

Summary
Tuberculosis (TB), caused by Mycobacterium tuberculosis (Mtb) latently infects approximately one-fourth of the world’s population. The immune mechanisms that govern progression from latent (LTBI) to active pulmonary TB (PTB) remain poorly defined. Experimentally Mtb-infected non-human primates (NHP) mirror the disease observed in humans and recapitulate both PTB and LTBI. We characterized the lung immune landscape in NHPs with LTBI and PTB using high-throughput technologies. Three defining features of PTB in macaque lungs include the influx of plasmacytoid dendritic cells (pDCs), an Interferon (IFN)-responsive macrophage population, and activated T cell responses. In contrast, a CD27+ Natural killer (NK) cell subset accumulated in the lungs of LTBI macaques. This NK cell population was also detected in the circulation of LTBI individuals. This comprehensive analysis of the lung immune landscape will improve the understanding of TB immunopathogenesis, providing potential targets for therapies and vaccines for TB control.
Keywords: tuberculosis, lung, immune protection, granulomas, single cell technologies, NK cells, pDCs, type I IFNs
Graphical Abstract

eTOC Blurb
Esaulova, Das, Singh et al. characterize the lung landscape during tuberculosis disease and control using single cell and conventional technologies. Their results delineate NK cells as a feature of protection during latency, and the presence of pDCs, IFN-responsive macrophages and activated T cells as a feature of disease.
Introduction
Tuberculosis (TB), caused by the bacterium Mycobacterium tuberculosis (Mtb), infects approximately one-fourth of the world’s population and kills approximately 1.5 million individuals each year ((WHO), 2018). Although the majority of infected persons are asymptomatic, latent TB infection (LTBI) can revert to active disease in 5–10% of infected individuals. The immune mechanisms that govern progression from latent to active pulmonary TB (PTB) remain poorly defined. A clear understanding of immune factors correlating with disease progression, as well as protection during TB is necessary for developing new immunotherapies to promote immune control of Mtb. Nonhuman primates (NHP) are a pre-clinical model for studying TB disease where granulomas in NHPs with TB disease mirror the morphology and physiology observed in human TB disease (Bucsan et al., 2019b). This model involves exposing Indian rhesus macaques to Mtb aerosols, which mimics the natural mode of exposure (Mehra et al., 2012). Based on the dose of Mtb infection, this model can recapitulate features of both PTB and LTBI (Gopal et al., 2013). Indeed, we showed that immune correlates of disease in the lungs of NHP with PTB overlap significantly with the immune correlates observed in the blood of human TB progressors (Ahmed et al., 2020). However, at the current time, the events occurring in the lung at the single-cell level in hosts that control disease or progress to TB disease are unknown. The use of single-cell technologies including RNA-sequencing (scRNA-seq) allows in-depth and unbiased profiling of immune cell populations in animal models and humans in both healthy and diseased states (Gubin et al., 2018; Jaitin et al., 2015; Szabo et al., 2019). Because scRNA-seq can define the transcriptomic heterogeneity of a complex community of cells and assign unbiased identity classifications to cell populations, it is well-suited for the study of complex inflammatory diseases such as TB. Therefore, in the present study, we have probed the immune landscape in NHPs with LTBI and PTB using high-throughput single-cell technologies including scRNA-seq and Time of flight cytometry (CyTOF), and validated our findings with conventional techniques such as flow cytometry and immunofluorescence (IF). Our discoveries about the immune landscape of TB latency and disease show the presence of distinct myeloid and lymphoid populations, including the accumulation of plasmacytoid DCs (pDCs), an Interferon (IFN)-responsive macrophage population, and activated T cell responses in the lungs of macaques and humans with PTB, contributing to the inflammation and disease without mediating substantial protection. Importantly, macaques and individuals with LTBI exhibited increased lung and circulating NK cells respectively, upregulated CD27 expression with mycobacterial stimulation, and expressed cytolytic effectors, including granzyme and perforin. Together, our study characterizes the lung landscape associated with control or disease progression during TB and significantly improves our overall understanding of Mtb infection and TB disease immunopathogenesis.
Results
Establishment of TB disease spectrum in rhesus macaques.
To interrogate the immune lung landscape of rhesus macaques with PTB and TB latency, we included rhesus macaques that were left uninfected (controls), or macaques with PTB disease as well as macaques with LTBI (Fig 1A). As shown previously (Ahmed et al., 2020; Gopal et al., 2013; Slight et al., 2013), latent infection is established in the majority of rhesus macaques upon delivery of 10 CFU of Mtb strain CDC1551, and PTB disease is uniformly established upon delivery of higher doses of Mtb CDC1551 (~200–1000 CFU) by the aerosol route. While all Mtb-infected macaques demonstrated tuberculin test (PPD) positivity after Mtb infection (data not shown), macaques with PTB but not LTBI showed clinical symptoms of TB disease including weight loss, increased serum CRP protein levels and associated lung TB pathology (Fig 1B). Additionally, macaques with PTB had increased lung inflammation and formed large granulomas, while LTBI macaques had decreased inflammation and formed significantly smaller granulomas (Fig S1A, B). As previously shown (Slight et al., 2013), macaques with LTBI demonstrated well-formed and large B cell lymphoid follicles within inducible Bronchus associated tissue (iBALT) located near the airway, within which CD3+ T cells localized effectively (Fig S1C, D). In contrast, macaques with PTB had significantly smaller B cell lymphoid follicles localized around the rims of granulomas (Granuloma associated lymphoid tissue, GrALT) (Fig S1C, D), with CD3+ T cells that surrounded the GrALT but not effectively infiltrating it. Macaques with PTB were sacrificed upon reaching pre-specified community-established endpoints (see STAR Methods), while macaques with LTBI were sacrificed at 5–6 months post-infection. Lung biopsies and corresponding peripheral blood mononuclear cells (PBMCs) were collected at the time of sacrifice and processed as below (Table S1).
Figure 1. Study outline of rhesus macaques with LTBI and PTB.
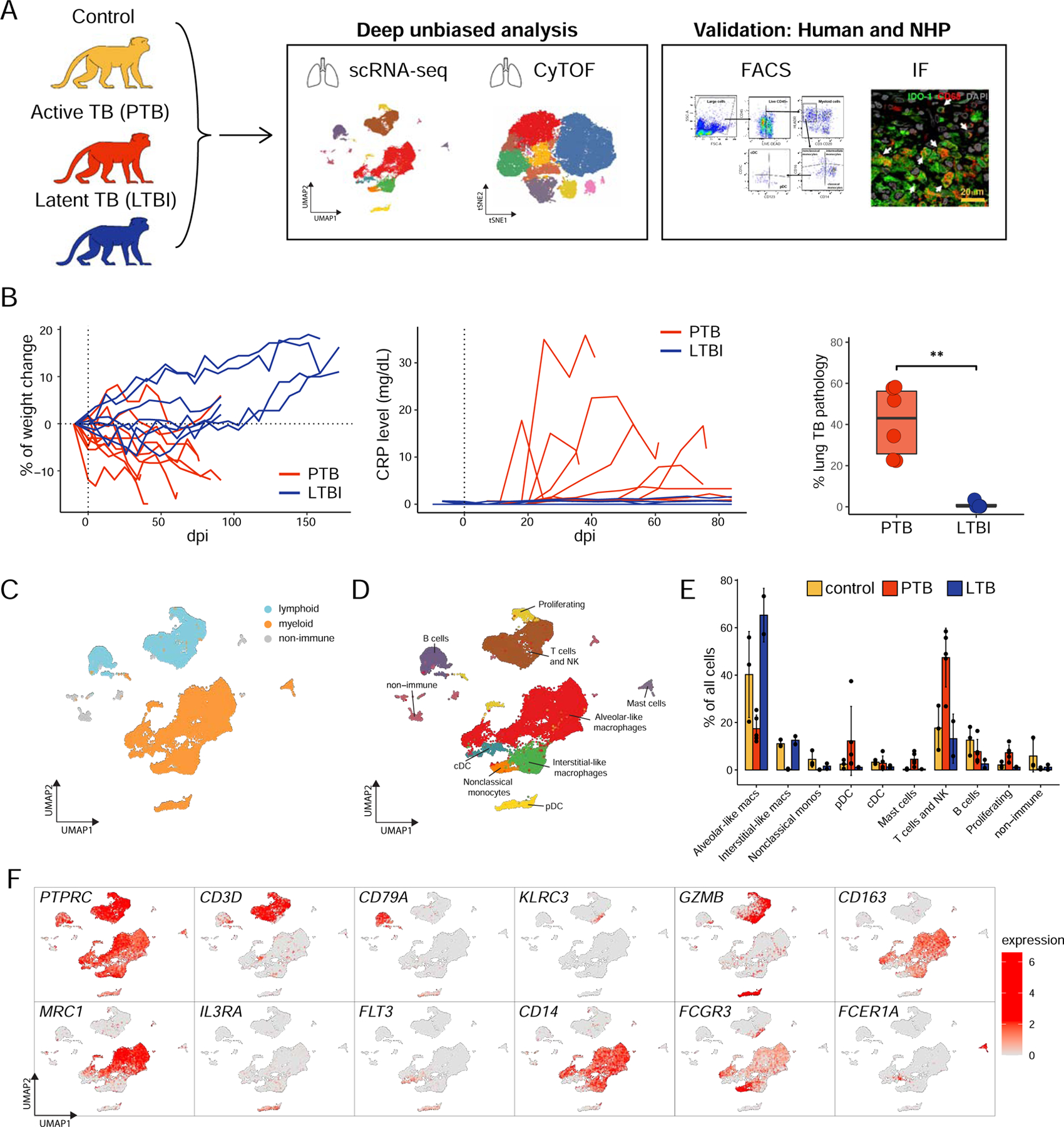
A. Single cell suspensions of lung biopsy from uninfected rhesus macaques (control), macaques with PTB at terminal end-point, or LTBI at 6 months were subjected to scRNA-seq or CyTOF analysis as described in STAR methods.
B. Clinical correlates of infection including changes in the percentage of body weight, changes in serum CRP (mg/dL) levels, and post-necropsy percent of lung pathology in Mtb-infected LTBI and PTB macaques. Data represented as mean ± SD, **p < 0.01, unpaired Student’s T-test.
C-D. UMAP plots of cells from all scRNA-seq samples together, colored according to (C) lymphoid/myeloid classification or (D) cell types. Control, n=3; PTB, n=5; LTBI, n=2.
E. Proportion of each cell type across samples.
F. UMAP plots with the expression of markers, characterizing main immune populations. See also Fig S1, Table S1 and Table S2.
Investigating the landscape of immune cells in the lung during TB disease and latency at the single cell level.
To delineate the lung landscape at the single-cell level, we first performed 3’ 10X scRNA-seq on dematricized single-cells that underwent CD45+ enrichment from the lungs of uninfected (control, n=3), PTB (n=5), and LTBI (n=2) macaques. Sequencing yielded from 300 to 7400 cells per sample (Table S2, STAR Methods). The mean number of cells per sample in control (5002) and LTBI (5381) samples were higher than in PTB samples (1437). The majority of the cells were immune cells due to the enrichment of CD45+ cells. Both lymphoid and myeloid cell populations were present in considerable proportions across all conditions (Fig 1C, Fig S1E). We identified 8 major cell types based on canonical gene expression: T cells (CD3D+), NK cells (KLRC3+GZMB+), B cells (CD79A+), macrophages (CD163+), cDCs and pDCs (FLT3+), non-classical monocytes (CD14−CD16+) and mast cells (FCER1A+) (Fig 1D–F). Macrophages and T cells were consistently the most abundant cell types across all groups.
Lymphoid cells.
We first focused on 10,676 lymphoid cells across all three conditions. Re-clustering yielded 12 subpopulations: 8 clusters of T cells, an NK cell cluster, B cell cluster, and a plasmablast cell cluster (Fig 2A–B), differently impacted in PTB and LTBI macaques (Fig 2C–D, Fig S2A, E). Based on the enrichment of corresponding transcriptional signatures, T cell populations were defined as naïve, effector memory, regulatory, activated, and proliferating subpopulations (Fig 2A–B). Major differences in lung T cells were observed between control and Mtb-infected macaques, especially related to its T cell activation state. T cells from control and LTBI macaques were predominantly represented by naïve CD4+/CD8+ T cells and effector memory CD4+/CD8+ T cells, whereas PTB lungs were enriched in activated CD4+/CD8+ T cells. Specifically, activated T cells expressed high levels of IFNG, CD69, LAG3, granzymes and were enriched in the activation (LAG3, ZBED2, CCL5, ICOS), Th1 (TBX21, GNLY, TNF, BHLHE40) as well as Th17 (RORC, CCR6, STAT3, IL23) gene signatures (Fig 2C–D, Fig S2C). Interestingly, naïve T cells were separated into two clusters, one of which was present in control and LTBI lungs, and the other was present in the PTB lungs. Both clusters expressed IL7R, TCF7, and LEF1 genes and had a low expression of activation markers. Naïve T cells from PTB were marked by the expression of heat shock proteins, including HSP1A and DNAJB1 (Fig. 2C). Additionally, the naïve T cells from the PTB condition are enriched with activation genes, such as NKG7, GZMB, S100A4, GZMK, CCL5, KLRD1, GZMA, GZMH, NKG2D, S100A11, IKZF3, TNF (Fig S2D), although not as uniformly enriched as in the activated CD4/CD8+ T cells. These results demonstrate the presence of activated T cell responses as a distinct feature of TB disease.
Figure 2. Lung lymphoid cell dynamics in macaques with LTBI and PTB by scRNA-seq describe the presence of activated T cells in PTB.
