

Abstract
The physiological role of histone deacetylase 11 (HDAC11), the newest member of the HDAC family, remained largely unknown until the discovery of its regulatory function in immune cells. Among them, the regulation of cytokine production by antigen-presenting cells and the modulation of the suppressive ability of myeloid-derived suppressor cells (MDSCs) (Sahakian et al. Mol Immunol 63: 579–585, 2015; Wang et al. J Immunol 186: 3986–3996, 2011; Villagra et al. Nat Immunol 10: 92–100, 2009). Our earlier data has demonstrated that HDAC11, by interacting at the chromatin level with the IL-10 promoter, downregulates il-10 transcription in both murine and human APCs in vitro and ex vivo models (Villagra et al. Nat Immunol 10: 92–100, 2009). However the role of HDAC11 in other cell types still remains unknown. Here we present several methods that can potentially be used to identify the functional role of HDAC11, assigning special attention to the evaluation of immunological parameters.
Keywords: Histone deacetylase 11, Chromatin immunoprecipitation, qRT-PCR, PCR, Flow cytometry, Phenotyping, Genotyping
1. Introduction
In the last decade, epigenetic modifications have captured special attention due to their multifaceted functionality. Noteworthy among the epigenetic modifiers, histone acetyltransferases (HATs) and histone deacetylases (HDACs) have gained particular notice as it has been demonstrated by many that these epigenetic modifiers can be targeted by highly selective small-molecule inhibitors. This characteristic raised a renewed interest in these epigenetic modifiers as potential therapeutic targets, including cancer and immune-related diseases. Functionally, HDACs alter chromatin by acetylation/deacetylation of histone tails resulting in transcriptionally active or inactive chromatin, respectively—ultimately leading to expression and/or suppression of numerous genes [1]. These proteins have been shown to play an important role in the regulation of genes involved in the inflammatory response [1–3] and cancer [4, 5], where they promote loss or gain of cellular functions, not naturally given to a particular cell type. Our approach has been to first identify the role of certain HDACs in the transcriptional regulation of specific components of immune-related pathways, which can be evaluated individually by the analysis of the anti/pro-inflammatory responses against singular stimuli.
HDAC11 was discovered in 2002 [6], and because of its shared characteristic with class I and class II HDACs it was classified in a new and independent group known as class IV HDACs, where it is the only member. Interestingly, most of the reported functions for this deacetylase are related to the immune function [1, 7, 8]. Therefore, in this manuscript, our focus is to present detailed methods for examining the function of HDAC11 in immune cells.
2. Materials
2.1. Animal Models
C57BL/6 (WT) mice were purchased from Jackson laboratories, Tg-HDAC11-eGFP [9] reporter mice were provided by Nathaniel Heintz through the Mutant Mouse Regional Centers, and HDAC11KO was kindly supplied by Dr. Edward Seto at H. Lee Moffitt Cancer Center. Mice were kept in pathogen-free conditions and handled in accordance with approved protocols by the Institutional Animal Care and Use Committee (IACUC) at the University of South Florida.
2.2. General Reagents and Buffers
10× PBS 7.4 pH: In 900 mL ddH2O, add 80 g NaCl, 2 g KCl, 14.4 g Na2HPO4, and 2.4 g KH2PO4. pH to 7.4 with HCl and bring volume up to 1 L (autoclave and filter).
Tissue culture-treated plates: 6, 12, and 24 wells.
Ethanol (99.98 %, ACS/USP/Kosher grade).
Mouse Biotin Selection/Mouse CD4+ T cell isolation Kit.
5 M NaCl: Dissolve 58.44 g of NaCl in 80 mL ddH2O, bring volume up to 100 mL (autoclave and filter).
1 M Tris–HCl pH 8.0: In 80 mL ddH2O, add 12.11 g of Tris base, use HCl to adjust pH value to 8.0, then bring volume up to 100 mL (autoclave and filter).
0.5 M EDTA: In 80 mL ddH2O, add 18.61 g of disodium EDTA·2H2O, use NaOH to adjust pH value to 8.0, then bring volume up to 100 mL (autoclave and filter).
10 % SDS: Dissolve 10 g of SDS in 80 mL of ddH2O, and then bring volume up to 100 mL (autoclave and filter).
TE buffer: 496 mL ddH2O, 5 mL 1 M Tris–HCl pH 8.0, 1 mL 0.5 M EDTA pH 8.0 (autoclave and filter).
Mouse tail lysis buffer: In water, consists of 10 mM Tris–HCl PH 8.0, 50 mM NaCL, 25 mM EDTA PH 8.0, 0.5 % SDS. Proteinase K has to be added to lysis buffer right before use.
Heat-activated Taq DNA polymerase.
1× Ammonium-chloride-potassium (ACK) lysis buffer: In 900 mL of ddH2O, add 8.3 g NH4Cl, 1 g KHCO3, and 200 μL of 0.5 M EDTA. Adjust pH to 7.2–7.4 and bring the total volume to 1 L. Autoclave and filter the solution.
FACS buffer: In 980 mL of 1× PBS, add 20 mL heat-inactivated FBS.
1× 4′,6-Diamidino-2-phenylindole (DAPI) viability dye: Dissolve 10 mg of DAPI with 360.6 μL for a 100 mM stock solution, aliquot into 20 μL fractions in 200 μL tubes. Store at −80 °C for long-term storage or −20 °C for short term. For a 10× stock for viability solution dilute to 10 μM in FACS buffer, add 1 μL of 100 mM DAPI stock solution per 10 mL of FACS buffer.
Fc block: Anti-CD32 and anti-CD16 functional grade with a final antibody concentration of 0.5 μg/mL each.
Flow cytometer compensation beads.
SYBR Green Supermix.
TRIzol reagent.
Chloroform (99 %).
