

Abstract
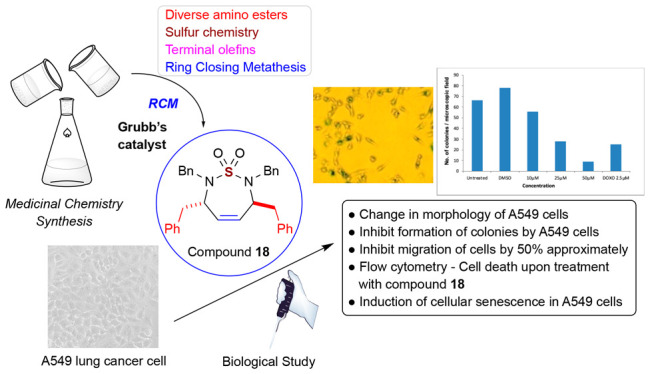
The sulfamide functional group has been extensively employed in organic synthesis to discover probes and drugs in various applications such as cancer, human immunodeficiency virus (HIV), virus, and diabetes. Herein, we describe the synthesis of 7-membered symmetric and unsymmetric sulfamide compounds and their biological evaluation through the National Cancer Institute (NCI) panel of 60 human tumor cell lines (NCI-60) and the mechanism of action study. The results of a study from the NCI-60 cell line exhibited that many synthesized cyclic sulfamide compounds inhibited breast cancer (MDA-MB-468). The mechanism of action study of a representative compound 18 showed the inhibition of proliferation and apoptosis in A549 lung cancer cells.
Keywords: Anticancer activity, Cyclic sulfamide, NCI-60 cell line, A549 lung cancer
The sulfamide is a broadly accepted functional moiety in medicinal chemistry for the design and identification of various biologically active compounds. Sulfamide-based inhibitors have been designed for various enzymes such as carbonic anhydrides (CAs),1−6 protease,7−10 γ-secretase,10 serine-protease,11 thrombin,12 metalloproteinase carboxypeptidase A (CPA),13 matrix metalloproteinase (MMP),13−15 steroid sulfatase (STS),16,17 protein tyrosine phosphatase inhibitors,17,18 etc. Also, there has been a report on the anticancer activity of sulfonamide-based compounds.19 In many cases, a free or substituted sulfamide moiety contributes to binding to the active site of a protein showing better affinity and exhibiting higher aqueous solubility and bioavailability. For example, cyclic sulfamide inhibitors interact with the catalytic aspartic acid residue of HIV protease.7,8,19,20 Cyclic sulfamides have been applied in medicinal chemistry to identify several valuable compounds as shown in Figure 1. Prominently, cyclic sulfamides have been used as a structural template applicable for the inhibitor development such as HIV, metalloprotease, serine proteases, and γ-secretase. Compound I was discovered by Merck as a γ-secretase targeting therapy in Alzheimer’s disease (AD) in 2005.21 The series of this cyclic sulfamide was one of the structural substitute analogues to the acyclic sulfonamide derivatives which were reported in the same year.22 Next, compound II, an orally bioavailable Factor Xa (FXa) inhibitor, was developed by the Yamanouchi Pharmaceutical Co. Ltd. (currently known as Astellas Pharma Inc.).23,24 FXa, a serine protease, is involved in blood clotting by cleaving prothrombin into active thrombin. Novel 5-membered cyclosulfamide compound III as a Norwalk virus inhibitor was developed via structure–activity relationship (SAR) studies.25N-Cyclopropyl sulfamide compound IV having a fused 6-membered ring system is another example of γ-secretase inhibitors studied by Merck for the treatment of AD.26 The Korea Institute of Science and Technology (KIST) has developed carbapenem compounds V containing a pendant cyclic sulfamide, which was found to display potent antibacterial activity.27
Figure 1.

Representative examples of biologically active cyclic sulfamides.
As discussed above, sulfamides have been shown to provide biological activities against different enzymes and have been developed for various diseases including cancer. However, cyclic sulfamides have not been actively investigated in cancer drug development. With this need, we set our objective in this study to explore the anticancer activity of seven-membered sulfamide compounds. Due to the intrinsic properties of this highly polarized moiety when attached to an organic scaffold, it is an attractive consideration to incorporate the sulfamide moiety for inducing desired physiochemical properties such as enhanced water solubility, better bioavailability, etc., to the druglike compounds. Encouraged by the possibilities offered by this moiety, we synthesized a set of new seven-member cyclic sulfamide compounds and investigated their potential as anticancer agents. The structural modifications were based on the assumption that the variation of physicochemical properties and thereby different biological activity could be achieved with a change in substituents. Herein we are describing the results of our detailed studies.
