

Abstract
In Rosaceae species, two gametophytic self-incompatibility (GSI) mechanisms are described, the Prunus self-recognition system and the Maleae (Malus/Pyrus/Sorbus) non-self- recognition system. In both systems the pistil component is a S-RNase gene, but from two distinct phylogenetic lineages. The pollen component, always a F-box gene(s), in the case of Prunus is a single gene, and in Maleae there are multiple genes. Previously, the Rosa S-locus was mapped on chromosome 3, and three putative S-RNase genes were identified in the R. chinensis ‘Old Blush’ genome. Here, we show that these genes do not belong to the S-locus region. Using R. chinensis and R. multiflora genomes and a phylogenetic approach, we identified the S-RNase gene, that belongs to the Prunus S-lineage. Expression patterns support this gene as being the S-pistil. This gene is here also identified in R. moschata, R. arvensis, and R. minutifolia low coverage genomes, allowing the identification of positively selected amino acid sites, and thus, further supporting this gene as the S-RNase. Furthermore, genotype–phenotype association experiments also support this gene as the S-RNase. For the S-pollen GSI component we find evidence for multiple F-box genes, that show the expected expression pattern, and evidence for diversifying selection at the F-box genes within an S-haplotype. Thus, Rosa has a non-self-recognition system, like in Maleae species, despite the S-pistil gene belonging to the Prunus S-RNase lineage. These findings are discussed in the context of the Rosaceae GSI evolution. Knowledge on the Rosa S-locus has practical implications since genes controlling floral and other ornamental traits are in linkage disequilibrium with the S-locus.
Subject terms: Evolution, Genetics, Plant sciences
Introduction
Gametophytic self-incompatibility (GSI), the most common pre-zygotic self-incompatibility genetic mechanism, prevents self-fertilization between genetically related individuals, where the genotype of the haploid pollen determines its incompatibility type1. This mechanism is determined by a single locus, the S-locus, and in the most frequent eudicot system2, the S-pistil gene codes for a protein with ribonuclease activity, called S-RNase3–5, and the S-pollen gene (s) code(s) for a F-box protein(s)6–26. In incompatible crosses, the cytotoxic S-RNases lead to pollen tube RNA degradation, that causes the cessation of pollen tube elongation. Two mechanisms of pollen recognition have been proposed, the self and non-self-recognition systems6–26. In the self-recognition mechanism, described in the Prunus genus of the Rosaceae family6, 7, 11, 17, 24, 27, only one F-box protein is the S-pollen GSI specificity determinant, called SFB, that interacts with the self S-RNase (reviewed by28). In this system, as expected, the S-RNase and SFB genes show patterns of co-evolution29. In the non-self-recognition mechanism, characterized in Solanaceae, Plantaginaceae, and in the Rosaceae Maleae tribe (Malus/Pyrus/Sorbus), there are multiple F-box proteins, called SLFs in Solanaceae and Plantaginaceae, and SFBBs in Maleae13–16, 18–26, 30–32, that interact with all S-RNases but the self-S-RNase15, 16, 18, 20–26, 30–32. Even though different recognition interactions occur due to different evolutionary paths, the S-RNase retained the same feature of cytotoxicity to self-pollen in both mechanisms28, 30.
Based on phylogenetic inferences, the S-RNase GSI system evolved once in core eudicots33–37, and has been the subject of multiple duplication events during evolution. In Rosaceae, different duplicates have been recruited for GSI function in Prunus and Malus/Pyrus/Sorbus24, 37. Convergent evolution has also been proposed for the evolution of the S-pollen determinant since the Prunus S-pollen gene, belongs to a different gene lineage than that of the Malus/Pyrus/Sorbus S-pollen genes. Therefore, it is not surprising the presence of different recognition systems in Prunus (self-recognition6, 7, 11, 12, 17, 24, 27, 28, 38) and in Malus/Pyrus/Sorbus (non-self-recognition mechanism13, 16, 18–20, 24, 25). How the two systems evolved is still debated. The observation that Prunus S-RNase and SFB lineage genes are present in Fragaria species (an outgroup), suggests that the ancestral Rosaceae S-locus was of the self-recognition type, and that the Malus S-locus region has a “de novo” evolution24. An alternative hypothesis postulates that the divergence between SFB and SFBB genes occurred early in the establishment of eudicots, and that Prunus species started using SFB gene as a S-pollen factor around the time of the Prunus divergence39. Under this evolutionary hypothesis, the common ancestor of Prunus and Malus/Fragaria would present a non-self-recognition mechanism. In this work, by characterizing the Rosa S-locus region we clarify the Rosaceae S-locus ancestral state.
Rose (Rosa sp., Rosaceae) species are known by their ornamental value and the production of essential oils used in perfume and cosmetic industry. There are about 200 species in this genus, but only 10 species (R. canina, R. chinenesis ‘Old Blush’ OB), R. foetida, R. gallica, R. gigantea, R. moschata, R. multiflora, R. phoenicia, R. rugosa, and R. wichuraiana ‘Basye’s Thornless’) have contributed for most of the modern species, throughout processes of hybridization and polyploidization40. Based on the absence of seed set after self-pollination, the presence of a GSI system has been postulated for most Rosa diploid species41–43. Analyses of one of the R. chinensis genomes44, revealed three putative S-RNase genes (SRNase26, SRNase30, and SRNase36) located on chromosome 3, as a candidate region of the Rosa S-locus44. In this work we show that this region is not the S-locus. This is the reason why segregation analyses using SRNase30 as a marker, shows co-segregation with the S-locus at a distance of 4.2 cM44. Nevertheless, these segregation analyses showed that the S-locus region is located on chromosome 3, as previously reported45. According to chromosome location, we also exclude a putative S-pollen gene –RrSLF, cloned from R. rugosa pollen RNA, that in phylogenetic analyses clusters with Prunus SFB46, since the orthologous R. chinensis gene is located on chromosome 2.
Rosa diploid genomes are of relatively small size (560 Mb47, 48), and for R. chinensis ‘Old Blush’44, 49, and R. multiflora50 there are assembled genomes publicly available (https://www.ncbi.nlm.nih.gov/sra). Here, we use these datasets to perform phylogenetic analyses to identify the Rosa S-locus genes. Transcriptomic analyses using R. chinensis pistil and ovary, stamen, leaf, stem, and root tissue (available in SRA database), support the identification of the Rosa S-locus genes. To show that the Rosa S-RNase gene shows evidence of positive selection, we used low coverage Rosa genomes (R. moschata, R. laevigata, R. rugosa, R. persica, R. xanthina, R. minutifolia, R. odorata, R. arvensis, and R. majalis) available at SRA database, to identify further S-RNase alleles. Furthermore, we use R. arvensis genotype–phenotype association experiments to confirm that the identified gene is the S-RNase. We also identify F-box genes in the vicinity of the S-RNase, to determine which GSI system is present in Rosa. Expression, phylogenetic analyses, polymorphism levels, and evidence for selection favoring diversification of F-box genes within an S-haplotype, suggest multiple F-box genes as the S-pollen component. Rosaceae GSI evolution is discussed in the context of these findings.
