

Abstract
The low temperature syntheses of AuTe2 and Ag2Te starting from the elements were investigated in the ionic liquids (ILs) [BMIm]X and [P66614]Z ([BMIm]+=1‐butyl‐3‐methylimidazolium; X = Cl, [HSO4]−, [P66614]+ = trihexyltetradecylphosphonium; Z = Cl−, Br−, dicyanamide [DCA]−, bis(trifluoromethylsulfonyl)imide [NTf2]−, decanoate [dec]−, acetate [OAc]−, bis(2,4,4‐trimethylpentyl)phosphinate [BTMP]−). Powder X‐ray diffraction, scanning electron microscopy, and energy‐dispersive X‐ray spectroscopy revealed that [P66614]Cl is the most promising candidate for the single phase synthesis of AuTe2 at 200 °C. Ag2Te was obtained using the same ILs by reducing the temperature in the flask to 60 °C. Even at room temperature, quantitative yield was achieved by using either 2 mol % of [P66614]Cl in dichloromethane or a planetary ball mill. Diffusion experiments, 31P and 125Te‐NMR, and mass spectroscopy revealed one of the reaction mechanisms at 60 °C. Catalytic amounts of alkylphosphanes in commercial [P66614]Cl activate tellurium and form soluble phosphane tellurides, which react on the metal surface to solid telluride and the initial phosphane. In addition, a convenient method for the purification of [P66614]Cl was developed.
Keywords: ionic liquid; purification of ionic liquids; silver telluride, tellurium; 125Te-NMR
Ag2Te and AuTe2 were obtained from the elements in the ionic liquid [P66614]Cl at room temperature. The reaction is driven by a dissolved tellurium species that reacts with the noble metal. In commercial [P66614]Cl, tellurium is reduced by trialkylphosphane impurities. A purification protocol was developed to suspend the influence of the trialkylphosphane. In the purified medium, a different tellurium species appears.
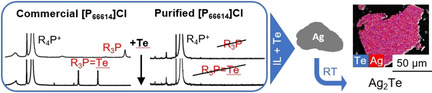
1. Introduction
Metal tellurides are narrow bandgap semiconductors. Numerous studies in recent decades revealed additional physical properties with potential for application, such as their efficient thermoelectric power conversion and photocatalytic activity under radiation ranging from ultraviolet (UV) to near‐infrared (NIR).[ 1 , 2 ] Gold ditelluride AuTe2, naturally found as the mineral calaverite, is discussed as a candidate for thermoelectric applications and is typically synthesized by reacting the elements at 600 °C, followed by annealing at 400 °C.[ 3 , 4 ] Alternatively, the compound can be accessed at 250 °C by wet‐chemical hot‐injection using solutions of highly reactive starting materials TeCl4 and HAuCl4 ⋅ 2H2O in dioctylether/oleylamine. [5]
Silver telluride, Ag2–δTe, in its different phases is an ion conductor, a thermoelectric, a topological insulator and has a large magnetoresistance that can be exploited in magnetic field sensors.[ 6 , 7 , 8 , 9 ] The textbook example for the synthesis of Ag2Te is the reaction of solid silver with tellurium vapor at 475 °C. [10] Thin films are accessible by vacuum deposition of tellurium on chlorine activated silver films. [11] For the synthesis of Ag2Te nanomaterials, which are especially interesting for thermoelectric applications, solvothermal methods are used.[ 6 , 12 , 13 , 14 , 15 , 16 , 17 ] Silver telluride nanowires have been synthesized by the composite‐hydroxide‐mediated (CHM) method at 180 to 225 °C, starting from AgNO3 and elemental tellurium in NaOH/KOH with hydrazine hydrate and ethylenediamine (en) in a Teflon vessel. [6] Ag2Te−Ag nanocomposites are accessible at room temperature by reacting AgCl and tellurium in an aqueous NaOH solution with NaBH4 as reducing agent. The addition of NaCl enhances the solubility of AgCl, which leads to smaller silver grains. [12] Na2TeO3 is often used as a tellurium source because of its good solubility. In a solvothermal co‐reduction approach using a Teflon‐lined stainless steel autoclave at 240 °C, more than 20 μm long nanowires formed from an aqueous solution of AgNO3 with the disodium salt of ethylenediaminetetraacetic acid (Na2(EDTA) ⋅ 2H2O), ethylene glycol (EG), and NaOH. [13] Crooked nanowires were synthesized in the microwave with EG as the solvent, starting from AgNO3, Na2TeO3 and NaOH at 190 °C within 15 min. [14] Nanowires are also accessible through an ammonium‐hydroxide‐buffered hydrothermal synthesis by using AgNO3 as silver source and hydrazine hydrate (N2H4⋅H2O) as reducing agent. [18] Many more hydrothermal approaches with AgNO3 have been published. Nanowires or nanorods can be synthesized by starting with tellurium nanowires (TeNWs) and N2H4 as reducing agent at room temperature or nanotubes with NaTeO3 instead of a tellurium template and NH3 at 120 °C.