

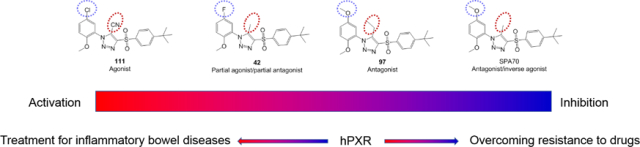
Abstract
Pregnane X receptor (PXR) plays roles in detoxification and other physiologic processes. PXR activation may enhance drug metabolism (leading to adverse drug reactions) or inhibit inflammation. Therefore, PXR agonists, antagonists, and inverse agonists may serve as research tools and drug candidates. However, a specific PXR modulator with an associated structure–activity relationship (SAR) is lacking. Based on the scaffold of specific human PXR (hPXR) antagonist SPA70 (10), we developed 81 SPA70 analogs and evaluated their receptor binding and cellular activities. Interestingly, analogs with subtle structural differences displayed divergent cellular activities, including agonistic, dual inverse agonistic and antagonistic, antagonistic, and partial agonistic/partial antagonistic activities (as in compound 111, 10, 97, and 42, respectively). We generated a pharmacophore model that represents 81 SPA70 analogs, and docking models that correlate strong interactions between the compounds and residues in the AF-2 helix with agonistic activity. These compounds are novel chemical tools for studying hPXR.
Graphical Abstract
INTRODUCTION
Nuclear receptors (NRs) are major targets for drug discovery and play key roles in development, homeostasis, and diseases such as obesity, diabetes, and cancers.1 NRs are transcription factors, and the NR superfamily has 48 members in humans and 49 in mice.1–3 These receptors bind and respond to steroids and other signaling molecules, such as vitamin D3, thyroid hormones, and retinoids.4 NRs have a modular structure that contains an N-terminal regulatory domain, a DNA-binding domain (DBD), a hinge region, a ligand binding domain (LBD), and a C-terminal domain.5–8
The pregnane X receptor (PXR, NR1I2) belongs to the NR subfamily 1I (NR1I), which includes two other members: the constitutive androstane receptor (CAR, NR1I3) and the vitamin D receptor (VDR, NR1I1).9–11 PXR and CAR were originally characterized as major xenobiotic receptors that regulate the expression of a broad spectrum of target genes encoding phase I and phase II drug-metabolizing enzymes (DMEs) and drug transporters.12–13 In humans, the major isoform of PXR consists of 434 amino acids and is predominantly expressed in the liver and small intestine and, to a lesser extent, in the kidneys, lungs, and breast.14–15 PXR forms a heterodimer with retinoid X receptor (RXR). Because of its large and flexible ligand binding pocket, many compounds of various sizes and chemical structures can promiscuously bind to PXR.13, 16–17 Agonist-bound PXR recruits co-activators, such as steroid receptor coactivator 1 (SRC-1), to enhance the expression of many target genes encoding proteins such as cytochrome P450 (CYP) 3A4 (CYP3A4), CYP2B6, CYP2C9, UDP glucuronosyltransferase family 1 member A1 (UGT1A1), and transporters (OCT1, Mrp2, Mrp3, Oatp1a, and Oatp4).18–21 In addition to enhancing the expression of proteins involved in drug metabolism, PXR also regulates other physiologic processes, such as cell growth, glucose metabolism, and lipid metabolism.22–25 Moreover, the activation of PXR can be inhibitory to other signaling pathways, such as the nuclear factor kappa B (NF-κB) pathway, and this might provide a treatment option for inflammatory bowel diseases (IBD).13, 26
The diverse functions of PXR in regulating multiple physiologic and pathologic processes suggest that PXR ligands with activating (i.e., agonist) or inhibitory (i.e., antagonist or inverse agonist) cellular functions are needed for specific applications.17 For example, PXR modulates the expression levels of proteins involved in the biotransformation, metabolism, and elimination of xenobiotics and endobiotics.21 Because of the ligand promiscuity, PXR can be activated by many drugs, causing enhanced metabolism and excretion of other drugs and possibly leading to drug toxicity, drug resistance, or other adverse drug reactions.27–28 PXR antagonists or inverse agonists are needed to reverse such adverse drug effects. Specifically, PXR antagonists or inverse agonists would be useful for overcoming resistance to anticancer drugs.29–30 As a proof of principle, increased sensitivity of colorectal cancer cells to irinotecan in the presence of sulforaphane has been reported.31,13 However, in the case of IBD, a PXR agonist is needed to inhibit the activity of NF-κB.13, 26
Numerous ligands have been reported for the human PXR (hPXR). The vast majority of these are agonists, with only a few antagonists having been discovered.32,17 Most of the reported hPXR agonists are diverse in structure with different known bioactivities (and are, therefore, not specific for hPXR). They include drugs [e.g., rifampicin (1) and dexamethasone (2)], marine sulfated sterols [e.g., solomonsterol A (3)], synthetic chemicals [e.g., T0901317 (4)], and many herbal compounds [e.g., praeruptorin A (5)] (Figure 1).33 Among the few compounds known to act as hPXR antagonists, such as sulforaphane (6), ketoconazole (7), metformin (8), A-792611 (9), and SPA70 (10) (Figure 1),34 only SPA70 (10) is highly specific for hPXR10, as compared to other NRs tested, such as human CAR (hCAR), human VDR (hVDR), human farnesoid X receptor (hFXR) and mouse PXR (mPXR).
Figure 1.
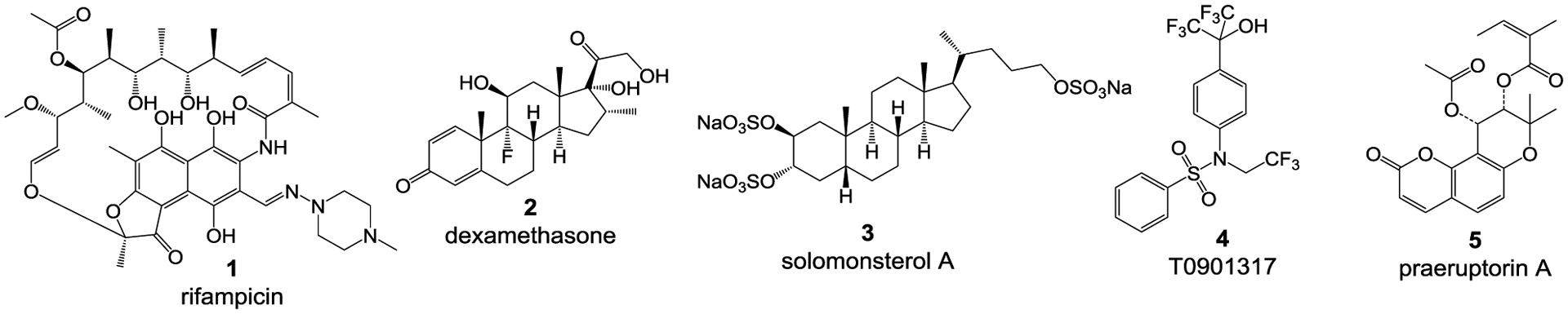
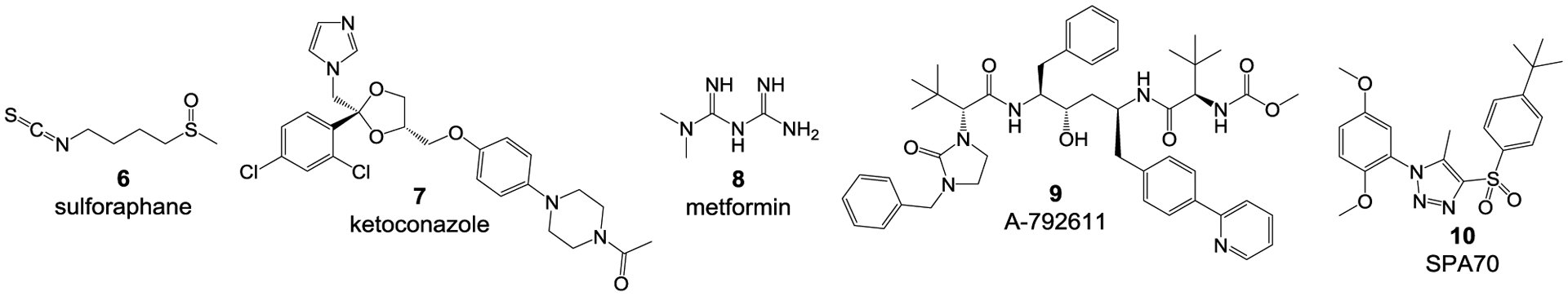
Structures of representative reported hPXR agonists rifampicin (1), dexamethasone (2), solomonsterol A (3) , T0901317 (4), praeruptorin A (5) and antagonists sulforaphane (6), ketoconazole (7), metformin (8), A-792611 (9), and SPA70 (10).
When we identified SPA70 (10) as the first specific PXR antagonist, we also discovered that subtle chemical modification of SPA70 (10) convert it to an agonist SJB7,10 suggesting that the SPA70 (10) scaffold might be an important pharmacophore for developing structurally related but functionally different PXR modulators. However, the structure–activity relationships (SARs) of SPA70 (10) and its analogs need further illustration. Therefore, in this study, we designed and synthesized SPA70 analogs and categorized them as agonists (represented by compound 111), antagonists (represented by compound 97), dual antagonists and inverse agonists (represented by SPA70, 10), or partial agonists and partial antagonists (represented by compound 42) based on their cellular activities by using a combination of chemistry, biology, and molecular modeling approaches. These structurally related but functionally different PXR modulators based on a unique scaffold provide the PXR research field with valuable chemical tools as well as with potential candidates for clinical applications.
SPA70 Analog Design
To perform extensive SAR studies based on the SPA70 (10) scaffold, we adopted a stepwise strategy. We structurally modified SPA70 (10) at four regions (Figure 2A): the 4-(tert-butyl)phenyl group (region A), the methoxyl group (region B), the hydrogen group (region C), and the 5-methyl-1H-1,2,3-triazole ring (region D).
Figure 2.
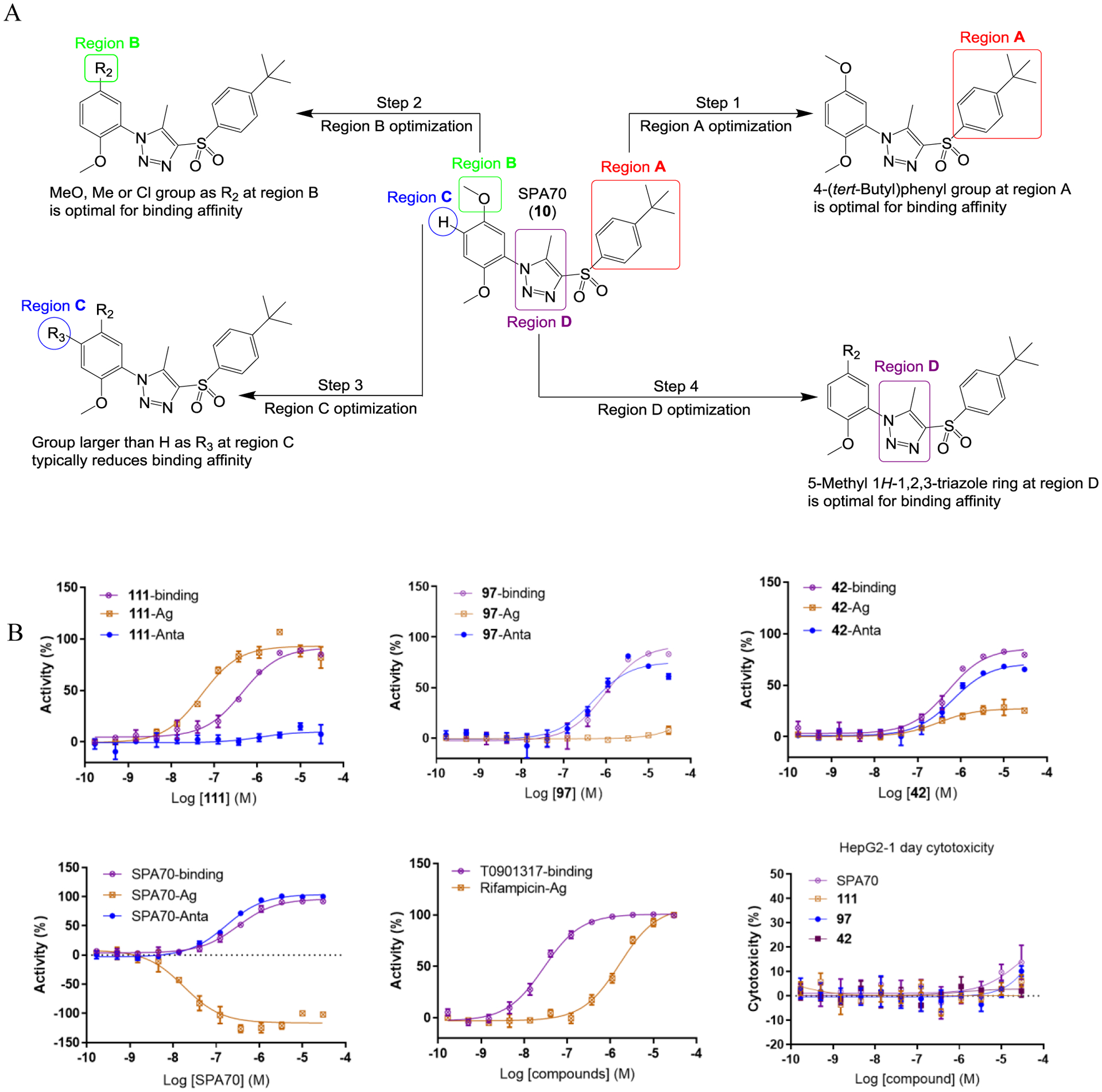
Design and activities of hPXR modulators. (A) Design of the PXR modulators by a step-by-step structural optimization approach starting from the SPA70 scaffold. (B) Representative dose response curves of agonist (111), antagonist (97), partial agonist and partial antagonist (42), and dual antagonist and inverse agonist [SPA70, (10)]. hPXR TR-FRET assay was used to determine binding inhibitory activity. HepG2 hPXR-CYP3A4-luciferase stable cells were used to test compounds in either agonistic mode (“Ag”, test compound alone for activation of hPXR; %Activity = %Activation. Note that SPA70 is an inverse agonist that decreased the basal activity of hPXR) or antagonistic mode (“Anta”, test compound in the presence of 5 μM rifampicin for inhibition of rifampicin-mediated activation of hPXR; %Activity = %Inhibition), and also in an 1-day cytotoxicity assay. Dose curves for reference compounds (SPA70, rifampicin and T0901317) were shown.
First, in the region A, the 4-(tert-butyl) in SPA70 was modified to an H, Me, Cl, HO, MeO, COOH, NH2, COOMe, CMe2OH, CMe2OMe or CMe2OButyl group. In addition, the 4-(tert-butyl) phenyl group in SPA70 was modified to a benzyl group (Step 1). We found that the 4-(tert-butyl)phenyl group maintained the highest hPXR binding affinity (Table 1) with most modifications yielded agonists (compounds 21 to 30) and the 4-alkoxypropylphenyl modifications in compound 31 and 32 created analogs with inverse agonistic activities. For region B, halogens, alkyl groups, hydrogen, cyano, methoxycarbonyl, aminocarbonyl, or hydroxyalkyl groups were introduced to replace the methoxy group in SPA70 (Step 2). We found that a methyl or chloro group can maintain an optimal hPXR binding activities with compounds ranged from to be an agonist, partial agonist, inverse agonist to an antagonist (Table 2). For region C, the hydrogen group was replaced with various halogens, alkyl groups, methoxy, cyano, acetyl, or methoxycarbonyl group (Step 3). Groups larger than hydrogen tend to reduce the compounds’ hPXR binding affinities, and most of the modifications produced agonists (Table 3). Finally, the 5-methyl-1H-1,2,3-triazole ring of the region D was replaced with differently substituted triazoles or an imidazole ring with varied substitutes (Step 4). The 5-methyl-1H-1,2,3-triazole ring system (SPA70) retained the highest binding affinity, antagonistic and inverse agonistic activity, compared to compounds with modifications of the 1H-1,2,3-triazole ring system with 5-ethyl, 5-NH2, 5-demethyl, 5-CN or 5-acetyl substitution or the 1H-imidazole ring system to have agonistic, partial agonistic /partial antagonistic, dual antagonistic and inverse agonistic activities (Table 4). Figure 2B shows representative dose responsive curves of agonist (111), antagonist (97), dual inverse agonist and antagonist (SPA70), and weak partial agonist/partial antagonist (42). SPA70 (10) is highly selective for hPXR with only marginal cytotoxicity in the cell models used as previously reported.10 Compounds 42 and 111 showed no cytotoxicity and 97 displayed very marginal cytotoxicity. To evaluate the selectivity of 111, 97 and 42, we tested their activity for two other members of the NR1I superfamily (hCAR and hVDR), as well as hFXR and mPXR. Compounds 42, 97 and 111 did not affect the activity of mPXR, hCAR, hVDR and hFXR (Figure S1 in Supporting Information), suggesting their selectivity for hPXR.
Table 1.
Structures and hPXR activities of SPA70 (10) analogs with modifications at region A in SPA70 (10)
 | |||||
|---|---|---|---|---|---|
| Compound | Region A | Bindinga (IC50/μM) (%inhibition at 10 μM) | Agonistb (EC50/μM) (%activation at 10 μM) | Inverse agonistc (IC50/μM) (%inhibition at 10 μM) | Antagonistd (IC50/μM) (%inhibition at 10 μM) |
| T0901317 (4) | 0.036 ± 0.006 (100 ± 1)% |
NTe | NT | NT | |
| Rifampicin (1) | NT | 1.4 ± 0.3 (100 ± 4)% |
NA | NT | |
| SPA70 (10) |  |
0.19 ± 0.03 (95 ± 2)% |
NAf | 0.024 ± 0.007 (100 ± 3)% |
0.25 ± 0.09 (100 ± 1)% |
| 21 |  |
2 ± 1 (92 ± 8)% |
2.5 ± 0.4 (105 ± 5)% |
NA | NA |
| 22 |  |
3.8 ± 0.7 (104 ± 6)% |
2.1 ± 0.4 (90 ± 11)% |
NA | NA |
| 23 | 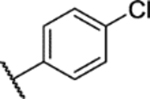 |
1.9 ± 0.4 (102 ± 2)% |
2.3 ± 0.3 (99 ± 12)% |
NA | NA |
| 24 | 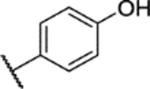 |
8.0 ± 0.1 (95 ± 1)% |
22 ± 6 (138 ± 19)% |
NA | NA |
| 25 | 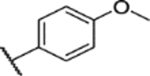 |
3.0 ± 0.6 (104 ± 5)% |
1.68 ± 0.08 (83 ± 14)% |
NA | NA |
| 26 | 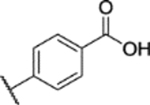 |
NA | NA | NA | NA |
| 27 | 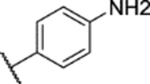 |
5 ± 1 (89 ± 7)% |
7 ± 1 (74 ± 10)% |
NA | NA |
| 28 | 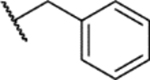 |
1.0 ± 0.4 (90 ± 6)% |
3.4 ± 0.6 (130 ± 10)% |
NA | NA |
| 29 | 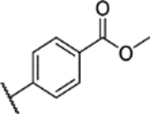 |
8.7 ± 0.8 (89 ± 1)% |
NA | NA | NA |
| 30 | 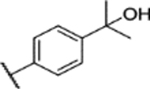 |
9.2 ± 0.7 (80 ± 11)% |
NA | NA | NA |
| 31 | 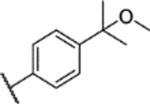 |
3.7 ± 0.7 (96 ± 4)% |
NA | 0.4 ± 0.1 (103 ± 3)% |
2.3 ± 0.5 (109 ± 5)% |
| 32 | 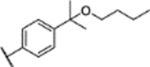 |
2.1 ± 0.7 (86 ± 3)% |
NA | 0.25 ± 0.05 (102 ± 4)% |
1.35 ± 0.06 (105 ± 2)% |
T0901317 (4) at 10 μM as 100% inhibition and DMSO (0.3%) as 0% inhibition for the hPXR binding assay;
Rifampicin (1) at 10 μM as 100% activation and DMSO (0.3%) as 0% activation for the cell-based hPXR agonistic assay;
SPA70 (10) at 10 μM as 100% inhibition and DMSO (0.3%) as 0% inhibition for the cell-based hPXR inverse agonistic assay;
SPA70 (10) at 10 μM with rifampicin (1, 5 μM) as 100% inhibition and rifampicin alone (1, 5 μM) as 0% inhibition for the cell-based hPXR antagonistic assay;
NT: not tested;
NA: no IC50 value could be experimentally determined within the concentration range tested (highest concentration tested at 30 μM).
Table 2.
Structures and hPXR activities of SPA70 analogs with modifications at region B in SPA70
 | |||||
|---|---|---|---|---|---|
| Compound | Region B (R2) |
Bindinga (IC50/μM) (%inhibition at 10 μM) | Agonistb (EC50/μM) (%activation at 10 μM) | Inverse agonistc (IC50/μM) (%inhibition at 10 μM) | Antagonistd (IC50/μM) (%inhibition at 10 μM) |
| T0901317 (4) | 0.036 ± 0.006 (100 ± 1)% |
NTe | NT | NT | |
| Rifampicin (1) | NT | 1.4 ± 0.3 (100 ± 4)% |
NA | NT | |
| SPA70 (10) | MeO | 0.19 ± 0.03 (95 ± 2)% |
NAf | 0.024 ± 0.007 (100 ± 3)% |
0.25 ± 0.09 (100 ± 1)% |
| 41 | H | 0.53 ± 0.09 (103 ± 2)% |
0.8 ± 0.3 (56 ± 8)% |
NA | 1.0 ± 0.2 (53 ± 4%)% |
| 42 | F | 0.4 ± 0.1 (80 ± 6)% |
0.24 ± 0.09 (37 ± 7)% |
NA | 0.7 ± 0.1 (70 ± 3)% |
| 43 | Cl | 0.25 ± 0.05 (94 ± 2)% |
NA | 0.27 ± 0.03 (62 ± 2)% |
0.31 ± 0.09 (96 ± 5)% |
| 44 | Br | 0.40 ± 0.03 (96 ± 3)% |
NA | 0.05 ± 0.03 (33 ± 4)% |
0.24 ± 0.03 (99 ± 2)% |
| 45 | Me | 0.8 ± 0.2 (98 ± 1)% |
NA | NA | 2 ± 2 (85 ± 12)% |
| 46 | CF3 | 0.41 ± 0.04 (90 ± 6)% |
NA | NA | 1.01 ± 0.06 (82 ± 4)% |
| 47 | CMe3 | 0.3 ± 0.2 (87 ± 1)% |
0.38 ± 0.06 (113 ± 8)% |
NA | NA |
| 48 | CN | 0.4 ± 0.1 (98 ± 4)% |
NA | 0.05 ± 0.04 (34 ± 3)% |
0.45 ± 0.07 (96 ± 1)% |
| 50 | CH2OH | 1.4 ± 0.7 (95 ± 2)% |
0.81 ± 0.02 (82 ± 5)% |
NA | 2.0 ± 0.3 (64 ± 7)% |
| 51 | CONH2 | 1.1 ± 0.1 (95 ± 1)% |
2.44 ± 0.02 (182 ± 16)% |
NA | NA |
| 54 (R) | 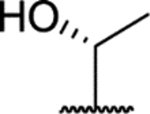 |
1.1 ± 0.1 (94 ± 2)% |
0.44 ± 0.04 (123 ± 22)% |
NA | NA |
| 55 (S) | 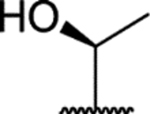 |
1.3 ± 0.4 (95 ± 1)% |
0.64 ± 0.01 (112 ± 11)% |
NA | NA |
T0901317 (4) at 10 μM as 100% inhibition and DMSO (0.3%) as 0% inhibition for the hPXR binding assay;
Rifampicin (1) at 10 μM as 100% activation and DMSO (0.3%) as 0% activation for the cell-based hPXR agonistic assay;
SPA70 (10) at 10 μM as 100% inhibition and DMSO (0.3%) as 0% inhibition for the cell-based hPXR inverse agonistic assay;
SPA70 (10) at 10 μM with rifampicin (1, 5 μM) as 100% inhibition and rifampicin alone (1, 5 μM) as 0% inhibition for the cell-based hPXR antagonistic assay;
NT: not tested;
NA: no IC50 value could be experimentally determined within the concentration range tested (highest concentration tested at 30 μM).
Table 3.
Structures and hPXR activities of SPA70 analogs with modifications at region C (the para-position to the 1H-1,2,3-triazole on the left phenyl group in SPA70)
 | ||||||
|---|---|---|---|---|---|---|
| Compound | R2 | Region C (R3) | Bindinga (IC50/μM) (%inhibition at 10 μM) | Agonistb (EC50/μM) (%activation at 10 μM) | Inverse agonistc (IC50/μM) (%inhibition at 10 μM) |
Antagonistd (IC50/μM) (%inhibition at 10 μM) |
| T0901317 (4) | 0.036 ± 0.006 (100 ± 1)% |
NTe | NT | NT | ||
| Rifampicin (1) | NT | 1.4 ± 0.3 (100 ± 4)% | NA | NT | ||
| SPA70 (10) |
MeO | H | 0.19 ± 0.03 (95 ± 2)% |
NAf | 0.024 ± 0.007 (100 ± 3)% |
0.25 ± 0.09 (100 ± 1)% |
| 66 | MeO | F | 4 ± 2 (87 ± 7)% |
1 ± 1 (24 ± 1)% |
NA | 2 ± 2 (72 ± 13)% |
| 71 | MeO | Cl | 0.9 ± 0.3 (73 ± 7)% |
0.6 ± 0.2 (63 ± 13)% |
NA | NA |
| 67 | MeO | Br | 0.7 ± 0.3 (46 ± 14)% |
0.6 ± 0.3 (91 ± 12)% |
NA | NA |
| 72 | MeO | Me | 0.5 ± 0.1 (73 ± 6)% |
0.6 ± 0.1 (75 ± 11)% |
NA | 1.6 ± 0.4 (30 ± 5)% |
| 73 | MeO | Et | 0.7 ± 0.2 (65 ± 8)% |
0.8 ± 0.1 (125 ± 10)% |
NA | NA |
| 74 | MeO | isoPro | 1.0 ± 0.5 (68 ± 4)% |
0.9 ± 0.2 (110 ± 9)% |
NA | NA |
| 75 | MeO | MeO | 1.3 ± 0.2 (90 ± 1)% |
1.05 ± 0.04 (124 ± 11)% |
NA | NA |
| 76 | MeO | CN | 0.36 ± 0.02 (36 ± 8)% |
1.1 ± 0.3 (88 ± 15)% |
NA | NA |
| 77 | MeO | COOMe | 2.3 ± 0.2 (72 ± 5)% |
3.4 ± 0.1 (78 ± 14)% |
NA | NA |
| 78 | MeO | COMe | 0.9 ± 0.1 (48 ± 4)% |
0.5 ± 0.2 (97 ± 10)% |
NA | NA |
| 79 | Me | F | 0.6 ± 0.2 (94 ± 4)% |
NA | NA | 0.7 ± 0.2 (81 ± 2)% |
| 68 | Me | Cl | 0.7 ± 0.1 (71 ± 4)% |
0.8 ± 0.2 (51 ± 13)% |
NA | 2.0 ± 0.2 (57 ± 2)% |
| 69 | Me | Br | 2.2 ± 0.3 (77 ± 11)% |
1.3 ± 0.3 (87 ± 17)% |
NA | NA |
| 80 | Me | Me | 1.22 ± 0.04 (84 ± 1)% |
0.7 ± 0.2 (52 ± 8)% |
NA | 1.3 ± 0.1 (60 ± 4)% |
| 81 | Me | Et | 1.0 ± 0.4 (76 ± 3)% |
0.9 ± 0.3 (138 ± 18)% |
NA | NA |
| 82 | Me | isoPro | 2 ± 1 (77 ± 5)% |
0.83 ± 0.02 (122 ± 8%) |
NA | NA |
|
83 (SJB7) |
Me | MeO | 0.84 ± 0.02 (89 ± 4)% |
0.28 ± 0.02 (112 ± 13)% |
NA | NA |
| 84 | Me | CN | 1.07 ± 0.03 (48 ± 4)% | 1.6 ± 0.3 (58 ± 8)% |
NA | 4 ± 2 (33 ± 9)% |
| 85 | Me | COOMe | 1.5 ± 0.1 (64 ± 1)% |
1.6 ± 0.3 (75 ± 11)% |
NA | 6 ± 1 (32 ± 3)% |
| 86 | Me | COMe | 1.4 ± 0.1 (80 ± 4)% |
0.4 ± 0.1 (77 ± 12)% |
NA | 9 ± 2 (111 ± 10)% |
| 87 | Cl | Cl | 0.4 ± 0.2 (75 ± 11)% |
NA | NA | 1.3 ± 0.7 (102 ± 12)% |
| 70 | Cl | Br | 0.55 ± 0.02 (67 ± 7)% |
0.6 ± 0.3 (26 ± 4)% |
NA | 3 ± 2 (38 ± 7)% |
| 88 | Cl | Me | 1.2 ± 0.6 (87 ± 7)% |
1 ± 1 (33 ± 1)% |
NA | 2.3 ± 0.6 (64 ± 2)% |
| 89 | Cl | Et | 1.5 ± 0.4 (59 ± 4)% |
0.7 ± 0.4 (58 ± 8)% |
NA | NA |
| 90 | Cl | isoPro | 1.1 ± 0.3 (73 ± 2)% |
1.8 ± 0.6 (99 ± 12)% |
NA | NA |
| 91 | Cl | MeO | 0.47 ± 0.08 (80 ± 1)% |
0.25 ± 0.06 (77 ± 7)% |
NA | NA |
| 92 | Cl | CN | 0.6 ± 0.2 (40 ± 8)% |
1.0 ± 0.4 (49 ± 7)% |
NA | 3.4 ± 0.4 (44 ± 5)% |
| 93 | Cl | COOMe | 0.7 ± 0.6 (35 ± 9)% |
1.9 ± 0.1 (62 ± 1)% |
NA | 10 ± 3 (51 ± 2)% |
| 94 | Cl | COMe | NA | 0.26 ± 0.09 (65 ± 4)% |
NA | NA |
T0901317 (4) at 10 μM as 100% inhibition and DMSO (0.3%) as 0% inhibition for the hPXR binding assay;
Rifampicin (1) at 10 μM as 100% activation and DMSO (0.3%) as 0% activation for the cell-based hPXR agonistic assay;
SPA70 (10) at 10 μM as 100% inhibition and DMSO (0.3%) as 0% inhibition for the cell-based hPXR inverse agonistic assay;
SPA70 (10) at 10 μM with rifampicin (1, 5 μM) as 100% inhibition and rifampicin alone (1, 5 μM) as 0% inhibition for the cell-based hPXR antagonistic assay;
NT: not tested;
NA: no IC50 value could be experimentally determined within the concentration range tested (highest concentration tested at 30 μM).
Table 4.