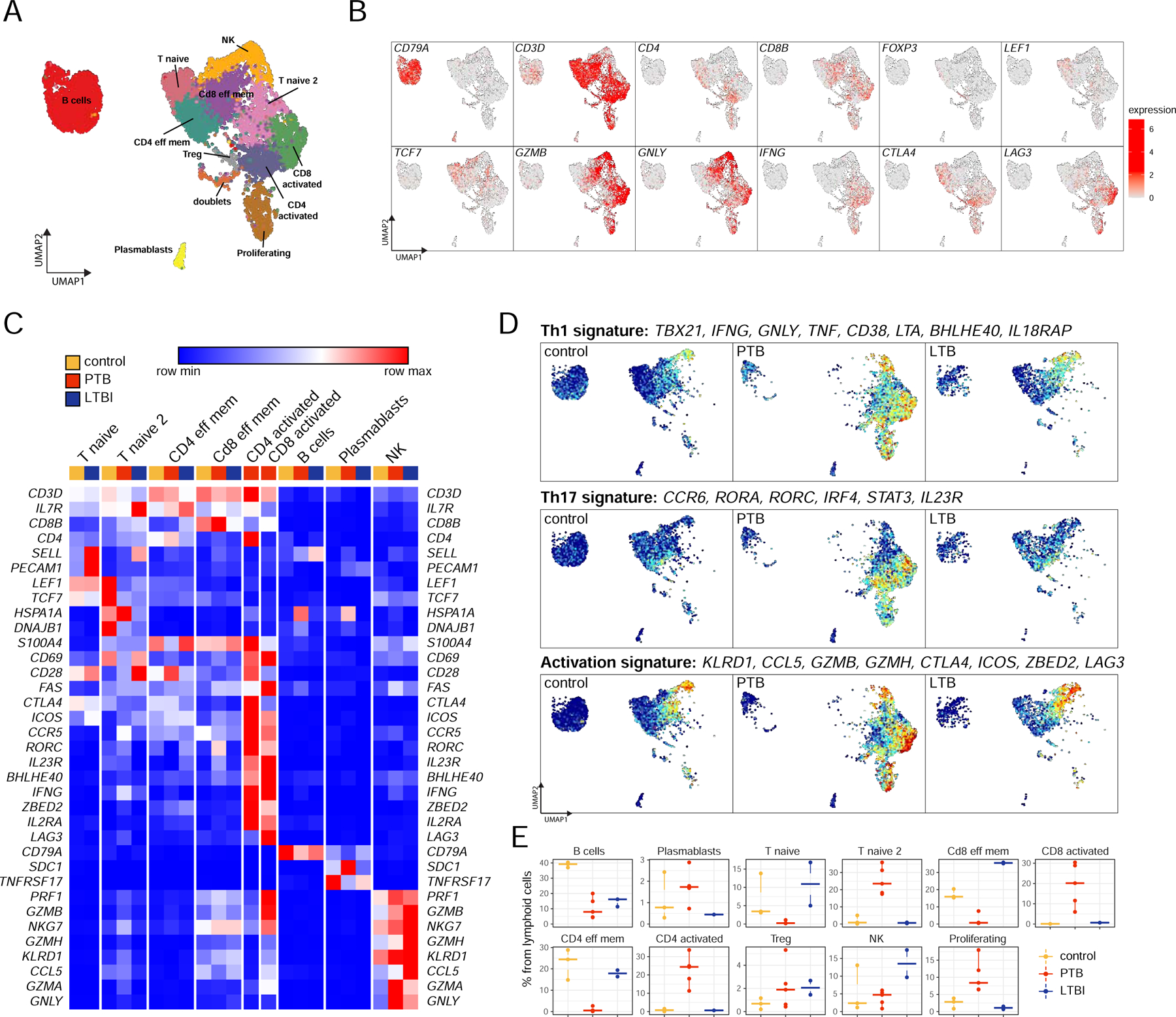
Control, n=3; PTB, n=5; LTBI, n=2.
A. UMAP plot of lymphoid cells across all samples, colored according to identified clusters.
B. UMAP plots with the expression of markers, characterizing main lymphoid populations.
C. Heatmap of normalized expression of selected genes in each lymphoid cluster per condition. Only sample-cluster combinations that contain more than 2% of cells from the corresponding sample are included.
D. UMAP plots of mean gene expression from Th1, Th17, and activation signatures, split by condition.
E. Cell proportion of each cluster per condition per sample.
See also Fig S2.
B cells were the largest represented lymphoid population in uninfected lungs (~40% of lymphoid cells), and the numbers decreased to 5–20% in LTBI and PTB lungs (Fig 2E). Among innate lymphoid cell populations, cytotoxic granzyme B (GZMB)+ perforin (PRF1)+ NK cells were increased in LTBI macaque lungs, reaching ~10–17% of total lymphoid cells (Fig 2E). They expressed classic NK genes associated with cytotoxicity such as granulysin (GNLY), PRF1, and GZM (A, B, K, and H) (Fig 2C). The majority of NK cells were CD16+ (FCGR3 gene). These results project a key protective role for NK-cell mediated cytotoxicity during TB latency in the lung compartment.
Myeloid cells.
We identified 20, 212 myeloid cells across all conditions (Fig 3A). We noted significant remodeling of all myeloid populations that differed between lungs with PTB and LTBI (Fig 3A, D, Fig S3A–C). We did not detect neutrophils by scRNA-seq, possibly due to the limitations associated with the 10X technology. As shown previously (Cai et al., 2014), the majority of myeloid cells in the lungs of healthy macaques were macrophages, either alveolar-like (AM) or interstitial-like (IM) (Fig 3A, B). In macaques, using expression of CD163 and MRC1 (CD206) genes (Cai et al., 2014) three populations can be classified, which expressed distinct transcriptional profiles that are previously not described for macaque macrophages (Fig 3C). To accommodate macrophage diversity, we refer to these populations by their prominently expressed markers rather than by conventional AM or IM classification. The first population referred to as “CD163+MRC1+ macrophages”, AM-like population, exhibited other classic markers of AMs, such as MARCO, MERTK, and APOE (Fig 3C) and constituted 20–80% of myeloid cells in lungs of control and LTBI macaques, while this population is almost absent from PTB macaque lungs. The second cluster of AM-like macrophages present in the lungs of control and LTBI macaques expressed CD163+MRC1+ but also had high expression of TREM2, C1Q genes, and TREM176A/B (Fig 3C). The “CD163+MRC1+TREM2+ macrophage” population was well represented in the lungs of healthy macaques and not previously described. A third population named “IFN-responsive macrophages” was CD163+MRC1low and exclusively present in the lungs of macaques with PTB, incorporating 15–60% of myeloid cells in this group (Fig 3D). This macrophage subpopulation exhibited a strong signature of response to type 1 and 2 IFNs (Fig 3E), high expression of the IFN-dependent chemokines, CXCL9–11 (Marshall et al., 2017) (Fig 3C), and expressed S100A8 and S100A9, proinflammatory proteins produced in the serum of PTB patients (Gopal et al., 2013; Scott et al., 2020). This macrophage population also expressed IDO1, which is produced within the macaque PTB granulomas (Gautam et al., 2018; Mehra et al., 2013). These results were consistent with increased protein expression of CXCL9–10, CXCL-1 (Gro-alpha) and CCL-4 [Macrophage inflammatory protein-1b (MIP-1β)] in bronchoalveolar lavage (BAL) of PTB macaques when compared with levels in BAL of LTBI macaques (Fig S3D). Thus, during TB disease, macrophages are key IFN-responders and express suppressive molecules such as IDO1. IM-like CD163+MRC1− macrophages constituted 15–25% of myeloid cells in the lungs of control and LTBI animals and were significantly decreased in macaques with PTB (Fig 3D).
Figure 3. Lung myeloid cell dynamics in macaques with LTBI and PTB by scRNA-seq describe the presence of pDCs and IFN-responsive macrophage population.
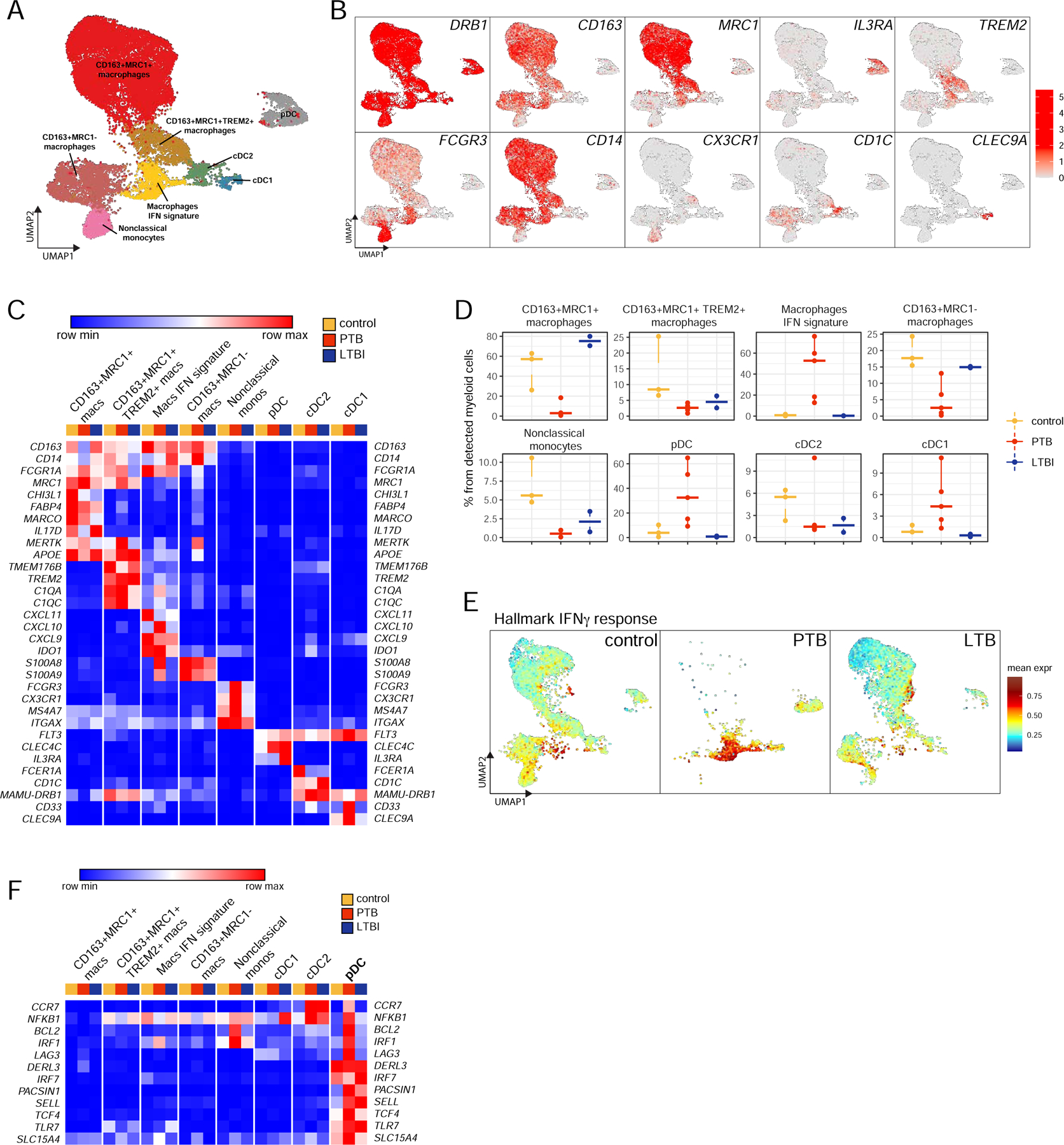
Control, n=3; PTB, n=5; LTBI, n=2.
A. UMAP plot of myeloid cells across all samples, colored according to identified clusters.
B. UMAP plots with the expression of markers, characterizing main myeloid populations in macaques.
C. Heatmap of normalized expression of selected genes in each myeloid cluster per condition.
D. Cell proportion of each cluster per condition.
E. UMAP plots of mean expression for the genes from the “Hallmark Interferon Gamma Response” pathway split by the condition.
F. Heatmap of normalized expression of genes, enriched in pDC population, per sample per condition.
See also Fig S3.
Dendritic cells (DCs), another predominant cell type in the lungs of macaques with PTB were represented by an increase in pDCs, which comprised ~30% of all myeloid cells (Fig 3D). This pDC population was smaller in the lungs of control and LTBI macaques, where pDCs comprised less than 10% of the myeloid cell population. pDCs expressed classic markers, such as IL3RA (CD123) and CLEC4C as well as the pDC master regulatory transcription factor TCF4 (Grajkowska et al., 2017). Additionally, pDCs in PTB macaque lungs expressed genes associated with microbial sensing and induction of type I IFNs such as IRF1, IRF7, TLR7, SLC15A4, and PACSIN1 as well as genes associated with secretion of IFNs such as DERL3 (Fig 3F). Although IFN transcriptional modules were not detected in lung pDCs in PTB macaques, IRF1 controls IFN expression in DCs (Schmitz et al., 2007), while IRF7 is the master regulator of IFN production in pDCs (Honda et al., 2005a; Honda et al., 2005b). pDCs in the TB macaque lungs also expressed CCR7, a chemokine receptor that is strongly upregulated upon exposure to TLR ligands (Seth et al., 2011). Importantly, pDCs in PTB macaque lung also express BCL2, a pro-survival factor in pDCs (Carrington et al., 2015), LAG3 which regulates pDC homeostasis (Workman et al., 2009) and CD62L (SELL gene), an adhesion molecule involved in trafficking of pDCs to high endothelial venules (HEVs) (Fig 3F). Besides pDCs, we identified a population of DCs that are FLT3+CD1C+ (cDC2) and FLT3+CLEC9A+ (cDC1). cDC1 cells were also enriched in PTB macaque lungs (Fig 3D). These results suggest the involvement of pDC in PTB disease and host response to Mtb infection.
We also identified a population of non-classical monocytes (CD163−MRC1−MAMU-DRB+ITGAX+CD14−CD16+) which constituted 5–10% of myeloid cells in control lungs and was decreased in Mtb-infected lungs, both LTBI and PTB. In the healthy macaque lung, this population has not been previously described, thus allowing for the characterization of monocyte populations in the primate lung.
Characterization of immune cell populations in the lungs of macaques with LTBI and PTB.
To independently assess major quantitative and qualitative changes occurring in immune cells following Mtb infection, we profiled the expression of 26 common surface or intracellular markers by mass cytometry (CyTOF) in lung samples of macaques namely, control (n=3), PTB (n=5), LTBI (n=3).
Lymphoid cells in the lung.
Using markers relevant for the lymphoid populations we were able to identify CD4+ and CD8+ naïve, activated/memory T cells, B cells, and NK cells (Fig 4A). Despite heterogeneity across individual animals, lung samples from the same disease group clustered together by the MDS projection plot (Fig 4B). Analysis of lymphoid subpopulations by CyTOF confirmed multiple findings from the scRNA-seq analysis. First, we were able to confirm the distinct presence of large numbers of NK cells in LTBI macaque lungs (Fig 4C, D). Strikingly, a feature of LTBI lymphoid cells is the expression of CD27 marker, expressed on a subset of NK cells, CD8+, and CD4+ cells, and these subsets were absent from the lungs of control and PTB macaques (Fig 4E, Fig S4A). This finding was validated in Mtb-infected individuals where the frequency of circulating NK cells was higher in individuals with LTBI than individuals with PTB (Fig 4F, gating strategy in Fig S4B, Table S3). In addition, although the frequency and mean fluorescence intensity (MFI) of circulating CD27+ NK cells were not different in Mtb-infected LTBI and PTB individuals (Fig 4G, H), the expansion of CD27+ NK cells in LTBI was significantly higher after in vitro exposure to a cell wall extract of Mtb (Fig 4I). CD27+ NK cells from individuals with PTB also expanded after such stimulation, but not to the extent of expansion in LTBI, while CD27+ NK cells did not expand in healthy controls (HC) (Fig 4I, Fig S4B). CD27+ NK cells from both LTBI and PTB showed enhanced expression of CD69, a classical activation and tissue-homing marker, with respect to their CD27− counterparts (data not shown), suggesting that CD27+ NK cells are activated to exert effector functions. We sorted CD27+ NK cells from PBMCs of healthy controls and individuals with LTBI and measured the relative mRNA expression of effector molecules. While no IL22 mRNA expression was detected in sorted CD27+ NK cells (Fig 4J), we found an increased trend for IFNG mRNA expression (Fig 4K), and significantly increased GZMB and PRF1 mRNA expression in CD27+ NK cells sorted from LTBI individuals, when compared with expression in HCs (Fig 4L, M). Finally, we observed significantly higher localization of NK cells within the parenchyma of LTBI macaque lungs, when compared with fewer NK cells which localized within the granulomatous lesions of macaques with PTB or in control lungs (Fig 4N, Fig S4C). These findings together support the presence of NK cells, especially a CD27+ subset in mediating protection and Mtb control during TB latency.
Figure 4. CyTOF and flow profiling of lymphoid cells validates the presence of lung NK cells in LTBI macaques and circulating NK cells in individuals with LTBI.