[ 15 , 16 , 17 ] A broad variety of water‐free syntheses were also reported. Sonochemical synthesis of nanocrystals is possible in en from AgNO3 and tellurium. [19] Nanocrystalline rod‐like crystals of Ag2Te were synthesized at room temperature starting with AgCl and tellurium in en/hydrazine hydrate mixtures. [20] Dendritic nanostructures can be synthesized at room temperature using AgNO3, TeO2, and N2H4⋅H2O in en without stirring or hollow microspheres forming under the same conditions with continuous stirring. [21] Thin films are accessible by electrodeposition in DMSO from NaNO3, AgNO3, and TeCl4. Trialkylphosphans (R 3P) like trioctylphosphane (TOP) are often used surfactants for the activation of tellurium or Ag. For example, TOP−Te (solution of tellurium in TOP) is used as the tellurium source in a hot injection method at 170 °C with dodecanethiol as the ligand and oleylamine as the reducing agent in order to gain nanoparticles. [1] A modification with additional Ag−TOP, extract of silver with TOP from an aqueous AgNO3 solution, as the silver source is also possible. [22] Recently, a surfactant‐free synthesis in an aqueous solution of AgNO3 was also developed using NaHTe as the active tellurium species. [23] A synthesis with elemental silver and tellurium as the starting materials at room temperature (RT) is also possible in liquid NH3 at 7 atm using a Teflon‐in‐glass sealable thick‐walled Schlenk tube. [24]
These versatile methods still have some disadvantages that can be circumvented using ionic liquids (IL). The ability of ILs to dissolve hard‐to‐dissolve elements can reduce the energy consumption for telluride syntheses in comparison to high temperature vapor deposition methods and avoids the use of toxic and/or expensive chemicals. [25] Because of their negligible vapor pressure there is no need for autoclaves, which are necessary for the solvothermal synthesis. In recent studies, it was discovered that almost insoluble chalcogens (Ch = Se, Te) can be activated by ionic liquids. Zhang et al. demonstrated that Ch react with the phosphonium cation at 220 °C and form PR 3−Ch (R = C4, C6, C14 alkyl chains). These intermediate trialkylphosphane chalcogens can be employed as precursors for the synthesis of metal chalcogenides, e. g. Ag2Te, with silver dicyanamide (Ag[DCA]) as the silver source (yield 51 %). [26] Unfortunately, the decomposition of the ionic liquid prohibits its recycling for multiple reactions and necessitates the use of an autoclave for the reaction. In this study, we investigated the activation of tellurium and the formation of gold telluride AuTe2 and silver tellurides (Ag2Te and Ag5‐δTe3) in [BMIm]X (X = Cl−, [HSO4]−) and [P66614]Z (Z = Cl−, Br−, [DCA]−, [NTf2]−, [dec]−, [OAc]−,[BTMP]−). For Ag2Te we describe a convenient one‐pot synthesis at temperatures down to RT that preserves the IL without the need for an autoclave.
2. Results and Discussion
2.1. Synthesis of Gold and Silver Tellurides in ILs
At first, the influence of different ILs on the formation of AuTe2 was analyzed at 200 °C. We used commercially available, easy to synthesize ILs that have the potential for upscaling the process. Two cations were tested, [BMIm]+ and [P66614]+, which both can form room temperature ionic liquids (RTILs) and have proved as suitable media for inorganic synthesis. [25] We then focused on phosphonium based ILs because they show higher thermal stability than the imidazolium salts. [27] Anions for [P66614]+ were Cl−, Br−, [DCA]−, [NTf2]−, [dec]−, [OAc]−, [BTMP]− and for [BMIm]+, Cl−, and [HSO4]−. Tellurium heated for 16 h to 200 °C in ILs with the anions [dec]−, [OAc]−, [DCA]−, and [BTMP]− showed a purple reaction solution and, after separation of the ILs and addition of protic solvents like EtOH, a black precipitate of tellurium (powder X‐ray diffraction, PXRD, in the supporting information, S1) was formed. When a mixture of tellurium and gold powder were treated in the same way, no significant yield of AuTe2 could be obtained in these ILs (Figure 1). However, the synthesis of single phase (according to PXRD) AuTe2 was achieved in ILs with chloride anions.
Figure 1.
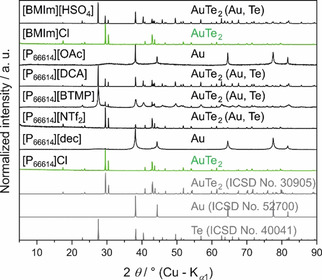
PXRD patterns of the solid products after the reaction of Au and Te in various ILs for 16 h at 200 °C. Single phase synthesis of AuTe2 was achieved in [BMIm]Cl and [P66614]Cl. Bottom: Pattern of AuTe2 simulated on the basis of the Inorganic Crystal Structure Database No. ICSD‐30905, pattern of Au simulated on the basis of the Inorganic Crystal Structure Database No. ICSD‐52700 and pattern of Te simulated on the basis of the Inorganic Crystal Structure Database No. ICSD‐40041.
In scanning electron microscope (SEM) studies of the product from [P66614]Cl, the particles appear to be porous agglomerates of a nanocrystalline powder (Figure 2). Energy‐dispersive X‐ray (EDX) spectroscopy confirmed that small particles have a homogenous distribution of gold and tellurium in the molar ratio of 1 : 2. Some of the larger particles, however, enclose aggregates of unreacted gold grains in the core. Their diameters (S2) fit to the starting gold material (0.3 to 0.8 microns). Possible reasons for this could be wetting problems of gold by the IL or passivation of gold by the formed shell of AuTe2, combined with insufficient mechanical crushing by the magnetic stirring bar.
Figure 2.

EDX map of the solid reaction products from the synthesis in [P66614]Cl after 16 h at 200 °C. Left: Au+2 Te; particle with unreacted gold in a shell of AuTe2. Right: 2 Ag+Te; even large particles show homogenous distribution of silver and tellurium in the molar ratio 2 : 1.
