

Summary
Studying the microbial composition of internal organs and their associations with disease remains challenging due to the difficulty of acquiring clinical biopsies. We designed a statistical model to analyze the prevalence of species across sample types from The Cancer Genome Atlas (TCGA), revealing that species equiprevalent across sample types are predominantly contaminants, bearing unique signatures from each TCGA-designated sequencing center. Removing such species mitigated batch effects and isolated the tissue-resident microbiome, which was validated by original matched TCGA samples. Gene copies and nucleotide variants can further distinguish mixed-evidence species. We, thus, present The Cancer Microbiome Atlas (TCMA), a collection of curated, decontaminated microbial compositions of oropharyngeal, esophageal, gastrointestinal, and colorectal tissues. This led to the discovery of prognostic species and blood signatures of mucosal barrier injuries and enabled systematic matched microbe-host multi-omic analyses, which will help guide future studies of the microbiome’s role in human health and disease.
Graphical abstract
In Brief
Dohlman et al. present The Cancer Microbiome Atlas, a public database of decontaminated, tissue-resident microbial profiles of TCGA gastrointestinal cancer tissues. As these profiles are matched to specific TCGA tissue samples, this work allows identification of prognostic species and provides a resource for performing multi-omic, pan-cancer analyses of host-microbe interactions.
Introduction
The human body supports an ecosystem of 10–100 trillion microorganisms (Luckey, 1972; Sender et al., 2016), representing 500–1,000 unique species per individual (Human Microbiome Project Consortium, 2012; Qin et al., 2010). Perturbations to this ecosystem, termed dysbiosis, can impact human health: microbial alterations have been implicated in a variety of health conditions, including obesity, diabetes, inflammatory bowel disease, cancer, and other diseases (Elinav et al., 2019; Iliev and Leonardi, 2017; Levy et al., 2017; Schirmer et al., 2019). While public microbiome projects such as the Human Microbiome Project (HMP) and MetaHIT have helped bring tremendous insights into the diversity and function of human flora, these databases are dominated by tissue swabs and stool samples that do not necessarily reflect the microbial composition of internal organs (Grice et al., 2008; Prast-Nielsen et al., 2019). Collection of clinical biopsies specifically dedicated to microbial profiling remains difficult despite many disease-related host-microbe interactions occurring at the epithelium of internal body sites.
Next-generation sequencing (NGS) is frequently used to profile biopsied human tissue samples at a broad range of body sites and disease states, and these sequencing datasets contain a significant number of sequencing reads of microbial origin (Kostic et al., 2011; Poore et al., 2020; Robinson et al., 2017). Large sequencing projects can, thus, be mined to promote understanding of host-microbe interactions in both healthy and diseased human tissue. To that end, the bioinformatics tool PathSeq (Kostic et al., 2011) was used to identify enrichment of Fusobacterium nucleatum in The Cancer Genome Atlas (TCGA) colorectal cancer (CRC) tumors (Kostic et al., 2013; Kostic et al., 2012). Since then, dozens of research articles explored the role of F. nucleatum in tumorigenesis, finding associations with stage, survival, metastasis, and even drug response (Bullman et al., 2017; Flanagan et al., 2014; Yu et al., 2017). More broadly, sequencing data from TCGA has been used ad hoc to screen for viral and bacterial presence in stomach adenoma (Cancer Genome Atlas Research Network, 2014) and cervical cancer (Cancer Genome Atlas Research Network et al., 2017) specifically, as well as viromes (Tang et al., 2013) and bacteriomes (Robinson et al., 2017). Recently, analysis of TCGA sequencing data has been used to demonstrate the potential for bloodborne microbial DNA to diagnose certain cancers (Poore et al., 2020). Given that even low-biomass tumors contain tissue-specific microbiomes (Nejman et al., 2020), analysis of microbial DNA and RNA in TCGA sequencing data has great potential for diagnostic applications, as well as for exploring host-microbe interactions along molecular and clinical axes.
However, few actionable microbiota targets like F. nucleatum have emerged from such analyses. When examining a subset of TCGA sequencing data, previous analyses (Robinson et al., 2017) found that microbial reads from a number of species were the result of contamination, and that distinguishing contamination from tissue-embedded microbes remained an outstanding challenge for use of this dataset. Indeed, while concerns over contamination are less pressing for samples with high microbial biomass such as stool or swabs, microbiome studies on low-biomass samples suffer from contamination during sample collection and DNA extraction. Contamination can originate from the laboratory environment, including nucleic acid extraction kits (Davis et al., 2018; Eisenhofer et al., 2019; Glassing et al., 2016). Thus, controlling for contamination in these datasets is a crucial step that must precede downstream analyses of host-microbe interactions. Samples for multi-institutional projects are acquired, processed, and sequenced at different sites, each of which may introduce its own contaminants that influence the extracted microbial profiles, impede reproducibility, and complicate discovery of microbial biomarkers. Thus, a number of strategies have been deployed to identify contamination in TCGA sequencing data, through examination of batch effects, sample analyte concentrations, and through manual curation (Poore et al., 2020; Robinson et al., 2017). To date, such analyses have never been validated by original TCGA tissue or blood samples, nor have decontaminated TCGA microbiome datasets been made readily available.
Sequencing data in TCGA provide a unique opportunity for identifying tissue-specific microbiota, since matched tissue and blood samples from various cancer types are processed and sequenced in parallel using various sequencing platforms at designated centers (Choi et al., 2017). Using an unbiased statistical model comparing the prevalence of microbial species in tissue and blood samples, we isolated the tissue-resident microbiome in TCGA sequencing data. We found that species equally prevalent across tissue types and blood samples are mostly artifacts or contaminants that (1) bear unique signatures from the designated TCGA sequencing center and (2) comprise more than half of all detectible microbial sequencing reads in many tissue samples. With gene- and nucleotide-level resolution, our model is also capable of normalizing read counts for “mixed-evidence” cases, in which sequencing reads aligning a given species may come from a combination of endogenous and contaminant microbiota. To validate our approach, we obtained original matched CRC tissue and plasma samples that were previously sequenced by TCGA and performed 16S rRNA amplicon sequencing. This independently confirmed not only the absence of putative contaminants but also that the tissue-resident, computationally decontaminated microbial profiles that we extracted from TCGA sequencing data matched the microbial composition of the original tissue samples.
Finally, we ran the vetted decontamination algorithm to establish the TCMA database, which users can access from an interactive website (https://tcma.pratt.duke.edu). The database contains curated tissue-resident microbial profiles for 4,937 sequencing runs on 3,689 unique samples from 1,772 patients representing 5 TCGA projects and 21 anatomic sites with tissue-resident populations. As proof-of-principle, we used TCMA to identify two bacterial co-abundance groups in CRC tissue, including species enriched in CRC tumors compared with matched adjacent normal tissue and species prognostic of patient survival. TCMA enabled a matched microbe-host transcriptomic, proteomic, and epigenetic analysis that identified associations between microbes and host gene expression patterns and pathways. Finally, by comparing TCMA-curated blood samples of CRC and brain cancer (BC) patients, we identified a bacterial signature associated with colorectal mucosal barrier injury unique to CRC blood samples. Thus, TCMA constitutes a powerful resource for validation and hypothesis generation in future studies of host-microbe interactions relevant to cancer.
Results
WGS and WXS harbor colorectal bacterial reads distinct from blood and brain
To explore the microbial populations of sequenced TCGA tissue, we began by analyzing multi-platform sequencing data for 730 tissue and 555 blood samples from 617 CRC (TCGA projects COAD/READ) patients and for 958 tissue and 914 blood samples from 923 BC (TCGA projects GBM/LGG) patients. For several thousand whole-genome sequencing (WGS) and whole-exome sequencing (WXS) experiments, we retrieved raw sequencing data from the TCGA database and extracted and mapped high-quality reads of bacterial origin using PathSeq (Kostic et al., 2011). We found that microbial reads were more abundant and more diverse in solid tissue than in matched blood samples from CRC patients; in contrast, the abundance and diversity of microbial reads were no greater in BC tissue than matched blood samples (Figures 1A and S1A). Furthermore, CRC tissue had more abundant and diverse microbiota than BC tissue (Figures 1B and S1B), consistent with the notion that blood and brain tissue are more sterile than colorectal tissues. Notably, microbial reads were also more abundant and diverse in blood samples from CRC patients than in those of BC patients (Figures 1B and S1B).
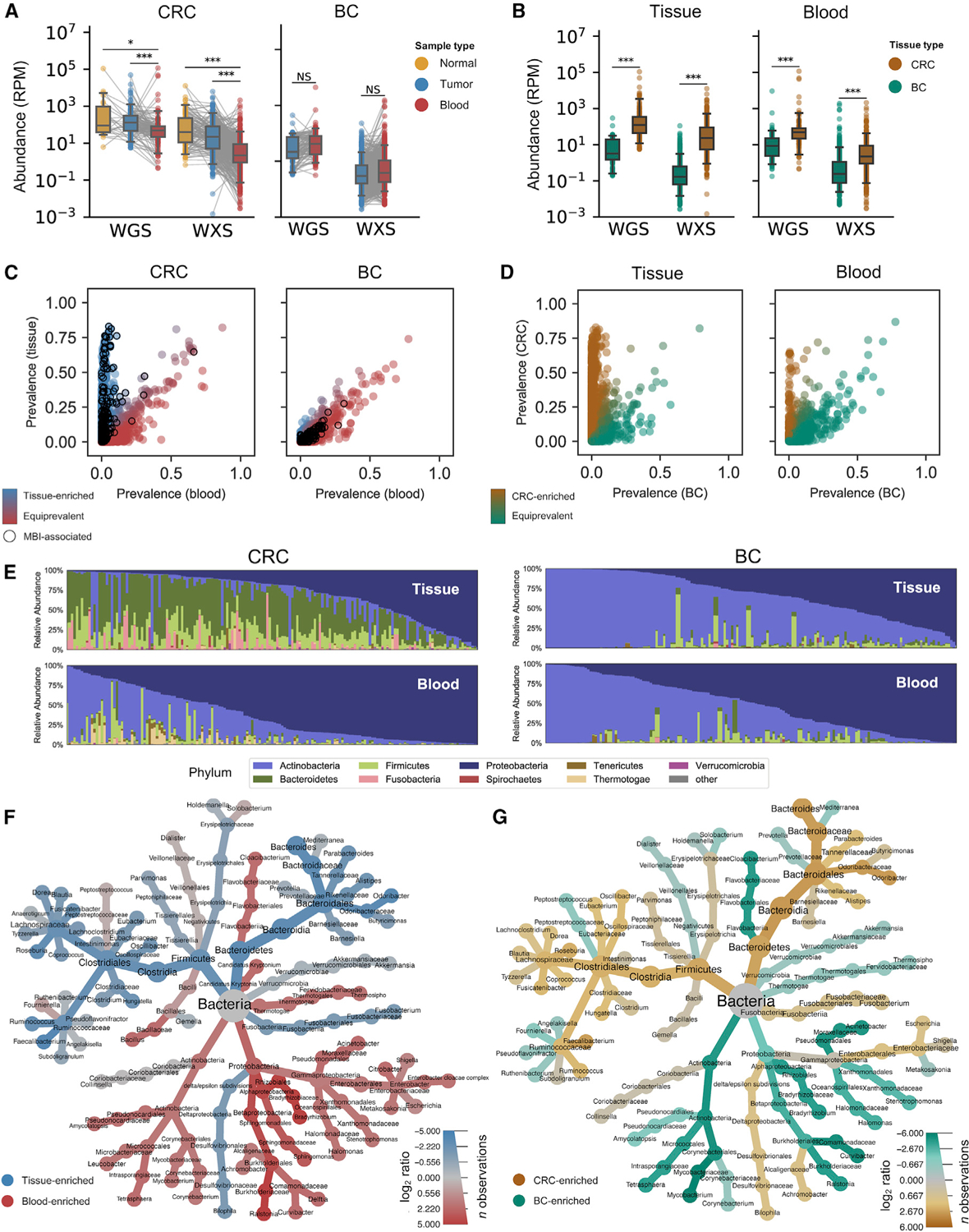
Figure 1. WGS and WXS harbor colorectal bacterial reads distinct from blood and brain.
See also Figure S1
(A) Matched analysis of bacterial sequencing reads per million (RPM) in normal tissue (yellow), tumor tissue (blue), and blood (red) from CRC and BC patients in TCGA. Significance is given by paired, one-sided t tests.
(B) Abundance data from (A) but comparing solid tissue (pooled tumor and normal) with blood samples from BC (green) and CRC (brown) patients. Significance is given by one-sided t tests.
(C) Comparison of bacterial species prevalence in WGS data for CRC blood and CRC tissue samples reveals populations of tissue-enriched species (blue) and species that are equiprevalent in blood and tissue (red). Black circles denote species associated with MBI.
(D) Comparison of bacterial species prevalence in WGS data for CRC and BC samples reveals populations of CRC-enriched species (brown) and species that are equiprevalent in CRC and BC (green).
(E) Relative abundance of bacterial phyla in WGS data for tissue (top) and blood (bottom) samples from CRC (left) and BC (right) patients.
(F and G) Heat tree comparing relative abundance of bacteria in WGS data for (F) matched blood samples (red) versus tissue samples (blue) and (G) CRC tissue (brown) versus BC tissue (green).