Results
R. chinensis and R. multiflora Rosaceae S-RNase lineage genes
S-RNases code for basic proteins (isoelectric point (IP) above 8)5, 24, 33, 34, 37, that present two conserved amino acid patterns (pattern 1 and 236), are expressed in pistils (where the rejection of self-pollen occurs during the growth of pollen tubes), stigma, and flowering buds, and present signs of diversifying selection5, 24, 33, 34, 36, 37. Of the 80 S-RNase like sequences identified in R. chinensis (55) and R. multiflora (25) genomes (Table 1; Supplementary Table S1), 41 code for putative proteins with a IP above 7 (Table 1; Supplementary Table S2). Phylogenetic analyses of these sequences, and 34 reference sequences from24 (Supplementary File S1), revealed that the sequences labeled Rchinensis3_16, Rchinensis2_32_2, and Rchinensis1_1-Rchinensis2_12, do not belong to the Rosaceae S-RNase lineage (Fig. 1). None of the Rosa S-RNase lineage sequences cluster with Malus/Pyrus S-RNases. Two groups of sequences Rmultiflora_4, Rmultiflora_8, and Rchinensis1_3-Rchinensis2_27 (gene 1), and Rchinensis2_16-Rchinensis3_17-Rchinensis4_40 (gene 2), cluster with Prunus S-RNases (Fig. 1), suggesting that one of them may represent the Rosa S-RNase gene. In R. chinensis these sequences are located on chromosome 3, where the S-locus has been identified44, 45. These sequences code for putative proteins with IP above 8, they have two putative introns, and amino acid pattern 2 conserved, features typical of S-RNases. Gene 1 sequences also present high levels of synonymous polymorphism (27.9%) as the S-RNases36. Although in this group of sequences pattern 1 shows a violation at the first pattern position (Y instead of [FST]), an identical pattern is observed in Solanaceae for the S-RNase Nicotiana tomentosiformis XP_018625910_1 (this sequence clusters with other Solanaceae S-RNases in phylogenetic analyses; data not shown). Because gene 2 is represented by one sequence, we cannot address levels of diversity. Nevertheless, the presence of identical sequences in the two R. chinensis genomes analyzed, suggests low levels of diversity. The putative SRNase26, SRNase30, and SRNase36 present IP below 8, and the first two sequences cluster with Prunus and Malus S-lineage 1 genes, and SRNase36 clusters with the Malus S-lineage 2 gene. In Prunus and Malus GSI, these sequences are not involved in specificity determination.
Table 1.
Summary of the S-RNase like sequences identified in the R. chinensis, and R. multiflora genomes.
| Rosa genome | Number of sequences | ||||||
|---|---|---|---|---|---|---|---|
| Total | Excluding pseudogenes | Presenting | |||||
| Both motifs | Both motifs and IP ≥ 7 | Only 2nd motif | Only 2nd motif and IP > 7 | Only 1st motif | |||
| R. chinensis Assembly-CDS (Rchinensis1) | 12 | 12 | 10 | 5 | − | − | − |
| R. chinensis Assembly-ORF (Rchinensis2) | 33 | 26 | 26 | 19 | − | − | − |
| R. chinensis GDR-CDS (Rchinensis3) | 18 | 18 | 17 | 11 | − | − | − |
| R. chinensis GDR-ORF (Rchinensis4) | 44 | 22 | 22 | 13 | − | − | − |
| R. multiflora | 34 | 25 | 18 | 14 | 4 | 4 | 3 |
Figure 1.

Bayesian phylogenetic tree, showing the relationship of the R. chinensis and R. multiflora S-RNase like sequences with Fragaria, Prunus, and Malus S-RNase lineage genes. For the R. chinensis sequences the chromosomal (Chr) location is given. The tree was rooted with MDP0000267606A T2- RNase, not involved in GSI24. In bold are the Rosa sequences that cluster with Prunus S-RNases, that could represent the S-RNase gene. Numbers below the branches represent posterior credibility values above 70.
Rchinensis1_3–Rchinensis2_27 is expressed in pistil and ovary tissue, like the S-RNases
The S-RNase gene is highly expressed in pistils, in stigmas and styles of flowers at anthesis, but also shows low expression in entire flower buds. It should be noted that S-RNase lineage genes, can also show a similar pattern of expression, since this expression is inferred to be the ancestral expression of the S-RNase lineage genes24. Therefore, it is not surprising that SRNase30, and SRNase36, show expression in pistil and ovary (Fig. 2), as previously reported44. SRNase26 shows no expression in the tissues here analyzed, as well as those used in44. Rchinensis1_3-Rchinensis2_27, shows a similar expression to SRNase30, but with high levels of expression in pistil and ovary, and low expression in stamen (Fig. 2). Rchinensis2_16-Rchinensis3_17-Rchinensis4_40 is not expressed in the tissues here analyzed. These results are compatible with Rchinensis1_3-Rchinensis2_27 gene, being the S-pistil gene determining GSI specificity.
Figure 2.
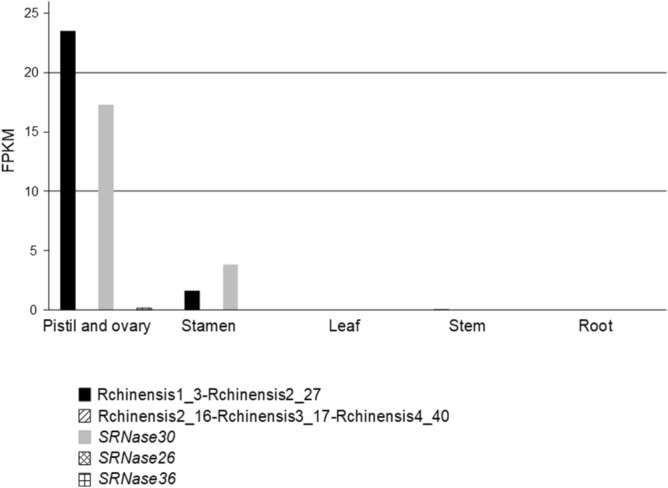
R. chinensis expression levels (FPKM) for the two Prunus S-RNase lineage genes, as well as the three previous putative S-RNases, in pistil and ovary, stamen, leaf, stem, and root tissues.
Rosa S-RNases show evidence for positively selected amino acid positions
To address if the Rosa sequences here identified as the S-locus pistil gene are the subject of diversifying selection, a feature of the S-RNase gene, we first identified and annotated sequences similar to S-RNases from other self-incompatible Rosa genomes with low coverage (R. moschata, R. laevigata, R. rugosa, R. persica, R. xanthina, R. minutifolia, R. odorata, R. arvenses, and R. majalis; Supplementary Table S1). Phylogenetic analyses using sequences presenting an IP above 7 covering at least the exon where motif 2 is located (Supplementary Fig. S1A,B; Supplementary File S2), together with the Rosa S-RNase sequences here identified, and the reference sequences from24, revealed four additional Rosa S-RNases (Rarvensis_11, Rminutifolia_20, Rmoschata17_14, and Rmoschata17_30; Supplementary Fig. S1A,B). Similar analyses with the sequences covering the motif 1, revealed four sequences (Rodorata07_39; Rmoschata08_12; Rminutifolia_7; and Rarvensis_6; Supplementary Fig. S2; Supplementary File S3) that also cluster with Rosa S-RNases sequences. Because the phylogenetic relationship of sequences Rarvensis_11 and Rarvensis_6 is similar, they may represent two exons of the same gene from the same S-haplotype, and thus were treated as such. The same applies to sequences Rminutifolia_20 and Rminutifolia_7, as well as sequences Rmoschata17_14 and Rmoschata17_34. Using these sequences together with Rchinensis1_3-Rchinensis2_27, Rmultiflora_4, and Rmultiflora_8, we identify 21 amino acid sites under positive selection (results available at http://bpositive.i3s.up.pt/ in the project named Rosa S-locus genes; BP2018000004) by performing codeML51 analyses. These amino acid sites are, in principle, responsible for GSI specificity24, 35, 52.The location of these sites at the predicted 3D structure is mostly around the active site pocket region (Fig. 3), as observed in other Rosaceae and Solanaceae species25, 52.