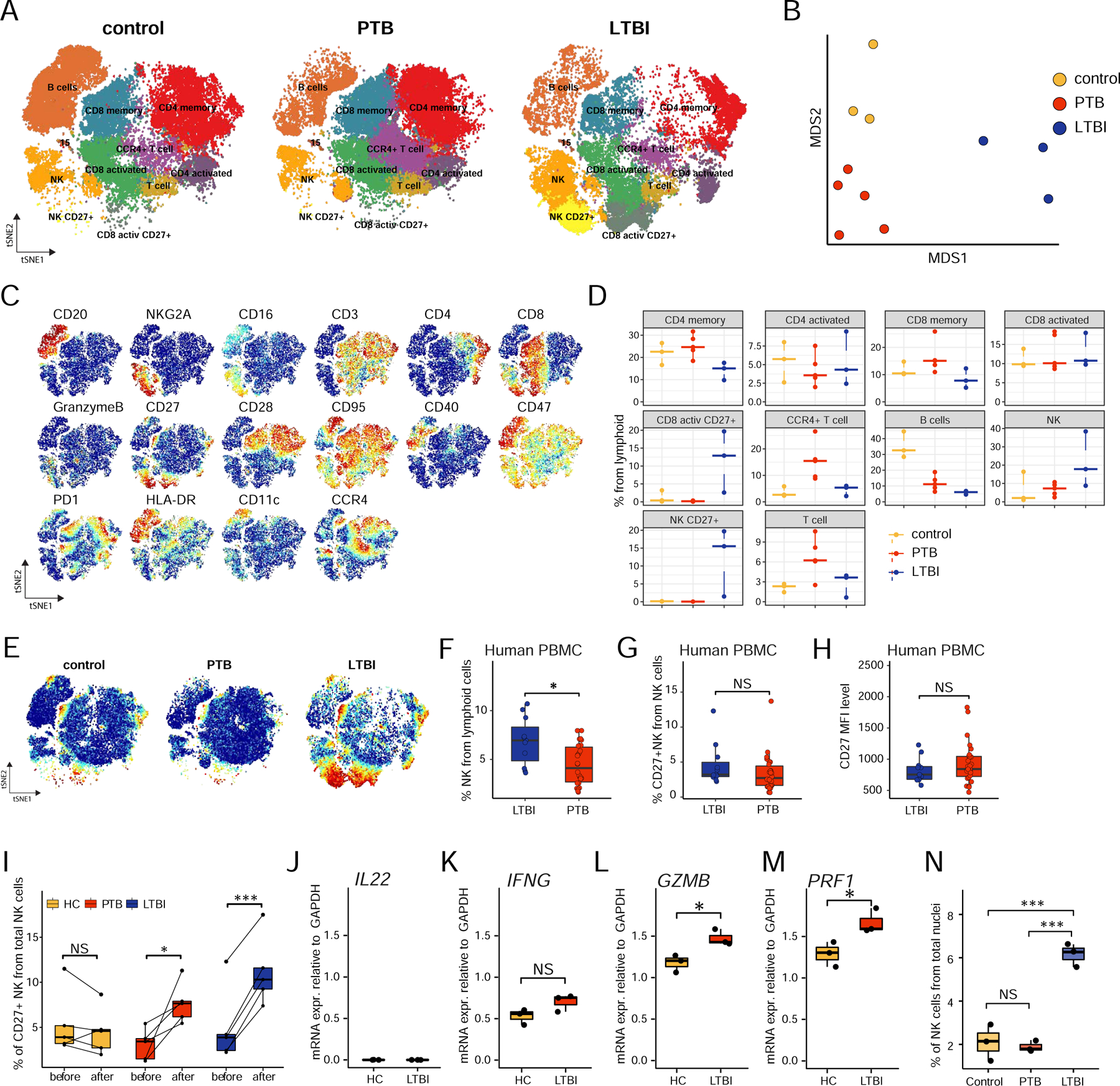
A. tSNE plots of lymphoid cells across all samples, colored according to identified clusters. Control, n=3; PTB, n=5; LTBI, n=3.
B. MDS projection for all CyTOF samples, colored by the condition. For each sample / mean marker expression was used to perform MDS.
C. tSNE plots with the expression of all markers used to perform tSNE and identify clusters for lymphoid cells.
D. Cell proportion of each cluster per sample per condition.
E. tSNE plots with the expression of CD27 marker by the condition.
F. Percentage of NK cells as determined by flow cytometry in PBMCs from LTBI (n=11) and PTB (n=25) patients.
G. Percentage of CD27+ NK cells as determined by flow cytometry in PBMCs from LTBI (n=11) and PTB (n=25) patients.
H. Relative expression of CD27+ in NK cells in terms of mean fluorescence intensity (MFI) in PBMCs from LTBI (n=11) and PTB (n=25) patients by flow cytometry.
I. Percentage of CD27+ NK cells from healthy controls (HC, n=5), LTBI (n=5), and PTB patients (n=5) before and after in vitro stimulation with Mtb cell wall protein.
J-M. mRNA expression of IL22 (J), IFNG (K), GZMB (L), and PRF1 (M) relative to GADPH in the purified CD27+ NK cells from HC (n=3) and LTBI (n=3) were quantified by RT–PCR.
N. Formalin-fixed paraffin embedded sections from macaque lung (control; n=3, PTB: n=3 and LTBI; n=3) were stained with NKG2a, CD3, CD20, CD14, CD66abce and DAPI and the morphometrical quantification of NKG2a+ CD3−, CD20−, CD14− cells across samples.
Data represented as mean ± SD, *p < 0.05, **p < 0.01, ***p < 0.001, NS = not significant, either by unpaired Student’s T-test (F-H, K-M), by Student’s T-test with Holm correction (I) or by one-way ANOVA with Tukey post-hoc test (N).
Using CyTOF, we confirmed the marked reduction in B cell population in lungs of macaques with PTB (~10%), with a further decrease in lungs of macaques with LTBI (less than 5%) when compared with control lungs (~30–40% of lymphoid cells) (Fig 4C–D). For discrimination of activation on T cells, we used CD28 and CD95 (FAS gene) surface markers (Pitcher et al., 2002), thereby CD28−CD95+ T cells were considered activated T cells (effector or effector memory cells). We observed little difference in the size of this population between control, PTB, and LTBI animals compared to scRNA-seq analysis (Fig 4D and Fig 2E). We visualized these markers on scRNA-seq lymphoid populations (Fig S2B) and, despite the partial overlap between CyTOF and scRNA-seq populations we found that the latter was able to distinguish T cell populations at much higher resolution due to the number of genes profiled.
Myeloid cells in the lung.
We used 14 markers to cluster myeloid cells from the lung using CyTOF. Neutrophils were the most abundant myeloid cell type (Fig 5A–D). Our results show a decrease in CD163+CD206+ AMs and CD163+CD206− IMs in the lungs of macaques of PTB when compared with control and LTBI lungs (Fig 5A–D). CyTOF (Fig 5A–D) confirmed the accumulation of increased pDCs in the lungs of macaques with PTB lungs. Furthermore, pDCs localized within granulomas of human PTB patients, and were significantly represented within lung granulomas of PTB macaques when compared with LTBI or control macaques (Fig 5E, F). Also, pDCs measured by flow cytometry were significantly increased in the lungs of macaques with PTB when compared with LTBI (Fig 5G, Fig S4D). This was mirrored by significant depletion of peripheral pDCs in PTB macaques when compared with the control or LTBI macaques (Fig 5H, Gating strategy shown in Fig S4E). The decrease in peripheral pDCs in macaques with PTB is associated with significantly increased accumulation of peripheral classical monocytes when compared with control or LTBI macaques (Fig 5I). There were no significant differences in the peripheral non-classical monocytes between control, LTBI, or PTB macaques (Fig 5J, Gating strategy shown in Fig S4E). Finally, we confirmed that peripheral pDCs were specifically depleted in PBMCs of individuals with PTB when compared with pDCs in LTBI or HCs (Fig 5K). Together, these results describe the distinct accumulation of pDCs in the lung during PTB disease in macaques and humans, likely by recruitment from the periphery.
Figure 5. CyTOF and flow cytometry profiling of myeloid cells validates the accumulation of pDCs in macaque lungs with PTB.
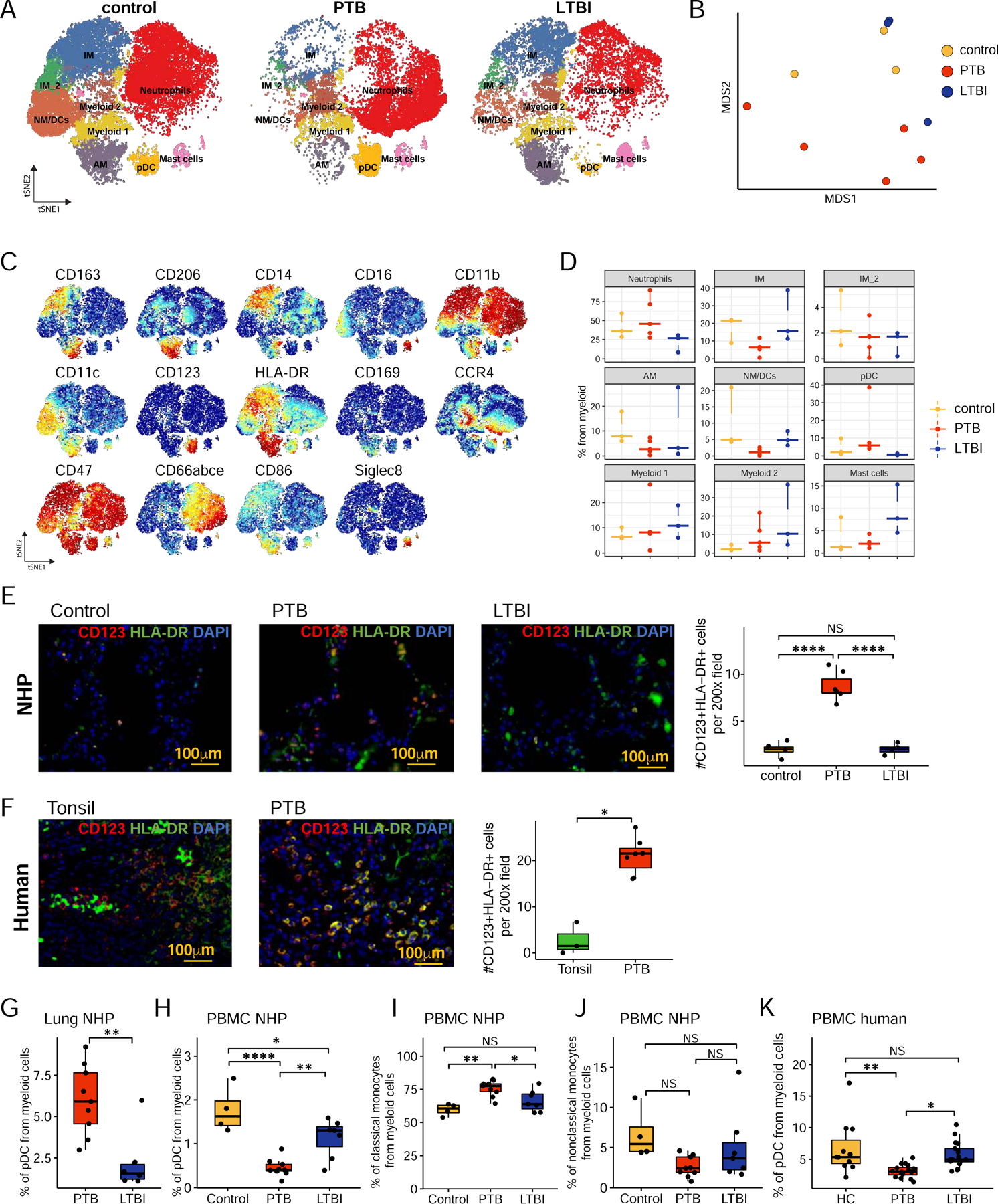
A. tSNE plots of myeloid cells across all samples, colored according to identified clusters. Control, n=3; PTB, n=5; LTBI, n=3.
B. MDS projection of CyTOF samples, colored by the condition. For each sample, mean marker expression was used to perform MDS.
C. tSNE plots with the expression of all markers used to perform tSNE and identify clusters for myeloid cells.
D. Cell proportion of each cluster per condition.
E and F. Immunohistochemistry staining of CD123, HLA-DR, and DAPI on formalin-fixed paraffin-embedded sections from macaque lung (control; n=4, PTB: n=6 and LTBI; n=4) and human tissues (tonsils, n=3 and lung tissue from individuals with PTB, n=6). The right panel displays the morphometrical quantification of CD123+HLA-DR+ cell numbers across subjects.
G. Percentage of pDC in lung single cell suspension from PTB (n=9) and LTBI (n=6) macaques by flow cytometry.
H-J. Percentage of pDC (H), classical (I) and non-classical (J) monocytes in PBMCs from control (n=4), PTB (n=9) and LTBI (n=6) macaques by flow cytometry.
K. Percentage of pDC was determined in PBMCs from HC (n=11), LTBI (n=17) and PTB (n=16) patients by flow cytometry.
Data represented as mean ± SD, *p < 0.05, **p < 0.01, ***p < 0.001, NS = not significant, either by unpaired Student’s T-test (F, G) or by one-way ANOVA with Tukey post-hoc test (E, H-K).
To further validate our findings on myeloid cells characterized in PTB, we found that IFN-responsive macrophages co-expressing CD163 and S100A8 or IDO-1 localized within the suppressive rims bordering necrotic human TB granulomas (Fig 6A,B,D,E). We also found co-localization of CD163+ macrophages expressing C1Q (a marker of TREM2-expressing AMs) within the suppressive rims of human necrotic TB granulomas (Fig 6C, E). These results were mirrored by the increased localization of CD163+ S100A8+ IFN-responsive macrophages within the granulomas of macaques with PTB (Fig 6F). Furthermore, in macaques with PTB, we found that CD68+ macrophages expressed IDO-1 and CD163, but did not co-express high levels of CD206 (Fig S5A). We did not find IDO-1 expression within CD66-expressing neutrophils in macaques with PTB (Fig S5A). We then measured CD163+ CD206− macrophages expressing CD38 and CD274, classical IFN-responsive markers in the lungs of PTB or LTBI macaques (Jablonski et al., 2015). Our results show that in the lungs of PTB, CD163+ CD206−macrophage subset expressing either CD274 or CD38 was higher when compared with this subset in LTBI lungs (Fig. 6G, Gating strategy shown in Fig S5B). Furthermore, no significant differences in accumulation of AMs, IMs/classical monocytes, and non-classical monocytes were found in the lungs of PTB and LTBI by flow cytometry (Fig S5C). Finally, consistent with the localization of C1Q-expressing CD163 macrophages within human TB granulomas, we found that C1Q-expressing CD163+CD206−macrophage population was significantly higher in lungs of macaques with PTB, when compared with LTBI (Fig S5D).
Figure 6. IFN-responsive macrophages are present within granuloma structures in humans and macaques during PTB.