Structures and hPXR activities of SPA70 analogs with modifications at region D (the 1H-1,2,3-triazole component in SPA70)
 | ||||||
|---|---|---|---|---|---|---|
| Compound | R2 | Region D | Bindinga (IC50/μM) (%inhibition at 10 μM) | Agonistb (EC50/μM) (%activation at 10 μM) | Inverse agonistc (IC50/μM) (%inhibition at 10 μM) |
Antagonistd (IC50/μM) (%inhibition at 10 μM) |
| T0901317 (4) | 0.036 ± 0.006 (100 ± 1) % |
NTe | NT | NT | ||
| Rifampicin (1) | NT | 1.4 ± 0.3 (100 ± 4)% |
NA | NT | ||
| SPA70 (10) |
MeO | 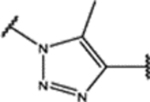 |
0.19 ± 0.03 (95 ± 2)% |
NAf | 0.024 ± 0.007 (100 ± 3)% |
0.25 ± 0.09 (100 ± 1)% |
| 97 | MeO | 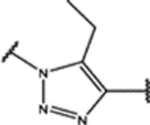 |
0.8 ± 0.2 (94 ± 1)% |
NA | NA | 0.500 ± 0.007 (82 ± 5)% |
| 98 | Me | 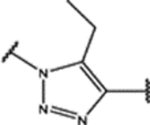 |
1.0 ± 0.3 (90 ± 10)% |
NA | NA | 0.8 ± 0.1 (81 ± 5)% |
| 99 | Cl | 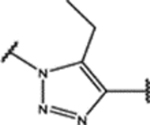 |
0.7 ± 0.1 (93 ± 3)% |
NA | NA | 0.96 ± 0.07 (81 ± 4)% |
| 100 | MeO | 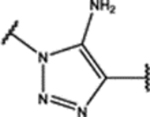 |
0.18 ± 0.05 (85 ± 2)% |
0.43 ± 0.05 (53 ± 6)% |
NA | 1.7 ± 0.1 (52 ± 4)% |
| 101 | Me | 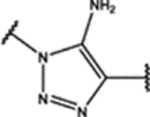 |
0.28 ± 0.05 (88 ± 6)% |
0.16 ± 0.07 (73 ± 10)% |
NA | 3.3 ± 0.4 (46 ± 5)% |
| 102 | Cl | 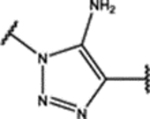 |
0.5 ± 0.1 (93 ± 1)% |
0.110 ± 0.008 (42 ± 13)% |
NA | 4.4 ± 0.5 (78 ± 1)% |
| 103 | MeO | 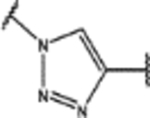 |
1.4 ± 0.3 (98 ± 1)% |
1.4 ± 0.2 (35 ± 13)% |
NA | 3.4 ± 0.3 (71 ± 3)% |
| 104 | Me | 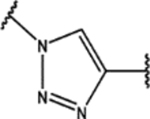 |
0.5 ± 0.2 (98 ± 5)% |
0.6 ± 0.2 (69 ± 9)% |
NA | 5 ± 1 (44 ± 9)% |
| 105 | Cl | 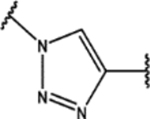 |
0.48 ± 0.05 (91 ± 1)% |
0.77 ± 0.09 (65 ± 11)% |
NA | 10.5 ± 0.1 (57 ± 11)% |
| 109 | MeO | 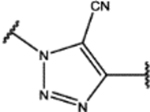 |
0.3 ± 0.1 (97 ± 1)% |
0.26 ± 0.06 (88 ± 7)% |
NA | 2.4 ± 0.2 (38 ± 2)% |
| 110 | Me | 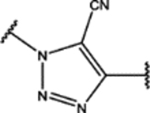 |
0.21 ± 0.06 (97 ± 1)% |
0.16 ± 0.03 (131 ± 23)% |
NA | NA |
| 111 | Cl | 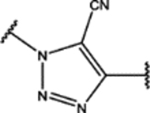 |
0.061 ± 0.008 (90 ± 2)% |
0.08 ± 0.04 (105 ± 18)% |
NA | NA |
| 112 | MeO | 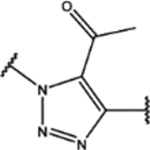 |
0.7 ± 0.3 (91 ± 2)% |
1.9 ± 0.7 (135 ± 18)% |
NA | NA |
| 113 | Me | 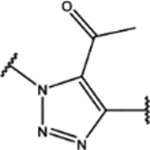 |
0.26 ± 0.09 (94 ± 3)% |
0.5 ± 0.2 (125 ± 24)% |
NA | NA |
| 114 | Cl | 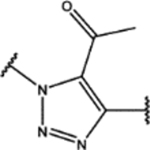 |
0.4 ± 0.3 (96 ± 4)% |
0.20 ± 0.06 (104 ± 11)% |
NA | NA |
| 122 | MeO |  |
2.5 ± 0.8 (87 ± 13)% |
4 ± 2 (67 ± 3)% |
NA | NA |
| 123 | Me | 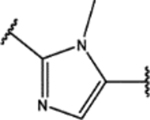 |
1.6 ± 0.6 (92 ± 3)% |
0.99 ± 0.05 (74 ± 7)% |
NA | NA |
| 124 | Cl | 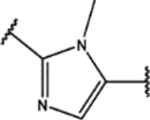 |
0.55 ± 0.02 (60 ± 1)% |
1.21 ± 0.05 (81 ± 9)% |
NA | NA |
| 128 | MeO | 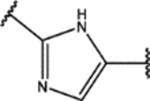 |
3.08 ± 0.02 (68 ± 16)% |
0.7 ± 0.3 (86 ± 4)% |
NA | NA |
| 129 | Me | 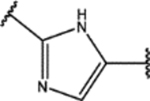 |
1.4 ± 0.6 (77 ± 13)% |
0.7 ± 0.2 (102 ± 12)% |
NA | NA |
| 130 | Cl | 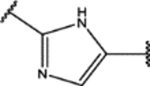 |
1.3 ± 0.5 (81 ± 16)% |
0.36 ± 0.07 (71 ± 10)% |
NA | NA |
| 140 | MeO | 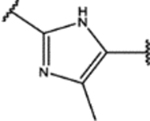 |
2.0 ± 0.4 (84 ± 17)% |
6 ± 2 (86 ± 9)% |
NA | NA |
| 145 | Cl | 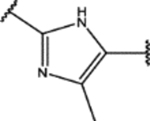 |
0.9 ± 0.4 (81 ± 16)% |
0.5 ± 0.3 (100 ± 10)% |
NA | NA |
| 150 | Me | 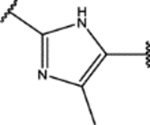 |
0.8 ± 0.3 (82 ± 13)% |
1.8 ± 0.4 (106 ± 17)% |
NA | NA |
| 155 | MeO | 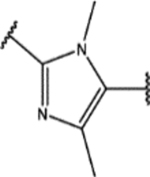 |
2.1 ± 0.4 (80 ± 13)% |
1.2 ± 0.3 (97 ± 10)% |
NA | NA |
| 156 | Me | 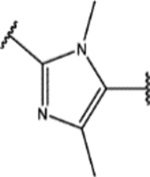 |
0.4 ± 0.1 (73 ± 2)% |
1.0 ± 0.3 (89 ± 9)% |
NA | NA |
| 157 | Cl | 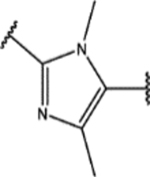 |
0.35 ± 0.03 (77 ± 4)% |
1.2 ± 0.5 (84 ± 9)% |
NA | NA |
T0901317 (4) at 10 μM as 100% inhibition and DMSO (0.3%) as 0% inhibition for the hPXR binding assay;
Rifampicin (1) at 10 μM as 100% activation and DMSO (0.3%) as 0% activation for the cell-based hPXR agonistic assay;
SPA70 (10) at 10 μM as 100% inhibition and DMSO (0.3%) as 0% inhibition for the cell-based hPXR inverse agonistic assay;
SPA70 (10) at 10 μM with rifampicin (1, 5 μM) as 100% inhibition and rifampicin alone (1, 5 μM) as 0% inhibition for the cell-based hPXR antagonistic assay;
NT: not tested;
NA: no IC50 value could be experimentally determined within the concentration range tested (highest concentration tested at 30 μM).
Chemistry
To explore the SAR based on the SPA70 (10) scaffold, a total of 81 analogs (Tables 1 to 4), including SPA70, were designed based on the stepwise SAR assessment approach. Among them 77 were newly synthesized (as summarized in Schemes 1 to 8), 3 (compounds 41, 43 and 45) were obtained from ChemDiv (San Diego, CA), and SPA70 (10) was previously report.10 The chemistry of 16 analogs, including SPA70 (10), compounds 66, 67, 73 to 78, 80, 83, 84, 87 and 90 to 92 has been reported previously10 together with extensive biological characterization of SPA70 (10). To synthesize analogs with a 1-phenyl-4-(phenylsulfonyl)-1H-1,2,3-triazole scaffold (compounds 21 to 32, 42, 44, 46 to 48, 50, 51, 54, 55, 66 to 94, 97 to 105, 109 to 114) (Figure 3A), we followed a published protocol (Schemes 1 to 4).35 To synthesize analogs with a 2-phenyl-5-(phenylsulfonyl)-1H-imidazole scaffold (compounds 122 to 124, 128 to 130, 140, 145, 150, and 155 to 157) (Figure 3B), appropriate substituted imidazole chemicals were used as the starting materials (Schemes 5 to 8).
Scheme 1.
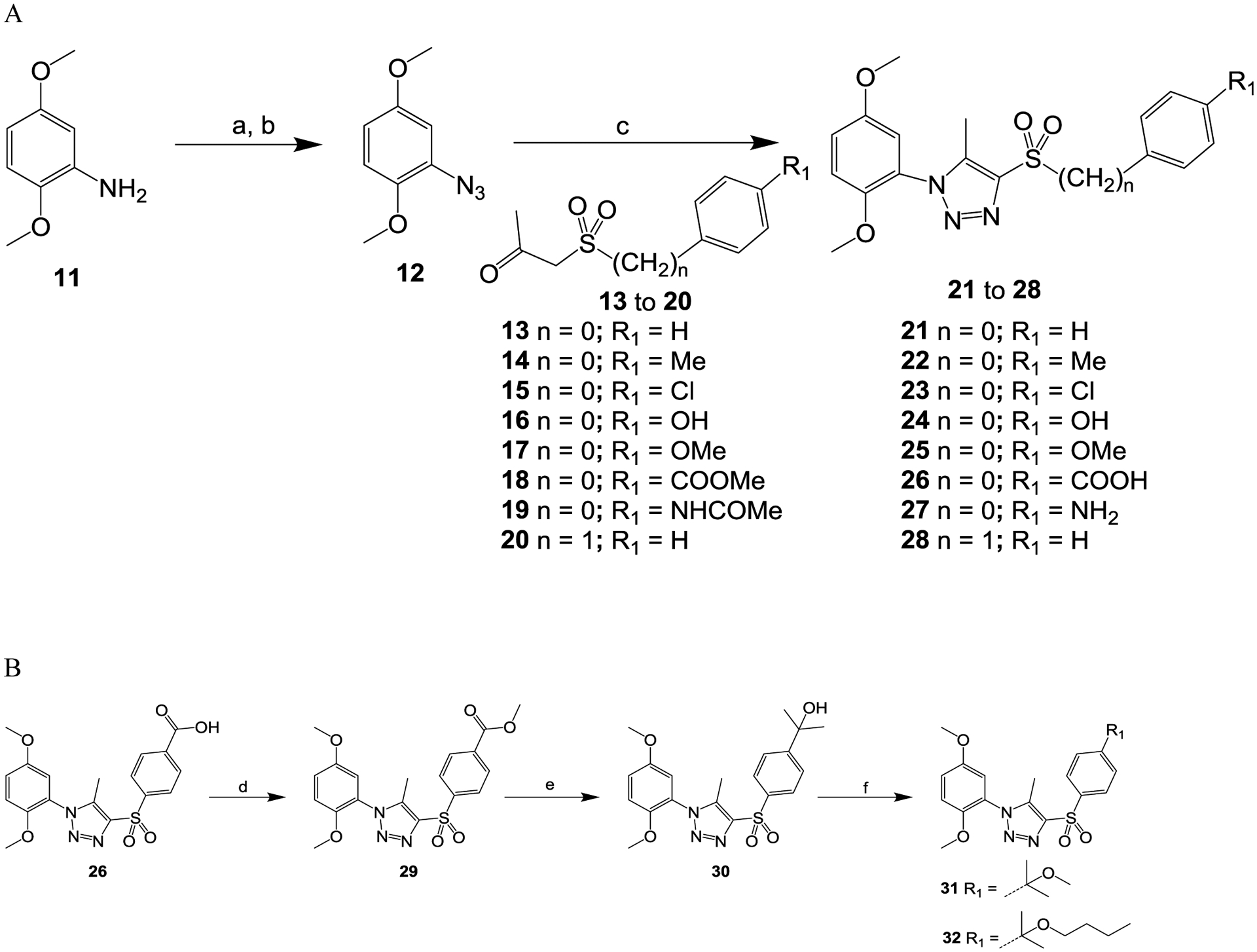
Reagents and conditions: (a) concentrated HCl, NaNO2, H2O, 0 °C, 15 min; (b) NaN3, H2O, room temperature, 2 h; (c) MeONa, MeOH, 60 °C, overnight; (d) MeOH, H2SO4, reflux, overnight; (e) methylmagnesium bromide, THF, −78 °C to room temperature, 2 h; (f) BiBr3, MeOH or n-BuOH, CCl4, room temperature, 3 h.
Scheme 8.
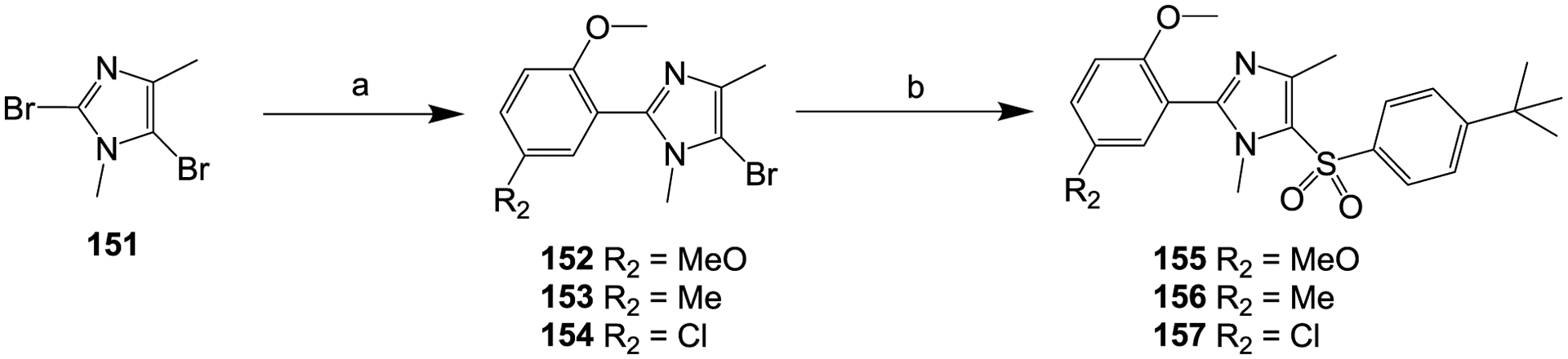
Reagents and conditions: (a) 2-methoxy-5-substituted-phenylboronic acid, Pd(dppf)Cl2, K2CO3, dioxane, H2O, N2, 100 °C, 3 h; (b) sodium 4-(tert-butyl)benzenesulfinate, CuI, DMF, N2, 65 °C, 18 h.
Figure 3.
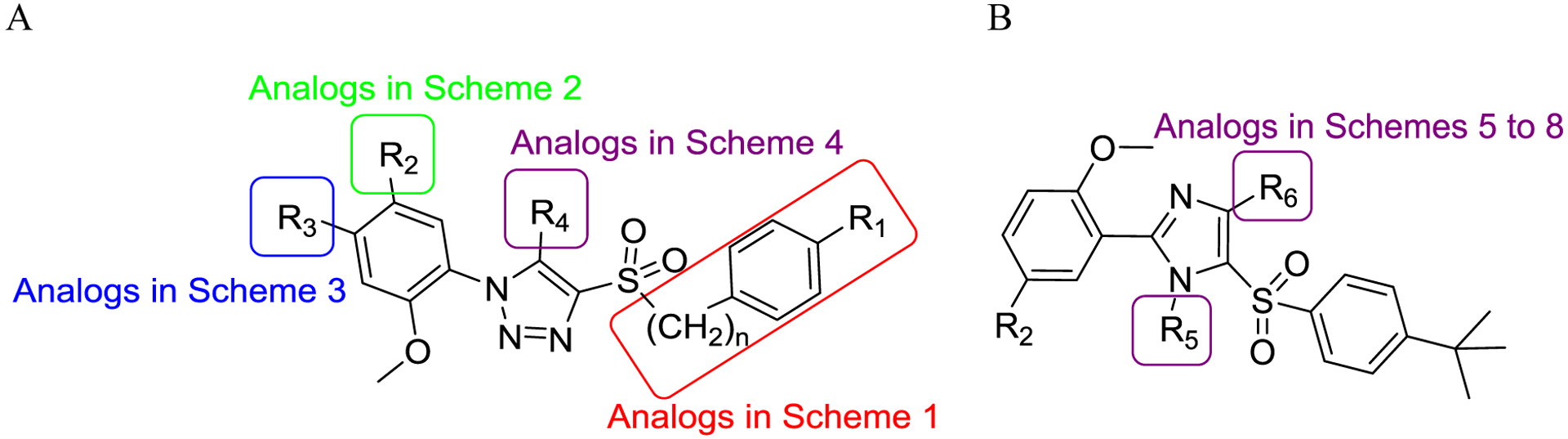
(A) Scaffold representation of SPA70 (10) analogs summarized in the synthetic schemes 1 to 4. (B) Scaffold representation of SPA70 (10) analogs summarized in the synthetic schemes 5 to 8.
Scheme 4.
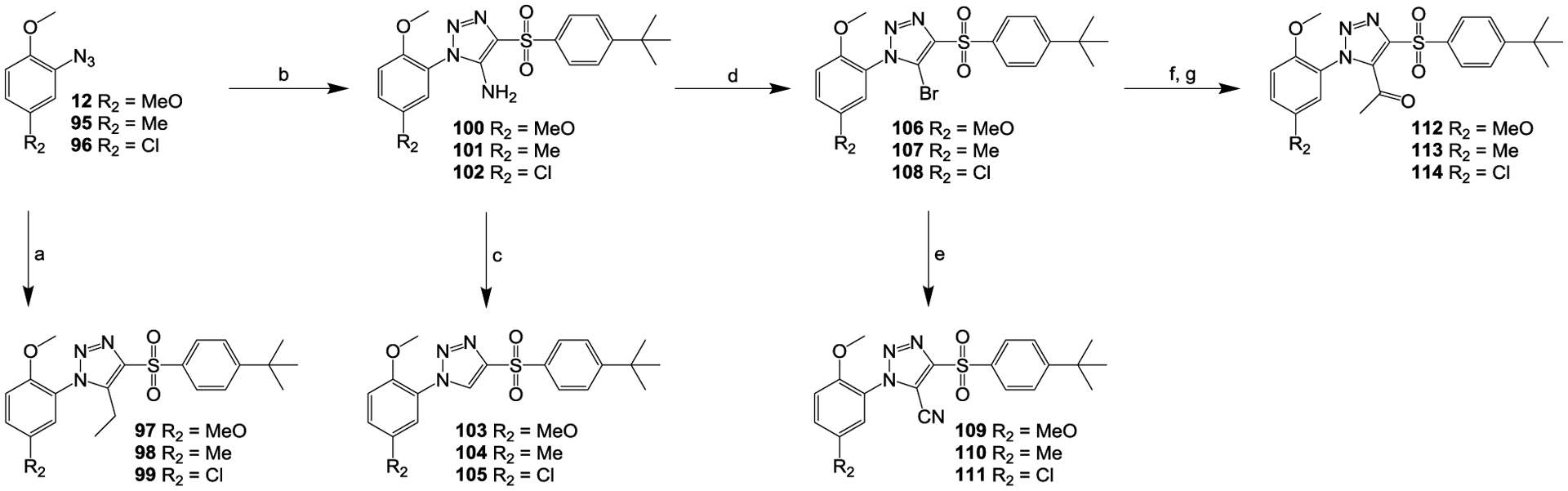
Reagents and conditions: (a) MeONa, 1-((4-(tert-butyl)phenyl)sulfonyl)butan-2-one, MeOH, 60 °C, overnight; (b) MeONa, 2-((4-(tert-butyl)phenyl)sulfonyl)acetonitrile, MeOH, room temperature, overnight; (c) EtOH, H2SO4, NaNO2, 50 °C, 4 h; (d) t-BuNO2, CuBr2, MeCN, N2, room temperature, 3 h; (e) KCN, CuCN, DMF, N2, 65 °C, 19 h; (f) tributyl(1-ethoxyvinyl)stannane, Pd(PPh3)4, toluene, 65 °C, 3 h; (g) conc. HCl, dioxane, room temperature, 1 h.
Scheme 5.
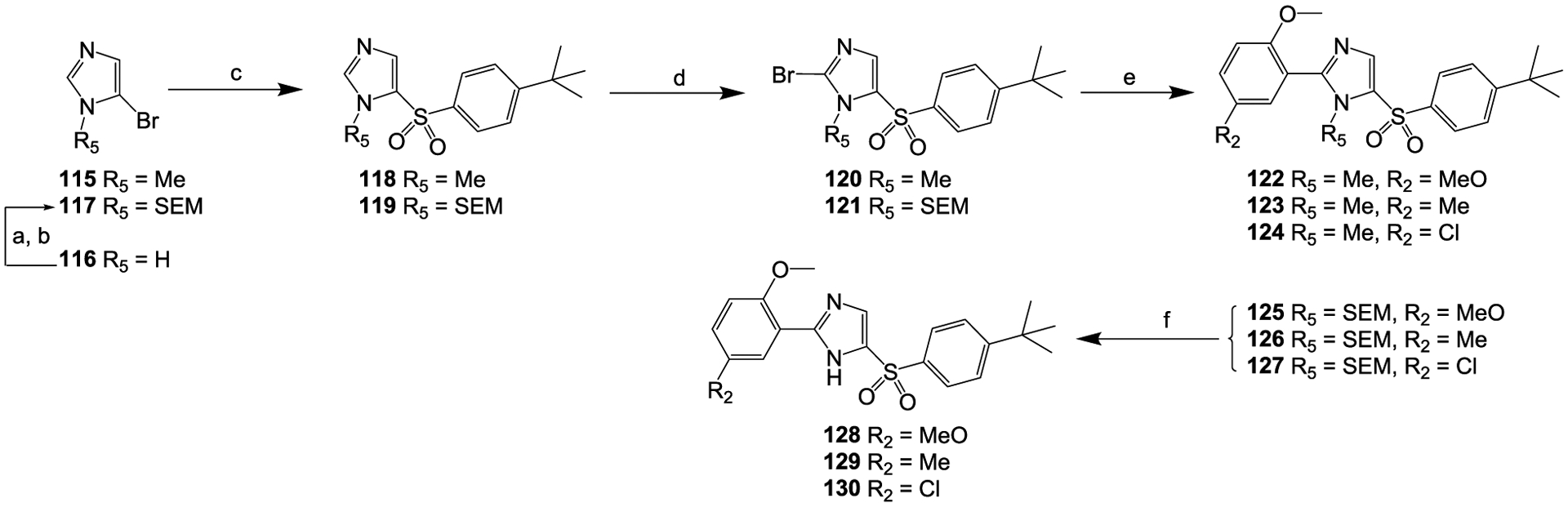
Reagents and conditions: (a) NaH, THF, 0 °C, 15 min; (b) SEMCl, 0 °C, 1 h; (c) sodium 4-(tert-butyl)benzenesulfinate, CuI, DMF, N2, 65 °C, 18 h; (d) NBS, AIBN, CCl4, 60 °C, 18 h; (e) 2-methoxy-5-substituted-phenylboronic acid, Pd(dppf)Cl2, K2CO3, dioxane, H2O, N2, 90 °C, 4 h; (f) TBAF, THF, N2, 60 °C, 2 h.
As depicted in Scheme 1A, compounds 21 to 28 were prepared by various corresponding substitutions of 1-(sulfonyl)propan-2-one (13 to 20) with 2-azido-1,4-dimethoxybenzene (12) which was generated from commercially available 2,5-dimethoxyaniline (11) via diazotization. Compounds 26 and 27 were prepared from the corresponding compounds 18 and 19 by a hydrolysis reaction with compound 12. Compound 29 was prepared from compound 26 via esterification and was further converted to compound 30 via methylation with methylmagnesium bromide. Proper etherification of compound 30 yielded 31 and 32 (Scheme 1B).36
Compounds 42, 44, 46 and 47 (Scheme 2A) were prepared by following a published protocol.35 Briefly, proper substituted phenyl amines (compounds 33 to 36) were first converted to the corresponding aryl azides via diazotization. The resulted aryl azides (compounds 37 to 40) were then converted to the product triazoles (compounds 42, 44, 46 and 47) by coupling with 1-((4-(tert-butyl)phenyl)sulfonyl)propan-2-one. 41, 43 and 45 were purchased from ChemDiv (San Diego, CA). Compound 44 was further converted to compound 48 via a Suzuki coupling37 with ZnCN in the presence of Pd(PPh3)4. As shown in Scheme 2B, compound 44 was carbonylated38 with CO in the presence of methanol to produce the methyl ester compound 49. Compound 49 was further selectively reduced39 to the alcohol compound 50 in the presence of LiBH4 or converted to compound 51 by direct amidation40 in the presence of ammonia methanol under pressurized conditions. The acetophenone derivative compound 52 was prepared from compound 44 by a palladium-mediated coupling with tributyl-(1-ethoxy-vinyl)-stannane. Reduction of compound 52 generated compound 53,39 and a follow up separation by supercritical fluid chromatography (SFC) using Phenomenex-Cellulose-2 column yielded compounds 54 and 55 (Scheme 2C).
Scheme 2.
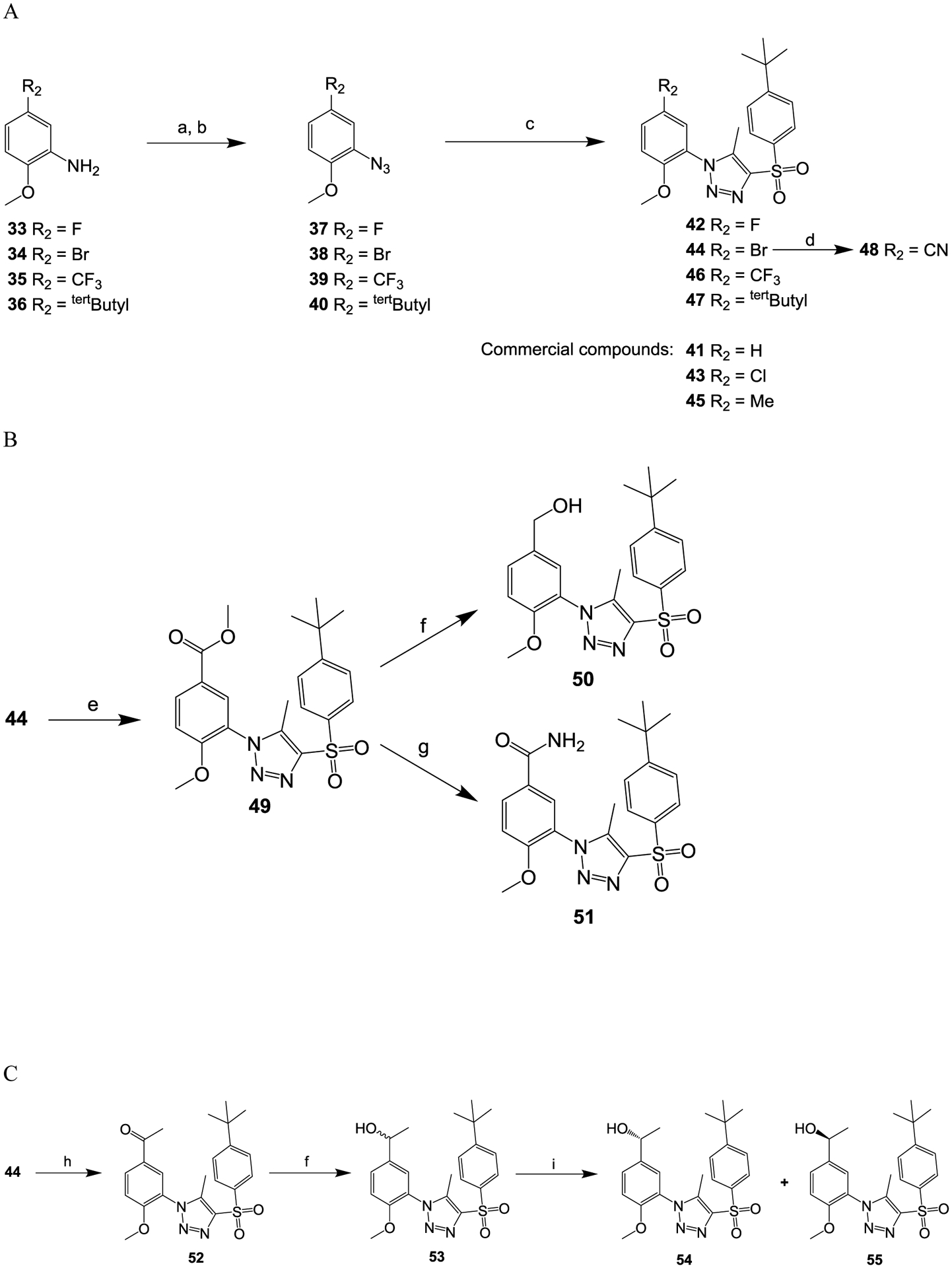
Reagents and conditions: (a) concentrated HCl, NaNO2, H2O, 0 °C, 15 min; (b) NaN3, H2O, room temperature, 2 h; (c) MeONa, 1-((4-(tert-butyl)phenyl)sulfonyl)propan-2-one, MeOH, 60 °C, overnight; (d) Pd(Ph3P)4, ZnCN, DMF, 90 °C, overnight; (e) CO(50 psi), Pd(dppf)2Cl2, MeOH, 80 °C, 16 h; (f) LiBH4, THF; (g) NH3, MeOH; (h) tributyl(1-ethoxyvinyl)stannane, Pd(PPh3)2Cl2, dioxane; (i) supercritical fluid chromatography (SFC).
Compounds 66 to 70 were prepared using the same protocol as used for the synthesis of compound 42 but with different starting materials 56 to 60 (Scheme 3A). The bromide-containing compounds 67, 69 and 70 were then converted to compounds 71 to 78 (from compound 67) (Scheme 3B), compounds 79 to 86 (from compound 69) (Scheme 3C) and compounds 87 to 94 (from compound 70) (Scheme 3D) with a typical 1- or 2-step reaction. Specifically, compounds 71 and 87 were prepared from compounds 67 and 70, respectively, via bromo-lithium exchange41 and chlorination with hexachloroethane;42 compounds 72, 80 and 88 were prepared by Suzuki coupling37 between the respective bromide-containing compounds 67, 69 and 70 and methylboronic acid in the presence of a palladium catalyst; compounds 73, 81 and 89 were prepared, respectively, from the bromide-containing compounds 67, 69 and 70 reacted with ethenylSn(Bu)3 in the presence of a palladium catalyst, Pd(PPh3)4, to form the respective ethenyl compounds43 then subjected to catalytic hydrogenation44 to convert the ethenyl group to an ethyl group; compounds 74, 82 and 90 were prepared, respectively, from the bromide-containing compounds 67, 69 and 70 via Suzuki coupling37 with 4,4,5,5-tetramethyl-2-(prop-1-en-2-yl)-1,3,2-dioxaborolane in the presence of Pd(PPh3)4 followed by catalytic hydrogenation;44 compounds 75, 83, and 91 were synthesized, respectively, from compounds 67, 69 and 70 via copper-catalyzed methoxylation;45 compounds 76, 84, and 93 were prepared, respectively, from the bromide-containing compounds 67, 69 and 70 by palladium-catalyzed cyanation46 under microwave irradiation; compounds 76, 84, and 93 were further converted to the ketone compounds 78, 86 and 94, respectively, by reacting them with the Grignard reagent methylmagnesium chloride;47 compounds 77, 85 and 93 were prepared by palladium-catalyzed carbalkoxylation of the bromide-containing compounds 67, 69 and 70, respectively, in the presence of pressured carbon monoxide;41 compound 79 was prepared from compound 69 via bromo-lithium exchange41 followed by fluorination48 with N-fluorobenzenesulfonimide.
Scheme 3.
