

Abstract
Genome-wide association studies (GWAS) identify genomic variants associated with human traits and diseases. Most trait-associated variants are located within cell type-specific enhancers, but the molecular mechanisms governing phenotypic variation are less understood. Here, we show that many enhancer-variants associated with red blood cell (RBC) traits map to enhancers that are co-bound by lineage-specific master transcription factors (MTFs) and signaling transcription factors (STFs) responsive to extracellular signals. The majority of enhancer variants resides on STF and not MTF motifs, perturbing DNA-binding by various STFs (BMP/TGFβ-directed SMADs or WNT-induced TCFs) and affecting target gene expression. Engineered human blood cells and expression quantitative trait loci (eQTL) analyses verify that disrupted STF binding leads to altered gene expression. Our results propose that the majority of the RBC trait-associated variants that reside on TF binding sequences falls in STF target sequences, suggesting that the phenotypic variation of RBC traits could stem from altered responsiveness to extracellular stimuli.
Introduction
A significant fraction of worldwide mortality is attributed to erythrocyte-related disorders1–7. Variation in red blood cell (RBC) traits is linked to mortality rates not related to primary hematologic disease1,3,6. Genome-wide association studies (GWAS) have identified numerous variable genomic regions associated with human traits and diseases, including RBC traits8–21. RBC-trait-associated single nucleotide polymorphisms (SNPs) rarely affect DNA binding of “master transcription factors” (MTFs), such as GATA2 and GATA1 even though they are often in close proximity to MTF target sequences 15,22,23. Additional mechanisms by which RBC-SNPs result in the phenotypic variability of human genetic traits remain to be discovered.
Environmental factors contribute to the phenotypic manifestation of complex human genetic traits1,3,6. Under stress conditions, growth factors and small molecules activate signaling pathways24–26 that converge on signal-induced effector transcription factors (“signaling transcription factors”, STFs) to control gene expression. By coordinating with MTFs, the same STFs may be active in multiple cell types but exert tissue-specific functions27,28. Hence, alterations in STF target sequences may lead to aberrant responses to various signals.
Here, we observed that human erythroid-trait-associated non-coding SNPs are enriched in a small subset of enhancers co-bound by MTFs and STFs, that we named “Transcriptional Signaling Centers (TSCs)”. Our study suggests that such SNPs alter the DNA-binding of various STFs significantly more frequently than that of blood-MTFs, leading to gene expression changes induced by extracellular signaling and consequently impacting red cell phenotypes.
Results
MTFs and STFs control cell type-specific gene expression
To understand how signaling impacts human erythropoiesis, we sought to identify genomic regions responsive to exogenous signals using in vitro erythroid differentiation of human hematopoietic progenitor cells (CD34+)29 (Extended Data Fig. 1a). By performing H3K27ac ChIP-seq to identify active enhancers30, ATAC-seq to determine chromatin accessibility31 and RNA-seq to quantify gene expression in these cells at various stages of differentiation (day 0 before differentiation induction and 6 hours, 3, 4, and 5 days after induction of erythroid differentiation), we observed two expression clusters before and after day 3 (D3), suggesting that CD34+ cells commit to an erythroid fate around D3 in this system (Extended Data Fig. 1 –f; Supplementary Table 1 represents genome-wide RNA expression values). Thus, we considered genes that are highly expressed before D3 as progenitor genes and after D3 as erythroid genes.
Fig. 1 |. BMP-SMAD1 signaling impacts human erythroid differentiation.
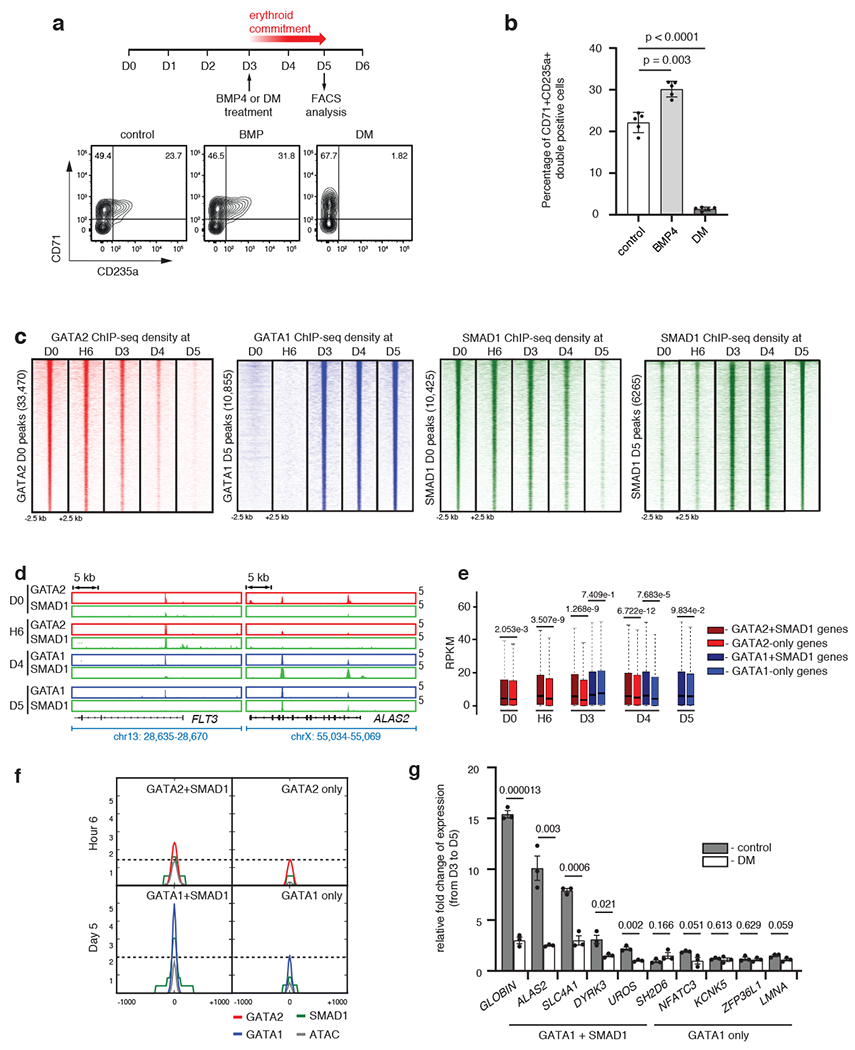
a, Representative FACS plots for CD71 and CD235a on BMP4-or dorsomorphin-treated CD34+ cells. b, Bar plots comparing the percentage of CD34+CD235a+ double positive cells from 1a. Mean ± SEM shown. (n=5; 5 biologically independent experiments). Two-sided Students-t test used. c, Region heatmaps depicting signal of ChIP-seq reads for GATA2, GATA1 and SMAD1 at D0, H6, D3, D4 and D5 of differentiation. Signal intensities around +/− 2.5 kb of the peak center are shown. d, Representative gene tracks for a progenitor-specific gene (FLT3) and an erythroid-specific gene (ALAS2) showing binding of each transcription factor at D0, H6, D4 and D5. e, RPKM expression distribution of genes bound either by GATA+SMAD1 or by GATA-alone at respective stages. Boxplots represent median RPKM as the thickest line, first and third quartile as the box, and 1.5 times interquartile range as whiskers. Two-sided Wilcoxon Rank-Sum tests used. f, Metagene plots comparing median signal intensities for ChIP-seq and ATAC-seq at regions co-bound by GATA2/1+SMAD1 versus GATA2/1 alone. Signal intensities around +/− 1 kb of the peak center shown. g, Change of expression of genes bound by GATA1+SMAD1 (HBB, ALAS2, SLC4A1, DYRK3 and UROS) or by GATA1-alone (SH2D6, NFATC3, KCNK5, ZFP36L1 and LMNA) after continuous dorsomorphin treatment for two days starting from D3. Mean ± SEM shown. (n=3; 3 biologically independent experiments). Two-sided Students-t test used.
Next, we investigated genomic occupancy of MTFs and STFs during erythroid differentiation. We chose GATA2 and GATA1 as exemplar progenitor and erythroid MTFs, respectively. To choose an STF, we tested the effect of BMP-SMAD signaling in our system, due to its importance in stress erythropoiesis28,32–35. Induction of BMP signaling by recombinant BMP4 or abrogation by dorsomorphin affected the efficiency of erythroid commitment (Fig. 1a, b), so we chose SMAD1 as an exemplar erythropoietic STF.
During differentiation, genomic occupancy of GATA2, identified by ChIP-seq, steadily decreased and GATA1 occupancy progressively increased while SMAD1 gradually re-localized to new genomic sites (Fig. 1c). SMAD1 binding at progenitor stages (D0-D3) or erythroid stages (D3-D5) overlapped markedly with MTFs of the respective stages (Fig. 1c, d; Extended Data Fig. 1g). We then identified the GATA2+SMAD1 co-occupied or GATA2-only genomic sites at D0, H6 and D3 and the GATA1+SMAD1 or GATA1-only genomic regions at D3, D4 and D5 and assigned them to the predicted target genes (Supplementary Table 2). Notably, GATA-only sites lack SMAD1 binding but possibly display binding of other MTFs besides GATA36,37. Ingenuity Pathway Analysis showed that genes co-bound by GATA1+SMAD1 are enriched for erythroid functions, whereas genes co-bound by GATA2+SMAD1 are enriched for progenitor functions (Extended Data Fig. 1h), indicating that GATA+SMAD1 co-bound regions regulate stage-specific genes. Next, by comparing expression between genes co-occupied by GATA+SMAD1 and genes occupied by GATA-alone, we found that genes proximal to co-occupied regions showed significantly higher expression (Fig. 1e). Overlap of stage-matched ATAC-seq and ChIP-seq data demonstrated that co-bound regions exhibit enhanced chromatin accessibility compared to regions where GATA factors bind without SMAD1 (Fig. 1f). Additionally, inhibition of BMP signaling by dorsomorphin significantly decreased expression of erythroid genes such as GLOBIN, ALAS and SLC4A1 that are co-bound by SMAD1+GATA1 at D5 but not of genes proximal to regions where GATA1 binds alone (Fig. 1g).
SMAD1+GATA regions are enriched for cell type-specific MTFs
To investigate the features that distinguish co-bound from MTF-only regions, we performed comparative motif analysis. This analysis showed over-representation of progenitor MTF sequence motifs (e.g. PU.1 and FLI1 motifs38,39) in the GATA2+SMAD1 regions at the H6 relative to GATA2-only regions, and erythroid factor motifs like EKLF/KLF1 and NFE motifs40,41 in GATA1+SMAD1 co-bound regions at D5-erythrocyte stage relative to GATA1-only regions (Extended Data Fig. 2a, b). Indeed, binding of PU.1 overlapped with GATA2+SMAD1 co-bound regions at D0 while GATA1+SMAD1 co-bound regions overlapped with KLF1 at D5. We observed at least 2.5-fold enrichment of PU.1 and KLF1 at co-occupied regions compared to the GATA-only regions at D0 and D5, respectively (Fig. 2a, b; Supplementary Table 3a). Additionally, genomic regions where stage-specific MTFs co-localize with SMAD1 are proximal to stage-specific genes, are located in open chromatin regions, and are enriched for H3K27ac (Fig. 1f, 2b; Extended Data Fig. 2c).
Fig. 2 |. Stage-specific MTFs are enriched in SMAD1+GATA co-bound regions.

a, Relative enrichment of PU.1 and KLF1 binding at GATA2/1+SMAD1 versus GATA2/1-only sites at D0 and D5. Two-sided Fisher’s exact test used. b, Representative gene tracks at FLI1 and ALAS2 at D0 and D5 showing occupancy of indicated transcription factors relative to ATAC-seq and H3K27ac signal. c, Binding intensities (mean reads per million per bin) of PU.1 and SMAD1 are shown, comparing control and PU.1 overexpressing cells. d. Representative gene tracks at CXCR4 and CXCL2 with peak intensities of PU.1 and SMAD1 in PU.1 over-expressing versus control cells. e, Binding intensities (average reads per bin) of PU.1, SMAD1 and GATA2 are shown, comparing control and PU.1 knockdown cells. f. Representative gene tracks at CD84 and WDR92 showing peak intensities of PU.1, SMAD1 and GATA2 in PU.1 knock down cells compared to control cells.
To examine the importance of binding of stage-specific MTFs within the SMAD1+GATA co-bound regions, we investigated the change of SMAD1 binding upon overexpression of PU.1 in K562 cells after BMP stimulation. PU.1 overexpressing cells showed increased binding of PU.1 in several genomic regions with a concomitant increase of SMAD1 binding within many of these regions, indicating that PU.1 can direct genomic localization of SMAD1 (Fig. 2c, d). We also confirmed that loss of PU.1 in K562 cells decreased PU.1 and SMAD1 occupancy within PU.1/SMAD1/GATA2 co-bound genomic regions while GATA2 binding did not diminish to the same extent (Fig. 2e). However, loss of PU.1 and SMAD1 binding could happen in the same or different cells. Overall, MTFs such as PU.1, enriched at GATA+SMAD1 sites, can recruit SMAD1 after stimulation to co-bound genomic regions, which likely behave as BMP-responsive enhancers.
Transcriptional Signaling Centers (TSCs)
Next, we sought to determine whether SMAD1+GATA co-bound regions could serve as docking sites for other STFs. We performed ChIP-seq for SMAD2 upon TGF-b stimulation42 and for TCF7L2 upon WNT stimulation28 at D0. Indeed, we observed co-localization of such STFs at GATA2-bound, ATAC-seq and H3K27ac signal-enriched enhancers, also co-occupied by SMAD1 upon BMP stimulation (Fig. 3a, b). 4,549 genomic regions, representing 25% of the total SMAD1-bound peaks, were co-occupied by SMAD1/2 and TCF7L2 (Extended Data Fig. 3a, Supplementary Table 3b). We reasoned that enhancers where combinations of STFs would converge with hematopoietic MTFs after induction by environmental stimuli are likely signal-responsive, and named them “Transcriptional Signaling Centers (TSCs)” (Fig. 3c).
Fig. 3 |. SMAD1+GATA co-bound enhancer regions form Transcriptional Signaling Centers (TSCs).