Comparative analysis of microbial reads between CRC tissue and blood samples revealed two distinct groups of bacterial species: those enriched in tissue and those equally prevalent in tissue and blood (Figures 1C and S1C). Species were seldom more prevalent in blood than in tissue. Species that were more prevalent in CRC tissue than in CRC blood included many species known to be associated with mucosal barrier injury (MBI) (CDC, 2019), whereas the group equally present in CRC tissue and blood contained very few such species (Figures 1C and S1C). By comparison, nearly all species detected in samples from BC patients were equiprevalent in tissue and blood, with few enriched in tissue (Figures 1C and S1C). Similar comparative analyses of tissue and blood from CRC and BC patients showed significant populations of disease-enriched species for CRC but few for BC (Figures 1D and S1D). We then repeated this comparative prevalence analysis using samples from ovarian cancer (OVC; TCGA project OV; Figures S1E and S1F).
The microbial composition of CRC tissue samples was also markedly distinct from that of matched blood samples or BC tissue samples. Among the most dominant phyla in CRC tissue were Bacteroidetes and Firmicutes, which were relatively absent in blood and brain tissue samples (Figures 1E and S1G). Next, we compared the relative abundance of bacterial taxa in CRC tissue versus matched blood samples (Figure 1F) and CRC tissue versus BC tissues (Figure 1G) from different donors. These analyses were largely consistent, with taxa from Bacteroidetes, Firmicutes, and Fusobacteria clades being consistently overrepresented in CRC tissue, compared with Proteobacteria and Actinobacteria, which accounted for a relatively greater fraction of reads in CRC blood samples and BC tissue samples. Genera that were relatively more abundant in CRC blood samples or BC tissue samples compared with CRC tissue samples consistently included Acinetobacter, Mycobacterium, and Ralstonia, among others (Figures 1F and 1G). Metagenomic profiling of TCGA samples using Kraken2 (Wood et al., 2019) largely recapitulated PathSeq results (Figures S1H–S1J).
Together, these comparative analyses were capable of distinguishing species enriched in CRC tissues from those with similar prevalence across different blood samples and disease types. The analyses confirmed that bacteria in CRC tissues were (1) more diverse and abundant and (2) were enriched for mucosa-related species.
Species equiprevalent in tissue and blood are predominantly contaminants
Besides species enriched in CRC tissues, a significant number of detected species were equally prevalent in blood, CRC tissue, BC tissue, and OV tissue (Figures 1C, 1D, and S1C–S1F). While compromised epithelial barrier function may allow the translocation of microorganisms to the bloodstream at low levels (Chelakkot et al., 2018), we expected that such species would be prevalent at much lower levels in blood than in CRC tissue. To analyze equiprevalent species and determine their origin, we first examined a set of 70 bacterial genera known to be enriched in negative controls of metagenomic sequencing experiments (Eisenhofer et al., 2019). Overall, genera in this “common contaminant” set were more prevalent in blood samples (p = 4.45e–10; Figures 2B and 2A) than genera not in the list.
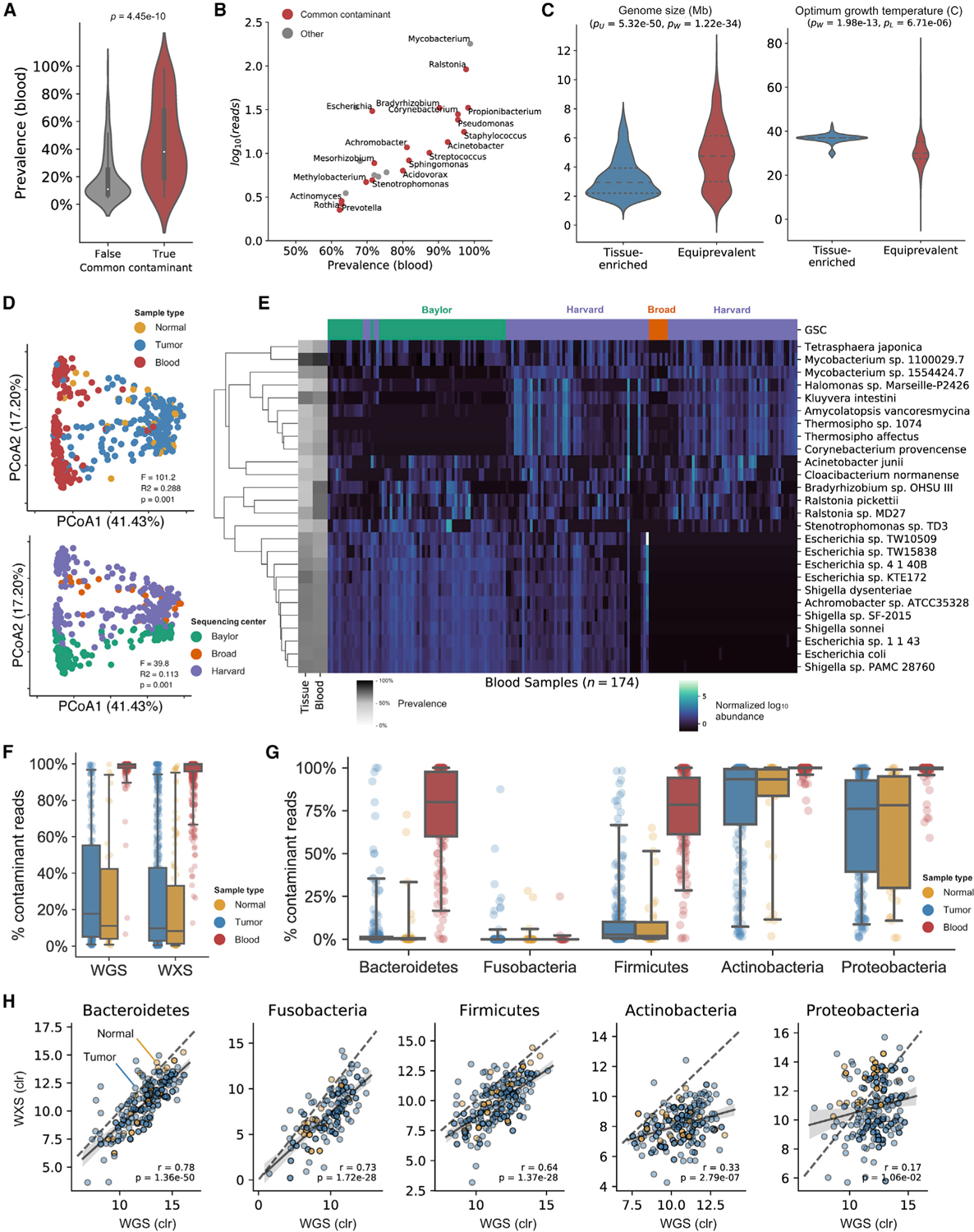
Figure 2. Most equiprevalent taxa are common contaminants and associated with particular sequencing centers.
See also Figure S2
(A) Genera commonly found in negative controls of metagenomic sequencing experiments (Eisenhofer et al., 2019) are highly prevalent in blood samples.
(B) Prevalence of common contaminants in blood correlates with absolute abundance.
(C) Genome size and temperature tolerance of equiprevalent species are differential (pW, Wilcoxon’s test) and more variable (pL, Levine’s test) than tissue-enriched species.
(D) PCoA of WGS data for CRC samples reveals considerable variation between blood samples and tissue samples along the first axis of variation and batch effects along the second axis.
(E) Heatmap clustering of bacterial species’ abundance in blood samples demonstrates the presence of center-specific contamination. The left vertical axis shows each species’ prevalence (gray).
(F) The fraction of all bacterial reads that is contamination in normal (yellow), tumor (blue), and blood (red) samples from CRC patients.
(G) The fraction of bacterial reads that is contamination in WGS data of normal (yellow), tumor (blue), and blood (red) from CRC patients, broken down by the five most prevalent phyla.
(H) Correlations between centered log ratio (CLR)-transformed relative abundances of WGS and WXS data for the five most prevalent phyla in tissue samples. Phyla contributing the most contaminant reads have the lowest correlation between assays.
Species equiprevalent in CRC tissue and blood were also considerably more genetically and phenotypically diverse than species enriched in CRC with respect to their G-C content, genome size, and optimal growth conditions. Conversely, CRC-enriched species were much less tolerant to extreme growth conditions, with optimal temperature, pH, and NaCl levels more closely resembling those of human homeostasis (Figures 2C and S2B). Together, these results suggest that the equiprevalent group may contain contaminant species, which have larger genomes—a signature of “generalist” bacteria that must endure more variable and unstable environmental conditions than gut microbiota (Sriswasdi et al., 2017).
Equiprevalent species are associated with particular sequencing centers
Principle coordinate analysis (PCoA) of UniFrac distances between CRC samples demonstrated that the primary axis of variation in TCGA microbiome data could be attributed to differences between blood samples and tissue samples (41.43%) (Figures 2D and S2C). Interestingly, the second axis of microbial variation was determined by the sequencing center at which the samples were processed (17.20%), regardless of sample type. All TCGA samples (tissue and blood) were harvested at a tissue source site and then sent to designated genome sequencing centers (Figure S2D). While the first PCoA axis captured differences in the presence of tissue-enriched species that are more abundant in CRC tissue than in blood, the second axis captured species found in both tissue and blood samples at similar levels, many of which were associated with sequencing center (Figures 2D, S2E, and S2F). We then examined the abundance of equiprevalent species in blood samples and found significant clustering according to the sequencing centers at which the samples were processed (Figures 2E and S2F). For comparison, we performed the same analysis on tissue and blood samples from BC patients, which revealed no discernible variation between tissue and blood samples, but rather significant clustering by sequencing center (Figures S2G and S2H).
Therefore, the majority of species equiprevalent in tissue and blood are not endogenous but are mostly artifacts introduced during processing and profiling at respective sequencing centers. For ease of description, we will refer to equiprevalent species as “contaminants” and tissue-enriched species as “tissue-resident” for the remainder of the article. However, the equiprevalent population may still contain biologically relevant species that are detected in both tissue and blood.
A generalizable model for isolating tissue-resident microbiota in TCGA tumor samples
Based on the comparative analyses of prevalence in tissue and blood, we developed a generalizable statistical model to distinguish tissue-resident microbiota from contaminant species across cancer types in TCGA. Of the 1,136 bacterial species detectable in more than 5% of CRC tissue samples, this model classified 769 species as tissue-resident (67.69%) and 367 species as contaminants (32.31%). Tissue-resident populations identified by comparing prevalence in tissue and blood were largely consistent with prevalence comparisons of CRC tissue with BC tissue, as well as analogous comparisons made with WXS data (Figure S2I). The model was used to perform binary classifications for all observed species. For each sample in the cohort, and at each subsequent taxonomic level, we then used species-level classifications to design a mixture model estimating the fraction of contaminant read counts within a given clade. This method provides a generalizable approach for decomposing observed microbial populations into their tissue-resident and contaminant fractions on a sample-by-sample basis at multiple taxonomic levels.
Proteobacteria and Actinobacteria contribute the largest fraction of contaminant reads
As expected, the removal of contaminant species resulted in a reduction in the number of bacterial reads in all sample types. Specifically, bacterial species classified as contaminants accounted for a median of 16.27% bacterial read counts in tissue but varied considerably (Figure 2F). Contaminants consistently dominated blood samples, with a median of 99.45% of detected WGS reads being the result of contamination. The phyla Proteobacteria and Actinobacteria contributed the greatest fraction of contaminant reads in WGS data, with medians of 76.67% and 80.95% of reads, respectively, found in CRC tissue samples being the result of contamination (Figure 2G). By contrast, only small fractions of Firmicutes (1.70%), Bacteroidetes (0.02%), and Fusobacteria (0.00%) reads were predicted to be the result of contamination (Figure 2G). Contamination rates were largely similar for WXS and across sequencing centers (Figures S2J and S2K).
Additionally, correlation between the normalized relative abundances of taxa in matched WGS and WXS samples was predictive of contamination rates. Correlations between Bacteroidetes, Fusobacteria, and Firmicutes abundances in WGS and WXS were consistently high, in contrast to Actinobacteria and Proteobacteria (Figure 2H). For blood samples, the normalized relative abundances of these five phyla were wholly uncorrelated between matched WGS and WXS (Figure S2L). Overall, these results show that significant fractions of the bacterial reads in WGS data for CRC tissue and blood samples are the result of contamination from Actinobacteria and Proteobacteria species.
Detecting tissue-resident and contaminant species with gene-level resolution
For species designated as tissue-resident (e.g., B. vulgatus) or as contamination (e.g., A. junii), we subsequently explored the extent to which microbial genes could be reliably detected in TCGA sequencing data. Using annotated genomes to search for gene-level assignments, we found that for many such species, sequencing alignments provided coverage of the full microbial genome. As expected, gene prevalence profiles of tissue and blood samples largely recapitulated those of species-level assignments (Figures 3A, 3B, S3A, and S3B). For tissue-resident species, the gene prevalence distribution was much lower in blood samples than tissue, while for contaminants, the gene prevalence distribution of blood and tissue samples were nearly identical (Figures 3D, 3E, S3C, and S3D); genome coverage was greater in tissue than blood for tissue-resident species but identical for contaminant species (Figures 3G, 3H, S3E, and S3F). Likewise, genome coverage in tissue samples was nearly equal at Harvard and Baylor for tissue-resident species but not for contaminant species (Figures S3G and S3H). These results suggest that gene- and nucleotide-level analyses of microbial sequencing reads may be leveraged to help distinguish contamination from tissue-resident populations.
