

Abstract
This renin‐angiotensin system (RAS) interpretation is focused on differences in tissue dependence on RAS and on the topological hierarchy that allows mediators to act only on downstream tissues.
Dependence of tissues on RAS: Tested by expectation maximization clustering of the RNA human tissue expression (https://biogps.org/). ACE and vasoconstrictive AT1R clustered with the prorenin receptor. ACE2 and dilatory MAS1 clustered with nine RAS‐related genes, highly expressed in: Liver; Cardiac_Myocytes; Skeletal_Muscle; Uterus; Kidney; Lung; Small_Intestine; Smooth_Muscle.
RAS and stress accumulation: While prorenin is active after binding to its receptor, binding of soluble renin increases its enzymatic activity several times. Increased renin secretion multiplies the overall capacity for producing Ang I, leading to hypertension and increased vascular resistance.
Coronavirus infection and comorbidities: Cardiorespiratory failure during infection is linked to the previously altered RAS role in lungs and myocardium. Reduced vasodilation by ACE2 lead to vasoconstriction and suboptimal tissue perfusion patterns. Also see the video abstract here https://www.youtube.com/watch?v=Jf0Iped-Mws
Keywords: allostasis, arterial hypertension, Corona viruses, renin‐angiotensin‐aldosterone system, SARS
Binding to prorenin receptor increases renin activity several times. Increased renin secretion enhances tissue Ang I production due to prorenin receptor binding. Availability of ACE, ACE2, AT1R and MAS1 determine vascular resistance and tissue perfusion.
Corona infections reduce ACE2 activity and lead to cardiopulmonary failure via the ACE and AT1R path.
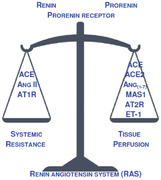
Abbreviations
- ACE
angiotensin‐I‐converting enzyme
- ACE2
angiotensin‐I‐converting enzyme
- Ang I
angiotensin I
- Ang II
angiotensin II
- Ang(1‐7)
angiotensin (1‐7)
- Ang(1‐9)
angiotensin(1‐9)
- AT1R
angiotensin II receptor, type 1
- AT2R
angiotensin II receptor, type 2
- EM
type of clustering based on the expectation–maximization algorithm
- ET‐1
Endothelin‐1
- MAS1
MAS1 proto‐oncogene, receptor for Ang(1‐7)
- RAS
renin‐angiotensin systems
- TSP1
thrombospondin 1
INTRODUCTION
Physiology and clinical medicine accept certain concepts, paradigms, constructed to explain health issues. Generally accepted interpretations of medical issues are presented in medical textbooks. Although the Occam's razor favors the simplest description as the most probably correct, new data emerge continuously and ad complexity to the prevailing interpretations.
A recent example of emerging complexity is the role of Angiotensin Convertase Enzyme 2 (ACE2) in patients with coronavirus infections that target this enzyme.[ 1 , 2 , 3 ] Despite existence of an extensive literature on the importance of ACE2 as a negative regulator of RAS, many health professionals remain less familiar with this component of the renin‐angiotensin system (RAS). If we look at RAS descriptions in four contemporary editions of physiological textbooks,[ 4 , 5 , 6 , 7 ] very little information can be found about ACE2 and other dilatory RAS components. Our textbooks seem to lack more details on ACE2, prorenin, prorenin receptors or AT2R molecules. This means that the prevailing RAS interpretation is possibly oversimplified and focused on the RAS induced vasoconstriction. This discrepancy suggests that a revised, more complex paradigm of RAS actions be needed.
ACE2 targeting coronavirus infections and age‐related health issues
For the two latest important Corona viruses, SARS‐CoV and SARS‐CoV‐2, similar patterns of age dependent mortality have been reported. SARS‐CoV epidemic in 2003 showed that the case‐fatality ratio in Hong Kong increased with age as in other parts of the world: 14.7% in patients <45 years of age, 21.4% between 45 and 65 years and 63.9% in patients more than 64 years, suggesting that SARS deaths were associated with the health issues of older age.[ 2 ]
For the COVID‐19 pandemic, the reported case fatality ratio in China was 1.38% (with 95% credible interval: 1.23 to 1.53), with substantially higher ratios in older age groups (0.32% (0.27 to 0.38) in those aged <60 years vs. 6.4% (5.7 to 7.2) in those aged >59 years), up to 13.4% (11.2 to 15.9) in those older than 79 years. For international cases age stratified mortality was 1.4% (0.4 to 3.5) for <60 years, 4.5% (1.8 to 11.1) for >59 years.[ 3 ]
Although, cytokine dysregulation at older age is known to result in a progressive tendency toward to a proinflammatory phenotype, linked to atherosclerosis, diabetes, Alzheimer's disease, rheumatoid arthritis, cancer, and aging itself,[ 8 ] this cannot be the whole answer to this puzzle since the described age‐dependent mortality is not so profoundly present in other potentially lethal viral infections. Evident differences in mortality rates, dependent on preexistent health issues and age, suggest that the ACE2 function might be more important in infected elderly patients.
The same ACE2 enzyme must have a physiological role in maintaining cardiopulmonary function in healthy individuals of any age. It seems plausible that this normal role is often somehow altered in patients with arterial hypertension, diabetes and other diseases, all conditions more prevalent in older age. This seems directly related to the functional deterioration of the circulatory system, often detectable in senior individuals. Animal and human studies unequivocally show that sympathetic activation participates in the development, maintenance and progression of arterial hypertension,[ 9 ] often combined with obesity and type 2 diabetes.[ 10 ] The preexistence of adrenergic overdrive seems linked to arterial hypertension, tachycardia, increased levels of renin‐angiotensin system activation, insulin resistance etc.[ 11 , 12 ]
How complex are interactions among RAS components?
The “classic” interpretation[ 4 , 5 , 6 , 7 , 13 ] is that RAS maintains arterial pressures and the circulating volume by arteriolar constriction and regulation of ion and fluid traffic in kidneys, colon and sweat glands. Kidneys are considered to play the central role since they secrete the initial mediator, renin and are also among target organs of Angiotensin II (Ang II). Our interpretation of the juxtaglomerular apparatus (JGA) actions is complex.[ 4 , 5 , 6 , 7 ] Activation of an individual JGA causes two effects, the afferent arteriole of that nephron is dilated by paracrine actions, while the secreted renin will increase availability of vasoconstrictive Ang II in circulation.
The consequence is that Ang II constricts arterioles of various tissues, including efferent arterioles in both kidneys.[ 4 ] This means that the dilatory action on the individual afferent arteriole affects only that nephron, while the unselective Ang II induced constriction affects various organs, including both kidneys. One might argue that the paracrine action on the afferent arteriole maintains the function of that individual nephron within the acceptable range, although its full effect requires the assistance of the Ang II in constricting efferent arterioles. In other words, the regulation of individual neprons cannot suffice without the general RAS vasoconstrictive action that maintains the peripheral arterial pressure and the average resistance of efferent arterioles.
In short, the “classical” RAS interpretation[ 13 , 14 , 15 ] is focused on angiotensinogen, renin, Ang I, ACE and Ang II that induces vasoconstriction, secretion of aldosterone[ 4 , 5 ] and vasopressin.[ 14 ] The last two actions maintain the extracellular fluid volume and osmolarity, thus preventing effects of hypovolemia on tissue perfusion and arterial pressure maintenance. Descriptions found in textbooks[ 4 , 5 , 6 , 7 ] rarely mention several other RAS components:
‐ Circulating renin molecules form complexes with renin‐binding protein (reninBP),[ 16 , 17 , 18 ] whose physiological functions seem vague.
‐ Beside renin, prorenin is another peptide secreted by various tissues,[ 19 , 20 , 21 , 22 ] enzymatically inactive unless bound to specific prorenin receptors.
-
‐ Prorenin receptor can bind circulating renin and prorenin molecules[ 23 ]:
‐ Prorenin is active only after binding to its receptor, as a membrane bound enzyme of renin activity that provides Ang I to downstream tissues.