To shed more light on this, passivation experiments were carried out, in which a gold flake and tellurium powder were placed in [P66614]Cl and heated to 200 °C without stirring. After 16 h, the gold flake was covered with a dark and hard layer, which withstands mechanical scratching with a needle (S3). SEM reveals a rough surface, in contrast to the unchanged smooth surface of a gold flake which was exposed to the IL without tellurium. EDX shows the formation of a telluride layer with the molar ratio Au : Te = 1 : 2 (S4). This indicates that elemental tellurium is activated and forms a mobile species in the chloride IL, which then reacts on the surface of the gold particles.
To test whether only tellurium is forming mobile species, diffusion experiments were performed. Powders of tellurium or gold were placed at opposite ends of a H‐shaped tube with central fritted glass filter. Both were carefully covered with [P66614]Cl, until the horizontal connection between the two sides was completely filled and the two elements were connected via the liquid phase. The tube was heated in an oil bath at 200 °C. After several hours, the IL on the tellurium side turned yellow, indicating the formation of a mobile tellurium species. Over time, the coloring expanded to the gold side. After three days, the gold had lost its metallic luster (S5). After one week, the solids were separated from the IL and washed with dichloromethane (DCM). The PXRD pattern of the brown powder that formed on the gold side showed only the reflections of gold. Obviously, the layer of AuTe2 on the gold surface was not thick enough to be detected with PXRD. The powder on the tellurium side was unreacted tellurium.
The results of the synthesis and the passivation and diffusion experiments can be merged into a model of the reaction. Wetting problems lead to an aggregation of the gold particles when gold powder is used. The chloride IL activates tellurium. The mobile tellurium species reacts on the surface of the gold particles forming a AuTe2 layer. Further reaction necessitates diffusion through this solid shell. If the agglomerates of gold are large, the reaction remains incomplete at the given reaction time and temperature.
When silver was used in the diffusion experiment, the same color change of the IL occurred, but the color on the silver side became never as deep as for the gold system. After two days, the surface of the silver powder had turned black and after one week, black needles had grown on the silver surface into the liquid phase (S6). These needles consisted of pure Ag2Te, as shown by PXRD. In the passivation experiment, the silver flake largely disintegrates within 16 hours to form a dendritic dark grey material (S7). The phase formation proceeds much faster than in the gold system. Seemingly, there are no passivation effects, which might be attributed to the mobility of silver cations in Ag2Te.
Starting from the silver powder, the influence of different ILs on the formation of Ag2Te was examined. The ILs are the same as in the gold system but the systems showed a completely different behavior. Ag2Te forms in the halide ILs, as well as in all other used ILs at 200 °C, yet the chloride ILs worked best. EDX showed a homogeneous distribution of silver and tellurium (molar ratio Ag : Te = 2 : 1) as shown in Figure 2, in even larger particles as for the gold system. In [BMIm][HSO4], the tellurium‐richer Ag5–δTe3 phase formed as main product. In [OAc]− or [DCA]− containing ILs, the formed material contained residuals of unreacted silver. Reaction mixtures that contained [OAc]−, [DCA]−, or [dec]− showed an intense red to purple color, indicating a different mobile tellurium species as observed in the other ILs.
At 200 °C, the used ILs decompose under formation of trialkylphoshane species (R 3P). [27] The Lewis bases R 3P are known to react with tellurium.[ 26 , 28 ] Reducing the reaction temperature to 105 °C led to single phase telluride in [P66614]Cl and [BMIm]Cl as well as to an extension of the reaction time necessary for full conversion from 4 h to 16 h. In [P66614]Cl, full conversion to Ag2Te is achieved even at 60 °C (Figure 3). This result is independent of the typical (phosphane) impurities of commercially available [P66614]Cl. Below this temperature, the increasing viscosity of the IL becomes an issue as stirring and diffusion are hindered and agglomeration of metal particles occurs, which results in residuals of unreacted silver. Addition of 5 mol % [P66614]Cl to [P66614][NTf2], which has a lower viscosity but gives no reaction products at room temperature, resulted in only partial conversion of the silver. Using a mixture of 2 mol % of [P66614]Cl in the inert solvent dichloromethane (DCM), however, yielded single phase Ag2Te after 16 h at RT (Figure 3).
Figure 3.
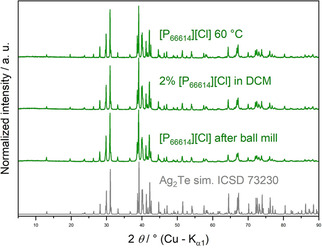
PXRD patterns of Ag2Te from 2 Ag+Te with [P66614]Cl. 60 °C is the lowest temperature at which the reaction with a magnetic stirrer in neat IL was successful. At RT, dilution the IL with the inert solvent DCM or using a planetary ball mill was successful in the synthesis of single phase Ag2Te. Bottom: Pattern of Ag2Te simulated on the basis of the Inorganic Crystal Structure Database No. ICSD‐73230.
In an alternative approach, we charged a planetary ball mill with the two elements and [P66614]Cl. A planetary ball mill was chosen because the high viscosity of the IL prevents the ball's movement in most vibrating ball mills at the lowest frequency at which the ball mills work steadily. Over the entire reaction time, the maximum temperature was 38.6 °C. The product was single phase Ag2Te (Figure 3). Since the width of the X‐ray reflections does not differ significantly for the products of all three methods, their crystalline domains appear to be of a similar size.
2.2. Mechanism of the Activation of Tellurium in [P66614]Cl at 60 °C
As shown by the single phase synthesis of silver telluride and the diffusion experiments, [P66614]Cl activates tellurium even at room temperature. We used in‐situ liquid‐phase 31P and 125Te‐NMR spectroscopy to gain further insight into the activation mechanism.