Figure 3.
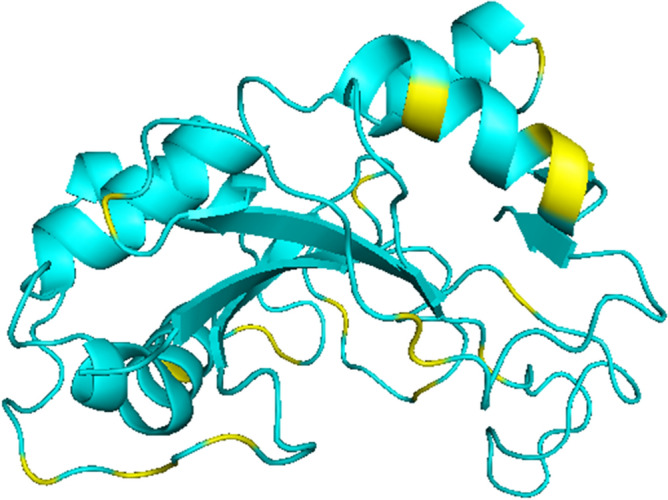
Positively selected amino acid sites, highlighted in yellow on the predicted 3D structure of the R. chinensis S-RNase.
R. arvensis genotype–phenotype association experiments
For 12 R. arvensis accessions, obtained from a breeding program of siblings53, S-haplotype was deduced either through hand-pollinated tests (Supplementary Fig. S3A,B), allowing to perform co-segregation experiments of the S2-allele. An amplification product with the expected size (300 bp; see Material and Methods) was obtained in six (Ose (S1 S2), Url (S4 S5), E200 (Ose x Wid (S3 S6); S2 S3), E404 (E200 x Ose; S1 S2), E435 (E200 x Wid; S2 S6), and E893 (E459 x Url; S1 S5) individuals. The sequence of these amplification products, reveled two types of identical sequences. One obtained from Ose, E200, E404, and E435 individuals, those individuals having the S2-RNase (GenBank acc. Numbers MW452856–MW452859). The other sequence type was obtained from Url and E893, those individuals having the S5-RNase (GenBank acc. numbers MW452860 and MW452861). These results support co-segregation of the S-locus with the S2-RNase genotypes here surveyed. Therefore, the S-RNase gene here identified is on the S-locus region.
Identification of the Rosa S-pollen F-box genes
The R. rugosa S-locus F-box gene (RrSLF; KY446808), expressed in pollen tissue and phylogenetically related with Prunus SFB gene, was reported as a putative Rosa S-pollen gene46. It should be noted that other F-box genes not involved in S-pollen specify determination are also expressed in pollen, as well as in other tissues24. The expression of RrSLF gene in other tissues has not, however, been addressed46. This gene has 99% homology at nucleotide level with a R. chinensis sequence (CM009583.1) located on chromosome 2, but the Rosa S-locus is located on chromosome 3 (44, 45, and this work). Furthermore, all S-pollen genes described in Rosaceae, Solanaceae, and Plantaginaceae are intronless genes, and the R. chinensis RrSLF (PRQ51373) and R. multiflora (Rmu_sc0016061.1;BDJD01015883.1) orthologous genes have one intron. The orthologous gene has been identified in all low coverage Rosa genomes, except R. majalis, covering the entire coding region. Low levels of divergence (0.036 for synonymous and 0.009 for non-synonymous divergence respectively, after Jukes and Cantor correction; N = 10) are obtained for this gene. This is in contrast with the Prunus SFB gene, that presents levels of variability above 20%6, 7, 11, 36. Therefore, RrSLF is not the S-pollen gene.
In R. chinensis chromosome 3 there are 30 SFB/SLFL/SFBB like genes (called Fbox − and + according to the 5′ or 3′ position relative to the S-RNase, respectively; Supplementary Table S3; Supplementary File S4). In the two R. multiflora scaffolds where the S-RNase is located, there are five such genes (Supplementary Table S3). The phylogenetic analyses of the R. chinensis and R. multiflora SFB/SLFL/SFBB like genes together with Prunus SFB and SLFL genes, M. domestica S1-SFBBs, Petunia and Nicotiana SLF sequences, and A. thaliana F-box/kelch-repeat, shown in Fig. 4 (Supplementary File S4), revealed that the Rchinensis_F-box + 13 gene clusters with Prunus SFB gene. This gene is expressed in all tissues here analyzed (Fig. 5), but the S-pollen gene(s) are mainly expressed in anthers /pollen only7, 10, 12–16, 21, 23–25, and thus is unlikely to be the S-pollen gene. Furthermore, using a 1188 bp gene region obtained from eight Rosa genomes (R. arvensis, R. laevigata, R. moschata, R. xantina, R. rugosa, R. odorata, R. multiflora, and R. chinensis), low average levels of divergence (0.0321 for synonymous and 0.0128 for non-synonymous divergence respectively, after Jukes and Cantor correction) are observed. Moreover, this gene is the neighbor F-box gene of the T2-RNase Rchinensis1_8-Rchinensis2_25, that does not cluster with Prunus or Maleae S-RNases (Fig. 1). Therefore, Rchinensis_F-box + 13 gene is not involved in S-pollen GSI specificity determination.
Figure 4.