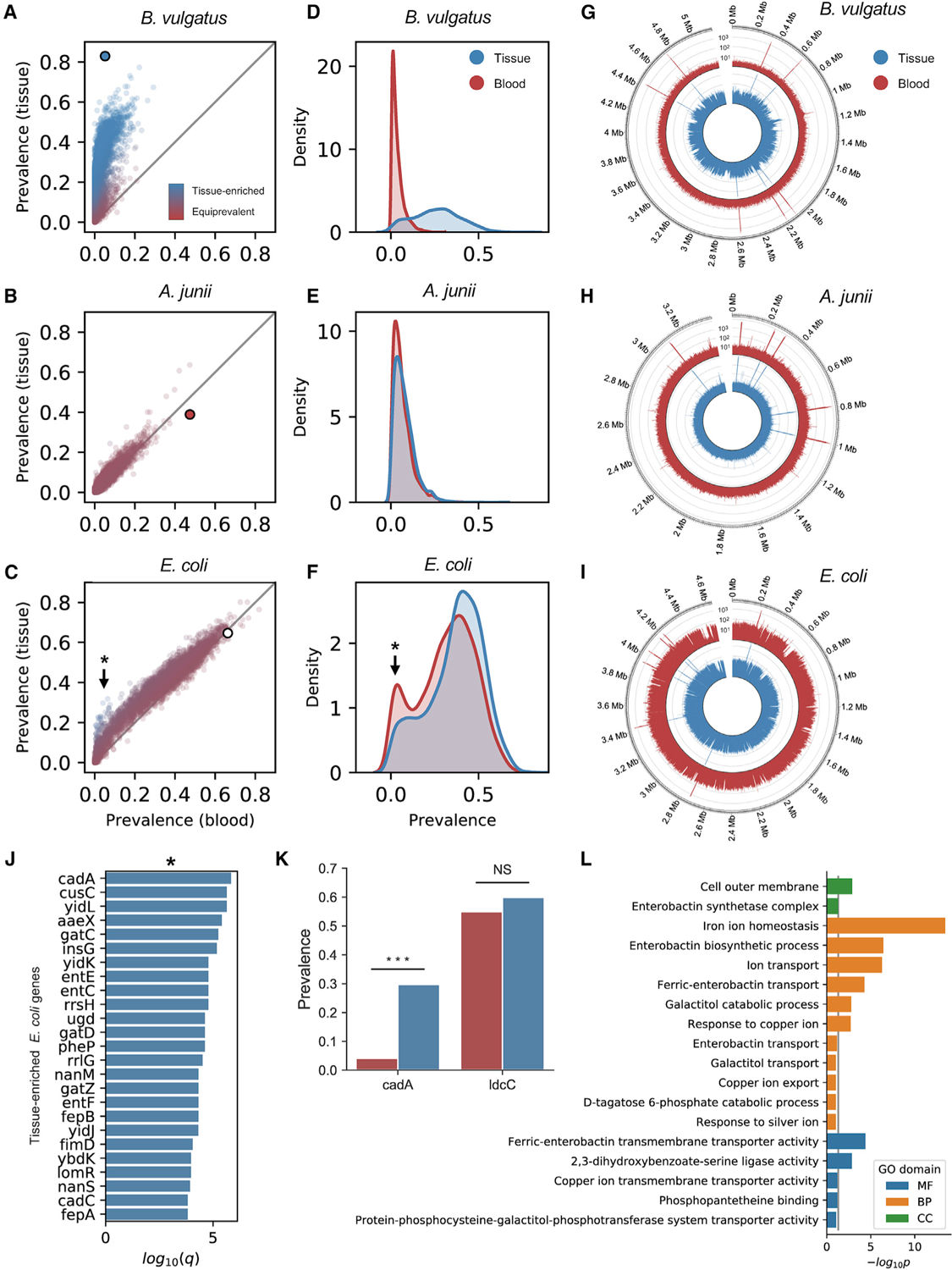
Figure 3. Detecting tissue-resident and contaminant species with gene-level resolution.
See also Figure S3
(A–C) Prevalence of genes belonging to B. vulgatus (A; tissue-resident), A. junii (B; contaminant), and E. coli (C; mixed-evidence) in blood versus tissue. The large dot indicates species-level prevalence.
(D–F) Kernel-density estimate of gene prevalence in blood (red) and tissue (blue) for B. vulgatus (A), A. junii (B), and E. coli (F).
(G–I) Coverage of WGS reads aligning to genomes of B. vulgatus (G), A. junii (H), and E. coli (I) in blood (red) and tissue (blue).
(J) Top 25 E. coli genes most significantly enriched in tissue.
(K) Comparison of the prevalence of E. coli genes, cadA and ldcC, in blood (red) and tissue (blue).
(L) Results of GO pathway analysis of tissue-enriched E. coli genes.
*Indicates tissue-enriched E. coli genes
Distinguishing tissue-resident Escherichia reads from contamination
An outstanding challenge in controlling contamination is the problem of mixed-evidence cases in which detected sequencing reads come from an unknown combination of endogenous and contaminant sources (Poore et al., 2020; Robinson et al., 2017). For example, although Escherichia coli is ubiquitous among human microbiomes, species-level E. coli reads were present in tissue (64.68%) and blood (66.29%) at nearly equal rates and were strongly associated with sequencing center (Figure 2E). We therefore explored whether gene-level read alignments could provide greater resolution and could be used to estimate the fraction of sequencing reads resulting from contamination versus endogenous microbiota. To test this, we mapped microbial sequencing reads from TCGA tissue and blood samples to genes in the annotated E. coli genome.
Overall, reads aligning to E. coli genes in tissue and blood samples were detected at up to the same rates as species-level E. coli alignments (Figure 3C) and had similar genome coverage (Figure 3I). However, a small number of E. coli genes displayed a signature analogous to tissue-resident microbiota in our species-level prevalence analysis (Figure 3C). Moreover, we observed bimodality in the blood prevalence of E. coli genes (Figure 3F), suggesting the presence of distinct tissue-resident and contaminant E. coli populations. We identified a set of 119 E. coli genes significantly enriched in tissue samples (q < 0.01; Figure 3J), several of which have credible reasons for being enriched in tissue samples. The top candidate, cadA (q = 4.44E-9), is a gene encoding one of two lysine decarboxylases (Kikuchi et al., 1997; Yamamoto et al., 1997) produced by E. coli; the other is ldcC, which is not enriched in tissue samples (q = 0.16) (Figures 3K and S3J). While ldcC encodes a gene that is constitutively expressed, cadA transcription is induced under conditions of anaerobic growth at low pH and its gene product displays greater thermostability and acid tolerance (Lemonnier and Lane, 1998). Additionally, genes in the pks island encoding colibactin were significantly more prevalent in tissue than blood, matching previous reports that E. coli strains expressing this gene are associated with CRC tissues (Figure S3L) (Arthur et al., 2012).
Discrepancies in intraspecies genome content may be explained by adaptive gene loss, an evolutionary mechanism whereby bacteria dispense with genes that are unnecessary for their environmental conditions (Koskiniemi et al., 2012; Mira et al., 2001). Pathway analysis (Huang da et al., 2009) revealed that tissue-enriched E. coli genes were significantly associated with processes including iron-ion homeostasis, enterobactin biosynthesis, ion transport, ferric-enterobactin transport, and copper-ion response (p < 0.01) (Figure 3L). Iron (Fe3+) and copper (Cu2+) are abundant in the host and can be toxic to E. coli in acidic, aerobic conditions; therefore, strains of E. coli must tightly regulate intracellular concentrations of these metals and undergo selection to do so (Porcheron et al., 2013; Rensing and Grass, 2003). Given that hypothetically bloodborne E. coli would also have to contend with high concentrations of copper and iron, enrichment of these genes and processes in tissue relative to blood suggests that the majority of E. coli reads detected in blood samples are not endogenous but rather the result of contamination. However, tissue-enriched genes such as cadA and others serve as benchmarks for distinguishing the two.
Tissue-enriched sequencing reads can be identified with nucleotide precision
We then examined microbial sequencing reads at nucleotide-level resolution. Given that gene-level alignments helped resolve mixed-evidence cases, we explored whether bacterial sequence variants, such as SNPs, could be used in a similar fashion. Variant prevalence across CRC tissue and blood samples largely recapitulated the results from species- and gene-level profiles (Figures S3L–S3M). Interestingly, we also found populations of apparent tissue-enriched and equiprevalent variants in E. coli genomes, suggesting that analyses of sequence variants may prove useful in distinguishing between endogenous and contaminant sequencing reads in mixed-evidence cases.
Decontamination removes sequencing center artifacts
Removing contamination affected all samples, but samples with low bacterial abundance a priori were the most affected (Figure 4A), consistent with observations that low-biomass samples are the most profoundly affected by contamination (Eisenhofer et al., 2019; Glassing et al., 2016). Decontamination also regularized the relative abundance profiles of CRC tissue samples, most prominently by the removal of contaminant Actinobacteria and Proteobacteria reads (Figure 4B).
Figure 4. Decontamination removes sequencing center artifacts and original TCGA tissue and blood samples validate tissue-resident microbial compositions and equiprevalent species as contaminants, see also Figure S4; Table S1.

(A) Abundance of WGS bacteria before and after decontamination. Samples with no reduction in bacterial reads lie along the gray line. Experiments with low microbial biomass a priori are disproportionally affected by decontamination.
(B) Relative abundance of bacterial phyla in tissue samples before and after decontamination, sorted by their a priori abundance of Actinobacteria.
(C) PCoA of the decontaminated, tissue-resident microbial component reveals retention of variation related to sample type but not sequencing center.
(D) PCoA of the contaminant microbial component reveals retention of variation related to sequencing center but not sample type.
(E and F) Prevalence of bacterial species in tissue samples sequenced at Baylor versus Harvard (E) before and (F) after removing contamination.
(G) Comparison of weighted UniFrac distances before and after removing contamination among all tissues (left) and specifically matched tissues sequenced at both Baylor and Harvard (right).
(H) Design of the validation experiment. Data are represented as mean ± 95% CI.
(I) Bacterial diversity of 16S rRNA-seq results from tissue (blue), plasma (red), and controls (bottom panel).
(J) Relative abundances in 16S results for tissue compared with tissue samples sequenced using WGS at Harvard and Baylor, before and after contamination.
Despite being naive to sequencing center, our prevalence-based model for decomposing the TCGA microbiome data into tissue-resident and contaminant fractions also mitigated center-related batch effects. Prior to removing contamination, TCGA microbiome data clustered by both sample type and sequencing center (Figure 2D). However, unsupervised clustering of the tissue-resident component extracted from the original TCGA sequencing data showed no dependency on sequencing center and maintained variation related to sample type (Figure 4C). Examining the contamination component, we found the opposite: samples no longer clustered by sample type but rather organized exclusively according to sequencing center (Figure 4D). These results reflected the removal of species that were uniquely prevalent in tissue samples from either Baylor or Harvard (Figures 4E, 4F, and S4A).
Finally, our algorithm greatly increased the similarity between the microbial populations in patient-matched tissue samples sequenced at both Harvard and Baylor, while maintaining diversity among samples overall (Figure 4G). Thus, our prevalence-based model is able to homogenize matched samples sequenced at different centers and mitigate sequencing center artifacts.
Original TCGA tissue and blood samples validate tissue-resident microbial compositions and equiprevalent species as contaminants
To benchmark our analysis, we obtained five primary CRC tumor samples and matched plasma samples from an original TCGA tissue provider (Table S1). These samples were specifically chosen to ensure that each tissue and plasma sample was profiled by WGSat both Baylor and Harvard. For controls, we also procured three plasma samples from healthy individuals and spiked one plasma sample with E. coli. We then used 16S amplicon sequencing to validate that the bacterial composition of the original TCGA samples resembled the decontaminated compositions extracted from TCGA sequencing data for matched tumor samples (Figure 4H).
We found that the original TCGA tumor samples contained ample bacterial diversity and read counts (Figures 4I and S4B) and that their bacterial composition largely recapitulated the decontaminated, tissue-resident microbial population our decontamination model extracted from TCGA WGS data on matched samples (Figures 4J and S4D). In addition to increasing the similarity between WGS and 16S validation results (Figure S4C), decontamination greatly improved the concordance between microbial compositions of matched WGS experiments performed at Harvard and Baylor (Figure 4G). Despite detecting a large number of bacterial sequencing reads in tumor samples, bacterial diversity of CRC plasma was not significantly greater than healthy plasma (p = 0.30) or water controls (p = 0.44). Moreover, the 16S bacterial composition of original TCGA plasma samples was distinct from the bacterial composition of WGS data from the same samples (Figures S4E and S4F), supporting the notion that the majority of bacterial reads detected in TCGA blood samples are contamination introduced during DNA extraction and sequencing, rather than at the time of procurement. These validation results demonstrate that our model accurately identifies and removes contaminants and that computational decontamination produced profiles that represent the true microbial composition of tissue.
Colorectal tissue microbiomes cluster into Fusobacterium and Bacteroides co-abundance groups
Having validated the contamination-adjusted microbial profiles, we sought to leverage the decontaminated TCMA dataset to investigate whether certain subgroups of microbiota were more likely to be found together in tissue from CRC patients. Using the bootstrapping procedure, SparCC (Friedman and Alm, 2012), we found two anticorrelated co-abundance groups (Figures 5A–5C, S5A, and S5B): the “Fusobacterium cluster” contained Porphyromonas, Prevotella, Peptostreptococcus, and Campylobacter, among other species that were associated with tumor samples; the second “Bacteroides cluster” is larger and contained a highly correlated set of microbes, including Parabacteroides, Clostridium, and Alistipes. This group may represent a more normal/healthy microbiome, as several of these species were positively associated with normal tissue samples. Taxa in the Fusobacterium cluster were significantly associated with colorectal neoplasms, while taxa in the Bacteroides cluster were associated with C. difficile infection, irritable bowel syndrome, and cirrhosis (q < 0.05). These co-abundance groups may represent two distinct “enterotypes” of CRC tissue microbiomes.