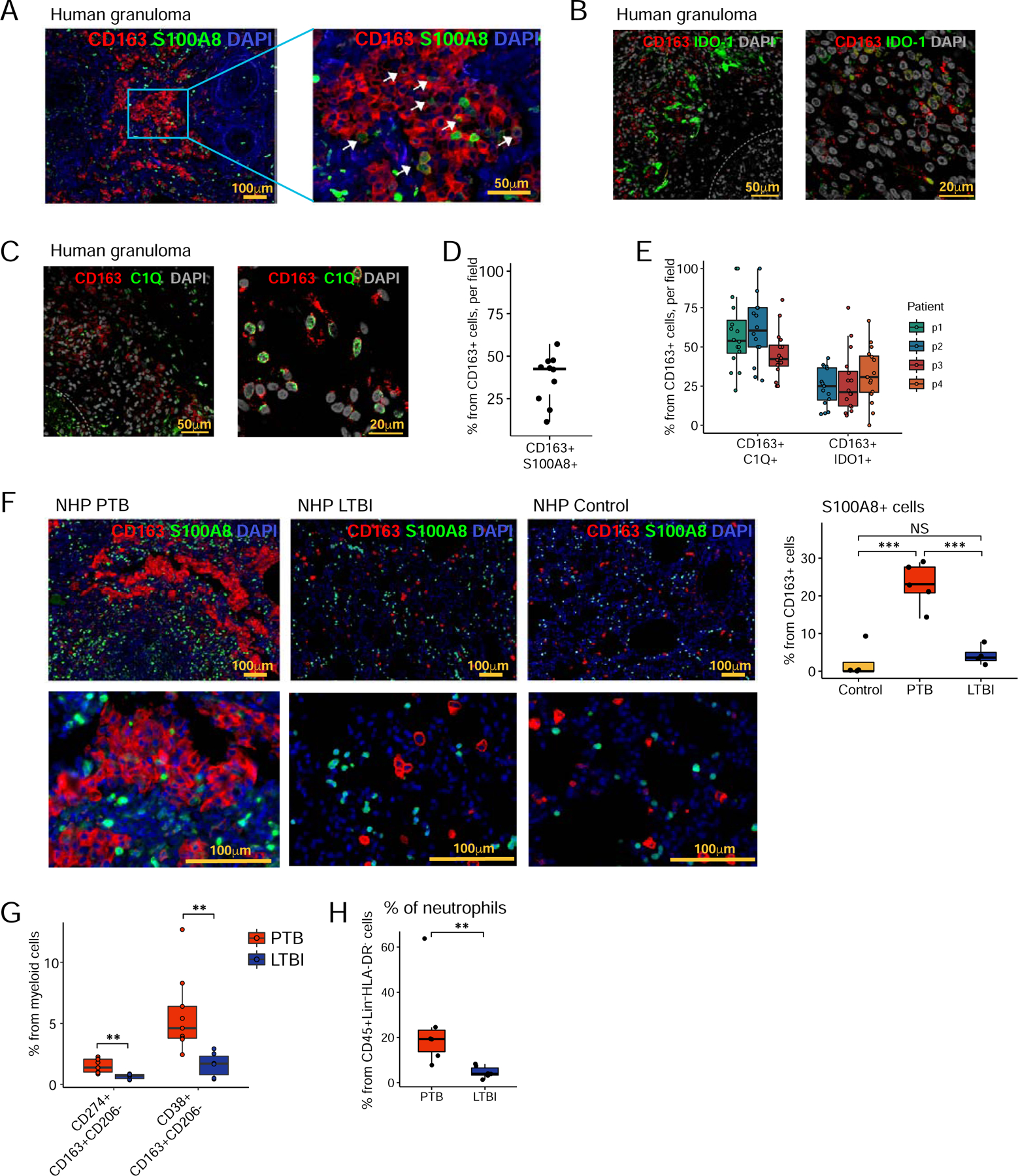
A - C. Immunohistochemistry staining of CD163 and S100A8 (A), CD163 and IDO-1 (B), CD163 and C1Q (C), and DAPI on formalin-fixed paraffin-embedded sections from human lung tissue with PTB, (n=3–4).
D. Morphometrical quantification of CD163+S100A8+ cells from a human TB granuloma. N=10 granulomas/patient. Data represented as mean ± SD.
E. Morphometrical quantification of CD163+IDO-1+ and CD163+C1Q+ cells from human TB granulomas. Data represented as mean ± SD.
F. Immunohistochemistry staining of CD163, S1008, and DAPI on formalin-fixed paraffin-embedded sections from macaque lung (control; n=4, PTB: n=6 and LTBI; n=4). Right panel display morphometrical quantification of CD163+S100A8+ cells across subjects. Data represented as mean ± SD, NS = not significant. ***p < 0.001, by one-way ANOVA with Tukey post-hoc test.
G and H. Percentage of CD163+CD206−CD274+, CD163+CD206−CD38+ cells and neutrophils (CD45+Lin−HLADR−) were determined by flow cytometry in lung single cell suspension from PTB (n=6–9) and LTBI (n=6) macaque. Data represented as mean ± SD, **p < 0.01, by Student’s T-test with Holm correction (F) or by Student’s T test (G).
See also Fig S5.
One of the most abundant populations of myeloid cells by CyTOF was neutrophils, comprising 25–75% of cells. Additionally, we also found neutrophil accumulation well represented in PTB macaque lungs by flow cytometry (Fig 6H, Gating strategy shown in Fig S5E).
Finally, we carried out a linear correlation analysis between the different disease readouts and immune cell characteristics identified in this study. We found a significant positive correlation between lung Mtb CFU and CRP levels, as well as Mtb CFU and lung pathology, suggesting that an increase in Mtb burden was associated with increased TB disease (Fig S6A). There was a significant linear inverse correlation between the increased size of B cell follicles within granulomas and decreased lung Mtb CFU, further supporting a protective role for iBALT in Mtb control. We did not find a positive correlation between the presence of neutrophils or pDC populations and Mtb CFU or disease pathology in Mtb-infected macaques (Fig S6B,C). In contrast, we identified a significant positive correlation between the presence of IFN-responsive macrophages and an increase in Mtb CFU as well as increased disease pathology in Mtb-infected macaques (Fig S6B,C). This further highlights the functional role that IFN-responsive macrophages likely play in mediating disease progression and promoting susceptibility during PTB.
Discussion
We now understand that Mtb responses to host conditions (Veatch and Kaushal, 2017) and the resulting TB disease (Lin and Flynn, 2018) exists as a spectrum. However, within the spectrum of TB disease, the immune parameters that mediate control or allow progression of disease are not well defined. A comprehensive approach to characterize the immune landscape of the lung during Mtb control and progression will fill these gaps in our understanding. In the current study, we have used single-cell technologies in combination with conventional technologies, to provide in-depth analysis of the immune landscape in the lungs of healthy, LTBI and PTB macaques. We found that during Mtb control in TB latency, NK cells, especially those expressing CD27+ are increased. In contrast, during TB disease and uncontrolled Mtb growth, we report the presence of pDCs, IFN-responsive macrophage population, as well as accumulation of activated T cells within the lung. Together, these findings provide detailed insights into the lung immune landscape of TB disease and control that was previously unknown, thereby identifying immune pathways that can be targeted for therapeutics and vaccines for TB.
Blood transcriptional profiles of patients with PTB and Mtb-infected individuals who progressed to PTB are dominated by a type I IFN-inducible gene signature and expression correlated with the severity of lung disease (Berry et al., 2010; Zak DE, 2016). While the NHP model of TB recapitulates key aspects of the human TB disease (Bucsan et al., 2019b), we recently provided proof-of-principle that bulk RNA sequencing of lungs from macaques with PTB disease also express the IFN-inducible transcriptional signature seen in the blood of TB progressors (Ahmed et al., 2020). Additionally, lungs of macaques with PTB by bulk sequencing mirrored the overexpression of IFN response pathways, innate immune system, myeloid cell activation pathways such as TLR cascades, and signaling by interleukins as seen in the blood of human TB progressors (Ahmed et al., 2020). The scRNA-seq analysis from the lungs of PTB macaques as described here mechanistically allows us to delineate the distinct cellular sources of IFN responses in the lungs during PTB. While IFN-inducible transcripts were overrepresented in blood neutrophils and monocytes in PTB patients and TB progressors (Berry et al., 2010; Zak DE, 2016), our current analysis describes that in addition to monocytes in the blood, locally in the lung, pDCs and T cells are important sources of type I and type II IFN responses, respectively. In mice, polyinosinic: polycytidylic acid (poly I:C) treatment resulted in type I IFN responses that promoted the recruitment of lung permissive myeloid populations and exacerbated TB disease (Antonelli et al., 2010). The lung macrophage population in PTB macaques exhibit classic IFN response signature including IFN-inducible genes CXCL9–11, S100A8, S100A9 and IDO-1, key mediators expressed during TB disease (Berry et al., 2010; Gopal et al., 2013; Mehra et al., 2013) and within the TB granuloma (Gopal et al., 2013; Mehra et al., 2013). Therefore, it is possible that this macrophage population is an early responder to type I IFNs and recruits Mtb-permissive myeloid cells such as neutrophils and myeloid-derived suppressor cells through S100A8/9-dependent mechanisms (Scott et al., 2020), while mediating a T cell suppressive environment through IDO-1-dependent pathway during PTB disease (Gautam et al., 2018). Such mechanisms would be required to limit pulmonary pathology, but clearly also allow Mtb to persist. Our published studies have shown that IDO-1 is particularly enriched in the macrophage-rich inner layer of the PTB granuloma and increased IDO-1 expression correlates with higher Mtb burden (Gautam et al., 2018; Mehra et al., 2013). Importantly, suppression of IDO-1 activity in PTB reduced bacterial burden, pathology, and clinical signs of TB disease, leading to increased host survival (Gautam et al., 2018). Indeed, we show that macrophages co-expressing CD163 and IDO-1 also localize within the suppressive rim of necrotic granulomas in macaques and humans with PTB, where they be actively involved in preventing Mtb-specific lymphocytes from reaching the infected macrophages within the necrotic centers, promoting Mtb persistence (Gautam et al., 2018). In this regard, expression of IDO-1 was found to be one of the defining features of granulomas obtained from individuals with PTB, by the use of an innovative multiplexed imaging modality, Multiplexed Ion Beam Imaging by Time-of-Flight (MIBI-TOF) (McCaffrey et al., 2020). Additionally, recruitment of inflammatory monocytes from the circulation into the lung may allow for differentiation into the IFN-responding macrophage population. This is consistent with the observed increase in classical monocytes in the PBMCs of macaques with PTB. Traditionally, CD206+ (MRC1+) macrophages have been considered to be long-lived “lung-resident” cells that do not undergo rapid turnover (Cai et al., 2014; Kuroda et al., 2018). These “lung-resident” cells were unlikely to be IFN-responsive. Instead, we report that the CD163+CD206low subpopulation, which is likely recruited to the lung in response to infection, appears to have IFN-responsive properties (expressing significantly higher CD274 and CD38 subsets in macaques with PTB relative to LTBI). Our results demonstrate a high level of complexity of myeloid populations in the lungs of Mtb-infected macaques. Based on these results, we suggest a scenario where macrophages act as early responders to IFN responses upon Mtb infection, propagating the downstream amplification of myeloid cell recruitment, likely suppression/localization of IFN-γ-producing T cells within granulomas, propagation of Mtb growth, and initiation and progression of TB pathology. This hypothesis is supported by our published studies where inhibiting IDO-1 activity in macaques with PTB altered granuloma organization and increased proliferating T cell translocation to the lesion core (Gautam et al., 2018). pDCs are highly effective in sensing intracellular DNA and RNA mainly via TLR7 and TLR9, and can rapidly produce type I and III IFNs. Key findings emerge from our discovery that pDCs are present in PTB macaque granuloma. TLR7 is highly expressed especially on pDCs, while TLR9 is expressed on other cells including pDCs in PTB macaques. TLR7 recognizes single-stranded RNA from pathogens, TLR9 recognizes bacterial and viral CpG DNA motifs (Barton, 2007). While the role of TLR7 deficient mice has not been tested in Mtb infection, TLR9 deficient mice are not much more susceptible to Mtb infection (Bafica et al., 2005). These results suggest that pDCs engage TLR7 for the induction of IFN responses. Using single-cell cytokine analysis with scRNA-seq profiling recently revealed no evidence for a pre-existing subset of type I IFN-producing human pDCs (Wimmers et al., 2018). However, human pDCs were primed for and produced large quantities of IFNs within hours following CpG stimulation or modulation of the microenvironment (Wimmers et al., 2018). These results are consistent with the presence of regulatory factors IRF1 and especially the expression of IRF7 in lung pDCs in PTB macaques, suggesting that these pDCs are also primed for IFN production (Wimmers et al., 2018). IRF7 is a crucial regulator of type I IFN production following infections, whereby activation of TLR7 and TLR9 can result in IRF7 phosphorylation and translocation into the nucleus to trigger signaling cascades for rapid type I IFN production (Honda et al., 2005a; Honda et al., 2005b). Other unique features of lung pDCs in macaques with PTB are the expression of CCR7 and CD62L. We and others have described that CCL19 but not CCL21 mRNA is induced in the Mtb-infected lung (Kahnert et al., 2007; Khader et al., 2011) and that HEVs are featured in human PTB granulomas (Slight et al., 2013). Thus, is it possible that pDCs can traffic and localize within TB granulomas as shown here via CD62L interactions with the HEVs and/or CCR7-CCL19 mediated interactions. BCL2 expression is reported to function as a prosurvival factor in pDCs (Carrington et al., 2015), suggesting that BCL2 expression in lung pDCs during TB disease can prolong their survival in the lung, thus amplifying the inflammatory milieu. Finally, a key feature of pDCs in the lungs of PTB macaques is the expression of LAG3, which is critical for the regulation of pDC homeostasis. Activated pDCs have a ~70-fold increase of LAG3 mRNA expression when compared with activated T cells (Workman et al., 2009), suggesting they could be primary mediators of an immunosuppressive environment (Graydon et al., 2019). We also showed that LAG3 expression is induced significantly in the lungs of macaques with PTB and correlates with increased bacterial burden, and LAG3 overproduction can diminish T cell responses (Phillips et al., 2017). Together, these results project pDCs as a key immune cell in the lungs of PTB macaques where they are primed for rapid type I IFN production, prolonged survival, and may contribute to suppression and exhaustion of T cell responses. Finally, circulating pDCs have been reportedly depleted in PTB patients when compared with healthy controls (Lu et al., 2017). These results are consistent with our data from humans and macaques where peripheral pDCs are depleted specifically during PTB, suggesting that pDCs are actively recruited to the Mtb-inflamed lung from the circulation to propagate the type I IFN signaling cascade. Whether pDCs are the cause or the effect of an exacerbated type I IFN response during Mtb infection in the lung should be mechanistically addressed in future studies. Indeed, while we show enrichment of IDO+ and S100A8+-expressing IFN-responsive macrophages and pDCs in human TB lesions, comparison to healthy human lung or lung tissue from individuals with LTBI is not possible due to paucity of such samples. Thus, our quantitation of the accumulation of IFN-responsive macrophages and pDCs within PTB lesions and significant absence in macaques with LTBI and healthy lung, confirm our findings in a relevant model which closely reflects human TB disease. Future experiments delineating the time kinetics in the lungs of macaques with PTB will shed light on the early role of pDCs and IFN-responsive macrophages in the lungs with respect to inflammatory responses and initiating TB disease.