In a first step, we investigated the impurities in the commercially available [P66614]Cl with a specified purity of > 95 % with 31P‐NMR and mass spectrometry, as impurities could play a major role in the tellurium activation. Our results are consistent with a similar analysis by Deferm et al. [27] In the 31P‐NMR spectrum (Figure 4), [P66614]+ yields a signal at 33.29 ppm. However, several further signals indicate the presence of other phosphorus‐containing species. Tetraalkylphosphonium salts are usually synthesized in a quaternization reaction of tertiary phosphanes by the nucleophilic addition of haloalkanes RX (X = Cl, Br, I). [29] The 31P spectrum provides evidence for tertiary phosphanes at −32.21 ppm and secondary phosphanes at −69.52 ppm. The mass spectrum confirms the presence of trihexylphosphane as protonated molecules (M+Z) (m/z: 287.286 [M+H]) and dihexyltetradecylphosphane (m/z: 399.411 [M+H]) as well as hexyltetradecylphosphane (m/z: 315.317 [M+H]). It is not possible to distinguish between trihexylphosphane and dihexyltetradecylphosphane solely by 31P‐NMR spectroscopy, because the influence of the alkyl chain length on the chemical shift of the central phosphorus is negligible when the number of carbon atoms exceeds four. [30] Tertiary phosphanes can be provided by a free radical addition of gaseous phosphane to α‐olefins. [31] Although the addition at the α‐carbon atom is preferred, the phosphane can also react with the β‐carbon atom, giving a branched alkyl phosphane like 1‐methylpentyl‐di‐n‐hexylphosphane, which was also detected at −18.90 ppm. When the branched phosphane is present during the quaternization, a branched phosphonium cation like 1‐methylpentyl‐di‐n‐hexyl(tetradecyl)phosphonium can be expected as a side product. The resonance of the branched phosphonium cation is indeed found at 37.11 ppm. Due to their high reactivity, phosphanes are sensitive to oxygen. A broad signal at 45.38 ppm indicates the presence of phosphane oxides. Due to the broadening, it is not possible to distinguish between different phosphane oxides. However, mass spectroscopy was able to detect the sodium adducts of trihexylphosphane oxide (M+Na) (m/z: 325.26 [M+Na]) and dihexylphosphane oxide (m/z: 241.169 [M+Na]). The signal at 12.03 ppm splits into a doublet when no proton decoupling is used during the measurement (J = 482 Hz). The chemical shift and coupling constant indicate that the resonance originates from the protonated tertiary phosphanes. [30]
Figure 4.
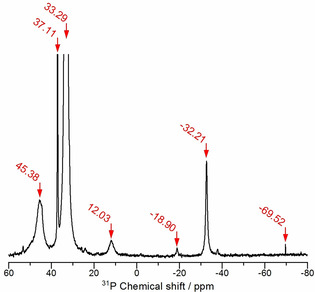
Section of the 31P‐NMR spectrum of commercial [P66614]Cl. The frame is zoomed in on one of the weak signals of the impurities. The signal at 33.29 ppm is the resonance of the [P66614]+ cation.
It is known that tertiary phosphanes (R 3P) can be oxidized by elemental tellurium and form organophosphorus(V) tellurides. [28] After the treatment of [P66614]Cl with tellurium for 16 h at 60 °C, the resonances of all tertiary and secondary phosphanes disappeared in the 31P‐NMR spectrum. In parallel, two new signals show up at 10.50 ppm and −7.58 ppm. Next to the latter one, two satellites were visible that might be an overlapping doublet, indicating the spin‐spin coupling between phosphorus and tellurium with a coupling constant of 1672 Hz. It is plausible that the new signals originate from the oxidation products of phosphane impurities by the elemental tellurium. Because of the low natural abundance of the NMR‐sensitive isotope of tellurium 125Te and the low concentration of the dissolved tellurium species, it is difficult to find the corresponding signal in the 125Te‐NMR spectrum. When 125Te‐enriched tellurium is used, the signals in the 31P‐NMR spectrum at 10.50 ppm and −7.58 ppm completely split into doublets (Figure 5).
Figure 5.
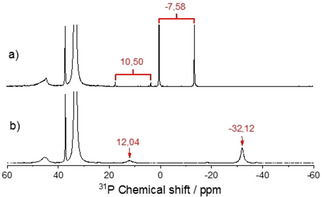
31P‐NMR spectra of the IL (a) of [P66614]Cl after the reaction with tellurium, and (b) after the synthesis of Ag2Te.
The 125Te NMR spectrum also reveals two doublets (Figure 6). The first one at −829.87 ppm has the same coupling constant of 1672 Hz as the doublet at −7.58 ppm in the 31P‐NMR spectrum. This proves the covalent P−Te bond and the formation of trihexylphosphane telluride. The second doublet in the 125Te‐spectrum at −903.34 ppm has a coupling constant of 1705 Hz. This is identical to the coupling constant of the doublet at 10.50 ppm in the 31P spectrum. These resonances originate from the oxidation product of the branched trialkylphosphonium cation with elemental telluride. These 31P‐ and 125Te‐NMR results reveal that the oxidation products of tertiary phosphanes are present as mobile tellurium species in tetraalkylphosphonium based ionic liquids. They probably act as intermediates in the formation of metal tellurides.
Figure 6.
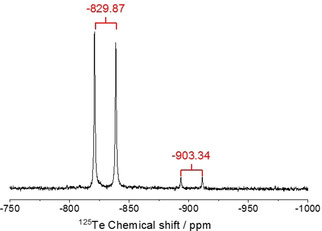
125Te‐NMR spectrum of the IL [P66614]Cl after the reaction with tellurium.