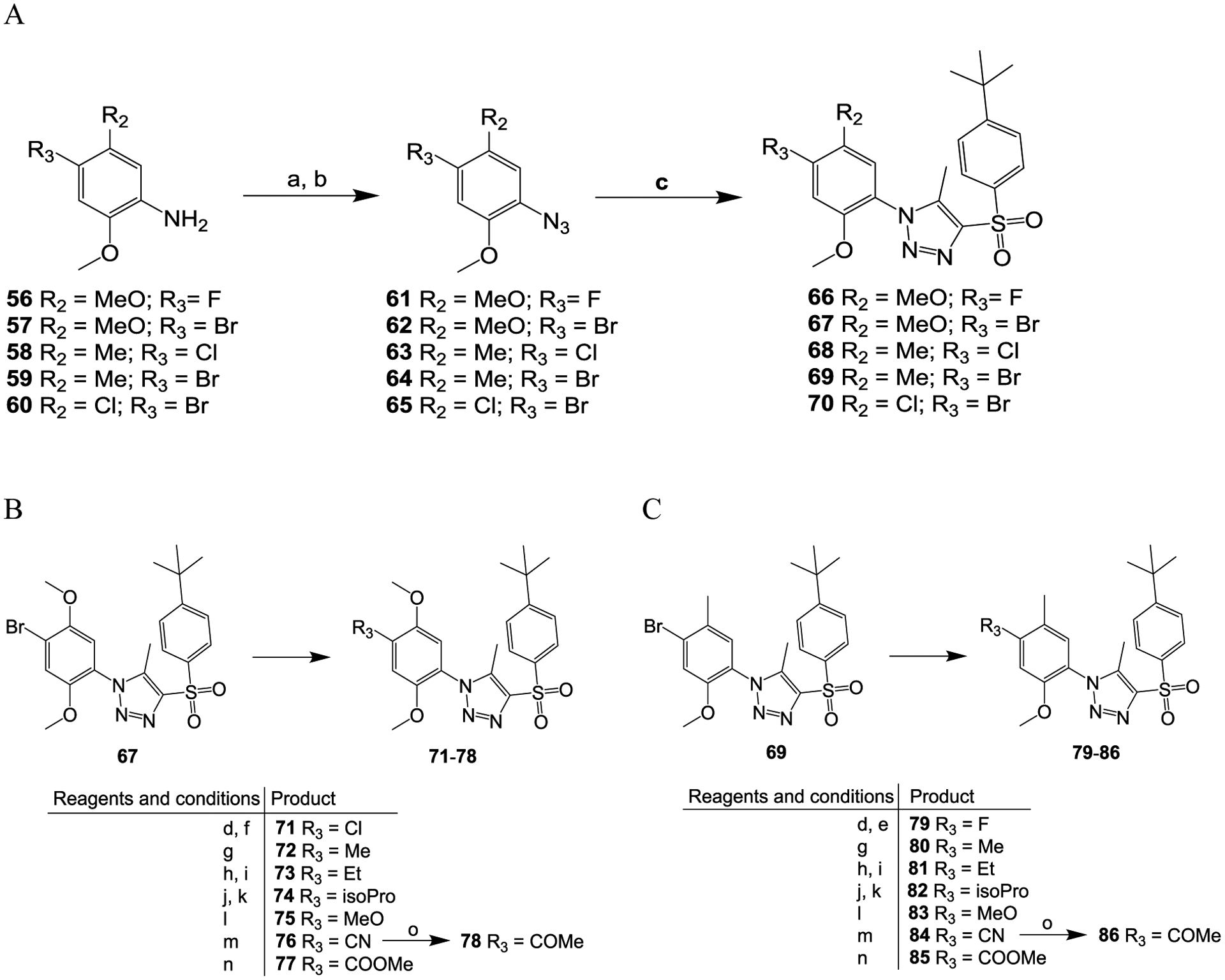
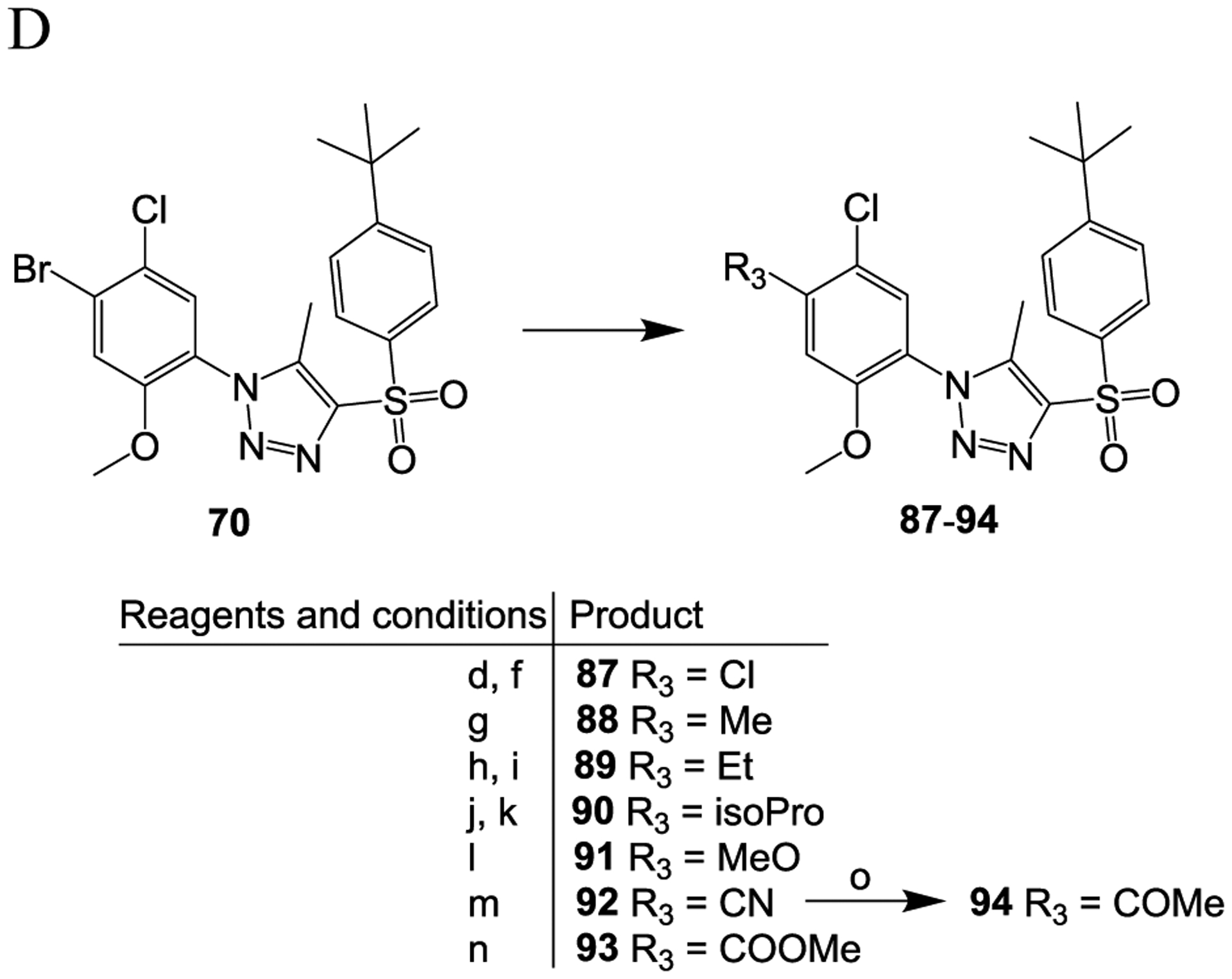
Reagents and conditions: (a) concentrated HCl, NaNO2, H2O, 0 °C, 15 min; (b) NaN3, H2O, room temperature, 2 h; (c) MeONa, 1-((4-(tert-butyl)phenyl)sulfonyl)propan-2-one, MeOH, 60 °C, overnight; (d) n-BuLi, THF, −78 °C, 5 min; (e) NFSI, THF, −78 °C, 1 h; (f) C2Cl6, THF, −78 °C, 1 h; (g) MeB(OH)2, Pd(PPh3)4, Na2CO3, dioxane, H2O, 120 °C, 5 h; (h) ethenylSn(Bu)3, Pd(PPh3)4, toluene, 120 °C, 5 h; (i) H2, 10% Pd/C, MeOH, THF, room temperature, 3 h; j) 4,4,5,5-tetramethyl-2-(prop-1-en-2-yl)-1,3,2-dioxaborolane, Pd(PPh3)4, Na2CO3, dioxane, H2O, 120 °C, 5 h; (k) H2, 10% Pd/C, MeOH, THF, room temperature, 3 h; (l) MeONa, CuCl2, DMF, 130 °C, overnight; m) CO (1.5 MPa), Pd(dppf)Cl2, MeOH, 80 °C, 24 h; (n) Zn(CN)2, Pd(dppf)Cl2, DMF, microwave, 160 °C, 15 min; (o) MeMgBr, THF, reflux, 15 h.
As shown in Scheme 4, compounds 97 to 99 were synthesized from the corresponding aryl azides 12, 95 and 96 by reacting with 1-((4-(tert-butyl)phenyl)sulfonyl)butan-2-one.35 When reacting with 2-((4-(tert-butyl)phenyl)sulfonyl)acetonitrile, the aryl azides 12, 95 and 96 were converted to amino compounds 100 to 102,35 which were further converted to corresponding compounds 103 to 105 via deamination.49 Bromide compounds 106 to 108 were prepared by substitutive deaminations50 of corresponding compounds 100 to 102 in the presence of t-BuNO2 and CuBr2 and were further converted to respective compounds 109 to 111, via the Rosenmund–von Braun reaction51 in the presence of CuCN or to corresponding compounds 112 to 114, via palladium-catalyzed coupling with tributyl(1-ethoxyvinyl)stannane43 and subsequent hydrolysis and isomerization.52
Compounds 122 to 124 and 128 to 130 were prepared with properly substituted 5-bromo-1H-imidazoles (115 and 117) as the starting materials (Scheme 5); compound 117 was prepared from compound 116 by de-protonation followed by SEM protection. The substituted 5-bromo-1H-imidazoles (115 and 117) were first converted to the 5-(phenylsulfonyl)-1H-imidazoles 118 and 119 via Cu(I)-catalyzed coupling with arylsulfinic salts,53 then the compounds 118 and 119 were further converted to the 2-bromo-5-(phenylsulfonyl)-1H-imidazoles 120 and 121 with the bromination reagent NBS.54 Compounds 122 to 124 and 125 to 127 were prepared by Suzuki coupling between 2,5-disubstituted-phenylboronic acids and respective compound 120 and compound 121 in the presence of Pd(dppf)Cl2 as the catalyst.55 Compounds 128 to 130 were subsequently obtained by removing the SEM protecting group in compounds 125–127, respectively.56
The syntheses of compounds 140 and 145 are summarized in Scheme 6. With 4-methyl-1H-imidazole (compound 131) as the starting material, the active proton on the nitrogen of the 4-methyl-1H-imidazole was first protected with an SEM protecting group,56 and a pair of isomers, compounds 132 and 133, were isolated. Brominations54 of compounds 132 and 133 with NBS as the brominating agent yielded two pairs of separable isomers: (1) compounds 134 and 135 and (2) compounds 136 and 137. Compounds 134 and 135 were converted to compound 138 via Cu(I)-catalyzed coupling reactions with arylsulfinic salts.53 Further bromination54 of compound 138 yielded the bromide compound 139, which was then converted to compound 140 via a Suzuki coupling55 with (2,5-dimethoxyphenyl)boronic acid in the presence of Pd(dppf)Cl2 as the catalyst. In contrast, Suzuki coupling55 of the isomers 136 and 137 with 2-methoxy-5-chlorophenylboronic acid in the presence of Pd(dppf)Cl2 as the catalyst produced isomers 141 and 142 which were further converted to another pair of bromide isomers, compounds 143 and 144, via NBS-mediated brominations.54 Compound 145 was then prepared by a substitution reaction between isomers 143 and 144 and sodium 4-(tert-butyl)benzenesulfinate, with CuI as the catalyst.53 In the preparations of compounds 140 and 145, a separated de-protection of the SEM protecting group step was not involved because the SEM group was removed during the Cu(I)-mediated coupling reactions.
Scheme 6.
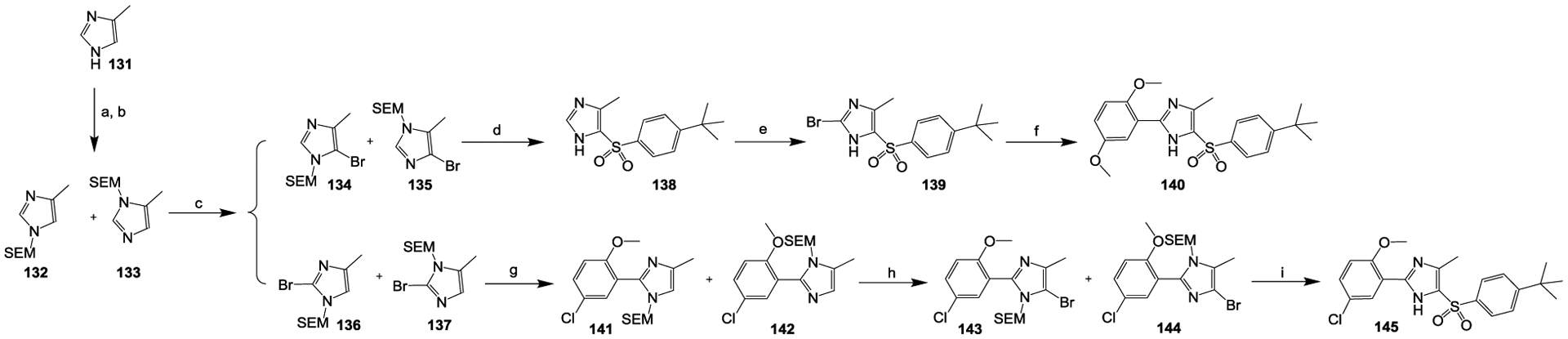
Reagents and conditions: (a) NaH, THF, 0 °C, 15 min; (b) SEMCl, 0 °C, 1 h; (c) NBS, MeCN, 0 °C → room temperature, 1 h; (d) sodium 4-(tert-butyl)benzenesulfinate, CuI, DMF, N2, 65 °C, 18 h; (e) NBS, MeCN, room temperature, 17 h; (f) (2,5-dimethoxyphenyl)boronic acid, Pd(dppf)Cl2, K2CO3, dioxane, H2O, N2, 100 °C, 6 h; (g) 2-methoxy-5-chlorophenylboronic acid, Pd(dppf)Cl2, K2CO3, dioxane, H2O, N2, 65 °C, 6 h; (h) NBS, MeCN, −30 °C → room temperature, 0.5 h; (i) sodium 4-(tert-butyl)benzenesulfinate, CuI, DMF, N2, 65 °C, 18 h.
The preparation of compound 150 is summarized in Scheme 7, with 2,5-dibromo-4-methyl-1H-imidazole (compound 146) as the starting material. Compound 146 was first converted to the SEM protected compound 147.56 Compound 147 was then converted to compound 148 via a Pd(dppf)Cl2-mediated Suzuki coupling55 with 2-methoxy-5-methyl-phenylboronic acid. Cu(I)-mediated coupling53 of compound 148 with the arylsulfinic salt yielded compound 149, which was then converted to compound 150 via SEM de-protection.56
Scheme 7.
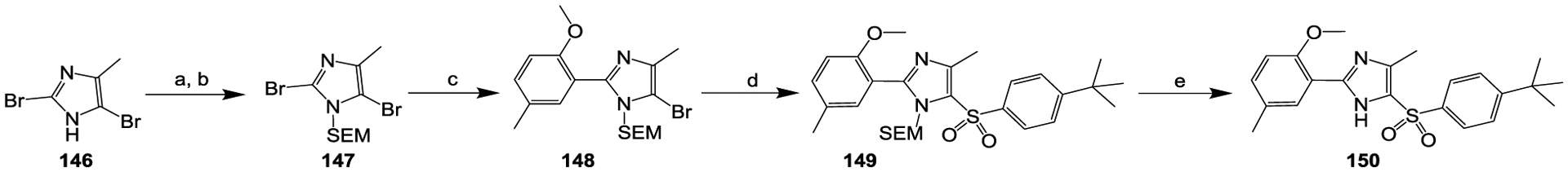
Reagents and conditions: (a) NaH, THF, 0 °C, 15 min; (b) SEMCl, 0 °C, 1 h; (c) 2-methoxy-5-methyl-phenylboronic acid, Pd(dppf)Cl2, K2CO3, dioxane, H2O, N2, 100 °C, 3 h; (d) sodium 4-(tert-butyl)benzenesulfinate, CuI, DMF, N2, 65 °C, 18 h; (e) TBAF, THF, N2, 60 °C, 2 h.
Compounds 155 to 157 were prepared using 2,5-dibromo-1,4-dimethyl-1H-imidazole (compound 151) as the starting material (Scheme 8). Briefly, compound 151 was first converted to compounds 152–154 via Pd(dppf)Cl2-mediated Suzuki couplings55 with appropriate 2-methoxy-5-substituted-phenylboronic acid and then to compounds 155 to 157 via Cu(I)-mediated couplings53 with sodium 4-(tert-butyl)benzenesulfinate.
RESULTS AND DISCUSSION
PXR Binding Activity, Cellular PXR functional Activity, and Cytotoxicity of SPA70 Analogs
The analogs of SPA70 (10) were tested for their hPXR binding activities, cell-based hPXR functional activities (i.e., agonistic, inverse agonistic, and antagonistic activities), and cytotoxic activities, along with the control compounds T0901317 (4), rifampicin (1), SPA70 (10) and DMSO. The final DMSO concentration was 0.3% in all assays. The hPXR binding activities were measured by using the PXR TR-FRET assay as previously described57 with minor modifications with T0901317 (4, 10 μM) as the positive control (100% inhibition) and DMSO as the negative control (0% inhibition). The agonistic, inverse agonistic and antagonistic activities of the compounds were determined in HepG2 cells stably expressing hPXR and a luciferase reporter under the control of a CYP3A4 promoter with the steadylite HTS reagent (PerkinElmer Life and Analytical Sciences, Boston, MA).58 In the agonistic assay, rifampicin (1, 10 μM) was used as the positive control (100% activation) and DMSO as the negative control (0% activation) for agonists, and SPA70 (10, 10 μM) was used as the positive control (100% inhibition) and DMSO as the negative control (0% inhibition) for inverse agonists. In the antagonistic assay, SPA70 (10, 10 μM) with rifampicin (1, 5 μM) was used as the positive control (100% inhibition) and rifampicin (1, 5 μM) alone as the negative control (0% inhibition). Compound cytotoxicity was determined in the same HepG2 stable cells under the same conditions as reporter activity assays (agonistic, inverse agonistic and antagonistic assays), but using a CellTiter-Glo luminescent cell viability assay (Promega Corporation, Madison, WI). DMSO without cells was used as the positive control (100% inhibition) and DMSO with cells as the negative control (0% inhibition). SPA70 (10) and most of its analogs showed no cytotoxicity [except for compound 86, which had low cytotoxicity (with an IC50 of 16.5 μM)], indicating that the reporter activity was not interfered by compound cytotoxicity.
First, we examined the effect of different substitutions at region A [the right 4-(tert-butyl)phenyl group region] in SPA70 (10) on hPXR activities. A total of 12 analogs were designed and prepared. The structures and activities of these analogs are summarized in Table 1. Replacing the tert-butyl in SPA70 (10) by other groups substantially decreased the binding activity, which ranged from 1 to 10 μM (IC50 value), except COOH in compound 26 which almost abolished the binding activity. The rank order of compound binding activity (from high to low) with various substitutions is Benzyl (in compound 28) > Cl (in compound 23) > H (in compound 21) ~ CMe2OButyl (in compound 32) > MeO (in compound 25) > CMe2OMe (in compound 31) ~ Me (in compound 22) > NH2 (in compound 27) > OH (in compound 24) > COOMe (in compound 29) > CMe2OH (in compound 30) >> COOH (in compound 26). The tert-butyl in SPA70 occupies a mostly hydrophobic cavity lined by residues that include Leu-209, Val-211, Phe-288, Trp-299, Tyr-306, Met-323 and Leu-324. Therefore, any substitution that minimizes hydrophobic interactions with these residues would result in decreased binding activity. In the cell-based functional assays, replacing the tert-butyl with a smaller H (in compound 21), Me (in compound 22), Cl (in compound 23), OH (in compound 24), MeO (in compound 25), or NH2 (in compound 27) group converted SPA70 (10, a dual inverse agonist and antagonist) to an agonist. Replacing the tert-butyl in SPA70 (10) with a larger CMe2OMe (in compound 31) or CMe2OButyl (in compound 32) group decreased the binding activity and both inverse agonistic and antagonistic activities. Hydrophilic substitution of OH (in compound 24), COOH (in compound 26), NH2 (in compound 27), COOMe (in compound 29), or CMe2OH (in compound 30) drastically decreased in the hPXR binding activities and made these analogs weak hPXR agonists. In contrast, hydrophobic modifications had less of an effect on reducing hPXR binding activities. For example, the hydrophobic modification of Cl in compound 23 (which had an hPXR binding IC50 of 1.91 μM) had less impact than the hydrophilic modification of OH in compound 24 (which had an hPXR binding IC50 of 8.03 μM); and the relatively hydrophobic MeO in compound 25 (which had an hPXR binding IC50 of 2.96 μM) had less impact than the hydrophilic COOH in compound 26 (which had an hPXR binding IC50 greater than 30 μM). Inserting a methylene group between the sulfonyl group and the right phenyl ring in compound 21 (which had an hPXR binding IC50 of 2.1 μM) to generate compound 28 (which had an hPXR binding IC50 of 1.02 μM) can even slightly increase the hPXR binding activity. It appears that the tert-butyl group on the 1-phenyl-4-(phenylsulfonyl)-1H-1,2,3-triazole scaffold is critical for an optimal binding activity and maintaining inverse agonistic and antagonistic activities of SPA70 (10). Changing this tert-butyl group to another smaller, larger, hydrophilic, or hydrophobic group reduced the binding activity of SPA70 (10) and decreased the inverse agonistic and antagonistic activities or converted it to an agonist.
We then investigated the effect of modifying the meta-methoxy to the left phenyl group of the 1H-1,2,3-triazole in SPA70 (10) (region B) on hPXR activities. A total of 12 analogs with the meta-position modified were designed and prepared. The structures and activities of these analogs are summarized in Table 2.
When the meta-methoxy group in SPA70 (10) was replaced with an H atom, the resulting compound (41) had only slightly decreased binding activity but became an agonist, with an hPXR agonistic EC50 of 0.76 μM. Although the EC50 of 0.76 μM made compound 41 a stronger agonist than rifampicin (which had an hPXR agonistic EC50 of 1.44 μM), the observed activity (% activation at 10 μM) of 41 (56%) is lower than that of rifampicin (1, 100%). It is, therefore, not surprising that 41 displayed weaker antagonistic activity (with an antagonistic IC50 of 1.0 μM) than that of SPA70 (10, with an antagonistic IC50 of 0.25 μM). The antagonistic activities (at 10 μM) of 41 and SPA70 (10) were 53% and 100%, respectively. Therefore, compound 41 behaved as an hPXR partial agonist and partial antagonist.59
Halogen modifications of the meta-methoxy group in SPA70 (10) to F, Cl and Br resulted in the compounds 42, 43 and 44, respectively, all of which retained similar hPXR binding activities to SPA70 (10) with 42 became a hPXR partial agonist/partial antagonist whereas 43 and 44 remained to be hPXR dual inverse agonist and antagonist.
When the meta-methoxy group in SPA70 (10) was replaced with a Me or CF3 group, the resulting compounds 45 and 46 maintained strong binding activities (respective hPXR binding IC50 of 0.79 μM and 0.41 μM) and they were pure hPXR antagonist (corresponding hPXR antagonistic IC50 of 2.21 μM and 1.01 μM) with no hPXR agonistic or inverse agonistic activity. Compound 45 has a Me group at the meta-methoxy position, whereas compound 46 has a CF3 group at the same position; however, compound 46 binds better to hPXR and is a stronger hPXR antagonist than compound 45. Because CF3 is similar to Me in size but more hydrophobic, it appears that hydrophobic modifications at the meta-methoxy position can increase hPXR binding and antagonistic activities.
When the meta-methoxy group in SPA70 (10) was replaced with a tert-butyl group, the resulting compound 47 still had strong binding activity (with an hPXR binding IC50 of 0.34 μM) but became a strong agonist (with an hPXR agonistic EC50 of 0.38 μM). Compound 48, which has a CN group in place of the meta-methoxy group in SPA70, remained to be a strong dual inverse agonist (with an IC50 of 0.045 μM) and antagonist (with an IC50 of 0.45 μM).
Introducing hydrophilic groups at the meta-methoxy position (CH2OH in compound 50, CONH2 in compound 51, R-HOMeCH in compound 54, and S-HOMeCH in compound 55) generated analogs with decreased binding activities but with agonistic activities.
Compounds SPA70 (10) (with a meta-MeO group), 43 (with a meta-Cl group), and 45 (with a meta-Me group) all had high hPXR binding activities (with binding IC50s of 0.19, 0.79, and 0.25 μM, respectively), but none of them had hPXR agonistic activity. SPA70 (10) and 43 were potent hPXR inverse agonists with respective IC50s of 0.024 and 0.27 μM. SPA70 (10), 43 and 45 were potent hPXR antagonists with respective IC50 values of 0.25, 2.21, and 0.31 μM. The MeO, Me, and Cl groups were chosen for the meta-position in the subsequent SAR exploration of hPXR inhibition based on SPA70 (10).
With the region B optimized with an MeO, Me, or Cl group in the previous step, we further explored the influence of different substitutions at the para-hydrogen atom position (region C, the para-position on the left phenyl group to the 1H-1,2,3-triazole in SPA70) on the hPXR activities. These substituents were halogens (F, Cl, or Br), small-size alkyl groups (Me, Et, or isoPro), nitrogen-containing cyano groups, or oxygen atom-containing MeO, COOMe, or COMe groups. A total of 29 analogs with modification at the region C were designed and prepared. The structures and activities of these analogs are summarized in Table 3, with the 29 compounds divided into three groups based on region B modifications of MeO, Me, or Cl.
Analogs with modifications from a proton to another group at this para-position except compound 94 retained hPXR binding activities, although the hPXR binding activities were weaker than that of SPA70 (which had an hPXR binding IC50 of 0.19 μM), but none of them preserved the hPXR inverse agonistic activity as observed in SPA70. Only the analogs with para-F (in compound 79, with an hPXR antagonistic IC50 of 0.66 μM) or para-Cl (in compound 87, with an hPXR antagonistic IC50 of 1.25 μM) modification retained hPXR antagonistic activities without exhibiting hPXR agonistic activity, although their hPXR antagonistic activities were weaker than that of SPA70 (which has an hPXR antagonistic IC50 of 0.25 μM). Eleven analogs (compounds 66, 72, 68, 80, 84, 85, 86, 70, 88, 92 and 93) were weak hPXR agonists and weak hPXR antagonists. The other 16 analogs (71, 67, 73, 74-78, 69, 81–83, 89–91, and 94) were strong hPXR agonists. Therefore, replacing the para-position proton with other groups resulted in analogs that lacked hPXR inverse agonistic or antagonistic activity but had hPXR agonistic activity.
Finally, we explored the 1H-1,2,3-triazole region (Region D) in SPA70. Twenty-seven compounds with two types of modifications (Figure 4, Modification 1 and Modification 2) were designed, synthesized, and evaluated. Their structures and biological activities are summarized in Table 4.
Figure 4.
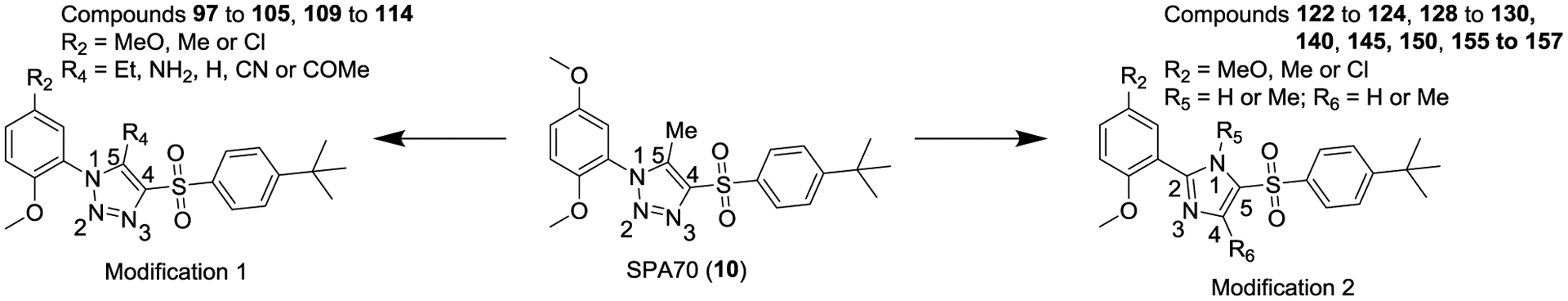
Compounds listed in Table 4 with two type of modifications from SPA70.
For the first type of modification (Modification 1 in Figure 4), the Me group at the 5-position of the 1H-1,2,3-triazole ring was replaced with an Et, NH2, H, CN, or COMe group. Replacing the Me group with an Et group (compounds 97 to 99) resulted in analogs with slightly reduced binding affinities but maintained hPXR antagonistic activities. Replacing the Me group with an NH2 group (compounds 100 to 102) or an H atom (compounds 103 to 105) reduced the hPXR binding activities very marginally but reduced the hPXR antagonistic activities drastically and introduced weak hPXR agonistic activities. Replacing the Me group with a CN group (compounds 109 to 111) did not affect the hPXR binding activities but introduced potent hPXR agonistic activities and abolished hPXR antagonistic activities with compound 111 having a hPXR agonistic EC50 of 0.081 μM. However, compound 109, in which a CN group was substituted at the 5-Me position of the 1H-1,2,3-triazole region and a meta-MeO group was on the left phenyl group to the 1H-1,2,3-triazole of SPA70, was a weak dual hPXR antagonist and agonist.
For the second type of modification (Modification 2 in Figure 4), the middle 1H-1,2,3-triazole ring was replaced with an imidazole ring, but a substitution pattern similar to that in SPA70 (10) was maintained to preserve the similar extended confirmation observed in SPA70 (10). The substitution patterns of SPA70 (10) and the representative analog 155 are illustrated in Figure 5. Instead of the middle 1H-1,2,3-triazole ring as in SPA70 (10), compounds 128 to 130 have an imidazole ring that joins the left substituted phenyl group and the right substituted phenyl sulfonyl group. Similarly, compounds 122 to 124 have a 1-Me-substituted imidazole ring, compounds 140, 145, and 150 have a 4-Me-substituted imidazole ring, and compounds 155 to 157 have a 1,4-diMe-substituted imidazole ring. All these analogs with an imidazole ring in the place of the 1H-1,2,3-triazole ring in SPA70 were hPXR agonists, although compounds 122 to 124 with a 1-Me-substituted imidazole ring displayed very low antagonistic activities at very high concentrations.
Figure 5.
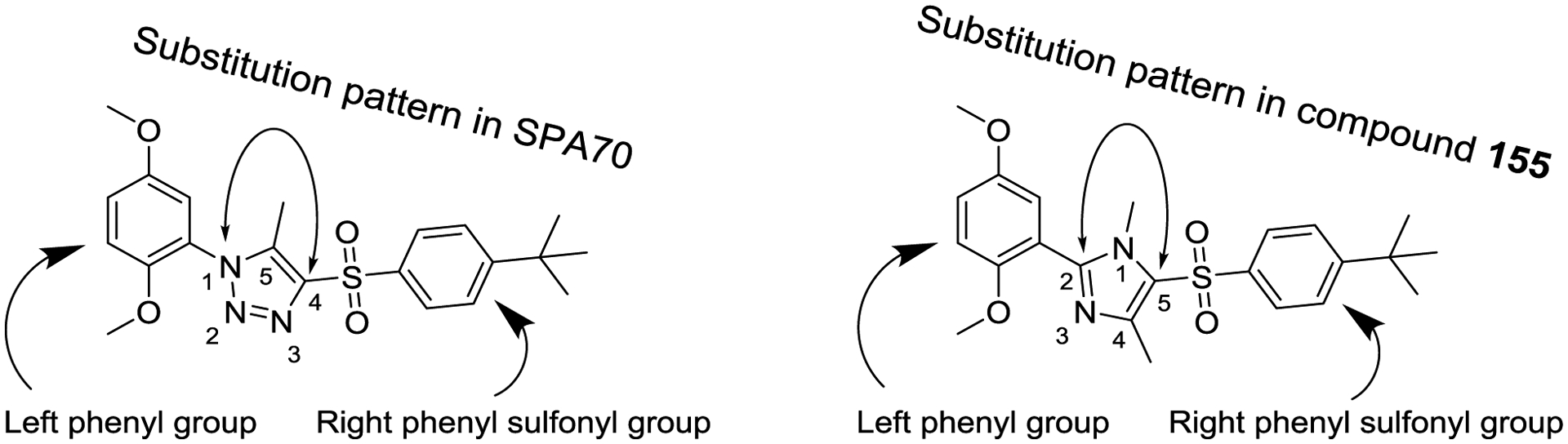
Substitution patterns of SPA70 and analog compound 155.
In Summary, to explore the SARs of hPXR modulators based on the scaffold of SPA70 (10), which is a specific hPXR antagonist10, 60 and an inverse agonist, we designed, synthesized, and evaluated a total of 81 SPA70 analogs. Interestingly, these structurally similar SPA70 analogs differed drastically in their hPXR cellular functional activities, including dual antagonists/inverse agonists (represented by SPA70 itself), strong pure antagonists (represented by compound 97), strong agonists (represented by compound 111), and weak partial agonists/partial antagonists (represented by compound 42). The subtly structural changes resulted in drastic changes in functional activities: replacing the 5-Me at the middle 1H-1,2,3-triazole ring of SPA70 (which is a dual antagonist/inverse agonist) with a 5-Et made compound 97 a pure antagonist; replacing the 5-Me at the middle 1H-1,2,3-triazole ring with a 5-CN and substituting a meta-Cl at the left phenyl ring made compound 111 an agonist; and substituting a meta-F at the left phenyl ring made compound 42 a partial agonist/partial antagonist (Figure 2B and Figure 6). A relatively straightforward SAR could not be easily derived based on the structure and observed activity.
Figure 6.