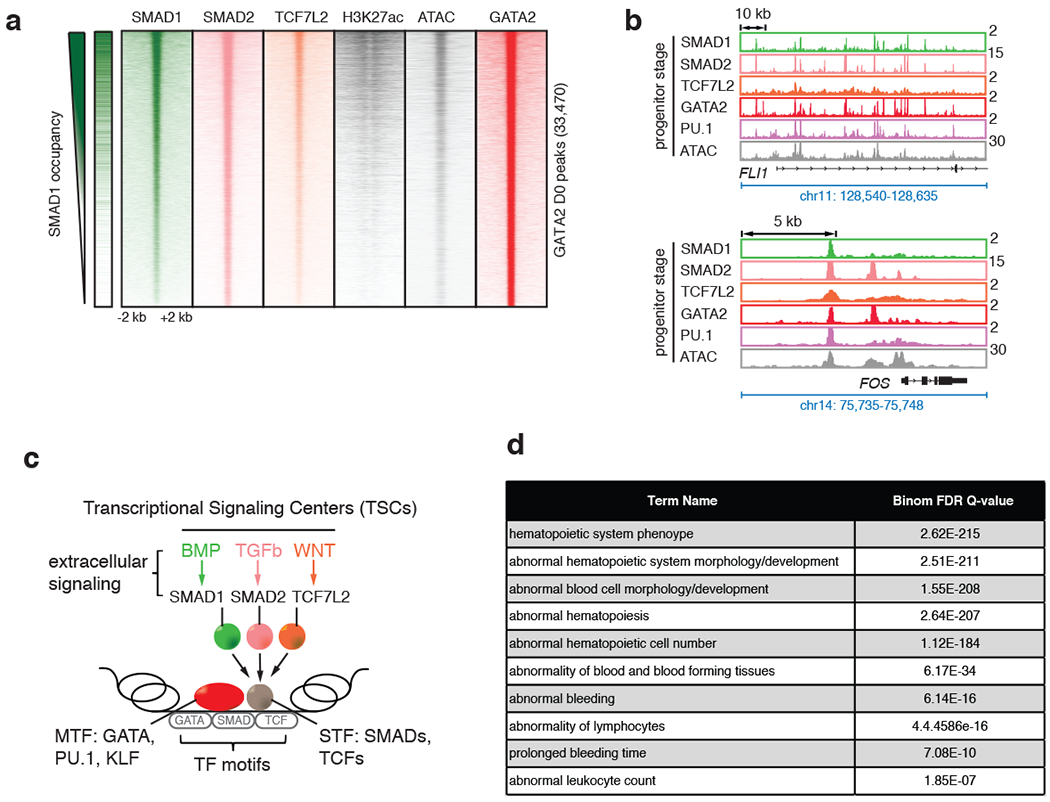
a, Signal heatmaps representing ChIP-Seq coverage of putative enhancers comprising GATA2 peaks, demonstrating co-occupancy by multiple STFs (SMAD1, green; SMAD2, magenta; and TCF7L2, orange) and MTF (GATA2, red) in progenitor CD34+ cells at D0 upon stimulation with BMP4, TGFβ and WNT signaling, respectively. Regions are considered occupied if they pass a significant coverage cutoff, shown as a binary green/white for SMAD1 heatmap on the left. GATA2 peak numbers obtained at D0 are represented in the Y-axis (33,470). H3K27ac and ATAC-seq heatmaps are also included. b, Representative gene tracks showing peak intensities of SMAD1 (BMP signaling, green), SMAD2 (TGFβ signaling, magenta) and TCF7L2 (WNT signaling, orange), with the master transcription factor GATA2 (red) and ATAC-seq signals at FLI1 and FOS genes at D0. c, Schematic representation of “transcriptional signaling centers (TSCs)”. TSCs are genomic regions that are co-occupied by multiple STFs induced by the respective signaling pathways. TSCs could be signal-specific leading to specific combinations of STFs co-occupying a given region with stage-specific MTFs. d, Human and mouse phenotypes associated with the peaks that are co-bound by SMAD1, SMAD2 and TCF7L2 upon stimulation with BMP4, TGFβ and WNT, respectively, identified using GREAT analysis.
While other STFs besides SMAD1 could define classes of TSCs, given the importance of BMP-SMAD1 signaling during stress hematopoiesis32–35, we focused on SMAD1-bound TSCs. GREAT analysis43 of genes associated with SMAD1+TCF7L2+SMAD2 co-bound regions showed enrichment for blood functions (Fig. 3d), suggesting that SMAD1, under BMP stimulation, could serve as a marker for TSCs during erythroid differentiation. Accordingly, we created a list of progenitor enhancers (merging ATAC-seq and H3K27ac ChIP-seq) and progenitor TSCs (overlapping enhancers with GATA2/SMAD1 ChIP-seq) by combining the data-points, D0 and H6. Similarly, erythroid enhancers and TSCs were identified by combining D4 and D5 ATAC-seq and ChIP-seq data (Supplementary Table 4). These analyses showed that TSCs represent a small fraction of ATAC/H3K27ac-positive active enhancers at each differentiation stage (7.2-21.7% of all the active enhancers, Extended Data Fig. 3b).
Perturbed STF binding at a TSC affects gene expression
To determine the functional consequences of STF occupancy within a TSC, we mutated STF or MTF binding sites within a representative TSC. We identified a TSC that was co-bound by GATA2, SMAD1 and PU.1 in both progenitor CD34+ (D0) and K562 erythro-leukemia cells and that was located within 5 kb from the nearest expressed gene, LHFPL2 (Fig. 4a). Perturbation of either the GATA, PU.1 or SMAD1 motifs in K562 or the human umbilical cord blood-derived erythroid progenitor (HUDEP2) cell line44 (Extended Data Fig. 4a, b) showed that, similar to loss of MTF binding sites (PU.1 and GATA), perturbations of binding sites of the STF SMAD1, led to downregulation of the LHFPL2 gene under BMP stimulation, while expression of two flanking genes (AP3B1 and SCAMP1) remained relatively unaltered (Fig. 4b, c; Extended Data Fig. 4c). Upon differentiation of HUDEP2 cells, perturbation of the same MTF or STF motifs within the LHFPL2 TSC led to a significantly decreased percentage of mature CD71low, CD235a+ erythroid cells (Fig. 4d, e). Mutation of the SMAD1 motif within the LHFPL2 TSC led to decreased occupancy of SMAD1 but not PU.1 (Fig. 4f). However, PU.1 knockdown in K562 cells led to decreased SMAD1 occupancy while GATA2 binding remain relatively unaltered (Fig. 4g). These results predict that, within a TSC, MTFs can direct the binding of an STF but not vice versa, at least in this specific TSC while STF binding can be at least as important as an MTF in controlling gene expression.
Fig. 4 |. STFs and MTFs at TSCs control gene expression.
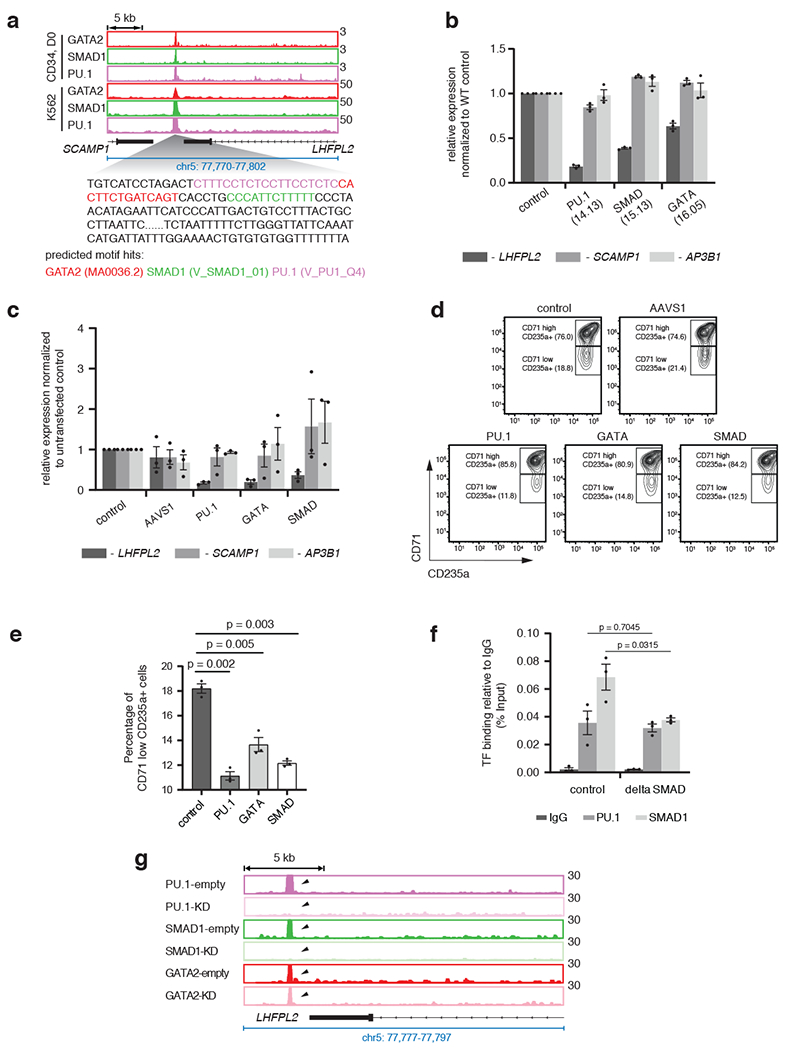
a, Overlap of occupancy of PU.1, GATA2 and SMAD1 at a representative TSC near LHFPL2 gene in progenitor CD34+ (D0) and K562 cells. Location of PU.1, GATA and SMAD1 motifs within the TSC are shown. b, Relative alteration of expression of LHFPL2, SCAMP1 and AP3B1 due to mutation of respective transcription factor motifs in specific K562 clones, as indicated. Mean ± SEM shown. (n=3; 3 biologically independent experiments). Two-sided Students-t test used. c, Relative change of expression of LHFPL2, SCAMP1 and AP3B1 in bulk edited HUDEP2 cells transduced with sgRNAs targeting PU.1, SMAD1 and/or GATA motifs in comparison with non-transduced cells or cells transduced with a control (AAVS1). Mean ± SEM shown. (n=3; 3 biologically independent experiments). Two-sided Students-t test used. d, Representative flow cytometry plots for CD71 and CD235a for HUDEP2 cell bulk cultures from 4c. Percentage distribution of cells within (CD71high, CD235a+) and (CD71low, CD235a+) compartments are shown. e, Bar plots comparing the percentage of CD71low, CD235a+ cells from 4d. Mean ± SEM shown. (n=3; 3 biologically independent experiments). Two-sided Students-t test used. f, Alteration of binding of PU.1 and SMAD1 in K562 cells with mutation of SMAD motif. Mean ± SEM shown. (n=3; 3 biologically independent experiments). Two-sided Students-t test used. g, Gene tracks at LHFPL2 TSC showing peak intensities of PU.1, SMAD1 and GATA2 in PU.1 knock down cells compared to control cells.
SNPs affecting RBC traits are enriched within TSCs
Since SNPs are primarily located in non-coding genomic regions45–50, we wondered whether TSCs harbor non-coding GWAS variants associated with RBC traits. We compiled a set of SNPs from thirteen published GWAS studies associated with seven erythrocyte traits: hemoglobin concentration (Hb), hematocrit or packed cell volume (Hct or PCV), mean corpuscular volume (MCV), mean corpuscular hemoglobin (MCH), mean corpuscular hemoglobin concentration (MCHC), red blood cell count (RBC) and red blood cell distribution width (RDW)4,14–20,51–55. To increase the likelihood of including functional SNPs, we used 1,270 lead SNPs for individual traits/region, together with the co-inherited SNPs in high linkage disequilibrium with them (LD r2 ≥ 0.6, as suggested by previous studies56–61, designated here as lead+LD SNPs). Altogether, 29,069 lead and LD SNPs with at least two usable alleles across 924 loci associated with the seven RBC traits were used (Supplementary Table 5a, b). Out of 1,270 lead SNPs and 29,069 lead+LD RBC-trait SNPs, 353 and 3,318 SNPs were located in enhancers defined by ATAC-seq and H3K27ac ChIP-seq data (Extended Data Fig. 5a, Supplementary Table 5c, d). To confirm that our criteria of selecting SNPs enriched for potentially functional variants, we used RegulomeDB62 and found a significant enrichment in SNPs with predicted effects on gene regulation (RegulomeDB score ≤ 4; 51.1% of ATAC+H3K27ac SNPs compared to 19.6% of all SNPs) (Fig. 5a; Supplementary Table 3c).
Fig. 5 |. RBC-trait SNPs enriched within TSCs predominantly reside in STF motifs.
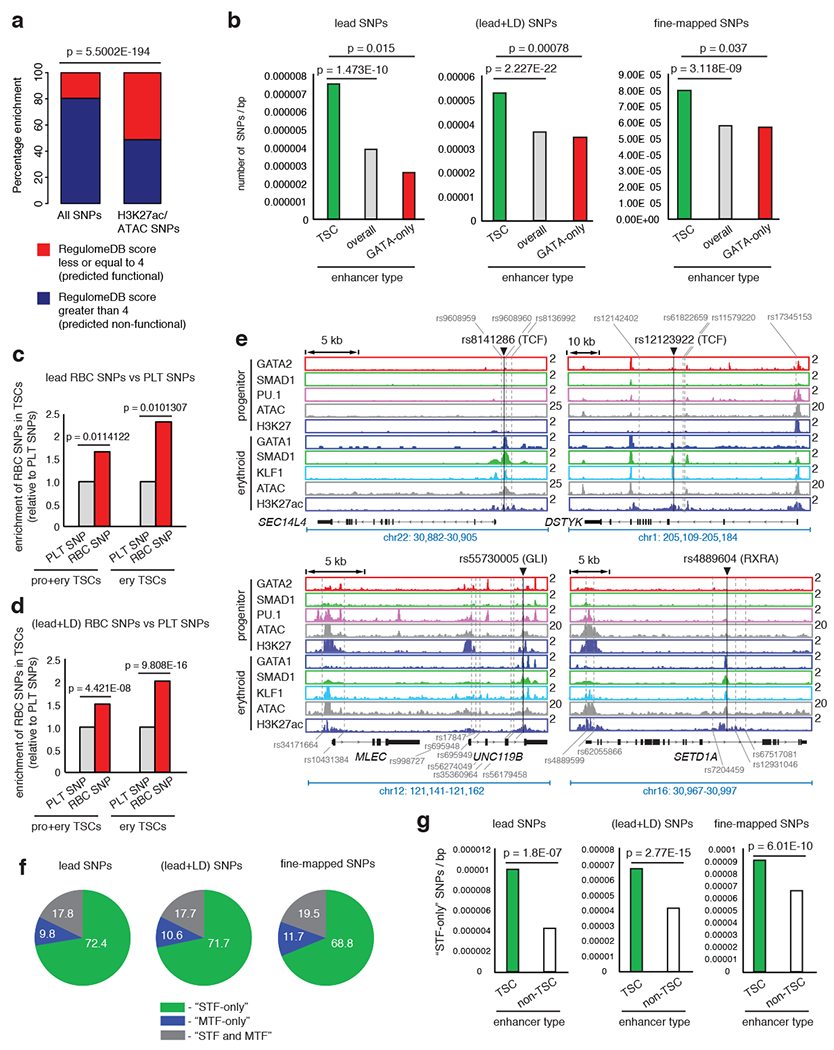
a, Enrichment of predicted functional SNPs in non-exonic open enhancer regions versus all SNPs. 2X2 chi-square significance tests used. b, Enrichment of SNPs within TSCs versus all and GATA-only enhancers. 2X2 chi-square significance values shown. p values for permutation tests obtained by shuffling SNP positions in TSCs <0.0001 for all SNP-types; in GATA-only enhancers: lead SNPs, p = 0.9166; lead+LD SNPs, p=1; fine-mapped SNPs, p = 1. p values by permuting the TSC/non-TSC labels of enhancers <0.0001 for all SNP-types. c and d, Enrichment of lead and lead+LD RBC-trait SNPs, relative to platelet-trait SNPs within progenitor+erythroid and erythroid-only TSCs. 2X2 chi-square significance tests used. e, Example RBC-trait SNPs (black line) localized within stage-specific TSCs. Binding sites of STFs at these SNPs shown. Additional SNPs with significant LD within enhancers shown (grey dashed lines). f, Distribution of lead, lead+LD or fine-mapped SNPs at “STF-only”, “MTF-only” or “STF and MTFs” motifs. g, Enrichment of SNPs overlapping STF-only motif-hits within TSC versus non-TSC enhancers. 2X2 chi-square significance values shown. p values calculated by randomly permuting SNP positions showing the enrichment of STF-only SNPs in TSCs: lead SNPs, p<0.0001; lead+LD SNPs, p<0.0001; fine-mapped SNPs, p<0.0001. p values calculated by randomly permuting labels of enhancers as TSC/non-TSC: lead SNPs, p<0.0001; lead+LD SNPs, p<0.0001; fine-mapped SNPs, p<0.0001. p values calculated by randomly permuting positions of STF motif hits: lead SNPs, p=0.0194; lead+LD SNPs, p<0.0001; fine-mapped SNPs, p=0.0033.
