

Abstract
Generating and characterizing immunoreagents to enable studies of novel emerging viruses is an area where ensembles of synthetic genes, recombinant antibody pipelines and modular antibody-reporter fusion proteins can respond rapidly. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) continues to spread through the global population causing widespread morbidity, mortality, and socioeconomic chaos. Using SARS-CoV-2 as our model and starting with a gBlocks™ encoded nucleocapsid (N) gene, we purified recombinant protein from E. coli, to serve as bait for selecting semi-synthetic nanobodies from our Nomad single-pot library. Clones were isolated in days and first fused to Gaussia luciferase to determine EC50 in the tens of nM range, and second fused to the ascorbate peroxidase derivative APEX2 for sensitive detection of SARS-CoV-2 infected cells. To generate inherently fluorescent immunoreagents, we introduce novel periplasmic sdAb fusions made with mNeonGreen and mScarlet-I which were produced at milligram amounts. The fluorescent fusion proteins enabled concise visualization of SARS-CoV-2 N in the cytoplasm but not nucleus 24 h post infection, akin to the distribution of SARS-CoV N, thereby validating these useful imaging tools. SdAb reactivity appeared specific to SARS-CoV-2 with very much weaker binding to SARS-CoV, and no noticeable cross-reactivity to a panel of overexpressed human codon optimized N proteins from other CoV. High periplasmic expression levels and in silico immortalization of the nanobody constructs guarantees a cost-effective and reliable source of SARS-CoV-2 immunoreagents. Our proof-of-principle study should be applicable to known and newly emerging CoV to broaden the tools available for their analysis and help safeguard human health in a more proactive than reactive manner.
Keywords: fluorescent protein, coronavirus, nucleocapsid, nanobody, SARS-CoV-2, emerging pathogen
Graphical Abstract
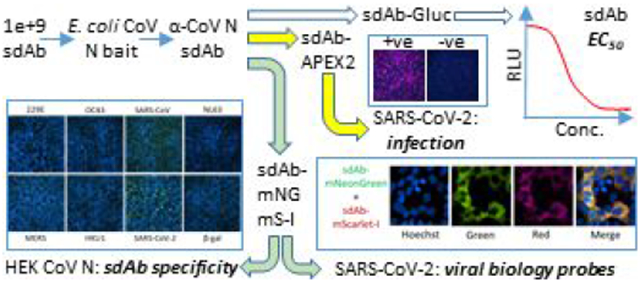
INTRODUCTION
Synthetic biology is enabling countermeasure development to emerging infectious diseases to begin before a pathogen is actually available onsite in the laboratory. Once pathogen genomes are made publically available from an outbreak researchers, the world over, can use gene synthesis to assemble vaccine candidates(1) and component proteins for structural/biological studies(2) and antibody development. The latter process, can leverage single-pot repertoires of affinity scaffolds to isolate binders within days (for review see (3)) which can then be fused to reporter proteins to enable single-step pathogen recognition. Here, we wished to apply the single-pot antibody reporter fusion pipeline to severe acute respiratory syndrome coronavirus (SARS-CoV-2) which causes coronavirus disease 2019 (COVID-19)(4). Our objective was to develop a basic toolkit to generate and characterize highly specific immunoreagents that would facilitate recognition of target antigen, infected cells, and enable single-step high resolution fluorescence microscopy. Critically, the process needed to be as seamless as possible with guaranteed compatibility between antibody expression and reporter protein expression rather than case-by-case optimization and mixing-and-matching. If successful, the proof-of-principle study would indicate the processes could be applied to other CoV to develop a broader immunoreagent toolkit for studying viral biology, viral classification and pandemic preparedness.
Our pipeline begins with the single-pot phage display repertoire “Nomad”, a semi-synthetic assembly of nanobodies (VHH, variable domains of heavy chain only antibodies; sdAb, single-domain antibodies). (5) The diversity of the initial 1e+6 sdAb gene repertoire had been expanded in vitro by a combination of complementarity determining region (CDR) shuffling and random mutagenesis to yield a productive 1e+9 member library. Cycles of “panning” are performed over the course of a week where the desired sdAb are enriched from the vast array of non-binders by antigen capture, elution and phage amplification in E. coli. SdAb are legendary in addressing pathogen crevices,(6) viral canyons,(7) cryptic viral concave(8) and planar/convex epitopes.(9)
Our previous success in targeting nucleoprotein (NP) of species of the genera Ebolavirus and Marburgvirus with Nomad lead us to target the functionally equivalent viral genome chaperone protein of SARS-CoV-2 which is nucleocapsid (N). The N protein is multifunctional and is the most abundant viral component in the infected cell and virus particles and exhibits overall structural conservation throughout the CoV family, though with varying degrees of amino acid variation (for review see(10)). Newly synthesized N protein assembles into dimers and then oligomers to envelope the 26-30 kb positive sense single stranded RNA genome to form the ribonucleocapsid which is packaged into virus particles and makes a highly avid target for antigen sandwich assays. Though infected hosts do make antibodies against N,(11) the interaction does not appear to be a cyclical process of surveillance-selection-escape, as occurs with the external viral spike (S) antigen responsible for receptor binding and cell membrane fusion.(12, 13) Consequently, N amino acid diversity fluctuates far less through time and across geographies making it a convenient marker for classifying past, contemporary and (potentially) future CoV yet to emerge. N protein is present in large amounts in serum early during CoV infection, and has been used as a biomarker of infection in SARS(14-16), Middle Eastern respiratory syndrome (MERS)(17) and COVID-19.(18) Recombinant CoV N proteins can be produced in both prokaryotic(19) and eukaryotic cells(20, 21), in the absence of other viral factors, allowing materials to be made for sdAb selection and affinity/specificity characterization.
Our first reporter protein was Gaussia luciferase (Gluc) initially developed by Tannous and colleagues (22) which we had previously fused to sdAb to enable equilibrium concentration at 50 % (EC50 ) determinations to be made by ELISA by leveraging the high sensitivity and large dynamic range of the enzyme.(8, 9) Gluc has five disulphide bonds(23) and so requires the oxidizing environment of the periplasm to fold efficiently, which is convenient for sdAb fusions since phage display naturally transits antibodies through the periplasmic compartment to enable their own disulphide bond(s) to form. Our second reporter was the soybean ascorbate peroxidase derivative APEX2 initially developed by Lam and colleagues(24) for proximity labelling in living cells after intracellular expression. Normally cytosolic, we had substituted a lone surface exposed cysteine for serine to enable the enzyme to be efficiently produced in the periplasm to allow haem cofactor incorporation through the E. coli outer membrane. When fused to sdAb specific for filoviral NP, the system enabled recognition of filoviral infected cells using fluorescent substrate Amplex™ UltraRed or colorimetric substrate 3,3’-diaminobenzidine (DAB) and conveniently enabled Western blotting using DAB.(25) While the catalytic turnover of APEX2 allows for very rapid and sensitive staining, the substrate is costly, and the reaction can be difficult to control leading to diffuse staining that is not optimal for high resolution fluorescence microscopy.
Consequently, we sought fluorescent proteins (FPs) that were amenable to periplasmic expression to generate inherently fluorescent sdAb fusion proteins more suited to imaging. It is generally accepted that most fluorescent proteins are produced at high levels in the reducing environment of the E. coli cytoplasm. In contrast, all sdAb that we have encountered (n=>200) are produced at high levels in the periplasm and, therefore, to engineer a generic system for high level production of sdAb-FP fusions in E. coli we must overcome this “compartment conundrum”. The difficulties in producing large amounts of recombinant antibody green fluorescent protein (GFP) fusions in the periplasm were first encountered over twenty years ago.(26) GFP was subsequently improved to balance rates of chromophore oxidation and beta-barrel formation in a superfolding version (sfGFP)(27) that is more tolerant of passenger proteins. However, while low levels of sfGFP have been secreted to enable bacterial physiology to be explored (for review see(28)), the overexpression and high level production required for pure immunoreagent development appeared to languish. Consequently, the field moved towards screening for sdAb that were soluble as cytosolic GFP fusions,(29) using sdAb that serendipitously fold well in the cytosol for fusions with mCherry(30) or enhancing cytoplasmic sdAb solubility by providing helper proteins in trans for fusions with enhanced GFP (eGFP).(31) Though the twin-arginine translocation pathway has been used for exporting pre-folded GFP(32) or mCherry(33) this route would still rely on a sdAb passenger capable of being folded in the cytosol or selected to do so. Having explored sfGFP and mCherry as secreted sdAb fusion partners without too much success, we found ourselves wondering if other members of the FP palette were more amenable to overproduction in the periplasm. The periplasmic antibiotic resistance and protein-protein interaction studies of Meiresonne and colleagues(34, 35), where negative impacts of FP export toxicity on cell physiology were considered, indicated that mNeonGreen and mScarlet-I may well be good candidates for E. coli. Monomeric (m) NeonGreen is the brightest green/yellow fluorescent protein so far described and was engineered from the tetrameric LanYFP (Allele Biotechnology) from the cephalocordate Branchiostoma lanceolatum.(36) mNeonGreen is around 1.5 to 3 times as bright as the commonly used yellow and green FP’s, has an excitation maximum at 506 nm and emission maximum at 517 nm, and is very photostable. mScarlet was evolved from a consensus red FP scaffold sequence with a redesigned barrel outer surface to discourage multimerization, and is around 3.5 times brighter than mCherry making it the brightest red FP known.(37) mScarlet-I is a faster maturing mutant of parental mScarlet, though with a reduced quantum yield (0.54 instead of 0.70) and has an excitation maximum at 569 nm and an emission maximum at 593 nm.