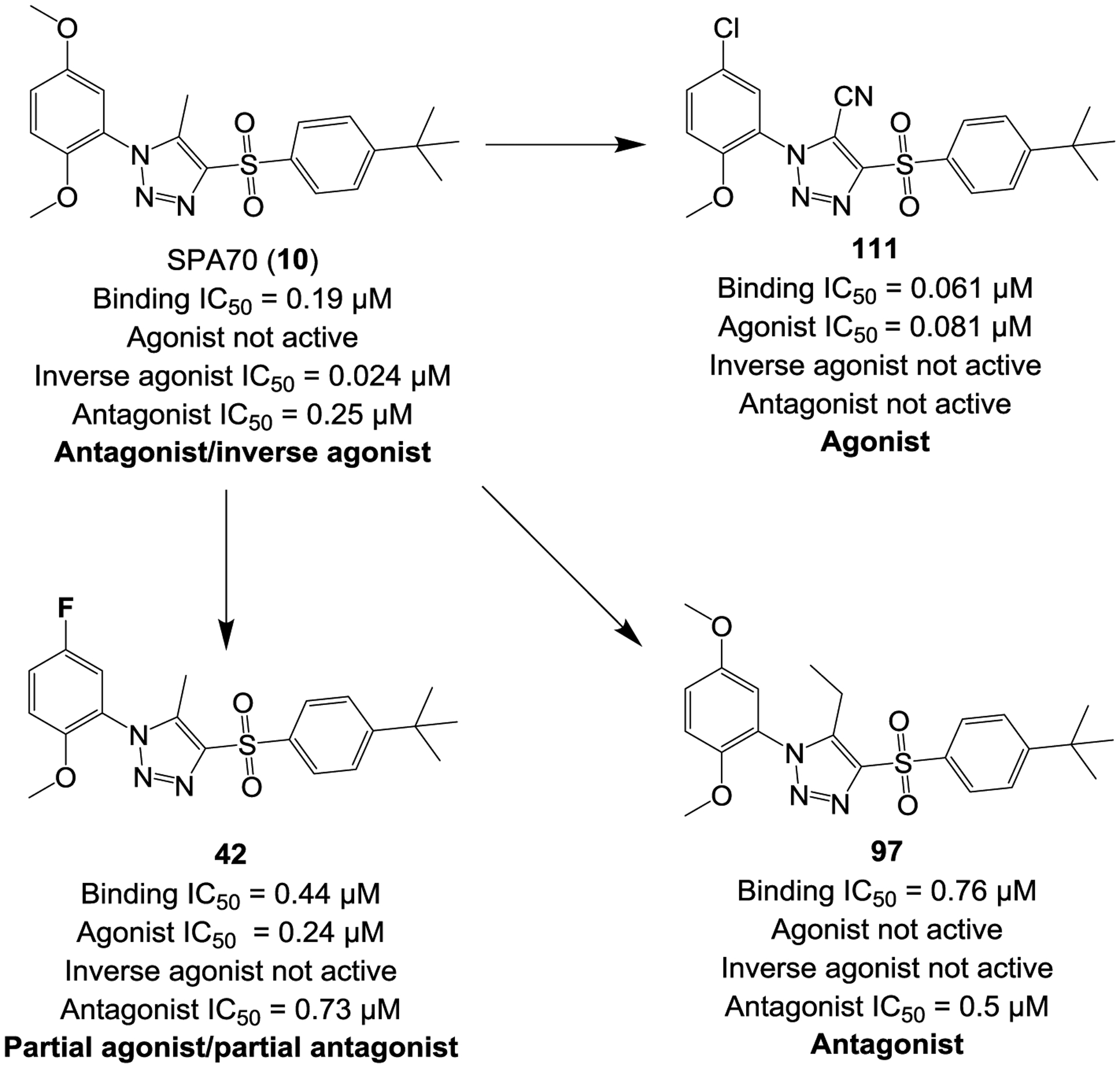
Subtle structural changes in SPA70 (10) (which is an antagonist/inverse agonist) generate an agonist (111), antagonist (97), or a partial agonist/partial antagonist (42).
To summarize the activities of 81 analogs, SPA70 (10) is the most potent inverse agonist and antagonist, with IC50 values of 0.19 μM for binding activity, 0.024 μM for inverse agonistic activity and 0.25 μM for antagonistic activity. Among the 81 SPA70 analogs, compound 111 binds hPXR with the highest affinity (with an IC50 value of 0.061 μM) and displays an agonistic EC50 of 0.091 μM, which made it a higher-affinity binder than SPA70 (SPA70 to 111 IC50 ratio: 3) and a much more potent hPXR agonist than rifampicin (rifampicin to 111 EC50 ratio: 16). Compound 97 is the most potent pure antagonist (with an IC50 value of 0.5 μM) with a high binding activity (with an IC50 value of 0.76 μM). Compound 42 maintained high hPXR binding affinity (with an IC50 value of 0.44 μM) but had reduced antagonistic activity (with an IC50 value of 0.73 μM) when compared to SPA70, and gained weak partial agonistic activity (with an EC50 of 0.24 μM and % activation of 37% at 10μM). The hPXR activities of these representative analogs of SPA70 (10), compound 111, 97 and 42 were summarized in Figure 2B and Figure 6. The tert-butyl group (region A) is essential for the binding and antagonistic activity. Replacing the methoxy group at region B affects both agonistic and antagonistic activities. The hydrogen at region C strongly affects inverse agonistic activity: when hydrogen was replaced by various other groups, all analogs lost their inverse agonistic activities. For the 5-methyl-1H-1,2,3-triazole ring at region D, the methyl group strongly affects compound activity, and changing the triazole to the other ring introduced agonistic activity.
Pharmacophore Model of SPA70 Analogs
To obtain an idea of the representative ligand features and their corresponding spatial orientations of SPA70 (10) analogs, we generated a pharmacophore model. Pharmacophore models take ligand input and, by analyzing important features such as atoms capable of acting as hydrogen bond acceptors, generate a single model representing multiple ligands. We successfully generated a six-feature pharmacophore for 80 SPA70 analogs. The resulting model is composed of three aromatic ring features, a hydrogen bond donor feature, and two hydrogen bond acceptor features. Upon atomic alignment, each of the SPA70 analogs fits our model, highlighting its quality as a true representation of these compounds. Each of the compounds fits the allowance spheres (which allow for 2-Å freedoms). The general spatial distribution of features was as follows: an aromatic feature within the right aromatic ring in the structure of SPA70 (10), two hydrogen bond acceptors each placed at the sulfonamide groups, an aromatic feature within the 1H-1,2,3-triazole ring, an aromatic feature within the left aromatic ring in the structure of SPA70 (10), and a hydrogen bond donor feature placed over the oxygen atoms of the meta-position of the 1H-1,2,3-triazole ring. We sanity-checked our model by testing it with SPA70 (10), which was not used as input for the pharmacophore, but it fits the model very well. This pharmacophore represents a model that could be used to guide future analog design, to perform virtual compound screening, or to predict the PXR biological activity of ligands.
Molecular Modeling Study of SPA70 Analogs
It is intriguing that SPA70 (10) analogs with very similar scaffolds can modulate the cellular activity of hPXR to produce divergent outcomes. The reported co-crystal structure of the hPXR LBD with SJB7 (83) indicates that the agonist interacts with residues in the AF-2 helix, stabilizing it in the active conformation primed for coactivator recruitment (Figure 8A),10 which is a common feature in the agonism of hPXR and of other ligand-activated NRs.13, 17 However, it was concluded that SPA70 antagonizes hPXR because it lacks strong interactions with the AF-2 helix and, thus, fails to fixate it in the active form.10 To understand the mode of action of the SPA70 analogs reported in this study, computational docking experiments were performed using compounds 111, 42, and 97 as representatives (Figure 8 and Figure S2 in Supporting Information). In the agonist analog compound 111, the chloro substituent (represented as a green sphere) is in close proximity to the AF-2 helix (represented as a salmon-red surface) (Figure 8B), which is similar to the interactions provided by the methoxy group (white sphere) of SJB7 (Figure 8A) that is necessary for the AF-2 helix to have a favorable configuration for coactivator engagement with the hPXR LBD. It is possible that the cyano group in the triazole ring of compound 111 can form a hydrogen bond with Ser-247. The ability to form interactions with Ser-247 could be a feature that causes triazole-containing analogs to incline towards agonistic behavior. Conversely, compounds 42 (Figure 8C) and 97 (Figure 8D) do not provide strong contacts with the AF-2 helix; this resembles the antagonistic mechanism of SPA70 and leads to the observed reduced activity in hPXR. The docking studies using these three representative cases support the notion that strong interactions between the analogs and residues in the AF-2 helix favor agonism, whereas the absence of such interactions prevents the AF-2 helix from maintaining an orientation that is responsive for coactivator binding.
Figure 8.
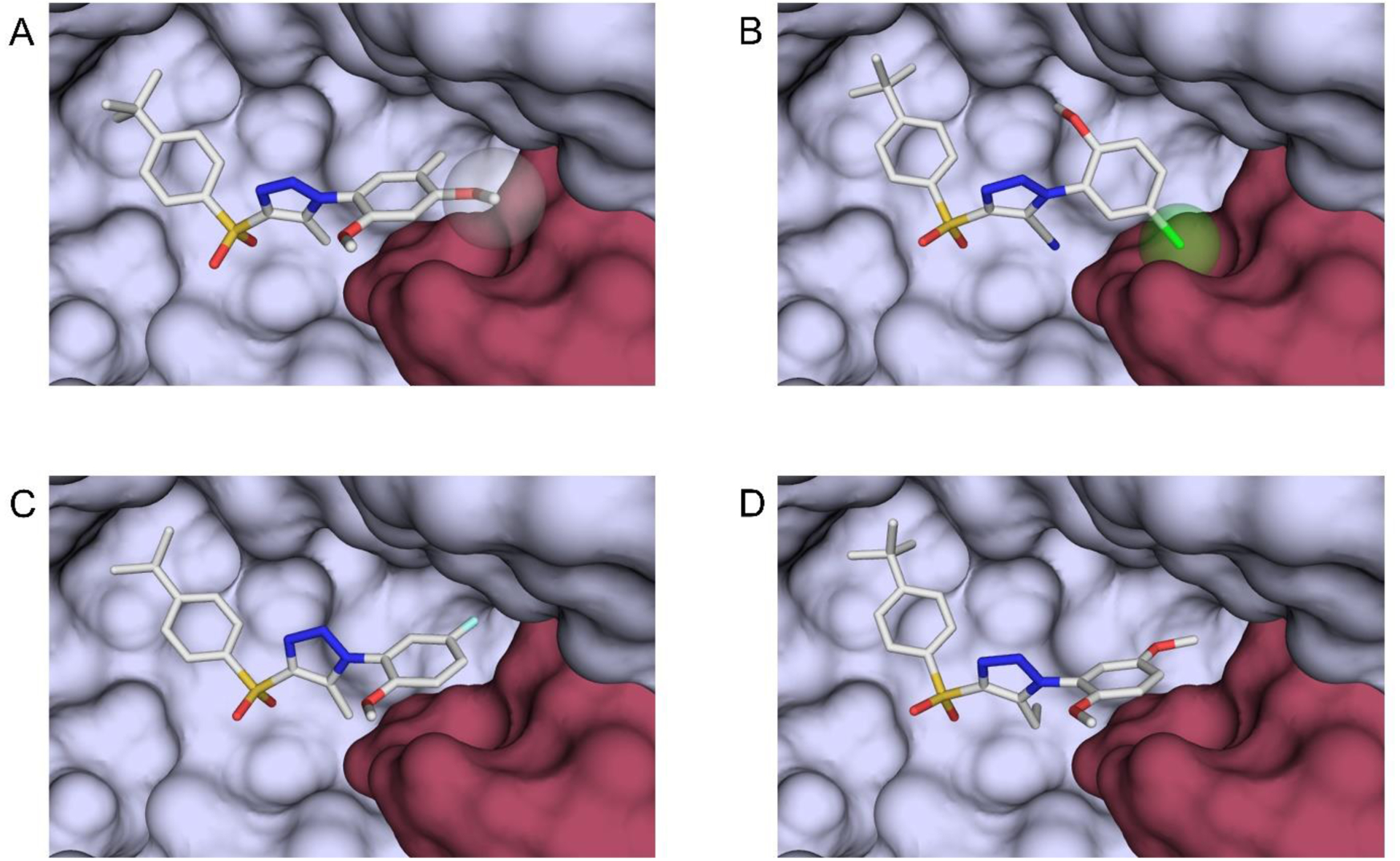
(A) For comparison, a crystal structure of the hPXR LBD (PDB code: 5X0R) shows the agonist SJB7 residing in the ligand-binding pocket (surface representation in light blue), with the methoxy group (white sphere) interacting with residues in the AF-2 helix (salmon red surface). (B) Docking studies reveal interactions between the chloro substituent of compound 111 (green sphere) and the AF-2 helix. (C, D) The docking poses of compounds 42 (C) and 97 (D) are similar to those of SJB7 and compound 111, with the important distinction that no major contacts are observed between these ligands and the AF-2 helix. Color scheme: carbon, white; oxygen, red; nitrogen, blue; sulfur, yellow; chlorine, green; fluorine, light blue.
Conclusions
In this study, 81 SPA70 analogs were synthesized (or obtained commercially) and evaluated for their hPXR binding, agonistic, inverse agonistic, and antagonistic activities in a SAR-guided stepwise approach. These SPA70 analogs have subtle structural differences but divergent cellular activities, with the latter including pure antagonistic activities (represented by compound 97), agonistic activities (represented by compound 111), dual inverse agonistic and antagonistic activities (represented by SPA70), and weak partial agonistic/partial antagonistic activities (represented by compound 42). The unique SAR of the SPA70 analogs could not be easily derived from the direct structure and activity observations. Therefore, we generated a model that represents 80 SPA70 analogs. The model was tested by using SPA70 (an antagonist): although SPA70 was not used as input for the pharmacophore, it fits the model very well, thus validating the model, which could be used to guide future analog design and to predict the hPXR cellular activity of ligands. We also performed docking studies, and the results are consistent with strong interactions between the compounds and residues in the AF-2 helix with agonistic activity. This set of 81 structurally related but functionally divergent compounds is a novel toolset for investigating the function of hPXR. The SAR data gained from these 81 analogs provides a basis for guiding future efforts to develop additional hPXR modulators. To our knowledge, our effort is the first chemistry endeavor in developing hPXR modulators with varying activities based on a novel chemical scaffold previously discovered through a large-scale compound-screening campaign.
Experimental Section
General Methods for Chemistry
General procedures: Organic reagents were purchased from commercial suppliers unless otherwise noted and were used without further purification. All solvents were analytical or reagent grade. All reactions were carried out in flame-dried glassware under argon or nitrogen. Flash column chromatography was performed by using silica gel 60 (200 – 400 mesh, Sigma-Aldrich, St. Louis, MO) and was carried out under moderate pressure, with columns of an appropriate size packed and eluted with appropriate eluents. All reactions were monitored by performing thin-layer chromatography (TLC) on precoated plates (silica gel HLF) (Analtech, Newark, Delaware). TLC spots were visualized by exposure to iodine vapor or by irradiation with UV light (Sigma-Aldrich, St. Louis, MO). Organic solvents were removed under vacuum with a rotary evaporator (BUCHI Corporation, New Castle, DE). The purities of the final compounds were determined by using a Waters Acquity UPLC MS system with a C18 column in a 2-min gradient (H2O + 0.1% formic acid → acetonitrile + 0.1% formic acid) and detectors of PDA (215–400 nm), ELSD, and Acquity SQD ESI-positive MS (Waters Corporation, Milford, MA). Preparative TLC separation was performed by using self-casted preparative TLC plates with Sigma-Aldrich silica gel 60 (200–400 mesh) on 20 cm × 20 cm glass plates. The reaction products were purified by using a Dionex APS 3000 dual purification/analytical LC/PDA/MS system with a C18 column in a 15-min gradient (H2O with 0.05% NH3·H2O → acetonitrile) and ESI-positive MS (vendor, city, state). High-resolution mass spectra were determined by using a Waters Acquity UPLC system with a C18 column (H2O + 0.1% formic acid → acetonitrile + 0.1% formic acid gradient over 2.5 min) and Xevo G2Q-TOF ESI-positive MS in resolution mode. Compounds were internally normalized to leucine-enkephalin lock solution, with a calculated error of <3 ppm. All final compounds used for SAR studies have purity at 95% or greater. All 1H NMR spectra and 13C NMR spectra were recorded on a Bruker ULTRASHIELD 400 plus or Bruker Ascend 500 NMR spectrometer (Bruker BioSpin Corporation, Billerica, MA). In the compound spectra, the chemical shift values are expressed in parts per million (ppm) relative to tetramethylsilane as the internal standard. Coupling constants (J) are reported in hertz (Hz). NMR spectra, high resolution mass spectrometry, and HPLC purity analyses of final compounds are shown in Supporting Information.
2-Azido-1,4-dimethoxybenzene (12).
NaNO2 (13.80 g, 200 mmol) in water (200 mL) was added dropwise to a solution of 11 (2,5-dimethoxyaniline, 30.6 g, 200 mmol) in concentrated HCl (48 mL) at 0 °C, and the reaction mixture was then stirred for 15 min at 0 °C. Then hexane (40 mL) was added to the reaction. A solution of NaN3 (14.30 g, 220 mmol) in water (40 mL) was added dropwise to the reaction solution at 0 °C and stirred for 2 h at ambient temperature. The reaction mixture was extracted with hexane/EtOAc = 1:1 (200 mL × 3). The organic layer was washed with water and brine, dried with anhydrous MgSO4, and concentrated with a Rotavapor to give product 12 (26.3 g, 73% yield). The resulting aryl azide 12 was used for the next step without further purification.
1-(2,5-Dimethoxyphenyl)-5-methyl-4-(phenylsulfonyl)-1H-1,2,3-triazole (21).
MeONa (0.216 g, 4.00 mmol) and 1-(phenylsulfonyl)propan-2-one (13, 0.396 g, 2.000 mmol) were added to a solution of compound 12 (0.358 g, 2 mmol) in MeOH. The mixture was stirred at 60 °C overnight. The reaction mixture was then poured into water (100 mL) and extracted with EtOAc (100 mL × 3). The EtOAc layer was washed with water, dried with anhydrous Na2SO4, and concentrated with a Rotavapor. The residue was purified by silica gel chromatography (0% to 100% EtOAc in hexane) to give product 21 (217 mg, 30% yield, 98.4% purity). 1H NMR (400 MHz, chloroform-d) δ 8.17–8.06 (m, 2H), 7.68–7.50 (m, 3H), 7.11–6.97 (m, 2H), 6.86 (d, J = 3.0 Hz, 1H), 3.76 (s, 3H), 3.72 (s, 3H), 2.45 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 153.69, 147.82, 143.84, 141.11, 138.72, 133.69, 129.27, 127.86, 123.73, 117.91, 113.73, 113.40, 56.35, 55.99, 8.98. ESI-TOF HRMS: m/z 360.1013 (C17H18N3O4S+H+ requires 360.1013).
Compounds 22 to 28 were synthesized by using 14 to 20, respectively, as the starting material, along with compound 12. The syntheses were performed using the procedure described for 21.
1-(2,5-Dimethoxyphenyl)-5-methyl-4-tosyl-1H-1,2,3-triazole (22)
42% yield, 95.8% purity. 1H NMR (400 MHz, chloroform-d) δ 8.04–7.96 (m, 2H), 7.39–7.32 (m, 2H), 7.07 (dd, J = 9.1, 3.0 Hz, 1H), 7.00 (d, J = 9.1 Hz, 1H), 6.86 (d, J = 3.0 Hz, 1H), 3.77 (s, 3H), 3.73 (s, 3H), 2.45 (s, 3H), 2.43 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 153.68, 147.85, 144.73, 144.19, 138.41, 138.18, 129.89, 127.94, 123.81, 117.87, 113.74, 113.38, 56.35, 55.99, 21.67, 8.97. ESI-TOF HRMS: m/z 374.1169 (C18H19N3O4S+H+ requires 374.1169).
4-((4-Chlorophenyl)sulfonyl)-1-(2,5-dimethoxyphenyl)-5-methyl-1H-1,2,3-triazole (23)
31% yield, 98.8% purity. 1H NMR (400 MHz, chloroform-d) δ 8.10–8.01 (m, 1H), 7.57–7.49 (m, 1H), 7.08 (dd, J = 9.2, 3.0 Hz, 0H), 7.01 (d, J = 9.2 Hz, 0H), 6.87 (d, J = 3.0 Hz, 0H), 3.78 (s, 1H), 3.74 (s, 3H), 2.45 (s, 1H). 13C NMR (101 MHz, CDCl3) δ 153.72, 147.80, 143.56, 140.45, 139.52, 138.79, 129.60, 129.42, 123.65, 117.97, 113.71, 113.39, 56.36, 56.00, 8.98. ESI-TOF HRMS: m/z 394.0618 (C17H16ClN3O4S+H+ requires 394.0623).
4-((1-(2,5-Dimethoxyphenyl)-5-methyl-1H-1,2,3-triazol-4-yl)sulfonyl)phenol (24)
20% yield, 97.5% purity. 1H NMR (400 MHz, DMSO-d6) δ 10.73 (s, 1H), 7.88–7.83 (m, 2H), 7.26 (d, J = 9.1 Hz, 1H), 7.20 (dd, J = 9.1, 3.0 Hz, 1H), 7.17 (d, J = 2.9 Hz, 1H), 7.01–6.96 (m, 2H), 3.74 (s, 3H), 3.73 (s, 3H), 2.35 (s, 3H). 13C NMR (101 MHz, DMSO) δ 162.53, 153.04, 147.57, 143.81, 137.93, 130.41, 129.90, 123.02, 117.95, 116.12, 113.89, 56.32, 55.85, 8.49. ESI-TOF HRMS: m/z 376.0965 (C17H17N3O5S+H+ requires 376.0962).
1-(2,5-Dimethoxyphenyl)-4-((4-methoxyphenyl)sulfonyl)-5-methyl-1H-1,2,3-triazole (25)
44% yield, 98.9% purity. 1H NMR (400 MHz, chloroform-d) δ 8.10–8.01 (m, 2H), 7.07 (dd, J = 9.0, 2.9 Hz, 1H), 7.03 (d, J = 2.0 Hz, 1H), 7.00 (d, J = 9.3 Hz, 2H), 6.87 (d, J = 3.0 Hz, 1H), 3.89–3.86 (m, 3H), 3.77 (d, J = 0.5 Hz, 3H), 3.73 (s, 3H), 2.45 (d, J = 0.5 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 163.81, 153.69, 147.87, 144.56, 138.07, 132.65, 130.21, 123.86, 117.86, 114.47, 113.74, 113.38, 56.35, 55.99, 55.70, 8.96. ESI-TOF HRMS: m/z 390.1118 (C18H19N3O5S+H+ requires 390.1118).
4-((1-(2,5-Dimethoxyphenyl)-5-methyl-1H-1,2,3-triazol-4-yl)sulfonyl)benzoic acid (26)
22% yield, 99.5% purity. 1H NMR (400 MHz, DMSO-d6) δ 13.46 (s, 1H), 8.20 (d, J = 8.7 Hz, 2H), 8.15 (d, J = 8.7 Hz, 2H), 7.27 (d, J = 9.1 Hz, 1H), 7.23–7.19 (m, 1H), 7.17 (d, J = 3.0 Hz, 1H), 3.75 (s, 3H), 3.73 (s, 3H), 2.40 (s, 3H). 13C NMR (101 MHz, DMSO) δ 165.91, 153.17, 147.67, 144.14, 142.24, 139.42, 135.76, 130.49, 127.55, 123.02, 118.13, 114.08, 113.97, 56.46, 55.93, 8.54. ESI-TOF HRMS: m/z 404.0912 (C18H17N3O6S+H+ requires 404.0911).
4-((1-(2,5-Dimethoxyphenyl)-5-methyl-1H-1,2,3-triazol-4-yl)sulfonyl)aniline (27)
30% yield, 96.0% purity. 1H NMR (400 MHz, Chloroform-d) δ 7.85 (d, J = 8.3 Hz, 2H), 7.06 (dd, J = 9.1, 3.0 Hz, 1H), 6.99 (d, J = 9.1 Hz, 1H), 6.86 (d, J = 3.0 Hz, 1H), 6.70 (d, J = 8.3 Hz, 2H), 3.76 (s, 3H), 3.72 (s, 3H), 2.42 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 153.66, 151.44, 147.88, 145.08, 137.72, 130.10, 128.96, 123.89, 117.84, 114.27, 113.74, 113.38, 56.35, 56.00, 8.95. ESI-TOF HRMS: m/z 375.1124 (C17H18N4O4S+H+ requires 375.1122).
4-(Benzylsulfonyl)-1-(2,5-dimethoxyphenyl)-5-methyl-1H-1,2,3-triazole (28)
24% yield, 98.5% purity. 1H NMR (400 MHz, DMSO-d6) δ 7.40–7.31 (m, 3H), 7.27 (d, J = 9.2 Hz, 1H), 7.22 (d, J = 3.0 Hz, 1H), 7.20–7.16 (m, 2H), 7.05 (d, J = 3.0 Hz, 1H), 4.72 (s, 2H), 3.78 (s, 3H), 3.75 (s, 3H), 1.78 (s, 3H). 13C NMR (101 MHz, DMSO) δ 153.12, 147.65, 140.30, 139.97, 131.07, 128.44, 128.29, 128.23, 123.13, 117.88, 114.12, 113.80, 61.47, 56.44, 55.89, 7.57. ESI-TOF HRMS: m/z 374.1172 (C18H19N3O4S+H+ requires 374.1169).
Methyl 4-((1-(2,5-dimethoxyphenyl)-5-methyl-1H-1,2,3-triazol-4-yl)sulfonyl)benzoate (29)
Sulfuric acid (98%) (1 mL) was added to compound 26 (200 mg, 0.496 mmol) in MeOH (10 mL). The mixture was stirred at reflux overnight. The reaction mixture was then cooled to room temperature, and water (100 mL) was added. Saturated Na2CO3 solution was added to adjust the reaction mixture to pH >7. The reaction solution was extracted with EtOAc (100 mL × 3). The combined EtOAc solution was dried with anhydrous Na2SO4 and concentrated with a Rotavapor. The residue was purified by column chromatography (0% to 100% EtOAc in hexane) to give product 29 (177 mg, 71% yield, 96.9% purity). 1H NMR (400 MHz, DMSO-d6) δ 8.22 (d, J = 8.2 Hz, 1H), 8.17 (d, J = 8.6 Hz, 1H), 7.27 (d, J = 9.1 Hz, 0H), 7.21 (dd, J = 9.2, 3.1 Hz, 0H), 3.91 (s, 2H), 3.75 (s, 2H), 3.73 (s, 2H), 2.39 (s, 3H). 13C NMR (101 MHz, DMSO) δ 164.95, 153.16, 147.66, 144.49, 142.13, 139.49, 134.43, 130.43, 127.70, 123.00, 118.14, 114.09, 113.96, 56.47, 55.94, 52.68, 8.55. ESI-TOF HRMS: m/z 418.1072 (C19H19N3O6S+H+ requires 418.1068).
2-(4-((1-(2,5-Dimethoxyphenyl)-5-methyl-1H-1,2,3-triazol-4-yl)sulfonyl)phenyl)propan-2-ol (30)
A solution of MeMgBr (6.7 mL, 20 mmol) in anhydrous diethyl ether was added dropwise to a solution of compound 29 (835 mg, 2 mmol) in anhydrous THF (30 mL) at 0 °C under a nitrogen atmosphere. The reaction mixture was stirred at room temperature for 6 h then cooled to −10 °C, quenched with water (1 mL), and filtered. The solution was then poured into water and extracted with EtOAc (50 mL × 3). The combined EtOAc layer was washed with water, dried with anhydrous Na2SO4, and concentrated with a rotavapor. The residue was purified by column chromatography to give product 30 (663 mg, 79% yield, 96.9% purity). 1H NMR (400 MHz, chloroform-d) δ 8.12–8.02 (m, 2H), 7.71–7.65 (m, 2H), 7.07 (dd, J = 9.2, 3.0 Hz, 1H), 7.01 (d, J = 9.1 Hz, 1H), 6.87 (d, J = 3.0 Hz, 1H), 3.77 (s, 3H), 3.74 (s, 3H), 2.46 (d, J = 0.6 Hz, 3H), 1.60 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 155.26, 153.70, 147.84, 143.99, 139.28, 138.61, 127.93, 125.49, 123.78, 117.91, 113.74, 113.38, 72.52, 56.36, 56.00, 31.75, 9.00. ESI-TOF HRMS: m/z 418.1430 (C20H23N3O5S + H+ requires 418.1431).
1-(2,5-Dimethoxyphenyl)-4-((4-(2-methoxypropan-2-yl)phenyl)sulfonyl)-5-methyl-1H-1,2,3-triazole (31)
A mixture of compound 30 (0.125 g, 0.3 mmol) and BiBr3 (0.135 g, 0.300 mmol) in CCl4 (3 mL) at room temperature was stirred vigorously for 15 min. A solution of methanol (9.61 mg, 0.300 mmol) in CCl4 (3 mL) was then slowly added and the stirring continued. When the compound 30 starting material had been depleted (as determined by LCMS monitoring), the reaction mixture was filtered and the solvent in the solution was removed with a Rotavapor to provide the raw product, which was purified by silica gel chromatography with EtOAc and hexane (0% to 100% EtOAc in hexane) to give the purified product 31 (97.2 mg, 75% yield, 98.5% purity). 1H NMR (400 MHz, DMSO-d6) δ 8.07–7.95 (m, 2H), 7.77–7.66 (m, 2H), 7.27 (d, J = 8.8 Hz, 1H), 7.24–7.18 (m, 2H), 3.74 (s, 3H), 3.73 (s, 3H), 3.02 (s, 3H), 2.39 (s, 3H), 1.47 (s, 6H). 13C NMR (101 MHz, DMSO) δ 153.04, 147.56, 142.83, 139.02, 138.89, 127.35, 126.89, 122.92, 118.03, 113.89, 76.31, 56.34, 55.86, 50.19, 27.32, 8.57. ESI-TOF HRMS: m/z 432.1588 (C21H25N3O5S+ H+ requires 432.1588).
4-((4-(2-Butoxypropan-2-yl)phenyl)sulfonyl)-1-(2,5-dimethoxyphenyl)-5-methyl-1H-1,2,3-triazole (32)
The preparation of 32 was similar to that of 31 except that butan-1-ol was used instead of methanol as part of the starting material. 114.2 mg, 80% yield, 97.8% purity. 1H NMR (400 MHz, DMSO-d6) δ 8.04–7.97 (m, 2H), 7.74–7.65 (m, 2H), 7.27 (d, J = 9.0 Hz, 1H), 7.25–7.16 (m, 2H), 3.74 (s, 3H), 3.73 (s, 3H), 3.14 (t, J = 6.5 Hz, 2H), 2.39 (s, 3H), 1.53–1.40 (m, 8H), 1.32 (h, J = 7.2 Hz, 2H), 0.84 (t, J = 7.3 Hz, 3H). 13C NMR (101 MHz, DMSO) δ 153.59, 153.04, 147.55, 142.83, 138.96, 138.89, 127.29, 126.78, 122.92, 118.03, 113.91, 113.89, 75.84, 61.77, 56.34, 55.86, 31.90, 27.77, 18.85, 13.76, 8.57. ESI-TOF HRMS: m/z 474.2059 (C24H31N3O5S+ H+ requires 474.2057).
Compounds 37 to 40 were synthesized by using respective compounds 33 to 36 as the starting material in accordance with the procedure described for compound 12, and the resulting aryl azides were used without further purification.
4-((4-(tert-Butyl)phenyl)sulfonyl)-1-(5-fluoro-2-methoxyphenyl)-5-methyl-1H-1,2,3-triazole (42)
MeONa (108 mg, 2 mmol) and 1-((4-(tert-butyl)phenyl)sulfonyl)propan-2-one (254 mg, 1 mmol) were added to a solution of compound 37 (167 mg, 1 mmol) in MeOH. The mixture was stirred at 60 °C overnight. The reaction mixture was then poured into water and extracted with EtOAc (50 mL × 3). The combined EtOAc solution was washed with water, dried with anhydrous Na2SO4, and concentrated with a Rotavapor. The residue was purified by preparative HPLC to give product 42 (150 mg, 37% yield, 98.2% purity). 1H NMR (400 MHz, chloroform-d) δ 8.09–7.97 (m, 2H), 7.67–7.53 (m, 2H), 7.30–7.20 (m, 1H), 7.10 (ddd, J = 7.8, 3.1, 1.4 Hz, 1H), 7.04 (dd, J = 9.2, 4.5 Hz, 1H), 3.78 (s, 3H), 2.46 (s, 3H), 1.34 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 157.72, 157.36, 154.95, 150.31, 150.28, 144.36, 138.50, 137.92, 127.76, 126.35, 123.86, 123.77, 118.94, 118.71, 116.10, 115.84, 113.21, 113.12, 56.43, 35.29, 31.06, 8.99. ESI-TOF HRMS: m/z 404.1441 (C20H22FN3O3S + H+ requires 404.1439).
Compounds 44, 46 and 47 were synthesized by using corresponding compounds 38, 39 and 40 as the starting material in accordance with the procedure described for 42.
1-(5-Bromo-2-methoxyphenyl)-4-((4-(tert-butyl)phenyl)sulfonyl)-5-methyl-1H-1,2,3-triazole (44)
204 mg, 44% yield, 98.8% purity. 1H NMR (400 MHz, Chloroform-d) δ 8.05 – 7.98 (m, 2H), 7.63 (dd, J = 8.9, 2.5 Hz, 1H), 7.59–7.54 (m, 2H), 7.46 (d, J = 2.4 Hz, 1H), 6.98 (d, J = 8.9 Hz, 1H), 3.79 (s, 3H), 2.45 (s, 3H), 1.34 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 157.72, 153.10, 144.37, 138.46, 137.91, 135.17, 131.36, 127.74, 126.35, 124.61, 113.82, 112.64, 56.26, 35.29, 31.06, 8.98. ESI-TOF HRMS: m/z 464.0641 (C20H22BrN3O3S + H+ requires 464.0638).