NK cells are increasingly recognized as a key component of the innate immune response to Mtb and link between innate and adaptive immunity (Khader et al., 2019). Recent analysis across multiple cohorts show higher numbers of cytotoxic NK cells in LTBI individuals when compared with uninfected individuals (Roy Chowdhury et al., 2018). The circulating NK cells decreased during the progression from LTBI to PTB disease and returned to the baseline post TB treatment, suggesting a protective role for NK cells during Mtb infection (Roy Chowdhury et al., 2018). Despite the emerging role of circulating NK cells in protective immunity to TB, our current findings show the accumulation and localization of CD27+ NK cells in the lungs of macaque hosts that control Mtb infection. CD27+ NK cells can distinguish antigens, expand, and survive long-term to generate memory-like responses and promote vaccine-induced protective immunity against Mtb infection (Venkatasubramanian et al., 2017). CD27 is a member of the TNF receptor superfamily and is considered a marker of memory/mature NK cells. CD27+ NK subset appears to be more reactive than CD27− NK cells with respect to cytotoxicity, cytokine production, proliferation, and function (Inngjerdingen et al., 2011). Our human studies further support the idea that NK cells are protective during TB, as we demonstrate the increased percentage of circulating NK cells in LTBI individuals, and the increased expansion of CD27+ NK cells upon mycobacterial stimulation. Potential mechanisms by which NK cells can contribute to immunity against Mtb infection are by lysing Mtb-infected human monocytes and AMs (Vankayalapati et al., 2005; Venkatasubramanian et al., 2017), upregulating CD8+ T cell responses (Vankayalapati et al., 2005), as well as directly killing Mtb through perforin and granulysin (Lu et al., 2014). These results are consistent with the results from our study describing the expression of GZMB1, PRF1, GNLY in NK cells found in LTBI macaque lungs. Additionally, sorted human CD27+ NK cells from individuals with LTBI express higher levels of PRF1 and GZMB1, suggesting cytotoxic potential as a key mechanism by which NK cells mediate protection. Our previous studies have established a protective role for the presence of organized iBALT structures harboring well-organized B cell follicles within the lungs of LTBI macaques (Gopal et al., 2013; Kaushal et al., 2015; Slight et al., 2013). These results are consistent with the data presented here, where macaques with LTBI have enhanced areas of iBALT and with infiltration of T cells, despite having considerably less inflammation in the lung. This is in sharp contrast to the smaller B cell lymphoid follicles which are associated with the rim of necrotic granulomas in the lungs of PTB macaques, which we have termed as “GrALT”. Whether NK cells localize within or near the iBALT structures in the LTBI lung is an area of future research. Additionally, while iBALT structures harboring B cell follicles are featured in LTBI lungs as shown before (Slight et al., 2013) and here, our scRNA-seq analysis shows that the relative numbers of B cells in LTBI lungs are lower when compared with control and PTB lungs. One possibility is that despite LTBI lungs having fewer B cells, these B cells are effectively localized into the lymphoid structure, likely due to chemokine mediated signals possibly by early NK cell-produced cytokines such as IFN-γ and IL-22 (Venkatasubramanian et al., 2017). BCG revaccination of adults with LTBI induces long-lived BCG-reactive NK cell responses (Suliman et al., 2016). Furthermore, in mice during Mtb infection after BCG vaccination, memory-like CD27+ NK cells expand and provide protection against challenge with Mtb (Venkatasubramanian et al., 2017). Thus, NK cells are a prime target for vaccine-responses against Mtb and should be actively pursued in vaccine development strategies against TB.
The role of T cells, especially the interplay between Th1 and Th17 cells in protective immunity to TB is complex and likely multi-faceted. Th1 and Th17 responses are associated with protection during primary and vaccine-induced T cell responses (Cooper et al., 1993; Gopal et al., 2014; Khader et al., 2007). The scRNA-seq analysis in our study has established the accumulation of activated T cells, including Th1 and Th17 responses and associated markers of exhaustion in the lungs of PTB macaques. Thus, while Th1 and Th17 responses are activated, amplified, and accumulate in the lung, whether these cells are correctly localized near Mtb-infected macrophages and mediate Mtb control is not fully known. It is possible that a suppressive environment created by IFN-responsive macrophages and pDCs including the production of IDO-1 and LAG3 actively limit T cell protective mechanisms and should be explored in future studies. Alternatively, dysregulated IL-17 and IFN-γ responses can also result in disease induction and propagation of inflammation signals (Cruz et al., 2010; Nandi and Behar, 2011). In the blood of PTB patients, Th1 and Th17 responses are reduced when compared to LTBI individuals (Scriba et al., 2008). The absence of Th1/Th17 cells from the circulation of PTB patients could indicate recruitment of these cells to the inflammatory milieu in the lung, where these cells can contribute to early protection but also mediate downstream inflammation during disease progression.
A significant hurdle in rapidly adopting single cell technologies such as scRNA-seq for deciphering the TB immune landscape includes the safety and technical adaptations needed to processing these technologies within the biohazard containment level 3. An additional hurdle is the isolation of live single-cells from pathological conditions with evidence of severe necrosis such as PTB. In the current study, standardization of the protocol reported here has effectively overcome both of these hurdles. Our scRNA-seq analysis demonstrated the importance of profiling more than a thousand genes per cell to accurately assign population identities, such as for multiple subpopulations of macrophages and previously uncharacterized non-classical monocytes in the lung. However, there are a few limitations of the use of scRNA-seq in this study. First, we were not able to profile neutrophils, that are well known to play a role in response to TB (Berry et al., 2010), by scRNA-seq, either because of low RNA content or the presence of RNases that can inhibit reaction inside 10X GEMs. Additionally, it is also possible that freeze-thaw of banked macaque lung samples may result in the loss of neutrophils and fragile immune cells with a potential impact on the transcriptomes of different cell types. 10X scRNA-seq profiles ~2,000 the most expressed genes per cell, which limits our profiling and description of the immune population’s function. Needless, even though we did not detect IFNα/β transcripts in pDCs and IL-17A in Th17 cells, we were able to make the inference about their function through other transcriptional signatures. Additionally, our scRNA-seq data on healthy macaque lung can also serve as a useful tool for identifying key markers for CyTOF to profile the immune population’s response to TB in a macaque setting. This study also utilized more male macaques than female macaques due to the limited availability of female macaques in our studies. However, we have validated all the key findings from macaques in the human cohorts where both sexes are well represented. Furthermore, while the sample size used in the scRNA-seq analysis is a limitation of the current study, we believe that this hypothesis-generating tool has fully served its purpose and allowed us to identify unique populations of immune cells in PTB compared to LTBI disease states, especially with respect to pDCs and IFN-responsive macrophages. Then, we were able to complement our discoveries by CyTOF analysis to alleviate the effects of post-translational modifications and neutrophil loss. Finally, based on the results from the single cell technologies, we were able to validate our key findings using conventional tools such as flow cytometry and IF in a larger cohort of macaque and human samples.
In conclusion, we carried out an in-depth characterization of the lung immune landscape during latency and TB disease. Our results have demonstrated the presence of unique immune populations that are associated with Mtb control and TB disease, which could potentially be harnessed for the design of more effective vaccines as well as therapeutics for TB treatment.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Shabaana A. Khader (sakhader@wustl.edu).
Materials Availability
This study did not generate any new unique reagents.
Data and Code Availability
The accession number for the raw and processed data for single cell RNA sequencing and CyTOF generated during this study is GEO: GSE149758. The additional accession number is Synapse: syn21988273.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Non-human primate model
All animal procedures were approved by the Tulane National Primate Research Center (TNPRC) Institutional Animal Care and Use Committee and performed in strict accordance with the NIH, USDA, and AALAAC guidelines. Specific-pathogen-free, retrovirus-free, mycobacteria-naive, adult rhesus macaques were bred and housed at the TNPRC and they were assigned into three groups. One group of macaques remained uninfected (n=5). The second (~ 10 CFU) (n=7) (LTBI) and third (~1000 CFU) group (n=9) (PTB) of animals were aerosol-challenged with Mtb CDC1551 using a custom head-only dynamic inhalation system housed within a class III biological safety cabinet as described extensively before (Bucsan et al., 2019a; Foreman et al., 2020; Foreman et al., 2016; Kaushal et al., 2015; Mehra et al., 2015). Humane endpoints were predefined in the animal use protocol and applied as a measure of the reduction of discomfort. Criteria for euthanasia included presentation of four or more of the following conditions: (i) body temperatures consistently greater than 2°F compared to pre-infection values for 3 or more weeks in a row; (ii) loss of body weight for 15% or more; (iii) serum CRP values higher than 10 mg/ml for 3 or more consecutive weeks since CRP is a marker for systemic inflammation and exhibits a high degree of correlation with active TB in macaques; (iv) Thoracic radiographs (CXR) values higher than 2 on a scale of 0–4; with a score of 0 denoting normal lung and a score of 4 denoting severe tuberculous pneumonia and pathology. (v) respiratory discomfort leading to vocalization; (vi) significant or complete loss of appetite; and (vii) detectable bacilli in BAL samples. At necropsy, lung, spleen, and liver tissues were collected and processed, as previously described, using two sections of pulmonary tissue that represented every lung lobe in at least one sample; CFU was determined per gram of tissue. We also evaluated CFU from individual lung granulomas isolated at necropsy, as described earlier. At necropsy, the lung tissue for all macaques was randomly sampled from each lung lobe by blinded pathologists using a grid (Luciw et al., 2011). No priority was given to granulomatous or non-granulomatous tissues and single-cell preparation was carried out as a common pool from random lung sections from all lobes for each animal. Despite the random selection of the lung sections, it is expected that the fraction of granulomatous tissue/cells sampled will be higher for macaques with PTB which exhibit more disease and pathology, than LTBI macaques. All information about animal’s sex, age, infection time, and necropsy date is listed in Supplementary Table S1.
Human study
We obtained blood samples from individuals with PTB and LTBI that attended the TB clinic of the Instituto Nacional de Enfermedades Respiratorias “Ismael Cosío Villegas” (INER), in Mexico City. The PTB group included patients with positive results in sputum smear microscopy, sputum/BAL culture, and GeneXpert MTB/RIF test (Cepheid, CA, USA), according to the ATS/IDSA/CDC guidelines (n=25). LTBI in healthy close-contacts of PTB patients was confirmed by positive results in QuantiFERON®-TB Gold Plus (QFT®-Plus) test (QIAGEN, Hilden, Germany) (n=17).
For histological studies, lung sections were obtained from participants with PTB from the Tuberculosis Outpatient Clinic at the National Institute of Respiratory Diseases (INER) in Mexico City. Samples were obtained from participants before anti-Mtb treatment.
All participants or their legal guardians provided written informed consent to participate in the study. Blood samples were processed and stored according to the Mexican Constitution law NOM-012-SSA3-2012, which establishes criteria for the execution of clinical research projects in humans. The current study was reviewed and approved by the Institutional Review Board of the INER (project number B04-15). Information about the donor’s sex, age, and TB infection status is listed in Supplementary Table S3.
METHOD DETAILS
In vitro stimulation of CD27+ NK cells.
Human peripheral blood mononuclear cells (PBMCs) were isolated by centrifugation gradient using Ficoll-Paque™ PLUS (GE Healthcare-Life Sciences, PA, USA). PBMCs from LTBI and PTB individuals were plated at a density of 2.5 X 106 cells per ml in RPMI-1640 supplemented with 1% FBS and cultured with 25 μg/ml Mtb H37Rv cell wall at 37°C, 5% CO2, during 48hrs. For some experiments, PBMCs from healthy donors were used and culture in the conditions described above.
Flow cytometry on circulating NK cells.
Freshly isolated human PBMCs were incubated with fluorochrome-labeled anti-human CD3 (AB_2562046), CD14 (AB_830677), CD19 (AB_314236), CD56 (AB_604101), and CD27 antibodies. Cells were acquired using a BD FACSTM Aria II cytometer (BD Biosciences, USA) and gated based on their forward and side scatter characteristics, as well as on fluorescence minus one (FMO) controls for each specific marker using the FACSDiva software. Human NK cells were defined as CD3−CD14−CD56+. The frequency of specific cell types was calculated using Flow Jo (Flow Jo, LLC, Ashland, OR).
Isolation of peripheral human CD27+ NK cells.
Untouched NK cells were enriched from single-cell suspensions of PBMCs obtained from three LTBI individuals and three healthy controls. For this purpose, we used a commercial kit of antibodies coupled to magnetic beads (NK Cell Isolation Kit, human, Miltenyi Biotec, Germany), magnetic columns (LS Column, Miltenyi Biotec, Germany), and a magnetic separator (MidiMACS™ Separator, Miltenyi Biotec, Germany), following the manufacturer’s instructions. Enriched NK cells were incubated with fluorochrome-labeled anti-human CD3 (AB_2562046), CD14 (AB_830677), CD19 (AB_314236), CD56 (AB_604101), and CD27 antibodies, as described above. After gating and death cell exclusion, CD27+ and CD27− NK cells fractions were sorted using a BD FACSTM Aria II cytometer (BD Biosciences, USA). The purity of sorted NK cells was confirmed by flow cytometry and was between 92–100%. Isolated CD27+ NK cells were immediately transferred into tubes containing TRIzol® reagent (Zymo Research, USA) and frozen until use. RNA from isolated CD27+ and CD27− NK cells was extracted using the Direct-zol RNA Miniprep kit (Zymo Research, USA). cDNA was synthesized using ABI reverse transcription reagents (Thermo Fisher) on a Bio-Rad DNA Engine Thermal Cycler. Gene expression was assessed using primers from ABI and run on a Viia7 Real-Time PCR System (Life Technologies, Thermo Fisher). The expression of genes of interest relative to housekeeping gene GAPDH was calculated.
Isolation of lung single-cells from macaques.
At necropsy, lung tissues were collected and digested with DNase I and Liberase at 37°C for 30–45 mins. Digested lung tissue was gently dispersed by passage through a 70 μm pore size nylon tissue strainer (Falcon; BD Biosciences); the resultant single-cell suspension was treated with ACK lysis buffer to remove any residual RBC, washed twice, and counted, as previously described (Foreman et al., 2016; Gautam et al., 2018; Kaushal et al., 2015; Kuroda et al., 2018; Mehra et al., 2015). Cells prepared in this way were stored at −70°C and then used for downstream processing of scRNA-seq and CyTOF. Due to cell damage during freezing and limited cell recovery, not all samples passed QC for scRNA-seq and/or CyTOF (Table S2).
Single cell RNA: Library generation and sequencing.
scRNA-seq was done according to the manufacturer instructions (10X genomics). Briefly, after quickly thawing the frozen lung single-cell suspension in a water bath, 2X106 cells were taken for downstream processing. Cell suspensions were enriched for CD45 by using NHP CD45-microbeads according to the manufacturer’s instruction (Milteny Biotec). After then, CD45+ enriched lung single cell suspensions were subjected to droplet-based massively parallel scRNA-seq using Chromium Single Cell 3’ (v3) Reagent Kit in the BSL-3 laboratory as per manufacturer’s instructions (10x Genomics). Briefly, cell suspensions were loaded at 1,000 cells/μl to capture 10,000 cells/lane. The 10x Chromium Controller generated GEM droplets, where each cell was labeled with a specific barcode, and each transcript was labeled with a unique molecular identifier (UMI) during reverse transcription. The barcoded cDNA was isolated and removed from the BSL-3 space for library generation. The cDNA underwent 11 cycles of amplification, followed by fragmentation, end repair, A-tailing, adapter ligation, and sample index PCR as per the manufacturer’s instructions. Libraries were sequenced on a NovaSeq S4 (200 cycles) flow cell, targeting 50,000 read pairs/cell.
Single cell RNA-seq: data processing.