RESULTS AND DISCUSSION
Production of Prokaryotic Recombinant N Bait Protein.
In stark contrast to our previous sdAb selections where we had employed live vaccinia and filovirus preparations as baits, here we used a single recombinant CoV antigen before SARS-CoV-2 was available. A synthetic gene encoding N derived from the original Wuhan isolate of SARS-CoV-2 deposited in Genbank by Wu and colleagues(4) was expressed using the T7 expression system within E. coli BL21(DE3). Our usual mild cytosolic expression induction regime (0.1 mM IPTG for 3 h at 25 °C in glucose free media) caused N protein cultures to stall perhaps, reflecting the inherent ability of N to bind RNA and interfere with homeostasis. To obtain sufficient biomass we incubated glucose containing cultures for 24 h at 25 °C to rely on gentler expression through gradual de-repression via glucose depletion. The approach was effective at generating 2.7 mg of relatively pure protein from 6 g wet weight of cells following bead beating and IMAC (Figure 1A) which was more than sufficient for our studies requiring a few hundred μg for phage panning, ELISA screens and titrations. Although we attempted to further purify the protein using size exclusion chromatography (SEC) it appeared in the void volume (data not shown) suggesting it was multimeric.
Figure 1.

Recombinant N bait protein, sdAb sequences derived from panning and their preliminary binding characteristics. (A) Coomassie stained SDS-PAGE analysis of IMAC pure recombinant N protein derived from E. coli expression. Molecular weight markers in kDa; 1, lysate pre-IMAC; 2, flow-through; 3-7, alternate peak fractions; N predicted mwt. 46.7 kDa. (B) Predicted amino acid sequences of the two sdAb selected on N highlighting the complementarity determining regions (CDRs). These Nomad derived clones have additional EPKTPKPQPAASGAEFAAA sequence after FR4 encoding the long hinge, Sfi I and Not I restriction sites. (C) Titration of sdAb-Gluc fusion proteins over constant amount of purified NP protein to determine the EC50 +/− SD shown in the legend. Data points are the means of two separate experiments with error bars representing +/− SD. (D) Western blot of HEK293T lysates following transient expression of recombinant N or control beta-galactosidase (β-gal) genes in pcDNA 48 h post transfection, probed with sdAb-APEX2 fusions and developed with DAB. Molecular weight markers are in kDa.
Selection of N Specific sdAb.
Four rounds of panning of our Nomad sdAb library on passively immobilized N protein yielded two similar sdAb clones occurring at equivalent frequency following sequencing of 24 clones (8 from rounds 2-4) (Figure 1B). Since each round of panning occurs through the course of 2 days, we were obtaining N reactive sdAb within 4 days at round 2 with an additional 3 days required to deconvolute the polyclonal population to monoclonal sdAb. We are obliged to send out our sequencing and this is probably the longest bottleneck in the entire process depending on the speed of the carrier and how busy the sequencing facilities are at any given time. While amino acid variation was apparent in framework regions 1 (Q1K, A14T) and 2 (T70S), the absence of variation in the complementarity determining regions (CDRs) means the two clones are likely to have equivalent binding characteristics.
Characterizing the Interaction Between sdAb and N Using Gluc and APEX2 Fusion Proteins.
By producing fusions of each sdAb with downstream Gluc we were able to determine the EC50 for the interaction between antibody and antigen to be in the tens of nM range with sdAb WuN3 slightly more potent than sdAb WuN1 (Figure 1C). These crude affinity measurements are on par with previous sdAb isolated from this library and were encouraging enough to warrant further study of reactivity and specificity. Thus, we assembled sdAb-APEX2 fusion proteins to probe Western blots of HEK293T lysates following transient expression of human codon optimized SARS-CoV-2 N in order to gauge their ability to recognize mammalian cell derived protein (WuN). Each sdAb-APEX2 fusion recognizes a species migrating at the position expected for N (predicted molecular weight of untagged N is 45.6 kDa) that is not present in the negative control β-galactosidase (β-gal) expressing cells (Figure 1D). Recognition of the HEK293T N protein by sdAb originally selected on E. coli derived N antigen indicates that binding of sdAb is not dependent on post-translational modifications specific to eukaryotic cells. Furthermore, recognition of denatured N protein on a membrane indicates the antibody-antigen interaction is not overly dependent on conformational information within the epitope. That the sdAb also show no non-specific background binding to the myriad of host cell proteins also present on the Western blot gave us the initial green light to pursue cell immunostaining.
SdAb-APEX2 Cell Immunostaining.
Both of the sdAb-APEX2 fusions were able to recognize recombinant N protein within transiently expressing HEK293T cells with no background signal apparent on control transfected cells when development was with either Amplex™ UltraRed or DAB (Figure 2A). By this time in our study, SARS-CoV-2 had been made available to US researchers by BEI Resources based on an isolate from Washington state USA-WA1/2020 to enable live-virus studies of the sdAb to be pursued. Signal strength was greatly improved when probing Vero cells 24 h post-infection by SARS-CoV-2 providing a higher number of cells producing N and, likely higher amounts of N per cell in this time frame. SARS-CoV-2 is a fast replicator and using a multiplicity of infection (moi) of 3, titers in supernatants climb from 1e+2 pfumL−1 at 4h to 1e+6 pfumL−1 by 24 h(38). Since we are using a much lower moi (approximately 1e-2 to 1e-3) we have a mix of infected and uninfected cells within each well 24 h post-infection. Wells of uninfected Vero cells also showed no capacity to bind the sdAb-APEX2 proteins indicating signals correlate to viral infection. The slightly improved performance of the sdAb WuN3 clone over the sdAb WuN1 clone in EC50, Western blot probing and immunostaining lead us to just work with the former in most of the subsequent experiments. As can be seen with a broadly distributed and highly expressed antigen like N the APEX2 imaging at high magnification can be a little difficult to interpret in terms of localization with both DAB and Amplex™ UltraRed development (Figure 2B) and prompted us to search for FP alternatives.
Figure 2.

Employing sdAb-APEX2 fusions to recognize recombinant or viral N protein within mammalian cells. (A) APEX2 fusion proteins of the WuN1 and WuN3 sdAb used as probes at 100 nM for recombinant N protein or β-gal transiently expressed in HEK293T cells 24 h post transfection, or SARS-CoV-2 infected or uninfected Vero cells 24 h post infection. Signal development used either Amplex™ UltraRed and fluorescent microscopy or DAB and bright field (10 x magnification). (B) 150x magnification of the SARS-CoV-2 infected cells developed with fluorescent or colorimetric substrate.
SdAb-FP Fusion Production.
Our initial studies of periplasmic sdAb-FP expression actually employed anti-filoviral sdAb as exemplified by anti-Marburgvirus NP sdAb A(8) fusion protein purification from the periplasm of a standard 500 mL shake-flask culture. The productivity potential of sdAb fusions to mNeonGreen, mScarlet-I and even a superfolding mTurquoise2 optimized for the periplasm (sfTq2ox) is clearly evident in contrast to sfGFP and mCherry when signaled to the periplasm using the dsbA signal (Figure 3A). The urgency of the COVID-19 outbreak lead us to put the filoviral studies to one side and to mobilize the mNeonGreen and mScarlet-I genes with long flexible linkers downstream of the anti-WuN sdAb in place of the APEX2 gene (Figure 3B). The mNeonGreen and mScarlet-I fusions appear well expressed and monomeric, albeit at lower levels relative to the APEX2 fusions used as our benchmark (Figure 3C). It is noteworthy that using either dsbA or pelB signal mediated secretion indicates these two FP are quite forgiving in their requirements for export to the periplasm thereby increasing the chances of compatibility with the full-range of slow or fast folding affinity scaffolds(39). The sdAb-BAP is a poor producer relative to the other three fusions (as a benchmark, sdAb with just a C-terminal His6 tag yield peaks of around the 2000 mAu mark from a 500 mL culture). Each of the main fusion proteins of interest are fairly pure protein preparations though the mScarlet-I maybe more prone to degradation than one would like (Figure 3D). To our knowledge, this is the first time sdAb-FP fusions have been overproduced in the periplasmic compartment at a useful scale and provides an effective route to milligram amounts of protein (Figure 3E). If we have 2 mL of 1.75 mg of a 45 kDa sdAb-FP fusion it provides us with 8,000 aliquots of 50 μL staining reagent at 100 nM, making it very cost effective for microscopy and high throughput drug screens of wild-type virus for example. By overcoming the compartment conundrum and producing high levels of soluble sdAb-FP fusions we bypass complex and bespoke refolding regimes or chemical conjugations and provide a facile means to generically refit clones from periplasmic antibody repertoires with FP functionality.