We have devoted our efforts to the development of synthetic methods for the sulfur-containing small molecule compounds in various ways.28−30 The synthesis of amino ester-derived C2-symmetric and unsymmetric sulfamides and further synthesis are described in Scheme 1. Amino esters are useful chiral auxiliary groups that are employed in medicinal chemistry synthesis and have been utilized in a wide range of areas such as peptide synthesis, asymmetric synthesis, and medicinal chemistry. The construction of chiral carbons in different positions of cyclic sulfamide was effectively possible by the employment of amino ester. Condensation of a slight excess of amino ester HCl salt with SO2Cl2 in CH2Cl2 at 0 °C provided sulfamides 3 and 4 in high yield. For the monobenzylation, 1 equiv of benzyl chloride was consumed to afford 7 which was used for next benzylation with p-methoxybenzyl chloride to furnish unsymmetric sulfamide 8. Benzylation using benzyl bromide (BnBr) or benzyl chloride (BnCl) and reduction by the addition of lithium aluminum hydride (LAH) carefully into a reaction mixture at 0 °C were the straightforward method to generate primary alcohol intermediates 9–11. When Dess–Martin periodinane or Swern oxidation reagent is added into analogues 9–11, two hydroxyl groups were transformed to the aldehydes to give intermediates 12–14. Under the Wittig reaction condition,31,32 the reaction of 5–7 using n-butyl lithium and Ph3P+CH3Br– afforded analogues 18–20, respectively. For the construction of seven-membered ring 18–20 from dienes 15–17, we explored ring closing metathesis (RCM) utilizing Grubbs catalyst II (3–6 mol %) in heating benzene. The highlight of our synthetic approach represent the innovative way to construct cyclic sulfamide compounds.
Scheme 1. Synthesis of Cyclic Sulfamide Compounds.

SO2Cl2, Et3N, CH2Cl2.
Benzyl bromide (BnBr) or benzyl chloride (BnCl), K2CO3, CH3CN.
PMBCl, K2CO3, CH3CN.
LiAlH4, THF.
Swern oxidation or Dess–Martin periodinane.
Ph3P+CH3Br–, n-BuLi, THF.
Grubbs Cat-II, benzene.
OsO4, NMO, H2O/acetone.
m-CPBA, CH2Cl2.
VCl3(THF)3, CH2Cl2, Zn; PMBCl = p-methoxybenzyl chloride, THF = tetrahydrofuran, m-CPBA = m-chloroperoxybenzoic acid, DIAD = diisopropyl azodicarboxylate, TFA = trifluoroacetic acid, NMO = N-methylmorpholine.
Next, we performed dihydroxylation and epoxidation from 18 and 19 which gave dihydroxyl compound 21 and 22 and epoxy sulfamide 23 and 24. Reaction of the aldehyde 12 employing Pinacol coupling with a vanadium(II) reagent, [V2Cl3(THF)6]192[Zn2Cl6],33 produced a single diastereomer 25. The nuclear Overhauser effect (NOE) study allowed us to determine the stereochemistry of each chiral center around a seven-membered ring as shown in Scheme 1.
Synthesis of seven-membered cyclic sulfamide compound 34 was conducted from the reaction of chlorosulfonyl isocyanate (CSI), l-valine methyl ester, and t-butanol to generate Boc-group protected sulfamide intermediate 26 in good yields. Sulfamide 27 was then successfully obtained by the reaction of methyl (S)-2-hydroxy-4-methylpentanoate and sulfamide 26 under the Mitsunobu reaction condition.34,35 The Mitsunobu reaction is a well-known method to invert the configuration of alcohols. Benzylation on the sulfamide nitrogen gave sulfamide 28 in 77% yield, followed by the deprotection of the Boc group using TFA at room temperature to furnish intermediate 29 in high yield. It was washed with saturated aqueous NaHCO3 solution to remove TFA salt and further benzylation constructed meso sulfamide 30. Two methyl ester groups were transformed into primary hydroxyl groups by the LAH reduction to furnish compound 31. This diol compound was converted to the unstable dialdehyde compound 32 through the Swern oxidation at low temperature (−78 °C). Wittig reaction of 32 generated terminal olefin compound 33 which was converted to cyclized meso sulfamide 34 by RCM using the second-generation Grubbs catalyst in refluxing benzene (see Scheme 2). Other compounds shown in this paper were synthesized as in the cited methods in the Supporting Information. Compounds 18–46 including the Cancer Chemotherapy National Service Center (NSC) numbers are illustrated in Table 1.
Scheme 2. Synthesis of C2-Symmetric Cyclic Sulfamide 34.

Methyl (S)-2-hydroxy-4-methylpentanoate, PPh3, DIAD, THF.
BnCl or BnBr, K2CO3, CH3CN.
TFA, CH2Cl2.
BnCl or BnBr, K2CO3, CH3CN.
LiAlH4, THF.
