

Apramycin is an aminoglycoside antibiotic that has been traditionally used in veterinary medicine. Recently, it has become an attractive candidate to repurpose in the fight against multidrug-resistant pathogens prioritized by the World Health Organization.
KEYWORDS: aminoglycoside-modifying enzymes, antibiotic resistance, apramycin
ABSTRACT
Apramycin is an aminoglycoside antibiotic with the potential to be developed to combat multidrug-resistant pathogens. Its unique structure evades the clinically widespread mechanisms of aminoglycoside resistance that currently compromise the efficacy of other members in this drug class. Of the aminoglycoside-modifying enzymes that chemically alter these antibiotics, only AAC(3)-IVa has been demonstrated to confer resistance to apramycin through N-acetylation. Knowledge of other modification mechanisms is important to successfully develop apramycin for clinical use. Here, we show that ApmA is structurally unique among the previously described aminoglycoside-modifying enzymes and capable of conferring a high level of resistance to apramycin. In vitro experiments indicated ApmA to be an N-acetyltransferase, but in contrast to AAC(3)-IVa, ApmA has a unique regiospecificity of the acetyl transfer to the N2′ position of apramycin. Crystallographic analysis of ApmA conclusively showed that this enzyme is an acetyltransferase from the left-handed β-helix protein superfamily (LβH) with a conserved active site architecture. The success of apramycin will be dependent on consideration of the impact of this potential form of clinical resistance.
INTRODUCTION
Waksman’s tandem discoveries of streptomycin and neomycin over 65 years ago ushered in the clinical use of the aminoglycoside antibiotics (AGs) for the treatment of bacterial infections (1, 2). Since then, a variety of AGs have found clinical success. All AGs have a six-membered aminocyclitol core that serves to distinguish subfamilies of the class. For example, the 4,6-deoxstreptamine antibiotics tobramycin, gentamicin, and amikacin are particularly effective against Gram-negative pathogens. These antibiotics offer improved oto- and nephrotoxicity profiles over the 4,5-deoxstreptamine-containing scaffolds such as neomycin (3, 4). Most AGs act through noncovalent binding to the small ribosomal subunit in a fashion that disrupts the proofreading property of translation (5). The result is subversion of the genetic code followed by the production of aberrant proteins and peptides, resulting in cell death.
Soon after the introduction of AGs in clinical practice, acquired resistance mediated by mobile genetic elements was reported (6). Over the past decades, a plethora of AG resistance genes have been identified, many of them moving through bacterial populations by lateral gene transfer (7). In addition to the upregulation of efflux systems and mutations in outer membrane porins occurring in some bacteria such as Pseudomonas aeruginosa (8), two general mechanisms of resistance dominate in pathogens, drug inactivation and target modification. Both mechanisms result in a decreased affinity of the AG for its ribosomal target (9, 10). Inactivation of AGs occurs via chemical modification by one of the following three classes of aminoglycoside-modifying enzymes (AMEs): O-phosphorylation (APHs), O-adenylylation (ANTs), or N-acetylation by aminoglycoside acetyltransferases (AACs) (11–13). Modification of the drug target is the most recent form of aminoglycoside resistance to emerge in the clinic (14). The 16S rRNA methyltransferases (RMTases) are responsible for the N7 methylation of G1405 (e.g., Escherichia coli numbering; ArmA, Rmt family) or N1 methylation of A1408 (14, 15) (e.g., NpmA, KamB) within the 16S rRNA, respectively, conferring resistance to only 4,6-disubstituted or all 4,5- and 4,6-disubstituted AGs. RMTases are increasingly found in carbapenem-resistant isolates, greatly limiting therapeutic options for infections caused by these bacteria (16).
The broad mechanistic and genetic diversity of AG resistance impacts the use of existing drugs and dampens enthusiasm in the discovery of new members of this family of antibiotics, despite their highly desirable bactericidal activity toward Gram-positive and Gram-negative pathogens. In response to this challenge, the group at Achaogen embarked on an effort to deliver a next-generation semisynthetic AG that was not susceptible to common AMEs. The result of this effort is plazomicin (Zemdri) approved by the FDA in 2018, which retains antibiotic activity in the presence of most AMEs (17, 18). However, plazomicin remains ineffective against isolates coexpressing many RMTases, resistance genes that were not in significant circulation when the development program was launched (19). This fact emphasizes the potential for rapid dissemination of new resistance elements in the clinic that may move more rapidly through bacterial populations than the drug development process.
Apramycin is an unusual AG where the deoxystreptamine (DOS) aminocyclitol ring is monosubstituted and is linked to an octadiose element (Fig. 1) (20). Apramycin has been used in veterinary medicine for decades but more recently has been found to exhibit broad activity against WHO-prioritized multidrug-resistant (MDR) pathogens such as carbapenemase-producing Enterobacteriaceae and Acinetobacter baumannii (3, 21–24). The unique monosubstitution of apramycin’s DOS ring prevents both inactivation by a majority of common AMEs and resistance due to target alteration by N7 RMTases (24). This characteristic makes apramycin particularly attractive as a candidate next-generation AG for clinical use in humans (25).
FIG 1.

Apramycin’s advantage to overcome aminoglycoside resistance in the clinic. (A) Aminoglycoside resistance elements explored in this study (Table 1; Fig. S1). Inner circle represents the two main modes of aminoglycoside resistance, drug inactivation and target modification. Outer circle highlights the individual enzymes (Fig. S1), colored based on chemical modification made to the target or antibiotic. (B) Apramycin’s unique monosubstitution of the DOS ring with the octadiose element limits the number of inactivating mechanisms. Lack of substitution at C6 allows avoidance of clinically relevant 16S rRNA methyltransferases.
NMR assignments of acetyl-apramycin. Download Table S1, DOCX file, 0.02 MB (16.1KB, docx) .
Copyright © 2021 Bordeleau et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Apramycin resistance elements identified while screening within the aminoglycoside resistance library expressed in E. coli BW25113 ΔtolCΔbamB. (A) Aminoglycoside resistance glycerol library layout. N-acetyltransferase, blue; O-phosphotransferase, gold; O-nucleotidyltransferase, yellow; bifunctional acetyltransferase, pink; 16S rRNA methyltransferase, red. Each gene was under the control of the Pbla promoter. (B) Control growth of library on cation-adjusted Mueller Hinton agar supplemented with no drug. Colony growth highlighted with a circle were those found to confer apramycin resistance. (C) 4 μg/ml apramycin; (D) 32 μg/ml apramycin; (E) 256 μg/ml apramycin. Download FIG S1, JPG file, 1.2 MB (1.2MB, jpg) .
{kind=link}
Copyright © 2021 Bordeleau et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Prior to the introduction of this antibiotic into the clinic, knowledge of the apramycin resistome is important. The N-acetyltransferase AAC(3)-IVa, a common selectable antibiotic resistance marker for molecular biology studies in actinomycetes, confers high-level apramycin resistance and is occasionally found in clinical isolates of Enterobacteriaceae (26–28). AAC(1) has been reported to be associated with apramycin resistance; however, the sequence of this gene is unavailable and the original isolates lost (A. Lovering, personal communication) (29, 30). ApmA is another acetyltransferase linked to apramycin resistance. ApmA was first reported in 2011 from bovine and porcine methicillin-resistant Staphylococcus aureus (MRSA) isolates (31, 32). The gene was found as the sole resistance element on a smaller plasmid as well as a larger antimicrobial multiresistance plasmid that also contained heavy metal resistance genes and potential virulence elements (32, 33). AAC(3)-IVa adopts the structural fold for the more recently studied AAC(3) enzymes belonging the Antibiotic_NAT family (PDB ID 6MN4). Primary sequence alignment suggests that ApmA does not share this three-dimensional structure. Given the growing interest in apramycin as a drug candidate, we have investigated ApmA’s activity toward apramycin and determined its three-dimensional structure. We identify ApmA as a member of the left-handed β-helix (LβH) superfamily, similar to chloramphenicol and streptogramin O-acetyltransferase resistance enzymes. This is the first report of an AG resistance element with this protein fold and reflects the diversity and enzymatic opportunism of antibiotic resistance.