4-((4-(tert-Butyl)phenyl)sulfonyl)-1-(2-methoxy-5-(trifluoromethyl)phenyl)-5-methyl-1H-1,2,3-triazole (46).
222 mg, 49% yield, 98.7% purity. 1H NMR (400 MHz, Chloroform-d) δ 8.12–7.98 (m, 2H), 7.82 (ddd, J = 8.8, 2.4, 0.8 Hz, 1H), 7.62 (d, J = 2.3 Hz, 1H), 7.60–7.55 (m, 2H), 7.19 (d, J = 8.8 Hz, 1H), 3.89 (s, 3H), 2.47 (s, 3H), 1.34 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 157.80, 156.34, 144.56, 138.49, 137.84, 129.75, 129.72, 127.78, 126.37, 126.24, 126.21, 124.65, 123.93, 123.86, 123.59, 123.25, 121.95, 112.53, 56.45, 35.30, 31.05, 8.99. ESI-TOF HRMS: m/z 454.1408 (C21H22F3N3O3S + H+ requires 454.1407).
1-(5-(tert-Butyl)-2-methoxyphenyl)-4-((4-(tert-butyl)phenyl)sulfonyl)-5-methyl-1H-1,2,3-triazole (47)
76 mg, 40% yield, 95.24% purity. 1H NMR (400 MHz, DMSO-d6) δ 7.99–7.94 (m, 2H), 7.74–7.68 (m, 2H), 7.64 (dd, J = 8.8, 2.5 Hz, 1H), 7.51 (d, J = 2.5 Hz, 1H), 7.24 (d, J = 8.8 Hz, 1H), 3.77 (s, 3H), 2.37 (s, 3H), 1.31 (s, 9H), 1.27 (s, 9H). 13C NMR (101 MHz, DMSO) δ 157.33, 151.26, 143.71, 142.98, 138.68, 137.90, 129.17, 127.22, 126.65, 125.44, 122.28, 112.43, 56.04, 35.03, 34.05, 31.03, 30.67, 8.64. ESI-TOF HRMS: m/z 442.2157 (C24H31N3O3S + H+ requires 442.2159).
3-(4-((4-(tert-Butyl)phenyl)sulfonyl)-5-methyl-1H-1,2,3-triazol-1-yl)-4-methoxybenzonitrile (48)
Zinc cyanide (0.887 g, 7.56 mmol) and tetrakis(triphenylphosphine)palladium(0) (0.336 g, 0.291 mmol) were added to a degassed solution of 44 (2.7 g, 5.81 mmol) in DMF (40 mL) under argon. The reaction mixture was heated at 90 °C for overnight. After being cooled, the reaction mixture was diluted with EtOAc (200 mL) and the suspension was filtered with Celite and concentrated with a Rotavapor. The residue was purified by silica gel chromatography (hexane-EtOAc 100/0 to 0/100) to give product 48 (1.72 g, 72% yield, 98.1% purity). 1H NMR (400 MHz, chloroform-d) δ 8.07–7.98 (m, 2H), 7.86 (dd, J = 8.7, 2.1 Hz, 1H), 7.65 (d, J = 2.1 Hz, 1H), 7.61–7.55 (m, 2H), 7.19 (d, J = 8.8 Hz, 1H), 3.91 (s, 3H), 2.46 (s, 3H), 1.34 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 157.91, 157.26, 144.71, 138.40, 137.70, 136.69, 132.54, 127.80, 126.40, 124.50, 117.10, 113.20, 105.15, 56.66, 35.32, 31.06, 9.00. ESI-TOF HRMS: m/z 411.1479 (C21H22N4O3S + H+ requires 411.1486).
Methyl 3-(4-((4-(tert-butyl)phenyl)sulfonyl)-5-methyl-1H-1,2,3-triazol-1-yl)-4-methoxybenzoate (49)
To a suspension of 44 (1.00 g, 2.15 mmol) and Et3N (872 mg, 8.61 mmol, 1.20 mL) in MeOH (10 mL) and THF (10 mL) was added Pd(dppf)Cl2 (157 mg, 215 μmol) under argon. The reaction mixture was purged with CO and then stirred at 80 °C for 16 h under CO (50 psi). The mixture was filtered with Celite, and the cake was washed with CH2Cl2 (100 mL). The solvent in the organic layer was removed with a Rotavapor. The residue was purified by column chromatography on silica gel (petroleum ether: EtOAc = 1:0–0:1) and triturated in MeOH (10 mL) to yield compound 49 (0.83 g, 1.59 mmol, 94.8% yield) as a brown solid. 1H NMR (CDCl3, 400 MHz): δ 8.24 (dd, 1H, J1 = 8.8 Hz, J2 = 2.0 Hz), 8.04 (d, 2H, J = 8.8 Hz), 8.00 (d, 1H, J = 2.0 Hz), 7.58 (d, 2H, J = 8.4 Hz), 7.13 (d, 1H, J = 8.8 Hz), 3.89 (s, 3H), 3.88 (s, 3H), 2.44 (s, 3H), 1.34 (s, 9H).
(3-(4-((4-(tert-Butyl)phenyl)sulfonyl)-5-methyl-1H-1,2,3-triazol-1-yl)-4-methoxyphenyl)methanol (50).
LiBH4 (196 mg, 9.02 mmol) was added to a solution of compound 49 (0.2 g, 451 μmol) in THF (5 mL) and MeOH (5 mL) in portions at 0–10 °C, then the mixture was stirred at 25 °C for 16 h. The mixture was quenched with water (20 mL), and the organic solvents were removed with a Rotavapor. The water layer was extracted with CH2Cl2 (30 mL × 2). The combined CH2Cl2 solution was dried with anhydrous Na2SO4, and the solvent was removed with a Rotavapor. The residue was purified by silica gel column chromatography (petroleum ether: EtOAc = 1:0–0:1) to yield compound 50 (170 mg, 409 μmol, 90.7% yield, 99.3% purity) as a pale yellow solid. 1H NMR (CDCl3, 400 MHz): δ 8.03 (dd, 2H, J1 = 6.8 Hz, J2 = 2.0 Hz), 7.58–7.51 (m, 3H), 7.34 (d, 1H, J = 2.4 Hz), 7.06 (d, 1H, J = 8.8 Hz), 4.68 (d, 2H, J = 3.6 Hz), 3.80 (s, 3H), 2.49 (s, 3H), 1.84 (br.s, 1H), 1.34 (s, 9H). 13C NMR (CDCl3, 100 MHz): δ 157.62, 153.15, 144.17, 138.46, 138.02, 134.07, 130.87, 127.72, 127.23, 126.33, 123.56, 112.27, 63.93, 56.04, 35.29, 31.07, 9.01. ESI-TOF HRMS: m/z 416.1636 (C21H25N3O4S + H+ requires 416.1639).
3-(4-((4-(tert-Butyl)phenyl)sulfonyl)-5-methyl-1H-1,2,3-triazol-1-yl)-4-methoxybenzamide (51)
To a solution of compound 49 (380 mg, 857 μmol) in MeOH (5 mL) and THF (5 mL) was added NH3 (in water, 292 mg, 17.1 mmol, 5 mL), then the mixture was stirred at 130 °C for 48 h in a sealed tube. LCMS monitoring showed the compound 49 starting material was consumed almost completely. The solvent in the mixture was removed with a Rotavapor. The crude was purified by silica gel column chromatography (CH2Cl2: MeOH = 1:0–10:1) and triturated in MeOH (3 mL) to yield compound 51 (60 mg, 140.02 μmol, 16.34% yield, 97.9% purity) as a yellow solid. 1H NMR (DMSO, 400 MHz): δ 8.16 (d, 2H, J = 6.4 Hz), 8.00–7.95 (m, 4H), 7.72 (m, 2H), 7.41 (s, 2H), 3.86 (s, 3H), 2.45 (s, 3H), 2.39 (s, 3H), 1.30 (s, 9H). 13C NMR (CDCl3, 100MHz): δ 166.34, 157.85, 156.16, 143.62, 139.36, 138.22, 132.64, 128.45, 127.63, 127.32, 127.12, 122.71, 113.16, 57.02, 35.49, 31.12, 8.98. ESI-TOF HRMS: m/z 429.1591 (C21H24N4O4S + H+ requires 429.1591).
1-(3-(4-((4-(tert-Butyl)phenyl)sulfonyl)-5-methyl-1H-1,2,3-triazol-1-yl)-4-methoxyphenyl)ethan-1-one (52)
Tributyl(1-ethoxyvinyl)stannane (855 mg, 2.37 mmol, 799 μL) and Pd(PPh3)2Cl2 (75.6 mg, 107 μmol) were added to a solution of compound 44 (1.00 g, 2.15 mmol) in dioxane (10 mL) under N2 protection. The mixture was stirred at 90 °C for 16 h and cooled to 25 °C. HCl solution (2M, 10 mL) was then added to the mixture and it was stirred for 2 h at 25 °C. The mixture was diluted with water (50 mL) and extracted with CH2Cl2 (50 mL × 3). The combined CH2Cl2 solution was then washed with water (50 mL) and dried with anhydrous Na2SO4. The remaining solvent was removed under reduced pressure with a Rotavapor. The residue was purified by silica gel column chromatography (petroleum ether: EtOAc =1:0–4:1) to yield compound 52 (900 mg, 2.11 mmol, 97.7% yield) as a yellow solid. 1H NMR (CDCl3, 400 MHz): δ 8.18 (dd, 1H, J1 = 8.8 Hz, J2 = 2.4 Hz), 8.05 (dd, 2H, J1 = 6.8Hz, J2 = 2.0 Hz), 7.92 (d, 1H, J = 2.4 Hz), 7.58 (d, 2H, J = 8.8 Hz), 7.15 (d, 1H, J = 8.8 Hz), 3.90 (s, 3H), 2.56 (s, 3H), 2.45 (s, 3H), 1.34 (s, 9H).
(R)-1-(3-(4-((4-(tert-Butyl)phenyl)sulfonyl)-5-methyl-1H-1,2,3-triazol-1-yl)-4-methoxyphenyl)ethan-1-ol (54) and (S)-1-(3-(4-((4-(tert-butyl)phenyl)sulfonyl)-5-methyl-1H-1,2,3-triazol-1-yl)-4-methoxyphenyl)ethan-1-ol (55)
NaBH4 (106 mg, 2.81 mmol) was added in portions to a solution of compound 52 (0.4 g, 935 μmol) in THF (8 mL) and MeOH (8 mL) at 0–5 °C. The mixture was then stirred for 3 h at 0–5 °C and quenched with water (20 mL). The organic solvent was removed under reduced pressure. The water layer was extracted with CH2Cl2 (20 mL × 2). The combined organic layer was dried with anhydrous Na2SO4, and the solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography (petroleum ether: EtOAc = 0:1 to 5:1) to yield compound 53 (290 mg, 326 μmol, 70.0% yield) as a white solid. Compound 53 was further separated into the faster-eluting isomer 54 (140 mg, 326 μmol, 95.8% purity) and the slower-eluting isomer 55 (130 mg, 303 μmol, 98.0% purity) as a white solid by SFC (column: Phenomenex-Cellulose-2 (250 mm*30 mm, 10 μm particle size); mobile phase: [0.1% NH3*H2O ETOH]; B%: 45%–45%,min).
Compound 54: 1H NMR (CDCl3, 400 MHz): δ 8.04 (d, 2H, J = 8.8 Hz), 7.58–7.52 (m, 3H), 7.34 (d, 1H, J = 2.0 Hz), 7.05 (d, 1H, J = 8.8 Hz), 4.91–4.89 (m, 1H), 3.80 (s, 3H), 2.45 (s, 3H), 1.82 (br.s, 1H), 1.48 (d, 3H, J = 6.8 Hz), 1.34 (s, 9H). 13C NMR (CDCl3, 100MHz): δ 157.60, 152.95, 144.16, 139.01, 138.45, 138.05, 129.37, 127.72, 126.31, 125.72, 123.48, 112.22, 69.12, 56.03, 35.27, 31.06, 25.30, 9.01. ESI-TOF HRMS: m/z 430.1794 (C22H26N3O4S + H+ requires 430.1795).
Compound 55: 1H NMR (CDCl3, 400 MHz): δ 8.04 (d, 2H, J = 8.4 Hz), 7.58–7.52 (m, 3H), 7.34 (d, 1H, J = 2.0 Hz), 7.05 (d, 1H, J = 8.4 Hz), 4.91–4.89 (m, 1H), 3.80 (s, 3H), 2.45 (s, 3H), 1.81 (d, 1H, J = 4.0 Hz), 1.48 (d, 3H, J = 6.4 Hz), 1.34 (s, 9H).13C NMR (CDCl3, 100MHz): δ 157.61, 152.92, 144.14, 139.03, 138.48, 138.04, 129.39, 127.70, 126.32, 125.72, 123.45, 112.22, 69.09, 56.02, 35.27, 31.06, 25.29, 9.01. ESI-TOF HRMS: m/z 430.1794 (C22H26N3O4S + H+ requires 430.1795).
Compounds 61 to 65 were synthesized by using compounds 56 to 60 as the respective starting materials in accordance with the procedure described for compound 21. The resulting aryl azides 61 to 65 were used for the next step without further purification.
Compounds 66 to 70 were synthesized by using respective compounds 61 to 65 as the starting material. The syntheses were performed in accordance with the procedure described for compound 42 except that components of the starting material were replaced as appropriate.
4-((4-(tert-Butyl)phenyl)sulfonyl)-1-(4-fluoro-2,5-dimethoxyphenyl)-5-methyl-1H-1,2,3-triazole (66)
The preparation of 66 was similar to that of 42 except that 61 was used as part of the starting material. 98 mg, 23% yield, 98.2% purity. 1H NMR (CDCl3, 400 MHz): δ (ppm) δ 8.03 (d, 2H, J = 8.4 Hz), 7.56 (d, 2H, J = 8.4 Hz), 6.94 (d, 1H, J = 8.4 Hz), 6.86 (d, 1H, J =12.4 Hz), 3.82 (s, 3H), 3.73 (s, 3H), 2.43 (s, 3H), 1.32 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 157.42, 154.07, 152.09, 148.09, 148.01, 143.03, 140.96, 140.87, 138.95, 137.84, 127.24, 126.71, 118.05, 118.02, 114.60, 114.57, 102.66, 102.48, 56.90, 56.89, 56.83, 56.81, 35.07, 30.70, 8.54. ESI-TOF HRMS: m/z 434.1546 (C21H24FN3O4S + H+ requires 434.1550).
1-(4-Bromo-2,5-dimethoxyphenyl)-4-((4-(tert-butyl)phenyl)sulfonyl)-5-methyl-1H-1,2,3-triazole (67)
The preparation of 67 was similar to that of 42 except that 62 was used as part of the starting material. 207 mg, 42% yield, 98.6% purity. 1H NMR (CDCl3, 400 MHz): δ (ppm) 8.56 (d, 2H, J = 8.8 Hz), 8.03 (d, 2H, J = 8.8 Hz), 7.29 (s, 1H), 6.87 (s, 1H), 3.82 (s, 3H), 3.75 (s, 3H), 2.45 (s, 3H), 1.32 (s, 9H13C NMR (126 MHz, CDCl3) δ (ppm) 157.712, 150.435, 147.781, 144.405, 138.529, 137.945, 127.796, 126.330, 122.750, 117.770, 115.065, 112.032, 57.012, 56.655, 35.291, 31.063, 9.021. ESI-TOF HRMS: m/z 494.0747 (C21H24BrN3O4S + H+ requires 494.0749).
4-((4-(tert-Butyl)phenyl)sulfonyl)-1-(4-chloro-2-methoxy-5-methylphenyl)-5-methyl-1H-1,2,3-triazole (68)
The preparation of 68 was similar to that of 42 except that 63 was used as part of the starting material. 26 mg, 6% yield, 97.5% purity. 1H NMR (CDCl3, 400 MHz): δ (ppm) 8.02 (d, 2H, J = 8.8 Hz), 7.55 (d, 2H, J = 8.8 Hz), 7.17 (s, 1H), 7.06 (s, 1H), 3.76 (s, 3H), 2.42 (s, 3H), 2.31 (s, 3H), 1.32 (s, 9H). 13C NMR (101 MHz, DMSO) δ 157.41, 152.29, 143.21, 138.79, 137.93, 136.73, 130.43, 128.20, 127.15, 126.60, 121.58, 113.87, 56.74, 35.01, 30.68, 18.33, 8.47. 13C NMR ESI-TOF HRMS: m/z 434.1303 (C21H24ClN3O3S + H+ requires 434.1305).
1-(4-Bromo-2-methoxy-5-methylphenyl)-4-((4-(tert-butyl)phenyl)sulfonyl)-5-methyl-1H-1,2,3-triazole (69)
The preparation of 69 was similar to that of 42 except that 64 was used as part of the starting material. 70 mg, 14.7% yield, 97.8% purity. 1H NMR (CDCl3, 400 MHz): δ (ppm) 8.02 (dd, 2H, J = 8.8 Hz, 2 Hz), 8.55 (dd, 2H, J = 8.8 Hz, 2 Hz), 7.24 (s, 1H), 7.17 (s, 1H), 3.76 (s, 3H), 2.42 (s, 3H), 2.34 (s, 3H), 1.32 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 157.86, 152.50, 143.59, 139.25, 138.23, 130.60, 130.52, 127.95, 127.64, 127.13, 122.38, 117.24, 57.16, 57.15, 35.50, 31.12, 21.66, 8.97. ESI-TOF HRMS: m/z 478.0790 (C21H24BrN3O3S + H+ requires 478.0800).
1-(4-Bromo-5-chloro-2-methoxyphenyl)-4-((4-(tert-butyl)phenyl)sulfonyl)-5-methyl-1H-1,2,3-triazole (70)
The preparation of 70 was similar to that of 42 except that 65 was used as part of the starting material. 75 mg, 75 mg, 15.1% yield, 99.5% purity. 1H NMR (CDCl3, 400 MHz): δ (ppm) 8.03 (d, 2H, J = 8.4 Hz), 8.57 (d, 2H, J = 8.4 Hz), 7.43 (s, 1H), 7.34 (s, 1H), 3.82 (s, 3H), 2.45 (s, 3H), 1.34 (s, 9H). 13C NMR (101 MHz, DMSO) δ 157.39, 153.00, 143.18, 139.04, 137.82, 129.65, 127.15, 126.54, 125.49, 124.77, 123.05, 118.41, 57.17, 34.97, 30.63, 8.41. ESI-TOF HRMS: m/z 498.0257 (C20H21BrClN3O3S + H+ requires 498.0254).
4-((4-(tert-Butyl)phenyl)sulfonyl)-1-(4-fluoro-2-methoxy-5-methylphenyl)-5-methyl-1H-1,2,3-triazole (79)
n-BuLi (0.4 mL, 1 mmol) was added dropwise to a solution of compound 69 (477 mg, 1 mmol) in THF (5 mL) at −78 °C with stirring. The mixture was stirred for 5 min, then NFSI (409 mg, 1.3 mmol) in THF (2 mL) was added. This was followed by stirring for 1 more hour. The reaction was quenched with aqueous NH4Cl and the mixture extracted with EtOAc (30 mL × 3). The combined EtOAc layer was washed with water, dried with anhydrous Na2SO4, and concentrated with a Rotavapor. The residue was purified by preparative HPLC to yield product 79 (30 mg, 7% yield, 99.7% purity). 1H NMR (CDCl3, 400 MHz): δ (ppm) 8.02 (d, 2H, J = 8.4 Hz), 7.55 (d, 2H, J = 8.4 Hz), 7.13 (d, 1H, J = 7.6 Hz), 6.74 (d, 1H, J = 10.4 Hz), 3.74 (s, 3H), 2.42 (s, 3H), 2.21 (d, 3H, J = 1.2 Hz), 1.32 (s, 9H). 13C NMR (101 MHz, DMSO) δ 163.50, 161.04, 157.38, 153.24, 153.12, 143.13, 138.81, 137.96, 131.00, 130.92, 127.14, 126.58, 118.79, 116.73, 116.54, 101.31, 101.03, 56.73, 35.01, 30.68, 13.00, 12.97, 8.46. ESI-TOF HRMS: m/z 418.1606 (C21H24FN3O3S + H+ requires 418.1600).
4-((4-(tert-Butyl)phenyl)sulfonyl)-1-(4-chloro-2,5-dimethoxyphenyl)-5-methyl-1H-1,2,3-triazole (71)
n-BuLi (0. 24 mL, 0.6 mmol) was added dropwise to solution of compound 67 (300 mg, 0.6 mmol) in THF (5 mL) at −78 °C with stirring. The mixture was stirred for 5 min, and then C2Cl6 (430 mg, 1.8 mmol) in THF (2 mL) was added. This was followed by stirring for 1 more hour. The reaction was quenched with aqueous NH4Cl and the mixture extracted with EtOAc (30 mL × 3). The combined EtOAc layer was washed with water, dried with anhydrous Na2SO4, and concentrated with a Rotavapor. The residue was purified by preparative HPLC to yield product 71 (30 mg, 11% yield, 99.8% purity). 1H NMR (CDCl3, 400 MHz): δ (ppm) 7.98 (d, 2H, J = 8.4 Hz), 7.51 (d, 2H, J = 8.4 Hz), 7.07 (s, 1H), 6.85 (s, 1H), 3.78 (s, 3H), 3.69 (s, 3H), 2.39 (s, 3H), 1.27 (s, 9H). 13C NMR (101 MHz, DMSO) δ 157.37, 148.70, 147.71, 143.11, 138.80, 137.88, 127.15, 126.56, 124.55, 121.68, 115.03, 113.39, 56.95, 56.89, 34.97, 30.63, 8.43. ESI-TOF HRMS: m/z 450.1247 (C21H24ClN3O4S + H+ requires 450.1254).
4-((4-(tert-Butyl)phenyl)sulfonyl)-1-(4,5-dichloro-2-methoxyphenyl)-5-methyl-1H-1,2,3-triazole (87)
The preparation of compound 87 (38 mg, 14% yield, 95.5% purity) was similar to that of compound 71 except that compound 70 was used as the starting material in place of compound 67. 1H NMR (CDCl3, 400 MHz): δ (ppm) 7.96 (d, 2H, J = 8.4 Hz), 7.51 (d, 2H, J = 8.4 Hz), 7.38 (s, 1H), 7.11 (s, 1H), 3.75 (s, 3H), 2.39 (s, 3H), 1.27 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 157.89, 153.66, 143.58, 139.63, 138.17, 135.55, 130.45, 127.68, 127.13, 123.06, 122.91, 115.85, 57.67, 35.50, 31.12, 8.95. ESI-TOF HRMS: m/z 454.0757 (C20H21Cl2N3O3S + H+ requires 454.0759).
4-((4-(tert-Butyl)phenyl)sulfonyl)-1-(2,5-dimethoxy-4-methylphenyl)-5-methyl-1H-1,2,3-triazole (72)
MeB(OH)2 (50 mg, 0.8 mmol), Pd(PPh3)4 (30 mg, 0.03 mmol) and aqueous Na2CO3 solution (0.5 mL, 1 mol) were added to a solution of compound 67 (0.3 g, 0.6 mmol) in dioxane (5 mL). The reaction mixture was then stirred for 5 h at 120 °C. The reaction mixture was then poured into water and extracted with EtOAc (30 mL × 3). The combined EtOAc layer was washed with water, dried with anhydrous Na2SO4, and concentrated with a Rotavapor. The residue was purified by preparative HPLC to yield product 72 (50 mg, 19% yield, 98.2% purity). 1H NMR (CDCl3, 400 MHz): δ (ppm) 7.98 (d, 2H, J = 8.8 Hz), 7.50 (d, 2H, J = 8.8 Hz), 6.82 (s, 1H), 6.69 (s, 1H), 3.70 (s, 3H), 3.66 (s, 3H), 2.38 (s, 3H), 2.22 (s, 3H), 1.27 (s, 9H). 13C NMR (101 MHz, DMSO) δ 157.35, 151.21, 147.15, 143.02, 138.67, 138.04, 130.45, 127.16, 126.58, 120.37, 115.34, 110.83, 56.40, 56.13, 35.00, 30.68, 16.17, 8.52. ESI-TOF HRMS: m/z 430.1799 (C22H27N3O4S + H+ requires 430.1800).
4-((4-(tert-Butyl)phenyl)sulfonyl)-1-(2-methoxy-4,5-dimethylphenyl)-5-methyl-1H-1,2,3-triazole (80)
The preparation of compound 80 (150 mg, 60.5% yield, 100% purity) was similar to that of compound 72 except that compound 69 was used as the starting material in place of compound 67. 1H NMR (CDCl3, 400 MHz): δ (ppm) 8.02 (d, 2H, J = 8.8 Hz), 7.55 (d, 2H, J = 8.8 Hz), 7.03 (s, 1H), 6.82 (s, 1H), 3.73 (s, 3H), 2.41 (s, 3H), 2.31 (s, 3H), 2.19 (s, 3H), 1.32 (s, 9H). 13C NMR (101 MHz, DMSO) δ 157.33, 151.32, 143.04, 141.30, 138.58, 138.04, 128.93, 128.76, 127.11, 126.57, 120.16, 114.09, 56.06, 35.00, 30.68, 19.66, 18.01, 8.50. ESI-TOF HRMS: m/z 414.1859 (C22H27N3O3S + H+ requires 414.1851).
4-((4-(tert-Butyl)phenyl)sulfonyl)-1-(5-chloro-2-methoxy-4-methylphenyl)-5-methyl-1H-1,2,3-triazole (88)
The preparation of compound 88 (135 mg, 51.9% yield, 99.5% purity) was similar to that of compound 72 except that compound 70 was used as the starting material in place of compound 67. 1H NMR (CDCl3, 400 MHz): δ (ppm) 8.01 (d, 2H, J = 8.8 Hz), 7.55 (d, 2H, J = 8.8 Hz), 7.29 (s, 1H), 6.92 (s, 1H), 3.76 (s, 3H), 2.43 (s, 6H), 1.32 (s, 9H). 13C NMR (101 MHz, DMSO) δ 157.33, 152.36, 143.05, 140.46, 138.90, 137.92, 128.34, 127.12, 126.53, 124.37, 121.43, 115.46, 56.49, 34.96, 30.64, 19.89, 8.45. ESI-TOF HRMS: m/z 434.1298 (C21H24ClN3O3S + H+ requires 434.1305).
4-((4-(tert-Butyl)phenyl)sulfonyl)-1-(4-ethyl-2,5-dimethoxyphenyl)-5-methyl-1H-1,2,3-triazole (73)
EthenylSn(Bu)3 (385 mg, 1.2 mmol) and Pd(PPh3)4 (70 mg, 0.06 mmol) was added to a solution of compound 67 (0.3 g, 0.6 mmol) in toluene (5 mL). The reaction mixture was stirred for 5 h at 120 °C then poured into water and extracted with EtOAc (50 mL × 3). The combined EtOAc layer was washed with water, dried with anhydrous Na2SO4, and concentrated with a Rotavapor. The residue was purified by preparative HPLC to yield the product 4-((4-(tert-butyl)phenyl)sulfonyl)-1-(2,5-dimethoxy-4-vinylphenyl)-5-methyl-1H-1,2,3-triazole (85 mg, 32% yield). m/z 442 (M+H)+.
Next, 10% Pd/C (10 mg) was added to a solution of 4-((4-(tert-butyl)phenyl)sulfonyl)-1-(2,5-dimethoxy-4-vinylphenyl)-5-methyl-1H-1,2,3-triazole (85 mg, 0.20 mmol) in MeOH/THF (5 mL/5mL). Bubbled with H2 gas, the reaction mixture was stirred for 3 h at room temperature. It was then filtered, and the filtrate was concentrated with a Rotavapor. The residue was purified by preparative HPLC to yield product 73 (65 mg, 76% yield, 99.2% purity). 1H NMR (CDCl3, 400 MHz): δ (ppm) 7.98 (d, 2H, J = 8.8 Hz), 7.50 (d, 2H, J = 8.8 Hz), 6.81 (s, 1H), 6.70 (s, 1H), 3.69 (s, 3H), 3.67 (s, 3H), 2.65~2.59 (m, 2H), 2.39 (s, 3H), 1.27 (s, 9H), 1.14 (t, 3H, J = 7.6 Hz). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 157.85, 151.17, 147.75, 143.42, 139.20, 138.37, 136.70, 127.69, 127.17, 120.71, 114.22, 111.57, 56.80, 56.78, 56.63, 56.61, 35.53, 31.17, 23.60, 14.50, 9.09. ESI-TOF HRMS: m/z 444.1945 (C23H29N3O4S + H+ requires 444.1957).
4-((4-(tert-Butyl)phenyl)sulfonyl)-1-(4-ethyl-2-methoxy-5-methylphenyl)-5-methyl-1H-1,2,3-triazole (81)
The preparation of compound 81 (60 mg, 23.4% yield, 99.0% purity) was similar to that of compound 73 except that compound 69 was used as the starting material in place of 67. 1H NMR (CDCl3, 400 MHz): δ (ppm) 7.97 (d, 2H, J = 8.4 Hz), 7.49 (d, 2H, J = 8.4 Hz), 6.99 (s, 1H), 6.77 (s, 1H), 3.68 (s, 3H), 2.63~2.57 (m, 2H), 2.37 (s, 3H), 2.18 (s, 3H), 1.27 (s, 9H), 1.18 (t, 3H, J = 7.2 Hz). 13C NMR (101 MHz, DMSO) δ 157.33, 151.59, 146.91, 143.04, 138.58, 138.03, 129.25, 128.15, 127.12, 126.57, 120.19, 112.55, 56.05, 35.00, 30.68, 25.95, 17.45, 14.04, 8.53. ESI-TOF HRMS: m/z 428.1990 (C23H29N3O3S + H+ requires 428.2008).
4-((4-(tert-Butyl)phenyl)sulfonyl)-1-(5-chloro-4-ethyl-2-methoxyphenyl)-5-methyl-1H-1,2,3-triazole (89)
The preparation of compound 89 (41 mg, 15.3% yield, 98.6% purity) was similar to that of compound 73 except that compound 70 was used as the starting material in place of compound 67. 1H NMR (CDCl3, 400 MHz): δ (ppm) 7.97 (d, 2H, J = 8.4 Hz), 7.50 (d, 2H, J = 8.4 Hz), 7.25 (s, 1H), 6.86 (s, 1H), 3.73 (s, 3H), 2.77~2.71 (m, 2H), 2.37 (s, 3H), 1.23 (s, 9H), 1.19 (t, 3H, J = 6.0 Hz). 13C NMR (101 MHz, DMSO) δ 157.33, 152.64, 145.86, 143.04, 138.90, 137.90, 128.70, 127.12, 126.52, 123.68, 121.47, 114.22, 56.49, 34.96, 30.64, 26.54, 13.71, 8.46. ESI-TOF HRMS: m/z 448.1453 (C22H26ClN3O3S + H+ requires 448.1461).
4-((4-(tert-Butyl)phenyl)sulfonyl)-1-(4-isopropyl-2,5-dimethoxyphenyl)-5-methyl-1H-1,2,3-triazole (74)
4,4,5,5-Tetramethyl-2-(prop-1-en-2-yl)-1,3,2-dioxaborolane (340 mg, 2 mmol), Pd(PPh3)4 (117 mg, 0.1 mmol) and Na2CO3 aq. (0.5 mL, 1 mol) were added to a solution of compound 67 (500 mg, 1.0 mmol). The reaction mixture was stirred for 5 h at 120 °C then poured into water and extracted with EtOAc (50 mL × 3). The combined EtOAc layer was washed with water, dried with anhydrous Na2SO4, and concentrated with a Rotavapor. The residue was purified by preparative HPLC to yield the product 4-((4-(tert-butyl)phenyl)sulfonyl)-1-(2,5-dimethoxy-4-(prop-1-en-2-yl)phenyl)-5-methyl-1H-1,2,3-triazole (65 mg, 24% yield). m/z 456 (M+H)+.
Next, 10% Pd/C (10 mg) was added to a solution of 4-((4-(tert-butyl)phenyl)sulfonyl)-1-(2,5-dimethoxy-4-(prop-1-en-2-yl)phenyl)-5-methyl-1H-1,2,3-triazole (65 mg, 0.24 mmol) in MeOH/THF (5 mL/5mL). Bubbled with H2 gas, the reaction mixture was stirred for 3 h at room temperature then filtered, and the filtrate was concentrated with a Rotavapor. The residue was purified by preparative HPLC to yield product 74 (100 mg, 91% yield, 99.1% purity). 1H NMR (CDCl3, 400 MHz): δ (ppm) 8.03 (d, 2H, J = 8.4 Hz), 7.55 (d, 2H, J = 8.4 Hz), 6.90 (s, 1H), 6.75 (s, 1H), 3.74 (s, 3H), 3.72 (s, 3H), 3.38~3.31 (m, 1H), 2.45 (s, 3H), 1.32 (s, 9H), 1.22 (d, 6H, J = 6.8 Hz). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 157.38, 150.16, 147.51, 142.95, 140.60, 138.73, 137.90, 127.23, 126.70, 120.18, 111.36, 110.94, 56.36, 56.34, 56.28, 56.26, 35.07, 30.70, 26.85, 22.26, 8.65. ESI-TOF HRMS: m/z 458.264 (C24H31N3O4S + H+ requires 458.2113).