Oxalyl chloride, DMSO, Et3N, CH2Cl2,-78 °C.
Ph3P+CH3Br–, n-BuLi, THF.
Grubbs Catalyst-II, benzene, reflux.
Table 1. Compounds Submitted to the NCI-60 Cell Lines.

The standard cell culture protocol of the National Cancer Institute panel of 60 human tumor cell lines (NCI-60) and for drug sensitivity analysis in these cell lines has been described previously in the hereby cited literature.36−38 All cultured cells were incubated in a humidified incubator with 5% CO2 in the air at 37 °C. To confirm the absence of contamination, the cell lines were tested for Mycoplasma periodically. The NCI-60 cancer cell lines were maintained in RPMI 1640 medium with 5% fetal bovine serum (FBS) and 2 mM l-glutamine (Lonza, Walkersville, MD). Cells were distributed into 96-well microtiter plates at a proper density and maintained for 1 day with no drug. Some of the plates were then administered to decide the cell density at time zero. Concisely, the panel is systematized into nine subpanels on behalf of diverse histologies: leukemia, melanoma, and cancers of lung, colon, kidney, ovary, breast, prostate, and central nervous system. All test compounds prepared in DMSO were added to cultivated cells at five concentrations with 10-fold dilutions, the highest being 10–4 M and the others being 10–5, 10–6, 10–7, and 10–8 M. Control cells were treated with DMSO alone and incubated under standard culture conditions for 48 h. The percentage growth inhibition was determined relative to cells without drug treatment and the time zero control. After the termination of the assay by addition of cold trichloroacetic acid, and the cells were fixed and stained with the treatment of sulforhodamine B. The bound stain is solubilized, and the absorbance detection of samples can be read in an automated microplate reader. 50% growth inhibition (GI50, cytostatic parameter) is the concentration for 50% max inhibition of cell proliferation, and it was calculated from time zero, control growth, and the five-concentration level absorbance. Inhibitory concentrations (LC50, cytotoxic parameter) are the concentration of drug or chemical that kills 50% of cells with a one-time exposure and represent the average of two independent experiments. Cell lines with a horizontal bar pointing to the left have a Gl50 that is greater than the mean and are more resistant than the mean, whereas cell lines with a horizontal bar pointing to the right have a Gl50 that is less than the mean and are more sensitive. The in vitro NCI-60 cell screening is a two-stage process started with the evaluation of the submitted compound at a single high dose (one-dose screen, 10–5 μM) in the full cell panel. Compounds showing compelling growth inhibition in the one-dose screen are evaluated for five-dose screening (five concentration levels). Only the compounds exhibiting more than 60% of growth inhibition in at least 8 tumor cell lines were selected for further testing, and the others were assumed inactive. All 22 compounds were initially evaluated at a single dose of 10 μM. Only the compounds exhibiting more than 60% of growth inhibition in at least 8 tumor cell lines were selected for further testing, and the others were assumed inactive. All 22 compounds were initially evaluated at a single-dose screening. Four of our compounds showed significant antiproliferative activity at 10 μM. The results of the single-dose screen are given in the Supporting Information. Subsequently, the antiproliferative activity of the most active four compounds was determined at different concentrations of 10-fold scale such as 100, 10, 1.0, 0.1, and 0.01 μM. The mean GI50 and LC50 values against a 60 cell line panel are summarized in Table S1 (please refer to the Supporting Information).
These compounds showed varied patterns against all cell lines. Compounds 25 showed nearly the same value for GI50, total growth inhibition (TGI), and LC50 suggesting potential toxicity and, therefore, should not be considered for further investigation. Compounds 21 and 23 showed a small differential range between cytostatic markers (GI50, TGI) and the cytotoxicity marker (LC50) suggesting a narrow therapeutic index. However, compound 18 displayed a very interesting selective cytotoxicity pattern against several cell lines. It was particularly potent at a nanomolar concentration against RPMI-8226 (leukemia), HCT-116 (colon cancer), SF-295 (CNS cancer), OVCAR-4 (ovarian cancer), PC-3 (prostate cancer), and MDA-MB-468 (breast cancer) cancer cell lines. Also, comparing the cytostatic data (GI50, TGI) and cytotoxic data (LC50), it is evident that compound 18 showed a remarkable difference between cytostatic indicators (GI50, TGI) and the cytotoxic indicator (LC50) suggesting a broad therapeutic index. In an earlier study,19 sulfonamide had shown the antiproliferative properties against A549 nonsmall cell lung cancer (NSCLS) cells in vitro. Therefore, to understand the mode of action, we further investigated the mechanism using A549. The five-dose graph and dose–response curves of 18 for the individual disease wise subpanel of NCI-60 cells are presented in Figure S1 (please refer to the Supporting Information).