‐ Binding of circulating renin molecules to the prorenin receptor makes them immobile and increase their enzymatic activity for several times.[ 23 ]
‐ There are two Ang I converting enzymes, ACE and ACE2.[ 1 , 13 , 15 ] This means that Ang I can be converted into vasoconstrictive angiotensin II (Ang II), vasodilatory Ang(1–7) and Ang(1–9) of little biological activity that can be further converted into vasodilatory Ang(1–7).
‐ AT1R and AT2R are receptors of Ang II, while Ang(1–7) binds to the MAS1 receptor.[ 15 ] Ang II that binds to AT1R mediates arteriolar vasoconstriction, while binding to AT2R receptors induce diverse actions, not directly related to the vascular resistance.[ 13 , 15 , 24 ]
Tissue exposure to Angiotensin peptides is also linked to the secretion of paracrine and endocrine mediators (aldosterone, vasopressin and Endothelin‐1 (ET‐1). ET‐1 binds to two types of receptors (type A (ET1RA) and type B (ET1RB), its secretion depends on Ang II exposure and strain‐induced stress.[ 25 ]
Cardiopulmonary tissues are under stress of repetitive changes in the cell shape due to the heart action and breathing. Their tissue structure integrity strongly depends on thrombospondin‐1 (TSP1), an adhesive glycoprotein that mediates cell‐to‐cell and cell‐to‐matrix interactions. It is among key inhibitors of angiogenesis, binds to fibrinogen, fibronectin, laminin, type V collagen and integrins.[ 26 , 27 , 28 ] A direct pulmonary link between these two mediators seems to exist, since it has been reported that loss‐of‐function thrombospondin‐1 mutations were found in familial pulmonary hypertension,[ 29 ] a clinical condition treatable with bosentan, an ET‐1 receptor blocker.[ 30 ]
Two distinct paths seem to exist for circulating Ang I molecules[ 15 ]:
-
‐ If Ang I encounter ACE as the first converting enzyme:
‐ Ang II is formed, able to bind to the AT1R receptor that results in vasonconstriction, or to the AT2R receptor for other, diverse actions.
‐ The excess Ang II that did not bind to receptors can be converted by ACE2 to Ang(1–7) that binds to MAS1 receptors to induce vasodilation.
‐ If Ang I encounter ACE2 as the first converting enzyme, it is converted to inactive Ang(1–9), possibly later converted by ACE into vasodilatory Ang(1–7).
These relations suggest that Ang II‐mediated vasoconstriction via AT1R receptors is possible only in the time gap between the moment when Ang I encountered ACE, and the moment when the new Ang II interacts with ACE2. This might be an important restriction against recirculating of unspent Ang II from peripheral tissue into the pulmonary circuit.
Can RNA expression data of RAS components in human tissues provide new information?
As an attempt to estimate whether various tissues differ in their dependence on the RAS actions, the available data of RNA expression (https://biogps.org/) in 176 samples of human cells, tissues or whole organs[ 31 , 32 ] were used. The next step was to exclude all data regarding single cell types, since the basic idea was to compare tissues, structures and organs in their RNA patterns. This intervention has left 82 tissue/organ samples. Among them, gonads, placenta, retina and few other tissue samples showed atypical expression patterns of certain RAS components, thus suggesting that in these organs, RAS has some unique local role, not related to perfusion. After exclusion of these samples, the remaining 60 samples were used for further analysis.
Since several datasets existed for some RAS components, often with different distributions of expression values, further analyses were done with the dataset that showed strongest linear correlations with other RAS genes (listed in Table 1). This move was based on the idea that higher linear correlations suggest that tested data share similar expression information regarding the expression of RAS genes in 60 samples of human tissues.
Table 1.
The results of ANOVA for continuous variables, calculated for two EM clusters that divide 60 tissue samples, selected from the data of RNA expression (https://biogps.org/).[ 31 , 32 ]
| RNA expression | ANOVA for continuous variables | ||||||
|---|---|---|---|---|---|---|---|
| RAS component gene | Dataset name | Between sum of squares | df | Within sum of squares | df | F | P value |
| “Constriction” EM clusters from Figure 1 (18 tissue samples of high and 42 samples of low expression) | |||||||
| ProreninRec | 201442_s_at | 37.7520714 | 1 | 69.8697619 | 58 | 31.33859 | 0.00000 |
| ACE | 209749_s_at | 159.075627 | 1 | 202.094206 | 58 | 45.65389 | 0.00000 |
| AT1R | 208016_s_at | 1172.95753 | 1 | 1411.7723 | 58 | 48.18875 | 0.00000 |
| “Perfusion” EM clusters from Figure 2 (21 tissue samples of high and 39 samples of low expression) | |||||||
| Renin | 206367_at | 20 | 1 | 58 | 58 | 20.26553 | 0.00003 |
| ReninBP | 206617_s_at | 31 | 1 | 121 | 58 | 14.67651 | 0.00032 |
| ProreninRec | 201442_s_at | 24 | 1 | 84 | 58 | 16.39999 | 0.00015 |
| ACE | 209749_s_at | 50 | 1 | 311 | 58 | 9.27032 | 0.00350 |
| AT2R | 207293_s_at | 5 | 1 | 22 | 58 | 12.07863 | 0.00097 |
| ACE2 | 222257_s_at | 299 | 1 | 2413 | 58 | 7.18629 | 0.00955 |
| MAS1 | 208210_at | 40 | 1 | 296 | 58 | 7.75939 | 0.00721 |
| ET‐1 | 218995_s_at | 52415 | 1 | 385990 | 58 | 7.87601 | 0.00681 |
| ET‐1 RecA | 216235_s_at | 826 | 1 | 1700 | 58 | 28.19492 | 0.00000 |
| ET‐1 RecB | 204271_s_at | 32603 | 1 | 102602 | 58 | 18.43008 | 0.000068 |
| TSP1 | 201110_s_at | 1798934 | 1 | 20770724 | 58 | 5.02333 | 0.028850 |
“Constriction” EM clusters use only three genes, while “perfusion” clusters use ten genes. Only expressions of ACE and of prorenin receptors were found important in both instances of EM clustering.
Data for the 12 genes of RAS components expressed in 60 tissue samples were then organized in a spreadsheet used by StatSoft, Inc. (2011) STATISTICA (data analysis software system), version 10. www.statsoft.com. The basic idea that some tissues are more active in the expression of RAS components was tested by the method of expectation maximization (EM) clustering.[ 33 ] The v‐fold cross‐validation algorithm for automatically determining the number of clusters in the data (provided by the Statistica program) was applied during the clustering. Results of EM clustering are considered consistent if detected clusters were significantly different in expression data for all included genes, as shown in Table 1.
Expected and unexpected results of EM clustering
All included tissue samples express RAS components and related mediators, clearly suggesting that RAS, as an overall body regulator of circulation, is not limited to the components involved in the “classic” RAS paradigm.
When trying to find associations between ACE and the vasoconstrictive angiotensin II receptor 1 (AT1R) with other datasets, the only consistent clustering was found with the dataset of prorenin receptor, suggesting that expression of only these three molecules is linked to the regulation of overall vascular resistance. Tissue samples divided in two clusters, listed in Table 2, are shown in Figure 1, as a distribution of gene expression mean values. Cluster 2 in Figure 1 showed consistently higher expression data of the three genes, suggesting that organs or tissues of this cluster produce an increased quantity of vasoconstrictive RAS components, in comparison to the remaining cluster 1. As shown in Table 1, all three tested genes showed significant difference in RNA expression values between these two clusters. Tissue samples that form these two clusters are listed in Table 2 as “constriction” clusters of high and low expression. Tissues from the “constriction” cluster of high expression (listed in Table 2 and shown as the cluster 2 in Figure 1) are known to have very different perfusion rates, some of them also highly variable.
Table 2.