Figure 5. Colorectal tissue microbiomes cluster into Fusobacterium and Bacteroides co-abundance groups predictive of host tissue molecular environment.
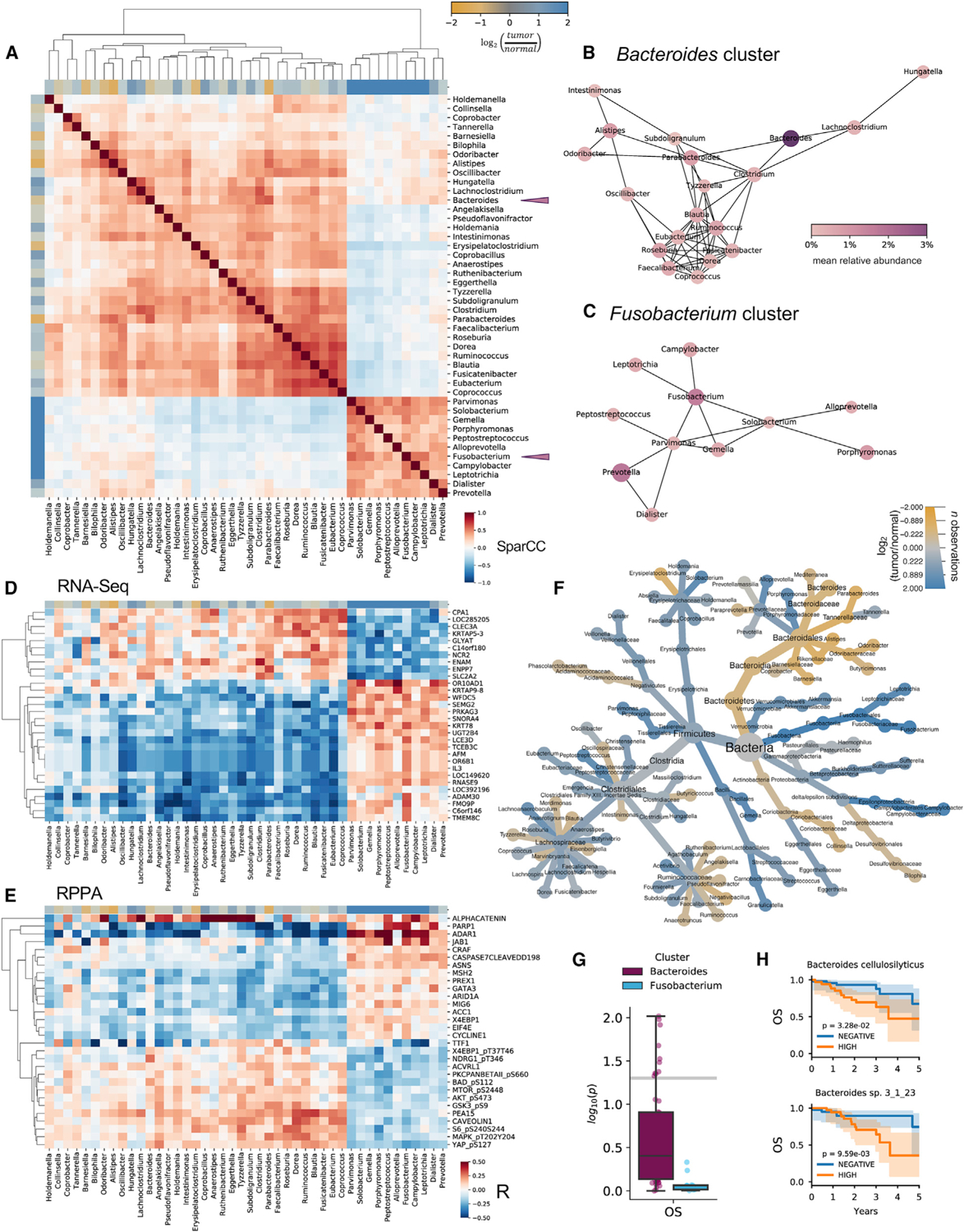
(A) Heatmap clustering of correlations between bacterial genera reveals anticorrelated clusters of genera, characterized by Bacteroides and Fusobacterium (purple triangles). Axes are colored according to species’ association with tumor (blue) or matched adjacent normal tissue (yellow).
(B and C) (B) Bacteroides- and (C) Fusobacterium-associated co-abundance networks. Node size is proportional to the prevalence of the genera in tissue samples, and node hue is proportional to abundance.
(D and E) Co-abundance groups are predictive of gene expression (D; RNA-seq) and protein expression (E; RPPA).
(F) Heat tree comparing bacterial taxa abundance in tumor samples (blue) or matched normal tissue (yellow).
(G) Survival analysis p values of species in the Bacteroides and Fusobacterium co-abundance groups.
(H) OS curves for Bacteroides spp.
Bacterial co-abundance groups are predictive of the host tissue molecular environment
Next, we evaluated whether the bacterial co-abundance groups we identified had discernible effects on host gene expression or regulation. Microbiota and host cells are known to engage with one another through a complex variety of molecular interactions. Host-derived nutrients and dietary macromolecules are utilized by microorganisms as a food source, while microbial byproducts including short-chain fatty acids (SCFAs) are known to modulate gene expression, cell differentiation, and inflammatory response (Donohoe et al., 2011; Furusawa et al., 2013).
The TCGA database contains a dense cube of molecular profiling data, including matched genetic, epigenetic, transcriptional, and proteomic assays performed on thousands of samples. Thus, the ability to compare microbial profiles with matched host molecular profiles represents an unprecedented opportunity for querying host-microbe interactions in various tissue types. As proof-of-principle, we used batch-normalized RPPA, mRNA-seq, miRNA-seq, and methylation 27-K data from TCGA to compute correlations between features in these datasets with genera in the Fusobacterium and Bacteroides clusters identified previously (Figures 5D, 5E, S5C, and S5D). For each of these assays, we found that these bacterial co-abundance groups were predictive of host gene expression patterns. For instance, in RPPA protein expression data we found that ADAR1 and PARP1 expression appeared to distinguish these co-abundance groups (Figure 5E). The protein ADAR1 is upregulated by inflammatory mediators such as TNF-alpha and IFN-gamma (Yang et al., 2003) and regulates pathogen detection and autoinflammation by discriminating self from non-self RNA (Chung et al., 2018), while PARP1 regulates DNA repair and is activated by Helicobacter pylori in gastric cancer (Nossa et al., 2009). Independently, we found that ADAR1 expression correlated with expression of PARP1, TNF-alpha, and IFN-gamma in TCGA RNA-seq data for CRC (Figure S5E). These results suggest that genes regulating inflammation and pathogen response may distinguish the Fusobacterium and Bacteroides co-abundance groups in CRC. More broadly, these analyses illustrate the utility of TCMA as a unique resource for comparing microbial and multi-omic host profiles from matched tissue samples.
Matched tumor-normal analysis reveals species associated with colorectal neoplasms
The TCGA database contains detailed annotations on each tissue donor, including statistics on tumor stage, size, morphology, and location, as well information on patient survival, treatment history, and therapeutic response. To identify microbes predictive of pathological and prognostic characteristics of CRC tissue, we used matched normal tissue and primary tumor samples to perform a paired comparison of tissue-resident microbes (Figures 5F, S5F, and S5G). This analysis identified 37 species that were significantly enriched in either normal (n = 14) or tumor (n = 23) samples (p < 0.05) (Table S2).
The species most significantly associated with CRC tumors compared with matched normal tissue was F. nucleatum (p = 1.82E-3), which is known to promote intestinal tumorigenesis. Overall, approximately half of tumor-associated species belonged to the genus Fusobacterium, including F. hwasookii, F. massiliense, and a number of unclassified Fusobacterium spp. (p < 0.01). Non-Fusobacterium species associated with CRC tumors included P. micra, S. moorei, and P. stomatis (p < 0.05), several of which belonged to the Fusobacterium co-abundance group (Figure S5A) and have previously been implicated in CRC (Kostic et al., 2013; Purcell et al., 2017; Warren et al., 2013). Other species, including several Campylobacter spp. did not have extant links to the disease. Of these, C. ureolyticus (Log2FC = 1.97; p = 2.19e–2) is an emerging gastrointestinal pathogen implicated in inflammatory bowel disease and colitis (Bullman et al., 2013; O’Donovan et al., 2014), prompting further examination. C. ureolyticus abundance correlated with expression of several genes, including CAMK2D and UGDH (Figures S5J, S5H, and S5I), and several genes expressed by C. ureolyticus were significantly associated with tumor samples compared with normal tissue (Figure S5J). Additionally, C. ureolyticus was associated with worse progression-free interval (PFI) in recurrent CRC patients (Figure S5K).
Taxa that were significantly more abundant in adjacent normal tissue compared with matched tumor tissue were dominated by Bacteroides and Parabacteroides spp. (p < 0.05) (Figure S5G), many of which belonged to the Bacteroides co-abundance group (Figure S5A). By leveraging patient-matched tumor and normal tissue samples, TCMA may thus be used identify bacterial associations with CRC and other gastrointestinal cancers.
Survival analysis reveals candidate microbial biomarkers predictive of clinical outcomes
Using survival data collected by the PanCanAtlas (Liu et al., 2018), we next examined whether co-abundance groups were predicative of overall survival (OS). For each species, we used a log-rank test to assess its individual prognostic value. Interestingly, species in the Bacteroides co-abundance group were generally more prognostic of survival than the Fusobacterium co-abundance group (Figure 5G). We found over a dozen Bacteroides spp. that were prognostic of survival, including B. cellulosilyticus and several unclassified Bacteroides spp. (Figures 5H and S5L). These findings demonstrate the utility of TCMA for the identification of prognostic microbial biomarkers relevant to CRC and other cancers.
Microbial presence in CRC tissue is predictive of host immunogenic response, inflammatory cancer pathways, and cell-cell adhesion
Next, we explored whether the 37 species that we identified as significantly associated with either tumor or normal tissue samples had identifiable effects on host gene expression or related biological pathways. Comparing normalized abundances of these species with matched mRNA expression data from 159 CRC tumor samples, we computed correlations and found transcriptional patterns that were associated with both tumor- and normal tissue-associated species (Figures 6A and S6A). Given the observed differences in the transcriptional correlations of tumor- and normal tissue-associated bacteria, we subsequently performed gene-set enrichment analysis (Subramanian et al., 2005) to identify biological pathways associated with the abundance of these species.
Figure 6. Microbial presence in CRC tissue is predictive of host gene expression pathways and MBI See also Figure S6.
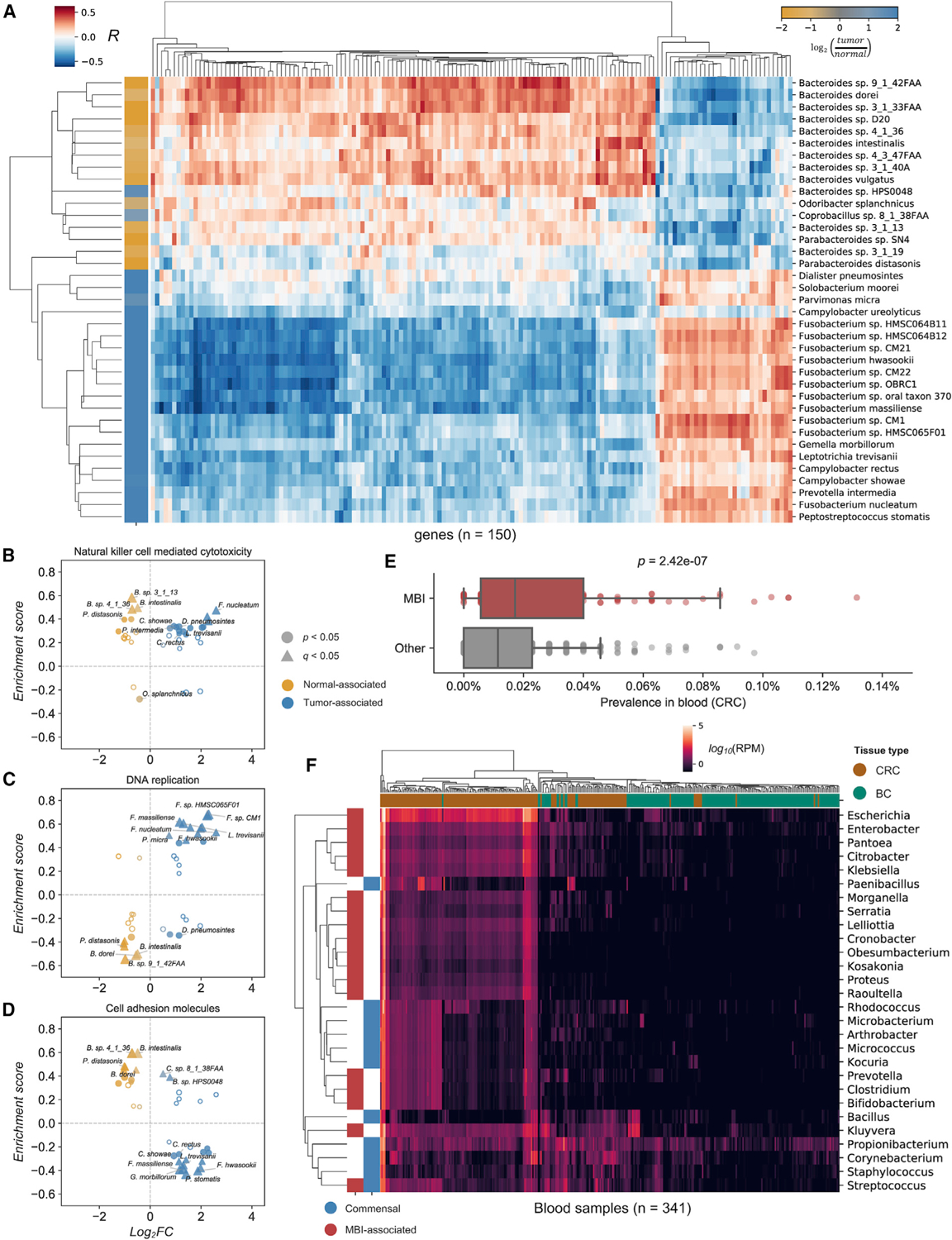
(A) Correlation between host gene expression (columns) and CLR-transformed species abundances (rows). Rows are colored according to each species’ association with tumor (blue) or normal tissue (yellow).