The log GI50, TGI (cytostatic parameter), and LC50 (cytotoxic parameter) for the active compounds 18, 21, 23 and 25 against the 60 cell lines are summarized in Table 2, along with the log median values (median value of multiple experiments), log delta values (the maximum sensitivity over the mean), and the log range values (the maximum difference between the least sensitive and the most sensitive cell lines).39,40 These parameters offer important information about potency and selectivity of anticancer agents. Large values of the delta and range imply high selectivity for some histological cancers over others.41
Table 2. Cytostatic (GI50 and TGI) and Cytotoxic (LC50) Parameters of Active Compounds.
| compounds |
|||||
|---|---|---|---|---|---|
| 18 (NSC 764190) | 21 (NSC 751486) | 23 (NSC 751478) | 25 (NSC 764189) | ||
| log GI50 | median | –4.94 | –5.49 | –5.31 | –5.30 |
| delta | 1.63 | 0.58 | 1.25 | 0.00 | |
| range | 2.57 | 0.85 | 1.44 | 0.00 | |
| log TGI | median | –4.00 | –5.10 | –5.12 | –5.30 |
| delta | 0.22 | 0.22 | 0.00 | 0.00 | |
| range | 0.22 | 0.62 | 0.00 | 0.00 | |
| log LC50 | median | –4.00 | –4.80 | –5.12 | –5.30 |
| delta | 0.00 | 0.18 | 0.00 | 0.00 | |
| range | 0.00 | 0.28 | 0.00 | 0.00 | |
Inhibitory effects of 18 on the proliferation of A549 lung epithelial cancer cells were evaluated by treating the overnight grown cells with test compound (1 μM). As shown in Figure 2A, upon treatment with test compound, A549 cells lost morphology and appeared as dead cells with no further proliferation. In a time-dependent experiment with A549 cells, 50% growth inhibition was observed with 18 after 36 h (Figure 2B).
Figure 2.

(A) A549 cells were treated with compound 18 (same as LSC-JHJ-III-128-13) at μM or DMSO. On exposure of A549 cells to 18, the change in morphology of cells was observed. (B) The A549 cell was treated with compound 18 at 10 μM or DMSO. On exposure of A549 cells to compound, 50% growth inhibition is observed after 36 h.
As we observed the antiproliferative activity of 18 against A549 cells and the effect of compound 18 on anchorage-independent growth A549 cells, a soft agar-based colony-forming assay was performed. The colonies stained in soft agar plates have clearly confirmed that the compound 18 inhibits the formation of colonies by cancer cells. In these assays, a dose-dependent inhibition on clonogenic growth by A549 cells by 18 indicates the efficacy of 18 toward inhibition of A549 cell growth (Figure 3A,B).
Figure 3.
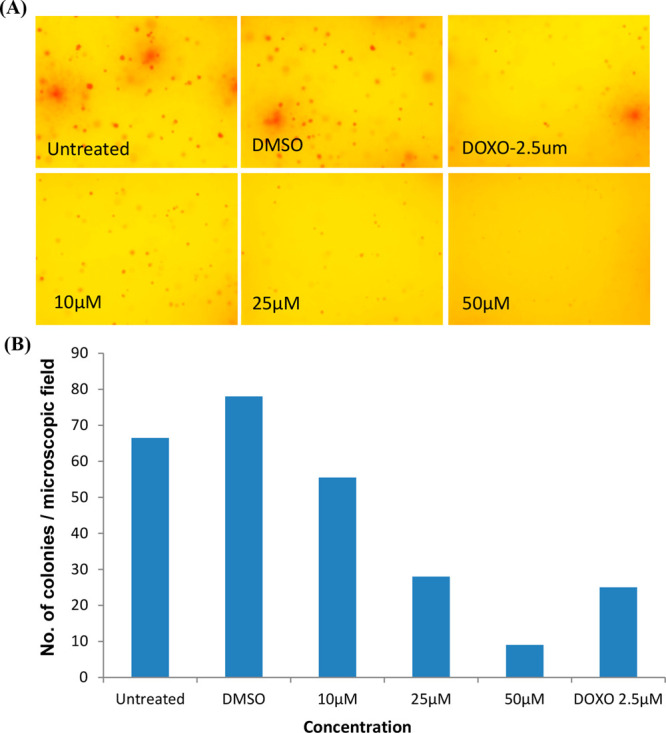
Effect of compound 18 on the anchorage-independent growth of A549 cells. In clonogenic assays, cells were allowed to grow in soft agar containing 18 (0–50 μM) for 9 days to form colonies. (A) Images of the colony-forming assay and (B) number of colonies stained with crystal violet.