Distribution of 60 human tissue samples, selected from the data of RNA expression (https://biogps.org/),[ 31 , 32 ] according to two instances of EM clustering, presented as two pairs of clusters of high and low RNA expression for tested RAS and related genes
| Distribution of 60 tissue samples based on two EM clustering | “Perfusion” EM clusters from Table 1 and Figure 2, based on: Renin, ReninBP, ProreninRec, ACE, AT2R, ACE2, MAS1, ET‐1, ET‐1 RecA, ET‐1 RecB, TSP1 | |||
|---|---|---|---|---|
| High expression | Low expression | Total number of tissue samples | ||
|
“Constriction” EM clusters from Table 1 and Figure 1, based on: ProreninRec. ACE, AT1R |
High expression |
High expression of both clusterings (4 samples): Liver.2 Prostate.1 CardiacMyocytes.1 CardiacMyocytes.2 |
High expression of the “constriction” clustering (14 samples): Adipocyte.1 Adipocyte.2 AdrenalCortex.1 Adrenalgland.1 Adrenalgland.2 Appendix.2 Bonemarrow.2 CD105+_Endothelial.2 Pancreas.1 PancreaticIslet.2 Salivarygland.1 Tongue.1 Tongue.2 Tonsil.1 |
18 |
| Low expression |
High expression of the “perfusion” clustering (17 samples): Appendix.1 Liver.1 PancreaticIslet.1 SkeletalMuscle.2 Thyroid.2 Uterus.1 Uterus.2 Heart.1 Kidney.1 Kidney.2 Lung.1 Lung.2 SkeletalMuscle.1 Small_intestine.1 Small_intestine.2 SmoothMuscle.1 SmoothMuscle.2 |
Low expression of both clusterings (25 samples): AdrenalCortex.2 AtrioventricularNode.1 AtrioventricularNode.2 BDCA4+_DentriticCells.1 BDCA4+_DentriticCells.2 Bonemarrow.1 BronchialEpithelialCells.1 BronchialEpithelialCells.2 CD105+_Endothelial.1 Colon.1 Colon.2 Heart.2 Pancreas.2 Prostate.2 Salivarygland.2 Skin.1 Skin.2 Thymus.1 Thymus.2 Thyroid.1 Tonsil.2 Trachea.1 Trachea.2 Wholebrain.1 Wholebrain.2 |
42 | |
| Total number of tissue samples | 21 | 39 | 60 | |
Coronavirus infections targeting ACE2 molecules can then be particularly damaging in tissues that showed high expression of “perfusion” clustering genes in both tested samples (shown in gray cells): liver; cardiac myocytes; skeletal muscle; uterus; kidney; lung; small intestine and smooth muscle.
FIGURE 1.
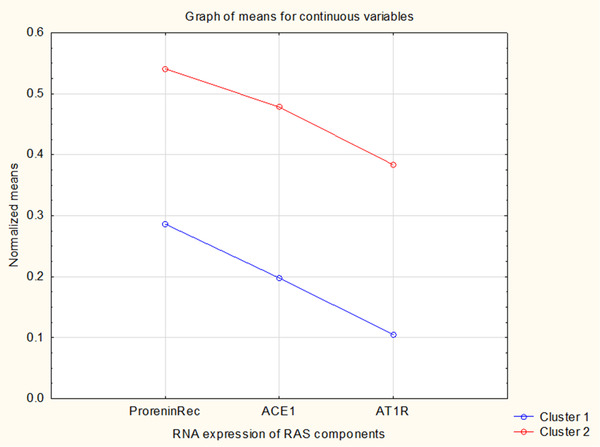
Based on only three genes linked to the vasoconstrictory RAS actions, EM clustering identified two clusters of RNA expression for 60 human tissue samples. ANOVA significance is shown in Table 1 and involved tissues are listed in Table 2
For expression data of dilatory ACE2 and MAS1 receptors, consistent clustering was found with datasets of Renin, ReninBP, Prorenin Receptor, ACE, AT2R, ET1, ET1RA, ET1RB and TSP1. Two detected clusters, listed in Table 2, are shown in Figure 2 as a distribution of mean gene expression values. Cluster 1 showed consistently higher expression of tested genes, suggesting that organs or tissues of this cluster produce an increased quantity of RAS components and related mediators, in comparison to the remaining cluster 2. Tissues from the “perfusion” cluster of high expression (shown as the cluster 1 in Figure 2) are known for their high perfusion rates, often also highly variable, so they require precise circulatory regulation, even in their smallest parts.
FIGURE 2.
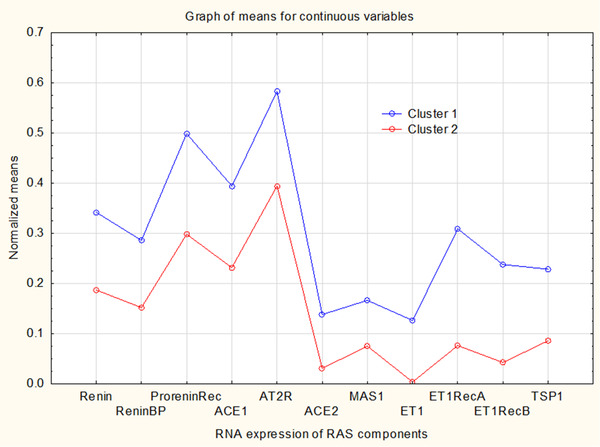
Based on ten genes linked to the vasodilatory RAS actions, EM clustering identified two clusters of RNA expression for 60 human tissue samples. ANOVA significance is shown in Table 1 and involved tissues are listed in Table 2
This leads us to an unexpected finding of these two clusterings. Beside ACE, both constrictive and dilatory RAS actions seem dependent on the tissue expression of prorenin receptors, suggesting that their presence might play the central role between RAS induced vascular constriction and dilation.
Tissue samples shown to belong to the low expression “perfusion” cluster and to the low “constriction” cluster (listed in Table 2) might be expected to have their perfusion controlled mainly by RAS components and mediators brought to them by blood from cardiopulmonary and other sources of high expression of RAS components. Tissues that belonged to this setting by the both tested samples (Table 2) are: Atrioventricular Node; BDCA4 ± DentriticCells; Bronchial Epithelial Cells; Colon; Skin; Thymus; Trachea; Whole Brain, mostly of stable and moderate perfusion rates.
Coronavirus infections targeting ACE2 molecules can be expected to be particularly damaging in tissues that showed high expression of “perfusion” clusterings genes in both tested samples (Table 2): Liver; Cardiac Myocytes; Skeletal Muscle; Uterus; Kidney; Lung; Small Intestine; Smooth Muscle. Reduced ACE2 actions due to coronavirus infection leads to vasoconstriction that might compromise local perfusion patterns in affected tissues.
SHOULD AN EXPANDED RAS PARADIGM INCLUDE TISSUE PERFUSION, BESIDE GENERAL CIRCULATION?
Proposition I: maintenance of the systemic vascular resistance is the “classic” RAS paradigm
Our systemic circulation can be divided in two parts, the high pressure segment, of aorta, arteries and arterioles and the low pressure segment of capillary beds and veins.[ 4 , 5 , 6 , 7 , 34 ] The former acts as a complex elastic chamber of a limited outflow, filled with pressurized fluid. Blood is incompressible, so it does not contain the pressure energy by itself. Instead of that, the pumping energy of the left ventricle is conducted and distributed by the blood along arteries as pulse waves, while some energy remains stored in the elasticity of arterial walls. The former is measurable as the systolic pressure, while the elastic tension of arterial walls, produced by the previous heart stroke, results in the diastolic pressure.