(B–D) Comparison of differentially abundant species and their association with tissue type (x axis) versus enrichment score (y axis) for KEGG terms (A) “natural killer cell-mediated cytotoxicity” (B), “DNA replication” (C), and “cell adhesion molecules” (D).
(E) Bacterial species implicated in MBI are more prevalent in decontaminated blood samples than other species.
(F) Bacterial genera implicated in MBI (red) are more abundant in CRC blood (brown) than BC blood (green), in contrast to some commensal species (blue).
Pathway analysis revealed that (1) genes correlated with the abundance of bacterial species were consistently enriched for the activation of immune system pathways and processes, irrespective of their association with tumor or normal tissue (Figures 6B and S6B) and (2) processes related to inflammatory cancer pathways and cell-cell adhesion were enriched among genes correlated with tumor-associated and normal tissue-associated species, respectively (Figures 6C, 6D, S6C, and S6D). Specifically, both tumor- and normal tissue-associated species were enriched for processes relating to intestinal IgA production, antigen presentation, natural killer cell-mediated cytotoxicity, cytokine signaling, and primary immunodeficiency, suggesting near-universal activation of an immunogenic transcriptional response to the presence of these bacteria (Figures 6B and S6B).
We also found that pathways including DNA replication, DNA repair, oxidative phosphorylation, p53 signaling, and ribosome activity were all negatively enriched among normal tissue-associated species, and positively enriched among tumor-associated species, particularly for Fusobacterium spp. (Figures 6C and S6C). Conversely, genes involved in the regulation of cellular adhesion were positively enriched among normal tissue-associated species and negatively enriched among tumor-associated species (Figures 6D and S6D). Together, these results indicate that within this cohort of CRC tumor samples, tumor-associated species may be associated with proinflammatory, neoplastic transformations and loss of epithelial integrity.
Microbial presence in CRC blood samples indicate mucosal barrier injury
As shown in Figures 1B and S1B, bacteria were significantly more abundant and diverse in blood samples from CRC patients than from BC patients (p < 0.01). The presence of transient, endogenous microbial DNA in the bloodstream has been reported in primary CRC patients, often before diagnosis, and may even be predictive of tumor stage and location (Abdulamir et al., 2011; Poore et al., 2020). Loss of mucosal barrier function is a common feature of CRC and other chronic inflammatory conditions and may lead to microbial translocations from CRC tumors to the lamina propria and bloodstream (Oshima and Miwa, 2016; Yu, 2018).
To explore this possibility, we examined the abundance of subsets of bacterial species and genera designated as common commensal (n = 407) or MBI-associated (n = 693) (CDC, 2019). Examining MBI-associated species among decontaminated blood samples within the CRC cohort, we found that species associated with MBI were considerably more prevalent than those that were not (p = 2.42e–7; Figure 6E). We then compared the abundance of MBI-associated genera with that of common commensals and discovered that genera associated with MBI were frequently more abundant in the blood of CRC patients than BC patients (Figures 6F and S6E). These results point toward the potential utility of bloodborne bacterial DNA from MBI-associated organisms as a potential biomarker for CRC.
Contamination-adjusted tissue microbiome profiles for all gastrointestinal cancers in TCGA
Having successfully identified the CRC tissue-associated microbial component in the TCGA dataset, we analyzed samples from other cancer types to search for tissue-resident microbiota. All sequencing datasets contained some bacterial reads but as expected, they were most abundant in gastrointestinal cancers (Figure 7A). In particular, tissue samples from head and neck cancer (HNSC), colon cancer (COAD), rectal cancer (READ), esophageal cancer (ESCA), and stomach cancer (STAD) had the greatest number of bacterial reads prior to decontamination, whereas uveal melanoma (UVM), lung squamous-cell (LUSC), and glioblastoma had the fewest.
Figure 7. Contamination-adjusted tissue microbiome profiles for all gastrointestinal cancers in TCGA.
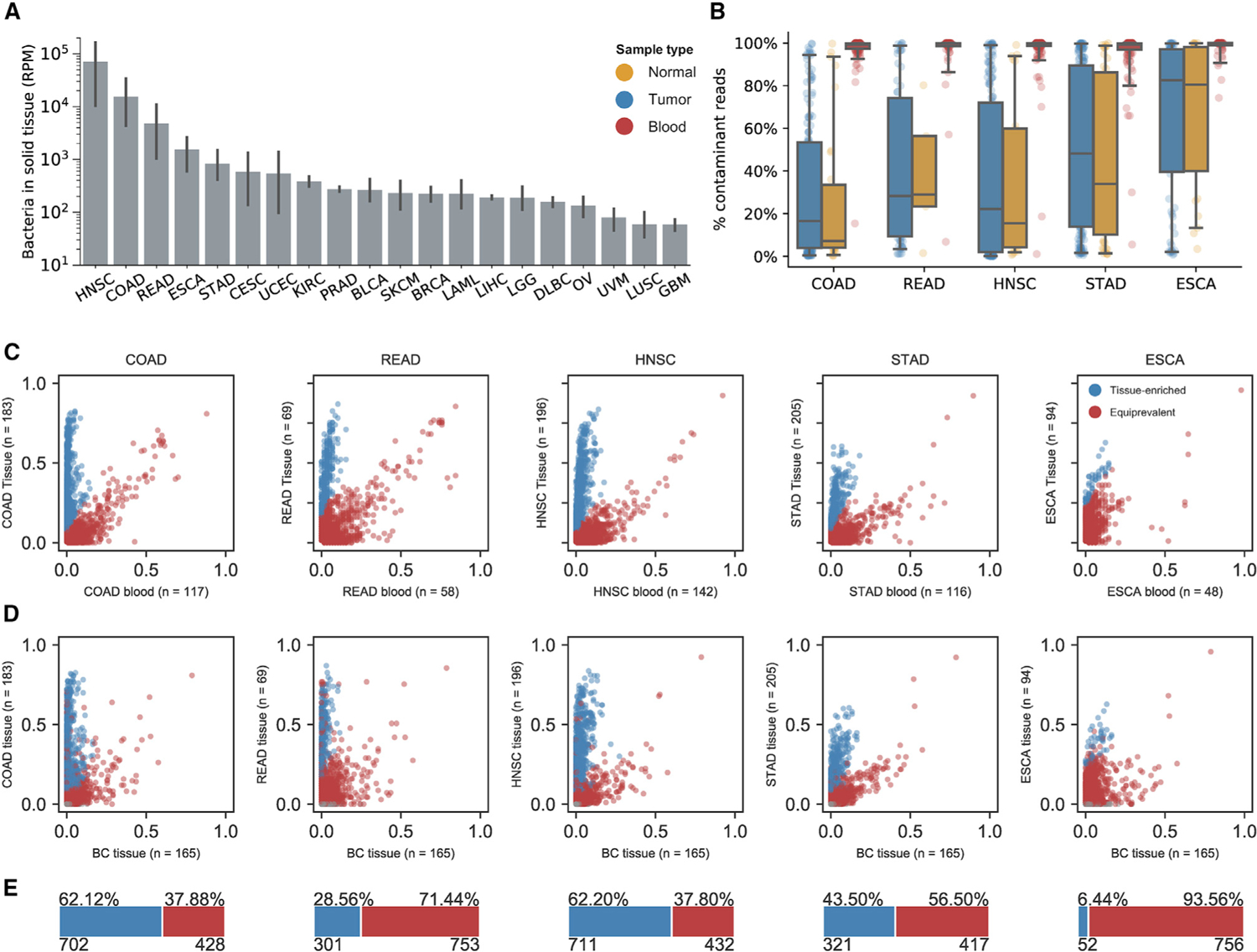
See also Figure S7
(A) Pan-cancer abundance of bacteria in solid tissue samples from TCGA projects prior to decontamination. Data are represented as mean ± 95% CI.
(B) Estimated fraction of contaminant reads for sequencing experiments on tumor (blue), normal (yellow), and blood (red) samples for each sequencing project in TCMA.
(C) Classification of tissue-resident (blue) and contaminant (red) species across TCGA gastrointestinal tissues by comparison of prevalence in blood and tissue.
(D) Labeling of tissue-resident (blue) and contaminant (red) species across gastrointestinal tissues by comparison of prevalence in brain tissue and disease-specific tissue, using classification from (C).
(E) Estimated proportions of tissue-resident (blue) and contaminant (red) species for each TCGA project.
Given the abundance of microbial reads in gastrointestinal cancer types, we used our prevalence-based approach to determine whether tissue-resident microbiota were present and estimate the fraction of contaminant reads in each sample type (Figure 7B). In addition to COAD and READ, we found a strong signature of tissue-resident species in HNSC, STAD, and ESCA projects by comparing species prevalence in tissue with blood and brain samples (Figures 7C and 7D), and we estimated the fraction of minimally detectable species that were tissue-resident or contaminants (Figure 7E). For each of these cancer types, we applied our decomposition model to isolate tissue-resident populations and establish TCMA. Few statistically significant tissue-resident populations in bladder (BLCA), breast (BRCA), uterine (UCEC), cervical (CESC), or prostate (PRAD) cancers could be detected (Figures S7A and S7B). The microbial biomass in these tissues is known to be magnitudes less than that of gastrointestinal tissues despite similar levels of contamination (p = 0.56), hence it may be more challenging to distinguish the few tissue-resident species from the over-whelming proportion of contaminants.
Discussion
By comparing and integrating data from multiple NGS platforms and various sample types, we isolated and experimentally validated the tissue-resident component of these datasets, thus producing a public resource of computationally decontaminated microbial profiles in TCGA tissue samples. This examination of equiprevalence provides a blueprint for future analyses of sequencing data for metagenomic profiling of tissue-resident microbiota. Putative contaminant species are more likely to originate from a single source and are also expected to demonstrate a lesser degree of intraspecies genetic variation, meaning that additional analyses of gene- and nucleotide-level prevalence may be helpful for controlling contamination, as we demonstrated for mixed-evidence cases such as E. coli. Prevalence-based analyses are likely to supplement standard batch-correction tools, which control technical variation but do not explicitly model contamination. More statistically rigorous tools that leverage prevalence and other technical variables to explicitly define observed metagenomic data as some linear combination of endogenous and contaminant read counts, may therefore be warranted.
The ability to retroactively remove contaminant species from NGS sequencing datasets will greatly expand the breadth and accessibility of metagenomic profiles for downstream analyses. Multi-institutional initiatives such as TCGA and GTEx have collected tens of thousands of tissue samples for sequencing, many of which are from internal organs and tissue types known to harbor microbiota. Most of these samples have been characterized extensively along genetic, epigenetic, transcriptional, and proteomic axes or provide detailed clinical profiles on patient donors. Meanwhile, a growing body of evidence suggests that alterations to the microbiome are associated with cancer development, progression, and drug response (Gopalakrishnan et al., 2018; Sivan et al., 2015; Viaud et al., 2013). Therefore, obtaining robust profiles of the microbial composition of human tissues in these sequencing databases will provide new insights into multi-omic host-microbe interactions in human tissue samples that would otherwise be difficult to acquire and analyze.
As proof-of-principle, we used TCMA to identify two dominant clusters of tissue-resident bacteria in CRC samples, as well as their associated molecular expression patterns and prognostic significance. Pathway analysis of matched transcriptional data demonstrated that tumor-associated species were positively correlated with cancer-related inflammatory pathways and negatively associated with cellular adhesion machinery. Specifically, enrichment of ribosome, p53 signaling, DNA repair, oxidative phosphorylation, and cellular adhesion pathways may point to a previously described mechanism wherein inflammatory cytokines downregulate p53 by stimulating ribosome biogenesis in colonic epithelial cells, leading to downregulation of E-cadherin and epithelial-mesenchymal transition (Brighenti et al., 2014). Given the established ability of many of these species to induce inflammation (Kostic et al., 2013), stimulate cytokine activity (Gemmell and Seymour, 1993), and modulate E-cadherin (Rubinstein et al., 2013) in colonocytes, the contribution of these species to this inflammatory cancer pathway necessitates further exploration.
Beyond CRC, the TCMA database will allow interrogations of pan-cancer relationships between the microbiome and tumor development. In most cases, the role of microbiota in cancer is context-specific. For example, H. pylori is known to advance gastric cancers but seemingly offer a protective effect in esophageal adenocarcinoma (Islami and Kamangar, 2008). However, certain pathogenic processes, such as chronic inflammation, altered metabolic states, and abrogation of viral latency display commonality across cancers (Plottel and Blaser, 2011). Since TCGA samples were collected and analyzed with common methodologies, the decontaminated metagenomic profiles for thousands of tissue samples presented here provide an ideal platform for examining host-microbe relationships that span cancer types, in contrast to meta-analyses, which must integrate data from disparate sources. Thus, in addition to providing a methodology for comprehensively identifying and removing contamination, TCMA represents an unprecedented resource for exploring the role of tissue-resident microbiota in various cancer types and identifying predictive microbial biomarkers.