RESULTS
The apramycin resistome is limited to four known genes.
The resistome of apramycin was evaluated through susceptibility testing against our in-house antibiotic resistance platform consisting of a panel of isogenic strains of Escherichia coli BW25113 ΔtolC ΔbamB, each expressing unique aminoglycoside resistance elements (34). Apramycin susceptibility was surveyed against 27 aminoglycoside resistance elements, consisting of 11 AACs, 11 APHs, two ANTs, and four RMTases. Gene expression levels were under the control of the constitutive promoter Pbla. A control strain not expressing an aminoglycoside resistance element was used as a reference for apramycin potency. We found only the four previously reported apramycin resistance elements to confer resistance (Fig. S1 in the supplemental material). The two N1-A1408-directed RMTases, NpmA and KamB, and the two acetyltransferases, AAC(3)-IVa and ApmA, each confer a high level of resistance to apramycin (≥64 μg/ml; Table 1). ApmA remains the only member of these apramycin resistance elements uncharacterized with respect to its structure and function.
TABLE 1.
Apramycin resistance elements identified through susceptibility testing of E. coli BW25113 ΔtolC ΔbamB expressing aminoglycoside resistance enzymes
| Aminoglycoside resistance mechanism | Resistance gene | E. coli BW25113 ΔtolC ΔbamB MIC (μg/ml) |
|---|---|---|
| None | None | 4 |
| Drug modification | aac(3)-IVa | ≥512 |
| apmA | 64 | |
| Target modification | kamB | ≥512 |
| npmA | ≥512 |
ApmA acetylates apramycin at 2′-NH2.
Purified recombinant ApmA was used to produce acetylated apramycin in vitro to determine the regiospecificity of acetyl group transfer. Characterization of the acetylated product was carried out using high-resolution electrospray ionization-mass spectrometry (HR-ESI-MS), which revealed a single acetylation of the apramycin core (mass increase of 42.0 Da) (Table 2).
TABLE 2.
HR-ESI-MS analysis of ApmA-catalyzed acetylated aminoglycosides in positive ion mode
| Modified aminoglycoside | Molecular formula | Exact mass [M+H] |
|
|---|---|---|---|
| Calculated | Observed | ||
| Apramycin | C21H42N5O11 | 540.2875 | 540.2891 |
| N-acetyl-apramycin | C23H45N5O12 | 582.2986 | 582.2989 |
Further characterization of the regiospecificity of acetyl transfer was accomplished using a combination of one- and two-dimensional nuclear magnetic resonance spectroscopy (1D and 2D NMR) of purified, ApmA-inactivated apramycin. (Table S1; Fig. S4 to S8) Acetyl-apramycin NMR spectra were compared with those reported in the literature for the apramycin-free base (35). We noted significant deshielding of the 2′ proton from 3.02 ppm in apramycin to 4.06 ppm in the acetylated product in the 1H NMR. This change is a result of the effect of a carbonyl group attached to a neighboring atom (N2′). In the 13C NMR, two new carbon shifts were observed at 21.8 ppm and at 172.89 ppm, corresponding to a methyl group carbon and carbonyl carbon chemical shifts. Lastly, heteronuclear multiple bond correlation spectra (HMBC) revealed correlations between the carbonyl carbon and methyl protons, as well as their correlation with the 2′ proton (Fig. 2). These data are consistent with acetylation of apramycin by ApmA at the 2′-NH2 of ring I.
FIG 2.

HMBC correlations. Correlations between carbonyl carbon of the acetyl group, methyl protons, and 2′ proton are indicated with the arrows.
ApmA is an N-acetyltransferase from the left-handed β-helix protein superfamily.
Primary sequence analysis of ApmA identified a seven-hexapeptide repeat motif, suggesting it is a member of the LβH superfamily of acetyltransferases that includes the xenobiotic acetyltransferases (XAT) subclass of LβHs, responsible for the inactivation of streptogramin group A antibiotics (36, 37) (Vat) and chloramphenicol (38, 39) (CATB). Crystals of the apoenzyme and ApmA in complex with apramycin or acetyl-CoA were obtained and their structures solved to resolutions of 2.08 Å, 1.85 Å, and 2.30 Å, respectively (Table S2). Analysis of these complexes showed ApmA to be a trimeric protein and confirmed it to be a member of the LβH superfamily.
X-ray diffraction data collection and refinement statistics. Download Table S2, DOCX file, 0.02 MB (16.3KB, docx) .
Copyright © 2021 Bordeleau et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The overall structure of ApmA is consistent with the canonical XAT architecture (36): it consists of a C-terminal region comprised of three α-helices (residues 232 to 274), a central LβH domain where the hexapeptide motif is repeated seven times (residues 83 to 231), and an insert located in the center of the LβH fold, characterized by two α-helices (residues 132 to 176). Unlike other XATs, ApmA contains an additional N-terminal region, comprised of four β-sheets and two α-helices (residues 1 to 82) (Fig. 3A and B). The LβH domains of three neighboring chains together form a tunnel to shuttle the pantothenate arm of acetyl-CoA into the apramycin binding pocket (Fig. 4A). The N-terminal region appears to play a role in distorting the first two β-strands from the 7-stranded β-sheet of the LβH domain relative to their position in VatA and XAT (PDB ID 2XAT) (Fig. S2). These β-strands are twisted nearly 90° from their position in VatA and XAT and significantly alter the shape and volume of the substrate binding pocket (Fig. S2).
FIG 3.

Structural comparison of ApmA to XAT subclass of LβH superfamily. (A) Domain architecture represented in ApmA monomer. (B) Structural superimposition of ApmA and VatA (PDB ID 4HUS) in complex with apramycin and virginiamycin, respectively (substrates not shown). ApmA chains are colored in red, blue, and green, and VatA chains are all colored in black.
FIG 4.

LβH domain creates a tunnel for acetyl-CoA binding. (A) Surface view of ApmA-acetyl-CoA complex superimposed on ApmA-apramycin complex. Chains are colored red, green, and blue. Apramycin and acetyl-CoA are shown in sticks. (B) Active site of superimposed ApmA substrate complexes highlighting residues suspected to be involved in apramycin binding and acetylation.