Table 3 estimates changes in the vascular resistance for a normal person while resting and during exercise, and for a hypertensive patient, while resting and during fever. It is based on published data on human circulation.[ 34 ] The cardiac output adapts to the ever changing integrated resistance of all arterial vessels. Calculated resistance values suggest that organs adapt their vascular resistance to maintain normal perfusion rates and hydrostatic capillary pressure. The arterial hydrostatic pressure reserve is an important survival feature that quickly compensates prompt changes in the overall vascular resistance. Running, quick changes in body posture, heavy work, fighting, bleeding, dehydration, all these settings require an increased heart output without decreasing the arterial pressure. This is achievable by making the overall peripheral circulation restricted and thus maintaining the diastolic pressure reserve in the arterial tree by the arteriolar tone controlled via nerves (catecholamines) and circulating mediators (Ang II, Ang(1–7), nitric oxide, ET‐1 etc.).
Table 3.
Changes in the vascular resistance in various organs are calculated for a normal person (while resting and during exercise) and for a hypertensive patient (resting and during fever)
| Normotensive person | Older hypertensive patient with a large allostatic load | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| During rest (ΔP = 97 mmHg) | Exercise (ΔP = 100 mmHg) | During rest (ΔP = 110 mmHg) | Fever (ΔP = 110 mmHg) | |||||||
| Circulatory circuit | Organs | Mass | Blood flow | Vascular resistance | Blood flow | Vascular resistance | Blood flow | Vascular resistance | Blood flow | Vascular resistance |
| kg | l/min | MPas/m3 | l/min | MPas/m3 | l/min | MPas/m3 | l/min | MPas/m3 | ||
| Peripheral circuit (data in grey cells from the ref. 34) | Liver | 2.6 | 1.50 | 517.33 | 1.50 | 533.33 | 1.50 | 586.67 | 1.50 | 586.67 |
| Kidneys | 0.3 | 1.26 | 615.87 | 1.26 | 634.92 | 1.26 | 698.41 | 1.26 | 698.41 | |
| Brain | 1.4 | 0.75 | 1034.67 | 0.75 | 1066.67 | 0.75 | 1173.33 | 0.75 | 1173.33 | |
| Skin | 3.6 | 0.46 | 1676.03 | 0.46 | 1727.86 | 0.46 | 1900.65 | 2.50 | 352.00 | |
| Skeletal muscle | 31 | 0.84 | 923.81 | 8.00 | 100.00 | 0.84 | 1047.62 | 2.00 | 440.00 | |
| Heart muscle | 0.3 | 0.25 | 3104.00 | 0.75 | 1066.67 | 0.25 | 3520.00 | 0.75 | 1173.33 | |
| Rest of the body | 23.8 | 0.34 | 2302.67 | 0.34 | 2373.89 | 0.34 | 2611.28 | 0.34 | 2611.28 | |
| Whole body | 63 | 5.40 | 143.70 | 13.06 | 61.26 | 5.40 | 162.96 | 9.10 | 96.74 | |
| Pulmonary circuit (ΔP = 9 mmHg) | 1.3 | 5.40 | 13.33 | 13.06 | 5.51 | 5.40 | 13.33 | 9.10 | 7.91 | |
The cardiac output adapts to the ever changing integrated resistance of all vessels. Calculated resistance values suggest that organs adapt their vascular resistance to maintain normal perfusion rates and hydrostatic capillary pressure. This means that increased allostatic load can lead to hypertension that will be compensated by increased resistance. This builds up the pressure reserve in the arterial tree that might improve acute physical responses and accelerate atherosclerosis. Pulmonary resistance needs to be protected from vasoconstrictors in the systemic circulation in febrile patients with allostatic loads. Otherwise, the pulmonary circuit might become the circulatory bottleneck, leading to compromised perfusion patterns and blood oxygenation in lungs
Proposition II: regulation of tissue perfusion patterns is the main role for both RAS branches
At the tissue level, precapillary arteries and arterioles have to sufficiently permissive to prevent ischemia, particularly in tissues with highly variable perfusion rates, like skeletal muscles. Regional circulation within any organ has to allow sufficiently uniform perfusion of capillary beds that all require similar pressure gradients for proper fluid exchange across capillary walls.[ 4 , 5 , 6 , 7 ] Despite arterial hypertension or hypotension, the capillary hydrostatic pressure remains balanced with the colloid osmotic pressure of plasma.
All mediators in the arterial blood can act only downstream from the point of entry, so we can expect that local perfusion regulators need to enter, or to be produced upstream from the respective arterioles, somewhere in small arteries that distribute blood toward precapillary arterioles.
Here proposed basic assumption is that due to the arterial tree complexity, any local tissue perfusion mechanism has to be positioned within the domain of a single functionally terminal artery to prevent interference with other, neighboring arterial vessels, and the mechanism needs to be situated upstream from precapillary arterioles. For this purpose, functional terminal arteries fit well. These vessels control perfusion of tissues that lack the already existent anastomoses, able to supply oxygenated blood from other arteries.[ 35 ] They are expected to exist in the brain, heart, liver, kidneys, spleen and intestines.
Between functionally terminal arteries and precapillary arterioles, narrow distributive arterial vessels inevitably reduce hydrostatic pressure since their resistance depends on their length, on the fourth potential of their radius and possibly on their shape as vessels. In more demanding tissues, like those from the first cluster in Figure 1, uniform perfusion patterns can be achieved if local vasoconstrictors and vasodilators are precisely introduced into the bloodstream at strategic points somewhere along regional and small arteries that distribute blood toward arterioles.
Based on previously described presumptions, the membrane‐bound prorenin receptors might be the key component of the local tissue perfusion regulation. Prorenin or renin binding to available membrane receptors provides a local source of Ang I for downstream enzymes. If so, stable tissue perfusion and acceptable hydrostatic capillary pressure would depend on the downstream expression of ACE/ACE2 in distributive vessels and of AT1R/AT2R/MAS1 receptors in vascular smooth muscle cells.
This notion would mean that all tissues reach in prorenin receptors are probable candidates for the local perfusion control based on downstream tissue expression of ACE and ACE2. Optimization of capillary pressure for small tissue areas depends on the pressure drop along distributive vessels, proportional to their distance from the terminal artery:
‐ Tissue parts closer to the pressure source need more arteriolar constriction, so more ACE than ACE2 in distributive vessels and more AT1R on downstream arterioles are expected.
‐ In tissues more remote from the arterial pressure source, sufficient perfusion needs more dilation, so less ACE than ACE2 molecules are expected in distributive vessels to provide sufficient amounts of Ang(1–7) and diminish the Ang II exposure. The expected downstream receptors include MAS1, AT2R and some AT1R.
This proposition requires that demanding tissues precisely express ACE and ACE2 in distributive vessels and AT1R/AT2R/MAS1 receptors in downstream arterioles, both according to the local RAS role. One example of distinct tissue specific expression of ACE and ACE2 was reported in the porcine eye.[ 36 ] ACE activity was markedly higher in the ciliary body than in retina, whereas ACE2 activities in the ciliary body and retina were at the same level. In the vitreous body ACE activity was manifold compared to that of ACE2.
Tissues with some ACE and ACE2 expression, but with little prorenin receptors, depends more on the renin presence in circulation to provide enough Ang I that can be locally converted to Angiotensin peptides. Their action downstream from converting enzymes depends on the local expression of AT1R/AT2R/MAS1 receptors in vascular smooth muscle cells.
ACE and ACE2 molecules are also enzymatically shed from membranes and act as soluble enzymes and ligands with diverse actions,[ 37 , 38 ] increased soluble ACE levels in the venous blood are among diagnostic tests for sarcoidosis. A possible role for the soluble ACE and ACE2 molecules might be to alter availability of Angiotensin peptides in the venous blood. Only ACE and ACE2 together can transform the remaining Ang I and Ang II into Ang(1–7) and thus reduce constriction in downstream tissues.
Proposition III: the central role of tissue scattered prorenin receptors is to provide stable local sources of Ang I
Acute stress can temporarily increase renin secretion from kidneys via adrenergic beta receptors. Renin binding to reninBP possibly ensure that any temporary excess of renin does not almost exclusively saturate prorenin receptors in highly perfused tissues, since this might disrupt their tissue perfusion, normally regulated by the enzymatic activity of bound prorenin molecules. Since complexes of renin with reninBP can spontaneously separate and thus liberate renin molecules anywhere in circulation, the available pool of reninBP can slowly increase the Ang I availability in all tissues, not just in the most perfused. This role is very similar to the role of sex hormone binding globulin in providing sex hormone actions in skin and other less perfused tissues.