STAR★methods
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Xiling Shen (xiling.shen@duke.edu).
Materials availability
The Cancer Genome Atlas (TCGA) collected biospecimens and associated clinical information from human subjects, under informed consent and authorization of local institutional review boards. All TCGA sequencing data were accessed from the Genomic Data Commons (GDC) portal in accordance with the TCGA Data Use Certification Agreement and under authorization of Duke’s campus institutional review board. Original TCGA tissue and plasma samples were acquired from Indivumed, a third-party vendor. Healthy patient samples used for validation analyses were collected and analyzed under authorization of Duke’s campus institutional review board. Primers used from 16S sequencing were acquired from IDT. This study did not generate new unique reagents.
Data and code availability
The TCMA database can be accessed via the website (https://tcma.pratt.duke.edu). The accession number for the data reported in this paper is https://doi.org/10.7924/r4rn36833. The TCGA sequencing data and associated aliquot, sample, and patient metadata on which this work was based were accessed from the GDC API. Molecular profiling data and clinical endpoints used for the survival analysis were obtained from the PanCanAtlas publication page. Scripts used for this work are available on request. More detailed information about data and code can be found in the Key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological Samples | ||
| Original TCGA tissue and plasma samples | Indivumed | N/A |
| Healthy plasma samples | Duke Hospital | N/A |
| Critical Commercial Assays | ||
| MagAttract PowerSoil DNA KF Kit | Qiagen | Cat# 27000-4-KF |
| QIAamp UCP Pathogen Mini Kit | Qiagen | Cat# 50214 |
| Deposited Data | ||
| TCGA WGS bam files | GDC API | https://api.gdc.cancer.gov/ |
| TCGA WXS bam files | GDC API | https://api.gdc.cancer.gov/ |
| TCGA sequencing metadata | GDC API | https://api.gdc.cancer.gov/ |
| TCGA sample metadata (biotab) | GDC web portal | https://portal.gdc.cancer.gov/ |
| TCGA patient metadata | PanCanAtlas | https://gdc.cancer.gov/about-data/publications/pancanatlas |
| TCGA clinical data resource outcomes | PanCanAtlas | https://gdc.cancer.gov/about-data/publications/pancanatlas |
| Human and microbe reference genomes | PathSeq bundle | ftp://gsapubftp-anonymous@ftp.broadinstitute.org/bundle/pathseq/ |
| Human and microbe reference genomes | PathSeq bundle | ftp://gsapubftp-anonymous@ftp.broadinstitute.org/bundle/pathseq/ |
| Species and genera designated as commensal or MBI-associated (NHSN Organism List) | (CDC-NHSN, 2019) | https://www.cdc.gov/nhsn/pdfs/pscmanual/pcsmanual_current.pdf (Chapter 4 p.33) |
| Genera designated as common contaminants | (Eisenhofer et al., 2019) | Table 1 |
| Genome for B. vulgatus | GenBank | CP000139.1 |
| Genome for A. junii | GenBank | NZ_CP019041.1 |
| Genome for E. coli | GenBank | U00096.3 |
| Genome for C. provencense | GenBank | NZ_CP024988.1 |
| Genome for F. nucleatum | GenBank | AE009951.2 |
| Pks gene cluster (colibactin) | GenBank | AM229678.1 |
| Greengenes classifier (gg-13-8-99-515-806-nb-classifier.qza) | (DeSantis et al., 2006) | https://docs.qiime2.org/ |
| TCGA mRNA-seq data | PanCanAtlas | https://gdc.cancer.gov/about-data/publications/pancanatlas |
| TCGA miRNA-seq data | PanCanAtlas | https://gdc.cancer.gov/about-data/publications/pancanatlas |
| TCGA RPPA data | PanCanAtlas | https://gdc.cancer.gov/about-data/publications/pancanatlas |
| TCGA DNA Methylation 27K data | PanCanAtlas | https://gdc.cancer.gov/about-data/publications/pancanatlas |
| PARADIGM Pathway inference matrix | PanCanAtlas | https://gdc.cancer.gov/about-data/publications/pancanatlas |
| Gene sets for GSEA (KEGG, GO) | MSigDB v7.1 | https://www.gsea-msigdb.org/gsea/msigdb/genesets.jsp |
| TCMA tissue-resident profiles (COAD, READ, HNSC, ESCA, STAD) | This paper | https://doi.org/10.7924/r4rn36833 |
| 16S sequencing results | This paper | N/A |
| Oligonucleotides | ||
| Primers for 16S analysis FWD:GTGYCAGCMGCCGCGGTAA REV:GGACTACNVGGGTWTCTAAT |
IDT | N/A |
| Software and Algorithms | ||
| GATK 4.0.3 (PathSeq & HaplotypeCaller) | (Kostic et al., 2011; Poplin et al., 2017) | https://github.com/broadinstitute/gatk/ |
| SAMtools 1.9 | (Li et al., 2009) | http://samtools.sourceforge.net/ |
| phyloseq 1.30.0 | (McMurdie and Holmes, 2013) | https://github.com/joey711/phyloseq |
| metacoder 0.3.3 | (Foster et al., 2017) | https://grunwaldlab.github.io/metacoder_documentation/ |
| gffread 0.11.6 | (Pertea and Pertea, 2020) | https://github.com/gpertea/gffread |
| STAR 2.7.3a | (Dobin et al., 2013) | https://github.com/alexdobin/STAR/ |
| subread 1.6.4 | (Liao et al., 2014) | http://subread.sourceforge.net/ |
| deepTools 3.3.0 | (Ramírez et al., 2014) | https://github.com/deeptools/deepTools |
| bedtools 2.29.0 | (Quinlan and Hall, 2010) | https://github.com/arq5x/bedtools2 |
| circos 0.69.8 | (Krzywinski et al., 2009) | http://circos.ca/software/download/ |
| IGV 2.4.14 | (Thorvaldsdottir et al., 2013) | http://software.broadinstitute.org/software/igv/ |
| QIIME2 2019.7 | (Bolyen et al., 2019) | https://qiime2.org/ |
| SparCC | (Friedman and Alm, 2012) | https://bitbucket.org/yonatanf/sparcc/src/default/ |
| MicroPattern | (Ma et al., 2017) | http://www.cuilab.cn/micropattern |
| lifelines 0.23.8 | (Davidson-Pilon et al., 2020) | https://github.com/CamDavidsonPilon/lifelines/tree/0.24.6 |
| GSEA 4.0.3 | (Subramanian et al., 2007) | https://www.gsea-msigdb.org/gsea/ |
| Other | ||
| Patient metadata for TCGA validation samples | This paper | Table S1 |
| Tumor- and normal tissue-associated taxa | This paper | Table S2 |
Method details
Acquisition and metagenomic profiling of TCGA sequencing data
The raw TCGA bam files and metadata associated with each sequencing run were obtained from the NCI Genomic Data Commons (GDC) via the GDC’s application programming interface (API). Specifically, WXS data were accessed from the GDC data repository and WGS data were accessed from the GDC’s legacy archive. Overall, we acquired bam files from 19,409 sequencing runs (WGS: n = 4,608; WXS: n = 15,066) for all TCGA cancer types with WGS or WXS data available. Sample-specific metadata were obtained from the GDC web portal, and patient-specific metadata were obtained from the PanCanAtlas publication page.
All WGS and WXS data from TCGA samples were screened for microbial content using the PathSeq pipeline (Kostic et al., 2011), which is made available as part of the Broad Institute’s Genome Analysis Toolkit (GATK 4.0). The PathSeq analysis was performed using prebuilt human and microbial reference genomes and the NCBI taxonomy database from the PathSeq resource bundle, which were accessed via ftp from the Broad Institute in December 2017. PathSeq was used with default settings, with the exception of the minimum clipped read length, which was set to 50 to minimize the false positive rate. All sequencing data were analyzed on a local high-performance computing (HPC) cluster, which is comprised of 60 compute nodes, 1,512 CPU cores, and approximately 15TB of RAM.
Unambiguously aligned sequencing reads for bacteria at each taxonomic level were aggregated for available WGS and WXS data from 22 TCGA sequencing projects representing a total of 19,409 sequencing runs (4,608 WGS and 15,066 WXS). Total read counts for TCGA input bam files and PathSeq output bam files were calculated using SAMtools’ flagstats function for RPM normalization. Total bacterial abundance values were then normalized to the total read count (in millions) of the input bam files. Aggregated PathSeq results and associated metadata for each sequencing run were then deposited as phyloseq objects (McMurdie and Holmes, 2013) for downstream analyses in R.
Decomposition of observed TCGA microbial profiles into tissue-resident and contaminant fractions
The classification of tissue-resident microbiota for each TCGA project was performed at the species-level using WGS sequencing data. To assess whether a species deviated significantly from equiprevalence and identify a tissue-resident population, we found the most generalizable criteria combined a statistical test of proportions with a hard cutoff on blood prevalence. Species were defined as tissue-resident if they were prevalent in fewer than 20% of blood samples and significantly more prevalent in tissue than blood by a one-sided Fisher exact test (q < 0.05).
Fisher’s test offered two major benefits: (1) it performs well for low prevalence cases, meaning that it naturally removed low-prevalence species which could not be statistically distinguished from contamination, and (2) it is sensitive to the group sample sizes provided (tissue and blood), making it sufficiently generalizable across sequencing projects which had varying numbers of tissue and blood samples. Because of the very large total number of detectible species (n = 11,745 in CRC), we used FDR-correction to adjust for multiple tests (q < 0.05). The second filter was hard cutoff on blood prevalence (<20%). This was effective for classifying high-prevalence species, which were statistically more prevalent in tissue than blood but still detectible in more blood samples than was plausible for endogenous blood-borne bacteria. Ultimately, for CRC data the second cutoff was only relevant for four species from the Enterobacteriaceae family, which likely represent mixed-evidence species. Finally, we defined “detectible” as having more than one sequencing read aligning to a given taxon (≥2 reads). Singletons (taxa with a single read) are known to frequently be sequencing artifacts or false positives and are commonly removed to reduce noise in downstream metagenomic analyses.
Many reads are not aligned at the species level. For example, unambiguous genus-level alignments are not necessarily equal to the sum of unambiguous species-level alignments from species within that genus. Therefore, in order to preserve read counts at taxonomic ranks above species-level, we adjusted read counts to reflect that a given clade could be comprised of a combination of contaminant and tissue-resident species. The decomposition of observed metagenomic data (K) into tissue-resident (T) and contaminant (C) components for a given taxon in a given sample can be described using a mixture with two components of the form K = mT + nC, where m and n represent the estimated fractions of tissue-resident or contaminant sequencing reads belonging to a given taxon, respectively (such that m + n = 1). For all taxa above the species-level, we assigned m and n using the relative fractions of unambiguously aligned sequencing reads from species classified as tissue-resident or contaminant within the corresponding clade. For taxa with fewer than 5 unambiguously assigned reads, we imputed mixtures from other sequencing runs processed on the same plate or center. Defining these mixtures thus allowed us to propagate the classification of tissue-resident species to higher taxonomic ranks on a sample-by-sample basis while preserving read counts that were unambiguously aligned above the species level.
Gene-level sequencing analysis of representative species
For each of bacterial genomes of interest, the fasta sequences and gff3 files were downloaded from GenBank. The gff3 files were converted to gtf using GffRead (Pertea and Pertea, 2020). The fasta and gtf files were then analyzed with STAR (Dobin et al., 2013) genomeGenerate to make genome files for the alignment process. For each sequencing run, the output bam file from PathSeq was used to align to annotated bacterial genomes. Using subread’s featureCounts (Liao et al., 2014), the output from the STAR aligner was then used to determine the read counts for each gene. For the genome coverage analysis, outputs from STAR were sorted and converted to bam files using SAMtools (Li et al., 2009). Deeptools (Ramírez et al., 2014) bamCoverage function was then used to generate the bedgraph files using RPKM normalization. The files were intersected using bedtools (Quinlan and Hall, 2010). The log10 read counts of each of the samples were summed and divided by the total number of samples per track. These genome tracks were then plotted using the Circos software (Krzywinski et al., 2009). For visualization of cadA and ldcC alignments, counts from each sample and bin (10bp) were summed and divided by the total number of samples per track. The bedgraph files were converted to bigwig using UCSC bedGraphToBigWig, then bigwigs were plotted using The Integrated Genomics Viewer (IGV) (Thorvaldsdóttir et al., 2013). Each track was scaled to the max bin height within the viewable region.
Nucleotide-level analysis of bacterial sequence variants
For each bacterial genome in the PathSeq reference, we screened each BAM file from the PathSeq output (COAD-READ, WGS) for sequence variants using the GATK HaplotypeCaller pipeline (Poplin et al., 2017). Variant calling and quality filtering parameters were chosen according to previously described methodology for bacterial sequencing data (Bush et al., 2020). The output VCF files were then converted to TSV format and were aggregated across sequencing runs. We defined each sequencing variant as a unique combination of genome accession ID, nucleotide position, reference base, and alternative base, producing a total of 3,445,630 unique variants across all genomes and sequencing datasets. For downstream analysis, this total was further filtered to select 143,215 (4%) features that were present in at least three sequencing runs. Strain-level genome accession IDs were then mapped to NCBI taxonomy IDs and associated lineage using the ete3 python package, then aggregated by species and genus for comparative prevalence analysis.