We then inquired whether enhancer-associated SNPs are primarily located in TSCs. We assessed the number of SNPs located within TSCs out of the 353 lead enhancer SNPs or the 3,318 (lead+LD) enhancer SNPs (Supplementary Table 5e, f) and compared the number of SNPs in TSCs to the number of SNPs in overall enhancers or in GATA2/1-only enhancers, normalized to the size of each region type in base pairs (bp). We found that enhancer-variants are significantly enriched within TSCs (Fig. 5b, Supplementary Table 3d). We also analyzed an independent list of fine-mapping based SNPs associated with sixteen different blood traits 23. Indeed, fine-mapping-based SNPs (with posterior probability value, PP > 0.01) are significantly enriched in TSCs compared to overall enhancers or enhancers that are occupied by only GATA2/1 during differentiation (Fig. 5b, Supplementary Table 3d). Taken together, these results show that enhancer-variants are significantly enriched within TSCs.
To test if SNPs linked to erythroid traits and not the traits of other lineages are enriched in erythroid TSCs, we compiled SNPs linked to platelet traits as controls. We used 786 platelet trait loci regions associated with 575 lead and 22,158 (lead+LD) SNPs (LD r2 ≥ 0.6) with at least two usable alleles19 (Supplementary Table 5g–j). By comparing lead RBC-trait SNPs to lead platelet trait SNPs, or (lead+LD) RBC-trait SNPs versus (lead+LD) platelet trait SNPs, we observed that RBC-trait SNPs are significantly enriched within erythroid TSCs (Fig. 5c–e; Extended Data Fig. 5b, Supplementary Table 3e). In conclusion, RBC trait SNPs, but not platelet trait SNPs, are primarily enriched within erythroid TSCs.
Many RBC trait SNPs are located within STF motif hits
We then asked whether non-coding RBC trait SNPs could modulate transcription by altering the binding of transcription factors (TFs). We predicted TF motif hits (Methods) and created lists of predicted binding sites of hematopoietic MTFs and generic STFs (Supplementary Table 6; Supplementary Note). We calculated the number of enhancer-associated SNPs appearing in STF or MTF motif hits. We categorized the motifs as “STF-only” or “MTF-only” (recognized by STFs or MTFs, respectively, but not both) and “STFs and MTFs” (motif hits recognized by either STFs or MTFs). While 72.4% of lead SNPs within MTF or STF motif hits overlap “STF-only” motif hits, only 9.8% overlap “MTF-only” motif hits and 17.8% reside on ambiguous, “STF and MTF” motif hits (Fig. 5f). Similar conclusions were true for (lead+LD) (Fig. 5f) and enhancer-associated fine-mapped SNPs (PP > 0.01) overlapping TF motif hits (Fig. 5f). We then investigated whether the SNPs within STF motif hits are enriched in TSCs compared to non-TSC enhancers. Using either the lead, (lead+LD) or the fine-mapped SNPs, we compared the number of SNPs in STF motif hits between TSCs and non-TSC enhancers, normalized to the total number of basepairs in each region type. Indeed, TSCs show a significant enrichment for SNPs associated with STF motif-hits relative to non-TSC enhancers (Fig. 5g; Supplementary Table 3f). Thus, the majority of enhancer-associated RBC-trait SNPs that overlap TF binding sequences, are found in STF binding sites, and such STF-SNPs are significantly over-represented within TSCs.
Functional alteration of STF-DNA binding by RBC SNPs
We hypothesized that STF-SNPs may lead to differential STF occupancy within TSCs, resulting in altered gene expression under stimulation. Thus, we analyzed protein binding microarray (PBM) datasets63 to identify RBC-trait SNPs that affect binding of STFs within TSCs (Extended Data Fig. 6a). Using previously published PBM data for several STFs (Supplementary Table 7)64,65, we compared the binding of in vitro expressed SMAD, between the two alleles of SNPs located in open chromatin enhancer regions as a proof of principle. Since SMAD1 PBM data were not available, we analyzed a mouse SMAD3 PBM dataset66 (the 69.61% identity of the MH1 DNA-binding domain sequence to the human SMAD1 MH1 DNA-binding domain strongly argues that the TFs share similar sequence specificity65). Analysis of PBM data identified examples like rs737092, where the change of the T to C allele significantly diminishes SMAD binding but causes little change in GATA binding between the two alleles of rs737092, despite its close proximity to the GATA motif15 (Fig. 6a,b; Extended Data Fig. 6b). This result argues for the existence of RBC trait-associated SNPs that could perturb STF-DNA binding without significantly altering the binding of a hematopoietic MTF.
Fig. 6 |. Protein binding microarray (PBM) identifies RBC trait-associated SNPs that perturb STF-DNA binding.
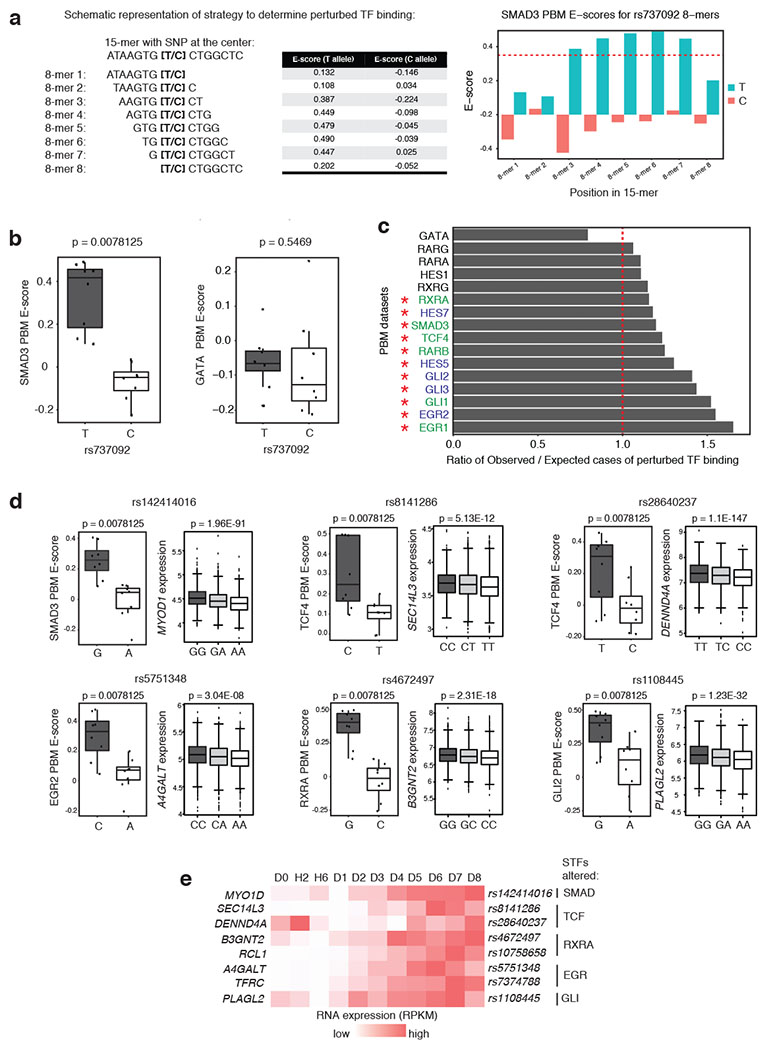
a, Schematic representation of perturbed transcription factor binding analysis strategy using PBM data. Red dashed line indicates universal PBM 8-mer enrichment (E) score = 0.35, which is used as a threshold for specific binding by a TF. b, Boxplots representing SMAD3 and the average GATA PBM E-scores of rs737092. Two-sided Wilcoxon signed-rank tests used. c, Bar plots depicting the ratio of observed versus expected number of perturbed TF binding events. Red dashed line indicates ratio = 1. Red asterisks indicate STFs with significantly greater than expected numbers of perturbed TF binding events (Q-value < 0.05 after Benjamini-Hochberg-adjusted empirical p-values. STFs indicated in green are expressed during CD34+ differentiation (RPKM > 1). STFs in blue have close paralogs that are expressed during erythroid differentiation. d, Example SNPs showing perturbed STF binding from PBM analysis and corresponding expression distribution of the most significantly altered nearby gene in homozygous and heterozygous individuals obtained from FHS eQTL analysis. Boxplots represent the median as the thickest line, the first and third quartile as the box, and 1.5 times the interquartile range as the whiskers. Two-sided Wilcoxon signed-rank tests used for PBM boxplots. Two-sided test with linear model for EffectAlleleDosage used for eQTL analysis with Benjamini-Hochberg adjusted P-values. e, Heatmap depicting the expression of the most significantly altered nearby gene (from FHS eQTL analysis), during normal erythroid differentiation.
We then analyzed our list of enhancer-associated SNPs for their predicted effects on STF binding and gene expression. For this purpose, universal PBM 8-mer enrichment (E) score datasets were downloaded from the UniPROBE and CIS-BP64,65 databases. (Supplementary Table 765–69). Of the 3,318 enhancer-associated (lead+LD) variants that included indels, we focused our analysis on the 3,263 single nucleotide substitutions (Supplementary Note). We considered perturbed binding events for GATA family MTFs, by using an averaged GATA binding profile from available GATA family PBM datasets64, for comparison against several STFs. We found that several STFs, including SMAD3, TCF4, RXRA, GLI1/2/3 and EGR1/2, showed a greater-than-expected frequency of perturbed binding events in this set of RBC-trait SNPs (Benjamini-Hochberg adjusted empirical p-value < 0.05), while GATA binding appeared to be perturbed less frequently than expected (Fig. 6c, d; Extended Data Fig. 6c). Inclusion of fine-mapped variants in PBM analysis further supported this conclusion (Supplementary Note, Supplementary Table 8). To further investigate the effects of STF-binding-altering SNPs on downstream gene expression, we coupled the PBM approach with eQTL analysis using microarray gene expression profiles of peripheral blood, isolated from participants in the Framingham Heart Study (FHS)70. In several instances, where the SNP resulted in a significant decrease in STF binding, the SNP was also identified as a cis-eQTL in the FHS dataset leading to a dose-dependent expression reduction of a proximal gene (Fig. 6d; Extended Data Fig. 6c). 86 out of the 115 transcripts from the FHS cis-eQTL gene list, and 108 out of 148 transcripts from the FHS cis-eQTL exon list showed at least one cis-eQTL SNP that also affected STF binding. Our RNA-seq results verify that those genes show a steady increase in expression during erythroid differentiation (Fig. 6e). Notably, loss of STF binding induced by a SNP allele could also lead to increased expression of associated genes. Overall, SNP-mediated modulation of STF-DNA binding results in expression alteration of relevant trait-associated genes.
STF-SNPs can perturb DNA binding and gene expression
To validate whether alternative alleles of representative SNPs govern STF occupancy and gene expression, we investigated the effects of SMAD1 binding at the MCV-associated SNP rs9467664 (T>A), residing on a SMAD target sequence within a TSC proximal to HIST1H4A, which shows increased expression during erythroid differentiation (Extended Data Fig. 7a, b). Electrophoretic Mobility Shift Assays showed that oligonucleotides harboring the T but not the A allele could efficiently bind SMAD1 (Extended Data Fig. 7c, d). eQTL analysis from the FHS showed that the A allele is significantly associated with reduced levels of HIST1H4A mRNA compared to the T allele (Extended Data Fig. 7e), further supporting our hypothesis that alteration of SMAD1 binding by an RBC trait-associated SNP may have significant effects on gene expression.
Fig. 7 |. STF-SNPs perturb STF-DNA binding and abrogate signal responsiveness.
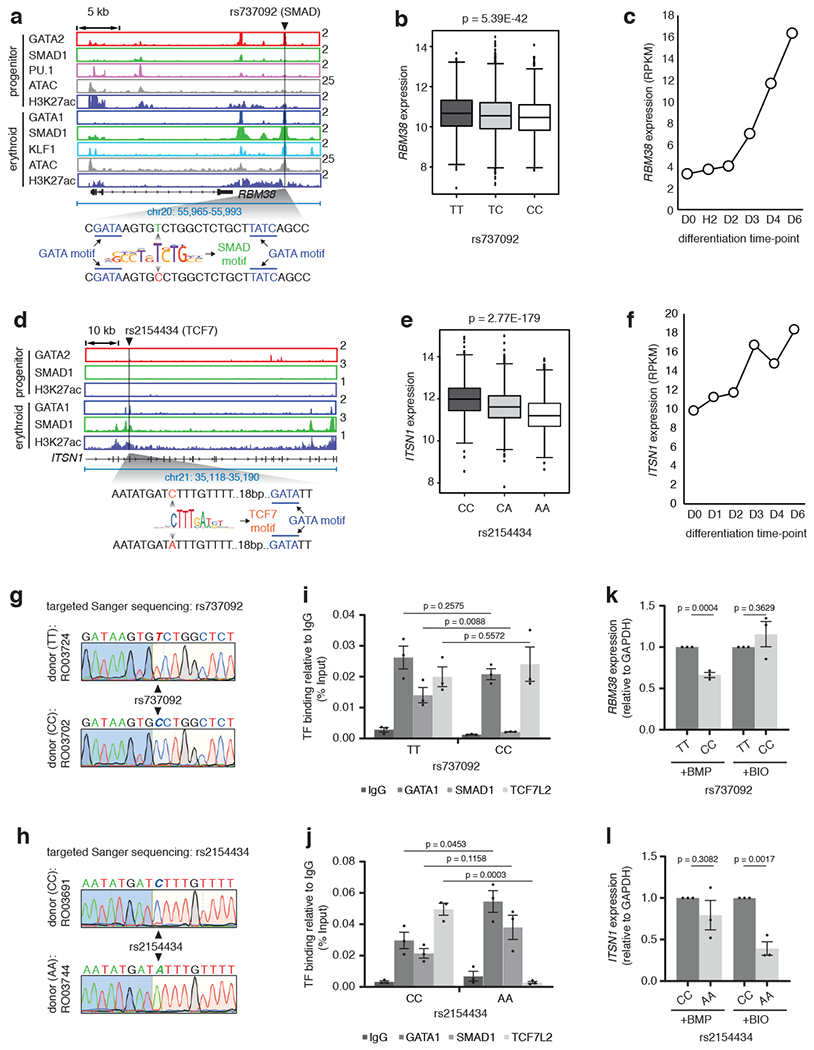
a, Gene tracks at RBM38 depicting the erythroid-specific TSC containing rs737092 at SMAD motif. b, RBM38 eQTL analysis for rs737092: boxplots represent median RBM38 expression as the thickest line, the first and third quartile as the box, and 1.5 times the interquartile range as whiskers. Two-sided test with linear model for EffectAlleleDosage used: effect estimate (β)=−0.05211; T-statistics=−13.6994, R2=0.034622; log10(P-value)=−41.2683 , log10(Benjamin-Hochberg’s FDR)=−38.6118. c, Expression RPKM values for the RBM38 gene at different stages of CD34+ erythroid differentiation. d, ITSN1 eQTL analysis for rs2154434: boxplots represent the median ITSN1 expression as the thickest line, the first and third quartile as the box, and 1.5 times the interquartile range as the whiskers. Two-sided test with linear model for EffectAlleleDosage used: effect estimate (β)=−0.0486; T-statistics=−29.7008, R2=0.144255; log10(P-value)=−178.558, log10(Benjamin-Hochberg’s FDR)=−175.322. f, Expression RPKM values for ITSN1 at stages of CD34+ erythroid differentiation. g and h, Sanger sequencing chromatograms of individual donors for SNPs rs737092 and rs2154434. Donor numbers indicated. i and j, Binding alteration of GATA1, SMAD1 and TCF7L2 for alternative alleles of rs737092 and rs2154434. Mean ± SEM shown. (n=3; 3 biologically independent experiments). Two-sided Students-t test used. k and l, qPCR analysis comparing the expression of RBM38 and ITSN1, relative to GAPDH, for alternative alleles of rs737092 and rs2154434, respectively, under BMP and BIO treatment. Mean ± SEM shown. (n=3; 3 biologically independent experiments). Two-sided Students-t test used.