Figure 3.

Identification of fluorescent proteins capable of being overexpressed in the periplasm as sdAb fusions, their relative purity, and productivity. (A) Size exclusion chromatogram of the initial panel of FP fused to an anti-Marburgvirus NP sdAb following IMAC purification from the periplasmic compartment. sfGFP fusion (black), mNeonGreen fusion (green), sfTq2ox fusion (cyan), mCherry fusion (yellow) mScarlet-I (red). (B) Schematic of the E. coli periplasmic expression vector cassettes used to generate the anti-WuN sdAb fusion proteins used in the immunoprobing with the parental APEX2 gene being substituted for the D1D2-mNeonGreen and D1D2-mScarlet-I genes from the anti-Marburgvirus NP sdAb-FP fusions. Ptac, tac promoter; pelB, pectate lyase signal sequence; D1D2, M13 gene III linker; His6, polyhistidine tag; tlpp, terminator. (C) Size exclusion chromatograms of the sdAbWuN3 fusion proteins following IMAC purification from the periplasmic compartment. sdAb-BAP (black), APEX2 fusion (yellow), mNeonGreen fusion (green), mScarlet-I fusion (red), ovalbumin (OVA, blue), bovine serum albumin (BSA, pink). (D) Coomassie stained SDS-PAGE gel of the purified proteins from both sdAbWuN1 and sdAbWuN3 fusion protein expressions. (E) Table of purified protein yields from one 500 mL expression culture for each of the main constructs.
Calibrating sdAb-FP Immunostaining of Recombinant N.
We were unsuccessful at detecting strong immunofluorescent signal using mNeonGreen sdAb probes on HEK293T cells transfected with pcDNA-WuN without increasing the settings to the extent that background green became apparent. We therefore employed a handful of home-made expression vectors to see if we could raise the levels of N expression to achieve immunofluorescence, and in so doing, afford a simple means of gauging the relative potency of sdAb-FP versus sdAb-APEX2 based detection. The work of Mariati and colleagues(40) in varying various post-transcriptional regulatory elements before or after the gene of interest was the foundation for our panel of constructs (Figure 4A). We used a sandwich ELISA to first monitor the presence of N antigen from the soluble HEK293T cell fraction through capture using sdAb-BAP and tracing using phage displayed sdAb. A gradation of expression levels from the constructs was apparent with the main drivers of overexpression being the adenovirus tripartite leader and intervening sequence (AdTri IVS)(41) or cytomegalovirus (CMV) intron A leader(42) both exceeding the mutant woodchuck hepatitis post-transcriptional regulatory element (HPREzb)(43), though the latter appeared to have an additive effect when combined with either of the leaders (Figure 4B). If we use 1e+8 RLU from 25 μL of pcDNA lysate as our baseline, all but puma3 required 1 to 0.4 μL to generate an equivalent signal (representing 25-62.5-fold increases), while puma3 required approximately 5 μL (representing a 5-fold increase). Western blotting and probing with sdAb-APEX2 confirmed the gradation of levels (Figure 4C) as did probing the transiently expressing cells with the sdAb-mNeonGreen fusion protein (Figure 4D), clearly showing the relationship between the abundance of N and the degree of staining. Immunofluorescence detection on a par with APEX2 was enabled at the high end of the expression spectrum, requiring increases of ten-fold or more of target protein. Despite the lower sensitivity of mNeonGreen relative to APEX2 we were encouraged to pursue probing of virus infected cells with the sdAb-FP fusions.
Figure 4.

Correlating expression levels of recombinant N by ELISA and Western blot with sdAb-mNeonGreen cell staining. (A) Schematic of the expression cassettes of the vectors used to elevate recombinant N protein levels for sdAb-mNeonGreen mediated detection in HEK293T cells. hCMVie, human cytomegalovirus promoter-enhancer; BGH, bovine growth hormone polyadenylation signals; AdTri-IVS, adenovirus tripartite leader with intervening sequence; CMV intron A, cytomegalovirus intron A; mutant HPREzb, woodchuck hepatitis virus post-transcriptional regulatory element. (B) ELISA titration of RIPA lysates from transient expressions of N protein, or control β-gal, captured using oriented sdAb as captor and phage displayed sdAb as tracer. The transfections and sandwich assay were performed twice and plots are the mean with error bars representing +/− SD. (C) Western blotting of the RIPA lysates of the pcDNA or puma1-5 vectors expressing WuN, or β-gal control, probed with sdAb-APEX2 and developed with SuperSignal West Pico chemiluminescent substrate. Molecular weight markers are indicated in kDa. (D) Transient transfections of control, parental pcDNA or puma1-5 vectors probed with 100 nM of the Wu3 sdAb-mNeonGreen fusion protein 24 h post-transfection (10x magnification).
SdAb-FP Immunostaining of Virus Infected Cells.
The sdAb-FP fusion proteins are specific for virus infected Vero cells and show no background binding on wells of uninfected cells (Figure 5A and 5B). In all of the single and double stained images (Figure 5C) the differentiation between nucleus and cytoplasm appears very clear highlighting the advantages of spatially concise registers not reliant on substrate turnover as with APEX2. There appears to be negligible staining of the nucleus and nucleoli, suggesting that within the 24 h time period, SARS-CoV-2 N does not appear to accumulate in this compartment akin to the situation with SARS-CoV(20, 21). In contrast, several other CoV N accumulate in the nucleoli which is thought to divert biosynthetic resources from the dividing nucleus to the cytoplasm, which is the main locale for CoV virus replication.
Figure 5.

Employing the sdAb-FP fusion proteins to probe SARS-CoV-2 infected cells. Immunoprobing Vero cells 24 h post-infection with SARS-CoV-2 or uninfected Vero cells with (A) 100 nM sdAb-mNeonGreen or (B) 100 nM sdAb-mScarlet-I or (C) a mix of both at 100 nM each. First panels represent Hoechst staining, middle are the individual channels and lastly is the merge (150x magnification).
CoV Specificity and Cross-reactivity.
In order to gauge the usefulness of these immunoreagents as tools in a diagnostic scenario we must define whether they are specific to SARS-CoV-2 or if they have reactivities to endemic CoV which may confound accurate identification as has been noted previously for SARS-CoV(44-46). Until 2003 only two endemic human CoV (HCoV-OC43 and HCoV-229E) were known, both isolated in the 1960’s(47, 48). Both viruses are normally associated with seasonal mild “cold-like” diseases of healthy immunocompetent individuals, though in infants, the elderly, and people with underlying health conditions the infections can be more severe. In 2003 the emergence of SARS-CoV in Guangdong, China(49) reignited interest in CoV family. SARS-CoV spread globally with over 8,000 cases and a 10 % case fatality rate, though rigorous contact tracing followed by home quarantine extinguished the outbreak. Phylogenetic studies revealed the virus originated in bats though probably spilled over to humans from palm civets and raccoon dogs serving as intermediate hosts in exotic meat markets in China(50).
The SARS-CoV outbreak motivated several searches for CoV in animal species different from those already known to harbor major veterinary threats including pigs and fowl. Additionally, in 2004, HCoV-NL63 was isolated from a 7-month old child with bronchitis and conjunctivitis in Holland(51) and HCoV-HKU1 was then isolated from a patient in Hong Kong suffering from pneumonia(52). The origin of HCoV-NL63 is thought to be bats while HCoV-HKU1 is thought to be rodents, but both intermediate hosts are unknown(53). A decade later MERS-CoV emerged in humans(54) and is also thought to originate in bats, though spills over from dromedary camels . The virus still circulates in these countries since camels are a mainstay of travel and food, and the virus has infected over a 1000 people with a fatality rate of around 35 % with disease being especially severe in elderly males with comorbidities. Most recently, in late 2019, SARS-CoV-2 emerged within China(55) to cause the current global pandemic with, at the time of writing, 77,530,799 cases including 1,724,904 million deaths according to the World Health Organization COVID-19 Dashboard. While the precise origin and ancestry of the virus is still under study(56), it’s global distribution suggests a trajectory toward becoming the 5th endemic HCoV.