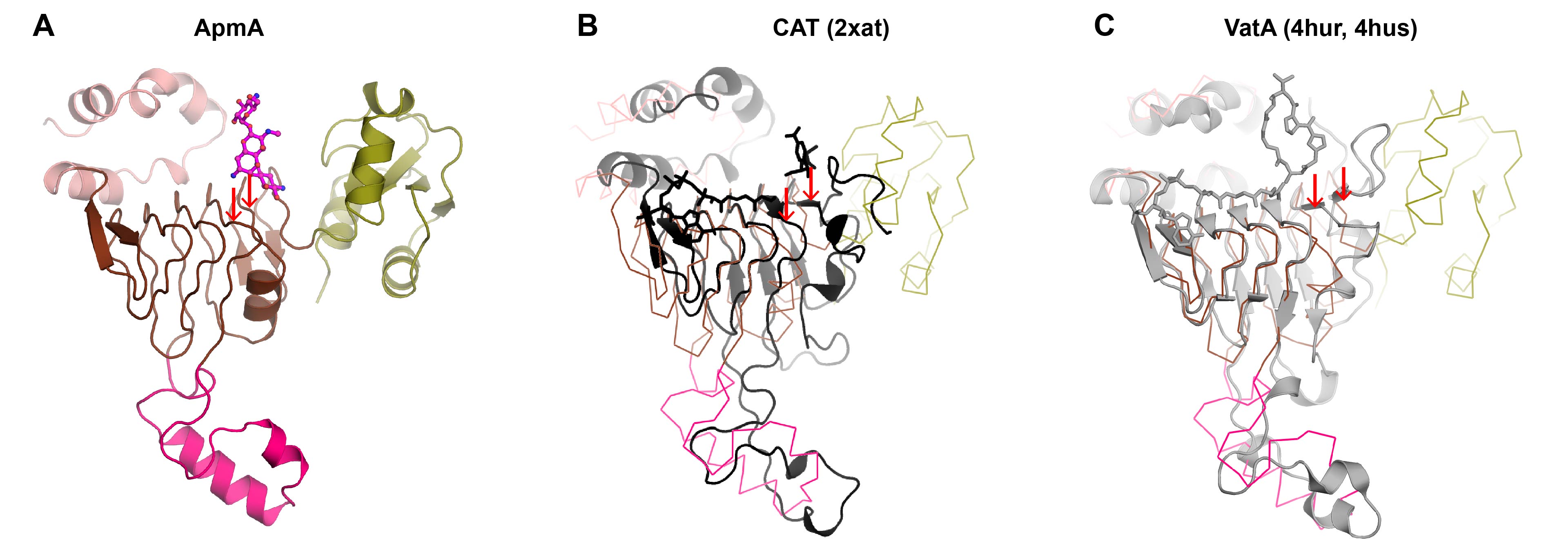
Impact of the N-terminal region on the substrate binding pocket of ApmA. Red arrows indicate the first two β-strands from the 7-stranded β-sheet of the LβH domain. Superimpositions of ApmA in complex with apramycin (A), CAT (B), and VatA (C). Download FIG S2, JPG file, 0.3 MB (365.9KB, jpg) .
{kind=link}
Copyright © 2021 Bordeleau et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Three acetyl-acceptor binding pockets were identified with the positioning of the central DOS ring consistent with other AMEs, which typically are lined with aspartate and glutamate residues (40). In the ApmA-apramycin complex, the N3 atom of the AG’s aminocyclitol ring is coordinated by two aspartic acids (Asp144 and Asp145) in a helix within the C-terminal domain and one glutamic acid (Glu85) position the N1 atom. Ring III of apramycin interacts with several residues from one of the helices of the inserted loop region of the LβH domain (Gln147, Gln159, and Tyr172). The 6′-OH of ring I interacts with N113 from within the LβH domain (Fig. 4A and 4B). Consistent with the NMR structure of 2′-acetyl-apramycin, we observed the 2′-NH2 of the antibiotic positioned for acetylation. Upon superimposition of the two binary complexes (root mean square deviation [RMSD], 0.29 Å), the 2′-NH2 lies between 3.1 to 3.7 Å from the carbonyl carbon of the acetyl moiety (Fig. 4B). Tyr102 participates in a network of hydrogen bonds between the 2′-NH2 and Asp144 that may be important for antibiotic binding. The imidazole side chain from His135 is within 2.8 to 3.1 Å of apramycin’s 2′-NH2. Consistent with other XAT enzymes, the N1 of this imidazole is hydrogen bonded to a carbonyl of the peptide backbone (Val142) (Fig. 4B). It is suspected that interaction increases the basicity of imidazole’s N2 to help in a role as a general base (37, 38). Superimposition of ApmA with VatA, in complex with their antibiotic substrates, demonstrates that the His135 of ApmA is geometrically equivalent to the catalytic histidine for VatA (His87) (36) (Fig. 5). Tyr102 of ApmA was also found in the same position of Tyr42 of VatA, a residue responsible for stabilizing the oxyanion intermediate for O-acetylation (36). To further assess the significance of His135 at the sequence level, we gathered reference sequences for Vats and CATBs from the Comprehensive Antibiotic Resistance Database (CARD) (41) to construct a multiple sequence alignment. Our analysis revealed that His135 of ApmA aligns with the conserved catalytic histidine found across all Vat and CATB sequences, essential for the O-acetylation of their respective substrates (36) (Fig. S3). We next generated the alanine mutant of His135 to evaluate the impact of this substitution on ApmA’s detoxification of apramycin through cell-based assays. Upon expression of the His135Ala mutant in E. coli, resistance to apramycin remained within 2-fold of when the wild-type enzyme was expressed (32 to 64 μg/ml). These results suggest the role of this histidine in the acetylation of apramycin does not hold the same significance as for the function of other XATs and requires further investigation.
FIG 5.

Superimposition of active site for VatA (PDB ID 4HUS) onto ApmA. Important catalytic residues for VatA and the equivalent residues found in ApmA are identified.
Sequence relationship of ApmA to the LβH superfamily. Full multiple sequence alignment of ApmA with CATs and Vats obtained from the Comprehensive Antibiotic Resistance Database. Amino acids are colored based on percent identity. Traditional hexapeptide motif (i, i + 1, and i + 4) of the LβH domain is highlighted in red. Conserved catalytic histidine of the XAT subclass is highlighted in pink. Download FIG S3, JPG file, 2.8 MB (2.9MB, jpg) .
{kind=link}
Copyright © 2021 Bordeleau et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
1H NMR for 2′-acetyl-apramycin. Download FIG S4, PDF file, 0.1 MB (93.1KB, pdf) .
Copyright © 2021 Bordeleau et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
DISCUSSION
Apramycin’s atypical structure in comparison with other AGs has garnered considerable attention for its potential as a next-generation AG antibiotic. The development of derivatives, termed apralogs, has focused on retaining apramycin’s low ototoxic potential while increasing potency coupled with evading resistance to apramycin through AAC(3)-IV-mediated modification (42, 43). Molecular modeling of 3-acetyl-apramycin bound to the ribosome indicates that reduced binding to the target results from a steric clash between the 16S rRNA phosphate backbone and the amide positioned at C3 of apramycin (24). We show that ApmA is an acetyltransferase that instead modifies apramycin at the N2′ position of the octadiose element to confer high-level drug resistance. However, N2′ does not make direct contacts with the 16S rRNA and could spatially accommodate the acetyl group. We suggest that acetylation at this position reduces the affinity of the overall molecule for its target. The N2′ participates in an intramolecular interaction with O5 of the 2-DOS ring. Intramolecular interactions between AG sugar rings have been suggested to play an important role in the recognition and binding of AGs to the 16S rRNA (44). The octadiose element of apramycin participates in important hydrogen bonds with A1408 of the 16S rRNA, creating a glycoside-adenine pseudo-base pair (43, 45, 46). Acetylation of N2′ would disrupt the orientation of the 2-DOS and octadiose ring. The carbonyl would also have the potential to create new intramolecular interactions that alter the configuration of the molecule. Unfavorable intermolecular interactions would also be introduced if the carbonyl group approaches the negatively charged phosphate backbone, creating an electronic clash. Lastly, the overall charge of apramycin will be impacted by the replacement of the primary amine with an amide. The removal of the positively charged amine would further hinder its ability to interact with the negatively charged RNA (Fig. 6).
FIG 6.