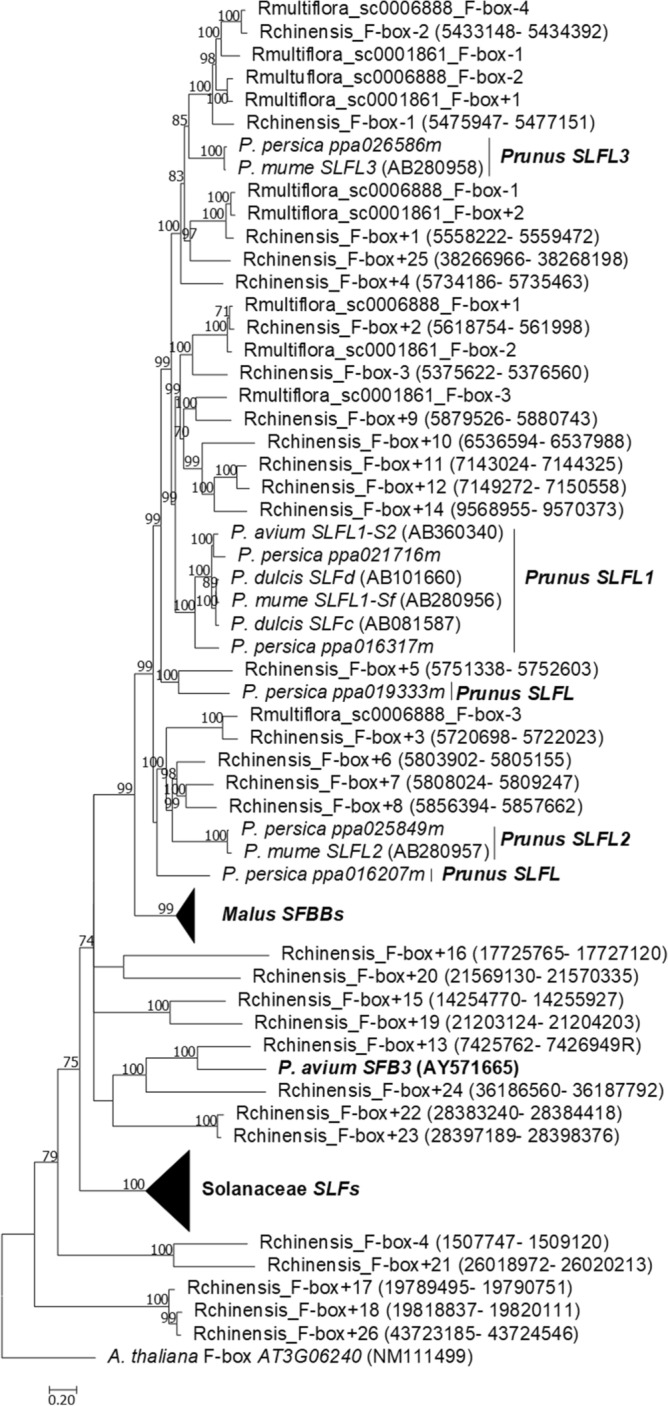
Bayesian phylogenetic tree, showing the relationship of the SFB/SLFL/SFBB like genes (called Fbox − and + according to the 5′ or 3′ position relative to the S-RNase) from R. chinensis chromosome 3, R. multiflora sc0006888, and R. multiflora sc0001861, with M. domestica SFBBs, Prunus SFBs and SLFLs, and Solanaceae SLFs. The tree was rooted with A. thaliana F-box/kelch-repeat, not involved in GSI. Numbers below the branches represent posterior credibility values above 70.
Figure 5.

R. chinensis expression levels (FPKM) for the F-box genes located on chromosome 3, in pistil and ovary, stamen, leaf, stem, and root tissues.
The F-box genes in the vicinity of the R. chinensis and R. multiflora S-RNase gene cluster, with high support, with different Prunus SLF genes (Fig. 4), that are a sister group of Malus SFBBs24. In R. chinensis 14 of these genes (Rchinensis_F-box-3 up to Rchinensis_F-box + 11) show expression in stamen only, compatible with being S-pollen genes (Fig. 5). Furthermore, the R. multiflora orthologs of R. chinensis are not in the same order relatively to the S-RNase gene (Fig. 4), showing that this region is highly rearranged, as observed in the Malus/Pyrus/Sorbus S-locus region13, 16, 18–20, 24, 25. This is surprising since we identified the Rosa S-RNase as belonging to the Prunus S-lineage, and in Prunus there is a single S-pollen gene. In Prunus the S-pollen gene presents levels of diversity similar to the S-RNase gene11, 29. Therefore, we also determined levels of synonymous and non-synonymous divergence for the F-box genes surrounding the R. chinensis S-RNase (Rchinensis_F- box-1 and Rchinensis_F- box + 1, used as query in a blastn to identify contigs containing the orthologous genes in the low coverage Rosa genomes). The low levels of synonymous and non-synonymous divergence for Rchinensis_F-box-1 (0.070 and 0.005 respectively), and Rchinensis_F- box + 1 (0.112 and 0.02129 respectively) are incompatible with the hypothesis that one of them is determining Rosa S-pollen specificity. Therefore, multiple F-box genes must be involved in Rosa pollen GSI specificity determination, as in Malus/Pyrus/Sorbus and Solanaceae species. Using the 14 R. chinensis F-box sequences in the vicinity of the S-RNase, that are expressed in stamen, we find evidence for positively selected amino acid sites, as expected if these genes are involved in S-pollen specificity determination (results available at http://bpositive.i3s.up.pt/ under project Rosa S-locus genes; BP2018000004). On the predicted 3D structure, these amino acid sites are located in the same regions (Fig. 6) as those observed for Petunia S-pollen genes26. Evidence for positive selection is also observed for the five F-box genes of the R. multiflora scaffold sca0006888 (results available at http://bpositive.i3s.up.pt/ under project Rosa S-locus genes; BP2018000004). Therefore, the data suggests that Rosa S-pollen specificity is determined by multiple F-box genes, like in Malus/Pyrus/Sorbus and Solanaceae species.
Figure 6.
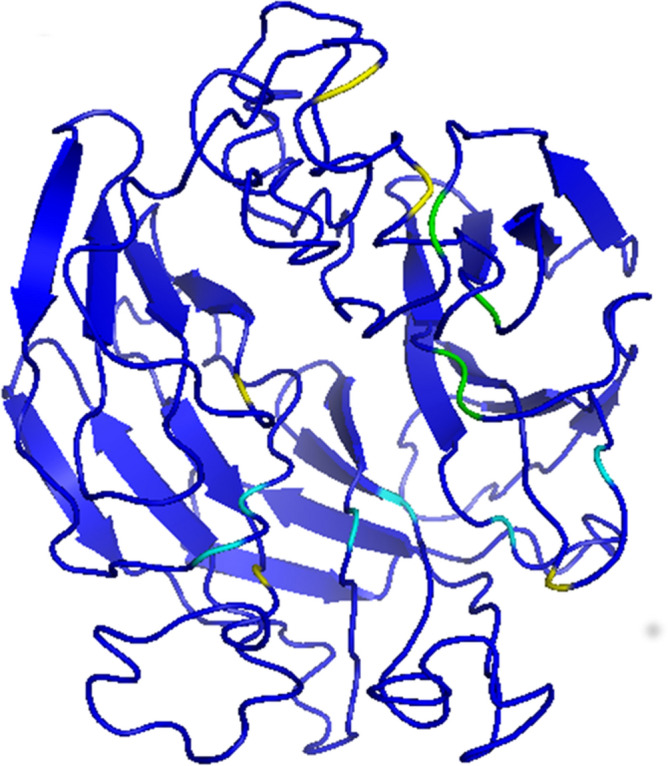
Positively selected amino acid sites on the predicted 3D structure of the R. chinensis F-box located in the 5` region of the S-RNase. In yellow are highlighted those sites that appear as positively selected in the two datasets analyzed (14 and 13 R. chinensis F-box genes in the vicinity of the S-RNase that are expressed in stamen), in green those that are located in a region not analyzed when 14 F-box genes are considered, and in blue those that change due to alignment gaps in the two datasets analyzed.