The Cell Ranger Single-Cell Software 3.0 available on the 10X website was used to perform sample demultiplexing. We aligned resulting fastq files on mmul10 genome (Genebank, https://www.ncbi.nlm.nih.gov/assembly/GCF_003339765.1/), with the addition of Ensembl mmul8 mitochondrial genes for GTF file) with cellranger count. For each sample, the recovered-cells parameter was set to 10,000 cells that we expected to recover for each library.
We used R package Seurat 3 (Stuart et al., 2019) for downstream analysis of count matrixes that we got as output from cellranger count. We filtered cells that (1) had more than 12.5% (samples KP77, KR71), 20% (samples KM67, KN08, LD09, LE99, 33935, KP87), 25% (samples GP50, HV02), 30% (samples KN21, 31810) of mitochondrial gene content, (2) had less than 725 detected genes. We chose different thresholds for mitochondrial gene content due to its different distributions between samples. Data was log-normalized with a scale factor of 104. The most variable genes were detected by the FindVariableFeatures function and used for subsequent analysis. Latent variables (number of UMI’s and mitochondrial content) were regressed out using a negative binomial model (function ScaleData). Principle component analysis (PCA) was performed with the RunPCA function. A UMAP dimensionality reduction was performed on the scaled matrix (with most variable genes only) using the first 20 PCA components to obtain a two-dimensional representation of the cell states. For clustering, we used the functions FindNeighbors (20 PCA) and FindClusters (resolution 0.25). To identify marker genes, we used FindAllMarkers to compare cluster against all other clusters, and FindMarkers to compare selected clusters. For each cluster, only genes that were expressed in more than 15% of cells with at least 0.15-fold differences were considered. For heatmap representation, the mean expression of markers inside each cluster for each sample was used. Heatmaps were built with Phantasus software (https://artyomovlab.wustl.edu/phantasus/). To re-analyze myeloid and lymphoid subpopulations, we pool clusters that we identified as belonging to the myeloid or lymphoid origin. After that, we re-run PCA, tSNE, and clustering to get a better resolution for the analysis.
CyTOF: antibody titration.
Most metal conjugated antibodies in the panel were available from Fluidigm. Antibodies that were not available from Fluidigm or that had to be placed on metals unavailable through Fluidigm were conjugated by labeling purified antibodies with the Maxpar Antibody Labeling kit (Fluidigm) according to the manufacturer’s protocol. The Key Resource table lists the antibody clone, manufacturer, and metal isotope. All antibodies were titrated for use by initially utilizing dilutions of 5–10 non-overlapping molecular weight antibodies on non-experimental macaque specimens avoiding +1 and +16 channels for each antibody. Abundant proteins can create “spillover” into molecular weight gates that are 1 or 2 values higher or lower than the actual isotopically pure metal. Manufacturer recommendations typically result in too high of a concentration for abundant surface proteins on cells like AMs that are being evaluated with high sensitivity antibodies, like HLA-DR. Also, over time the isotopic metals can become oxidized and the oxidized isotope is also detected by the mass spectrometer. This is referred to as +16 interference as the oxidized isotope has a molecular weight that is one-oxygen greater than the initial isotope. In the study of macrophages, these titration steps are important as most manufacturers have titrated the antibodies to detect these surface markers in cells like monocytes that produce much lower quantities of many of the proteins of interest. All antibodies with a positive signal (1 log over the background in any cell type) were kept in the final panel.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | RRID | Catalog Number |
|---|---|---|---|
| Antibodies | |||
| CD11b, 144Nd, Anti-Human CD11b/ Mac-1 (ICRF44) | Fluidigm | AB_2714152 | Cat# 3144001B |
| CD11c, 146Nd, Anti-Human CD11c (3.9) | Fluidigm | Cat# 3146014B | |
| CD20, 147Sm, Anti-Human CD20 (2H7) | Fluidigm | Cat# 3147001B | |
| CD16, 148Nd, Anti-Human CD16 (3G8) | Fluidigm | AB_2661791 | Cat# 3148004B |
| CD86, 150Nd, Anti-Human CD86/ B7.2 (IT2.2) | Fluidigm | AB_2687852 | Cat# 3150020B |
| CD123, 151Eu, Anti-Human CD123/ IL-3R (6H6) | Fluidigm | AB_2661794 | Cat# 3151001B |
| CD95, 152Sm, Anti-Human CD95/ Fas (DX2) | Fluidigm | Cat# 3152017B | |
| CD194(CCR4), 153Eu, Anti-Human CD194/ CCR4 (L291H4) | Fluidigm | Cat# 3153030A | |
| CD163, 154Sm, Anti-Human CD163 (GHI/61) | Fluidigm | AB_2661797 | Cat# 3154007B |
| CD279(PD1), 155Gd, Anti-Human CD279/ PD-1 (EH12.2H7) | Fluidigm | AB_2811087 | Cat# 3155009B |
| CD4, 156Gd, Anti-Human CD4 (L200) | BDBiosciences | AB_393787 | Cat# 550625 |
| CD169, 158Gd, Anti-Human CD169 (7-239) | Fluidigm | Cat# 3158027B | |
| CD28, 160Gd, Anti-Human CD28 (CD28.2) | Fluidigm | AB_2868400 | Cat# 3160003B |
| CD8, 162Dy, Anti-Human CD8 (RPA-T8) | Fluidigm | AB_2811089 | Cat# 3162015B |
| Siglec-8, 164Dy, Anti-Human Siglec 8 (7C9) | Fluidigm | Cat# 3164017B | |
| CD40, 165Ho, Anti-Human CD40 (5C3) | Fluidigm | Cat# 3165005B | |
| CD27, 167Er, Anti-Human CD27 (L128) | Fluidigm | AB_2811093 | Cat# 3167006B |
| CD206, 168Er, Anti-Human CD206/ MMR (15-2) | Fluidigm | AB_2661805 | Cat# 3168008B |
| NKG2A, 169Tm, Anti-Human CD159a/NKG2A (Z199) | Fluidigm | AB_2756426 | Cat# 3169013B |
| CD3, 170Er, Anti-Human CD3 (UCHT1) | Fluidigm | AB_2811085 | Cat# 3170001B |
| GranzymeB, 171Yb, Anti-Human Granzyme B (GB11) | Fluidigm | AB_2687652 | Cat# 3171002B |
| CD66abce(CAECAM-1), 173Yb, Anti-Human CD66abce | Miltenyi Biotec | AB_2751886 | Cat# 130-119-848 |
| HLA-DR, 174Yb, Anti-Human HLA-DR (L243) | Fluidigm | AB_2665397 | Cat# 3174001B |
| CD14, 175Lu, Anti-Human CD14 (M5E2) | Fluidigm | AB_2811083 | Cat# 3175015B |
| CD45, 176Yb, Anti-NHP CD45 (D058-1283), conjugated in lab with Maxpar kit | BDBiosciences | AB_394433 | Cat# 552566 |
| CD47, 209Bi, Anti-Human CD47 (CC2C6) | Fluidigm | Cat# 3209004B | |
| CD45, BUV395, Anti-NHP CD45 (D058-1283) | BD Biosciences | Cat# 564099 | |
| HLA-DR, PE-Cy7, Anti-Human HLA-DR (L243 ) | BD Biosciences | AB_399973 | Cat# 335795 |
| HLA-DR, APC-Cy7, Anti-Human HLA-DR (L243) | BD Biosciences | AB_399974 | Cat# 335796 |
| CD3, AF700, Anti-Human CD3 (SP34-2) | BD Biosciences | AB_396938 | Cat# 557917 |
| CD20, AF700, Anti-Human CD20 (2H7) | BD Biosciences | AB_1727447 | Cat# 560631 |
| CD163, PE, Anti-Human CD163 (GHI/61) | BD Biosciences | AB_396296 | Cat# 556018 |
| CD206, BV650, Anti-Human CD206 (19.2) | BD Biosciences | AB_2740298 | Cat# 740598 |
| CD16, BV510, Anti-Human CD16 (3G8) | BD Biosciences | AB_2744296 | Cat# 563830 |
| CD14, BV711, Anti-Human CD14 (M5E2) | Biolegend | AB_2562909 | Cat# 301838 |
| CD1c, BV421, Anti-Human CD1c (L161) | Biolegend | AB_10962909 | Cat# 331526 |
| CD123, PerCP-Cy5.5, Anti-Human CD123 (7G3) | BD Biosciences | AB_1645547 | Cat# 558714 |
| C1q, FITC, Anti-human C1q FITC | Abcam | AB_304387 | Cat# ab4223 |
| CD274, BV 711, Anti-human CD274 (29E.2A3) | Biolegend | AB_2565764 | Cat# 329722 |
| CD38, APC, Anti-Human CD38 (OKT10) | NHP Reagent Resource | AB_2819277 | Cat# PR-3801 |
| CD66abce, PE, Anti-Human CD66abce (TET2) | Miltenyi Biotec | AB_2733834 | Cat# 130-117-811 |
| CD3, FITC, Anti-human CD3 (UCHT1) | Biolegend | AB_2562046 | Cat# 300440 |
| CD14, FITC, Anti-human CD14 (HCD14) | Biolegend | AB_830677 | Cat# 325604 |
| CD19, FITC, Anti-human CD19 (HIB19) | Biolegend | AB_314236 | Cat# 302206 |
| CD56, PE, Anti-human CD56 (HCD56) | Biolegend | AB_604101 | Cat# 318306 |
| CD27, AF700, Anti-human CD27 (O323) | Biolegend | Cat# 302814 | |
| NKG2a, Unconjugated, Anti-human NKG2a (Polyclonal) | Invitrogen | AB_11154787 | Cat# PA5-21949 |
| CD3, PE, Anti-Human CD3 (SP34-2) | BD Biosciences | AB_394342 | Cat# 552127 |
| CD14, PE, Anti-Human CD14 (M5E2) | BD Biosciences | AB_10924593 | Cat# 561707 |
| CD20, PE, Anti-Human CD20 (LT20) | Miltenyi Biotec | AB_2726143 | Cat# 130-113-374 |
| CD123, Anti‑Human IL3RA / CD123 (BSB‑59) | Lifespan Biosciences | Cat# LS‑C311924 | |
| Ig G, Cy3, Donkey Anti-mouse Ig G | Jackson ImmunoResearch Laboratories | AB_2340817 | Cat# 715-166-151 |
| HLADR, Anti-human HLADR (YE2/36-HLK) | Lifespan Biosciences | AB_11188152 | Cat# LS-B7229 |
| IgG, AF 488, Donkey Anti-rat IgG | Thermo Fisher Scientific | AB_141709 | Cat# A-21208 |
| CD163, Anti-human CD163 (10D6) | Thermo Fisher Scientific | AB_10982556 | Cat# MA5-11458 |
| S100/A8, Anti-human S100/A8 ( EPR3554) | Abcam | AB_2050283 | Cat# ab92331 |
| Ig G, FITC, donkey Anti-rabbit Ig G | Jackson ImmunoResearch Laboratories | AB_2340597 | Cat# 711-096-152 |
| CD3, Anti-human CD3 (M-20) | Santa Cruz | AB_631128 | Cat# sc1127 |
| RORγt, Anti-human RORγt (6F3.1) | Milipore Sigma-Aldrich | AB_11205416 | Cat#MABF81 |
| CD20, Anti -Human CD20 (L26) | Lifespan Biosciences | AB_1664671 | LS‑B2605 |
| IgG, biotin, donkey Anti-rabbit IgG | Jackson ImmunoResearch Laboratories | Cat#711-065-162 | |
| Streptavidin-Alexa Fluor 680 | ThermoFisher Scientific | Cat# S32358 | |
| CD20, Anti-human CD20 (L26) | GeneTex | AB_368330 | Cat# GTX29475 |
| Ig G, FITC, donkey Anti-mouse Ig G | Jackson ImmunoResearch Laboratories | AB_2340792 | Cat# 715-095-150, |
| IDO1, Unconjugated, Anti-Human IDO1(Polyclonal) | Millipore Sigma | AB_1846220 | Cat# HPA023072 |
| CD66abce, FITC, Anti-Human CD66abce (TET2) | Miltenyi Biotec | AB_2727588 | Cat# 130-116-522 |
| CD163, Unconjugated, Anti-Human CD163 (EDHu-1) | Bio-Rad laboratories | AB_2074540 | Cat# MCA1853 |
| CD68, Unconjugated, Anti-Human CD68 (KP1) | Thermo Fisher Scientific | AB_10987212 | Cat# MA513324 |
| CD206, Alexa Fluor 488, Anti-Human CD206 (15-2) | BioLegend | AB_571875 | Cat# 321114 |
| Goat anti-rabbit IgG (H+L), Alexa Fluor 488, (Polyclonal) | Thermo Fischer Scientific | AB_143165 | Cat# A11008 |
| goat anti-mouse IgG (H+L), Alexa Fluor 555, (Polyclonal) | Thermo Fischer Scientific | AB_141822 | Cat# A21422 |
| goat anti-rabbit IgG (H+L), Alexa Fluor 555, (Polyclonal) | Thermo Fischer Scientific | AB_774740 | Cat # A32732 |
| donkey anti-goat IgG (H+L), Alexa Fluor 568, (Polyclonal) | Thermo Fischer Scientific | AB_142581 | Cat# A11057 |
| goat anti-rabbit IgG (H+L), Alexa-Fluor 647, (Polyclonal) | Thermo Fischer Scientific | AB_2535813 | Cat# A21245 |
| donkey anti-rabbit IgG (H+L), Alexa-Fluor 647, (Polyclonal) | Jackson ImmunoResearch Laboratories | AB_2340625 | Cat# 711-606-152 |
| Bacterial and Virus Strains | |||
| Mycobacterium tuberculosis | BEI resources | ||
| Biological Samples | |||
| Human samples | Instituto Nacional de Enfermedades Respiratorias “Ismael Cosío Villegas” (INER) | Refer to Table 3 for identifiiers | |
| Chemicals, Peptides, and Recombinant Proteins | |||
| Critical Commercial Assays | |||
| Deposited Data | |||
| scRNA-seq, raw and analyzed data | This paper | GEO: GSE149758, Synapse: syn21988273 | |
| CyTOF, raw and analyzed data | This paper | Synapse: syn21988273 | |
| Experimental Models: Cell Lines | |||
| Experimental Models: Organisms/Strains | |||
| Rhesus macaque (Macaca mulatta) | Tulane National Primate Research Center and Sothwest National primate Center | Refer to Table 1 for identifiers | |
| Oligonucleotides | |||
| Human GAPDH | Thermo Fisher Scientific | Cat# Hs02786624_g1 | |
| Human IL-22 | Thermo Fisher Scientific | Cat# Hs01574154_m1 | |
| Human Perforin PRF1 | Thermo Fisher Scientific | Cat# Hs00169473_m1 | |
| Human Granzyme B | Thermo Fisher Scientific | Cat# Hs00188051_m1 | |
| Human IFN gamma | Thermo Fisher Scientific | Cat# Hs00989291_m1 | |
| Recombinant DNA | |||
| Software and Algorithms | |||
| Cellranger 3.1 | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest | |
| Seurat 3 | Stuart et al., 2019 | Seurat 3 (CRAN) | |
| ggplot2 | tidyverse | https://cloud.r-project.org/package=ggplot2 | |
| Cytobank | Cytobank | https://www.cytobank.org | |
| Rphenograph | Levine et al., 2015 | https://github.com/JinmiaoChenLab/Rphenograph | |
| CATALYST | Chevrier et al., 2018 | https://bioconductor.org/packages/CATALYST/ | |
| Phantasus | https://artyomovlab.wustl.edu/phantasus | ||
| Other | |||
CyTOF: antibody labeling of frozen cells.