If renin availability is chronically increased, surplus of renin molecules can bind to available unoccupied prorenin receptors. Binding immobilizes renin molecules and strongly augments their enzymatic activity,[ 23 ] thus locally increasing the Ang I presence, particularly in tissues rich in prorenin receptors.
If enough ACE is available in tissues, more Ang II molecules form and arterioles with AT1R receptors constrict, due to the Ang II binding to AT1R. The cardiovascular system adapts to this increase in arteriolar resistance by providing higher arterial pressure. Then, due to arteriolar constriction, the arterial hydrostatic pressure reserve remains elevated, while tissue perfusion remains at normal pressure, thus providing adequate capillary fluid exchange and oxygenation.
POSSIBLE REASONS FOR ALTERED RAS ACTIONS DURING CORONAVIRUS INFECTION
Allostasis as a compensated alteration of homeostasis
Disorders of circulation and metabolism, more common among elderly patients, seem directly to link mortality to the concept of allostasis, often defined as a condition of altered physiological balance that maintains body functions despite age‐accumulated dysfunctions, disorders or injuries.[ 39 ] Allostasis is an emerging pathophysiological and clinical concept, expanding beyond physiological homeostasis. If homeostasis is described as the state of steady internal, physical and chemical conditions maintained by any living systems, allostasis is a dynamic process that allows cells, tissues, organs or the whole body to adapt to the accumulated physiological, anatomical or behavioral changes,[ 39 , 40 ] until a new functional balance has been reached.
The main difference might be that homeostasis is often understood as a set of mechanisms that alter functions toward optimal condition, whatever environmental changes and individual health history. In allostasis, any functional setting is shaped by previous events, so the current state of allostatic adaptations results from a slow, lifelong development, imposed mainly by environmental factors. The concept of allostasis is often applied by using the allostatic load, defined as a burden of changes that results in the accumulation of various adaptations, important for the survival of an individual.[ 41 ]
Combination of arterial hypertension and chronic stress can still be found in textbooks as psychogenic hypertension.[ 42 ] This adaptation to increased circulatory pressures is limited to the arteries, since beyond the constricted arterioles, capillary hydrostatic pressure remains within the normal values. Only the arterial hydrostatic pressure reserve is elevated, as an evolutionary precaution, in case it might be suddenly needed. Plasma catecholamines display important metabolic effects at concentrations slightly above the normal resting levels,[ 43 ] by increasing total cholesterol, triglycerides and insulin, lowering HDL cholesterol, leading to insulin resistance and glucose intolerance. Sympathetic stimulation increases renin secretion from kidneys.[ 44 ] Beside beta blockers, other antihypertensive drugs like ACE inhibitors and Ang II receptor blockers, also show sympathoinhibitory actions.[ 43 , 45 ]
Unlike animals, humans are mainly challenged by social conflicts that produce a specific type of allostatic load, rarely solvable by the “fight or flight” response.[ 40 , 41 ] Social stress can be acute, intermittent or continuous and as such, it imposes an important health risk, since it alters exposure to stress hormones and cytokines, activities of the autonomic nervous system and RAS:
‐ In posttraumatic stress disorder (PTSD) patients at rest showed a baseline autonomic hyperarousal state comparable to that seen in the control subjects reaction to the stress model.[ 46 ] PTSD patients may get and maintain prolonged conditioned responses to various stressors during their lifespan, or become sensitized to reminders of past traumas.[ 47 ] Increased prevalence of the metabolic syndrome was found among police officers with severe PTSD symptoms.[ 48 ]
‐ Occupational stress is often associated with hypertension, cardiovascular disease[ 49 ] and metabolic syndrome.[ 50 ] Occupational noise can induce transitory hypertension in younger and permanent hypertension in older workers.[ 51 , 52 , 53 ] Severe occupational noise‐induced hearing loss is associated with the presence of type II diabetes.[ 54 ] These data suggest that chronic stress exposure can often lead to cardiovascular, metabolic or psychic disorders, all parts of the allostatic load.
The accumulated stress increases during the lifetime, so these stress‐related health issues are more common in elderly patients, possibly also due to remembering traumatic events. Indirect evidence for this proposition can be found in reports describing changes in plasma renin activity during therapy with beta blockers. In a retrospective analysis of patients with moderate to severe chronic heart failure (16 pts. without beta‐blockers or ACE‐inhibitors; 324 pts. with ACE‐inhibitors and 31 pts with beta‐blockers and ACE‐inhibitors), plasma renin levels were significantly lower in patients treated adjunctively with beta‐blockers.[ 55 ]
Among available beta blockers, nebivolol is a selective ß1‐receptor antagonist with vasodilating properties. In 24 patients with essential hypertension, nebivolol significantly reduced plasma renin, angiotensin II and aldosterone concentrations.[ 56 ] It was also reported among 805 hypertensive patients that plasma renin activity increased with valsartan therapy (54%–73%) and decreased with nebivolol (51%–65%) and the combination treatment (17%–39%). Beside that, plasma aldosterone decreased with valsartan (11%–22%); nebivolol (20%–26%), and 35% with maximum combination dose.[ 57 ] This clearly suggests that the activation of ß1‐receptors is important in the control of renin secretion in hypertensive patients and can relieve increased renin activity during treatment with AT1R blockers.
Are there separate types of patients with chronically increased renin secretion?
Physiological textbooks describe two disorders characterized by chronically increased renin secretion.[ 4 , 5 , 6 , 7 ] One example is the renovascular (Goldblatt's) hypertension and the second is diabetic nephropathy. Both disorders are not rare, but also not too common. Table 4 shows comparison of these two disorders with the psychogenic hypertension [ 42 ] and proposes therapeutic effect of nebivolol, a selective β1 receptor antagonist, on blood levels of renin, Ang I or Ang II, as a possible tool for detecting patients whose hypertension is mainly caused by the accumulated stress.
Table 4.
A possible approach to distinguishing among types of disorders associated with chronically increased renin secretion from kidneys
| Possible types of increased renin secretion | Renovascular or Goldblatt's hypertension | Diabetic nephropathy | “Psychogenic hypertension” | Comments | |
|---|---|---|---|---|---|
| The basic mechanism | Some nephrons are perfused under suboptimal pressure due to stenotic renal arteries | In all nephrons JGAs are activated due to increased sodium absorption in proximal tubules caused by glucose overload | Sympathetic stimulation of β1 receptors activates JGA in many nephrons | Renovascular hypertension is unique in its regional nature, the hypoperfused kidney alters arterial pressure, systemic resistance and kidney performance | |
| Kidney perfusion | normal or modestly decreased | increased | slightly decreased | Diabetic nephropathy forces both kidneys to supranormal perfusion and filtration, diuresis often relieves hypertension. ACE inhibitors or AT1R blockers can normalize filtration, but perfusion usually remains increased | |
| Kidney filtration rate | normal or modestly decreased | increased | normal | ||
| Arterial hypertension | always | in some cases | always | ||
| Systemic vascular resistance | increased | increased | increased | ||
| RAS components | Medication | Effects of medication on plasma levels of RAS components | |||
| high renin | ACE inhibitors | no reduction | no reduction | no reduction | Pressure normalization in all three types due to the reduced exposure of AT1R receptors in arterioles to the circulating Ang II |
| high Ang I | ACE inhibitors | no reduction | no reduction | no reduction | |
| high Ang II | ACE inhibitors | decreased AT II production by ACE | decreased AT II production by ACE | decreased AT II production by ACE | |
| high renin | AT1R blockers | no reduction | no reduction | no reduction |
Pressure normalization in all three types due to the blocked AT1R activation. Increased availability of AT II allows more interactions with AT2R and other receptors |
| high Ang I | AT1R blockers | no reduction | no reduction | no reduction | |
| high Ang II | AT1R blockers | no reduction | no reduction | no reduction | |
| high renin | Nebivolol | no reduction | no reduction | reduced | Pressure normalization by blocking β1 receptors only in the “psychogenic hypertension” |
| high Ang I | Nebivolol | no reduction | no reduction | reduced | |
| high Ang II | Nebivolol | no reduction | no reduction | reduced | |
Renovascular hypertension and Diabetic nephropathy are based on refs 4, 5, 6, 7, the proposed “Psychogenic hypertension” on refs 42, 56 and 57. Changes in plasma levels of renin, Ang I and Ang II, before and after the introduction of nebivolol might help in detection of patients with “Psychogenic hypertension”, due to an increased allostatic load.