Discussion
In Rosa, there are very important traits of horticultural interest such as flower development, architecture, senescence, scent biosynthesis and emission, ease of reproduction, and resistance to biotic and abiotic stresses, that have been selected only once during the history of rose selection, and incorporated into many rose varieties44, 45, 48. Indeed, only 10 species have contributed to the genetic make-up of most of the modern rose cultivars, and some old and popular cultivars, such as ‘Old Blush’ have dominated the history of rose selection40. This Chinese rose from the Song dynasty (960–1279) conveys several desirable characters such as recessive reblooming habit, recessive lack of prickles (stem) and dominant flower doubleness44, 45, 48, all co-segregating with the S-locus region. The characterization of the S-locus region here performed is thus, very important in order to help breeding selection, and the control of genetic diversity.
The Rosa S-locus is composed of a S-RNase gene that belongs to the Prunus lineage (Fig. 1). This gene shows all expected features of a S-pistil gene since it shows expression in pistil and ovary (Fig. 2), evidence for diversifying selection (Fig. 3), and co-segregation with the S-locus. In Fragaria, that also belongs to the Rosoideae subfamily (the two genera have been diverging for about 50 million years54), a Prunus lineage S-RNase gene has been also reported as the putative S-pistil gene24. This suggests that the Rosoideae GSI system could be of the self-recognition type, with one S-pollen gene, as in Prunus (24 and references therein). Nevertheless, the Rosa F-box gene that clusters with the Prunus SFB gene (Fig. 4) is not the neighbor of the S-RNase gene, as observed in Prunus, and presents expression (Fig. 5) and polymorphism levels incompatible with being the S-pollen gene. In Rosa there are multiple F-box genes in the vicinity of the S-RNase, that show expression compatible with being the S-pollen gene (Fig. 5), and evidence for diversifying selection for F-box genes within a S-haplotype (Fig. 6). These are the expected features of S-pollen genes in a non-self-recognition system, as reported in Malus/Sorbus/Pyrus13, 16, 18–20, 24, 25 and Solanaceae species15, 22, 23, 26, 30–32. Therefore, the Rosa S-locus region encompasses a large region, as reported for other species presenting non-self recognition systems, and this could explain why in rose several traits of horticultural interest are linked to the S-locus. It should be noted that the Rosa S-locus region may be highly rearranged, since we could not align the contig containing the S-RNase gene from R. chinensis with those from R. multiflora as well as the two contigs containing the S-RNase gene from R. multiflora. This observation has been previously reported for the two available R. chinensis genomes, that could not be aligned in this region (conceivably carrying two different S-haplotypes; see Fig. 1 of55). Pollen transcriptome analyses of multiple S-haplotypes are, however needed to determine how many F-box genes in the vicinity of the Rosa S-RNase are involved in pollen GSI specificity, as performed in Malus25 and Petunia21.
Since multiple S-pollen genes are determining Rosa S-pollen specificity, as in Maleae species, this suggests that the ancestral Rosaceae GSI system was of the non-self-recognition system type. This implies that during evolution the Malus S-RNase lineage, that does not cluster with the Rosa S-RNase, has been recruited de novo from a duplicate of the ancestral S-RNase gene. Moreover, it implies that the Prunus S-pollen gene, that does not cluster with Rosa S-pollen genes, evolved de novo from an unrelated F-box gene.
Low levels of variation have been reported in roses but the S-locus region, being large and under balancing selection, may help retain a substantial fraction of the variability. Indeed, the evolution within the genus Rosa, occurred via interspecific hybridization, allopolyploidization and genetic reticulation among sympatric species56. In Europe, the recent post-glaciation period expedited the process of speciation, as the northward extension of the biotopes increased. The diploid R. arvensis holds a special place in this process, since, although it belongs to the synstylae group, has a substantial genomic promiscuity with the polyploid caninae group56, 57, that may also help maintain diversity levels. The inbred strains that are being established for R. arvensis53 are a starting point to investigate the diversity of the S-locus in European wild roses. Mutants breaking down self-incompatibility, will also help refine the S-locus effect on roses molecular diversity.
Methods
Identification of putative S-RNase like sequences in Rosa genomes
The annotations (CDS) of two R. chinensis genomes (available at NCBI RefSeq database (www.ncbi.nlm.nih.gov) and GDR database (https://www.rosaceae.org) Supplementary Table S1) were downloaded, and the sequences showing similarity with reference S-RNases assigned as Rchinensis1 and Rchinensis3, respectively. Moreover, the corresponding genome sequences were downloaded as a FASTA file, and the S-RNase sequences here annotated labeled as Rchinensis2 and Rchinensis4, respectively. R. multiflora genome (NCBI assembly database, Supplementary Table S1) was also downloaded as a FASTA file, since annotations are not available for this species. To find and extract protein encoding segments larger than 100 bp, we have used getorf, using the emboss Docker image available at pegi3s Bioinformatics Docker images project (htttps:pegi3s.github.io/dockerfiles). Then, we selected the protein encoding segments that show similarity with reference S-RNase sequences, using tblastx (Expect value (e) < 0.05), as implemented in SEDA58, 59, using as query Prunus S3-RNase (AJ298312), Malus Sh-RNase (AB032247), and Fragaria putative S-RNase (gi561957436, gi561674690 and gi561985884)24. Based on this information, we manually annotated the corresponding genome region to identify the exons of each gene. For each putative gene we obtained the corresponding amino acid sequence to calculate IP, using ExPASy software60.
We used the same protocol for the genomes here assembled using the short reads of nine Rosa genomes downloaded from NCBI SRA database (Supplementary Table S1). In this case, we used FastQC to evaluate read quality, and Cutadapt to trim reads61, and ABySS 2.062 for the de novo assembly, using the Docker images available at pegi3s Bioinformatics Docker images project (htttps://pegi3s.github.io/dockerfiles).
Identification of the F-box genes located in R. chinensis chromosome 3 and R. multiflora scaffolds where the S-RNase is located
The protocol presented for the S-RNase like sequences was also used to obtain F-box genes located in R. chinensis chromosome 3, and for the two R. multiflora scaffolds where the S-RNase gene has been identified, using as query P. avium SFB3 (AAT72121.1), P. avium SLFL1 (BAG12295.1), and M. domestica SFBB3-beta (BAF47180.1).
Phylogenetic analyses
Phylogenetic analyses of the R. chinensis and R.multiflora S-RNase like sequences and F-box like genes were performed using sequences aligned with MUSCLE alignment algorithm, as implemented in ADOPS63. Only codons with a support value above 2 were used for phylogenetic reconstruction. Bayesian trees were obtained using MrBayes 3.1.264 as implemented in the ADOPS pipeline63. The Generalised Time-Reversible (GTR) model of sequence evolution was implemented in the analyses, allowing for among-site rate variation and a proportion of invariable sites. Third codon positions were allowed to have a gamma distribution shape parameter different from that of first and second codon positions. Two independent runs of 1,000,000 generations with four chains each, were carried out. The average standard deviation of split frequencies was always ~ 0.01 and the potential scale reduction factor for every parameter was ~ 1.00, showing that convergence was achieved. Trees were sampled every 100th generation and the first 5000 samples were discarded (burn-in). The tree was converted to Newick format using the Format Conversion website (http://phylogeny.lirmm.fr/phylo_cgi/data_converter.cgi) and edited using Mega765.