Isopropyl alcohol (99.5 %, ACS grade plus).
cDNA synthesis using RNA strand.
Rabbit anti-HDAC11 antibody.
Biotin-conjugated normal rabbit IgG-B.
Protein A agarose beads.
37 % Formaldehyde solution.
1.25 M Glycine: 9.38 g Glycine in 100 mL water. Filter.
1.0 M KCl: 7.46 g KCl in 100 mL water. Autoclave.
1.0 M MgCl2: 20.33 g MgCl2 in 100 mL water. Autoclave.
5.0 M NaCl: 29.22 g NaCl in 100 mL water. Autoclave.
2.0 M LiCl: 8.48 g LiCl in 100 mL water. Autoclave.
20 % SDS: Dissolve 20 g of SDS in 80 mL of ddH2O, bring volume up to 100 mL.
Phenol/chloroform/isoamyl alcohol pH 6.7, phenol: chloroform:isoamyl alcohol = 25:24:1.
Commercially available PCR Purification Kit.
Cross-linking buffer: 2.7 mL 37 % formaldehyde was added into 97.3 mL 1× PBS pH 7.2, to reach 1 % formaldehyde final concentration in a 100 mL final volume.
Stop buffer: 10 mL 1.25 M Glycine was added into 90 mL 1× PBS pH 7.2 to reach a 0.125 M glycine final concentration in a 100 mL final volume. It is preferred to prepare cross-linking buffer and stop buffer freshly before each experiment. Add protease inhibitor cocktail tablets just before use.
Lysis buffer: In mQwater, consists of 50 mM HEPES pH 7.8, 3 mM MgCl2, 20 mM KCl, 0.25 % Triton X-100, 0.5 % IGEPAL.
Wash buffer: In water, consists of 10 mM Tris–HCl pH 8.0, 1 mM EDTA pH 8.0, 200 mM NaCl.
Sonication buffer: In water, consists of 50 mM HEPES pH 7.8, 140 mM NaCl, 1 mM EDTA, 0.5 % SDS, 1 % Triton X-100.
Dilution buffer I: In water, consists of 50 mM HEPES pH 7.8, 140 mM NaCl, 1 mM EDTA pH 8.0, 1 % Triton X-100.
Dilution buffer II: In water, consists of 50 mM HEPES pH 7.8, 500 mM NaCl, 1 mM EDTA pH 8.0, 1 % Triton X-100.
LiCl buffer: In water, consists of 20 mM Tris–HCl pH 8.0, 250 mM LiCl, 1 mM EDTA pH 8.0, 0.5 % Triton X-100.
TE buffer: In water, consists of 10 mM Tris–HCl pH 8.0, 1 mM EDTA pH 8.0.
Elution buffer: In water, consists of 50 mM Tris–HCl pH 8.0, 1 mM EDTA pH 8.0, 1 % SDS.
All buffers were filtered, and kept cold. Protease inhibitor cocktail tablets were added before use (items 34–43).
2.3. Cell Culture
All cells were cultured and maintained in RPMI supplemented with 10 % FBS, 2.05 mM l-glutamine, 100 IU/L penicillin, and 100 IU/L streptomycin. Culture conditions were set at 5 % CO2 and 37 °C in serologic plates and flasks.
2.4. Equipment
Mini vortex.
Tube-rocker (for 15 or 50 mL tubes).
Water bath tube-top sonicator (for chromatin shearing).
Ultracentrifuge (for 1.5 mL tubes).
Refrigerated centrifuge (for 15 or 50 mL tubes).
Tube shaker/rotator (for 1.5 mL tubes).
Quantitative real-time PCR thermal cycler system.
PCR thermal cycler system.
Dounce homogenizer, 7 mL.
Flow cytometer (with a minimum of 14-color parameter).
3. Methods
3.1. PCR Genotyping of HDAC11 Knockout (HDAC11KO) Tg-HDAC11 eGFP Reporter (Tg-HDAC11-eGFP) Mice
The Tg-HDAC11 eGFP reporter mouse model was developed by engineering a bacterial artificial chromosome (BACs) insertion containing a promoter region of HDAC11 and a terminal eGFP reporter (Tg-HDAC11-eGFP) [10]. This model allows us to follow the dynamic changes in HDAC11 transcriptional activation, by the expression of eGFP. HDAC11KO mice were established using Lox/CRE technology to remove a floxed exon 3, a portion of the histone deacetylase 11 with catalytic function.
3.1.1. Mouse Tail Genomic DNA Preparation
Cut 0.2 cm piece of HDAC11 knockout (HDAC11KO) mouse tail and put the tail snip in 1.5 mL Eppendorf tube.
Add 400 μL LYSIS BUFFER and 10 μL Proteinase K (0.5 mg/mL working concentration, 20 mg/mL stocking concentration) into each tube and incubate at 55–60 °C overnight.
Add 200 μL 5 M NaCl to each tube and vortex immediately for 10 s.
Centrifuge sample tubes at maximum speed for 20–30 min at room temperature.
Transfer the supernatant (about 500–600 μL) to a new 1.5 mL Eppendorf tube.
Add 1 mL room-temperature ethanol (100 %) to each tube, and invert for several times until stringy DNA precipitate is visible.
Centrifuge sample tubes at 14,000 × g for 1 min, and discard the supernatant.
Wash the pellets once with 70 % ethanol. Centrifuge sample tubes at 14,000 × g for 1 min, and discard the supernatant.
Dry the pellets for 10–15 min on the bench top.
Add 50–100 μL TE buffer to the pellet and dissolve at 4 °C overnight.
3.1.2. PCR
1. PCR reaction:
For each 25 μL PCR reaction, use 50 ng of mouse genomic DNA, 1 μL of HDAC11 promoter mouse genotyping primer forward and 1 μL of eGFP mouse genotyping primer reverse (see Table 1). As shown in Table 2, each mouse colony has to be tested with its own specific set of primers.