To test whether perturbed STF binding impairs signal-induced gene expression, we selected a second SNP rs737092 (T>C). This SNP resides in an erythroid-specific TSC co-bound by GATA1, SMAD1 and KLF1 within an H3K27ac-positive open chromatin region. The SNP is present within a SMAD motif flanked by two GATA motifs (Fig. 7a). PBM analysis showed that this SNP perturbed SMAD binding without altering GATA binding. rs737092 was identified in a previously published massively parallel reporter assay (MPRA) study as functional, regulating the expression of RBM3815, which was confirmed by eQTL analysis from the FHS study. Finally, RBM38 is significantly more highly expressed in a population with the T but not the C allele and its expression is steadily increased during differentiation in our dataset (Fig. 7b, c). We obtained a CRISPR-Cas9 modified K562 cell line15, where the SMAD1 motif within the RBM38 TSC is mutated together with the upstream but not the downstream GATA motif (Extended Data Fig. 7f). ChIP-qPCR assays for SMAD1 binding under BMP stimulation showed significant abrogation of SMAD1 binding in these cells but GATA1 binding remained relatively unchanged presumably due to compensation from the other flanking GATA motif (Extended Data Fig. 7g). As a control, the binding of the WNT responsive factor TCF7L228 to its motif in the same TSC after WNT pathway stimulation with BIO71 was not affected (Extended Data Fig. 7g). Concomitantly, the expression of RBM38 was significantly reduced in the mutant cells under BMP but not under BIO treatment (Extended Data Fig. 7h). We then cloned the actual RBM38 TSC with either the T or the C allele upstream of the firefly luciferase gene15. The T allele, which retains SMAD binding, showed higher increase in luciferase expression under BMP stimulation relative to no stimulation or dorsomorphin treatment (Extended Data Fig. 7i). These results suggest that abrogation of the SMAD1 motif in the RBM38 TSC that harbors the rs737092 SNP diminished SMAD1 binding and compromised BMP responsiveness.
Effect of STF-SNPs within TSCs in primary human samples
We then decided to investigate the effects of RBC trait SNPs in primary human peripheral blood CD34+ cells. We first validated that knockdown of SMAD1 in CD34+ cells impaired activation of RBM38 under BMP stimulation (Extended Data Fig. 7j, k). Next, we screened eighteen human donors and identified individuals with homozygous alleles for preselected TSC-associated SNPs - rs737092 (T>C) and rs2154434 (C>A) (minor allele frequencies for rs737092 and rs2154434 are 47.9% and 42.9%, respectively). Similar to rs737092, rs2154434 is also located within an erythroid TSC during erythroid differentiation (Fig. 7d), and we observed a dose-dependent decrease of ITSN1 expression in FHS when the C allele is replaced by the A allele (Fig. 7e). ITSN1 also increases expression during erythroid differentiation (Fig. 7f). Individual donors with homozygous genotypes for alternative alleles of rs737092 and rs2154434 were confirmed by PCR and sequencing (Fig. 7g, h). We next evaluated transcription factor binding and BMP4-responsiveness of the alleles in donor CD34+ cells. rs737092 should affect SMAD but not TCF7L2 binding when the T is replaced by the C allele. Indeed, ChIP-PCR performed in BMP4-treated CD34+ cells with rs737092 alleles, differentiated for 5 days, showed attenuated SMAD1 binding under T to C change (Fig. 7i). TCF7L2 binding under BIO stimulation or GATA1 binding did not change significantly (Fig. 7i). In contrast, rs2154434 should primarily disrupt the DNA-binding of TCF7L2 but not of SMAD1. Indeed, we observed disrupted TCF7L2 but not SMAD1 or GATA1 binding upon BIO stimulation when the C allele was replaced by the A allele (Fig 7j). We also tested the allele-specific mRNA expression of RBM38 and ITSN1 for the alleles of rs737092 and rs2154434 after acutely stimulating CD34+ cells with BMP4 and BIO, respectively at D5 of differentiation for 2 h. Change from T to C allele mediated by the rs737092 SNP led to decreased expression of RBM38 under BMP but not BIO treatment (Fig. 7k). Similarly, ITSN1 expression was downregulated primarily under WNT stimulation when the C allele of rs2154434 was replaced by the A allele (Fig. 7l). These results suggest that RBC trait associated SNPs, overlapping TF binding sites, often abrogate DNA binding of STFs and not of MTFs to affect gene expression by respective signaling pathways in primary human samples.
Discussion
The majority of GWAS-associated variants linked to human genetic traits and diseases are non-coding45–50. Using genetic fine-mapping of sixteen traits associated with blood, Ulirsch et al. showed that SNPs are often located within open chromatin regions enriched for lineage-specific MTF motifs23. Although blood trait-associated GWAS SNPs are often found in close proximity to MTF motifs, the majority does not disrupt their binding sites directly15,22,23. Here, utilizing functional and computational approaches, we show that the alteration of STF binding induced by SNPs within TSCs, which represent a subset of enhancers co-occupied by both MTFs and STFs, may drive a disproportionate fraction of phenotypic variability of human RBCs. Importantly, using several systems, including primary CD34+ cells isolated from human donors with specific SNP alleles, we show that SNPs altering STF binding can modulate the induction of adjacent genes by respective signaling pathways (Supplementary Note).
It is important to understand why allele-specific effects of SNPs residing in STFs are more common than in MTF motifs. We speculate that SNPs affecting MTF binding can drastically affect expression of genes essential for development, and thus be less likely to be favored by natural selection. STF-SNPs on the other hand can cause expression variability leading to tolerable phenotypic changes in RBC traits and thereby escape evolutionary pressure. Accordingly, we evaluated the published prediction scores from NCboost72, which predict the pathogenicity of a variant occurring at non-coding positions of the genome based on evolutionary signals. The predicted pathogenicity of altering bases in MTF motifs appears significantly higher than that caused by alterations in STF motif hits (data not shown). Thus, an STF-SNP can render enhancers and their regulated genes sub-optimally responsive to one or more signaling pathways during episodic stresses such as infections or environmental changes. The abnormal response to periodic stress signals could contribute to tissue damage and disease over time. Such altered signaling events over time could lead to “signalopathies”, ultimately resulting in phenotypic variation and susceptibility to a spectrum of human genetic diseases (Extended Data Fig. 8).
Online Methods
Expansion and differentiation of CD34+ cells
Human CD34+ cells, isolated from peripheral blood of granulocyte colony-stimulating factor-mobilized healthy volunteers, were purchased from the Fred Hutchinson Cancer Research Center. The cells were maintained and differentiated as previously described28,73. Briefly the cells were expanded in StemSpan medium (Stem Cell Technologies Inc.) supplemented with StemSpan CC100 cytokine mix (Stem Cell Technologies Inc.) and 2% P/S for a total of 6 days. After six days of expansion the cells were stimulated for 2 h with rhBMP4 (R&D) at a final concentration of 25 ng/ml and harvested for performing all the experiments corresponding to D0 time point. For studying differentiated cells after day 6 of expansion, cells were reseeded in differentiation medium (StemSpan SFEM Medium with 2% P/S, 20 ng/ml SCF, 1 U/ml Epo, 5 ng/ml IL-3, 2 mM dexamethasone, and 1 mM b-estradiol), at a density of 0.5–13 106 cells/ml. Prior to harvesting at H2, H6, D1-D8 the cells were treated with 25ng/ml hrBMP4 for 2hrs.
For testing the effects of BMP4 and dorsomorphin, cells at the beginning of the third day of differentiation were treated with either 25 ng/ml hrBMP4 or 20 μM dorsomorphin until the beginning of the fifth day of differentiation. At D5, cells were isolated for flow cytometry and qPCR analysis. Cells treated with DMSO were used for control experiments.
Flow cytometry analysis
Control and treated stage-matched CD34+ cells or CD34+ cells at different stages of differentiation were washed in PBS and stained with propidium iodide (PI), 1:60 APC-conjugated CD235a (eBioscience, clone HIR2, 17-9987-42), 1:60 FITC-conjugated CD71 (eBioscience, OKT9, 11-0719-42), 1:60 PE-conjugated CD41a (eBioscience, HIP8, 12-0419-42) and 1:60 PE-conjugated CD11b (eBioscience, ICRF44, 12-011842). BD Bioscience LSR II flow cytometer was used to record raw FACS data, which were analyzed subsequently using FlowJo (v10.3).
Next-generation sequencing
Methodologies for all the massively parallel sequencing assays (ChIP-seq, RNA-seq and ATAC-seq) are described in the Supplementary Note. Overall quality control of each dataset is represented in Supplementary Table 9. Supplementary Table 10 describes counts and genomic span of individual TF-bound regions along with counts of associated genes, as obtained from ChIP-seq and RNA-seq data. The ChIP-seq and ATAC-seq peaks/enriched regions obtained from D0, H6, D3, D4 and D5 are shown in Supplementary Tables 11–15.
qPCR analysis
RNA was extracted from CD34+ cells without any treatment or treated with hrBMP4 or dorsomorphin at the specified developmental stages using TRIZOL extraction (Invitrogen), followed by RNeasy column purification (QIAGEN). First strand cDNA synthesis was performed using the Superscript VILO (Invitrogen) and equivalent amounts of starting RNA from all samples. The cDNA was analyzed with the Light Cycler 480 II SYBR green master mix (Applied Biosystems), and the QuantStudio 12K Flex (Applied Biosystems). All samples were prepared in triplicate. The PCR cycle conditions used are: (a) 95° C for 5 min, (b) [95° C for 10 sec, 54° C for 10 sec, 72° C for 15 sec] X 40 cycles. The analysis of Ct values were performed using 2Λ-ΔΔT method 74. The PCR primer-pairs used can be found in Supplementary Table 16.
Generation of CRISPR clones in K562 and checking the expression with qPCR
pSpCas9(BB)-2A-GFP (PX458) (a gift from Feng Zhang, Addgene plasmid # 48138),75 was used to generate mutations at the LHFPL2 transcriptional signaling center. gRNAs were designed using CHOPCHOP tool76 or the CRISPR design tool from the Zhang lab77. The sequences of gRNAs selected are schematized in Extended Data Fig. 3. The gRNAs were cloned in pSpCas9(BB)-2A-GFP (PX458) and verified by sequencing according to the instructions by Cong et al77. For the generation of mutant cell lines, 20 μg of each gRNA that was cloned into pSpCas9(BB)-2A-GFP (PX458), was electroporated into K562 cells. 48 hours after, single fluorescent cells were FACS sorted into 96-well plates. Oligonucleotide sequences corresponding to individual gRNAs (to target PU1, GATA and SMAD1 motif) used for cloning can be found in Supplementary Table 16.
Genome editing and differentiation of HUDEP2 cells
HUDEP2 cells were cultured as previously described78. Cas9-expressing HUDEP2 cells in expansion cultures were transduced with sgRNAs targeting AAVS1 as negative control79, LHFPL2, or the PU.1 motif, GATA motif or SMAD1 motif in the corresponding signaling center. The same gRNAs that were validated in K562 cells were used in this experiment. 24 hours after transduction, cells were transferred to a “growth phase” erythroid differentiation medium containing stem cell factor and doxycycline for 3 days. Puromycin was added to this medium to select for sgRNA transduced cells. Then cells were transferred to a “maturation phase” erythroid differentiation medium containing doxycycline for 4 days. After 4 days in this medium, an aliquot of cells was collected and processed for RNA isolation to determine LHFPL2 expression. The remaining cells were transferred to erythroid differentiation medium without doxycycline for 2 days, and erythroid differentiation status was assessed on the final day by cell surface marker staining, using anti-CD71-PeCy7 (eBioscience, #25-0719-42) and anti-CD235a-APC (eBioscience, # 17-9987-42) antibodies, and flow cytometry.
Identifying human blood donors with homozygous SNP alleles
Genomic DNA from CD34+ cells isolated from peripheral blood of individual donors were extracted using the DNeasy Blood & Tissue kit (Qiagen, 69506) as per manufacturer’s protocol. The PCR amplification of each TSC region was carried out using the Q5 High-Fidelity 2X Master Mix (M0492S) and primers used can be found in Supplementary Table 16.
siRNA-mediated SMAD1 knock down
SMAD1 knock down was performed upon nucleofecting siRNA for SMAD1 during the expansion of CD34+ cells (using Amexa 4D-Nucleofector kit from Lonza, V4XP-3024, as per manufacturer’s protocol). We used confirmed SMAD1 siRNA from Dharmacon (onTARGETplus, SMARTpool, L-012723-00-0005) and a standard non-targeting siRNA as control (D-001810-10-05). Three different treatment doses for SMAD1 siRNA were used – 25nM, 50nM and 100nM. Control siRNA was used at 100 nM concentration. After confirming SMAD1 knockdown, we differentiated CD34+ cells to erythroid lineage and kept them under BMP stimulation from D3 onwards. Expression of RBM38 RNA and protein were verified at D5.
Luciferase Reporter Assay
Firefly luciferase reporter constructs (pGL4.24) were made by separately cloning each of the alleles of interest centered in 426 nucleotides of genomic context upstream of the minimal promoter using BglII and Xhol sites. The firefly constructs (500ng) were co-transfected with a pRL-SV40 Renilla luciferase construct (50ng) into 100,000 K562 cells using Lipofectamine LTX (Invitrogen, Ref: 15338-030). After 48 hours, luciferase activity was measured by Dual-Glo Luciferase assay system (Promega, Ref: E2940) according to manufacturer’s protocol. 24 hours before luciferase activity measurement, cells were treated with 25ng/mL rhBMP4. The sequences of the constructs are in Supplementary Data Table 16.
Electrophoretic Gel Mobility Shift Assay (EMSA):
G1ER and G1ER-S1FB murine hematopoietic progenitor cells80 were differentiated for 24 hours with beta-estradiol and treated with doxycycline to express FLAG-SMAD1. Two hours prior to collecting the cell extracts, cells treated with 25 ng/ml rhBMP4 to activate the BMP pathway. Cell extracts were made using the Pierce IP lysis buffer (Thermo Scientific, 87788) according to manufacturer’s protocol. EMSAs were performed using the Lightshift Chemiluminescent kit (Thermo Scientific, 20148) according to manufacturer’s instructions. Briefly, binding reactions were performed with 10μg of protein, 20 fmol of biotinylated DNA probe, 1X binding buffer, 5% glycerol, 500ng of poly (dl-dC), 50mM KCl and 1.5 mM MgCl2. Reactions were incubated for 30 min at room temperature. Cold competitor reactions contained 4 pmol non-biotinylated probe. Then the reactions were run on a 10% polyacyrlamide/TBE non-denaturing gel (Biorad Mini-PROTEAN Precast, 456-5034). The DNA probes used for this study can be found in Supplementary Data Table 16.