To begin exploring the epitope of the sdAb and whether this is shared or unique among CoV we overexpressed the N protein of each of the reference virus sequences from human codon optimized genes in HEK293T cells to serve as viral surrogates. Here we used a 72 h time-point to enable accumulation of recombinant proteins to very high levels to maximize the chances of detecting cross-reactivities. Each species of N was well represented as judged by Coomassie stained bands around the 40-50 kDa molecular weight range and visualization of the transfection control β-gal monomer at 116 kDa (Figure 6A) and provides us with a direct means of confirming overexpression without the use of tags which may alter the biology of N. HCoV-OC43 N appears to have two overexpressed products in the expected range while HKU1 appears to have a full-length and an additional shorter/degradation product. Western blotting using the sdAb-APEX-2 fusion protein revealed very strong signal for the SARS-CoV-2 N but only a weak signal for the SARS-CoV N, despite the excessive amounts of recombinant N being present (Figure 6B). No cross-reactivity was apparent to the collection of other recombinant CoV N. At the 48 h time-point (data not shown) with slightly less N proteins present, no reactivity was observable on the SARS-CoV lane yet very strong signal was still present on the SARS-CoV-2 sample. Immunostaining of cells 24 h post–transfection with the more sensitive sdAb-APEX2 fusion revealed strong staining of the SARS-CoV-2 N as expected and weaker staining of SARS-CoV N (Figure 6C) which was mirrored by the less sensitive sdAb-mNeonGreen probing of the cells 72 h post-transfection (Figure 6D). In both immunostaining regimes, no reactivity was observed to the other CoV N or negative control β-gal transfected cells. Collectively, the data suggest that the sdAb is highly specific for SARS-CoV-2 N with very low cross-reactivity to SARS-CoV N and undetectable cross-reactivity with the other major CoV N, making the APEX2 and FP fusions useful tools in being able to distinguish the pandemic SARS-CoV-2 from other CoV. With 40 amino acid differences between SARS-CoV-2 (420 aa) and SARS-CoV (423 aa) fairly evenly distributed throughout the protein we were surprised at the ability of a non-immune sdAb to be so discriminating. Future domain mapping and structural studies should enable us to define the epitope and predict the likelihood of “epitope-erosion” through viral evolution as we have done previously for Marburgvirus(8) and Ebolavirus.(9)
Figure 6.

Assessing the cross-reactivity of the anti-WuN sdAb with non-SARS-CoV-2 N. (A) Coomassie blue stained SDS-PAGE gel of whole cell lysates of HEK293T cells harvested 72 h post transfection with puma5 vectors expressing each of the N genes derived from the reference CoV sequence plus the original Wuhan SARS-CoV-2 N. Predicted molecular weights are; HCoV-229E N, 43.4 kDa; HCoV-OC43 N, 49.5 kDa; SARS-CoV N, 46.0 kDa; HCoV-NL63 N, 42.3 kDa; MERS-CoV N, 45.1 kDa; HCoV-HKU1, 49.1 kDa; SARS-CoV-2, 45.6 kDa. Molecular weight markers are indicated in kDa. (B) Western blot of the same lysate samples probed with 100 nM sdAb-APEX2 followed by chemiluminescent development. (C) Immunostaining HEK293T cells 24 h post transfection with the puma5 N vectors with 100 nM sdAb-APEX2 followed by development with Amplex™ UltraRed. (D) Immunostaining 72 h post transfection with 100 nM sdAb-mNeonGreen.
CONCLUSION
The paucity of publically available and free immunoreagents to CoV is hampering research advances(57) e.g. BEI Resources currently have a handful of sera listed to date, 50 μL available once a year. While others in the field may use privately held conventional anti-CoV N antibodies for diagnostics, and virus hunting(58) sdAb single-pot repertoires offer a recombinant alternative that bypasses lengthy, costly and unethical(59) animal immunizations and are not dependent on cryopreservation of hybridomas which is increasingly expensive and can fail. Milligram amounts of our sdAb-reporter fusions can be made in E. coli for relatively little expense, in contrast to monoclonal antibodies that require mammalian cell culture for high yields, immediately decreasing the financial bar to engage in CoV immuno-detection. Single-step immunoprobing also bypasses the expense involved in purchasing labelled secondary antibodies and accelerates the imaging process further. With sdAb sequences immortalized in silico in Genbank for all to use, unlike most IgG generated by pharma, we further broaden technology access to all.
As tools for studying viral biology and with optimization of DAB staining, the sdAb-APEX2 conjugates should combine the best of APEX2’s proximity labelling abilities with higher resolution than a much larger IgG probe(60) and should bypass artefacts that might arise using in vivo expressed gene fusions. Selection pressures on viral replication rates typically lead to viral genomes and their encoded proteins being quite compact, multi-functional and limited in how tolerant they are of fusions to reporters without compromising their ability to function correctly. Meanwhile, the sdAb-FP fusions offer a non-catalytic alternative to APEX2 that may enable lucrative opportunities to be realized using single molecule super-resolution microscopy techniques being advanced for pin-point localization of the target antigen. The judicious choice of two monomeric FP scaffolds to act as downstream substitutes for APEX2 provides a straightforward and generic avenue for others in the field to fuse their recombinant antibodies of interest to mNeonGreen and m-Scarlet-I and interrogate other biological processes. It is noteworthy that mNeonGreen and mScarlet-I also make an ideal couple for Förster energy resonance transfer (FRET)(61) which should enable suitably paired recombinant antibody protein fusions to be developed into in vivo FRET probes and in vitro wash-less optical biosensors for rapid antigen detection.
Our proof-of-principle success in being able to smoothly generate and characterize SARS-CoV-2 N specific molecular probes should be applicable to other CoV to expand the repertoire of tools available for studying this important family of viruses that continues to pose epidemic and pandemic threats.
MATERIALS AND METHODS
General Cloning.
Recombinant DNA methods were according to established procedures and employed commercially available reagents; Phusion High-Fidelity DNA Polymerase (Thermo Fisher, Waltham, MA); restriction enzymes and β-agarase (New England BioLabs, Beverly, MA); T4 DNA ligase, CIP and T4 PNK (Roche, Nutley, NJ); GTG low melting temperature agarose for in gel cloning, (Lonza, Walkersville, MD); oligonucleotides (Integrated DNA Technologies, Coralville, IA). Assemblies involving cloning and PCR amplification were sequenced through the inserts and junctions to verify the desired construct. Cloning was typically carried out in XL1-Blue cells unless otherwise stated. Full details of cloning, oligonucleotides, maps and sequences of resulting constructs are available on request.
Cytoplasmic Production of N Protein in E. coli.
The N gene from the SARS-CoV-2 Wuhan reference sequence (Genbank NC_045512.2) was synthesized as an E. coli codon optimized gBlocks™ (IDT, Coralville, IA) with a C-terminal SG3H6 tag. The gene was cloned into pE(8), a home-made high copy pUC based T7 expression vector with symmetrical lacO via Nde I and Hind III, and sequenced. The plasmid was mobilized to BL21(DE3) + pRARE and grown at 30 °C overnight in 50 mL starter cultures of TB with 2 % glucose, 200 μgmL−1 ampicillin, and 30 μgmL−1 chloramphenicol until saturation. Cultures were poured into 450 mL TB with 2 % glucose (no antibiotics) and grown with vigorous aeration in 2500 mL Bellco baffled flasks for 24 h at 25 °C. Cultures were centrifuged and the pellets drained of excess media and stored at −80 °C until ready for bead beating (BioSpec Products, Bartlesville, OK). Once thawed, the pellets were resuspended in 20 mL 1 x immobilized metal affinity chromatography (IMAC) buffer (20 mM M Na2HPO4, 0.5 M NaCl, 20 mM imidazole, 0.1 % Tween-20, pH 7.5) plus a cOmplete™ EDTA free protease inhibitor tablet (Roche) and added to a 15 mL chamber filled halfway with 0.1 mm glass beads. The chamber was topped off with 1xIMAC buffer to remove any air bubbles and the cell/bead mixture was blended on ice within a 4 °C fridge for a total of 12 min with 6 cycles of 2 min on and 2 min off, cooling on ice in between. Once contents settled, the cell debris was transferred to a 50 mL conical tube and centrifuged at 3,000 rpm for 15 min at 4 °C (Beckman Allegra 6R, swing out). The supernatant was decanted into a new 50 mL tube and centrifuged at 9,500 rpm for 15 min at 4 °C (Sorvall RC 6+, F13 FiberLite rotor). The supernatant was again decanted and centrifuged. The supernatant was filtered through a 32 mm diameter 0.8/0.2 μm filter (Pall, Port Washington, NY) and applied to a 1 mL HisTrapHP column equilibrated in 1xIMAC buffer. Protein was eluted with a 0-500 mM imidazole gradient in 1xIMAC buffer. Protein was analyzed by SDS-PAGE to assess purity. Fractions were pooled, made to 15 % glycerol, quantified by UV adsorption and aliquoted for storage at −80 °C.