Table 1.
| Subpopulation | Surface markers |
|---|---|
| Test A: Common myeloid progenitors (CMP) | CD3−,CD19−,NK1.1−,CD11B−,Ly6G−,CD45+, CD117High, CD127− |
| Test A: Myeloblasts | CD45int, SSClow, CD117+,Ly6G− |
| Test A: Promyelocytes | CD45int, SSChigh, CD117+, Ly6G− |
| Test B: Triple-negative DCs | CD3−, CD19−,NK1.1−, CD11C+, CD11B−, CD8−, CD4− |
| Test B: Double-negative DCs | CD3−, CD19−, NK1.1−, CD11C+, CD11B+, CD8−, CD4− |
| Test B: CD4+ DCs | CD3−, CD19−, NK1.1−, CD11C+, CD11B+, CD8−, CD4+ |
| Test B: CD8+ DCs | CD3−, CD19−, NK1.1−, CD11C+, CD11B+, CD8+, CD4− |
| Test C: Classical monocytes | CD3−, Nk1.1−, CD115+, CD45R−, MHCII−, Ly6C−, CD43− |
| Test C: Nonclassical monocytes | CD3−, Nk1.1−, CD115+, CD45R, MHCII−, Ly6C+, CD43+ |
| Test D: Neutrophils | CD3−,CD19−,NK1.1−,CD11C−,CD11B+, Ly6G+ |
| Test D: Eosinophils | CD3−,CD19−,NK1.1,CD11C−,CD11B+,Ly6G−,SSChigh,Ly6Clow/− |
Table 2.
Primers for HDAC11 knockout mouse genotyping and HDAC11 exon qRT-PCR
| Purpose | Primer name | Sequence |
|---|---|---|
| Genotyping | Primer 1 forward | CTGTGGAGGGAGAGTTGCTC |
| Genotyping | Primer 1 reverse | GTTGAGATAGCGCCTCGTGT |
| Genotyping | Primer 2 reverse | GTGGACAGGACAAGGGCTAA |
| Genotyping | H11 promoter forward | GAGGCGTGTGTTCTGCGTA |
| Genotyping | eGFP reverse | GTAGGTCAGGGTGGTCACGA |
| qRT-PCR | H11 exon 3 forward | AGAGAAGCTGCTGTCCGATG |
| qRT-PCR | H11 exon 3/4 reverse | AGGACCACTTCAGCTCGTTG |
| qRT-PCR | Mm_Hdac11_1_SG | (Qiagen) Exons 8–9 |
The HDAC11KO mouse genotyping primers were designed as 1 forward primer and 2 reverse primers by our laboratory as described. Primer 1 forward is designed to be located at upstream of HDAC11 exon 3 and primer 2 reverse is located at downstream of HDAC11 exon 3; primer 1 reverse is located within HDAC11 exon 3. During PCR, 3 primers were added to each reaction. For a wild-type mouse, primer 1 forward and primer 1 reverse will produce a DNA product of 437 base pair indicating the existence of exon 3. Meanwhile, primer 1 forward and primer 2 reverse will produce another DNA product which is over 780 base pair. A homozygous HDAC11KO mouse with a missing exon 3 will produce a DNA product of 278 base pair made from primer 1 forward and primer 1 reverse. A heterozygous HDAC11 knockout mouse, both DNA fragments of 437 base pair and 278 base pair will be observed on a 1.5 % agarose gel (Fig. 1).
Fig. 1.
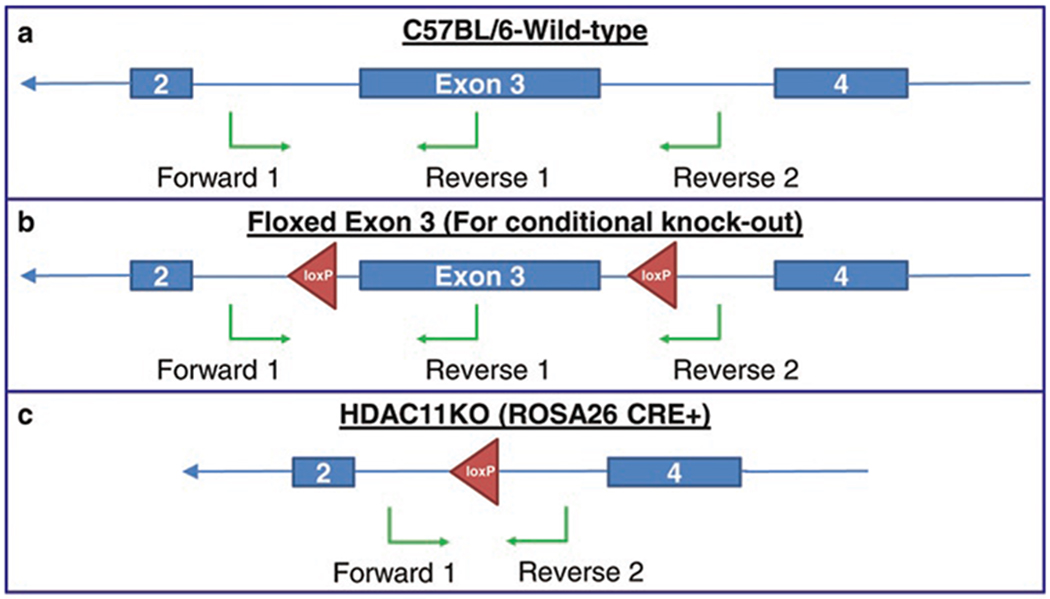
Total and conditional HDAC11KO genotyping. Three genotyping primers were specifically designed to identify the loxP sites downstream and upstream of exon 3 and one primer found within the exon. Schematic representations of the three genotyping primer locations on a C57BL/6 wild-type HDAC11 allele (a), a conditional HDAC11KO allele (b), and a total HDAC11KO allele are displayed above. Genotyping PCR will result in a 437 bp fragment for a wild-type allele (a), a 471 bp fragment for a loxP allele (b), and a 278 bp fragment for a total HDAC11KO allele (c)
The Tg-HDAC11-eGFP mouse genotyping primers were designed by our laboratory. Briefly, HDAC11 promoter forward is designed to be located within the 3′ end of the HDAC11 promoter sequence and eGFP reverse is located within the first 200 bp of the 5′ end of the eGFP coding sequence. For a wild-type mouse, no DNA product is produced. For a homozygous or heterozygous Tg-HDAC11-eGFP mouse, a DNA product of approximately 400 base pair will be observed on a 1.5 % agarose gel.