Identification of RBC trait-associated SNPs and related analyses
Lists of SNPs associated with blood traits were compiled from multiple studies, as referred in the results section. We selected 1000 Genomes European populations (CEU, TSI, FIN, GBR, and IBS) for our study and filtered for SNPs associated with MCV, HGB, RBC#, MCH, HTC, MCHC, and RDW as phenotypes. In total, 1,325 lead SNPs associated with any of the above RBC parameters were obtained. Using the lead GWAS SNP for each region, in order to increase the likelihood of including the functional SNPs from a reported hit, we also included highly associated SNPs with the lead SNP (with linkage disequilibrium LD r2 ≥ 0.6), which we included in the “Lead + LD” SNP list. We selected SNPs based on the LD threshold of r2 > 0.6 using 1000 Genomes European populations (CEU, TSI, FIN, GBR, and IBS). Only SNPs with an “rs” identifier in dbSNP version 142 were considered. SNPs can have multiple allele pairs that show differential association with traits. To account for this possibility, we broke out each allele pair for each SNP. We removed any SNP from the analysis that has different alleles reported in the publication and in the dbSNP database. Such alleles were represented as “NA” alleles for a given SNP. Only allele pairs that had two non-NA alleles were designated as “usable alleles” and were retained for the final analysis. Accordingly, 29,069 lead and LD SNPs with at least two usable alleles, across 924 loci associated with the seven RBC traits, were used to initiate the study. Unless otherwise reported, numbers of SNPs reported refer to the positions of SNPs, i.e. two allele pairs of the same SNP are reported once. We used the same approach and criteria for selecting the platelet trait-associated GWAS SNPs from Astle et al., 201619 to use as negative controls. RBCs and platelets share origins from megakaryocyte and erythroblast progenitor cells, suggesting platelet trait SNPs as the ideal negative control for our study. We used 786 quantitative trait loci regions associated with 575 lead and 22,158 (lead+LD) platelet trait SNPs (LD r2 ≥ 0.6) with at least two usable alleles. Positions of these SNPs relative to the hg19 revision of the human reference genome were taken from the UCSC genome browser track containing dbSNP version 142. Fine-mapped SNPs for blood traits were downloaded from Ulirsch et al., 201923 and were converted to BED format for downstream analyses using reported positions. Fine-mapped SNPs were filtered for those with a PP>0.01, which was the threshold used in the initial publication of these trait-associated SNPs, resulting in 54,255 SNP-trait associations and 39,822 SNPs with unique positions and identifiers, and that are associated with at least one trait. SNP-enhancer or SNP-TSC overlap was determined using bedtools intersect. SNP-motif hit overlap was determined using bedtools intersect. The lists of all the SNPs that fall within overall enhancers and within TSCs are mentioned in Supplementary Table 5.
To predict whether either allele of a given SNP was likely to be bound by a transcription factor of interest, we built sequences containing either allele in context. Each allele for each SNP passing the above filters were used to create short, generally 41nt-long DNA fragments that contain hg19 reference genome sequence upstream and downstream of the SNP position, i.e. 20nt of reference sequence upstream, one allele of the SNP, 20nt of reference sequence downstream. Alleles of variants called SNPs that were greater than 1 bp in length generated sequences longer than 41nt, but the vast majority of short sequences was 41nt. Each ~41nt sequence was scanned for presence of predicted transcription factor-binding sequences using FIMO 4.11.481 with a reference motif library that included multiple motif position-weight matrices. Based on our lists of sTFs and mTFs, we identified all non-redundant PWMs from the CIS-BP database build 2.0065 that had been inferred from protein-binding microarray (PBM) and SELEX experiments. These PWMs learned from in vitro experiments were selected to focus on direct TF binding (versus motifs inferred from e.g., ChIP-seq, which may include information about tethering TFs). We used this set of PWMs as our motif dictionary for FIMO scans of open, non-exonic regions for identifying motif hits, and this list is available as Supplementary Table 6. Motif hits that overlapped the SNP position in the 41nt sequence were retained and used for comparison between risk and reference alleles, i.e. the SNP was required to overlap the motif hit. Thus, we also required that, for a SNP to be associated with a motif hit, the motif hit directly overlap the center of the region, i.e. the SNP’s position. The construction of 41bp sequences centered on the SNP itself, allowed for the SNP to appear at the extreme ends of longer motifs, such as motifs from heterodimeric TF binding. Unique SNP IDs were the unit used for counting.
To test whether our H3K27ac ChIP-seq/ATAC-seq based approach enriches for “functional” SNPs, we used RegulomeDB62. A RegulomeDB score ≤ 4 was used to predict SNPs with the minimal functional evidences. This resulted in 5,695 RBC SNPs out of total 29,069 SNPs with two usable alleles.
Motif occurrence identification
Positions of predicted motif occurrences were determined across the hg19 revision of the human reference genome using FIMO81 with default parameters and a position weight matrix reference library built as described above. The numbers of base pairs contained within each category of motif occurrence were calculated after collapsing all occurrences of either STFs motifs or MTF motifs using bedtools merge82. SNPs overlapping motif occurrences were determined using bedtools intersect.
Determining significance of enrichment in SNPs
To determine the relative enrichment of SNPs in pairs of region types when accounting for the collective size of regions, we used multiple statistical analyses, including 2x2 chi-square tests and permutation tests.
2x2 Chi-square tests compared the numbers of SNPs falling in two categories and the number of base pairs in the collective region type after collapsing. Note that the 2x2 Chi-square tests assume observations are independent, which is not always the case in this biological system, especially when multiple SNPs in LD with each other are interrogated. Hence, we performed additional simulation analysis to determine significance of our observations.
SNP position permutation tests were performed using 10000 iterations of SNPs from the three lists described above (Lead, Lead+LD, fine-mapped) shuffled randomly within specified region types using bedtools shuffle.
To determine the enrichment of SNPs in signaling TF motif hits in enhancers using SNP position permutation, SNPs were randomly shuffled in all enhancers as defined above (bedtools shuffle -incl), and the resulting positions were used to construct 41bp sequences that were scanned by FIMO as described above for signaling TF motif occurrences in either allele (described in detail above). Shuffled SNPs that fell in enhancers were interrogated for whether the sequences they created are likely motif occurrences for signaling TFs or master TFs, and occurrence-overlapping SNPs were counted. The corresponding p value represents the number of random permutations that meet or exceed the actual observed count.
To determine the enrichment of SNPs in TSCs vs. non-TSC enhancers using SNP position permutation, SNPs were randomly shuffled within enhancers as defined above and interrogated for whether they overlap the subset of enhancers defined as TSCs. The corresponding p value represents the fraction of 10,000 random permutations that meet or exceed the actual observed count.
To determine the enrichment of SNPs in signaling TF motif hits within TSCs vs. signaling TF motif hits within non-TSC enhancers using SNP position permutation, SNPs were randomly shuffled within enhancers as defined above, interrogated for whether they fall within TSCs, and whether they are predicted to fall within motif occurrences at their original (read: not shuffled) position. The corresponding p value represents the fraction of 10000 random permutations that meet or exceed the actual observed count.
Enhancer labeling permutation tests were performed by selecting a random subset of enhancers to represent TSCs to test whether SNPs are unusually concentrated in actual TSCs above background. Note that the number of observed successes differs in this approach from that of above, as the SNP position permutation analysis used both alleles of each SNP to determine if the sequence created during shuffling was recognizable by specified TFs. 7421 of 81636 enhancers across the system were randomly selected each of 10,000 iterations using the Unix utility shuf. The numbers of trait-associated SNPs from each of the three lists that are contained in each random TSC subset were tallied. The corresponding p value represents the number of random permutations that meet or exceed the actual observed count.
Motif hit permutation tests were performed by randomly shuffling the positions of unambiguous STF motif hits that fall within enhancers across all enhancer loci using bedtools shuffle -incl. Note that the number of observed successes differs in this approach from that of above, as the SNP position permutation analysis used both alleles of each SNP to determine if the sequence created during shuffling was recognizable by specified TFs. The corresponding p value represents the number of random permutations that meet or exceed the actual observed count of SNPs in their real position overlapping permuted STF motif hits.
Expression analysis from Framingham Heart Study (FHS)
Minor allele frequencies in different ethnic groups were looked up from Hapmap CEU, YRI, or CHB population data through http://snp-nexus.org/83–85. Expression QTLs (eQTLs) were queried using R or Perl scripting based on our selected SNP lists from data-set downloaded from https://grasp.nhlbi.nih.gov/Updates.aspx86 (GRASP 2.0.0.0 Expression QTLs), and data-set downloaded from Framingham Heart Study population (FHS whole blood eQTL results) ftp://ftp.ncbi.nlm.nih.gov/eqtl/original submissions/FHS eQTL/70,87. For FHS whole blood eQTL results, we only focus on significant eQTLs (peer validated results up to a logFDR value of −4.0, at the levels of genes and exons respectively), and report the cis-eQTL with best p-value in each region, or all of the significant cis and trans-eQTLs for our selected SNPs as a reference.
Statistical analysis
The detailed methodologies used for statistical tests and the resulting significance values obtained comparing the control and test groups are described in the relevant methods sections, figures, figure legends and in the Supplementary Data Table 3. Biological replicates and observed data-point variations are mentioned wherever applicable. All statistical analyses were carried out using the statistical computing/graphics software R and GraphPad Prism 8.
Data Availability Statement
The massively parallel sequencing data associated with this manuscript have been uploaded to GEO under the accession numbers GSE74483 and GSE104574 and are currently open to public. The web links for the publicly available databases used in this study are: UniPROBE: http://thebrain.bwh.harvard.edu/uniprobe/, CIS-BP: http://cisbp.ccbr.utoronto.ca/, FHS: https://www.ncbi.nlm.nih.gov/proiects/gap/cgi-bin/study.cgi?study_id=phs000007.v30.p11, RegulomeDB: https://regulomedb.org/regulome-search/, HEMMER: http://hmmer.org/, EMBOSS Needle: https://www.ebi.ac.uk/Tools/psa/emboss_needle/. dbSNP: https://www.ncbi.nlm.nih.gov/snp/?cmd=search. Links to all the PBM datasets used are available in Supplementary Table 7.
Code Availability
Custom codes used in this study are available at https://bitbucket.org/abrahamb/workspace/projects/TSC. The code and data files for the PBM analyses are available at https://github.com/BulykLab/RBCSNPs_2020.
Extended Data
Extended Data Fig. 1: Human CD34+ cells commit to an erythroid fate around day 3 (D3) during differentiation.
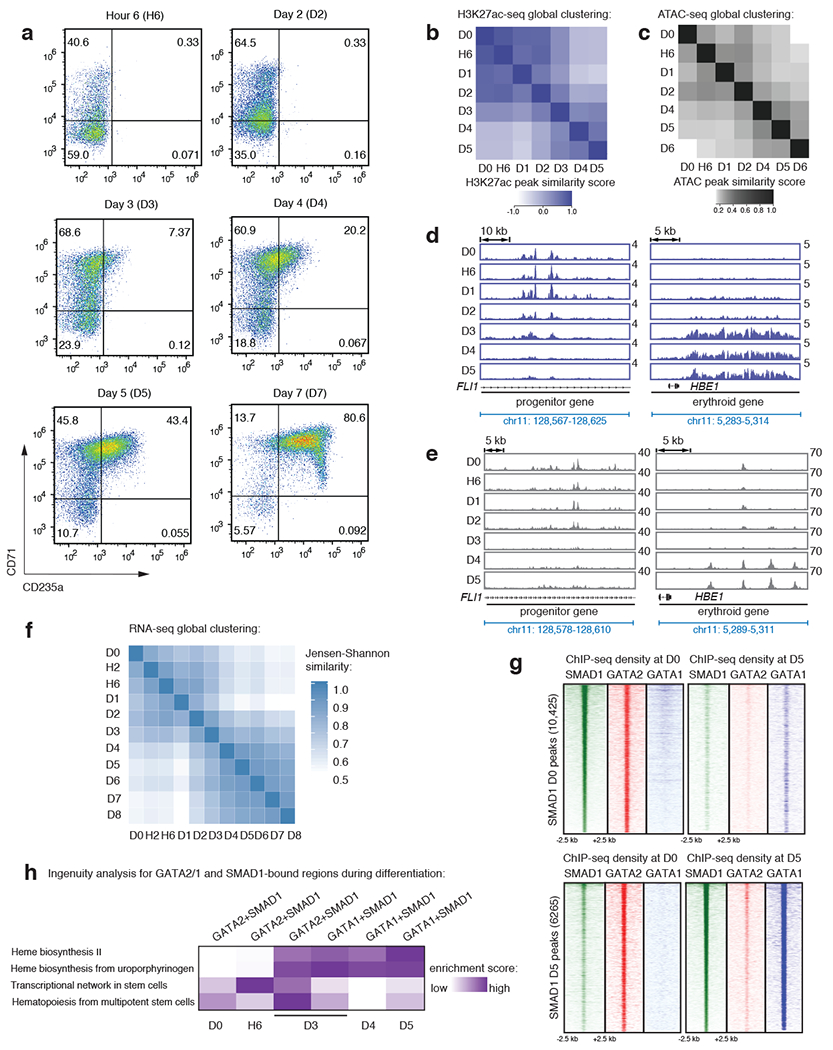
a, Representative FACS plots for the erythroid markers CD71 and CD235a on CD34+ cells after induction of erythroid differentiation at hour 6 (H6), day 2 (D2), day 3 (D3), day 4 (D4), day 5 (D5) and day 7 (D7). b-c, Heatmaps depicting correlation of peaks from H3K27ac ChIP-seq (violet) (b) or ATAC-seq (grey) (c) obtained from distinct differentiation time-points, as indicated on axes. The progenitor and erythroid timepoint clusters separate cell identities into groups before and after D3, supporting our separation of time points into progenitor and erythroid before and after D3. d-e, Gene tracks showing H3K27ac ChIP-seq signal (violet-upper panel) (d) or ATAC-seq signal (grey-lower panel) (e) at FLI1 (a progenitor gene) and at the β-globin locus control region (LCR; a genomic region that should be activated only after erythroid commitment) at different differentiation stages, as indicated. D0 = progenitor CD34+ cells before induction of differentiation; H6 = 6 hours after differentiation; and D1 through D5 = 1, 2, 3, 4 and 5 days after differentiation. Y-axis represents reads-permillion. f, Heatmap depicting correlation of gene expression profiles of all the protein-coding RNAs from D0 through D8 of erythroid differentiation. Progenitor and erythroid clusters separate around D3. D0 = progenitor CD34+ cells before induction of differentiation; H2 and H6 = 2 and 6 hours after differentiation; and D1 through D8 = 1, 2, 3, 4, 5, 6, 7 and 8 days after differentiation. g, Signal heatmaps comparing ChIP-seq read densities of SMAD1 (green), GATA2 (red), and GATA1 (blue) at SMAD1 peaks identified at D0 (upper panel) and D5 (lower panel). Each plot represents signal intensities centered around +/− 2.5 kb of the peaks showed in Y-axis, as indicated. Peak numbers are indicated in the parenthesis in the Y-axis. h, Ingenuity pathway analysis (IPA) for GATA2+SMAD1 bound genes at D0, H6, D3 and D4 and GATA1+SMAD1 bound genes at D3, D4, D5 identifying differentiation stage-specific biological properties, as indicated.
Extended Data Fig. 2: Comparative TF motif enrichment and H3K27ac signal density analysis surrounding GATA+SMAD1 versus GATA-only regions.