After the reaction of equivalent amounts of tellurium and silver in [P66614]Cl, the IL was again analyzed with 31P‐NMR spectroscopy. The resonances of the tertiary, secondary and protonated phosphane reoccur in the spectrum (Figure 5 b). A possible reaction mechanism might be the initial reduction of elemental tellurium by the phosphane impurities in the IL. The trialkylphosphane telluride then reacts with the elemental silver to form Ag2Te and restores the phosphanes. Since the phosphanes are restored after the reaction, only catalytic amounts are necessary.
To test whether the tellurium activation is based only on the phosphane impurities or further activation processes are involved, we repeated our experiment with purified [P66614]Cl. Most of the purification protocols in literature rely on liquid extraction with hexane or heptane.[ 27 , 32 ] Using these protocols, we always experienced a partial blend of the IL and the extraction solvent, and furthermore, incomplete extraction of the phosphane impurities. We thus developed a protocol to purify the IL by adsorption on silica gel and stepwise elution. First, the impurities are mobilized by the almost nonpolar solvent diethyl ether. Then the phosphonium cations and the chloride ions are remobilized by the polar solvent acetonitrile. After the eluent had been removed, purified [P66614]Cl remained with a yield of 30 to 40 %. The organic phase of the first step can be reused to repeat the process.
The 31P spectra of several batches of purified [P66614]Cl showed no evidence for secondary or tertiary phosphanes in the region from 10 ppm to −150 ppm (S8). However in some of the batches, the resonance of 1‐methylpentyl‐di‐n‐hexyl(tetradecyl) phosphonium (next to the main signal of [P66614]Cl) as well as resonances at 44.7 ppm and 53.4 ppm are still present in the spectra. These resonances have chemical shifts similar to phosphane oxides, although no phosphane oxides were found in mass spectra. The additional signals might originate from other phosphonium cations with more than one branched alkyl group that are formed by the addition of phosphane to the β‐carbon of α‐olefins. This would also explain the similarity of their elution behavior with that of [P66614]+.
The reaction with tellurium and the synthesis of Ag2Te were repeated with purified [P66614]Cl at 100 °C and 60 °C. After the treatment with elemental tellurium, no visible change of the IL is noticed and 125Te and 31P‐NMR spectra gave no evidence for phosphane tellurides in the IL. The 31P‐NMR spectrum of the IL remained unchanged, even after the (successful) synthesis of Ag2Te. This means, the phosphonium cation remains inert during the syntheses at these moderate temperatures. An alternative mobile tellurium species must exist since a quantitative yield of Ag2Te was nevertheless obtained in the purified IL at 60 °C. The lack of resonances in the 125Te‐NMR spectrum leads to the assumption that their concentration is lower than the detection limit.
3. Conclusions
In this article, we demonstrated the activation of tellurium and the formation of silver tellurides in [BMIm]X (X = Cl−, [HSO4]−) and [P66614]Z (Z = Cl−, Br−, [DCA]−, [NTf2]−, [dec]−, [OAc]−, [BTMP]−) at low temperatures. [P66614]Cl has shown to be the best choice for the quantitative synthesis of AuTe2 at 200 °C and Ag2Te down to 60 °C. In comparison to the results of Zhang et al., the described method has decreased the reaction temperature for the synthesis of Ag2Te even down to room temperature. No evidence for a decomposition of the IL or a reaction between the IL cation and the elemental tellurium is found. This is essential for the recyclability of the IL. We found that one possible reaction mechanism is the catalytic tellurium activation by phosphane impurities in commercially available [P66614]Cl. The phosphanes reduce elemental tellurium and form phosphane tellurides. The latter react with solid silver, whereby Ag2Te forms and the phosphane is recovered. The unexpected finding that phosphane‐free [P66614]Cl also allows the quantitative synthesis of Ag2Te at 60 °C implies an additional activation mechanism independent from phosphane, which is yet unknown.
Experimental Section
The starting materials and the products were handled in an argon‐filled glove‐box (MBraun UNILab) with p(O2)/p 0<1 ppm and p(H2O)/p 0<1 ppm. Tellurium (Chempur, 99.9999 %, bar) was distilled and reduced in a hydrogen flow at 400 °C. Gold powder (ABCR, 99.8 %, spherical, 0.3 to 0.5 microns), gold flakes (Chempur 99.99 %), silver powder (−200 mesh), and Ag flakes (Roth, 99 %, 0.1 mm thickness) were used without further treatment. DCM (Fisher Scientific, HPLC grade, amylene stabilized) and EtOH (Fisher Scientific, absolute 99.8 %, Certified AR for Analysis) were degassed and dried in an MBraun SPS. Enriched 125Te (99.7 %) for NMR experiments was purchased from Coretechnet. Ionic liquids were purchased from IoLiTec and dried at 110 °C for 16 h at about 10−3 mbar. Further purification (see below) was applied only for special purpose. [P66614]Cl>95 %, [P66614]Br>95 %, [P66614][DCA]>95 %, [P66614][BTMP]>90 %, [P66614][NTf2] 99 %, [P66614][dec]>95 % and [BMIm]Cl 99 %, [BMIm][HSO4] 98 %.
Synthesis of [P66614][OAc]. [P66614][OAc] was synthesized by anion metathesis using potassium acetate and undried [P66614]Cl. 20.8 g of the chloride IL (40 mmol) was dissolved in 20 mL absolute EtOH, combined under vigorous stirring with 50 mL ethanolic solution of (excess) potassium acetate (4.9 g, 50 mmol) and stirred for 16 h at room temperature. Precipitated potassium chloride was filtered off. After removal of the solvent on a rotary evaporator, during which more salt precipitated, the IL was filtered again and afterwards dried under dynamic vacuum (about 10−3 mbar).