2. PCR program:
A thermocycler with gradient ability is required for this protocol. The step programming description can be found in Table 3.
Table 3.
Universal touchdown genotyping PCR
| Step | Temperature (°C) | Time | Actions per step |
|---|---|---|---|
| 1 | 94 | 3 minutes | |
| 2 | 94 | 30 seconds | |
| 3 | 70 | 30 seconds | Decrease 0.5 °C per cycle |
| 4 | 72 | 30 seconds | |
| 5 | Go to step 2 40 times | ||
| 6 | 94 | 30 seconds | |
| 7 | 50 | 30 seconds | |
| 8 | 72 | 30 seconds | |
| 9 | Go to step 6 15 times | ||
| 10 | 72 | 2 minutes | |
| 11 | 4 | Infinite | Hold |
This technique has been used as a standard operating procedure by our lab and collaborators pertaining genotyping of these mouse colonies [7, 8].
3.2. Phenotyping of Tg-HDAC11-eGFP Reporter Mice
In this mouse model, expression of HDAC11 message is determined by flow cytometry analysis of eGFP reporter gene expression [10]. Only mice with a positive eGFP/HDAC11 PCR band are used as breeders and are subsequently checked for eGFP expression via flow cytometry. Breeders and pups, of age, have a submandibular bleed (SMB) performed to determine eGFP expression prior to breeding or experimental design.
3.2.1. Peripheral Blood Preparation
A submandibular bleed of an anesthetized mouse is performed to obtain, at minimum, 100 μL of blood in a heparinized tube and rocked.
The peripheral blood (PB) is subsequently transferred to a 5 mL cytometry tube.
The PB is lysed with 1 mL of ACK lysis buffer for 3 min.
The mix is then diluted with 3 mL of PBS and centrifuged for 3 min at 300 × g.
Remove the supernatant and wash the resultant pellet twice with PBS (see Note 1).
The resultant pellet is reconstituted in 500 μL of FACS buffer and run on a flow cytometry machine collecting for forward scatter (FSC), side scatter (SSC), eGFP, and a viability dye, typically DAPI or propidium iodide.
10,000 events of the granulocyte population, as determined in the FSC and SSC plot, are collected and all events are saved (see Note 2).
3.3. Multiparameter Flow Cytometric Analysis to Dynamically Visualize the HDAC11 Expression Profile in Myelopoiesis
3.3.1. Extraction of Bone Marrow Cells
Bone marrow aspirate (BMA) was removed from Tg-HDAC11-eGFP using the following steps:
Collect mouse bone marrow cells by flushing the tibias and femurs with 10 mL ice-cold PBS.
Centrifuge the cells at 500 × g for 5 min at 4 °C.
Remove supernatant, reconstitute the cells in 3 mL ACK lysis buffer, and incubate at room temperature for 4 min.
Add ice-cold PBS and centrifuge the cells at 500 × g for 5 min at 4 °C.
Remove supernatant. Reconstitute the cells in 10 mL ice-cold PBS. Let cell suspension go through a 45 μm strainer to remove remaining large chunks.
Centrifuge single-cell suspension at 500 × g for 5 min at 4 °C.
Remove supernatant. Reconstitute cell pellet in FACS buffer for future use.
3.3.2. Flow Cytometry (Staining and Analysis)
Cells were labeled (as indicated in the chart below) with distinct cell surface markers that have been extensively described as specific for the cellular subpopulation under analysis (Fig. 2). Always create one test more than required for pipette error (i.e., seven samples would require a master mix for eight samples).
A single-cell suspension of BMA is created at 3 × 106 cells per mL in FACS buffer. Note: At minimum 1 mL is required for the following study.
2 μL of BD FC block is added per mL of BMA suspension and incubated for 15 min at room temperature.
- 100 μL of each sample is added to a fresh 5 mL cytometry tube labeled for each test below:
- Common myeloid progenitors (CMPs).
- Dendritic cells (DCs).
- Granulocytes.
- Monocytes.
- Control tube (remaining mix).
400 μL of fresh FACS buffer is added per tube and centrifuged at 300 × g for 5 min (except for control tubes).
- Create the following control tubes for each sample type tested for each test:
- Fluorescence minus one (FMO) for each type of fluorescence tested (five tubes for CMPs, DCs, and monocytes and three tubes for granulocytes).
- Unstained sample.
- Viability stain only (DAPI).
Create master mixes of the antibodies for each test to be run and bring the master mix volume up to equal a volume appropriate for 50 μL per tube (plus extra tube) (see Table 4 for each test requirements).
Reconstitute each test tube with 50 μL of the appropriate test master mix.
FMO control tubes will be labeled “(Test Name) FMO—(Name of Fluoresence)” and all the antibodies EXCEPT the one listed will be added to the tube in the same concentration as listed in each test plus 50 μL of FACS buffer (i.e., the tube labeled CMP FMO—APC would contain all antibodies of the CMP test except APC).