Impact of 2′ acetylation on ribosomal binding of apramycin. Crystal structure of apramycin bound to the ribosome (PDB ID 4AQY). Important interactions between N3 and the ribosome for recognition of the 2-DOS ring are indicated. Intramolecular interaction between N2′ and O5 is highlighted. Acetylation at position N3 creates steric clash with the phosphate backbone. Acetylation of the N2′ position is predicted to create an electronic clash with the negatively charged backbone of A1492 and G1491.
The regiospecificity of the acetyl transfer phenotypically assigns ApmA to the AAC(2′) family of AMEs. However, unlike ApmA, AAC(2′) enzymes are members of the GNAT superfamily (19) and are found chromosomally encoded in Providencia stuartii and Mycobacterium tuberculosis (47, 48). Our analysis of AG resistance shows that AAC(2′)-Ia from P. stuartii does not confer resistance toward apramycin, making ApmA the first AAC(2′) enzyme documented to do so. Furthermore, initial reports of apmA found the sequence mobilized on plasmids from Staphylococci isolates from bovine and swine (31–33). The most recent report of the gene has also identified apmA in Campylobacter isolated from pork, demonstrating an expansion of host and crossover to Gram-negative pathogens (49).
The findings of this study highlight the adaptation of a common protein fold to generate new functions in antibiotic resistance. Concerns that other XAT enzymes capable of conferring resistance to other classes of antibiotics were discussed over 20 years ago (50). Antibiotic acetyltransferases are products of convergence of function, with CoA-dependent acetylation spanning several protein scaffolds (39, 51–55). Our crystallographic data demonstrate ApmA belongs to the LβH superfamily of acetyltransferases, which had not previously been linked to the detoxification of AG antibiotics. The substrate specificity of the LβH scaffold has expanded to accommodate AGs, reminiscent of other sugar-containing substrates of LβH N-acetyltransferases involved in O-antigen biosynthesis (56, 57). Previously, XATs were observed to only O-acetylate and their respective antibiotic substrates with the help of a conserved catalytic histidine. There are also LβH N-acetyltransferases that contain a histidine in the active site and have been shown to be important in the acetylation of their respective nucleotide-linked sugar substrates (56, 58). We show that the His135Ala ApmA mutant is still capable of N-acetylating apramycin to confer an equivalent level of resistance to when the wild-type protein is expressed. This suggests that the molecular mechanism of acetyl transfer is similar to that of the GCN5 family AAC(6′) enzymes where no catalytic base is necessary. Here, acyl transfer occurs due to the alignment and proximity of acetyl-CoA and the nucleophilic receptor amine of the antibiotic (59).
Overall, this work has the potential to aid in the identification of apralogs with reduced susceptibility to ApmA. Furthermore, the distribution of apmA should be monitored for further transfer into clinical isolates. This unique AME threatens not only the success of apramycin’s introduction into the clinic but may impact other AGs susceptible to its modification as well.
MATERIALS AND METHODS
Bacterial strains and apmA cloning.
apmA (GenBank accession no. FN806789.3) was synthesized as a gBlock by Integrated DNA Technologies (IDT) for cloning into pGDP3 (34) and pET19b-TEV using NdeI and XhoI restriction sites. The pET19b-TEV plasmid consists of an N-terminal His10 tag cleavable by a tobacco etch virus protease (TEV). The pGDP3 construct of apmA was transformed into hyperpermeable, efflux-deficient mutant E. coli BW25113 ΔtolC ΔbamB (for antimicrobial susceptibility testing). Construct of pET19b-TEV:apmA was transformed into BL21(DE3)-Gold competent cells (for crystallography). All constructs were verified through Sanger sequencing at the Mobix sequencing facility, McMaster University.
Site-directed mutagenesis.
Nucleotide substitutions (indicated by bold text in primers) were introduced in apmA with PCR site-directed mutagenesis by primer extension (60) using pGDP3:apmA as a template and the following mutagenic oligonucleotide primers to produce apmA:H135A: forward, 5′-AGAGATCCATGCGAACGCTCAGTTAAACATGACCTTTG-3′, and reverse, 5′-AAGGTCATGTTTAACTGAGCGTTCGCATGGATCTCTG-3′.
The final construct was verified through Sanger sequencing at the Mobix sequencing facility, McMaster University.
Antimicrobial susceptibility testing.
Screening against our in-house antibiotic resistance platform was carried out as previously described (34). Apramycin susceptibility testing of E. coli BW25113 ΔtolC ΔbamB expressing apmA and apmA(H135A) was completed in triplicate following the Clinical and Laboratory Standards Institute (CLSI) protocols for the broth microdilution method (61). Strains were cultured in cation-adjusted Mueller-Hinton broth (CAMHB) in a 96-well format. Plates were incubated in a shaking incubator at 37°C for 18 h.
Protein expression and purification.
For acetylated product characterization, E. coli BL21(DE3) Rosetta-gami pLysS transformed with pET19b-TEV constructs of apmA were grown in autoinduction medium supplemented with selection antibiotics for 3 days at 25°C and 180 rpm. Cells were collected by centrifugation at 6,400 × g, 4°C, and resuspended in binding buffer (25 mM HEPES [pH 7.5], 300 mM NaCl, and 10 mM imidazole). Resuspended cells were lysed using a continuous cell disrupter at 20,000 lb/in2, 4°C, followed by centrifugation at 30,000 × g to remove cell debris. ApmA was purified from the lysate by nickel-nitrilotriacetic acid (Ni-NTA) affinity chromatography (Qiagen) at 4°C. A 2-ml volume of Ni-NTA resin was preequilibrated with binding buffer and incubated with the lysate for 1 h on ice prior to purification. The resin was washed with wash buffer (25 mM HEPES [pH 7.5], 300 mM NaCl, and 25 mM imidazole), and proteins were eluted with a 4-stepwise gradient (25%, 50%, 75%, and 100%) of elution buffer (25 mM HEPES [pH 7.5], 300 mM NaCl, and 250 mM imidazole). Elutions were dialyzed overnight against dialysis buffer (25 mM HEPES [pH 7.5], 300 mM NaCl). SDS-PAGE gel electrophoresis was performed to assess sample purity. To prepare stock solutions, concentrated ApmA was diluted to a final concentration ranging from 30 μM to 150 μM in dialysis buffer. Protein dilutions were flash frozen in liquid nitrogen and stored at −80°C.
For crystallography studies, native ApmA was expressed in Gold competent E. coli BL21(DE3). A 3-ml overnight culture was diluted into 1 liter of LB media containing selection antibiotics and grown at 37°C with shaking. Expression of selenomethionine-derivatized ApmA was carried out in M9 minimal media following the manufacturer’s instructions (Shanghai Medicilon). Expression was induced with isopropyl β-d-1-thiogalactopyranoside (IPTG) at 17°C when the optical density at 600 nm (OD600) reached 0.6 to 0.8. The overnight cell culture was then collected by centrifugation at 7,000 × g. Cells were resuspended in binding buffer (100 mM HEPES [pH 7.5], 500 mM NaCl, 5 mM imidazole, and 5% glycerol [vol/vol]) and lysed with a sonicator, and cell debris was removed by centrifugation at 30,000 × g. The cell lysate was loaded on a 4-ml Ni-NTA column (Qiagen) preequilibrated with 250 ml of binding buffer. The resin was washed with wash buffer (100 mM HEPES [pH 7.5], 500 mM NaCl, 30 mM imidazole, and 5% glycerol [vol/vol]), and the proteins were eluted with elution buffer (100 mM HEPES [pH 7.5], 500 mM NaCl, 250 mM imidazole, and 5% glycerol [vol/vol]). The His10-tagged proteins were then subjected to overnight TEV cleavage using 50 μg of TEV protease per mg of His10-tagged protein in binding buffer and dialyzed overnight against the binding buffer. The His10-tag and TEV were removed by running the protein again over the Ni-NTA column. The tag-free proteins were dialyzed against dialysis buffer (50 mM HEPES [pH 7.5], 500 mM NaCl) overnight, and the purity of the protein was analyzed by SDS-polyacrylamide gel electrophoresis.
