knudsen TB, Martin MT, et al. Real-time growth kinetics measuring hormone mimicry for ToxCast chemicals in T-47D human ductal carcinoma cells. Chem Res Toxicol 2013;26:1097–1107. [DOI] [PubMed] [Google Scholar]
- 76.Xing JZ, Zhu L, Gabos S, Xie L. Microelectronic cell sensor assay for detection of cytotoxicity and prediction of acute toxicity. Toxicol In Vitro 2006;20:995–1004. [DOI] [PubMed] [Google Scholar]
- 77.Haji M, Kato K, Nawata H, Ibayashi H. Age-Related Changes in the Concentrations of Cytosol Receptors for Sex Steroid Hormones in the Hypothalamus and Pituitary Gland of the Rat. Brain Res 1981;204:373–386. [DOI] [PubMed] [Google Scholar]
- 78.Knudsen TB, Houck KA, Sipes NS, Singh AV, Judson RS, Martin MT, et al. Activity profiles of 309 ToxCast™ chemicals evaluated across 292 biochemical targets. Toxicology 2011;282:1–15. [DOI] [PubMed] [Google Scholar]
- 79.O’Keefe JA, Handa RJ. Transient elevation of estrogen receptors in the neonatal rat hippocampus. Brain Res Dev Brain Res 1990;57:119–127. [DOI] [PubMed] [Google Scholar]
- 80.Sipes NS, Martin MT, Kothiya P, Reif DM, Judson RS, Richard AM, et al. Profiling 976 ToxCast chemicals across 331 enzymatic and receptor signaling assays. Chem Res Toxicol 2013;26:878–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.MacDonald ML, Lamerdin J, Owens S, Keon BH, Bilter GK, Shang Z, et al. Identifying off-target effects and hidden phenotypes of drugs in human cells. Nat Chem Biol 2006;2:329–337. [DOI] [PubMed] [Google Scholar]
- 82.Yu H, West M, Keon BH, Bilter GK, Owens S, Lamerdin J, et al. Measuring Drug Action in the Cellular Context Using Protein-Fragment Complementation Assays. Assay Drug Dev Technol 2003;1:811–822. [DOI] [PubMed] [Google Scholar]
- 83.Stossi F, Bolt MJ, Ashcroft FJ, Lamerdin JE, Melnick JS, Powell RT, et al. Defining estrogenic mechanisms of bisphenol A analogs through high throughput microscopy-based contextual assays. Chem Biol 2014;21:743–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Martin MT, Dix DJ, Judson RS, Kavlock RJ, Reif DM, Richard AM, et al. Impact of Environmental Chemicals on Key Transcription Regulators and Correlation to Toxicity End Points within EPA’s ToxCast Program. Chem Res Toxicol 2010;23:578–590. [DOI] [PubMed] [Google Scholar]
- 85.United States Environmental Protection Agency. Use of High Throughput Assays and Computational Tools; Endocrine Disruptor Screening Program; Notice of Availability and Opportunity for Comment . Fed Regist 2015;80:35350–35355. [Google Scholar]
- 86.Zhu BT, Lee AJ. NADPH-dependent metabolism of 17β-estradiol and estrone to polar and nonpolar metabolites by human tissues and cytochrome P450 isoforms. Steroids 2005;70:225–244. [DOI] [PubMed] [Google Scholar]
- 87.Schrager S, Potter BE. Diethylstilbestrol exposure. Am Fam Physician 2004;69:2395–2400. [PubMed] [Google Scholar]
- 88.Greenberger LM, Annable T, Collins KI, Komm BS, Lyttle CR, Miller CP, et al. A new antiestrogen, 2-(4-hydroxy-phenyl)-3-methyl-1-[4-(2-piperidin-1-yl-ethoxy)-benzyl]-1H- indol-5-ol hydrochloride (ERA-923), inhibits the growth of tamoxifen-sensitive and -resistant tumors and is devoid of uterotropic effects in mice and rats. Clin Cancer Res 2001;7:3166–3177. [PubMed] [Google Scholar]
- 89.Riggs BL & Hartmann LC Selective Estrogen-Receptor Modulators — Mechanisms of Action and Application to Clinical Practice. N Engl J Med 2003;348:618–629. [DOI] [PubMed] [Google Scholar]
- 90.Stump AL, Kelley KW, Wensel TM. Bazedoxifene: A third-generation selective estrogen receptor modulator for treatment of postmenopausal osteoporosis. Ann Pharmacother 2007;41:833–839. [DOI] [PubMed] [Google Scholar]
- 91.Zhu H, Tropsha A, Fourches D, Varnek A, Papa E, Gramatica P, et al. Combinatorial QSAR modeling of chemical toxicants tested against Tetrahymena pyriformis. J Chem Inf Model 2008;48:766–784. [DOI] [PubMed] [Google Scholar]
- 92.Jaworska J, Nikolova-Jeliazkova N, Aldenberg T. QSAR applicability domain estimation by projection of the training set in descriptor space: A review. Altern Lab Anim 2005;33:445–459. [DOI] [PubMed] [Google Scholar]
- 93.Organization for Economic Co-operation and Development. OECD Principles for the Validation, for Regulatory Purposes, of (Quantitative) Structure-Activity Relationship Models. 2004. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.