Single-fluorescence stain controls are made using UltraComp beads, and apply the same volume of each antibody used to a separate tube labeled for each type of fluorescence, in each test. One drop of UltraComp beads is added to each of these tubes.
All tubes are incubated in the dark for 1 h at room temperature and subsequently washed with 450 μL FACS buffer and centrifuged for 5 min at 300 × g and supernatant is removed. Note: Unnecessary to wash single-color controls.
Reconstitute all the test tubes and the DAPI control tube in a 1× DAPI staining mix—do not do this to the control tubes. Run samples on flow cytometer.
-
When running samples, gate out dead cells, doublets, and aggregates and collect until you have 10,000 cells of the smallest population of interest; save all events.
Previous work done in our lab has already confirmed the co-expression of HDAC11 message and eGFP message using qRT-PCR using HDAC11 and eGFP primers (data not shown).
Fig. 2.

Dynamic expression of HDAC11 in various compartments of myelopoiesis. Schematic and analytic visualization of HDAC11 message in myelopoiesis. Using HDAC11-eGFP transgenic mouse, in a multiparameter flow cytometric analysis (FACS-Aria (BD) device). In this figure, expression eGFP protein coincides with transcriptional activation of HDAC11
Table 4.
Flow cytometry master mixes
| A | uL per test | CD Marker or Designation | Fluorescent molecule | C | uL per test | CD Marker or Designation | Fluorescent molecule |
|---|---|---|---|---|---|---|---|
| Common Myeloid Progenitor | 1 | CD3 | eFluor 450 | Monocytes (Classical and Non-Classical) | 1 | CD3 | eFluor 450 |
| 1 | CD19 | v450 | 1 | CD19 | v450 | ||
| 1 | NK1.1 | v450 | 1 | NK1.1 | v450 | ||
| 1 | NKG2D | v450 | 1 | NKG2D | v450 | ||
| 1 | CD11b | v450 | 1 | CD115 | APC | ||
| 1 | Ly6g | v450 | 1 | Ly6c | AlexaFlour700 | ||
| 1 | CD45 | PE-Cy7 | 1 | CD43 | PE | ||
| 1 | CD34 | AlexaFlour700 | 1 | MHCII | PE-Cy7 | ||
| 1 | CD127 | PerCP-Cy5.5 | 8uL per well | ||||
| 1 | CD117 (c-Kit) | APC | |||||
| 10uL per well | |||||||
| B | uL per test | CD Marker or Designation | Fluorescent molecule | D | uL per test | CD Marker or Designation | Fluorescent molecule |
| Dendritic Cells | 1 | CD3 | eFluor 450 | Granulocytes (Neutrophils/Eosinophils) | 1 | CD3 | eFluor 450 |
| 1 | CD19 | v450 | 1 | CD19 | v450 | ||
| 1 | NK1.1 | v450 | 1 | NK1.1 | v450 | ||
| 1 | NKG2D | v450 | 1 | NKG2D | v450 | ||
| 1 | CD11b | PE | 1 | CD11b | APC | ||
| 1 | CD11c | APC | 1 | Ly6g | PE | ||
| 1 | CD4 | PerCP | 6uL per well | ||||
| 1 | CD8 | AlexaFlour700 | |||||
| 8uL per well | |||||||
See Note 7
Table 1. This protocol has been used in our laboratory to study the effect of HDAC11 in MDSC function and differentiation [11] as well as in other compartments of hematopoiesis. The flow cytometry technique described above has been the standard operating procedure for most of the flow data published in our recent manuscripts [12–15].
3.4. Quantitative Real-Time PCR of HDAC11KO Mouse Model
HDAC11KO mice were established by excision of the catalytic region of this gene (exon 3). Therefore in this model, if the qRT-PCR primers are not designed in the exon 3 region, the results are truncated HDAC11 amplicons. Hence, we designed a special HDAC11 primer set that was within the exon 3 region of this gene.
3.4.1. Total mRNA Isolation and cDNA Synthesis
Peritoneal neutrophils were harvested from C57BL/6 and HDAC11 knockout mice.
Collect cells in 15 mL conical and wash once with PBS. Pellets were transferred into a new 1.5 mL Eppendorf tube.
Add 1 mL TRIzol reagent to each sample, and lyse for 10 min at room temperature.
Add 0.2 mL chloroform to each 1 mL sample. Shake vigorously by hand for 15 s and rest at room temperature for 3 min.
Centrifuge the samples at 12,000 × g for 15 min at 4 °C.
Transfer supernatant to a new 1.5 mL Eppendorf tube. Add 0.5 mL of isopropyl alcohol to each sample. Incubate at room temperature for 10 min.
Centrifuge the samples at 12000 × g for 10 min at 4 °C.
Discard supernatant. Wash pellet once with cold 75 % ethanol. Gently invert the samples 4–5 times.
Centrifuge the samples at 7500 × g for 8 min at 4 °C.
Discard supernatant. Let the pellet dry at room temperature for 10 min.
Add 20–40 μL DEPC-treated water to each sample.
Iscript™ cDNA synthesis kit was used following the manufacturer’s protocol. In 20 μL volume for each reaction, use 1 μg total mRNA.
3.4.2. Quantitative Real-Time PCR
-
For each qRT-PCR reaction, add 0.5 μL cDNA from each C57BL/6 or HDAC11 mouse sample, 0.5 μL of each HDAC11 exon 3 forward and reverse primer (see Table 2), 5 μL Bio-Rad SYRB green super mix, and 3.5 μL DEPC-treated water.
For cells from HDAC11 knockout mice, no HDAC11 exon 3 can be detected by qRT-PCR.
For qRT-PCR program (see Table 5), 58–60 °C melting temperature was used based on the HDAC11 exon 3 primer gradient study (Fig. 3).