A simple model of a cardiopulmonary malfunction in coronavirus infections
It is here assumed that the development of cardiorespiratory failure during infection with ACE2 targeting Corona viruses is linked to the previously altered expression of ACE/ACE2 enzymes and of AT1R/AT2R/MAS1 receptors in lungs, the heart muscle and possibly other tissues more dependable on the dilatory RAS activity.
The proposed description of the altered RAS setting in hypertensive patients is simple:
-
‐ In hypertensive patients with chronically increased renin activity (due to the allostatic load, or to the reduced nephron performance), peripheral tissues protect themselves from the increased perfusion pressure by increased expression of ACE in distributive vessels and more AT1R than MAS1 receptors in downstream arterioles. This is a necessary step to protect capillaries from chronically increased arterial pressure.
‐ The prevailing arteriolar constriction becomes an allostatic adaptation to the increased arterial pressure, potentially helpful in cases of acute danger or hypovolemia. In this setting, the downstream capillary bed remains under the normal perfusion pressure due to the arteriolar constriction.
‐ If myocardial ACE2 presence is not diminished by the coronavirus infection, the venous blood from the heart muscle, directly entering the right side of the heart via Coronary sinus and local Thebesian veins [ 58 ]reach in Ang(1–7) and thus protects lungs from the Ang II exposure by binding of Ang(1–7) to the pulmonary MAS1 receptors.
Described circulatory changes in a hypertensive patient result in circulation becoming more dependent on precise expression of ACE, ACE2, AT1R and MAS1. In this way, arterial hypertension, chronic stress, allostatic load or other preexisting conditions can make myocardial and pulmonary perfusion patterns more dependent on the local protective ACE2 action.
Coronavirus infections that target pulmonary and myocardial cells with membrane‐bound ACE2 molecules can alter the role of Angiotensin peptides in hypertensive patients:
‐ Any febrile condition acts as an acute stress that increases the renin secretion, more renin molecules bind to free prorenin receptors and thus much more Ang I molecules enter pulmonary circulation since renin bound on prorenin receptors is several times more active and immobile.[ 23 ]
-
‐ When coronavirus infection has diminished the cardiopulmonary ACE2 activity:
‐ Diminished distribution of cells with ACE2 molecules increases irregularities in pulmonary and myocardial perfusion patterns, while overall blood flow rates are reduced due to the increased Ang II exposure that constricts arterioles.
‐ In peripheral areas of the lungs and the heart muscle, perfusion becomes suboptimal due to reduced number of ACE2 enzymes, so less Ang(1–7) is formed and the downstream dilation is not induced.
‐ More Ang II is formed and the surplus can longer remain in circulation since both cellular and soluble ACE2 presence is diminished. The plasma half‐life of circulating Ang II molecules is extended. Some of them reenter the pulmonary circuit and compromise pulmonary circulation by vasoconstriction and by increasing local ET‐1 secretion that leads to pulmonary hypertension.
The unaffected constrictive ACE actions during coronavirus infection prevail and slowly exhaust the cardiopulmonary reserve. Based on this line of thoughts, it seems plausible that administration of recombinant human soluble ACE2,[ 59 ] might help COVID‐19 patients by competitive inhibition of virus binding to the cellular ACE2 and by enzymatic transformation of the circulating Ang II into vasodilatory Ang(1–7). The latter might shorten the Ang II survival in blood.
It was reported that among twelve COVID‐19 patients with pneumonia, that the viral load was crucial in determining the disease severity.[ 60 ] This study demonstrated that the combinations of the hypoalbuminemia, lymphopenia, and high concentrations of CRP and LDH upon hospital admission may predict more severe acute lung injury. Ang II levels in plasma were markedly increased, suggesting the RAS dysregulation.
On the other hand, in a report of RAS activity in 31 COVID‐19 patients of various illness severity and 27 controls, plasma levels of RAS components (Ang I, Ang II, Ang(1–7), Ang(1–5), ACE2 and (Ang I + Ang II) as the Plasma renin activity).[ 61 ] The control group and the COVID‐19 group were each divided in three subgroups (one with ACE inhibitors, one with AT1R blockers, and one without both medications). No major changes in RAS activity in plasma including ACE2 were found in COVID‐19 patients. Plasma ACE2 activity was increased only in COVID‐19 patients treated with an ACE inhibitor.
Data of these two studies suggest that in COVID‐19 patients, a single measurement of the circulating ACE2 and other RAS components cannot detect dysregulated RAS activation, except in patients that later developed pneumonia.[ 60 ]
A dangerous dysbalance between RAS branches can develop in any patient, along infection, so more than a single measurement might be needed. As proposed in Table 4, dynamic changes of plasma values of RAS components might be more informative in the introduction of nebivolol therapy. The same approach might prove predictive of complications during coronavirus infection.
CONCLUSIONS AND OUTLOOK
The presented interpretation follows several reviews that point at the prorenin receptor as a RAS component of many roles.[ 18 , 19 , 23 ] The proposed description is in several points compatible with published concepts,[ 56 , 62 ] that also consider RAS as composed of two opposing arms or branches: the pressor arm, acting via the AT1R receptor and the vasodepressor arm of complex actions that include vasodilatory, antiproliferative, antifibrotic, and antithrombotic actions, mainly via the MAS receptor.
The main difference is that the presented description is focused on differences in tissue dependence on the RAS actions and on the topological hierarchy that allows mediators to act selectively on downstream tissues or organs.
It is here assumed that RAS actions in our body are centered on the tissue presence of prorenin receptors, as a unique setting when two independent and counteracting RAS arms use the same receptor, with one key difference: the binding of prorenin starts its enzymatic function, while binding of soluble and functional renin increases its activity several times.[ 23 ] Since in healthy individuals renin secretion is limited in quantity and duration due to the precise JGA activation mechanisms, most of the prorenin receptors are expected to be occupied by prorenin molecules. In patients with chronically increased renin secretion due to renovascular JGA activation, chronic stress or diabetic nephropathy, circulating renin binds to unoccupied prorenin receptors and thus increases the overall tissue capacity for producing Ang I. These changes adapt the arterial tree to arterial hypertension due to an increased cardiac afterload and vascular resistance.
CONFLICT OF INTEREST
The author declares no conflict of interest.
ACKNOWLEDGMENT
This paper was financed through grant VIF2018MEFOS02 from the Croatian Ministry of Science, Education and Sport.
Kurbel, S . (2021). The renin‐angiotensin system in COVID‐19: Why ACE2 targeting by coronaviruses produces higher mortality in elderly hypertensive patients? BioEssays, 43, e2000112. 10.1002/bies.202000112
DATA AVAILABILITY STATEMENT
Data about RNA expression in samples of human tissues, available at Biogps (http://biogps.org/) were used. Names of the used datasets are listed in Table 1.