Each cryovial of cells was quickly thawed at 37°C and transferred to 10 ml of RPMI plus media (RPMI, 10% FBS, 10U/ml heparin, 25U/ml benzonase). Cells were pelleted (300g, 10 mins, Room temperature: RT), supernatant aspirated, and resuspended in 1 ml CyTOF PBS (10X Rockland PBS, Rockland Immunochemicals, Limerick, PA diluted to 1X in MaxPar Water, Fluidigm Corporation, South San Francisco, CA). Cell number and viability were determined by trypan dye exclusion method manual counting (Table S1).
Cells suspended in CyTOF PBS were labeled with 1.25 μl of 1mM stock of Cell-ID Cisplatin (1.25 μM final concentration, Fluidigm) for 1 min at RT. Staining was quenched after the incubation with the addition of 5 ml of MaxPar Cell Staining Buffer (CSB, Fluidigm). Cells were pelleted (300g, 10 mins, RT), supernatant aspirated and resuspended in 50 μl in CSB.
The antibody cocktail for the macaque panel was diluted in CSB to the previously determined titrated concentration (Resource Table). Sufficient volume was prepared so that each sample was labeled with 50 μl of the antibody cocktail. For these experiments, 2 separate staining cocktails were utilized for cell surface and intracellular antigens. 50 μl of the cell suspension was pipetted into a 5 ml round-bottom polypropylene tube, followed by 50 μl of the cell surface antibody cocktail. This was incubated 1 hr at 4°C. Following the incubation, the cells were washed twice with 2 ml CSB, pelleted (500g, 5 mins, 4°C), and supernatant aspirated. Cells were resuspended in 2 ml 4% paraformaldehyde (16% solution, EM grade, Electron Microscopy Sciences, Hatfield, PA diluted in CyTOF PBS) and incubated for 40 mins at RT. Following fixation, cells were washed with 2 ml CSB, pelleted, and supernatant aspirated. Cells were incubated with 50 μl CSB and 50 μl intracellular antibody cocktail in fix and perm buffer and incubated for 1 hr at 4°C. Cells were washed with CSB, pelleted and supernatant aspirated.
Because specimens could contain Mtb and additional fixation (Cells were resuspended in 2 ml 4% paraformaldehyde and incubated for 40 mins at RT) was performed as previous experience had determined some viable Mtb after the existing fixation steps. Although this lessens staining intensity, the unshielded nature of the cytometer requires this for safety.
MaxPar Intercalator-IR (Fluidigm) was prepared in 1% formalin to a final concentration of 41.7nM (3000X dilution of 125 μM stock). 1 ml of solution was added to each tube. Cells were incubated in the dark at 4°C. The following day, cells were washed twice with 2 ml CSB, then counted. Cells were subsequently washed with 2 ml MaxPar Water, pelleted and supernatant aspirated. The final cell concentration was adjusted to 1×106 cells/ml in MaxPar Water with EQ calibration beads (Fluidigm) and filtered into cell strainer cap tubes. Data were then acquired on CyTOF. Prior experiments on formalin alone revealed no free metals that interfered with CyTOF.
CyTOF: data processing.
Individual samples were manually gated using Cytobank to exclude normalization beads, cell debris, dead cells, and doublets for the identification of CD45+ cisplatin-negative cells for further downstream analyses. We used viSNE(Amir el et al., 2013) from Cytobank for dimensionality reduction and exported data for downstream analysis in R. We normalized data using the CATALYST package (Chevrier et al., 2018) with arcsinh-transformation (cofactor of five). Data were clustered with Phenograph package(Levine et al., 2015), using k = 110. Visualization was done using the R package ggplot2. Similarly to the single cell RNA-seq, we separated lymphoid and myeloid cells to process them separately.
Flow cytometry.
Flow cytometry was performed as previously described on single-cell preparation from macaque lung collected at the endpoint or from macaque PBMC or human PBMCs. A comprehensive list of antibodies used in these experiments is provided in the Key Resource table. Briefly, after thawing the frozen cells, the cells were first stained in 96-well U-bottom plates in the dark using the LIVE/DEAD™ Fixable Near-IR or LIVE/DEAD™ Fixable Aqua Dead Cell Stain Kit (ThermoFisher Scientific) for 15 minutes followed by washing as per the manufacturer’s protocol. Then the cells were stained for surface markers (Resource Table) for 20 minutes at 4°c followed by washing and fixation with 1% formalin. FMO controls and single color control were also prepared from the available samples. Samples were run on a four-laser BD Fortessa-X20 flow cytometer and analyzed using FlowJo (FlowJo LLC.).
Multiplex cytokine analysis of BAL supernatant.
Rhesus macaques BAL fluids were analyzed for multiple cytokines and chemokines by Luminex using established protocols for Old World primates (Giavedoni, 2005).
Histology
FFPE lung sections of macaques with PTB or LTBI were stained with hematoxylin and eosin, and inflammatory features were evaluated by light microscopy. Inflammatory lesions were outlined with the automated tool of the Zeiss Axioplan 2 microscope (Carl Zeiss) and the percentage of inflammation was calculated by dividing the inflammatory area by the total area of individual lung lobes.
B cell follicle quantitation
FFPE sections from individuals with and PTB and macaques with LTBI and PTB were processed to remove paraffin and then hydrated in 96% alcohol and phosphate-buffered saline. Antigens were retrieved with a DakoCytomation Target Retrieval Solution (Dako) and non-specific binding was blocked using 5% (v/v) normal donkey serum and Fc block (BD Pharmingen). Sections were then probed with primary goat anti-CD3ε (AB_631128, clone M-20, Santa Cruz Biotechnology), mouse anti-human RORγt (AB_11205416, clone 6F3.1, Millipore Sigma-Aldrich), and rabbit anti-human CD20 (AB_1664671, LS-B2605-125, Lifespan Biosciences). Slides were incubated with primary antibodies overnight at room temperature in a humid chamber. The next day, slides were briefly washed in PBS, and primary antibodies were revealed with Alexa Fluor 568-conjugated donkey anti-goat IgG (AB_142581, A11057, ThermoFisher Scientific), FITC-conjugated donkey anti-mouse IgG (AB_2340792,715-095-150, Jackson ImmunoResearch Laboratories), biotin-conjugated donkey anti-rabbit (711-065-162, Jackson ImmunoResearch Laboratories). Finally, streptavidin Alexa Fluor 680 (S32358, ThermoFisher Scientific) was added to the slides to visualize CD20 for human or non-human primate sections. Slides were washed in PBS and mounted with Vectashield antifade mounting medium with DAPI (AB_2336790, Vector Laboratories, H-1200). For B cell follicles analyses, follicles were outlined with the automated tool of the Zeiss Axioplan 2 microscope (Zeiss), and the average size of the B cell follicles was calculated in μm2.
Quantitation of immune cell types histologically within lung tissue.
To detect pDCs, 5 μm lung sections of non-human primates and PTB patients were incubated with mouse anti-human IL3RA/CD123 (clone BSB-59, Lifespan Biosciences) and rat-anti human HLA-DR (AB_11188152, clone YE2/36-HLK, LS-B7229, Lifespan Biosciences). Slides were incubated with primary antibodies overnight at RT in a humid chamber. The next day, slides were briefly washed in PBS, and primary antibodies were revealed with Cy3 donkey anti-mouse IgG (AB_2340817, 715-166-151, Jackson ImmunoResearch Laboratories), Alexa Fluor 488 donkey anti-rat IgG (AB_141709, A-21208, Thermo Fisher Scientific), Alexa fluor 568 donkey anti-goat IgG (AB_142581, A-11057, Thermo Fisher Scientific), FITC donkey anti-mouse IgG (AB_2340792, 715-095-150, Jackson ImmunoResearch Laboratories) and Alexa Fluor 647 donkey anti-rabbit IgG (AB_2340625, 711-606-152, Jackson ImmunoResearch Laboratories). 5 μm sections were stained with mouse monoclonal antibodies specific for CD163 (AB_10982556, MA5-11458, Thermo Fisher Scientific) and rabbit anti-S100A/8 (AB_2050283, EPR3554, Abcam). Primary antibodies were detected with Cy3-donkey anti-mouse IgG (AB_2340817, 715-166-151, Jackson ImmunoResearch Laboratories) and FITC donkey anti-rabbit IgG (AB_2340597, 711-096-152, Jackson ImmunoResearch Laboratories). Slides were washed in PBS and mounted with Vectashield antifade mounting medium with DAPI (Vector Laboratories, H-1200). pDCs were counted in 3−5 random 200X fields in each slide. Representative 200X images were taken with an Axioplan Zeiss Microscope and recorded with a Hamamatsu camera. For the detection of NK cells, a rabbit polyclonal NKG2a antibody (AB_11154787, Thermo Fischer Scientific, 1:100, 37°C for 2 h) was used. PE-conjugated mouse anti-human CD3 (AB_394342, clone SP34–2, 1:10, BD Biosciences); mouse anti-human CD14, (AB_10924593, clone ME52, 1:5, BD Biosciences), mouse anti-human CD66abce (AB_2727588, clone TET-2, 1:20, Miltenyi Biotech) and mouse anti-human CD20 (AB_2726143, clone LT20, 1:20, Miltenyi Biotech) antibodies were used for identification of T-cells, monocytes/ macrophages, neutrophils and B-cells respectively. Other primary antibodies used in this study includes-rabbit polyclonal IDO-1 antibody (AB_1846220, 1:1000, Millipore Sigma), mouse anti-human CD163 (AB_2074540, Clone EdHu-1, 1:40, Bio-Rad laboratories), mouse CD68 (AB_10987212, 1:50, Thermo Fisher Scientific), Alexa Fluor 488-mouse anti-human CD206 (AB_571875, clone 15–2, 1:10, Biolegend), rabbit polyclonal C1q-FITC conjugated (AB_304387, 1:10, Abcam). Goat anti-rabbit IgG (H+L), Alexa Fluor 488 conjugate (AB_143165); goat anti-mouse IgG (H+L), Alexa Fluor 555 conjugate (AB_141822); goat anti-rabbit IgG (H+L), Alexa Fluor 555 conjugate (AB_774740); goat anti-rabbit IgG (H+L), Alexa-Fluor 647 conjugate secondary antibodies (AB_2535813) (Thermo Fisher Scientific, 1:500) were used for labeling primary antibodies in these experiments. DAPI (1:5000, Thermo Fisher Scientific) was used to stain the nuclei, and mounting was done with the Prolong Diamond Antifade mountant (Thermo Fisher Scientific). Zeiss LSM 800 confocal microscope was used for the acquisition of the images of the stained sections.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Analyses.
All the statistical analysis was done using R package. No methods were applied to determine assumptions of the statistical approach. Student’s T-test was used for analysis between two groups, with Holms correction in case of multiple comparisons. One-way ANOVA with a post-hoc Tukey test was used to compare means between multiple groups. For correlation analysis Pearson’s correlation coefficient was used. All of the statistical details of experiments can be found in the figure legends including the statistical tests used, exact value of n (number of animals or human samples used). Data represented as mean± SD (standard deviation). *p < 0.05, **p < 0.01, ***p < 0.001, NS = not significant.
Supplementary Material
Supplementary Table 1: Description of macaque samples, clinical parameters, and annotation of samples used in multiple analyses in the study. Related to Figure 1.
Supplementary Table 2: Description of macaque samples and QC values from scRNA-seq analysis. Related to Figure 1.
Supplementary Table 3: Description of human sample demographic that are part of this study. Related to Figure 4.
Highlights.
Characterization of the lung landscape of tuberculosis (TB) latency and disease.
Lung CD27+ NK cells are a key feature associated with protection during TB latency.
TB disease features lung pDCs, IFN-responsive macrophages, and activated T cells.
Acknowledgement.
This work was supported by Washington University in St.Louis School of Medicine, NIH grant HL105427, AI111914-02, AI134236-02 and AI123780 to S.A.K. and D.K., the Department of Molecular Microbiology, Washington University School of Medicine, and Stephen I. Morse Fellowship to S.D. E.E. was supported by Shawn Hu and Angela Zeng Graduate Fellowship. J.A.C.P was supported by the National Council of Science and Technology of Mexico (CONACyT, CVU 737347). J.R.-M. was supported by funds of the Department of Medicine, University of Rochester, and NIH grant U19 AI91036. None of the authors have conflicts to report.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interest. The authors declare no competing interests.
References
- (WHO), W.H.O. (2018). Global tuberculosis report 2018. https://wwwwhoint/tb/publications/global_report/en/.