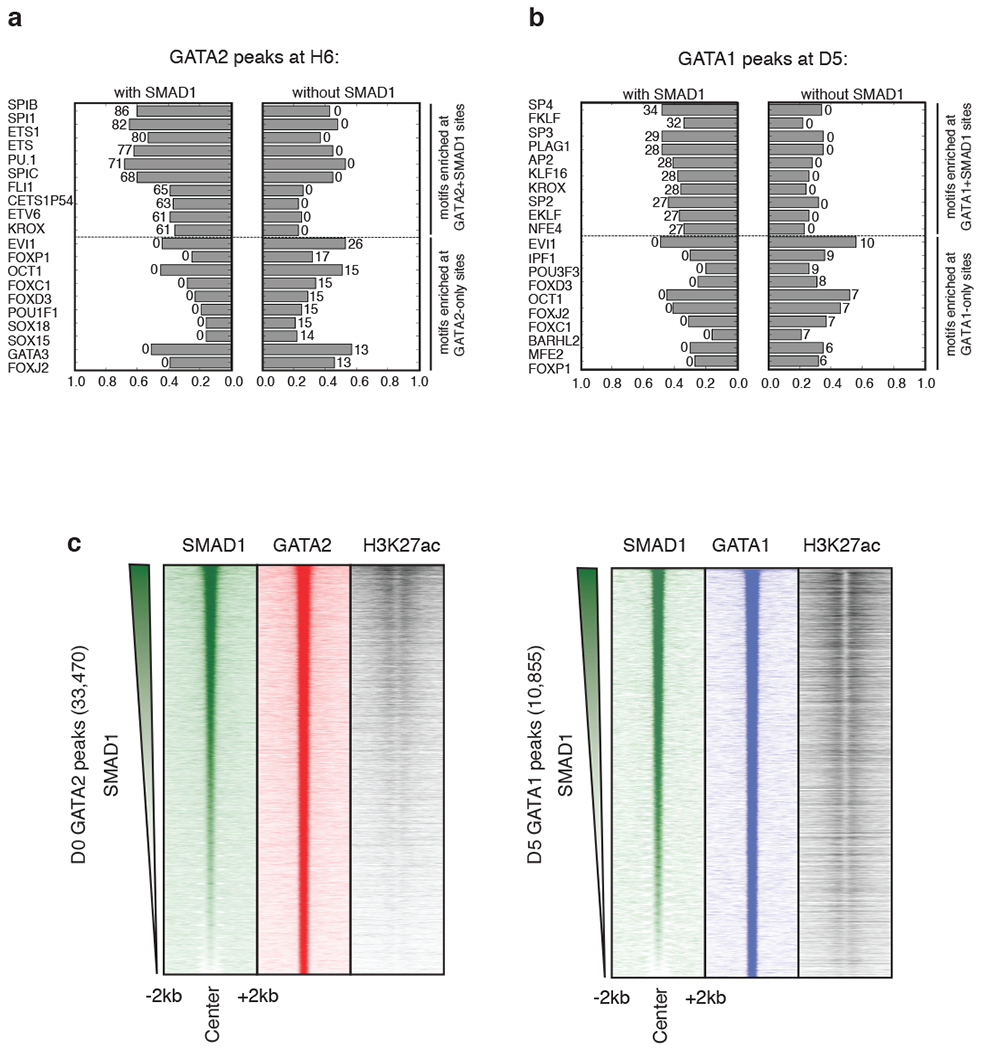
Bar charts depicting the enrichment of transcription factor motif hits at regions co-bound by GATA+SMAD1 (left) versus by GATA only (right) at H6 (a) and D5 (b). Length of the bar indicates the fraction of peaks containing a given motif hit, and the number associated with the bar represents the corresponding -log10(p-value) obtained from the hyper-geometric test to assess the significance of motif enrichment. For both (a) and (b), top and bottom of the ranked lists are shown. c, (left panel) Region heatmaps depicting signal of ChIP-seq reads for D0 SMAD1, GATA2 and H3K27ac at 33,470 GATA2 bound peaks at D0. The peaks are ranked by the SMAD1 intensity across the row. Each plot represents signal intensities around +/− 2 kb of the peak center. (right panel) Region heatmaps depicting signal of ChIP-seq reads for D5 SMAD1, GATA1 and H3K27ac at 10,855 GATA1 bound peaks at D5. The peaks are ranked by the SMAD1 intensity across the row. Each plot represents signal intensities around +/− 2 kb of the peak center.
Extended Data Fig. 3: TSCs are a small subset of overall enhancers as defined by SMAD1 and GATA co-occupancy.
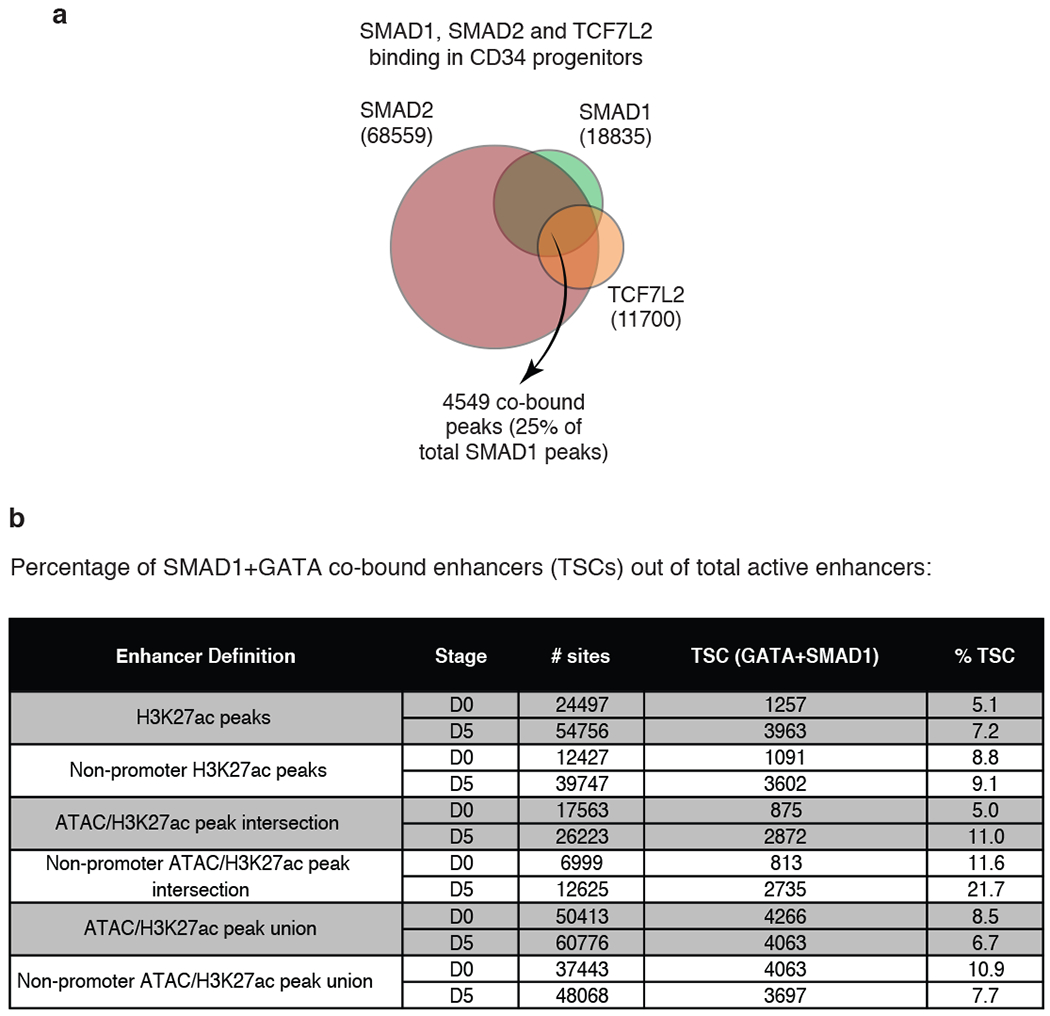
a, Venn diagram representing genomic regions co-occupied by different STFs - SMAD1, SMAD2 and TCF7L2 in progenitor CD34+ cells upon stimulation with BMP4, TGFβ and WNT signaling, respectively. The genomic regions bound by all three factors are 4549. The other numbers refer to the total number of peaks bound by each factor, as indicated Regions are considered occupied if they pass a significant coverage cutoff. b, Table representing different strategies to define enhancers using H3K27ac ChIP-seq and/or ATAC-seq. The proportion of enhancers that can be classified as TSCs (GATA+SMAD1 co-bound) at progenitor (D0) or erythroid (D5) stages are as indicated.
Extended Data Fig. 4: Targeting a TSC near the LHFPL2 gene by CRISPR-CAS9.
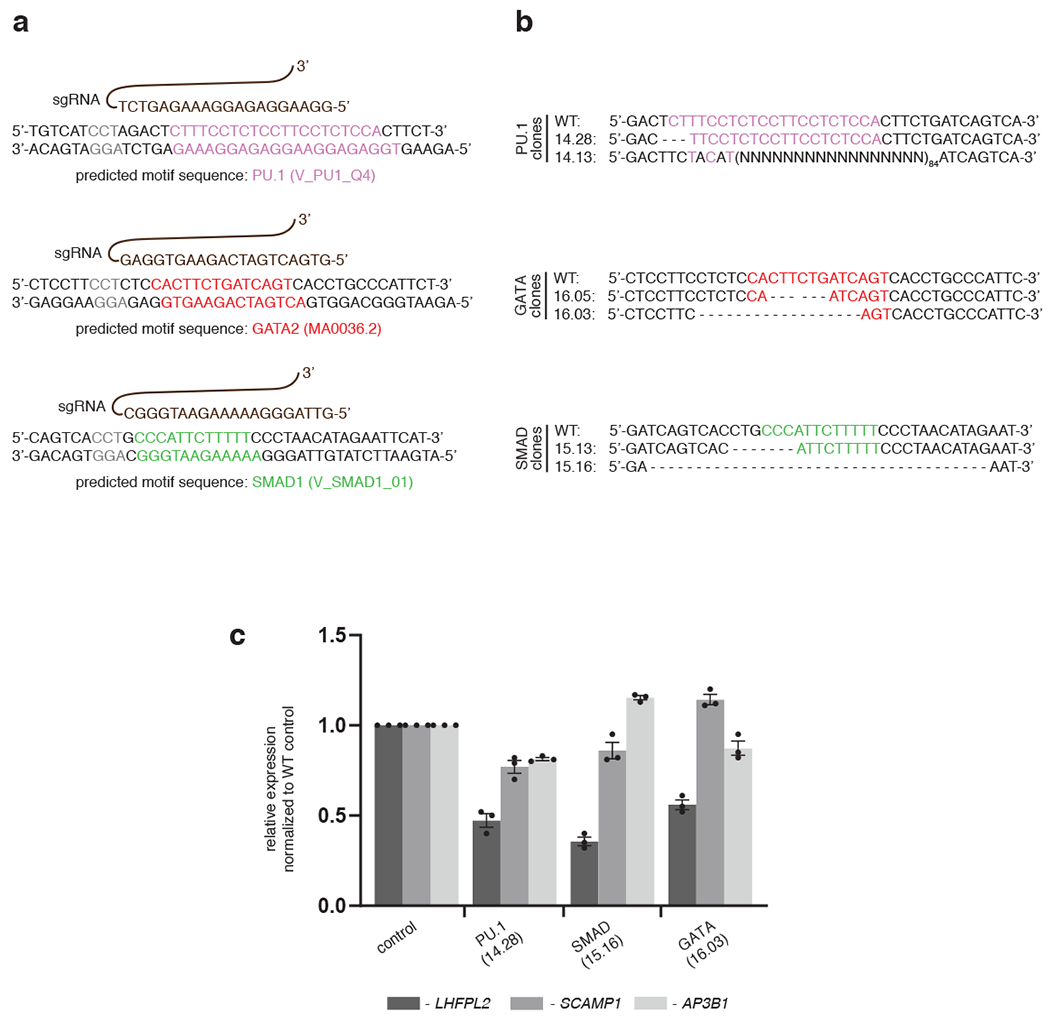
a, sgRNAs (shown in brown) targeting specific sequences near PU.1 (pink), GATA (red) and SMAD1 (green) motif-hits within the TSCs. b, Genomic sequences of the specific CRISPR-edited clones are compared against wild-type genomic sequence. Clones 14.13, 16.03 and 15.16 appear to target multiple motifs (i.e. PU.1 and partial GATA; partial PU.1 and partial GATA; SMAD1 and partial GATA). c, qPCR results depicting relative expression of LHFPL2 (black bar) and two other flanking genes (SCAMP1, grey bar and AP3B1, white bar) are shown in different CRISPR clones compared to the WT K562 cells, as indicated. Data represent mean ± SEM from three replicate observations.
Extended Data Fig. 5: Approach for interrogating enhancer-associated RBC-trait SNPs showing that SNPs targeting STF motif-hits are localized within TSCs.
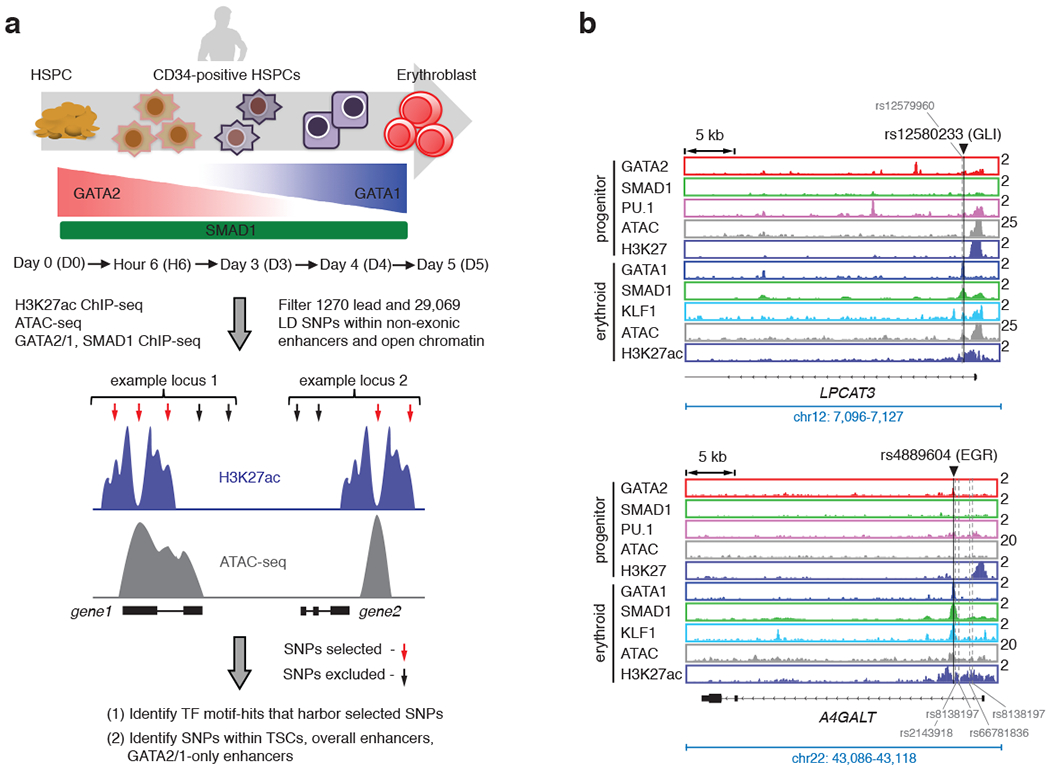
a, Schematic diagram of the strategy used to identify SNPs that may alter activity of transcriptional enhancers during human erythroid differentiation. Human CD34+ cells from mobilized peripheral blood were differentiated towards erythrocytes. Genomic experiments were performed at D0, H6, D3, D4 and D5. 1270 lead RBC-trait SNPs and additional SNPs that are in linkage disequilibrium with lead SNPs, with LD score r2 ≥ 0.6 (total number of SNPs = 29,069), were first overlapped with genomic regions that are defined as non-exonic enhancer (represented as violet tracks) and open chromatin peaks (represented as grey tracks) in our study. SNPs that fall within such regions (indicated with red arrows) were used to carry out motif hit analysis, and were overlapped either with TSCs, or overall enhancers or GATA-only enhancers. b, Gene tracks showing RBC-trait SNPs that are located within stage-specific TSCs are shown. The binding of GATA2 (red), GATA1 (blue), SMAD1 (green), PU.1 (pink) and KLF1 (light blue) and the peaks of H3K27ac (violet) and ATAC-seq (grey) are shown in progenitor and differentiated stages. The black lines on the gene tracks indicate the position of representative SNPs (rs12580233 - associated with RDW, rs4889604 – associated with MCV and RBC). The potential STF motifs that these SNPs could perturb (e.g. GLI, EGR) are as indicated. Y-axis indicates reads per million. For each representative SNP that resides in a TSC, the other associated SNPs in significant LD that fall within H3K27ac/ATAC-positive enhancers are also indicated with grey dashed lines.