Acquisition and analysis of original TCGA tissue and plasma samples
For validation of TCMA we obtained original, matched tissue and plasma samples from a total of five CRC patients from Indivumed, an original TCGA tissue provider. Plasma samples from three healthy subjects were obtained from patients at Duke University Hospital. For tissue samples, microbial DNA was extracted from tissue samples using the MoBio PowerMag Soil DNA isolation kit (Qiagen Cat# 27000-4-KF), following the Earth Microbiome Project (EMP) protocol (http://www.earthmicrobiome.org/) (Marotz et al., 2017). Microbial DNA was extracted from plasma using the QIAamp UCP Pathogen Mini Kit (Qiagen Cat# 50214) following a protocol developed by Jiang et. al (Jiang, 2018). Briefly, plasma samples were pre-treated with proteinase K, followed by lysing and spin-down through QIA amp UCP spin column. After washing with AW1 and AW2 buffers, microbial DNA was eluted in 50uL buffer AVE for downstream 16S library preparation and sequencing.
Bacterial compositions of isolated DNA samples were determined by amplification of the V4 variable region of the 16S rRNA gene by polymerase chain reaction using the forward primer 515 and reverse primer 806, following the EMP protocol. These primers were obtained from IDT and carry unique barcodes that allow for multiplexed sequencing. Equimolar 16S rRNA PCR products from all samples were quantified and pooled prior to sequencing. Sequencing was performed on a 250bp PE MiSeq lane at the Duke University Center for Genomic and Computational Biology sequencing core. The 16S sequencing results were analyzed using QIIME2 (Bolyen et al., 2019). Paired-end sequencing reads (250bp) were demultiplexed, denoised, and forward reads were trimmed at 10bp from the left and at 240bp on the right, while reverse reads were trimmed at 10bp from the left and at 220bp on the right. Taxonomic assignments were performed using the GreenGenes database with 99% OTUs at all taxonomic levels (DeSantis et al., 2006). Read counts for all observed taxa were summed over all assigned operational taxonomic units.
Estimation of bacterial co-abundance groups and associated molecular signatures
Compositional effects in microbiome data often complicate the calculation of correlations between microbiota; we therefore used SparCC (Friedman and Alm, 2012) to estimate taxa that are coabundant. This method relies on a bootstrapping procedure to control for spurious results common in microbiome survey data. Following the filtering criteria recommended in the SparCC paper, we removed samples with fewer than 500 reads and taxa with an average abundance of fewer than 2 reads per sample prior to calculating correlations. We ran SparCC with default parameters on decontaminated CRC tissue sequencing data for 100 iterations to identify coabundant taxa. The results of MicroPattern (Ma et al., 2017) pathway and disease-association enrichment analysis were obtained by identifying the top 20 genera most correlated with each of Fusobacterium and Bacteroides.
To estimate molecular signatures associated with these co-abundance groups, we collected batch-normalized molecular profiling data from the PanCanAtlas publication page, including RPPA, miRNA-seq, mRNA-seq, and Methylation 27K experiments performed on matched TCGA samples. Prior to calculating Pearson correlations between matched samples, we performed preliminary normalizations on both molecular profiling data and decontaminated tissue profiles. A log10 transform was used to ensure RNA-seq and miRNA-seq expression profiles were normally distributed. The RPPA and Methylation 27K data were left unchanged. The relative abundances of decontaminated CRC tissue profiles were normalized using pseudocaounts and a centered log-ratio (CLR) transform.
Identification of tumor- and normal tissue-associated microbiota
Microbes associated with tumor samples or matched normal tissue were calculated in R, using a custom paired analysis function written for metacoder (Foster et al., 2017). We filtered decontaminated microbial compositions in TCMA by selecting taxa using filtering criteria suggested by the PhyloSeq preprocessing tutorial. Such filters are standard when preparing for downstream metagenomic analyses as they remove low-abundance and low-prevalence taxa which frequently have small means and large coefficients of variation, contributing unnecessary noise for downstream differential abundance comparisons. After adding pseudocounts, we calculated the relative abundance of microbiota for each sequencing run. Across all patients with matched tumor and normal tissue, we then calculated the median log2 ratio between the relative abundance of each taxa in each tissue type. Significance values were calculated using Wilcoxon’s rank-sums test and corrected for false discovery rate. Taxa with significant p-values (p < 0.05) were selected for downstream analysis.
Survival analysis
We performed our survival analyses using a log-rank test, using the lifelines survival analysis python package (Davidson-Pilon et al., 2020). Relative abundances of decontaminated bacterial compositions for all tissue samples belonging to a given patient were used for both models. Data on patient survival, disease-free interval, and progression-free interval were collected from the PanCanAtlas’ clinical follow-up data (Liu et al., 2018). For log-rank tests, patients were segregated into two groups: one with taxa relative abundance below the bottom quartile (“low” or “absent”), and one with above the top quartile (“high”). In many cases, particularly for species-level alignments, the bottom quartile was zero and therefore may include more than a quarter of patients. To ensure quartiles were non-equal, taxa that were present in fewer than 25% of samples were excluded from the analysis. The CPH test was performed with default parameters and 10-fold cross-validation.
Pathway analysis of species associated with tumors or adjacent normal tissue
We used GSEA (Subramanian et al., 2005) to analyze gene expression pathways associated with species of interest. For each species, we defined a continuous phenotype (cls) using CLR-transformed abundance values of decontaminated TCMA data. Using RNA-seq expression data from the PanCanAtlas as the expression dataset and gene lists obtained from MSigDB v7.1 (KEGG, GO Biological Process, GO Molecular Function), we ran GSEA for 1000 iterations. Analysis was performed for 158 matched tumor samples, as well as for each subset with pathological stage (I: n = 33; II: n = 60; III: n = 44; IV: n = 19) within this cohort.
Quantification and statistical analysis
All statistical tests between unmatched groups were performed using a Wilcoxon rank-sums test (p-value), and all statistical tests between matched groups were performed using a Wilcoxon signed-rank test (p-value) unless otherwise specified. Statistical tests of prevalence were performed using a one-sided Fisher’s exact test. Statistical tests of variance for microbial compositions were performed using PERMANOVA. Statistical tests for survival analyses were performed using the log-rank test. For multiple tests, the false discovery rate (FDR; q-value) was calculated using the Benjamini-Hochberg method. All analyses were performed in python 3.7.1 and R 3.6.1. P-values are indicated as follows: *, <0.05; **, <0.01; ***, <0.001.
Supplementary Material
Highlights.
Decontaminated microbial compositions for 3,689 gastrointestinal cancer samples
Resolved “mixed-evidence” species with gene and nucleotide resolution
Identified prognostic species and blood signatures of mucosal barrier injury
Enabled matched multi-omic, pan-cancer analyses of host-microbe interactions
Acknowledgments
The authors would like to thank Mark Walker and Daniel Somers for technical guidance in deploying the PathSeq pipeline as well as Mark Turner for help with developing the TCMA website. We also thank Sayan Mukherjee, Lingchong You, Lawrence David, Justin Silverman, Kimberly Roche, and Darien Lombardi for constructive discussion and advice. We are grateful to Caroline Connor for reviewing and editing the manuscript. This study was supported by NIH R35GM122465, DK119795, and DARPA W911NF1920111.
Footnotes
Supplemental information
Supplemental Information can be found online at https://doi.org/10.1016/j.chom.2020.12.001.
Declaration of interests
The authors declare no competing interests.
References
- Abdulamir AS, Hafidh RR, and Abu Bakar F (2011). The association of Streptococcus bovis/gallolyticus with colorectal tumors: the nature and the underlying mechanisms of its etiological role. J. Exp. Clin. Cancer Res 30, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur JC, Perez-Chanona E, Mühlbauer M, Tomkovich S, Uronis JM, Fan TJ, Campbell BJ, Abujamel T, Dogan B, Rogers AB, et al. (2012). Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 338, 120–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol 37, 852–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brighenti E, Calabrese C, Liguori G, Giannone FA, Treré D, Montanaro L, and Derenzini M (2014). Interleukin 6 downregulates p53 expression and activity by stimulating ribosome biogenesis: a new pathway connecting inflammation to cancer. Oncogene 33, 4396–4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullman S, Lucid A, Corcoran D, Sleator RD, and Lucey B (2013). Genomic investigation into strain heterogeneity and pathogenic potential of the emerging gastrointestinal pathogen Campylobacter ureolyticus. PLoS One 8, e71515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullman S, Pedamallu CS, Sicinska E, Clancy TE, Zhang X, Cai D, Neuberg D, Huang K, Guevara F, Nelson T, et al. (2017). Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science 358, 1443–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush SJ, Foster D, Eyre DW, Clark EL, De Maio N, Shaw LP, Stoesser N, Peto TEA, Crook DW, and Walker AS (2020). Genomic diversity affects the accuracy of bacterial single-nucleotide polymorphism-calling pipelines. GigaScience 9, giaa007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network (2014). Comprehensive molecular characterization of gastric adenocarcinoma. Nature 513, 202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network; Albert Einstein College of Medicine; Analytical Biological Service; Barretos Cancer Hospital; Baylor College of Medicine; Beckman Research Institute of City of Hope; Buck Institute for Research on Aging; Canada’s Michael Smith Genome Sciences Center; Harvard Medical School; Helen F. Graham Cancer Center &Research Institute at Christiana Care Health Services, et al. (2017). Integrated genomic and molecular characterization of cervical cancer. Nature 543, 378–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CDC (2019). NHSN organism list. In National Healthcare Safety Network (NHSN) patient safety component manual (Centers for Disease Control and Prevention).
- Chelakkot C, Ghim J, and Ryu SH (2018). Mechanisms regulating intestinal barrier integrity and its pathological implications. Exp. Mol. Med 50, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JH, Hong SE, and Woo HG (2017). Pan-cancer analysis of systematic batch effects on somatic sequence variations. BMC Bioinformatics 18, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung H, Calis JJA, Wu X, Sun T, Yu Y, Sarbanes SL, Dao Thi VL, Shilvock AR, Hoffmann HH, Rosenberg BR, and Rice CM (2018). Human ADAR1 prevents endogenous RNA from triggering translational shutdown. Cell 172, 811–824.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson-Pilon C, Kalderstam J, Jacobson N, Zivich P, Kuhn B, Williamson M, Sean-Reed JK, Fiore-Gartland A, Datta D, and Moneda L (2020). CamDavidsonPilon/lifelines: 0.24.6 (Zenodo).