The phylogenetic analyses of the low coverage Rosa genomes (Supplementary Table S1) sequences were performed with Mega765, using ClustalW alignment algorithm, Neighbor-Joining method, bootstrap test with 10,000 replicates, the p-distance method for computing the evolutionary distances, and pairwise deletion since sequences can have different sizes (Supplementary Table S2).
Expression of R. chinensis S-RNase like, and F-box genes located on chromosome 3 in pistil and ovary, stamen, leaf, stem and root transcriptomes
To estimate expression of the Rosa S- RNase like sequences located on chromosome 3, we use RNA-seq data from R. chinensis pistil and ovary, stamen, leaf, stem, and root transcriptomes (Supplementary Table S4). We used FastQC to evaluate read quality, and Cutadapt to trim reads61. FPKM values were estimated using the RSEM method, as implemented in Trinity66, using the R. chinensis Refseq CDS, and the R. chinensis S-RNase like and F-box sequences located on chromosome 3.
R. arvensis genotype–phenotype association experiments
Genomic DNA was extracted from leaves of 12 R. arvensis individuals, for which the haplotypes were predicted according to their parents and progeny (Supplementary Fig. S3A,B), using the method of67. PCRs were performed using the genomic DNA and primers RA-F 5′ GGAAGCCARACTGAAGAT 3′ and RA-R 5´AGCATCACAGTYTCGATCA 3′, designed for conserved regions of the putative Rosa S-RNase sequences here identified. Standard amplification conditions were 35 cycles of denaturation at 94 °C for 30 s, 52 °C for 30 s for primer annealing, and primer extension at 72 °C for 2 min. The amplification products with the expected size (353 bp) for individuals Ose, E200, E404, Url, and E893 were cloned using the TA cloning kit (Invitrogen, Carlsbad, CA). For each individual, the insert of 16 colonies was cut separately with DdeI, and HinfI restriction enzymes, and only one restriction pattern was observed, and thus three colonies only were sequenced. The ABI PRISM BigDye cycle-sequencing kit (Perkin Elmer, Foster City, CA), and specific primers, or the primers for the M13 forward and reverse priming sites of the pCR2.1 vector, were used to prepare the sequencing reactions. Sequencing runs were performed by STABVIDA (Lisboa, Portugal).
Identification of positively selected amino acid sites, their location on the crystal structure, and polymorphism levels
For the six sequences identified as putative Rosa S-RNases, we inferred positively selected amino acid sites, using codeML51, as implemented in ADOPS63, using Muscle as the alignment method. Such analyses where also performed for the 14 R. chinensis F-box sequences that are in the vicinity of the S-RNase, that cluster with Prunus SLFL, and that are expressed in stamen. Since the inclusion of Rchinensis_F-box-3 gene sequence excludes from the analyses a large fraction of the 3´region, we performed these analyses after removing this sequence. codeML analyses were also performed for five R. multiflora sc0006888 F-box sequences in the vicinity of the S-RNase. The details of the analyses can be seen at the B + database (bpositive.i3s.up.pt68; Rosa S-locus genes BP2018000004). Model comparisons were M2a-M1a and M8-M7. We consider as positively selected those amino acid sites that show a probability higher than 90% for both naive empirical Bayes (NEB) or Bayes empirical Bayes (BEB) methods.
To visualize these positions in the 3D structure, for the S-RNase (translation of Rchinensis1_3–Rchinensis2_27; Supplementary File S1) we first identified the signal peptide using SignalIP (http://www.cbs.dtu.dk/services/SignalP/) website tool available at ExPASy60. After removing the signal peptide, the 3D structure was modeled by I-Tasser69, and the model with the highest C-score value used. The same methodology was used for the putative S-pollen gene (translation of R chinensis_F box-1; Supplementary File S4), but in this case after removing the F-box domain (the first 60 amino acid positions). All structural images were produced using PyMOL (The PyMOL Molecular Graphics System, Version 1.7.4 Schrödinger, LLC.).
Levels of polymorphism were obtained with DnaSp70.
Supplementary Information
Acknowledgements
This work was financed by the National Funds through FCT—Fundação para a Ciência e a Tecnologia, I.P., under the project UIDB/04293/2020, and the Centre National de la Recherche Scientifique (CNRS).
Author contributions
J.V., and C.P.V. designed the research. P. H. collected the plant material and designed the R. arvensis genetics. J.V., J. P., A.G., J. L., S. R., and C.P.V. performed the genomic and transcriptomic analyses. J. P., and C.P.V. performed the molecular work. All authors reviewed and approved the manuscript.
Data availability
All data generated or analyzed during this study are included in this published article (and its Supplementary Data File S1–S4). R. arvensis S2- and S5-RNase are deposited in GenBank, acc. Numbers MW452856-MW452861.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-83243-8.
References
- 1.De Nettancourt D. Incompatibility in angiosperms. Berlin: Springer; 1977. [Google Scholar]
- 2.Igic B, Lande R, Kohn JR. Loss of self-incompatibility and its evolutionary consequences. Int. J. Plant Sci. 2008;169:93–104. doi: 10.1086/523362. [DOI] [Google Scholar]
- 3.Roalson EH, McCubbin AG. S-RNases and sexual incompatibility: Structure, functions, and evolutionary perspectives. Mol. Phylogenet. Evol. 2003;29:490–506. doi: 10.1016/s1055-7903(03)00195-7. [DOI] [PubMed] [Google Scholar]
- 4.McClure B. Darwin's foundation for investigating self-incompatibility and the progress toward a physiological model for S-RNase-based SI. J. Exp. Bot. 2009;60:1069–1081. doi: 10.1093/jxb/erp024. [DOI] [PubMed] [Google Scholar]
- 5.Nowak MD, Davis AP, Anthony F, Yoder AD. Expression and trans-specific polymorphism of self-incompatibility RNases in coffea (Rubiaceae) PLoS ONE. 2011;6:e21019. doi: 10.1371/journal.pone.0021019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Entani T, et al. Comparative analysis of the self-incompatibility (S-) locus region of Prunus mume: identification of a pollen-expressed F-box gene with allelic diversity. Genes Cells. 2003;8:203–213. doi: 10.1046/j.1365-2443.2003.00626.x. [DOI] [PubMed] [Google Scholar]
- 7.Ushijima K, et al. Structural and transcriptional analysis of the self-incompatibility locus of almond: identification of a pollen-expressed F-box gene with haplotype-specific polymorphism. Plant Cell. 2003;15:771–781. doi: 10.1105/tpc.009290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qiao H, et al. The F-box protein AhSLF-S2 controls the pollen function of S-RNase-based self-incompatibility. Plant Cell. 2004;16:2307–2322. doi: 10.1105/tpc.104.024919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tsukamoto T, Ando T, Watanabe H, Marchesi E, Kao TH. Duplication of the S-locus F-box gene is associated with breakdown of pollen function in an S-haplotype identified in a natural population of self-incompatible Petunia axillaris. Plant Mol. Biol. 2005;57:141–153. doi: 10.1007/s11103-004-6852-6. [DOI] [PubMed] [Google Scholar]
- 10.Cheng J, Han Z, Xu X, Li T. Isolation and identification of the pollen-expressed polymorphic F-box genes linked to the S-locus in apple (Malus × domestica) Sex. Plant Reprod. 2006;19:175. doi: 10.1007/s00497-006-0034-4. [DOI] [Google Scholar]
- 11.Nunes MD, Santos RA, Ferreira SM, Vieira J, Vieira CP. Variability patterns and positively selected sites at the gametophytic self-incompatibility pollen SFB gene in a wild self-incompatible Prunus spinosa (Rosaceae) population. New Phytol. 2006;172:577–587. doi: 10.1111/j.1469-8137.2006.01838.x. [DOI] [PubMed] [Google Scholar]
- 12.Hua Z, Meng X, Kao TH. Comparison of Petunia inflata S-Locus F-box protein (Pi SLF) with Pi SLF like proteins reveals its unique function in S-RNase based self-incompatibility. Plant Cell. 2007;19:3593–3609. doi: 10.1105/tpc.107.055426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sassa H, et al. S locus F-box brothers: multiple and pollen-specific F-box genes with S haplotype-specific polymorphisms in apple and Japanese pear. Genetics. 2007;175:1869–1881. doi: 10.1534/genetics.106.068858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wheeler D, Newbigin E. Expression of 10 S-class SLF-like genes in Nicotiana alata pollen and its implications for understanding the pollen factor of the S locus. Genetics. 2007;177:2171–2180. doi: 10.1534/genetics.107.076885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kubo K, et al. Collaborative non-self recognition system in S-RNase-based self-incompatibility. Science. 2010;330:796–799. doi: 10.1126/science.1195243. [DOI] [PubMed] [Google Scholar]
- 16.Minamikawa M, et al. Apple S locus region represents a large cluster of related, polymorphic and pollen-specific F-box genes. Plant Mol. Biol. 2010;74:143–154. doi: 10.1007/s11103-010-9662-z. [DOI] [PubMed] [Google Scholar]
- 17.Tsukamoto, T., Hauck, N. R., Tao, R., Jiang, N. & Iezzoni, A. F. Molecular and genetic analyses of four nonfunctional S haplotype variants derived from a common ancestral S haplotype identified in sour cherry (Prunus cerasus L.). Genetics184, 411–427. 10.1534/genetics.109.109728 (2010). [DOI] [PMC free article] [PubMed]
- 18.Kakui H, et al. Sequence divergence and loss-of-function phenotypes of S locus F-box brothers genes are consistent with non-self recognition by multiple pollen determinants in self-incompatibility of Japanese pear (Pyrus pyrifolia) Plant J. 2011;68:1028–1038. doi: 10.1111/j.1365-313X.2011.04752.x. [DOI] [PubMed] [Google Scholar]
- 19.Okada K, et al. Related polymorphic F-box protein genes between haplotypes clustering in the BAC contig sequences around the S-RNase of Japanese pear. J. Exp. Bot. 2011;62:1887–1902. doi: 10.1093/jxb/erq381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aguiar B, et al. Patterns of evolution at the gametophytic self-incompatibility Sorbus aucuparia (Pyrinae) S pollen genes support the non-self recognition by multiple factors model. J. Exp. Bot. 2013;64:2423–2434. doi: 10.1093/jxb/ert098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Williams JS, Der JP, dePamphilis CW, Kao TH. Transcriptome analysis reveals the same 17 S-locus F-box genes in two haplotypes of the self-incompatibility locus of Petunia inflata. Plant Cell. 2014;26:2873–2888. doi: 10.1105/tpc.114.126920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Williams JS, et al. Four previously identified Petunia inflata S-locus F-box genes are involved in pollen specificity in self-incompatibility. Mol. Plant. 2014;7:567–569. doi: 10.1093/mp/sst155. [DOI] [PubMed] [Google Scholar]
- 23.Kubo K, et al. Gene duplication and genetic exchange drive the evolution of S-RNase-based self-incompatibility in Petunia. Nat. Plants. 2015;1:14005. doi: 10.1038/nplants.2014.5. [DOI] [PubMed] [Google Scholar]
- 24.Aguiar B, et al. Convergent evolution at the gametophytic self-incompatibility system in Malus and Prunus. PLoS ONE. 2015;10:e0126138. doi: 10.1371/journal.pone.0126138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pratas MI, et al. Inferences on specificity recognition at the Malusxdomestica gametophytic self-incompatibility system. Sci. Rep. 2018;8:1717. doi: 10.1038/s41598-018-19820-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vieira J, et al. Predicting specificities under the non-self gametophytic self-incompatibility recognition model. Front Plant. Sci. 2019;10:879. doi: 10.3389/fpls.2019.00879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ikeda K, et al. Primary structural features of the S haplotype-specific F-box protein, SFB Prunus. Sex. Plant Reprod. 2004;16:235–243. doi: 10.1007/s00497-003-0200-x. [DOI] [Google Scholar]
- 28.Matsumoto, D. & Tao, R. Distinct Self-recognition in the Prunus S-RNase-based gametophytic self-incompatibility system. Hortic. J.85. 10.2503/hortj.MI-IR06 (2016).
- 29.Tsukamoto, T. et al. Genetic and molecular characterization of three novel S-haplotypes in sour cherry (Prunus cerasus L.). J. Exp. Bot.59, 3169–3185. 10.1093/jxb/ern172 (2008). [DOI] [PMC free article] [PubMed]
- 30.Sun P, Li S, Lu D, Williams JS, Kao TH. Pollen S-locus F-box proteins of Petunia involved in S-RNase-based self-incompatibility are themselves subject to ubiquitin-mediated degradation. Plant J. 2015;83:213–223. doi: 10.1111/tpj.12880. [DOI] [PubMed] [Google Scholar]
- 31.Li J, et al. Electrostatic potentials of the S-locus F-box proteins contribute to the pollen S specificity in self-incompatibility in Petunia hybrida. Plant J. 2017;89:45–57. doi: 10.1111/tpj.13318. [DOI] [PubMed] [Google Scholar]
- 32.Wu L, et al. Use of domain-swapping to identify candidate amino acids involved in differential interactions between two allelic variants of type-1 S-locus F-box protein and S3-RNase in petunia inflata. Plant Cell Physiol. 2018;59:234–247. doi: 10.1093/pcp/pcx176. [DOI] [PubMed] [Google Scholar]
- 33.Igic B, Kohn JR. Evolutionary relationships among self-incompatibility RNases. Proc. Natl. Acad. Sci. USA. 2001;98:13167–13171. doi: 10.1073/pnas.231386798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Steinbachs JE, Holsinger KE. S-RNase-mediated gametophytic self-incompatibility is ancestral in eudicots. Mol. Biol. Evol. 2002;19:825–829. doi: 10.1093/oxfordjournals.molbev.a004139. [DOI] [PubMed] [Google Scholar]
- 35.Vieira J, Morales-Hojas R, Santos RA, Vieira CP. Different positively selected sites at the gametophytic self-incompatibility pistil S-RNase gene in the Solanaceae and Rosaceae (Prunus, Pyrus, and Malus) J. Mol. Evol. 2007;65:175–185. doi: 10.1007/s00239-006-0285-6. [DOI] [PubMed] [Google Scholar]
- 36.Vieira J, Fonseca NA, Vieira CP. An S-RNase-based gametophytic self-incompatibility system evolved only once in eudicots. J. Mol. Evol. 2008;67:179–190. doi: 10.1007/s00239-008-9137-x. [DOI] [PubMed] [Google Scholar]
- 37.Ramanauskas K, Igic B. The evolutionary history of plant T2/S-type ribonucleases. PeerJ. 2017;5:e3790. doi: 10.7717/peerj.3790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tao R, Iezzoni AF. The S-RNase-based gametophytic self-incompatibility system in Prunus exhibits distinct genetic and molecular features. Sci. Hortic. 2010;124:423–433. doi: 10.1016/j.scienta.2010.01.025. [DOI] [Google Scholar]
- 39.Akagi T, Henry IM, Morimoto T, Tao R. Insights into the prunus-specific S-RNase-based self-incompatibility system from a genome-wide analysis of the evolutionary radiation of S locus-related F-box genes. Plant Cell Physiol. 2016;57:1281–1294. doi: 10.1093/pcp/pcw077. [DOI] [PubMed] [Google Scholar]
- 40.Gudin S. Rose: genetics and breeding. Plant Breed. Rev. 2000;17:159–189. doi: 10.1002/9780470650134.ch3. [DOI] [Google Scholar]
- 41.Cole P, Melton B. Self-compatibility and cross-compatibility relationships among genotypes and between ploidy of the rose. J. Am. Soc. Hortic. Sci. 1986;111:122–125. [Google Scholar]
- 42.Heslop-Harrison Y, Shivanna KR. The receptive surface of the angiosperms stigma. Ann. Bot. 1977;41:1233–1258. doi: 10.1093/oxfordjournals.aob.a085414. [DOI] [Google Scholar]
- 43.Ueda Y, Akimoto S. Cross- and self-compatibility in various species of the Genus Rosa. J. Hortic. Sci. Biotechnol. 2001;76:392–395. doi: 10.1080/14620316.2001.11511382. [DOI] [Google Scholar]
- 44.Hibrand Saint-Oyant, L. et al. A high-quality genome sequence of Rosa chinensis to elucidate ornamental traits. Nat. Plants4, 473–484. doi:10.1038/s41477-018-0166-1 (2018). [DOI] [PMC free article] [PubMed]
- 45.Debener T, et al. Genetic and molecular analyses of key loci involved in self incompatibility and floral scent in roses. Acta Hort. 2010;870:183–190. doi: 10.17660/ActaHortic.2010.870.24. [DOI] [Google Scholar]
- 46.Wei Y, et al. Cloning and bioinformatics analysis of Rosa rugose S Locus F-Box Gene (RrSLF) Am. J. Plant Sci. 2017;8:1550–1559. doi: 10.4236/ajps.2017.87107. [DOI] [Google Scholar]
- 47.Yokoya K, Roberts AV, Mottley J, Lewis R, Brandham PE. Nuclear DNA amounts in roses. Ann. Bot. 2000;85:557–561. doi: 10.1006/anbo.1999.1102. [DOI] [Google Scholar]
- 48.Bendahmane M, Dubois A, Raymond O, Bris ML. Genetics and genomics of flower initiation and development in roses. J. Exp. Bot. 2013;64:847–857. doi: 10.1093/jxb/ers387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Raymond O, et al. The Rosa genome provides new insights into the domestication of modern roses. Nat. Genet. 2018;50:772–777. doi: 10.1038/s41588-018-0110-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nakamura N, et al. Genome structure of Rosa multiflora, a wild ancestor of cultivated roses. DNA Res. 2018;25:113–121. doi: 10.1093/dnares/dsx042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 52.Vieira J, Ferreira PG, Aguiar B, Fonseca NA, Vieira CP. Evolutionary patterns at the RNase based gametophytic self-incompatibility system in two divergent Rosaceae groups (Maloideae and Prunus) BMC Evol. Biol. 2010;10:200. doi: 10.1186/1471-2148-10-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Heitzler P. Rosa arvensis as a possible genetic model. Acta Hortic. 2019;1232:25–32. doi: 10.17660/ActaHortic.2019.1232.5. [DOI] [Google Scholar]
- 54.Xiang Y, et al. Evolution of Rosaceae fruit types based on nuclear phylogeny in the context of geological times and genome duplication. Mol. Biol. Evol. 2017;34:262–281. doi: 10.1093/molbev/msw242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smulders MJM, et al. In the name of the rose: A roadmap for rose research in the genome era. Hortic. Res. 2019;6:65. doi: 10.1038/s41438-019-0156-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhu ZM, Gao XF, Fougere-Danezan M. Phylogeny of Rosa sections Chinenses and Synstylae (Rosaceae) based on chloroplast and nuclear markers. Mol. Phylogenet. Evol. 2015;87:50–64. doi: 10.1016/j.ympev.2015.03.014. [DOI] [PubMed] [Google Scholar]
- 57.De Cock, K. Genetic diversity of wild roses (Rosa spp.) in Europe, with an in-depth morphological study of Flemish populations. Ghent University. Faculty of Bioscience Engineering, Ghent, Belgium. (2008).
- 58.López-Fernández H, et al. Bioinformatics protocols for quickly obtaining large-scale data sets for phylogenetic inferences. Interdiscip. Sci. Comput. Life Sci. 2019;11:1–9. doi: 10.1007/s12539-018-0312-5. [DOI] [PubMed] [Google Scholar]
- 59.López-Fernández H, et al. SEDA: a desktop tool suite for FASTA files processing. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020 doi: 10.1109/TCBB.2020.3040383. [DOI] [PubMed] [Google Scholar]
- 60.Artimo P, et al. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 2012;40:W597–603. doi: 10.1093/nar/gks400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.61Martin, M. CUTADAPT removes adapter sequences from high-throughput sequencing reads. EMBnet.journal17, doi:10.14806/ej.17.1.200 (2011).
- 62.Jackman, S. D. et al. ABySS 2.0: resource-efficient assembly of large genomes using a Bloom filter. Genome Res.27, 768–777. 10.1101/gr.214346.116 (2017). [DOI] [PMC free article] [PubMed]
- 63.Reboiro-Jato D, et al. ADOPS–Automatic Detection Of Positively Selected Sites. J Integr Bioinform. 2012;9:200. doi: 10.2390/biecoll-jib-2012-200. [DOI] [PubMed] [Google Scholar]
- 64.Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754–755. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- 65.Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol.33, 1870–1874. 10.1093/molbev/msw054 (2016). [DOI] [PMC free article] [PubMed]
- 66.Haas BJ, et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013;8:1494–1512. doi: 10.1038/nprot.2013.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ingram GC, et al. Dual role for fimbriata in regulating floral homeotic genes and cell division in Antirrhinum. EMBO J. 1997;16:6521–6534. doi: 10.1093/emboj/16.21.6521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vazquez N, et al. Large scale analyses and visualization of adaptive amino acid changes projects. Interdiscip. Sci. 2018;10:24–32. doi: 10.1007/s12539-018-0282-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yang J, et al. The I-TASSER Suite: protein structure and function prediction. Nat. Methods. 2015;12:7–8. doi: 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003;19:2496–2497. doi: 10.1093/bioinformatics/btg359. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article (and its Supplementary Data File S1–S4). R. arvensis S2- and S5-RNase are deposited in GenBank, acc. Numbers MW452856-MW452861.