Purification of P66614Cl. The first step of P66614Cl purification was a neutralization of the hydrogen chloride impurities. The IL was stirred with an equivalent volume of 0.1 m NaOH solution for 2 h. Following the separation, the IL phase was washed several times with water. This step might cause foaming during the mixing of IL and water. For the following column chromatography, the IL was adsorbed on silica (Macherey‐Nagel, Silica 60 M, 0.04–0.063 mm), which then was dispersed in diethyl ether and filled in the chromatography column. The column (diameter 34 mm) contained 5 cm of the same silica gel dispersed in diethyl ether. The prepared column was eluted with 100 mL diethyl ether. The obtained eluate was rinsed again on the column, which was then eluted again with 200 mL diethyl ether, giving the fraction with the impurities. The subsequent elution with acetonitrile give the fraction with the purified IL. The acetonitrile was removed by vacuum evaporation in a rotary evaporator, and the remaining IL was dried at 110 °C for 16 h under dynamic vacuum (about 10−3 mbar).
Synthesis setup. In general, the synthesis of the metal tellurides was carried out in 5 mL round‐bottom flasks equipped with a magnetic stirring bar (PTFE lined, cylindrical, 12.5 mm×4.5 mm). The flasks were filled with the elements and 1 g of IL under argon atmosphere in a glovebox, sealed with a glass stopper and high‐vacuum silicon grease and placed in an oil bath. After the mixture was stirred (minimum 350 rpm is necessary) for 16 h at various temperatures, the flask was removed from the bath and allowed to cool to room temperature. After the reaction the samples were brought into a glovebox again followed by the manual separation of the IL by a pipette. The samples were washed with dried DCM three times, except [dec]− and [HSO4]− ILs, which are insoluble in DCM and were thus washed with EtOH. Afterwards the powders were dried in a vacuum drying chamber at room temperature outside the glovebox.
Synthesis of AuTe2. Gold powder (0.2212 mmol, 43.56 mg) and tellurium (2 equiv. 0.4423 mmol, 56.44 mg) were freshly ground in an agate mortar before use. The black precipitate resulting from the reaction at 200 °C in [P66614]Cl or [BMIm]Cl was pure AuTe2 (PXRD). SEM/EDX shows that larger particles may contain enclosures of unreacted gold. Quantitative yield (100 mg) of single phase AuTe2 was achieved.
Passivation experiment. A gold flake (0.7077 mmol, 139.40 mg) was flattened mechanically in an agate mortar. The gold flake and tellurium (1.4155 mmol, 180.61 mg) were overlayed with 1 g of [P66614]Cl in a 5 mL glass flask. Reaction without stirring leads to an opaque brown‐greyish surface layer on the gold flake without any change in morphology of the flake.
Diffusion experiment. For diffusion experiments H‐shaped tubes with central fritted glass filter, to prevent mixing of the solid starting materials and to assure connection only via the liquid phase, was used. Gold (0.33 mmol, 65.34 mg) and tellurium (0.66 mmol, 84.66 mg) were placed in opposite ends of the H‐shaped tubes. [P66614]Cl was then carefully added on both sides till the horizontal connection is completely filled and both parts are only connected via the liquid phase. The apparatus was kept upright in a glass beaker and mounted in an oil bath, were it was kept at 200 °C for one week. Afterwards the apparatus was moved inside a glovebox were the IL was separated on both sides from the solids. The residues were washed with DCM.
Synthesis of Ag2Te. Silver powder (0.58 mmol, 62.83 mg) and tellurium (0.2913 mmol, 37.17 mg) were freshly ground in an agate mortar before use. The black precipitate resulting from the reaction at 200 °C was pure Ag2Te in [P66614]Cl, [P66614][NTf2], [P66614][BTMP], [P66614][dec], or [BMIm]Cl. In [P66614]Cl, or [BMIm]Cl quantitative yield (100 mg) was achieved.
At a reaction temperature of 105 °C only in [P66614]Cl, or [BMIm]Cl quantitative yield (100 mg) was achieved.
At reaction temperatures below 60 °C only [P66614]Cl can be used due to the melting point of [BMIm]Cl (T m = 65 °C).
Passivation experiment. For passivation experiments the starting materials were silver flakes which were cut out of a silver foil (stored in air; 0.1205 mmol, 13 mg) by a scissor and 0.5 equiv. of tellurium (0.06 mmol, 7.69 mg). Both were placed in a 5 mL glass flask that was sealed after addition of the IL and heated to 200 °C for 1 h, 2 h, 4 h and 16 h.
Diffusion experiment. The procedure is the same as for diffusion experiments in the gold system, with silver (1.17 mmol, 125.68 mg) and tellurium (0.58 mmol, 74.34 mg) in [P66614]Cl at 200 °C for one week.
DCM with 2 mol % [P66614]Cl: Silver (1.164 mmol, 125.6 mg), tellurium (0.582 mmol, 74.33 mg) freshly ground in an agate mortar, were placed in an 25 mL Schlenk tube equipped with a magnetic stirring bar with 0.5 g (0.963 mmol) of [P66614]Cl, sealed with a septa and placed on a magnetic stirring plate. After the addition of 3 mL (46.979 mmol) dry DCM, under dynamic argon flow, by using a syringe in the Schlenk technique and turning on the stirring (350 rmp), the reaction mixture turned to dark grey after 1 min. The reaction mixture was left stirring for 16 h at room temperature. The black precipitate was washed three times with DCM and characterized by PXRD. Single phase Ag2Te in quantitative yield was obtained.