Phage Panning.
Each well of an 8 well high binding ELISA strip was coated with 100 μL of 1 μg mL−1 N protein in phosphate buffered saline (PBS) overnight at 4 °C. Wells were washed three times to brimming with PBS and blocked for 1 h with 2 % Carnation non-fat dried milk in PBS (MPBS). Phagemids representing 1000 clones of each member of our Nomad semi-synthetic library(5) were applied in eight aliquots of 100 μL MBPS for 1 h with shaking and then left static for 1 h. Wells were then washed with PBS containing 0.1 % Tween-20 (PBST) followed by PBS; washings were 10, 20, 40, 80 with each buffer for each for rounds 1 through 4 respectively. Phagemids were eluted with 100 μL 100 mM triethylamine per well for 10 min followed by pooling and neutralization with 400 μL 1M Tris-HCl pH 7.5. 600 μL of the neutralized eluate was added to 10 mL of mid exponential phase XL1-Blue* and incubated at 37 °C for 30 min before titrating an aliquot while the rest was gently pelleted and plated on 15 cm diameter dishes of 2xYT plus 2 % glucose, 200 μgml−1 ampicillin and 30 μgmL−1 tetracycline. (*Mid-exponential phase cells were grown from cryopreserved aliquots for 1 h at 37 °C). Overnight growth on plates at 37 °C was followed by scraping large plates for glycerol stocking and liquid culture (2xYT plus 2 % glucose with ampicillin and tetracycline) in baffled flasks at 37 °C for M13K07 superinfection, selection with 70 μgmL−1 kanamycin, induction with addition of IPTG to 1 mM, and overnight growth at 30 °C. Aliquots of the cultures were clarified by microfuge and kept at −80 °C for polyclonal ELISA while 800 μL was mixed with 200 μL of 10 % MPBS and used for the next panning round. Polyclonal ELISA was used to monitor antigen specific enrichment of clones and monoclonal ELISA was used to deconvolute positive phage. DNA sequencing was then used to identify unique clones.
SdAb and sdAb Fusion Vectors.
The anti-N sdAb genes, were mobilized to pecan126(62) to create the biotinylated sdAb for use as captor and the phage displayed sdAb to use as tracer. SdAb were mobilized to pecan 287 (a tac promoter version of the lac based pecan35(8)) to create the sdAb-Gluc fusions, and to pecan278(25) to create the sdAb-APEX2 fusions. A sdAb fusion vector for another job (pecan326) bearing the dsbA signal sequence and anti-Marburgvirus NP sdAb A with a downstream PCR amplified fragment encoding the long flexible linker between domains 1 and 2 of M13 gene III served as an intermediate to receive PCR fragments encoding sfGFP(27) (Nde I and Kpn I sites removed by silent codon mutagenesis) and mCherry (Genbank AY678264, Pst I and Nco I removed), or gBlocks™ encoding mNeonGreen (Genbank KC295282.1, Nco I removed), mScarlet-I (Genbank KY021424.1, Not I site removed), or sfmTurquoise2ox (Addgene sequence #117960). Genes were inserted via Spe I and Hind III and all encoded a C-terminal G4SH6 tag. The mNeonGreen and mScarlet-I genes were subsequently transferred to the anti-WuN sdAb construct pecan278 along with D1D2, replacing the APEX2 gene via Not I and Hind III to create pecan367 (expressing sdAb-mNeonGreen) and pecan373 (expressing sdAb-mScarlet-I). Sequences of the major components used in this study are presented in the supplementary information.
Periplasmic Production of sdAb, sdAb-Gluc, sdAb-APEX2, sdAb-FP proteins.
Plasmids were mobilized into E. coli Tuner + pRARE for recombinant protein production and were grown in 50 mL starter cultures of terrific broth (TB) plus 2 % glucose at 30 °C overnight with 200 μgmL−1 ampicillin and 30 μgmL−1 chloramphenicol in 250 mL Bellco baffled flasks. For APEX2 fusions, a stock solution of hemin (Sigma, St. Luois, MO) was prepared by dissolving 32.5 mg in 500 μL of 1.4 M ammonium acetate water (Sigma) and vortexing which was then added to 450 mL of glucose free TB (no antibiotics) while swirling in 2500 mL Bellco baffled flasks. The saturated overnight cultures were poured in to the large flasks (to yield 1 mM final concentration of hemin) and shaken for 3 h at 25 °C. Expression was induced by addition of IPTG to 1 mM for 3 h at 25 °C, the cells pelleted, drained, and weighed. Cells were osmotically shocked (63) by resuspension in 14 mL ice-cold 0.75 M sucrose in 100 mM Tris-HCl pH 7.5, addition of 1.4 mL of 1 mgmL−1 hen egg lysozyme (Sigma), followed by drop-wise addition of 28 mL of 1 mM EDTA pH 7.5 and swirling on ice for 15 min. Two mL of 0.5 M MgCl2 was added, swirling continued for 15 min and cells pelleted. The 45 mL supernatant (osmotic shockate) was mixed with 5 mL of 10xIMAC buffer (0.2 M Na2HPO4, 5 M NaCl, 0.2 M imidazole, 1 % Tween-20, pH 7.5), followed by 0.5 mL of High Performance Ni Sepharose (GE Healthcare, Pittsburgh, PA) and the suspension gently mixed on ice for 1 h. Resin was pelleted at 3,000 rpm for 5 min (Beckman Allegra 6R swing out rotor) and washed twice with 50 mL of 1xIMAC solution before elution with 2 mL 0.5 M imidazole in 1xIMAC buffer, pH 7.4 in Poly-Prep® columns (Bio-Rad, Hercules, CA). Proteins were concentrated in Amicon 10 kDa ultrafiltration devices (Millipore, Billerica, MA) to 200 μL for separation by SEC on a Superdex 200 Increase 10/300 GL column (GE Healthcare) operating in PBS. Preparations were made to 50 % glycerol and aliquoted for long-term storage at −80 °C. Proteins were quantified by UV adsorption and analyzed by SDS-PAGE and Coomassie blue staining for impurities.
sdAb-Gluc Titrations to Determine EC50.
Duplicate wells of ELISA plates were coated overnight at 4 °C with 100 μL of 1 μgmL−1 N protein in PBS or 100 μL of 1 μgmL−1 bovine serum albumin (BSA) control antigen. Plates were washed three times with PBS and then blocked by filling to brimming with MPBS for 1 h. 100 μL of sdAb-Gluc dilutions in MBPS were applied to duplicate wells for 1 h. Wells were washed to brimming three times with PBST and two times with PBS. Signals were developed with injection of coelenterazine (NanoLight™ Technology, Pinetop, AZ) in lucky buffer (10 mM Tris, 1 mM EDTA, 500 mM NaCl, pH 7.4) and emissions collected using a luminometer (Turner BioSystems, Sunnyvale, CA) with a 2 s integration. The experiment was performed 2 times and curves are the plots of two mean RLU of sdAb-Gluc on N minus the corresponding mean of control sdAb-Gluc on the BSA coat with error bars representing +/− SD. The EC50 y value was calculated for curves that plateaued using the equation [RLUmin + (RLUmax-RLUmin)/2]. The corresponding x values were calculated using one observed point greater and one less than the y EC50 using the trend function in Excel and the two values averaged and presented +/− SD nM.
Mammalian Expression Vectors for Recombinant N.
A human codon optimized version of the Wuhan SARS-CoV-2 N gene was obtained as a gBlocks™ and cloned into pcDNASfiNco-(64) via Sfi I. For elevated N levels, the gene was mobilized to a series of pcDNASfiNco-derivatives varying in their post-transcriptional control elements; puma1, adenovirus tripartite leader plus murine immunoglobulin leader sequences(65); puma2, cytomegalovirus intron A(8); puma3, woodchuck hepatitis virus post-transcriptional regulatory element minus the X-protein open reading frame (HPREzb); puma4, puma1 with HPREzb(25); puma5, puma2 with HPREzb. Genes encoding human codon optimized versions of N from other CoV were obtained as plasmid clones from IDT and were mobilized to puma5 via Sfi I. Genbank reference genomes used to source the parental N genes were; HCoV-229E, NC_002645.1; HCoV-OC43, NC_006213.1; SARS-CoV Tor, NC_004718.3; HCoV-NL63, NC_005831.2; HCoV-HKU1, NC_006577.2; MERS-CoV, NC_019843.3.
Production of N in Mammalian Cells.