Spectroscopic characterization of ApmA-catalyzed acetylated apramycin.
ApmA-catalyzed acetylated apramycin was produced from 50-ml in vitro reactions (50 mM HEPES, pH 7.5) consisting of 500 μM aminoglycoside, 500 μM acetyl-CoA, and 1 μM ApmA. Reaction mixtures were incubated at room temperature until acetylated products (mass increase of 42.0 Da) were detected by liquid chromatography (LC)/ESI-MS. Enzymes were removed by centrifugation using an Amicon Ultra-15 centrifugal filter and the flowthrough subsequently concentrated. Acetyl-apramycin was purified from the concentrate using AG50W-X8 strong cation resin. The resin was preequilibrated with 1% NH4OH and washed with H2O until a neutral pH was obtained. Fractions containing acetylated products were identified by LC/ESI-MS, followed by detailed analysis with NMR and HR-ESI-MS. LC/ESI-MS data were acquired using a QTrap 2000 (Applied Biosystems) system equipped with an Agilent 1100 LC interface. HR-ESI-MS data were acquired using an Agilent 1290 ultraperformance liquid chromatography (UPLC) separation module and quadrupole time of flight (Q-TOF) G6550A mass detector in positive ion mode. NMR analysis was completed using a Bruker AVIII 700 MHz instrument in deuterated water as the solvent. The chemical shifts are reported in parts per million.
Crystallization and structure determination.
All crystals were grown at room temperature using the vapor diffusion sitting drop method with 0.5 μl of protein solution mixed with 0.5 μl of reservoir solution. Crystals were grown using the following reservoir solutions: ApmA plus apramycin complex (0.2 M CaCl, 20% polyethylene glycol 3350 [PEG 3350], and 5 mM apramycin) and ApmA plus acetyl-CoA complex (0.1 M citric acid, pH 3.6, 30% PEG 200, 5 mM apramycin, and 2 mM acetyl-CoA). All crystals were cryoprotected with Paratone-N or ethylene glycol and then flash frozen in liquid nitrogen prior to diffraction data collection. Diffraction data were collected at the Advanced Photon Source, Argonne National Laboratory, beamlines 19-ID or 21-ID. All data were processed by HKL-3000. The ApmA plus apramycin structure was solved using the single anomalous diffraction (SAD) method, and this was used to solve all additional complexes by MR. Structure refinement was performed using Phenix.refine plus manual building with Coot. The presence of substrate molecules was identified by building into the Fo-Fc difference density after the initial rounds of refinement.
Sequence and structural analysis.
PyMOL (62) was used to identify potential interacting residues (≤4.0 Å in distance) of ApmA with substrates apramycin and acetyl-CoA. Structural superimpositions with VatA (PDB ID 4HUS) were constructed using the cealign function in PyMol. XAT representative sequences were obtained from the CARD (41). ApmA and Vat sequences were aligned with the Expresso (63–65) function of T-Coffee (66, 67) to build a profile hidden Markov model (HMMER 3.3.1) (68). All sequences were aligned with the resulting HMM profile and visualized using Jalview (69).
Data availability.
PDB validation reports for crystal structures obtained in this study have been submitted with the manuscript. Accession numbers are as follows: 7JM0 (ApmA apoenzyme), 7JM1 (ApmA complex with acetyl-CoA), and 7J6M2 (ApmA complex with apramycin).
13C NMR for 2′-acetyl-apramycin. Download FIG S5, PDF file, 0.1 MB (66.2KB, pdf) .
Copyright © 2021 Bordeleau et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
1H-1H correlation spectroscopy (COSY) NMR for 2′-acetyl-apramycin. Download FIG S6, PDF file, 0.1 MB (124.7KB, pdf) .
Copyright © 2021 Bordeleau et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
1H-13C heteronuclear single quantum coherence (HSQC) for 2′-acetyl-apramycin. Download FIG S7, PDF file, 0.1 MB (98.6KB, pdf) .
Copyright © 2021 Bordeleau et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
1H-13C HMBC for 2′-acetyl-apramycin. Download FIG S8, PDF file, 0.1 MB (106.7KB, pdf) .
Copyright © 2021 Bordeleau et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
ACKNOWLEDGMENTS
We thank Zdzislaw Wawrzak, Life Sciences Collaborative Access Team, Advanced Photon Source, Argonne National Laboratory, for diffraction data collection.
Crystal structures solved in this work were funded in whole or in part with U.S. federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under contract No. HHSN272201700060C (Center for Structural Genomics of Infectious Diseases (CSGID); http://csgid.org). This research was funded by a Canadian Institutes of Health Research grant (FRN-148463), the Ontario Research Fund, and by a Canada Research Chair to G.D.W.
Footnotes
Citation Bordeleau E, Stogios PJ, Evdokimova E, Koteva K, Savchenko A, Wright GD. 2021. ApmA is a unique aminoglycoside antibiotic acetyltransferase that inactivates apramycin. mBio 12:e02705-20. https://doi.org/10.1128/mBio.02705-20.
Contributor Information
Marcelo Tolmasky, California State University, Fullerton.
Karen Bush, Indiana University Bloomington.