- Davis NM, Proctor DM, Holmes SP, Relman DA, and Callahan BJ (2018). Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 6, 226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, and Andersen GL (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol 72, 5069–5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donohoe DR, Garge N, Zhang X, Sun W, O’Connell TM, Bunger MK, and Bultman SJ (2011). The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab 13, 517–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhofer R, Minich JJ, Marotz C, Cooper A, Knight R, and Weyrich LS (2019). Contamination in low microbial biomass microbiome studies: issues and recommendations. Trends Microbiol 27, 105–117. [DOI] [PubMed] [Google Scholar]
- Elinav E, Garrett WS, Trinchieri G, and Wargo J (2019). The cancer microbiome. Nat. Rev. Cancer 19, 371–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan L, Schmid J, Ebert M, Soucek P, Kunicka T, Liska V, Bruha J, Neary P, Dezeeuw N, Tommasino M, et al. (2014). Fusobacterium nucleatum associates with stages of colorectal neoplasia development, colorectal cancer and disease outcome. Eur. J. Clin. Microbiol. Infect. Dis 33, 1381–1390. [DOI] [PubMed] [Google Scholar]
- Foster ZS, Sharpton TJ, and Grünwald NJ (2017). Metacoder: an R package for visualization and manipulation of community taxonomic diversity data. PLoS Comput. Biol 13, e1005404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman J, and Alm EJ (2012). Inferring correlation networks from genomic survey data. PLOS Comput. Biol 8, e1002687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, Nakanishi Y, Uetake C, Kato K, Kato T, et al. (2013). Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504, 446–450. [DOI] [PubMed] [Google Scholar]
- Gemmell E, and Seymour GJ (1993). Interleukin 1, interleukin 6 and transforming growth factor-beta production by human gingival mononuclear cells following stimulation with Porphyromonas gingivalis and Fusobacterium nucleatum. J. Periodont. Res 28, 122–129. [DOI] [PubMed] [Google Scholar]
- Glassing A, Dowd SE, Galandiuk S, Davis B, and Chiodini RJ (2016). Inherent bacterial DNA contamination of extraction and sequencing reagents may affect interpretation of microbiota in low bacterial biomass samples. Gut Pathog 8, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, Prieto PA, Vicente D, Hoffman K, Wei SC, et al. (2018). Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 359, 97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grice EA, Kong HH, Renaud G, Young AC, NISC Comparative Sequencing Program, Bouffard GG, Blakesley RW, Wolfsberg TG, Turner ML, and Segre JA (2008). A diversity profile of the human skin microbiota. Genome Res 18, 1043–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W., Sherman BT, and Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Human Microbiome Project Consortium (2012). Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliev ID, and Leonardi I (2017). Fungal dysbiosis: immunity and interactions at mucosal barriers. Nat. Rev. Immunol 17, 635–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islami F, and Kamangar F (2008). Helicobacter pylori and esophageal cancer risk: a meta-analysis. Cancer Prev. Res. (Phila) 1, 329–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W (2018). A protocol for quantizing total bacterial 16S rDNA in plasma as a marker of microbial translocation in vivo. Cell. Mol. Immunol 15, 937–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi Y, Kojima H, Tanaka T, Takatsuka Y, and Kamio Y (1997). Characterization of a second lysine decarboxylase isolated from Escherichia coli. J. Bacteriol 179, 4486–4492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koskiniemi S, Sun S, Berg OG, and Andersson DI (2012). Selection-driven gene loss in bacteria. PLoS Genet 8, e1002787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, Michaud M, Clancy TE, Chung DC, Lochhead P, Hold GL, et al. (2013). Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 14, 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostic AD, Gevers D, Pedamallu CS, Michaud M, Duke F, Earl AM, Ojesina AI, Jung J, Bass AJ, Tabernero J, et al. (2012). Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res 22, 292–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostic AD, Ojesina AI, Pedamallu CS, Jung J, Verhaak RG, Getz G, and Meyerson M (2011). PathSeq: software to identify or discover microbes by deep sequencing of human tissue. Nat. Biotechnol 29, 393–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, and Marra MA (2009). Circos: an information aesthetic for comparative genomics. Genome Res 19, 1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemonnier M, and Lane D (1998). Expression of the second lysine decarboxylase gene of Escherichia coli. Microbiology 144, 751–760. [DOI] [PubMed] [Google Scholar]
- Levy M, Kolodziejczyk AA, Thaiss CA, and Elinav E (2017). Dysbiosis and the immune system. Nat. Rev. Immunol 17, 219–232. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, Kovatich AJ, Benz CC, Levine DA, Lee AV, et al. (2018). An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell 173, 400–416.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luckey TD (1972). Introduction to intestinal microecology. Am. J. Clin. Nutr 25, 1292–1294. [DOI] [PubMed] [Google Scholar]
- Ma W, Huang C, Zhou Y, Li J, and Cui Q (2017). Micropattern: a web-based tool for microbe set enrichment analysis and disease similarity calculation based on a list of microbes. Sci. Rep 7, 40200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marotz C, Amir A, Humphrey G, Gaffney J, Gogul G, and Knight R (2017). DNA extraction for streamlined metagenomics of diverse environmental samples. BioTechniques 62, 290–293. [DOI] [PubMed] [Google Scholar]
- McMurdie PJ, and Holmes S (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8, e61217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mira A, Ochman H, and Moran NA (2001). Deletional bias and the evolution of bacterial genomes. Trends Genet 17, 589–596. [DOI] [PubMed] [Google Scholar]
- Nejman D, Livyatan I, Fuks G, Gavert N, Zwang Y, Geller LT, Rotter-Maskowitz A, Weiser R, Mallel G, Gigi E, et al. (2020). The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 368, 973–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nossa CW, Jain P, Tamilselvam B, Gupta VR, Chen LF, Schreiber V, Desnoyers S, and Blanke SR (2009). Activation of the abundant nuclear factor poly(ADP-ribose) polymerase-1 by Helicobacter pylori. Proc. Natl. Acad. Sci. USA 106, 19998–20003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donovan D, Corcoran GD, Lucey B, and Sleator RD (2014). Campylobacter ureolyticus: a portrait of the pathogen. Virulence 5, 498–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima T, and Miwa H (2016). Gastrointestinal mucosal barrier function and diseases. J. Gastroenterol 51, 768–778. [DOI] [PubMed] [Google Scholar]
- Pertea G, and Pertea M (2020). GFF Utilities: GffRead and GffCompare [version 1; peer review: 3 approved. F1000Res 9, 304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plottel CS, and Blaser MJ (2011). Microbiome and malignancy. Cell Host Microbe 10, 324–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poore GD, Kopylova E, Zhu Q, Carpenter C, Fraraccio S, Wandro S, Kosciolek T, Janssen S, Metcalf J, Song SJ, et al. (2020). Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature 579, 567–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poplin R, Ruano-Rubio V, DePristo MA, Fennell TJ, Carneiro MO, Van der Auwera GA, Kling DE, Gauthier LD, Levy-Moonshine A, Roazen D, et al. (2017). Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv 10.1101/201178v3. [DOI]
- Porcheron G, Garénaux A, Proulx J, Sabri M, and Dozois CM (2013). Iron, copper, zinc, and manganese transport and regulation in pathogenic Enterobacteria: correlations between strains, site of infection and the relative importance of the different metal transport systems for virulence. Front. Cell. Infect. Microbiol 3, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prast-Nielsen S, Tobin AM, Adamzik K, Powles A, Hugerth LW, Sweeney C, Kirby B, Engstrand L, and Fry L (2019). Investigation of the skin microbiome: swabs vs. biopsies. Br. J. Dermatol 181, 572–579. [DOI] [PubMed] [Google Scholar]
- Purcell RV, Visnovska M, Biggs PJ, Schmeier S, and Frizelle FA (2017). Distinct gut microbiome patterns associate with consensus molecular sub-types of colorectal cancer. Sci. Rep 7, 11590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. (2010). A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Dündar F, Diehl S, Grüning BA, and Manke T (2014). deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res 42, W187–W191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rensing C, and Grass G (2003). Escherichia coli mechanisms of copper homeostasis in a changing environment. FEMS Microbiol. Rev 27, 197–213. [DOI] [PubMed] [Google Scholar]
- Robinson KM, Crabtree J, Mattick JS, Anderson KE, and Dunning Hotopp JC (2017). Distinguishing potential bacteria-tumor associations from contamination in a secondary data analysis of public cancer genome sequence data. Microbiome 5, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, and Han YW (2013). Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/beta-catenin signaling via its FadA adhesin. Cell Host Microbe 14, 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirmer M, Garner A, Vlamakis H, and Xavier RJ (2019). Microbial genes and pathways in inflammatory bowel disease. Nat. Rev. Microbiol 17, 497–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sender R, Fuchs S, and Milo R (2016). Revised estimates for the number of human and bacteria cells in the body. PLoS Biol 14, e1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, Benyamin FW, Lei YM, Jabri B, Alegre M-L, et al. (2015). Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 350, 1084–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriswasdi S, Yang CC, and Iwasaki W (2017). Generalist species drive microbial dispersion and evolution. Nat. Commun 8, 1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang KW, Alaei-Mahabadi B, Samuelsson T, Lindh M, and Larsson E (2013). The landscape of viral expression and host gene fusion and adaptation in human cancer. Nat. Commun 4, 2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdóttir H., Robinson JT, and Mesirov JP (2013). Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform 14, 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillére R, Hannani D, Enot DP, Pfirschke C, Engblom C, Pittet MJ, et al. (2013). The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science 342, 971–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren RL, Freeman DJ, Pleasance S, Watson P, Moore RA, Cochrane K, Allen-Vercoe E, and Holt RA (2013). Co-occurrence of anaerobic bacteria in colorectal carcinomas. Microbiome 1, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood DE, Lu J, and Langmead B (2019). Improved metagenomic analysis with Kraken 2. Genome Biol 20, 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y, Miwa Y, Miyoshi K, Furuyama J. i., and Ohmori H (1997). The Escherichia coli ldcC gene encodes another lysine decarboxylase, probably a constitutive enzyme. Genes Genet. Syst 72, 167–172. [DOI] [PubMed] [Google Scholar]
- Yang JH, Luo X, Nie Y, Su Y, Zhao Q, Kabir K, Zhang D, and Rabinovici R (2003). Widespread inosine-containing mRNA in lymphocytes regulated by ADAR1 in response to inflammation. Immunology 109, 15–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu LC (2018). Microbiota dysbiosis and barrier dysfunction in inflammatory bowel disease and colorectal cancers: exploring a common ground hypothesis. J. Biomed. Sci 25, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T, Guo F, Yu Y, Sun T, Ma D, Han J, Qian Y, Kryczek I, Sun D, Nagarsheth N, et al. (2017). Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Cell 170, 548–563.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The TCMA database can be accessed via the website (https://tcma.pratt.duke.edu). The accession number for the data reported in this paper is https://doi.org/10.7924/r4rn36833. The TCGA sequencing data and associated aliquot, sample, and patient metadata on which this work was based were accessed from the GDC API. Molecular profiling data and clinical endpoints used for the survival analysis were obtained from the PanCanAtlas publication page. Scripts used for this work are available on request. More detailed information about data and code can be found in the Key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological Samples | ||
| Original TCGA tissue and plasma samples | Indivumed | N/A |
| Healthy plasma samples | Duke Hospital | N/A |
| Critical Commercial Assays | ||
| MagAttract PowerSoil DNA KF Kit | Qiagen | Cat# 27000-4-KF |
| QIAamp UCP Pathogen Mini Kit | Qiagen | Cat# 50214 |
| Deposited Data | ||
| TCGA WGS bam files | GDC API | https://api.gdc.cancer.gov/ |
| TCGA WXS bam files | GDC API | https://api.gdc.cancer.gov/ |
| TCGA sequencing metadata | GDC API | https://api.gdc.cancer.gov/ |
| TCGA sample metadata (biotab) | GDC web portal | https://portal.gdc.cancer.gov/ |
| TCGA patient metadata | PanCanAtlas | https://gdc.cancer.gov/about-data/publications/pancanatlas |
| TCGA clinical data resource outcomes | PanCanAtlas | https://gdc.cancer.gov/about-data/publications/pancanatlas |
| Human and microbe reference genomes | PathSeq bundle | ftp://gsapubftp-anonymous@ftp.broadinstitute.org/bundle/pathseq/ |
| Human and microbe reference genomes | PathSeq bundle | ftp://gsapubftp-anonymous@ftp.broadinstitute.org/bundle/pathseq/ |
| Species and genera designated as commensal or MBI-associated (NHSN Organism List) | (CDC-NHSN, 2019) | https://www.cdc.gov/nhsn/pdfs/pscmanual/pcsmanual_current.pdf (Chapter 4 p.33) |
| Genera designated as common contaminants | (Eisenhofer et al., 2019) | Table 1 |
| Genome for B. vulgatus | GenBank | CP000139.1 |
| Genome for A. junii | GenBank | NZ_CP019041.1 |
| Genome for E. coli | GenBank | U00096.3 |
| Genome for C. provencense | GenBank | NZ_CP024988.1 |
| Genome for F. nucleatum | GenBank | AE009951.2 |
| Pks gene cluster (colibactin) | GenBank | AM229678.1 |
| Greengenes classifier (gg-13-8-99-515-806-nb-classifier.qza) | (DeSantis et al., 2006) | https://docs.qiime2.org/ |
| TCGA mRNA-seq data | PanCanAtlas | https://gdc.cancer.gov/about-data/publications/pancanatlas |
| TCGA miRNA-seq data | PanCanAtlas | https://gdc.cancer.gov/about-data/publications/pancanatlas |
| TCGA RPPA data | PanCanAtlas | https://gdc.cancer.gov/about-data/publications/pancanatlas |
| TCGA DNA Methylation 27K data | PanCanAtlas | https://gdc.cancer.gov/about-data/publications/pancanatlas |
| PARADIGM Pathway inference matrix | PanCanAtlas | https://gdc.cancer.gov/about-data/publications/pancanatlas |
| Gene sets for GSEA (KEGG, GO) | MSigDB v7.1 | https://www.gsea-msigdb.org/gsea/msigdb/genesets.jsp |
| TCMA tissue-resident profiles (COAD, READ, HNSC, ESCA, STAD) | This paper | https://doi.org/10.7924/r4rn36833 |
| 16S sequencing results | This paper | N/A |
| Oligonucleotides | ||
| Primers for 16S analysis FWD:GTGYCAGCMGCCGCGGTAA REV:GGACTACNVGGGTWTCTAAT |
IDT | N/A |
| Software and Algorithms | ||
| GATK 4.0.3 (PathSeq & HaplotypeCaller) | (Kostic et al., 2011; Poplin et al., 2017) | https://github.com/broadinstitute/gatk/ |
| SAMtools 1.9 | (Li et al., 2009) | http://samtools.sourceforge.net/ |
| phyloseq 1.30.0 | (McMurdie and Holmes, 2013) | https://github.com/joey711/phyloseq |
| metacoder 0.3.3 | (Foster et al., 2017) | https://grunwaldlab.github.io/metacoder_documentation/ |
| gffread 0.11.6 | (Pertea and Pertea, 2020) | https://github.com/gpertea/gffread |
| STAR 2.7.3a | (Dobin et al., 2013) | https://github.com/alexdobin/STAR/ |
| subread 1.6.4 | (Liao et al., 2014) | http://subread.sourceforge.net/ |
| deepTools 3.3.0 | (Ramírez et al., 2014) | https://github.com/deeptools/deepTools |
| bedtools 2.29.0 | (Quinlan and Hall, 2010) | https://github.com/arq5x/bedtools2 |
| circos 0.69.8 | (Krzywinski et al., 2009) | http://circos.ca/software/download/ |
| IGV 2.4.14 | (Thorvaldsdottir et al., 2013) | http://software.broadinstitute.org/software/igv/ |
| QIIME2 2019.7 | (Bolyen et al., 2019) | https://qiime2.org/ |
| SparCC | (Friedman and Alm, 2012) | https://bitbucket.org/yonatanf/sparcc/src/default/ |
| MicroPattern | (Ma et al., 2017) | http://www.cuilab.cn/micropattern |
| lifelines 0.23.8 | (Davidson-Pilon et al., 2020) | https://github.com/CamDavidsonPilon/lifelines/tree/0.24.6 |
| GSEA 4.0.3 | (Subramanian et al., 2007) | https://www.gsea-msigdb.org/gsea/ |
| Other | ||
| Patient metadata for TCGA validation samples | This paper | Table S1 |
| Tumor- and normal tissue-associated taxa | This paper | Table S2 |