Lysates were generated by PEI transfections of plasmid DNA. Human embryonic kidney (HEK) 293T cells (ATCC, Manassas, VA) were grown in Dulbecco’s modified Eagle’s medium (DMEM) with 4.5 gL−1 glucose, L-glutamine, sodium pyruvate (Corning cellgro), 5 % fetal bovine serum (Corning, Corning, NY) and penicillin/ streptomycin (complete medium) at 37 °C and 10 % CO2 with humidity. Cells were seeded in complete medium 16-18 h prior to transfection.
Lysates for Western blotting were initially generated by transfection of pcDNA WuN plasmid or a β-gal control plasmid (also employed for staining to monitor transfection efficiency). Cells were seeded at 5e+6 per 10 cm diameter plate in 25 mL complete media. 105 μL DNA (approx. 10 μg) was combined with 41 μL PEI in 2.5 mL serum free DMEM and equilibrated for 15 min before being added to the cells. At 48 h post transfection, cells were washed gently with 10 mL cold Tris buffered saline (TBS) twice, then 4 mL of ice-cold radio immunoprecipitation buffer (RIPA; 10 mM Tris–HCl pH 7.8, 150 mM NaCl, 1 mM EDTA, 1 % NP-40) containing protease inhibitors was added and left at 4 °C for 30 min. The soluble fraction was separated from the insoluble fraction by microcentrifugation (5415D microcentrifuge, Eppendorf, Hauppauge, NY, USA) at 15,000 rpm for 10 min at 4 °C and the supernatant collected. Glycerol was added to a final concentration of 15 %, aliquoted and stored at −80 °C. Samples were heated at 100 °C for 5 min before 20 μL was loaded on to SDS-PAGE Laemmli gels. Gels were semi-dry transferred to Immobilon P and the membrane blocked in MPBS for 1 h prior to probing with 100 nM of the sdAb-APEX2 fusions for 1 h. Following washing three times with PBST for 5 min each and twice with PBS for 5 min each, membranes were developed with DAB and washed in water used to stop the reaction.
To provide lysates representing the soluble fraction for the WuN sandwich ELISA, cells were seeded at 7.5 e+5 cells per well in a 6-well plate in 3 mL of complete medium. 12.5 μL DNA (approx. 1.5 μg) was combined with 5 μL PEI in 300 μL serum free DMEM, equilibrated for 15 min prior to being added to the cells. At 48 h post transfection, cells were washed gently with 2 mL cold TBS twice, then 500 μL of ice-cold RIPA buffer and processed as above. Ten μL of the sample was also subjected to SDS-PAGE and Western blotting using SuperSignal West Pico Chemiluminescent Substrate development and image capture using a ChemiDoc™ (BioRad).
To provide lysates of the repertoire of CoV N, each well of a transiently transfected 6-well plate as above was lysed in 500 μL of Laemmli sample buffer with reducing agent and briefly sonicated 72 h post transfection. All samples were stored at −20 °C before further processing. Samples were heated at 100 °C for 5 min before 20 μL was loaded on to SDS-PAGE Laemmli gels for either Coomassie Blue staining or Western blotting.
Recombinant N Sandwich ELISA.
100 μL of neutravidin at 1 μgmL−1 in PBS was used to coat duplicate wells of Costar white high binding ELISA plates overnight at 4 °C. After washing 3x with PBS to brimming, wells were filled to brimming with Bioplex buffer (PBS, 2 % w/v BSA and 0.05 % Tween-20) for an hour. Block was replaced with 100 μL of 100 nM biotinylated sdAb from pecan126 preparations in Bioplex buffer for 10 min with gentle shaking. Wells were washed 3x with PBST, 2x with PBS, blocked with 400 μL MPBS. The MPBS block was removed, and dilutions of HEK293T cells transiently expressing N and lysed in RIPA buffer were added to the wells in MBPS to 100 μL and plates shaken for 10 min. Following aspiration and washing 3x with PBST and 2x with PBS, 1 μL of phage displayed sdAb preparation (from 40 mL scale culture precipitated to 1 mL) in 100 μL MPBS was applied and the plates shaken for 10 min. After washing as above, 100 μL of 1/2500 dilution of anti-M13 HRP (GE Healthcare) in MPBS was added and plates shaken for 10 min. Following washing, signals were developed with SuperSignal ELISA Pico chemiluminescent substrate (Thermo-Fisher) with 2 s integration using the luminometer, and the duplicates averaged. The assay was performed once more, using a different set of transfections on a different occasion to create a graph representing the average of the two plots with maximum and minimum bars representing +/− SD.
Immunoprobing Cells Expressing Recombinant N.
Wells of 8 chamber μ-slides (ibidi, Fitchburg, WI) were coated for 10 min with 200 μL of a 1/10th dilution in water of 0.1 % poly-L-lysine (Millipore Sigma, St. Louis, MO) and then washed 3 times with PBS. Eighteen h prior to transfection, cells were seeded at 7.5e+4 in 300 μL medium per chamber. Miniprep DNA (approx. 125 ng in 1.25 uL) and 500 ng linear polyethylenimine (PEI, 1mgmL−1, approx. 0.5 μL) were mixed in 15 μL serum free DMEM to make a final volume of 30 μL, and allowed to equilibrate for 20 min at room temperature and then added to cells. After 24 (or 72 h for the CoV N repertoire), slides were washed with warm serum free DMEM twice. Slides were fixed with 10 % buffered formalin for 1 h at 4 °C. Slides were washed three times with PBS before permeabilizing with 0.1 % Triton X-100 in PBS for 10 min. Slides were washed 3 times with PBS and blocked with 2 % BSA, 0.05 % Tween-20 in PBS for 1 h. Block was removed and then cells were probed with 100 nM APEX fusion proteins in 200 μL of 2 % BSA, 0.05 % Tween-20 in PBS for 15 min. Cells were then washed 3 times with PBST, then twice with PBS and developed with metal enhanced DAB tablet solution (Sigma) for 1 min before washing with PBS to stop the reaction. For fluorescent APEX2 staining, cells were probed with 100 nM of sdAb-APEX fusions and development was with 200 μL of PBS containing 50 μM Amplex™ UltraRed and 10 mM H2O2 for 10 min. Counterstaining was with Hoechst for 10 min followed by 2 PBS washes. Slides were viewed using an Eclipse Ti confocal microscope (Nikon) with NIS Elements Imaging Software and ImageJ within Fiji was used to process images. For the fluorescent protein sdAb fusions, probing was performed as above at 100 nM.
Immunoprobing Virus Infected cells.
Vero-E6 cells (ATCC) were seeded at approx. 5e+4 in 100 μL complete DMEM per well in ibidi 96 well cell culture plates to be confluent 24 h later. Medium was replaced with 150 μL fresh complete DMEM containing 80 pfu SARS-CoV-2 USA-WA1/2020 (BEI Resources, Manasas, VA – p6 stock in Tx Biomed strain collection) within the BSL-4 laboratory. The plate was left at 37 °C with 5 % CO2 and humidity for 24 h, immersed in 10 % buffered formalin for 18 h at 4 °C and removed from the laboratory. Probing and visualization was performed as for the recombinant N immunoprobing experiments after removing the fixative and washing several times with PBS.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported through NIH awards R01AI112851 and R21AI152200.
Footnotes
SUPPORTING INFORMATION
Nucleotide sequences of the sdAb-mNeonGreen, mScarlet-I, Gluc and APEX2 components.
Conflicts of interest: None.