REFERENCES
- 1.Schatz A, Bugle E, Waksman SA. 1944. Streptomycin, a substance exhibiting antibiotic activity against Gram-positive and Gram-negative bacteria. Exp Biol Med 55:66–69. doi: 10.3181/00379727-55-14461. [DOI] [PubMed] [Google Scholar]
- 2.Waksman SA, Lechevalier HA. 1949. Neomycin, a new antibiotic active against streptomycin-resistant bacteria, including tuberculosis organisms. Science 109:305–307. doi: 10.1126/science.109.2830.305. [DOI] [PubMed] [Google Scholar]
- 3.Hu Y, Liu L, Zhang X, Feng Y, Zong Z. 2017. In vitro activity of neomycin, streptomycin, paromomycin and apramycin against carbapenem-resistant Enterobacteriaceae clinical strains. Front Microbiol 8:2275. doi: 10.3389/fmicb.2017.02275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hobbie SN, Akshay S, Kalapala SK, Bruell CM, Shcherbakov D, Bottger EC. 2008. Genetic analysis of interactions with eukaryotic rRNA identify the mitoribosome as target in aminoglycoside ototoxicity. Proc Natl Acad Sci U S A 105:20888–20893. doi: 10.1073/pnas.0811258106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davies J, Gorini L, Davis BD. 1965. Misreading of RNA codewords induced by aminoglycoside antibiotics. Mol Pharmacol 1:93–106. [PubMed] [Google Scholar]
- 6.Davies J 1995. Vicious circles: looking back on resistance plasmids. Genetics 139:1465–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Courvalin P 1994. Transfer of antibiotic resistance genes between gram-positive and gram-negative bacteria. Antimicrob Agents Chemother 38:1447–1451. doi: 10.1128/aac.38.7.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pang Z, Raudonis R, Glick BR, Lin T-J, Cheng Z. 2019. Antibiotic resistance in Pseudomonas aeruginosa: mechanisms and alternative therapeutic strategies. Biotechnol Adv 37:177–192. doi: 10.1016/j.biotechadv.2018.11.013. [DOI] [PubMed] [Google Scholar]
- 9.Davies J, Wright GD. 1997. Bacterial resistance to aminoglycoside antibiotics. Trends Microbiol 5:234–240. doi: 10.1016/S0966-842X(97)01033-0. [DOI] [PubMed] [Google Scholar]
- 10.Schaenzer AJ, Wright GD. 2020. Antibiotic resistance by enzymatic modification of antibiotic targets. Trends Mol Med 26:768–782. doi: 10.1016/j.molmed.2020.05.001. [DOI] [PubMed] [Google Scholar]
- 11.Ramirez MS, Tolmasky ME. 2010. Aminoglycoside modifying enzymes. Drug Resist Updat 13:151–171. doi: 10.1016/j.drup.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shaw KJ, Rather PN, Hare RS, Miller GH. 1993. Molecular genetics of aminoglycoside resistance genes and familial relationships of the aminoglycoside-modifying enzymes. Microbiol Rev 57:138–163. doi: 10.1128/MR.57.1.138-163.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wright GD 1999. Aminoglycoside-modifying enzymes. Curr Opin Microbiol 2:499–503. doi: 10.1016/s1369-5274(99)00007-7. [DOI] [PubMed] [Google Scholar]
- 14.Wachino J, Shibayama K, Kurokawa H, Kimura K, Yamane K, Suzuki S, Shibata N, Ike Y, Arakawa Y. 2007. Novel plasmid-mediated 16S rRNA m1A1408 methyltransferase, NpmA, found in a clinically isolated Escherichia coli strain resistant to structurally diverse aminoglycosides. Antimicrob Agents Chemother 51:4401–4409. doi: 10.1128/AAC.00926-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Macmaster R, Zelinskaya N, Savic M, Rankin CR, Conn GL. 2010. Structural insights into the function of aminoglycoside-resistance A1408 16S rRNA methyltransferases from antibiotic-producing and human pathogenic bacteria. Nucleic Acids Res 38:7791–7799. doi: 10.1093/nar/gkq627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wachino J, Arakawa Y. 2012. Exogenously acquired 16S rRNA methyltransferases found in aminoglycoside-resistant pathogenic Gram-negative bacteria: an update. Drug Resist Updat 15:133–148. doi: 10.1016/j.drup.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 17.Aggen JB, Armstrong ES, Goldblum AA, Dozzo P, Linsell MS, Gliedt MJ, Hildebrandt DJ, Feeney LA, Kubo A, Matias RD, Lopez S, Gomez M, Wlasichuk KB, Diokno R, Miller GH, Moser HE. 2010. Synthesis and spectrum of the neoglycoside ACHN-490. Antimicrob Agents Chemother 54:4636–4642. doi: 10.1128/AAC.00572-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Armstrong ES, Miller GH. 2010. Combating evolution with intelligent design: the neoglycoside ACHN-490. Curr Opin Microbiol 13:565–573. doi: 10.1016/j.mib.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 19.Cox G, Ejim L, Stogios PJ, Koteva K, Bordeleau E, Evdokimova E, Sieron AO, Savchenko A, Serio AW, Krause KM, Wright GD. 2018. Plazomicin retains antibiotic activity against most aminoglycoside modifying enzymes. ACS Infect Dis 4:980–987. doi: 10.1021/acsinfecdis.8b00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O'Connor S, Lam LK, Jones ND, Chaney MO. 1976. Apramycin, a unique aminocyclitol antibiotic. J Org Chem 41:2087–2092. doi: 10.1021/jo00874a003. [DOI] [PubMed] [Google Scholar]
- 21.Kang AD, Smith KP, Eliopoulos GM, Berg AH, McCoy C, Kirby JE. 2017. In vitro apramycin activity against multidrug-resistant Acinetobacter baumannii and Pseudomonas aeruginosa. Diagn Microbiol Infect Dis 88:188–191. doi: 10.1016/j.diagmicrobio.2017.03.006. [DOI] [PubMed] [Google Scholar]
- 22.Meyer M, Freihofer P, Scherman M, Teague J, Lenaerts A, Böttger EC. 2014. In vivo efficacy of apramycin in murine infection models. Antimicrob Agents Chemother 58:6938–6941. doi: 10.1128/AAC.03239-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hao M, Shi X, Lv J, Niu S, Cheng S, Du H, Yu F, Tang Y-W, Kreiswirth BN, Zhang H, Chen L. 2020. In vitro activity of apramycin against carbapenem-resistant and hypervirulent Klebsiella pneumoniae isolates. Front Microbiol 11:425. doi: 10.3389/fmicb.2020.00425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Juhas M, Widlake E, Teo J, Huseby DL, Tyrrell JM, Polikanov YS, Ercan O, Petersson A, Cao S, Aboklaish AF, Rominski A, Crich D, Böttger EC, Walsh TR, Hughes D, Hobbie SN. 2019. In vitro activity of apramycin against multidrug-, carbapenem- and aminoglycoside-resistant Enterobacteriaceae and Acinetobacter baumannii. J Antimicrob Chemother 74:944–952. doi: 10.1093/jac/dky546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Böttger EC, Crich D. 2020. Aminoglycosides: time for the resurrection of a neglected class of antibacterials? ACS Infect Dis 6:168–172. doi: 10.1021/acsinfecdis.9b00441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chaslus-Dancla E, Glupczynski Y, Gerbaud G, Lagorce M, Lafont JP, Courvalin P. 1989. Detection of apramycin resistant Enterobacteriaceae in hospital isolates. FEMS Microbiol Lett 61:261–265. doi: 10.1111/j.1574-6968.1989.tb03634.x. [DOI] [PubMed] [Google Scholar]
- 27.Magalhaes MLB, Blanchard JS. 2005. The kinetic mechanism of AAC(3)-IV aminoglycoside acetyltransferase from Escherichia coli. Biochemistry 44:16275–16283. doi: 10.1021/bi051777d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davies J, O'Connor S. 1978. Enzymatic modification of aminoglycoside antibiotics: 3-N-acetyltransferase with broad specificity that determines resistance to the novel aminoglycoside apramycin. Antimicrob Agents Chemother 14:69–72. doi: 10.1128/aac.14.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lovering AM, White LO, Reeves DS. 1987. AAC1: a new aminoglycoside-acetylating enzyme modifying the Cl aminogroup of apramycin. J Antimicrob Chemother 20:803–813. doi: 10.1093/jac/20.6.803. [DOI] [PubMed] [Google Scholar]