- Ahmed M, Thirunavukkarasu S, Rosa BA, Thomas KA, Das S, Rangel-Moreno J, Lu L, Mehra S, Mbandi SK, Thackray LB, et al. (2020). Immune correlates of tuberculosis disease and risk translate across species. Sci Transl Med 12, eaay0233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir el AD, Davis KL, Tadmor MD, Simonds EF, Levine JH, Bendall SC, Shenfeld DK, Krishnaswamy S, Nolan GP, and Pe’er D (2013). viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat Biotechnol 31, 545–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonelli LR, Gigliotti Rothfuchs A, Goncalves R, Roffe E, Cheever AW, Bafica A, Salazar AM, Feng CG, and Sher A (2010). Intranasal Poly-IC treatment exacerbates tuberculosis in mice through the pulmonary recruitment of a pathogen-permissive monocyte/macrophage population. J Clin Invest 120, 1674–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bafica A, Scanga C, Feng C, Leifer C, Cheever A, and Sher A (2005). TLR9 regulates Th1 responses and cooperates with TLR2 in mediating optimal resistance to Mycobacterium tuberculosis. J Exp Med 202, 1715–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton GM (2007). Viral recognition by Toll-like receptors. Semin Immunol 19, 33–40. [DOI] [PubMed] [Google Scholar]
- Berry A, Matthews L, Jangani M, Plumb J, Farrow S, Buchan N, Wilson PA, Singh D, Ray DW, and Donn RP (2010). Interferon-inducible factor 16 is a novel modulator of glucocorticoid action. FASEB J 24, 1700–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucsan AN, Chatterjee A, Singh DK, Foreman TW, Lee TH, Threeton B, Kirkpatrick MG, Ahmed M, Golden N, Alvarez X, et al. (2019a). Mechanisms of reactivation of latent tuberculosis infection due to SIV coinfection. J Clin Invest 129, 5254–5260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucsan AN, Mehra S, Khader SA, and Kaushal D (2019b). The current state of animal models and genomic approaches towards identifying and validating molecular determinants of Mycobacterium tuberculosis infection and tuberculosis disease. Pathog Dis 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y, Sugimoto C, Arainga M, Alvarez X, Didier ES, and Kuroda MJ (2014). In vivo characterization of alveolar and interstitial lung macrophages in rhesus macaques: implications for understanding lung disease in humans. J Immunol 192, 2821–2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrington EM, Zhang JG, Sutherland RM, Vikstrom IB, Brady JL, Soo P, Vremec D, Allison C, Lee EF, Fairlie WD, et al. (2015). Prosurvival Bcl-2 family members reveal a distinct apoptotic identity between conventional and plasmacytoid dendritic cells. Proc Natl Acad Sci U S A 112, 4044–4049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevrier S, Crowell HL, Zanotelli VRT, Engler S, Robinson MD, and Bodenmiller B (2018). Compensation of Signal Spillover in Suspension and Imaging Mass Cytometry. Cell systems 6, 612–620.e615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper AM, Dalton DK, Stewart TA, Griffin JP, Russell DG, and Orme IM (1993). Disseminated tuberculosis in interferon gamma gene-disrupted mice. J Exp Med 178, 2243–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz A, Fraga AG, Fountain JJ, Rangel-Moreno J, Torrado E, Saraiva M, Pereira DR, Randall TD, Pedrosa J, Cooper AM, et al. (2010). Pathological role of interleukin 17 in mice subjected to repeated BCG vaccination after infection with Mycobacterium tuberculosis. J Exp Med 207, 1609–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foreman TW, Bucsan AN, Mehra S, Peloquin C, Doyle LA, Russell-Lodrigue K, Gandhi NR, Altman J, Day CL, Ernst JD, et al. (2020). Isoniazid and Rifapentine Treatment Eradicates Persistent Mycobacterium tuberculosis in Macaques. Am J Respir Crit Care Med 201, 469–477. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Foreman TW, Mehra S, LoBato DN, Malek A, Alvarez X, Golden NA, Bucsan AN, Didier PJ, Doyle-Meyers LA, Russell-Lodrigue KE, et al. (2016). CD4+ T-cell-independent mechanisms suppress reactivation of latent tuberculosis in a macaque model of HIV coinfection. Proc Natl Acad Sci U S A 113, E5636–5644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam US, Foreman TW, Bucsan AN, Veatch AV, Alvarez X, Adekambi T, Golden NA, Gentry KM, Doyle-Meyers LA, Russell-Lodrigue KE, et al. (2018). In vivo inhibition of tryptophan catabolism reorganizes the tuberculoma and augments immune-mediated control of Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 115, E62–e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giavedoni LD (2005). Simultaneous detection of multiple cytokines and chemokines from nonhuman primates using luminex technology. J Immunol Methods 301, 89–101. [DOI] [PubMed] [Google Scholar]
- Gopal R, Monin L, Slight S, Uche U, Blanchard E, Fallert Junecko A, B., Ramos-Payan R, Stallings CL, Reinhart TA, Kolls JK, et al. (2014). Unexpected Role for IL-17 in Protective Immunity against Hypervirulent Mycobacterium tuberculosis HN878 Infection. PLoS Pathog 10, e1004099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopal R, Monin L, Torres D, Slight S, Mehra S, McKenna KC, Fallert Junecko BA, Reinhart TA, Kolls J, Baez-Saldana R, et al. (2013). S100A8/A9 proteins mediate neutrophilic inflammation and lung pathology during tuberculosis. Am J Respir Crit Care Med 188, 1137–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grajkowska LT, Ceribelli M, Lau CM, Warren ME, Tiniakou I, Nakandakari Higa S, Bunin A, Haecker H, Mirny LA, Staudt LM, et al. (2017). Isoform-Specific Expression and Feedback Regulation of E Protein TCF4 Control Dendritic Cell Lineage Specification. Immunity 46, 65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graydon CG, Balasko AL, and Fowke KR (2019). Roles, function and relevance of LAG3 in HIV infection. PLoS Pathog 15, e1007429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubin MM, Esaulova E, Ward JP, Malkova ON, Runci D, Wong P, Noguchi T, Arthur CD, Meng W, Alspach E, et al. (2018). High-Dimensional Analysis Delineates Myeloid and Lymphoid Compartment Remodeling during Successful Immune-Checkpoint Cancer Therapy. Cell 175, 1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K, Ohba Y, Yanai H, Negishi H, Mizutani T, Takaoka A, Taya C, and Taniguchi T (2005a). Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature 434, 1035–1040. [DOI] [PubMed] [Google Scholar]
- Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, et al. (2005b). IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 434, 772–777. [DOI] [PubMed] [Google Scholar]
- Inngjerdingen M, Kveberg L, Naper C, and Vaage JT (2011). Natural killer cell subsets in man and rodents. Tissue Antigens 78, 81–88. [DOI] [PubMed] [Google Scholar]
- Jablonski KA, Amici SA, Webb LM, Ruiz-Rosado Jde D, Popovich PG, Partida-Sanchez S, and Guerau-de-Arellano M (2015). Novel Markers to Delineate Murine M1 and M2 Macrophages. PLoS One 10, e0145342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaitin DA, Keren-Shaul H, Elefant N, and Amit I (2015). Each cell counts: Hematopoiesis and immunity research in the era of single cell genomics. Seminars in immunology 27, 67–71. [DOI] [PubMed] [Google Scholar]
- Kahnert A, Hopken UE, Stein M, Bandermann S, Lipp M, and Kaufmann SH (2007). Mycobacterium tuberculosis triggers formation of lymphoid structure in murine lungs. J Infect Dis 195, 46–54. [DOI] [PubMed] [Google Scholar]
- Kaushal D, Foreman TW, Gautam US, Alvarez X, Adekambi T, Rangel-Moreno J, Golden NA, Johnson AM, Phillips BL, Ahsan MH, et al. (2015). Mucosal vaccination with attenuated Mycobacterium tuberculosis induces strong central memory responses and protects against tuberculosis. Nature communications 6, 8533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khader SA, Bell GK, Pearl JE, Fountain JJ, Rangel-Moreno J, Cilley GE, Shen F, Eaton SM, Gaffen SL, Swain SL, et al. (2007). IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat Immunol 8, 369–377. [DOI] [PubMed] [Google Scholar]
- Khader SA, Divangahi M, Hanekom W, Hill PC, Maeurer M, Makar KW, Mayer-Barber KD, Mhlanga MM, Nemes E, Schlesinger LS, et al. (2019). Targeting innate immunity for tuberculosis vaccination. J Clin Invest 129, 3482–3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khader SA, Guglani L, Rangel-Moreno J, Gopal R, Fallert Junecko BA, Fountain JJ, Martino C, Pearl JE, Tighe M, Lin Y. y., et al. (2011). IL-23 is required for long-term control of Mycobacterium tuberculosis and B cell follicle formation in the infected lung. J Immunol 187, 5402–5407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda MJ, Sugimoto C, Cai Y, Merino KM, Mehra S, Arainga M, Roy CJ, Midkiff CC, Alvarez X, Didier ES, et al. (2018). High Turnover of Tissue Macrophages Contributes to Tuberculosis Reactivation in Simian Immunodeficiency Virus-Infected Rhesus Macaques. J Infect Dis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine JH, Simonds EF, Bendall SC, Davis KL, Amir el AD, Tadmor MD, Litvin O, Fienberg HG, Jager A, Zunder ER, et al. (2015). Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis. Cell 162, 184–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin PL, and Flynn JL (2018). The End of the Binary Era: Revisiting the Spectrum of Tuberculosis. J Immunol 201, 2541–2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu CC, Wu TS, Hsu YJ, Chang CJ, Lin CS, Chia JH, Wu TL, Huang TT, Martel J, Ojcius DM, et al. (2014). NK cells kill mycobacteria directly by releasing perforin and granulysin. J Leukoc Biol 96, 1119–1129. [DOI] [PubMed] [Google Scholar]
- Lu YB, Xiao DQ, Liang KD, Zhang JA, Wang WD, Yu SY, Zheng BY, Gao YC, Dai YC, Jia Y, et al. (2017). Profiling dendritic cell subsets in the patients with active pulmonary tuberculosis. Mol Immunol 91, 86–96. [DOI] [PubMed] [Google Scholar]
- Luciw PA, Oslund KL, Yang XW, Adamson L, Ravindran R, Canfield DR, Tarara R, Hirst L, Christensen M, Lerche NW, et al. (2011). Stereological analysis of bacterial load and lung lesions in nonhuman primates (rhesus macaques) experimentally infected with Mycobacterium tuberculosis. Am J Physiol Lung Cell Mol Physiol 301, L731–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall A, Celentano A, Cirillo N, McCullough M, and Porter S (2017). Tissue-specific regulation of CXCL9/10/11 chemokines in keratinocytes: Implications for oral inflammatory disease. PLoS One 12, e0172821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaffrey EF, Donato M, Keren L, Chen Z, Fitzpatrick M, Jojic V, Delmastro A, Greenwald NF, Baranski A, Graf W, et al. (2020). Multiplexed imaging of human tuberculosis granulomas uncovers immunoregulatory features conserved across tissue and blood. bioRxiv, 2020.2006.2008.140426. [Google Scholar]
- Mehra S, Alvarez X, Didier PJ, Doyle LA, Blanchard JL, Lackner AA, and Kaushal D (2013). Granuloma Correlates of Protection Against Tuberculosis and Mechanisms of Immune Modulation by Mycobacterium tuberculosis. J Infect Dis 207, 1115–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehra S, Foreman TW, Didier PJ, Ahsan MH, Hudock TA, Kissee R, Golden NA, Gautam US, Johnson AM, Alvarez X, et al. (2015). The DosR Regulon Modulates Adaptive Immunity and is Essential for M. tuberculosis Persistence. Am J Respir Crit Care Med [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehra S, Golden NA, Stuckey K, Didier PJ, Doyle LA, Russell-Lodrigue KE, Sugimoto C, Hasegawa A, Sivasubramani SK, Roy CJ, et al. (2012). The Mycobacterium tuberculosis stress response factor SigH is required for bacterial burden as well as immunopathology in primate lungs. J Infect Dis 205, 1203–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandi B, and Behar SM (2011). Regulation of neutrophils by interferon-{gamma} limits lung inflammation during tuberculosis infection. J Exp Med 208, 2251–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips BL, Gautam US, Bucsan AN, Foreman TW, Golden NA, Niu T, Kaushal D, and Mehra S (2017). LAG-3 potentiates the survival of Mycobacterium tuberculosis in host phagocytes by modulating mitochondrial signaling in an in-vitro granuloma model. PLoS One 12, e0180413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitcher CJ, Hagen SI, Walker JM, Lum R, Mitchell BL, Maino VC, Axthelm MK, and Picker LJ (2002). Development and homeostasis of T cell memory in rhesus macaque. J Immunol 168, 29–43. [DOI] [PubMed] [Google Scholar]
- Roy Chowdhury R, Vallania F, Yang Q, Lopez Angel CJ, Darboe F, Penn-Nicholson A, Rozot V, Nemes E, Malherbe ST, Ronacher K, et al. (2018). A multi-cohort study of the immune factors associated with M. tuberculosis infection outcomes. Nature 560, 644–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz F, Heit A, Guggemoos S, Krug A, Mages J, Schiemann M, Adler H, Drexler I, Haas T, Lang R, et al. (2007). Interferon-regulatory-factor 1 controls Toll-like receptor 9-mediated IFN-beta production in myeloid dendritic cells. Eur J Immunol 37, 315–327. [DOI] [PubMed] [Google Scholar]
- Scott NR, Swanson RV, Al-Hammadi N, Domingo-Gonzalez R, Rangel-Moreno J, Kriel BA, Bucsan AN, Das S, Ahmed M, Mehra S, et al. (2020). S100A8/A9 regulates CD11b expression and neutrophil recruitment during chronic tuberculosis. J Clin Invest 130, 3098–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scriba TJ, Kalsdorf B, Abrahams DA, Isaacs F, Hofmeister J, Black G, Hassan HY, Wilkinson RJ, Walzl G, Gelderbloem SJ, et al. (2008). Distinct, Specific IL-17- and IL-22-Producing CD4+ T Cell Subsets Contribute to the Human Anti-Mycobacterial Immune Response. J Immunol 180, 1962–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth S, Oberdorfer L, Hyde R, Hoff K, Thies V, Worbs T, Schmitz S, and Forster R (2011). CCR7 essentially contributes to the homing of plasmacytoid dendritic cells to lymph nodes under steady-state as well as inflammatory conditions. J Immunol 186, 3364–3372. [DOI] [PubMed] [Google Scholar]
- Slight SR, Rangel-Moreno J, Gopal R, Lin Y, Fallert Junecko BA, Mehra S, Selman M, Becerril-Villanueva E, Baquera-Heredia J, Pavon L, et al. (2013). CXCR5+ T helper cells mediate protective immunity against tuberculosis. J Clin Invest 123, 712–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suliman S, Geldenhuys H, Johnson J, Hughes J, Smit E, Murphy M, Toefy A, Lerumo L, Hopley C, Pienaar B, et al. (2016). Bacillus Calmette-Guérin (BCG) Revaccination of Adults with Latent Mycobacterium tuberculosis Infection Induces Long-Lived BCG-Reactive NK Cell Responses. Journal of immunology (Baltimore, Md : 1950) 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo PA, Levitin HM, Miron M, Snyder ME, Senda T, Yuan J, Cheng YL, Bush EC, Dogra P, Thapa P, et al. (2019). Single-cell transcriptomics of human T cells reveals tissue and activation signatures in health and disease. Nat Commun 10, 4706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vankayalapati R, Garg A, Porgador A, Griffith DE, Klucar P, Safi H, Girard WM, Cosman D, Spies T, and Barnes PF (2005). Role of NK cell-activating receptors and their ligands in the lysis of mononuclear phagocytes infected with an intracellular bacterium. J Immunol 175, 4611–4617. [DOI] [PubMed] [Google Scholar]
- Veatch AV, and Kaushal D (2017). Opening Pandora’s Box: Mechanisms of Mycobacterium tuberculosis Resuscitation. Trends Microbiol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatasubramanian S, Cheekatla S, Paidipally P, Tripathi D, Welch E, Tvinnereim AR, Nurieva R, and Vankayalapati R (2017). IL-21-dependent expansion of memory-like NK cells enhances protective immune responses against Mycobacterium tuberculosis. Mucosal Immunol 10, 1031–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimmers F, Subedi N, van Buuringen N, Heister D, Vivie J, Beeren-Reinieren I, Woestenenk R, Dolstra H, Piruska A, Jacobs JFM, et al. (2018). Single-cell analysis reveals that stochasticity and paracrine signaling control interferon-alpha production by plasmacytoid dendritic cells. Nat Commun 9, 3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman CJ, Wang Y, El Kasmi KC, Pardoll DM, Murray PJ, Drake CG, and Vignali DA (2009). LAG-3 regulates plasmacytoid dendritic cell homeostasis. J Immunol 182, 1885–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zak DE, P.-N.A., Scriba TJ, Thompson E, Suliman S, Amon LM, Mahomed H, Erasmus M, Whatney W, Hussey GD, Abrahams D, Kafaar F, Hawkridge T, Verver S, Hughes EJ, Ota M, Sutherland J, Howe R, Dockrell HM, Boom WH, Thiel B, Ottenhoff THM, Mayanja-Kizza H, Crampin AC, Downing K, Hatherill M, Valvo J, Shankar S, Parida SK, Kaufmann SHE, Walzl G, Aderem A, Hanekom WA, for other members of the ACS and GC6–74 cohort study teams (2016). A prospective blood RNA signature for tuberculosis disease risk: a prospective cohort study. The Lancet [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1: Description of macaque samples, clinical parameters, and annotation of samples used in multiple analyses in the study. Related to Figure 1.
Supplementary Table 2: Description of macaque samples and QC values from scRNA-seq analysis. Related to Figure 1.
Supplementary Table 3: Description of human sample demographic that are part of this study. Related to Figure 4.
Data Availability Statement
The accession number for the raw and processed data for single cell RNA sequencing and CyTOF generated during this study is GEO: GSE149758. The additional accession number is Synapse: syn21988273.