Planetary Ball Mill: Silver (1.164 mmol, 125.6 mg), tellurium (0.582 mmol, 74.33 mg) and 6 mL of [P66614]Cl were filled into a planetary ball mill container (Fritsch Pulverisette 7 premium line; 20 mL container and ten 10 mm balls of yttrium‐stabilized zirconia) inside the glovebox. Afterwards the sealed ball mill containers were placed in the ball mill, mounted in a laboratory fume hood outside the glovebox. The milling program comprised 135 cycles (almost 16 h), each with 2 min. at 200 rpm followed by 5 min. break for cooling the system. Temperature was measured by a sensor (Fritsch MillControl), which was in direct contact with the reaction mixture (T max = 38.6 °C). The obtained dark grey suspension was washed three times with 10 mL of DCM and centrifuged with 4000 rpm for 5 min in a glovebox. 200 mg (quantitative yield) of pure Ag2Te were obtained.
Powder X‐ray Diffraction. Characterization of solid reaction products were done by powder X‐ray diffraction (PXRD) at 296(1) K on an X'Pert Pro MPD diffractometer (PANalytical), equipped with a curved Ge(111) monochromator, using Cu Kα 1 radiation (λ = 154.056 pm).
SEM/EDX Measurements. Powder samples for SEM/EDX were fixed in two compound epoxy polymer pellet in which graphite was dispersed. The pellets were wet‐ground with a MetaServ 250 (Buehler, silicon carbide grinding paper) and subsequently polished with a VibroMet 2 (Buehler, MasterPrep alumina suspension 0.05 μm). The polished pellets were fixed on the sample holders with a graphite pad and sputtered with carbon. Needle shaped crystals and flakes were glued to a carbon pad. Scanning electron microscopy (SEM) was performed by using an SU8020 instrument (Hitachi) with a triple detector system for secondary and low‐energy backscattered electrons (U e = 3 kV). The compositions of the samples were determined by quantitative EDX (U e = 20 kV) analysis using an SU8020 instrument (Hitachi) equipped with a Silicon Drift Detector X–MaxN (Oxford). In light of the preparation method, the elements C and O (both part of the polymer) and Al (from polishing suspension) were omitted in EDX quantifications.
NMR Spectroscopy. For the liquid‐state NMR experiments a Bruker Avance Neo 300 MHz spectrometer with a 5 mm high‐resolution probe was used. All samples were prepared and sealed with polytetrafluoroethylene caps in low pressure/vacuum 5 mm NMR tubes from Deutero in the glove box under argon atmosphere. The samples of the ionic liquid after the solvation of tellurium were filtered, using syringe filters with the pore width 0.45 μm. 125Te enriched tellurium (>99.7 %, Cortechnet) was used as starting material in the synthesis and experiments that were analyzed by 125Te‐NMR‐spectroscopy. The filtered ILs were not diluted with a deuterated solvent, instead capillaries filled with DMSO‐d6 were added to the IL inside the NMR tube to ensure field‐frequency lock. The 31P‐NMR spectra were recorded at the transmitter frequency of 121.49 MHz using 128 scans, a relaxation delay of 10 s and a pulse length of 8.3 μs. The chemical shifts in the 31P‐NMR spectra were referenced relative to H3PO4. The 125Te‐NMR spectra were recorded at the transmitter frequency 94.65 MHz using 16000 to 32000 scans. In order to shorten the relaxation time and increase the excitation bandwidths, a 30° instead of a 90° pulse with a pulse length of 3.33 μs and relaxation delay of 5 s was used. To scan a large chemical shift range from 7000 to −4000 ppm the transmitter frequency offset was varied in steps of 104159.35 Hz. The chemical shift was measured relative to Me2Te (δ = 0 ppm). For calibration the secondary external standard Te(OH)6 (δ = 707 ppm) was used [33]
HPLC‐MS. For the HPLC‐MS measurement a 1260 infinity HPLC coupled with 6538 UHD Accurate Mass Q‐TOF LC/MS was used. The samples were prepared in the glove box in glass vials and dissolved in dry acetonitrile after withdrawal. For separation, a Zorbax Extend‐C18 1.8 μm (2.1×50 mm) column was used at 25 °C. After injection of 1.00 μl the elution started with a mixture of 40 % acetonitrile for mobile phase A and 60 % water for mobile phase B at a flow rate of 0.5 ml/min from 0 to 10 min. Afterwards a linear gradient was applied from 40 % to 100 % A, from 10 to 20 min, hold from 20 to 50 min and back from 100 % to 40 %, from 50 to 51 min. These conditions were maintained for another 5 min. For the electrospray ionization we used the following parameter: nebulizer pressure 50 psig, fragmentor voltage 200 V, capillary voltage 4 kV, drying gas temperature 350 °C and gas flow 10 L/min.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We gratefully acknowledge the help with column chromatography and planetary ball mill by Dr. Kai Schwedtmann and Christian Vogt (both TU Dresden). This work was supported by the Deutsche Forschungsgemeinschaft (DFG) within the Priority Program SPP 1708.