References
- 1.Dormitzer PR, Suphaphiphat P, Gibson DG, Wentworth DE, Stockwell TB, Algire MA, Alperovich N, Barro M, Brown DM, Craig S, Dattilo BM, Denisova EA, De Souza I, Eickmann M, Dugan VG, Ferrari A, Gomila RC, Han L, Judge C, Mane S, Matrosovich M, Merryman C, Palladino G, Palmer GA, Spencer T, Strecker T, Trusheim H, Uhlendorff J, Wen Y, Yee AC, Zaveri J, Zhou B, Becker S, Donabedian A, Mason PW, Glass JI, Rappuoli R, and Venter JC (2013) Synthetic generation of influenza vaccine viruses for rapid response to pandemics, Sci Transl Med 5, 185ra168. [DOI] [PubMed] [Google Scholar]
- 2.Hsieh CL, Goldsmith JA, Schaub JM, DiVenere AM, Kuo HC, Javanmardi K, Le KC, Wrapp D, Lee AG, Liu Y, Chou CW, Byrne PO, Hjorth CK, Johnson NV, Ludes-Meyers J, Nguyen AW, Park J, Wang N, Amengor D, Lavinder JJ, Ippolito GC, Maynard JA, Finkelstein IJ, and McLellan JS (2020) Structure-based design of prefusionstabilized SARS-CoV-2 spikes, Science 369, 1501–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Muyldermans S. (2020) A guide to: generation and design of nanobodies, FEBS J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG, Hu Y, Tao ZW, Tian JH, Pei YY, Yuan ML, Zhang YL, Dai FH, Liu Y, Wang QM, Zheng JJ, Xu L, Holmes EC, and Zhang YZ (2020) A new coronavirus associated with human respiratory disease in China, Nature 579, 265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goldman ER, Anderson GP, Liu JL, Delehanty JB, Sherwood LJ, Osborn LE, Cummins LB, and Hayhurst A (2006) Facile generation of heat-stable antiviral and antitoxin single domain antibodies from a semisynthetic llama library, Anal Chem 78, 8245–8255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stijlemans B, Conrath K, Cortez-Retamozo V, Van Xong H, Wyns L, Senter P, Revets H, De Baetselier P, Muyldermans S, and Magez S (2004) Efficient targeting of conserved cryptic epitopes of infectious agents by single domain antibodies. African trypanosomes as paradigm, J Biol Chem 279, 1256–1261. [DOI] [PubMed] [Google Scholar]
- 7.Strauss M, Schotte L, Thys B, Filman DJ, and Hogle JM (2016) Five of five VHHs neutralizing poliovirus bind the receptor-binding site, J Virol 90, 3496–3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garza JA, Taylor AB, Sherwood LJ, Hart PJ, and Hayhurst A (2017) Unveiling a Drift Resistant Cryptotope within Marburgvirus Nucleoprotein Recognized by Llama Single-Domain Antibodies, Front Immunol 8, 1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sherwood LJ, Taylor AB, Hart PJ, and Hayhurst A (2019) Paratope Duality and Gullying are Among the Atypical Recognition Mechanisms Used by a Trio of Nanobodies to Differentiate Ebolavirus Nucleoproteins, J Mol Biol 431, 4848–4867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McBride R, van Zyl M, and Fielding BC (2014) The coronavirus nucleocapsid is a multifunctional protein, Viruses 6, 2991–3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burbelo PD, Riedo FX, Morishima C, Rawlings S, Smith D, Das S, Strich JR, Chertow DS, Davey RT, and Cohen JI (2020) Sensitivity in Detection of Antibodies to Nucleocapsid and Spike Proteins of Severe Acute Respiratory Syndrome Coronavirus 2 in Patients With Coronavirus Disease 2019, J Infect Dis 222, 206–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ren L, Zhang Y, Li J, Xiao Y, Zhang J, Wang Y, Chen L, Paranhos-Baccala G, and Wang J (2015) Genetic drift of human coronavirus OC43 spike gene during adaptive evolution, Sci Rep 5, 11451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wong AHM, Tomlinson ACA, Zhou D, Satkunarajah M, Chen K, Sharon C, Desforges M, Talbot PJ, and Rini JM (2017) Receptor-binding loops in alphacoronavirus adaptation and evolution, Nat Commun 8, 1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Di B, Hao W, Gao Y, Wang M, Wang YD, Qiu LW, Wen K, Zhou DH, Wu XW, Lu EJ, Liao ZY, Mei YB, Zheng BJ, and Che XY (2005) Monoclonal antibody-based antigen capture enzyme-linked immunosorbent assay reveals high sensitivity of the nucleocapsid protein in acute-phase sera of severe acute respiratory syndrome patients, Clin Diagn Lab Immunol 12, 135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li YH, Li J, Liu XE, Wang L, Li T, Zhou YH, and Zhuang H (2005) Detection of the nucleocapsid protein of severe acute respiratory syndrome coronavirus in serum: comparison with results of other viral markers, J Virol Methods 130, 45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lau SK, Woo PC, Wong BH, Tsoi HW, Woo GK, Poon RW, Chan KH, Wei WI, Peiris JS, and Yuen KY (2004) Detection of severe acute respiratory syndrome (SARS) coronavirus nucleocapsid protein in sars patients by enzyme-linked immunosorbent assay, J Clin Microbiol 42, 2884–2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Y, Chan KH, Kang Y, Chen H, Luk HK, Poon RW, Chan JF, Yuen KY, Xia N, Lau SK, and Woo PC (2015) A sensitive and specific antigen detection assay for Middle East respiratory syndrome coronavirus, Emerg Microbes Infect 4, e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li T, Wang L, Wang H, Li X, Zhang S, Xu Y, and Wei W (2020) Serum SARS-COV-2 Nucleocapsid Protein: A Sensitivity and Specificity Early Diagnostic Marker for SARS-COV-2 Infection, Front Cell Infect Microbiol 10, 470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szelazek B, Kabala W, Kus K, Zdzalik M, Twarda-Clapa A, Golik P, Burmistrz M, Florek D, Wladyka B, Pyrc K, and Dubin G (2017) Structural Characterization of Human Coronavirus NL63 N Protein, J Virol 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.You J, Dove BK, Enjuanes L, DeDiego ML, Alvarez E, Howell G, Heinen P, Zambon M, and Hiscox JA (2005) Subcellular localization of the severe acute respiratory syndrome coronavirus nucleocapsid protein, J Gen Virol 86, 3303–3310. [DOI] [PubMed] [Google Scholar]
- 21.Rowland RR, Chauhan V, Fang Y, Pekosz A, Kerrigan M, and Burton MD (2005) Intracellular localization of the severe acute respiratory syndrome coronavirus nucleocapsid protein: absence of nucleolar accumulation during infection and after expression as a recombinant protein in vero cells, J Virol 79, 11507–11512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tannous BA, Kim DE, Fernandez JL, Weissleder R, and Breakefield XO (2005) Codon-optimized Gaussia luciferase cDNA for mammalian gene expression in culture and in vivo, Mol Ther 11, 435–443. [DOI] [PubMed] [Google Scholar]
- 23.Wu N, Kobayashi N, Tsuda K, Unzai S, Saotome T, Kuroda Y, and Yamazaki T (2020) Solution structure of Gaussia Luciferase with five disulfide bonds and identification of a putative coelenterazine binding cavity by heteronuclear NMR, Sci Rep 10, 20069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lam SS, Martell JD, Kamer KJ, Deerinck TJ, Ellisman MH, Mootha VK, and Ting AY (2015) Directed evolution of APEX2 for electron microscopy and proximity labeling, Nat Methods 12, 51–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sherwood LJ, and Hayhurst A (2019) Periplasmic Nanobody-APEX2 Fusions Enable Facile Visualization of Ebola, Marburg, and Mengla virus Nucleoproteins, Alluding to Similar Antigenic Landscapes among Marburgvirus and Dianlovirus, Viruses 11, E364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Griep RA, van Twisk C, van der Wolf JM, and Schots A (1999) Fluobodies: green fluorescent single-chain Fv fusion proteins, J Immunol Methods 230, 121–130. [DOI] [PubMed] [Google Scholar]
- 27.Pedelacq JD, Cabantous S, Tran T, Terwilliger TC, and Waldo GS (2006) Engineering and characterization of a superfolder green fluorescent protein, Nat Biotechnol 24, 79–88. [DOI] [PubMed] [Google Scholar]
- 28.Dammeyer T, and Tinnefeld P (2012) Engineered fluorescent proteins illuminate the bacterial periplasm, Comput Struct Biotechnol J 3, e201210013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olichon A, and Surrey T (2007) Selection of genetically encoded fluorescent single domain antibodies engineered for efficient expression in Escherichia coli, J Biol Chem 282, 36314–36320. [DOI] [PubMed] [Google Scholar]
- 30.Buser DP, Schleicher KD, Prescianotto-Baschong C, and Spiess M (2018) A versatile nanobody-based toolkit to analyze retrograde transport from the cell surface, Proc Natl Acad Sci U S A 115, E6227–E6236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mazzega E, Beran A, Cabrini M, and de Marco A (2019) In vitro isolation of nanobodies for selective Alexandrium minutum recognition: A model for convenient development of dedicated immuno-reagents to study and diagnostic toxic unicellular algae, Harmful Algae 82, 44–51. [DOI] [PubMed] [Google Scholar]
- 32.Pradel N, Santini CL, Bernadac A, Shih YL, Goldberg MB, and Wu LF (2007) Polar positional information in Escherichia coli spherical cells, Biochem Biophys Res Commun 353, 493–500. [DOI] [PubMed] [Google Scholar]