- 30.Hedges RW, Shannon KP. 1984. Resistance to apramycin in Escherichia coli isolated from animals: detection of a novel aminoglycoside-modifying enzyme. J Gen Microbiol 130:473–482. doi: 10.1099/00221287-130-3-473. [DOI] [PubMed] [Google Scholar]
- 31.Fessler AT, Kadlec K, Schwarz S. 2011. Novel apramycin resistance gene apmA in bovine and porcine methicillin-resistant Staphylococcus aureus ST398 isolates. Antimicrob Agents Chemother 55:373–375. doi: 10.1128/AAC.01124-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kadlec K, Feßler AT, Couto N, Pomba CF, Schwarz S. 2012. Unusual small plasmids carrying the novel resistance genes dfrK or apmA isolated from methicillin-resistant or -susceptible staphylococci. J Antimicrob Chemother 67:2342–2345. doi: 10.1093/jac/dks235. [DOI] [PubMed] [Google Scholar]
- 33.Feßler AT, Zhao Q, Schoenfelder S, Kadlec K, Brenner Michael G, Wang Y, Ziebuhr W, Shen J, Schwarz S. 2017. Complete sequence of a plasmid from a bovine methicillin-resistant Staphylococcus aureus harbouring a novel ica-like gene cluster in addition to antimicrobial and heavy metal resistance genes. Vet Microbiol 200:95–100. doi: 10.1016/j.vetmic.2016.07.010. [DOI] [PubMed] [Google Scholar]
- 34.Cox G, Sieron A, King AM, De Pascale G, Pawlowski AC, Koteva K, Wright GD. 2017. A common platform for antibiotic dereplication and adjuvant discovery. Cell Chem Biol 24:98–109. doi: 10.1016/j.chembiol.2016.11.011. [DOI] [PubMed] [Google Scholar]
- 35.Eneva GI, Spassov SL, Haimova MA, Sandström J. 1992. Complete 1H and 13C NMR assignments for apramycin, sisomicin and some N- and N,O-polyacetylated aminoglycosides. Magn Reson Chem 30:841–846. doi: 10.1002/mrc.1260300908. [DOI] [Google Scholar]
- 36.Stogios PJ, Kuhn ML, Evdokimova E, Courvalin P, Anderson WF, Savchenko A. 2014. Potential for reduction of streptogramin A resistance revealed by structural analysis of acetyltransferase VatA. Antimicrob Agents Chemother 58:7083–7092. doi: 10.1128/AAC.03743-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sugantino M, Roderick SL. 2002. Crystal structure of Vat(D): an acetyltransferase that inactivates streptogramin group A antibiotics. Biochemistry 41:2209–2216. doi: 10.1021/bi011991b. [DOI] [PubMed] [Google Scholar]
- 38.Beaman TW, Sugantino M, Roderick SL. 1998. Structure of the hexapeptide xenobiotic acetyltransferase from Pseudomonas aeruginosa. Biochemistry 37:6689–6696. doi: 10.1021/bi980106v. [DOI] [PubMed] [Google Scholar]
- 39.White PA, Stokes H, Bunny KL, Hall RM. 1999. Characterisation of a chloramphenicol acetyltransferase determinant found in the chromosome of Pseudomonas aeruginosa. FEMS Microbiol Lett 175:27–35. doi: 10.1111/j.1574-6968.1999.tb13598.x. [DOI] [PubMed] [Google Scholar]
- 40.Bassenden AV, Rodionov D, Shi K, Berghuis AM. 2016. Structural analysis of the tobramycin and gentamicin clinical resistome reveals limitations for next-generation aminoglycoside design. ACS Chem Biol 11:1339–1346. doi: 10.1021/acschembio.5b01070. [DOI] [PubMed] [Google Scholar]
- 41.Alcock BP, Raphenya AR, Lau TT, Tsang KK, Bouchard M, Edalatmand A, Huynh W, Nguyen AV, Cheng AA, Liu S, Min SY, Miroshnichenko A, Tran HK, Werfalli RE, Nasir JA, Oloni M, Speicher DJ, Florescu A, Singh B, Faltyn M, Hernandez-Koutoucheva A, Sharma AN, Bordeleau E, Pawlowski AC, Zubyk HL, Dooley D, Griffiths E, Maguire F, Winsor GL, Beiko RG, Brinkman FS, Hsiao WW, Domselaar GV, McArthur AG. 2020. CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res 48:D517–D525. doi: 10.1093/nar/gkz935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Quirke JCK, Rajasekaran P, Sarpe VA, Sonousi A, Osinnii I, Gysin M, Haldimann K, Fang Q-J, Shcherbakov D, Hobbie SN, Sha S-H, Schacht J, Vasella A, Böttger EC, Crich D. 2020. Apralogs: apramycin 5-O-glycosides and ethers with improved antibacterial activity and ribosomal selectivity and reduced susceptibility to the aminoacyltransferase (3)-IV resistance determinant. J Am Chem Soc 142:530–544. doi: 10.1021/jacs.9b11601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matt T, Ng CL, Lang K, Sha S-H, Akbergenov R, Shcherbakov D, Meyer M, Duscha S, Xie J, Dubbaka SR, Perez-Fernandez D, Vasella A, Ramakrishnan V, Schacht J, Bottger EC. 2012. Dissociation of antibacterial activity and aminoglycoside ototoxicity in the 4-monosubstituted 2-deoxystreptamine apramycin. Proc Natl Acad Sci U S A 109:10984–10989. doi: 10.1073/pnas.1204073109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vicens Q, Westhof E. 2003. RNA as a drug target: the case of aminoglycosides. Chembiochem 4:1018–1023. doi: 10.1002/cbic.200300684. [DOI] [PubMed] [Google Scholar]
- 45.Han Q, Zhao Q, Fish S, Simonsen KB, Vourloumis D, Froelich JM, Wall D, Hermann T. 2005. Molecular recognition by glycoside pseudo base pairs and triples in an apramycin-RNA complex. Angew Chem Int Ed Engl 44:2694–2700. doi: 10.1002/anie.200500028. [DOI] [PubMed] [Google Scholar]
- 46.Pfister P, Hobbie S, Vicens Q, Böttger EC, Westhof E. 2003. The molecular basis for A-site mutations conferring aminoglycoside resistance: relationship between ribosomal susceptibility and X-ray crystal structures. Chembiochem 4:1078–1088. doi: 10.1002/cbic.200300657. [DOI] [PubMed] [Google Scholar]
- 47.Swiatlo E, Kocka FE. 1987. Inducible expression of an aminoglycoside-acetylating enzyme in Providencia stuartii. J Antimicrob Chemother 19:27–30. doi: 10.1093/jac/19.1.27. [DOI] [PubMed] [Google Scholar]
- 48.Hegde SS, Javid-Majd F, Blanchard JS. 2001. Overexpression and mechanistic analysis of chromosomally encoded aminoglycoside 2′-N-acetyltransferase (AAC(2′)-Ic) from Mycobacterium tuberculosis. J Biol Chem 276:45876–45881. doi: 10.1074/jbc.M108810200. [DOI] [PubMed] [Google Scholar]
- 49.Fabre A, Oleastro M, Nunes A, Santos A, Sifré E, Ducournau A, Bénéjat L, Buissonnière A, Floch P, Mégraud F, Dubois V, Lehours P. 2018. Whole-genome sequence analysis of multidrug-resistant Campylobacter isolates: a focus on aminoglycoside resistance determinants. J Clin Microbiol 56:1–12. doi: 10.1128/JCM.00390-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Murray IA, Shaw WV. 1997. O-acetyltransferases for chloramphenicol and other natural products. Antimicrob Agents Chemother 41:1–6. doi: 10.1128/AAC.41.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wybenga-Groot LE, Draker K, Wright GD, Berghuis AM. 1999. Crystal structure of an aminoglycoside 6’-N-acetyltransferase: defining the GCN5-related N-acetyltransferase superfamily fold. Structure 7:497–507. doi: 10.1016/S0969-2126(99)80066-5. [DOI] [PubMed] [Google Scholar]
- 52.Stogios PJ, Kuhn ML, Evdokimova E, Law M, Courvalin P, Savchenko A. 2017. Structural and biochemical characterization of Acinetobacter spp. aminoglycoside acetyltransferases highlights functional and evolutionary variation among antibiotic resistance enzymes. ACS Infect Dis 3:132–143. doi: 10.1021/acsinfecdis.6b00058. [DOI] [PubMed] [Google Scholar]
- 53.Biswas T, Houghton JL, Garneau-Tsodikova S, Tsodikov OV. 2012. The structural basis for substrate versatility of chloramphenicol acetyltransferase CATI. Protein Sci 21:520–530. doi: 10.1002/pro.2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morar M, Wright GD. 2010. The genomic enzymology of antibiotic resistance. Annu Rev Genet 44:25–51. doi: 10.1146/annurev-genet-102209-163517. [DOI] [PubMed] [Google Scholar]