Table 5.
Quantitative RT-PCR
| Step | Temperature (°C) | Time | Actions per step |
|---|---|---|---|
| 1 | 95 | 3 minutes | |
| 2 | 95 | 30 seconds | |
| 3 | 60 | 30 seconds | Read |
| 4 | Go to step 2 40 times | ||
| 5 | 95 | 1 minute | |
| 6 | 58 | 10 seconds |
Melt Curve (Start Temp.) Read 0.5°C/cycle to End Temp. |
| 7 | 95 | Melt curve (End Temp.) |
Fig. 3.

Verification of HDAC11KO mice using qRT-PCR analysis. Peritoneal neutrophils (PNs) from 3 C57BL/6 (left) and HDAC11 KO (right) mice were harvested at 18 h post-thioglycolate injection at 5 %. Expression of HDAC11 mRNA was measured by qRT-PCR analysis
This protocol has been the standard operating procedure for studies performed in the HDAC11KO mice and this procedure has been instrumental in the preparation of our latest manuscript identifying the regulatory role on HDAC11 in neutrophil function [16].
3.5. Chromatin Immunoprecipitation (ChIP) to Determine the Regulatory Role of HDAC11
3.5.1. Cells Cross-Linking
Sample Preparation
Approximately 20 × 106 cells from C57BL/6 and HDAC11 knockout mice per condition will be optimal.
Cells were collected in 50 mL conical tubes and washed twice with 10 mL 1× PBS.
Remove PBS by centrifuging at 1500 × g for 5 min at room temperature.
Gently reconstitute cells in 20 mL of cross-linking buffer, set up the sample tubes on a tube-rocker or orbital rotator, and incubate for 10 min at room temperature (see Note 3).
Centrifuge samples at 1500 × g for 5 min, at room temperature, and remove the supernatant.
Cells were gently reconstituted in 20 mL of stop buffer, and then incubated for 10 min on a tube-rocker at room temperature (step 4).
Centrifuge the samples at 1500 × g for 5 min at 4 °C, and discard supernatant.
Reconstitute cells in 20 mL of PBS, gently invert tubes several times, spin at 1500 × g for 5 min at 4 °C, and discard supernatant.
Repeat step 8 if needed, and aliquot cells before centrifuge.
Proceed with chromatin preparation. Cell pellets from step 9 can be stored at −80 °C until further use.
3.5.2. Chromatin Preparation
Sample Preparation
Remove sample tubes containing frozen cell pellets from −80 °C and allow to thaw on ice.
Prepare Dounce homogenizer (7 mL), set on ice.
Reconstitute each pellet of 20 × 106 cells in 2 mL of lysis buffer. Gently pipette up and down until completely homogenized. Incubate on ice for 10 min.
Transfer cells to the Dounce homogenizer and apply 25 strokes using the loose pestle (see Note 4). Transfer the nuclei to a new 15 mL conical and centrifuge at 1500 × g for 5 min at 4 °C. Then discard supernatant.
Reconstitute nuclei pellet with 2 mL of wash buffer. Gently pipette up and down until completely homogenized. Incubate on ice for 10 min.
Pellet the nuclei by centrifuging at 1500 × g for 5 min at 4 °C. Discard supernatant.
-
Add 600 μL sonication buffer to each tube, and gently pipette up and down until homogenized. Divide the sample by transferring 300 μL of nuclei sample to each 1.5 mL Eppendorf tube and proceed to the sonication section.
Samples have to be kept on ice at all times.
Sample Sonication
For this specific protocol we used a Bioruptor™ XL from Diagenode. However, any bath sonicator with a timing programming will be suitable. Briefly, the water bath was precooled to 4 °C with crushed ice before each sonication cycle and a thin layer of crushed ice was kept in the water bath for each cycle to avoid rapid temperature increase during sonication.
For each sonication cycle, use a program with eight pulses of 30 s followed by 30 s of resting time at 20 KHz frequency (300 W). To obtain 250 base pair to 500 base pair DNA fragments for the mouse peritoneal neutrophils, approximately eight cycles are required for each sample. However, this parameter has to be set for each cellular type (see Note 5). Centrifuge the samples at 16,000 × g for 10 min at 4 °C. Transfer the supernatant to a new 1.5 mL Eppendorf tube (approximately 600 μL supernatant).
Take an aliquot (20 μL) from each sample and proceed to the sample shear check section. Either the rest of the samples can be stored at −80 °C or continue with immunoprecipitation.
Sample Shear Check
To each tube add 20 μL sample aliquot, 47.2 μL H2O, and 2.8 μL of 5 M NaCl to reach a final concentration of 200 mM.
Incubate at 65 °C for 4 h or overnight.
Add RNase A to a final concentration of 20 μg/mL to each sample aliquot; incubate at 37 °C for 30 min.
Add Proteinase K to a final concentration of 100 μg/mL to each sample aliquot; incubate at 42 °C for 2 h.
Add 200 μL phenol/chloroform/isoamyl alcohol pH 6.7 to each sample aliquot, vortex for 15 s, and spin down at 16,000 × g for 5 min.
Transfer 15 μL of the supernatant from the top layer; add 3 μL of 5× DNA loading dye with light xylene cyanol only.
Run samples on 1.5 % agarose gel at 40 V for 1.5 h.
3.5.3. Chromatin Immunoprecipitation
Sample Pre-clearance
Add 30 μL of protein A agarose beads to each sample, and incubate on a tube rotator for 1 h at 4 °C.
Centrifuge the samples at 6000 × g for 5 min at 4 °C.
Transfer the supernatant to a new 1.5 mL Eppendorf tube. Take an aliquot of 50 μL from each sample as input. Input samples can be stored at −80 °C before reverse cross-linking.