Extended Data Fig. 6: Analysis of protein binding microarray (PBM) 8-mer data identifies several RBC trait-associated SNPs that perturb STF-DNA binding.
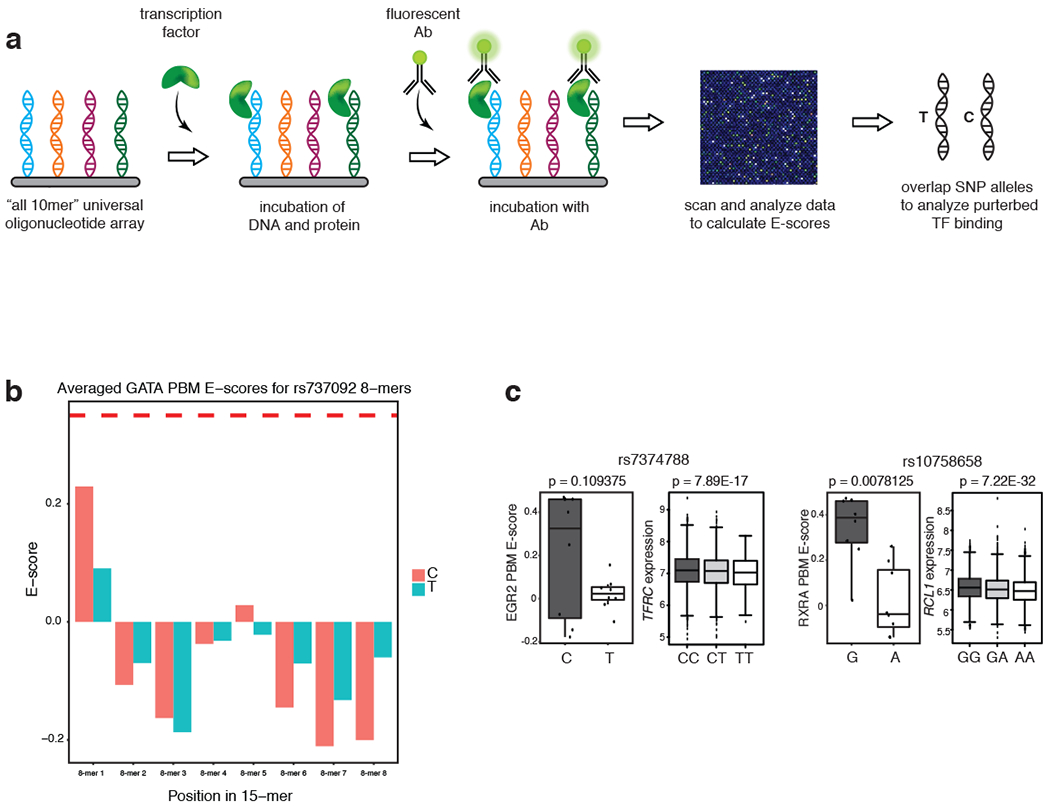
a, Schematic representation of the strategy to identify SNPs that alter STF binding utilizing protein binding microarrays. b, The bar charts for the GATA average PBM dataset for rs737092. The p-value is computed using the Wilcoxon signed-rank test. c, Additional examples of SNPs showing perturbed STF binding from PBM analysis (left) and corresponding distribution of expression values of the most significantly altered nearby gene in homozygous and heterozygous individuals obtained from FHS eQTL analysis (right). rs7374788 (MCH, MCV, MCHC, RDW) shows altered binding of EGR1; rs10758658 (MCV, MCH, RBC) modulates binding of RXRA, respectively. Y-axis values for PBM boxplots represent universal PBM enrichment (E) scores. The p-values are computed using Wilcoxon signed-rank tests. Individual genotypes and the cis-eQTL gene/exon obtained from the FHS dataset are as indicated.
Extended Data Fig. 7: RBC-trait SNPs perturb STF-DNA binding.
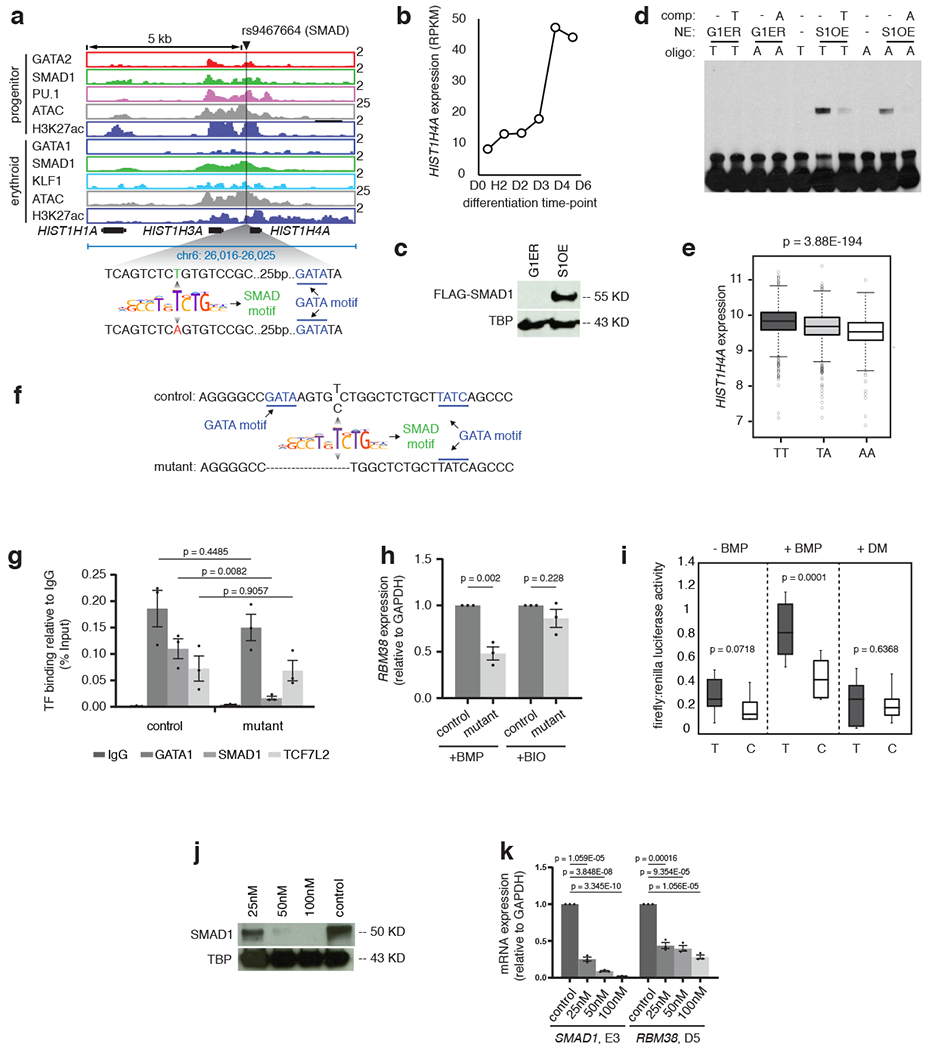
a, Binding of GATA2 (red), GATA1 (blue), SMAD1 (green), PU.1 (pink) and KLF1 (light blue) and the peaks of H3K27ac (violet) and ATAC-seq (grey) near HIST1H4A gene are shown in progenitor and differentiated stages. The black line indicates the position of SNP rs9467664. The zoomed in DNA sequence highlights the position of T and A allele relative to the SMAD motif (green) and the nearby GATA motif (blue). Y-axis indicates reads per million. b, RPKM values are shown for the gene HIST1H4A at different stages of CD34+ erythroid differentiation, as indicated. c, Western blot showing the expression of FLAG-SMAD1 protein after treating the SMAD1 overexpressing G1ER cells (S1-FB) with doxycycline (+DOX) for 24 hours. G1ERrepresents the protein extracts from the control parental cell-line. TATA binding protein(TBP) was used as loading control. d, Representative gel-shift assay with the A or T allele of rs9467664. Competitor oligonucleotides have been used in each case to show binding specificity, as indicated and G1ER extracts were used as negative control for the binding assays. S1OE = SMAD1 overexpressing clone. e, HIST1H4A eQTL analysis for the SNP rs9467664 using genotype and gene expression data from the Framingham Heart Study (FHS). Boxplots represent the distribution of HIST1H4Agene expression in individuals with either the TT, TA or AA genotype along with the significance value, as indicated. f, Schematic representation of a K562 clone with altered sequence around rs737092. The deletion is evident in the lower sequence (mutant). g, ChIP-qPCR quantification comparing the binding of GATA1 (blue),SMAD1 (green) and TCF7L2 (orange) between the control WT K562 cells and the K562 cells with RBM38 enhancer deletion. Binding of each factor under each condition is shown with respect to IgG, represented as percentage input (grey). The t-test significance values comparing samples are as indicated. h, qPCR analysis comparing the expression of RBM38 relative to GAPDH between control and mutant K562 clones under BMP and BIO treatment. i, Ratio of firefly and renilla luciferase values without stimulation or with BMP stimulation or with dorsomorphin (DM) stimulation of cells stably transfected with enhancer constructs containing either the T allele or C allele of rs737092. The t-test significance values under each condition are indicated. j, Western blot comparing expression of SMAD1 protein between control shRNA treated CD34+cells and cells treated with 25, 50 and 50 nM SMAD1 shRNA 5 days after differentiation. TATA binding protein (TBP) was used as loading control. k, qPCR analysis comparing the expression of SMAD1 at day 3 of CD34+ expansion (SMAD1,E3) and RBM38 after 5 days of differentiation (RBM38, D5) between control shRNA,25, 50 and 100 nM SMAD1 shRNA treated CD34+ cells. The t-test significance values under each condition are indicated.
Extended Data Fig. 8: A model explaining how human genetic variation within TSCs induces RBC trait phenotypes.

A combination of STFs with MTF drives optimal gene expression-regulation by the TSC. The normal signal-induced expression of a red blood cell gene is perturbed due to a SNP that either eliminates an existing STF binding event or creates a new STF binding site in a critical signaling center. This can lead to a lack of response to an episodic signaling pathway, initiated by an exogenous stressor, and eventually lead to phenotypic variability.
Supplementary Material
Acknowledgement
We are grateful to Prof. Stuart Orkin, Amy Wagers and Cristina Santoriello for the critical reading and editing of our manuscript. We thank HHMI high-throughput sequencing facility at Children’s Hospital Boston for generating the genome-wide raw sequencing data. We acknowledge Drs. Vijay G. Sankaran and Satish Nandakumar for providing us the luciferase plasmids containing non-coding alleles of RBM38 gene and the enhancer mutant for RBM38 gene in K562 cells. This work was supported by the following grants from L.I.Z. – R01 HL04880, P015PO1HL32262-32, 5P30 DK49216, 5R01 DK53298, 5U01 HL10001-05, R24 DK092760, 1R24OD017870-01 and funding by the Max Planck Society, The Fritz Thyssen Stiftung (Az 10.17.1.026MN), a Marie Curie Career Integration Grant (631432 Bloody Signals) and the Deutsche Forschungsgemeinschaft DFG under Germany’s Excellence Strategy (CIBSS-EXC-2189-Project-ID-390939984) and the Deutsche Forschungsgemeinschaft, Research Training Group 322977937/GRK2344 “MeInBio – BioInMe” to E.T, the Hope Funds for Cancer Research Grillo-Marxuach Family Fellowship and the American Lebanese Syrian Associated Charities (B.J.A.). R.A.Y. is supported by NIH grants GM123511, CA213333, and CA155258. K.H.K. is supported by an A*STAR National Science Scholarship. M.L.B. is supported by the following NIH grant, R21 HG010200. S.K.G. is supported by R01HL139672, R01HL122684, and R01HL086694. D.E.B. was supported by NHLBI (P01HL32262 and DP2HL137300).
The Framingham Heart Study is funded by National Institutes of Health contract N01-HC-25195. The eQTL work for this investigation was funded by the Division of Intramural Research, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD (D. Levy, Principal Investigator). The analytical component of this project was funded by the Division of Intramural Research, National Heart, Lung, and Blood Institute, and the Center for Information Technology, National Institutes of Health, Bethesda, MD. The views expressed in this manuscript are those of the authors and do not necessarily represent the views of the National Heart, Lung, and Blood Institute; the National Institutes of Health; or the U.S. Department of Health and Human Services.
Footnotes
Competing Interests Statement
L.I.Z. is founder and stockholder of Fate, Inc. and Scholar Rock, Camp4 therapeutics and a scientific advisor for Stemgent. R.A.Y. is a founder and shareholder of Syros Pharmaceuticals, Camp4 Therapeutics, Omega Therapeutics, and Dewpoint Therapeutics. B.J.A. is a shareholder in Syros Pharmaceuticals. M.L.B. is a co-inventor on patents on PBM technology. The other authors declare no competing interests.