- 55.Xu Z, Stogios PJ, Quaile AT, Forsberg KJ, Patel S, Skarina T, Houliston S, Arrowsmith C, Dantas G, Savchenko A. 2017. Structural and functional survey of environmental aminoglycoside acetyltransferases reveals functionality of resistance enzymes. ACS Infect Dis 3:653–665. doi: 10.1021/acsinfecdis.7b00068. [DOI] [PubMed] [Google Scholar]
- 56.Thoden JB, Reinhardt LA, Cook PD, Menden P, Cleland WW, Holden HM. 2012. Catalytic mechanism of perosamine N-acetyltransferase revealed by high-resolution X-ray crystallographic studies and kinetic analyses. Biochemistry 51:3433–3444. doi: 10.1021/bi300197h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thoden JB, Cook PD, Schäffer C, Messner P, Holden HM. 2009. Structural and functional studies of QdtC: an N-acetyltransferase required for the biosynthesis of dTDP-3-acetamido-3,6-dideoxy-α-d-glucose. Biochemistry 48:2699–2709. doi: 10.1021/bi802313n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Craggs PD, Mouilleron S, Rejzek M, De Chiara C, Young RJ, Field RA, Argyrou A, De Carvalho LPS. 2018. The mechanism of acetyl transfer catalyzed by Mycobacterium tuberculosis GlmU. Biochemistry 57:3387–3401. doi: 10.1021/acs.biochem.8b00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Draker K, Wright GD. 2004. Molecular mechanism of the Enterococcal aminoglycoside 6‘-N-acetyltransferase’: role of GNAT-conserved residues in the chemistry of antibiotic inactivation. Biochemistry 43:446–454. doi: 10.1021/bi035667n. [DOI] [PubMed] [Google Scholar]
- 60.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 61.Clinical and Laboratory Standards Institute. 2018. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically, 11th ed CLSI M07-A10 Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 62.Delano WL 2002. The PyMOL molecular graphics system. DeLano Scientific, Palo Alto, CA. [Google Scholar]
- 63.Armougom F, Moretti S, Poirot O, Audic S, Dumas P, Schaeli B, Keduas V, Notredame C. 2006. Expresso: automatic incorporation of structural information in multiple sequence alignments using 3D-Coffee. Nucleic Acids Res 34:W604–W608. doi: 10.1093/nar/gkl092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.O'Sullivan O, Suhre K, Abergel C, Higgins DG, Notredame C. 2004. 3DCoffee: combining protein sequences and structures within multiple sequence alignments. J Mol Biol 340:385–395. doi: 10.1016/j.jmb.2004.04.058. [DOI] [PubMed] [Google Scholar]
- 65.Poirot O, Suhre K, Abergel C, O'Toole E, Notredame C. 2004. 3DCoffee@igs: a web server for combining sequences and structures into a multiple sequence alignment. Nucleic Acids Res 32:W37–W40. doi: 10.1093/nar/gkh382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Di Tommaso P, Moretti S, Xenarios I, Orobitg M, Montanyola A, Chang J-M, Taly J-F, Notredame C. 2011. T-Coffee: a web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res 39:W13–W17. doi: 10.1093/nar/gkr245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Notredame C, Higgins DG, Heringa J. 2000. T-coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol 302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 68.Eddy SR 2020. HMMER v3.3.1 - biological sequence analysis using profile hidden Markov models. http://hmmer.org/.
- 69.Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. 2009. Jalview version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics 25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
NMR assignments of acetyl-apramycin. Download Table S1, DOCX file, 0.02 MB (16.1KB, docx) .
Copyright © 2021 Bordeleau et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Apramycin resistance elements identified while screening within the aminoglycoside resistance library expressed in E. coli BW25113 ΔtolCΔbamB. (A) Aminoglycoside resistance glycerol library layout. N-acetyltransferase, blue; O-phosphotransferase, gold; O-nucleotidyltransferase, yellow; bifunctional acetyltransferase, pink; 16S rRNA methyltransferase, red. Each gene was under the control of the Pbla promoter. (B) Control growth of library on cation-adjusted Mueller Hinton agar supplemented with no drug. Colony growth highlighted with a circle were those found to confer apramycin resistance. (C) 4 μg/ml apramycin; (D) 32 μg/ml apramycin; (E) 256 μg/ml apramycin. Download FIG S1, JPG file, 1.2 MB (1.2MB, jpg) .
Copyright © 2021 Bordeleau et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
X-ray diffraction data collection and refinement statistics. Download Table S2, DOCX file, 0.02 MB (16.3KB, docx) .
Copyright © 2021 Bordeleau et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Impact of the N-terminal region on the substrate binding pocket of ApmA. Red arrows indicate the first two β-strands from the 7-stranded β-sheet of the LβH domain. Superimpositions of ApmA in complex with apramycin (A), CAT (B), and VatA (C). Download FIG S2, JPG file, 0.3 MB (365.9KB, jpg) .
Copyright © 2021 Bordeleau et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Sequence relationship of ApmA to the LβH superfamily. Full multiple sequence alignment of ApmA with CATs and Vats obtained from the Comprehensive Antibiotic Resistance Database. Amino acids are colored based on percent identity. Traditional hexapeptide motif (i, i + 1, and i + 4) of the LβH domain is highlighted in red. Conserved catalytic histidine of the XAT subclass is highlighted in pink. Download FIG S3, JPG file, 2.8 MB (2.9MB, jpg) .
Copyright © 2021 Bordeleau et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
1H NMR for 2′-acetyl-apramycin. Download FIG S4, PDF file, 0.1 MB (93.1KB, pdf) .
Copyright © 2021 Bordeleau et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
13C NMR for 2′-acetyl-apramycin. Download FIG S5, PDF file, 0.1 MB (66.2KB, pdf) .
Copyright © 2021 Bordeleau et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
1H-1H correlation spectroscopy (COSY) NMR for 2′-acetyl-apramycin. Download FIG S6, PDF file, 0.1 MB (124.7KB, pdf) .
Copyright © 2021 Bordeleau et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
1H-13C heteronuclear single quantum coherence (HSQC) for 2′-acetyl-apramycin. Download FIG S7, PDF file, 0.1 MB (98.6KB, pdf) .
Copyright © 2021 Bordeleau et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
1H-13C HMBC for 2′-acetyl-apramycin. Download FIG S8, PDF file, 0.1 MB (106.7KB, pdf) .
Copyright © 2021 Bordeleau et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Data Availability Statement
PDB validation reports for crystal structures obtained in this study have been submitted with the manuscript. Accession numbers are as follows: 7JM0 (ApmA apoenzyme), 7JM1 (ApmA complex with acetyl-CoA), and 7J6M2 (ApmA complex with apramycin).
13C NMR for 2′-acetyl-apramycin. Download FIG S5, PDF file, 0.1 MB (66.2KB, pdf) .
Copyright © 2021 Bordeleau et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
1H-1H correlation spectroscopy (COSY) NMR for 2′-acetyl-apramycin. Download FIG S6, PDF file, 0.1 MB (124.7KB, pdf) .
Copyright © 2021 Bordeleau et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
1H-13C heteronuclear single quantum coherence (HSQC) for 2′-acetyl-apramycin. Download FIG S7, PDF file, 0.1 MB (98.6KB, pdf) .
Copyright © 2021 Bordeleau et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
1H-13C HMBC for 2′-acetyl-apramycin. Download FIG S8, PDF file, 0.1 MB (106.7KB, pdf) .
Copyright © 2021 Bordeleau et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.