Toward identifying NCEs with novel anticancer potential, understanding their effects on migration properties of cancer cells is also important. In vitro scratch or wound healing assays are the suitable assay to evaluate the effect of test compounds on cell migration for epithelial or mesenchymal cells. As shown in Figure 4, the results confirm that compound 18 (NSC764190) inhibited migration of cells by 50% approximately even at the lowest tested concentration whereas, in DMSO plates, cells showed migration the same as the control untreated assays (Figure 4, upper left panel).
Figure 4.

Effect of compound 18 (NSC 764190) on migration of the A549 cell. The representative images of the in vitro scratch assay are shown here. After creating scratches of A549 cell monolayers, cells growing in media containing the test compound (0–50 μM) or DMSO vehicle were analyzed with a phase-contrast microscope, and images are presented. Compound 18 inhibited migration of A549 cells.
As we observed the effect of 18 on proliferation and migration of A549 cells, we next determined whether 18 induces apoptosis and/or inhibits cell proliferation. Hence, we determined cell death and cell cycle progression using flow cytometry. After postexposure of A549 cells to 18 for 48 h, propidium iodide (PI) was applied to measure cell cycle progression and cell death.
The flow cytometer results showed an increase in SubG1 population, indicating that compound 18 is inducing cell death (Figure 5).
Figure 5.

Senescence induced by compound 18 was quantified using SA-β-gal-staining. SA-β-gal positive cells are indicated with the arrow mark. The number of SA-β-gal positive cells increased with increasing concentration of compound indicating that compound 18 induces senescence of A549 cells.
To gain additional insight into the mechanisms by which 18 treatment induces cell death and inhibits proliferation, we have investigated caspase activation and senescence in A549 cells treated with the compound. A significant increase in the caspase activity could not be observed in compound treated cells compared to the control (data not shown). Therefore, we assessed the endogenous level of β-galactosidase as an indicator for senescence in A549 cells upon treatment with 18. Interestingly, we could observe more cells with an increased level of SA-β-gal in cells upon treatment with the compound, and the number of cells positive for SA-β-gal also increased with increasing concentration. These results support the hypothesis that the exposure of cells to compound 18 leads to the induction of cellular senescence in A549 cells for cell cycle arrest.
In conclusion, a set of seven-member cyclic sulfamide has been synthesized and tested for potential anticancer activity against nine different cancer panels. Generally, compounds showed selectively sensitivity against a host of cell lines. Almost every compound is strongly inhibited on breast cancer (MDA-MB-468). A mechanism study with a representative compound 18 (NSC764190) using A549 (nonsmall cell lung cancer) cells suggests inhibition caused by apoptosis.
Acknowledgments
This investigation was generously supported by partial funds provided by the Petroleum Research Fund (PRF-AC, administered by the ACS), the National Institutes of Health (National Institute of General Medical Sciences RO1-GM58103), and a minority supplement (RO1-GM58103-S1, MdSJ). The authors thank Justin Douglas and Sarah Neuenswander in the University of Kansas Nuclear Magnetic Resonance (NMR) Laboratory and Dr. Todd Williams for high-resolution mass spectrometry (HRMS) analysis. Support for the NMR instrumentation was provided by NIH shared instrumentation grants P20 GM103418, S10RR024664, and S10 OD016360 and National Science Foundation (NSF) academic research infrastructure grants 9512331, 9977422, and 0320648. We also acknowledge the IICT communication number IICT/Pubs./2020/370.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00460.
Characterization data for the synthesized compounds, copies of their NMR spectra, and biochemical assays protocol (PDF)
Author Contributions
# S.V.M. is the lead author.
The authors declare no competing financial interest.