References (for main text)
- 1.Evans DM, Frazer IH & Martin NG Genetic and environmental causes of variation in basal levels of blood cells. Twin Res 2, 250–257 (1999). [DOI] [PubMed] [Google Scholar]
- 2.Guindo A, Fairhurst RM, Doumbo OK, Wellems TE & Diallo DA X-linked G6PD deficiency protects hemizygous males but not heterozygous females against severe malaria. PLoS Med 4, e66, doi: 10.1371/journal.pmed.0040066 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lin JP et al. Evidence for linkage of red blood cell size and count: genome-wide scans in the Framingham Heart Study. Am J Hematol 82, 605–610, doi: 10.1002/ajh.20868 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Lo KS et al. Genetic association analysis highlights new loci that modulate hematological trait variation in Caucasians and African Americans. Hum Genet 129, 307–317, doi: 10.1007/s00439-010-0925-1 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tishkoff SA et al. Haplotype diversity and linkage disequilibrium at human G6PD: recent origin of alleles that confer malarial resistance. Science 293, 455–462, doi: 10.1126/science.1061573 (2001). [DOI] [PubMed] [Google Scholar]
- 6.Whitfield JB & Martin NG Genetic and environmental influences on the size and number of cells in the blood. Genet Epidemiol 2, 133–144, doi: 10.1002/gepi.1370020204 (1985). [DOI] [PubMed] [Google Scholar]
- 7.Koury MJ Abnormal erythropoiesis and the pathophysiology of chronic anemia. Blood Rev 28, 49–66, doi: 10.1016/j.blre.2014.01.002 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Edwards SL, Beesley J, French JD & Dunning AM Beyond GWASs: illuminating the dark road from association to function. Am J Hum Genet 93, 779–797, doi: 10.1016/j.ajhg.2013.10.012 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guo MH et al. Comprehensive population-based genome sequencing provides insight into hematopoietic regulatory mechanisms. Proc Natl Acad Sci U S A 114, E327–E336, doi: 10.1073/pnas.1619052114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gusev A et al. Partitioning heritability of regulatory and cell-type-specific variants across 11 common diseases. Am J Hum Genet 95, 535–552, doi: 10.1016/j.ajhg.2014.10.004 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Melnikov A et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat Biotechnol 30, 271–277, doi: 10.1038/nbt.2137 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nandakumar SK, Ulirsch JC & Sankaran VG Advances in understanding erythropoiesis: evolving perspectives. Br J Haematol 173, 206–218, doi: 10.1111/bjh.13938 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patwardhan RP et al. Massively parallel functional dissection of mammalian enhancers in vivo. Nat Biotechnol 30, 265–270, doi: 10.1038/nbt.2136 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Polfus LM et al. Whole-Exome Sequencing Identifies Loci Associated with Blood Cell Traits and Reveals a Role for Alternative GFI1B Splice Variants in Human Hematopoiesis. Am J Hum Genet 99, 785, doi: 10.1016/j.ajhg.2016.08.002 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ulirsch JC et al. Systematic Functional Dissection of Common Genetic Variation Affecting Red Blood Cell Traits. Cell 165, 1530–1545, doi: 10.1016/j.cell.2016.04.048 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van der Harst P et al. Seventy-five genetic loci influencing the human red blood cell. Nature 492, 369–375, doi: 10.1038/nature11677 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ganesh SK et al. Multiple loci influence erythrocyte phenotypes in the CHARGE Consortium. Nat Genet 41, 1191–1198, doi: 10.1038/ng.466 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Rooij FJ et al. Genome-wide Trans-ethnic Meta-analysis Identifies Seven Genetic Loci Influencing Erythrocyte Traits and a Role for RBPMS in Erythropoiesis. Am J Hum Genet 100, 51–63, doi: 10.1016/j.ajhg.2016.11.016 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Astle WJ et al. The Allelic Landscape of Human Blood Cell Trait Variation and Links to Common Complex Disease. Cell 167, 1415–1429.e1419, doi: 10.1016/j.cell.2016.10.042 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chami N et al. Exome Genotyping Identifies Pleiotropic Variants Associated with Red Blood Cell Traits. Am J Hum Genet 99, 8–21, doi: 10.1016/j.ajhg.2016.05.007 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pankratz N et al. Genome-wide association study for susceptibility genes contributing to familial Parkinson disease. Hum Genet 124, 593–605, doi: 10.1007/s00439-008-0582-9 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Levo M et al. Unraveling determinants of transcription factor binding outside the core binding site. Genome Res 25, 1018–1029, doi: 10.1101/gr.185033.114 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ulirsch JC et al. Interrogation of human hematopoiesis at single-cell and single-variant resolution. Nat Genet 51, 683–693, doi: 10.1038/s41588-019-0362-6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dent P et al. Stress and radiation-induced activation of multiple intracellular signaling pathways. Radiat Res 159, 283–300 (2003). [DOI] [PubMed] [Google Scholar]
- 25.Gaki GS & Papavassiliou AG Oxidative stress-induced signaling pathways implicated in the pathogenesis of Parkinson’s disease. Neuromolecular Med 16, 217–230, doi: 10.1007/s12017-014-8294-x (2014). [DOI] [PubMed] [Google Scholar]
- 26.Uchida K et al. Activation of stress signaling pathways by the end product of lipid peroxidation. 4-hydroxy-2-nonenal is a potential inducer of intracellular peroxide production. J Biol Chem 274, 2234–2242 (1999). [DOI] [PubMed] [Google Scholar]
- 27.Mullen AC et al. Master transcription factors determine cell-type-specific responses to TGF-beta signaling. Cell 147, 565–576, doi: 10.1016/j.cell.2011.08.050 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trompouki E et al. Lineage regulators direct BMP and Wnt pathways to cell specific programs during differentiation and regeneration. Cell 147, 577–589, doi: 10.1016/j.cell.2011.09.044 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sankaran VG et al. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science 322, 1839–1842, doi: 10.1126/science.1165409 (2008). [DOI] [PubMed] [Google Scholar]
- 30.Bannister AJ & Kouzarides T Regulation of chromatin by histone modifications. Cell Res 21, 381–395, doi: 10.1038/cr.2011.22 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Buenrostro JD, Giresi PG, Zaba LC, Chang HY & Greenleaf WJ Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10, 1213–1218, doi: 10.1038/nmeth.2688 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lenox LE, Perry JM & Paulson RF BMP4 and Madh5 regulate the erythroid response to acute anemia. Blood 105, 2741–2748, doi: 10.1182/blood-2004-02-0703 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Lenox LE, Shi L, Hegde S & Paulson RF Extramedullary erythropoiesis in the adult liver requires BMP-4/Smad5-dependent signaling. Exp Hematol 37, 549–558, doi: 10.1016/j.exphem.2009.01.004 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McReynolds LJ, Tucker J, Mullins MC & Evans T Regulation of hematopoiesis by the BMP signaling pathway in adult zebrafish. Exp Hematol 36, 1604–1615, doi: 10.1016/j.exphem.2008.08.005 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Porayette P & Paulson RF BMP4/Smad5 dependent stress erythropoiesis is required for the expansion of erythroid progenitors during fetal development. Dev Biol 317, 24–35, doi: 10.1016/j.ydbio.2008.01.047 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hnisz D et al. Convergence of developmental and oncogenic signaling pathways at transcriptional super-enhancers. Mol Cell 58, 362–370, doi: 10.1016/j.molcel.2015.02.014 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Whyte WA et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319, doi: 10.1016/j.cell.2013.03.035 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fisher RC & Scott EW Role of PU.1 in hematopoiesis. Stem Cells 16, 25–37, doi: 10.1002/stem.160025 (1998). [DOI] [PubMed] [Google Scholar]
- 39.Li Y, Luo H, Liu T, Zacksenhaus E & Ben-David Y The ets transcription factor Fli-1 in development, cancer and disease. Oncogene 34, 2022–2031, doi: 10.1038/onc.2014.162 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shivdasani RA & Orkin SH Erythropoiesis and globin gene expression in mice lacking the transcription factor NF-E2. Proc Natl Acad Sci U S A 92, 8690–8694, doi: 10.1073/pnas.92.19.8690 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Siatecka M & Bieker JJ The multifunctional role of EKLF/KLF1 during erythropoiesis. Blood 118, 2044–2054, doi: 10.1182/blood-2011-03-331371 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakao A et al. TGF-beta receptor-mediated signalling through Smad2, Smad3 and Smad4. EMBO J 16, 5353–5362, doi: 10.1093/emboj/16.17.5353 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McLean CY et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 28, 495–501, doi: 10.1038/nbt.1630 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kurita R et al. Establishment of immortalized human erythroid progenitor cell lines able to produce enucleated red blood cells. PLoS One 8, e59890, doi: 10.1371/journal.pone.0059890 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang F & Lupski JR Non-coding genetic variants in human disease. Human molecular genetics 24, R102–110, doi: 10.1093/hmg/ddv259 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Visscher PM et al. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am J Hum Genet 101, 5–22, doi: 10.1016/j.ajhg.2017.06.005 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cohen AJ et al. Hotspots of aberrant enhancer activity punctuate the colorectal cancer epigenome. Nat Commun 8, 14400, doi: 10.1038/ncomms14400 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Corradin O et al. Combinatorial effects of multiple enhancer variants in linkage disequilibrium dictate levels of gene expression to confer susceptibility to common traits. Genome Res 24, 1–13, doi: 10.1101/gr.164079.113 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morrow JJ et al. Positively selected enhancer elements endow osteosarcoma cells with metastatic competence. Nat Med 24, 176–185, doi: 10.1038/nm.4475 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Scacheri CA & Scacheri PC Mutations in the noncoding genome. Curr Opin Pediatr 27, 659–664, doi: 10.1097/MOP.0000000000000283 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meta-analysis of rare and common exome chip variants identifies S1PR4 and other loci influencing blood cell traits. Nat Genet 48, 867–876, doi: 10.1038/ng.3607 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen Z et al. Genome-wide association analysis of red blood cell traits in African Americans: the COGENT Network. Human molecular genetics 22, 2529–2538, doi: 10.1093/hmg/ddt087 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li C et al. Genome-Wide Association Study Meta-Analysis of Long-Term Average Blood Pressure in East Asians. Circulation. Cardiovascular genetics 10, e001527, doi: 10.1161/circgenetics.116.001527 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Paul DS et al. Maps of open chromatin highlight cell type-restricted patterns of regulatory sequence variation at hematological trait loci. Genome Res 23, 1130–1141, doi: 10.1101/gr.155127.113 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Paul DS et al. Maps of open chromatin guide the functional follow-up of genome-wide association signals: application to hematological traits. PLoS genetics 7, e1002139, doi: 10.1371/journal.pgen.1002139 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Amos CI et al. The OncoArray Consortium: A Network for Understanding the Genetic Architecture of Common Cancers. Cancer Epidemiol Biomarkers Prev 26, 126–135, doi: 10.1158/1055-9965.EPI-16-0106 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fachal L et al. Fine-mapping of 150 breast cancer risk regions identifies 191 likely target genes. Nat Genet 52, 56–73, doi: 10.1038/s41588-019-0537-1 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fritsche LG et al. Association of Polygenic Risk Scores for Multiple Cancers in a Phenome-wide Study: Results from The Michigan Genomics Initiative. Am J Hum Genet 102, 1048–1061, doi: 10.1016/j.ajhg.2018.04.001 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jansen IE et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet 51, 404–413, doi: 10.1038/s41588-018-0311-9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lin JR et al. Integrated Post-GWAS Analysis Sheds New Light on the Disease Mechanisms of Schizophrenia. Genetics 204, 1587–1600, doi: 10.1534/genetics.116.187195 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vicente CT et al. Long-Range Modulation of PAG1 Expression by 8q21 Allergy Risk Variants. Am J Hum Genet 97, 329–336, doi: 10.1016/j.ajhg.2015.06.010 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Boyle AP et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res 22, 1790–1797, doi: 10.1101/gr.137323.112 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu N et al. Direct Promoter Repression by BCL11A Controls the Fetal to Adult Hemoglobin Switch. Cell 173, 430–442 e417, doi: 10.1016/j.cell.2018.03.016 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hume MA, Barrera LA, Gisselbrecht SS & Bulyk ML UniPROBE, update 2015: new tools and content for the online database of protein-binding microarray data on protein-DNA interactions. Nucleic Acids Res 43, D117–122, doi: 10.1093/nar/gku1045 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Weirauch MT et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell 158, 1431–1443, doi: 10.1016/j.cell.2014.08.009 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Badis G et al. Diversity and complexity in DNA recognition by transcription factors. Science 324, 1720–1723, doi: 10.1126/science.1162327 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Barrera LA et al. Survey of variation in human transcription factors reveals prevalent DNA binding changes. Science 351, 1450–1454, doi: 10.1126/science.aad2257 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mariani L, Weinand K, Vedenko A, Barrera LA & Bulyk ML Identification of Human Lineage-Specific Transcriptional Coregulators Enabled by a Glossary of Binding Modules and Tunable Genomic Backgrounds. Cell Syst 5, 654, doi: 10.1016/j.cels.2017.12.011 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Peterson KA et al. Neural-specific Sox2 input and differential Gli-binding affinity provide context and positional information in Shh-directed neural patterning. Genes Dev 26, 2802–2816, doi: 10.1101/gad.207142.112 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Joehanes R et al. Integrated genome-wide analysis of expression quantitative trait loci aids interpretation of genomic association studies. Genome Biol 18, 16, doi: 10.1186/s13059-016-1142-6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tran FH & Zheng JJ Modulating the wnt signaling pathway with small molecules. Protein Sci 26, 650–661, doi: 10.1002/pro.3122 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Caron B, Luo Y & Rausell A NCBoost classifies pathogenic non-coding variants in Mendelian diseases through supervised learning on purifying selection signals in humans. Genome Biol 20, 32, doi: 10.1186/s13059-019-1634-2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Method-only References
- 73.Sankaran VG, Orkin SH & Walkley CR Rb intrinsically promotes erythropoiesis by coupling cell cycle exit with mitochondrial biogenesis. Genes Dev 22, 463–475, doi: 10.1101/gad.1627208 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Livak KJ & Schmittgen TD Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408, doi: 10.1006/meth.2001.1262 (2001). [DOI] [PubMed] [Google Scholar]
- 75.Ran FA et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308, doi: 10.1038/nprot.2013.143 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Montague TG, Cruz JM, Gagnon JA, Church GM & Valen E CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res 42, W401–407, doi: 10.1093/nar/gku410 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cong L et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823, doi: 10.1126/science.1231143 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vinjamur DS & Bauer DE Growing and Genetically Manipulating Human Umbilical Cord Blood-Derived Erythroid Progenitor (HUDEP) Cell Lines. Methods Mol Biol 1698, 275–284, doi: 10.1007/978-1-4939-7428-3_17 (2018). [DOI] [PubMed] [Google Scholar]
- 79.Canver MC et al. Integrated design, execution, and analysis of arrayed and pooled CRISPR genome-editing experiments. Nat Protoc 13, 946–986, doi: 10.1038/nprot.2018.005 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gregory T et al. GATA-1 and erythropoietin cooperate to promote erythroid cell survival by regulating bcl-xL expression. Blood 94, 87–96 (1999). [PubMed] [Google Scholar]
- 81.Grant CE, Bailey TL & Noble WS FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018, doi: 10.1093/bioinformatics/btr064 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Quinlan AR & Hall IM BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842, doi: 10.1093/bioinformatics/btq033 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chelala C, Khan A & Lemoine NR SNPnexus: a web database for functional annotation of newly discovered and public domain single nucleotide polymorphisms. Bioinformatics 25, 655–661, doi: 10.1093/bioinformatics/btn653 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dayem Ullah AZ, Lemoine NR & Chelala C SNPnexus: a web server for functional annotation of novel and publicly known genetic variants (2012 update). Nucleic Acids Res 40, W65–70, doi: 10.1093/nar/gks364 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dayem Ullah AZ, Lemoine NR & Chelala C A practical guide for the functional annotation of genetic variations using SNPnexus. Brief Bioinform 14, 437–447, doi: 10.1093/bib/bbt004 (2013). [DOI] [PubMed] [Google Scholar]
- 86.Leslie R, O’Donnell CJ & Johnson AD GRASP: analysis of genotype-phenotype results from 1390 genome-wide association studies and corresponding open access database. Bioinformatics 30, i185–194, doi: 10.1093/bioinformatics/btu273 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Splansky GL et al. The Third Generation Cohort of the National Heart, Lung, and Blood Institute’s Framingham Heart Study: design, recruitment, and initial examination. Am J Epidemiol 165, 1328–1335, doi: 10.1093/aje/kwm021 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The massively parallel sequencing data associated with this manuscript have been uploaded to GEO under the accession numbers GSE74483 and GSE104574 and are currently open to public. The web links for the publicly available databases used in this study are: UniPROBE: http://thebrain.bwh.harvard.edu/uniprobe/, CIS-BP: http://cisbp.ccbr.utoronto.ca/, FHS: https://www.ncbi.nlm.nih.gov/proiects/gap/cgi-bin/study.cgi?study_id=phs000007.v30.p11, RegulomeDB: https://regulomedb.org/regulome-search/, HEMMER: http://hmmer.org/, EMBOSS Needle: https://www.ebi.ac.uk/Tools/psa/emboss_needle/. dbSNP: https://www.ncbi.nlm.nih.gov/snp/?cmd=search. Links to all the PBM datasets used are available in Supplementary Table 7.