Immunoprecipitation
Dilute 50 μL of each sample after preclearing with 450 μL dilution buffer I, to 500 μL final volume, one dilution per antibody, into a 1.5 mL Eppendorf tube.
During the first dilution tube of each sample, add a mixture of 5 μL (5 μg) rabbit-anti-HDAC11 antibody from Sigma and 20 μL (4 μg) rabbit-anti-HDAC11 antibody from BioVision. In the second dilution tube of each sample, add 9 μg normal rabbit IgG-B.
Set the sample tubes on a tube rotator overnight, at 4 °C, and cover the tubes with aluminum foil to avoid light exposure (see Note 6). Add 50 μL of protein A agarose beads to each sample, and incubate on a tube rotator for 4 h at 4 °C.
Sample Wash
After immunoprecipitation, centrifuge the samples at 6000 × g for 5 min at 4 °C, and discard supernatant.
-
Wash the pellet with 1 mL of each buffer in the following order:
Twice with dilution buffer I.
Twice with dilution buffer II.
Twice with LiCl buffer.
Twice with TE buffer.
For each wash, centrifuge at 6000 × g for 5 min at 4 °C.
Elute and Reverse Cross-Linking
Discard supernatant from the last wash. Add 100 μL of elution buffer to each sample and incubate at 65 °C for 15 min. The immuno-complexes will be in the soluble fraction.
Centrifuge the samples at maximum speed for 20 s. Transfer supernatant to a new 1.5 mL Eppendorf tube.
Remove the input sample tubes from −80 °C and thaw on ice. Add 50 μL of elution buffer to each input tube to reach a final volume of 100 μL.
Add 4.16 μL 5 M NaCl (200 mM final concentration) to each IP and input sample tubes. Reverse cross-linking at 65 °C for 6 h or overnight.
DNA Purification
Add RNase A to a final concentration of 20 μg/mL to each sample; incubate at 37 °C for 30 min.
Add Proteinase K to a final concentration of 100 μg/mL to each sample; incubate at 42 °C for 2 h.
-
Use Qiagen™ DNA purification kit to purify each DNA sample.
To obtain a higher amount of DNA, researchers can elute DNA twice from each column; use 50 μL elution buffer each time to obtain a higher amount of DNA.
Use 2 μL DNA for each qRT-PCR reaction. This is in a triplicate reaction, and the purified DNA must be kept in TE buffer pH 8.0.
It is recommended to determine the primer efficiency and melting temperature before running PCR on the ChIP samples.
This protocol has been used as a standard operating procedure by our lab and collaborators [8]. A prototypical experimental result is shown in Fig. 4. Briefly, we evaluated the recruitment of HDAC11 to the il10 promoter after stimulation of mouse macrophages with LPS (see Note 7).
Fig. 4.
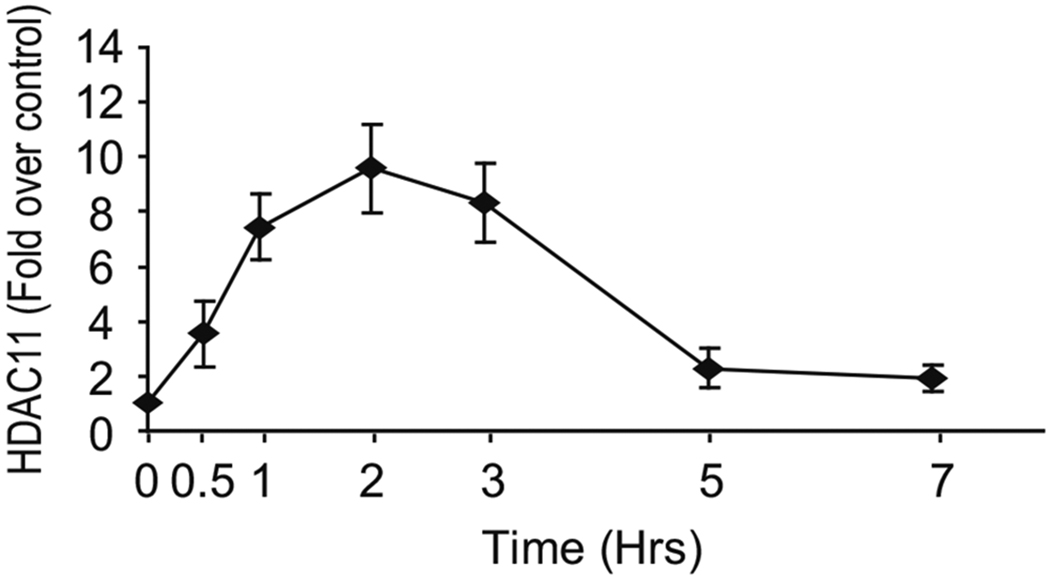
ChIP analysis of HDAC11 in IL-10 promoter region. HDAC11 is recruited to the IL-10 gene promoter in macrophages under LPS stimulation. Macrophages were treated with LPS at 1.0 g/mL, and then harvested at 0 or 0, 0.5, 1, 2, 3, 5, and 7 h after treatment. Cells were then harvested and proceed to ChIP analysis using anti-HDAC11 cocktail and anti-rabbit IgG as described. Quantitative real-time PCR analysis was performed in the −87 base pair to −7 base pair region of the mouse gene IL-10 promoter. The recruitment of HDAC11 to IL-10 promoter was observed at 0.5 h and reached to a peak by 2 h after LPS stimulation [8] (representative figure from previously published studies)
Acknowledgments
The authors gratefully acknowledge the flow cytometry core facilities at H. Lee Moffitt Cancer Center and their extended technical support for our project.
Footnotes
If the resultant cell pellet is still robustly red in color, the ACK lysis treatment has to be repeated. Two lysis cycles are sufficient to remove enough red blood cells for analysis. This is directly related to the initial volume of red blood cells added to each tube.