Supplementary Material
References
- Gruner B.; Brynda J.; Das V.; Sicha V.; Stepankova J.; Nekvinda J.; Holub J.; Pospisilova K.; Fabry M.; Pachl P. Metallacarborane sulfamides: unconventional, specific, and highly selective inhibitors of carbonic anhydrase IX. J. Med. Chem. 2019, 62 (21), 9560–9575. 10.1021/acs.jmedchem.9b00945. [DOI] [PubMed] [Google Scholar]
- Özgeriş B.; Göksu S.; Köse L. P.; Gülçin I.; Salmas R. E.; Durdagi S.; Tümer F.; Supuran C. T. Acetylcholinesterase and carbonic anhydrase inhibitory properties of novel urea and sulfamide derivatives incorporating dopaminergic 2-aminotetralin scaffolds. Bioorg. Med. Chem. 2016, 24 (10), 2318–2329. 10.1016/j.bmc.2016.04.002. [DOI] [PubMed] [Google Scholar]
- Supuran C. T.; Scozzafava A.; Casini A. Carbonic anhydrase inhibitors. Med. Res. Rev. 2003, 23 (2), 146–189. 10.1002/med.10025. [DOI] [PubMed] [Google Scholar]
- Supuran C. T.; Scozzafava A.; Conway J.. Carbonic anhydrase: its inhibitors and activators; CRC press, 2004; Vol. 1. [Google Scholar]
- Pastorekova S.; Parkkila S.; Pastorek J.; Supuran C. Carbonic anhydrases: current state of the art, therapeutic applications and future prospects. J. Enzyme Inhib. Med. Chem. 2004, 19 (3), 199–229. 10.1080/14756360410001689540. [DOI] [PubMed] [Google Scholar]
- Maryanoff B. E.; McComsey D. F.; Lee J.; Smith-Swintosky V. L.; Wang Y.; Minor L. K.; Todd M. J. Carbonic Anhydrase-II Inhibition. What are the True Enzyme–Inhibitory Properties of the Sulfamide Cognate of Topiramate?. J. Med. Chem. 2008, 51 (8), 2518–2521. 10.1021/jm7015649. [DOI] [PubMed] [Google Scholar]
- Bäckbro K.; Löwgren S.; Österlund K.; Atepo J.; Unge T.; Hultén J.; Bonham N. M.; Schaal W.; Karlén A.; Hallberg A. Unexpected binding mode of a cyclic sulfamide HIV-1 protease inhibitor. J. Med. Chem. 1997, 40 (6), 898–902. 10.1021/jm960588d. [DOI] [PubMed] [Google Scholar]
- Hultén J.; Andersson H. O.; Schaal W.; Danielson H. U.; Classon B.; Kvarnström I.; Karlén A.; Unge T.; Samuelsson B.; Hallberg A. Inhibitors of the C 2-symmetric HIV-1 protease: nonsymmetric binding of a symmetric cyclic sulfamide with ketoxime groups in the P2/P2 ’side chains. J. Med. Chem. 1999, 42 (20), 4054–4061. 10.1021/jm991054q. [DOI] [PubMed] [Google Scholar]
- Akıncıoğlu A.ın; Kocaman E.; Akıncıoğlu H.; Salmas R. E.; Durdagi S.; Gulcin I.; Supuran C. T.; Goksu S. The synthesis of novel sulfamides derived from β-benzylphenethylamines as acetylcholinesterase, butyrylcholinesterase and carbonic anhydrase enzymes inhibitors. Bioorg. Chem. 2017, 74, 238–250. 10.1016/j.bioorg.2017.08.012. [DOI] [PubMed] [Google Scholar]
- Hultén J.; Bonham N. M.; Nillroth U.; Hansson T.; Zuccarello G.; Bouzide A.; Åqvist J.; Classon B.; Danielson U. H.; Karlén A. Cyclic HIV-1 protease inhibitors derived from mannitol: synthesis, inhibitory potencies, and computational predictions of binding affinities. J. Med. Chem. 1997, 40 (6), 885–897. 10.1021/jm960728j. [DOI] [PubMed] [Google Scholar]
- Ilies M. A.; Supuran C. T.; Scozzafava A. Therapeutic applications of serine protease inhibitors. Expert Opin. Ther. Pat. 2002, 12 (8), 1181–1214. 10.1517/13543776.12.8.1181. [DOI] [Google Scholar]
- Groutas W. C.; Schechter N. M.; He S.; Yu H.; Huang P.; Tu J. Human chymase inhibitors based on the 1, 2, 5-thiadiazolidin-3-one 1, 1 dioxide scaffold. Bioorg. Med. Chem. Lett. 1999, 9 (15), 2199–2204. 10.1016/S0960-894X(99)00377-7. [DOI] [PubMed] [Google Scholar]
- Park J. D.; Kim D. H. Sulfamide derivatives as transition state analogue inhibitors for carboxypeptidase A. Bioorg. Med. Chem. 2004, 12 (9), 2349–2356. 10.1016/j.bmc.2004.02.012. [DOI] [PubMed] [Google Scholar]
- Purohit A.; Woo L.; Chander S.; Newman S.; Ireson C.; Ho Y.; Grasso A.; Leese M.; Potter B.; Reed M. Steroid sulphatase inhibitors for breast cancer therapy. J. Steroid Biochem. Mol. Biol. 2003, 86 (3–5), 423–432. 10.1016/S0960-0760(03)00353-4. [DOI] [PubMed] [Google Scholar]
- Winum J. Y.; Scozzafava A.; Montero J. L.; Supuran C. T. Therapeutic potential of sulfamides as enzyme inhibitors. Med. Res. Rev. 2006, 26 (6), 767–792. 10.1002/med.20068. [DOI] [PubMed] [Google Scholar]
- Nelson F. C.; Zask A. The therapeutic potential of small molecule TACE inhibitors. Expert Opin. Invest. Drugs 1999, 8 (4), 383–392. 10.1517/13543784.8.4.383. [DOI] [PubMed] [Google Scholar]
- Winum J.-Y.; Scozzafava A.; Montero J.-L.; Supuran C. T. The sulfamide motif in the design of enzyme inhibitors. Expert Opin. Ther. Pat. 2006, 16 (1), 27–47. 10.1517/13543776.16.1.27. [DOI] [PubMed] [Google Scholar]
- Coppola G.; Davies J.; Jewell C.; Li Y.-C.; Wareing J.; Sperbeck D.; Stams T.; Topiol S.; Vlattas I.. Cyclic sulfamide derivatives and methods of use; US Patent US20040023974A1, 2004.
- Schaal W.; Karlsson A.; Ahlsén G.; Lindberg J.; Andersson H. O.; Danielson U. H.; Classon B.; Unge T.; Samuelsson B.; Hultén J. Synthesis and comparative molecular field analysis (CoMFA) of symmetric and nonsymmetric cyclic sulfamide HIV-1 protease inhibitors. J. Med. Chem. 2001, 44 (2), 155–169. 10.1021/jm001024j. [DOI] [PubMed] [Google Scholar]
- Oehme D. P.; Wilson D. J.; Brownlee R. T. Effect of Structural Stress on the Flexibility and Adaptability of HIV-1 Protease. J. Chem. Inf. Model. 2011, 51 (5), 1064–1073. 10.1021/ci2000677. [DOI] [PubMed] [Google Scholar]
- Sparey T.; Beher D.; Best J.; Biba M.; Castro J. L.; Clarke E.; Hannam J.; Harrison T.; Lewis H.; Madin A.; Shearman M.; Sohal B.; Tsou N.; Welch C.; Wrigley J. Cyclic sulfamide gamma-secretase inhibitors. Bioorg. Med. Chem. Lett. 2005, 15 (19), 4212–4216. 10.1016/j.bmcl.2005.06.084. [DOI] [PubMed] [Google Scholar]
- Lewis S. J.; Smith A. L.; Neduvelil J. G.; Stevenson G. I.; Lindon M. J.; Jones A. B.; Shearman M. S.; Beher D.; Clarke E.; Best J. D.; Peachey J. E.; Harrison T.; Castro J. L. A novel series of potent gamma-secretase inhibitors based on a benzobicyclo[4.2.1]nonane core. Bioorg. Med. Chem. Lett. 2005, 15 (2), 373–378. 10.1016/j.bmcl.2004.10.062. [DOI] [PubMed] [Google Scholar]
- Hirayama F.; Koshio H.; Ishihara T.; Watanuki S.; Hachiya S.; Kaizawa H.; Kuramochi T.; Katayama N.; Kurihara H.; Taniuchi Y.; Sato K.; Sakai-Moritani Y.; Kaku S.; Kawasaki T.; Matsumoto Y.; Sakamoto S.; Tsukamoto S. Design, synthesis and biological activity of YM-60828 derivatives: Potent and orally-bioavailable factor Xa inhibitors based on naphthoanilide and naphthalensulfonanilide templates. Bioorg. Med. Chem. 2002, 10 (8), 2597–2610. 10.1016/S0968-0896(02)00106-2. [DOI] [PubMed] [Google Scholar]
- Hirayama F.; Koshio H.; Katayama N.; Ishihara T.; Kaizawa H.; Taniuchi Y.; Sato K.; Sakai-Moritani Y.; Kaku S.; Kurihara H.; Kawasaki T.; Matsumoto Y.; Sakamoto S.; Tsukamoto S. Design, synthesis and biological activity of YM-60828 derivatives. Part 2: Potent and orally-bioavailable factor Xa inhibitors based on benzothiadiazine-4-one template. Bioorg. Med. Chem. 2003, 11 (3), 367–381. 10.1016/S0968-0896(02)00462-5. [DOI] [PubMed] [Google Scholar]
- Dou D. F.; Tiew K. C.; He G. J.; Mandadapu S. R.; Aravapalli S.; Alliston K. R.; Kim Y.; Chang K. O.; Groutas W. C. Potent inhibition of Norwalk virus by cyclic sulfamide derivatives. Bioorg. Med. Chem. 2011, 19 (20), 5975–5983. 10.1016/j.bmc.2011.08.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw D.; Best J.; Dinnell K.; Nadin A.; Shearman M.; Pattison C.; Peachey J.; Reilly M.; Williams B.; Wrigley J.; Harrison T. 3,4-Fused cyclohexyl sulfiones as gamma-secretase inhibitors. Bioorg. Med. Chem. Lett. 2006, 16 (11), 3073–3077. 10.1016/j.bmcl.2005.12.083. [DOI] [PubMed] [Google Scholar]
- Kim S. J.; Park H. B.; Lee J. S.; Jo N. H.; Yoo K. H.; Baek D.; Kang B. W.; Cho J. H.; Oh C. H. Novel 1 beta-methylcarbapenems having cyclic sulfonamide moieties: Synthesis and evaluation of in vitro antibacterial activity. Eur. J. Med. Chem. 2007, 42 (9), 1176–1183. 10.1016/j.ejmech.2007.02.001. [DOI] [PubMed] [Google Scholar]
- Jun J. H.; Javed S.; Ndi C. N.; Hanson P. R.. Synthesis of P-, S-, Si-, B-, and Se-heterocycles via ring-closing metathesis. In Synthesis of Heterocycles by Metathesis Reactions; Springer, 2015; pp 319–379. [Google Scholar]