M. A. Grasser, T. Pietsch, E. Brunner, T. Doert, M. Ruck, ChemistryOpen 2021, 10, 117.
References
- 1. Zhou W., Zhao W., Lu Z., Zhu J., Fan S., Ma J., Hng H. H., Yan Q., Nanoscale 2012, 4, 3926–3931. [DOI] [PubMed] [Google Scholar]
- 2. Jiang W., Wu Z., Zhu Y., Tian W., Liang B., Appl. Surf. Sci. 2018, 427, 1202–1216. [Google Scholar]
- 3. Perumal S., Roychowdhury S., Biswas K., J. Mater. Chem. C 2016, 4, 7520–7536. [Google Scholar]
- 4. Charoenphakdee A., Kurosaki K., Harnwunggmoung A., Muta H., Yamanaka S., J. Alloys Compd. 2010, 496, 53–55. [Google Scholar]
- 5. Sakata T., Mott D. M., Maenosono S., J. Mater. Res. 2013, 28, 2106–2112. [Google Scholar]
- 6. Li F., Hu C., Xiong Y., Wan B., Yan W., Zhang M., J. Phys. Chem. C 2008, 112, 16130–16133. [Google Scholar]
- 7. Zhang W., Yu R., Feng W., Yao Y., Weng H., Dai X., Fang Z., Phys. Rev. Lett. 2011, 106, 15–18. [DOI] [PubMed] [Google Scholar]
- 8. Xu R., Husmann A., Rosenbaum T. F., Saboungi M. L., Enderby J. E., Littlewood P. B., Nature 1997, 390, 57. [Google Scholar]
- 9. Zhao Y., Yang W., Schnyders H. S., Husmann A., Zhang G., Ren Y., Price D. L., Mao H. K., Saboungi M. L., Phys. Rev. B 2018, 98, 205126. [Google Scholar]
- 10. Hönigschmid O., Sachtleben R., Wintersberger K., Z. Anorg. Allg. Chem. 1933, 212, 242–256. [Google Scholar]
- 11. Chen R., Xu D., Guo G., Gui L., Electrochim. Acta 2004, 49, 2243–2248. [Google Scholar]
- 12. Lin F. H., Liu C. J., Green Chem. 2016, 18, 5288–5294. [Google Scholar]
- 13. Xiao F., Chen G., Wang Q., Wang L., Pei J., Zhou N., J. Solid State Chem. 2010, 183, 2382–2388. [Google Scholar]
- 14. Pei J., Chen G., Jia D., Yu Y., Sun J., Xu H., Qiu Z., New J. Chem. 2014, 38, 59–62. [Google Scholar]
- 15. Samal A. K., Pradeep T., J. Phys. Chem. C 2009, 113, 13539–13544. [Google Scholar]
- 16. Zuo P., Zhang S., Jin B., Tian Y., Yang J., J. Phys. Chem. C 2008, 112, 14825–14829. [Google Scholar]
- 17. Qin A., Fang Y., Tao P., Zhang J., Su C., Inorg. Chem. 2007, 46, 7403–7409. [DOI] [PubMed] [Google Scholar]
- 18. Chang Y., Guo J., Tang Y. Q., Zhang Y. X., Feng J., Ge Z. H., CrystEngComm 2019, 21, 1718–1727. [Google Scholar]
- 19. Li B., Xie Y., Liu Y., Huang J., Qian Y., J. Solid State Chem. 2001, 158, 260–263. [Google Scholar]
- 20. Jiang Y., Wu Y., Yang Z., Xie Y., Qian Y., J. Cryst. Growth 2001, 224, 1–4. [Google Scholar]
- 21. Dong G. H., Zhu Y. J., CrystEngComm 2012, 14, 1805–1811. [Google Scholar]
- 22. Cadavid D., Ibáñez M., Shavel A., Durá O. J., López De La Torre M. A., Cabot A., J. Mater. Chem. A 2013, 1, 4864–4870. [Google Scholar]
- 23. Wang H., Liu X., Zhang B., Huang L., Yang M., Zhang X., Zhang H., Wang G., Zhou X., Han G., Chem. Eng. J. 2020, 393, 124763. [Google Scholar]
- 24. Henshaw G., Parkin I. P., Shaw G. A., J. Chem. Soc. Dalton Trans. 1997, 231–236. [Google Scholar]
- 25. Wolff A., Pallmann J., Boucher R., Weiz A., Brunner E., Doert T., Ruck M., Inorg. Chem. 2016, 55, 8844–8851. [DOI] [PubMed] [Google Scholar]
- 26. Zhang T., Schwedtmann K., Weigand J. J., Doert T., Ruck M., Chem. Eur. J. 2018, 24, 9325–9332. [DOI] [PubMed] [Google Scholar]
- 27. Deferm C., Van den Bossche A., Luyten J., Oosterhof H., Fransaer J., Binnemans K., Phys. Chem. Chem. Phys. 2018, 20, 2444–2456. [DOI] [PubMed] [Google Scholar]
- 28. Nordheider A., Woollins J. D., Chivers T., Chem. Rev. 2015, 115, 10378–10406. [DOI] [PubMed] [Google Scholar]
- 29. Bradaric C. J., Downard A., Kennedy C., Robertson A. J., Zhou Y., Green Chem. 2003, 5, 143–152. [Google Scholar]
- 30. Quin L., Williams A., Practical Interpretation of P-31 NMR Spectra and Computer Assisted Structure Verification, Advanced Chemistry Development, Toronto, Canada, 2004. [Google Scholar]
- 31. Rauhut M. M., Currier H. A., Semsel A. M., Wystrach V. P., J. Org. Chem. 1961, 26, 5138–5145. [Google Scholar]
- 32. Ramnial T., Taylor S. A., Bender M. L., Gorodetsky B., Lee P. T. K., Dickie D. A., McCollum B. M., Pye C. C., Walsby C. J., Clyburne J. A. C., J. Org. Chem. 2008, 73, 801–812. [DOI] [PubMed] [Google Scholar]
- 33. Totsch B. W., Peringer P., Sladky F., J. Chem. Soc. Chem. Commun. 1981, 841–842. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary