

Abstract
The European LeukemiaNet (ELN) recommendations for diagnosis and management of acute myeloid leukemia (AML) have become an important tool to assess patients’ prognosis and guide treatment. We tested the prognostic impact of the 2017 ELN classification in a large cohort of 863 AML patients aged <60 years similarly treated on Cancer and Leukemia Group B/Alliance for Clinical Trials in Oncology studies. Based on multivariable models within each ELN genetic-risk group, we identified additional gene mutations that may refine the 2017 ELN risk classification. BCOR- or SETBP1-mutated Favorable-risk patients with non-core-binding-factor AML and IDH-mutated Adverse-risk patients had Intermediate-risk outcomes. Outcomes of NPM1/WT1 co-mutated patients and those of ZRSR2-mutated patients resembled outcome of Adverse-risk patients. Moreover, FLT3-ITDhigh allelic ratio conferred adverse rather than Intermediate-risk irrespective of the NPM1 mutation status, and DNMT3A mutations associated with very poor survival. Applying these refinements reclassified 9% of current Favorable risk patients and 53% of current Intermediate-risk patients to the Adverse-risk group, with similar poor survival as current Adverse-risk patients. Furthermore, 4% of current Favorable-risk patients and 9% of Adverse-risk patients were reclassified to the Intermediate-risk group.
Introduction
The first edition of the European LeukemiaNet (ELN) recommendations for diagnosis and management of acute myeloid leukemia (AML) in adults,1 authored by a panel of international experts, was published in 2010 and was incorporated into patient care. The 2010 ELN recommendations contained a standardized system for reporting cytogenetic and molecular alterations in studies that correlated such genetic findings with treatment outcomes of AML patients to facilitate meaningful comparisons among clinical studies.1 Subsequently, several studies have established the clinical utility of the 2010 ELN genetic-risk classification.2–4 The need for such a classification has been evident because of the heterogeneity of outcomes of AML patients, with only 35–40% of patients aged <60 years achieving long-term survival.5 Importantly, the increasing knowledge about molecular features of AML, including gene mutations, gene-expression changes and epigenetic modifications, highlights the heterogeneity of the disease and helps identify patients that are likely to do well or, conversely, do poorly with standard chemotherapy.6–11 Moreover, these molecular markers may be potential targets for anti-leukemic therapies and outcome of these patients may change with time following introduction of new agents.12,13 Following the advances in understanding of the molecular landscape of AML, the ELN expert panel revised their classification in 2017, and, among other changes, added additional gene markers to the 2017 ELN genetic-risk categories.14
The aim of our study was to test how well the 2017 ELN classification stratifies AML patients into genetic-risk categories in, to our knowledge, the hitherto largest group of de novo adult AML patients aged <60 years with centrally reviewed cytogenetics who were treated with intensive cytarabine/daunorubicin-based chemotherapy. In addition, the patients were molecularly analyzed using a panel of 81 cancer- and leukemia-associated genes, which enabled us to investigate additional genetic markers that might refine the current ELN classification. Given the integration of the 2017 ELN classification into practice and study protocols, improvements to the classification may lead to earlier identification of patients that are unlikely to respond to standard treatment and may need different induction therapy, or of patients that are likely to experience relapse after initial response and need additional postremission therapy in first complete remission (CR).
Patients and methods
Patients, treatment, and cytogenetic studies
Pretreatment bone marrow (BM) or blood samples containing ≥20% leukemic blasts were obtained from 863 adults aged <60 years who were diagnosed with de novo AML (excluding acute promyelocytic leukemia). Patients with AML evolving from an antecedent hematologic disorder and those with treatment-related AML were excluded. The patients were treated on Cancer and Leukemia Group B (CALGB) trials15–22 described in the Supplementary Information. Because most CALGB treatment protocols did not allow performing allogeneic stem cell transplantation (allo-SCT) in first CR on study and patients receiving allo-SCT had to be taken off protocol, which resulted in either lack of or incomplete follow-up data, we excluded all patients who received allo-SCT from the current study. CALGB is now part of Alliance for Clinical Trials in Oncology (Alliance). Cytogenetic analyses of pretreatment BM and/or blood samples were performed by CALGB/Alliance-approved institutional laboratories, and results confirmed by central karyotype review.23 Patients provided study-specific written informed consent to participate in treatment studies, CALGB 8461 (cytogenetic studies), CALGB 9665 (leukemia tissue banking) and CALGB 20202 (molecular studies). Study protocols were in accordance with the Declaration of Helsinki and approved by the institutional review boards at each center.
Statistical analysis
Definitions of the clinical endpoints–CR, disease-free survival (DFS) and overall survival (OS)–are provided in the Supplementary Information. Clinical and biological characteristics were compared using the Fisher’s exact and Wilcoxon rank-sum tests for categorical and continuous variables, respectively. For CR, we calculated P-values using Fisher’s exact test. For time-to-event analyses, we calculated survival estimates using the Kaplan-Meier method and compared groups using the log-rank test (P-values presented in the Figures with Kaplan-Meier curves).24 A limited backward selection technique was used to build the final multivariable models for achievement of CR, DFS and OS.25 We used logistic regression for modeling CR and Cox proportional hazard regression for modeling DFS and OS for univariable and multivariable outcome analyses. For a given gene mutation to be considered as a possible addition to the current risk group-defining markers, it had to significantly impact on outcome in at least one of the multivariable analyses. Furthermore, the CR rate or 3-year DFS or OS rates of patients carrying such mutation would have to be similar to those of patients belonging to a different risk-group in the 2017 ELN classification. The dataset was locked on January 10, 2019. Data collection and statistical analyses were performed by the Alliance Statistics and Data Center using SAS 9.4 and TIBCO Spotfire S+ 8.2.
Molecular analyses
The mutational status of 80 protein-coding genes was determined centrally at The Ohio State University by targeted amplicon sequencing using the MiSeq platform (Illumina, San Diego, CA; see Supplementary Information for details).26 The presence or absence of FLT3 internal tandem duplications (FLT3-ITD), as well as quantification of the FLT3-ITD to FLT3 wild-type allelic ratio (low/no vs high defined as ratio ≥0.5), were determined as previously described.27 In addition, biallelic CEBPA mutations were determined by Sanger sequencing,28 bringing the total number of genes analyzed in our study to 81.
Results
Clinical characteristics and outcome of AML patients according to the 2017 ELN classification
The pretreatment characteristics of 863 patients classified into the 2017 ELN genetic-risk groups are depicted in Table 1. The median age of all patients was 45 years (range, 17–59 years) and 45% of patients were women. Almost one-half (49%) of the patients belonged to the ELN Favorable-risk group, whereas 22% and 29% of patients belonged to the Intermediate- and Adverse-risk groups, respectively.
Table 1.
Pretreatment clinical characteristics and outcome of younger patients with de novo acute myeloid leukemia assigned to the genetic-risk groups according to the 2017 ELN classification
| Characteristic | All patients n=863 | Favorable-risk n=423 | Intermediate-risk n=189 | Adverse-risk n=251 | P-valuea |
|---|---|---|---|---|---|
| Age, years | 0.54 | ||||
| Median | 45 | 44 | 44 | 46 | |
| Range | 17–59 | 17–59 | 17–59 | 17–59 | |
| Sex, n (%) | 0.01 | ||||
| Male | 473 (55) | 226 (53) | 92 (49) | 155 (62) | |
| Female | 390 (45) | 197 (47) | 97 (51) | 96 (38) | |
| Race, n (%) | 0.27 | ||||
| White | 723 (85) | 360 (87) | 154 (82) | 209 (85) | |
| Nonwhite | 124 (15) | 53 (13) | 33 (18) | 38 (15) | |
| Hemoglobin, g/dl | 0.37 | ||||
| Median | 9.2 | 9.2 | 9.4 | 9.1 | |
| Range | 2.3–25.1 | 2.3–25.1 | 2.9–14.4 | 4.6–14.8 | |
| Platelet count, x109/l | 0.07 | ||||
| Median | 53 | 49 | 56 | 58 | |
| Range | 4–648 | 4–648 | 9–445 | 8–341 | |
| WBC count, x109/l | 0.01 | ||||
| Median | 24.3 | 24.2 | 28.6 | 20.7 | |
| Range | 0.4–475.0 | 0.4–475.0 | 0.9–308.8 | 0.6–320.0 | |
| Blood blasts, % | 0.05 | ||||
| Median | 56 | 52 | 62 | 56 | |
| Range | 0–99 | 0–97 | 0–98 | 0–99 | |
| Bone marrow blasts, % | <0.001 | ||||
| Median | 66 | 63 | 74 | 67 | |
| Range | 0–97 | 2–97 | 10–96 | 0–97 | |
| Extramedullary involvement, n (%) | 222 (27) | 127 (31) | 46 (26) | 49 (20) | 0.001 |
| Complete remission, n (%) | 655 (76) | 389 (92) | 145 (77) | 121 (48) | <0.001 |
| Disease-free survival | <0.001 | ||||
| Median, years | 1.3 | 4.7 | 0.8 | 0.7 | |
| % Disease-free at 3 years (95% CI) | 38 (34–42) | 53 (48–57) | 22 (16–29) | 10 (5–16) | |
| Overall survival | <0.001 | ||||
| Median, years | 2.0 | 12.4 | 1.4 | 0.9 | |
| % Alive at 3 years (95% CI) | 44 (40–47) | 64 (59–68) | 31 (25–38) | 19 (14–24) | |
Abbreviations: CI, confidence interval; n, number; ELN, European LeukemiaNet; WBC, white blood cell.
P-values for categorical variables are from Fisher’s exact test, P-values for continuous variables are from the Wilcoxon rank sum test and they are comparing the three risk groups: Favorable, Intermediate and Adverse. P-values for the time to event variables are from the log-rank test and they are comparing the three risk groups: Favorable, Intermediate and Adverse.
Patients classified in the Adverse-risk group were predominantly male (male vs female, 62% vs 38%, P=0.01), whereas the Favorable- and Intermediate-risk groups had similar male to female ratios (53% vs 47% and 49% vs 51%, respectively). Adverse-risk group patients presented with lower white blood cell (WBC) counts compared with Favorable- and Intermediate-risk patients (Adverse-risk, median WBC 20.7 ×109/l vs 24.2 and 28.6 ×109/l respectively, P=0.01; Table 1).
With respect to clinical outcome, the CR rate of our patient cohort was 76%. There were 38% of patients disease-free and 44% of patients alive 3 years after diagnosis, with median DFS and OS of 1.3 years and 2.0 years, respectively. As expected, the patient outcomes differed according to the ELN genetic-risk groups to which the patients were assigned (Table 1). The CR rate of Favorable-risk patients was 92%, compared with 77% and 48% CR rates of Intermediate-risk and Adverse-risk patients (P<0.001). Fifty-three percent of patients belonging to the Favorable-risk group were disease-free and 64% were alive 3 years after diagnosis, compared with 22% disease-free and 31% alive patients in the Intermediate-risk group, and only 10% and 19%, respectively, of patients classified in the Adverse-risk group (both P<0.001).
Mutational landscape of AML patients
We detected 2354 mutations, with an average of 3 mutations per patient (range, 0–9). The frequencies of gene mutations detected in ≥2% of patients in the entire cohort are provided in Supplementary Table S1. Forty-four genes were mutated in ≥2% of patients in one of the ELN groups (Supplementary Table S1). In addition to the ELN risk group-defining mutations [Favorable-risk, biallelic CEBPA mutations and NPM1 mutations with no FLT3-ITD or low FLT3-ITD allelic ratio (FLT3-ITDlow); Intermediate-risk, NPM1 and FLT3-ITD with high allelic ratio (FLT3-ITDhigh); Adverse-risk, FLT3-ITDhigh and NPM1 wild-type, RUNX1, ASXL1 (in the absence of favorable genetic features) and TP53],14 the frequencies of several additional gene mutations differed between the ELN genetic-risk groups (Supplementary Table S1, Figure 1).
Figure 1.
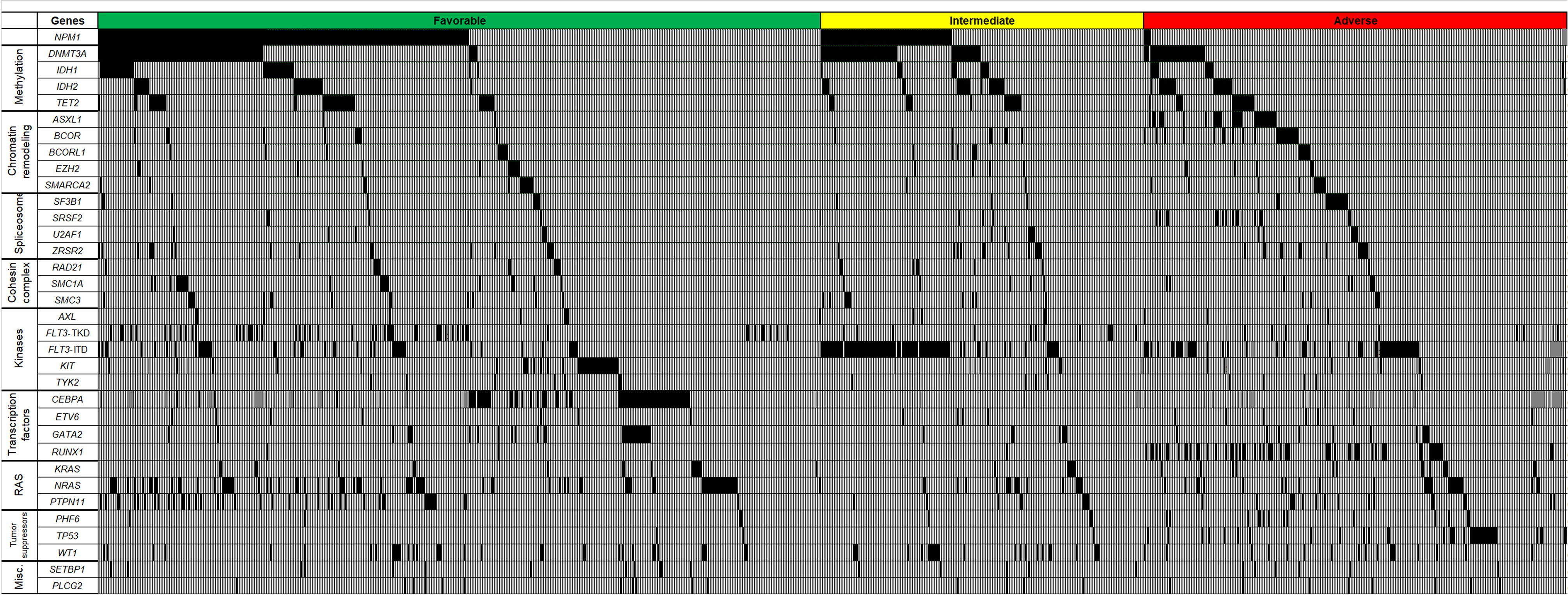
Oncoprint of genes most frequently mutated in younger acute myeloid leukemia patients categorized into genetic-risk groups according to the 2017 European LeukemiaNet (ELN) classification (green color, Favorable-risk; yellow, Intermediate-risk; red, Adverse-risk). Each column represents an individual patient. Black color indicates genes found to be mutated, light gray indicates wild-type status of the gene, and dark gray indicates missing values.
In the Favorable-risk group, DNMT3A and NRAS mutations were frequently found, in 24% and 20% of patients, respectively (Supplementary Table S1, Figure 1). In the Intermediate-risk group, mutations in DNMT3A were the most frequent mutations (33%) that were observed aside from the group-defining mutations outlined above, followed by mutations in IDH2 and WT1, which were present in 13% and 12% of Intermediate-risk patients, respectively. In the Adverse-risk group, mutations in DNMT3A (found in 14% of patients), NRAS (found in 11% of patients) as well as IDH2 and BCOR (both detected in 9% of patients) were the most frequent besides risk-group defining mutations indicated above.
We also assessed frequencies of mutations categorized into functional groups in the 2017 ELN genetic-risk categories (Supplementary Table S2). RAS pathway mutations were more frequently found in the Favorable-risk group than in the Intermediate-risk and Adverse-risk groups (Favorable, 35%, Intermediate, 19%, Adverse, 24%; P<0.001, 3-way comparison). Mutations in kinase and methylation-related genes were most frequently observed in the Intermediate-risk group (Favorable, 33% and 39%, Intermediate, 56% and 51%, Adverse, 35% and 27%, respectively; P<0.001 for each 3-way comparison). Mutations in genes encoding for spliceosomes, transcription factors and tumor suppressors (Favorable, 8%, 23% and 10%, Intermediate, 11%, 9% and 14%, Adverse, 20%, 36% and 27%, respectively; P<0.001 for each 3-way comparison) were more common in the Adverse-risk group compared with the Favorable and Intermediate-risk groups.
Impact of gene mutations on the outcome of patients classified according to the 2017 ELN genetic-risk groups
We next evaluated whether any of the detected gene mutations associated with better or worse outcomes within each 2017 ELN genetic-risk group and might potentially be used to refine the current ELN classification. We performed univariable and multivariable outcome analyses for the achievement of CR, DFS and OS within each of the 2017 ELN genetic-risk groups and identified gene mutations within each group that associated with outcome in multivariable analyses (Table 2). Within the Favorable-risk group outcome analyses were performed separately for core-binding factor AML (CBF-AML) patients and non-CBF-AML patients.
Table 2.
Multivariable outcome analyses of younger patients with de novo acute myeloid leukemia assigned to the genetic-risk groups according to the 2017 ELN genetic risk classification
| Variables in final model | Complete remission | Disease-free survival | Overall survival | |||
|---|---|---|---|---|---|---|
| OR (95% CI) | Pa | HR (95% CI) | Pb | HR (95% CI) | Pb | |
| Favorable-risk groupc | ||||||
| Non-CBF-AML patients | ||||||
| BCOR, mutated vs wild-type | 2.61 (1.14-6.00) | 0.02 | 2.57 (1.12-5.89) | 0.03 | ||
| IDH1, mutated vs wild-type | 1.67 (1.07-2.59) | 0.02 | 1.81 (1.17-2.80) | 0.007 | ||
| PTPN11, mutated vs wild-type | 1.54 (1.02-2.33) | 0.04 | ||||
| SETBP1, mutated vs wild-type | 2.27 (1.10-4.66) | 0.03 | 2.38 (1.20-4.73) | 0.01 | ||
| WT1, mutated vs wild-type | 0.28 (0.10-0.75) | 0.01 | 1.91 (1.14-3.20) | 0.01 | 2.78 (1.72-4.50) | <0.001 |
| ZRSR2, mutated vs wild-type | 0.24 (0.07-0.89) | 0.03 | 2.12 (1.03-4.37) | 0.04 | 2.48 (1.31-4.69) | 0.005 |
| Age, 10-year increase | 1.21 (1.03-1.42) | 0.02 | ||||
| WBC count, 50-unit increase | 1.19 (1.06-1.33) | 0.004 | ||||
| CBF-AML patients | ||||||
| KIT, mutated vs wild-type | 3.31 (1.94-5.63) | <0.001 | 2.12 (1.18-3.81) | 0.01 | ||
| Intermediate-risk groupc | ||||||
| DNMT3A, mutated vs wild-type | 3.02 (1.99-4.59) | <0.001 | 1.85 (1.31-2.62) | <0.001 | ||
| WT1, mutated vs wild-type | 0.31 (0.12-0.81) | 0.02 | 2.15 (1.17-3.94) | 0.01 | 1.98 (1.23-3.16) | 0.005 |
| Age, 10-year increase | 1.31 (1.12-1.52) | <0.001 | ||||
| Hemoglobin level, 1-unit increase | 1.24 (1.02-1.51) | 0.03 | 0.85 (0.77-0.94) | 0.002 | ||
| Platelet count, 50-unit increase | 0.79 (0.68-0.93) | 0.004 | 0.86 (0.75-0.98) | 0.03 | ||
| Sex, male vs female | 0.33 (0.15-0.70) | 0.004 | ||||
| Adverse-risk groupc | ||||||
| IDH2, mutated vs wild-type | 0.57 (0.34-0.95) | 0.03 | ||||
| NRAS, mutated vs wild-type | 3.32 (1.80-6.11) | <0.001 | 1.74 (1.15-2.61) | 0.008 | ||
| TET2, mutated vs wild-type | 1.94 (1.18-3.18) | 0.009 | ||||
| Age, 10-year increase | 1.18 (1.04-1.33) | 0.008 | ||||
| Hemoglobin level | 1.24 (1.05-1.46) | 0.01 | ||||
| Platelet count, 50-unit increase | 0.81 (0.69-0.96) | 0.01 | ||||
| Race, white vs nonwhite | 0.32 (0.15-0.72) | 0.005 | ||||
| WBC count, 50-unit increase | 1.20 (1.06-1.37) | 0.006 | ||||
Variables in the table are in alphabetical order and separated by gene mutations and other clinical characteristics. Multivariable analyses were done separately for CBF- and non-CBF patients within the Favorable-risk group.
Abbreviations: AML, acute myeloid leukemia, CI, confidence interval; CBF, core-binding factor; CR, complete remission; DFS, disease-free survival; ELN, European LeukemiaNet; HR, hazard ratio; OR, odds ratio; OS, overall survival; WBC, white blood cell.
NOTE. An OR <1 (>1) means lower (higher) CR rate for first category listed of a dichotomous variable or higher values of a continuous variable. A HR >1 (<1) corresponds to a higher (lower) risk for first category listed of a dichotomous variable or higher values of a continuous variable. A limited backward selection technique was used to build the final model for achievement of CR, DFS and OS. Variables considered in the model were variables that were significant at the likelihood ratio test P-value <0.20 from the univariable models (detailed in the Supplementary Information).
P-values for CR attainment were determined by logistic regression.
P-values for DFS and OS were determined using Cox proportional hazards regression.
Please see Supplementary Material for variables considered in the multivariable models
Within the non-CBF-AML patients of the Favorable-risk group, the presence of WT1 and ZRSR2 mutations associated with lower odds of achieving a CR (P=0.01, odds ratio [OR]=0.28; P=0.03, OR=0.24, respectively). Mutations in BCOR (P=0.02, hazard ratio [HR]=2.61), IDH1 (P=0.02, HR=1.67), PTPN11 (P=0.04, HR=1.54), SETBP1 (P=0.03, HR=2.27), WT1 (P=0.01, HR=1.91) and ZRSR2 (P=0.04, HR=2.12) associated with a worse DFS, whereas mutations in BCOR (P=0.03, HR=2.57), IDH1 (P=0.007, HR=1.81), SETBP1 (P=0.01, HR=2.38), WT1 (P<0.001, HR=2.78), and ZRSR2 (P=0.005, HR=2.48) associated with a shorter OS. Within the CBF-AML patients of the Favorable-risk group, the presence of KIT mutations associated with shorter DFS (P<0.001, HR=3.31) and OS (P=0.01, HR=2.12).
In the Intermediate-risk group, mutations in WT1 associated with lower odds of achieving a CR (P=0.02, OR=0.31). Mutations in DNMT3A and WT1 associated with a shorter DFS (P<0.001, HR=3.02; and P=0.01, HR=2.15, respectively) and OS (P<0.001, HR=1.85; and P=0.005, HR=1.98, respectively). Among patients belonging to the Adverse-risk group, those harboring IDH2 mutations had longer OS (P=0.03, HR=0.57) than patients with wild-type IDH2. The presence of NRAS mutations associated with reduced DFS (P<0.001, HR=3.32) and OS (P=0.008, HR=1.74), and TET2-mutated patients had shorter OS (P=0.009, HR=1.94) than patients without these mutations. Not a single NRAS- or TET2-mutated patient belonging to the Adverse-risk group was disease-free after one year, and no patient was still alive 3 years after diagnosis of AML.
Gene mutations identified in multivariable models may refine the 2017 ELN classification
To test whether inclusion of the gene mutations identified in the multivariable models might refine the ELN genetic risk classification, we compared the outcomes of patients harboring these gene mutations with the outcomes of patients belonging to another ELN risk-group (e.g., CR, DFS and OS of Favorable-risk patients with a given mutation were compared with the CR, DFS and OS of patients in the Intermediate- and Adverse-risk categories). In the non-CBF-AML Favorable-risk group, the CR rates of patients harboring mutations in WT1 were similar to the CR rates of patients belonging to the Intermediate group (Figure 2a; WT1-mutated Favorable-risk vs Intermediate-risk, 75% vs 77%, P=0.81). With respect to DFS, non-CBF-AML Favorable-risk patients with mutations in BCOR, SETBP1, or WT1 had similar DFS to that of the Intermediate-risk patients (3-year rates, BCOR-mutated Favorable-risk vs Intermediate-risk, 29% vs 22%; P=0.72; SETBP1-mutated Favorable-risk vs Intermediate-risk, 20% vs 22%, P=0.88; WT1-mutated Favorable-risk vs Intermediate-risk, 24% vs 22%, P=0.72). The DFS of patients with ZRSR2 mutations actually resembled those of the Adverse-risk group (ZRSR2-mutated Favorable-risk vs Adverse-risk, 11% vs 10%, P=0.33).
Figure 2.

Line graphs showing the impact of gene mutations found to be prognostically significant in multivariable models for a Non-core-binding factor acute myeloid leukemia (non-CBF-AML) patients in the 2017 European LeukemiaNet (ELN) Favorable-risk group, b CBF-AML Favorable-risk patients, c Intermediate-risk patients and d Adverse-risk patients on complete remission rates (left panel), 3-year disease-free survival rates (middle panel) and 3-year overall survival rates (right panel). The percentages are depicted for both wild-type (wt) and mutated (mut) patients, relative to the median rates according to the ELN genetic-risk classification (blue vertical bars). * mutated versus Intermediate-risk, P≤0.05 (adjusted for multiple comparisons); ** mutated versus Adverse-risk, P≤0.05 (adjusted for multiple comparisons).
With respect to OS, again the 3-year rates of Favorable-risk BCOR-, SETBP1- and WT1-mutated patients closely resembled those of Intermediate-risk patients (3-year rates, BCOR-mutated 38% vs 32%, P=0.91; SETBP1-mutated 45% vs 32%, P=0.67; WT1-mutated 29% vs 32%, P=0.69), whereas ZRSR2-mutated patients had OS similar to OS of the Adverse-risk group (23% vs 19%, P=0.46). Among the CBF-AML patients in the Favorable-risk group, those harboring KIT mutations had a DFS similar to DFS of the Intermediate-risk patients (3-year rates, 24% vs 22%, P=0.62; Figure 2b). However, the OS of KIT-mutated Favorable-risk CBF-AML patients was significantly better than OS of Intermediate-risk patients (3-year rates, 53% vs 32%, P=0.01), suggesting that the former should remain classified as having Favorable-risk.
In the ELN Intermediate-risk group, patients harboring WT1 mutations had CR rates, DFS and OS that were similar to those of ELN Adverse-risk patients (CR rates, 58% vs 48%, P=0.40; 3-year DFS rates, 14% vs 10%, P=0.80; 3-year OS rates, 19% vs 19%, P=0.99; Figure 2c). Of the 14 WT1-mutated patients who achieved a CR, all but one experienced relapse of their disease. Furthermore, DNMT3A-mutated patients in the ELN Intermediate-risk group had DFS and OS that resembled those of Adverse-risk patients (3-year DFS rates, 10% vs 10%, P=0.67; 3-year OS rates, 19% vs 19%, P=0.28). Patients classified in the ELN Adverse-risk group who harbored IDH2 mutations had outcome similar to that of Intermediate-risk patients with regard to CR rates (65% vs 77%, P=0.30), DFS (20% vs 22%, P=0.48) and OS (30% vs 32%, P=.89; Figure 2d), which supports their inclusion in the Intermediate-risk group.
High allelic ratio of FLT3-ITD confers short DFS and OS irrespective of co-occurring NPM1 mutation
The inclusion of FLT3-ITD allelic ratio as a criterion for genetic-risk group assignment (with only high allelic ratio being considered as an adverse prognosticator) was one of the major changes in the 2017 ELN classification update. Patients with mutated NPM1 without FLT3-ITD or with FLT3-ITDlow are now classified as having Favorable genetic risk, whereas wild-type NPM1 patients without FLT3-ITD or with FLT3-ITDlow, and NPM1-mutated patients with FLT3-ITDhigh belong to the Intermediate-risk group. Finally, patients who harbor FLT3-ITDhigh without concomitant NPM1 mutation are classified as having Adverse-risk. To test the validity of these criteria in our data, we assessed the prognostic impact of NPM1 mutations within the FLT3-ITD-negative/FLT3-ITDlow patient group, as well as the impact of NPM1 mutations on outcome of FLT3-ITDhigh patients.
Among FLT3-ITD-negative/FLT3-ITDlow patients, those harboring a NPM1 mutation had a higher CR rate (88% vs 74%, P<0.001), and a longer OS (3-year OS rates, 60% vs 45%, P<0.001) than patients with wild-type NPM1 (Figure 3a). There was no significant difference in DFS between NPM1-mutated and NPM1 wild-type patients. Within the FLT3-ITDhigh cohort, patients harboring a NPM1 mutation had higher CR rates than those without a NPM1 mutation (81% vs 51%, P=0.003). However, there was no significant difference in either DFS or OS between patients with and without NPM1 mutations (Figure 3b), indicating that the negative prognostic impact of FLT3-ITDhigh may outweigh the positive prognostic impact of NPM1 mutations with respect to those survival endpoints.
Figure 3.

a Disease-free survival (DFS, upper graph) and overall survival (OS, lower graph) of NPM1-mutated (blue line) and NPM1-wild-type (red line) patients who were either FLT3-ITD-negative or harbored FLT3-ITDlow. b DFS (upper graph) and OS (lower graph) of NPM1-mutated (blue line) and NPM1-wild-type (red line) patients who harbored FLT3-ITDhigh.
Co-occurrence of WT1 and NPM1 mutations confers especially poor outcome
We analyzed the prognostic impact of combinations of mutations in the WT1 and NPM1 genes in patients classified in the non-CBF-AML Favorable-risk or Intermediate-risk groups. Patients with WT1 mutations had lower CR rates than those with wild-type WT1 regardless of whether they had NPM1 mutations (WT1-mutated/NPM1-mutated, 69%; WT1-mutated/NPM1 wild-type, 68%; WT1 wild-type/NPM1-mutated, 90%; WT1 wild-type/NPM1 wild-type, 83%; P=0.001). However, the adverse prognostic impact of WT1 mutations with respect to DFS and OS was dependent on coexisting NPM1 mutations (Figure 4). The outcome of WT1-mutated/NPM1-mutated patients was, unexpectedly, much worse than that of WT1-mutated/NPM1-wild-type patients: the former had 3-year DFS rates of 5% versus 46% (P=0.008) and 3-year OS rates of 9% versus 47% (P=0.002). Thus, in addition to the FLT3-ITD high allelic ratio, the coexistence of WT1 and NPM1 mutations considerably alters the positive prognostic impact of NPM1 mutations, making patients harboring such a combination of mutations candidates for more aggressive treatment.
Figure 4.

Kaplan-Meier curves depicting the disease-free (left panel) and overall survival (right panel) of NPM1-mutated/WT1-mutated patients (red line), NPM1-mutated/WT1 wild-type patients (blue line), NPM1 wild-type/WT1-mutated patients (black line), and NPM1 wild-type/WT1 wild-type patients (green line).
DNMT3A mutations negatively impact DFS and OS, but not achievement of CR, in Intermediate-risk patients
Our investigation of the association of DNMT3A mutations with outcome of patients belonging to the Intermediate-risk group revealed that DNMT3A-mutated patients had a CR rate of 83% (compared with 73% of DNMT3A wild-type patients, P=0.21), but that 85% of those DNMT3A-mutated patients who achieved a CR relapsed. Both DFS (3-year rates, 10% vs 29%, P<0.001), and OS (3-year rates, 19% vs 37%, P=0.002) of DNMT3A-mutated patients were shorter than those of DNMT3A wild-type patients. Consequently, DNMT3A-mutated patients without Favorable-risk markers should be considered as having Adverse-risk because of their high relapse rate and poor DFS and OS, despite high likelihood of achieving a CR. Thus, early allo-SCT may be useful for these DNMT3A-mutated patients.
Suggested refinement of the 2017 ELN classification by inclusion of additional molecular markers
Based on the results of our multivariable analyses, as well as analysis of prognostic significance of co-occurring mutations, we propose possible refinement of criteria used for risk group assignment (Table 3). We propose that only those NPM1-mutated non-CBF-AML patients who harbor neither FLT3-ITDhigh nor a WT1 mutation should be considered as having a Favorable risk. In fact, FLT3-ITDhigh patients should always be considered as Adverse-risk, regardless of their NPM1 mutation status. In addition, we identified ZRSR2 mutations as a new marker associated with Adverse-risk outcome. Furthermore, DNMT3A mutations in the absence of Favorable-risk markers were associated with very short DFS and OS rates, and may also be considered as Adverse-risk.
Table 3.
Proposed refinement of the 2017 European LeukemiaNet (ELN) risk classification by additional gene mutations
| Risk category | Genetic abnormality |
|---|---|
| Favorable | t(8;21)(q22;q22.1); RUNX1-RUNX1T1 |
| inv(16)(p13.1q22) or t(16;16)(p13.1;q22); CBFB-MYH11 | |
| Mutated NPM1 without FLT3-ITD or with FLT3-ITDlow | |
| Mutated NPM1 without WT1 mutation | |
| Biallelic mutated CEBPA | |
| Intermediate | Mutated BCORa (without adverse-risk genetic lesions) |
| Mutated SETBP1a(without adverse-risk genetic lesions) | |
| Mutated IDH2b(without adverse-risk genetic lesions) | |
| Wild-type NPM1 without FLT3-ITD or with FLT3-ITDlow (without adverse-risk genetic lesions) | |
| t(9;11)(p21.3;q23.3); MLLT3-KMT2A | |
| Cytogenetic abnormalities not classified as favorable or adverse | |
| Adverse | t(6;9)(p23;q34.1); DEK-NUP214 |
| t(v;11q23.3); KMT2A rearranged | |
| t(9;22)(q34.1;q11.2); BCR-ABL1 | |
| inv(3)(q21.3q26.2) or t(3;3)(q21.3;q26.2); GATA2,MECOM (EVI1) | |
| −5 or del(5q); −7; −17/abn(17p) | |
| Complex karyotype, monosomal karyotype | |
| FLT3-ITDhigh (irrespective of NPM1 mutation status)c | |
| Mutated NPM1 and mutated WT1a | |
| Mutated DNMT3Aa,b | |
| Mutated RUNX1b | |
| Mutated ASXL1b | |
| Mutated TP53 | |
| Mutated ZRSR2a |
Indicated in red color are gene mutations identified in our models whose outcomes resembled those of the groups that they were now added to, and that may refine the current risk stratification
Markers impacting on disease-free and overall survival, but not on the achievement of a complete remission.
These markers should not be used as an adverse prognostic marker if they co-occur with favorable-risk AML subtypes.
FLT3-ITDhigh is defined as ≥0.5 as per ELN guidelines.
Patients originally classified in the non-CBF-AML 2017 Favorable-risk group who harbored mutations in BCOR or SETBP1, and Adverse-risk patients with IDH2 mutations had outcomes that resembled those of patients currently classified in the Intermediate-risk group.
Application of the aforementioned changes to the 2017 ELN non-CBF-AML Favorable-risk group resulted in reclassification of 19 (4%) patients to the revised Intermediate-risk group, and 33 patients (9%) to the revised Adverse-risk group. Notably, of the 189 patients originally classified in the Intermediate-risk group, more than half were reclassified as having Adverse-risk when using the suggested refinement (n=100, 53%). The re-classification of the majority of Intermediate-risk patients was due to either the presence of FLT3-ITDhigh (n=70) and/or DNMT3A mutation (n=60), while the presence of either WT1 or ZRSR2 mutations each accounted for the change of 12% of patients. Conversely, 23 (9%) patients were transferred from the 2017 ELN Adverse-risk group to the modified Intermediate-risk group. Clinical outcome of patients in the refined 2017 ELN classification is shown in Supplementary Table S3, and the Kaplan-Meier curves are shown in Figure 5a. For comparison, Figure 5b contains Kaplan-Meier curves depicting DFS and OS of patients classified according to the original 2017 ELN classification.
Figure 5.

a Kaplan-Meier curves depicting the disease-free (DFS, left panel) and overall survival (OS, right panel) of younger (aged <60 years) adult patients with acute myeloid leukemia classified into proposed Favorable-risk (blue line), Intermediate-risk (black) and Adverse-risk (red) 2017 European LeukemiaNet (ELN) groups after the proposed refinement of the ELN classification. b DFS (left graph) and OS (right graph) of younger patients classified into the original ELN genetic-risk groups.
As a result of the aforementioned reclassification of patients, the 3-year DFS and OS rates of 371 patients constituting the revised Favorable-risk group were slightly better than the respective DFS (3-year rates: 57% vs 53%) and OS (3-year rates: 69% vs 64%) of 423 patients classified in the original 2017 ELN Favorable-risk group. Likewise, the outcome of 131 patients in the new Intermediate-risk group also improved in comparison with that of the original Intermediate-risk group (n=189) with regard to both DFS (3-year rates: 32% vs 22%) and OS (3-year rates: 41% vs 31%). In contrast, DFS (10% vs 10%) and OS (19% vs 19%) of the original (n=251) and revised (n=361) Adverse-risk groups were identical (Table 1 and Supplementary Table S3).
Discussion
The determination of genetic risk in AML patients is a moving target. First, because new gene mutations constantly add new information to the prognostic landscape.6,29–32 Second, a risk classification is based on a standardized treatment received by the analyzed patient group, which means that molecular markers previously considered as adverse or favorable may lose their prognostic significance when novel targeted therapies are used, as exemplified by FLT3 inhibitors,33 or tyrosine kinase inhibitors targeting KIT mutations in CBF-AML.34
Continuous updating of genetic-risk classifications is of special importance for patients that are classified as having Favorable risk, because they respond relatively well to standard chemotherapy, and thus are not being considered for allo-SCT in first CR nor for alternative treatment strategies. Our analysis of a relatively large panel of recurrent mutations in adult AML suggests several gene mutations and mutation combinations as possible important refinements of the existing ELN classification.
Our data suggest WT1 mutations as a possibly important additional marker that should be considered in NPM1-mutated patients, since the presence of both these mutations negatively affects the patients’ DFS and OS. The presence of WT1 mutations has been associated with poor outcomes of AML patients in most,35–38 but not all,39 studies. However, our data suggest that WT1 mutations should be considered in the context of NPM1 mutation status, since it was coexistence of both mutations, not the presence of WT1 mutations alone, that was associated with the worst outcome. Although NPM1-mutated patients receiving standard chemotherapy who also harbored a WT1 mutation had a CR rate of 69% and thus might be considered good candidates for standard consolidation treatment, only 5% of those patients remained disease-free and only 9% were alive 3 years after diagnosis. However, given the relatively small numbers of patients with both NPM1 and WT1 mutations, these findings should be corroborated by future studies. This suggests that patients with WT1 and NPM1 mutations may be good candidates for early allo-SCT. Similarly, DNMT3A-mutated patients, who currently belong to the Intermediate-risk group, have a high likelihood of responding to initial treatment, but 84% of them experienced relapse of their disease and had short DFS and OS similar to patients classified in the Adverse-risk group.
Our findings that the presence of a high FLT3-ITD allelic ratio outweighs the positive prognostic impact of NPM1 mutations, and should be considered as portending Adverse-risk regardless of co-occurring mutations, is in agreement with Boddu et al.40 who found no significant difference in survival of mutated vs wild-type NPM1 patients when comparing patients with high vs those with low FLT3-ITD allelic ratio. However, in contrast to Boddu et al.40 we did see an impact of NPM1 mutations in the cohort of patients with FLT3-ITDlow/no. Interestingly, our analyses also suggested a prognostic importance of less common gene mutations in the BCOR, SETBP1 and ZRSR2 genes. While Papaemmanuil et al.6 suggested that AML with chromatin remodeling–spliceosome mutations constitutes a distinct subgroup, very little is known about the possible prognostic impact of SETBP1 mutations in AML.41 Thus, our results showing prognostic impact of less common gene mutations should be confirmed by further studies.
The only patients reclassified from a prognostically worse ELN risk group to a better one were Adverse-risk patients harboring IDH2 mutations whose outcome was similar to that of Intermediate-risk patients. Most patients were FLT3-ITDlow/no, and a complex karyotype was detected in only 7 of 23 (30%) patients in this group. The association of IDH2 mutations with an improved OS was previously reported by Patel et al.7 both in the entire cohort of AML patients they analyzed and in patients with Intermediate-risk. However, this suggested risk-group refinement by IDH2, as well as the known importance of FLT3-ITDhigh, have to be considered in view of the FDA-approved inhibitors (e.g., addition of midostaurin to induction chemotherapy for FLT3-mutated patients, and/or FLT3- or IDH-directed targeted therapies), which may only be the beginning of changes in current risk-stratification approaches in the era of increasing targeted therapy options.
Importantly, our study did not include testing for minimal residual disease (MRD). Given the substantial body of evidence about the impact of MRD on survival of AML patients, this represents a notable limitation to our analyses, which needs to be addressed in future studies. In summary, our study provides a comprehensive analysis of prognostic markers in younger adults with AML treated with standard chemotherapy without allo-SCT within the framework of the 2017 ELN genetic-risk classification. Our results suggest a refinement of this classification by including additional gene mutations, which has led to identification of patients that may need more intensive treatment.
Supplementary Material
Acknowledgments
The authors are grateful to the patients who consented to participate in these clinical trials and the families who supported them; to Donna Bucci and Christopher Manring, and the CALGB/Alliance Leukemia Tissue Bank at The Ohio State University Comprehensive Cancer Center, Columbus, OH, for sample processing and storage services, Lisa J. Sterling and Christine Finks for data management and Jason Hsu for his statistical input. This paper is dedicated to the memory of the senior author (CDB) who unexpectedly died during final completion of this manuscript.
Support: Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Numbers U10CA180821 and U10CA180882 (to the Alliance for Clinical Trials in Oncology), https://acknowledgments.alliancefound.org;
UG1CA233338, U24CA196171, UG1CA233180, UG1CA189824, UG1CA189850, P30CA016058, the Coleman Leukemia Research Foundation, Pelotonia (A-KE), American Society of Hematology Scholar Award (AK-E), NIH R35 CA197734 (JCB) and by an allocation of computing resources from The Ohio Supercomputer Center. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflict of interest The authors declare no conflicts of interest.
Supplementary information is available at the journal’s website.
ClinicalTrials.gov Identifier: NCT00006363 (CALGB-19808); ClinicalTrials.gov Identifier: NCT00651261 (CALGB-10603); and ClinicalTrials.gov Identifier: NCT00416598 (CALGB-10503).
References
- 1.Döhner H, Estey EH, Amadori S, Appelbaum FR, Büchner T, Burnett AK et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 2010; 115: 453–474. [DOI] [PubMed] [Google Scholar]
- 2.Röllig C, Bornhäuser M, Thiede C, Taube F, Kramer M, Mohr B et al. Long-term prognosis of acute myeloid leukemia according to the new genetic risk classification of the European LeukemiaNet recommendations: evaluation of the proposed reporting system. J Clin Oncol 2011; 29: 2758–2765. [DOI] [PubMed] [Google Scholar]
- 3.Mrózek K, Marcucci G, Nicolet D, Maharry KS, Becker H, Whitman SP et al. Prognostic significance of the European LeukemiaNet standardized system for reporting cytogenetic and molecular alterations in adults with acute myeloid leukemia. J Clin Oncol 2012; 30: 4515–4523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alpermann T, Kern W, Schnittger S, Schmid C, Kreuzer KA, Serve H et al. Evaluation of the proposed reporting system of the European LeukemiaNet and recommendations for prognosis of acute myeloid leukemia. Leuk Res 2013; 37: 197–200. [DOI] [PubMed] [Google Scholar]
- 5.Dӧhner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med 2015; 373: 1136–1152. [DOI] [PubMed] [Google Scholar]
- 6.Papaemmanuil E, Gerstung M, Bullinger L, Gaidzik VI, Paschka P, Roberts ND et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med 2016; 374: 2209–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patel JP, Gönen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med 2012; 366: 1079–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo myeloid leukemia. N Engl J Med 2013; 368: 2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Metzeler KH, Herold T, Rothenberg-Thurley M, Amler S, Sauerland MC, Görlich D et al. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood 2016; 128: 686–698. [DOI] [PubMed] [Google Scholar]
- 10.Eisfeld A-K, Kohlschmidt J, Mrózek K, Blachly JS, Walker CJ, Nicolet D et al. Mutation patterns identify adult patients with de novo acute myeloid leukemia aged 60 years and older who respond favorably to standard chemotherapy: an analysis of Alliance studies. Leukemia 2018; 32: 1338–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mrózek K, Eisfeld A-K, Kohlschmidt J, Carroll AJ, Walker CJ, Nicolet D et al. Complex karyotype in de novo acute myeloid leukemia: typical and atypical subtypes differ molecularly and clinically. Leukemia 2019; 33: 1620–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stein EM, DiNardo CD, Pollyea DA, Fathi AT, Roboz GJ, Altman JK et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017; 130: 722–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DiNardo CD, Stein EM, de Botton S, Roboz GJ, Altman JK, Mims AS et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med 2018; 378: 2386–2398. [DOI] [PubMed] [Google Scholar]
- 14.Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Büchner T et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017; 129: 424–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kolitz JE, George SL, Marcucci G, Vij R, Powell BL, Allen SL et al. P-glycoprotein inhibition using valspodar (PSC-833) does not improve outcomes for patients under age 60 years with newly diagnosed acute myeloid leukemia: Cancer and Leukemia Group B study 19808. Blood 2010; 116: 1413–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blum W, Sanford BL, Klisovic R, DeAngelo DJ, Uy G, Powell BL et al. Maintenance therapy with decitabine in younger adults with acute myeloid leukemia in first remission: A phase 2 Cancer and Leukemia Group B study (CALGB 10503). Leukemia 2017; 31: 34–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kolitz JE, George SL, Dodge RK, Hurd DD, Powell BL, Allen SL et al. Dose escalation studies of cytarabine, daunorubicin, and etoposide with and without multidrug resistance modulation with PSC-833 in untreated adults with acute myeloid leukemia younger than 60 years: final induction results of Cancer and Leukemia Group B study 9621. J Clin Oncol 2004; 22: 4290–4301. [DOI] [PubMed] [Google Scholar]
- 18.Mayer RJ, Davis RB, Schiffer CA, Berg DT, Powell BL, Schulman P et al. Intensive postremission chemotherapy in adults with acute myeloid leukemia. N Engl J Med 1994; 331: 896–903. [DOI] [PubMed] [Google Scholar]
- 19.Moore JO, George SL, Dodge RK, Amrein PC, Powell BL, Kolitz JE et al. Sequential multiagent chemotherapy is not superior to high-dose cytarabine alone as postremission intensification therapy for acute myeloid leukemia in adults under 60 years of age: Cancer and Leukemia Group B study 9222. Blood 2005; 105: 3420–3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stone RM, Mandrekar SJ, Sanford BL, Laumann K, Geyer S, Bloomfield CD et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med 2017; 377: 454–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moore JO, Dodge RK, Amrein PC, Kolitz J, Lee EJ, Powell B et al. Granulocyte-colony stimulating factor (filgrastim) accelerates granulocyte recovery after intensive postremission chemotherapy for acute myeloid leukemia with aziridinyl benzoquinone and mitoxantrone: Cancer and Leukemia Group B study 9022. Blood 1997; 89: 780–788. [PubMed] [Google Scholar]
- 22.Schiffer CA, Davis RB, Schulman P, Cooper B, Coyle T, Lee E et al. Intensive post remission therapy of acute myeloid leukemia (AML) with cytoxan/etoposide (CY/VP16) and diazaquone/mitoxantrone (AZQ/MITO). Blood 1991; 78(suppl): 460 (abstract 1829). [Google Scholar]
- 23.Mrózek K, Carroll AJ, Maharry K, Rao KW, Patil SR, Pettenati MJ et al. Central review of cytogenetics is necessary for cooperative group correlative and clinical studies of adult acute leukemia: the Cancer and Leukemia Group B experience. Int J Oncol 2008; 33: 239–244. [PMC free article] [PubMed] [Google Scholar]
- 24.Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc 1958; 53: 457–481. [Google Scholar]
- 25.Vittinghoff E, Glidden DV, Shiboski SC, McCulloch CE. Regression Methods in Biostatistics: Linear, Logistic, Survival and Repeated Measures Models. Springer: New York, NY, USA, 2005. [Google Scholar]
- 26.Eisfeld A-K, Mrózek K, Kohlschmidt J, Nicolet D, Orwick S, Walker CJ et al. The mutational oncoprint of recurrent cytogenetic abnormalities in adult patients with de novo acute myeloid leukemia. Leukemia 2017; 31: 2211–2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Whitman SP, Archer KJ, Feng L, Baldus C, Becknell B, Carlson BD et al. Absence of the wild-type allele predicts poor prognosis in adult de novo acute myeloid leukemia with normal cytogenetics and the internal tandem duplication of FLT3: a Cancer and Leukemia Group B study. Cancer Res 2001; 61: 7233–7239. [PubMed] [Google Scholar]
- 28.Marcucci G, Maharry K, Radmacher MD, Mrózek K, Vukosavljevic T, Paschka P et al. Prognostic significance of, and gene and microRNA expression signatures associated with, CEBPA mutations in cytogenetically normal acute myeloid leukemia with high-risk molecular features: a Cancer and Leukemia Group B study. J Clin Oncol 2008; 26: 5078–5087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grimwade D, Ivey A, Huntly BJP. Molecular landscape of acute myeloid leukemia in younger adults and its clinical significance. Blood 2016; 127: 29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meyer SC, Levine RL. Translational implications of somatic genomics in acute myeloid leukaemia. Lancet Oncol 2014; 15: e382–e394. [DOI] [PubMed] [Google Scholar]
- 31.Abdel-Wahab O, Levine RL. Mutations in epigenetic modifiers in the pathogenesis and therapy of acute myeloid leukemia. Blood 2013; 121: 3563–3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsai C-H, Hou H-A, Tang J-L, Liu C-Y, Lin C-C, Chou W-C et al. Genetic alterations and their clinical implications in older patients with acute myeloid leukemia. Leukemia 2016; 30: 1485–1492. [DOI] [PubMed] [Google Scholar]
- 33.Perl AE, Martinelli G, Cortes JE, Neubauer A, Berman E, Paolini S et al. Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML. NEJM 2019; 381:1728–1740. [DOI] [PubMed] [Google Scholar]
- 34.Paschka P, Marcucci G, Ruppert AS, Mrózek K, Chen H, Kittles RA et al. Adverse prognostic significance of KIT mutations in adult acute myeloid leukemia with inv(16) and t(8;21): a Cancer and Leukemia Group B study. J Clin Oncol 2006; 24: 3904–3911. [DOI] [PubMed] [Google Scholar]
- 35.Paschka P, Marcucci G, Ruppert AS, Whitman SP, Mrózek K, Maharry K et al. Wilms’ tumor 1 gene mutations independently predict poor outcome in adults with cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol 2008; 26: 4595–4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Virappane P, Gale R, Hills R, Kakkas I, Summers K, Stevens J et al. Mutation of the Wilms’ tumor 1 gene is a poor prognostic factor associated with chemotherapy resistance in normal karyotype acute myeloid leukemia: the United Kingdom Medical Research Council Adult Leukaemia Working Party. J Clin Oncol 2008; 26: 5429–5435. [DOI] [PubMed] [Google Scholar]
- 37.Renneville A, Boissel N, Zurawski V, Llopis L, Biggio V, Nibourel O et al. Wilms tumor 1 gene mutations are associated with a higher risk of recurrence in young adults with acute myeloid leukemia: a study from the Acute Leukemia French Association. Cancer 2009; 115: 3719–3727. [DOI] [PubMed] [Google Scholar]
- 38.Becker H, Marcucci G, Maharry K, Radmacher MD, Mrózek K, Margeson D et al. Mutations of the Wilms tumor 1 gene (WT1) in older patients with primary cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. Blood 2010; 116: 788–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gaidzik VI, Schlenk RF, Moschny S, Becker A, Bullinger L, Corbacioglu A et al. Prognostic impact of WT1 mutations in cytogenetically normal acute myeloid leukemia: a study of the German-Austrian AML Study Group. Blood 2009; 113: 4505–4511. [DOI] [PubMed] [Google Scholar]
- 40.Boddu PC, Kadia TM, Garcia-Manero G, Cortes J, Alfayez M, Borthakur G et al. Validation of the 2017 European LeukemiaNet classification for acute myeloid leukemia with NPM1 and FLT3-internal tandem duplication genotypes. Cancer 2019; 125: 1091–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thol F, Suchanek KJ, Koenecke C, Stadler M, Platzbecker U, Thiede C et al. SETBP1 mutation analysis in 944 patients with MDS and AML. Leukemia 2013; 27: 2072–2075. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.