- Jun J. H.; Kumar V.; Dexheimer T. S.; Wedlich I.; Nicklaus M. C.; Pommier Y.; Malhotra S. V. Synthesis, anti-cancer screening and tyrosyl-DNA phosphodiesterase 1 (Tdp1) inhibition activity of novel piperidinyl sulfamides. Eur. J. Pharm. Sci. 2018, 111, 337–348. 10.1016/j.ejps.2017.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun J. H.; Dougherty J. M.; del Sol Jiménez M. a.; Hanson P. R. New strategies to symmetric and unsymmetric cyclic sulfamide analogs of DMP 323: a ’sulfur linchpin’/RCM approach. Tetrahedron 2003, 59 (45), 8901–8912. 10.1016/j.tet.2003.04.001. [DOI] [Google Scholar]
- Bergmann J.; Löfstedt C.; Ivanov V. D.; Francke W. Identification and assignment of the absolute configuration of biologically active methyl-branched ketones from Limnephilid caddis flies. Eur. J. Org. Chem. 2001, 2001 (16), 3175–3179. 10.1002/1099-0690(200108)2001:16<3175::AID-EJOC3175>3.0.CO;2-4. [DOI] [Google Scholar]
- Petroski R.; Weisleder D.. Improved preparation of Sap bettle (Coleoptera: Nitidulidae) aggregation pheromones; ACS Symposium Series; American Chemical Society: 2000; Vol. 800, pp 231–237.
- Yasui Y. [V2Cl3 (thf) 6] 2 [Zn2Cl6]: Reagent for a Highly Selective Pinacol-Coupling Reaction. Aust. J. Chem. 2002, 55 (10), 685–685. 10.1071/CH02170. [DOI] [Google Scholar]
- Regaınia Z.; Abdaoui M.; Aouf N.-E.; Dewynter G.; Montero J.-L. Synthesis of 1, 2, 5-thiadiazolidines 1, 1-dioxides (cyclosulfamides) starting from amino acids and chlorosulfonyl isocyanate. Tetrahedron 2000, 56 (3), 381–387. 10.1016/S0040-4020(99)01025-X. [DOI] [Google Scholar]
- Dewynter G.; Aouf N.; Regainia Z.; Montero J.-L. Synthesis of pseudonucleosides containing chiral sulfahydantoins as aglycone (II). Tetrahedron 1996, 52 (3), 993–1004. 10.1016/0040-4020(95)00932-9. [DOI] [Google Scholar]
- Shoemaker R. H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6 (10), 813–823. 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
- Shoemaker R. H.; Monks A.; Alley M. C.; Scudiero D. A.; Fine D. L.; McLemore T. L.; Abbott B. J.; Paull K. D.; Mayo J. G.; Boyd M. R. Development of human tumor cell line panels for use in disease-oriented drug screening. Prog. Clin. Biol. Res. 1988, 276, 265–286. [PubMed] [Google Scholar]
- Holbeck S. L.; Collins J. M.; Doroshow J. H. Analysis of Food and Drug Administration–approved anticancer agents in the NCI60 panel of human tumor cell lines. Mol. Cancer Ther. 2010, 9 (5), 1451–1460. 10.1158/1535-7163.MCT-10-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd M. R.; Paull K. D. Some practical considerations and applications of the National Cancer Institute in vitro anticancer drug discovery screen. Drug Dev. Res. 1995, 34 (2), 91–109. 10.1002/ddr.430340203. [DOI] [Google Scholar]
- Nakatsu N.; Yoshida Y.; Yamazaki K.; Nakamura T.; Dan S.; Fukui Y.; Yamori T. Chemosensitivity profile of cancer cell lines and identification of genes determining chemosensitivity by an integrated bioinformatical approach using cDNA arrays. Mol. Cancer Ther. 2005, 4 (3), 399–412. [DOI] [PubMed] [Google Scholar]
- Kumar A.; Kumar V.; Alegria A. E.; Malhotra S. V. N-Hydroxyethyl-4-aza-didehydropodophyllotoxin derivatives as potential antitumor agents. Eur. J. Pharm. Sci. 2011, 44 (1–2), 21–26. 10.1016/j.ejps.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.