100 % of the granulocyte population will express eGFP and ~35–50 % (depending on the age of the mouse) of the lymphocyte population, also determined by FSC versus SSC, will express eGFP. We chose mice with eGFP expression greater than 103 mean fluorescent intensity unit with the aforementioned expression pattern as HDAC11-eGFP-positive mice.
Both chromatin shearing efficiency and immunoprecipitation efficiency can be affected by formaldehyde concentration and cross-linking time. In this protocol, we are using 1 % formaldehyde and 10-min incubation time for mouse peritoneal neutrophils. Cross-linking time and formaldehyde percentage should be optimized for other cell types, such as mouse T cells or cell lines [17, 18].
The Dounce homogenizer has to be set on ice at all times. It can be rinsed with cold PBS 3–5 times between different samples. The number of strokes could vary and should be optimized according to different cell types or cell lines. Briefly, take an aliquot of cells and stain with trypan blue and check under the microscope. Adjust stroke times until researchers can see that over 90 % of the cells were stained but have kept a round nuclei. Decrease the number of strokes if broken nuclei were observed.
Sonication efficiency varies according to different cell types and equipment. It is recommended to optimize this step by a pilot experiment using a number of different sonication cycles on different cell types or cell lines. For some types of sonicator, it is recommended to check the water bath temperature and replace ice after each cycle, or the water bath could be kept on a heat-adjusted platform to maintain a 4 °C environment.
Using a mixture of two different HDAC11 antibodies has been confirmed with good immunoprecipitation efficiency on macrophages [8] and mouse peritoneal neutrophils (manuscript in submission). It is recommended to optimize IP antibody use for different cell types or cell lines.
HDAC11 has been observed to be differentially expressed in various compartments of myelopoiesis. This expression of HDAC11 appears to coincide with the granularity of cell subtype. HDAC11 also appears to have a role in the fate of cell differentiation in the process of myelopoiesis.
References
- 1.Villagra A, Sotomayor EM, Seto E (2010) Histone deacetylases and the immunological network: implications in cancer and inflammation. Oncogene 29(2):157–173 [DOI] [PubMed] [Google Scholar]
- 2.Foster SL, Hargreaves DC, Medzhitov R (2007) Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature 447(7147):972–978 [DOI] [PubMed] [Google Scholar]
- 3.Woan KV, Sahakian E, Sotomayor EM, Seto E, Villagra A (2012) Modulation of antigen-presenting cells by HDAC inhibitors: implications in autoimmunity and cancer. Immunol Cell Biol 90(1):55–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miremadi A, Oestergaard MZ, Pharoah PDP, Caldas C (2007) Cancer genetics of epigenetic genes. Hum Mol Genet 16(R1):R28–R49 [DOI] [PubMed] [Google Scholar]
- 5.Schreiber RD, Old LJ, Smyth MJ (2011) Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science 331(6024):1565–1570 [DOI] [PubMed] [Google Scholar]
- 6.Gao L, Cueto MA, Asselbergs F, Atadja P (2002) Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J Biol Chem 277(28):25748–25755 [DOI] [PubMed] [Google Scholar]
- 7.Sahakian E, Powers JJ, Chen J et al. (2014) Histone deacetylase 11: a novel epigenetic regulator of myeloid derived suppressor cell expansion and function. Mol Immunol 63(2):579–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheng F, Lienlaf M, Perez-Villarroel P et al. (2014) Divergent roles of histone deacetylase 6 (HDAC6) and histone deacetylase 11 (HDAC11) on the transcriptional regulation of IL10 in antigen presenting cells. Mol Immunol 60(1):44–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gong S, Zheng C, Doughty ML et al. (2003) A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature 425(6961):917–925 [DOI] [PubMed] [Google Scholar]
- 10.Heintz N (2001) BAC to the future: the use of bac transgenic mice for neuroscience research. Nat Rev Neurosci 2(12):861–870 [DOI] [PubMed] [Google Scholar]
- 11.Sahakian E, Powers JJ, Chen J et al. (2015) Histone deacetylase 11: a novel epigenetic regulator of myeloid derived suppressor cell expansion and function. Mol Immunol 63(2):579–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang H, Cheng F, Woan K et al. (2011) Histone deacetylase inhibitor LAQ824 augments inflammatory responses in macrophages through transcriptional regulation of IL-10. J Immunol 186(7):3986–3996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Villagra A, Cheng F, Wang HW et al. (2009) The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nat Immunol 10(1):92–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng F, Lienlaf M, Wang HW et al. (2014) A novel role for histone deacetylase 6 in the regulation of the tolerogenic STAT3/IL-10 pathway in APCs. J Immunol 193(6): 2850–2862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng F, Wang H, Horna P et al. (2012) Stat3 inhibition augments the immunogenicity of B-cell lymphoma cells, leading to effective antitumor immunity. Cancer Res 72(17): 4440–4448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eva Sahakian JC, Powers John J., Chen Xianghong, Maharaj Kamira, Deng Susan L., Lienlaf Maritza, Wang Hong Wei, Sodré Andressa L., Distler Allison, Xing Limin, Perez-Villarroel Patricio, Wei Sheng, Villagra Alejandro, Seto Ed, Sotomayor Eduardo M., Horna Pedro and Pinilla-Ibarz Javier (2015) Essential regulatory role for histone deacetylase 11 (HDAC11) In neutrophil function. J Leukoc Biol. Under 2nd revision [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee TI, Johnstone SE, Young RA (2006) Chromatin immunoprecipitation and microarray-based analysis of protein location. Nat Protoc 1(2):729–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nelson JD, Denisenko O, Bomsztyk K (2006) Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat Protoc 1(1): 179–185 [DOI] [PubMed] [Google Scholar]