4-((4-(tert-Butyl)phenyl)sulfonyl)-1-(4-isopropyl-2-methoxy-5-methylphenyl)-5-methyl-1H-1,2,3-triazole (82)
The preparation of compound 82 (140 mg, 15.9% yield for two steps, 100% purity) was similar to that of compound 74 except that compound 69 was used as the starting material in place of compound 67. 1H NMR (CDCl3, 400 MHz): δ (ppm) 7.97 (d, 2H, J = 8.4 Hz), 7.50 (d, 2H, J = 8.4 Hz), 6.98 (s, 1H), 6.89 (s, 1H), 3.74 (s, 3H), 3.41~3.34 (m, 1H), 2.40 (s, 3H), 1.27 (s, 9H), 1.22 (d, 6H, J = 6.8 Hz). 13C NMR (101 MHz, DMSO) δ 157.33, 151.85, 151.35, 143.04, 138.58, 138.03, 129.41, 127.48, 127.12, 126.57, 120.08, 109.44, 56.04, 35.00, 30.68, 29.39, 22.71, 17.59, 8.56. ESI-TOF HRMS: m/z 442.2159 (C24H31N3O3S + H+ requires 442.2164).
4-((4-(tert-Butyl)phenyl)sulfonyl)-1-(5-chloro-4-isopropyl-2-methoxyphenyl)-5-methyl-1H-1,2,3-triazole (90)
The preparation of compound 90 (31 mg, 3.4% yield for two steps, 100% purity) was similar to that of compound 74 except that compound 70 was used as the starting material in place of compound 67. 1H NMR (CDCl3, 400 MHz): δ (ppm) 7.97 (d, 2H, J = 8.4 Hz), 7.50 (d, 2H, J = 8.4 Hz), 7.25 (s, 1H), 6.83 (s, 1H), 3.70 (s, 3H), 3.13~3.06 (m, 1H), 2.38 (s, 3H), 2.21 (s, 3H), 1.27 (s, 9H), 1.19 (d, 6H, J = 6.8 Hz). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 157.84, 153.31, 150.31, 143.44, 139.44, 138.24, 129.31, 127.67, 127.12, 123.63, 121.75, 111.68, 56.95, 56.93, 35.50, 31.13, 22.56, 9.03. ESI-TOF HRMS: m/z 462.1609 (C23H28ClN3O3S + H+ requires 462.1618).
4-((4-(tert-Butyl)phenyl)sulfonyl)-5-methyl-1-(2,4,5-trimethoxyphenyl)-1H-1,2,3-triazole (75)
MeONa (130 mg, 2.4 mmol) and CuCl2 (119 mg, 1.2 mmol) were added to a solution of compound 67 (300 mg, 0.6 mmol) in DMF (10 mL). The reaction mixture was stirred overnight at 130 °C then poured into water and extracted with EtOAc (100 mL × 3). The combined EtOAc layer was washed with water, dried with anhydrous Na2SO4, and concentrated with a Rotavapor. The residue was purified by preparative HPLC to yield product 75 (36 mg, 13.4% yield, 97.3% purity). 1H NMR (CDCl3, 400 MHz): δ (ppm) 7.98 (d, 2H, J = 8.4 Hz), 7.50 (d, 2H, J = 8.4 Hz), 6.75 (s, 1H), 6.55 (s, 1H), 3.90 (s, 3H), 3.75 (s, 3H), 3.69 (s, 3H), 2.38 (s, 3H), 1.27 (s, 9H). 13C NMR (126 MHz, CDCl3) δ (ppm) 157.540, 151.902, 148.419, 144.045, 143.281, 138.671, 138.116, 127.720, 126.252, 115.010, 111.554, 97.536, 56.610, 56.566, 56.352, 35.241, 31.037, 8.975. ESI-TOF HRMS: m/z 446.1745 (C22H27N3O5S + H+ requires 446.1749).
4-((4-(tert-Butyl)phenyl)sulfonyl)-1-(2,4-dimethoxy-5-methylphenyl)-5-methyl-1H-1,2,3-triazole (83)
The preparation of compound 83 (30 mg, 11.7% yield, 100% purity) was similar to that of compound 75 except that compound 69 was used as the starting material in place of compound 67. 1H NMR (CDCl3, 400 MHz): δ (ppm) 7.97 (d, 2H, J = 8.4 Hz), 7.49 (d, 2H, J = 8.4 Hz), 6.97 (s, 1H), 6.44 (s, 1H), 3.84 (s, 3H), 3.71 (s, 3H), 2.36 (s, 3H), 2.08 (s, 3H), 1.27 (s, 9H). 13C NMR (126 MHz, CDCl3) δ (ppm) 159.386, 156.463, 152.089, 142.885, 137.533, 137.198, 128.544, 126.686, 125.241, 118.416, 114.416, 94.310, 55.028, 54.753, 34.241, 30.048, 14.140, 7.942. ESI-TOF HRMS: m/z 430.1790 (C22H27N3O4S + H+ requires 430.1800).
4-((4-(tert-Butyl)phenyl)sulfonyl)-1-(5-chloro-2,4-dimethoxyphenyl)-5-methyl-1H-1,2,3-triazole (91)
The preparation of compound 91 (60 mg, 22% yield, 100% purity) was similar to that of compound 75 except that compound 70 was used as the starting material in place of compound 67. 1H NMR (CDCl3, 400 MHz): δ (ppm) 8.01 (d, 2H, J = 8.4 Hz), 7.55 (d, 2H, J = 8.4 Hz), 7.31 (s, 1H), 6.58 (s, 1H), 3.97 (s, 3H), 3.80 (s, 3H), 2.42 (s, 3H), 1.32 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 157.90, 157.80, 154.46, 143.33, 139.55, 138.30, 129.52, 127.64, 127.10, 115.78, 112.55, 98.73, 57.33, 57.31, 57.19, 57.17, 35.49, 31.13, 8.98. ESI-TOF HRMS: m/z 450.1239 (C21H24ClN3O4S + H+ requires 450.1254).
4-(4-((4-(tert-Butyl)phenyl)sulfonyl)-5-methyl-1H-1,2,3-triazol-1-yl)-2,5-dimethoxybenzonitrile (76)
Zn(CN)2 (423 mg, 3.6 mmol) and Pd(dppf)Cl2 (88 mg, 0.12 mmol) were added to a solution of 67 (600 mg, 1.2 mmol) in DMF (10 mL). The reaction mixture was stirred for 15 min at 160 °C under microwave radiation. It was then poured into water and extracted with EtOAc (100 mL × 3). The combined EtOAc layer was washed with water, dried with anhydrous Na2SO4, and concentrated with a Rotavapor. The residue was purified by preparative HPLC to yield product 76 (280 mg, 53% yield, 98.7% purity). 1H NMR (CDCl3, 400 MHz): δ (ppm) 8.63 (d, 2H, J = 8.8 Hz), 8.09 (d, 2H, J = 8.8 Hz), 7.31 (s, 1H), 7.05 (s, 1H), 3.94 (s, 3H), 3.85 (s, 3H), 2.53 (s, 3H), 1.39 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 157.52, 155.08, 147.42, 143.29, 138.98, 137.71, 127.45, 127.28, 126.75, 117.79, 115.35, 113.55, 103.04, 57.21, 57.19, 57.03, 57.01, 35.08, 30.69, 8.56. ESI-TOF HRMS: m/z 441.1588 (C22H24N4O4S + H+ requires 441.1596).
4-(4-((4-(tert-Butyl)phenyl)sulfonyl)-5-methyl-1H-1,2,3-triazol-1-yl)-5-methoxy-2-methylbenzonitrile (84)
The preparation of compound 84 (52 mg, 10.2% yield, 98.9% purity) was similar to that of compound 76 except that compound 69 was used as the starting material in place of compound 67. 1H NMR (CDCl3, 400 MHz): δ (ppm) 7.97 (d, 2H, J = 8.4 Hz), 7.51 (d, 2H, J = 8.4 Hz), 7.25 (s, 1H), 7.19 (s, 1H), 3.77 (s, 3H), 2.46 (s, 3H), 2.39 (s, 3H), 1.27 (s, 9H). ESI-TOF HRMS: m/z 425.1649 (C22H24N4O3S + H+ requires 425.1647).
4-(4-((4-(tert-Butyl)phenyl)sulfonyl)-5-methyl-1H-1,2,3-triazol-1-yl)-2-chloro-5-methoxybenzonitrile (92)
The preparation of compound 92 (60 mg, 11.3% yield, 97.3% purity) was similar to that of compound 76 except that compound 70 was used as the starting material in place of compound 67. 1H NMR (CDCl3, 400 MHz): δ (ppm) 8.02 (d, 2H, J = 8.4 Hz), 7.57 (d, 2H, J = 8.4 Hz), 7.53 (s, 1H), 7.34 (s, 1H), 3.87 (s, 3H), 2.47 (s, 3H), 1.33 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 158.00, 153.38, 143.80, 139.76, 138.11, 130.61, 127.77, 127.75, 127.44, 127.19, 119.62, 115.86, 115.64, 57.95, 57.93, 35.55, 31.16, 9.01. ESI-TOF HRMS: m/z 445.1091 (C21H21N4O3S + H+ requires 445.1101).
Methyl 4-(4-((4-(tert-butyl)phenyl)sulfonyl)-5-methyl-1H-1,2,3-triazol-1-yl)-2,5-dimethoxybenzoate (77)
A mixture of compound 67 (250 mg, 0.5 mmol) and Pd(dppf)Cl2 (41 mg, 0.05 mmol) in MeOH (10 mL) was stirred at 80 °C under CO (1.5 MPa) for 24 h. The reaction mixture was then cooled to room temperature and concentrated with a Rotavapor. The residue was purified by preparative HPLC to yield product 77 (80 mg, 33% yield, 97.6% purity). 1H NMR (CDCl3, 400 MHz): δ (ppm) 7.98 (d, 2H, J = 8.8 Hz), 7.51 (d, 2H, J = 8.8 Hz), 7.44 (s, 1H), 6.92 (s, 1H), 3.87 (s, 3H), 3.78 (s, 3H), 3.73 (s, 3H), 2.41 (s, 3H), 1.27 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ (ppm) 165.40, 157.45, 151.54, 146.93, 143.17, 138.86, 137.77, 127.25, 126.72, 125.57, 123.49, 114.37, 113.78, 56.78, 56.57, 52.43, 35.06, 30.68, 8.56. ESI-TOF HRMS: m/z 474.1687 (C23H27N3O6S + H+ requires 474.1699).
Methyl 4-(4-((4-(tert-butyl)phenyl)sulfonyl)-5-methyl-1H-1,2,3-triazol-1-yl)-5-methoxy-2-methylbenzoate (85).
The preparation of compound 85 (54 mg, 23.6% yield, 100% purity) was similar to that of compound 77 except that compound 69 was used as the starting material in place of compound 67. 1H NMR (CDCl3, 400 MHz): δ (ppm) 8.02 (d, 2H, J = 8.4 Hz), 7.75–7.78 (m, 3H), 7.21 (s, 1H), 3.93 (s, 3H), 3.81 (s, 3H), 2.52 (s, 3H), 2.44 (s, 3H), 1.32 (s, 9H). 13C NMR (101 MHz, DMSO) δ 166.48, 157.43, 151.30, 143.31, 138.77, 137.90, 133.27, 131.42, 131.37, 127.16, 126.60, 125.23, 113.94, 56.40, 52.33, 35.01, 30.67, 19.46, 8.51. ESI-TOF HRMS: m/z 458.1741 (C23H27N3O5S + H+ requires 458.1749).
Methyl 4-(4-((4-(tert-butyl)phenyl)sulfonyl)-5-methyl-1H-1,2,3-triazol-1-yl)-2-chloro-5-methoxybenzoate (93)
The preparation of compound 93 (57 mg, 44% yield, 97.9% purity) was similar to that of compound 77 except that compound 70 was used as the starting material in place of compound 67. 1H NMR (CDCl3, 400 MHz): δ (ppm) 8.01 (d, 2H, J = 8.4 Hz), 7.56 (d, 2H, J = 8.4 Hz), 7.50 (s, 1H), 7.44 (s, 1H), 3.96 (s, 3H), 3.84 (s, 3H), 2.45 (s, 3H), 1.32 (s, 9H). 13C NMR (101 MHz, DMSO) δ 164.75, 157.41, 152.51, 143.26, 139.08, 137.81, 134.06, 130.47, 127.16, 126.55, 125.58, 122.55, 114.81, 56.91, 52.87, 34.97, 30.63, 8.45. ESI-TOF HRMS: m/z 478.1184 (C22H24ClN3O5S + H+ requires 478.1203).
1-(4-(4-((4-(tert-Butyl)phenyl)sulfonyl)-5-methyl-1H-1,2,3-triazol-1-yl)-2,5-dimethoxyphenyl)ethan-1-one (78)
A solution of MeMgBr (0.2 mL, 1.5 mmol) in THF was added to a solution of compound 76 (100 mg, 0.2 mmol) in THF (30 mL). The reaction was allowed to proceed under reflux for 15 h. The reaction mixture was then cooled to room temperature. HCl (1 mL, 1N) was added, and the mixture was stirred for a further 2 h. The reaction mixture was then poured into water and extracted with EtOAc (100 mL × 3). The combined EtOAc layer was washed with water, dried with anhydrous Na2SO4, and concentrated with a Rotavapor. The residue was purified by preparative HPLC to yield product 78 (60 mg, 58% yield, 99.9% purity). 1H NMR (CDCl3, 400 MHz): δ (ppm) 7.99 (d, 2H, J = 8.8 Hz), 7.52 (d, 2H, J = 8.8 Hz), 7.41 (s, 1H), 6.93 (s, 1H), 3.81 (s, 3H), 3.74 (s, 3H), 2.60 (s, 3H), 2.42 (s, 3H), 1.28 (s, 9H). 13C NMR (126 MHz, CDCl3) δ (ppm) 198.288, 157.763, 152.973, 147.278, 144.500, 138.558, 137.894, 130.457, 127.789, 126.987, 126.338, 113.884, 112.572, 56.414, 56.371, 35.291, 31.908, 31.055, 9.065. ESI-TOF HRMS: m/z 458.1739 (C23H27N3O5S + H+ requires 458.1749).
1-(4-(4-((4-(tert-Butyl)phenyl)sulfonyl)-5-methyl-1H-1,2,3-triazol-1-yl)-5-methoxy-2-methylphenyl)ethan-1-one (86)
The preparation of compound 86 (80 mg, 90.7% yield, 99.8% purity) was similar to that of compound 78 except that compound 84 was used as the starting material in place of compound 77. 1H NMR (CDCl3, 400 MHz): δ (ppm) 8.02 (d, J = 8.4 Hz, 2H), 7.55 (d, J = 8.4 Hz, 2H), 7.20–7.24 (m, 2H), 3.82 (s, 3H), 2.60 (s, 3H), 2.44 (s, 3H), 2.42 (s, 3H), 1.33 (s, 9H). 13C NMR (101 MHz, DMSO) δ 201.51, 157.38, 151.37, 143.25, 141.76, 138.69, 137.91, 131.14, 128.91, 127.14, 126.56, 124.26, 112.94, 56.48, 34.97, 30.64, 30.07, 18.94, 8.50. ESI-TOF HRMS: m/z 442.1784 (C23H27N3O4S + H+ requires 442.1800).
1-(4-(4-((4-(tert-Butyl)phenyl)sulfonyl)-5-methyl-1H-1,2,3-triazol-1-yl)-2-chloro-5-methoxyphenyl)ethan-1-one (94)
The preparation of compound 94 (53 mg, 57.5% yield, 100% purity) was similar to that of compound 78 except that compound 92 was used as the starting material in place of compound 77. 1H NMR (CDCl3, 400 MHz): δ (ppm) 8.02 (d, J = 8.8 Hz, 2H), 7.56 (d, J = 8.8 Hz, 2H), 7.24 (s, 1H), 7.21 (s, 1H), 3.82 (s, 3H), 2.70 (s, 3H), 2.46 (s, 3H), 1.33 (s, 9H). 13C NMR (101 MHz, DMSO) δ 199.40, 157.40, 152.71, 143.23, 142.49, 139.04, 137.83, 130.18, 127.17, 126.55, 124.77, 120.30, 113.16, 56.92, 34.97, 30.64, 30.48, 8.47. ESI-TOF HRMS: m/z 462.1243 (C22H24ClN3O4S + H+ requires 462.1254).
4-((4-(tert-Butyl)phenyl)sulfonyl)-1-(2,5-dimethoxyphenyl)-5-ethyl-1H-1,2,3-triazole (97)
MeONa (108 mg, 2 mmol) and compound 1261 (179 mg, 1 mmol) were added to a solution of 1-((4-(tert-butyl)phenyl)sulfonyl)butan-2-one (268 mg, 1 mmol) in MeOH. The mixture was stirred at 60 °C overnight. H2O (15 mL) was then added, and the mixture was extracted with EtOAc (15 mL × 3). The combined EtOAc organic layers were dried with anhydrous Na2SO4 and concentrated with a Rotavapor. The residue was purified by preparative HPLC to yield product 97 (55 mg, 13% yield, 96.0% purity). 1H NMR (CDCl3, 400 MHz): δ (ppm) 8.05 (d, 2H, J = 8.4 Hz), 7.57 (d, 2H, J = 8.4 Hz), 7.06–7.09 (m, 1H), 6.99–7.01 (m, 1H), 6.84 (d, 1H, J = 2.8 Hz), 3.77 (s, 3H), 3.72 (s, 3H), 2.89–2.91 (m, 2H), 1.34 (s, 9H), 1.09 (t, 3H, J = 7.6 Hz). 13C NMR (101 MHz, DMSO) δ 157.38, 153.12, 147.96, 143.26, 142.68, 137.86, 127.25, 126.55, 123.17, 118.19, 114.19, 113.97, 56.32, 55.93, 34.98, 30.65, 16.06, 12.60. ESI-TOF HRMS: m/z 430.1798 (C22H27N3O4S + H+ requires 430.1800).
4-((4-(tert-Butyl)phenyl)sulfonyl)-5-ethyl-1-(2-methoxy-5-methylphenyl)-1H-1,2,3-triazole (98)
The preparation of compound 98 (51 mg, 12.3% yield, 99.88% purity) was similar to that of compound 97 except that compound 9561 was used as the starting material in place of compound 12. 1H NMR (CDCl3, 400 MHz): δ (ppm) 7.99 (d, 2H, J = 8.8 Hz), 7.50 (d, 2H, J = 8.8 Hz), 7.25 (d, 1H, J = 8.4 Hz), 7.02 (s, 1H), 6.89 (d, 1H, J = 8.4 Hz), 3.67 (s, 3H), 2.81 (s, 2H), 2.26 (s, 3H), 1.27 (s, 9H), 1.02 (t, 3H, J = 7.6 Hz). 13C NMR (101 MHz, DMSO) δ 157.37, 151.72, 143.18, 142.70, 137.86, 133.00, 130.29, 128.86, 127.19, 126.54, 122.56, 112.81, 56.00, 34.97, 30.64, 19.53, 16.04, 12.63. ESI-TOF HRMS: m/z 414.1848 (C22H27N3O3S + H+ requires 414.1851).
4-((4-(tert-Butyl)phenyl)sulfonyl)-1-(5-chloro-2-methoxyphenyl)-5-ethyl-1H-1,2,3-triazole (99).
The preparation of compound 99 (94 mg, 21.7% yield, 99.7% purity) was similar to that of compound 97 except that compound 9661 was used as the starting material in place of compound 12. 1H NMR (CDCl3, 400 MHz): δ (ppm) 8.04 (d, 2H, J = 8.4 Hz), 7.57 (d, 2H, J = 8.4 Hz), 7.49–7.52 (m, 1H), 7.30 (s, 1H), 7.02 (d, 1H, J = 8.8 Hz), 3.78 (s, 3H), 2.88–2.90 (m, 2H), 1.34 (s, 9H), 1.09 (t, 3H, J = 7.6 Hz). 13C NMR (101 MHz, DMSO) δ 157.42, 153.10, 143.47, 142.77, 137.78, 132.54, 128.60, 127.25, 126.54, 124.30, 123.77, 114.67, 56.50, 34.98, 30.64, 16.04, 12.61. ESI-TOF HRMS: m/z 434.1298 (C21H24ClN3O3S + H+ requires 434.1305).
4-((4-(tert-Butyl)phenyl)sulfonyl)-1-(2,5-dimethoxyphenyl)-1H-1,2,3-triazol-5-amine (100)
MeONa (2.2 g, 40 mmol) and compound 1261 (4.74 g, 20 mmol) were added to a solution of 2-((4-(tert-butyl)phenyl)sulfonyl)acetonitrile (3.58 g, 20 mmol) in MeOH. The mixture was stirred overnight at room temperature, and the precipitate that formed was collected by filtration. The crude product was washed with water and MeOH and purified with recrystallization to yield product 100 (5.12 g, 62% yield, 99.8% purity). 1H NMR (CDCl3, 400 MHz): δ (ppm) 7.98 (d, 2H, J = 8.4 Hz), 7.53 (d, 2H, J = 8.4 Hz), 7.01 (s, 2H), 6.95 (s, 1H), 5.20 (s, 2H), 3.80 (s, 3H), 3.74 (s, 3H), 1.31 (s, 9H). 13C NMR (101 MHz, DMSO) δ 156.54, 153.14, 148.41, 144.68, 139.43, 126.32, 126.29, 124.16, 122.26, 117.67, 114.11, 114.01, 56.25, 55.84, 34.88, 30.68. ESI-TOF HRMS: m/z 417.1582 (C20H24N4O4S + H+ requires 417.1596).
4-((4-(tert-Butyl)phenyl)sulfonyl)-1-(2-methoxy-5-methylphenyl)-1H-1,2,3-triazol-5-amine (101)
The preparation of compound 101 (60 mg, 15% yield, 96.8% purity) was similar to that of compound 100 except that compound 95 was used as the starting material in place of compound 12 and on a smaller scale (1 mmol). 1H NMR (CDCl3, 400 MHz): δ (ppm) 7.88 (d, 2H, J = 8.4 Hz), 7.63 (d, 2H, J = 8.4 Hz), 7.33 (d, 1H, J = 8.4 Hz), 7.17 (s, 1H), 7.12 (d, 1H, J = 8.4 Hz), 6.39 (s, 2H), 3.70 (s, 3H), 2.23 (s, 3H), 1.26 (s, 9H). 13C NMR (101 MHz, DMSO) δ 156.53, 152.12, 144.63, 139.44, 132.28, 129.98, 128.92, 126.28, 124.20, 121.67, 112.92, 55.88, 34.88, 30.68, 19.57. ESI-TOF HRMS: m/z 401.1647 (C20H24N4O3S + H+ requires 401.1647).
4-((4-(tert-Butyl)phenyl)sulfonyl)-1-(5-chloro-2-methoxyphenyl)-1H-1,2,3-triazol-5-amine (102)
The preparation of compound 102 (60 mg, 14.3% yield, 99.0% purity) was similar to that of compound 100 except that compound 96 was used as the starting material in place of compound 12 and on a smaller scale (1 mmol). 1H NMR (CDCl3, 400 MHz): δ (ppm) 7.96 (d, 2H, J = 8.4 Hz), 7.53 (d, 2H, J = 8.4 Hz), 7.38–7.45 (m, 2H), 7.02 (d, 1H, J = 9.2 Hz), 5.18 (br, 2H), 3.86 (s, 3H), 1.31 (s, 9H). 13C NMR (101 MHz, DMSO) δ 156.54, 153.59, 144.88, 139.41, 131.72, 128.64, 126.31, 126.28, 124.04, 124.01, 122.97, 114.66, 56.32, 34.88, 30.68. ESI-TOF HRMS: m/z 421.646 (C19H21ClN4O3S + H+ requires 421.651).
4-((4-(tert-Butyl)phenyl)sulfonyl)-1-(2,5-dimethoxyphenyl)-1H-1,2,3-triazole (103)
NaNO2 (83 mg, 1.2 mmol) was added to a solution of compound 100 (416 mg, 1 mmol) in EtOH (20 mL) and H2SO4 (2 mL, 98%). The reaction mixture was then stirred at 50 °C for 4 h. H2O (25 mL) was then added, and the mixture was extracted with EtOAc (25 mL × 3). The combined EtOAc layers were dried with anhydrous Na2SO4 and concentrated with a Rotavapor. The residue was purified by preparative HPLC to yield product 103 (98 mg, 24% yield, 99.8% purity). 1H NMR (CDCl3, 400 MHz): δ (ppm) 8.74 (s, 1H), 8.03 (d, 2H, J = 8.4 Hz), 7.55 (d, 2H, J = 8.4 Hz), 7.37 (d, 1H, J = 2.8 Hz), 6.95–7.02 (m, 2H), 3.86 (s, 3H), 3.77 (s, 3H), 1.31 (s, 9H). 13C NMR (101 MHz, DMSO) δ 157.52, 153.11, 147.71, 146.00, 137.33, 129.55, 127.39, 126.59, 124.88, 117.08, 114.31, 111.73, 56.67, 55.91, 34.98, 30.63. ESI-TOF HRMS: m/z 402.1477 (C20H23N3O4S + H+ requires 402.1487).
4-((4-(tert-Butyl)phenyl)sulfonyl)-1-(2-methoxy-5-methylphenyl)-1H-1,2,3-triazole (104)
The preparation of compound 104 (50 mg, 10.8% yield, 98.3% purity) was similar to that of compound 103 except that compound 101 was used as the starting material in place of compound 100. 1H NMR (CDCl3, 400 MHz): δ (ppm) 8.67 (s, 1H), 8.03 (d, 2H, J = 8.8 Hz), 7.53–7.58 (m, 3H), 7.21–7.24 (m, 1H), 6.96 (d, 1H, J = 8.4 Hz), 3.86 (s, 3H), 2.32 (s, 3H), 1.31 (s, 9H). 13C NMR (101 MHz, DMSO) δ 157.48, 149.82, 147.66, 137.34, 131.90, 130.14, 129.49, 127.33, 126.58, 126.40, 124.29, 112.99, 56.28, 34.98, 30.62, 19.61. ESI-TOF HRMS: m/z 386.1537 (C20H23N3O3S + H+ requires 386.1538).
4-((4-(tert-Butyl)phenyl)sulfonyl)-1-(5-chloro-2-methoxyphenyl)-1H-1,2,3-triazole (105)
The preparation of compound 105 (96 mg, 19.8% yield, 98.6% purity) was similar to that of compound 103 except that compound 102 was used as the starting material in place of compound 100. 1H NMR (CDCl3, 400 MHz): δ (ppm) 8.70 (s, 1H), 8.02 (d, 2H, J = 8.8 Hz), 7.83 (d, 1H, J = 2.4 Hz), 7.55 (d, 2H, J = 8.8 Hz), 7.38–7.41 (m, 1H), 7.03 (d, 1H, J = 9.2 Hz), 3.92 (s, 3H), 1.31 (s, 9H). 13C NMR (101 MHz, DMSO) δ 157.55, 151.16, 147.75, 137.20, 131.32, 129.74, 127.37, 126.59, 126.05, 125.36, 124.22, 114.78, 56.72, 34.99, 30.62. ESI-TOF HRMS: m/z 406.0986 (C19H20ClN3O3S + H+ requires 406.0992).
4-((4-(tert-Butyl)phenyl)sulfonyl)-1-(2,5-dimethoxyphenyl)-1H-1,2,3-triazole-5-carbonitrile (109)
t-BuNO2 (683 mg, 6.64 mmol) was added dropwise to a solution of compound 100 (2.3 g, 5.53 mmol) and CuBr2 (1.85 g, 8.3 mmol) in MeCN (25 mL) under N2 and at room temperature. The reaction mixture was stirred under these conditions for 3 h. H2O (25 mL) was then added, and the mixture was extracted with EtOAc (25 mL × 2). The combined EtOAc layers were dried with anhydrous Na2SO4 and concentrated with a Rotavapor. The residue was purified by silica gel chromatography (petroleum ether/EtOAc = 10/1 to 6/1) to yield product 106 (1.0 g, 37.7% yield). 1H NMR (CDCl3, 400 MHz): δ (ppm) 8.06 (d, J = 8.0 Hz, 2H), 7.57 (d, J = 8.0 Hz, 2H), 6.84–7.00 (m, 3H), 3.77 (s, 3H), 3.74 (s, 3H), 1.33 (s, 9H). m/z 480 (M+H)+.
A suspension of compound 106 (1.0 g, 2.1 mmol), KCN (1.36 g, 21 mmol), and CuCN (934 mg, 10.5 mmol) in DMF (15 mL) was heated at 65 °C for 19 h under N2. EtOAc (30 mL) was then added to the reaction mixture, and the mixture was washed with brine, dried with anhydrous Na2SO4, and concentrated with a Rotavapor. The residue was purified by preparative HPLC to yield product 109 (180 mg, 20.1% yield, 96.3% purity). 1H NMR (CDCl3, 400 MHz): δ (ppm) 8.11 (d, J = 6.8 Hz, 2H), 7.61 (d, J = 6.8 Hz, 2H), 7.00–7.13(m, 3H), 3.85 (s, 3H), 3.78 (s, 3H), 1.34 (s, 9H). 13C NMR (101 MHz, DMSO) δ 158.90, 153.06, 151.19, 146.74, 135.30, 128.02, 127.06, 122.30, 119.25, 115.16, 114.36, 113.37, 106.33, 56.59, 55.98, 35.16, 30.57. ESI-TOF HRMS: m/z 427.1422 (C21H22N4O4S + H+ requires 427.1440).
4-((4-(tert-Butyl)phenyl)sulfonyl)-1-(2-methoxy-5-methylphenyl)-1H-1,2,3-triazole-5-carbonitrile (110)
The preparation of compound 107 was similar to that of compound 106 except that compound 101 was used as the starting material in place of compound 100 [m/z 464 (M+H)+]. A suspension of compound 107 (400 mg, 0.86 mmol), KCN (559 mg, 8.6 mmol), and CuCN (382 mg, 4.3 mmol) in DMF (6 mL) was heated at 65 °C for 19 h under N2. EtOAc (15 mL) was then added to the reaction mixture, and the mixture was washed with brine, dried with anhydrous Na2SO4, and concentrated with a Rotavapor. The residue was purified by preparative HPLC to yield product 110 (53 mg, 15.0% yield, 99.5% purity). 1H NMR (CDCl3, 40 0MHz): δ (ppm) 8.09 (d, J = 8.4 Hz, 2H), 7.61 (d, J = 8.4 Hz, 2H), 7.36 (d, J = 8.4 Hz, 1H), 7.24 (s, 1H), 7.01(d, J = 8.4 Hz, 1H), 3.90 (s, 3H), 2.33 (s, 3H), 1.33 (s, 9H). 13C NMR (101 MHz, DMSO) δ 158.84, 151.20, 150.51, 135.34, 134.01, 130.51, 127.99(3C), 127.02(2C), 121.90, 115.15, 113.12, 106.37, 56.28, 35.15, 30.56, 19.58. ESI-TOF HRMS: m/z 411.1490 (C21H22N4O3S + H+ requires 411.1491).
4-((4-(tert-Butyl)phenyl)sulfonyl)-1-(5-chloro-2-methoxyphenyl)-1H-1,2,3-triazole-5-carbonitrile (111)
The preparation of compound 108 was similar to the preparation of compound 106 except that compound 102 was used as the starting material in place of compound 100. 1H NMR (CDCl3, 400 MHz): δ (ppm) 8.06 (d, J = 8.4 Hz, 2H), 7.57 (d, J = 8.4 Hz, 2H), 7.52 (d, J = 8.4 Hz, 1H), 7.30 (s, 1H), 7.01(d, J = 8.4 Hz, 1H), 3.79 (s, 3H), 1.33 (s, 9H). m/z 484 (M+H)+. A suspension of compound 108 (600 mg, 1.24 mmol), KCN (807 mg, 12.4 mmol), and CuCN (331 mg, 3.72 mmol) in DMF (7 mL) was heated at 65 °C for 19 h under N2. EtOAc (30 mL) was then added to the reaction mixture, and the mixture was washed with brine, dried with anhydrous Na2SO4, and concentrated with a Rotavapor. The residue was purified by preparative HPLC to yield product 111 (160 mg, 30.0% yield, 99.8% purity). 1H NMR (CDCl3, 400 MHz): δ (ppm) 8.10 (d, J = 8.4 Hz, 2H), 7.62 (d, J = 8.4 Hz, 2H), 7.54 (t, J1 = 8.8 Hz, J2 = 2.0 Hz, 1H), 7.46 (d, J = 2.0 Hz, 1H), 7.09 (d, J = 8.8 Hz, 1H), 3.90 (s, 3H), 1.33 (s, 9H). 13C NMR (101 MHz, DMSO) δ 158.93, 151.89, 151.28, 135.22, 133.48, 128.04, 127.79, 127.05, 124.40, 122.80, 115.31, 115.11, 106.20, 56.83, 35.16, 30.56. ESI-TOF HRMS: m/z 431.0950 (C20H19ClN4O3S + H+ requires 431.0944).