- 33.Skoog K, Soderstrom B, Widengren J, von Heijne G, and Daley DO (2012) Sequential closure of the cytoplasm and then the periplasm during cell division in Escherichia coli, J Bacteriol 194, 584–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meiresonne NY, van der Ploeg R, Hink MA, and den Blaauwen T (2017) Activity-Related Conformational Changes in d,d-Carboxypeptidases Revealed by In Vivo Periplasmic Forster Resonance Energy Transfer Assay in Escherichia coli, MBio 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meiresonne NY, Consoli E, Mertens LMY, Chertkova AO, Goedhart J, and den Blaauwen T (2019) Superfolder mTurquoise2(ox) optimized for the bacterial periplasm allows high efficiency in vivo FRET of cell division antibiotic targets, Mol Microbiol 111, 1025–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shaner NC, Lambert GG, Chammas A, Ni Y, Cranfill PJ, Baird MA, Sell BR, Allen JR, Day RN, Israelsson M, Davidson MW, and Wang J (2013) A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum, Nat Methods 10, 407–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bindels DS, Haarbosch L, van Weeren L, Postma M, Wiese KE, Mastop M, Aumonier S, Gotthard G, Royant A, Hink MA, and Gadella TW Jr. (2017) mScarlet: a bright monomeric red fluorescent protein for cellular imaging, Nat Methods 14, 53–56. [DOI] [PubMed] [Google Scholar]
- 38.Ogando NS, Dalebout TJ, Zevenhoven-Dobbe JC, Limpens R, van der Meer Y, Caly L, Druce J, de Vries JJC, Kikkert M, Barcena M, Sidorov I, and Snijder EJ (2020) SARS-coronavirus-2 replication in Vero E6 cells: replication kinetics, rapid adaptation and cytopathology, J Gen Virol 101, 925–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Steiner D, Forrer P, Stumpp MT, and Pluckthun A (2006) Signal sequences directing cotranslational translocation expand the range of proteins amenable to phage display, Nat Biotechnol 24, 823–831. [DOI] [PubMed] [Google Scholar]
- 40.Mariati, Ho SC, Yap MG, and Yang Y (2010) Evaluating post-transcriptional regulatory elements for enhancing transient gene expression levels in CHO K1 and HEK293 cells, Protein Expr Purif 69, 9–15. [DOI] [PubMed] [Google Scholar]
- 41.Huang MT, and Gorman CM (1990) Intervening sequences increase efficiency of RNA 3' processing and accumulation of cytoplasmic RNA, Nucleic Acids Res 18, 937–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chapman BS, Thayer RM, Vincent KA, and Haigwood NL (1991) Effect of intron A from human cytomegalovirus (Towne) immediate-early gene on heterologous expression in mammalian cells, Nucleic Acids Res 19, 3979–3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zanta-Boussif MA, Charrier S, Brice-Ouzet A, Martin S, Opolon P, Thrasher AJ, Hope TJ, and Galy A (2009) Validation of a mutated PRE sequence allowing high and sustained transgene expression while abrogating WHV-X protein synthesis: application to the gene therapy of WAS, Gene Ther 16, 605–619. [DOI] [PubMed] [Google Scholar]
- 44.Sun ZF, and Meng XJ (2004) Antigenic cross-reactivity between the nucleocapsid protein of severe acute respiratory syndrome (SARS) coronavirus and polyclonal antisera of antigenic group I animal coronaviruses: implication for SARS diagnosis, J Clin Microbiol 42, 2351–2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Patrick DM, Petric M, Skowronski DM, Guasparini R, Booth TF, Krajden M, McGeer P, Bastien N, Gustafson L, Dubord J, Macdonald D, David ST, Srour LF, Parker R, Andonov A, Isaac-Renton J, Loewen N, McNabb G, McNabb A, Goh SH, Henwick S, Astell C, Guo JP, Drebot M, Tellier R, Plummer F, and Brunham RC (2006) An Outbreak of Human Coronavirus OC43 Infection and Serological Cross-reactivity with SARS Coronavirus, Can J Infect Dis Med Microbiol 17, 330–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee HK, Lee BH, Seok SH, Baek MW, Lee HY, Kim DJ, Na YR, Noh KJ, Park SH, Kumar DN, Kariwa H, Nakauchi M, Heo SJ, and Park JH (2010) Production of specific antibodies against SARS-coronavirus nucleocapsid protein without cross reactivity with human coronaviruses 229E and OC43, J Vet Sci 11, 165–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tyrrell DA, and Bynoe ML (1965) Cultivation of a Novel Type of Common-Cold Virus in Organ Cultures, Br Med J 1, 1467–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hamre D, and Procknow JJ (1966) A new virus isolated from the human respiratory tract, Proc Soc Exp Biol Med 121, 190–193. [DOI] [PubMed] [Google Scholar]
- 49.Ksiazek TG, Erdman D, Goldsmith CS, Zaki SR, Peret T, Emery S, Tong S, Urbani C, Comer JA, Lim W, Rollin PE, Dowell SF, Ling AE, Humphrey CD, Shieh WJ, Guarner J, Paddock CD, Rota P, Fields B, DeRisi J, Yang JY, Cox N, Hughes JM, LeDuc JW, Bellini WJ, and Anderson LJ (2003) A novel coronavirus associated with severe acute respiratory syndrome, N Engl J Med 348, 1953–1966. [DOI] [PubMed] [Google Scholar]
- 50.Guan Y, Zheng BJ, He YQ, Liu XL, Zhuang ZX, Cheung CL, Luo SW, Li PH, Zhang LJ, Guan YJ, Butt KM, Wong KL, Chan KW, Lim W, Shortridge KF, Yuen KY, Peiris JS, and Poon LL (2003) Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China, Science 302, 276–278. [DOI] [PubMed] [Google Scholar]
- 51.van der Hoek L, Pyrc K, Jebbink MF, Vermeulen-Oost W, Berkhout RJ, Wolthers KC, Wertheim-van Dillen PM, Kaandorp J, Spaargaren J, and Berkhout B (2004) Identification of a new human coronavirus, Nat Med 10, 368–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Woo PC, Lau SK, Chu CM, Chan KH, Tsoi HW, Huang Y, Wong BH, Poon RW, Cai JJ, Luk WK, Poon LL, Wong SS, Guan Y, Peiris JS, and Yuen KY (2005) Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia, J Virol 79, 884–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Corman VM, Muth D, Niemeyer D, and Drosten C (2018) Hosts and Sources of Endemic Human Coronaviruses, Adv Virus Res 100, 163–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, and Fouchier RA (2012) Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia, N Engl J Med 367, 1814–1820. [DOI] [PubMed] [Google Scholar]
- 55.Gralinski LE, and Menachery VD (2020) Return of the Coronavirus: 2019-nCoV, Viruses 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lau SKP, Luk HKH, Wong ACP, Li KSM, Zhu L, He Z, Fung J, Chan TTY, Fung KSC, and Woo PCY (2020) Possible Bat Origin of Severe Acute Respiratory Syndrome Coronavirus 2, Emerg Infect Dis 26, 1542–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Agnihothram S, Gopal R, Yount BL Jr., Donaldson EF, Menachery VD, Graham RL, Scobey TD, Gralinski LE, Denison MR, Zambon M, and Baric RS (2014) Evaluation of serologic and antigenic relationships between middle eastern respiratory syndrome coronavirus and other coronaviruses to develop vaccine platforms for the rapid response to emerging coronaviruses, J Infect Dis 209, 995–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Woo PCY, Lau SKP, Chen Y, Wong EYM, Chan KH, Chen H, Zhang L, Xia N, and Yuen KY (2018) Rapid detection of MERS coronavirus-like viruses in bats: potential for tracking MERS coronavirus transmission and animal origin, Emerg Microbes Infect 7, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gray AC, Bradbury ARM, Knappik A, Pluckthun A, Borrebaeck CAK, and Dubel S (2020) Animal-derived-antibody generation faces strict reform in accordance with European Union policy on animal use, Nat Methods 17, 755–756. [DOI] [PubMed] [Google Scholar]
- 60.Martell JD, Deerinck TJ, Lam SS, Ellisman MH, and Ting AY (2017) Electron microscopy using the genetically encoded APEX2 tag in cultured mammalian cells, Nat Protoc 12, 1792–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McCullock TW, MacLean DM, and Kammermeier PJ (2020) Comparing the performance of mScarlet-I, mRuby3, and mCherry as FRET acceptors for mNeonGreen, PLoS One 15, e0219886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sherwood LJ, and Hayhurst A (2012) Hapten mediated display and pairing of recombinant antibodies accelerates assay assembly for biothreat countermeasures, Sci Rep 2, 807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Neu HC, and Heppel LA (1965) The release of enzymes from Escherichia coli by osmotic shock and during the formation of spheroplasts, J Biol Chem 240, 3685–3692. [PubMed] [Google Scholar]
- 64.Sherwood LJ, and Hayhurst A (2013) Ebolavirus nucleoprotein C-termini potently attract single domain antibodies enabling monoclonal affinity reagent sandwich assay (MARSA) formulation, PLoS One 8, e61232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Darling TL, Sherwood LJ, and Hayhurst A (2017) Intracellular Crosslinking of Filoviral Nucleoproteins with Xintrabodies Restricts Viral Packaging, Front Immunol 8, 1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.