REFERENCES
- 1. Gheblawi, M. , Wang, K. , Viveiros, A. , Nguyen, Q. , Zhong, J. ‐ C. , Turner, A. J. , … Oudit, G. Y. (2020). Angiotensin‐Converting Enzyme 2: SARS‐CoV‐2 Receptor and regulator of the renin‐angiotensin system. Circ. Res., 126, 1456–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chan‐Yeung, M. , & Xu, R. H. (2003). SARS: epidemiology. Respirology, 8, S9–S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Verity, R. , Okell, L. C. , Dorigatti, I. , Winskill, P. , Whittaker, C. , Imai, N. , … Ferguson, N. M. (2020). Estimates of the severity of coronavirus disease 2019: a model‐based analysis. Lancet Infect. Dis., 20, 669–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hall, J. (2016). Guyton And Hall Textbook Of Medical Physiology [13Th Ed.], Elsevier, Amman, Jordan. [Google Scholar]
- 5. Barrett, K. , & Ganong, W. (2019). Ganong's Review Of Medical Physiology, Mcgraw‐Hill Medical, New York. [Google Scholar]
- 6. Boron W. F., Boulpaep E. L., Eds. (2017). Medical Physiology, Elsevier, Philadelphia, PA. [Google Scholar]
- 7. Costanzo, L. S. (2018). Physiology, Elsevier, Philadelphia, PA. [Google Scholar]
- 8. Rea, I. M. , Gibson, D. S. , McGilligan, V. , McNerlan, S. E. , Alexander, H. D. , & Ross, O. A. (2018). Age and age‐related diseases: Role of inflammation triggers and cytokines. Front. Immunol., 9, 586. 10.3389/fimmu.2018.00586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Grassi, G. (2004). Counteracting the sympathetic nervous systemin essential hypertension. Curr. Opin. Nephrol. Hypertens., 13, 513–519. [DOI] [PubMed] [Google Scholar]
- 10. Zimmet, P. , Boyko, E. , Collier, G. , & Courten, M. (1999). Etiology of the metabolic syndrome: Potential role of insulin resistance, leptin resistance, and other players. Ann. N. Y. Acad. Sci., 892, 25–44. [DOI] [PubMed] [Google Scholar]
- 11. Grassi, G. (2006). Sympathetic Overdrive and Cardiovascular Risk in the Metabolic Syndrome. Hypertension Res., 29, 839–847. [DOI] [PubMed] [Google Scholar]
- 12. Egan, B. (2003). Insulin resistance and the sympathetic nervous system. Curr. Hypertens. Rep., 5, 247–254. [DOI] [PubMed] [Google Scholar]
- 13. Li, X. C. , Zhang, J. , & Zhuo, J. L. (2017). The vasoprotective axes of the renin‐angiotensin system: Physiological relevance and therapeutic implications in cardiovascular, hypertensive and kidney diseases. Pharmacological Res., 125, 21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fyhrquist, F. , & Saijonmaa, O. (2008). Renin‐angiotensin system revisited. J. Intern. Med. 264, 224–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chappell, M. C. (2007). Emerging Evidence for a Functional Angiotensin‐Converting Enzyme 2‐Angiotensin‐(1‐7)‐Mas Receptor Axis. Hypertension, 50, 596–599. [DOI] [PubMed] [Google Scholar]
- 16. Inoue, H. , Takahashi, S. , Fukui, K. , & Miyake, Y. (1991). Genetic and Molecular Properties of Human and Rat Renin‐Binding Proteins with Reference to the Function of the Leucine Zipper Motif. The Journal of Biochem., 110, 493–500. [DOI] [PubMed] [Google Scholar]
- 17. Takahashi, S. , Kumagai, M. , Shindo, S. , Saito, K. , & Kawamura, Y. (2000). Renin Inhibits N‐Acetyl‐d‐Glucosamine 2‐Epimerase (Renin‐Binding Protein). J. Biochem., 128, 951–956. [DOI] [PubMed] [Google Scholar]
- 18. Knoll, A. (1997). Human Renin Binding Protein: Complete Genomic Sequence and Association of an Intronic T/C Polymorphism with the Prorenin Level in Males. Hum. Mol. Genet., 6, 1527–1534. [DOI] [PubMed] [Google Scholar]
- 19. Nguyen, G. , & Danser, A. H. J. (2008). Prorenin and (pro)renin receptor: a review of available data from in vitro studies and experimental models in rodents. Experimental Physiol., 93, 557–563. [DOI] [PubMed] [Google Scholar]
- 20. Peters, J. (2017). The (pro)renin receptor and its interaction partners. Pflugers Arch ‐ Eur J Physiol, 469, 1245. [DOI] [PubMed] [Google Scholar]
- 21. Ramkumar, N. , & Kohan, D. E. (2019). The (pro)renin receptor: an emerging player in hypertension and metabolic syndrome. Kidney Int., 95, 1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xu, Q. , Jensen, D. D. , Peng, H. , & Feng, Y. , (2016). The critical role of the central nervous system (pro)renin receptor in regulating systemic blood pressure. Pharmacol. Ther., 164, 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nguyen, G. , Delarue, F. , Burcklé, C. , Bouzhir, L. , Giller, T. , & Sraer, J. ‐ D. (2002). Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J. Clin. Invest., 109, 1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gerdin, A. (2010). The Sanger Mouse Genetics Programme: high throughput characterisation of knockout mice. Acta Ophthalmologica, 88, 0. [Google Scholar]
- 25. Cheng, C. ‐ P. , Ukai, T. , Onishi, K. , Ohte, N. , Suzuki, M. , Zhang, Z. ‐ S. , … Little, W. C. (2001) The role of ANG II and endothelin‐1 in exercise‐induced diastolic dysfunction in heart failure. Am. J. Physiol.: Heart Circ. Physiol., 280, H1853. [DOI] [PubMed] [Google Scholar]
- 26. Matsuo, Y. , Tanaka, M. , Yamakage, H. , Sasaki, Y. , Muranaka, K. , Hata, H. , … Satoh‐Asahara, N. (2015). Thrombospondin 1 as a novel biological marker of obesity and metabolic syndrome. Metabolism, 64, 1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bonnefoy, A. , Hantgan, R. , Legrand, C. , & Frojmovic, M. M. (2000). A Model of Platelet Aggregation Involving Multiple Interactions of Thrombospondin‐1, Fibrinogen, and GPIIbIIIa Receptor. J. Biol. Chem., 276, 5605. [DOI] [PubMed] [Google Scholar]
- 28. Maile, L. A. , Allen, L. B. , Hanzaker, C. F. , Gollahon, K. A. , Dunbar, P. , & Clemmons, D. R. (2010) Glucose Regulation of Thrombospondin and Its Role in the Modulation of Smooth Muscle Cell Proliferation. Experim. Diabetes Res., 2010, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Maloney, J. P. , Stearman, R. S. , Bull, T. M. , Calabrese, D. W. , Tripp‐Addison, M. L. , Wick, M. J. , … Loyd, J. E. (2012) Loss‐of‐function thrombospondin‐1 mutations in familial pulmonary hypertension. Am. J. Physiol.‐Lung Cell. Mol. Physiol. 302, L541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Channick, R. N. , Simonneau, G. , Sitbon, O. , Robbins, I. M. , Frost, A. , Tapson, V. F. , … Rubin, L. J. (2001). Effects of the dual endothelin‐receptor antagonist bosentan in patients with pulmonary hypertension: a randomised placebocontrolled study. Lancet, 358, 1119. [DOI] [PubMed] [Google Scholar]
- 31. Wu, C. , Jin, X. , Tsueng, G. , Afrasiabi, C. , & Su, A. I. (2015) BioGPS: building your own mash‐up of gene annotations and expression profiles. Nucleic Acids Res., 44, D313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Su, A. I. , Wiltshire, T. , Batalov, S. , Lapp, H. , Ching, K. A. , Block, D. , … Hogenesch, J. B. (2004). A gene atlas of the mouse and human protein‐encoding transcriptomes. Proc. Natl. Acad. Sci. USA, 101, 6062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Celeux, G. , & Govaert, G. (1992). A classification EM algorithm for clustering and two stochastic versions. Computational Statistics Data Anal., 14, 315–332. [Google Scholar]
- 34. Bard, P. (1961). Medical Physiology, Mosby, St. Louis, Mo.