1-(4-((4-(tert-Butyl)phenyl)sulfonyl)-1-(2,5-dimethoxyphenyl)-1H-1,2,3-triazol-5-yl)ethan-1-one (112)
A suspension of compound 106 (400 mg, 0.835 mmol), tributyl(1-ethoxyvinyl)stannane (361 mg, 1.0 mmol), and Pd(PPh3)4 (96 mg, 0.0835 mmol) in toluene (8 mL) was heated at 65 °C for 3 h under N2. After the reaction mixture had cooled, concentrated HCl/dioxane (8 mL) was added and the solution was stirred at room temperature for 1 h. H2O (15 mL) was then added, and the mixture was extracted with EtOAc (15 mL × 3). The combined EtOAc organic layers were dried with anhydrous Na2SO4 and concentrated with a Rotavapor. The residue was purified by preparative HPLC to yield product 112 (55 mg, 14.9% yield, 96.6% purity). 1H NMR (CDCl3, 400 MHz): δ (ppm) 8.04 (d, J = 8.4 Hz, 2H), 7.58 (d, J = 8.4 Hz, 2H), 6.96–7.04 (m, 3H), 3.76 (s, 3H), 3.71 (s, 3H), 2.76 (s, 3H), 1.34 (s, 9H). 13C NMR (101 MHz, DMSO) δ 189.85, 158.15, 153.21, 145.85, 144.90, 137.59, 136.30, 127.88, 126.73, 123.64, 117.86, 113.82, 112.99, 56.11, 55.92, 35.08, 30.67, 30.61. VESI-TOF HRMS: m/z 444.1586 (C22H25N3O5S + H+ requires 441.1593).
1-(4-((4-(tert-Butyl)phenyl)sulfonyl)-1-(2-methoxy-5-methylphenyl)-1H-1,2,3-triazol-5-yl)ethan-1-one (113)
A suspension of compound 107 (300 mg, 0.64 mmol), tributyl(1-ethoxyvinyl)stannane (281 mg, 0.78 mmol), and Pd(PPh3)4 (74 mg, 0.064 mmol) in toluene (6 mL) was heated at 65 °C for 3 h under N2. After the reaction mixture had cooled to room temperature, concentrated HCl/dioxane (7 mL) was added and the solution was stirred at room temperature for 1 h. H2O (15 mL) was then added, and the mixture was extracted with EtOAc (10 mL × 3). The combined EtOAc organic layers were dried with anhydrous Na2SO4 and concentrated with a Rotavapor. The residue was purified by preparative HPLC to yield product 113 (80 mg, 29.3% yield, 99.5% purity). 1H NMR (CDCl3, 400 MHz): δ (ppm) 8.04 (d, J = 8.4 Hz, 2H), 7.58 (d, J = 8.4 Hz, 2H), 7.23–7.24 (m, 2H), 6.88 (d, J = 8.4 Hz, 1H), 3.75 (s, 3H), 2.75 (s, 3H), 2.32 (s, 3H), 1.34 (s, 9H). 13C NMR (101 MHz, DMSO) δ 189.96, 158.13, 149.56, 144.87, 137.53, 136.32, 132.76, 130.52, 127.83, 127.50, 126.73, 123.08, 112.66, 55.81, 35.07, 30.64, 30.61, 19.59. ESI-TOF HRMS: m/z 428.1637 (C22H25N3O4S + H+ requires 428.1644).
1-(4-((4-(tert-Butyl)phenyl)sulfonyl)-1-(5-chloro-2-methoxyphenyl)-1H-1,2,3-triazol-5-yl)ethan-1-one (114)
A suspension of compound 108 (350 mg, 0.72 mmol), tributyl(1-ethoxyvinyl)stannane (314 mg, 0.87 mmol), and Pd(PPh3)4 (83 mg, 0.072 mmol) in toluene (6 mL) was heated at 65 °C for 3 h under N2. After the reaction mixture had cooled to room temperature, concentrated HCl/dioxane (6 mL) was added and the solution was stirred at room temperature for 1 h. H2O (15 mL) was added and the mixture was extracted with EtOAc (10 mL × 3). The combined EtOAc organic layers were dried with anhydrous Na2SO4 and concentrated with a Rotavapor. The residue was purified by preparative HPLC to give the product 114 (35 mg, 10.9% yield, 95.2% purity). 1H NMR (CDCl3, 400 MHz): δ (ppm) 8.02 (d, J = 8.4 Hz, 2H), 7.59 (d, J = 8.4 Hz, 2H), 7.42–7.49 (m, 2H), 6.93 (d, J = 8.4 Hz, 1H), 3.77 (s, 3H), 2.81 (s, 3H), 1.34 (s, 9H). 13C NMR (101 MHz, DMSO) δ 189.75, 158.22, 151.05, 145.09, 137.38, 136.16, 132.21, 127.92, 127.23, 126.75, 124.48, 124.29, 114.53, 56.37, 35.08, 30.74, 30.60. ESI-TOF HRMS: m/z 448.644 (C21H22ClN3O4S + H+ requires 448.648).
5-((4-(tert-Butyl)phenyl)sulfonyl)-2-(2,5-dimethoxyphenyl)-1-methyl-1H-imidazole (122)
A solution of compound 115 (1.6 g, 10 mmol), compound sodium 4-(tert-butyl)benzenesulfinate (3.3 g, 15 mmol), and CuI (1.9 g, 10 mmol) in DMF (20 mL) was heated at 65 °C for 18 h under N2. The reaction mixture was then cooled to room temperature and filtered. H2O (100 mL) was added to the filtrate, and the mixture was extracted with EtOAc (100 mL × 3). The combined organic layers were dried with anhydrous Na2SO4 and concentrated with a Rotavapor. The residue was purified by silica gel chromatography (petroleum ether/EtOAc = 10/1 to 2/1) to yield compound 118 (1.18 g, 42% yield). 1H NMR (CDCl3, 400 MHz): δ (ppm) 7.83 (d, 2H, J = 8.4 Hz), 7.72 (s, 1H), 7.53 (d, 2H, J = 8.4 Hz), 7.48 (s, 1H), 3.71 (s, 3H), 1.31 (s, 9H). m/z 279 (M+H)+.
A solution of compound 118 (1.1 g, 4 mmol), NBS (1.06 g, 6 mmol), and AIBN (16.4 mg, 0.1 mmol) in CCl4 (30 mL) was stirred at 60 °C for 18 h. The reaction mixture was cooled to room temperature and concentrated with a Rotavapor. The residue was purified by preparative TLC (petroleum ether/EtOAc = 2/1) to yield compound 120 (270 mg, 19% yield). m/z 357 (M+H)+.
A solution of compound 120 (72 mg, 0.2 mmol), 2,5-dimethoxyphenylboronic acid (73 mg, 0.4 mmol), Pd(dppf)Cl2 (8 mg), and K2CO3 (138 mg, 1 mmol) in dioxane (3 mL) and H2O (0.6 mL) was heated at 90 °C for 4 h under N2. After the reaction mixture was cooled to room temperature, H2O (5 mL) was added and the mixture was extracted with EtOAc (5 mL × 3). The combined organic layers were dried with anhydrous Na2SO4 and concentrated with a Rotavapor. The residue was purified by preparative HPLC to yield compound 122 (30 mg, 36% yield, 97.8% purity). 1H NMR (CDCl3, 400 MHz): δ (ppm) 7.83–7.85 (m, 2H), 7.79 (s, 1H), 7.52–7.54 (m, 2H), 6.97–7.00 (m, 1H), 6.89–6.90 (m, 1H), 6.84–6.87 (m, 1H), 3.70 (s, 3H), 3.66 (s, 3H), 3.54 (s, 3H), 1.28 (s, 9H). 13C NMR (101 MHz, DMSO) δ 157.22, 153.03, 151.01, 150.18, 137.87, 135.65, 129.99, 127.00, 126.66, 118.16, 117.40, 116.81, 113.04, 55.96, 55.61, 34.96, 32.43, 30.62. ESI-TOF HRMS: m/z 415.1689 (C22H26N2O4S + H+ requires 415.1691).
5-((4-(tert-Butyl)phenyl)sulfonyl)-2-(2-methoxy-5-methylphenyl)-1-methyl-1H-imidazole (123)
The preparation of compound 123 (20 mg, 25% yield, 96.9% purity) was similar to that of compound 122 except that 2-methoxy-5-methyl-phenylboronic acid was used in place of 2,5-dimethoxyphenylboronic acid. 1H NMR (CDCl3, 400 MHz): δ (ppm) 7.86 (d, 2H, J = 8.8 Hz), 7.79 (s, 1H), 7.54 (d, 2H, J = 8.8 Hz), 7.19–7.24 (m, 2H), 6.83 (d, 1H, J = 8.4 Hz), 3.70 (s, 3H), 3.49 (s, 3H), 2.27 (s, 3H), 1.32 (s, 9H). 13C NMR (101 MHz, DMSO) δ 157.15, 154.83, 150.67, 137.96, 135.87, 132.22, 132.06, 129.80, 129.52, 126.93, 126.63, 117.50, 111.65, 55.55, 34.95, 32.33, 30.62, 19.65. ESI-TOF HRMS: m/z 399.1737 (C22H26N2O3S + H+ requires 399.1742).
5-((4-(tert-Butyl)phenyl)sulfonyl)-2-(5-chloro-2-methoxyphenyl)-1-methyl-1H-imidazole (124)
The preparation of compound 124 (23 mg, 27.3% yield, 98.3% purity) was similar to that of compound 122 except that 2-methoxy-5-chloro-phenylboronic acid was used in place of 2,5-dimethoxyphenylboronic acid. 1H NMR (CDCl3, 400 MHz): δ (ppm) 7.82–7.85 (m, 3H), 7.55 (d, 2H, J = 8.4 Hz), 7.41–7.43 (m, 1H), 7.30 (d, 1H, J = 2.0 Hz), 6.89 (d, 1H, J = 9.2 Hz), 3.73 (s, 3H), 3.57 (s, 3H), 1.29 (s, 9H). 13C NMR (101 MHz, DMSO) δ 157.26, 155.89, 148.96, 137.81, 135.84, 131.54, 131.19, 130.26, 127.00, 126.66, 124.29, 119.41, 113.68, 56.05, 34.96, 32.49, 30.62. ESI-TOF HRMS: m/z 419.1184 (C21H23ClN2O3S + H+ requires 419.1196).
5-((4-(tert-Butyl)phenyl)sulfonyl)-2-(2,5-dimethoxyphenyl)-1H-imidazole (128)
A solution of compound 11762 (2.76 g, 10 mmol), compound sodium 4-(tert-butyl)benzenesulfinate (2.2 g, 10 mmol), and CuI (1.9 g, 10 mmol) in DMF (50 mL) was heated at 65 °C for 18 h under N2. The reaction mixture was then cooled to room temperature and filtered. H2O (100 mL) was added to the filtrate, and the mixture was extracted with EtOAc (100 mL × 3). The combined organic layers were dried with anhydrous Na2SO4 and concentrated with a Rotavapor. The residue was purified by silica gel chromatography (petroleum ether/EtOAc = 10/1 to 2/1) to yield chemical 119 (551 mg, 14% yield). 1H NMR (CDCl3, 400 MHz): δ (ppm) 7.98 (d, 2H, J = 8.4 Hz), 7.77 (s, 1H), 7.65 (s, 1H), 7.55 (d, 2H, J = 8.4 Hz), 5.31 (s, 2H), 3.52 (t, 2H, J = 8.0 Hz), 1.34 (s, 9H), 0.92 (t, 2H, J = 8.0 Hz), 0.00 (s, 9H). m/z 395 (M+H)+.
A solution of compound 119 (394 mg, 1 mmol), NBS (354 g, 2 mmol), and AIBN (16.4 mg, 0.1 mmol) in CCl4 (20 mL) was stirred at 60 °C for 18 h. The mixture was then cooled to room temperature and concentrated with a Rotavapor. The residue was purified by preparative TLC (petroleum ether/EtOAc = 2/1) to yield compound 121 (206 mg, 44% yield). 1H NMR (CDCl3, 400 MHz): δ (ppm) 7.96 (d, 2H, J = 8.4 Hz), 7.78 (s, 1H), 7.54 (d, 2H, J = 8.4 Hz), 5.28 (s, 2H), 3.56 (t, 2H, J = 8.0 Hz), 1.33 (s, 9H), 0.93 (t, 2H, J = 8.0 Hz), 0.00 (s, 9H). m/z 473 (M+H)+.
A solution of compound 121 (94 mg, 0.2 mmol), 2,5-dimethoxyphenylboronic acid (73 mg, 0.4 mmol), Pd(dppf)Cl2 (8 mg), and K2CO3 (138 mg, 1 mmol) in dioxane (3 mL) and H2O (0.6 mL) was heated at 90 °C for 4 h under N2. After the reaction mixture was cooled to room temperature, H2O (5 mL) was added and the mixture was extracted with EtOAc (5 mL×3). The combined organic layers were dried with anhydrous Na2SO4 and concentrated with a Rotavapor. The residue was purified by preparative TLC (petroleum ether/EtOAc = 2/1) to yield compound 125 (98 mg, 92% yield). m/z 531 (M+H)+.
A solution of compound 125 (98 mg, 0.184 mmol) and TBAF (500 mg) in THF (2 mL) was heated at 60 °C for 2 h under N2. After being cooled to room temperature, the reaction mixture was diluted with H2O (10 mL) and extracted with EtOAc (30 mL × 2). The combined organic layers were dried with anhydrous Na2SO4 and concentrated with a Rotavapor. The residue was purified by preparative HPLC to yield compound 128 (68 mg, 92% yield, 99.4% purity). 1H NMR (CDCl3, 400 MHz): δ (ppm) 7.98 (d, 2H, J = 8.4 Hz), 7.85 (d, 1H, J = 1.6 Hz), 7.74 (s, 1H), 7.49 (d, 2H, J = 8.8 Hz), 6.89 (s, 2H), 3.94 (s, 3H), 3.81 (s, 3H), 1.28 (s, 9H). 13C NMR (101 MHz, DMSO) δ 156.21, 153.17, 150.42, 145.17, 140.20, 138.70, 126.93, 126.09, 122.89, 117.68, 116.29, 113.27, 113.23, 56.01, 55.62, 34.81, 30.66. ESI-TOF HRMS: m/z 401.1529 (C21H24N2O4S + H+ requires 401.1535).
5-((4-(tert-Butyl)phenyl)sulfonyl)-2-(2-methoxy-5-methylphenyl)-1H-imidazole (129)
The preparation of compound 128 (34 mg, 48.1% yield, 99.5% purity) was similar to that of compound 128 except that 2-methoxy-5-methyl-phenylboronic acid was used in place of 2,5-dimethoxyphenylboronic acid. 1H NMR (CDCl3, 400 MHz): δ (ppm) 10.70 (s, 1H), 8.15 (s, 1H), 7.98 (d, 2H, J = 8.0 Hz), 7.73 (s, 1H), 7.49 (d, 2H, J = 8.4 Hz), 7.12 (d, 1H, J = 7.2 Hz), 6.86 (d, 1H, J = 8.4 Hz), 3.95 (s, 3H), 2.30 (s, 3H), 1.28 (s, 9H). 13C NMR (101 MHz, DMSO) δ 156.17, 154.15, 145.49, 140.16, 138.76, 131.21, 129.54, 128.70, 126.91, 126.09, 122.69, 116.80, 111.85, 55.64, 34.81, 30.67, 19.84. ESI-TOF HRMS: m/z 385.1575 (C21H24N2O3S + H+ requires 385.1586).
5-((4-(tert-Butyl)phenyl)sulfonyl)-2-(5-chloro-2-methoxyphenyl)-1H-imidazole (130)
The preparation of compound 130 (55 mg, 74% yield, 100% purity) was similar to that of compound 128 except that 2-methoxy-5-chloro-phenylboronic acid was used in place of 2,5-dimethoxyphenylboronic acid. 1H NMR (CDCl3, 400 MHz): δ (ppm) 10.68 (s, 1H), 8.32 (d, 1H, J = 2.4 Hz), 7.98 (d, 2H, J = 8.4 Hz), 7.75 (d, 1H, J = 1.6 Hz), 7.50 (d, 2H, J = 8.4 Hz), 7.24–7.28 (m, 1H), 6.90 (d, 1H, J = 8.8 Hz), 3.98 (s, 3H), 1.29 (s, 9H). 13C NMR (101 MHz, DMSO) δ 156.29, 154.91, 143.94, 140.55, 138.58, 130.17, 127.51, 126.98, 126.14, 124.60, 123.16, 118.81, 114.03, 56.12, 34.82, 30.66. ESI-TOF HRMS: m/z 405.1032 (C20H21ClN2O3S + H+ requires 405.1039).
5-Bromo-1-SEM-4-methyl-1H-imidazole (134), 4-bromo-1-SEM-5-methyl-1H-imidazole (135), 2-bromo-1-SEM-4-methyl-1H-imidazole (136), and 2-bromo-1-SEM-5-methyl-1H-imidazole (137).
NaH (4.8 g, 0.12 mol) was added to a solution of compound 131 (8.2 g, 0.1 mol) in THF (100 mL) at 0 °C. The mixture was stirred at 0 °C for 15 min, and SEMCl (19.9 g, 0.12 mol) was added to it dropwise. The reaction mixture was stirred for another hour at 0 °C and then quenched with H2O (50 mL) and extracted with EtOAc (100 mL × 3). The combined organic layers were dried with anhydrous Na2SO4 and concentrated with a Rotavapor. The residue was purified by silica gel chromatography (petroleum ether/EtOAc = 10/1 to 1/1) to yield compounds 132 and 133 (17.5 g, 83% yield). m/z 213 (M+H)+.
NBS (12 g, 67.9 mmol) in MeCN (100 mL) was added dropwise to a mixture of compounds 132 and 133 (16 g, 75.5 mmol) in MeCN (100 mL) at 0 °C. The reaction mixture was stirred at room temperature for 1 h, quenched with H2O (200 mL), and then extracted with EtOAc (100 mL × 3). The combined organic layers were dried with anhydrous Na2SO4 and concentrated with a Rotavapor. The residue was purified by silica gel chromatography (petroleum ether/EtOAc = 10/1 to DCM/MeOH = 1/1) to yield compounds 134, 135 (9.2 g, 42.0% yield) and 136, 137 (1.3 g, 5.9% yield). 1H NMR (CD3OD, 400 M Hz): δ (ppm) 7.45 (s, 1H), 5.22 (s, 2H), 3.47–3.55 (m, 2H), 2.27 (s, 2H), 0.91–0.96 (m, 2H), 0.02 (s, 9H). m/z 291 (M+H)+.
5-((4-(tert-Butyl)phenyl)sulfonyl)-2-(2,5-dimethoxyphenyl)-4-methyl-1H-imidazole (140)
A solution of compounds 134 and 135 (9.2 g, 31.7 mmol), sodium 4-(tert-butyl)benzenesulfinate (9.06 g, 41.2 mmol), and CuI (6.6 g, 34.9 mmol) in DMF (100 mL) was heated at 65 °C for 18 h under N2. After the reaction mixture had cooled to room temperature, it was filtered and H2O (100 mL) was added to the filtrate. The mixture was then extracted with EtOAc (100 mL × 3). The combined organic layers were dried with anhydrous Na2SO4 and concentrated with a Rotavapor. The residue was purified by silica gel chromatography (petroleum ether/EtOAc = 10/1 to DCM/MeOH = 15/1) to yield compound 138 (100 mg, 1.14% yield). m/z 279 (M+H)+.
NBS (64 mg, 0.36 mmol) was added to compound 138 (100 mg, 0.36 mmol) in MeCN (3 mL), and the solution was stirred at room temperature for 17 h. H2O (5 mL) was then added to the reaction mixture, and the mixture was extracted with EtOAc (5 mL × 3). The combined organic layers were dried with anhydrous Na2SO4 and concentrated with a Rotavapor. The residue was purified by preparative HPLC to yield compound 139 (100 mg, 78.1% yield). m/z 257 (M+H)+.
A solution of compound 139 (100 mg, 0.28 mmol), 2,5-dimethoxyphenylboronic acid (61 mg, 0.336 mmol), Pd(dppf)Cl2 (10 mg), and K2CO3 (1.38 g, 4.26 mmol) in dioxane (3 mL) and H2O (0.6 mL) was heated at 100 °C for 6 h under N2. After the reaction mixture had cooled to room temperature, H2O (5 mL) was added and the mixture was extracted with EtOAc (5 mL × 3). The combined organic layers were dried with anhydrous Na2SO4 and concentrated with a Rotavapor. The residue was purified by preparative HPLC to yield compound 140 (50 mg, 43.1% yield, 99.4% purity). 1H NMR (CDCl3, 400 MHz): δ (ppm) 7.88 (d, J = 8.4 Hz, 2H), 7.76 (s, 1H), 7.44 (d, J = 8.4 Hz, 2H), 6.85 (s, 2H), 3.90 (s, 3H), 3.75 (s, 3H), 2.55 (s, 3H), 1.24 (s, 9H). 13C NMR (101 MHz, DMSO) δ 155.94, 153.14, 150.27, 142.77, 139.65, 134.60, 133.64, 126.53, 126.02, 118.03, 115.90, 113.40, 113.10, 56.00, 55.60, 34.78, 30.68, 10.32. ESI-TOF HRMS: m/z 415.1692 (C22H26N2O4S + H+ requires 415.1691).
5-((4-(tert-Butyl)phenyl)sulfonyl)-2-(5-chloro-2-methoxyphenyl)-4-methyl-1H-imidazole (145)
A solution of compounds 136 and 137 (1 g, 3.45 mmol), 2-methoxy-5-chlorophenylboronic acid (770 mg, 4.14 mmol), Pd(dppf)Cl2 (100 mg), and K2CO3 (0.95 g, 6.9 mmol) in dioxane (10 mL) and H2O (2 mL) was heated at 65 °C for 6 h under N2. After the reaction mixture had cooled to room temperature, H2O (10 mL) was added and the mixture was extracted with EtOAc (10 mL × 3). The combined organic layers were dried with anhydrous Na2SO4 and concentrated with a Rotavapor. The residue was purified by preparative HPLC to yield compounds 141 and 142 (300 mg, 25% yield). m/z 353 (M+H)+.
NBS (151 mg, 0.85 mmol) was added to a solution of 141 and 142 (300 mg, 0.85 mmol) in MeCN (5 mL) at −30 °C. The reaction mixture was then stirred at room temperature for 0.5 h. The reaction was quenched with H2O (5 mL), and the mixture was extracted with EtOAc (10 mL × 3). The combined organic layers were dried with anhydrous Na2SO4 and concentrated with a Rotavapor. The residue was purified by preparative HPLC to yield compounds 143 and 144 (260 mg, 85.6% yield). m/z 358 (M+H)+.
A solution of compounds 143 and 144 (260 mg, 0.73 mmol), sodium 4-(tert-butyl)benzenesulfinate (208 mg, 0.94 mmol), and CuI (153 mg, 0.8 mmol) in DMF (5 mL) was heated at 65 °C for 18 h under N2. After the reaction mixture had cooled to room temperature, the reaction was quenched with H2O (10 mL) and the mixture was extracted with EtOAc (10 mL × 3). The combined organic layers were dried with anhydrous Na2SO4 and concentrated with a Rotavapor. The residue was purified by preparative HPLC to yield compound 145 (15 mg, 4.9% yield, 100% purity). 1H NMR (CDCl3, 400 MHz): δ (ppm) 10.21 (s, 1H), 8.24 (s, 1H), 7.91 (d, J = 8.4 Hz, 2H), 7.44 (d, J = 8.4 Hz, 2H), 7.17 (s, 1H), 6.83 (d, J = 8.4 Hz, 1H), 3.90 (s, 3H), 2.60 (s, 3H), 1.24 (s, 9H). 13C NMR (101 MHz, DMSO) δ 156.02, 154.74, 141.53, 139.51, 135.03, 133.94, 129.85, 127.45, 126.55, 126.07, 124.53, 119.11, 113.93, 56.10, 34.78, 30.67, 10.29. ESI-TOF HRMS: m/z 419.1195 (C21H23ClN2O3S + H+ requires 419.1196).
5-((4-(tert-Butyl)phenyl)sulfonyl)-2-(2-methoxy-5-methylphenyl)-4-methyl-1H-imidazole (150)
NaH (200 mg, 5 mmol) was added to a stirred solution of compound 146 (952 mg, 4 mmol) in THF (10 mL) at 0 °C and the reaction mixture was stirred at 0 °C for 15 min. SEMCl (797 mg, 4.8 mmol) was then added and the reaction was stirred at 0 °C for a further hour. The reaction was then quenched with H2O (30 mL), and the mixture was extracted with EtOAc (30 mL × 3). The combined organic layers were dried with anhydrous Na2SO4 and concentrated with a Rotavapor. The residue was purified by silica gel chromatography (petroleum ether/EtOAc = 10/1 to 1/1) to yield compound 147 (1.2 g, 82% yield). m/z 369 (M+H)+.
A solution of compound 147 (1.2 g, 3.3 mmol), 2-methoxy-5-methyl-phenylboronic acid (664 mg, 4 mmol), Pd(dppf)Cl2 (81.6 mg, 0.1 mmol), and K2CO3 (1.38 g, 10 mmol) in dioxane (15 mL) and H2O (5 mL) was heated at 100 °C for 3 h under N2. After the reaction mixture had cooled to room temperature, the reaction was quenched with H2O (30 mL) and the mixture was extracted with EtOAc (50 mL × 2). The combined organic layers were dried with anhydrous Na2SO4 and concentrated with a Rotavapor. The residue was purified by preparative TLC (petroleum ether/EtOAc = 5/1) to yield compound 148 (450 mg, 33% yield). 1H NMR (CDCl3, 400 MHz): δ (ppm) 7.37 (s, 1H), 7.32 (d, 1H, J = 8.4 Hz), 6.95 (d, 1H, J = 8.4 Hz), 5.23 (s, 2H), 3.86 (s, 3H), 3.21 (t, 2H, J = 8.4 Hz), 2.39 (s, 3H), 2.41 (s, 3H), 0.79 (t, 2H, J = 8.4 Hz), 0.00 (s, 9H). m/z 411 (M+H)+.
A solution of compound 148 (410 mg, 1 mmol), sodium 4-(tert-butyl)benzenesulfinate (440 mg, 2 mmol), and CuI (190 mg, 1 mmol) in DMF (10 mL) was heated at 65 °C for 18 h under N2. After the reaction mixture had cooled to room temperature, it was filtered. H2O (20 mL) was added to the filtrate, which was then extracted with EtOAc (30 mL × 3). The combined organic layers were dried with anhydrous Na2SO4 and concentrated with a Rotavapor. The residue was purified by preparative TLC (petroleum ether/EtOAc = 2/1) to yield compound 149 (25 mg, 5% yield). m/z 529 (M+H)+.
A solution of compound 149 (25 mg, 0.05 mmol) and TBAF (500 mg) in THF (2 mL) was heated at 60 °C for 2 h under N2. After the reaction mixture had cooled to room temperature, the reaction was quenched with H2O (10 mL) and the mixture was extracted with EtOAc (30 mL × 2). The combined organic layers were dried with anhydrous Na2SO4 and concentrated with a Rotavapor. The residue was purified by preparative HPLC to yield compound 150 (11 mg, 55% yield, 97.0% purity). 1H NMR (CDCl3, 400 MHz): δ (ppm) 8.04 (s, 1H), 7.90 (d, 2H, J = 8.4 Hz), 7.53 (d, 2H, J = 8.8 Hz), 7.22 (s, 1H), 6.91 (d, 1H, J = 8.8 Hz), 4.01 (s, 3H), 2.58 (s, 3H), 2.29 (s, 3H), 1.30 (s, 9H). 13C NMR (101 MHz, DMSO) δ 155.90, 154.00, 143.08, 139.72, 134.49, 133.48, 130.91, 129.44, 128.73, 126.47, 126.03, 117.10, 111.75, 55.62, 34.77, 30.68, 19.85, 10.31. ESI-TOF HRMS: m/z 399.1732 (C22H26N2O3S + H+ requires 399.1742).
5-((4-(tert-Butyl)phenyl)sulfonyl)-2-(2,5-dimethoxyphenyl)-1,4-dimethyl-1H-imidazole (155)
A solution of compound 151 (1.0 g, 3.97 mmol), 2,5-dimethoxyphenylboronic acid (722 mg, 3.97 mmol), Pd(dppf)Cl2 (100 mg), and K2CO3 (1.1 g, 7.94 mmol) in dioxane (10 mL) and H2O (2 mL) was heated at 100 °C for 3 h under N2. After the reaction mixture had cooled to room temperature, the reaction was quenched with H2O (15 mL) and the mixture was extracted with EtOAc (15 mL × 3). The combined organic layers were dried with anhydrous Na2SO4 and concentrated with a Rotavapor. The residue was purified by preparative TLC (petroleum ether/EtOAc = 2/1) to yield compound 152 (500 mg, 40.6% yield). m/z 311 (M+H)+.
A solution of compound 152 (500 mg, 1.6 mmol), sodium 4-(tert-butyl)benzenesulfinate (704 mg, 3.2 mmol), and CuI (337 mg, 1.77 mmol) in DMF (6 mL) was heated at 65 °C for 18 h under N2. The reaction mixture was cooled to room temperature and filtered. H2O (10 mL) was added to the filtrate, which was then extracted with EtOAc (10 mL × 3). The combined organic layers were dried with anhydrous Na2SO4 and concentrated with a Rotavapor. The residue was purified by preparative HPLC to yield compound 155 (100 mg, 14.6% yield, 97.3% purity). 1H NMR (CDCl3, 400 MHz): δ (ppm) 7.85 (d, J = 8.8 Hz, 2H), 7.61 (d, J = 8.8 Hz, 2H), 6.93–7.13 (m, 3H), 3.76 (s, 3H), 3.74 (s, 3H), 3.65 (s, 3H), 2.75 (s, 3H), 1.34 (s, 9H). 13C NMR (101 MHz, DMSO) δ 157.30, 153.07, 151.12, 147.38, 143.59, 138.70, 126.74, 126.51, 124.75, 118.02, 116.72, 116.69, 113.12, 56.03, 55.70, 34.99, 33.43, 30.65, 13.75. ESI-TOF HRMS: m/z 429.1831 (C23H28N2O4S + H+ requires 429.1848).
5-((4-(tert-Butyl)phenyl)sulfonyl)-2-(2-methoxy-5-methylphenyl)-1,4-dimethyl-1H-imidazole (156)
A solution of compound 151 (1.0 g, 3.97 mmol), 2-methoxy-5-methyl-phenylboronic acid (659 mg, 3.97 mmol), Pd(dppf)Cl2 (100 mg), and K2CO3 (1.1 g, 7.94 mmol) in dioxane (10 mL) and H2O (2 mL) was heated at 100 °C for 3 h under N2. After the reaction mixture had cooled to room temperature, the reaction was quenched with H2O (15 mL) and the mixture was extracted with EtOAc (15 mL × 3). The combined organic layers were dried with anhydrous Na2SO4 and concentrated with a Rotavapor. The residue was purified by preparative TLC (petroleum ether/EtOAc = 2/1) to yield compound 153 (400 mg, 34.2% yield). 1H NMR (CDCl3, 400 MHz): δ (ppm) 7.23–7.24 (m, 1H), 7.18 (d, J = 8.8 Hz, 1H), 6.83 (d, J = 8.8 Hz, 1H), 3.77 (s, 3H), 3.38 (s, 3H), 2.28 (s, 3H), 2.22 (s, 3H). m/z 295 (M+H)+.
A solution of compound 153 (400 mg, 1.36 mmol), sodium 4-(tert-butyl)benzenesulfinate (598 mg, 2.72 mmol), and CuI (284 mg, 1.5 mmol) in DMF (6 mL) was heated at 65 °C for 18 h under N2. The reaction mixture was then cooled to room temperature and filtered. H2O (10 mL) was added to the filtrate, which was then extracted with EtOAc (10 mL × 3). The combined organic layers were dried with anhydrous Na2SO4 and concentrated with a Rotavapor. The residue was purified by preparative HPLC to yield compound 156 (80 mg, 14.3% yield, 100% purity). 1H NMR (CDCl3, 400 MHz): δ (ppm) 7.80 (d, J = 8.4 Hz, 2H), 7.56 (d, J = 8.4 Hz, 2H), 7.30 (d, J = 8.8 Hz, 1H), 7.19 (s, 1H), 6.84 (d, J = 8.8 Hz, 1H), 3.71 (s, 3H), 3.60 (s, 3H), 2.70 (s, 3H), 2.22 (s, 3H), 1.29 (s, 9H). 13C NMR (101 MHz, DMSO) δ 157.19, 154.92, 147.83, 143.73, 138.75, 132.73, 132.00, 129.59, 126.69, 126.41, 124.50, 116.05, 111.73, 55.64, 34.96, 33.32, 30.63, 19.64, 13.80. ESI-TOF HRMS: m/z 413.1887 (C23H28N2O3S + H+ requires 413.1899).
5-((4-(tert-Butyl)phenyl)sulfonyl)-2-(5-chloro-2-methoxyphenyl)-1,4-dimethyl-1H-imidazole (157)
A solution of compound 151 (1.0 g, 3.97 mmol), 2-methoxy-5-chloro-phenylboronic acid (738 mg, 3.97 mmol), Pd(dppf)Cl2 (100 mg), and K2CO3 (1.1 g, 7.94 mmol) in dioxane (10 mL) and H2O (2 mL) was heated at 100 °C for 3 h under N2. After the reaction mixture had cooled to room temperature, the reaction was quenched with H2O (15 mL) and the mixture was extracted with EtOAc (15 mL × 3). The combined organic layers were dried with anhydrous Na2SO4 and concentrated with a Rotavapor. The residue was purified by preparative TLC (petroleum ether/EtOAc = 2/1) to yield compound 154 (500 mg, 40.0% yield). m/z 315 (M+H)+.