- 35. Mulvany, M. J. , & Aalkjaer, C. (1990). Structure and function of small arteries. Physiological Rev., 70, 921. [DOI] [PubMed] [Google Scholar]
- 36. Luhtala, S. , Vaajanen, A. , Oksala, O. , Valjakka, J. , & Vapaatalo, H. (2009). Activities of Angiotensin‐Converting Enzymes ACE1 and ACE2 and Inhibition by Bioactive Peptides in Porcine Ocular Tissues. J. Ocul. Pharmacol. Ther., 25. [DOI] [PubMed] [Google Scholar]
- 37. Hamming, I. , Cooper, M. , Haagmans, B. , Hooper, N. , Korstanje, R. , Osterhaus, A. , … van Goor, H. (2007). The emerging role of ACE2 in physiology and disease. J. Pathol., 212, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Danilov, S. M. , Gordon, K. , Nesterovitch, A. B. , Lünsdorf, H. , Chen, Z. , Castellon, M. , … Sturrock, E. D. (2011). An Angiotensin I‐Converting Enzyme Mutation (Y465D) Causes a Dramatic Increase in Blood ACE via Accelerated ACE Shedding. PLoS One, 6, e25952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. McEwen, B. (1993). Stress and the IndividualMechanisms Leading to Disease. Arch. Intern. Med., 153, 2093–2101. [PubMed] [Google Scholar]
- 40. McEwen, B. , & Wingfield, J. (2003). The concept of allostasis in biology and biomedicine. Hormones and Behavior, 43, 2–15. [DOI] [PubMed] [Google Scholar]
- 41. Goldstein, D. S. , & Kopin, I. J. (2018). Linking stress, catecholamine autotoxicity, and allostatic load with neurodegenerative diseases: a focused review in memory of Richard Kvetnansky. Cell. Mol. Neurobiol., 38(1), 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nalivaiko, E. (2011). Animal models of psychogenic cardiovascular disorders: what we can learn from them and what we cannot. Clin. Exp. Pharmacol. Physiol., 38, 115–125. [DOI] [PubMed] [Google Scholar]
- 43. Kjeldsen, S. E. , Rostrup, M. , Moan, A. , Mundal, H. H. , Gjesdal, K. , & Eide, I. K. (1992). The sympathetic nervous system may modulate the metabolic cardiovascular syndrome in essential hypertension. J. Cardiovasc. Pharmacol., 20, S32–S39. [PubMed] [Google Scholar]
- 44. Mitchell, G. F. (2008). Effects of central arterial aging on the structure and function of the peripheral vasculature: implications for end‐organ damage. J. Appl. Physiol., 105, 1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Grassi, G. (2004). Counteracting the sympathetic nervous systemin essential hypertension. Curr. Opin. Nephrol. Hypertens., 13. [DOI] [PubMed] [Google Scholar]
- 46. Cohen, H. , Kotler, M. , Matar, M. , Kaplan, Z. , Loewenthal, U. , Miodownik, H. , & Cassuto, Y. (1998). Analysis of heart rate variability in posttraumatic stress disorder patients in response to a trauma‐related reminder. Biol. Psychiatry, 44, 1054–1059. [DOI] [PubMed] [Google Scholar]
- 47. Shalev, A. , Peri, T. , Gelpin, E. , Orr, S. , & Pitman, R. (1997). Psychophysiologic assessment of mental imagery of stressful events in israeli civilian posttraumatic stress disorder patients. Comprehensive Psychiatry, 38. [DOI] [PubMed] [Google Scholar]
- 48. Almadi, T. , Cathers, I. , & Chow, C. (2013). Associations among work‐related stress, cortisol, inflammation, and metabolic syndrome. Psychophysiology, 50, 821–830. [DOI] [PubMed] [Google Scholar]
- 49. Kawada, T. (2015). Job stress and the metabolic syndrome with special reference to sex and age. Int. J. Cardiol., 194, 63–64. [DOI] [PubMed] [Google Scholar]
- 50. Chandola, T. , Brunner, E. , Marmot, M. (2006). Chronic stress at work and the metabolic syndrome: prospective study. BMJ, 332, 521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tomei, F. , Fantini, S. , Tomao, E. , Baccolo, T. , & Rosati, M. (2000). Hypertension and Chronic Exposure to Noise. Archives Environ. Health: An Inter. J., 55, 319–325. [DOI] [PubMed] [Google Scholar]
- 52. Fogari, R. , Zoppi, A. , Corradi, L. , Marasi, G. , Vanasia, A. , & Zanchetti, A. (2001). Transient but not sustained blood pressure increments by occupational noise. An ambulatory blood pressure measurement study. J. hypertension, 19(6), 1021–1027. [DOI] [PubMed] [Google Scholar]
- 53. Fogari, R. , Zoppi, A. , Vanasia, A. , Marasi, G. , & Villa, G. (1994). Occupational noise exposure and blood pressure. J. hypertension, 12(4), 475–480. [PubMed] [Google Scholar]
- 54. Ishii, E. , Talbott, E. , Findlay, R. , D'Antonio, J. , & Kuller, L. (1992). Is NIDDM a risk factor for noise‐induced hearing loss in an occupationally noise exposed cohort?. Sci. Total Environ., 127, 155–165. [DOI] [PubMed] [Google Scholar]
- 55. Teisman, A. C. H. , van Veldhuisen, D. J. , Boomsma, F. , de Kam, P. J. , Tjeerdsma, G. , … van Gilst, W. H. (2000). Chronic beta‐blocker treatment in patients with advanced heart failure. Int. J. Cardiol., 73, 7–12. [DOI] [PubMed] [Google Scholar]
- 56. Mose, F. H. , Jensen, J. M. , Therwani, S. , Mortensen, J. , Hansen, A. B. , Bech, J. N. , & Pedersen, E. B. (2015). Effect of nebivolol on renal nitric oxide availability and tubular function in patients with essential hypertension. Br. J. Clin. Pharmacol., 80(3), 425–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Giles, T. D. , Bakris, G. , Oparil, S. , Weber, M. A. , Li, H. , … Ferguson, W. G. (2015). Correlations of plasma renin activity and aldosterone concentration with ambulatory blood pressure responses to nebivolol and valsartan, alone and in combination, in hypertension. J. Am. Soc. Hypertens., 9, 845–854. [DOI] [PubMed] [Google Scholar]
- 58. Ansari, A. (2001). Anatomy and clinical significance of ventricular Thebesian veins. Clin. Anat., 14, 102–110. [DOI] [PubMed] [Google Scholar]
- 59. Sullivan, R. D. , Mehta, R. M. , Tripathi, R. , Reed, G. L. , & Gladysheva, I. P. (2019). Renin Activity in Heart Failure with Reduced Systolic Function—New Insights. IJMS, 20, 3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Liu, Y. , Yang, Y. , Zhang, C. , Huang, F. , Wang, F. , Yuan, J. , … Liu, L. (2020). Clinical and biochemical indexes from 2019‐nCoV infected patients linked to viral loads and lung injury. Sci. China Life Sci., 63, 364–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kintscher, U. , Slagman, A. , Domenig, O. , Röhle, R. , Konietschke, F. , Poglitsch, M. , & Möckel, M. (2020). Plasma Angiotensin Peptide Profiling and ACE (Angiotensin‐Converting Enzyme)‐2 Activity in COVID‐19 Patients Treated With Pharmacological Blockers of the Renin‐Angiotensin System. Hypertension, 76, e34–e36. 10.1161/hypertensionaha.120.15841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cerrato, B. D. , Frasch, A. P. , Nakagawa, P. , Longo‐Carbajosa, N. , Peña, C. , Höcht, C. , & Gironacci, M. M. , (2012). Angiotensin‐(1–7) upregulates central nitric oxide synthase in spontaneously hypertensive rats. Brain Res., 1453, 1–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data about RNA expression in samples of human tissues, available at Biogps (http://biogps.org/) were used. Names of the used datasets are listed in Table 1.