A solution of compound 154 (500 mg, 1.6 mmol), sodium 4-(tert-butyl)benzenesulfinate (704 mg, 3.2 mmol), and CuI (337 mg, 1.77 mmol) in DMF (6 mL) was heated at 65 °C for 18 h under N2. The reaction mixture was then cooled to room temperature and filtered. H2O (10 mL) was added to the filtrate, which was then extracted with EtOAc (10 mL × 3). The combined organic layers were dried with anhydrous Na2SO4 and concentrated with a Rotavapor. The residue was purified by preparative HPLC to yield compound 157 (150 mg, 21.7% yield, 99.4% purity). 1H NMR (CDCl3, 400 MHz): δ (ppm) 7.85 (d, J = 8.4 Hz, 2H), 7.60 (d, J = 8.4 Hz, 2H), 7.49 (d, J = 8.8 Hz, 1H), 7.38 (s, 1H), 6.94 (d, J = 8.8 Hz, 1H), 3.78 (s, 3H), 3.63 (s, 3H), 2.72 (s, 3H), 1.34 (s, 9H). 13C NMR (101 MHz, DMSO) δ 157.12, 155.93, 146.60, 144.57, 138.88, 131.69, 131.14, 126.67, 126.36, 124.66, 124.32, 118.87, 113.68, 56.09, 34.96, 33.29, 30.64, 14.16. ESI-TOF HRMS: m/z 433.1341 (C22H25ClN2O3S+ H+ requires 433.1347).
Biology
DMEM (Dulbecco’s Modified Eagle Medium), phenol red–free DMEM, Tb-anti-GST (Terbium-anti–Glutathione S-Transferase), GST hPXR-LBD, Tris (pH 7.5, 1 M), and dithiothreitol (DTT, 1 M) were purchased from Thermo Fisher Scientific (Atlanta, GA). MgCl2 (1 M) was purchased from Boston BioProducts (Ashland, MA). Rifampicin and bovine serum albumin (BSA) were purchased from Sigma (St. Louis, MO). T0901317 was purchased from Cayman Chemical (Ann Arbor, MI). Steadylite HTS reagent and 384-well white tissue culture–treated plates were purchased from PerkinElmer Life and Analytical Sciences (Boston, MA). 384-well black low-volume assay plates was purchased from Corning Incorporated (Tewksbury, MA). BODIPY FL vindoline was synthesized in house as previously reported.57 CellTiter-Glo Luminescent Cell Viability Assay reagent was purchased from Promega (Madison, WI). Fetal bovine serum (FBS) and charcoal/dextran-treated FBS were purchased from Hyclone (Logan, UT).
PXR Time-resolved fluorescence resonance energy transfer (TR-FRET) assay.
The PXR TR-FRET assay was performed as previously described57 with minor modifications. Briefly, BODIPY FL vindoline (5 μL/well, 400 nM) was first dispensed to 384-well low volume black assay plates, then 10 μL/well of indicated concentrations of tested compounds, or DMSO (negative control), or 10 μM T0901317 (positive control) were added. Finally, 5 μL/well of 20 nM Tb-anti-GST and 20 nM GST-hPXR-LBD was added. The final concentrations (in 20 μL of final assay volume per well) for the assay components are: BODIPY FL vindoline concentration (100 nM); Tb-anti-GST antibody (5 nM); GST-hPXR-LBD (5 nM); DMSO (0.3%); T0901317 (from 0.17 nM to 30 μM with 1-to-3 dilutions for 12 concentration levels); compounds (from 0.17 nM to 30 μM with 1-to-3 dilutions for 12 concentration levels). In addition, 0.3% DMSO alone or T0901317 (10 μM, with buffer, diluted from 6 μL of 3.33 mM DMSO stock to 1 mL assay volume) was also included in each plate to serve as corresponding negative or positive controls. With all assay components added, the assay plates were shaken at 900 rpm (80 g) on an IKA MTS 2/4 digital microtiter plate shaker for 1 min then briefly centrifuged at 1,000 rpm (201 g) for 30 s in an Eppendorf 5810 centrifuge equipped with an A-4–62 swing-bucket rotor (Eppendorf AG, Hamburg, Germany). The plates were then protected from light exposure and incubated for 60 min. After incubation, the TR-FRET signal from each well was collected with a PHERAstar FS Microplate Reader (BMG Labtech, Durham, NC). The %Inhibition for each well was calculated using Equation (1),
| (1) |
where TR-FRET SignalChemical is the TR-FRET signal from the respective chemical-treated well, TR-FRET SignalPositive is the TR-FRET signal from the positive control well, and TR-FRET SignalNegative is the TR-FRET signal from the negative control well as indicated.
Luciferase Reporter Assays.
For the luciferase assays for agonist, inverse agonist and antagonist using HepG2 hPXR-CYP3A4-luciferase stable cells,58 the cells were harvested and resuspended in phenol red–free DMEM supplemented with 5% charcoal/dextran-treated FBS at 0.25 million cells/mL. Tested compounds, rifampicin or SPA70 were serially diluted (in phenol red–free DMEM supplemented with 5% charcoal/dextran-treated FBS and 0.9% DMSO) and dispensed (10 μL/well) to 384-well white tissue culture-treated plates. In addition, DMSO (0.9% in phenol red–free DMEM supplemented with 5% charcoal/dextran-treated FBS, as negative control), rifampicin (30 μM in phenol red–free DMEM supplemented with 5% charcoal/dextran-treated FBS and 0.9% DMSO, as positive control for agonist) and SPA70 (30 μM in phenol red–free DMEM supplemented with 5% charcoal/dextran-treated FBS and 0.9% DMSO, as positive control for inverse agonist) were dispensed (10 μL/well) to the plates. HepG2 hPXR-CYP3A4-luciferase cell suspension (20 μL) in phenol red–free DMEM supplemented with 5% charcoal/dextran-treated FBS at 0.25 million cells/mL was then added. The final concentrations for tested compounds, rifampicin and SPA70 ranged from 0.17 nM to 30 μM with 1-to-3 dilutions for 12 concentration levels. The final concentrations: DMSO at 0.3% as negative control, SPA70 at 10 μM as positive control for inverse agonist, and rifampicin at 10 μM as positive control for agonist (agonistic mode). The chemical-treated cell plates were then incubated at 37°C in a humidified atmosphere containing 5% CO2 for 24 h, followed by a luciferase assay with steadylite HTS reagent (PerkinElmer Life and Analytical Sciences, Boston, MA) by following the manufacturer’s instruction. The luminescence signals from each plate were collected with an Envision HTS Multilabel Plate Reader (Model 2102; PerkinElmer Life and Analytical Sciences, Boston, MA). The activities of individual chemical tested were normalized to the corresponding positive and negative controls to generate the %Activation in the agonistic mode: %Activation = 100% × (compound signal - DMSO signal)/(10 μM rifampicin signal - DMSO signal); %Inhibition for inverse agonists: %Inhibition = 100% × (compound signal - DMSO signal)/(10 μM SPA70 signal - DMSO signal).
The luciferase assays for antagonists (antagonistic mode) were performed in the presence of 5 μM of rifampicin. For assay controls, rifampicin alone (at 5 μM) was used as 0% inhibition control, and rifampicin (at 5 μM) with SPA70 (at 10 μM) as 100% inhibition control. The activity of individual chemical tested was normalized to the corresponding positive and negative controls to generate the %Inhibition in the antagonistic assay: %Inhibition = 100% × (5 μM rifampicin signal - compound signal)/(5 μM rifampicin signal –10 μM SPA70 signal).
Cytotoxicity Assays.
The cytotoxicity assays were performed under conditions similar to the luciferase reporter assays, except that the CellTiter-Glo Luminescent Cell Viability Assay reagent (Promega) was used instead of the steadylite HTS reagent. The luminescence signals for each plate were collected using the Envision HTS Multilabel Plate Reader. Signal from cells treated with DMSO served as negative control (0% Cytotoxicity), and signals from assay medium with DMSO but without cells served as positive control (100% cytotoxicity). The cytotoxicity (% Relative Cytotoxicity) for each well was calculated using Equation (2),
| (2) |
where LumiNegative is the luminescence signal from the negative control, LumiChemical is the luminescence signal from test chemical, and LumiPositive is the luminescence signal from the positive control.
Dose Response Data Analysis.
In all the assays (the luciferase reporter, cytotoxicity, and TR-FRET assays), compounds were tested in quadruplicate for at least three times, and each data point represents the mean ± standard deviation (SD). For dose response data analysis, the relative luciferase signals (from the luciferase assays), the relative cytotoxicity (from the cytotoxic assays), or the %inhibition (from the PXR TR-FRET assays) of each chemical at the corresponding concentrations were plotted using GraphPad PRISM software (version 8.1.1, GraphPad Software, San Diego, CA) to generate the dose response curves and derive IC50 or EC50 values (if applicable) via the built-in sigmoidal dose-response fitting equation.
Pharmacophore Modeling.
SPA70 analogs were loaded into Maestro software (Schrödinger, release 2019–1) and prepared with the Ligprep function. Specifically, the OPLS3e forcefield63 was used on each ligand without changing the ionization states or generating tautomers. Specified chiralities were retained. Ligands were next input into the Develop Pharmacophore Model panel and the “find best alignment and common features” option was selected. Models with up to six features were generated, and the top-scoring pharmacophore was selected for follow-up with ligand alignment.
Computational Docking Studies.
The chemical structures of compounds 111, 42, and 97 were prepared using the Ligprep module from Schrodinger v11.6: the structures were converted to 3D format, hydrogen atoms were added, and ionization states at pH 7 were generated64 before docking to the hPXR LBD (PDB code: 5X0R) using the GLIDE module.65 Chain B was processed using the Protein Preparation Wizard in the Maestro suite in order to assign correct hetero atom bond order, remove water molecules, and add the protonation state of the protein.66 The docking experiments were conducted using the Ligand Docking protocol (SP-mode) in the Maestro suit, setting the grid to encompass the ligand binding site of the hPXR LBD. The resulting poses were ranked based on scores provided by GLIDE.67
Supplementary Material
Figure 7.
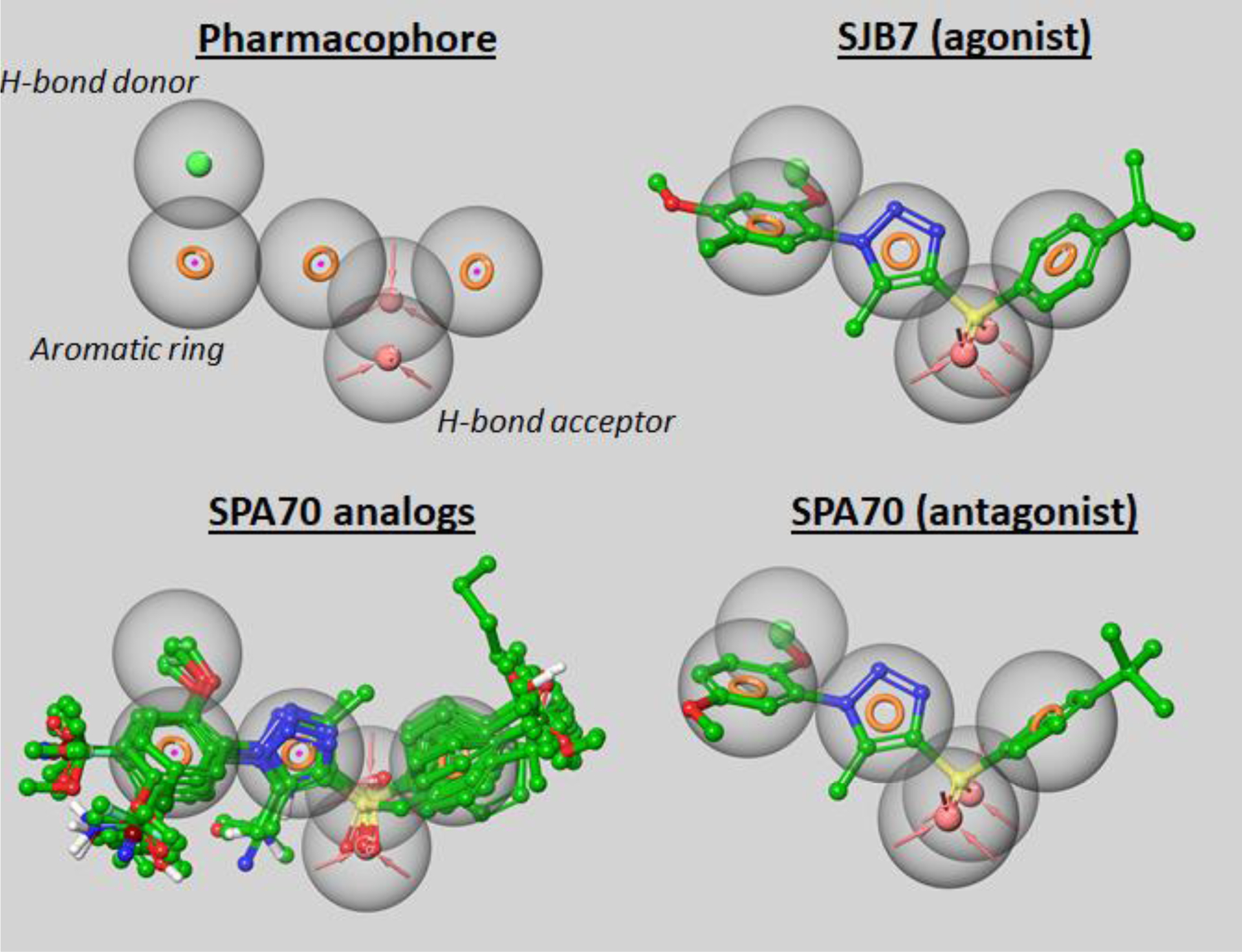
A pharmacophore model of SPA70 analogs.
Acknowledgments
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number R35GM118041. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank ALSAC for support; WuXi AppTec for technical assistance in chemical synthesis; Dr. Keith A. Laycock in the Department of Scientific Editing for editing the manuscript; and members of the Chen laboratory for valuable discussions.
Abbreviations
- PXR
pregnane X receptor
- hPXR
human PXR
- SAR
structure–activity relationship
- NR
nuclear receptor
- DBD
DNA-binding domain
- LBD
ligand binding domain
- CAR
constitutive androstane receptor
- VDR
vitamin D receptor
- DME
drug-metabolizing enzyme
- RXR
retinoid X receptor
- SRC-1
steroid receptor coactivator 1
- CYP
cytochrome P450
- UGT1A1
UDP glucuronosyltransferase family 1 member A1
- NF-κB
the nuclear factor kappa B
- TLC
thin-layer Chromatography
- FT
Fourier transform
- LC-MS
liquid chromatography–mass spectrometry
- DMEM
Dulbecco’s Modified Eagle Medium
- BSA
bovine serum albumin.
Footnotes
Supporting Information
The supporting information is available free of charge via the Internet at:
Figure S1 displaying the selectivity of 42, 97 and 111; Figure S2 displaying the residues interacting with the ligands that resulted from docking studies; 13C NMR and 1H NMR spectra, high resolution mass spectrometry, and HPLC purity analyses of final compounds 21-32, 42, 44, 46-48, 50, 51, 54, 55, 66-94, 97-105, 109-114, 122-124, 128-130, 140, 145, 150, 155-157 (PDF).
Molecular formula strings (CSV).
PDB coordinates (pdb).
The authors have the following patent related to this manuscript:
Chen T, Lin W, Wang Y. 1,4,5-Substituted 1,2,3-Triazole Analogues as Antagonists of the Pregnane X Receptor. International Patent Application published as WO/2017/165139, 2017; US Patent Application published as US 2019/0077770 A1, 2019. US patent No. 10,550,091 B2 issued, 2020.
References
- 1.Gronemeyer H; Gustafsson JA; Laudet V Principles for Modulation of the Nuclear Receptor Superfamily. Nat. Rev. Drug Discovery 2004, 3, 950–964. [DOI] [PubMed] [Google Scholar]
- 2.Robinson-Rechavi M; Carpentier AS; Duffraisse M; Laudet V How Many Nuclear Hormone Receptors Are There in the Human Genome? Trends Genet. 2001, 17, 554–556. [DOI] [PubMed] [Google Scholar]
- 3.Zhang ZD; Burch PE; Cooney AJ; Lanz RB; Pereira FA; Wu JQ; Gibbs RA; Weinstock G; Wheeler DA Genomic Analysis of the Nuclear Receptor Family: New Insights into Structure, Regulation, and Evolution from the Rat Genome. Genome Res. 2004, 14, 580–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang PX; Chandra V; Rastinejad F Structural Overview of the Nuclear Receptor Superfamily: Insights into Physiology and Therapeutics. Annu. Rev. Physiol 2010, 72, 247–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kumar R; Thompson EB The Structure of the Nuclear Hormone Receptors. Steroids 1999, 64, 310–319. [DOI] [PubMed] [Google Scholar]
- 6.Klinge CM Estrogen Receptor Interaction with Co-activators and Co-repressors. Steroids 2000, 65, 227–251. [DOI] [PubMed] [Google Scholar]
- 7.Warnmark A; Treuter E; Wright APH; Gustafsson JA Activation Functions 1 and 2 of Nuclear Receptors: Molecular Strategies for Transcriptional Activation. Mol. Endocrinol 2003, 17, 1901–1909. [DOI] [PubMed] [Google Scholar]
- 8.Weatherman RV; Fletterick RJ; Scanlan TS Nuclear-receptor Ligands and Ligand-binding Domains. Annu. Rev. Biochem 1999, 68, 559–581. [DOI] [PubMed] [Google Scholar]
- 9.Liang DD; Li LH; Lynch C; Diethelm-Varela B; Xia MH; Xue FT; Wang HB DL5050, a Selective Agonist for the Human Constitutive Androstane Receptor. ACS Med. Chem. Lett 2019, 10, 1039–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin WW; Wang YM; Chai SC; Lv LL; Zheng J; Wu J; Zhang QJ; Wang YD; Griffin PR; Chen TS SPA70 is a Potent Antagonist of Human Pregnane X Receptor. Nat. Commun 2017, 8, 741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moore DD; Kato S; Xie W; Mangelsdorf DJ; Schmidt DR; Xiao R; Kliewer SA International Union of Pharmacology. LXII. The NR1H and NR1I Receptors: Constitutive Androstane Receptor, Pregnene X Receptor, Farnesoid X Receptor Alpha, Farnesoid X Receptor Beta, Liver X Receptor Alpha, Liver X Receptor Beta, and Vitamin D Receptor. Pharmacol. Rev 2006, 58, 742–759. [DOI] [PubMed] [Google Scholar]
- 12.Chen YK; Tang Y; Guo CX; Wang JH; Boral D; Nie DT Nuclear Receptors in the Multidrug Resistance through the Regulation of Drug-metabolizing Enzymes and Drug Transporters. Biochem. Pharmacol 2012, 83, 1112–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chai SC; Wright WC; Chen T Strategies for Developing Pregnane X Receptor Antagonists: Implications from Metabolism to Cancer. Med. Res. Rev 2020, 40, 1061–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sandanaraj E; Lal S; Selvarajan V; Ooi LL; Wong ZW; Wong NS; Ang PCS; Lee EJD; Chowbay B PXR Pharmacogenetics: Association of Haplotypes with Hepatic CYP3A4 and ABCB1 Messenger RNA Expression and Doxorubicin Clearance in Asian Breast Cancer Patients. Clin. Cancer Res 2008, 14, 7116–7126. [DOI] [PubMed] [Google Scholar]
- 15.Carnahan VE; Redinbo MR Structure and Function of the Human Nuclear Xenobiotic Receptor PXR. Curr. Drug Metab 2005, 6, 357–367. [DOI] [PubMed] [Google Scholar]
- 16.Buchman CD; Chai SC; Chen T A Current Structural Perspective on PXR and CAR in Drug Metabolism. Expert Opin. Drug Metab. Toxicol 2018, 14, 635–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chai SC; Cherian MT; Wang YM; Chen TS Small-molecule Modulators of PXR and CAR. Biochim. Biophys. Acta 2016, 1859, 1141–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smutny T; Mani S; Pavek P Post-translational and Post-transcriptional Modifications of Pregnane X Receptor (PXR) in Regulation of the Cytochrome P450 Superfamily. Curr. Drug Metab 2013, 14, 1059–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eloranta JJ; Kullak-Ublick GA Coordinate Transcriptional Regulation of Bile Acid Homeostasis and Drug Metabolism. Arch. Biochem. Biophys 2005, 433, 397–412. [DOI] [PubMed] [Google Scholar]
- 20.Tompkins LM; Wallace AD Mechanisms of Cytochrome P450 Induction. J. Biochem. Mol. Toxicol 2007, 21, 176–181. [DOI] [PubMed] [Google Scholar]
- 21.Pavek P Pregnane X Receptor (PXR)-Mediated Gene Repression and Cross-Talk of PXR with Other Nuclear Receptors via Coactivator Interactions. Front. Pharmacol 2016, 7, 456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Banerjee M; Robbins D; Chen TS Targeting Xenobiotic Receptors PXR and CAR in Human Diseases. Drug Discovery Today 2015, 20, 618–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oladimeji PO; Chen T PXR: More Than Just a Master Xenobiotic Receptor. Mol. Pharmacol 2018, 93, 119–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mackowiak B; Hodge J; Stern S; Wang H The Roles of Xenobiotic Receptors: Beyond Chemical Disposition. Drug Metab. Dispos 2018, 46, 1361–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Corcoran E Long March to Approval. Sci. Am 1988, 259, 136–138. [DOI] [PubMed] [Google Scholar]
- 26.Xie W; Tian Y Xenobiotic Receptor Meets NF-kappaB, a Collision in the Small Bowel. Cell Metab. 2006, 4, 177–178. [DOI] [PubMed] [Google Scholar]
- 27.Brewer CT; Chen TS PXR Variants: the Impact on Drug Metabolism and Therapeutic Responses. Acta Pharm. Sin. B 2016, 6, 441–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Willson TM; Kliewer SA PXR, CAR and Drug Metabolism. Nat. Rev. Drug Discovery 2002, 1, 259–266. [DOI] [PubMed] [Google Scholar]
- 29.Chen T Overcoming Drug Resistance by Regulating Nuclear Receptors. Adv. Drug Delivery Rev 2010, 62, 1257–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mani S; Dou W; Redinbo MR PXR Antagonists and Implication in Drug Metabolism. Drug Metab. Rev 2013, 45, 60–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raynal C; Pascussi JM; Legueline G; Breuker C; Kantar J; Lallemant B; Poujol S; Bonnans C; Joubert D; Hollande F; Lumbroso S; Brouillet JP; Evrard A Pregnane X Receptor (PXR) Expression in Colorectal Cancer Cells Restricts Irinotecan Chemosensitivity Through Enhanced SN-38 Glucuronidation. Mol. Cancer 2010, 9, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chai SC; Lin WW; Li YT; Chen TS Drug Discovery Technologies to Identify and Characterize Modulators of the Pregnane X Receptor and the Constitutive Androstane Receptor. Drug Discovery Today 2019, 24, 906–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu CS; Huang M; Bi HC PXR- and CAR-mediated Herbal Effect on Human Diseases. Biochim. Biophys. Acta 2016, 1859, 1121–1129. [DOI] [PubMed] [Google Scholar]
- 34.Staudinger JL Clinical Applications of Small Molecule Inhibitors of Pregnane X Receptor. Mol. Cell. Endocrinol 2019, 485, 61–71. [DOI] [PubMed] [Google Scholar]
- 35.Pokhodylo NT; Matiychuk VS; Obushak MD (Arylsulfonyl)acetones and -acetonitriles: New Activated Methylenic Building Blocks for Synthesis of 1,2,3-Triazoles. Synthesis 2009, 14, 2321–2323. [Google Scholar]
- 36.Boyer B; Keramane EM; Roque JP; Pavia AA BiBr3, an Efficient Catalyst for the Benzylation of Alcohols: 2-phenyl-2-propyl, a New Benzyl-type Protecting Group. Tetrahedron Lett. 2000, 41, 2891–2894. [Google Scholar]
- 37.Bennett MJ; Betancort JM; Boloor A; Kaldor SW; Stafford JA; Veal JM Heterocyclic Compounds as Bromodomain Inhibitors and Their Preparation. WO2015058160A1, 2015. [Google Scholar]
- 38.Albaneze-Walker J; Bazaral C; Leavey T; Dormer PG; Murry JA Improved Carbonylation of Heterocyclic Chlorides and Electronically Challenging Aryl Bromides. Org. Lett 2004, 6, 2097–2100. [DOI] [PubMed] [Google Scholar]
- 39.Antczak MI; Zhang Y; Wang C; Doran J; Naidoo J; Voruganti S; Williams NS; Markowitz SD; Ready JM Inhibitors of 15-Prostaglandin Dehydrogenase To Potentiate Tissue Repair. J. Med. Chem 2017, 60, 3979–4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ai T; Willett R; Williams J; Ding R; Wilson DJ; Xie J; Kim DH; Puertollano R; Chen L N-(1-Benzyl-3,5-dimethyl-1H-pyrazol-4-yl)benzamides: Antiproliferative Activity and Effects on mTORC1 and Autophagy. ACS Med. Chem. Lett 2017, 8, 90–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen JB; Lei GX; Xu WH; Jin XL; Shao MC; Tang YQ Synthesis and Crystal-Structure of Isomerized Butadiene(Dicarbonyl)(Ethoxyarylcarbene)Iron Complexes. J. Organomet. Chem 1985, 286, 55–67. [Google Scholar]
- 42.Slocum DW; Reece TL; Sandlin RD; Reinscheld TK; Whitley PE Metalations Utilizing Aryllithiums; Ortho-functionalization of p-bromoanisole (pBrA). Tetrahedron Lett. 2009, 50, 1593–1595. [Google Scholar]
- 43.Mckean DR; Parrinello G; Renaldo AF; Stille JK Synthesis of Functionalized Styrenes Via Palladium-Catalyzed Coupling of Aryl Bromides with Vinyl Tin Reagents. J. Org. Chem 1987, 52, 422–424. [Google Scholar]
- 44.Beattie D; Beer D; Bradley ME; Bruce I; Charlton SJ; Cuenoud BM; Fairhurst RA; Farr D; Fozard JR; Janus D; Rosethorne EM; Sandham DA; Sykes DA; Trifilieff A; Turner KL; Wissler E An Investigation into the Structure-activity Relationships Associated with the Systematic Modification of the Beta(2)-adrenoceptor Agonist Indacaterol. Bioorg. Med. Chem. Lett 2012, 22, 6280–6285. [DOI] [PubMed] [Google Scholar]
- 45.Aalten HL; Vankoten G; Grove DM; Kuilman T; Piekstra OG; Hulshof LA; Sheldon RA The Copper Catalyzed Reaction of Sodium Methoxide with Aryl Bromides - a Mechanistic Study Leading to a Facile Synthesis of Anisole Derivatives. Tetrahedron 1989, 45, 5565–5578. [Google Scholar]
- 46.Maligres PE; Waters MS; Fleitz F; Askin D A Highly Catalytic Robust Palladium Catalyzed Cyanation of Aryl Bromides. Tetrahedron Lett. 1999, 40, 8193–8195. [Google Scholar]
- 47.Ortiz-Marciales M; Tirado LM; Colon R; Ufret ML; Figueroa R; Lebron M; DeJesus M; Martinez J; Malave T N-tert-butyldimethylsilyl Imines as Intermediates for the Synthesis of Amines and Ketones. Synth. Commun 1998, 28, 4067–4075. [Google Scholar]
- 48.Furuya T; Kaiser HM; Ritter T Palladium-mediated Fluorination of Arylboronic Acids. Angew. Chem., Int. Ed. Engl 2008, 47, 5993–5996. [DOI] [PubMed] [Google Scholar]
- 49.Guo S; Pan R; Guan Z; Li P; Cai L; Chen S; Lin A; Yao H Synthesis of Indole-Fused Polycyclics via Rhodium-Catalyzed Undirected C-H Activation/Alkene Insertion. Org. Lett 2019, 21, 6320–6324. [DOI] [PubMed] [Google Scholar]
- 50.Doyle MP; Siegfried B; Dellaria JF Alkyl Nitrite Metal Halide Deamination Reactions .2. Substitutive Deamination of Arylamines by Alkyl Nitrites and Copper(Ii) Halides - Direct and Remarkably Efficient Conversion of Arylamines to Aryl Halides. J. Org. Chem 1977, 42, 2426–2431. [Google Scholar]
- 51.Rosenmund KW; Struck E Halogen Attached to a Ring Carbon Atom and Its Replacement by Other Substituents. I. Replacement of the Halogen by the Carboxyl Group. Ber. Dtsch. Chem. Ges. B 1919, 52B, 1749–1756. [Google Scholar]
- 52.Mo J; Xu L; Xiao J Ionic Liquid-promoted, Highly Regioselective Heck Arylation of Electron-rich Olefins by Aryl Halides. J. Am. Chem. Soc 2005, 127, 751–760. [DOI] [PubMed] [Google Scholar]
- 53.Peng Y Cu(I)-catalysed Coupling of Arylsulfinic Salts with Aryl Bromides. J. Chem. Res 2014, 38, 447–449. [Google Scholar]
- 54.Oconnell JF; Parquette J; Yelle WE; Wang W; Rapoport H Convenient Synthesis of Methyl 1-Methyl-2,4-Dibromo-5-Imidazolecarboxylate. Synthesis 1988, 10, 767–771. [Google Scholar]
- 55.Sutherland HS; Blaser A; Kmentova I; Franzblau SG; Wan BJ; Wang YH; Ma ZK; Palmer BD; Denny WA; Thompson AM Synthesis and Structure-activity Relationships of Antitubercular 2-Nitroimidazooxazines Bearing Heterocyclic Side Chains. J. Med. Chem 2010, 53, 855–866. [DOI] [PubMed] [Google Scholar]
- 56.Recnik LM; Abd El Hameid M; Haider M; Schnurch M; Mihovilovic MD Selective Sequential Cross-Coupling Reactions on Imidazole towards Neurodazine and Analogues. Synthesis 2013, 45, 1387–1405. [Google Scholar]
- 57.Lin WW; Liu JY; Jeffries C; Yang L; Lu Y; Lee RE; Chen TS, Development of BODIPY FL Vindoline as a Novel and High-Affinity Pregnane X Receptor Fluorescent Probe. Bioconjugate Chem. 2014, 25, 1664–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lin W; Bwayi M; Wu J; Li Y; Chai SC; Huber AD; Chen T, CITCO Directly Binds to and Activates Human Pregnane X Receptor. Mol. Pharmacol 2020, 97, 180–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhu BT Mechanistic Explanation for the Unique Pharmacologic Properties of Receptor Partial Agonists. Biomed. Pharmacother 2005, 59, 76–89. [DOI] [PubMed] [Google Scholar]
- 60.Lin WW; Goktug AN; Wu J; Currier DG; Chen TS High-Throughput Screening Identifies 1,4,5-Substituted 1,2,3-Triazole Analogs as Potent and Specific Antagonists of Pregnane X Receptor. Assay Drug Dev. Technol 2017, 15, 383–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shrestha JP; Fosso MY; Bearss J; Chang CWT Synthesis and Anticancer Structure Activity Relationship Investigation of Cationic Anthraquinone Analogs. Eur. J. Med. Chem 2014, 77, 96–102. [DOI] [PubMed] [Google Scholar]
- 62.Abeywardane A; Farmer B; Farrow NA; Gao DA; Heim-Riether A; Keenan LLS; Mugge IA; Taylor SJ; Xiong Z; Yu Y; Zhang Q Preparation of Heteroaryl Substituted Indole Compounds Useful as MMP-13 Inhibitors. WO2010045188A1, 2010. [Google Scholar]
- 63.Harder E; Damm W; Maple J; Wu C; Reboul M; Xiang JY; Wang L; Lupyan D; Dahlgren MK; Knight JL; Kaus JW; Cerutti DS; Krilov G; Jorgensen WL; Abel R; Friesner RA OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput 2016, 12, 281–296. [DOI] [PubMed] [Google Scholar]
- 64.Sastry GM; Adzhigirey M; Day T; Annabhimoju R; Sherman W Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comput.-Aided Mol. Des 2013, 27 (3), 221–234. [DOI] [PubMed] [Google Scholar]
- 65.Halgren TA; Murphy RB; Friesner RA; Beard HS; Frye LL; Pollard WT; Banks JL Glide: a New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem 2004, 47, 1750–1759. [DOI] [PubMed] [Google Scholar]
- 66.Kumar S; Singh J; Narasimhan B; Shah SAA; Lim SM; Ramasamy K; Mani V Reverse Pharmacophore Mapping and Molecular Docking Studies for Discovery of GTPase HRas as Promising Drug Target for Bis-pyrimidine Derivatives. Chem. Cent. J 2018, 12, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tahlan S; Kumar S; Ramasamy K; Lim SM; Shah SAA; Mani V; Narasimhan B In-silico Molecular Design of Heterocyclic Benzimidazole Scaffolds as Prospective Anticancer Agents. MC Chem. 2019, 13, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.